

Abstract
All bacteria must compete for growth niches and other limited environmental resources. These existential battles are waged at several levels, but one common strategy entails the transfer of growth inhibitory protein toxins between competing cells. These antibacterial effectors are invariably encoded with immunity proteins that protect cells from intoxication by neighboring siblings. Several effector classes have been described, each designed to breach the cell envelope of target bacteria. Although effector architectures and export pathways tend to be clade specific, phylogenetically distant species often deploy closely related toxin domains. Thus, diverse competition systems are linked through a common reservoir of toxin-immunity pairs that is shared via horizontal gene transfer. These toxin-immunity protein pairs are extraordinarily variable in sequence, and this polymorphism underpins an important mechanism of self/nonself discrimination in bacteria. This review focuses on the structures, functions and delivery mechanisms of polymorphic toxin effectors that mediate bacterial competition.
Keywords: colicins, contact-dependent growth inhibition (CDI), MafB, type VI secretion system (T6SS), Esx secretion system (ESS), outer-membrane exchange (OME)
INTRODUCTION
Toxin-immunity protein pairs play a prominent role in prokaryotic biology. In this context, a “toxin” is a small protein, or protein domain, that inhibits bacterial cell growth. Bacteriocins were the first antibacterial toxins to be characterized (27). Many species release bacteriocins to kill competitors in the environment. Bacteriocinogenic cells also produce specific immunity proteins, which neutralize toxicity and protect against auto-inhibition. In this manner, bacteriocin release provides a competitive growth advantage to some strains. In the 1980s, a different class of small toxin-antitoxin gene pairs were found to contribute to plasmid maintenance. The antitoxins of these systems are rapidly degraded by cytoplasmic proteases, which in turn exerts a selective pressure to retain the plasmid for continued antitoxin synthesis. These latter toxin-antitoxin systems are also commonly encoded in the genomes of eubacteria and archaea, where they are thought to be important regulators of cell growth (53; 113).
In 2005, a new competitive mechanism that requires direct contact between toxin-producing and target bacteria was discovered. This “contact-dependent growth inhibition” (CDI) phenomenon is mediated by CdiA proteins exported by a subclass of type V secretion systems (T5SS) (4). Like bacteriocins, CdiA effectors are encoded with specific immunity proteins that neutralize toxicity. With time, it became clear that bacteria utilize a broad spectrum of toxin delivery strategies. The potent bactericidal activity of the type VI secretion system (T6SS) was first reported in 2010 (57; 83); followed rapidly by predictions of other antibacterial mechanisms (56; 105; 157; 158). These predictions have been verified for Neisserial MafB proteins (6; 65) and Esx-like secretion systems (ESS) in Gram-positive bacteria (25; 146). Further, some type IV secretion systems (T4SS) deliver toxins directly into target bacteria (129), and antibacterial WapA and SitA effectors have been characterized from Bacillus subtilis and Myxococcus xanthus, respectively (73; 139). It is now clear that inter-cellular toxin transfer is a fundamental and ubiquitous aspect of prokaryotic biology.
POLYMORPHIC EFFECTOR FAMILIES
Polymorphic effectors are a diverse assemblage of proteins linked by their common function in inter-bacterial competition. Effector classes vary in size, sequence and structure, but share similarly organized multi-domain architectures. Each class is characterized by conserved N-terminal regions that guide export and/or mediate delivery into target bacteria (Fig. 1a). Neisseria MafB (6; 65), Myxococcus SitA (139) and Bacillus WapA (73) proteins rely on Sec-dependent secretion, whereas CdiA carries an additional transport domain required for T5SS-dependent export across the outer membrane (52). T6SSs export valine-glycine repeat G (VgrG) and hemolysin-coregulated proteins (Hcp), which sometimes carry toxin domains (34). Several other T6SS effectors have Pro-Ala-Ala-Arg (PAAR) repeat domains, which enable export via direct interactions with VgrG (125). The ESS selects substrates through N-terminal coiled-coil domains that contain Trp-Xaa-Gly (WXG) or Leu-Xaa-Gly (LXG) motifs (48; 157). For each effector class, the extreme C-terminal domain is responsible for bactericidal activity. Toxin domain sequences can vary dramatically, even between closely related strains, and therefore the “polymorphic” appellation describes the diversity of toxic cargos. Thus, all polymorphic effectors have the capacity to deliver a variety of toxin domains. This modularity was first described for E-type colicins, which use homologous receptor-binding (R) and translocation (T) domains to transfer distinct DNase, rRNase and tRNase nucleases into target bacteria (Fig. 1b) (27).
Figure 1. Polymorphic effector families.
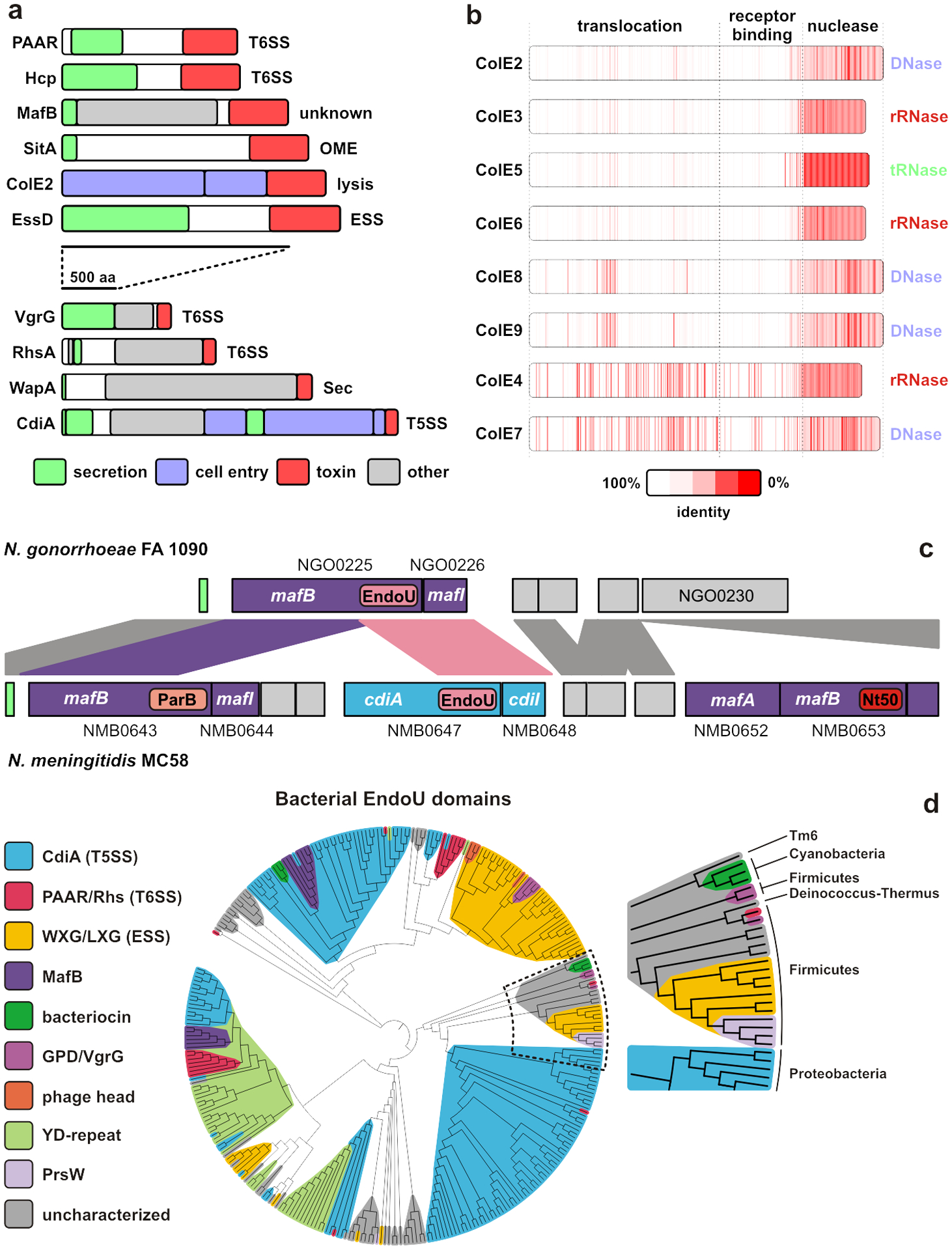
a) Polymorphic effector classes that function in bacterial competition. b) E-type colicin domain structure and homology. c) Alignment of maf genomic islands from N. gonorrhoeae FA 1090 and N. meningitidis MC58. Ordered locus identifiers are given and sequences encoding predicted ParB, EndoU and Ntox50 (Nt50) toxin domains are indicated. d) Distribution of EndoU (PF14436) RNase domains across effector classes. Abbreviations: ESS, Esx-like secretion system; Hcp, hemolysin-coregulated protein; OME, outer membrane exchange; T5SS, type V secretion system; T6SS, type VI secretion system.
Because polymorphic effectors are antibacterial, they are invariably produced with immunity proteins that prevent self-intoxication. Immunity proteins are usually encoded immediately downstream of the effector gene, and this organization enables toxin-immunity pairs to be fused to various effector classes. For example, CdiA proteins from Chromobacterium violaceum (NCBI ID: WP_011136418.1) and Burkholderia ubonensis (WP_080434302.1) carry toxins that are >50% identical to the RNase domains of ColE4 and ColE5, respectively. Similarly, Paracoccus saliphilus encodes a PAAR-Rhs effector-immunity pair (WP_141225802.1/WP_084203033.1) that shares ~49% identity with the ColE8 DNase domain and immunity protein. These observations suggest that horizontally acquired toxin-immunity pairs are integrated as modules into secretion systems. Comparison of the maf genomic island-2 from N. meningitidis MC58 and N. gonorrhoeae FA 1090 illustrates this phenomenon (65). The nucleotide sequence encoding the MafB toxin domain and MafI immunity protein in FA 1090 is the same as that of a cdiA-cdiI derived gene pair in the MC58 island (Fig. 1c). Thus, MafB and CdiA effectors can apparently deliver identical EndoU RNase domains. The MC58 island contains additional fragmented effector-immunity pairs (6; 65), which are also a common feature of cdiA, rhs and wapA loci (7; 73; 105). These “orphaned” toxin-immunity sequences likely represent ancestral modules displaced by more recently acquired cassettes. Horizontal gene transfer also enables closely-related bacteria to deploy distinct T6SS effector arsenals (37; 122; 136). The versatility of polymorphic toxins is exemplified by the aforementioned EndoU (PF14436) family, which is found at the C-terminus of CdiA, MafB, Rhs, WXG/LXG and several uncharacterized effectors across diverse phyla (Fig. 1d) (65; 88; 157).
TOXIN ACTIVITIES
The identification of polymorphic toxin activities remains a significant challenge. Aravind and colleagues have predicted biochemical activities for dozens of toxin families (62; 157; 158), but the C-terminal domains of many effectors remain unannotated. Despite their extraordinary variability, polymorphic toxins use only a handful of activities to disrupt critical molecular structures in target bacteria.
Ionophores: pore-forming toxins
Bacterial cells maintain a proton gradient across the cytoplasmic membrane, and this electrochemical proton-motive force (pmf) powers nutrient uptake, flagellar rotation and ATP synthesis. Therefore, pore-forming toxins that dissipate the pmf are potent inhibitors of bacterial growth. Many bacteriocins carry a stereotypical pore-forming domain composed of a hydrophobic α8-α9 hairpin core surrounded by eight amphipathic α-helices (100; 148). This domain remains soluble as it diffuses to target cells, but upon encountering a polarized membrane, the α8-α9 hairpin everts from the core and inserts into the lipid bilayer to initiate pore formation. Diphtheria toxin uses a related 10-helix domain to breach the eukaryotic plasma membrane (99), and the VasX T6SS effector from V. cholerae apparently uses it to form pores in both eukaryotic and prokaryotic membranes (89; 90). Though unannotated, these pore-forming domains are commonly carried by PAAR proteins in enterobacteria (e.g. WP_095441387.1) and Pseudomonas species (e.g. WP_087089637.1). T6SSs also deploy pore-forming toxins that are distinct from the 10-helix domain. Tse4 from P. aeruginosa contains four transmembrane helices, with the C-terminal helix containing a predicted glycine-zipper motif (74). Ssp6 from S. marcescens is yet another novel pore-forming toxin built from two short transmembrane helices (86). Finally, CdiA from E. coli EC93 dissipates the pmf in target cells (5). This latter domain contains two predicted glycine-zipper motifs, but lacks hydrophobic α-helices, suggesting a mechanism distinct from other pore-forming toxins.
