

Abstract
The addition of new neurons to existing neural circuits in the adult brain remains of great interest to neurobiology because of its therapeutic implications. The premier model for studying this process has been the hippocampal dentate gyrus in mice, where new neurons are added to mature circuits during adulthood. Notably, external factors such as an enriched environment (EE) and exercise markedly increase hippocampal neurogenesis. Here, we demonstrate that EE acts by increasing fibroblast growth factor receptor (FGFR) function autonomously within neurogenic cells to expand their numbers in adult male and female mice. FGFRs activated by EE signal through their mediators, FGFR substrate (FRS), to induce stem cell proliferation, and through FRS and phospholipase Cγ to increase the number of adult-born neurons, providing a mechanism for how EE promotes adult neurogenesis.
SIGNIFICANCE STATEMENT How the environment we live in affects cognition remains poorly understood. In the current study, we explore the mechanism underlying the effects of an enriched environment on the production of new neurons in the adult hippocampal dentate gyrus, a brain area integral in forming new memories. A mechanism is provided for how neural precursor cells in the adult mammalian dentate gyrus respond to an enriched environment to increase their neurogenic output. Namely, an enriched environment acts on stem and progenitor cells by activating fibroblast growth factor receptor signaling through phospholipase Cγ and FGF receptor substrate proteins to expand the pool of precursor cells.
Keywords: adult neurogenesis, dentate gyrus, environmental enrichment, exercise, fibroblast growth factor receptors, neural stem cells
Introduction
Neurogenesis in adults continues to generate much interest because of its impact on cognition and its ability to be influenced by external factors. The rodent hippocampal dentate gyrus (DG) includes a neurogenic niche, the subgranular zone (SGZ), where radial glia-like stem cells (RGLs) generate new granule neurons into adulthood (Ming and Song, 2011; Gonçalves et al., 2016). Remarkably, neurogenesis in the DG is strongly promoted by an enriched environment (EE) and exercise, which increase proliferation and survival of neurogenic cells and improves cognition (Benarroch, 2013).
Brain-derived neurotrophic factor (BDNF) signaling has been a leading candidate for mediating at least some of the changes observed in the adult neurogenic lineage in response to environmental factors (Liu and Nusslock, 2018). For example, deletion of the BDNF receptor TrkB in adult neural stem cells blocks the accelerated differentiation of neuroblasts induced by the antidepressant ketamine (Ma et al., 2017). However, while deletion of TrkB during development reduces the effects of exercise on adult neurogenesis (Li et al., 2008), direct evidence demonstrating that BDNF–TrkB signaling is required in adult neural stem cells or progenitor cells to mediate the early expansion of precursors in response to factors such as environmental enrichment and exercise is lacking. BMPs have also been implicated in modulating the impact of environment on increasing the number of new neurons in the adult DG, but in this case by repressing rather than enhancing neurogenesis (Gobeske et al., 2009). The CX3CR1 fractalkine receptor acting in microglia can nonautonomously promote expansion of neural precursors in response to exercise (Vukovic et al., 2012). Yet despite these findings, since the discovery of the positive effects of EE and exercise on adult hippocampal neurogenesis (Kempermann et al., 1997; van Praag et al., 1999), little progress has been made in identifying the molecular pathways operating within early neurogenic precursors to intrinsically promote their expansion in response to external conditions.
Although the fibroblast growth factor (FGF) signaling pathway plays roles throughout developmental neurogenesis, the possibility that FGF2, the most studied FGF ligand, plays a role in adult hippocampal neurogenesis remains unclear (Mudò et al., 2009). Nevertheless, the FGF receptors (FGFRs) are necessary and sufficient to promote neurogenesis in the adult DG under standard housing conditions (Kang and Hébert, 2015). Deletion of all three of the expressed Fgfr genes, Fgfr1, Fgfr2, and Fgfr3, in adult stem cells results in a dramatic loss of neurogenesis in the DG. Conversely, and similar to the effects of EE and exercise, increasing FGFR activity in neurogenic cells expands their numbers as well as those of their descendants in the DG (Kang and Hébert, 2015). Despite this requirement for FGFRs in neurogenesis, the role of these receptors in regulating neurogenesis in response to environmental influences is unclear. In support of a possible role, acute stress increases FGF2 expression in astrocytes and hippocampal cell proliferation, while neutralization of FGF2 abolishes this proliferative response (Kirby et al., 2013). Based on these previous findings, we hypothesized that EE acts via FGFRs to enhance adult hippocampal neurogenesis.
Here, we tested this hypothesis by deleting the three Fgfr genes in adult neural precursors and measuring the impact that EE has on neurogenesis in the mutants. As is standard in the field, our paradigm for studying the effects of EE includes voluntary exercise because the mice actively run while exploring their complex surroundings. Hence, for simplicity we refer to an EE as one that includes exercise. We demonstrate that the expansion of neurogenic cells in response to EE depends on increasing FGFR activity. Moreover, we show that FGFR activity operates through FGFR substrate (FRS) proteins to expand early neurogenic precursors, and, through FRS, phopholipase-Cγ and CREB to increase the number of late progenitors. Therefore, this study provides essential new insights into the mechanism by which EE increases hippocampal neurogenesis.
Materials and Methods
Animals and tamoxifen administration.
All experiments were approved by the Albert Einstein College of Medicine Institutional Animal Care and Use Committee. A maximum of five mice were housed in each cage in a 12 h light/dark cycle (8:00 A.M. to 8:00 P.M.) facility with free access to water and food. Mice with the Nestin-CreERT2 allele (Balordi and Fishell, 2007) were crossed with mice with conditional floxed Fgfr alleles, Fgfr1fx (Trokovic et al., 2003); Fgfr2fx (Yu et al., 2003), Fgfr3fx (Su et al., 2010), Fgfr1PLCγ (Partanen et al., 1998), and Fgfr1ΔFRS (Hoch and Soriano, 2006), to generate mutants and littermate controls. Fgfr1T2A-H2B-GFP; Fgfr2T2A-H2B-mCherry mice (Molotkov et al., 2017) were used to assess FGFR1 and FGR2 expression in the DG. Both males and females were used and showed no obvious differences; therefore, data for both were pooled. Two-month-old mutants and Cre– control littermates were administered an intraperitoneal injection of tamoxifen (5 mg/35 g body weight) dissolved in corn oil, once a day, every other day for a total of five doses, except see Figures 4 and 11C–I where all mice were of the same CreERT2;Fgfr1fx/fx;Fgfr2fx/fx;Fgfr3fx/fx genotype and only animals labeled as Fgfr-3KO mice received tamoxifen, while controls received corn oil. Mice were anesthetized with KetaSet and xylazine, perfused with 4% paraformaldehyde (PFA) 5 d or 2, 3, or 5 weeks post-tamoxifen injection for analysis, and were postfixed in 4% PFA overnight.
Figure 4.

FGFRs mediate the effects of EE on neurogenic cell expansion. All mice are of the same genotype (Nestin-CreERT2;Fgfr1fx/fx;Fgfr2fx/fx;Fgfr3fx/fx) and were administered either tamoxifen (Tam; +) or vehicle corn oil (–). A–G, Experimental timeline (A) for B–G. B–E, Immunostains of the DG of control (Ctrl; B, D) and Fgfr-3KO (C, E) mice in home cages (HCs; B, C) or EE (D, E). BrdU, White; DCX, red; DAPI, blue. Scale bar, 50 µm. F, Number of BrdU+DCX+ for mice in home cages (blue) or EE (red). Main effect of housing: F(1,29) = 127.1, p < 0.0001; main effect of genotype: F(1,29) = 39.97, p < 0.0001; interaction: F(1,29) = 39.36, p < 0.0001; ****p < 0.0001, **p = 0.0065 and 0.0086. G, Number of DCX+ immature neurons/late progenitor. Main effect of housing: F(1,29) = 91.94, p < 0.0001; main effect of genotype: F(1,29) = 1.171, p = 0.2882; interaction: F(1,29) = 0.8057, p = 0.3768; ****p < 0.0001. H–O, Experimental timeline (H) for I–O. I–L Immunostains of the DG of control (I, K) and Fgfr-3KO (J, L) mice in home cages (I, J) and EE (K, L). BrdU, White; DCX, red; GFAP, green; DAPI, blue. Scale bar, 50 µm. M, Number of BrdU+DCX– RGL/early progenitor cells. Main effect of housing: F(1,63) = 2874, p < 0.0001; main effect of genotype: F(1,63) = 8.604, p = 0.0047; interaction: F(1,63) = 2.366, p = 0.1290; *p = 0.0179, **p = 0.0012, ***p = 0.0002, ****p < 0.0001. N, Number of DCX+ cells. Main effect of housing: F(1,63) = 93.86, p < 0.0001; main effect of genotype: F(1,63) = 22.16, p < 0.0001; interaction: F(1,63) = 1.749, p = 0.0001; *p = 0.0111, ****p < 0.0001. O, Number of BrdU+DCX+ cells. Main effect of housing: F(1,63) = 28.26, p < 0.0001; main effect of genotype: F(1,63) = 13.39, p = 0.0005; interaction: F(1,63) = 0.1461, p = 0.7036; **p = 0.0028, 0.0013, and 0.0038, ****p < 0.0001. Each circle represents a mouse. Numbers of mice are indicated in each bar. Values are the mean ± SEM, using two-way ANOVA with Tukey's post hoc test.
