

Abstract
Elevated synchronized oscillatory activity in the beta band has been hypothesized to be a pathophysiological marker of Parkinson's disease (PD). Recent studies have suggested that parkinsonism is closely associated with increased amplitude and duration of beta burst activity in the subthalamic nucleus (STN). How beta burst dynamics are altered from the normal to parkinsonian state across the basal ganglia–thalamocortical (BGTC) motor network, however, remains unclear. In this study, we simultaneously recorded local field potential activity from the STN, internal segment of the globus pallidus (GPi), and primary motor cortex (M1) in three female rhesus macaques, and characterized how beta burst activity changed as the animals transitioned from normal to progressively more severe parkinsonian states. Parkinsonism was associated with an increased incidence of beta bursts with longer duration and higher amplitude in the low beta band (8–20 Hz) in both the STN and GPi, but not in M1. We observed greater concurrence of beta burst activity, however, across all recording sites (M1, STN, and GPi) in PD. The simultaneous presence of low beta burst activity across multiple nodes of the BGTC network that increased with severity of PD motor signs provides compelling evidence in support of the hypothesis that low beta synchronized oscillations play a significant role in the underlying pathophysiology of PD. Given its immersion throughout the motor circuit, we hypothesize that this elevated beta-band activity interferes with spatial–temporal processing of information flow in the BGTC network that contributes to the impairment of motor function in PD.
SIGNIFICANCE STATEMENT This study fills a knowledge gap regarding the change in temporal dynamics and coupling of beta burst activity across the basal ganglia–thalamocortical (BGTC) network during the evolution from normal to progressively more severe parkinsonian states. We observed that changes in beta oscillatory activity occur throughout BGTC and that increasing severity of parkinsonism was associated with a higher incidence of longer duration, higher amplitude low beta bursts in the basal ganglia, and increased concurrence of beta bursts across the subthalamic nucleus, globus pallidus, and motor cortex. These data provide new insights into the potential role of changes in the temporal dynamics of low beta activity within the BGTC network in the pathogenesis of Parkinson's disease.
Keywords: burst coupling, low beta burst, MPTP, nonhuman primate, Parkinson's disease, temporal dynamics
Introduction
Exaggerated beta band (8–35 Hz) oscillatory activity in the basal ganglia–thalamocortical (BGTC) network is hypothesized to be a pathophysiological marker of Parkinson's disease (PD; Wichmann et al., 1994; Nini et al., 1995; Brown, 2003, 2006; Chen et al., 2007; Hammond et al., 2007; Kühn et al., 2009; Devergnas et al., 2014). Support for this hypothesis comes from the observations that suppression of local field potential (LFP) activity in the beta band in the subthalamic nucleus (STN) and globus pallidus (GP) after the administration of levodopa or during deep brain stimulation (DBS) is positively correlated with the improvement of motor symptoms (Brown et al., 2004; Kühn et al., 2008; Oswal et al., 2016; Trager et al., 2016; Wang et al., 2018). In other studies, however, the correlation between beta power and symptom severity was not consistent across subjects (Weinberger et al., 2006; Rosa et al., 2011; Devergnas et al., 2014; Connolly et al., 2015; Muralidharan et al., 2016), contributing to a continued debate about the role of beta oscillations in the pathogenesis of PD.
Beta oscillations in the motor network often occur as transient events, or bursts, and play an important role in encoding movement information in the healthy nondiseased state (Murthy and Fetz, 1992, 1996; Bartolo and Merchant, 2015; Feingold et al., 2015; Shin et al., 2017; Little et al., 2019). Recent studies suggest that the critical disease-specific pathophysiological feature of parkinsonism is not necessarily a static, continuous elevation of beta power but rather a change in the temporal dynamics of burst activity (Tinkhauser et al., 2017a; Torrecillos et al., 2018). In PD patients, the suppression of prolonged beta bursts in the basal ganglia was positively correlated with motor improvement during DBS or the administration of levodopa (Tinkhauser et al., 2017a,b; Deffains et al., 2018; Lofredi et al., 2019). It was hypothesized that prolonged beta bursts across multiple nodes produce concurrent beta bursts at other nodal points in the BGTC circuit and thus pathologic synchronization across the motor network, leading to the motor deficits in PD (Tinkhauser et al., 2018). It remains unclear, however, how the temporal dynamics of beta burst activity evolve from the normal to parkinsonian state or how these dynamics are altered across multiple nodes in the BGTC network.
In this study, we begin to address these gaps in knowledge using the 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine (MPTP) nonhuman primate (NHP) model of PD. This model allows for the simultaneous recording of neuronal activity across multiple nodes in the BGTC network using a within-subject design that is not feasible in patient studies. We hypothesized that temporal dynamics of beta bursts (i.e., burst incidence, duration, and amplitude) in individual subcortical and cortical sites would evolve with increasing severity of PD (Fig. 1A), leading to greater overlap of beta burst activity and enhanced synchronization of neuronal activity across nodal points in the BGTC network (Fig. 1B). We tested this hypothesis by simultaneously recording LFP activity in the STN, internal segment of the GP (GPi), and primary motor cortex (M1) as the animal evolved from the normal to progressively more severe parkinsonian states, and by investigating the following: (1) the changes in incidence, duration, and amplitude of beta burst activity; and (2) the temporal relationship of the occurrence of beta bursts across subcortical and cortical nodes in the BGTC network.
Figure 1.
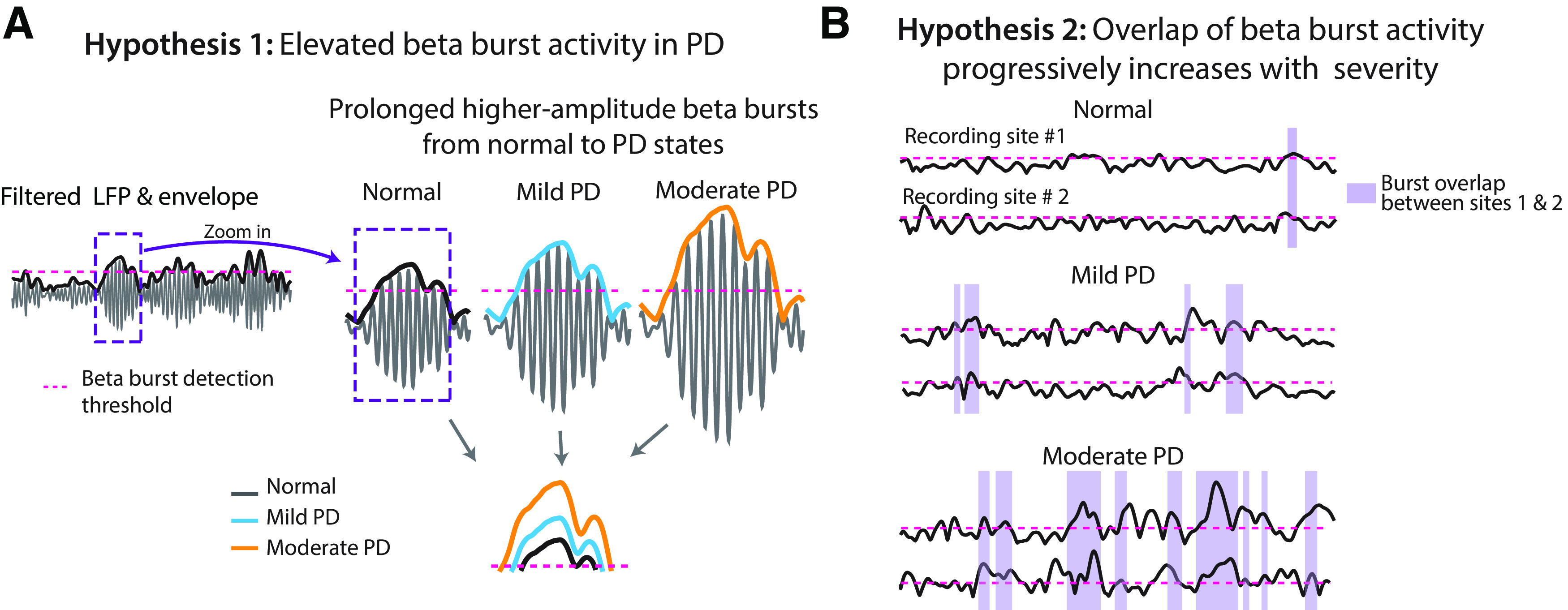
Beta burst dynamics in PD. A, Hypothesis 1: the amplitude and duration of beta bursts evolve with increasing severity of parkinsonism. The bottom plot showed an example of how we hypothesized beta bursts would change from the normal to different stages of severity in parkinsonism. B, Hypothesis 2: overlap of burst activity across structures in the BGTC network increases with increasing severity of PD.
Materials and Methods
Subjects
All procedures were approved by the University of Minnesota Institutional Animal Care and Use Committee and complied with US Public Health Service policy on the humane care and use of laboratory animals. Three adult female rhesus macaques (Macaca mulatta, animal P, 18 years of age; animal J, 16 years of age; and animal K, 13 years of age) were used in the study. Preoperative cranial computed tomography and 7T MRI images were coregistered in the Monkey Cicerone neurosurgical navigation program (Miocinovic et al., 2007) and used for surgical planning for the placement of a titanium cephalic chamber targeting the STN and GPi. After the cephalic chamber was placed, extracellular microelectrode mapping was used to confirm the location of target nuclei. Each animal was subsequently implanted in both the STN and GPi with eight-contact scaled down versions of human DBS leads (contact height, 0.5 mm; intercontact spacing, 0.5 mm; diameter, 0.625 mm; NUMED). Mapping and implantation methods are described in detail in a previous publication (Hashimoto et al., 2003). In a separate surgical procedure, a 96-channel Utah Microelectrode Array (Pt-Ir; depth, 1.5 mm; interelectrode spacing, 400 μm; Blackrock Microsystems) was placed in the arm area of the M1 of each animal using surgical methods described previously (Rousche and Normann, 1992; Maynard et al., 1997; Escobar et al., 2017). Pt/Ir reference wires were placed between the dura and skull adjacent to the array. M1 was identified intraoperatively based on sulcal landmarks (Fig. 2, top plots for each animal). All surgeries were performed using aseptic techniques under isoflurane anesthesia. DBS lead locations were verified histologically using frozen sagittal sections for animal P and coronal sections for animals J and K (40–50 μm thick) that were imaged and visualized in Avizo 3D (FEI) and 3D Slicer analysis software (https://www.slicer.org/; Fedorov et al., 2012; Fig. 2, bottom plots), together with microelectrode identification of target nuclei before implantation of the leads.
Figure 2.
