

Abstract
Interleukin-4 (IL-4) is an anti-inflammatory cytokine, which can be protective in inflammatory and neurologic disorders, and can alleviate pain. Classically, IL-4 diminishes pain by blocking the production of proinflammatory cytokines. Here, we uncovered that IL-4 induces acute antinociception by IL-4 receptor α (IL-4Rα)-dependent release of opioid peptides from M1 macrophages at injured nerves. As a model of pathologic pain, we used a chronic constriction injury (CCI) of the sciatic nerve in male mice. A single application of IL-4 at the injured nerves (14 d following CCI) attenuated mechanical hypersensitivity evaluated by von Frey filaments, which was reversed by co-injected antibody to IL-4Rα, antibodies to opioid peptides such as Met-enkephalin (ENK), β-endorphin and dynorphin A 1–17, and selective antagonists of δ-opioid, µ-opioid, and κ-opioid receptors. Injured nerves were predominately infiltrated by proinflammatory M1 macrophages and IL-4 did not change their numbers or the phenotype, assessed by flow cytometry and qRT-PCR, respectively. Macrophages isolated from damaged nerves by immunomagnetic separation (IMS) and stimulated with IL-4 dose dependently secreted all three opioid peptides measured by immunoassays. The IL-4-induced release of ENK was diminished by IL-4Rα antibody, intracellular Ca2+ chelator, and inhibitors of protein kinase A (PKA), phosphoinositide 3-kinase (PI3K), and ryanodine receptors. Together, we identified a new opioid mechanism underlying the IL-4-induced antinociception that involves PKA-mediated, PI3K-mediated, ryanodine receptor-mediated, and intracellular Ca2+-mediated release from M1 macrophages of opioid peptides, which activate peripheral opioid receptors in injured tissue.
SIGNIFICANCE STATEMENT Interleukin-4 (IL-4) is an anti-inflammatory cytokine, which can ameliorate pain. The IL-4-mediated effects are considered to mostly result from the inhibition of the production of proinflammatory mediators (e.g., IL-1β, tumor necrosis factor, prostaglandin E2). Here, we found that IL-4 injected at the injured nerves attenuates pain by releasing opioid peptides from the infiltrating macrophages in mice. The opioids were secreted by IL-4 in the intracellular Ca2+-dependent manner and activated local peripheral opioid receptors. These actions represent a novel mode of IL-4 action, since its releasing properties have not been so far reported. Importantly, our findings suggest that the IL-4–opioid system should be targeted in the peripheral damaged tissue, since this can be devoid of central and systemic side effects.
Keywords: analgesia, antinociception, calcium-mediated release, neuropathic pain, opioids, peripheral opioid receptors
Introduction
Interleukin-4 (IL-4) is an important anti-inflammatory cytokine produced by T helper 2 lymphocytes, mast cells, eosinophils, basophils, and macrophages. It is involved in the regulation of immune responses and has been shown to ameliorate arthritis, improve memory, exert anti-cancer and protective actions in neurologic disorders (encephalomyelitis, Alzheimer's disease, spinal cord injury) in animal models (van Roon et al., 2001; Gadani et al., 2012; Luzina et al., 2012), and decrease inflammation associated with psoriasis in humans (Ghoreschi et al., 2003).
IL-4 is also involved in the modulation of pain. Its blood levels were diminished in patients with chronic widespread pain and tended to be lower in patients with painful than with painless peripheral neuropathies (Üçeyler et al., 2006, 2007). IL-4 knock-out mice developed mechanical hypersensitivity (Üçeyler et al., 2011), whereas in wild-type mice, IL-4 injected into the paw attenuated mechanical hypersensitivity induced by carrageenan, bradykinin, or tumor necrosis factor (TNF; Cunha et al., 1999). IL-4 applied intraperitoneally reduced visceral nociception induced by acetic acid or zymosan in mice, and attenuated mechanical hypersensitivity in the zymosan-induced knee joint inflammation in rats (Vale et al., 2003). Additionally, viral vector delivery of IL-4 to the dorsal root ganglia or perineural IL-4 injection reduced mechanical and heat hypersensitivity in neuropathic pain models in rats or mice (Hao et al., 2006; Kiguchi et al., 2015). These IL-4-induced antinociceptive effects were proposed to result from the decreased production of proinflammatory mediators (TNF, IL-1β, prostaglandin E2) or increased synthesis of anti-inflammatory cytokine IL-10, presumably in macrophages (Cunha et al., 1999; Vale et al., 2003; Kiguchi et al., 2015).
Notably, accumulating evidence suggests interactions between IL-4 and the opioid system. IL-4 induced expression of opioid receptors in human blood immune cells and in rat neuronal cell cultures (Kraus et al., 2001; Börner et al., 2004). Additionally, the intracellular content of β-endorphin (END) and the mRNA levels of its precursor proopiomelanocortin (Pomc) were elevated in IL-4-stimulated rat lymph node lymphocytes in vitro (Busch-Dienstfertig et al., 2012). Furthermore, we have recently shown that IL-4 induced anti-inflammatory M2 macrophages in vitro (Pannell et al., 2016) and switched proinflammatory M1 macrophages into M2 macrophages at the injured nerves after chronic constriction injury (CCI) in mice in vivo (Celik et al., 2020). In both conditions, compared with M1 cells, the M2 cells contained higher levels of opioid peptides END, Met-enkephalin (ENK) and dynorphin A 1–17 (DYN), and mRNAs of their corresponding precursors Pomc, proenkephalin (Penk) and prodynorphin (Pdyn). This occurred following several days-lasting incubation of IL-4 with macrophages in cultures (Pannell et al., 2016) or following repetitive application of IL-4 at the injured nerves in vivo (Celik et al., 2020). Importantly, the repetitive perineural IL-4 injection resulted in persistent attenuation of mechanical hypersensitivity, which lasted beyond the IL-4 treatment, was independent of IL-4 receptors α (IL-4Rα), but was mediated by M2 macrophage-derived opioid peptides which activated opioid receptors (δ, µ, κ) at the nerves. These findings suggested that when macrophages are polarized by IL-4 into the M2 phenotype, they continuously produce and constitutively secrete opioids to exert antinociception (Celik et al., 2020). Interestingly, we also observed that single IL-4 injection at the injured nerves resulted in acute antinociception (Celik et al., 2020).
Here, we hypothesized that single perineural IL-4 application directly releases opioid peptides from macrophages to exert acute attenuation of pathologic pain, and examined the underlying intracellular mechanisms. This is novel since the typical effect of IL-4-induced IL-4Rα activation is the signaling via Janus kinases (JAKs) and transcription factors such as signal transducers and activators of transcription (STATs). The JAK–STAT pathway modulates gene expression and the corresponding protein production (Wills-Karp and Finkelman, 2008; Martinez et al., 2009; Busch-Dienstfertig et al., 2012; Gadani et al., 2012; Kiguchi et al., 2015), but the IL-4–IL-4Rα-mediated release processes have not yet been reported.
Materials and Methods
Animals
Experiments were approved by the State animal care committee (Landesamt für Gesundheit und Soziales, Berlin, Germany) and were performed according to the Guide for the Care and Use of Laboratory Animals adopted by the National Institutes of Health, and the ARRIVE guidelines (Kilkenny et al., 2010). Male C57BL/6J mice (22–32 g, 6–13 weeks old) were purchased from Janvier Laboratories. They were kept in groups of two to four per cage, with free access to food and water, in environmentally controlled conditions (12/12 h light/dark schedule, light on at 7 A.M.; 22–24°C; humidity 60–65%). After in vivo experiments and for tissue collection for ex vivo experiments, animals were killed with isoflurane overdose (AbbVie). All efforts were made to minimize animal suffering and to reduce their numbers.
Substances
For in vivo experiments the following substances were used: recombinant mouse IL-4 (R&D Systems), mouse antibody to IL-4Rα (anti-IL-4Rα; clone mIL4R-M1) and isotype-matched control IgG2aκ (BD PharMingen), mouse antibody to IL-10 (anti-IL-10; clone JES052A5) and isotype-matched control IgG1 (R&D Systems), rabbit antibodies to opioid peptides including ENK (anti-ENK; catalog #T-4293), END (anti-END; catalog #T-4044), DYN (anti-DYN; catalog #T-4267; Peninsula Laboratories) and control rabbit IgG (Sigma-Aldrich), opioid receptor antagonists including naloxone methiodide (NLXM; non-selective, peripherally restricted), D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP; µ-receptor selective), nor-binaltorphimine dihydrochloride (nor-BNI; κ-receptor selective; Sigma-Aldrich) and N,N-diallyl-Tyr-Aib-Aib-Phe-Leu hydrochloride (ICI 174,864; δ-receptor selective; Biozol).
For ex vivo enzyme immunoassay (EIA) experiments the following substances were used: IL-4 (see above), EGTA (extracellular Ca2+ chelator), 1-[6-[((17β)−3-methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-1H-pyrrole-2,5-dione [U73122; phospholipase C (PLC) inhibitor], 2-aminoethoxydiphenyl borate [2-APB; inositol 1,4,5-trisphosphate (IP3) receptor inhibitor], wortmannin [phosphatidylinositol 3-kinase (PI3K) inhibitor], dantrolene sodium salt (ryanodine receptor inhibitor; Sigma-Aldrich), 1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid tetrakis(acetoxymethyl ester; BAPTA-AM; intracellular Ca2+ chelator), and PKI 14–22 [protein kinase A (PKA) inhibitor; Tocris].
For in vitro radioligand binding experiments the following substances were used: IL-4 (see above), [3H]-[D-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin ([3H]-DAMGO; µ-receptor agonist), [3H]-diprenorphine (non-selective opioid receptor antagonist), [3H]-naloxone (non-selective opioid receptor antagonist (PerkinElmer), fentanyl citrate (µ-receptor agonist), trans-(±)3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl]-benzeneacetamide (U50,488; κ-receptor agonist; Sigma-Aldrich), and D-Pen2,D-Pen5-enkpephalin (DPDPE; δ-receptor agonist; Bachem).
Induction of neuropathy
Mice were intraperitoneally injected with ketamine (50 mg/kg; CP-Pharma); 20 min later, they were anesthetized by isoflurane and CCI was induced by exposing the sciatic nerve at the level of the right mid-thigh and placing three loose silk ligatures (4/0) around the nerve with ∼1 mm spacing; the ligatures were tied until they elicited a brief twitch in the respective hindlimb. The wound was closed with silk sutures (Labuz et al., 2009; Labuz and Machelska, 2013; Celik et al., 2016, 2020). After CCI, mice received 0.7 ml of novaminsulfon (500 mg/ml; 1 A Pharma) in drinking water (50 ml) for the next 24 h, according to the requirements of the ethics committee (Landesamt für Gesundheit und Soziales).