Phospholipases
Phospholipase toxins are commonly delivered by T6SSs, but are largely absent from other delivery systems (157). Russell et al. identified and characterized five families of T6SS lipase effectors (Tle1 - Tle5) (119). The first four clades use a common nucleophilic elbow motif to hydrolyze acyl chains from the A1 and/or A2 position. The Tle5 family contains duplicated HxKxxxxD motifs found in phospholipase D enzymes (119). Phospholipases are packaged for T6SS-dependent delivery in several ways. Tle1 toxins are fused to VgrG in Pseudomonas species (60; 157), and to Hcp in many Salmonella strains (80). Dickeya, Pectobacterium, Aeromonas, and Shewanella all encode PAAR proteins with predicted phospholipase A1 domains (73), and T6SS-associated Rhs effectors from Vibrio vulnificus (e.g. POB69978.1) carry Tle1 family toxins. Lastly, lipase toxins can be linked to VgrG indirectly through adaptor proteins (135).
Peptidoglycan: blocking synthesis and promoting its degradation
Polymorphic effectors use two strategies to disrupt the peptidoglycan cell wall and lyse target bacteria. ColM and related bacteriocins degrade the lipid II precursor of peptidoglycan biosynthesis. This activity generates dephosphorylated undecaprenol, which cannot be recycled to support further polymerization of the cell wall (49; 124; 156). Streptococcus intermedius deploys an LXG effector with the same phosphatase activity, though it has a novel fold unrelated to ColM (146). The second strategy entails enzymatic degradation of peptidoglycan. Pesticin is a muramidase bacteriocin produced by Yersinia pestis. Like lysozyme, pesticin hydrolyzes the β−1,4-glycosidic linkage between N-acetyl-muramic acid and N-acetyl-glucosamine (103; 143). Lytic muramidases have also been described for T6SSs. VgrG-3 from V. cholerae carries a C-terminal lysozyme-like domain (20), and Tse3 from Pseudomonas aeruginosa has similar activity (118). In addition, T6SSs deploy a variety of amidases that cleave peptide cross-links in the cell wall (118; 120). The C-terminal domain of P. aeruginosa VgrG2b is a metalloprotease that cleaves cross-links only at the division septum (151). T6SS Tse1 and Tae4 effectors from Pseudomonas aeruginosa and Enterobacter cloacae are NlpC/P60 family amidases that cleave peptide cross-links between D-Glu and m-diaminopimelic acid residues (120). The latter amidases lack the canonical multi-domain architecture of most polymorphic effectors, but they are sequence-diverse and rely on a specialized delivery system.
Nucleases and deaminases
Nucleases are perhaps the most common and versatile of the polymorphic toxin payloads (157). Most DNase toxins are members of either the HNH/EndoVII or PD-(D/E)XK superfamilies (157). HNH/EndoVII DNases contain a ββα-metal active-site motif first identified in phage T4 endonuclease VII and colicins (71; 72; 111). Zhang et al. have predicted that polymorphic effectors deploy 14 distinct clades of HNH/EndoVII DNases (157; 158). Few of these have been tested, though DNase activities have been verified for Tox-GHH2 (PF15635) (104) and Endonuclease NS_2 (PF13930) domains (25; 73; 97). PD-(D/E)XK phosphodiesterase domains are common in restriction endonucleases and DNA repair enzymes, but their extraordinarily plastic active sites make them difficult to identify through informatics (131). Zhang et al. identified 10 novel toxic restriction endonuclease (Ntox-REase) clades associated with polymorphic toxin systems (157), though only five are listed in the current Pfam database. Other PD-(D/E)XK toxins have been discovered through structure-function analyses (66; 92), suggesting that more family members remain to be characterized. The structure of the Tde DNase from Agrobacterium tumefaciens remains unknown. This T6SS effector was originally thought to have a novel RNase fold (157), but has since been shown to be specific for DNA (81).
Polymorphic RNase domains adopt a greater diversity of folds than the DNase toxins. Zhang et al. predicted that several novel toxin (Ntox) families have RNase activities (157; 158), though few have been examined experimentally (2; 13). Ntox21 adopts the barnase-EndoU-colicin D/E5-RelE (BECR) superfamily fold and cleaves 16S rRNA at the same position as ColE3 (13; 157). Ntox28 is an α-helical metal-dependent tRNA anticodon nuclease (42; 67). Ntox41 adopts the vertebrate RNase A fold, though it lacks the characteristic disulfide bonds of the superfamily (11). EndoU toxins contain a partial duplication the BECR core and are structurally similar to RNA processing enzymes found in vertebrates and coronaviruses (88; 158). Lastly, some Burkholderia CdiA proteins carry PD-(D/E)XK phosphodiesterase domains that cleave specific sites within tRNA molecules (68; 92; 139).
Deaminases are another major class of toxins associated with polymorphic effectors (157). These toxins have undergone a major radiation into at least nine clades (62). Surprisingly, this important class of enzymes has not yet been tested experimentally, and substrate specificities for the various clades remain unknown. In principle, these enzymes could act on adenine or cytosine in the context of DNA, RNA or nucleotides.
Nucleotide specific toxins
Mougous and colleagues first described the structure and activity of an Ntox46 (PF15538) domain that adopts the same ADP-ribosyltransferase (ART) fold as diphtheria toxin (147). However, this toxin does not ADP-ribosylate target proteins, but rather acts as an NAD(P)+ glycohydrolase (NADase). Thus, these enzymes block biosynthesis and greatly reduce ATP production by disrupting the flow of electrons to the respiratory chain. There are likely to be more unrecognized NADase toxins, because recent work from Whitney and coworkers has uncovered a new class associated with Rhs and LXG effectors (132). Moreover, the widespread Arg-Glu-Ser (RES) toxin domain (PF08808) is now known to degrade nicotinamide dinucleotides (128). However, there are toxic ART domains that ADP-ribosylate protein targets. Tre1 is a T6SS effector from Serratia proteamaculans that covalently modifies FtsZ to block cell division (133). Finally, RelA/SpoT hydrolases (RSH) are associated with some polymorphic effector classes (157). These enzymes classically synthesize and degrade the stringent factor alarmone (p)ppGpp, but recent work shows that toxic RSH domains inhibit target-cell growth by producing a novel (p)ppApp nucleotide (1).
TOXIN-ACTIVATING FACTORS
Polymorphic toxins typically hijack cell-envelope proteins to gain entry into target cells, but a subset also require co-factors to support enzymatic activity. ColM is unable to kill E. coli fkpA mutants, because they lack a peptidyl prolyl isomerase that refolds the phosphatase domain after translocation into the periplasm (54; 61). Similarly, Ssp2 and Ssp4 effectors of the Serratia marcescens T6SS rely on periplasmic oxidoreductases in target bacteria for proper disulfide formation after delivery (85). Thus, toxins that unfold during delivery can become reliant on chaperones. In addition, several CdiA toxins have evolved interactions with target-cell proteins to promote tRNase activities. The Ntox28 domain from uropathogenic E. coli is active only when bound to CysK, an enzyme that catalyzes the final step of L-cysteine biosynthesis (42; 67). This interaction is intriguing because the toxin mimics CysE – the physiological binding partner of CysK (42). Other BECR-fold tRNases collaborate with translation factors EF-Tu and EF-Ts to cleave substrate (51; 69; 87). These latter toxins recognize substrate only in the context of tRNA•EF-Tu•GTP ternary complexes, the formation of which is greatly accelerated by EF-Ts (69). The selective pressures driving interactions with EF-Tu could stem from biophysical constraints that limit the size of toxin payloads. In this model, economized domains are so small that they depend on binding partners to stabilize the nuclease fold and/or organize the catalytic center.
IMMUNITY PROTEINS
Immunity proteins prevent auto-intoxication, but are also required to protect bacteria from toxins delivered by neighboring siblings. Immunity proteins typically bind directly over the active sites of enzymatic toxins, though some nuclease toxins are neutralized by antitoxins that bind to “exosites” adjacent to the catalytic center (26; 71; 92). The Tri1 immunity protein is unique in that it actively hydrolyzes ADP-ribosyl groups added to FtsZ by its cognate toxin Tre1 (133; 157). The molecular basis of immunity to pore-forming toxins is unknown as there are no high-resolution structures of membrane-embedded pores. However, the latter immunity proteins are usually small and very hydrophobic, suggesting that they localize to the cytoplasmic membrane, where they could block pore opening or interfere with glycine-zipper oligomerization.
Structures of polymorphic toxin-immunity protein complexes have revealed that individual families are rapidly diversifying. As an example, colicin DNase domains generally share >50% pairwise identity, and their immunity proteins are ~47% identical on average. Much of this sequence divergence is concentrated at the complex binding interface (Fig. 2); and consequently, immunity proteins exhibit significantly lower affinity for “near-cognate” DNase domains (76). Because immunity function is essential to prevent auto-intoxication, this evolution must proceed through a series of reciprocal mutations that gradually sculpt the interface without catastrophically perturbing binding affinity (112). Nuclease colicins bind cognate immunity proteins with femtomolar dissociation constants, and this extraordinarily high affinity may facilitate divergence. Substitutions that reduce binding 1000-fold still yield complexes with nanomolar affinity, which is sufficient to protect against intoxication (76). Although this phenomenon has been explored most extensively with colicins, rapid evolution at the protein binding interface is a hallmark of all toxin-immunity pairs that mediate bacterial competition (51; 88; 93).
Figure 2. Diversification of toxin-immunity pairs.
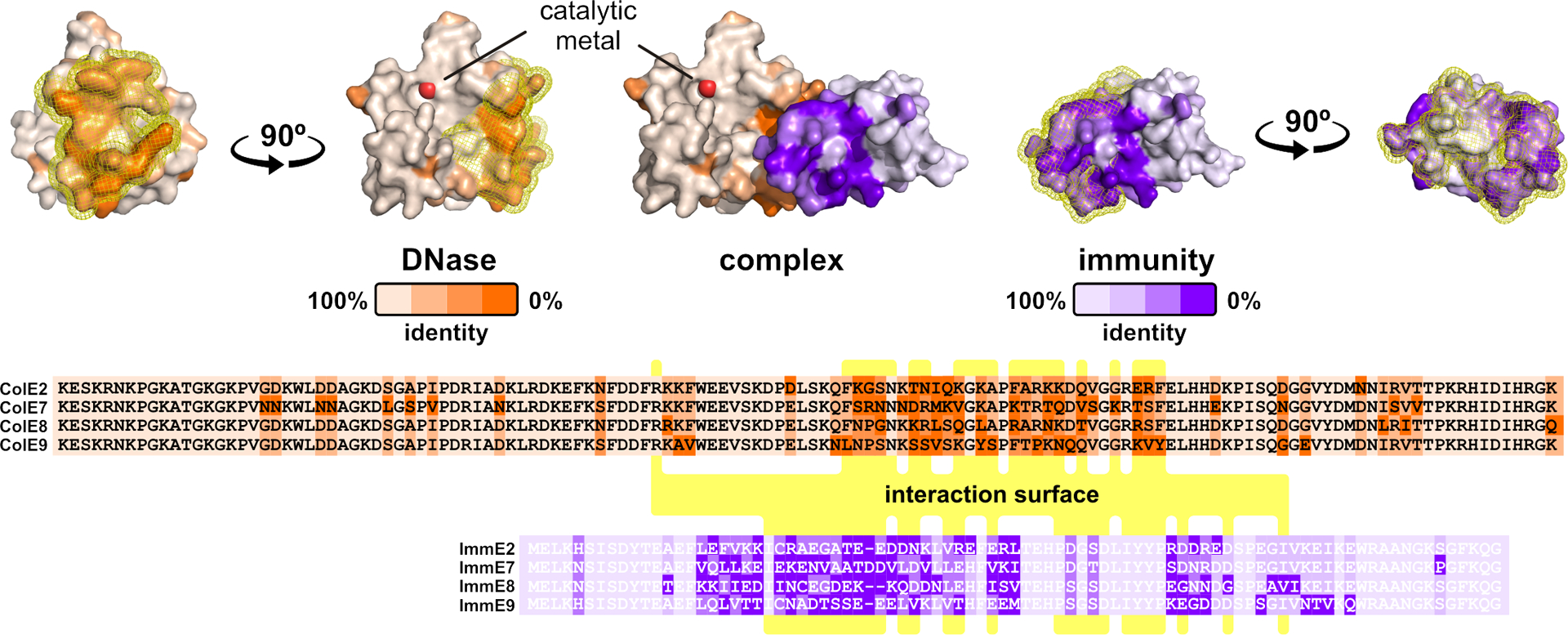
Sequence variation is mapped onto the crystal structure of the ColE2 DNase domain bound to its immunity protein. The interaction surface is highlighted with yellow mesh, and residues making direct contact are indicated with yellow in the alignments. The DNase catalytic center is indicated by the divalent metal ion.