Figure 11.

Phosphorylation of CREB, central to neural differentiation, is regulated by EE-induced FGFR signaling. Immunofluorescent staining for pERK showed no colocalization with DCX+ cells, only staining cells in the granule cell layer removed from the SGZ at 5 weeks post-tamoxifen (Tam) injection. A, A′, control (A) and enlarged view of boxed area in A (A′). B, B′, fr-3KO (B) and enlarged view of boxed area in B (B′). C–I, All mice are Nestin-CreER;Fgfr1fx/fx;Fgfr2fx/fx;Fgfr3fx/fx and were administered either tamoxifen (+) or corn oil vehicle (–). C–F, Immunostains for pCREB (green) and DCX (red) with DAPI (blue) of control and Fgfr-3KO mice in home cages (C, D) or in EE (E, F). G–I, Quantification of the fraction of DCX+ cells that are pCREB+. F, Control and Fgfr-3KO. Main effect of housing: F(1,16) = 0.1153, p = 0.7386; main effect of genotype: F(1,20) = 14.70, p = 0.0015; interaction: F(1,16) = 10.08, p = 0.0059; ***p = 0.0002. H, Control and FGFR-PLCγ. Main effect of housing: F(1,14) = 7.895, p = 0.0139; main effect of genotype: F(1,14) = 18.46, p = 0.0007; interaction: F(1,14) = 0.0866, p = 0.7729; ***p = 0.0009, *p = 0.0184, ***p = 0.0009. I, Control and FGFR-ΔFRS. Main effect of housing: F(1,14) = 2.286, p = 0.1528; main effect of genotype: F(1,14) = 2.105, p = 0.1688; interaction: F(1,14) = 0.1.375, p = 0.2606. Each circle represents a mouse (total number of mice is indicated in each bar). Two-way ANOVA with Tukey's post hoc test. Values are the mean ± SEM. HC, Home cage.
BrdU administration.
For labeling precursor cells in S-phase, bromodeoxyuridine (BrdU; 100 mg/kg body weight) was injected intraperitoneally twice daily for 2 or 3 consecutive days before collecting brain samples. For birthdating newly generated neurons, mice were injected with BrdU twice daily for 7 consecutive days starting the day after the last tamoxifen treatment. In no case did mice receiving BrdU exhibit weight loss or other overt signs of BrdU toxicity.
Enriched environment/exercise.
Mice were placed in EE for either 10 or 14 d (see Fig. 4, timelines). Males and females were placed in separate boxes. EE boxes were made of clear polycarbonate, 12 inches high, 45.5 inches long, and 21.5 inches wide, with two filtered air openings in the lid and nontransparent material on the bottom to prevent fear of heights. The boxes were placed on a stainless steel rack in a climate-controlled room on a 12 h light/dark cycle. Each box contained no more than 10 mice at a time, with two running wheels, three tunnels, a water and food station, two domes, standard bedding, and bedding squares. All toys and food stations were reorganized every day to promote exploration.
Immunohistofluorescent staining.
For cell counts, 30 µm vibratome (model VT1000S, Leica) sections were washed three times in PBS, blocked (10% normal goat serum, 1% bovine serum albumin, 100 mm glycine, 0.1% Triton X-100 in PBS) for 1 h at room temperature and incubated with primary antibodies overnight at 4˚C in 1% normal goat serum, 1% bovine serum albumin, and 0.1% Triton X-100 in PBS. For BrdU staining, floating sections were incubated with 2N HCl for 30 min at 37°C to denature DNA before the blocking step, washed in 0.1 m sodium borate, pH 8.5, for 20 min, washed with PBS, and blocked in goat serum. Samples were then washed in PBS and incubated with goat anti-mouse, anti-rabbit, anti-rat, or anti-guinea pig IgG(H + L) Alexa Fluor 488, 568, and 647 secondary antibodies (Thermo Fisher Scientific) for 1 h at room temperature. For phosphorylated (p)-ERK staining, brains were kept in 30% sucrose overnight, embedded in O.C.T. compound (catalog #4583, Sciagen) and frozen on dry ice. The 25 µm cryostat sections (catalog #CM3050S, Leica) were processed as above, except that sections were heated in 10 mm sodium citrate at pH 6.0 in a microwave oven for antigen retrieval, and the signal was amplified with a TSA (tyramide signal amplification) fluorescein system (catalog #NEL701001KT, PerkinElmer) according to the manufacturer recommendations. Secondary antibodies were goat anti-mouse HRP (1:1000; catalog #115–036-146, Jackson ImmunoResearch) and goat anti-rabbit HRP (1:1000; catalog #ab97080, Abcam). Sections for controls and mutants were matched along the anteroposterior axis for comparison. The primary antibodies used were as follows: mouse anti-NeuN (1:1000; catalog #MAB377, Millipore), rabbit anti-GFAP (1:1000; catalog #Z033401-2, Agilent), guinea pig anti-DCX (doublecortin; 1:4000; catalog #ab2253, Millipore), rat anti-BrdU (1:300; Accurate), rabbit anti-P-p44/p42 (p-ERK; 1:100; catalog #901S, Cell Signaling Technology), rabbit anti-GFAP (1:1000; catalog #Z0334, Dako), rabbit anti-p-CREB (Ser133; 1:100; catalog #9198S, Cell Signaling Technology), and rabbit anti-SOX2 (1:2000; catalog #ab5603, Millipore).
Imaging.
Sections were imaged by fluorescence microscopy (AxioSkop2, Zeiss) or confocal microscopy (Airyscan Confocal Microscope with Super-Resolution, model LSM880, Zeiss).
Cell counts.
A minimum of five 30 µm sections 180 µm apart were used to count cells along 450 µm of the dorsal blade in a minimum of four mice for each experimental group. Experimenter was blind to the genotypes for all cell counts. Counts were normalized to the length of the DG, as indicated in the figures. GFAP+ RGLs cells were quantified by counting the number of radial processes that spanned the granular layer of the DG (astrocytes were excluded from analysis on the basis of their stellate morphology and their lack of a radial process).
Neurosphere isolation and culture.
The 2-month-old Nestin-CreERT2;Fgfr1fx/fx;Fgfr2fx/fx;Fgfr3fx/fx mutant mice were anesthetized in isoflurane followed by cervical dislocation. The hippocampal dentate gyrus was dissected as previously described (Guo et al., 2012) with modifications. Briefly, the dentate gyrus was enzymatically dissociated using a Neural Tissue Dissociation Kit (P; catalog #130–092-628, Miltenyi Biotec) according to the manufacturer instructions. Primary neurospheres were cultured in proliferation medium containing the following: Neurobasal (catalog #21103–049, Thermo Fisher Scientific); B27 without vitamin A (catalog #12587–010, Thermo Fisher Scientific); GlutaMAX (catalog #35050061, Thermo Fisher Scientific); Penicillin-Streptomycin (catalog #15140–122, Thermo Fisher Scientific); 20 ng/ml FGF-2 (catalog #324831, EMD), and 20 ng/ml EGF (catalog #GF003, Millipore). FGF and EGF were replenished every 2 d. Cells were incubated at 5% CO2 and 37˚C. Neurospheres were passaged as previously described in the study by Azari et al. (2010) with modifications. Primary neurospheres were isolated 10–14 d after plating, spun down at 1000 rpm for 10 min, and dissociated with 0.125% trypsin-EDTA (catalog #25200–056, Thermo Fisher Scientific) for 15 min at 37˚C. Soybean trypsin inhibitor, 0.014% (w/v; catalog #17075–029, Thermo Fisher Scientific), was used to stop the reaction. Cells were spun down at 700 rpm for 5 min, dissociated in Advanced DMEM/F-12 (catalog #12634–010, Thermo Fisher Scientific); B27 without vitamin A; N-2 Supplement (100×; catalog #17502048, Thermo Fisher Scientific); GlutaMAX; Penicillin-Streptomycin; 20 ng/ml FGF-2; 20 ng/ml EGF; and heparin sodium salt (catalog #2106-10VL, Sigma-Aldrich). Cells were plated at 10,000 cells/cm2. FGF and EFG were replenished every 2 d. 4-Hydroxy-tamoxifen (1 μm; catalog #H7904, Sigma-Aldrich) or vehicle was added to neurosphere cultures 24 h before collection.
Passage 4 neurospheres were pelleted, washed twice with PBS, snap frozen on liquid nitrogen, and submitted to MS Bioworks for phosphoproteomics analysis.
Phosphoproteomics.