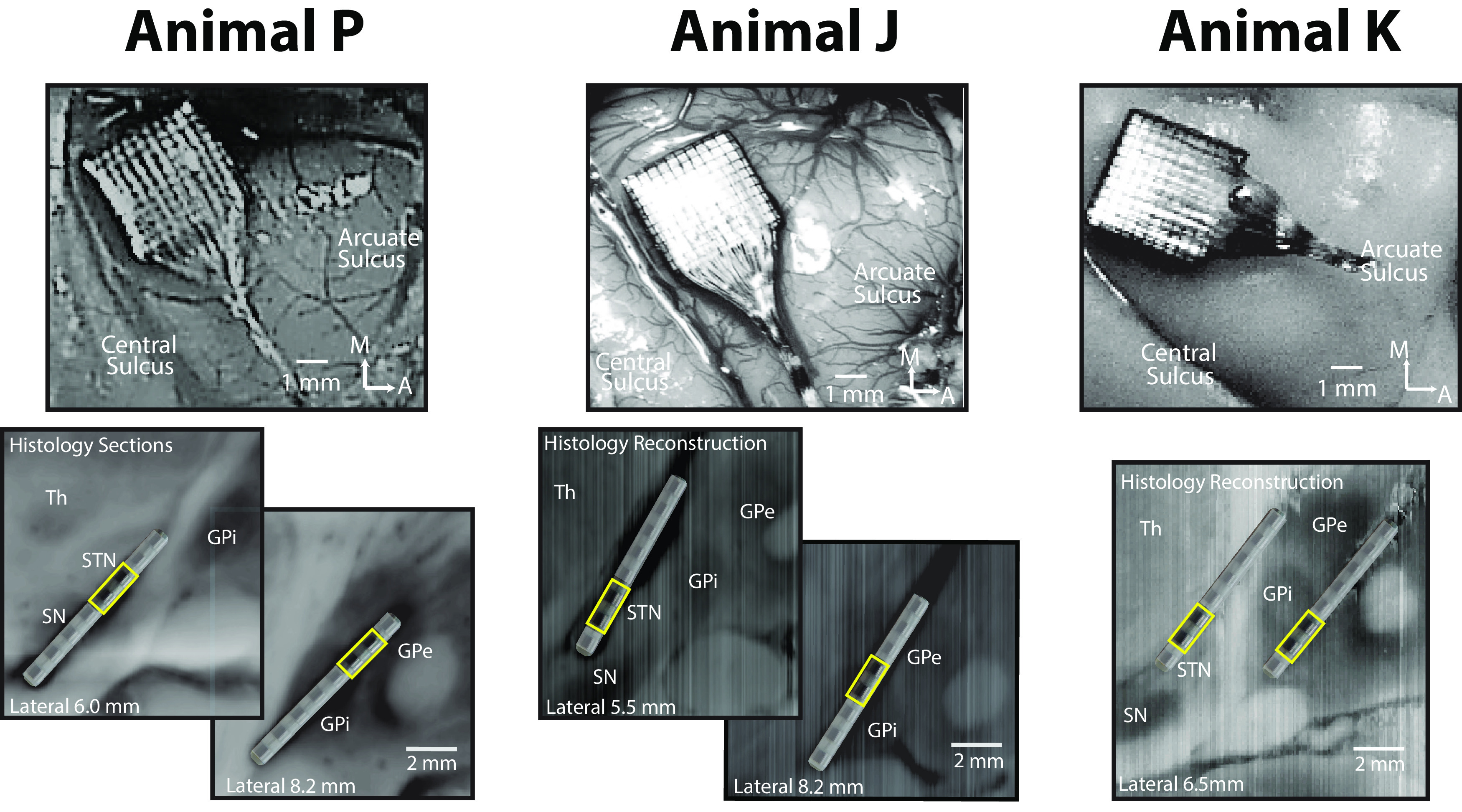
Recording locations in M1, STN, and GPi. Top plots, Utah Array locations in arm area of M1. The images of animals P and J were obtained intraoperatively; the image of animal K was obtained at the end of study after perfusion. Bottom plots, DBS lead locations in STN and GPi, verified with frozen sagittal sections for animal P and frozen coronal sections that were imaged and visualized in the sagittal plane for animals J and K. Bipolar LFPs from DBS contacts C4-5 for animal P, C1-2 for animal J, C1-2 for animal K in the STN, and C5-6, C3-4, C1-2 for animals P, J, and K, respectively, in the GP (all marked as yellow squares) were used for all measurements for each animal. STN and GPi contacts that were identified to be therapeutic for deep brain stimulation were selected for analysis, though stimulation was not a part of this study.
MPTP administration
Following completion of data collection in the normal state, animals were given injections (0.3–0.8 mg/kg) of the neurotoxin MPTP until animals were in a mild and subsequently in a moderate parkinsonian state, as defined below. For animal P, systemic intramuscular injections were given weekly or biweekly to induce mild and moderate parkinsonism (total doses, 5.35 and 6.3 mg/kg, respectively). For animal J, three systemic intramuscular injections over consecutive days (total dose, 1.0 mg/kg) were required to achieve mild parkinsonism. A moderate parkinsonian state was induced in this animal by one intracarotid injection (0.4 mg/kg). For animal K, six intramuscular injections (total dose, 1.8 mg/kg) were given to induce moderate parkinsonism.
Behavioral assessments
The severity of PD motor signs was assessed using a modified Unified Parkinson's Disease Rating Scale (mUPDRS), which rated axial motor symptoms (gait, posture, balance, and defense reaction) as well as upper and lower limb rigidity, bradykinesia, akinesia, and tremor on the hemi-body contralateral to the site of neural recordings using a 0–3 scale (0 = normal, 3 = severe, maximum total score = 42; Vitek et al., 2012; Connolly et al., 2015). Different severity states of PD were defined as follows: mild, mUPDRS score <18; moderate, 18–31; severe, ≥32. The mUPDRS scores were obtained throughout the recording period to ensure the stability of parkinsonian motor signs.
In addition, animals J and P were acclimated to perform a Klüver board reaching task to further characterize bradykinesia. The total movement times (reach, manipulation, and retrieval) in both normal and PD states were determined using a motion capture system (Motion Analysis) that tracked the position of the arm contralateral to the recording sites. Klüver board task data in normal and PD states were not available for animal K.
LFP recordings and signal processing
All data were obtained after a stable parkinsonian state was achieved beginning ∼1 month after the last MPTP injection. mUPDRS scores were obtained in every recording session to verify the severity state of the animal (except for one session in animal J in mild and moderate states). Neurophysiological data were collected during the resting state while the animal was seated in a primate chair with its head fixed, using a TDT (Tucker Davis Technologies) workstation operating at ∼24 kHz sampling rate. All analysis was performed using customized scripts in MATLAB (MathWorks 2016). Raw signals were first bandpass filtered (0.5–300 Hz) then downsampled (∼3 kHz). LFP activity within the STN and GPi were extracted via bipolar montage (i.e., signal subtraction) of adjacent contacts located within each target (Fig. 2, bottom row, yellow squares). A mean M1 LFP was obtained by averaging recordings from all 96-array channels, excluding noisy channels (n = channel 51, 91, and 96 for animals P, J, and K, respectively). LFPs were divided into individual time segments (15 s in duration) during which the animals maintained an awake and nonmovement state (see criteria below). To compare beta-band bursts across trials, recording sessions, states, and animals, and to account for shifts in signal power over time associated with recording settings (e.g., ground, references) and allow scale-free assessment of dynamic changes in beta synchronization, we normalized each LFP segment using z-scores and used these scores for subsequent analysis (Lofredi et al., 2019). The number of recording sessions, the time period (weeks, months) over which recordings took place, and the resulting number of data segments included in the analysis are as follows: animal P: normal, 14 recording sessions over a 2 month period (96 data segments); mild, 7 recording sessions over a 1.5 month period (86 data segments); moderate, 23 recording sessions over a 3 month period (459 data segments); animal J: normal, 13 recording sessions over a 1.5 month period (116 data segments); mild, 21 sessions over a 1 month period (277 data segments); moderate, 15 sessions over a 1 month period (153 data segments); and animal K: normal, 4 recording sessions over a 1 week period (23 data segments); moderate, 15 recording sessions over a 2 month period (140 data segments). The time between the end of mild state recordings and the start of moderate state recordings in animals P and J was 6 months and 1 month, respectively.
Time periods with movement artifacts were identified and excluded from further analysis using the following two methods: (1) by detecting abnormally high-amplitude broadband power in the time–frequency spectrogram (spectral analysis described below); and (2) identifying periods of movement using the aforementioned motion capture system that tracked the position of the arm contralateral to the recording sites. For animal K, motion capture data were not available; hence, only the abnormally high-amplitude broadband power was used.
We have observed that parkinsonian animals are particularly susceptible to periods of drowsiness and sleep during resting-state recordings, creating a potentially confounding variable if data are combined regardless of animal vigilance (Escobar et al., 2017). Periods of wakefulness and drowsiness/sleep were differentiated using a combination of eye monitoring and analysis of instantaneous power of low-frequency oscillations in the M1 using methods described in our previous publication (Escobar et al., 2017). Only low-frequency power was used to estimate the vigilance state of animal K in the normal state since eye video was not available for this animal in the normal state.
Power spectral density analysis
Power spectral densities (PSDs) were used to quantify power changes across disease states and brain structures, and to identify the peaks in the low and high beta frequency bands subsequently used for beta burst detection. In the present study, we define low and high beta bands as 8–20 and 21–35 Hz, respectively. This is based on previous reports that the peak frequency of beta-band activity in the NHP MPTP model is generally lower than that reported in humans together with our observations of distinct beta peaks occurring within these two frequency ranges in the parkinsonian NHP (Hammond et al., 2007; Stein and Bar-Gad, 2013; Connolly et al., 2015; Escobar et al., 2017; Wang et al., 2017; Hendrix et al., 2018). PSDs were computed for each normalized LFP time segment using Welch's method, with 214 points (frequency resolution, ∼0.1863 Hz) in the fast Fourier transform, a Hamming window of 1.34 s (one-quarter 214 points), zero padding (three-quarters 214 points), and an overlap of 50%. Total power in the low and high beta bands, calculated as the sum of PSD values within each frequency interval, were compared across normal, mild, and moderate parkinsonian states.
Within-structure beta burst detection and analysis
The method used to detect beta bursts in the LFP signals was similar to that used in a previous study (Tinkhauser et al., 2017a). For each animal and disease condition, an average PSD was calculated and a peak frequency in the low and high beta band was identified. Each normalized LFP segment was then bandpass filtered ±3 Hz around the beta peak frequency and the envelopes obtained with the Hilbert transform (Fig. 3). The average peak frequency across all states was used for this filtering. In some recording sites, particularly in the normal state, no clear peak was observed; in such cases, the peak frequency identified in the moderate PD state was used to determine the filter settings.
