

Abstract
Peripheral T‐cell lymphoma (PTCL) is a heterogeneous entity generally with a poor prognosis. Recent genomic analyses have characterized genomic alterations and described gene expression profiling and epigenetic mechanisms in PTCL, leading to reveal molecular pathophysiology in detail. One of several important findings is that heterogeneities exist in both the disease and in individuals. Among PTCL subtypes, adult T‐cell leukemia/lymphoma (ATLL) and peripheral T‐cell lymphoma, not otherwise specified (PTCL‐NOS) are common in Japan. ATLL is an incurable T‐cell malignancy induced by human T‐cell lymphotropic virus type 1 (HTLV‐1). The global genomics of ATLL can be summarized as alterations involving T‐cell receptor (TCR) signaling and immune escape mechanisms. This highlights the fact that ATLL is a viral‐mediated T‐cell malignancy. Interestingly, several previous studies have found that the genomics of ATLL differ according to geographical region and age at diagnosis, suggesting disease heterogeneity, though they share HTLV‐1 infection as initial disease hit. Clonal expansion of the cells acquired by somatic mutations in ATLL‐related genes is identified in a part of HTLV‐1 carriers who developed ATLL later. The risk for ATLL may be updated based on findings in detail. PTCL‐NOS is a heterogeneous disease type of T‐cell lymphoma that does not correspond to any other type of PTCL. Several studies have stratified PTCL‐NOS according to transcriptional, genomic, microenvironmental, and clinical aspects. These kinds of analysis from multiple aspects are useful to understand the heterogeneous group. These efforts will help guide suitable translational research to target PTCL.
Keywords: adult T‐cell leukemia, genomic landscape, heterogeneity, lymphoma, peripheral T‐cell lymphoma, premalignant status
Coexistence of CD28 fusions is highly characteristic of young Japanese adult T‐cell leukemia/lymphoma (ATLL). This suggests the existence of heterogeneity of ATLL.

Abbreviations
- ATLL
adult T‐cell leukemia/lymphoma
- HTLV‐1
human T‐cell lymphotropic virus type 1
- PTCL
peripheral T‐cell lymphoma
- PTCL‐NOS
peripheral T‐cell lymphoma, not otherwise specified
- PTTL
primary thyroid T‐cell lymphoma
- TCR
T‐cell receptor
1. INTRODUCTION
Peripheral T‐cell lymphoma (PTCL) is a heterogeneous disease entity with currently 30 subtypes, including those characterized as provisional. 1 To understand its pathophysiology, various methods such as gene expression, genome copy number analysis, and high‐throughput sequencing have been adopted around the world. The genomic landscapes of extremely rare PTCL types of hepatosplenic T‐cell lymphoma and enteropathy‐associated T‐cell lymphoma, both of which account for <5% of all PTCLs, have been characterized. 2 , 3 As such, virtually all genomic alterations in PTCL are currently known. In PTCL, as well as other malignancies, genomic alterations are summarized based on each disease entity, and analysis can reveal not only genomic alterations but also the heterogeneity of alterations in the disease.
Among PTCL subtypes, adult T‐cell leukemia/lymphoma (ATLL) and PTCL, not otherwise specified (PTCL‐NOS), are common in Japan. 4 , 5 ATLL is a T‐cell neoplasm caused by human T‐cell lymphotropic virus type 1 (HTLV‐1) infection, whereas PTCL‐NOS is a heterogeneous entity that does not correspond to any other type of PTCL. The present review summarizes the molecular pathophysiology of both diseases and describes their heterogeneity based mainly on findings from our previous studies. We will also provide directions for deeper understanding of the pathophysiology.
