

Abstract
The breast cancer susceptibility protein BRCA1 and its partner BRCA1-associated RING domain protein 1 (BARD1) form an E3-ubiquitin (Ub) ligase complex that acts as a tumor suppressor in mitotic cells. However, the roles of BRCA1–BARD1 in postmitotic cells, such as neurons, remain poorly defined. Here, we report that BRC-1 and BRD-1, the Caenorhabditis elegans orthologs of BRCA1 and BARD1, are required for adult-specific axon regeneration, which is positively regulated by the EGL-30 Gqα–diacylglycerol (DAG) signaling pathway. This pathway is downregulated by DAG kinase (DGK), which converts DAG to phosphatidic acid (PA). We demonstrate that inactivation of DGK-3 suppresses the brc-1 brd-1 defect in axon regeneration, suggesting that BRC-1–BRD-1 inhibits DGK-3 function. Indeed, we show that BRC-1–BRD-1 poly-ubiquitylates DGK-3 in a manner dependent on its E3 ligase activity, causing DGK-3 degradation. Furthermore, we find that axon injury causes the translocation of BRC-1 from the nucleus to the cytoplasm, where DGK-3 is localized. These results suggest that the BRC-1–BRD-1 complex regulates axon regeneration in concert with the Gqα–DAG signaling network. Thus, this study describes a new role for breast cancer proteins in fully differentiated neurons and the molecular mechanism underlying the regulation of axon regeneration in response to nerve injury.
SIGNIFICANCE STATEMENT BRCA1–BRCA1-associated RING domain protein 1 (BARD1) is an E3-ubiquitin (Ub) ligase complex acting as a tumor suppressor in mitotic cells. The roles of BRCA1–BARD1 in postmitotic cells, such as neurons, remain poorly defined. We show here that Caenorhabditis elegans BRC-1/BRCA1 and BRD-1/BARD1 are required for adult-specific axon regeneration, a process that requires high diacylglycerol (DAG) levels in injured neurons. The DAG kinase (DGK)-3 inhibits axon regeneration by reducing DAG levels. We find that BRC-1–BRD-1 poly-ubiquitylates and degrades DGK-3, thereby keeping DAG levels elevated and promoting axon regeneration. Furthermore, we demonstrate that axon injury causes the translocation of BRC-1 from the nucleus to the cytoplasm, where DGK-3 is localized. Thus, this study describes a new role for BRCA1–BARD1 in fully-differentiated neurons.
Keywords: axon regeneration, BRCA1-BARD1, Caenorhabditis elegans, diacylglycerol kinase
Introduction
Genetic susceptibility to breast cancer is caused largely by mutations in the BRCA1 and BRCA2 genes (Fackenthal and Olopade, 2007). These regulate a wide range of biological processes, including DNA damage repair by homologous recombination, gene silencing, cell cycle checkpoint, and centrosome duplication, all of which are relevant to the regulation of cell proliferation (Scully and Livingston, 2000; Moynahan and Jasin, 2010; Zhu et al., 2011; Li and Greenberg, 2012). BRCA1 exists primarily in a heterodimeric complex with the BRCA1-associated RING domain protein 1 (BARD1; Wu et al., 1996). It has been shown that this BRCA1–BARD1 complex possesses E3-ubiquitin (Ub) ligase activity, and this activity can be disrupted by cancer-derived mutations, underscoring the critical role of this enzymatic function in suppressing tumorigenesis (Baer and Ludwig, 2002). To date, intensive efforts have been devoted to understanding the tumor-suppressive functions of BRCA1–BARD1 and BRCA2 in mitotic cells. However, their roles in postmitotic cells, such as neurons, remain poorly understood at the molecular level.
Neurons are one type of postmitotic cell, specialized for transmitting information over long distances through axons. Although axons can be damaged by various internal and external stresses, neurons have a conserved system of regenerating axons postinjury, and failure of this system can cause sensory and motor paralysis. This axon's regenerative capacity is controlled by intrinsic neuronal signaling pathways (He and Jin, 2016). Upon axon injury, Ca2+ and cAMP levels rise in severed neurons, which drives various signaling pathways (Ghosh-Roy et al., 2010; Mar et al., 2014). For instance, cAMP elevation activates cAMP-dependent protein kinase A (PKA), which promotes axonal regeneration through phosphorylation of various downstream targets (Neumann et al., 2002; Bhatt et al., 2004; Gao et al., 2004). However, the intrinsic signaling pathways that regulate regeneration in the adult nervous system have yet to be fully elucidated.
The nematode Caenorhabditis elegans has recently emerged as an attractive model to dissect the mechanisms of axon regeneration in the mature nervous system (Yanik et al., 2004). Recent studies in C. elegans have identified many signaling molecules that promote or inhibit axon regeneration (Chen et al., 2011; Nix et al., 2014; Kim et al., 2018). We have previously demonstrated that the evolutionarily conserved JNK MAP kinase (MAPK) pathway, consisting of MLK-1 MAPKKK–MEK-1 MAPKK–KGB-1 JNK, drives the initiation of axon regeneration (Nix et al., 2011). Two different protein kinases act as MAP4Ks for MLK-1 in a manner specific for different life stages. The Ste20-related kinase MAX-2 phosphorylates and activates MLK-1 mainly at the L4 stage to promote axon regeneration (Pastuhov et al., 2016). On the other hand, the protein kinase C (PKC) ortholog TPA-1 can activate MLK-1 at the young adult stage, but not at the L4 stage (Pastuhov et al., 2012). The Gqα protein EGL-30 acts as a component upstream of TPA-1. EGL-30 activates the phospholipase Cβ (PLCβ) EGL-8, which in turn generates diacylglycerol (DAG), an activator of TPA-1, from phosphatidylinositol bisphosphate (Lackner et al., 1999). DAG kinases (DGKs) antagonize the EGL-30 pathway by converting DAG to phosphatidic acid (PA; Miller et al., 1999).
We have recently found that BRC-2, the C. elegans ortholog of BRCA2, acts as a regulator of axon regeneration (Shimizu et al., 2018). C. elegans also has two genes, brc-1 and brd-1, which encode orthologs of mammalian BRCA1 and BARD1, respectively (Fig. 1A; Boulton et al., 2004). BRC-1 and BRD-1 share extensive sequence and domain conservation with their mammalian counterparts, including RING and BRCT domains. Similar to mammalian BRCA1–BARD1, BRC-1 heterodimerizes with BRD-1 to form a complex having E3-Ub ligase activity (Polanowska et al., 2006). BRC-1–BRD-1 is involved in DNA repair at sites damaged by ionizing radiation. Our finding that BRC-2 is implicated in axon regeneration prompted us to explore the possibility that BRC-1 and BRD-1 also participate in this process.
Figure 1.

C. elegans BRC-1 and BRD-1. A, Structures of BRC-1 and BRD-1. Schematic diagrams of BRC-1, BRD-1, and their mammalian counterparts, BRCA1 and BARD1, are shown. RING finger domain is shown in red and BRCT domains in yellow. The bold lines underneath denote the extent of the deleted regions in the tm1145, dw1, and gk297 mutants. An asterisk indicates a premature stop codon caused by the km88 mutation. B, Isolation of brc-1 mutants. Genomic structure of the brc-1 gene is shown. Exons are indicated by boxes, and introns and untranslated regions are indicated by bars. Small and capital letters indicate nucleotides and the corresponding amino acids, respectively. The brc-1(km88) mutation is a 2-bp deletion, causing a frameshift (bold amino acids) and premature stop codon (*) in exon 2. C, Ring finger domain. Sequence alignment in the RING finger domain between BRCA1 and BRC-1 is shown. Identical and similar residues are highlighted with black and gray shading, respectively. The black arrow indicates the conserved isoleucine residue required for E3-Ub ligase activity.
In this study, we investigated the roles of BRC-1 and BRD-1 in axon regeneration. We found that the BRC-1–BRD-1 complex is required for axon regeneration after injury, specifically in the adult stage. We demonstrate that BRC-1–BRD-1 poly-ubiquitylates DGK-3, resulting in its degradation. Thus, BRC-1–BRD-1 enhances the EGL-30 signaling pathway by downregulating DGK-3 to promote axon regeneration. Furthermore, we show that PKA phosphorylates BRC-1, which causes the translocation of BRC-1 from the nucleus to the cytoplasm, where DGK-3 is localized. These results suggest that the BRC-1–BRD-1 complex regulates axon regeneration in concert with the Gqα–DAG signaling network. Thus, this study uncovers an unexpected role of BRC-1–BRD-1 in postmitotic neurons and suggests a molecular mechanism by which BRC-1–BRD-1 regulates axon regeneration in response to nerve injury.
Materials and Methods
C. elegans strains
The C. elegans strains used in this study are listed in Table 1. All strains were maintained on nematode growth medium plates and fed with bacteria of the OP50 strain by the standard method (Brenner, 1974).
Table 1.