Figure 3.
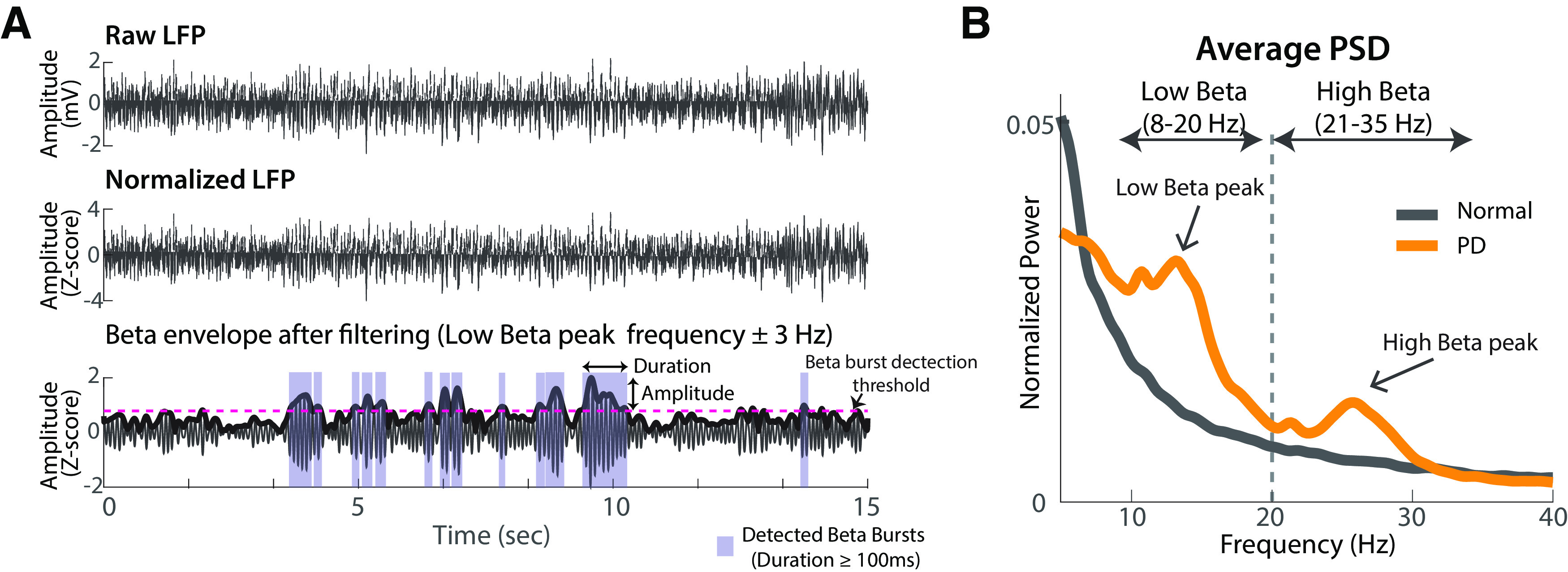
Beta burst detection. A, Beta burst detection method. The raw LFP recording (top) was first normalized as LFP z-scores (middle) and then bandpass filtered ±3 Hz around the beta peaks (bottom, low beta peak was selected for filtering as an example). The pink dashed line represented the average value of 75th percentile envelope amplitude of LFPs in all states as the threshold used to detect beta bursts. B, Beta peaks detection. The example averaged PSDs were extracted from the normal and moderate PD state, respectively, in GPi for animal P. The beta peaks were identified from the average PSD calculated from all LFP data segments. The peak frequencies identified in low and high beta bands were used for filtering (for details, see Materials and Methods).
Beta bursts were detected using an averaged threshold of 75th percentile of the beta envelope amplitude of all conditions for each structure (Tinkhauser et al., 2017a). The duration of each beta burst was defined as the time period in which the beta envelope exceeded this 75th percentile threshold level (Fig. 3A). Beta bursts with a duration <100 ms were excluded from further analysis (Tinkhauser et al., 2017a). Beta burst amplitude was defined as the maximum beta envelope amplitude during the beta burst. The incidence of beta bursts (time spent in beta bursts and number of bursts per 15 s data segment) and the distribution of durations for individual beta bursts (bin = 0.01 s) were computed for normal, mild, and moderate PD states.
Analysis of beta burst coupling across structures
We also investigated whether increasing disease severity was associated with changes in beta burst coupling across multiple nodes. The amount of overlap of beta burst activity across nodal points in the BGTC circuit, which represents a form of amplitude-to-amplitude coupling, was analyzed across subcortical and cortical recording sites. The time of overlap in beta burst activity for each pair of recording sites (i.e., M1-STN, M1-GPi, STN-GPi) as well as across all three recording structures (M1-STN-GPi) was calculated. We also calculated the overlap time because of chance using the same shuffling method used in the study by Tinkhauser et al. (2018). The overlap time was calculated after randomly shuffling beta bursts in each data segment in each structure. A total of 100 iterations of shuffling was performed, and the mean value of the overlap time of these iterations was calculated for each data segment and compared with the actual overlap time we observed.
In addition, the temporal relationship between beta bursts across each pair of sites was characterized by generating periburst time probability histograms (Fig. 4). The onset of a beta burst was first identified in one site (e.g., STN). Bursts within a ±1 s time window were then identified in a second site (e.g., GPi). By repeating this process across all bursts, a probability histogram was generated. This approach is analogous to the calculation of a perievent time histogram in spike train analysis, which reflects the probability of a spike occurring (analogous to a burst detected in the GPi) at time points around a reference event (analogous to a burst detected in the STN). The periburst time probability histogram was constructed using the ft_spikedensity function (sampling rate, 1000 Hz; window size, 100 ms) in the FieldTrip toolbox (Oostenveld et al., 2011) in MATLAB, and then normalized into the range of 0 ≤ probability ≤ 1; a larger probability value means a higher probability of concurrent bursts onsets across sites.
Figure 4.

Quantifying the temporal relationship of burst activity across recording sites. A, B, Simulated beta burst time series (top) and corresponding periburst time probability histogram with 95% confidence intervals (bottom) in pairs of sites with a low probability of concurrent bursts (A) and a high probability of concurrent bursts (B). The low-concurrence pair was composed of two independent time series with bursts occurring at random. The high-concurrence pair had 50% of burst onsets occurring simultaneously (B, top, highlighted by orange shaded areas). The histograms represent the probability of burst onset in site 2 relative to burst onset in site 1. The pink shaded area of the probability histogram in the lower panel in B indicates a significant increase in the probability histogram relative to the shuffled baseline probability (the average value indicated as Pbase), which represented the occurrence of concurrent bursts by chance, and reflects the significant temporal correlation between burst onsets in the high-concurrence pair of recording sites.
Further analysis was conducted to identify a significant increase in the periburst time probability histogram above the “baseline” level. The process described above was repeated but with randomly shuffling the times of beta burst onsets in one of the structures to generate a surrogate probability dataset, characterizing the distribution of periburst time probabilities that occurred by chance. The average value of the surrogate dataset was calculated, indicating the average baseline of the periburst time probability histogram (Pbase). A cluster-based statistical inference testing was then performed to determine whether the periburst time probability is significantly higher than the surrogate dataset (p < 0.05), using the ft_timelockanalysis function (montecarlo method, 1000 randomizations) in FieldTrip (Oostenveld et al., 2011). The significant increase in the probability histogram reflects a significant temporal relationship between bursts occurring in a pair of recording sites.
To illustrate this analysis method, we simulated burst time series and calculated periburst time probability histograms from two pairs of recording sites with a low and high likelihood of concurrent beta bursts, respectively. The pair of sites with low burst concurrence was composed of two time series with a total of 150 bursts in a 100 s period for each, with bursts occurring at random times independent between the two sites (Fig. 4A, top). The high-concurrence pair, consisting of 150 bursts in site 1 and 80 bursts in site 2 in a 100 s period, was constructed such that 50% of the bursts in site 2 occurred simultaneously with bursts in site 1 (Fig. 4B, top). A significant increase in the periburst time probability histogram is only observed for the pair of recording sites with high burst concurrence (Fig. 4B, bottom, shaded). It should be noted that the higher Pbase level in the low-concurrence pair (Fig. 4A, bottom, gray dashed line) reflects the higher number of bursts in the two sites, but the lack of a significant peak indicates no consistent temporal relationship between the occurrence of bursts in the two sites. In other words, an increase in the number of bursts within a given time period across sites (e.g., as is hypothesized to occur with onset of parkinsonism) would be reflected as an increase in Pbase; however, the analysis described here also identifies whether there is a significant relationship between the time at which bursts occur across sites, addressing the question of whether they occur simultaneously or whether there is a temporal relationship of one site to another (e.g., does burst onset in one site typically precede or follow burst onset at the other site). Additional simulation was also conducted to verify the influence of the changes of beta burst duration on the Pbase level of probability histogram. Our results indicated no significant difference of the Pbase levels across different pairs of data segments in which the beta bursts occurred with similar frequency.
For simplicity, probability histogram peaks with widths <60 ms (twice the period of the highest beta frequency analyzed, 35 Hz) were not considered in this study. Analysis based on burst onset times are shown in the present study; similar results were obtained using beta burst peak times to construct probability histograms.
Statistics
We conducted post hoc statistical analysis using nonparametric tests (Wilcoxon/Kruskal–Wallis tests (one-way test) to determine whether there were significant differences in beta power, burst incidence, and median beta burst duration between disease states, corrected for multiple comparisons (normal vs mild PD, normal vs moderate PD, mild PD vs moderate PD, p < 0.01). The effect size (r) of each significant difference was calculated to measure the magnitude of these differences (Pallant, 2011). It equals to the division results of the z value of the Wilcoxon/Kruskal–Wallis tests by the square root of the total number of observations, indicating a large difference between groups if r ≥ 0.5, medium difference for 0.3 ≤ r < 0.5, small difference for 0.1 ≤ r < 0.3, very small difference for r < 0.1. Similar tests were also performed on burst overlap time and temporal delay between beta bursts occurring in paired recording sites. In addition, the skewness and kurtosis of the distribution of the durations of beta bursts were used to further characterize the changes that occurred between the normal and PD states. Larger skewness and kurtosis reflect more asymmetric and heavier-tailed burst duration distribution, indicating more bursts with longer duration. Spearman's rank correlation coefficient was used to measure the strength of the monotonic relationship between the duration and amplitude of beta bursts. These tests were better suited than parametric tests because not all data were normally distributed.