Samples were lysed, and 500 μg of protein lysates were reduced and trypsinized before solid-phase extraction. Phosphopeptides were enriched using Titansphere TiO2 tips (GL Sciences) using the vendor protocol. Mass spectrometry was performed by MS Bioworks according to their standard open-access protocol. Data were processed with MaxQuant version 1.6.0.13 (Max Planck Institute for Biochemistry, Martinsried, Germany) for calibration, protein/peptide identification with the Andromeda database, identification and localization of phosphorylation sites, filtering of search results at the 1% false discovery rate, and calculation of peak areas for detected proteins and peptides. The MaxQuant output was further processed using Perseus version 1.6.0.7 (Max Planck Institute for Biochemistry). Data were also analyzed for phosphosites that yielded >1.5-fold difference using ingenuity pathway analysis for molecular pathways, upstream and downstream genes, as well as major signaling pathways.
Experimental design and statistical analyses.
The experimental designs and timelines are illustrated within each figure in which they are relevant. Data analysis was performed using GraphPad Prism version 8.4.2. Two-tailed unpaired t tests or one-way or two-way ANOVA followed by Tukey's or Sidak post hoc tests, were performed, as appropriate, with a p value of < 0.05 considered significant. All experiments were repeated at least three times with independent biological samples. Experimenter was blind to the genotype during all numerical analyses. Data were expressed as the mean ± SEM. The exact N (number of animals) values are reported in parentheses on the bar graphs, while p values are reported in the figure legends.
Results
In the absence of EE, FGFRs are required for neurogenesis
Fgfr1, Fgfr2, and Fgfr3 are expressed in neural precursors throughout development (Guillemot and Zimmer, 2011; Hébert, 2011), and we found using Fgfr1 and Fgfr2 reporter alleles (Molotkov et al., 2017) that coexpression extends to adulthood with 96% of SOX2+ cells in the DG expressing Fgfr1 and Fgfr2 (Fig. 1). Although there was no available Fgfr3 reporter line or reliable anti-FGFR3 antibody to detect endogenous levels of protein in the adult DG, previous single-cell sequencing and genetic studies suggest that Fgfr3 is also expressed and functional in most (or all) active adult dentate gyrus stem and progenitor cells (Kang and Hébert, 2015; Artegiani et al., 2017). Given that Fgfr genes generally compensate for each other's deletion (Hébert, 2011), we simultaneously targeted all three receptors.
Figure 1.

FGFR1 and FGFR2 are coexpressed in neural precursors of the DG. A, Immunofluorescent staining of the DG showing colocalization of SOX2+ cells with fluorescently labeled reporters for Fgfr1 and Fgfr2 expression. B, Enlargement of boxed area in A; arrows point to SOX2+ cells colabeled for FGFR1-GFP and FGFR2-mCherry. C, Quantification of SOX2+ cells colocalizing only with FGFR1 (green; 3.5%), only with FGFR2 (red; 0.6%), or both (orange; 95.9%). n = 5 mice.
Before testing whether FGFRs mediate the effects of EE, we determined the length of time needed after gene deletion to detect reductions in neurogenic cell numbers under standard housing conditions. We conditionally deleted the Fgfr genes with a tamoxifen-inducible Nestin-CreERT2 driver that targets neural precursors, which we define as including both stem cells and more differentiated progenitor cells (Balordi and Fishell, 2007). At 5 d post-tamoxifen injection, Nestin-CreERT2;Fgfr1fx/fx;Fgfr2fx/fx;Fgfr3fx/fx mutants, hereafter called Fgfr-3KO mice, and littermate controls did not differ in RGL, progenitor cell, or new neuron numbers (Figs. 2A–C,O, 3A–D). However, by 2 weeks post-tamoxifen injection, proliferative RGLs and early type 2a progenitors, identified as positive for the DNA synthesis marker BrdU, negative for DCX, and located in the SGZ were reduced in Fgfr-3KO mutants (Fig. 2D–F,N). Identification of proliferating RGLs by colabeling for GFAP, a marker for radial fibers, and BrdU was not possible because of their mutually exclusive subcellular locations (Fig. 2G,H). Proliferative late type 2b progenitors (DCX+BrdU+ cells) were also reduced in mutants by ∼45% at 2 weeks post-tamoxifen injection (Fig. 2E,F,P).
Figure 2.
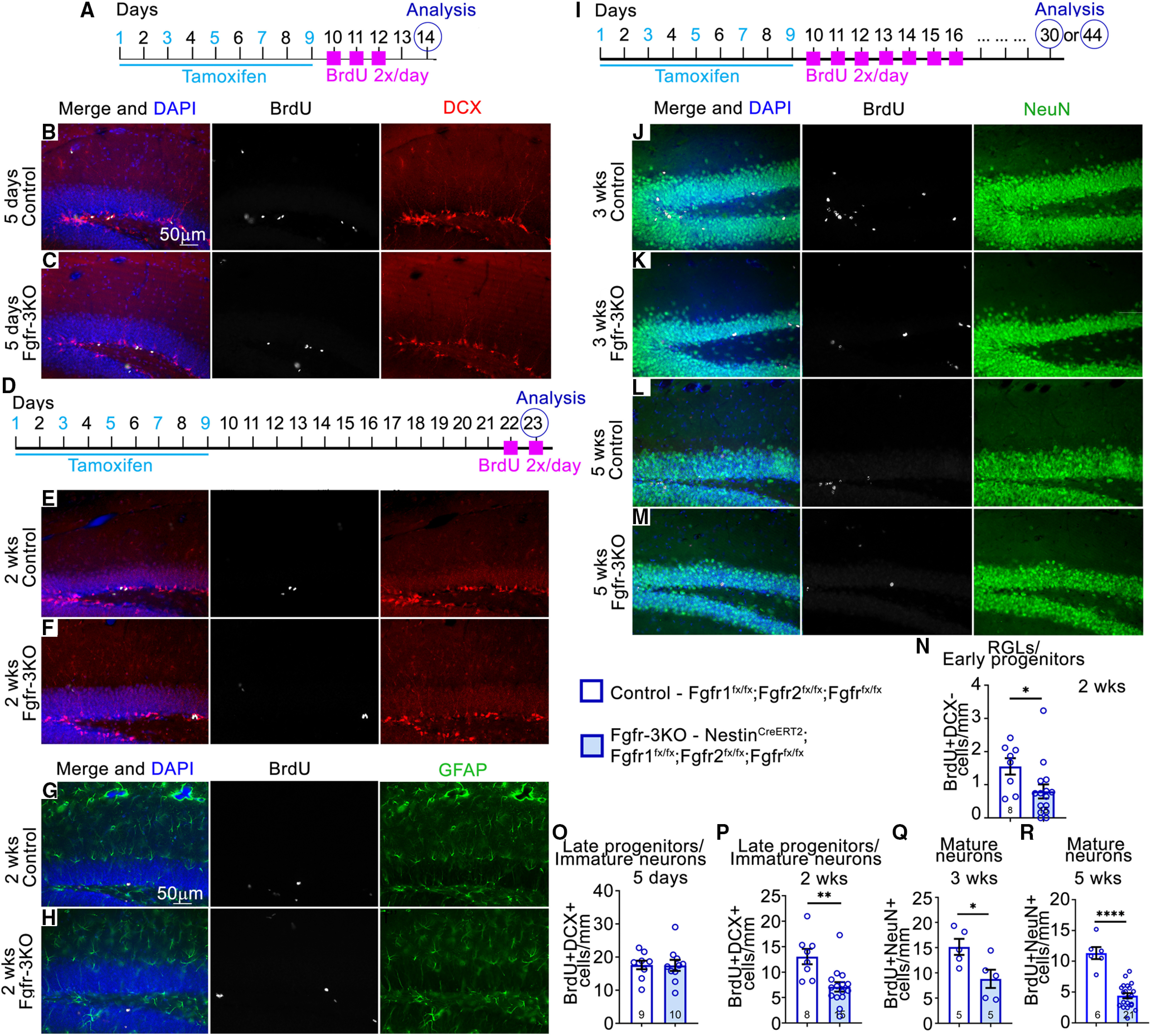
FGFRs are required early in the neurogenic cell lineage of the DG. A–R, Experimental timelines: A for B, C, O; D for E–H, N, P); and I for J–M, Q, R). B–M, Immunostains of the DG of mice in home cages, for control and Fgfr-3KO at 5 d (B, C), 2 weeks (E–H), 3 weeks (J, K), and 5 weeks (L, M) post-tamoxifen injection. In G, H: BrdU, white; DCX, red; DAPI, blue; GFAP, green; in J–M: NeuN, green. Scale bar, 50 µm. N–R, Number of BrdU+DCX– RGL/early progenitor cells 2 weeks post-tamoxifen injection (N), BrdU+DCX+ immature neurons/late progenitors 5 d post-tamoxifen injection (O), BrdU+DCX+ immature neurons/late progenitors 2 weeks post-tamoxifen injection (P), BrdU+NeuN+ mature neurons 3 weeks post-tamoxifen injection (Q), and BrdU+NeuN+ mature neurons 5 weeks post-tamoxifen injection (R). Two-tailed unpaired Student's t test: *p = 0.022 in N; **p = 0.006 in P; *p = 0.0303 in Q; ****p < 0.0001 in R. Each circle represents a mouse (total number of mice is indicated at the bottom of each bar). Values are the mean ± SEM. wks, Weeks.