Strains used in this study
| Strain | Genotype |
|---|---|
| KU501 | juIs76 II |
| KU88 | juIs76 II; brc-1(km88) III |
| KU1440 | juIs76 II; brc-1(km88) III; kmEx1440 [Punc-25::brc-1] |
| KU1441 | juIs76 II; brc-1(km88) III; kmEx1441 [Punc-25::brc-1(I23A)] |
| KU1442 | juIs76 II; brd-1(gk297) III |
| KU1443 | juIs76 II; brc-1(tm1145) brd-1(dw1) III |
| KU1444 | juIs76 II; kmEx1444 [Punc-25::brc-1 + Punc-25::brd-1 (line 1)] |
| KU1445 | juIs76 II; kmEx1445 [Punc-25::brc-1 + Punc-25::brd-1 (line 2)] |
| KU456 | egl-30(ad805) I; juIs76 II |
| KU457 | egl-30(tg26) I; juIs76 II |
| KU461 | juIs76 II; tpa-1(k501) IV |
| KU1446 | egl-30(ad805) I; juIs76 II; brc-1(tm1145) brd-1(dw1) III |
| KU1447 | egl-30(tg26) I; juIs76 II; brc-1(tm1145) brd-1(dw1) III |
| KU1448 | goa-1(n1134) I; juIs76 II; brc-1(tm1145) brd-1(dw1) III |
| KU1449 | eat-16(nj8) I; juIs76 II; brc-1(tm1145) brd-1(dw1) III |
| KU1450 | juIs76 II; brc-1(tm1145) brd-1(dw1) III; dgk-1(ok1462) X |
| KU89 | juIs76 II; dgk-3(km89) III |
| KU1451 | juIs76 II; brc-1(tm1145) brd-1(dw1) dgk-3(km89) III |
| KU1452 | juIs76 II; brc-1(tm1145) brd-1(dw1) dgk-3(km89) III; dgk-1(ok1462) X |
| KU1453 | juIs76 II; brc-1(tm1145) brd-1(dw1) dgk-3(km90) III; dgk-1(ok1462) X |
| KU1454 | juIs76 II; brc-1(tm1145) brd-1(dw1) dgk-3(km89) III; tpa-1(k501) IV |
| KU1455 | juIs76 II; brc-1(S266A) III |
| KU1456 | wpIs36 I; kmEx1456 [Punc-25::dgk-3::gfpnovo2] |
| KU1457 | wpIs36 I; brc-1(tm1145) brd-1(dw1) III; kmEx1456 [Punc-25::dgk-3::gfpnovo2] |
| KU1458 | wpIs36 I; kmEx1458 [Punc-25::gfpnovo2::brc-1] |
| KU1459 | wpIs36 I; kmEx1459 [Punc-25::gfpnovo2::brc-1(S266A)] |
| KU1343 | muIs32 II |
| KU1460 | muIs32 II; brc-1(tm1145) brd-1(dw1) III |
Plasmids
Punc-25::brc-1 and Punc-25::brd-1 were respectively generated by inserting brc-1 cDNA (isoform a) and brd-1 cDNA isolated from cDNA library into a pSC325 vector, respectively. Punc-25::gfpnovo2::brc-1 was generated by inserting the GFPnovo2 coding sequence isolated from the pSM-GFPnovo2 plasmid into Punc-25::brc-1. Punc-25::brc-1(I23A) and Punc-25::gfpnovo2::brc-1(S266A) were generated by oligonucleotide-directed PCR using Punc-25::brc-1 and Punc-25::gfpnovo2::brc-1 as templates, respectively, and the mutations were verified by DNA sequencing. Punc-25::dgk-3::gfpnovo2 was generated by inserting the dgk-3 cDNA and the GFPnovo2 coding sequence, which were isolated from a cDNA library and the pSM-GFPnovo2 plasmid, respectively, into the pSC325 vector. The T7-DGK-3, GFP-BRC-1, and BRD-1-RFP plasmids were generated by inserting the dgk-3, brc-1 and brd-1 cDNAs into the pCMV-T7, pEGFP-C1, and pTagRFP-N vectors, respectively. GFP-BRC-1(I23A) was generated by oligonucleotide-directed PCR using GFP-BRC-1 as a template, and the mutation was verified by DNA sequencing. The Pmyo-2::dsred-monomer, and HA-Ub plasmids were described previously (Hanafusa et al., 2011; Li et al., 2012).
Generation of the brc-1 and dgk-3 mutations using CRISPR–Cas9
The brc-1 mutations (km88 deletion and S266A point mutation) and the dgk-3 mutations (km89 insertion and km90 deletion) were obtained using the CRISPR–Cas9 system as described previously (Dokshin et al., 2018). The CRISPR RNAs [5′-UGGAAACAUGUGGACAGAAU-3′ for brc-1(km88), 5′-UUGCGAGUUCUCAAGAUCUU-3′ for brc-1(S266A), and 5′-UAUCACCGGAGCAAUUCUCG-3′ for dgk-3(km89, km90)] and the single-stranded donor template DNA [5′-ATCAGAGAAACCAGCGAATCGAAGAGTAgccTTTGCGAGTTCTCAAGATCTTGAAAACA\TAAAAATTATG-3′ for brc-1(S266A)] were synthesized (Integrated DNA Technologies; IDT), co-injected with the trans-activating CRISPR RNA (IDT), Streptococcus pyogenes Cas9 3NLS (IDT) protein, and the pRF4(rol-6d) plasmid into the KU501 [for brc-1(km88) and brc-1(S266A)] and KU1448 [for dgk-3(km89, km90)] strains. Each of the F1 animals carrying the transgene was transferred onto a new dish and used for single-worm PCR, followed by DNA sequencing to detect the mutations. The brc-1(km88) mutation is a 2-bp deletion in the brc-1 gene, causing a frameshift and premature stop codon in exon 2. The dgk-3(km89) mutation is a 20-bp insertion that contains an in-frame stop codon, thus terminating translation in the middle of exon 1. The dgk-3(km90) mutation is a 5-bp deletion, causing a frameshift and premature stop codon in exon 1.
Transgenic animals
Transgenic animals were obtained using the standard C. elegans microinjection method (Mello et al., 1991). Pmyo-2::dsred-monomer, Punc-25::brc-1, Punc-25::brc-1(I23A), Punc-25::brd-1, Punc-25::dgk-3::gfpnovo2, Punc-25::gfpnovo2::brc-1, and Punc-25::gfpnovo2::brc-1(S266A) plasmids were used in kmEx1440 [Punc-25::brc-1 (5 ng/µl) + Pmyo-2::dsred-monomer (5 ng/µl)], kmEx1441 [Punc-25::brc-1(I23A) (5 ng/µl) + Pmyo-2::dsred-monomer (5 ng/µl)], kmEx1444/kmEx1445 [Punc-25::brc-1 (25 ng/µl) + Punc-25::brd-1 (25 ng/µl) + Pmyo-2::dsred-monomer (5 ng/µl)], kmEx1456 [Punc-25::dgk-3::gfpnovo2 (5 ng/µl) + Pmyo-2::dsred-monomer (5 ng/µl)], kmEx1458 [Punc-25::gfpnovo2::brc-1 (10 ng/µl)+ Pmyo-2::dsred-monomer (5 ng/µl)], kmEx1459 [Punc-25::gfpnovo2::brc-1(S266A) (10 ng/µl) + Pmyo-2::dsred-monomer (5 ng/µl)], respectively. The juIs76, wpIs36, and muIs32 integrated arrays were described previously (Huang et al., 2002; Ch'ng et al., 2003; Firnhaber and Hammarlund, 2013).
Microscopy
Standard fluorescent images of transgenic animals were observed under an 100× objective of a Nikon ECLIPSE E800 fluorescent microscope and photographed with a Zyla CCD camera. Confocal fluorescent images were taken on a Zeiss LSM-800 confocal laser-scanning microscope with a 63× objective.
Axotomy
Axotomy and microscopy were performed as described previously (Li et al., 2012). Animals were subjected to axotomy at the young adult or L4 stage. The young adult stage was defined as a state in which the vulva is well developed and no eggs have formed yet. Imaged commissures that had growth cones or small branches present on the proximal fragment were counted as “regenerated.” Proximal fragments that showed no change after 24 h were counted as “no regeneration.” A minimum of 20 individuals with one to three axotomized commissures were observed for most experiments.
Measurements of regenerating axons
The length of regenerating axons for either D-type motor neurons or touch sensory posterior lateral microtubule (PLM) neurons was measured using the segmented line tool of ImageJ. Measurements were made from the site of injury to the tip of the longest branch of the regenerating axon. Axons that did not regenerate were excluded. Data were plotted using R (ver. 4.0.1) and R studio (ver. 1.3.959).
Immunoprecipitation
For immunoprecipitation, transfected COS-7 cells that were incubated with or without MG132 (Sigma; 10 μm) for 8 h were lysed in RIPA buffer [50 mm Tris–HCl, pH 7.4, 0.15 m NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mm EDTA, 1 mm dithiothreitol, 1 mm phenylmethylsulfonyl fluoride, phosphatase inhibitor cocktail 2 and 3 (Sigma), and protease inhibitor cocktail (Sigma)], followed by centrifugation at 15,000 × g for 12 min. A total of 10 µl (bed volume) of Dynabeads Protein G (Invitrogen) with anti-T7 antibody (PM022; MBL) was added to supernatant and the sample was rotated for 2 h at 4°C. The beads were then washed three times with ice-cold PBS and subjected to immunoblotting.
Immunoblotting
After cell extracts were subjected to SDS-PAGE, proteins were transferred to a polyvinylidene difluoride membrane (Hybond-P; GE Healthcare). The membranes were immunoblotted with anti-HA antibody (mouse 16B12; BioLegend), anti-T7 antibody (mouse T7-Tag; Merck; or rabbit PM022; MBL), anti-GFP antibody (mouse JL-8; Clontech), or anti-RFP antibody (rabbit AB233; Evrogen), and bound antibodies were visualized with horseradish peroxidase (HRP)-conjugated antibodies against rabbit or mouse IgG using an HRP chemiluminescent substrate reagent kit (Novex ECL; Invitrogen).