Results
Induction of progressive parkinsonian states
Following administration of MPTP, animals exhibited the cardinal motor signs of parkinsonism including rigidity, bradykinesia, and akinesia as reflected in the mUPDRS clinical ratings and summaries of reach task performance (Table 1). As is typical for MPTP-treated rhesus monkeys, minimal resting tremor was observed. For animal P, the induction of parkinsonism resulted in a very mild state, as reflected by a total mUPDRS score of 5.4/42. Subsequent MPTP injections produced a moderate PD state (24.0/42) with bradykinesia, akinesia, and axial signs as primary motor signs; rigidity was not pronounced in either PD state. Reach task movement times increased from normal to mild states (Table 1), but the animal was unable to perform the task in the moderate state. Animal J was more impaired than animal P in the mild state, exhibiting rigidity, bradykinesia, akinesia, and axial signs, resulting in a total mUPDRS of 15.0/42. The moderate PD state was associated primarily with increased axial scores (3.5 and 8.2 in mild and moderate PD states, respectively). Total movement time for the animal progressively increased from 1.17 s in normal to 1.96 and 2.30 s in mild and moderate PD states, respectively. Animal J was also the least impaired in the moderate state (total score, 18.9/42), while animal K was the most impaired with pronounced rigidity and axial symptoms (total mUPDRS, 27.5/42).
Table 1.
mUPDRS scores for all animals
| Motor symptoms (maximum score) | Animal P |
Animal J |
Animal K |
||
|---|---|---|---|---|---|
| Mild PD (n = 7) | Moderate PD (n = 23) | Mild PD (n = 20) | Moderate PD (n = 14) | Moderate PD (n = 15) | |
| Rigidity (6) | 1.1 ± 0.6 | 1.8 ± 0.3 | 2.7 ± 0.7 | 2.0 ± 0.4 | 4.2 ± 0.5 |
| Tremor (6) | 0.4 ± 0.4 | 1.1 ± 0.8 | 0.9 ± 0.7 | 0.1 ± 0.3 | 0.6 ± 0.3 |
| Bradykinesia (6)* | 1.5 ± 0.7 | 4.4 ± 0.5 | 3.3 ± 0.5 | 3.4 ± 0.8 | 4.3 ± 0.7 |
| Akinesia (6) | 1.4 ± 0.5 | 4.3 ± 0.4 | 3.4 ± 0.5 | 3.7 ± 0.3 | 4.6 ± 0.6 |
| Food retrieval (3) | 0.0 ± 0.0 | 2.5 ± 0.3 | 1.3 ± 0.4 | 1.6 ± 0.4 | 2.6 ± 0.3 |
| Axiala (15) | 1.0 ± 0.7 | 10.0 ± 1.2 | 3.5 ± 1.3 | 8.2 ± 0.9 | 11.2 ± 0.6 |
| Total (42) | 5.4 | 24.0 | 15.0 | 18.9 | 27.5 |
Values are the mean ± SD. mUPDRS, Modified Unified Parkinson's Disease Rating Scale for nonhuman primates; n, number of observations, except for axial scores: Animal P: mild PD state, n = 7; moderate PD state, n = 14; Animal J: mild PD state, n = 4; moderate PD state, n = 3; Animal K: moderate PD state, n = 5.
aAxial symptoms are an average of posture, gait, and balance scores.
*A reaching task was performed to further assess bradykinesia for animals P and J. Animal P was not able to perform the reaching task in the moderate state. Total movement time increased from 1.30 s in the normal state (n = 377) to 1.40 s in the mild PD state (n = 324); Wilcoxon/Kruskal–Wallis tests (one-way test), χ2 = 68.65, p < 0.0001. For animal J, the total movement time progressively increased from 1.17 s in the normal PD state (n = 470) to 1.96 s in the mild PD state (n = 273) and 2.30 s in the moderate PD state (n = 248). Wilcoxon/Kruskal–Wallis tests (one-way test), χ2 = 749.02, p < 0.0001, post hoc test for each pair comparison, p < 0.0001 for all pairs.
Parkinsonism altered the power of low beta-band oscillatory activity in the BGTC network
Significant changes in beta-band power were observed in subcortical and cortical structures with the induction of parkinsonism. Power in low beta band (8–20 Hz) increased in the STN and GPi but decreased in M1 in all parkinsonian states in all three animals (Fig. 5, summarized in Table 2, where significant increases or decreases in beta power under the different conditions are indicated with up or down arrows, respectively). Notably, in the STN of animals P and J, where both mild and moderate severity states were examined, there was a progressive increase in low beta-band power associated with each increase in the level of severity (Fig. 5B, top). High beta-band (21–35 Hz) activity was also altered in parkinsonism but did not show the same correlation to severity level with less consistency in the direction of change across animals and structures.
Figure 5.

Changes in power and peak frequency of beta-band oscillatory activity with increasing severity of parkinsonism. A, Power spectral density in beta band for each site and animal. B, Low (8–20 Hz) and high (21–35 Hz) beta-band power for each site. For animals P and J: Wilcoxon/Kruskal–Wallis tests (one-way test), χ2 ≥ 23.42, p < 0.0001, post hoc test for each pair comparison p < 0.005, ****r ≥ 0.5, ***0.3 ≤ r < 0.5, **0.1 ≤ r < 0.3; for animal K: Wilcoxon/Kruskal–Wallis tests (one-way test), χ2 ≥ 24.20, p < 0.005, ****r ≥ 0.5, ***0.3 ≤ r < 0.5.
Table 2.
Changes of beta-band oscillatory activity in parkinsonism (relative to normal state)
| Beta-band frequency | Structure | Animal P Mild/moderate PD | Animal J Mild/moderate PD | Animal K Moderate PD |
|---|---|---|---|---|
| Low beta band (8–20 Hz) | M1 | ↓/↓ | ↓/↓ | ↓ |
| STN | ↑/↑ | ↑/↑ | ↑ | |
| GPi | ↑/↑ | ↑/↑ | ↑ | |
| High beta band (21–35 Hz) | M1 | ↑/--- | ↑/↑ | ↓ |
| STN | ---/↑ | ↑/↑ | ↑ | |
| GPi | ↑/ ↑ | ↓/↓ | ↓ |
The down arrows, up arrows and dash lines indicate the reduction, increase and no change of the power of beta oscillatory activity in each structure of each animal, PD states compared to normal state, respectively. Bold rows reflect changes that were consistent across all animals.
Changes in temporal dynamics of burst activity in the low beta band were associated with the progression of parkinsonism
With the induction of parkinsonism, significant changes in the temporal dynamics of low beta-band burst activity occurred across the BGTC network. We observed an increase in low beta burst activity in the STN and GPi as reflected by increased time spent in beta bursts (time spent in beta bursts per 15 s data segment; Fig. 6A), whereas low beta burst activity was reduced in M1. Notably, these changes were significant in the mild PD state, and progressively increased in the STN and GPi with increasing severity of parkinsonism. The increase in total time spent in bursts in the STN and GPi in PD was reflected by increases in both the frequency (Fig. 6B) and duration (Fig. 6C) of individual bursts. The frequency of bursts per data segment was decreased in M1 (Fig. 6B). Greater skewness and kurtosis of the beta burst duration distributions were observed in STN and GPi, especially in moderate PD states. These results indicated that a greater number of low beta bursts occurred in PD and that the durations of these bursts were prolonged in STN and GPi in more severe PD states.
Figure 6.

Altered temporal dynamics of low beta burst activity with increasing PD severity. A, B, Total time spent in bursts (A) and frequency of burst occurrence per data segment (B). C, Distribution of beta burst duration (bin size =10 ms). Burst counts per bin in each state were normalized with the total burst counts as (burst counts per bin) / (total number of bursts * bin size). The median value of the distribution was compared across different states in the same structure, for animals P and J, Wilcoxon/Kruskal–Wallis tests (one-way test), χ2 ≥ 13.11, p ≤ 0.0014, post hoc test for each pair comparison. *Normal versus Mild, *normal versus moderate, and *mild versus moderate, p ≤ 0.0003; for animal K, Wilcoxon/Kruskal–Wallis tests (one-way test), χ2 ≥ 5.91. *Normal versus moderate, p < 0.02. Larger skewness and kurtosis in STN and GPi in PD states (especially moderate PD) reflected more asymmetric and heavier-tailed burst duration distribution, indicating more bursts with longer duration of bursts. D, Increased amplitude of beta bursts in STN and GPi in PD. For A, B, and D, for animals P and J, Wilcoxon/Kruskal–Wallis tests (one-way test), χ2 ≥ 21.49, p < 0.0001; post hoc test for each pair comparison, p < 0.005, ****r ≥ 0.5, ***0.3 ≤ r < 0.5, **0.1 ≤ r < 0.3, *r < 0.1. For animal K, Wilcoxon/Kruskal–Wallis tests (one-way test), χ2 ≥ 20.74, p < 0.005, ****r ≥ 0.5, ***0.3 ≤ r < 0.5, **0.1 ≤ r < 0.3, *r < 0.1. E, Monotonic correlation between the amplitude and duration of beta bursts. Examples were extracted from low beta results of animal P. The significance of their monotonic positive relationship was measured by Spearman's correlation coefficient (Spearman's rank correlation, *p < 0.0001).
While changes in burst duration and frequency likely contribute to the overall spectral power changes described previously (Fig. 5), the increased amplitude of bursts could also contribute. We found that changes in burst amplitude were similar to those for burst duration and frequency (Fig. 6D) and found a positive correlation between burst amplitude and duration across all animals in all structures (0.56 < Spearman's ρ < 0.76, p < 0.0001; Fig. 6E, examples from animal P). Higher amplitude and more frequent bursts were especially profound in the more severe parkinsonian state.
Changes of high beta burst temporal dynamics were not as consistent as those for low beta (data not shown). The time spent in bursts for high beta was only consistently increased in STN across all three animals in the moderate PD state. The duration, frequency, and amplitude of beta bursts did not show consistent changes across sites. Similar to low beta bursts, however, we found a monotonic positive relationship between amplitude and duration of high beta burst activity (0.51 < Spearman's ρ < 0.76, p < 0.0001) that was present for all structures across all three animals in normal and PD states.
