

Abstract
The pandemic, COVID-19, has spread worldwide and affected millions of people. There is an urgent need, therefore, to find a proper treatment for the novel coronavirus, Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), the causative agent. This paper focuses on identifying inhibitors that target SARS-CoV-2 proteases, PLPRO and 3CLPRO, which control the duplication and manages the life cycle of SARS-CoV-2. We have carried out detailed in silico Virtual high-throughput screening using Food and Drug Administration (FDA) approved drugs from the Zinc database, COVID-19 clinical trial compounds from Pubchem database, Natural compounds from Natural Product Activity and Species Source (NPASS) database and Maybridge database against PLPRO and 3CLPRO proteases. After thoroughly analyzing the screening results, we found five compounds, Bemcentinib, Pacritinib, Ergotamine, MFCD00832476, and MFCD02180753 inhibit PLPRO and six compounds, Bemcentinib, Clofazimine, Abivertinib, Dasabuvir, MFCD00832476, Leuconicine F inhibit the 3CLPRO. These compounds are stable within the protease proteins’ active sites at 20ns MD simulation. The stability is revealed by hydrogen bond formations, hydrophobic interactions, and salt bridge interactions. Our study results also reveal that the selected five compounds against PLPRO and the six compounds against 3CLPRO bind to their active sites with good binding free energy. These compounds that inhibit the activity of PLPRO and 3CLPRO may, therefore, be used for treating COVID-19 infection.
Keywords: SARS-CoV-2, COVID-19, PLPRO, 3CLPRO, Drug repositioning
Graphical abstract

1. Introduction
In December 2019, rare pneumonia, now called COVID-19, that emerged in China, has spread worldwide and affected millions of people (Huang et al., 2020; Wang et al., 2020). This novel SARS-CoV-2 can infect many hosts, including swine, bats and humans. It is also capable of human-to-human transmission (Li et al., 2020). The virus is characterized by fever, dry cough, tiredness and affects people by producing mild or no symptoms to death (Huang et al., 2020). This is a fast-evolving virus derived from the characteristic high genomic nucleotide replacement rates and recombination (de Wit et al., 2016). Before the outbreak of COVID-19, the world was hit by two different viruses in 2002 and 2012, arising from the same family coronaviridae, namely Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS). Since then, the hunt for the treatment of coronavirus diseases has been going on, though not with success (Peeri et al., 2020; Chen et al., 2020).
Coronaviruses, the large single-stranded positive-sense ribose nucleic acid (RNA) viruses, consist of 27–31 kb viral genome (Chen et al., 2020; Zhou et al., 2020). SARS-CoV-2 belongs to the β-genus of the coronaviridae family and shares <80% nucleotide identity and 89.10% nucleotide similarity with SARS-CoV genes. These viruses produce ~800 kDa of polypeptide chain upon transcription (Fehr and Perlman, 2015). The polypeptide chain, when cleaved by the proteolytic enzymes, generates various proteins. This cleavage is mediated by the papain-like protease (PLPRO) and 3-chymotrypsin-like protease (3CLPRO) (Anand et al., 2003). PLPRO cleaves the polyproteins at three sites, namely 181–182, 818–819 and 2763–2764, at the N-terminus and 3CLPRO cleaves the polyproteins at 11 sites to generate various non-structural proteins that are important for viral replication. 3CLPRO protease is believed to regulate the proteolytic processing of the two large viral replicase polyproteins, pp1a (450 kDa) and pplab (750 kDa), translated by the open reading frames (ORFs1a/b). The other ORFs at 3′- terminus encodes for the spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins (Chen et al., 2020; Zhou et al., 2020; Anand et al., 2003) (Fig. 1 ). The critical role played by PLPRO and 3CLPRO in replicating the virus, therefore, makes these two protease proteins potential targets for screening inhibitors for the virus, SARS-CoV-2 (Thiel et al., 2003; Ziebuhr, 2004; Ziebuhr et al., 2001). However, no inhibitor has been developed and approved by the FDA so far despite some efforts made in this direction recently (Ahamad et al., 2020; Kouznetsova et al., 2020).
Fig. 1.