In vitro kinase assays
GFP-BRC-1 proteins were immunopurified from transfected COS-7 cells using anti-GFP antibody (mouse M048-3; MBL). Kinase reactions were performed in a final volume of 20 µl in buffer consisting of 25 mm MOPS (pH 7.2), 12.5 mm glycerol phosphate, 25 mm MgCl2, 2 mm EDTA, 0.25 mm DTT, 200 μm ATP, and 0.4 µg of recombinant PKA (Carna Biosciences). Samples were incubated for 20 min at 30°C and the reactions were terminated by the addition of Laemmli sample buffer and boiling. Phosphorylation of BRC-1 was detected by immunoblotting with rabbit anti-phospho-PKA substrate antibody (100G7E; Cell Signaling).
Forskolin treatment
Treatment of animals with forskolin was performed as described previously (Ghosh-Roy et al., 2010). Forskolin (ab120058; Abcam) dissolved in DMSO was diluted in M9 media (500 mm). L4 stage worms were incubated in the forskolin solution (containing heat-killed OP50) for 12 h followed by fluorescent microscopic observation.
Quantification of DGK-3 poly-ubiquitylation
To compare differences in DGK-3 poly-ubiquitylation, band intensity minus background of HA (Ub) and T7 (DGK-3) was quantified in lanes 4 and 5 using the FUSION system (VILBER). The HA (Ub) value was divided by the corresponding T7 (DGK-3) value to determine a normalized HA (Ub) value for lanes 4 and 5. To compare DGK-3 poly-ubiquitylation levels between lanes 4 and 5, normalized HA (Ub) values in lane 5 were divided by the values in lane 4, and the derived ratios were plotted on a bar graph.
Quantitative measures of fluorescence intensity for DGK-3 degradation
Animals expressing mCherry and DGK-3::GFP in D-type motor neurons were imaged immediately after axotomy (0 h) and 8 h after axotomy of selected motor neuron axons. A LSM800 confocal microscope (Zeiss) was used to obtain a z-stack of fluorescent images for mCherry and DGK-3::GFP. Mean intensity of DGK-3::GFP and mCherry in cytoplasm of neurons with severed axons was measured by drawing a circular region of interest in the center of the cell and using the measure function of ImageJ. Background intensity was determined near analyzed cells. Relative DGK-3::GFP intensity (RIDGK-3) was obtained by dividing the background-subtracted value of GFP by the corresponding background-corrected value of mCherry, followed by dividing the value 8 h after axotomy by the corresponding value 0 h after axotomy. The RIDGK-3 values for wild-type and brc-1 brd-1 mutants were plotted and checked for significant differences by Wilcoxon rank-sum test using R (ver. 4.0.1) and R studio (ver. 1.3.959).
Quantitative measures of fluorescence intensity for BRC-1 localization
Animals expressing mCherry and GFP-BRC-1 in D-type motor neurons with or without forskolin treatment were imaged using a Nikon ECLIPSE E800 fluorescent microscope and Zyla CCD camera. Mean intensities of GFP-BRC-1 and mCherry were measured in the cytoplasm and nucleus of D-type neurons, respectively. Background intensity was determined by measuring the mean GFP (or mCherry) intensity of adjacent regions of the same size. Normalized cytoplasmic and nuclear GFP-BRC-1 values were calculated by dividing background-subtracted cytoplasmic or nuclear GFP-BRC-1 by the corresponding background-corrected mCherry intensity. To compare cytoplasmic and nuclear GFP-BRC-1, a cytoplasmic-to-nuclear ratio was calculated and plotted using R (ver. 4.0.1) and R studio (ver. 1.3.959).
Experimental design and statistical analyses
All experiments were not randomized and the investigators were not blinded to the group allocation during experiments and outcome assessment. No statistical methods were used to predetermine sample size. Data visualization was performed using Microsoft Excel 2016, R (ver. 4.0.1), and R studio (ver. 1.3.959). Statistical analysis was conducted as described previously (Pastuhov et al., 2012). Briefly, 95% confidence intervals were calculated using the modified Wald method, and the two-tailed p values were calculated using Fisher's exact test on GraphPad QuickCalcs (http://www.graphpad.com/quickcalcs/contingency1/). The Wilcoxon rank-sum test (two-tailed) was performed using R (ver. 4.0.1), R studio (ver. 1.3.959), and the R exactRankTests package.
Results
BRC-1 and BRD-1 are required for axon regeneration
To assess whether the BRCA1 ortholog BRC-1 is involved in axon regeneration, we used the CRISPR–Cas9 system to generate the null mutant brc-1(km88), which harbors a 2-bp deletion generating a premature stop codon in the second exon of the brc-1 gene (Fig. 1A,B). We first assayed regrowth following laser axotomy in GABA-releasing D-type motor neurons (Fig. 2A). In young adult wild-type animals, ∼70% of the axons initiated regeneration within 24 h after axon injury (Fig. 2A,B; Table 2). However, in brc-1(km88) mutants the frequency of axon regeneration was significantly reduced (Fig. 2B; Table 2). This indicates that BRC-1 is required for efficient axon regeneration following laser axotomy. To test whether BRC-1 can act in a cell-autonomous manner, we expressed the brc-1 cDNA from the unc-25 promoter in brc-1 mutants. We found that the axon regeneration defect of brc-1(km88) mutants was rescued by expression of brc-1 in D-type motor neurons (Fig. 2B; Table 2). These results demonstrate that BRC-1 functions cell autonomously in injured neurons.
Figure 2.

BRC-1 and BRD-1 are required for axon regeneration of D-type motor neurons. A, Representative D-type motor neurons in wild-type and brc-1(tm1145) brd-1(dw1) mutant animals 24 h after laser surgery. In wild-type animals, severed axons exhibited regenerated growth cones (yellow arrowheads). In brc-1(tm1145) brd-1(dw1) mutants, the proximal ends of axons failed to regenerate (white arrowheads). Scale bar: 10 µm. B, Percentages of axons that initiated regeneration 24 h after laser surgery at the young adult stage. The number of axons examined is shown. Error bars indicate 95% confidence intervals; **p < 0.01, ***p < 0.001, as determined by Fisher's exact test. NS, not significant. C, Length of regenerating axons 24 h after laser surgery. Data are presented as a box-plot representing median (thick line within the box) and interquartile range (edge of box) with individual data points. The number (n) of axons examined is shown. Statistical significance was determined by Wilcoxon rank-sum test.
Table 2.
Raw data for genotypes tested by axotomy
| Strain | Genotype (juIs76 background) | Age | Number of axons | Number of regenerations (% of total) | p value | Compared with |
|---|---|---|---|---|---|---|
| KU501a1 | Wild type | YA | 62 | 44 (71%) | - | - |
| KU88 | brc-1(km88) | YA | 59 | 23 (39%) | 0.0005 | KU501a1 |
| KU1440 | brc-1(km88); kmEx1440 [Punc-25::brc-1] | YA | 55 | 37 (67%) | 0.0029 | KU88 |
| KU1441 | brc-1(km88); kmEx1441 [Punc-25::brc-1(I23A)] | YA | 51 | 25 (49%) | 0.3374 | KU88 |
| KU1442 | brd-1(gk297) | YA | 48 | 14 (29%) | <0.0001 | KU501a1 |
| KU1443a | brc-1(tm1145) brd-1(dw1) | YA | 45 | 15 (33%) | 0.8231 | KU1442 |
| KU501a2 | Wild type(–kmEx1444) | YA | 49 | 30 (61%) | - | - |
| KU1444 | kmEx1444 [Punc-25::brc-1 + Punc-25::brd-1 (line 1)] | YA | 65 | 51 (78%) | 0.0604 | KU501a2 |
| KU501a3 | Wild type(–kmEx1445) | YA | 50 | 32 (64%) | - | - |
| KU1445 | kmEx1445 [Punc-25::brc-1 + Punc-25::brd-1 (line 2)] | YA | 51 | 39 (76%) | 0.1961 | KU501a3 |
| KU501b | Wild type | L4 | 57 | 40 (70%) | - | - |
| YA | 74 | 50 (68%) | - | - | ||
| KU1443b | brc-1(tm1145) brd-1(dw1) | L4 | 71 | 51 (72%) | 0.8471 | KU501b (L4) |
| YA | 53 | 19 (36%) | 0.0006 | KU501b (YA) | ||
| KU456 | egl-30(ad805) | L4 | 41 | 31 (76%) | 0.2688 | KU501b (L4) |
| YA | 50 | 20 (40%) | 0.0031 | KU501b (YA) | ||
| KU457 | egl-30(tg26) | YA | 30 | 21 (70%) | 1.0000 | KU501b (YA) |
| KU461 | tpa-1(k501) | L4 | 49 | 34 (69%) | 1.0000 | KU501b (L4) |
| YA | 47 | 17 (36%) | 0.0013 | KU501b (YA) | ||
| KU1446 | egl-30(ad805); brc-1(tm1145) brd-1(dw1) | YA | 50 | 17 (34%) | 1.0000 | KU1443b(YA) |
| 0.6790 | KU456(YA) | |||||
| KU1447 | egl-30(tg26); brc-1(tm1145) brd-1(dw1) | YA | 94 | 61(65%) | 0.0010 | KU1443b(YA) |
| KU501c | Wild type | YA | 60 | 37 (62%) | - | - |
| KU1443c | brc-1(tm1145) brd-1(dw1) | YA | 48 | 18 (38%) | 0.0197 | KU501c |
| KU1448 | goa-1(n1134); brc-1(tm1145) brd-1(dw1) | YA | 58 | 20 (34%) | 0.8395 | KU1443c |
| KU1449 | eat-16(nj8); brc-1(tm1145) brd-1(dw1) | YA | 51 | 13 (25%) | 0.2783 | KU1443c |
| KU1450 | brc-1(tm1145) brd-1(dw1); dgk-1(ok1462) | YA | 56 | 21 (38%) | 1.0000 | KU1443c |
| KU1452 | brc-1(tm1145) brd-1(dw1) dgk-3(km89); dgk-1(ok1462) | YA | 60 | 38 (63%) | 0.0115 | KU1443c |
| KU1453 | brc-1(tm1145) brd-1(dw1) dgk-3(km90); dgk-1(ok1462) | YA | 53 | 35 (66%) | 0.0053 | KU1443c |
| KU1451 | brc-1(tm1145) brd-1(dw1) dgk-3(km89) | YA | 45 | 30 (67%) | 0.0069 | KU1443c |
| KU1454 | brc-1(tm1145) brd-1(dw1) dgk-3(km89); tpa-1(k501) IV | YA | 59 | 25 (42%) | 0.6931 | KU1443c |
| KU89 | dgk-3(km89) | YA | 64 | 51 (80%) | 0.0310 | KU501c |
| KU501d | Wild type | YA | 55 | 38 (69%) | - | - |
| KU1455 | brc-1(S266A) | YA | 61 | 23 (38%) | 0.0008 | KU501d |
We next asked whether the BARD1 ortholog BRD-1 also participates in axon regeneration. We found that the brd-1(gk297) deletion (Fig. 1A) markedly reduced axon regrowth following laser injury (Fig. 2B; Table 2). Furthermore, we observed that the regeneration defect observed in brc-1(tm1145) brd-1(dw1) double mutants (Fig. 1A) was no greater than that seen in the single brd-1(gk297) mutant (Fig. 2B; Table 2), suggesting that BRC-1 and BRD-1 act in the same pathway. This suggests that BRC-1 and BRD-1 function as a complex to regulate axon regeneration.