Overlap of burst activity across sites in the BGTC circuit increases in parkinsonism
To assess the extent of coupling across sites within the BGTC circuit and its relationship to severity of disease, we measured the amount of time beta burst activity overlapped across each pair of structures in the different conditions (normal and PD states). LFP traces in Figure 7A illustrate the increase in overlap of low beta burst activity across nodal points in the BGTC circuit for subject J. The increase in overlap of beta activity between structures (M1-STN, M1-GPi, and STN-GPi) was present in all three animals with the induction of parkinsonism (Fig. 7B) and increased with severity with few exceptions (see below). The median overlap time between STN and GPi increased from normal to mild and moderate states: from 0.14 to 0.64, then 2.38 s per 15 s data segment for animal P (i.e., from 0.93% to 4.27% and 15.87%); from 0.24 to 0.97, then 3.67 s per segment for animal J (i.e., from 1.60% to 6.47% and 24.47%); from 0.08 to 3.10 s per segment for animal K (i.e., from 0.53% in normal to 20.67% in moderate). The overlap time for low beta bursts between the STN and GPi to M1, however, was more variable. While there was a consistent increase from the normal to moderate state in all three animals, a progressive increase in overall time across all three states (normal, mild, and moderate) only occurred for M1-STN in animal P and for M1-GPi in animal J (Fig. 7B).
Figure 7.
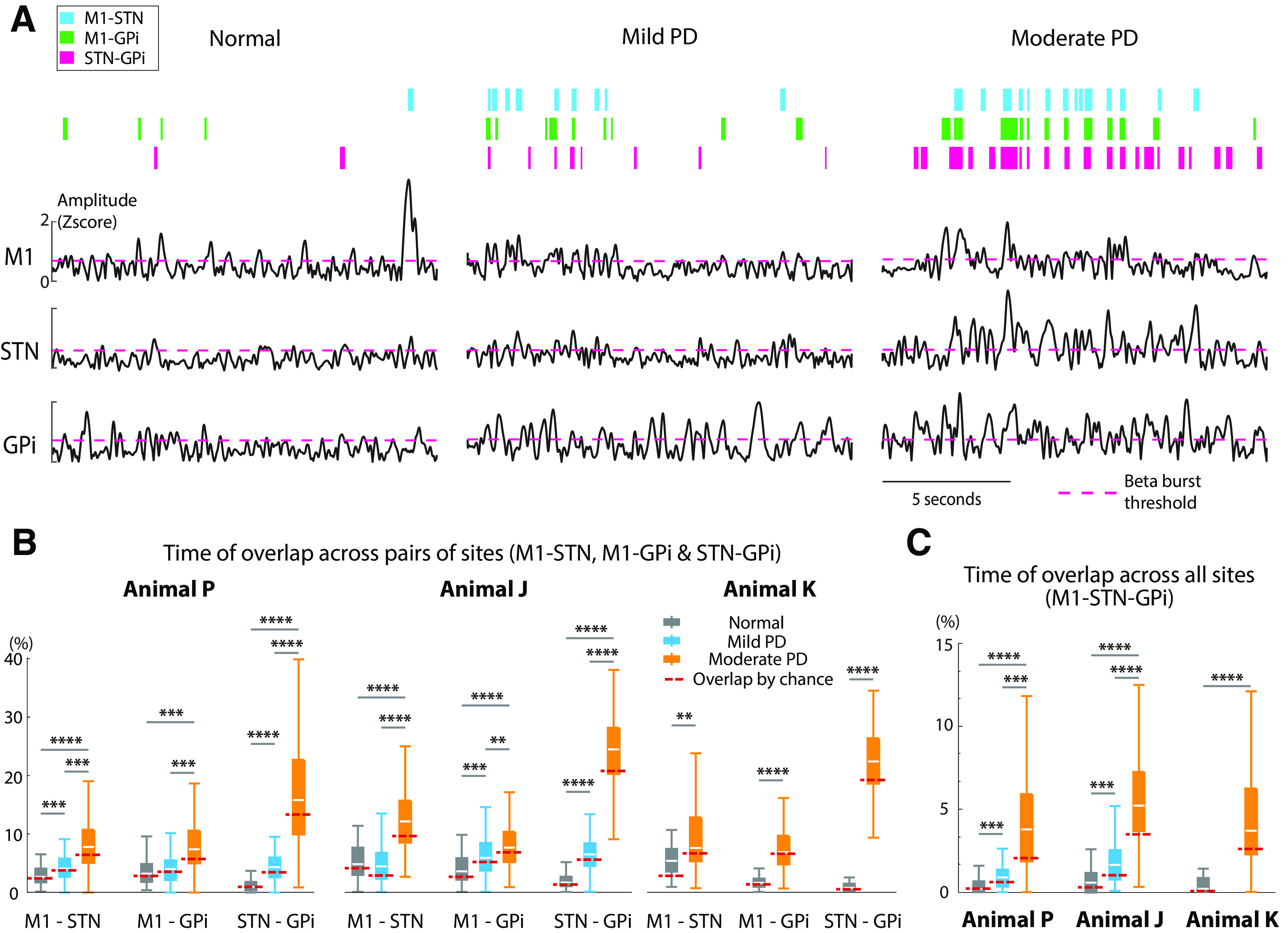
Beta burst overlap increases across structures with increasing severity of PD. A, Increased overlap of low beta bursts across structures from normal to mild and moderate PD states. Representative examples of low beta envelopes obtained from animal J. The pink dashed line indicates the threshold for beta burst detection. Colored bars on top represent burst overlap between a pair of recording sites, between M1 and STN (blue), M1 and GPi (green), and STN and GPi (pink). As the severity of parkinsonism increased, there was increased incidence of bursts that overlapped across sites. B, C, Increased time of overlap between each pair of structures (B) and across all three structures (C) with increasing severity of PD. The overlap time for per data segment across sites increased in the low beta band, indicating stronger amplitude–amplitude coupling across the network in PD states. Coupling between STN and GPi increased with severity of PD across animals P and J. For animals P and J, Wilcoxon/Kruskal–Wallis tests (one-way test), χ2 ≥ 14.07, p ≤ 0.0009; post hoc test for each pair comparison p < 0.005, ****r ≥ 0.5, ***0.3 ≤ r < 0.5, **0.1 ≤ r < 0.3; for animal K, Wilcoxon/Kruskal–Wallis tests (one-way test), χ2 ≥ 8.54, p < 0.005, ****r ≥ 0.5, **0.1 ≤ r < 0.3. The red dashed line represents the overlap time by chance for each state in each animal (see Materials and Methods). Comparing this time by chance to the actual overlap time with nonshuffled data, we observed no significant difference in most cases for animal P and K (Wilcoxon/Kruskal–Wallis tests (one-way test), χ2 ≤ 1.74, p > 0.18 for all pairs for animal P; for animal K, M1-STN χ2 = 9.94, p = 0.0016, r = 0.46; M1-GPi, STN-GPi, and M1-STN-GPi, χ2 ≤ 0.26, p > 0.61) and a significant shorter overlap time by chance for animal J (χ2 ≥ 4.27, p ≤ 0.04, r ≥ 0.14 for all pairs) in normal state. In mild PD state, significant shorter overlap time by chance was present for the pair of STN-GPi and M1-STN-GPi for animal P (M1-STN and M1-GPi, χ2 ≤ 1.86, p > 0.17; STN-GPi, χ2 = 4.75, p = 0.03, r = 0.17; M1-STN-GPi, χ2 = 5.24, p = 0.02, r = 0.17); and all pairs for animal J (M1-STN, STN-GPi, and M1-STN-GPi, χ2 ≥ 18.47, p < 0.0001, r ≥ 0.18; M1-GPi, χ2 = 5.98, p = 0.015, r = 0.10). Moreover, the overlap time by chance was significantly shorter in the moderate PD state for almost all conditions in all animals (the only exception was M1-GPi: χ2 = 1.03, p = 0.31 for animal K; animal P, χ2 ≥ 12.42, p ≤ 0.0004, r ≥ 0.12; and animal J, χ2 ≥ 4.27, p ≤ 0.04, r ≥ 0.12; for all pairs, animal K, M1-STN, STN-GPi, and M1-STN-GPi, χ2 ≥ 5.43, p ≤ 0.02, r ≥ 0.14), suggesting that the increased temporal overlap is not merely because of chance.
In the high beta band (data not shown here), the overlap time across subcortical and/or cortical structures generally increased in the PD states for animals P and J and reduced in animal K. These changes, however, were neither consistent across animals nor appeared to be associated with progression of parkinsonism.
We also measured the amount of time that beta burst activity overlapped across all three structures, M1, STN, and GPi, under the different conditions. For low beta, we observed a consistent increase from normal to PD and a progressive increase with severity of PD symptoms (Fig. 7C). Changes in the high beta band were neither consistent across animals nor appeared to be associated with the progression of parkinsonism (data not shown).
Furthermore, we determined whether the observed overlap times were significantly different from what would be expected by chance based on shuffled versions of the detected beta bursts (see Materials and Methods; Fig. 7 B,C, red dashed lines). In the normal state, there was minimal difference between the overlap observed and that expected by chance. In the mild state, however, observed overlap time was significantly greater than chance for all pairs in animal J and across STN-GPi and M1-STN-GPi for animal P. In the moderate PD states, across all animals and recording sites the observed overlap times were significantly larger than would be expected by chance (the only exception was M1-GPi for animal K).
Temporal relationship between low beta bursts across the BGTC network in parkinsonism
The results from the previous section indicated there was a greater overlap of low beta burst activity between pairs of structures across all three animals, suggestive of increased coupling in the low beta band across multiple nodal points throughout the BGTC network in parkinsonism. We further characterized this activity by examining the temporal relationship between the onset of bursts occurring in each pair of recording sites.
We found that with increasing severity of parkinsonism there was not only an increase in the number of low beta bursts detected in the STN and GPi, but there was also an increased tendency for burst onsets to occur at approximately the same time (time difference, <100 ms) across all sites. Representative LFP traces from STN and GPi in animal J are shown in Figure 8A, where the raster on top of each trace indicates burst onsets. This observation is further quantified by the periburst time probability histograms (see Materials and Methods) generated from all pairs of recording sites (M1-STN, M1-GPi, and STN-GPi) in all three animals (Fig. 8B). We found that the Pbase increased across both subcortical and cortical structures in PD (Fig. 8B, elevated horizontal dash lines in mild and moderate PD states). As shown in Figure 4 and described in the Materials and Methods, an increased number of detected bursts in two sites, but with no temporal relationship between the onset of those bursts, results in an increased Pbase. Therefore, this increase is to be expected given the increased frequency of beta bursts across the BGTC network with the induction of parkinsonism described above.
Figure 8.

Concurrence of low beta bursts across the BGTC network. A, An example of increased concurrence of low beta burst onsets across structures. Representative examples of low beta envelopes in STN and GPi obtained from animal J are presented. Burst onset is illustrated with a raster over the top of LFP activity. The pink dashed line indicates the threshold for beta burst detection. Bolded raster in orange and black represent burst onsets that occurred approximately the same time (time difference, <100 ms) across sites. As the severity of parkinsonism increased, there was increased incidence of concurrent bursts onsets across STN and GPi. B, Periburst probability histograms (including 95% confidence intervals) show increased concurrence of low beta burst onsets across structures in PD states. Significant elevation in the periburst probability histograms, relative to a baseline probability derived from shuffled burst times, are indicated by bold lines. Horizontal dash lines in each plot represented the corresponding Pbase values for normal and PD states.