Figure 3.
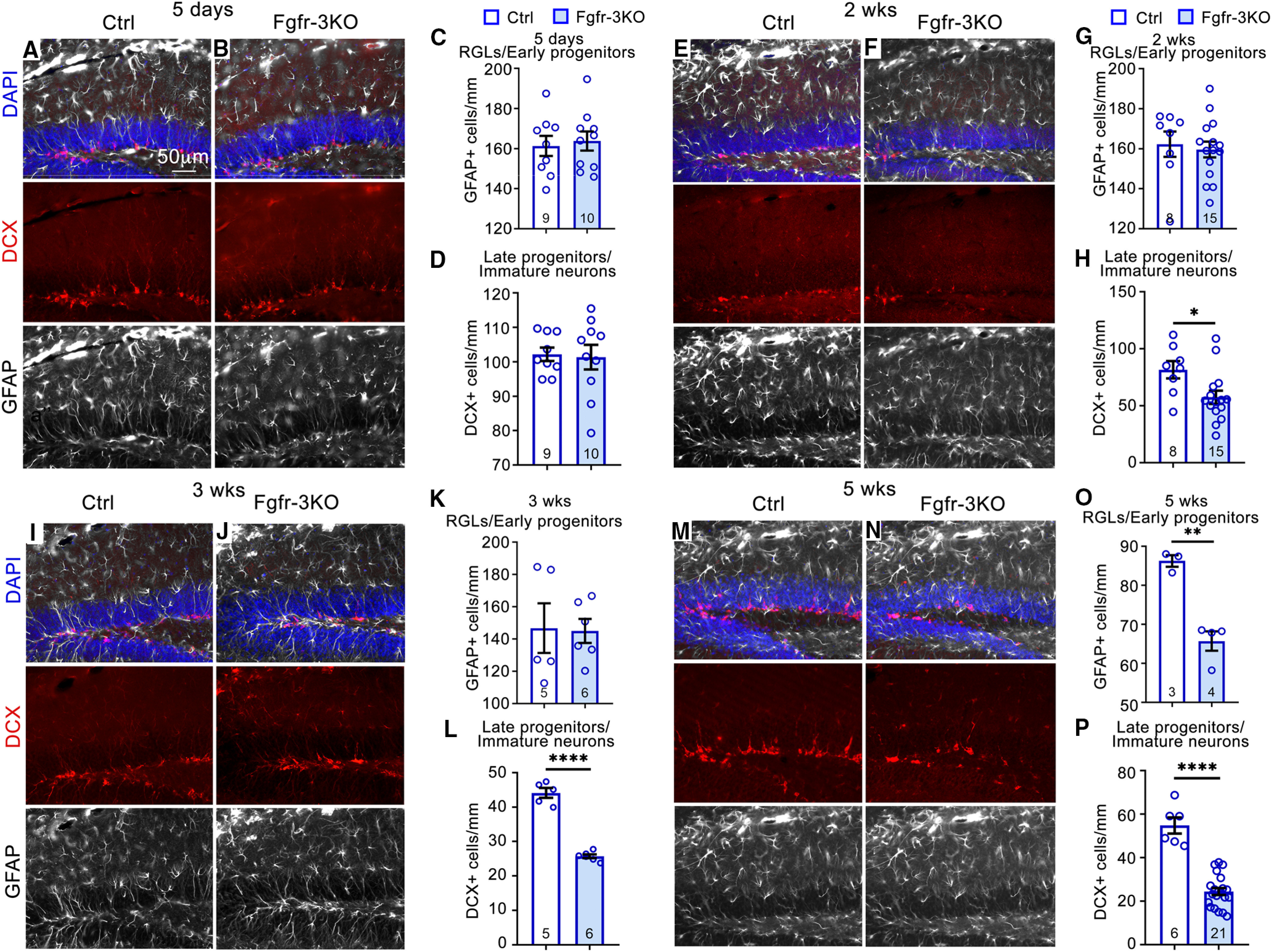
FGFRs are required to maintain DCX+ cells. A–N, Immunostains of the DG for control (Ctrl) and Fgfr-3KO mice kept in their home cages: 5 d (A, B), 2 weeks (G, H), 3 weeks (I, J), and 5 weeks (M, N) post-tamoxifen injection. DCX, Red; GFAP, white; DAPI, blue. Scale bar, 50 µm. C, D, Numbers of GFAP+ RGLs (C, G, K, O) and DCX+ cells (D, H, L, P) for control and Fgfr-3KO mice in home cages 5 d (C, D), 2 weeks (G, H), 3 weeks (K, L), and 5 weeks (O, P) post-tamoxifen injection. Two-tailed unpaired Student's t test: *p = 0.0211 in H; ****p < 0.0001 in L; **p = 0.0014 in O; and, ****p < 0.0001 in P. Each circle represents a mouse (total number of mice indicated at the bottom of each bar). Values are the mean ± SEM.
Total numbers of DCX+ cells (late progenitors and immature neurons) were also reduced by 2 weeks post-tamoxifen injection in mutants and remained low thereafter, decreasing to ∼60% by 5 weeks (Fig. 3E,F,H–J,L–N,P). In contrast, the total number of RGLs (GFAP+ cells in the SGZ with a radial process and without an astrocytic stellate morphology) was not affected until 5 weeks, at which time their numbers decreased (Fig. 3A–C,E–G,I–K,M–O), presumably because of their dependence on FGFR activity for maintenance (Kang and Hébert, 2015). The decreases in precursors resulted in 45% and 60% fewer new neurons (NeuN+ cells birthdated with BrdU) 3 and 5 weeks post-tamoxifen injection, respectively (Fig. 2I–M,Q,R). Together, these data indicate that in Fgfr-3KO mutants, it takes up to 2 weeks post-tamoxifen injection for the numbers of proliferating RGLs and type 2a progenitors, late progenitors, and total DCX+ cells to decrease, and up to 5 weeks for total RGL numbers to decrease.
FGFRs are required to mediate the effects of EE on neurogenic precursors
Expansion of neural precursors in the DG is heavily promoted by EE (Gonçalves et al., 2016; Gage, 2019). Since FGFRs are necessary and sufficient to promote neural precursor cell expansion in the DG (Figs. 2, 3; Kang and Hébert, 2015), we tested whether EE induces cell expansion through FGFRs. In a first experimental timeline, control and Fgfr-3KO mutants experienced EE for 10 d starting on the fourth day of a 9 d tamoxifen regimen (Fig. 4A). At 5 d post-tamoxifen injection (the last day of EE), there was a 3.5-fold increase in DCX+BrdU+ cells in control mice housed in EE versus home cages (Fig. 4B,D,F). However, in Fgfr-3KO mutants, the effect of EE on the number of DCX+BrdU+ cells was greatly stunted, with only a 1.6-fold increase (Fig. 4C,E,F). Since Fgfr-3KO mutants do not on their own exhibit a phenotype until 2 weeks post-tamoxifen injection (Figs. 2, 3), the stunted induction of neurogenesis under EE conditions in 5 d demonstrates that the EE effects require upregulating FGFR activity. In other words, for the EE effects to manifest, FGFR activity must likely be upregulated, not simply maintained.
The experimental timeline used above, in which mice begin to be exposed to EE before the completion of tamoxifen administration, meant that some effects of EE could have occurred before tamoxifen caused a more extensive loss of FGFRs. If so, then although at the time of analysis (the last day of EE) the immediate effect of EE on proliferating progenitors is reduced in the mutants (Fig. 4F), the longer-term effect on the total numbers of progenitors may not be detectable in mutants because of their expansion before sufficient loss of FGFRs. Indeed, with the experimental timeline used, the total numbers of DCX+ and GFAP+ cells were similar between controls and mutants (Figs. 4G, 5A–E).
Figure 5.