We investigated the effects of brc-1 and brd-1 on growth cone behavior, and found that the length of regenerated axons in brc-1(tm1145) brd-1(dw1) mutants was shorter than observed in wild-type animals (Fig. 2C). In contrast, when both brc-1 and brd-1 were overexpressed using the unc-25 promoter, regenerated axons were longer than those in wild-type animals (Fig. 2C). In fact, 28% (25/90) of regenerated axons reached the dorsal nerve cord of animals overexpressing brc-1 and brd-1 compared with 11% (7/62) in wild-type adult animals. Overexpression of brc-1/brd-1 appeared to increase the frequency of axon regeneration, but the difference was not statistically significant (Fig. 2B; Table 2). Thus, BRC-1–BRD-1 is required to initiate axon regeneration and control growth cone behavior.
Next, to determine whether the effect of BRC-1–BRD-1 complex on axon regeneration is specific to D-type motor neurons, we examined the effect of brc-1 and brd-1 on axon regeneration in glutaminergic touch sensory PLM neurons (Fig. 3A). Chen et al. (2011) previously performed a systematic mutant screen looking for defects in axon regeneration, and identified brd-1 as a positive regulator of axon regeneration in PLM neurons. Consistent with their finding, we found that brc-1(tm1145) brd-1(dw1) mutants were defective in axon regeneration in PLM neurons (Fig. 3A,B). These results suggest that BRC-1–BRD-1 is generally required by neurons for axon regeneration.
Figure 3.

BRC-1 and BRD-1 are required for axon regeneration of PLM sensory neurons. A, Representative PLM sensory neurons in wild-type and brc-1(tm1145) brd-1(dw1) mutant animals 24 h after laser surgery. Red arrowheads indicate cut sites. Scale bar: 10 µm. B, Length of PLM regrowth 24 h after laser surgery. Data are presented as a box-plot representing median (thick line within the box) and interquartile range (edge of box) with individual data points. The number (n) of axons examined is shown. Statistical significance was determined by Wilcoxon rank-sum test.
BRCA1 contains a RING finger domain that functions as an E3-Ub ligase in vitro. This activity is greatly increased when complexed with BARD1, which also harbors a RING domain (Fig. 1A; Baer and Ludwig, 2002). The Ile-26 residue in the BRCA1 RING domain is essential for its interaction with the E2-Ub conjugating enzyme but not for its interaction with BARD1, suggesting that BRCA1 is the critical subunit required for E3-Ub ligase activity. Accordingly, the I26A mutant, in which Ile-26 was replaced with alanine, is defective in E3-Ub ligase activity (Brzovic et al., 2003). Similar to mammalian BRCA1, BRC-1 possesses a RING domain with a conserved site, Ile-23, corresponding to the mammalian Ile-26 (Fig. 1C). To determine the importance of BRC-1 E3-Ub ligase activity in axon regeneration, we generated a mutant form of BRC-1 [BRC-1(I23A)] with Ile-23 mutated to alanine. We found that the I23A point mutation could not rescue the brc-1(km88) phenotype (Fig. 2B; Table 2). Taken together, these results suggest that the BRC-1–BRD-1 complex is required for axon regeneration in a manner dependent on its E3-Ub ligase activity.
BRC-1–BRD-1 functions in the EGL-30 Gqα signaling pathway to regulate axon regeneration
We have previously demonstrated that the CED-10 Rac type GTPase–MAX-2 and EGL-30 Gqα–TPA-1 PKC pathways regulate axon regeneration mainly at the L4 and young adult developmental stages, respectively (Pastuhov et al., 2012, 2016). It has been shown that max-2 is expressed in ventral cord neurons during early development, but not at the young adult stage (Lucanic et al., 2006). This suggests that TPA-1 takes the place of MAX-2 to activate MLK-1 in axon regeneration at the adult stage. Therefore, we examined the relationship between life stage and axon regeneration in brc-1(tm1145) brd-1(dw1) double mutants. We found that axon regeneration in brc-1(tm1145) brd-1(dw1) mutants was reduced only in young adult animals and not in L4 larvae, a phenotype similar to that observed in egl-30(ad805) loss-of-function and tpa-1(k501) mutants (Fig. 4A; Table 2; Pastuhov et al., 2012, 2016). Thus, the BRC-1–BRD-1 complex participates in axon regeneration specifically at the adult stage.
Figure 4.
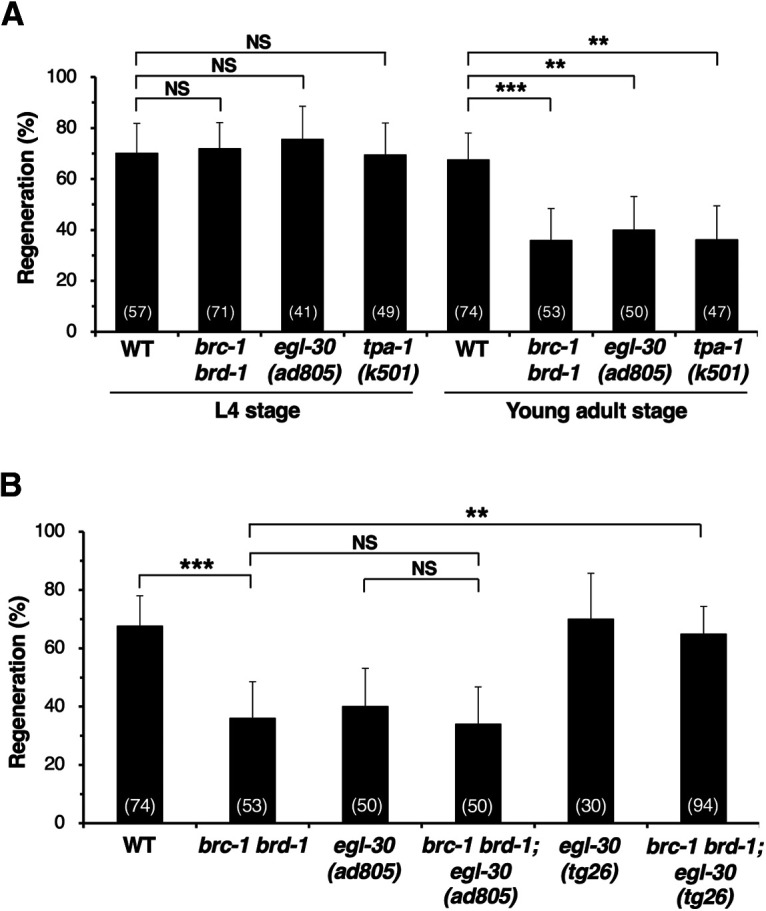
BRC-1–BRD-1 functions in the EGL-30 Gqα signaling pathway to regulate axon regeneration. A, Percentages of axons that initiated regeneration 24 h after laser surgery at the L4 or young adult stage. The number of axons examined is shown. Error bars indicate 95% confidence intervals; **p < 0.01, ***p < 0.001, as determined by Fisher's exact test. NS, not significant. B, Percentages of axons that initiated regeneration 24 h after laser surgery at the young adult stage. The number of axons examined is shown. Error bars indicate 95% confidence intervals; **p < 0.01, ***p < 0.001, as determined by Fisher's exact test. NS, not significant.