Evaluation of mechanical sensitivity (von Frey test)
Animals were habituated to the test cages daily (one to two times for 15 min), starting 6 d before nociceptive testing; they were individually placed in clear Plexiglas chambers located on a stand with anodized mesh (Model 410; IITC Life Sciences). To assess the sensitivity, calibrated von Frey filaments (Stoelting) in the range of 0.054 mN (0.0056 g) to 42.85 mN (4.37 g) were used. The filaments were applied until they bowed, for ∼3 s, to the plantar surface of hind paws. The up-down method was used to estimate 50% withdrawal thresholds (Chaplan et al., 1994). Testing began using a 2.74 mN (0.28 g) filament. If the animal withdrew the paw, the preceding weaker filament was applied. In the absence of withdrawal, the next stronger filament was applied. The maximum number of applications was 6–9, and the cutoff was 42.85 mN (4.37 g), according to our previous studies (Labuz et al., 2009; Labuz and Machelska, 2013; Celik et al., 2016, 2020).
Evaluation of heat sensitivity (Hargreaves test)
Before experiments, mice were habituated to the test cages daily (one to two times for 15 min), starting 6 d before nociceptive testing; they were placed in clear Plexiglas chambers positioned on a stand with a glass surface (Model 336; IITC Life Sciences). To examine heat sensitivity, radiant heat was applied to the plantar surface of hind paws from underneath the glass floor with a high intensity projector lamp bulb and paw withdrawal latency was evaluated using an electronic timer. The withdrawal latency was defined as the average of two measurements separated by at least 10 s. The heat intensity was adjusted to obtain baseline withdrawal latency of ∼10–12 s in uninjured paws, and the cutoff was set at 20 s to avoid tissue damage (Labuz and Machelska, 2013; Labuz et al., 2016; Celik et al., 2020).
In vivo treatments
Injections were performed perineurally at the site of nerve injury (CCI site; 30 µl) under brief isoflurane anesthesia. A polyethylene tube was placed 2 mm from the tip around the needle to ensure the same depth of needle insertion into the middle of the scar after CCI, as described earlier (Labuz et al., 2009; Labuz and Machelska, 2013; Celik et al., 2016, 2020). We have previously verified that volume of 30 µl injected at the CCI site remains at this site and covers ∼1 cm of the nerve, including the ligation site and sites proximal and distal to the ligation (Labuz et al., 2009). Furthermore, the antinociceptive effects of opioid receptor agonists applied at the CCI site were fully reversed by opioid receptor antagonists injected at this site. Intraplantar (into the hind paw innervated by the ligated nerve) or intrathecal application of opioid receptor antagonists did not alter the antinociceptive effects of agonists applied at the CCI site (Labuz and Machelska, 2013).
To evaluate the time course of IL-4-induced antinociception, IL-4 (50–400 ng) was injected at the CCI site on day 14 after CCI. In our previous studies we have demonstrated that CCI-induced hypersensitivity appears on day 1 and persists up to 14–26 d following CCI (Labuz et al., 2009, 2016; Labuz and Machelska, 2013; Pannell et al., 2016; Celik et al., 2020). Heat and mechanical sensitivities were measured before CCI, and on day 14 after CCI, before and 5–60 min after injections. To assess the contribution of IL-4Rα, opioid peptides, opioid receptors, and IL-10 to IL-4-induced antinociception, we used anti-IL-4Rα (1.5–12 µg), control IgG2aκ (6 µg), antibodies to opioid peptides anti-ENK (0.5–4 µg), anti-END (0.5-4 µg), anti-DYN (0.5-4 µg) and control IgG (2-4 µg), antagonists to opioid receptors NLXM (2.5–20 µg), ICI 174,864 (4–8 µg), CTOP (1–2 µg) and nor-BNI (10–20 µg), as well as anti-IL-10 (5–40 µg) and control IgG1 (20 µg). The doses of all substances listed above also include those tested in pilot experiments, which were performed to select the optimal doses reported in the results section, and to keep the animal numbers to minimum. Antibodies and antagonists were co-injected with IL-4 (200 ng) and mechanical sensitivity was measured before and 5 min after injections (the peak of IL-4-induced antinociception). IL-4 was dissolved in 0.1% bovine serum albumin (BSA; Sigma-Aldrich) in PBS (Biochrom). Other substances were dissolved in sterile water and diluted with 0.9% NaCl to obtain the desired concentrations. Control groups were treated with respective vehicle (PBS, 0.9% NaCl) or control IgG, and tested accordingly.
Immune cell isolation from injured nerves
On day 14 after CCI, mice were treated with vehicle (PBS) or IL-4 (200 ng) at the CCI site, and 5 min after the injections (i.e., at the peak of IL-4-induced antinociception) they were transcardially perfused with ice-cold PBS under terminal isoflurane anesthesia. The ligated part of the sciatic nerves, including the ligation site and sites distal and proximal to it (∼1 cm long), were isolated. The nerves were cut into small pieces, digested, filtered, centrifuged, and the cell pellets were re-suspended in RPMI 1640 with GlutaMax buffer (Invitrogen). The cells were stained with acridine orange/propidium iodide (Logos) to verify their viability, counted using Luna dual fluorescence cell counter (Celik et al., 2016, 2020), and used for flow cytometry, real-time quantitative reverse transcription PCR (qRT-PCR), and EIAs to measure intracellular levels of opioid peptides in experiments described below. For measurements of extracellular levels of opioid peptides (by EIA) and IL-10 (by ELISA), the cells were prepared in a similar way, except they were isolated from mice (on day 14 after CCI) not treated with vehicle or IL-4.
Flow cytometry
Two injured sciatic nerve fragments were pooled and single cell suspensions were prepared as described above. The cells were first labeled with LIVE/DEAD fixable aqua dead cell stain kit for 30 min on ice, according to the manufacturer's instructions (ThermoFisher Scientific), to exclude dead cells. The cells were then washed with ice-cold FACS buffer [2% fetal bovine serum (FBS)/PBS], followed by centrifugation and removal of the supernatant. Cells were stained for 10 min on ice with anti-mouse CD16/32 (Fc Block) to ensure antigen specific binding. To identify immune cell populations, the cells were labeled with anti-mouse CD45-APC-eFluor 780 (clone 30 F11; eBioscience) to identify hematopoietic cells, CD3-APC (clone 17A2; eBioscience) to identify T lymphocytes, Ly6g-FITC (clone 1A8; Biolegend) to identify neutrophils, and F4/80-PE (clone BM8; Biolegend) to identify macrophages. All antibodies were prepared in PBS containing 2% FBS. As staining controls, the fluorescence minus one (FMO), single stain, unstain, and all stain controls were included for accurate identification of immune cells, compensation, and voltage adjustments. To quantify the absolute number of cells, 50 µl of count bright absolute counting beads (ThermoFisher Scientific) was added to the solutions according to the manufacturer's recommendations before the flow cytometry analysis. Counting beads were gated on the forward scatter versus linear side scatter plot. The percentages of positively-stained cells determined over 10,000 events were analyzed using FACS Canto II (BD Biosciences), and fluorescence intensity was expressed in arbitrary units on a logarithmic scale. Single cells were gated on the forward scatter height versus forward scatter area density plot to exclude doublets and the data were analyzed using FlowJo software (version 10.1r5; TreeStar; Celik et al., 2020).
Immunomagnetic separation (IMS) of F4/80+ macrophages
F4/80+ macrophages were separated from immune cells isolated from injured nerve fragments (pooled from three animals) using EasySep release mouse PE positive selection kit (StemCell) according to the manufacturer's protocol. Briefly, immune cells were labeled with F4/80-PE antibody (clone BM8; Biolegend) for 20 min and magnetic particles (StemCell) for 10 min, and separated using an EasySep magnet (StemCell). Negatively stained, F4/80- cells were poured off, whereas the positively stained, F4/80+ cells remained in the tube. To acquire particle-free cells, bound magnetic particles were removed from the isolated F4/80+ cells using the release buffer (StemCell; Celik et al., 2020). The purity of F4/80+ separated cells (∼105 cells) was verified by flow cytometry, as described above, and it was 94–97%. The isolated F4/80+ macrophages were used for qRT-PCR, EIA, and ELISA experiments described below.
qRT-PCR
The IMS isolated F4/80+ macrophages were disrupted and homogenized in RLT buffer (QIAGEN) and kept at −80°C until further use. The total RNA was extracted from the cells using RNeasy micro kit following the manufacturer's protocol with on-column DNaseI treatment (QIAGEN). Quantity and quality of the total RNA were assessed by DS-11 spectrophotometer (DeNovix) and agarose gel electrophoresis (18s/28s rRNA). cDNA synthesis was done using Superscript IV VILO mastermix according to the manufacturer's protocol (ThermoFisher Scientific).
The qPCR was performed in duplicates using the TaqMan Fast Advanced Master Mix according to the manufacturer's instructions (ThermoFisher Scientific). The following TaqMan gene expression assays were used: (1) for M1 macrophage markers: Il-1β (Mm00434228_m1), Tnf (Mm00443258_m1), and inducible nitric oxide synthase (iNos; Mm00440502_m1); (2) for M2 macrophage markers: Il-10 (Mm01288386_m1), arginase-1 (Arg-1; Mm00475988_m1), and chitinase-like 3 protein (Ym1; Mm00657889_mH); (3) for opioid peptide precursors: Penk (Mm01212875_m1), Pomc (Mm00435874_m1), and Pdyn (Mm00457573_m1); and (4) for the hauskeeping gene Gapdh (Mm99999915_g1; ThermoFisher Scientific). Additional no-RT controls were run to test for impurities or genomic DNA contaminations. The amplification was conducted for 40 cycles and the mean Ct values of the genes of interest (Il-1β, Tnf, iNos, Il-10, Arg-1, Ym1, Penk, Pomc, Pdyn) and of the housekeeping gene Gapdh were calculated. The Ct values of the genes of interest were then normalized using the Ct value of the Gapdh of the same sample, and these normalized values were referred to as the ΔCt values. The ΔCt values were then used to compute the relative expression levels (i.e., fold change) of the genes of interest using the 2-ΔΔCt formula (Celik et al., 2016, 2020).
EIA and ELISA
The EIA was used to measure the intracellular and extracellular levels of opioid peptides, whereas ELISA was used to measure the extracellular levels of IL-10, in the IMS isolated F4/80+ macrophages. To determine the intracellular opioid peptide levels, the cell samples were thawed, resuspended in RPMI buffer, lysed by a freezing/thawing procedure (8 min at −80°C and 1 min at 50°C; repeated five times) followed by sonication (Ultra-Turrax T8; IKA Labortechnik), and supernatants were stored at −20°C until EIA measurements.