SARS-CoV-2 genome containing 16 non-structural proteins (Nsp) organized in individual with polyprotein 1ab (PP1ab). Red (PLPRO) and Blue (3CLPRO) triangles show the cleavage sites of the protease.
Recently several vaccine candidates have been developed. Some of them, subjected to large scale clinical trials, are being administered in several countries (Dai and Gao, 2021). COVID vaccines which have been approved by different regulatory authorities for use are Comirnaty, Moderna COVID-19, Covishield, Sputnik V, CoronaVac, BBIBP-CorV, EpiVacCorona, Convidicea, JNJ-78436735, Covaxin and CoviVac (https://www.raps.org/). Questions, however, have come up regarding the mutation rates of SARS-CoV-2, which could lead to immune evasion. Studies have also suggested that the mutations in the target proteins of the COVID-19 can be associated with drug resistance and vaccine inefficacy (Silveira et al., 2021). But then, a vaccine is not the end-all of the pandemic. Efforts to develop more potent therapeutics for COVID-19 should continue. There is an urgent need, therefore, to find alternatives like an effective inhibitor to prevent or treat this infection (Kim et al., 2020).
Considering this urgent need to find effective approaches to prevent COVID-19, fast-track approaches are being made today (Stasi et al., 2020; Weglarz-Tomczak et al., 2020). The two proteases, PLPRO and 3CLPRO, which play an essential role in processing the polyprotein translated from the viral RNA, are considered to be essential for the survival and growth of SARS-CoV-2 (Kanjanahaluethai et al., 2007; Oostra et al., 2008; van Hemert et al., 2008; Báez-Santos et al., 2014, 2015). Considering the crucial roles played by PLPRO and 3CLPRO protease proteins, these proteins can be used as potential targets to control the SARS-CoV-2 life cycle's duplication. In the present study, we downloaded the ZINC database (FDA compounds), Pubchem COVID-19 clinical trial compounds, NPASS, and Maybridge database natural compounds. After subjecting them to space filtering based on threshold physicochemical drug like properties, 2D similarity index and ADMET, we carried out virtual high throughput screening to develop inhibitors for SARS-CoV-2 proteases, PLPRO and 3CLPRO.
2. Materials and methods
2.1. Preparation of SARS-CoV-2 PLPRO and 3CLPROprotein structures
The structures of SARS-CoV-2 PLPRO and SARS-CoV-2 3CLPRO were retrieved from the protein data bank (PDB), PLPRO PDB ID 6w9c and 3CLPRO PDB ID 6lu7. The ligand and water molecules were removed from their structures. The protein structures were prepared by adding hydrogen atoms and energy minimization through UCSF Chimera molecular software (Pettersen et al., 2004).
2.2. pKa calculation
The protein stability and its conformation can be explained in terms of the ionizable residues' perturbed pKa values. The protonation state for the titratable residues at pH 6.0 was assigned according to structure-based pKa calculations, performed with the H++ web server (version 3.1) (http://biophysics.cs.vt.edu/H++) (Anandakrishnan et al., 2012). PLPRO and 3CLPRO protein crystal structures obtained from the PDB, 6w9c (2.70 Å) and 6lu7 (2.16 Å), were used for the study. Three different dielectric constants were selected for the protein (εint = 4, 10, and 20) to evaluate the results' consistency.
2.3. Prediction of the active binding site
In PLPRO (PDB structure 6w9c protein) there is no active site in the crystal structure. Its active sites were predicted through MetaPocket 2.0 meta server on the protein with five pockets threshold (Zhang et al., 2011). 3CLPRO (PDB structure 6lu7 protein) contains a mechanism-based inhibitor N3 and hence this binding site was selected for our study.
2.4. Preparing the selected database compounds
DrugBank FDA only (https://zinc.docking.org/catalogs/dbfda/), FDA-approved drugs (via DSSTOX) (http://zinc.docking.org/catalogs/fda/), PubChem COVID-19 clinical trial compounds (https://pubchemdocs.ncbi.nlm.nih.gov/covid-19), Natural compounds from NPASS database (Zeng et al., 2018) and Maybridge database (http://www.maybridge.com) were used for the virtual high throughput screening against PLPRO and 3CLPRO protease proteins (Irwin and Shoichet, 2005; Sterling and Irwin, 2015). ZINC database has a wide range of compounds. We downloaded the FDA-approved drugs, FDA-approved PubChem COVID-19 clinical trial compounds (Kim et al., 2019), compounds from the NPASS database and Maybridge database in 2D, SDF format. All the selected compounds were converted to 3D, mol2 format using the molconvert tool of InstJChem, ChemAxon software (http://www.chemaxon.com) (Kumar et al., 2019) and hydrogen atoms were added through ChemAxon command line.
2.5. Calculation of physicochemical properties
The drug-likeness of the downloaded compounds were assessed through Rule of 3 (Jhoti et al., 2013) and the Rule of 5 (Pollastri, 2010; Lipinski et al., 1997). This property of space filtering is based on the threshold drug-like physicochemical parameters of the compounds, namely Molecular weight (MW), sLogP, Hydrogen Bond Accepter (HBA), Hydrogen Bond Donor (HBD), Topological Polar Surface Area (TPSA), Aromatic Ring and Rotatable Bonds. InstJChem command-line software was used to calculate these physicochemical properties (http://www.chemaxon.com).
2.6. Compound screening using similarity Tc search
The hit compounds obtained from screening of the threshold physicochemical properties, were used for calculating the 2D similarity using ScreenMD (JChem Suite, ChemAxon) (http://www.chemaxon.com), which is based on chemical fingerprints (Sharma et al., 2020; Kumar et al., 2019). Tc of the selected compounds was calculated and the compounds were ranked according to the Tc similarity score. We assumed a database compound as an analog if the Tc is ≥ 0.75. Using this criterion, we determined the similarities between reference PubChem COVID-19 clinical trial compounds with NPASS database and Maybridge database compounds. This was followed by ranking according to the compounds' Tc values from the NPASS database and Maybridge database with significant similarities to the reference compounds. The similarity threshold (Tc ≥ 0.75) helped in the selection of similar compounds from the vast chemical space.
2.7. Absorption, distribution, metabolism, excretion and toxicity (ADMET) prediction
The selected hit compounds were subjected to computational predictions for exploring ADMET using DS-4.1 software. The prediction of ADMET properties is an important step in drug design and development (Han et al., 2019; Norinder and Bergström, 2006). Six mathematical models, such as plasma protein binding, human intestinal absorption (HIA), blood-brain barrier (BBB) penetration, Cytochrome P450 2D6 inhibition, aqueous solubility and hepatotoxicity were used to screen the compounds. The Toxicity Prediction by Komputer Assisted Technology (TOPKAT) model consists of mutagenicity, carcinogenicity, biodegradability and skin irritancy of the compounds.
2.8. Molecular docking
The selected FDA-approved drugs, PubChem COVID-19 clinical trial compounds, NPASS and Maybridge database compounds, were subjected to molecular docking using AutoDock Tools 4.6 (ADT) (Morris et al., 2009; Holt et al., 2008). Polar hydrogen was added to the protease protein, and charges were assigned through the Kollman charges method. The number of a grid point in xyz 72 Å × 76 Å × 76 Å (x, y, and z) and the grid box center 32.386, 33.998, 25.566 were assigned PLPRO with the spacing 0.375 Å. The number of grid points in xyz 86 Å × 100 Å × 100 Å (x, y, and z) and the grid box center −20.299, 22.277, 66.282 were assigned to 3CLPRO with the spacing 0.375 Å. All docking calculation parameters were kept as a default value. A total of 10 conformations were generated for each screened compound. Autogrid 4 was used to calculate grid maps and Autodock4 was used to perform docking procedures.
2.9. Compound selection based on clustering
After docking, the virtual compounds were further subjected to Clustering using ChemBioServer online tool based on the Hierarchical Clustering method (Karatzas et al., 2020). The hit compounds screened against PLPRO and 3CLPRO were used for Clustering through Soergel (Tc) Distance method and Ward linkage Clustering method. The top cluster compounds are considered as good hits and hence we selected the top one Clustering Threshold value.
2.10. Molecular dynamics (MD) simulation
GROMACS-4.6.5 was used for performing MD simulation (Abraham et al., 2015). Protein topology was generated using GROMOS96 54a7 force field (Schmid et al., 2011). PRODRUG was used to create topologies for the ligands (Schüttelkopf and Van Aalten, 2004). The complex structure was solved with simple point charge (spc 216) water by putting the protein-ligand complex in a triclinic box. The system was neutralized by adding Na+ and Cl−ions and relaxed through energy the minimization process. PME algorithm was used to estimate the electrostatic interaction. Initially, 1ns temperature and pressure equilibrium step was performed, followed by a 20ns production simulation. Using g_rms, g_rmsf, and g_gyrate, the root means square deviation (RMSD), root mean square fluctuation (RMSF) and Radius of gyration (Rg) were calculated. The formation of hydrogen bonds between the protein and the ligand were determined and the secondary structures of the protein and the ligand-protein complex were checked using do_dssp program.
2.11. Calculating the free energy of binding usingMM-PBSA
The MD simulation trajectories were subjected to Molecular Mechanics energies combined with Poisson-Boltzmann (MM-PBSA) and the binding free energies (BFE) were calculated using the formula,
where, ΔEele and ΔEvdW are electrostatic and van der Waals components, respectively, and ΔGpol and ΔGnonpol are polar and non-polar components, respectively (Genheden and Ryde, 2015). TΔS is the temperature and entropic contribution towards BFE. BFE plays a significant role in drug discovery, giving a quantitative estimation of the ligand's binding to the protein. We used the 20ns MD trajectory to calculate MM-PBSA by using the g_mmpbsa Tool (Kumari et al., 2014).
The interaction analysis and visualization of the protein-ligand complex were carried out through AutoDock ADT and PyMOL. Docking conformation and MD simulation results were generated using PyMOL and the 2D graphs of RMSD, RMSF, Rg along with the protein-solvent hydrogen bond interactions and protein-compound hydrogen bond interactions were generated using xmgrace.
3. Results
3.1. Preparation of the protease structures and analysis
The protein structures of SARS-CoV-2 PLPRO (6w9c) and 3CLPRO (6lu7) were retrieved from the PDB. The ligand and water molecules were removed and the proteins were subjected to energy minimization using AMBER ff14 S B force field through UCSF Chimera molecular software. Crystal structures of PLPRO (6w9c) and 3CLPRO (6lu7) are shown in Fig. 2 .
Fig. 2.