This result raised the possibility that BRC-1–BRD-1 functions in the EGL-30 signaling pathway. To investigate this possibility, we examined the genetic interactions of brc-1 and brd-1 with egl-30. We found that the defect in axon regeneration caused by the egl-30(ad805) mutation was not enhanced by introduction of the brc-1(tm1145) brd-1(dw1) mutations (Fig. 4B; Table 2). This result supports the possibility that BRC-1–BRD-1 and EGL-30 act in the same pathway. Moreover, a gain-of-function egl-30(tg26) mutation was able to suppress the brc-1 brd-1 phenotype (Fig. 4B; Table 2). These results suggest that BRC-1–BRD-1 promotes axon regeneration upstream of EGL-30. Alternatively, it is possible that BRC-1–BRD-1 enhances the EGL-30 pathway by inhibiting the action of a negative regulator of this signaling pathway.
BRC-1–BRD-1 enhances the EGL-30 signaling pathway by downregulating DGK-3
How does BRC-1–BRD-1 regulate the EGL-30 pathway in axon regeneration? The observation that BRC-1-associated E3-Ub ligase activity is required for axon regeneration (Fig. 2B) could suggest that some negative regulator of regeneration is inactivated by Ub-dependent protein degradation. It is known that GOA-1 Goα, the regulator of G-protein signaling (RGS) EAT-16, and DGK negatively regulate the EGL-30 pathway (Fig. 5A). We have previously demonstrated that the endocannabinoid anandamide inhibits axon regeneration via GOA-1, which antagonizes EGL-30 (Pastuhov et al., 2012). EAT-16 appears to negatively regulate EGL-30 by enhancing the rate of GTP hydrolysis (Chase et al., 2001). We examined whether BRC-1 promotes axon regeneration by downregulation of GOA-1 or EAT-16. However, we found that neither the goa-1(n1134) nor the eat-16(nj8) loss-of-function mutation suppressed the regeneration defect observed in brc-1(tm1145) brd-1(dw1) mutants (Fig. 5B; Table 2). Therefore, it is unlikely that GOA-1 or EAT-16 is a target for BRC-1–BRD-1-mediated degradation.
Figure 5.
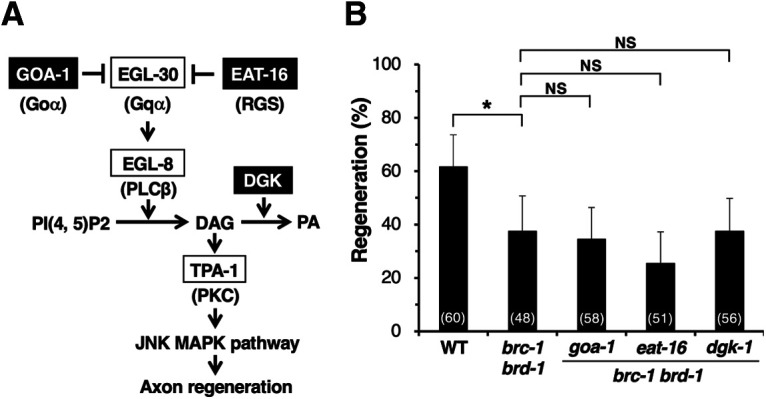
Effects of negative regulators in the EGL-30 pathway on BRC-1–BRD-1-mediated axon regeneration. A, The EGL-30 pathway regulating axon regeneration. EGL-30 Gqα activates EGL-8 PLCβ, which in turn generates DAG from phosphatidylinositol bisphosphate [PI(4, 5)P2]. DAG activates TPA-1 PKC, resulting in activation of the JNK pathway to promote axon regeneration. GOA-1 Goα and EAT-16 RGS antagonize EGL-30 signaling. DGK downregulates the EGL-30 pathway by converting DAG to PA. B, Percentages of axons that initiated regeneration 24 h after laser surgery at the young adult stage. The number of axons examined is shown. Error bars indicate 95% confidence intervals; *p < 0.05, as determined by Fisher's exact test. NS, not significant.
DGK negatively regulates the EGL-30 pathway by converting DAG, an activator of TPA-1, into PA (Fig. 5A; Lackner et al., 1999). Indeed, we have previously reported that DGK-1, an ortholog of mammalian DGKθ, acts as a negative regulator of axon regeneration (Alam et al., 2016). However, we found that the dgk-1(ok1462) null mutation also failed to suppress the brc-1(tm1145) brd-1(dw1) phenotype of defective axon regeneration (Fig. 5B; Table 2). As the C. elegans genome contains five genes encoding DGKs, dgk-1 to dgk-5, we considered the possibility that another DGK may be involved. Interestingly, Matsuki et al. (2006) recently reported that DGK-1 and DGK-3 function redundantly to reduce DAG levels and are required for olfactory adaptation. DGK-3 is an ortholog of mammalian DGKβ. To test whether dgk-1; dgk-3 double mutations could suppress the brc-1 brd-1 phenotype, we used CRISPR–Cas9 mutagenesis to generate two independent dgk-3(km89) and dgk-3(km90) null alleles (Fig. 6A) in the endogenous dgk-3 locus of brc-1(tm1145) brd-1(dw1); dgk-1(ok1462) mutants. We found that dgk-3(km89); dgk-1(ok1462) and dgk-3(km90); dgk-1(ok1462) mutations were able to suppress the brc-1(tm1145) brd-1(dw1) defect in axon regeneration (Fig. 6B; Table 2). To examine whether DGK-1 and DGK-3 redundantly regulate axon regeneration or whether DGK-3 does so alone, we constructed brc-1(tm1145) brd-1(dw1) dgk-3(km89) mutants. We found that the dgk-3(km89) single mutation was sufficient to suppress the brc-1 brd-1 defect (Fig. 6B; Table 2). Axon regeneration was significantly improved with dgk-3(km89) single mutants compared with wild-type animals (Fig. 6B; Table 2). These results suggest that BRC-1–BRD-1 promotes axon regeneration by negatively regulating DGK-3, thereby ensuring elevated DAG levels, which activates TPA-1. Consistent with this, the dgk-3(km89) mutation failed to suppress the defect in axon regeneration in brc-1 brd-1; tpa-1 mutants (Fig. 6B; Table 2).
Figure 6.

BRC-1–BRD-1 downregulates DGK-3 to promote axon regeneration. A, Isolation of dgk-3 mutants. Genomic structure of the dgk-3 gene is shown. The dgk-3(km89) mutation is a 20-bp insertion (red nucleotides) that contains an in-frame premature stop codon (*), thus terminating translation in the middle of exon 1. The dgk-3(km90) mutation is a 5-bp deletion, causing a frameshift (bold amino acids) and premature stop codon (*) in exon 1. B, Percentages of axons that initiated regeneration 24 h after laser surgery at the young adult stage. The number of axons examined is shown. Error bars indicate 95% confidence intervals; *p < 0.05, **p < 0.01, as determined by Fisher's exact test. NS, not significant.
BRC-1–BRD-1 poly-ubiquitylates DGK-3, leading to its degradation
The genetic analysis described above raised the possibility that BRC-1–BRD-1 could act as an E3-Ub ligase to mediate ubiquitylation of DGK-3, thus promoting its degradation. To test this hypothesis, we examined whether BRC-1–BRD-1 ubiquitylates DGK-3 in mammalian cell cultures. We co-expressed T7-tagged DGK-3 and HA-tagged Ub in COS-7 cells, immunoprecipitated cell lysates with anti-T7 antibody and immunoblotted with anti-HA antibody. We detected mono-ubiquitylation and weak poly-ubiquitylation of DGK-3 (Fig. 7A, lanes 1 and 2), suggesting that there is some endogenous E3-Ub ligase in COS-7 cells that can ubiquitylate DGK-3. We next evaluated whether BRC-1–BRD-1 could stimulate the ubiquitylation of DGK-3. T7-DGK-3 and HA-Ub were co-transfected with GFP-BRC-1 and BRD-1-RFP into COS-7 cells. We found that co-expression of BRC-1 and BRD-1 decreased the levels of poly-ubiquitylated DGK-3 (Fig. 7A, lane 3), suggesting that BRC-1–BRD-1 promotes the degradation of ubiquitylated DGK-3. Consistent with this possibility, when cells were treated with MG132, a specific inhibitor of the 26S proteasome, the level of poly-ubiquitylated DGK-3 clearly increased (Fig. 7A, lane 4). However, instead of wild-type BRC-1, co-expressing BRD-1 with mutant BRC-1(I23A), which is defective in E3-Ub ligase activity, resulted in decreased levels of poly-ubiquitylated DGK-3 in the presence of MG132 (Fig. 7A,B). These results suggest that BRC-1–BRD-1 controls DGK-3 protein levels through proteasome-mediated degradation.
Figure 7.

BRC-1–BRD-1 mediates poly-ubiquitylation of DGK-3. A, Poly-ubiquitylation of DGK-3 by BRC-1–BRD-1. COS-7 cells were transfected with T7-DGK-3, HA-Ub, GFP-BRC-1, and BRD-1-RFP, as indicated. Cells were incubated with or without MG132. Cell lysates were immunoprecipitated (IP) with anti-T7 antibody and immunoblotted (IB) with anti-HA and anti-T7 antibodies. Total lysates were analyzed by IB with anti-GFP and anti-RFP antibodies. The experiment was done in triplicate with similar results, shown here for trial #2. B, Comparison of DGK-3 poly-ubiquitylation levels. The DGK-3 poly-ubiquitylation experiment was performed three times and each bar represents the result of each trial (#1–#3). Data represent the percentage of normalized poly-ubiquitylated DGK-3 in lane 5 relative to that found in lane 4. The blots in lanes 4 and 5 of A from three trials are shown in the upper part. C, Interaction of DGK-3 with BRC-1. COS-7 cells were co-transfected with T7-DGK-3, GFP-BRC-1, and BRD-1-RFP, as indicated. Cells were then incubated with MG132. Cell lysates were immunoprecipitated (IP) with anti-T7 antibody and immunoblotted (IB) with anti-GFP and anti-T7 antibodies. Total lysates were analyzed by IB with anti-GFP antibody.