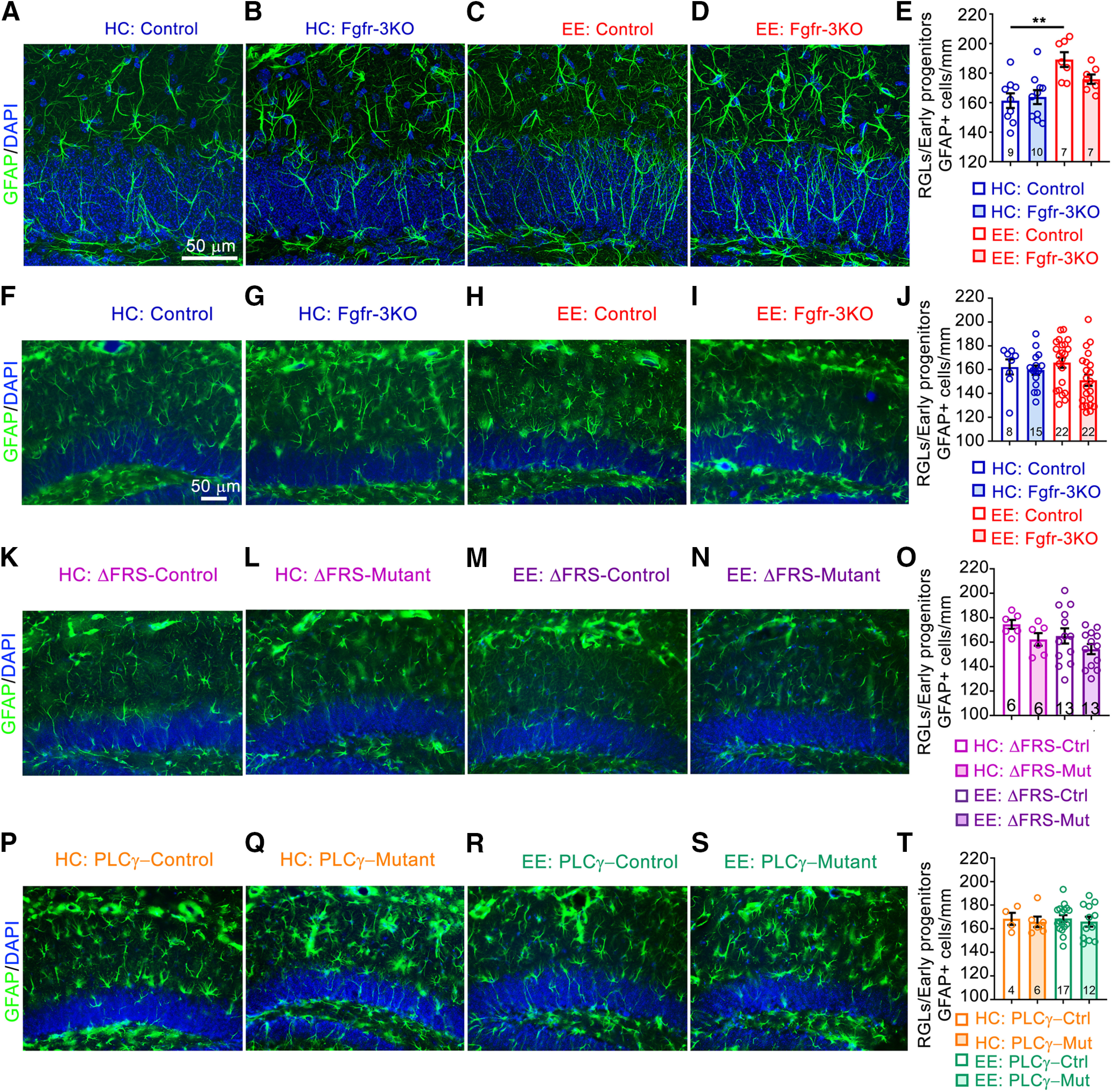
EE has only a transient effect on RGL cell numbers. A–D, Immunostains of the DG of mice kept in their home cages (HCs; A, B) or in an enriched environment (C, D), for control (Ctrl; A, C) and Fgfr-3KO (B, D) mice 5 d post-tamoxifen injection. GFAP, Green; DAPI, blue. E, Quantitation for A–D. Main effect of housing: F(1,29) = 17.56, p = 0.0002; main effect of genotype: F(1,29) = 1.280, p = 0.2671; interaction: F(1,29) = 2.700, p = 0.1111; **p = 0.0017. F–I, Immunostains of mice kept in their home cages (F, G) or an enriched environment (H, I) for Ctrl and Fgfr-3KO mice 2 weeks post-tamoxifen injection. GFAP, Green; DAPI, blue. Scale bar, 50 µm. J, Quantitation for F–I. K–N, Immunostains of mice kept in their home cages (K, L) or an enriched environment (M, N), for Ctrl (K, M) and Fgfr-ΔFRS (L, N) mice, after 2 weeks in enriched environment. GFAP, Green; DAPI, blue. Scale bar, 50 µm. O, Quantitation for K–N. P–S, Immunostains of mice kept in their home cages (P, Q) or an enriched environment (R, S), for Ctrl (P, R) and Fgfr-PLCγ mice (Q, S), 2 weeks post-tamoxifen injection. GFAP, Green; DAPI, blue. Scale bar, 50 µm. T, Quantitation for P–S. Each circle represents a mouse (total number of mice indicated at the bottom of each bar). Two-way ANOVA with Tukey's post hoc test. Values are the mean ± SEM. Scale bar, 50 µm. Mut, Mutant.
Therefore, to test whether FGFRs were required to mediate effects of EE on total precursor numbers (not just their proliferation; Fig. 4F), we devised a more protracted experimental timeline in which tamoxifen administration was completed before mice were placed in EE (Fig. 4H). In this case, not only were EE-induced increases in numbers of DCX+BrdU+ cells suppressed by the loss of FGFRs, but so were the numbers of total DCX+ cells (Fig. 4I–L,N,O). The induced increase in proliferating RGLs and type 2a progenitors (DCX–, BrdU+ cells in the SGZ) was also stunted (Fig. 4I–M), while total numbers of RGLs (GFAP+ cells with a radial fiber through the granule cell layer) were no longer different after EE with this protracted timeline even in controls (Fig. 5F–J), consistent with a transient effect of EE on the expansion of stem cells. Together, these findings demonstrate that FGFRs are required for expanding neurogenic precursors in response to EE.
As an alternative approach to identifying proliferative neural stem and progenitor cells, we also stained the DG soon after BrdU treatment for SOX2 and found similar results: the EE-induced increase in BrdU+SOX2+ cells was stunted in Fgfr-3KO mutants (Fig. 6A–F). To determine how many SOX2+ cells retained BrdU for extended periods, we stained the DG for SOX2 in mice that had been administered BrdU 30 d before collection. We found that although the number of SOX2+ cells with long-term retention of BrdU was very low, there were detectably fewer of them in Fgfr-3KO mice (Fig. 6G–J).
Figure 6.

Loss of FGFRs has a small effect on the quiescent stem cells. A–F, Experimental timeline (A) for B–F. A–F All mice are of the same Nestin-CreERT2;Fgfr1fx/fx;Fgfr2fx/fx;Fgfr3fx/fx genotype and were administered either tamoxifen (Tam; +) or corn oil (–). B–E. Immunostains of the DG of control (B, D) and Fgfr-3KO (C, E) mice in home cages (HCs; B, C) or EE (D, E). BrdU, Green; SOX2, red; DAPI, blue. Scale bar, 50 µm. F, Number of SOX2+BrdU+ cells for mice in home cages (blue) or EE (red); *p = 0.0279, **p = 0.0012, ****p < 0.0001. Main effect of housing: F(1,20) = 48.28, p < 0.0001; main effect of genotype: F(1,20) = 7.20, p = 0.0143; interaction: F(1,20) = 1.173, p = 0.2916. Each circle represents a mouse. Numbers of mice are indicated in each bar. Values are the mean ± SEM, using two-way ANOVA with Tukey's post hoc test. G, Experimental timeline for H–J. H–I Immunostains of the DG of control (H) and Fgfr-3KO (I) mice. SOX2, Red; BrdU, green; DAPI, blue. Scale bar, 50 µm. J, Number of SOX+BrdU+ cells. *p = 0.0129, two-tailed Student's t test, values are the mean ± SEM.
FGFRs mediate the effects of EE by signaling through FRS, phospholipase Cγ, and CREB
FGFRs can directly bind to and signal through FRS proteins and phospholipase Cγ (PLCγ; Goetz and Mohammadi, 2013). Given that a single wild-type allele of Fgfr1 in an otherwise FGFR-deficient background is largely sufficient to rescue decreased numbers of DCX+ cells in the DG (Fig. 7), we tested the requirement for FGFR signaling through FRS and PLCγ by swapping the single wild-type allele for either an Fgfr1ΔFRS allele (with which FGFR1 signaling through FRS is specifically blocked; Hoch and Soriano, 2006) or an Fgfr1Y766F al lele (with which signaling through PLCγ is specifically blocked; Partanen et al., 1998) in an other wise FGFR-deficient background. These Nestin-CreERT2;Fgfr1fx/ΔFRS;Fgfr2fx/fx;Fgfr3fx/fx mice, hereafter called Fgfr-ΔFRS mutants, Nestin-CreERT2;Fgfr1fx/Y766F;Fgfr2fx/fx;Fgfr3fx/fx mice, hereafter Fgfr-PLCγ mutants, and their respective control littermates underwent the same protracted treatment schedule as Fgfr-3KO mutants (Fig. 4H).
Figure 7.