To evaluate the IL-4-induced release of opioid peptides (ENK, END, DYN) and IL-10, the IMS isolated F4/80+ macrophages (105 cells) were incubated with vehicle (PBS) or IL-4 (0.1–1000 ng/ml) for 5 min. The intracellular mechanisms were investigated for the secretion of ENK as a representative opioid peptide. The macrophages were preincubated with anti-IL-4Rα (0.1–1000 ng/ml; 30 min), EGTA (3 mm; 30 min), BAPTA-AM (100 μm; 30 min), U73122 (50 μm; 30 min), 2-APB (100 μm; 15 min), PKI 14-22 (5-40 μm; 30 min), wortmannin (25–200 μm; 30 min), or dantrolene (75–600 μm; 30 min) followed by application of the most optimal dose of IL-4 (100 ng) for 5 min. Control groups were treated with vehicle (PBS). After treatments, macrophages were centrifuged (380 × g, 5 min, 4°C) and supernatants were stored at −20°C until EIA/ELISA measurements. The anti-IL-4Rα, PKI 14-22, wortmannin, and dantrolene were tested in several concentrations (listed above) in pilot experiments to find out the most effective concentrations for the final experiments. The concentrations of EGTA, BAPTA-AM, U73122, and 2-APB are based on our previous study (Celik et al., 2016). To verify cell viability, the remaining cell pellets were diluted in RPMI buffer, stained with acridine orange/propidium iodide and counted using Luna dual fluorescence cell counter. Some substances at higher concentrations decreased viable cell numbers in pilot experiments and therefore were not used in final experiments: anti-IL-4Rα (1000 ng/ml), wortmannin (200 μm), and dantrolene (600 μm). The viability of cells used for final analyses was 82–90%.
Additionally, samples containing IL-4 (100 ng), anti-IL-4Rα (100 ng/ml), PKI 14-22 (20 μm), wortmannin (100 μm), or dantrolene (300 μm; without immune cells) were accordingly prepared to examine whether they are recognized by antibodies to opioid peptides or IL-10 in EIA and ELISA, respectively. We have previously verified that this was not the case for opioid peptide antibodies and EGTA (3 mm), BAPTA-AM (100 μm), U73122 (50 μm), and 2-APB (100 μm; Celik et al., 2016).
The EIA measurements were performed according to the manufacturer's instructions using the kits for the corresponding opioid peptides, ENK (catalog #S-1419; Peninsula Laboratories), END (catalog #EK-022-06), and DYN (catalog #EK-021-03; Phoenix Pharmaceuticals). Briefly, samples were thawed and incubated for 2 h with biotinylated peptide and anti-ENK, anti-END, or anti-DYN. Streptavidin-horseradish peroxidase was added and the samples were incubated for 1 h. After washing, tetramethylbenzidine was added for 1 h, and the reaction was terminated by application of 2 N HCl. The ELISA measurements were performed according to the manufacturer's instructions using the IL-10 kit (catalog #BMS614; ThermoFisher Scientific). Briefly, samples were thawed and incubated for 3 h with biotinylated peptide and anti-mouse IL-10 coating antibody. Afterwards, the cells were washed and streptavidin-horseradish peroxidase was added for 1 h. Then the cells were washed, the TMB substrate solution was added for 10 min, and the reaction was terminated by application of the stop solution. For both EIA and ELISA, the absorbance was measured at 450 nm (Molecular Devices Spectra Max), according to the standard curve. All samples were measured in duplicates. To reduce animal numbers, whenever possible, the same controls were used in experiments with similar conditions and in parallel tested by EIA, and therefore, these controls are shared by some graphs (see legends to Figs. 7, 8).
Figure 7.
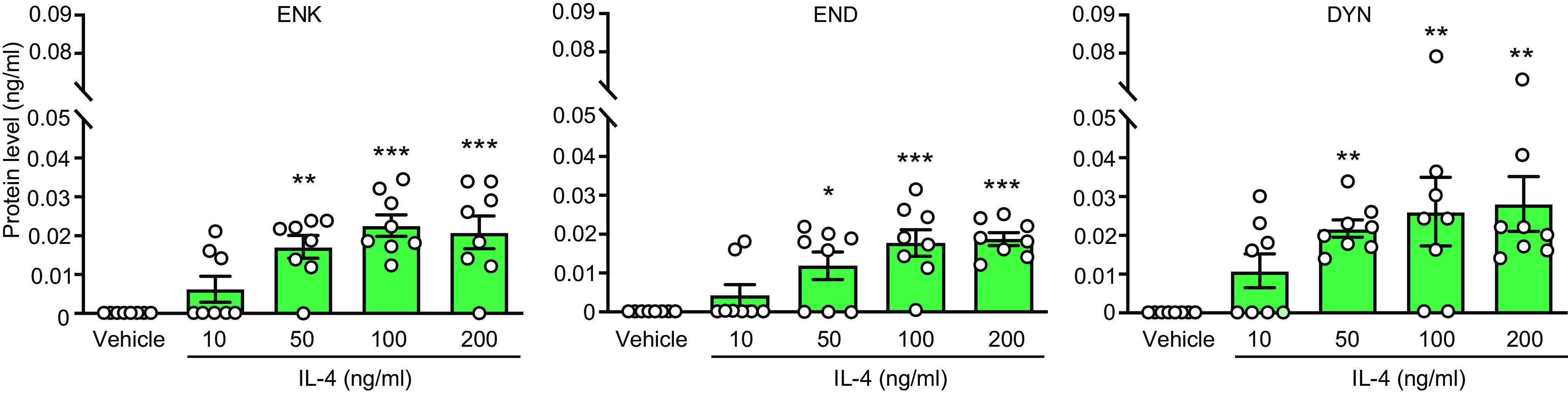
IL-4 induces opioid peptide release from M1 macrophages from injured nerves. Extracellular levels of ENK, END, and DYN secreted from F4/80+ macrophages isolated by IMS from injured nerves on day 14 after CCI and stimulated ex vivo (5 min) with IL-4 (10–200 ng/ml) or vehicle (PBS). Opioids were measured by EIA. The data in vehicle groups are the same in all graphs, to reduce animal numbers (see also Materials and Methods). ENK (F(4,35) = 10.82, p < 0.0001), END (F(4,35) = 9.781, p < 0.0001; one-way ANOVA), DYN (Kruskal–Wallis statistic = 16.87, p = 0.0021); *p < 0.05, **p < 0.01, ***p < 0.001 versus vehicle; one-way ANOVA and Bonferroni's test (ENK, END) or Kruskal–Wallis one-way ANOVA and Dunn's test (DYN). Data are presented as individual data points and mean ± SEM N = 8 samples per group.
Figure 8.
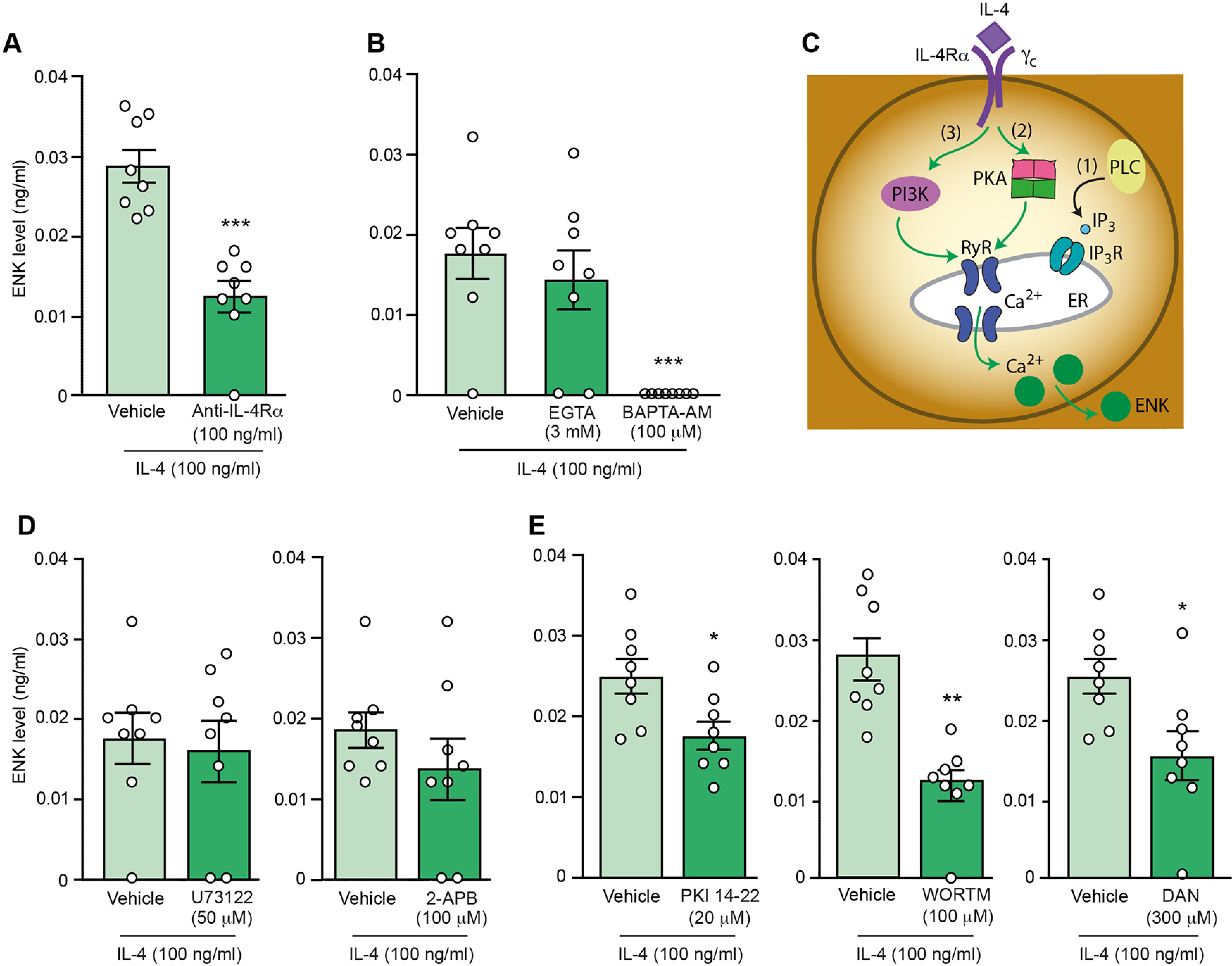
Intracellular mechanisms of IL-4-induced ENK release from M1 macrophages from injured nerves. A, Attenuation of IL-4-induced ENK secretion by anti-IL-4Rα; ***p = 0.0009 versus vehicle; Mann–Whitney test (U = 0.0). B, Inhibition of IL-4-induced ENK secretion by intracellular Ca2+ chelator BAPTA-AM (F(2,21) = 11.13, p < 0.0001) but not by extracellular Ca2+ chelator EGTA (F(2,21) = 0.287, p > 0.05); ***p < 0.001 versus vehicle; one-way ANOVA and Bonferroni's test. C, Schematic representation of pathways involved in the liberation of Ca2+ from ER and the identification of those involved in IL-4-induced ENK release. (1) PLC-induced IP3 generation, which activates IP3 receptors (IP3R); (2) PKA-induced activation of ryanodine receptors (RyR); (3) PI3K-mediated activation of RyR. The green and black arrows respectively indicate pathways involved or not involved in the IL-4-induced ENK secretion, investigated in D, E. D, No effects of PLC inhibitor U73122 (t = 0.3264, p = 0.748; unpaired t test; left) and IP3R inhibitor 2-APB (U = 20.5, p = 0.244; Mann–Whitney test; right) on IL-4-induced ENK secretion. E, Attenuation of IL-4-induced ENK secretion by PKA inhibitor PKI 14-22 (*p = 0.0181 vs vehicle; unpaired t test, t = 2.674; left), PI3K inhibitor wortmannin (WORTM; **p = 0.0013 vs vehicle; Mann–Whitney test, U = 1; middle), and RyR inhibitor dantrolene (DAN; *p = 0.0185 versus vehicle; unpaired t test, t = 2.665; right). F4/80+ macrophages were isolated by IMS from injured nerves on day 14 after CCI, ex vivo preincubated with chelators/inhibitors for 30 min (anti-IL-4Rα, EGTA, BAPTA-AM, U73122, PKI 14-22, wortmannin, dantrolene) or 15 min (2-APB) and then IL-4 (100 ng/ml) was added (5 min). Cells in vehicle groups were preincubated with vehicle (PBS) for the corresponding time before addition of IL-4. ENK extracellular levels were measured by EIA. The data in vehicle groups in B and for U73122 in D are the same, to reduce animal numbers (see also Materials and Methods). Similarly, in E, data in vehicle groups for PKI 14-22 and DAN are the same. Data are presented as individual data points and mean ± SEM N = 8 samples per group.