To determine whether DGK-3 interacts with BRC-1, COS-7 cells were transiently transfected with T7-DGK-3, GFP-BRC-1, and BRD-1-RFP, and then treated with MG132 to inhibit DGK-3 degradation. Co-immunoprecipitation experiments revealed an interaction between DGK-3 and BRC-1 (Fig. 7C, lane 1). Similar results were observed between DGK-3 and BRC-1(I23A) (Fig. 7C, lane 2). Therefore, the E3-Ub ligase activity of BRC-1 is not required to interact with DGK-3. These results indicate that BRC-1 interacts with and poly-ubiquitylates DGK-3 for degradation.
Next, we investigated whether BRC-1–BRD-1 regulates DGK-3 levels in animals by expressing GFP-fused DGK-3 in D-type motor neurons using the unc-25 promoter. In wild-type animals, DGK-3::GFP was uniformly distributed in the cytoplasm of D-type neuron cell bodies (Fig. 8A). Following axon laser ablation, fluorescence intensity of DGK-3::GFP in the cytoplasm of D-type neurons was significantly decreased (Fig. 8A,B). In contrast, we found that the brc-1(tm1145) brd-1(dw1) mutations resulted in significant stabilization of cytosolic DGK-3::GFP levels (Fig. 8A,B). Thus, BRC-1–BRD-1 is involved in axon injury-induced destabilization of DGK-3 in animals. These results suggest that increases in DGK-3 protein levels in brc-1 brd-1 mutants lead to a decrease in DAG levels, which eventually results in the inhibition of the TPA-1 PKC signaling pathway.
Figure 8.

BRC-1–BRD-1 promotes axotomy induced degradation of cytoplasmic DGK-3. A, Fluorescent images of wild-type and brc-1 brd-1 mutant animals expressing Punc-47::mcherry (D-type motor neuron, top) and Punc-25::dgk-3::GFP (bottom) are shown. Images were taken at 0 or 8 h after laser surgery. Red arrowheads indicate the tip of the severed axons. Yellow arrows indicate cell bodies corresponding to the severed axons and their magnification is shown in the insets. Scale bar: 10 µm. B, Quantification of DGK-3::GFP fluorescence levels in cytoplasm of D-type neurons. Relative DGK-3 intensity was calculated as a fraction of the relative DGK-3::GFP intensity 8 h after laser surgery divided by the corresponding value at 0 h postaxotomy. Data are presented as a box-plot representing median (thick line within the box) and interquartile range (edge of box) with individual data points. The number (n) of axons examined is shown. Statistical significance was determined by Wilcoxon rank-sum test.
PKA phosphorylation induces cytoplasmic localization of BRC-1
How is BRC-1 function regulated in axon regeneration? Upon axon severance, intracellular levels of cAMP increase and PKA is activated (Neumann et al., 2002; Bhatt et al., 2004). Interestingly, BRC-1 contains a PKA phosphorylation consensus motif (Arg-Arg-Xxx-Ser) at Ser-266 (Fig. 9A). We therefore asked whether PKA phosphorylates BRC-1 at this residue. We performed in vitro kinase assays with active PKA and immuno-purified GFP-BRC-1 from COS-7 cells. Western blot analysis using an antibody recognizing phosphorylated PKA substrates revealed PKA phosphorylation of GFP-BRC-1 (Fig. 9B, lanes 1 and 2). To determine whether PKA can phosphorylate BRC-1 at Ser-266, we generated a mutant form of BRC-1 [BRC-1(S266A)], in which Ser-266 was replaced with alanine. In vitro kinase assays revealed that the S266A mutation abolished the phosphorylation of BRC-1 by PKA (Fig. 9B, lane 3). These results demonstrate that PKA phosphorylates Ser-266 of BRC-1 in vitro. In order to address the physiological significance of this phosphorylation, we used the CRISPR–Cas9 system to engineer a non-phosphorylatable brc-1(S266A) mutant, replacing the codon encoding the Ser-266 residue with an alanine codon in the endogenous brc-1 locus. We found that axon regeneration was significantly reduced in brc-1(S266A) mutants (Fig. 9C; Table 2). This result indicates that Ser-266 phosphorylation is important for activation of the regeneration pathway by BRC-1.
Figure 9.

PKA phosphorylates BRC-1. A, A schematic diagram of BRC-1. RING finger domain is shown in red, and BRCT domains in yellow. The amino acid sequences around a PKA phosphorylation consensus site (underline) and a putative nuclear localization signal (red characters) are shown below. The Ser-266 residue is indicated by an asterisk. B, PKA phosphorylates BRC-1 at Ser-266 in vitro. In vitro phosphorylation of BRC-1 by PKA is shown. COS-7 cells were transfected with GFP-BRC-1 (WT) or GFP-BRC-1(S266A), and cell lysates were immunoprecipitated (IP) with anti-GFP antibody. The immunoprecipitates were subjected to in vitro kinase assay using active recombinant PKA. Phosphorylated BRC-1 was detected by immunoblotting (IB) with anti-phospho-PKA substrate rabbit monoclonal antibody. C, Percentages of axons that initiated regeneration 24 h after laser surgery at the young adult stage. The number of axons examined is shown. Error bar indicates 95% confidence interval; ***p < 0.001, as determined by Fisher's exact test.
We next examined how PKA-mediated phosphorylation might regulate BRC-1 in axon regeneration. Interestingly, the PKA phosphorylation site of BRC-1 is in a putative nuclear localization signal (NLS) sequence (Fig. 9A), raising the possibility that phosphorylation of BRC-1 might impact its localization. We investigated this possibility by monitoring GFP::BRC-1 localization during activation of PKA. Under normal conditions, GFP::BRC-1 was predominantly localized in the nucleus (Fig. 10A,B). Treatment of animals with forskolin is expected to cause an increase in cAMP levels by activating adenylyl cyclase and concomitantly PKA (Ghosh-Roy et al., 2010). We found that forskolin treatment strongly induced cytoplasmic localization of GFP::BRC-1 (Fig. 10A,B). In contrast, forskolin was unable to induce cytoplasmic accumulation of the GFP::BRC-1(S266A) mutant (Fig. 10A,B), suggesting that PKA phosphorylation of the Ser-266 site is required for the translocation of BRC-1 from the nucleus to the cytoplasm. Thus, by altering its subcellular localization, the phosphorylation of BRC-1 at Ser-266 can regulate axon regeneration.
Figure 10.

PKA phosphorylation induces cytoplasmic localization of BRC-1. A, Localization of BRC-1 in response to PKA activation. Fluorescent images of wild-type animals expressing Punc-47::mcherry (D-type motor neuron, top) and Punc-25::GFP::brc-1 or Punc-25::GFP::brc-1(S266A) (bottom) with or without forskolin treatment are shown. Red and yellow arrowheads indicate cell nucleus. Scale bar: 10 µm. B, Quantification of GFP::BRC-1 fluorescence levels in D-type neurons with or without forskolin treatment. The cytoplasmic-to-nuclear ratio of GFP::BRC-1 signal was calculated as a fraction of the relative GFP::BRC-1 intensity in the cytoplasm divided by the corresponding value in the nucleus. Data are presented as a box-plot representing median (thick line within the box) and interquartile range (edge of box) with individual data points. The number (n) of cell bodies examined is shown. Statistical significance was determined by Wilcoxon rank-sum test.
Discussion
BRCA1 and BRCA2 genes were identified as causative genes for early-onset hereditary breast cancer (Fackenthal and Olopade, 2007). BRCA-deficient cells use error-prone DNA-repair pathways, which cause increased genomic instability (Scully and Livingston, 2000; Moynahan and Jasin, 2010; Ceccaldi et al., 2016). However, recent studies have identified new functions of BRCA1 and BRCA2 in the regulation of transcription and RNA processing relevant to their tumor-suppressive activity (Kleiman et al., 2005). Previous studies have established that the C. elegans orthologs, BRC-1 (for BRCA1) and BRC-2 (for BRCA2), possess many functional similarities with their human counterparts, including DNA damage repair, homologous recombination, and meiosis (Martin et al., 2005; Polanowska et al., 2006; Adamo et al., 2008; Janisiw et al., 2018; Li et al., 2018). Therefore, C. elegans has proven to be a very useful model system for studying the function and signaling pathways of BRCA1 and BRCA2.
We have recently found that BRC-2 regulates axon regeneration of postdifferentiated GABAergic D-type motor neurons after injury through the Rho GTPase signaling pathway (Shimizu et al., 2018). In the present study, we find that BRC-1 is also involved in axon regeneration. In humans, BRCA1 exists mostly in a heterodimeric complex with its binding partner BARD1 (Wu et al., 1996). Similarly, BRC-1 forms a complex with the C. elegans BARD1 ortholog BRD-1, and BRD-1 is also required for the regeneration of severed axons. However, the site of action of BRC-1–BRD-1 in the regulation of axon regeneration is different from that of BRC-2. BRC-1–BRD-1 participates in adult-specific axon regeneration regulated by the EGL-30 Gqα signaling pathway. Activated EGL-30 signaling induces increased production of DAG, which in turn activates TPA-1 PKC (Lackner et al., 1999). TPA-1 phosphorylates and activates MLK-1 MAPKKK to promote axon regeneration (Pastuhov et al., 2012). DGK converts DAG to PA (Miller et al., 1999); thus, inactivation of DGK activity results in elevated DAG levels. The BRC-1–BRD-1 complex enhances the EGL-30 pathway by poly-ubiquitylating DGK-3, which results in its degradation through the 26S proteasome pathway (Fig. 11). Based on this possibility, the recovery of axon regeneration in brc-1 brd-1 mutants by gain-of-function egl-30 or dgk-3 deletion mutations could be a compensatory effect. The brc-1 brd-1 mutant is defective in DGK-3 degradation, resulting in reduced DAG levels. The egl-30 or dgk-3 mutation can suppress the brc-1 brd-1 deficiency by increasing DAG levels.