Crystal structures of SARS-CoV-2. (A) PLPRO and (B) 3CLPRO.
3.2. pKa calculation
The pKa values for the titratable residues of PLPRO and 3CLPRO proteases were calculated. The atomic resolution of PLPRO is 2.70 Å and the experimental pH is 7.5. The atomic resolution of 3CLPRO is 2.16 Å and the experimental pH is 6.0. Calculated pKa values are shown in Table S1. The titration curves are shown in Fig. 3 . The isoelectric point of PLPRO is 8.82, the total charge is 3.9 when Salinity is 0.15, External Dielectric is 80 and Internal Dielectric is 4. When the Internal Dielectric was changed to 10 and 20, different isoelectric points were obtained. The total charges obtained are shown in Fig. 3A. The isoelectric point of 3CLPRO is 5.45, the total charge is −2 when Salinity is 0.15, External Dielectric is 80 and the Internal Dielectric was 4. When the Internal Dielectric was changed to 10 and 20 the different isoelectric points and total charges were obtained as shown in Fig. 3B.
Fig. 3.

Titration Curves for PLPRO and 3CLPRO protease structures with different Internal Dielectric is 4, 10, and 20.
3.3. Active site identification in PLPRO and 3CLPRO
PLPRO contains four domains, namely the thumb domain, the palm domain, the fingers domain, and the extended ubiquitin-like domain (Ubl) as shown in Fig. 2A (Báez-Santos et al., 2015). An active site, the catalytic triad site, is present between the palm and thumb domains, which contains CYS111, TRP106, TYR112, HIS272, ASP286, GLY271, LEU162, ASP164, GLY271, TYR273, ASP302, PRO248, TYR264, TYR268 and THR301 (Arya et al., 2020; Kong et al., 2015; Báez-Santos et al., 2014). Fig. 4 A shows the top five binding active sites.
Fig. 4.

Top active site of protease enzyme: (A) PLPRO and (B) 3CLPRO.
3CLPRO contains two chymotrypsin-like β-domains, domain-I (1–99 residues), domain-II (100–185 residues) and domain-III (201–303 residues) which contains an α-helical structure (Fig. 2B). Domain-III and II are connected with a long loop having 185–200 amino acids. In Domain-I and II, the active site cleft is located along with the catalytic triad consisting of HIS41 and CYS145. The residues present in the loop connecting domain-I and II are VAL186, ASP187, ARG188 and GLN189 and the side-chain residues are HIS41, MET49 and MET165. Residues MET165, LEU167, GLN189, THR190 and GLN192, are present around domain-I and domain-II. Along with these, residues THR190, ALA191 and GLN192 also participate in the active site (Hsu et al., 2005; Li et al., 2016). Fig. 4B shows the top binding pockets for 3CLPRO. We selected the active pocket 1 for our study.
3.4. Calculation of physicochemical properties
The 4182 FDA approved drugs and the 321 compounds from PubChem COVID-19 clinical trials were not subjected to property space filtering based on physiochemical property calculation because these are FDA approved drugs. NPASS database natural product compounds of 30925 and the Maybridge database compounds of 53344 were, however, subjected to physicochemical property calculation. The downloaded 2D coordinates for all the compounds were converted to 3D coordinates. The selected compounds were then refined using the physicochemical threshold parameters. ChemAxon (command line) software was used to calculate the drug-likeness properties based on the Rule of 3 and Rule of 5. The results are given in Table S2.
Based on the physicochemical property threshold, the 30925 natural compounds from NPASS reduced to 9044 compounds and 53344 compounds from Maybridge database reduced to 24328 compounds. Calculated physicochemical properties are shown in Fig. S1 for the NPASS database and Fig. S2 for the Maybridge database. The red line shows the decided threshold boundary.
3.5. Compound screening using similarity tanimoto coefficient (tc)
Using PubChem COVID-19 clinical trial compounds as reference compounds, NPASS compounds (9044) and Maybridge compounds (24328) were screened to assess their inhibition potential against proteases PLPRO and 3CLPRO and the similarity index were calculated to filter the most similar compounds to PubChem COVID-19 clinical trial compounds, using ChemAxon ScreenMD program. Based on this screening, 659 NPASS compounds with 0.75 Tc value (Fig. 5 A) and 329 Maybridge compounds with 0.75 Tc value were selected for further study (Fig. 5B).
Fig. 5.

Calculation of Average Chemical Fingerprint (CF), Tanimoto 2D Similarity Tc value. (A) Average CF Tanimoto value for NPASS database, (B) Average CF Tanimoto value for Maybridge database. The Red line shows the threshold value.
3.6. ADMET prediction
The selected 659 compounds from the NPASS database and 329 compounds from the Maybridge database were than analyzed using the ADMET descriptors of DS4.1. ADMET plot, obtained by plotting the ADMET_PSA_2D vs. ADMET_AlogP98, describes BBB penetration and HIA level for the compounds (Fig. 6 ). The HIA, plasma protein binding, Cytochrome P450 2D6 inhibition and hepatotoxicity along with BBB were calculated. The selected compounds that pass the ADMET properties have no toxicity.
Fig. 6.

The plot of ADMET_PSA_2D vs. AlogP98 (the 95 and 99% confidence limit ellipses corresponding to the HIA and BBB models for ligands). (A) NPASS database, (B) Maybridge database.
3.7. Molecular docking
3.7.1. Protease PLPRO
NPASS compounds of 659, Maybridge compounds of 329 and all the FDA approval compounds of 4182 and PubChem COVID-19 clinical trial of 321 were subjected to docking. The binding energies (BE) for these compounds were calculated using the AutoDock tool and the selected compounds based on the decided cut-off value of −6.00 kcal/mol are shown in Fig. 7 . The violet color line shows the cut off range.
Fig. 7.

BE for virtual hit compounds. The violet line shows the cut-off range for the compounds selected. (A) FDA approved compounds, (B) PubChem COVID-19 clinical trial compounds, (C) NPASS database compounds and (D) Maybridge database compounds.
3.7.2. FDA approved drugs
Based on molecular docking carried out for the 4182 FDA approved compounds, 1429 compounds were selected which showed BE more than and equal to −6.00 kcal/mol (Fig. 7A). From the 1429 compounds, the top ten compounds were selected for interaction study. Among these ten compounds, Troglitazone, Conivaptan, Differin, Erivedge and Ergotamine bind at a region near the thumb region slightly above the catalytic triad region. All the other compounds bind at the Catalytic triad region (Fig. 8 ). Imatinib, Differin and Erivedge form pi-stacking with HIS175. Conivaptan forms pi-Stacking with TYR171. pi-Cation interactions are formed by Imatinib, Troglitazone, Differin, Erivedge with HIS175. The residue ARG166 forms pi-Cation interaction with Conivaptan and Estradiol Benzoate. Differin and Ergotamine form salt bridges with HIS175. Differin also forms salt bridge with HIS73. Fazadinium forms salt bridge with ASP164 and GLU167. BE, 2D structure, hydrogen bond and hydrophobic interactions obtained for the ten compounds are shown in Table S3 and the graphical binding interactions are shown in Table S4.
Fig. 8.