MECHANISMS OF TOXIN DELIVERY
Protein bacteriocins/colicins
Bacteriocins are diverse ribosome-synthesized antimicrobials that include small modified peptides and multi-domain proteins (27). The protein bacteriocins of γ-proteobacteria – and in particular the colicins of E. coli – are a paradigm for polymorphic effectors. Colicins are usually plasmid-encoded and are only expressed in response to extensive DNA damage and other stresses (Fig. 3a) (27; 79). In contrast, the downstream immunity genes are transcribed constitutively. Colicins lack signal sequences and typically require bacteriocin release proteins for export (106; 137). These small lipoproteins activate OmpLA phospholipase, which disrupts envelope integrity and allows cytoplasmic contents to leak from the cell (Fig. 3b) (39; 107). Nuclease colicins are released in complex with immunity proteins, because the latter are produced constitutively and have very high affinity for their cognate toxin domains. Notably, colicin release is lethal, and therefore the toxin producing cell obtains no competitive advantage. However, non-expressing sibling cells are immune and gain a significant growth advantage over susceptible competitors.
Figure 3. Colicins.
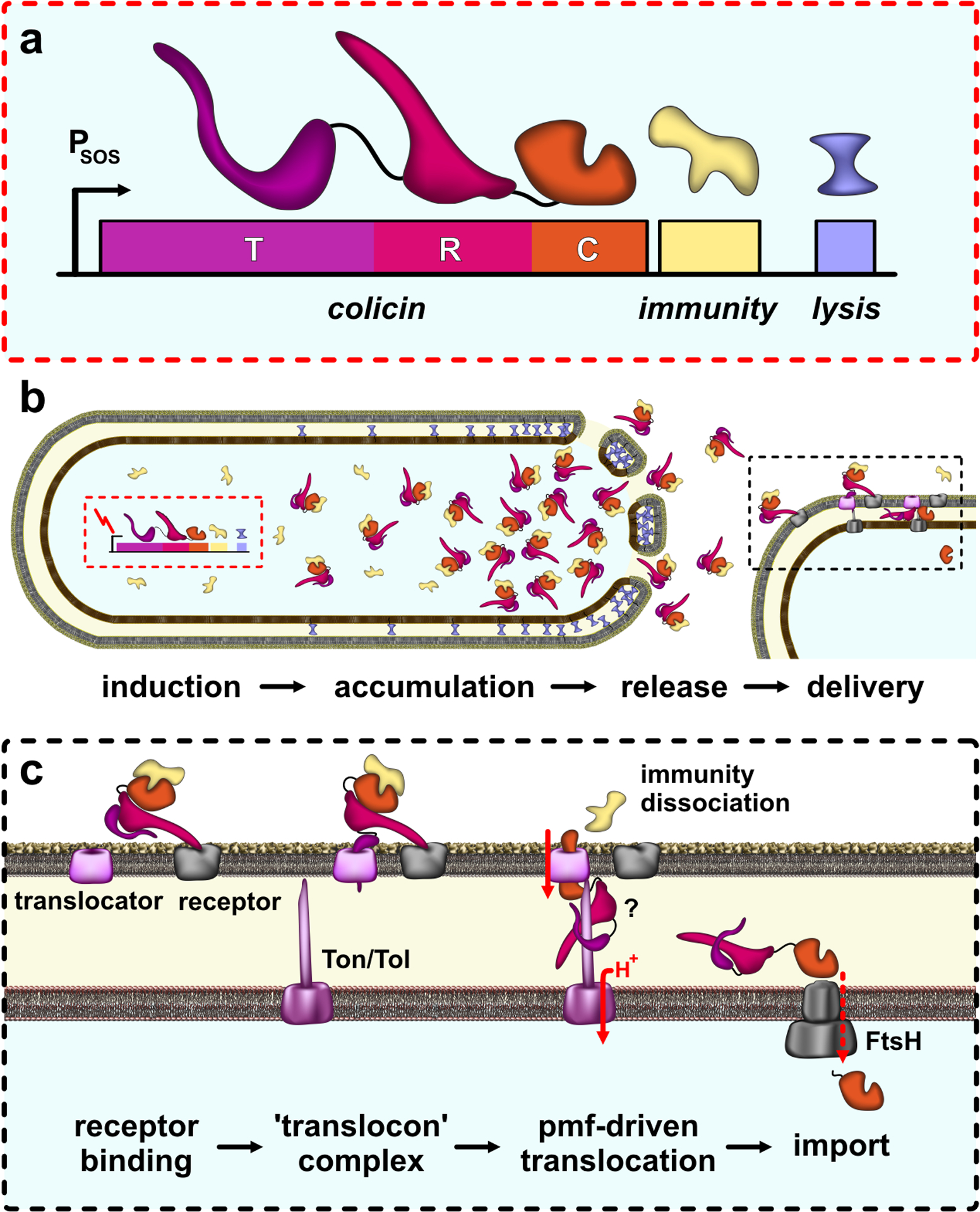
a) Colicin operon organization. The bacteriocin release protein is encoded by the lysis gene, which is separated from the upstream colicin and immunity cistrons by a transcription terminator. b) Colicin release. Induction of colicin expression allows read-through of the terminator, producing release proteins that disrupt the cell envelope to release colicin-immunity complexes. c) Colicin delivery. Colicins use R domain to recognize OMP receptors on target bacteria, then recruit a second OMP translocator using the T domain. Translocation is energized by the pmf through Tol or Ton systems. Nuclease domains require FtsH for import to the cytosol.
Colicins bind to target bacteria through specific cell-surface receptors, which are usually outer membrane proteins (OMP) that function in nutrient uptake. ColIa, ColD, and ColM recognize the iron-siderophore transporters Cir, FepA and FhuA (respectively), whereas E colicins use the vitamin B12 transporter BtuB as a receptor (27). In principle, these receptors could also be exploited as portals for toxin import, but colicins recruit a second OMP that serves as a “translocator” (Fig. 3B). Most E colicins thread the unstructured N-terminal segment of their T domains through the lumen of OmpF translocators (58; 153). The analogous region of ColIa appears to mimic a siderophore, enabling it to recruit a second Cir molecule for the translocation step (64). In each case, the colicin inserts an N-terminal peptide motif into the periplasm where it interacts with components of the Tol or Ton machineries. Tol maintains OM integrity and is particularly important during cell division (28). TolQ and TolR form an inner membrane complex that harnesses the proton gradient and conveys that energy to the OM through TolA. The Ton system powers the import of cobalamin and siderophores through TonB-dependent OM transporters (149). ExbB and ExbD convert pmf energy into conformational changes in TonB, which spans the periplasm to control transporters in the OM. Colicin translocation across the outer membrane depends on pmf-driven conformational changes in TolA and TonB, but it remains unclear whether the entire colicin molecule is imported to the periplasm. Cramer and coworkers have suggested that the ColE3 nuclease domain becomes destabilized after dissociation of the immunity protein, which in turn enables the toxin to translocate through the OmpF translocator (35; 154). Despite this uncertainty, it is clear that colicins unfold during import, and that immunity proteins must be actively dissociated from nuclease domains before translocation can proceed (59).
Once inside the periplasm, pore-forming domains insert into the cytoplasmic membrane and dissipate the proton gradient. Colicin nuclease domains must be further transported into the cytosol. This final translocation step depends on FtsH, which is an ATP-dependent metalloprotease that normally extracts misfolded proteins from the inner membrane for degradation (36). E colicin nuclease domains are strongly electropositive and bind to anionic phospholipids. Membrane binding destabilizes these domains, rendering them into molten globules that are presumably recognized as defective membrane proteins and retro-translocated into the cytosol by FtsH (94; 95; 144). The tRNase domain of ColD does not associate with membranes and instead relies on interactions with the LepB signal peptidase for cell entry (91). Nevertheless, ColD also depends on FtsH for import (91), suggesting that LepB brings the nuclease into close proximity to the membrane for delivery to FtsH. The final step of colicin nuclease delivery entails proteolytic cleavage to release the toxin domain into the cytosol (30; 38). FtsH appears to mediate this proteolytic processing (30), though it remains unclear how the nuclease domains avoid complete degradation during import.
Contact-dependent growth inhibition (CDI)/type V secretion systems (T5SS)
CdiB/CdiA two-partner secretion (TPS) systems are most common in β- and γ-proteobacteria, though several α-proteobacteria, fusobacteria and Negativicutes also contain cdi genes (2; 157). CdiB is an Omp85 β-barrel protein that transports CdiA across the outer membrane. CdiA proteins span from ~200 to over 700 kDa, but all share the same general domain architecture (Fig. 4a). The N-terminal half of CdiA is dominated by filamentous hemagglutinin-1 (FHA-1; PF05594) peptide repeats, which form a β-helical filament that projects from the inhibitor-cell surface (70; 116). FHA-1 domains range from ~300 to 4,600 residues, which corresponds to filament lengths of ~15 to over 100 nm (70). A second repeat domain composed of distinct FHA-2 (PF13332) repeats comprises much of the C-terminus. The two repeat regions surround a central receptor-binding domain (RBD), which functions in target-cell recognition (115; 117). The variable C-terminal (CdiA-CT) region is demarcated by a conserved peptide sequence, exemplified by the VENN motif in enterobacterial effectors (2). The CdiA-CT is frequently composed to two domains with distinct functions. The N-terminal “cytoplasm entry” domain mediates translocation across the target-cell inner membrane, and the extreme C-terminal domain has toxic activity (150).
Figure 4. Contact-dependent growth inhibition (CDI).
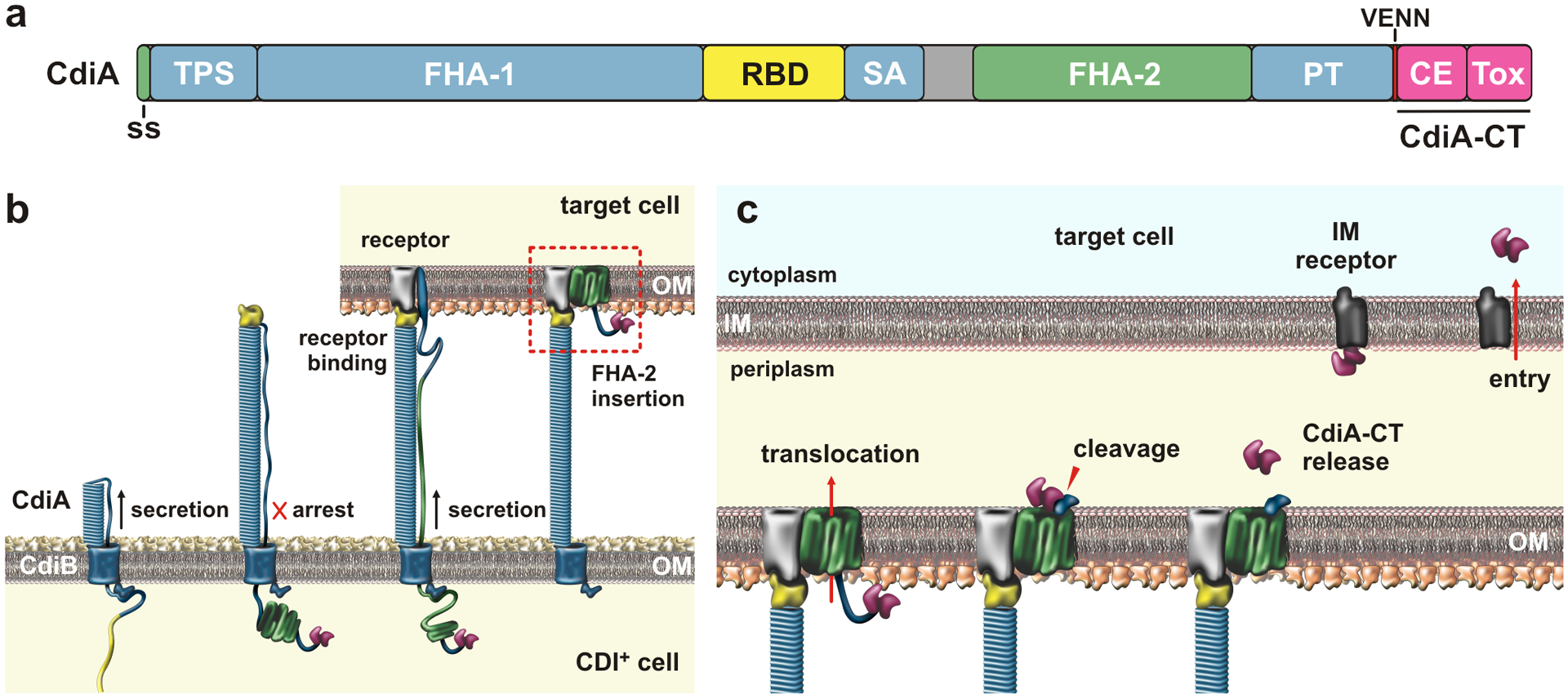
a) CdiA domain architecture. The CdiA-CT is composed of cytoplasm entry (CE) and toxin domains; Abbreviations: ss, signal sequence; TPS, two-partner secretion transport domain; RBD, receptor-binding domain; SA, secretion arrest domain; PT, pretoxin domain; and CE, cytoplasm entry domain. b) CdiA biogenesis. CdiB guides CdiA export across the outer membrane (OM). The C-terminal half of CdiA remains sequestered in the periplasm due to a programmed secretion arrest. CdiA export resumes upon binding receptor, and the FHA-2 domain integrates into the target-cell OM to form a toxin translocation conduit. c) Toxin delivery. Once transferred into the periplasm, the CdiA-CT is released for a second translocation step across the inner membrane (IM). The final step is mediated by cytoplasm entry domains, which interact with membrane proteins and translocate the C-terminal toxin domain in a pmf-dependent manner. Abbreviations: CE, cytoplasm entry domain; CT, C-terminal region; IM, inner membrane; OM, outer membrane; PT, pretoxin domain; RBD, receptor-binding domain; SA, secretion arrest domain; ss, signal sequence; Tox, toxin domain; TPS, two-partner secretion transport domain.