2. GENOMIC MUTATIONS OF ATLL: T‐CELL RECEPTOR (TCR) SIGNALING AND IMMUNE ESCAPE MECHANISMS
Because HTLV‐1 infection is common in several geographical regions, such as Japan and the Caribbean islands, ATLL is also common in these areas. In fact, ATLL is the most common type of malignant lymphoma in the Kyushu region of Japan. 4 Currently, approximately 10 to 20 million individuals are estimated to be infected with HTLV‐1 worldwide. 6 Approximately 5% of HTLV‐1 carriers develop ATLL, suggesting that alterations that synergistically act with HTLV‐1 are involved in the pathogenesis and development of ATLL. Although the median age of ATLL onset is close to 70 years, the infection also occurs in infants, which suggests that ATLL develops due to the accumulation of genomic abnormalities in HTLV‐1–infected cells over a long period of time. Several comprehensive genomic analyses of ATLL have been performed to characterize these alterations. 7 , 8 , 9 , 10 , 11 , 12 In particular, a study by Kataoka et al analyzed a large number of samples using high‐throughput sequencing, which revealed the molecular landscape in detail. Genomic analysis of ATLL characterized hallmark alterations of genes involving TCR signaling/NF‐κB signaling pathways. Alterations involving these pathways were identified in 90% of all ATLL cases analyzed. In addition, genomic alterations related to immune escape mechanisms have been frequently identified in ATLL. Somatic alterations in PD‐L1 (also known as CD274), HLA‐B, and CD58 found in ATLL can cause escape from host immunity. 9 , 10 , 13 These comprehensive genomic analyses also identified a wide variety of alterations that possibly drive the development of ATLL. In terms of ATLL development, synergy between these genomic alterations and their order of acquisition may play important roles.
3. VIRAL PRODUCTS OF HTLV‐1 RELATED TO ATLL
From the perspective of HTLV‐1, Tax is regarded to be an oncoprotein of HTLV‐1 14 , 15 and is required for viral replication. Tax is highly immunogenic, 16 , 17 and its expression has been shown to be reduced in clinical samples from ATLL patients. 14 Interestingly, Kataoka et al found that frequent alterations in TCR/NF‐κB signaling pathways in ATLL are recognized in genes of the Tax interactome, 10 suggesting that they can activate the TCR without Tax expression in ATLL. We recently reported important findings in this regard using pathological analyses. 18 In this study, we performed clinicopathological investigation of ATLL using in situ RNA analysis of Tax. Our analysis found that ATLL cases with high Tax expression exhibited substantially reduced human leukocyte antigen (HLA) class I and the component of B2M on the cell surface compared with those without Tax expression (Figure 1). In addition, Tax‐expressing ATLL cases exhibited an increase in PD‐1–positive cytotoxic T cells (CTLs). Collectively, Tax‐expressing cases reduced the escape of HLA class I from surrounding CTLs. Although we did not assess the mechanisms related to the reduction of HLA class I, it has been reported that genomic mutations and epigenetic modifications can reduce the expression. 10 , 13 An increase in PD‐1–positive CTL may be caused by chronic HTLV‐1 infection. Additionally, it is known that PD‐1 plays an inhibitory role during naive‐to‐effector CD8 T‐cell transition and that the PD‐1 pathway can also be modulated at this stage of T‐cell differentiation. 19 The induced PD‐1 expression in surrounding cells may also be associated with immune escape of ATLL cells. In our study, Tax expression in those with >400 signals per 1000 ATLL cells was a poor prognostic factor. In addition to Tax, HBZ is also recognized as an important oncovirus protein. 14 Unlike Tax, however, HBZ is expressed in virtually all cases of ATLL, 20 suggesting its important role in the development of ATLL. In fact, HBZ transgenic mice frequently develop ATLL. 21 Using an in vivo mouse model, we also showed that the synergistic effects of HBZ and the other genomic alterations identified in ATLL cases caused ATLL. 22 Furthermore, a recent study identified the importance of HBZ in directly inducing a transcriptional factor, BATF3. 23 ATLL cells depend on BATF3 and the related pathways are therapeutic target candidates. Although the importance of HBZ in this regard has been reported, our study revealed that some ATLL cases exhibited low HBZ expression with an advanced stage. In other words, cases with high HBZ expression were characterized by less skin involvement and tended to exhibit localized disease compared with those with low HBZ expression (Figure 1). 18 In terms of HLA class I expression, there were no marked differences based on HBZ status. These points should be addressed in other cohorts in future studies.
FIGURE 1.