(A) The ten docked potential inhibitors against SARS-CoV-2 PLPRO from the FDA approved compounds, (B) The 10 potential inhibitors bind to PLPRO, (C) The surface protein with ligands bound to the pocket.
3.7.3. PubChem COVID-19 clinical trial compounds
Based on the molecular docking carried out for the 321 PubChem COVID-19 clinical trial compounds, 57 compounds were selected with BE more than and equal to −6.00 kcal/mol (Fig. 7B). From the 57 compounds, top 10 compounds were selected for interaction study. Bemcentinib forms hydrophobic interactions with TYR154, ASN156, TYR171, VAL202 and hydrogen bonds with HIS73, ASN156, GLN174. Pacritinib, Doxycycline and Ketotifen form salt bridges with ASP164 and Imatinib forms salt bridge with GLU167. Niclosamide forms pi-stacking interactions with TYR264, Ketamine with TYR264 and Imatinib with HIS175. Niclosamide forms halogen bonds with THR301 and ASP302. All these selected compounds bind to the catalytic triad region except Imatinib (light pink) and Bemcentinib (gray color) which bind in between the thumb and catalytic triad region ( Fig. 9 ). The BE, 2D structure, hydrogen bond and hydrophobic interactions are shown in Table S5 and the graphical binding interactions are shown in Table S6.
Fig. 9.

(A) The ten docked potential inhibitors against SARS-CoV-2 PLPRO from PubChem COVID-19 clinical trial compounds, (B) Top 10 potential inhibitors bind to PLPRO protein, (C) The surface protein with ligand bound to the pocket.
3.7.4. NPASS hit compounds
Based on molecular docking carried out for 659 compounds from the NPASS database, 387 compounds were selected having BE more than and equal to six −6.00 kcal/mol (Fig. 7C). From the 387 compounds, the top ten compounds were selected for interaction study. Vallesiachotamine and 19-(R)-Acetoxytabersonine form pi-stacking interactions with TYR264. Lyscamine, Heyneaninehydroxyindolenine form pi-cation interaction with TYR264 and HIS175.19-(R)-Acetoxytabersonine forms salt bridge with ASP164, ARG166, ASP302. Talpinine, Lyscamine form salt bridge with ASP164. Alstilobanine C, 3-Deoxy Nagilactone C and 3-Deoxy Nagilactone C form salt bridge with ARG166. Heyneaninehydroxyindolenine forms salt bridge with ASP76 and HIS175. The NPASS database compounds bind to the catalytic triad region as shown in Fig. 10 . The BE, 2D structure, hydrogen bond and hydrophobic interactions are shown in Table S7 and the graphical representation of interactions is shown in Table S8.
Fig. 10.

(A) The ten docked compounds from NPASS database are potential inhibitors against SARS-CoV-2 PLPRO, (B) Top 10 potential inhibitor binds to PLPRO protein in the Catalytic triad region, (C) The surface protein with ligand bound to the pocket.
3.7.5. Maybridge hits compounds
Based on the molecular docking carried out for 329 Maybridge compounds, 143 compounds were selected based on BE more than and equal to −6.00 kcal/mol (Fig. 7D). From the 143 compounds, the top 10 compounds were selected for interaction study. MFCD02180753 forms pi-stacking interaction with TYR264, hydrophobic interactions with LEU162, ARG166, TYR264, THR301 and hydrogen bonds with LEU162, ASP164, TYR264 and TYR273. MFCD02180753 and MFCD02679250 form pi-stacking interactions with TYR264. MFCD04123932 forms pi-stacking interaction with HIS175. MFCD00552607 forms pi-cation interaction with ARG140. MFCD00552607 forms hydrophobic interaction with LEU101, GLN122, and hydrogen bond with GLU124, LYS126, ARG140. The Maybridge database compounds bind to the Catalytic triad region as shown in Fig. 11 . The BE, 2D structure, hydrogen bond and hydrophobic interactions are shown in Table S9 and the graphical representation of interactions is shown in Table S10.
Fig. 11.

(A) The ten docked potential inhibitors agents against SARS-CoV-2 PLPRO from the Maybridge database, (B) Top 10 potential inhibitor binds to PLPRO protein in the Catalytic triad region, (C) The surface protein with ligand bound to the pocket.
The compounds docked in PLPRO protease are present near to the catalytic triad and Palm region. We found the virtual hit compounds bind to this region and show interactions with different amino acids. Compounds which bind in the Catalytic triad region were selected and sorted out based on the clustering method.
3.7.6. Protease 3CLPRO
NPASS database compounds of 659, Maybridge database compounds of 329, all the 4182 compounds from FDA approved drugs and 321 compounds from PubChem COVID-19 clinical trial were subjected to docking. The BE for these compounds, calculated using AutoDock tool and the selected compounds based on the cut-off value by −8.00 kcal/mol are shown in Fig. 12 . The violet color shows the cut-off range.
Fig. 12.

BE for the virtual hit compounds. The violet line shows the cut-off range for compound selection, (A) FDA approved compounds, (B) PubChem COVID-19 clinical trial compounds, (C) NPASS Database compounds, and (D) Maybridge Database compounds.
3.7.7. FDA approved drugs
Based on molecular docking for the 4182 FDA approved drugs, 1000 compounds were selected which showed BE more than and equal to −8.00 kcal/mol (Fig. 12A). From the selected 1000 compounds, the top ten compounds were selected for interaction study. Paritaprevir forms hydrophobic interactions with HIS41, LEU141, GLU166, and GLN189 and hydrogen bonds with PHE140, HIS164, GLU166. Nintedanib forms hydrophobic interactions with HIS41, ASP187, GLN189, and hydrogen bonds with GLY143 and GLU166. Imatinib, Nintedanib and Ergotamine form salt bridge with GLU166. Imatinib and Lifitegrast form the pi-stacking interactions with HIS163 and HIS41. Lifitegrast forms the salt bridge with ASP187. Nilotinib forms halogen bond with GLN189. All the ten compounds bind to the active site between Domain-I and Domain-II, near to the long loop that connect Domain-III with Domain-II ( Fig. 13 ). The BE, 2D structure, hydrogen bond and hydrophobic interactions are shown in Table S11 and the graphical representation of interactions is shown in Table S12.
Fig. 13.

(A) The ten potential inhibitors against SARS-CoV-2 3CLPRO from FDA approved compounds, (B) Top 10 potential inhibitors bind to 3CLPRO protein near the long loop connected by domain-I and domain-II, (C) The surface protein with ligand bound to the pocket.
3.7.8. PubChem COVID-19 clinical trial compounds
Based on molecular docking carried out for the selected 321 PubChem COVID-19 clinical trial compounds, 48 compounds were selected having a BE more than and equal to −8.00 kcal/mol (Fig. 12B). From these 48 compounds, the top 10 compounds were selected for interaction study. Bemcentinib forms salt bridge with GLU166, hydrophobic interactions with HIS41, MET49, ASN142, MET165, GLU166, PRO168, GLN189 and hydrogen bonds with ASN142 and GLU166. Bemcentinib, Imatinib, and Abivertinib form salt bridge with GLU166. Ozanimod shows pi-Stacking interaction with HIS41. Amiodarone forms hydrophobic interactions with MET165, GLN189, and hydrogen bond with GLU166. The selected ten compounds bind in the active site between Domain-I and Domain-II, near the long loop that connect Domain-III and Domain-II shown in Red color (Fig. 14 ). The BE, 2D structure, hydrogen bond and hydrophobic interactions are shown in Table S13 and the graphical representation of interactions is shown in Table S14.
Fig. 14.