A single FGFR allele largely rescues the Fgfr-3KO phenotype. A–C, Immunostains of the DG of mice in home cages, control (A) Fgfr-3KO (B), and 3KO-FGFR1fx/+ mice (Nestin-CreERT2;Fgfr1fx/+;Fgfr2fx/fx;Fgfr3fx/fx; C). D, Total numbers of immature neurons/late progenitors for Control, Fgfr-3KO, and 3KO-Fgfr1fx/+, 5 weeks post-tamoxifen injection; F(2,14) = 35.84, p < 0.0001; ****p < 0.0001; ***p = 0.0006; **p = 0.0063. Each circle represents a mouse (total number of mice is indicated at the bottom of each bar). One-way ANOVA with Tukey's post hoc test. Values are the mean ± SEM.
Fgfr-ΔFRS mutants failed to mediate EE-induced RGL proliferation and reduced the effect of EE on total and proliferating DCX+ cells (Fig. 8). In contrast, Fgfr-PLCγ mutants mildly, if at all, blunted the effect of EE on RGL and DCX+ cell proliferation, and greatly inhibited the effect of EE on the total number of DCX+ cells (Fig. 9). As for Fgfr-3KO mutants, total GFAP+ RGL numbers in Fgfr-ΔFRS and Fgfr-PLCγ mutants and controls did not differ using the protracted timeline (Fig. 5K–T). Because RGL and early progenitor proliferation failed to be induced in response to EE in Fgfr-ΔFRS mutants, our findings suggest that FRS mediates the FGFR-dependent effects of EE on precursor proliferation, although they do not exclude the possibility that FGFR1–FRS signaling also mediates survival of late progenitors and immature neurons. On the other hand, since the proliferative effect of EE was mildly if at all affected in Fgfr-PLCγ mutants while the effect on increased DCX+ cells was greatly inhibited, our findings also suggest that PLCγ preferentially mediates survival rather than proliferation of DCX+ cells.
Figure 8.

FGFR signaling through FRS is required for the effects of EE on early and late precursors. A–H, Experimental timeline (A) for B–H. All mice received tamoxifen. B–E. Immunostains of the DG of mice in home cages (HCs; B, C), and EE (D, E), control (Ctrl; B, D), and Fgfr-ΔFRS (C, E). BrdU, White; DCX, red; DAPI, blue. Scale bar, 50 µm. F, Number of BrdU+DCX– RGL/early progenitor cells for control and Fgfr-ΔFRS mice in home cages (pink) and enriched environment (purple). Main effect of housing: F(1,33) = 5.241, p = 0.0286; main effect of genotype: F(1,33) = 2.976, p = 0.0939; interaction: F(1,33) = 0.0220, p = 0.0220; *p = 0.012 and 0.0384, **p = 0.0051. G, Number of DCX+ immature neurons/late progenitors. Main effect of housing: F(1,34) = 25.29, p < 0.0001; main effect of genotype: F(1,34) = 16.29, p = 0.0003; interaction: F(1,34) = 0.0438, p = 0.8355; **p = 0.0088, 0.0040, and 0.0033, ****p < 0.0001. H, Number of DCX+BrdU+ cells. Main effect of housing: F(1,33) = 20.93, p < 0.0001; main effect of genotype: F(1,33) = 3.980, p = 0.0541; interaction: F(1,33) = 0.165, p = 0.2804; *p = 0.0283, **p = 0.0064, ***p = 0.0003.
Figure 9.

FGFR signaling through PLCγ is required for the effects of EE on late precursors. A–H, Experimental timeline (A) for B–H. All mice received tamoxifen. B–E, Immunostains of the DG of mice in home cages (HCs; B, C); and EE (D, E), control (B, D), and Fgfr-PLCγ (C, E). BrdU, White; DCX, red; DAPI, blue. Scale bar, 50 µm. F, Number of BrdU+DCX– RGL/early progenitor cells for control and Fgfr-PLCγ mice in home cages (orange) and EE (green). Main effect of housing: F(1,35) = 11.47, p = 0.0018; main effect of genotype: F(1,35) = 1.076, p = 0.3068; interaction: F(1,35) = 0.00177, p = 0.9667; **p = 0.0071. G, Number of DCX+ immature neurons/late progenitors; main effect of housing: F(1,35) = 30.36, p < 0.0001; main effect of genotype: F(1,35) = 11.67, p = 0.0016; interaction: F(1,35) = 2.738, p = 0.1070; *p = 0.0324, ***p = 0.0002, ****p < 0.0001. H, Number of DCX+BrdU+ cells. Main effect of housing: F(1,35) = 12.87, p = 0.0010; main effect of genotype: F(1,35) = 5.421, p = 0.0258; interaction: F(1,35) = 0.0204, p = 0.8874; *p = 0.0404, ***p = 0.0003. Each circle represents a mouse (total number of mice is indicated at the bottom of each bar). Two-way ANOVA with Tukey's post hoc test. Values are the mean ± SEM.
To further understand the molecular pathways through which FGFR activity mediated by FRS and PLCγ promote DG neurogenesis, we performed a phosphoproteomics screen comparing SGZ-derived control and Fgfr-3KO neurospheres. Consistent with an active role for FGFRs in neurogenesis, the screen revealed 1047 differentially (>1.5-fold) phosphorylated protein sites (Fig. 10), a variety and number of which made identifying functionally relevant transducers or pathways indecipherable. Hence, we turned to a candidate approach. We initially examined ERK phosphorylation because it is a canonical mediator of extracellular receptor kinases, including FGFRs, in progenitor cells (Hébert, 2011; Ornitz and Itoh, 2015; Brewer et al., 2016; Mossahebi-Mohammadi et al., 2020). Surprisingly, p-ERK could not be detected in neural precursors, only in sparse granule cells away from the SGZ (Fig. 11A–B′). A phospho-protein central to early differentiation steps of DCX+ progenitors in the adult DG is pCREB (Nakagawa et al., 2002a,b; Merz et al., 2011). Under EE conditions, the fraction of DCX+ cells that were pCREB+ was reduced in Fgfr-3KO mutants compared with controls (Fig. 11C–G). Consistent with PLCγ mediating FGFR activity in DCX+ cells, the effects on pCREB in mutants were predominantly recapitulated in the Fgfr-PLCγ rather than Fgfr-ΔFRS mutants (Fig. 11H,I).
Figure 10.
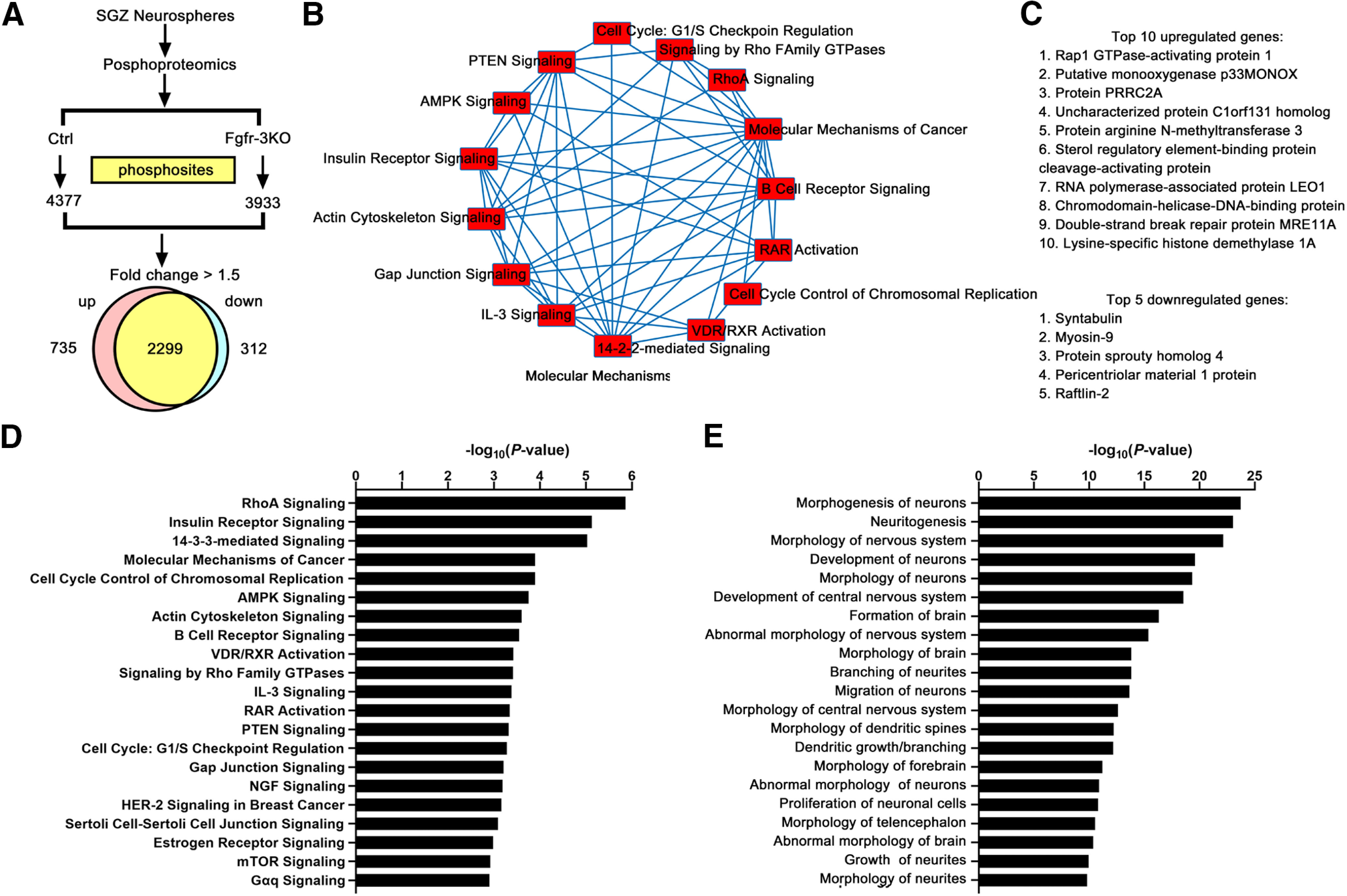
Phosphoproteomics analysis of Fgfr-3KO mice. A, Numbers of phosphosites in control and Fgfr-3KO SGZ neurospheres and the number of genes with phosphorylated sites that were upregulated or downregulated at least 1.5-fold. To reduce variability, neurosphere preparations with the same triple-mutant genotype were split in two, with one-half receiving 4-HO-tamoxifen for 24 h (Fgfr-3KO), and the other half receiving no 4-HO-tamoxifen (Control). B, Top, Fifteen molecular pathways with differentially phosphorylated proteins. C, Top 10 upregulated and top 5 downregulated genes. D, Major signaling pathways identified for all tissue types. E, Nervous system-specific categories with p values > 2.5. AMPK, Adenosine 5′ monophosphate-activated protein kinase; RAR, retinoic acid receptor; RXR, retinoic X receptor.