Notably, in PD states we also found significant peaks in the periburst probability histograms (Fig. 8B, bold lines; significance was calculated relative to the shuffled baseline; see Materials and Methods). A significant peak in the periburst probability histogram at a time difference near zero means the following: given a burst occurrence in one site, the likelihood of a burst occurring in the other site at approximately the same time is significantly above chance. We observed more significant peaks in the probability histograms at time differences close to zero in PD states (the only exception was for M1-GPi for animal K). Significant temporal relationships between burst onsets were consistently found in moderate PD states in all three animals for STN-GPi and were also evident in the mild state of PD (Fig. 8B, bottom, animals P and J).
Discussion
We observed significant changes in the incidence, amplitude, and duration of low beta-band burst activity following the administration of MPTP and the induction of parkinsonism. These changes were correlated to the severity of disease and were coherent in time across sites in the BGTC network. These data provide new insights into how changes in temporal dynamics of low beta activity within the BGTC network are linked to the development of PD motor signs.
Prolonged, higher-amplitude low beta bursts in the basal ganglia in PD
Recently, it has been suggested that alterations in the temporal dynamics of episodic beta burst activity in the form of prolonged and more frequent beta bursts, rather than elevations in the mean power of LFP beta oscillations, play a more significant role in the pathophysiology of PD (Tinkhauser et al., 2017a; Torrecillos et al., 2018). Support for this argument comes from the observation that improvement in motor signs during DBS or levodopa therapy was associated with shorter-duration beta bursts in the STN or GPi of PD patients (Tinkhauser et al., 2017a,b; Lofredi et al., 2019). Other studies are less supportive of this hypothesis, however, reporting that beta burst activity either did not change during DBS (Schmidt et al., 2020) or that DBS reduced only the amplitude but not the duration of beta bursts in GPi and M1 (Wang et al., 2018). Some studies also reported that levodopa reduced the duration but not the amplitude of GPi beta bursts in PD patients (Lofredi et al., 2019). The relative role of beta burst dynamics (e.g., duration, amplitude, incidence) in the development of PD, therefore, remains controversial.
Data collected in patients during DBS or levodopa therapy, while informative, may not reflect the state of activity that would be present in the naive nondiseased condition (Lofredi et al., 2019). An advantage of the present study was our ability to examine temporal features of beta bursts in a progressive model of PD using a within-subject design, comparing neural activity from the same recording sites in both naive and PD states. We found increases in the power, duration, amplitude, and incidence of low beta (8–20 Hz) burst activity in the STN and GPi with the induction of parkinsonism, suggestive of a potential pathophysiological role of all these features in the basal ganglia in PD. For example, increased burst amplitude may reflect an exaggerated synchrony that limits the amount of information local neural populations can transmit (Brittain and Brown, 2014), while prolonged burst durations could impede flexible switching between motor states, thereby leading to motor symptoms such as bradykinesia and akinesia (Feingold et al., 2015; Lofredi et al., 2019). Our results suggest that burst amplitude and duration are highly correlated, and whether one feature is functionally more important than the other is a question requiring further investigation. In either case, our findings provide a further rationale for targeting both the amplitude and duration of beta activity detected in the basal ganglia in disease-modifying therapies (e.g., as control variables for closed-loop DBS).
Whether beta-band activity in M1 is exaggerated in PD remains in dispute. Using epidural recordings over M1, Devergnas et al. (2014) found that the severity of parkinsonism in MPTP-treated nonhuman primates correlated with increases in spectral power at frequencies between 7.8 and 15.5 Hz in two of three animals, but either increases or decreases in power in frequency ranges of 15.6–23.3 and 23.4–35.1 Hz were observed across animals. An increase in power in M1 in the 15–30 Hz range has also been reported after 6-hydroxydopamine lesion in rats (Mallet et al., 2008; Lehmkuhle et al., 2009), while another study observed no beta-band changes (Brazhnik et al., 2012). Although some magnetoencephalography or electroencephalography studies reported an increase of beta power (13–30 Hz) within the sensorimotor cortex at rest in PD patients compared with healthy controls (Moazami-Goudarzi et al., 2008; Pollok et al., 2012), many others showed a decrease (Bosboom et al., 2006; Stoffers et al., 2007; Benz et al., 2014; Heinrichs-Graham et al., 2014; Stegemöller et al., 2016) or no difference (George et al., 2013). In this study, we observed from normal to PD states a reduction of low beta power (8–20 Hz) in M1 across all three animals. The power change in the low beta band was consistent with the increase of the disease severity in one animal (animal P). There was an increase of high beta power (21–35 Hz) in M1 in two animals (animals P and J) and a decrease in the third animal (animal K). These findings, together with the conflicting results from previous studies, suggest a less consistent relationship between the change of cortical beta activity and the development of parkinsonism compared with the low beta burst activity in the basal ganglia.
It should be noted, however, that in the present study the magnitude of changes in beta power and temporal features of basal ganglia beta burst activity cannot fully explain the changes in PD motor signs. For example, in animal J the difference between mUPDRS scores in mild and moderate PD states was relatively small compared with mild and moderate states in animal P (Table 1), yet the difference in beta power and burst characteristics was greater in animal J compared with animal P (Figs. 5B, 6A,B). Although there appears to be a relationship between these features of beta activity and parkinsonism, the power and temporal features of beta burst activity alone, at any one nodal point in the BGTC network may not be the entire story. Future investigation will be needed to clarify the pathophysiological roles of these and other features (e.g., high-frequency oscillations, phase–amplitude coupling, and functional and directional connectivity) within and across nodal points in the basal ganglia–thalamocortical network in PD.
Increased concurrence of beta burst activity across the BGTC network in parkinsonism
Coordinated synchrony across neuronal populations is considered essential for information processing and control of normal movement (van Wijk et al., 2012; Fries, 2015). Uncontrolled synchronization, however, has been argued to lead to involuntary movement (Vitek et al., 1999) or a lack of movement (Brown, 2007; Kühn et al., 2009). It has been suggested that in PD long-duration beta bursts in the STN reflect not just exaggerated local synchrony but also is associated with coupling in the beta band across structures throughout the motor circuit (Tinkhauser et al., 2018). Here we demonstrated that low beta burst activity not only increased in the BGTC network in PD, but that coupling across nodal points (i.e., STN, GPi, and M1) in the form of overlapping beta bursts progressively increased with severity of disease. These results provide further evidence supporting the pathophysiological role of exaggerated beta-band synchrony in the motor network in the development of parkinsonian motor signs.
Notably, low beta burst overlap between STN and GPi was consistently present across all animals in mild and moderate PD states and increased with the severity of PD in animals P and J. An increase of M1-STN and M1-GPi overlap was only consistently observed at moderate levels of severity. Although not conclusive, these findings provide compelling evidence to suggest that an evolution of coupling occurs across the network as symptoms evolve beginning in subcortical structures, and as coupling strengthens between these regions it increases between subcortical–cortical structures (e.g., M1-STN, M1-GPi). Indeed, significant temporal correlation of burst activity was present in the mild state across both M1-GPi and M1-STN only in animal J whose mild state was greater than animal P (Fig. 8B). Additional studies with neural recordings across more incremental states of PD severity will be useful to better elucidate how burst overlap evolves across the network and whether increased coupling between subcortical structures precedes coupling across subcortical–cortical structures.
Our findings align somewhat with those of Devergnas et al. (2014), who found STN-GPi and GPi-M1 coherence (in frequency ranges 7.8–15.5 and 15.6–23.3 Hz) correlated with parkinsonian severity in MPTP-treated monkeys. Contrary to our study, however, they observed no systematic changes in STN beta-band power or STN-M1 coherence. A potential explanation for disparities between studies may be differences in the methodologies. Recording locations in the STN could differ given that they collected LFPs from single microelectrodes that were inserted daily into the STN, while we performed recordings from scaled down DBS leads that were chronically implanted. The encephalographic signals they used and the average LFP across the microelectrode array in M1 arm area for analysis here could also contribute to differences in the spatial distribution and resolution of beta oscillatory activity.
Increased beta burst overlap time could result from the fact that there were more frequent, longer-duration bursts occurring at each site in PD states, increasing the likelihood of overlapping bursts across sites. The overlap time we observed in moderate PD states was significantly greater than what would be expected by chance, however, indicating that the increased temporal overlap is not explained solely by the findings of increased frequency and duration of beta bursts in each recording site. To further characterize this temporal overlap, the current study also addresses whether the beta burst occurrences in different sites are temporally correlated with each other. Our observation of higher baseline burst probability in PD states (Fig. 8B) reflects an increased likelihood of overlapping bursts across sites. Significant peaks in the periburst time probability histograms, however, indicate that beta bursts across the STN, GPi, and M1 tended to occur with a certain time delay (close to zero here). This would suggest that the enhanced coupling was not likely only because of chance as a result of bursts occurring more frequently or with longer durations, but from some common factor that affects each nodal point leading to the simultaneous onset of beta burst activity across the different structures. Whether or not the increased overlap time occurs by chance, compared with increased beta burst activity locally within a single site, episodes of elevated beta-band synchronization across multiple nodes in the BGTC network simultaneously may further reduce the information-encoding capacity of the circuit and lead to a greater impact on motor function.
Given the anatomic connectivity of these structures, a common input could be one mechanism that underlies this co-occurrence (Mathai and Smith, 2011). Previous studies have suggested cortical areas as a candidate source of the beta oscillations in the network (Lindenbach and Bishop, 2013), with exaggerated beta activity driven by changes in hyperdirect (cortical to STN) connectivity (Moran et al., 2011; Mathai et al., 2015; Sharott et al., 2018). Others suggest a striatal origin of the pathologic beta oscillations in PD (McCarthy et al., 2011). Our results of increased concurrent burst onsets do not provide definitive evidence for a common source for the induction of synchronization among these structures, as the information flow between nodal points in the network may not be reflected only by the temporal delay between bursts. Analysis tools capable of deriving frequency-dependent, direction-specific gains and phase lags, which quantify the degree of signal amplification and delays in circuit interconnections, will be necessary to further characterize the causal, directed, temporal relationships of the oscillatory activity across structures in the BGTC network.