Newly synthesized CdiA is directed to the periplasm by a Sec-dependent signal sequence (Fig. 4b). The TPS transport domain of CdiA is then bound by CdiB, which uses N-terminal polypeptide transport-associated (POTRA) domains to guide the effector through its outer membrane β-barrel (12). The lumen of CdiB is too narrow to accommodate a β-helix, so CdiA must be exported in an unfolded state. Thus, the filament likely forms as the FHA-1 peptide repeats emerge from CdiB, with the free energy of β-helix folding to power secretion (52). Shortly after the CdiA RBD is exported, secretion is halted at a Tyr/Pro-enriched region. As a result, the N-terminal half of CdiA forms a cell-surface filament, while the FHA-2 and CdiA-CT regions remain sequestered in the periplasm (Fig. 4b) (116). Because the RBD forms the distal tip of the filament, the TPS transport domain presumably remains associated with CdiB at the cell surface. Thus, CdiA forms a hairpin with the Tyr/Pro-enriched region adopting an extended conformation to span the length of the filament, linking the RBD to the periplasmic FHA-2 domain (116).
As with colicins, CDI toxin delivery is initiated by specific interactions with cell-surface receptors. Different classes of E. coli CdiA effectors recognize BamA (3), OmpC/OmpF (14; 142) and Tsx (115) as receptors. Receptor binding alleviates the export arrest through an unknown mechanism, and the FHA-2 domain is secreted directly onto the target-cell surface (Fig. 4c). FHA-2 becomes stably associated with the target-cell outer membrane and is thought to form a transmembrane conduit for toxin translocation (116). The energetics of outer membrane transport are not understood, but Tol and Ton have no role in CDI toxin delivery (114). Once inside the target-cell periplasm, the CdiA-CT is cleaved near the VENN motif for delivery to the cytosol (116). This final translocation step is mediated by the N-terminal cytoplasm entry domain of the CdiA-CT. Genetic evidence suggests that entry domains exploit inner membrane proteins to transfer nuclease domains into the cytosol (150). Entry domains are highly variable, and each class recognizes a specific integral membrane protein. Some of the hijacked proteins, like PtsG, MetI, RbsC and GltK/GltJ, are metabolite transporters, but transport function is not required for toxin import (150). CdiA-CT entry domains have low thermodynamic stability and are thought to adopt a molten globule-like conformation (9), which could facilitate insertion into the membrane. Finally, the target-cell pmf is required for CdiA-CT delivery into the cytoplasm (114), suggesting that the electrochemical gradient drives transport.
Type VI secretion systems (T6SS)
The T6SS is structurally and evolutionarily related to contractile bacteriophages. The T6SS ejects a spear shaped projectile that impales neighboring cells to deliver a payload of effector proteins. These systems were first characterized as virulence determinants (109), but many T6SSs function in inter-bacterial conflict. The T6SS apparatus is built from three sub-complexes that assemble sequentially during the secretion duty cycle (Fig. 5a). TssJ, TssL, and TssM form a multimeric, membrane-embedded complex that spans the cell envelope and provides the secretion conduit. TssJ is an outer-membrane lipoprotein (8), and TssL is an integral membrane protein that also anchors the sub-complex to the peptidoglycan cell wall (82). TssM spans the periplasm and binds TssJ near the inner leaflet of the outer membrane (46; 82). This trans-envelope complex serves as the docking site for the phage-like baseplate, which assembles independently in the cytoplasm (21). Trimeric VgrG is homologous to the gp27-gp5 tailspike phage T4 (75; 108; 134), and it forms the central hub around which the baseplate assembles. TssE, TssF and TssG form a hexagonal array surrounding VgrG, with TssK oligomers forming a bowl-shaped ring about the outer edge of the baseplate (45). TssK cavity interfaces with the cytoplasmic domains of TssM and TssL to connect the baseplate to the membrane complex (78; 159). Once engaged with the trans-envelope complex, the baseplate serves as the origin for assembly of the contractile sheath and inner tube complex (Fig. 5b). Hexameric Hcp rings stack onto VgrG to form a tube, which is surrounded by a latticed sheath of TssBC heterodimers (10; 19). Sheath and tube polymerization is coordinated by TssA, which caps the growing end of the tail (160). Once the sheath-tube complex extends across this cell (123), it rapidly contracts through a rigid-body rotation of TssBC heterodimers that causes the sheath to reduce its length in half with an attendant increase in diameter (29; 145). Because the sheath is anchored to the baseplate and trans-envelope complexes, contraction causes the distal end of the tail to move toward the membrane complex rapidly. The baseplate releases VgrG, freeing the spike complex to exit through open pore of the trans-envelope complex. In addition, the TssM ring must open from its resting diameter of 15 Å to allow passage of the Hcp tube (44). Following contraction, cytosolic components are disassembled and recycled for further rounds of secretion (18).
Figure 5. Type VI secretion systems.
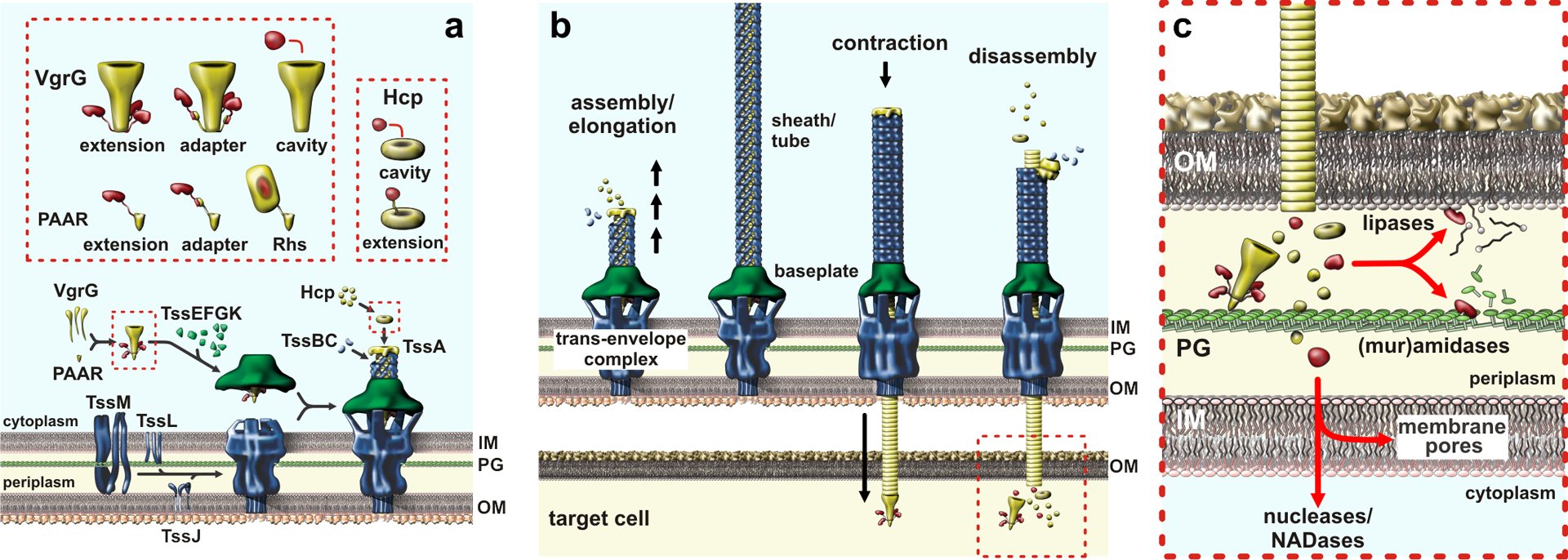
a) T6SS sub-complex assembly and effector packaging onto VgrG and Hcp. b) Sheath-tube assembly initiates at the baseplate and extends across the width of the cell. The sheath rapidly contracts, expelling the Hcp tube through the baseplate and trans-envelope complexes. The PAAR-tipped VgrG protein penetrates the OM of target bacteria and releases effector proteins. c) T6SS effector proteins activities. Nucleases and NADases are translocated into the cytoplasm through poorly understood pathways. Abbreviations: Hcp, hemolysin-coregulated protein; IM, inner membrane; NADase, NAD(P)+ glycohydrolase; OM, outer membrane; PG, peptidoglycan; Rhs, rearrangement hotspot; T6SS, type VI secretion system; VgrG, valine-glycine repeat G protein.
All known T6SS effectors are exported through interactions with either VgrG or Hcp (Fig. 5a). In some instances, toxin domains are fused to VgrG as C-terminal extensions from the β-spike (20; 55; 108; 151). Other toxic effectors are tethered to the VgrG spike through PAAR-repeat domains. PAAR and related DUF4150 domains form pyramidal structures that cap the end of the spike through β-complementation interactions (125). PAAR-containing effectors exhibit many different architectures, ranging from relatively small two-domain effectors to the large (~160 kDa) rearrangement hotspot (Rhs) proteins (17; 22; 33; 73; 104; 125). The latter effectors are characterized by RHS/YD-peptide repeats that form an expansive β-cage that encapsulates the C-terminal toxin domain (24). Other T6SS effectors are linked to the β-spike indirectly through DUF4123 adaptor proteins, which are encoded in operons with vgrG and effectors genes (77; 121; 135; 155). Because VgrG is sequestered within a ~17.5 nm chamber formed by TssK (32), effectors must be loaded onto the β-spike before the baseplate docks onto the trans-envelope complex. Electron cryotomography reveals that this chamber contains a mass consistent with VgrG-tethered effectors (29). Recent structural studies suggest that toxins are also loaded into the cavity formed by the N-terminal gp27-like domains of VgrG trimers (152). Hcp associated effectors are packaged within the lumen of the hexameric ring (127). Given that only a short segment of the tube is likely to be delivered into target bacteria, it is unclear whether every Hcp hexamer is loaded with an effector. Hcp associated effectors could also have affinity for the VgrG hub, which would ensure that toxin is preferentially loaded into the first hexamer of the nascent tube. It is also possible that effectors become depleted early in assembly, such that distal portions of the tube would be devoid of toxin. A subset of Hcp proteins carry fused C-terminal toxin domains with lipase, DNase and cell wall hydrolase activities (80). Given that the tube lumen can only accommodate one toxin domain, these latter effectors must form mixed hexamers with canonical Hcp.
T6SSs use mechanical force to breach the outer membranes of target bacteria (Fig. 5c). This process is presumably facilitated by the PAAR domain, which forms a sharp, metal-reinforced apex at the tip of the VgrG β-spike (125). However, VgrG also carries various globular toxin domains that dangle from the spike and potentially hinder translocation across the outer membrane. The initial impact of PAAR could create a membrane disruption, perhaps forming a pore large enough for toxin passage. Alternatively, the membrane could become hyper-fluidic as lipids are displaced by the PAAR domain. Regardless of the precise mechanism, non-covalently tethered toxins must resist shear forces associated with outer membrane passage. This is particularly important for PAAR-Rhs effectors, which auto-cleave their toxins and hold the released domains inside the β-cage structure (24; 43). Hcp encapsulated toxins are shielded from these forces, but the tube must presumably dissociate into individual rings after delivery to release its payload.