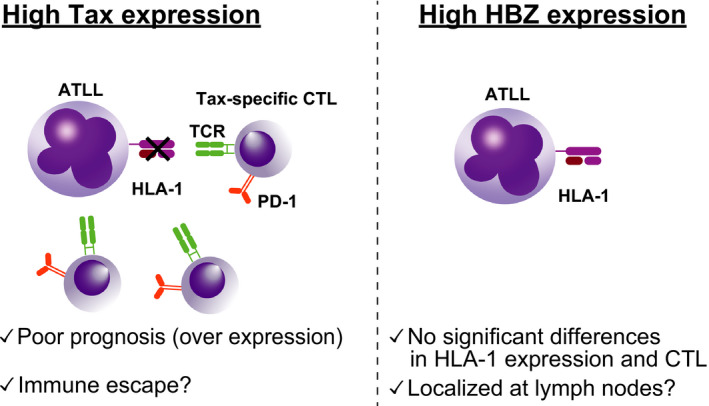
Differences between adult T‐cell leukemia/lymphoma (ATLL) with Tax (left) and HBZ expression (right). Tax expression is highly immunogenic, and activated cytotoxic T cells (CTLs) target the cells. In high–Tax‐expressing cases, ATLL cells lack human leukocyte antigen (HLA) class I and the component B2M on cell surfaces, resulting in the disruption of interaction with CTLs. More than 400 Tax signals have been identified as poor prognostic factors in ATLL. In contrast to Tax, there were no substantial differences in MHC class I and CTL based on HBZ expression
4. REGIONAL DIFFERENCES IN ATLL
HTLV‐1 is an endemic disease in some geographical regions, including Japan, the Caribbean, South America, the Middle East, and Australia 24 ; moreover, ATLL frequently develops in endemic areas. However, the incidence of ATLL decreased in endemic areas, whereas increased in nonendemic areas, though the total numbers of cases in the nonendemic areas are small. 25 It has been established that there are regional differences in the clinical pathology of ATLL. For example, ATLL patients in the Caribbean and South America are younger than those in Japan. 26 A study investigating genomic alterations of ATLL in North America 11 identified a distinct genomic landscape with frequent mutations related to the epigenome. Mutations in epigenetic or histone modifier genes were found in 57% of North American ATLL cases but only in 22% of Japanese cases. Alterations related to epigenetic genes have also been frequently found in several hematological malignancies such as acute myeloid leukemia, 27 although no regional differences have been reported. DNA methylation is an epigenetic event that negatively controls gene expression. A recent study investigating ATLL in Japanese subjects identified the accumulation of aberrant DNA methylation, which was correlated with development and progression. 28 Based on genomic findings from Japanese subjects with ATLL, it is believed that DNA hypermethylation status in ATLL cells is not related to genetic events. 10 Although the methylation pattern of ATLL in North American subjects remains unclear, gene expression profiling distinguished North American and Japanese cases, suggesting that they have different properties. One particular feature related to this difference may be the presence of alterations related to epigenetic factors that could drive global changes in gene expression. From a clinical standpoint, DNA methyltransferase inhibitors were active against North American ATLL cases harboring epigenetic alterations and Japanese ATLL. 11 , 28 This clearly suggests that Japanese ATLL cases highly depend on DNA hypermethylation status for the survival regardless of absence of genomic alterations that directly affect the methylation status. Identification of the causes of methylation in Japanese ATLL may also lead to elucidation of the therapeutic targets. Although frequency of mutations in epigenetic or histone modifier genes, especially EP300, were higher in North American ATLL (Figure 2A), several ATLL driver mutations including PRKCB and CCR4, which were frequently found in Japanese ATLL, have not been addressed in North American ATLL cases. Comparison of the frequencies of the driver mutations between these areas will further provide insights into molecular aspects of ATLL. In addition, genomic alterations of ATLL in other geographical regions remain unclear, and delineation will help us better understand the pathophysiology of ATLL.
FIGURE 2.