Cell culture, transfection, and membrane preparation
Wild-type human embryonic kidney 293 (HEK) cells (German Collection of Microorganisms and Cell Cultures; RRID:CVCL_0045) and HEK cells stably expressing FLAG-epitope-tagged rat µ-receptor (HEK-µ-receptor) were grown in 175-cm2 culture flasks and maintained in DMEM (Sigma-Aldrich) supplemented with 10% FBS and 1% penicillin/streptomycin without or with 0.1 mg/ml geneticin (Biochrome) at 37°C and 5% CO2. Depending on the confluence, the cells were passaged 1:3–1:10 every second or third day. Wild-type HEK cells were transiently transfected with 36 µg of plasmid encoding either human δ-receptor (vector: pcDNA3.1;OPRD100000) or HA-tagged human κ-receptor (vector: pcDNA3.1;OPRK10TN00; cDNA Resource Center) using X-treme GENE HP DNA reagent (Roche) according to the manufacturer's instructions. Forty-eight hours after transient transfection (HEK-δ-receptor and HEK-κ-receptor) or after reaching the confluence of a culture flask (HEK-µ-receptor), membrane preparation was performed. Cells were washed with ice-cold Trizma (50 mm, pH 7.4; Sigma-Aldrich), scratched with a cell scrapper, homogenized, and centrifuged twice at 42,000 × g for 20 min at 4°C. Protein concentration was determined using the Bradford method (Bradford, 1976).
Radioligand binding
To examine whether IL-4 binds to opioid receptors, we performed competitive radiolabeled binding assays. To detect binding to δ-receptors, we used HEK-δ-receptor cells and measured the displacement of [3H]-diprenorphine (1.5 nm, 25.1 Ci/mmol) by IL-4 (6, 3, 1.5, 0.75, 0.375, 0.1875, 0.09375, 0.04687, 0.02343 ng/µl) or DPDPE (δ-receptor selective; 5, 1, 0.5, 0.1, 0.05, 0.01, 0.005, 0.001, 0.0005 μm) as a positive control. To detect binding to µ-receptors, we used HEK-µ-receptor cells and measured the displacement of [3H]-DAMGO (4 nm, 53.7 Ci/mmol) by IL-4 (the same concentrations as above) or fentanyl (µ-receptor selective; 5, 1, 0.5, 0.1, 0.05, 0.01, 0.005, 0.001, 0.0005 μm) as a positive control. To detect binding to κ-receptors, we used HEK-κ-receptor cells and measured the displacement of [3H]-naloxone (5 nm, 58.2 Ci/mmol) by IL-4 (the same concentrations as above) or U50,488 (κ-receptor selective; 5, 1, 0.5, 0.1, 0.05, 0.01, 0.005, 0.001, 0.0005 μm) as a positive control.
A protein amount of 100 µg was incubated with the radioligands, DPDPE, fentanyl and U50,488, or IL-4 dissolved in 50 mm Trizma at pH 7.4 for 90 min at room temperature. Nonspecific binding was determined by the addition of 10 μm unlabeled naloxone. The measurements were done in duplicates. Data are expressed as [3H]-ligand-bound protein normalized to baseline (100%), and the baseline was defined as the [³H]-ligand binding in the absence of the competitor (opioid or IL-4).
Experimental design and statistical analyses
Animals were randomly placed in cages by an animal caretaker who was not involved in the study. In vivo experiments were performed by an experimenter blinded to the treatments. Substances were prepared in separate, coded vials (one per animal) by a person not involved in in vivo testing. The codes were broken after completion of experiments. Flow cytometry experiments were performed by an experimenter unaware of the sample assignment. A colleague not involved in these experiments coded flow cytometry samples; the codes were broken after completion of the analyses. The animal/sample size estimation was performed a priori using the G*Power 3.1.2 program. In all in vivo experiments, nine animals per group were used and tested on two to three different days. In all ex vivo experiments (flow cytometry, qRT-PCR, EIA/ELISA), eight independent biological samples were used per group and measured on two to three different days. In radioligand binding assays, each group consists of six independent experiments.
Statistical analyses were performed using GraphPad Prism software (version 5.02 for Windows; GraphPad Software Inc.). All data were assessed for normal distribution and equal variances by D'Agostino and Pearson or Kolmogorov–Smirnov tests. No animals or samples were excluded from the analysis. Nevertheless, the Grubbs' test was performed to identify potential outliers, as described in the results section. Two-sample comparisons of normally distributed data were made using paired t test for dependent or unpaired t test for independent data. Two-sample comparisons of nonnormally distributed data were made using Wilcoxon test for dependent or Mann–Whitney test for independent data. Two-way repeated-measures (RM) ANOVA followed by Bonferroni's test were used to compare two groups over time (more than two time points). Changes within one group over time were evaluated using one-way RM ANOVA followed by Bonferroni's test. Multiple comparisons at one time point were performed using one-way ANOVA followed by Bonferroni's test for normally distributed data or Kruskal–Wallis one-way ANOVA followed by Dunn's test for nonnormally distributed data. Differences were considered significant if p < 0.05. Detailed statistical analysis is presented in figure legends and Table 1. In binding experiments, means of values at each agonist concentration and each condition were used and the data were fitted by a three-parameter log (agonist) versus response nonlinear regression curve fit. Data are presented as individual data points and/or means ± SEM.
Table 1.
Detailed statistical evaluation of the data in Figure 1A,B
| Figure 1A, heat hypersensitivity | |
| Vehicle vs IL-4 50 ng | |
| Time | F(6,96) = 311.1; p < 0.0001 |
| Treatment | F(1,16) = 0.24; p = 0.633 |
| Time × treatment interaction | F(6,96) = 0.43; p = 0.856 |
| Vehicle vs IL-4 100 ng | |
| Time | F(6,96) = 331.89; p < 0.0001 |
| Treatment | F(1,16) = 2.15; p = 0.1616 |
| Time × treatment interaction | F(6,96) = 1.84; p = 0.0992 |
| Vehicle vs IL-4 200 ng | |
| Time | F(6,96) = 277.62; p < 0.0001 |
| Treatment | F(1,16) = 0.28; p = 0.6071 |
| Time × treatment interaction | F(6,96) = 1.70; p = 0.1299 |
| Figure 1B, mechanical hypersensitivity | |
| Vehicle vs IL-4 50 ng | |
| Time | F(6,96) = 176.6; p < 0.0001 |
| Treatment | F(1,16) = 6.76; p = 0.0193 |
| Time × treatment interaction | F(6,96) = 2.949; p = 0.011 |
| Vehicle vs IL-4 100 ng | |
| Time | F(6,96) = 165.0; p < 0.0001 |
| Treatment | F(1,16) = 17.4; p = 0.0007 |
| Time × treatment interaction | F(6,96) = 7.988; p < 0.001 |
| Vehicle vs IL-4 200 ng | |
| Time | F(6,96) = 160.5; p < 0.0001 |
| Treatment | F(1,16) = 57.83; p < 0.0001 |
| Time × treatment interaction | F(6,96) = 13.85; p < 0.0001 |
All data were analyzed be two-way RM ANOVA.
Results
IL-4 single application at injured nerves attenuates mechanical but not heat hypersensitivity
Fourteen days following CCI, mice developed heat hypersensitivity manifested by reduced withdrawal latencies to heat (assessed by the Hargreaves test; Fig. 1A) and mechanical hypersensitivity manifested by reduced mechanical von Frey withdrawal thresholds (Fig. 1B) compared with latencies and thresholds before CCI, in paws ipsilateral to the CCI. IL-4 (50–200 ng) applied perineurally at the injured nerve (CCI site) did not modify heat (Fig. 1A) but dose dependently attenuated mechanical hypersensitivity (Fig. 1B). Higher IL-4 dose (400 ng) was not more effective (1.4 ± 0.53 g; n = 3). The antinociceptive effects of the most effective IL-4 dose (200 ng) peaked at 5 min and returned to the preinjection thresholds at 30 min after injection (Fig. 1B). Vehicle administered at the CCI site did not significantly alter paw withdrawal latencies (Fig. 1A) or thresholds (Fig. 1B).
Figure 1.

IL-4 attenuates neuropathy-induced mechanical but not heat hypersensitivity. A, Time course of IL-4 effects on heat hypersensitivity measured by the Hargreaves test, and (B) on mechanical hypersensitivity measured by the von Frey test. IL-4 and vehicle (PBS) were injected at the CCI site. The effects were measured before CCI and on day 14 after CCI, before and 5–60 min after injections; +++p < 0.001 versus latencies or thresholds before CCI; one-way RM ANOVA and Bonferroni's test. For the clarity of the graphs, the statistical significance is only shown for the most effective IL-4 dose (200 ng); ***p < 0.001 versus vehicle (PBS); two-way RM ANOVA and Bonferroni's test. Detailed statistical analysis is described in Table 1. C, Reversibility of IL-4 (200 ng)-induced attenuation of mechanical hypersensitivity by anti-IL-4Rα (Kruskal–Wallis statistic = 14.01; p = 0.0029); ***p < 0.001 versus control IgG; Kruskal–Wallis one-way ANOVA and Dunn's test. Anti-IL-4Rα was co-injected with IL-4 at the CCI site and mechanical von Frey thresholds were measured 5 min after injections, on day 14 after CCI. Data are presented as individual data points and/or mean ± SEM N = 9 mice per group.