Whatever the mechanism underlying the increased co-occurrence of beta burst activity across multiple sites in the BGTC network, the correlation of these increases with severity of disease provides compelling evidence in support of a critical role of this activity in the pathophysiology of PD. While synchronized neuronal activity is important for aspects of normal brain function (Engel and Fries, 2010; Feingold et al., 2015), excessive coupling across sites can produce deleterious network-level signal processing that prevents or disrupts the transmission of information within the network (Hammond et al., 2007; Hanslmayr et al., 2012). Such activity would diminish the information-encoding space across the network, and thereby compromise motor function and, in the case of PD, lead to its cardinal motor features (i.e., akinesia, bradykinesia, and rigidity; Brittain and Brown, 2014; Torrecillos et al., 2018). While the above proposal suggests that information flow is disrupted or diminished by enhanced coupling, an alternative hypothesis is that increased coupling may lead to excessive flow of redundant information, leading to a loss of parallel processing that impairs the dynamic ability of the network to shift sets to select and implement motor commands (Cruz et al., 2011; Dorval et al., 2015). Future studies dedicated to understanding how beta burst activity encodes movement information, how the coupling of beta bursts affects the encoding capability of neuronal ensembles during the preparation and execution of movement, and how the spatial–temporal evolution of beta burst activity within the BGTC network disrupts information transfer in PD are necessary to elucidate the roles of beta burst interactions in the development of PD.
Conclusion
These findings provide support for the hypothesis that exaggerated beta-band coupling occurs across the BGTC circuit in PD, increases with severity of disease, and evolves from subcortical–subcortical local circuits in the basal ganglia to subcortical–cortical portions of the BGTC network. These data provide support for the role of pathologic network-wide coupling of synchronized oscillatory activity as a critical feature in the pathophysiology of PD and the potential impact of these changes on temporal dynamics of low beta-band oscillatory activity on the information coding capacity within the BGTC network.
Footnotes
J.L.V. serves as a consultant for Medtronic, Boston Scientific, and Abbott; and serves on the scientific advisory board for Surgical Information Sciences. The authors declare no other competing financial interests.
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grants R01 NS058945, R01 NS037019, R01 NS110613, P50 NS098573, R37 NS077657; and by the MnDRIVE (Minnesota's Discovery, Research and Innovation Economy) Brain Conditions Program and Engdahl Family Foundation.
References
- Bartolo R, Merchant H (2015) Oscillations are linked to the initiation of sensory-cued movement sequences and the internal guidance of regular tapping in the monkey. J Neurosci 35:4635–4640. 10.1523/JNEUROSCI.4570-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz N, Hatz F, Bousleiman H, Ehrensperger MM, Gschwandtner U, Hardmeier M, Ruegg S, Schindler C, Zimmermann R, Monsch AU, Fuhr P (2014) Slowing of EEG background activity in Parkinson's and Alzheimer's disease with early cognitive dysfunction. Front Aging Neurosci 6:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosboom JLW, Stoffers D, Stam CJ, van Dijk BW, Verbunt J, Berendse HW, Wolters ECh (2006) Resting state oscillatory brain dynamics in Parkinson's disease: an MEG study. Clin Neurophysiol 117:2521–2531. 10.1016/j.clinph.2006.06.720 [DOI] [PubMed] [Google Scholar]
- Brazhnik E, Cruz AV, Avila I, Wahba MI, Novikov N, Ilieva NM, McCoy AJ, Gerber C, Walters JR (2012) State-dependent spike and local field synchronization between motor cortex and substantia nigra in hemiparkinsonian rats. J Neurosci 32:7869–7880. 10.1523/JNEUROSCI.0943-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain J-S, Brown P (2014) Oscillations and the basal ganglia: motor control and beyond. Neuroimage 85:637–647. 10.1016/j.neuroimage.2013.05.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P (2003) Oscillatory nature of human basal ganglia activity: relationship to the pathophysiology of Parkinson's disease. Mov Disord 18:357–363. 10.1002/mds.10358 [DOI] [PubMed] [Google Scholar]
- Brown P (2006) Bad oscillations in Parkinson's disease. In: Parkinson's disease and related disorders (Riederer P, Reichmann H, Youdim MBH, Gerlach M, eds), pp 27–30. Vienna, Austria: Springer. [Google Scholar]
- Brown P (2007) Abnormal oscillatory synchronisation in the motor system leads to impaired movement. Curr Opin Neurobiol 17:656–664. 10.1016/j.conb.2007.12.001 [DOI] [PubMed] [Google Scholar]
- Brown P, Mazzone P, Oliviero A, Altibrandi MG, Pilato F, Tonali PA, Di Lazzaro V (2004) Effects of stimulation of the subthalamic area on oscillatory pallidal activity in Parkinson's disease. Exp Neurol 188:480–490. 10.1016/j.expneurol.2004.05.009 [DOI] [PubMed] [Google Scholar]
- Chen CC, Litvak V, Gilbertson T, Kühn A, Lu CS, Lee ST, Tsai CH, Tisch S, Limousin P, Hariz M, Brown P (2007) Excessive synchronization of basal ganglia neurons at 20 Hz slows movement in Parkinson's disease. Exp Neurol 205:214–221. 10.1016/j.expneurol.2007.01.027 [DOI] [PubMed] [Google Scholar]
- Connolly AT, Jensen AL, Bello EM, Netoff TI, Baker KB, Johnson MD, Vitek JL (2015) Modulations in oscillatory frequency and coupling in globus pallidus with increasing parkinsonian severity. J Neurosci 35:6231–6240. 10.1523/JNEUROSCI.4137-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz AV, Mallet N, Magill PJ, Brown P, Averbeck BB (2011) Effects of dopamine depletion on information flow between the subthalamic nucleus and external globus pallidus. J Neurophysiol 106:2012–2023. 10.1152/jn.00094.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deffains M, Iskhakova L, Katabi S, Israel Z, Bergman H (2018) Longer β oscillatory episodes reliably identify pathological subthalamic activity in Parkinsonism. Mov Disord 33:1609–1618. 10.1002/mds.27418 [DOI] [PubMed] [Google Scholar]
- Devergnas A, Pittard D, Bliwise D, Wichmann T (2014) Relationship between oscillatory activity in the cortico-basal ganglia network and parkinsonism in MPTP-treated monkeys. Neurobiol Dis 68:156–166. 10.1016/j.nbd.2014.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorval AD, Muralidharan A, Jensen AL, Baker KB, Vitek JL (2015) Information in pallidal neurons increases with Parkinsonian severity. Parkinsonism Relat Disord 21:1355–1361. 10.1016/j.parkreldis.2015.09.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel AK, Fries P (2010) Beta-band oscillations—signalling the status quo? Curr Opin Neurobiol 20:156–165. 10.1016/j.conb.2010.02.015 [DOI] [PubMed] [Google Scholar]
- Escobar D, Johnson LA, Nebeck SD, Zhang J, Johnson MD, Baker KB, Molnar GF, Vitek JL (2017) Parkinsonism and vigilance: alteration in neural oscillatory activity and phase-amplitude coupling in the basal ganglia and motor cortex. J Neurophysiol 118:2654–2669. 10.1152/jn.00388.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov A, Beichel R, Kalpathy-Cramer J, Finet J, Fillion-Robin J-C, Pujol S, Bauer C, Jennings D, Fennessy F, Sonka M, Buatti J, Aylward S, Miller JV, Pieper S, Kikinis R (2012) 3D slicer as an image computing platform for the quantitative imaging network. Magn Reson Imaging 30:1323–1341. 10.1016/j.mri.2012.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold J, Gibson DJ, DePasquale B, Graybiel AM (2015) Bursts of beta oscillation differentiate post performance activity in the striatum and motor cortex of monkeys performing movement tasks. Proc Natl Acad Sci U S A 112:13687–13692. 10.1073/pnas.1517629112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries P (2015) Rhythms for cognition: communication through coherence. Neuron 88:220–235. 10.1016/j.neuron.2015.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George JS, Strunk J, Mak-McCully R, Houser M, Poizner H, Aron AR (2013) Dopaminergic therapy in Parkinson's disease decreases cortical beta band coherence in the resting state and increases cortical beta band power during executive control. Neuroimage Clin 3:261–270. 10.1016/j.nicl.2013.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C, Bergman H, Brown P (2007) Pathological synchronization in Parkinson's disease: networks, models and treatments. Trends Neurosci 30:357–364. 10.1016/j.tins.2007.05.004 [DOI] [PubMed] [Google Scholar]
- Hanslmayr S, Staudigl T, Fellner M-C (2012) Oscillatory power decreases and long-term memory: the information via desynchronization hypothesis. Front Hum Neurosci 6:74. 10.3389/fnhum.2012.00074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Elder CM, Okun MS, Patrick SK, Vitek JL (2003) Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. J Neurosci 23:1916–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrichs-Graham E, Kurz MJ, Becker KM, Santamaria PM, Gendelman HE, Wilson TW (2014) Hypersynchrony despite pathologically reduced beta oscillations in patients with Parkinson's disease: a pharmaco-magnetoencephalography study. J Neurophysiol 112:1739–1747. 10.1152/jn.00383.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix CM, Campbell BA, Tittle BJ, Johnson LA, Baker KB, Johnson MD, Molnar GF, Vitek JL (2018) Predictive encoding of motor behavior in the supplementary motor area is disrupted in parkinsonism. J Neurophysiol 120:1247–1255. 10.1152/jn.00306.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn AA, Kempf F, Brücke C, Gaynor Doyle L, Martinez-Torres I, Pogosyan A, Trottenberg T, Kupsch A, Schneider G-H, Hariz MI, Vandenberghe W, Nuttin B, Brown P (2008) High-frequency stimulation of the subthalamic nucleus suppresses oscillatory activity in patients with Parkinson's disease in parallel with improvement in motor performance. J Neurosci 28:6165–6173. 10.1523/JNEUROSCI.0282-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn AA, Tsui A, Aziz T, Ray N, Brücke C, Kupsch A, Schneider G-H, Brown P (2009) Pathological synchronisation in the subthalamic nucleus of patients with Parkinson's disease relates to both bradykinesia and rigidity. Exp Neurol 215:380–387. 10.1016/j.expneurol.2008.11.008 [DOI] [PubMed] [Google Scholar]