(A) The ten docked against SARS-CoV-2 3CLPRO from PubChem COVID-19 clinical trial compounds, (B) Top 10 potential inhibitor bind to 3CLPRO protein near the long loop connected by domain-I and domain-II, (C) The surface protein with ligand bound to the pocket.
3.7.9. NPASS database hit compounds
Based on molecular docking carried out for the 659 NPASS database compounds, 137 compounds were selected having BE more than and equal to −8.00 kcal/mol (Fig. 12C). From these 137 compounds, the top 10 compounds were selected for interaction study. Serpentine and 3-Hydroxyglyantrypine form pi-stacking with HIS41. Leuconicine F forms pi-Cation interaction with HIS41. The selected top 10 compounds bind in the region between Domain-I and Domain-II, near the long loop connected to Domain-III and Domain-II shown in red color ( Fig. 15 ). The BE, 2D structure, hydrogen bond and hydrophobic interactions are shown in Table S15 and the graphical representation of interactions is shown in Table S16.
Fig. 15.

(A) The ten docked potential inhibitors against SARS-CoV-2 3CLPRO from NPASS database compounds (B) Top 10 potential inhibitors bind to the 3CLPRO protein near the long loop connected by domain-I domain-II, (C) The surface protein with ligand bound to the pocket.
3.7.10. Maybridge database hits compounds
Based on molecular docking carried out for 329 Maybridge compounds, 96 compounds were selected with BE more than and equal to −8.00 kcal/mol (Fig. 12D). From these 96 compounds, the top 10 compounds were selected for interaction study. MFCD03407257, MFCD04123932, MFCD00604704, MFCD00116061, MFCD00832476 and MFCD04123470 form pi-stacking interaction with HIS41. MFCD03407257 forms hydrogen bond interactions with HIS41, GLY143, GLU166 and hydrophobic interactions with HIS41, GLU166, ASP187. The hit compounds bind between Domain-I and Domain-II near the long loop connected to Domain-III and Domain-II, shown in red color ( Fig. 16 ). The BE, 2D structure, hydrogen bond and hydrophobic interactions are shown in Table S17 and the graphical representation of interactions is shown in Table S18.
Fig. 16.

(A) The ten potential inhibitors against SARS-CoV-2 3CLPRO from Maybridge database compounds, (B) Top 10 potential inhibitors bind to 3CLPRO protein near to the long loop connected by domain-I and domain-II, (C) The surface protein with ligand bound to the pocket.
Molecular docking studies confirmed that the hit compounds bind to the 3CLPRO protease active site between residues 140–145 and residues 163–166. These compounds bind to the catalytic diad at HIS41, CYS145 and GLU166. The docking score for all compounds from the selected database is summarized in Tables S3–S18.
3.8. Clustering analysis
Clustering analysis re-ranks the compounds and generates a filtered new list of compounds from docking that rank high for the receptor PLPRO and 3CLPRO binding. The top of the cluster are considered as better hits.
3.8.1. PLPRO clustering
We performed Clustering analysis using ChemBioServer online tool based on the Hierarchical Clustering using Soergel (Tc) Distance method and Ward Linkage clustering method. We found 10 Clusters for PLPRO (Fig. 17 A). We selected the top cluster, which contains 9 compounds. From these nine compounds, we selected five compounds based on MD simulation and free energy calculation.
Fig. 17.

(A) 10 Clusters found in PLPRO and (B) 8 Clusters found in 3CLPRO based on the Hierarchical Clustering method.
3.8.2. 3CLPRO clustering
In 3CLPRO Clustering, we found eight clusters (Fig. 17B). We selected the top cluster, which contains 11 compounds. From these 11 compounds, we selected six compounds based on MD simulation and free energy calculation.
3.9. MD trajectory analysis and binding free energy calculation
The structural and conformational stability of the protein-ligand complexes were checked during the 20ns simulation by calculating three properties, namely RMSD, RMSF, and Rg. We also checked the hydrogen bond interactions between the protein and ligand and between the protein and solvent.
3.9.1. MD simulation for PLPRO
The selected top five PLPRO- compound complexes were analyzed for 20ns simulation through RMSD, RMSF, and Rg ( Fig. 18 ). RMSD analysis shows the difference in the protein backbone from the initial structure (0ns) to the final confirmed structure (20ns). The complexes C-α backbone values were calculated after 20ns to check the stability of the system. Fluctuations were observed during the 20 ns simulation. Fig. 18A shows the RMSD plot for the selected five PLPRO-compound complexes. All the five complexes show more or less the same fluctuation as the protein without the ligand. All the five complexes except Bemecentinib-PLPRO show RMSD values between 0.1 nm and 0.3 nm. Bemecentinib-PLPRO complex shows an RMSD value of more than 0.3 nm up to the end of the simulation. The RMSD result reveals that the PLPRO-hit compound complexes are stable throughout the 20ns simulation period.
Fig. 18.

MD trajectory analysis for the selected top five compound-PLPRO complexes. (A) RMSD, (B) RMSF, (C) Rg, (D) Hydrogen bonds interaction between protein and solvent, (E) Hydrogen bond interaction between PLPRO and compounds.
PLPRO protease shows high RMSF in the loop, turns, and coils. The initial residues (0–75 residues) of the protein are not very stable. It shows fluctuation of more than 0.4 nm at 185–200 and 215–230 residues. Residues 262–275 show an increase in the RMSF value of less than 0.4 nm (Fig. 18B). All the five PLPRO-hit compound complexes have almost similar RMSF patterns. RMSF results of the five complexes confirm their strong binding to PLPRO protease.
Rg analysis was performed to judge the level of compactness of protein and the PLPRO-compound complexes during simulations. Lower Rg values characterize the compactness of the protein (Fig. 18C). Rg value at the start of the simulation for all the five PLPRO-compound complexes increases between 2.35 nm and 2.40 nm up to 500ps. After 500ps, it shows a slight decrease and becomes steady up to 20000ps (20ns) simulation between 2.30 and 2.40 nm. A constant Rg value between 2.30 and 2.40 nm shows the protein's constant compactness when compounds bind to the protein and show constant interaction during the simulation (Fig. 18C). The Rg results reveal that the selected compounds bind to PLPRO protein with good compactness.
The hydrogen bond interactions between PLPRO and the solvent (Fig. 18D) and between PLPRO-compound complexes (Fig. 18E) were also calculated. Hydrogen bond formations in PLPRO-compound complexes reveal that when the PLPRO-compound complex is simulated for 20ns, hydrogen bond formation is constant. The majority of the compounds form more than one hydrogen bond. The hydrogen bond between the protein and the solvent is constant between 540 and 640 numbers.
3.9.2. MD simulation for 3CLPRO
The selected top six 3CLPRO-compound complexes were analyzed for 20ns MD simulation through RMSD, RMSF and Rg (Fig. 19 ). RMSD plot shows fluctuations in 3CLPRO and 3CLPRO-compound backbone. At the initial simulation up to 5ns all the six 3CLPRO- compound complexes show the same fluctuation between 0.1 nm and 0.2 nm (Fig. 19A). When we compare the 3CLPRO and 3CLPRO-compound complexes, Clofazimine shows the highest RMSD value which is more than 0.2 nm between 5 and 5.5ns time and after 15ns it shows a fluctuation of more than 0.2 nm up to end of the simulation (20ns). The RMSD results reveal that the six 3CLPRO-compound complexes are stable throughout the simulation.
Fig. 19.