Transfer into the target-cell periplasm is sufficient for T6SS phospholipase and peptidoglycan amidase effectors, but nucleases and NADases must be delivered into the cytosol (Fig. 5c). Studies from the Mekalanos and Basler laboratories indicate that VgrG and Hcp are delivered directly into the cytoplasm of Vibrio (55; 141), but studies with VgrG associated phospholipase toxins from other bacteria suggest that T6SS effectors are generally deposited into the periplasm (47; 119). Work on the PAAR-containing Tse6 effector from P. aeruginosa indicates that its N-terminal transmembrane helices insert into the inner membrane of target bacteria, resulting in translocation of the NADase domain into the cytosol (110). Presumably the entire effector remains membrane-bound, perhaps positioning it to more effectively disrupt electron flow to the respiratory chain. Some PAAR-Rhs effectors carry similar N-terminal transmembrane segments, which could initiate fusion of the RHS β-cage with the membrane to release the encapsulated nuclease toxin into the cytosol. These hydrophobic helices must be prevented from inserting into membranes prior to export. Dimeric effector-associated gene (Eag) chaperones bind these helices and presumably help guide the effectors into the T6SS apparatus (33; 43; 147). Notably, some evolved VgrG (e.g. WP_000371003.1) and Hcp (e.g. WP_000502504.1) nuclease effectors contain central pyocin S translocation domains (PF06958) (15; 80). These translocation domains are not found in similar effectors that carry phospholipase and muramidase toxins (55; 80), suggesting they may be specifically required to deliver nucleases into the cytosol.
Neisserial MafB effectors
MafB proteins are polymorphic effectors encoded on five genomic islands in strains of N. meningitidis and N. gonorrhoeae (6; 65; 157). MafB carries a signal peptide and a conserved N-terminal DUF1020 (PF06255) domain that defines the family. MafB can be detected in culture supernatants when over-produced (65), but no mechanism has been established for export across the outer membrane. Tommassen and coworkers have proposed that MafA lipoproteins encoded on a subset of the maf islands may constitute a novel secretion pathway (6), though Jamet et al. found that MafA is not required for MafB secretion (65). The Nassif and Tommassen labs have both demonstrated MafB-dependent competition, and it appears that inhibition only occurs on solid media when effectors are expressed at endogenous levels (6; 65). This latter finding may indicate that toxin transfer is cell-contact or proximity dependent, though this has yet to be tested explicitly.
Esx secretion systems (ESS) in Gram-positive bacteria
The ESS was discovered in Mycobacterium tuberculosis, which uses the pathway to export EsxA and EsxB, which are ~100 residue peptides that form α-hairpins with a conserved WXG motif in the connecting turn. These particular WXG100 proteins are critical for M. tuberculosis pathogenesis (130), but related systems to function in iron uptake and competence (50; 126). Mark Pallen first recognized that Bacillus, Clostridium and Staphylococcus species encode WXG100 homologs, and hypothesized that Gram-positive bacteria harbor ESSs (98). These predictions were quickly verified for Staphylococcus aureus and Bacillus anthracis (23; 48). It was also recognized early on that the Gram-positive WXG100-like proteins carry extended C-terminal domains with homology with CdiA, Rhs and MafB proteins (48; 98). Zhang et al. formally hypothesized that these latter extended WXG proteins, and similar LXG-motif containing proteins, mediates inter-bacterial competition (157; 158).
ESS mediated export relies on at least six proteins. EsaA, EssB and EssA are integral membrane proteins that likely form the core export channel, and EsaB is a required cytosolic factor (23). Substrate selection is governed by membrane-bound EssC, which is an FtsK/SpoIIIE family AAA+ ATPase that also powers secretion (23; 63). LXG/WXG effectors heterodimerize with EsxA through their N-terminal α-hairpin domains in order to be exported through the ESS (25; 146). S. aureus also encodes a peptidoglycan hydrolase, EssH, which is required for effector release into the supernatant (16). This suggest that a channel must be excavated through the cell wall in some species. There are intervening cistrons between the chaperone and effector genes in Streptococcus intermedius (146), raising the possibility that additional proteins guide effectors to the core export machinery. The ESS of S. intermedius is capable of inhibiting various Gram-positive species during co-culture on solid media (146). There are conflicting reports on the role of the S. aureus ESS in bacterial competition. Missiakas and coworkers found that EssD/EsaD DNase effector was principally used against host cells to modulate immune responses (97), whereas Palmer and coworkers reported that over-expression of this effector provides a competitive advantage over non-immune target bacteria (25). The mechanistic details of WXG/LXG effector transfer between bacteria are almost completely unexplored. Mougous and coworkers showed that purified LXG effectors have no effect when added to susceptible bacteria, and that direct cell-to-cell contact is required for toxin transfer (146). It remains to be determined how ESS effectors are conducted across the thick cell wall of Gram-positive target bacteria, but some WXG/LXG effectors contain central domains that may mediate translocation across the cell membrane (146).
Myxobacterial outer-membrane exchange (OME)
Myxobacteria are soil bacteria that exhibit several cooperative behaviors, including social motility and fruiting body formation (96). These bacteria also have the remarkable ability to share outer membrane lipids and OMPs through outer-membrane exchange (OME) (101; 102). This process requires TraA and TraB proteins in both partners, and gliding motility is critical to properly align the cells for exchange. TraA is a cell-surface protein with a variable N-terminal domain that mediates homotypic interactions with compatible TraA receptors on partner cells (Fig. 6) (102). TraB contains a predicted β-barrel and C-terminal OmpA-like domain that presumably anchors it to the cell wall. Molecular flux is remarkably rapid during OME, with bi-directionally exchanged lipoproteins equilibrating between partner cells within minutes. The biophysical basis of exchange remains poorly understood, but the efficient transfer of lipid is consistent with membrane fusion. Although prodigious quantities of lipid and protein cargo are exchanged, TraA itself is not transferred, presumably due to its interaction with peptidoglycan-tethered TraB. Wall and co-workers have demonstrated that healthy bacteria can use OME to help siblings repair damage to their cell envelopes (138). Thus, traA is an example of a ‘greenbeard’ gene – a theoretical locus that imparts the ability to recognize and direct benefits toward other individuals that carry the same allele (84). Because OME only requires congruence at a single locus, valuable resources could be donated to non-sibling cells that happen to share compatible traA alleles. Myxobacteria mitigate this risk by delivering toxic SitA lipoproteins during OME (Fig. 6) (40; 139). SitA effectors carry several different classes of C-terminal nuclease domains (139; 140), which must ultimately be transferred into the cytoplasm. Translocation to cytoplasm has not been examined in detail, but it appears to require a specific integral membrane protein OmrA (41). Mutants lacking OmrA participate in OME, but are resistant to SitA-mediated swarm inhibition, suggesting that this integral membrane protein is hijacked for toxin import. Remarkably, SitA lipoproteins can be serially exchanged between multiple cells before they enter the cytoplasm (139), indicating that cell entry is slow relative to the frequency of OME. This time delay may help ensure that SitA toxins do not engage in futile auto-delivery. It is not known whether the lipid anchor must be removed before the toxin can be transported to the cytoplasm, but this could provide a mechanism to control the kinetics of toxin delivery.
Figure 6. Outer-membrane exchange.
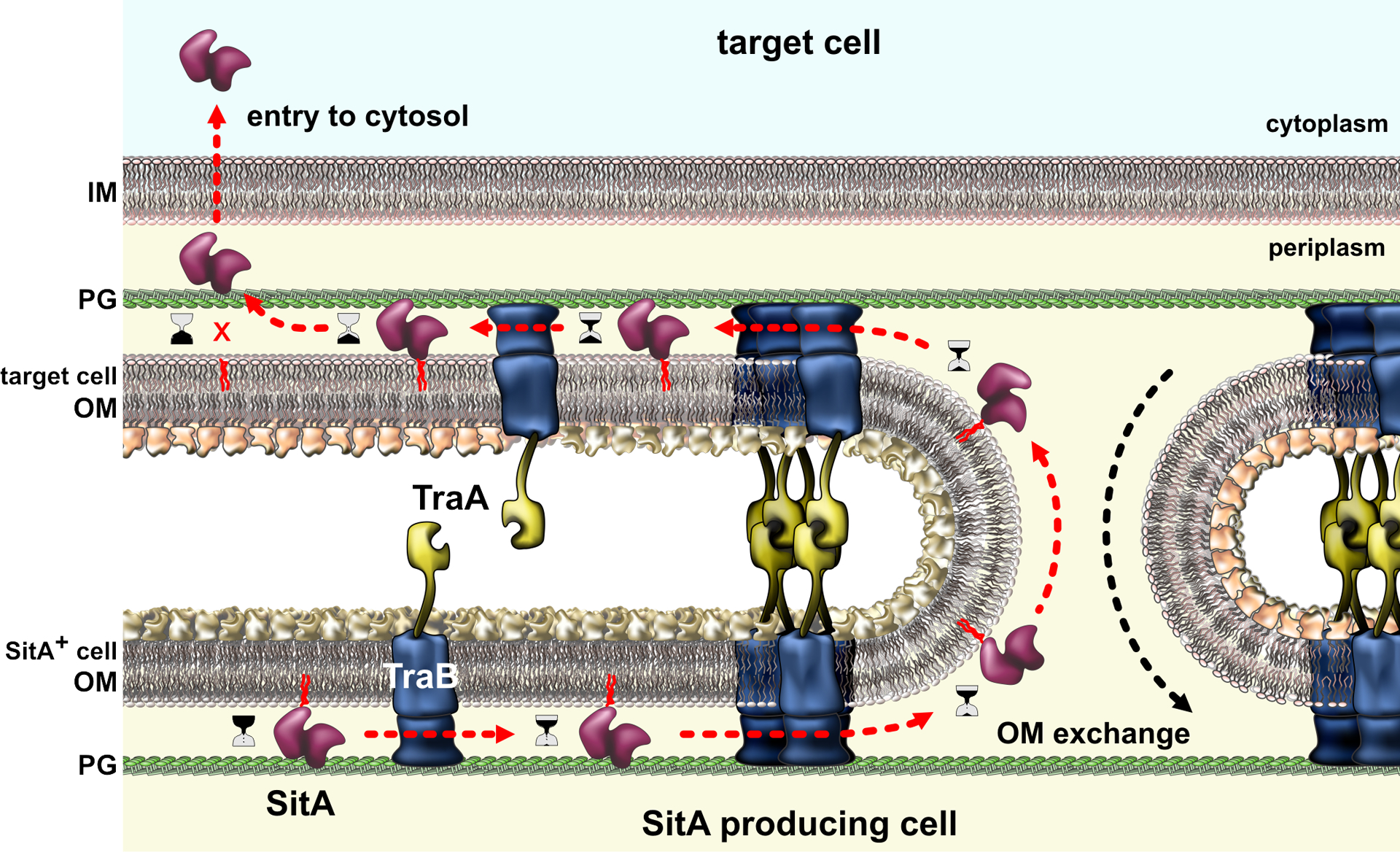
Myxobacteria with compatible TraA proteins engage in homotypic interactions that bring the cells into close approximation for outer-membrane fusion. OM lipids and proteins are freely exchanged between the cells, but TraA itself is not transferred due to interactions with TraB, which is tethered to the peptidoglycan (PG) cell wall. Polymorphic SitA effectors are among the many lipoproteins transferred during OME. SitA nucleases ultimately disengage from the OM and enter the cytoplasm through an uncharacterized mechanism. Abbreviations: IM, inner membrane; OM, outer membrane; PG, peptidoglycan.
CONCLUDING REMARKS
Polymorphic toxin delivery systems have emerged as important mediators of inter-bacterial competition and self/nonself recognition. In addition to the obvious ecological implications, polymorphic effector systems have garnered interest as platforms for novel antibacterial therapies. Indiscriminant use of antibiotics has fueled the emergence of multidrug resistant bacteria, and antibiotic-induced dysbioses can lead to catastrophic Clostridium difficile infections (31). New approaches are clearly needed to treat bacterial infections. While challenging to implement, strategies that utilize phages, bacteriocins or polymorphic toxin delivery systems may be the key to target specific bacterial pathogens. In principle, non-pathogenic strains could be armed to edit microbial communities precisely, removing unwanted residents, while leaving commensals unmolested.
Many toxins carried by polymorphic effectors can be delivered into both Gram-negative and Gram-positive target bacteria. The seeming ease with which these domains traverse cell envelopes belies the formidability of these barriers. The Gram-negative outer membrane excludes a host of small hydrophobic and hydrophilic compounds, yet five of the effector classes reviewed here are capable of translocating 15 kDa enzymatic domains across this structure. Toxin domains are clearly modular, but there is often bias in the cargoes carried by effector classes. E colicins are limited to three types of nuclease domains, presumably because the translocation pathway imposes strict biophysical constraints on the cargo. By contrast, CdiA effectors deliver the same DNase, rRNase and tRNases as colicins, but also carry more than 100 other toxin sequence types. Perhaps the advent of the cytoplasm entry domain allowed CdiA to expand its toxin repertoire. More work is needed to understand the mechanisms that govern protein retro-translocation into bacterial cells. Once these rules are established, perhaps we can fully engineer polymorphic effectors to shape microbial communities.