IL-4 (200 ng)-induced antinociception was dose dependently attenuated by co-injection of anti-IL-4Rα (1.5–6 µg) at the CCI site (Fig. 1C), suggesting the involvement of local IL-4Rα. There were no changes in the contralateral paws after any treatment in any test (data not shown).
Endogenous opioid peptides and opioid receptors at injured nerves contribute to IL-4-induced antinociception
We have recently shown that sustained analgesia produced by repetitive IL-4 injection at injured nerves, which persisted after discontinuation of IL-4 application, did not require IL-4Rα, but involved the endogenous opioid system (Celik et al., 2020). Therefore, here we investigated whether the opioid system contributes to the antinociception induced by a single perineural IL-4 injection. We found that this IL-4 (200 ng)-induced antinociception was diminished by co-injected opioid peptide antibodies anti-ENK (0.5–2 µg), anti-END (0.5–2 µg), and anti-DYN (0.5-4 µg; Fig. 2A), peripherally restricted, non-selective opioid receptor antagonist NLXM (2.5–10 µg; Fig. 2B), and antagonists selective at δ-receptors (ICI 174,864; 8 µg), µ-receptors (CTOP; 2 µg), and κ-receptors (norBNI; 20 µg; Fig. 2C). There were no alterations in the contralateral paws (data not shown). These results indicate that antinociception produced by a single perineural IL-4 application is mediated by IL-4Rα and involves opioid peptides and receptors at injured nerves.
Figure 2.

IL-4-induced antinociception is mediated by endogenous opioid peptides and peripheral opioid receptors. A, Reversibility of IL-4 (200 ng)-induced antinociception by opioid peptide antibodies, anti-ENK (F(3,32) = 10.78, p < 0.0001), anti-END (F(3,32) = 15.67, p < 0.0001), and anti-DYN (Kruskal–Wallis statistic = 27.14, p < 0.0001). B, Reversibility of IL-4 (200 ng)-induced antinociception by a peripherally-restricted opioid receptor antagonist NLXM (Kruskal–Wallis statistic = 29.05, p < 0.0001). C, Reversibility of IL-4 (200 ng)-induced antinociception by antagonists selective at δ-opioid (ICI 174,864; ICI), µ-opioid (CTOP), and κ-opioid (norBNI) receptors (F(3,32) = 44.44, p < 0.0001). Antibodies or antagonists were co-injected with IL-4 at the CCI site and mechanical von Frey thresholds were measured 5 min after injections, on day 14 after CCI; **p < 0.01, ***p < 0.001 versus control IgG (A) or vehicle (0.9% NaCl; B, C); one-way ANOVA and Bonferroni's test (A, anti-ENK and anti-END; C) or Kruskal–Wallis one-way ANOVA and Dunn's test (A, anti-DYN; B). Data are presented as individual data points and mean ± SEM N = 9 mice per group.
IL-4 does not bind to opioid receptors
To exclude the possibility that IL-4 directly activates opioid receptors, we performed binding analysis in HEK cells stably expressing µ-receptors or transiently transfected with δ-receptor or κ-receptor. We found that IL-4 failed to displace [3H]-diprenorphine, [3H]-[DAMGO], or [3H]-naloxone from δ-receptor, µ-receptor, or κ-receptor, respectively (Fig. 3A,C,E). In contrast, the binding of the radioligands was displaced by the corresponding selective agonists DPDPE, fentanyl, or U50,488 (Fig. 3B,D,F), confirming the reliability of the binding assays. Thus, whereas in experiments with the selective agonists, the sigmoidal dose-responses were obtained (Fig. 3B,D,F), in the IL-4 experiments, there was only a slight and inverse fitting for δ-receptors (Fig. 3A) and no fitting for µ-receptor and κ-receptor (Fig. 3C,E). An inverse dose–response fitting for δ-receptors in the presence of different concentrations of IL-4 indicates that IL-4 might strengthen the binding of the radioactively labeled ligand ([³H]-diprenorphine) to δ-receptors, but does not bind to the receptors itself. These data show that IL-4 does not bind to the opioid receptors.
Figure 3.

IL-4 does not bind to opioid receptors in HEK cells. A, C, E, IL-4 did not displace binding of [3H]-diprenorphine to δ-receptors (A), [3H]-DAMGO to µ-receptors (C), and of [3H]-naloxone to κ-receptors (E). B, D, F, In contrast, selective agonists of δ-receptors (DPDPE; B), µ-receptors (fentanyl; D), and κ-receptors (U50,488; F) displaced binding of [3H]-diprenorphine to δ-receptors (B), [3H]-DAMGO to µ-receptors (D), and of [3H]-naloxone to κ-receptors (F). Experiments were performed in HEK cells stably expressing µ-receptors or transiently transfected with δ-receptors or κ-receptors. [³H]-ligand binding in the absence of the competitor (opioid or IL-4) was set to 100% binding (baseline, depicted by dashed lines) and the data in the presence of the competitor were normalized accordingly and presented as mean ± SEM N = 6 independent experiments per group.
IL-4 single injection does not change immune cell numbers and the M1 macrophage phenotype at injured nerves
In the recent study we found that sustained analgesia produced by repetitive perineural IL-4 treatment was associated with increased macrophage counts and their switch from the proinflammatory M1 to the anti-inflammatory M2 phenotype (Celik et al., 2020). Here, we examined whether similar effects might occur after single perineural IL-4 application. Of note, increased immune cell extravasation following acute IL-4 treatment has been previously observed (Hickey et al., 1999; Ratthé et al., 2009). We performed the following ex vivo experiments at the peak of IL-4 (200 ng)-induced antinociception (5 min). Our flow cytometry analysis revealed that IL-4 did not alter the number of CD45+ immune cells, Ly6g+ neutrophils, and F4/80+ macrophages. Only the number of CD3+ T lymphocytes was statistically significantly elevated, but the effect was very small and because of one outlier identified by the Grubbs' test in the IL-4 group, so that the removal of this outlier resulted in a non-significant difference between vehicle (4.6 ± 0.6 × 103 cells; n = 8) and IL-4 groups (5.2 ± 0.5 × 103 cells; n = 7; p < 0.05; Mann–Whitney test). Notwithstanding, macrophages were the major immune cell population infiltrating injured nerves in both groups (Fig. 4).
Figure 4.
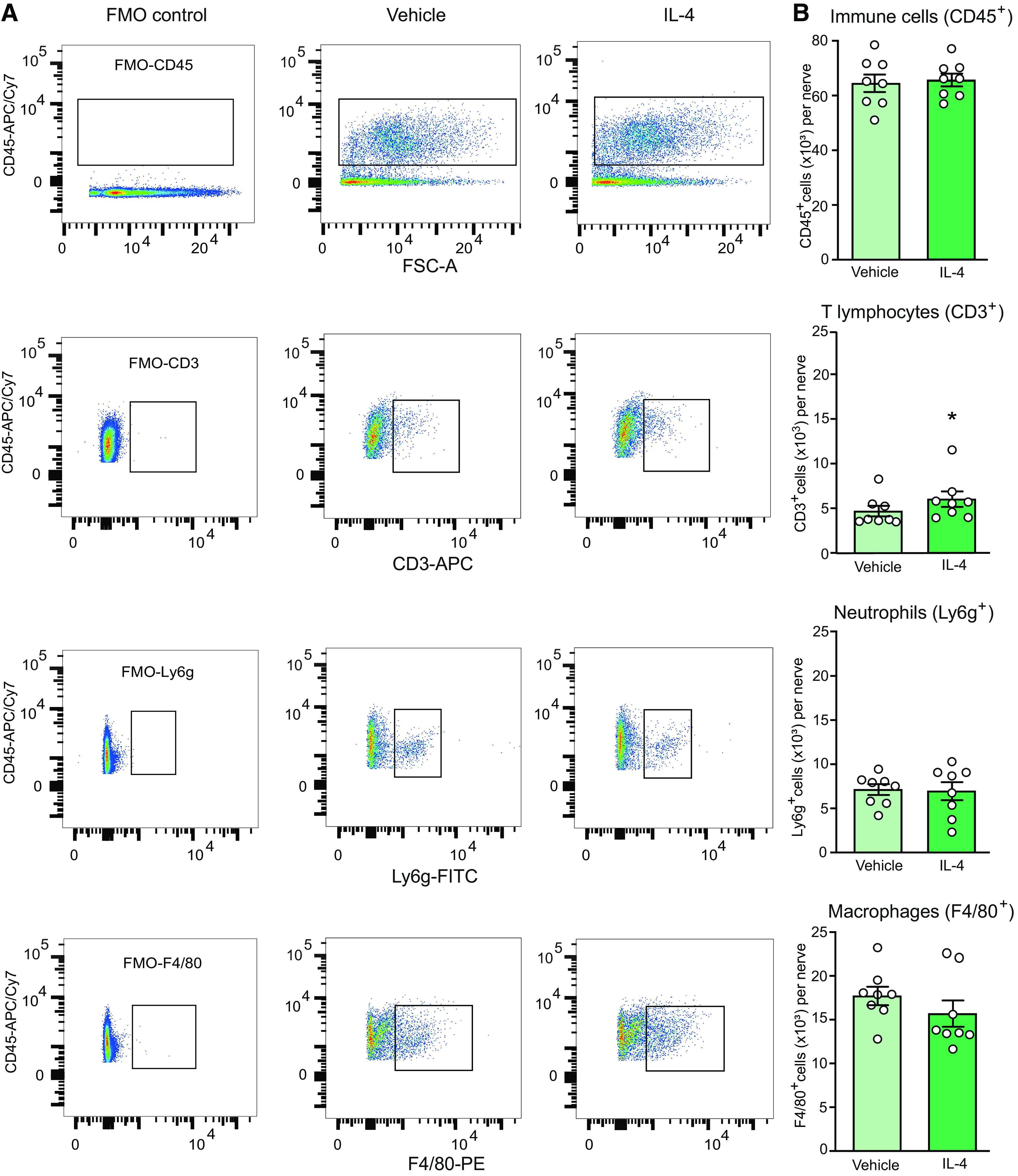
IL-4 does not substantially change immune cell number at injured nerves. A, Representative dot blots showing expression of all immune cells (CD45+), T lymphocytes (CD3+), neutrophils (Ly6g+), and macrophages (F4/80+). The blots represent the corresponding FMO negative controls (left), cells isolated from injured nerves after injection of vehicle (PBS; middle) or IL-4 (200 ng; right). Cells stained positive for the appropriate marker are shown inside the rectangular gates. B, Quantification of the corresponding cell populations shown in A; *p = 0.0499 versus vehicle; Mann–Whitney test (U = 13). There were no other significant differences: CD45+ (t = 0.3029, p = 0.7664), Ly6g+ (t = 0.1555, p = 0.8786), F4/80+ (t = 1.105, p = 0.2880); unpaired t test. Data are presented as individual data points and mean ± SEM N = 8 samples per group. Immune cells were isolated from the injured nerves 5 min after vehicle or IL-4 injection at the CCI site, on day 14 after CCI. The data were analyzed using flow cytometry and FlowJo software. The number of positively stained cells were calculated using absolute counting beads.