Figure 11.

Schematic model for the regulation of axon regeneration by BRC-1–BRD-1. Under normal conditions, BRC-1–BRD-1 is localized in the nucleus. In response to axon injury, PKA phosphorylation of BRC-1 Ser-266 induces the translocalization of BRC-1–BRD-1 to the cytoplasm. The BRC-1–BRD-1 complex poly-ubiquitylates DGK-3, resulting in its degradation. BRC-1–BRD-1 enhances the EGL-30 signaling pathway to promote axon regeneration.
In mammals, at least 10 DGK isoforms have been reported, and their expression patterns or interactors differ among isoforms (Topham and Epand, 2009; Ishisaka and Hara, 2014). C. elegans contains five dgk genes (dgk-1 to dgk-5), each encoding a different isoform corresponding to mammalian DGK. To date, DGK-1 and DGK-3 have been shown to play roles in the nervous system. The dgk-1 mutation enhances DAG signaling in several sensory and motor neurons (Miller et al., 1999; Matsuki et al., 2006), whereas DGK-3 functions in AFD sensory neurons and modulates thermotactic behavior (Biron et al., 2006). In AWC chemosensory neurons, DGK-1 and DGK-3 function redundantly to control olfactory adaptation (Matsuki et al., 2006). In this study, we observed that deletion of dgk-3 alone is sufficient to reverse the regeneration defect of brc-1 brd-1 mutants. In contrast, the dgk-1 single knockout in brc-1 brd-1 mutants has no effect. Therefore, BRC-1–BRD-1 specifically inhibits DGK-3; however, disruption of dgk-1 may cause an increase in DAG levels, resulting in suppression of the brc-1 brd-1 phenotype. Recently, we demonstrated that the C. elegans small GTPase RHO-1 promotes axon regeneration by inactivating DGK-1, leading to DAG upregulation in D-type motor neurons (Alam et al., 2016). Thus, these results suggest that deletion of dgk-1 does not further increase DAG level in D-type neurons of brc-1 brd-1 mutants because DGK-1 activity is already inhibited by RHO-1 during axon regeneration.
Since tumor-derived BRCA1 mutations eliminate E3-Ub ligase activity (Baer and Ludwig, 2002), it is clear that this activity of mammalian BRCA1–BARD1 is of critical functional importance. The identification of targets of BRCA1–BARD1-dependent ubiquitylation would inform our understanding of the role of BRCA1–BARD1 in tumorigenesis, however at present these targets are unknown. We show here that the E3-Ub ligase activity of BRC-1–BRD-1 is critical for its function in axon regeneration. We identify DGK-3 as a specific target for BRC-1–BRD-1-mediated ubiquitylation, which may suggest that mammalian BRCA1–BARD1 function in diverse cellular processes could involve ubiquitylation of DGKs. Indeed, the mammalian DGKζ, whose function is linked to cancer cell growth and survival, is poly-ubiquitylated and degraded through the proteasome system (Okada et al., 2012; Torres-Ayuso et al., 2015). It would be interesting to ask whether ubiquitylation of DGKζ is mediated by the BRCA1–BARD1 complex. Recently, Krishnan et al. (2018) reported that mammalian BRCA1 is also involved in axon regeneration of adult peripheral neurons. Axon injury triggers BRCA1-dependent DNA damage response signaling in the neuronal soma. In contrast to BRC-1 in C. elegans, BRCA1 is mainly localized in the cytoplasm, and axotomy induces translocation to the nucleus. As a result, BRCA1 supports the transcriptional program of injured neurons. Thus, the targets of BRCA1/BRC-1 in the regulation of axon regeneration may be different between mammals and C. elegans.
BRCA1–BARD1-dependent ubiquitylation events are regulated at sites of DNA damage. Human BRCA1 is directly phosphorylated by ATM and ATR kinases in response to DNA damage (Cortez et al., 1999), suggesting that this phosphorylation regulates the retention of BRCA1–BARD1 at sites of DNA damage. It is therefore plausible that axon injury regulates BRC-1–BRD-1-dependent ubiquitylation of DGK-3 through phosphorylation. Indeed, we show that PKA activated by axon injury phosphorylates BRC-1 at Ser-266 and this induces the translocation of BRC-1 from the nucleus to the cytoplasm. The Ser-266 site is located in a putative NLS sequence of BRC-1. Consistent with this, the BRC-1(S266A) mutant remains localized to the nucleus even with PKA activation. Our results suggest the following model for the control of BRC-1 localization (Fig. 11). Under normal conditions, BRC-1 is mostly localized in the nucleus. In response to axon injury, PKA phosphorylation induces the translocalization of BRC-1 to the cytoplasm. Because DGK-3 is present in the cytoplasm, we postulate that phosphorylation-dependent cytoplasmic accumulation of BRC-1 results in enhanced poly-ubiquitylation of DGK-3.
Footnotes
This work was supported by grants from the Ministry of Education, Culture and Science of Japan (S.I.P., N.H., and K.M.) and the Project for Elucidating and Controlling Mechanisms of Aging and Longevity from Japan Agency for Medical Research and Development, AMED, Grant Number JP20gm5010001 (to N.H.). T.S. was supported by a Japan Society for the Promotion of Science Research Fellowship. We thank Dr. Ikue Mori, Caenorhabditis Genetic Center (CGC), National Bio-Resource Project, and C. elegans Knockout Consortium for materials. Some strains were provided by the CGC, which is funded by the National Institutes of Health Office of Research Infrastructure Programs Grant P40 OD10440.
The authors declare no competing financial interests.
References
- Adamo A, Montemauri P, Silva N, Ward JD, Boulton SJ, La Volpe A (2008) BRC-1 acts in the inter-sister pathway of meiotic double-strand break repair. EMBO Rep 9:287–292. 10.1038/sj.embor.7401167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam T, Maruyama H, Li C, Pastuhov SI, Nix P, Bastiani M, Hisamoto N, Matsumoto K (2016) Axotomy-induced HIF-serotonin signalling axis promotes axon regeneration in C. elegans. Nat Commun 7:10388. 10.1038/ncomms10388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer R, Ludwig T (2002) The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr Opin Genet Dev 12:86–91. 10.1016/s0959-437x(01)00269-6 [DOI] [PubMed] [Google Scholar]
- Bhatt DH, Otto SJ, Depoister B, Fetcho JR (2004) Cyclic AMP-induced repair of zebrafish spinal circuits. Science 305:254–258. 10.1126/science.1098439 [DOI] [PubMed] [Google Scholar]
- Biron D, Shibuya M, Gabel C, Wasserman SM, Clark DA, Brown A, Sengupta P, Samuel AD (2006) A diacylglycerol kinase modulates long-term thermotactic behavioral plasticity in C. elegans. Nat Neurosci 9:1499–1505. 10.1038/nn1796 [DOI] [PubMed] [Google Scholar]
- Boulton SJ, Martin JS, Polanowska J, Hill DE, Gartner A, Vidal M (2004) BRCA1/BARD1 orthologs required for DNA repair in Caenorhabditis elegans. Curr Biol 14:33–39. 10.1016/j.cub.2003.11.029 [DOI] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzovic PS, Keeffe JR, Nishikawa H, Miyamoto K, Fox D, Fukuda M, Ohta T, Klevit R (2003) Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc Natl Acad Sci USA 100:5646–5651. 10.1073/pnas.0836054100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Rondinelli B, D'Andrea AD (2016) Repair pathway choices and consequences at the double-strand break. Trends Cell Biol 26:52–64. 10.1016/j.tcb.2015.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase DL, Patikoglou GA, Koelle MR (2001) Two RGS proteins that inhibit Gαo and Gαq signaling in C. elegans neurons require a Gβ5-like subunit for function. Curr Biol 11:222–231. 10.1016/S0960-9822(01)00071-9 [DOI] [PubMed] [Google Scholar]
- Chen L, Wang Z, Ghosh-Roy A, Hubert T, Yan D, O'Rourke S, Bowerman B, Wu Z, Jin Y, Chisholm AD (2011) Axon regeneration pathways identified by systematic genetic screening in C. elegans. Neuron 71:1043–1057. 10.1016/j.neuron.2011.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch'ng Q, Williams L, Lie YS, Sym M, Whangbo J, Kenyon C (2003) Identification of genes that regulate a left-right asymmetric neuronal migration in Caenorhabditis elegans. Genetics 164:1355–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Wang Y, Qin J, Elledge SJ (1999) Requirement of ATM-dependent phosphorylation of Brca1 in the DNA damage response to double-strand breaks. Science 286:1162–1166. 10.1126/science.286.5442.1162 [DOI] [PubMed] [Google Scholar]
- Dokshin GA, Ghanta KS, Piscopo KM, Mello CC (2018) Robust genome editing with short single-stranded and long, partially single-stranded DNA donors in Caenorhabditis elegans. Genetics 210:781–787. 10.1534/genetics.118.301532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fackenthal JD, Olopade OI (2007) Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat Rev Cancer 7:937–948. 10.1038/nrc2054 [DOI] [PubMed] [Google Scholar]