MD trajectory analysis for selected top six compound-3CLPROcomplexes. (A) RMSD, (B) RMSF, (C) Rg, (D) Hydrogen bonds formation between protein-solvent, and (E) Hydrogen bond formation between the 3CLPRO and compounds in complexes.
All the 3CLPRO-compound complexes show almost the same RMSF value (Fig. 19B). The changes in the RMSF values may be due to the loop, turns, and coils. All the six complexes show the same pattern of RMSF, but in some regions they show slightly more or less RMSF values. The RMSF fluctuations of the six 3CLPRO-compound complexes reveal that compounds strongly bind to the 3CLPRO active site.
Rg for all the compound-3CLPRO complexes is between 2.18 nm and 2.31 nm (Fig. 19C). At the initial stage up to 10ns, it shows the same pattern. After that, each complex shows a different Rg value. Comparing the Rg value between the six 3CLPRO-compound complexes and the protein, a slightly lower Rg value is observed for all complexes except 3CLPRO-Abivertinib and 3CLPRO-Clofazimine. Rg values of all the complexes show the compactness of the protein when the compounds bind to the 3CLPRO and all the complexes are stable with good compactness.
We also calculated the hydrogen bond formations between 3CLPRO and the compounds in the complexes and between and the protein and the solvent (Fig. 19D). Protein-solvent hydrogen bond formations are observed by the amino acids 580–650. The compounds that bind to the 3CLPRO form hydrogen bonds with the solvent in the 470–580 amino acid region in the entire 20ns simulation. When the compounds bind to 3CLPRO, an average of two hydrogen bonds are formed in the whole simulation (Fig. 19E).
3.10. Binding free energy (BFE) analyses
BFE calculation is an attractive and reliable approach to predict protein-ligand binding affinities in drug discovery processes. BFEs were calculated using MM-PBSA for the selected five PLPRO and six 3CLPRO protein-compound complexes and the data are shown in Table 1 . The data reveal that all the complexes show negative BFE indicating the stability of the complexes and thus good affinity between the ligand and the protein. Among all the PLPRO-compound complexes, MFCD00832476-protein shows the highest BFE of −64.11 kJ/mol and Ergotamine shows the lowest BFE of −200.060 kJ/mol. Bemcentinib, MFCD00832476 and MFCD02180753 protein complexes show better van der waals energy of −133.533 kJ/mol, −144.428 kJ/mol and −157.661 kJ/mol, respectively and electrostatic energy of −67.779 kJ/mol, −97.818 kJ/mol and −4.705 kJ/mol, respectively, SASA energy of −13.151 kJ/mol, −13.318 kJ/mol, −13.784 kJ/mol, respectively. BFE of Ergotamine (−200.060 kJ/mol) shows good binding affinity.
Table 1.
MM-PBSA Binding free energy for the five PLPRO–compound and six 3CLPRO–compound complexes.
| Compound | Van der Waal energy (kJ/mol) | Electrostatic energy (kJ/mol) | Polar solvation energy (kJ/mol) | SASA energy (kJ/mol) | Binding free energy (kJ/mol) |
|---|---|---|---|---|---|
| PLPRO Hit Compounds | |||||
Bemcentinib 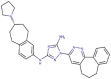
|
−133.533 | −67.779 | 135.999 | −13.151 | −78.464 |
Pacritinib 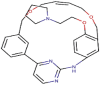
|
−161.261 | −5.222 | 72.476 | −13.373 | −107.380 |
Ergotamine 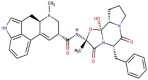
|
−164.065 | −278.233 | 258.320 | −16.082 | −200.060 |
MFCD00832476 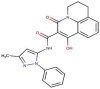
|
−144.428 | −97.818 | 191.451 | −13.318 | −64.114 |
MFCD02180753 
|
−157.661 | −4.705 | 59.149 | −13.784 | −117.001 |
| 3CLPRO Hit Compounds | |||||
Clofazimine 
|
−220.155 | −7.036 | 81.063 | −19.471 | −165.599 |
Bemcentinib 
|
−162.502 | −233.925 | 180.432 | −17.077 | −233.071 |
Abivertinib
|
−244.313 | −256.523 | 221.044 | −21.576 | −301.367 |
Dasabuvir 
|
−246.323 | −22.167 | 123.144 | −20.809 | −166.156 |
MFCD00832476 
|
−209.214 | −24.103 | 92.367 | −17.706 | −158.657 |
Leuconicine F 
|
−130.320 | −260.961 | 179.384 | −12.425 | −224.322 |
Among the all 3CLPRO-compound complexes, MFCD00832476 shows the highest BFE of −158.657 kJ/mol and Abivertinib shows the lowest BFE of −301.367 kJ/mol. Bemcentinib, MFCD00832476 and Leuconicine F complexes show high van der waals energy of −162.502 kJ/mol, −209.214 kJ/mol and-130.32 kJ/mol, respectively and electrostatic energy of −233.925 kJ/mol, −24.103 kJ/mol and −260.961 kJ/mol, respectively and SASA energy of −17 kJ/mol, −17.7 kJ/mol, −12.425 kJ/mol, respectively. All the five PLPRO and the six 3CLPRO complexes show good BFE.
4. Discussion
COVID-19 epidemic has made experts to realize that our options today to treat this life-threatening disease are unfortunately very limited. In spite of considerable research efforts carried out, as of today no drugs have been approved by FDA. Developing a new drug from preclinical identification to approval, however, takes nearly US$1000 million and 12 years of time. A few vaccines developed and approved by regulatory authorities are being administered in several countries. It is only proper, therefore, that the existing drugs and potential therapeutic compounds available in public domain are investigated based on detailed and thorough computational bioinformatic approach to repurpose them to treat COVID-19.
In the present study, we have carried in silico high throughput virtual screening of FDA approved drugs, COVID-19 clinical trial compounds, natural products from NPASS and Maybridge databases against PLPRO and 3CLPRO proteases which cleave the causative agent, SARS-CoV-2, to several non-structural proteins that are important for the viral replication. A few studies like the present one have been carried out by earlier investigators using only small numbers of compounds and limited step-by-step virtual screening techniques (Deep et al., 2021; Sajjan et al., 2021; Nejabat et al., 2021; Mitra et al., 2020). We believe that the present study is more detailed and thorough, as it is based on subjecting the existing drugs/compounds to filtering threshold physicochemical drug like properties and 2D similarity index followed by ADMET prediction, molecular docking, clustering, molecular dynamics simulation and binding free-energy calculation. It may be pointed out here that BFE calculation is an attractive and reliable approach to predict the binding affinities in drug discovery processes, which some of the earlier investigators have not carried out thoroughly like the present study. The study has revealed that five compounds, namely MFCD00832476, MFCD02180753, Bemcentinib, Pacritinib and Ergotamine inhibit PLPRO and six compounds, namely Bemcentinib, Clofazimine, Abivertinib, Dasabuvir, MFCD00832476 and Leuconicine F inhibit 3CLPRO. Interestingly, the results also reveal that Bemcentinib and MFCD00832476 are dual inhibitors of SARS-CoV-2 proteases, PLPRO and 3CLPRO. All these compounds are possible pharmacotherapeutic agents for treating COVID-19 disease. Among the compounds studied for BFE calculation, Ergotamine shows the lowest BFE and favourable binding affinity to PLPRO and Abivertinib shows the lowest BFE and favourable binding affinity for 3CLPRO. These two compounds are, therefore, more potential inhibitors for SARS-CoV-2 and hence good starting points/leads for designing and developing drugs for COVID-19. Our study is purely based on computer-based screening. As of now, we have not carried out any in vitro and in vivo studies. We are keen on sharing our results with our fellow scientists and hence this publication.