Discussion
In this study, we disrupt the function of all three expressed FGFR genes in neurogenic precursors to test the role of FGF signaling in mediating the effects of EE on adult hippocampal neurogenesis. Although we did not examine single and double Fgfr mutants, our finding that a single wild-type Fgfr1 allele largely compensates for the loss of all the other Fgfr alleles in Nestin-CreER-driven Fgfr-3KO mice is consistent with a need for disrupting both alleles of all three receptors before observing a phenotype (Fig. 7; Grońska-Pęski et al., 2021). Note that the phenotypic rescue in this study and our accompanying study by a single Fgfr allele also underscores the efficiency of the Nestin-CreER transgene in driving the loss of most or all FGFR activity in the triple homozygous Fgfr mutant mice. Our study thus demonstrates that EE-induced neurogenesis is dependent on FGFR activity, presumably through all three receptors.
The requirement for FGFR activity in mediating the effects of EE could in principle be because of either a need to maintain FGFR activity or, instead, a need to increase FGFR activity. The latter possibility appears more likely for two reasons. First, levels of FGF2 are upregulated in the hippocampal neurogenic niche with EE and exercise (Gómez-Pinilla et al., 1998; Molteni et al., 2002). And second, the dependence on FGFRs for increased EE-induced neurogenesis is observed within 5 d after tamoxifen-mediated deletion of the Fgfr alleles (Fig. 4), a time before a reduction in neurogenesis can be detected in Fgfr-3KO mice kept in their home cage (where a reduction in neurogenesis is only observed at 2 weeks post-tamoxifen injection; Figs. 2, 3). This suggests that at 5 d post-tamoxifen injection, despite efficient DNA recombination at the Fgfr loci, baseline levels of FGFR activity are still present (due presumably to mRNA or protein perdurance), but that an increase in activity would no longer be possible. Thus, an increase in FGFR activity in response to EE appears necessary to increase neurogenesis.
In an attempt to further define the precursors that respond to EE in an FGF-dependent manner, we first examined the numbers of SOX2+ cells that retained BrdU for 30 d as a potential marker of quiescent stem cells for mice in their home cages. This analysis showed that although the numbers of BrdU+SOX2+ cells 30 d after BrdU administration were detectably fewer in Fgfr-3KO mutants, they were very few in number to begin with (Fig. 6G–J). Given that SOX2 is not strictly a marker for proliferating neural precursors and is instead also expressed in hippocampal OPCs by single-cell sequencing (Artegiani et al., 2017) and by immunostaining (Zhang et al., 2018) as well as in S100+ astrocytes (Kang and Hébert, 2012), our use of SOX2 and long-term BrdU retention as markers for quiescent stem cells would likely overestimate their numbers, making it difficult to detect and interpret differences in the small numbers of cells in response to EE, even using other markers for long-term quiescence [e.g., BrdU labeling followed 30 d later by EdU (5-ethynyl-2'- deoxyuridine) labeling]. Hence, in this study, we failed to assess the role of FGF signaling in mediating a response specifically on quiescent stem cells. Nevertheless, our data demonstrate an FGF dependency for the overall response by RGLs and early progenitors to EE (Figs. 4, 8).
In examining the downstream effectors of FGFR activity in mediating the response to EE, we found that this response was attenuated in both the Fgfr-ΔFRS and Fgfr-PLCγ mutants. However, the phenotypes were different, with Fgfr-ΔFRS mutants revealing a role in promoting proliferation in response to EE, and Fgfr-PLCγ mutants revealing more of a role in survival (Figs. 8, 9). Therefore, the FGFR-dependent response of neurogenic cells to EE occurs at least in part through increased proliferation of precursor cells, but may also include a positive effect on cell survival at later stages of the neurogenic lineage when considerable cell death is known to occur (Gonçalves et al., 2016).
Exercise is generally thought to have a stronger proproliferation effect while enrichment is thought to have a stronger prosurvival effect on neurogenesis in the adult DG (Benarroch, 2013; Gonçalves et al., 2016). The established EE protocol used in our study makes it impossible to separate the effects of exercise from enrichment because the larger cage and exploration opportunities necessarily result in an increase in voluntary physical activity. Thus, our use of EE, which we defined up front as a combination of exercise and environmental enrichment, does not distinguish between the potential effects of exercise on cell proliferation and of enrichment on cell survival. Nevertheless, the differing effects observed on proliferation and survival in Fgfr-ΔFRS and Fgfr-PLCγ mutants are consistent with FGFRs mediating the effects of both exercise and enrichment on both cell proliferation and survival.
Interestingly, the EE-induced increase in phosphorylated CREB in DCX+ cells is lost in the Fgfr-PLCγ mutants but not the Fgfr-ΔFRS mutants (Fig. 11H,I), suggesting that Fgfr-PLCγ may signal through CREB to promote the survival of DCX+ cells, consistent with previous reports on the role of CREB in hippocampal neurogenesis (Jagasia et al., 2009). It is worth noting that basal levels of pCREB in the control mice for the different genotypes appeared to differ (Fig. 11G–I). These differences in basal pCREB could be strain specific since each of these multiallele lines were by necessity in mixed backgrounds, underscoring the importance of having littermate controls. Alternatively, the Fgfr1-PLCγ and Fgfr1-ΔFRS alleles, which are not conditional and therefore heterozygous throughout the mice, may have contributed to the observed baseline differences.
FGFRs are not likely to be the only receptors mediating the effects of EE by signaling through FRS, PLCγ, and CREB. TrkB activity, which also signals through these same effectors, has also been implicated in mediating the effects of EE. The TRK pathway may be nonredundantly required with the FGFR pathway to promote the full effects of EE, or the TRK pathway may preferentially act at later stages in the lineage to further promote dendritogenesis and neuronal survival (Wang et al., 2015). Further studies will be required to disentangle interactions among FGF, BDNF, and their downstream effectors in mediating the effects of EE. In sum, our findings indicate that EE induces stem and progenitor cell expansion in the DG largely through FGFR activation of FRS and PLCγ, which in turn act at least in part through CREB to increase neurogenesis.
Footnotes
This research was supported by The Einstein Training Program in Stem Cell Research from the Empire State Stem Cell Fund through New York State Department of Health Contract C34874GG (M.G.-P.), New York State Stem Cell Science Contract C32567GG (J.M.H.), and National Institutes of Health Grant MH-070596 (J.M.H.). We thank Philippe Soriano for providing Fgfr reporter mice.
The authors declare no competing financial interests.