- Firnhaber C, Hammarlund M (2013) Neuron-specific feeding RNAi in C. elegans and its use in a screen for essential genes required for GABA neuron function. PLoS Genet 9:e1003921. 10.1371/journal.pgen.1003921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Deng K, Hou J, Bryson JB, Barco A, Nikulina E, Spencer T, Mellado W, Kandel ER, Filbin MT (2004) Activated CREB Is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 44:609–621. 10.1016/j.neuron.2004.10.030 [DOI] [PubMed] [Google Scholar]
- Ghosh-Roy A, Wu Z, Goncharov A, Jin Y, Chisholm AD (2010) Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J Neurosci 30:3175–3183. 10.1523/JNEUROSCI.5464-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanafusa H, Ishikawa K, Kedashiro S, Saigo T, Iemura SI, Natsume T, Komada M, Shibuya H, Nara A, Matsumoto K (2011) Leucine-rich repeat kinase LRRK1 regulates endosomal trafficking of the EGF receptor. Nat Commun 2:158. 10.1038/ncomms1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Jin Y (2016) Intrinsic control of axon regeneration. Neuron 90:437–451. 10.1016/j.neuron.2016.04.022 [DOI] [PubMed] [Google Scholar]
- Huang X, Cheng HJ, Tessier-Lavigne M, Jin Y (2002) MAX-1, a novel PH/MyTH4/FERM domain cytoplasmic protein implicated in netrin-mediated axon repulsion. Neuron 34:563–576. 10.1016/S0896-6273(02)00672-4 [DOI] [PubMed] [Google Scholar]
- Ishisaka M, Hara H (2014) The roles of diacylglycerol kinases in the central nervous system: review of genetic studies in mice. J Pharmacol Sci 124:336–343. 10.1254/jphs.13r07cr [DOI] [PubMed] [Google Scholar]
- Janisiw E, Dello Stritto MR, Jantsch V, Silva N (2018) BRCA1-BARD1 associate with the synaptonemal complex and pro-crossover factors and influence RAD-51 dynamics during Caenorhabditis elegans meiosis. PLoS Genet 14:e1007653. 10.1371/journal.pgen.1007653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KW, Tang NH, Piggott CA, Andrusiak MG, Park S, Zhu M, Kurup N, Cherra IIS, Wu Z, Chisholm AD, Jin Y (2018) Expanded genetic screening in Caenorhabditis elegans identifies new regulators and an inhibitory role for NAD+ in axon regeneration. Elife 7:e39756. 10.7554/eLife.39756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman FE, Wu-Baer F, Fonseca D, Kaneko S, Baer R, Manley JL (2005) BRCA1/BARD1 inhibition of mRNA 3′ processing involves targeted degradation of RNA polymerase II. Genes Dev 19:1227–1237. 10.1101/gad.1309505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan A, Purdy K, Chandrasekhar A, Martinez J, Cheng C, Zochodne DW (2018) A BRCA1-dependent DNA damage response in the regenerating adult peripheral nerve milieu. Mol Neurobiol 55:4051–4067. 10.1007/s12035-017-0574-7 [DOI] [PubMed] [Google Scholar]
- Lackner MR, Nurrish SJ, Kaplan JM (1999) Facilitation of synaptic transmission by EGL-30 Gqα and EGL-8 PLCβ: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron 24:335–346. 10.1016/S0896-6273(00)80848-X [DOI] [PubMed] [Google Scholar]
- Li C, Hisamoto N, Nix P, Kanao S, Mizuno T, Bastiani M, Matsumoto K (2012) The growth factor SVH-1 regulates axon regeneration in C. elegans via the JNK MAPK cascade. Nat Neurosci 15:551–557. 10.1038/nn.3052 [DOI] [PubMed] [Google Scholar]
- Li ML, Greenberg RA (2012) Links between genome integrity and BRCA1 tumor suppression. Trends Biochem Sci 37:418–424. 10.1016/j.tibs.2012.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Saito TT, Martinez-Garcia TS, Deshong AJ, Nadarajan S, Lawrence KS, Checchi PM, Colaiacovo MP, Engebrecht J (2018) The tumor suppressor BRCA1-BARD1 complex localizes to the synaptonemal complex and regulates recombination under meiotic dysfunction in Caenorhabditis elegans. PLoS Genet 14:e1007701. 10.1371/journal.pgen.1007701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucanic M, Kiley M, Ashcroft N, L'etoile N, Cheng HJ (2006) The Caenorhabditis elegans P21-activated kinases are differentially required for UNC-6/netrin-mediated commissural motor axon guidance. Development 133:4549–4559. 10.1242/dev.02648 [DOI] [PubMed] [Google Scholar]
- Mar FM, Bonni A, Sousa MM (2014) Cell intrinsic control of axon regeneration. EMBO Rep 15:254–263. 10.1002/embr.201337723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JS, Winkelmann N, Petalcorin MIR, McIlwraith MJ, Boulton SJ (2005) RAD-51-dependent and -independent roles of a Caenorhabditis elegans BRCA2-related protein during DNA double-strand break repair. Mol Cell Biol 25:3127–3139. 10.1128/MCB.25.8.3127-3139.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki M, Kunitomo H, Iino Y (2006) Goα regulates olfactory adaptation by antagonizing Gqα-DAG signaling in Caenorhabditis elegans. Proc Natl Acad Sci USA 103:1112–1117. 10.1073/pnas.0506954103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V (1991) Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10:3959–3970. 10.1002/j.1460-2075.1991.tb04966.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KG, Emerson MD, Rand JB (1999) Goα and diacylglycerol kinase negatively regulate the Gqα pathway in C. elegans. Neuron 24:323–333. 10.1016/S0896-6273(00)80847-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Jasin M (2010) Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol 11:196–207. 10.1038/nrm2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI (2002) Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34:885–893. 10.1016/S0896-6273(02)00702-X [DOI] [PubMed] [Google Scholar]
- Nix P, Hisamoto N, Matsumoto K, Bastiani MJ (2011) Axon regeneration requires co-activation of p38 and JNK MAPK pathways. Proc Natl Acad Sci USA 108:10738–10743. 10.1073/pnas.1104830108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix P, Hammarlund M, Hauth L, Lachnit M, Jorgensen EM, Bastiani M (2014) Axon regeneration genes identified by RNAi screening in C. elegans. J Neurosci 34:629–645. 10.1523/JNEUROSCI.3859-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Hozumi Y, Tanaka T, Suzuki Y, Yanagida M, Araki Y, Evangelisti C, Yagisawa H, Topham MK, Martelli AM, Goto K (2012) DGKζ is degraded through the cytoplasmic ubiquitin-proteasome system under excitotoxic conditions, which causes neuronal apoptosis because of aberrant cell cycle reentry. Cell Signal 24:1573–1582. 10.1016/j.cellsig.2012.03.021 [DOI] [PubMed] [Google Scholar]
- Pastuhov SI, Fujiki K, Nix P, Kanao S, Bastiani M, Matsumoto K, Hisamoto N (2012) Endocannabinoid–Goα signalling inhibits axon regeneration in Caenorhabditis elegans by antagonizing Gqα–PKC–JNK signalling. Nat Commun 3:1136. 10.1038/ncomms2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastuhov SI, Fujiki K, Tsuge A, Asai K, Ishikawa S, Hirose K, Matsumoto K, Hisamoto N (2016) The core molecular machinery used for engulfment of apoptotic cells regulates the JNK pathway mediating axon regeneration in Caenorhabditis elegans. J Neurosci 36:9710–9721. 10.1523/JNEUROSCI.0453-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanowska J, Martin JS, Garcia-Muse T, Petalcorin MIR, Boulton SJ (2006) A conserved pathway to activate BRCA1-dependent ubiquitylation at DNA damage sites. EMBO J 25:2178–2188. 10.1038/sj.emboj.7601102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Livingston DM (2000) In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature 408:429–432. 10.1038/35044000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Pastuhov SI, Hanafusa H, Matsumoto K, Hisamoto N (2018) The C. elegans BRCA2-ALP/Enigma complex regulates axon regeneration via a Rho GTPase-ROCK-MLC phosphorylation pathway. Cell Rep 24:1880–1889. 10.1016/j.celrep.2018.07.049 [DOI] [PubMed] [Google Scholar]
- Topham MK, Epand RM (2009) Mammalian diacylglycerol kinases: molecular interactions and biological functions of selected isoforms. Biochim Biophys Acta 1790:416–424. 10.1016/j.bbagen.2009.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Ayuso P, Tello-Lafoz M, Mérida I, Ávila-Flores A (2015) Diacylglycerol kinase-ζ regulates mTORC1 and lipogenic metabolism in cancer cells through SREBP-1. Oncogenesis 4:e164. 10.1038/oncsis.2015.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, Yang MC, Hwang LY, Bowcock AM, Baer R (1996) Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet 14:430–440. 10.1038/ng1296-430 [DOI] [PubMed] [Google Scholar]
- Yanik MF, Cinar H, Cinar HN, Chisholm AD, Jin Y, Ben-Yakar A (2004) Neurosurgery: functional regeneration after laser axotomy. Nature 432:822. 10.1038/432822a [DOI] [PubMed] [Google Scholar]
- Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, Gage FH, Verma IM (2011) BRCA1 tumour suppression occurs via heterochromatin mediated silencing. Nature 477:179–184. 10.1038/nature10371 [DOI] [PMC free article] [PubMed] [Google Scholar]