5. Conclusion
There is an urgent need to find a suitable drug to treat COVID-19. In this connection, we have screened a total of 5491 compounds/drugs from selected databases using in silico high throughput screening to find inhibitors for SARS-CoV-2 proteases, PLPRO and 3CLPRO, so as to treat COVID-19 by controlling its lifecycle. Our studies reveal that five compounds, namely MFCD00832476, MFCD02180753, Bemcentinib, Pacritinib and Ergotamine that inhibit PLPRO and six compounds, namely Bemcentinib, Clofazimine, Abivertinib, Dasabuvir, MFCD00832476 and Leuconicine F that inhibit 3CLPRO, may be used for treating COVID-19. In particular, Ergotamine and Abivertinib are good leads for designing and developing drugs for COVID-19.
Funding
Authors Selvaraj Ayyamperumal (2019-5557/CMB-BMS) and Vyshnavi Tallapaneni [3/1/3(12)19-NCD II] wish to express their gratitude to the Indian Council of Medical Research for the award of Senior Research Fellowship.
CRediT authorship contribution statement
Dhananjay Jade: Conceptualization, Methodology, Software, Writing – original draft. Selvaraj Ayyamperumal: Conceptualization, Methodology, Software. Vyshnavi Tallapaneni: Data curation, Writing – original draft. Chandrasekar Moola Joghee Nanjan: Visualization, Investigation, Resources, Writing – review & editing. Sagar Barge: Software, Validation. Surender Mohan: Software, Validation. Moola Joghee Nanjan: Supervision, Visualization, Investigation, Writing – review & editing.
Declaration of competing interest
The authors declare no competing interests.
Acknowledgment
We thank JSS College of Pharmacy, Department of Pharmaceutical Chemistry, Ooty, Tamilnadu, India., Biochemistry and Drug Discovery Lab, Institute of Advanced Study in Science and Technology, Paschim Boragaon, Guwahati-35, Assam, India, for providing us with the resources and completion of our research.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejphar.2021.174082.
Appendix A. Supplementary data
The following is/are the supplementary data to this article:
References
- Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. Software. 2015;1:19–25. [Google Scholar]
- Ahamad S., Kanipakam H., Birla S., Ali M.S., Gupta D. Screening Malaria-box compounds to identify potential inhibitors against Sars-Cov-2 Mpro using molecular docking and dynamics simulations studies. Eur. J. Pharmacol. 2020:173664. doi: 10.1016/j.ejphar.2020.173664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3clpro) structure: basis for design of anti-sars drugs. Science. 2003;300:1763. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Anandakrishnan R., Aguilar B., Onufriev A.V. H++ 3.0: automating p K prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012;40:W537–W541. doi: 10.1093/nar/gks375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya R., Das A., Prashar V., Kumar M. Potential inhibitors against papain-like protease of novel coronavirus (Covid-19) from Fda approved drugs. Chemrxiv. 2020 [Google Scholar]
- Baez-Santos Y.M., John S.E.S., Mesecar A.D. The Sars-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez-Santos Y.M., Mielech A.M., Deng X., Baker S., Mesecar A.D. Catalytic function and substrate specificity of the papain-like protease domain of nsp3 from the Middle East respiratory syndrome coronavirus. J. Virol. 2014;88:12511–12527. doi: 10.1128/JVI.01294-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Liu Q., Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 2020;92:418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L., Gao G.F. Viral targets for vaccines against Covid-19. Nature reviews. Immunology. 2021;21(2):73–82. doi: 10.1038/s41577-020-00480-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wit E., Van Doremalen N., Falzarano D., Munster V.J. Sars and Mers: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016;14:523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deep Bhowmik, Sharma Ravi Datta, Prakash Amresh, Kumar Diwakar. Identification of nafamostat and Vr23 as covid-19 drug candidates by targeting 3clpro and plpro. J. Mol. Struct. 2021;130094 doi: 10.1016/j.molstruc.2021.130094. 0022-2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr A.R., Perlman S. Springer; 2015. Coronaviruses: an Overview of Their Replication and Pathogenesis. Coronaviruses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genheden S., Ryde U. The Mm/Pbsa and Mm/Gbsa methods to estimate ligand-binding affinities. Expet Opin. Drug Discov. 2015;10:449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Zhang J., Hu C.Q., Zhang X., Ma B., Zhang P. In silico Adme and toxicity prediction of ceftazidime and its impurities. Front. Pharmacol. 2019;10:434. doi: 10.3389/fphar.2019.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt P.A., Chaires J.B., Trent J.O. Molecular docking of intercalators and groove-binders to nucleic acids using Autodock and Surflex. J. Chem. Inf. Model. 2008;48:1602–1615. doi: 10.1021/ci800063v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M.F., Kuo C.J., Chang K.T., Chang H.C., Chou C.C., Ko T.P., Shr H.L., Chang G.G., Wang A.H., Liang P.H. Mechanism of the maturation process of Sars-Cov 3cl protease. J. Biol. Chem. 2005;280:31257–31266. doi: 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J.J., Shoichet B.K. Zinc− a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhoti H., Williams G., Rees D.C., Murray C.W. The'rule of three'for fragment-based drug discovery: where are we now? Nat. Rev. Drug Discov. 2013;12 doi: 10.1038/nrd3926-c1. 644-644. [DOI] [PubMed] [Google Scholar]
- Kanjanahaluethai A., Chen Z., Jukneliene D., Baker S.C. Membrane topology of murine coronavirus replicase nonstructural protein 3. Virology. 2007;361:391–401. doi: 10.1016/j.virol.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatzas E., Zamora J.E., Athanasiadis E., Dellis D., Cournia Z., Spyrou G.M. Chembioserver 2.0: an advanced web server for filtering, clustering and networking of chemical compounds facilitating both drug discovery and repurposing. Bioinformatics. 2020;36:2602–2604. doi: 10.1093/bioinformatics/btz976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim P.S., Read S.W., Fauci A.S. Therapy for early covid-19: a critical need. Jama. 2020;324:2149–2150. doi: 10.1001/jama.2020.22813. [DOI] [PubMed] [Google Scholar]
- Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B.A., Thiessen P.A., Yu B. Pubchem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47:D1102–D1109. doi: 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L., Shaw N., Yan L., Lou Z., Rao Z. Structural view and substrate specificity of papain-like protease from avian infectious bronchitis virus. J. Biol. Chem. 2015;290:7160–7168. doi: 10.1074/jbc.M114.628636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouznetsova V.L., Zhang A., Tatineni M., Miller M.A., Tsigelny I.F. Potential Covid-19 papain-like protease Plpro inhibitors: repurposing Fda-approved drugs. Peerj. 2020;8 doi: 10.7717/peerj.9965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R., Jade D., Gupta D. A novel identification approach for discovery of 5-Hydroxytriptamine 2a antagonists: combination of 2d/3d similarity screening, molecular docking and molecular dynamics. J. Biomol. Struct. Dyn. 2019;37:931–943. doi: 10.1080/07391102.2018.1444509. [DOI] [PubMed] [Google Scholar]
- Kumari R., Kumar R., Consortium O.S.D.D., Lynn A. g_mmpbsa A Gromacs tool for high-throughput Mm-Pbsa calculations. J. Chem. Inf. Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Li C., Teng X., Qi Y., Tang B., Shi H., Ma X., Lai L. Conformational flexibility of a short loop near the active site of the sars-3clpro is essential to maintain catalytic activity. Sci. Rep. 2016;6:20918. doi: 10.1038/srep20918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Guan X., Wu P., Wang X., Zhou L., Tong Y., Ren R., Leung K.S.M., Lau E.H.Y., Wong J.Y., Xing X., Xiang N., Wu Y., Li C., Chen Q., Li D., Liu T., Zhao J., Liu M., Tu W., Chen C., Jin L., Yang R., Wang Q., Zhou S., Wang R., Liu H., Luo Y., Liu Y., Shao G., Li H., Tao Z., Yang Y., Deng Z., Liu B., Ma Z., Zhang Y., Shi G., Lam T.T.Y., Wu J.T., Gao G.F., Cowling B.J., Yang B., Leung G.M., Feng Z. Early transmission dynamics in wuhan, China, of novel coronavirus–infected pneumonia. N. Engl. J. Med. 2020;382:1199–1207. doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Mitra K., Ghanta P., Acharya S., Chakrapani G., Ramaiah B., Doble M. Dual inhibitors of Sars-Cov-2 proteases: pharmacophore and molecular dynamics based drug repositioning and phytochemical leads. J. Biomol. Struct. Dyn. 2020:1–14. doi: 10.1080/07391102.2020.1796802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. Autodock4 and Autodocktools 4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejabat M., Ghodsi R., Hadizadeh F. Coumarins and quinolones as effective multiple targeted agents versus covid-19: an in silico study. Med. Chem. 2021 doi: 10.2174/1573406417666210208223924. [DOI] [PubMed] [Google Scholar]
- Norinder U., Bergström C.A. Prediction of admet properties. ChemMedChem: Chemistry Enabling Drug Discovery. 2006;1:920–937. doi: 10.1002/cmdc.200600155. [DOI] [PubMed] [Google Scholar]
- Oostra M., Hagemeijer M.C., Van Gent M., Bekker C.P.J., Te Lintelo E.G., Rottier P.J.M., De Haan C.A.M. Topology and membrane anchoring of the coronavirus replication complex: not all hydrophobic domains of nsp3 and nsp 6 are membrane spanning. J. Virol. 2008;82:12392–12405. doi: 10.1128/JVI.01219-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeri N.C., Shrestha N., Rahman M.S., Zaki R., Tan Z., Bibi S., Baghbanzadeh M., Aghamohammadi N., Zhang W., Haque U. The Sars, Mers and novel coronavirus (Covid-19) epidemics, the newest and biggest global health threats: what lessons have we learned? Int. J. Epidemiol. 2020;49:717–726. doi: 10.1093/ije/dyaa033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. Ucsf Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pollastri M.P. Overview on the rule of five. Current Protocols in Pharmacology. 2010;49 doi: 10.1002/0471141755.ph0912s49. 9.12.1-9.12.8. [DOI] [PubMed] [Google Scholar]
- Sajjan Rajpoot, Manikandan Alagumuthu, Mirza S. Baig. Dual targeting of 3clpro and Plpro of Sars-Cov-2: a novel structure-based design approach to treat Covid-19. Current Research in Structural Biology. 2021;3:9–18. doi: 10.1016/j.crstbi.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid N., Eichenberger A.P., Choutko A., Riniker S., Winger M., Mark A.E., Van Gunsteren W.F. Definition and testing of the Gromos force-field versions 54a7 and 54b7. Eur. Biophys. J. 2011;40:843–856. doi: 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- Schüttelkopf A.W., Van Aalten D.M. Prodrg: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Sharma R., Jade D., Mohan S., Chandel R., Sugumar S. In-silico virtual screening for identification of potent inhibitor for L2-β-lactamase from Stenotrophomonas maltophilia through molecular docking, molecular dynamics analysis study. J. Biomol. Struct. Dyn. 2020:1–15. doi: 10.1080/07391102.2020.1805365. [DOI] [PubMed] [Google Scholar]
- Silveira M.M., Moreira G., Mendonça M. Dna vaccines against covid-19: perspectives and challenges. Life Sci. 2021;267:118919. doi: 10.1016/j.lfs.2020.118919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasi C., Fallani S., Voller F., Silvestri C. Treatment for covid-19: an overview. Eur. J. Pharmacol. 2020;889:173644. doi: 10.1016/j.ejphar.2020.173644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling T., Irwin J.J. Zinc 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V., Ivanov K.A., Putics Á., Hertzig T., Schelle B., Bayer S., Weißbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. Mechanisms and enzymes involved in Sars coronavirus genome expression. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- Van Hemert M.J., Van Den Worm S.H.E., Knoops K., Mommaas A.M., Gorbalenya A.E., Snijder E.J. Sars-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathog. 2008;4 doi: 10.1371/journal.ppat.1000054. e1000054-e1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Horby P.W., Hayden F.G., Gao G.F. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470–473. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weglarz-Tomczak E., Tomczak J.M., Giurg M., Burda-Grabowska M., Brul S. biorxiv; 2020. Discovery of Potent Inhibitors of Plprocov2 by Screening Libraries of Selenium-Containing Compounds. [Google Scholar]
- Zeng X., Zhang P., He W., Qin C., Chen S., Tao L., Wang Y., Tan Y., Gao D., Wang B. Npass: natural product activity and species source database for natural product research, discovery and tool development. Nucleic Acids Res. 2018;46:D1217–D1222. doi: 10.1093/nar/gkx1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Li Y., Lin B., Schroeder M., Huang B. Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics. 2011;27:2083–2088. doi: 10.1093/bioinformatics/btr331. [DOI] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., Chen H.D., Chen J., Luo Y., Guo H., Jiang R.D., Liu M.Q., Chen Y., Shen X.R., Wang X., Zheng X.S., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J. Molecular biology of severe acute respiratory syndrome coronavirus. Curr. Opin. Microbiol. 2004;7:412–419. doi: 10.1016/j.mib.2004.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Thiel V., Gorbalenya A.E. The autocatalytic release of a putative Rna virus transcription factor from its polyprotein precursor involves two paralogous papain-like proteases that cleave the same peptide bond. J. Biol. Chem. 2001;276:33220–33232. doi: 10.1074/jbc.M104097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.