To assess the inflammatory status of macrophages, we analyzed by qRT-PCR the mRNAs of markers of M1 cells (Il-1β, Tnf, iNos) and M2 cells (Il-10, Arg-1, Ym1; Wills-Karp and Finkelman, 2008; Sica and Mantovani, 2012; Celik et al., 2020) in F4/80+ macrophages isolated by IMS from the injured nerves. We found that F4/80+ macrophages collected from vehicle-treated animals expressed considerably higher mRNA levels of M1 than of M2 cell markers (Fig. 5A), and there were no differences between vehicle-treated and IL-4-treated groups (Fig. 5B,C). Together, these data indicate that injured nerves are infiltrated by proinflammatory M1 macrophages and single perineural IL-4 application does not affect this state.
Figure 5.

Injured nerves are infiltrated by M1 macrophages and IL-4 does not alter their phenotype. A, Quantitative mRNA expression of M1 markers (Il-1β, Tnf, iNos) and M2 markers (Il-10, Arg-1, Ym1) in F4/80+ macrophages isolated by IMS from injured nerves of control animals [5 min after injection of vehicle (PBS) at the CCI site], on day 14 after CCI. Data were acquired by qRT-PCR, represent relative mRNA expression levels normalized to Gapdh, and are expressed as fold change versus Ym1. F(5,42) = 19.84, p < 0.001; **p < 0.01, *** p < 0.001 represent a significant difference between the respective M1 marker and each M2 marker; one-way RM ANOVA and Bonferroni's test. B, C, Quantitative mRNA expression of M1 (B) and M2 markers (C) in F4/80+ macrophages isolated by IMS from injured nerves 5 min after injection of vehicle (PBS) or IL-4 (200 ng) at the CCI site, on day 14 after CCI. Data were acquired by qRT-PCR, represent relative mRNA expression levels normalized to Gapdh, and are expressed as fold change versus vehicle. There were no significant differences between vehicle and IL-4 groups in any case: Il-1β (t = 0.1051, p = 0.9178), Tnf (t = 0.6008, p = 0.5576), iNos (t = 0.1279, p = 0.9), Il-10 (t = 0.5613, p = 0.5853), Arg-1 (t = 0.4268, p = 0.676), Ym1 (t = 1.168, p = 0.2622); unpaired t test. All data are presented as individual data points and mean ± SEM N = 8 samples per group.
IL-4 single application does not alter production, but induces the release of opioid peptides from M1 macrophages
We found previously that repetitive perineural IL-4 treatment induced M2 macrophages, which produced high amounts of opioids and released them constitutively, independently of IL-4Rα activation, to exert (long-lasting) analgesia that persisted after the discontinuation of IL-4 treatment (Celik et al., 2020). Here, we examined how opioids are used in response to single perineural IL-4 application, which apparently acted via M1 macrophages. By qRT-PCR we examined the mRNA levels of Penk, Pomc, and Pdyn (the precursors of the corresponding opioid peptides ENK, END, and DYN) in F4/80+ macrophages isolated by IMS from the injured nerves of animals treated perineurally with vehicle or IL-4 (200 ng). This analysis showed that there were no differences between vehicle-treated and IL-4-treated groups in the mRNA levels of Penk, Pomc, and Pdyn (Fig. 6A). For Pomc, the effect reminded comparable also after removing the outlier identified by the Grubbs' test in the vehicle group: 0.905 ± 0.122 (vehicle; n = 7) versus 0.921 ± 0.151 (IL-4; n =8; p = 0.937; unpaired t test). Interestingly, however, the intracellular content of ENK and END, but not DYN, was significantly lower in macrophages isolated from animals treated with IL-4 compared with those treated with vehicle (Fig. 6B). These findings suggest that single IL-4 injection does not affect production, but might induce the release of opioid peptides from M1 macrophages.
Figure 6.

IL-4 does not change production of opioid peptides but decreases their content in macrophages from injured nerves. A, Quantitative mRNA expression of opioid peptide precursors Penk, Pomc, and Pdyn in F4/80+ macrophages. Data were acquired by qRT-PCR, represent relative mRNA expression levels normalized to Gapdh, and are expressed as fold change versus vehicle. Penk (U = 26, p = 0.5737), Pomc (U = 29, p = 0.798; Mann–Whitney test), Pdyn (t = 1.085, p = 0.2961; unpaired t test). B, Intracellular content of opioid peptides ENK, END, and DYN in F4/80+ macrophages measured by EIA; *p = 0.0367 (ENK: t = 2.310) or *p = 0.0461 (END: t = 2.188); DYN (t = 0.6615, p = 0.5190) versus vehicle; unpaired t test. In all experiments, F4/80+ macrophages were isolated by IMS from injured nerves 5 min after injection of vehicle (PBS) or IL-4 (200 ng) at the CCI site, on day 14 after CCI. The data are shown as individual data points and mean ± SEM N = 8 samples per group.
In the following experiments, we specifically addressed the IL-4-induced secretion of opioid peptides. For this purpose, we isolated F4/80+ macrophages by IMS from injured nerves on day 14 after CCI and stimulated them ex vivo (5 min) with IL-4. We found that IL-4 (10–200 ng/ml) dose dependently released ENK, END, and DYN detected extracellularly by EIAs (Fig. 7).
IL-4-induced ENK release from M1 macrophages is mediated by IL-4Rα and intracellular Ca2+
Next, we investigated the IL-4Rα involvement and intracellular mechanisms underlying the IL-4-induced secretion of ENK as a representative opioid peptide. Similar to experiments depicted in Figure 7, we used F4/80+ macrophages isolated by IMS from injured nerves on day 14 after CCI and stimulated them ex vivo (5 min) with the most optimal IL-4 dose (100 ng/ml; Fig. 8). We found that IL-4-induced ENK release was diminished by anti-IL-4Rα (100 ng/ml; Fig. 8A). The effect reminded comparable and statistically significant also after removing the outlier (Grubbs' test) in the anti-IL-4Rα + IL-4 group: 0.028 ± 0.002 ng/ml (vehicle + IL-4; n = 8) versus 0.014 ± 0.001 ng/ml (anti-IL-4Rα + IL-4; n = 7; p < 0.0001; unpaired t test). Thus, IL-4-induced ENK release involves IL-4Rα.
Since exocytosis is regulated by calcium ions, we determined the source of Ca2+. The IL-4-induced ENK release was not affected by the extracellular Ca2+ chelator EGTA (3 mm), but was fully inhibited by the intracellular Ca2+ chelator BAPTA-AM (100 μm; Fig. 8B), suggesting the contribution of Ca2+ deriving from the intracellular stores. The major intracellular store of Ca2+ is endoplasmic reticulum (ER) and the pathways implicated in this Ca2+-mediated exocytosis include (1) PLC-induced IP3 generation, which activates IP3 receptors in ER (Celik et al., 2016); (2) PKA-induced activation of ER ryanodine receptors (Reiken et al., 2003); and (3) PI3K which also might involve the activation of ryanodine receptors leading to the Ca2+ mobilization (Pereira et al., 2017; Vogelaar et al., 2018; Fig. 8C). Our data show that IL-4-induced ENK release was not affected by inhibitors of PLC (U73122; 50 μm) and IP3 receptors (2-APB; 100 μm; Fig. 8D). In contrast, the ENK release was attenuated by inhibitors of PKA (PKI 14-22; 20 μm), PI3K (wortmannin; 100 μm), and ryanodine receptors (dantrolene; 300 μm; Fig. 8E). The effect of PI3K inhibitor was also comparable and statistically significant after removing the outlier (Grubbs' test) in the wortmannin + IL-4 group: 0.027 ± 0.002 ng/ml (vehicle + IL-4; n = 8) versus 0.013 ± 0.001 ng/ml (wortmannin + IL-4; n = 7; p = 0.0004; unpaired t test). Together, these findings suggest that M1 macrophages secreted ENK in response to IL-4 activation of IL-4Rα that led to the intracellular Ca2+ liberation, which involved PKA, PI3K, and ryanodine receptors (Fig. 8C).
Our specificity control experiments confirmed that the EIA opioid peptide antibodies did not recognize any of the substances in the concentrations used in final experiments, including IL-4, anti-IL-4Rα, PKI 14-22, wortmannin, dantrolene (data not shown), EGTA, BAPTA-AM, U73122, and 2-APB (Celik et al., 2016), and none of them affected immune cell viability (see Materials and Methods). We have also previously verified that ENK, END, and DYN antibodies specifically recognize their respective opioid peptides by testing ENK, Leu-ENK, END, and DYN in all three assays (Schreiter et al., 2012) and by performing release experiments using immune cells from mice lacking Penk, END, or Pdyn (Celik et al., 2016).
IL-10 is not involved in the IL-4-induced effects
Some studies suggested the involvement of IL-10 in the antinociceptive actions of IL-4 (Kiguchi et al., 2015). In contrast, we found that IL-4 (200 ng)-induced antinociception was not affected by a co-injection of anti-IL-10 (5–20 µg) at the CCI site (Fig. 9A). There were also no changes in the von Frey withdrawal thresholds in the contralateral paws (data not shown). Furthermore, IL-4 (10–200 ng/ml) did not induce the release of IL-10 from M1 macrophages isolated from the injured nerves (Fig. 9B). Thus, antinociception produced by a single perineural IL-4 application does not involve IL-10.
Figure 9.

IL-4-induced actions do not involve IL-10. A, IL-4 (200 ng)-induced antinociception is not reversed by anti-IL-10 (F(3,32) = 1.413, p = 0.2569; one-way ANOVA). N = 9 mice per group. IL-4 was co-injected with anti-IL-10 or control IgG at the CCI site and mechanical von Frey thresholds were measured 5 min after injections, on day 14 after CCI. B, IL-4 (10–200 ng/ml) does not induce IL-10 release from M1 macrophages from injured nerves (all values represent 0). F4/80+ macrophages were isolated by IMS from injured nerves on day 14 after CCI, ex vivo stimulated (5 min) with IL-4 or vehicle (PBS), and extracellular levels of IL-10 were measured by ELISA. N = 8 samples per group. All data are presented as individual data points and mean ± SEM.
Discussion
The main finding of our study is that single application of IL-4 at injured nerves produces acute antinociception mediated by IL-4Rα-dependent release of opioid peptides from M1 macrophages. This concept is supported by the following data: (1) IL-4-induced attenuation of mechanical hypersensitivity was reversed by blocking IL-4Rα, opioid peptides (ENK, END, DYN), or opioid receptors (δ, µ, κ) at the site of nerve injury; (2) injured nerves were predominately infiltrated by M1 macrophages and IL-4 did not alter their numbers or the phenotype; (3) IL-4 induced release of all three opioid peptides from M1 macrophages isolated from injured nerves; and (4) the IL-4-induced release of ENK (used as a representative opioid peptide) was mediated by IL-4Rα and intracellular Ca2+ likely secreted from ER, which involved activation of PKA, PI3K, and ryanodine receptors.