Finally, it is certain that more toxin delivery mechanisms remain to be discovered. Aravind and coworkers proposed that PrsW, TcdB/TcaC and phage head morphogenesis proteins function in inter-bacterial competition (157), but there are still no experimental studies that test these hypotheses. Database queries with known toxin sequences often recover proteins of unknown function, suggesting they are the effector arms of novel toxin-delivery systems. For example, EndoU RNase domains are fused to PAAR and VgrG-like proteins in Clostridium and Paenibacillus, though these species encode no other T6SS components. These genes may be non-functional dead-end products of horizontal gene transfer. Alternatively, perhaps Gram-positive bacteria harbor an unrecognized phage-derived competition apparatus. Given the rapidly accelerating pace of research into polymorphic effectors, we anticipate that several more inter-bacterial competition mechanisms will be described in the near future.
Acknowledgments
Research in the Hayes and Low laboratories is funded by NIH grants GM117373 and GM117930.
LITERATURE CITED
- 1.Ahmad S, Wang B, Walker MD, Tran HR, Stogios PJ, et al. 2019. An interbacterial toxin inhibits target cell growth by synthesizing (p)ppApp. Nature 575:674–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aoki SK, Diner EJ, de Roodenbeke CT, Burgess BR, Poole SJ, et al. 2010. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature 468:439–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aoki SK, Malinverni JC, Jacoby K, Thomas B, Pamma R, et al. 2008. Contact-dependent growth inhibition requires the essential outer membrane protein BamA (YaeT) as the receptor and the inner membrane transport protein AcrB. Mol. Microbiol 70:323–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aoki SK, Pamma R, Hernday AD, Bickham JE, Braaten BA, Low DA. 2005. Contact-dependent inhibition of growth in Escherichia coli. Science 309:1245–8 [DOI] [PubMed] [Google Scholar]
- 5.Aoki SK, Webb JS, Braaten BA, Low DA. 2009. Contact-dependent growth inhibition causes reversible metabolic downregulation in Escherichia coli. J. Bacteriol 191:1777–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arenas J, de Maat V, Caton L, Krekorian M, Herrero JC, et al. 2015. Fratricide activity of MafB protein of N. meningitidis strain B16B6. BMC Microbiol 15:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arenas J, Schipper K, van Ulsen P, van der Ende A, Tommassen J. 2013. Domain exchange at the 3´ end of the gene encoding the fratricide meningococcal two-partner secretion protein A. BMC genomics 14:622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aschtgen MS, Bernard CS, De Bentzmann S, Lloubes R, Cascales E. 2008. SciN is an outer membrane lipoprotein required for type VI secretion in enteroaggregative Escherichia coli. Journal of bacteriology 190:7523–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartelli NL, Sun S, Gucinski GC, Zhou H, Song K, et al. 2019. The Cytoplasm-Entry Domain of Antibacterial CdiA Is a Dynamic alpha-Helical Bundle with Disulfide-Dependent Structural Features. J Mol Biol 431:3203–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basler M, Pilhofer M, Henderson GP, Jensen GJ, Mekalanos JJ. 2012. Type VI secretion requires a dynamic contractile phage tail-like structure. Nature 483:182–6 [DOI] [PMC free article] [PubMed] [Google Scholar]; Showed the dynamic nature of T6SS assembly and contraction
- 11.Batot G, Michalska K, Ekberg G, Irimpan EM, Joachimiak G, et al. 2017. The CDI toxin of Yersinia kristensenii is a novel bacterial member of the RNase A superfamily. Nucleic Acids Res 45:5013–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baud C, Guerin J, Petit E, Lesne E, Dupre E, et al. 2014. Translocation path of a substrate protein through its Omp85 transporter. Nat Commun 5:5271. [DOI] [PubMed] [Google Scholar]
- 13.Beck CM, Morse RP, Cunningham DA, Iniguez A, Low DA, et al. 2014. CdiA from Enterobacter cloacae delivers a toxic ribosomal RNase into target bacteria. Structure 22:707–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beck CM, Willett JL, Cunningham DA, Kim JJ, Low DA, Hayes CS. 2016. CdiA Effectors from Uropathogenic Escherichia coli Use Heterotrimeric Osmoporins as Receptors to Recognize Target Bacteria. PLoS Pathog 12:e1005925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blondel CJ, Jimenez JC, Contreras I, Santiviago CA. 2009. Comparative genomic analysis uncovers 3 novel loci encoding type six secretion systems differentially distributed in Salmonella serotypes. BMC genomics 10:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bobrovskyy M, Willing SE, Schneewind O, Missiakas D. 2018. EssH Peptidoglycan Hydrolase Enables Staphylococcus aureus Type VII Secretion across the Bacterial Cell Wall Envelope. Journal of bacteriology 200:e00268–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bondage DD, Lin JS, Ma LS, Kuo CH, Lai EM. 2016. VgrG C terminus confers the type VI effector transport specificity and is required for binding with PAAR and adaptor-effector complex. Proc Natl Acad Sci U S A 113:E3931–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonemann G, Pietrosiuk A, Diemand A, Zentgraf H, Mogk A. 2009. Remodelling of VipA/VipB tubules by ClpV-mediated threading is crucial for type VI protein secretion. EMBO J 28:315–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brackmann M, Wang J, Basler M. 2018. Type VI secretion system sheath inter-subunit interactions modulate its contraction. EMBO Rep 19:225–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks TM, Unterweger D, Bachmann V, Kostiuk B, Pukatzki S. 2013. Lytic activity of the Vibrio cholerae type VI secretion toxin VgrG-3 is inhibited by the antitoxin TsaB. J Biol Chem 288:7618–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brunet YR, Zoued A, Boyer F, Douzi B, Cascales E. 2015. The Type VI Secretion TssEFGK-VgrG Phage-Like Baseplate Is Recruited to the TssJLM Membrane Complex via Multiple Contacts and Serves As Assembly Platform for Tail Tube/Sheath Polymerization. PLoS Genet 11:e1005545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burkinshaw BJ, Liang X, Wong M, Le ANH, Lam L, Dong TG. 2018. A type VI secretion system effector delivery mechanism dependent on PAAR and a chaperone-co-chaperone complex. Nat Microbiol 3:632–40 [DOI] [PubMed] [Google Scholar]
- 23.Burts ML, Williams WA, DeBord K, Missiakas DM. 2005. EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc Natl Acad Sci U S A 102:1169–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Busby JN, Panjikar S, Landsberg MJ, Hurst MR, Lott JS. 2013. The BC component of ABC toxins is an RHS-repeat-containing protein encapsulation device. Nature 501:547–50 [DOI] [PubMed] [Google Scholar]
- 25.Cao Z, Casabona MG, Kneuper H, Chalmers JD, Palmer T. 2016. The type VII secretion system of Staphylococcus aureus secretes a nuclease toxin that targets competitor bacteria. Nat Microbiol 2:16183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carr S, Walker D, James R, Kleanthous C, Hemmings AM. 2000. Inhibition of a ribosome-inactivating ribonuclease: the crystal structure of the cytotoxic domain of colicin E3 in complex with its immunity protein. Structure 8:949–60 [DOI] [PubMed] [Google Scholar]
- 27.Cascales E, Buchanan SK, Duche D, Kleanthous C, Lloubes R, et al. 2007. Colicin biology. Microbiol. Mol. Biol. Rev 71:158–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cascales E, Lloubes R, Sturgis JN. 2001. The TolQ-TolR proteins energize TolA and share homologies with the flagellar motor proteins MotA-MotB. Mol Microbiol 42:795–807 [DOI] [PubMed] [Google Scholar]
- 29.Chang YW, Rettberg LA, Ortega DR, Jensen GJ. 2017. In vivo structures of an intact type VI secretion system revealed by electron cryotomography. EMBO Rep 18:1090–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chauleau M, Mora L, Serba J, de Zamaroczy M. 2011. FtsH-dependent processing of RNase colicins D and E3 means that only the cytotoxic domains are imported into the cytoplasm. J Biol Chem 286:29397–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chavira A, Belda-Ferre P, Kosciolek T, Ali F, Dorrestein PC, Knight R. 2019. The Microbiome and Its Potential for Pharmacology. Handb Exp Pharmacol 260:301–26 [DOI] [PubMed] [Google Scholar]
- 32.Cherrak Y, Rapisarda C, Pellarin R, Bouvier G, Bardiaux B, et al. 2018. Biogenesis and structure of a type VI secretion baseplate. Nat Microbiol 3:1404–16 [DOI] [PubMed] [Google Scholar]
- 33.Cianfanelli FR, Alcoforado Diniz J, Guo M, De Cesare V, Trost M, Coulthurst SJ. 2016. VgrG and PAAR Proteins Define Distinct Versions of a Functional Type VI Secretion System. PLoS Pathog 12:e1005735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cianfanelli FR, Monlezun L, Coulthurst SJ. 2016. Aim, Load, Fire: The Type VI Secretion System, a Bacterial Nanoweapon. Trends Microbiol 24:51–62 [DOI] [PubMed] [Google Scholar]
- 35.Cramer WA, Sharma O, Zakharov SD. 2018. On mechanisms of colicin import: the outer membrane quandary. Biochem J 475:3903–15 [DOI] [PubMed] [Google Scholar]
- 36.Dalbey RE, Wang P, van Dijl JM. 2012. Membrane proteases in the bacterial protein secretion and quality control pathway. Microbiol Mol Biol Rev 76:311–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Maayer P, Venter SN, Kamber T, Duffy B, Coutinho TA, Smits TH. 2011. Comparative genomics of the Type VI secretion systems of Pantoea and Erwinia species reveals the presence of putative effector islands that may be translocated by the VgrG and Hcp proteins. BMC genomics 12:576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Zamaroczy M, Mora L. 2012. Hijacking cellular functions for processing and delivery of colicins E3 and D into the cytoplasm. Biochem Soc Trans 40:1486–91 [DOI] [PubMed] [Google Scholar]
- 39.Dekker N, Tommassen J, Verheij HM. 1999. Bacteriocin release protein triggers dimerization of outer membrane phospholipase A in vivo. Journal of bacteriology 181:3281–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dey A, Vassallo CN, Conklin AC, Pathak DT, Troselj V, Wall D. 2016. Sibling Rivalry in Myxococcus xanthus Is Mediated by Kin Recognition and a Polyploid Prophage. Journal of bacteriology 198:994–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dey A, Wall D. 2014. A genetic screen in Myxococcus xanthus identifies mutants that uncouple outer membrane exchange from a downstream cellular response. Journal of bacteriology 196:4324–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diner EJ, Beck CM, Webb JS, Low DA, Hayes CS. 2012. Identification of a target cell permissive factor required for contact-dependent growth inhibition (CDI). Genes Dev 26:515–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donato SL, Beck CM, Ruhe ZC, Cunningham DA, Singleton I, et al. 2020. The beta-encapsulation cage of rearrangement hotspot (Rhs) effectors is required for type VI secretion. Proc Natl Acad Sci U S A:in revision [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Durand E, Nguyen VS, Zoued A, Logger L, Pehau-Arnaudet G, et al. 2015. Biogenesis and structure of a type VI secretion membrane core complex. Nature 523:555–60 [DOI] [PubMed] [Google Scholar]
- 45.English G, Byron O, Cianfanelli FR, Prescott AR, Coulthurst SJ. 2014. Biochemical analysis of TssK, a core component of the bacterial Type VI secretion system, reveals distinct oligomeric states of TssK and identifies a TssK-TssFG subcomplex. Biochem J 461:291–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Felisberto-Rodrigues C, Durand E, Aschtgen MS, Blangy S, Ortiz-Lombardia M, et al. 2011. Towards a structural comprehension of bacterial type VI secretion systems: characterization of the TssJ-TssM complex of an Escherichia coli pathovar. PLoS Pathog 7:e1002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flaugnatti N, Le TT, Canaan S, Aschtgen MS, Nguyen VS, et al. 2016. A phospholipase A1 antibacterial Type VI secretion effector interacts directly with the C-terminal domain of the VgrG spike protein for delivery. Mol Microbiol 99:1099–118 [DOI] [PubMed] [Google Scholar]
- 48.Garufi G, Butler E, Missiakas D. 2008. ESAT-6-like protein secretion in Bacillus anthracis. Journal of bacteriology 190:7004–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghequire MGK, Buchanan SK, De Mot R. 2018. The ColM Family, Polymorphic Toxins Breaching the Bacterial Cell Wall. MBio 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gray TA, Clark RR, Boucher N, Lapierre P, Smith C, Derbyshire KM. 2016. Intercellular communication and conjugation are mediated by ESX secretion systems in mycobacteria. Science 354:347–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gucinski GC, Michalska K, Garza-Sanchez F, Eschenfeldt WH, Stols L, et al. 2019. Convergent Evolution of the Barnase/EndoU/Colicin/RelE (BECR) Fold in Antibacterial tRNase Toxins. Structure 27:1660–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guerin J, Bigot S, Schneider R, Buchanan SK, Jacob-Dubuisson F. 2017. Two-Partner Secretion: Combining Efficiency and Simplicity in the Secretion of Large Proteins for Bacteria-Host and Bacteria-Bacteria Interactions. Front Cell Infect Microbiol 7:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harms A, Brodersen DE, Mitarai N, Gerdes K. 2018. Toxins, Targets, and Triggers: An Overview of Toxin-Antitoxin Biology. Mol Cell 70:768–84 [DOI] [PubMed] [Google Scholar]
- 54.Helbig S, Patzer SI, Schiene-Fischer C, Zeth K, Braun V. 2011. Activation of colicin M by the FkpA prolyl cis-trans isomerase/chaperone. J. Biol. Chem 286:6280–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ho BT, Fu Y, Dong TG, Mekalanos JJ. 2017. Vibrio cholerae type 6 secretion system effector trafficking in target bacterial cells. Proc Natl Acad Sci U S A 114:9427–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holberger LE, Garza-Sanchez F, Lamoureux J, Low DA, Hayes CS. 2012. A novel family of toxin/antitoxin proteins in Bacillus species. FEBS Lett 586:132–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hood RD, Singh P, Hsu F, Guvener T, Carl MA, et al. 2010. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 7:25–37 [DOI] [PMC free article] [PubMed] [Google Scholar]; First demonstration of the antibacterial activity of T6SS