- Lehmkuhle MJ, Bhangoo SS, Kipke DR (2009) The electrocorticogram signal can be modulated with deep brain stimulation of the subthalamic nucleus in the hemiparkinsonian rat. J Neurophysiol 102:1811–1820. 10.1152/jn.90844.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach D, Bishop C (2013) Critical involvement of the motor cortex in the pathophysiology and treatment of Parkinson's disease. Neurosci Biobehav Rev 37:2737–2750. 10.1016/j.neubiorev.2013.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little S, Bonaiuto J, Barnes G, Bestmann S (2019) Human motor cortical beta bursts relate to movement planning and response errors. PLoS Biol 17:e3000479. 10.1371/journal.pbio.3000479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lofredi R, Neumann W, Brücke C, Huebl J, Krauss JK, Schneider G, Kühn AA (2019) Pallidal beta bursts in Parkinson's disease and dystonia. Mov Disord 34:420–424. 10.1002/mds.27524 [DOI] [PubMed] [Google Scholar]
- Mallet N, Pogosyan A, Sharott A, Csicsvari J, Bolam JP, Brown P, Magill PJ (2008) Disrupted dopamine transmission and the emergence of exaggerated beta oscillations in subthalamic nucleus and cerebral cortex. J Neurosci 28:4795–4806. 10.1523/JNEUROSCI.0123-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathai A, Smith Y (2011) The corticostriatal and corticosubthalamic pathways: two entries, one target. so what? Front Syst Neurosci 5:64. 10.3389/fnsys.2011.00064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathai A, Ma Y, Paré J-F, Villalba RM, Wichmann T, Smith Y (2015) Reduced cortical innervation of the subthalamic nucleus in MPTP-treated parkinsonian monkeys. Brain J Brain 138:946–962. 10.1093/brain/awv018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard EM, Nordhausen CT, Normann RA (1997) The Utah intracortical electrode array: a recording structure for potential brain-computer interfaces. Electroencephalogr Clin Neurophysiol 102:228–239. 10.1016/s0013-4694(96)95176-0 [DOI] [PubMed] [Google Scholar]
- McCarthy MM, Moore-Kochlacs C, Gu X, Boyden ES, Han X, Kopell N (2011) Striatal origin of the pathologic beta oscillations in Parkinson's disease. Proc Natl Acad Sci U S A 108:11620–11625. 10.1073/pnas.1107748108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miocinovic S, Zhang J, Xu W, Russo GS, Vitek JL, McIntyre CC (2007) Stereotactic neurosurgical planning, recording, and visualization for deep brain stimulation in non-human primates. J Neurosci Methods 162:32–41. 10.1016/j.jneumeth.2006.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazami-Goudarzi M, Sarnthein J, Michels L, Moukhtieva R, Jeanmonod D (2008) Enhanced frontal low and high frequency power and synchronization in the resting EEG of parkinsonian patients. Neuroimage 41:985–997. 10.1016/j.neuroimage.2008.03.032 [DOI] [PubMed] [Google Scholar]
- Moran RJ, Mallet N, Litvak V, Dolan RJ, Magill PJ, Friston KJ, Brown P (2011) Alterations in brain connectivity underlying beta oscillations in parkinsonism. PLoS Comput Biol 7:e1002124. 10.1371/journal.pcbi.1002124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan A, Jensen AL, Connolly A, Hendrix CM, Johnson MD, Baker KB, Vitek JL (2016) Physiological changes in the pallidum in a progressive model of Parkinson's disease: are oscillations enough? Exp Neurol 279:187–196. 10.1016/j.expneurol.2016.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Fetz EE (1992) Coherent 25- to 35-Hz oscillations in the sensorimotor cortex of awake behaving monkeys. Proc Natl Acad Sci U S A 89:5670–5674. 10.1073/pnas.89.12.5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Fetz EE (1996) Oscillatory activity in sensorimotor cortex of awake monkeys: synchronization of local field potentials and relation to behavior. J Neurophysiol 76:3949–3967. 10.1152/jn.1996.76.6.3949 [DOI] [PubMed] [Google Scholar]
- Nini A, Feingold A, Slovin H, Bergman H (1995) Neurons in the globus-pallidus do not show correlated activity in the normal monkey, but phase-locked oscillations appear in the MPTP model of Parkinsonism. J Neurophysiol 74:1800–1805. 10.1152/jn.1995.74.4.1800 [DOI] [PubMed] [Google Scholar]
- Oostenveld R, Fries P, Maris E, Schoffelen J-M (2011) FieldTrip: open source software for advanced analysis of MEG, EEG, and invasive electrophysiological data. Comput Intell Neurosci 2011:156869. 10.1155/2011/156869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswal A, Beudel M, Zrinzo L, Limousin P, Hariz M, Foltynie T, Litvak V, Brown P (2016) Deep brain stimulation modulates synchrony within spatially and spectrally distinct resting state networks in Parkinson's disease. Brain 139:1482–1496. 10.1093/brain/aww048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallant J (2011) Calculating effect size. In: SPSS Survival Manual: a step by step guide to data analysis using the SPSS program, Ed 4, pp 254–255. Crows Nest, NSW, Australia: Allen & Unwin. [Google Scholar]
- Pollok B, Krause V, Martsch W, Wach C, Schnitzler A, Südmeyer M (2012) Motor-cortical oscillations in early stages of Parkinson's disease. J Physiol 590:3203–3212. 10.1113/jphysiol.2012.231316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa M, Giannicola G, Servello D, Marceglia S, Pacchetti C, Porta M, Sassi M, Scelzo E, Barbieri S, Priori A (2011) Subthalamic local field beta oscillations during ongoing deep brain stimulation in Parkinson's disease in hyperacute and chronic phases. Neurosignals 19:151–162. 10.1159/000328508 [DOI] [PubMed] [Google Scholar]
- Rousche PJ, Normann RA (1992) A method for pneumatically inserting an array of penetrating electrodes into cortical tissue. Ann Biomed Eng 20:413–422. 10.1007/BF02368133 [DOI] [PubMed] [Google Scholar]
- Schmidt SL, Peters JJ, Turner DA, Grill WM (2020) Continuous deep brain stimulation of the subthalamic nucleus may not modulate beta bursts in patients with Parkinson's disease. Brain Stimul 13:433–443. 10.1016/j.brs.2019.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharott A, Gulberti A, Hamel W, Köppen JA, Münchau A, Buhmann C, Pötter-Nerger M, Westphal M, Gerloff C, Moll CKE, Engel AK (2018) Spatio-temporal dynamics of cortical drive to human subthalamic nucleus neurons in Parkinson's disease. Neurobiol Dis 112:49–62. 10.1016/j.nbd.2018.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Law R, Tsutsui S, Moore CI, Jones SR (2017) The rate of transient beta frequency events predicts behavior across tasks and species. eLife 6:e29086. 10.7554/eLife.29086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegemöller EL, Allen DP, Simuni T, MacKinnon CD (2016) Motor cortical oscillations are abnormally suppressed during repetitive movement in patients with Parkinson's disease. Clin Neurophysiol 127:664–674. 10.1016/j.clinph.2015.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein E, Bar-Gad I (2013) β oscillations in the cortico-basal ganglia loop during parkinsonism. Exp Neurol 245:52–59. 10.1016/j.expneurol.2012.07.023 [DOI] [PubMed] [Google Scholar]
- Stoffers D, Bosboom JLW, Deijen JB, Wolters EC, Berendse HW, Stam CJ (2007) Slowing of oscillatory brain activity is a stable characteristic of Parkinson's disease without dementia. Brain 130:1847–1860. 10.1093/brain/awm034 [DOI] [PubMed] [Google Scholar]
- Tinkhauser G, Pogosyan A, Little S, Beudel M, Herz DM, Tan H, Brown P (2017a) The modulatory effect of adaptive deep brain stimulation on beta bursts in Parkinson's disease. Brain 140:1053–1067. 10.1093/brain/awx010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinkhauser G, Pogosyan A, Tan H, Herz DM, Kühn AA, Brown P (2017b) Beta burst dynamics in Parkinson's disease OFF and ON dopaminergic medication. Brain 140:2968–2981. 10.1093/brain/awx252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinkhauser G, Torrecillos F, Duclos Y, Tan H, Pogosyan A, Fischer P, Carron R, Welter M-L, Karachi C, Vandenberghe W, Nuttin B, Witjas T, Régis J, Azulay J-P, Eusebio A, Brown P (2018) Beta burst coupling across the motor circuit in Parkinson's disease. Neurobiol Dis 117:217–225. 10.1016/j.nbd.2018.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrecillos F, Tinkhauser G, Fischer P, Green AL, Aziz TZ, Foltynie T, Limousin P, Zrinzo L, Ashkan K, Brown P, Tan H (2018) Modulation of beta bursts in the subthalamic nucleus predicts motor performance. J Neurosci 38:8905–8917. 10.1523/JNEUROSCI.1314-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager MH, Koop MM, Velisar A, Blumenfeld Z, Nikolau JS, Quinn EJ, Martin T, Bronte-Stewart H (2016) Subthalamic beta oscillations are attenuated after withdrawal of chronic high frequency neurostimulation in Parkinson's disease. Neurobiol Dis 96:22–30. 10.1016/j.nbd.2016.08.003 [DOI] [PubMed] [Google Scholar]
- van Wijk BCM, Beek PJ, Daffertshofer A (2012) Neural synchrony within the motor system: what have we learned so far? Front Hum Neurosci 6:252. 10.3389/fnhum.2012.00252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitek JL, Chockkan V, Zhang JY, Kaneoke Y, Evatt M, DeLong MR, Triche S, Mewes K, Hashimoto T, Bakay RA (1999) Neuronal activity in the basal ganglia in patients with generalized dystonia and hemiballismus. Ann Neurol 46:22–35. [DOI] [PubMed] [Google Scholar]
- Vitek JL, Zhang J, Hashimoto T, Russo GS, Baker KB (2012) External pallidal stimulation improves parkinsonian motor signs and modulates neuronal activity throughout the basal ganglia thalamic network. Exp Neurol 233:581–586. 10.1016/j.expneurol.2011.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD, de Hemptinne C, Miocinovic S, Ostrem JL, Galifianakis NB, San Luciano M, Starr PA (2018) Pallidal deep-brain stimulation disrupts pallidal beta oscillations and coherence with primary motor cortex in Parkinson's disease. J Neurosci 38:4556–4568. 10.1523/JNEUROSCI.0431-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Johnson LA, Jensen AL, Baker KB, Molnar GF, Johnson MD, Vitek JL (2017) Network-wide oscillations in the parkinsonian state: alterations in neuronal activities occur in the premotor cortex in parkinsonian nonhuman primates. J Neurophysiol 117:2242–2249. 10.1152/jn.00011.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger M, Mahant N, Hutchison WD, Lozano AM, Moro E, Hodaie M, Lang AE, Dostrovsky JO (2006) Beta oscillatory activity in the subthalamic nucleus and its relation to dopaminergic response in Parkinson's disease. J Neurophysiol 96:3248–3256. 10.1152/jn.00697.2006 [DOI] [PubMed] [Google Scholar]
- Wichmann T, Bergman H, DeLong MR (1994) The primate subthalamic nucleus. III. Changes in motor behavior and neuronal activity in the internal pallidum induced by subthalamic inactivation in the MPTP model of parkinsonism. J Neurophysiol 72:521–530. 10.1152/jn.1994.72.2.521 [DOI] [PubMed] [Google Scholar]