Antinociceptive effects of IL-4 have been proposed to result from the inhibition of proinflammatory cytokine production, suppression of iNOS, and attenuation of cyclooxygenase-2-dependent prostaglandin synthesis (Seitz et al., 1994; Onoe et al., 1996; Vale et al., 2003). Additionally, Kiguchi et al. (2015) suggested the contribution of macrophage-derived IL-10, since the repetitive application of IL-4 at injured nerves elevated the IL-10 mRNA expression in nerve homogenates and this effect correlated with the attenuation of neuropathy-induced hypersensitivity. In contrast, here we demonstrate that although M1 macrophages expressed IL-10 mRNA and protein, a single treatment with IL-4 did not alter the IL-10 mRNA levels and did not induce IL-10 release from M1 cells. Furthermore, blockade of IL-10 did not affect the IL-4-induced antinociception, clearly suggesting that IL-4 actions were independent of IL-10. Whereas the mode of IL-4 injection (repetitive vs single) might have contributed to these discrepancies, it needs to be noted that Kiguchi et al. (2015) have neither examined the presence of IL-10 in perineural macrophages nor the direct IL-10 involvement in IL-4-induced antinociception.
Here, we uncovered that the IL-4-induced antinociception was dependent on the opioid system. All three opioid peptides (ENK, END, DYN) and receptors (δ, μ, κ) at injured nerves are involved, since the perineural application of selective antibodies to each opioid peptide or antagonists of each opioid receptor diminished IL-4-induced antinociception. Additionally, this effect was inhibited by NLXM, an opioid receptor antagonist with limited blood-brain barrier permeability (Brown and Goldberg, 1985), suggesting the involvement of peripheral opioid receptors. This is relevant, because in contrast to opioid receptors in the brain, the activation of peripheral opioid receptors is devoid of serious side effects such as respiratory arrest, sedation, and addiction (Spahn et al., 2017; Machelska and Celik, 2018). The lack of attenuation of IL-4-induced antinociception by naloxone (a non-selective antagonist acting at peripheral and central opioid receptors) in the acetic acid-induced writhing model in the earlier study could be related to the use of only one, possibly insufficient naloxone dose (Vale et al., 2003). Thus, our comprehensive dose-dependent experiments unrevealed the contribution of peripheral opioid receptors activated by endogenous opioid peptides at injured nerves to the attenuation of mechanical hypersensitivity induced by a single perineural IL-4 injection. This notion is further supported by our in vitro data showing that IL-4 does not bind to opioid receptors in HEK cells.
The next step was to identify the cellular source of opioid peptides. We found that injured nerves (14 d following CCI) are predominately infiltrated by F4/80+ macrophages identified by flow cytometry, and that these cells display M1 phenotype characterized by substantially higher mRNA levels of proinflammatory markers (Il-1β, Tnf, iNos) compared with anti-inflammatory M2 cell markers (Il-10, Arg-1, Ym1) detected by qRT-PCR, in line with previous studies (Labuz et al., 2009, 2010; Celik et al., 2020). Single perineural IL-4 injection neither changed the number nor the phenotype of M1 macrophages. Furthermore, this IL-4 treatment did not alter the mRNA expression of opioid peptide precursors Penk, Pomc, and Pdyn, but interestingly, diminished the intracellular content of ENK and END in M1 macrophages. These data indicated that IL-4 might release opioid peptides from M1 cells. Indeed, our ex vivo experiments revealed a dose-dependent secretion of ENK, END, and DYN from these cells. Lack of changes in the opioid peptide production following single IL-4 application is not surprising, since such effects require several days-lasting in vitro incubation or repetitive in vivo treatment with IL-4 (Busch-Dienstfertig et al., 2012; Pannell et al., 2016; Celik et al., 2020). However, the IL-4-induced release actions have not been so far described.
Therefore, it was important to examine the mechanisms of IL-4-induced opioid peptide secretion. Using ENK as a representative opioid peptide, we found that its release was diminished by anti-IL-4Rα, confirming the contribution of IL-4Rα. Consistent with the regulated exocytosis, the IL-4-induced ENK release was dependent on Ca2+, apparently deriving from intracellular stores such as ER, since the effect was fully blocked by the intracellular Ca2+ chelator, but was unaffected by the extracellular Ca2+ chelator. Searching for the mechanism of Ca2+-mediated ENK secretion we first examined the PLC–IP3 receptor pathway, which has been shown to induce opioid peptide release from immune cells in response to activation of CXC chemokine receptors 2 or opioid receptors (Rittner et al., 2006; Celik et al., 2016). However, inhibitors of PLC and IP3 receptors did not affect the IL-4-induced ENK release, suggesting that this pathway is not involved. Apart from the IP3 receptors, the other important ER channels responsible for the intracellular Ca2+ mobilization are ryanodine receptors (Berridge, 1993), which have been found in immune cells such as dendritic cells, B and T lymphocytes, and monocytes (Hakamata et al., 1994; Sei et al., 1999; Hosoi et al., 2001; O'Connell et al., 2002). The ryanodine receptor activity can be enhanced by PKA-dependent phosphorylation (Hain et al., 1995), which resulted in the intracellular Ca2+ release (Patel et al., 1995), and IL-4 promoted the PKA activation in B lymphocytes (McKay et al., 2000). Additionally, IL-4 via IL-4Rα can activate PI3K (Kelly-Welch et al., 2003; O'Connor et al., 2007), which resulted in Ca2+ mobilization from the intracellular stores in neurons (Vogelaar et al., 2018), and might also involve ryanodine receptors (Pereira et al., 2017). In line with these data, we found that the IL-4-induced ENK secretion was diminished by inhibitors of PKA, PI3K, and ryanodine receptors. Hence, our findings suggest that IL-4 releases opioid peptides from M1 macrophages via IL-4Rα, which is dependent on PKA, PI3K, and ryanodine receptor activation, any likely underlies the IL-4-mediated attenuation of nerve injury-triggered mechanical hypersensitivity. Interestingly, these opioid-mediated actions of IL-4 in M1 macrophages are different from those in M2 macrophages. Thus, when macrophages were polarized by IL-4 into the M2 phenotype, the M2 cells released opioids constitutively, without involvement of IL-4Rα to ameliorate nerve injury-triggered mechanical pain (Pannell et al., 2016; Celik et al., 2020).
The finding that heat hypersensitivity was not attenuated by single IL-4 injection is in agreement with earlier studies showing that IL-4 knock-out mice displayed enhanced acute mechanical, but unaltered heat sensitivity (Üçeyler et al., 2011) and that repetitive IL-4 application at the CCI site in wild-type mice ameliorated mechanical, but not heat hypersensitivity (Celik et al., 2020). This might be related to findings that in contrast to exogenous opioid receptor agonists, the endogenous opioid peptides are less relevant to the attenuation of CCI-induced heat then mechanical hypersensitivity (Labuz et al., 2016; Pannell et al., 2016), but additional work is needed to mechanistically explain these issues. Thus, as the role of IL-4, opioid peptides, and immune cells in the modulation of different pain modalities remains to be clarified, it appears that opioid-dependent actions of IL-4 and macrophages are mainly beneficial in diminishing mechanical hypersensitivity. As we used male mice, it will also be important to examine females, since the involvement of macrophages in pain may be sex dependent (Luo et al., 2019).
In conclusion, in this study, we have identified a new opioid mechanism involved in antinociceptive effects of IL-4. Hence, activation of IL-4Rα by IL-4 in M1 cells leads to the acute, intracellular Ca2+-regulated opioid peptide secretion and transient attenuation of nerve injury-triggered mechanical pain. For a therapeutic benefit, M1 cells need to be switched to M2 cells, which continuously release opioid peptides resulting in long-lasting antinociception (Celik et al., 2020).
Footnotes
This work was supported by a the Deutsche Forschungsgemeinschaft Grant MA 6432/2-1 (to H.M.). We thank Nicole Vogel for technical assistance in binding experiments.
The authors declare no competing financial interests.