- 58.Housden NG, Hopper JT, Lukoyanova N, Rodriguez-Larrea D, Wojdyla JA, et al. 2013. Intrinsically disordered protein threads through the bacterial outer-membrane porin OmpF. Science 340:1570–4 [DOI] [PMC free article] [PubMed] [Google Scholar]; Structural characterization of the quaternary colicin-translocon complex
- 59.Housden NG, Loftus SR, Moore GR, James R, Kleanthous C. 2005. Cell entry mechanism of enzymatic bacterial colicins: porin recruitment and the thermodynamics of receptor binding. Proc Natl Acad Sci U S A 102:13849–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu H, Zhang H, Gao Z, Wang D, Liu G, et al. 2014. Structure of the type VI secretion phospholipase effector Tle1 provides insight into its hydrolysis and membrane targeting. Acta Crystallogr D Biol Crystallogr 70:2175–85 [DOI] [PubMed] [Google Scholar]
- 61.Hullmann J, Patzer SI, Romer C, Hantke K, Braun V. 2008. Periplasmic chaperone FkpA is essential for imported colicin M toxicity. Mol. Microbiol 69:926–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iyer LM, Zhang D, Rogozin IB, Aravind L. 2011. Evolution of the deaminase fold and multiple origins of eukaryotic editing and mutagenic nucleic acid deaminases from bacterial toxin systems. Nucleic Acids Res 39:9473–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jager F, Kneuper H, Palmer T. 2018. EssC is a specificity determinant for Staphylococcus aureus type VII secretion. Microbiology 164:816–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jakes KS, Finkelstein A. 2010. The colicin Ia receptor, Cir, is also the translocator for colicin Ia. Mol Microbiol 75:567–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jamet A, Jousset AB, Euphrasie D, Mukorako P, Boucharlat A, et al. 2015. A new family of secreted toxins in pathogenic Neisseria species. PLoS Pathog 11:e1004592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jana B, Fridman CM, Bosis E, Salomon D. 2019. A modular effector with a DNase domain and a marker for T6SS substrates. Nat Commun 10:3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson PM, Beck CM, Morse RP, Garza-Sanchez F, Low DA, et al. 2016. Unraveling the essential role of CysK in CDI toxin activation. Proc Natl Acad Sci U S A 113:9792–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson PM, Gucinski GC, Garza-Sanchez F, Wong T, Hung LW, et al. 2016. Functional Diversity of Cytotoxic tRNase/Immunity Protein Complexes from Burkholderia pseudomallei. J Biol Chem 291:19387–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jones AM, Garza-Sanchez F, So J, Hayes CS, Low DA. 2017. Activation of contact-dependent antibacterial tRNase toxins by translation elongation factors. Proc Natl Acad Sci U S A 114:E1951–E7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kajava AV, Cheng N, Cleaver R, Kessel M, Simon MN, et al. 2001. Beta-helix model for the filamentous haemagglutinin adhesin of Bordetella pertussis and related bacterial secretory proteins. Mol. Microbiol 42:279–92 [DOI] [PubMed] [Google Scholar]
- 71.Kleanthous C, Kuhlmann UC, Pommer AJ, Ferguson N, Radford SE, et al. 1999. Structural and mechanistic basis of immunity toward endonuclease colicins. Nat Struct Biol 6:243–52 [DOI] [PubMed] [Google Scholar]
- 72.Ko TP, Liao CC, Ku WY, Chak KF, Yuan HS. 1999. The crystal structure of the DNase domain of colicin E7 in complex with its inhibitor Im7 protein. Structure 7:91–102 [DOI] [PubMed] [Google Scholar]
- 73.Koskiniemi S, Lamoureux JG, Nikolakakis KC, t’Kint de Roodenbeke C, Kaplan MD, et al. 2013. Rhs proteins from diverse bacteria mediate intercellular competition. Proc Natl Acad Sci U S A 110:7032–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.LaCourse KD, Peterson SB, Kulasekara HD, Radey MC, Kim J, Mougous JD. 2018. Conditional toxicity and synergy drive diversity among antibacterial effectors. Nat Microbiol 3:440–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leiman PG, Basler M, Ramagopal UA, Bonanno JB, Sauder JM, et al. 2009. Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc Natl Acad Sci U S A 106:4154–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li W, Keeble AH, Giffard C, James R, Moore GR, Kleanthous C. 2004. Highly discriminating protein-protein interaction specificities in the context of a conserved binding energy hotspot. J Mol Biol 337:743–59 [DOI] [PubMed] [Google Scholar]
- 77.Liang X, Moore R, Wilton M, Wong MJ, Lam L, Dong TG. 2015. Identification of divergent type VI secretion effectors using a conserved chaperone domain. Proc Natl Acad Sci U S A 112:9106–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Logger L, Aschtgen MS, Guerin M, Cascales E, Durand E. 2016. Molecular Dissection of the Interface between the Type VI Secretion TssM Cytoplasmic Domain and the TssG Baseplate Component. J Mol Biol 428:4424–37 [DOI] [PubMed] [Google Scholar]
- 79.Lu FM, Chak KF. 1996. Two overlapping SOS-boxes in ColE operons are responsible for the viability of cells harboring the Col plasmid. Mol Gen Genet 251:407–11 [DOI] [PubMed] [Google Scholar]
- 80.Ma J, Pan Z, Huang J, Sun M, Lu C, Yao H. 2017. The Hcp proteins fused with diverse extended-toxin domains represent a novel pattern of antibacterial effectors in type VI secretion systems. Virulence 8:1189–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ma LS, Hachani A, Lin JS, Filloux A, Lai EM. 2014. Agrobacterium tumefaciens deploys a superfamily of type VI secretion DNase effectors as weapons for interbacterial competition in planta. Cell Host Microbe 16:94–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ma LS, Lin JS, Lai EM. 2009. An IcmF family protein, ImpLM, is an integral inner membrane protein interacting with ImpKL, and its walker a motif is required for type VI secretion system-mediated Hcp secretion in Agrobacterium tumefaciens. Journal of bacteriology 191:4316–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.MacIntyre DL, Miyata ST, Kitaoka M, Pukatzki S. 2010. The Vibrio cholerae type VI secretion system displays antimicrobial properties. Proc Natl Acad Sci U S A 107:19520–4 [DOI] [PMC free article] [PubMed] [Google Scholar]; First demonstration of the antibacterial activity of T6SS
- 84.Madgwick PG, Belcher LJ, Wolf JB. 2019. Greenbeard Genes: Theory and Reality. Trends Ecol Evol 34:1092–103 [DOI] [PubMed] [Google Scholar]
- 85.Mariano G, Monlezun L, Coulthurst SJ. 2018. Dual Role for DsbA in Attacking and Targeted Bacterial Cells during Type VI Secretion System-Mediated Competition. Cell Rep 22:774–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mariano G, Trunk K, Williams DJ, Monlezun L, Strahl H, et al. 2019. A family of Type VI secretion system effector proteins that form ion-selective pores. Nat Commun 10:5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Michalska K, Gucinski GC, Garza-Sanchez F, Johnson PM, Stols LM, et al. 2017. Structure of a novel antibacterial toxin that exploits elongation factor Tu to cleave specific transfer RNAs. Nucleic Acids Res 45:10306–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Michalska K, Quan Nhan D, Willett JLE, Stols LM, Eschenfeldt WH, et al. 2018. Functional plasticity of antibacterial EndoU toxins. Mol Microbiol 109:509–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Miyata ST, Kitaoka M, Brooks TM, McAuley SB, Pukatzki S. 2011. Vibrio cholerae requires the type VI secretion system virulence factor VasX to kill Dictyostelium discoideum. Infect Immun 79:2941–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miyata ST, Unterweger D, Rudko SP, Pukatzki S. 2013. Dual expression profile of type VI secretion system immunity genes protects pandemic Vibrio cholerae. PLoS Pathog 9:e1003752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mora L, Moncoq K, England P, Oberto J, de Zamaroczy M. 2015. The Stable Interaction Between Signal Peptidase LepB of Escherichia coli and Nuclease Bacteriocins Promotes Toxin Entry into the Cytoplasm. J Biol Chem 290:30783–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Morse RP, Nikolakakis KC, Willett JL, Gerrick E, Low DA, et al. 2012. Structural basis of toxicity and immunity in contact-dependent growth inhibition (CDI) systems. Proc Natl Acad Sci U S A 109:21480–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morse RP, Willett JL, Johnson PM, Zheng J, Credali A, et al. 2015. Diversification of beta-Augmentation Interactions between CDI Toxin/Immunity Proteins. J Mol Biol 427:3766–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mosbahi K, Walker D, James R, Moore GR, Kleanthous C. 2006. Global structural rearrangement of the cell penetrating ribonuclease colicin E3 on interaction with phospholipid membranes. Protein Sci 15:620–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mosbahi K, Walker D, Lea E, Moore GR, James R, Kleanthous C. 2004. Destabilization of the colicin E9 Endonuclease domain by interaction with negatively charged phospholipids: implications for colicin translocation into bacteria. J Biol Chem 279:22145–51 [DOI] [PubMed] [Google Scholar]
- 96.Munoz-Dorado J, Marcos-Torres FJ, Garcia-Bravo E, Moraleda-Munoz A, Perez J. 2016. Myxobacteria: Moving, Killing, Feeding, and Surviving Together. Front Microbiol 7:781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ohr RJ, Anderson M, Shi M, Schneewind O, Missiakas D. 2017. EssD, a Nuclease Effector of the Staphylococcus aureus ESS Pathway. Journal of bacteriology 199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pallen MJ. 2002. The ESAT-6/WXG100 superfamily -- and a new Gram-positive secretion system? Trends Microbiol 10:209–12 [DOI] [PubMed] [Google Scholar]
- 99.Parker MW, Pattus F. 1993. Rendering a membrane protein soluble in water: a common packing motif in bacterial protein toxins. Trends Biochem Sci 18:391–5 [DOI] [PubMed] [Google Scholar]
- 100.Parker MW, Pattus F, Tucker AD, Tsernoglou D. 1989. Structure of the membrane-pore-forming fragment of colicin A. Nature 337:93–6 [DOI] [PubMed] [Google Scholar]
- 101.Pathak DT, Wei X, Bucuvalas A, Haft DH, Gerloff DL, Wall D. 2012. Cell contact-dependent outer membrane exchange in myxobacteria: genetic determinants and mechanism. PLoS Genet 8:e1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pathak DT, Wei X, Dey A, Wall D. 2013. Molecular recognition by a polymorphic cell surface receptor governs cooperative behaviors in bacteria. PLoS Genet 9:e1003891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Patzer SI, Albrecht R, Braun V, Zeth K. 2012. Structural and mechanistic studies of pesticin, a bacterial homolog of phage lysozymes. J Biol Chem 287:23381–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pissaridou P, Allsopp LP, Wettstadt S, Howard SA, Mavridou DAI, Filloux A. 2018. The Pseudomonas aeruginosa T6SS-VgrG1b spike is topped by a PAAR protein eliciting DNA damage to bacterial competitors. Proc Natl Acad Sci U S A 115:12519–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Poole SJ, Diner EJ, Aoki SK, Braaten BA, T’Kint de Roodenbeke C, et al. 2011. Identification of functional toxin/immunity genes linked to contact-dependent growth inhibition (CDI) and rearrangement hotspot (Rhs) systems. PLoS Genet. 7:e1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pugsley AP, Schwartz M. 1983. Expression of a gene in a 400-base-pair fragment of colicin plasmid ColE2-P9 is sufficient to cause host cell lysis. Journal of bacteriology 156:109–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pugsley AP, Schwartz M. 1984. Colicin E2 release: lysis, leakage or secretion? Possible role of a phospholipase. EMBO J 3:2393–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pukatzki S, Ma AT, Revel AT, Sturtevant D, Mekalanos JJ. 2007. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc Natl Acad Sci U S A 104:15508–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, et al. 2006. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci U S A 103:1528–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Quentin D, Ahmad S, Shanthamoorthy P, Mougous JD, Whitney JC, Raunser S. 2018. Mechanism of loading and translocation of type VI secretion system effector Tse6. Nat Microbiol 3:1142–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Raaijmakers H, Vix O, Toro I, Golz S, Kemper B, Suck D. 1999. X-ray structure of T4 endonuclease VII: a DNA junction resolvase with a novel fold and unusual domain-swapped dimer architecture. EMBO J 18:1447–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Riley MA. 1998. Molecular mechanisms of bacteriocin evolution. Annu Rev Genet 32:255–78 [DOI] [PubMed] [Google Scholar]
- 113.Ronneau S, Helaine S. 2019. Clarifying the Link between Toxin-Antitoxin Modules and Bacterial Persistence. J Mol Biol 431:3462–71 [DOI] [PubMed] [Google Scholar]
- 114.Ruhe ZC, Nguyen JY, Beck CM, Low DA, Hayes CS. 2014. The proton-motive force is required for translocation of CDI toxins across the inner membrane of target bacteria. Mol Microbiol 94:466–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ruhe ZC, Nguyen JY, Xiong J, Koskiniemi S, Beck CM, et al. 2017. CdiA effectors use modular receptor-binding domains to recognize target bacteria. MBio 8:e00290–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ruhe ZC, Subramanian P, Song K, Nguyen JY, Stevens TA, et al. 2018. Programmed Secretion Arrest and Receptor-Triggered Toxin Export during Antibacterial Contact-Dependent Growth Inhibition. Cell 175:921–33 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]; Proposed that CdiA effectors build their own translocons in the outer membrane of target bacteria.