References
- Artegiani B, Lyubimova A, Muraro M, van Es JH, van Oudenaarden A, Clevers H (2017) A single-cell RNA sequencing study reveals cellular and molecular dynamics of the hippocampal neurogenic niche. Cell Rep 21:3271–3284. 10.1016/j.celrep.2017.11.050 [DOI] [PubMed] [Google Scholar]
- Azari H, Rahman M, Sharififar S, Reynolds BA (2010) Isolation and expansion of the adult mouse neural stem cells using the neurosphere assay. J Vis Exp. Advance online publication. Retrieved Nov 20, 2010. doi: 10.3791/2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balordi F, Fishell G (2007) Mosaic removal of hedgehog signaling in the adult SVZ reveals that the residual wild-type stem cells have a limited capacity for self-renewal. J Neurosci 27:14248–14259. 10.1523/JNEUROSCI.4531-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE (2013) Adult neurogenesis in the dentate gyrus: general concepts and potential implications. Neurology 81:1443–1452. 10.1212/WNL.0b013e3182a9a156 [DOI] [PubMed] [Google Scholar]
- Brewer JR, Mazot P, Soriano P (2016) Genetic insights into the mechanisms of FGF signaling. Genes Dev 30:751–771. 10.1101/gad.277137.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH (2019) Adult neurogenesis in mammals. Science 364:827–828. 10.1126/science.aav6885 [DOI] [PubMed] [Google Scholar]
- Gobeske KT, Das S, Bonaguidi MA, Weiss C, Radulovic J, Disterhoft JF, Kessler JA (2009) BMP signaling mediates effects of exercise on hippocampal neurogenesis and cognition in mice. PLoS One 4:e7506. 10.1371/journal.pone.0007506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz R, Mohammadi M (2013) Exploring mechanisms of FGF signalling through the lens of structural biology. Nat Rev Mol Cell Biol 14:166–180. 10.1038/nrm3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Pinilla F, So V, Kesslak JP (1998) Spatial learning and physical activity contribute to the induction of fibroblast growth factor: neural substrates for increased cognition associated with exercise. Neuroscience 85:53–61. 10.1016/s0306-4522(97)00576-9 [DOI] [PubMed] [Google Scholar]
- Gonçalves JT, Schafer ST, Gage FH (2016) Adult neurogenesis in the hippocampus: from stem cells to behavior. Cell 167:897–914. 10.1016/j.cell.2016.10.021 [DOI] [PubMed] [Google Scholar]
- Grońska-Pęski M, Mowrey W, Hébert JM (2021) FGFR regulation of dendrite elaboration in adult-born granule cells depends on intracellular mediator and proximity to the soma. Neuroscience 453:148–167. 10.1016/j.neuroscience.2020.10.024 [DOI] [PubMed] [Google Scholar]
- Guillemot F, Zimmer C (2011) From cradle to grave: the multiple roles of fibroblast growth factors in neural development. Neuron 71:574–588. 10.1016/j.neuron.2011.08.002 [DOI] [PubMed] [Google Scholar]
- Guo W, Patzlaff NE, Jobe EM, Zhao X (2012) Isolation of multipotent neural stem/progenitor cells from both the dentate gyrus and subventricular zone of a single adult mouse. Nat Protoc 7:2005–2012. 10.1038/nprot.2012.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert JM (2011) FGFs: neurodevelopment's Jack-of-all-Trades - how do they do it? Front Neurosci 5:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch RV, Soriano P (2006) Context-specific requirements for Fgfr1 signaling through Frs2 and Frs3 during mouse development. Development 133:663–673. 10.1242/dev.02242 [DOI] [PubMed] [Google Scholar]
- Jagasia R, Steib K, Englberger E, Herold S, Faus-Kessler T, Saxe M, Gage FH, Song H, Lie DC (2009) GABA-cAMP response element-binding protein signaling regulates maturation and survival of newly generated neurons in the adult hippocampus. J Neurosci 29:7966–7977. 10.1523/JNEUROSCI.1054-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W, Hébert JM (2012) A Sox2 BAC transgenic approach for targeting adult neural stem cells. PLoS One 7:e49038. 10.1371/journal.pone.0049038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W, Hébert JM (2015) FGF signaling is necessary for neurogenesis in young mice and sufficient to reverse its decline in old mice. J Neurosci 35:10217–10223. 10.1523/JNEUROSCI.1469-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby ED, Muroy SE, Sun WG, Covarrubias D, Leong MJ, Barchas LA, Kaufer D (2013) Acute stress enhances adult rat hippocampal neurogenesis and activation of newborn neurons via secreted astrocytic FGF2. Elife 2:e00362. 10.7554/eLife.00362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH (1997) More hippocampal neurons in adult mice living in an enriched environment. Nature 386:493–495. 10.1038/386493a0 [DOI] [PubMed] [Google Scholar]
- Li Y, Luikart BW, Birnbaum S, Chen J, Kwon CH, Kernie SG, Bassel-Duby R, Parada LF (2008) TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 59:399–412. 10.1016/j.neuron.2008.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PZ, Nusslock R (2018) Exercise-mediated neurogenesis in the hippocampus via BDNF. Front Neurosci 12:52. 10.3389/fnins.2018.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Zang T, Birnbaum SG, Wang Z, Johnson JE, Zhang CL, Parada LF (2017) TrkB dependent adult hippocampal progenitor differentiation mediates sustained ketamine antidepressant response. Nat Commun 8:1668. 10.1038/s41467-017-01709-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz K, Herold S, Lie DC (2011) CREB in adult neurogenesis–master and partner in the development of adult-born neurons? Eur J Neurosci 33:1078–1086. 10.1111/j.1460-9568.2011.07606.x [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H (2011) Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70:687–702. 10.1016/j.neuron.2011.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molotkov A, Mazot P, Brewer JR, Cinalli RM, Soriano P (2017) Distinct requirements for FGFR1 and FGFR2 in primitive endoderm development and exit from pluripotency. Dev Cell 41:511–526.e4. 10.1016/j.devcel.2017.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossahebi-Mohammadi M, Quan M, Zhang JS, Li X (2020) FGF signaling pathway: a key regulator of stem cell pluripotency. Front Cell Dev Biol 8:79. 10.3389/fcell.2020.00079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molteni R, Ying Z, Gómez-Pinilla F (2002) Differential effects of acute and chronic exercise on plasticity-related genes in the rat hippocampus revealed by microarray. Eur J Neurosci 16:1107–1116. 10.1046/j.1460-9568.2002.02158.x [DOI] [PubMed] [Google Scholar]
- Mudò G, Bonomo A, Di Liberto V, Frinchi M, Fuxe K, Belluardo N (2009) The FGF-2/FGFRs neurotrophic system promotes neurogenesis in the adult brain. J Neural Transm 116:995–1005. 10.1007/s00702-009-0207-z [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Kim JE, Lee R, Malberg JE, Chen JS, Steffen C, Zhang YJ, Nestler EJ, Duman RS (2002a) Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J Neurosci 22:3673–3682. 10.1523/JNEUROSCI.22-09-03673.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Kim JE, Lee R, Chen J, Fujioka T, Malberg J, Tsuji S, Duman RS (2002b) Localization of phosphorylated cAMP response element-binding protein in immature neurons of adult hippocampus. J Neurosci 22:9868–9876. 10.1523/JNEUROSCI.22-22-09868.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz D, Itoh N (2015) The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol 4:215–266. 10.1002/wdev.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partanen J, Schwartz L, Rossant J (1998) Opposite phenotypes of hypomorphic and Y766 phosphorylation site mutations reveal a function for Fgfr1 in anteroposterior patterning of mouse embryos. Genes Dev 12:2332–2344. 10.1101/gad.12.15.2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su N, Xu X, Li C, He Q, Zhao L, Li C, Chen S, Luo F, Yi L, Du X, Huang H, Deng C, Chen L (2010) Generation of Fgfr3 conditional knockout mice. Int J Biol Sci 6:327–332. 10.7150/ijbs.6.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trokovic R, Trokovic N, Hernesniemi S, Pirvola U, Weisenhorn DMV, Rossant J, McMahon AP, Wurst W, Partanen J (2003) FGFR1 is independently required in both developing mid- and hindbrain for sustained response to isthmic signals. EMBO J 22:1811–1823. 10.1093/emboj/cdg169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H, Kempermann G, Gage FH (1999) Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci 2:266–270. 10.1038/6368 [DOI] [PubMed] [Google Scholar]
- Vukovic J, Colditz MJ, Blackmore DG, Ruitenberg MJ, Bartlett PF (2012) Microglia modulate hippocampal neural precursor activity in response to exercise and aging. J Neurosci 32:6435–6443. 10.1523/JNEUROSCI.5925-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Chang X, She L, Xu D, Huang W, Poo MM (2015) Autocrine action of BDNF on dendrite development of adult-born hippocampal neurons. J Neurosci 35:8384–8393. 10.1523/JNEUROSCI.4682-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM (2003) Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130:3063–3074. 10.1242/dev.00491 [DOI] [PubMed] [Google Scholar]
- Zhang S, Zhu X, Gui X, Croteau C, Song L, Xu J, Wang A, Bannerman P, Guo F (2018) Sox2 is essential for oligodendroglial proliferation and differentiation during postnatal brain myelination and CNS remyelination. J Neurosci 38:1802–1820. 10.1523/JNEUROSCI.1291-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]