References
- Berridge MJ (1993) Inositol trisphosphate and calcium signalling. Nature 361:315–325. 10.1038/361315a0 [DOI] [PubMed] [Google Scholar]
- Börner C, Wöltje M, Höllt V, Kraus J (2004) STAT6 transcription factor binding sites with mismatches within the canonical 5'-TTC…GAA-3' motif involved in regulation of delta- and mu-opioid receptors. J Neurochem 91:1493–1500. 10.1111/j.1471-4159.2004.02846.x [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. 10.1006/abio.1976.9999 [DOI] [PubMed] [Google Scholar]
- Brown DR, Goldberg LI (1985) The use of quaternary narcotic antagonists in opiate research. Neuropharmacology 24:181–191. 10.1016/0028-3908(85)90072-3 [DOI] [PubMed] [Google Scholar]
- Busch-Dienstfertig M, Labuz D, Wolfram T, Vogel NV, Stein C (2012) JAK-STAT1/3-induced expression of signal sequence-encoding proopiomelanocortin mRNA in lymphocytes reduces inflammatory pain in rats. Mol Pain 8:83. 10.1186/1744-8069-8-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik MÖ, Labuz D, Henning K, Busch-Dienstfertig M, Ruff-Gaveriaux C, Kieffer B, Zimmer A, Machelska H (2016) Leukocyte opioid receptors mediate analgesia via Ca(2+)-regulated release of opioid peptides. Brain Behav Immun 57:227–242. 10.1016/j.bbi.2016.04.018 [DOI] [PubMed] [Google Scholar]
- Celik MÖ, Labuz D, Keye J, Glauben R, Machelska H (2020) IL-4 induces M2 macrophages to produce sustained analgesia via opioids. JCI Insight 5:e133093. 10.1172/jci.insight.133093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53:55–63. 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- Cunha FQ, Poole S, Lorenzetti BB, Veiga FH, Ferreira SH (1999) Cytokine-mediated inflammatory hyperalgesia limited by interleukin-4. Br J Pharmacol 126:45–50. 10.1038/sj.bjp.0702266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadani SP, Cronk JC, Norris GT, Kipnis J (2012) IL-4 in the brain: a cytokine to remember. J Immunol 189:4213–4219. 10.4049/jimmunol.1202246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K, Thomas P, Breit S, Dugas M, Mailhammer R, van Eden W, van der Zee R, Biedermann T, Prinz J, Mack M, Mrowietz U, Christophers E, Schlöndorff D, Plewig G, Sander CA, Röcken M (2003) Interleukin-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat Med 9:40–46. 10.1038/nm804 [DOI] [PubMed] [Google Scholar]
- Hain J, Onoue H, Mayrleitner M, Fleischer S, Schindler H (1995) Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. J Biol Chem 270:2074–2081. 10.1074/jbc.270.5.2074 [DOI] [PubMed] [Google Scholar]
- Hakamata Y, Nishimura S, Nakai J, Nakashima Y, Kita T, Imoto K (1994) Involvement of the brain type of ryanodine receptor in T-cell proliferation. FEBS Lett 352:206–210. 10.1016/0014-5793(94)00955-4 [DOI] [PubMed] [Google Scholar]
- Hao S, Mata M, Glorioso JC, Fink DJ (2006) HSV-mediated expression of interleukin-4 in dorsal root ganglion neurons reduces neuropathic pain. Mol Pain 2:6. 10.1186/1744-8069-2-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey MJ, Granger DN, Kubes P (1999) Molecular mechanisms underlying IL-4-induced leukocyte recruitment in vivo: a critical role for the alpha 4 integrin. J Immunol 163:3441–3448. [PubMed] [Google Scholar]
- Hosoi E, Nishizaki C, Gallagher KL, Wyre HW, Matsuo Y, Sei Y (2001) Expression of the ryanodine receptor isoforms in immune cells. J Immunol 167:4887–4894. 2001 10.4049/jimmunol.167.9.4887 [DOI] [PubMed] [Google Scholar]
- Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD (2003) Interleukin-4 and interleukin-13 signaling connections maps. Science 300:1527–1528. 10.1126/science.1085458 [DOI] [PubMed] [Google Scholar]
- Kiguchi N, Kobayashi Y, Saika F, Sakaguchi H, Maeda T, Kishioka S (2015) Peripheral interleukin-4 ameliorates inflammatory macrophage-dependent neuropathic pain. Pain 156:684–693. 10.1097/j.pain.0000000000000097 [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010) Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8:e1000412. 10.1371/journal.pbio.1000412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus J, Borner C, Giannini E, Hickfang K, Braun H, Mayer P, Hoehe MR, Ambrosch A, Konig W, Höllt V (2001) Regulation of mu-opioid receptor gene transcription by interleukin-4 and influence of an allelic variation within a STAT6 transcription factor binding site. J Biol Chem 276:43901–43908. 10.1074/jbc.M107543200 [DOI] [PubMed] [Google Scholar]
- Labuz D, Machelska H (2013) Stronger antinociceptive efficacy of opioids at the injured nerve trunk than at its peripheral terminals in neuropathic pain. J Pharmacol Exp Ther 346:535–544. 10.1124/jpet.113.205344 [DOI] [PubMed] [Google Scholar]
- Labuz D, Schmidt Y, Schreiter A, Rittner HL, Mousa SA, Machelska H (2009) Immune cell-derived opioids protect against neuropathic pain in mice. J Clin Invest 119:278–286. 10.1172/JCI36246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labuz D, Schreiter A, Schmidt Y, Brack A, Machelska H (2010) T lymphocytes containing β-endorphin ameliorate mechanical hypersensitivity following nerve injury. Brain Behav Immun 24:1045–1053. 10.1016/j.bbi.2010.04.001 [DOI] [PubMed] [Google Scholar]
- Labuz D, Celik MÖ, Zimmer A, Machelska H (2016) Distinct roles of exogenous opioid agonists and endogenous opioid peptides in the peripheral control of neuropathy-triggered heat pain. Sci Rep 6:32799. 10.1038/srep32799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Huh Y, Bang S, He Q, Zhang L, Matsuda M, Ji RR (2019) Macrophage toll-like receptor 9 contributes to chemotherapy-induced neuropathic pain in male mice. J Neurosci 39:6848–6864. 10.1523/JNEUROSCI.3257-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzina IG, Keegan AD, Heller NM, Rook GA, Shea-Donohue T, Atamas SP (2012) Regulation of inflammation by interleukin-4: a review of “alternatives”. J Leukoc Biol 92:753–764. 10.1189/jlb.0412214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machelska H, Celik MO (2018) Advances in achieving opioid analgesia without side effects. Front Pharmacol 9:1388. 10.3389/fphar.2018.01388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Helming L, Gordon S (2009) Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 27:451–483. 10.1146/annurev.immunol.021908.132532 [DOI] [PubMed] [Google Scholar]
- McKay CE, Hewitt EL, Ozanne BW, Cushley W (2000) A functional role for interleukin (IL)-4-driven cyclic amp accumulation in human b lymphocytes. Cytokine 12:731–736. 10.1006/cyto.1999.0627 [DOI] [PubMed] [Google Scholar]
- O'Connell PJ, Klyachko VA, Ahern GP (2002) Identification of functional type 1 ryanodine receptors in mouse dendritic cells. FEBS Lett 512:67–70. 10.1016/S0014-5793(01)03321-X [DOI] [PubMed] [Google Scholar]
- O'Connor JC, Sherry CL, Guest CB, Freund GG (2007) Type 2 diabetes impairs insulin receptor substrate-2-mediated phosphatidylinositol 3-kinase activity in primary macrophages to induce a state of cytokine resistance to IL-4 in association with overexpression of suppressor of cytokine signaling-3. J Immunol 178:6886–6893. 10.4049/jimmunol.178.11.6886 [DOI] [PubMed] [Google Scholar]
- Onoe Y, Miyaura C, Kaminakayashiki T, Nagai Y, Noguchi K, Chen QR, Seo H, Ohta H, Nozawa S, Kudo I, Suda T (1996) IL-13 and IL-4 inhibit bone resorption by suppressing cyclooxygenase-2-dependent prostaglandin synthesis in osteoblasts. J Immunol 156:758–764. [PubMed] [Google Scholar]
- Pannell M, Labuz D, Celik MÖ, Keye J, Batra A, Siegmund B, Machelska H (2016) Adoptive transfer of M2 macrophages reduces neuropathic pain via opioid peptides. J Neuroinflammation 13:262. 10.1186/s12974-016-0735-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JR, Coronado R, Moss RL (1995) Cardiac sarcoplasmic reticulum phosphorylation increases Ca2+ release induced by flash photolysis of nitr-5. Circ Res 77:943–949. 10.1161/01.res.77.5.943 [DOI] [PubMed] [Google Scholar]
- Pereira L, Bare DJ, Galice S, Shannon TR, Bers DM (2017) β-Adrenergic induced SR Ca2+ leak is mediated by an Epac-NOS pathway. J Mol Cell Cardiol 108:8–16. 10.1016/j.yjmcc.2017.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratthé C, Ennaciri J, Garcês Gonçalves DM, Chiasson S, Girard D (2009) Interleukin (IL)-4 induces leukocyte infiltration in vivo by an indirect mechanism. Mediators Inflamm 2009:193970. 10.1155/2009/193970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiken S, Lacampagne A, Zhou H, Kherani A, Lehnart SE, Ward C, Huang F, Gaburjakova M, Gaburjakova J, Rosemblit N, Warren MS, He KL, Yi GH, Wang J, Burkhoff D, Vassort G, Marks AR (2003) PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J Cell Biol 160:919–928. 10.1083/jcb.200211012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittner HL, Labuz D, Schaefer M, Mousa SA, Schulz S, Schäfer M, Stein C, Brack A (2006) Pain control by CXCR2 ligands through Ca2+-regulated release of opioid peptides from polymorphonuclear cells. FASEB J 20:2627–2629. 10.1096/fj.06-6077fje [DOI] [PubMed] [Google Scholar]
- Schreiter A, Gore C, Labuz D, Fournie-Zaluski MC, Roques BP, Stein C, Machelska H (2012) Pain inhibition by blocking leukocytic and neuronal opioid peptidases in peripheral inflamed tissue. FASEB J 26:5161–5171. 10.1096/fj.12-208678 [DOI] [PubMed] [Google Scholar]
- Sei Y, Gallagher KL, Basile AS (1999) Skeletal muscle type ryanodine receptor is involved in calcium signaling in human B lymphocytes. J Biol Chem 274:5995–6002. 10.1074/jbc.274.9.5995 [DOI] [PubMed] [Google Scholar]
- Seitz M, Loetscher P, Dewald B, Towbin H, Ceska M, Baggiolini M (1994) Production of interleukin-1 receptor antagonist, inflammatory chemotactic proteins, and prostaglandin E by rheumatoid and osteoarthritic synoviocytes–regulation by IFN-gamma and IL-4. J Immunol 152:2060–2065. [PubMed] [Google Scholar]
- Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122:787–795. 10.1172/JCI59643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn V, Del Vecchio G, Labuz D, Rodriguez-Gaztelumendi A, Massaly N, Temp J, Durmaz V, Sabri P, Reidelbach M, Machelska H, Weber M, Stein C (2017) A nontoxic pain killer designed by modeling of pathological receptor conformations. Science 355:966–969. 10.1126/science.aai8636 [DOI] [PubMed] [Google Scholar]
- Üçeyler N, Valenza R, Stock M, Schedel R, Sprotte G, Sommer C (2006) Reduced levels of antiinflammatory cytokines in patients with chronic widespread pain. Arthritis Rheum 54:2656–2664. 10.1002/art.22026 [DOI] [PubMed] [Google Scholar]
- Üçeyler N, Rogausch JP, Toyka KV, Sommer C (2007) Differential expression of cytokines in painful and painless neuropathies. Neurology 69:42–49. 10.1212/01.wnl.0000265062.92340.a5 [DOI] [PubMed] [Google Scholar]
- Üçeyler N, Topuzoğlu T, Schießer P, Hahnenkamp S, Sommer C (2011) IL-4 deficiency is associated with mechanical hypersensitivity in mice. PLoS One 6:e28205. 10.1371/journal.pone.0028205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale ML, Marques JB, Moreira CA, Rocha FA, Ferreira SH, Poole S, Cunha FQ, Ribeiro RA (2003) Antinociceptive effects of interleukin-4, -10, and -13 on the writhing response in mice and zymosan-induced knee joint incapacitation in rats. J Pharmacol Exp Ther 304:102–108. 10.1124/jpet.102.038703 [DOI] [PubMed] [Google Scholar]
- van Roon JA, Lafeber FP, Bijlsma JW (2001) Synergistic activity of interleukin-4 and interleukin-10 in suppression of inflammation and joint destruction in rheumatoid arthritis. Arthritis Rheum 44:3–12. 10.1002/1529-0131(200101)44:1<3::AID-ANR2>3.0.CO;2-U [DOI] [PubMed] [Google Scholar]
- Vogelaar CF, Mandal S, Lerch S, Birkner K, Birkenstock J, Bühler U, Schnatz A, Raine CS, Bittner S, Vogt J, Kipnis J, Nitsch R, Zipp F (2018) Fast direct neuronal signaling via the IL-4 receptor as therapeutic target in neuroinflammation. Sci Transl Med 10:eaao2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills-Karp M, Finkelman FD (2008) Untangling the complex web of IL-4–and IL-13–mediated signaling pathways. Sci Signal 1:pe55. 10.1126/scisignal.1.51.pe55 [DOI] [PMC free article] [PubMed] [Google Scholar]