- 117.Ruhe ZC, Townsley L, Wallace AB, King A, Van der Woude MW, et al. 2015. CdiA promotes receptor-independent intercellular adhesion. Mol Microbiol 98:175–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Russell AB, Hood RD, Bui NK, LeRoux M, Vollmer W, Mougous JD. 2011. Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475:343–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Russell AB, LeRoux M, Hathazi K, Agnello DM, Ishikawa T, et al. 2013. Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature 496:508–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Russell AB, Singh P, Brittnacher M, Bui NK, Hood RD, et al. 2012. A widespread bacterial type VI secretion effector superfamily identified using a heuristic approach. Cell Host Microbe 11:538–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Salomon D, Kinch LN, Trudgian DC, Guo X, Klimko JA, et al. 2014. Marker for type VI secretion system effectors. Proc Natl Acad Sci U S A 111:9271–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Salomon D, Klimko JA, Trudgian DC, Kinch LN, Grishin NV, et al. 2015. Type VI Secretion System Toxins Horizontally Shared between Marine Bacteria. PLoS Pathog 11:e1005128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Santin YG, Doan T, Journet L, Cascales E. 2019. Cell Width Dictates Type VI Secretion Tail Length. Current biology : CB 29:3707–13 e3 [DOI] [PubMed] [Google Scholar]
- 124.Schaller K, Holtje JV, Braun V. 1982. Colicin M is an inhibitor of murein biosynthesis. J. Bacteriol 152:994–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shneider MM, Buth SA, Ho BT, Basler M, Mekalanos JJ, Leiman PG. 2013. PAAR-repeat proteins sharpen and diversify the type VI secretion system spike. Nature 500:350–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Siegrist MS, Unnikrishnan M, McConnell MJ, Borowsky M, Cheng TY, et al. 2009. Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc Natl Acad Sci U S A 106:18792–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Silverman JM, Agnello DM, Zheng H, Andrews BT, Li M, et al. 2013. Haemolysin coregulated protein is an exported receptor and chaperone of type VI secretion substrates. Mol Cell 51:584–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Skjerning RB, Senissar M, Winther KS, Gerdes K, Brodersen DE. 2019. The RES domain toxins of RES-Xre toxin-antitoxin modules induce cell stasis by degrading NAD+. Mol Microbiol 111:221–36 [DOI] [PubMed] [Google Scholar]
- 129.Souza DP, Oka GU, Alvarez-Martinez CE, Bisson-Filho AW, Dunger G, et al. 2015. Bacterial killing via a type IV secretion system. Nat Commun 6:6453. [DOI] [PubMed] [Google Scholar]
- 130.Stanley SA, Raghavan S, Hwang WW, Cox JS. 2003. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci U S A 100:13001–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Steczkiewicz K, Muszewska A, Knizewski L, Rychlewski L, Ginalski K. 2012. Sequence, structure and functional diversity of PD-(D/E)XK phosphodiesterase superfamily. Nucleic Acids Res 40:7016–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tang JY, Bullen NP, Ahmad S, Whitney JC. 2018. Diverse NADase effector families mediate interbacterial antagonism via the type VI secretion system. J Biol Chem 293:1504–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ting SY, Bosch DE, Mangiameli SM, Radey MC, Huang S, et al. 2018. Bifunctional Immunity Proteins Protect Bacteria against FtsZ-Targeting ADP-Ribosylating Toxins. Cell 175:1380–92 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Uchida K, Leiman PG, Arisaka F, Kanamaru S. 2014. Structure and properties of the C-terminal beta-helical domain of VgrG protein from Escherichia coli O157. J Biochem 155:173–82 [DOI] [PubMed] [Google Scholar]
- 135.Unterweger D, Kostiuk B, Otjengerdes R, Wilton A, Diaz-Satizabal L, Pukatzki S. 2015. Chimeric adaptor proteins translocate diverse type VI secretion system effectors in Vibrio cholerae. EMBO J 34:2198–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Unterweger D, Miyata ST, Bachmann V, Brooks TM, Mullins T, et al. 2014. The Vibrio cholerae type VI secretion system employs diverse effector modules for intraspecific competition. Nat Commun 5:3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.van der Wal FJ, Luirink J, Oudega B. 1995. Bacteriocin release proteins: mode of action, structure, and biotechnological application. FEMS Microbiol Rev 17:381–99 [DOI] [PubMed] [Google Scholar]
- 138.Vassallo C, Pathak DT, Cao P, Zuckerman DM, Hoiczyk E, Wall D. 2015. Cell rejuvenation and social behaviors promoted by LPS exchange in myxobacteria. Proc Natl Acad Sci U S A 112:E2939–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Vassallo CN, Cao P, Conklin A, Finkelstein H, Hayes CS, Wall D. 2017. Infectious polymorphic toxins delivered by outer membrane exchange discriminate kin in myxobacteria. Elife 6 [DOI] [PMC free article] [PubMed] [Google Scholar]; Discovered that SitA lipoproteins are novel polymorphic effectors delivered through OME.
- 140.Vassallo CN, Wall D. 2019. Self-identity barcodes encoded by six expansive polymorphic toxin families discriminate kin in myxobacteria. Proc Natl Acad Sci U S A 116:24808–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Vettiger A, Basler M. 2016. Type VI Secretion System Substrates Are Transferred and Reused among Sister Cells. Cell 167:99–110 e12 [DOI] [PubMed] [Google Scholar]
- 142.Virtanen P, Waneskog M, Koskiniemi S. 2019. Class II contact-dependent growth inhibition (CDI) systems allow for broad-range cross-species toxin delivery within the Enterobacteriaceae family. Mol Microbiol 111:1109–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Vollmer W, Pilsl H, Hantke K, Holtje JV, Braun V. 1997. Pesticin displays muramidase activity. Journal of bacteriology 179:1580–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Walker D, Mosbahi K, Vankemmelbeke M, James R, Kleanthous C. 2007. The role of electrostatics in colicin nuclease domain translocation into bacterial cells. J Biol Chem 282:31389–97 [DOI] [PubMed] [Google Scholar]
- 145.Wang J, Brackmann M, Castano-Diez D, Kudryashev M, Goldie KN, et al. 2017. Cryo-EM structure of the extended type VI secretion system sheath-tube complex. Nat Microbiol 2:1507–12 [DOI] [PubMed] [Google Scholar]
- 146.Whitney JC, Peterson SB, Kim J, Pazos M, Verster AJ, et al. 2017. A broadly distributed toxin family mediates contact-dependent antagonism between gram-positive bacteria. Elife 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Whitney JC, Quentin D, Sawai S, LeRoux M, Harding BN, et al. 2015. An interbacterial NAD(P)(+) glycohydrolase toxin requires elongation factor Tu for delivery to target cells. Cell 163:607–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wiener M, Freymann D, Ghosh P, Stroud RM. 1997. Crystal structure of colicin Ia. Nature 385:461–4 [DOI] [PubMed] [Google Scholar]
- 149.Wiener MC. 2005. TonB-dependent outer membrane transport: going for Baroque? Curr Opin Struct Biol 15:394–400 [DOI] [PubMed] [Google Scholar]
- 150.Willett JL, Gucinski GC, Fatherree JP, Low DA, Hayes CS. 2015. Contact-dependent growth inhibition toxins exploit multiple independent cell-entry pathways. Proc Natl Acad Sci U S A 112:11341–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wood TE, Howard SA, Forster A, Nolan LM, Manoli E, et al. 2019. The Pseudomonas aeruginosa T6SS Delivers a Periplasmic Toxin that Disrupts Bacterial Cell Morphology. Cell Rep 29:187–201 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yang X, Clemens DL, Lee BY, Cui Y, Zhou ZH, Horwitz MA. 2019. Atomic Structure of the Francisella T6SS Central Spike Reveals a Unique alpha-Helical Lid and a Putative Cargo. Structure 27:1811–9 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zakharov SD, Eroukova VY, Rokitskaya TI, Zhalnina MV, Sharma O, et al. 2004. Colicin occlusion of OmpF and TolC channels: outer membrane translocons for colicin import. Biophys J 87:3901–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zakharov SD, Zhalnina MV, Sharma O, Cramer WA. 2006. The colicin E3 outer membrane translocon: immunity protein release allows interaction of the cytotoxic domain with OmpF porin. Biochemistry 45:10199–207 [DOI] [PubMed] [Google Scholar]
- 155.Zepeda-Rivera MA, Saak CC, Gibbs KA. 2018. A Proposed Chaperone of the Bacterial Type VI Secretion System Functions To Constrain a Self-Identity Protein. Journal of bacteriology 200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zeth K, Romer C, Patzer SI, Braun V. 2008. Crystal structure of colicin M, a novel phosphatase specifically imported by Escherichia coli. J Biol Chem 283:25324–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zhang D, de Souza RF, Anantharaman V, Iyer LM, Aravind L. 2012. Polymorphic toxin systems: Comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol Direct 7:18. [DOI] [PMC free article] [PubMed] [Google Scholar]; Proposed that LXG/WXG, PrsW and phage head morphogenesis proteins deliver toxins for bacterial competition
- 158.Zhang D, Iyer LM, Aravind L. 2011. A novel immunity system for bacterial nucleic acid degrading toxins and its recruitment in various eukaryotic and DNA viral systems. Nucleic Acids Res 39:4532–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zoued A, Cassaro CJ, Durand E, Douzi B, Espana AP, et al. 2016. Structure-Function Analysis of the TssL Cytoplasmic Domain Reveals a New Interaction between the Type VI Secretion Baseplate and Membrane Complexes. J Mol Biol 428:4413–23 [DOI] [PubMed] [Google Scholar]
- 160.Zoued A, Durand E, Brunet YR, Spinelli S, Douzi B, et al. 2016. Priming and polymerization of a bacterial contractile tail structure. Nature 531:59–63 [DOI] [PubMed] [Google Scholar]