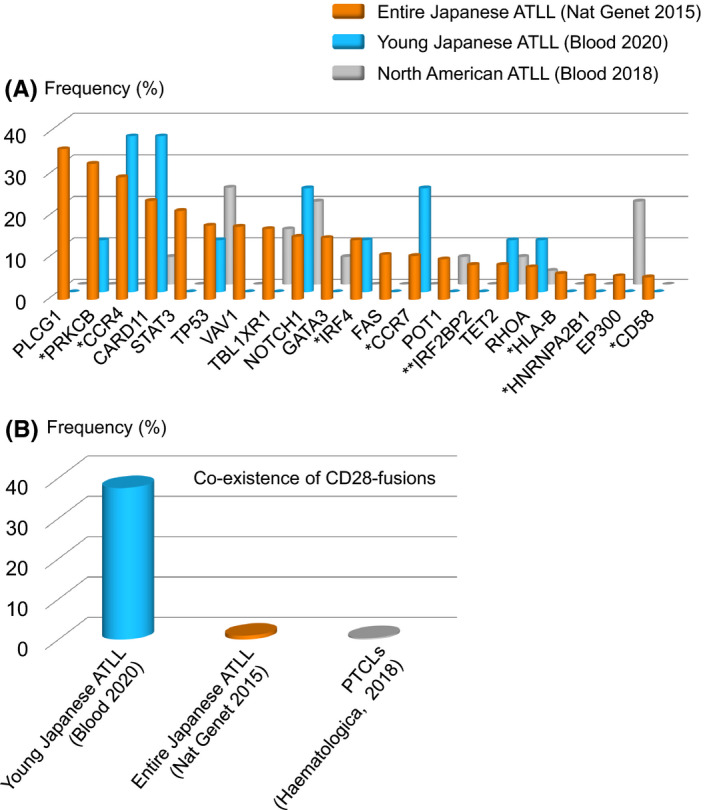
Frequency of genomic alterations in adult T‐cell leukemia/lymphoma (ATLL). A, Frequency of somatic mutations across the cohorts. Genes that were recurrently mutated in a previous report (Kataoka et al, Nat Genet 2015) were summarized. *PRKCB, CCR4, VAV1, IRF4, CCR7, HLA‐B, and HNRNPA2B1 were not evaluated in a North American ATLL cohort. **IRF2BP2 was not evaluated in young Japanese and North American ATLL cohorts. B, Frequency of co‐occurrence of CTLA4‐CD28 and ICOS‐CD28 fusions in peripheral T‐cell lymphoma (PTCL) including ATLL
5. GENOMIC LANDSCAPE OF ATLL IN YOUNGER INDIVIDUALS
As described above, ATLL frequently develops in the elderly, although it has been reported that the age at onset is gradually increasing. 5 , 29 HTLV‐1 is mainly transmitted from mother to child through breastfeeding. 30 , 31 , 32 It is, therefore, believed that ATLL develops due to the accumulation of genetic abnormalities over a long period of time after HTLV‐1 infection. In other cancers, younger age is associated with particular genetic alterations, which are considered to be “driver” genes. 33 , 34 , 35 , 36 , 37 , 38 We speculated that ATLL in younger patients would exhibit distinct genetic alterations and, accordingly, performed genomic analysis. We identified eight patients with ATLL who were <50 years of age at the time of diagnosis and defined them as young ATLL. First, we evaluated the prognosis of these young patients, and the overall survival was 14.5 months (95% confidence interval, 5.8 – not reached) from diagnosis. Genomic mutations were analyzed using a targeted sequencing approach. CCR4 and CARD11 were frequently mutated in young ATLL cases, although no genetic mutation(s) characteristic of these younger cases was observed (Figure 2A). This point may be due to the limited number of analyzed cases, and further studies using larger samples would characterize genomic mutations in young ATLL cases. PLCG1 is the most frequently mutated gene in all ATLL cases; however, the mutation has not been observed in younger patients. In addition, there were no young ATLL cases with a STAT3 mutation, which was also found in all ATLL cases. In terms of fusion genes, 38% (three of eight) of young patients had both CTLA4‐CD28 and ICOS‐CD28, and the frequency was higher in this cohort than in all ATLL cases, suggesting that these fusion genes are characteristic of young ATLL cases (Figure 2B). This is in contrast with previous studies in which cases with both fusions were identified in 1% of ATLL (1 of 105) and 0.7% of PTCL (2 of 273). 10 , 39 We suggest that CD28 fusions were associated with the onset of ATLL at a young age; this was also observed in 144 Japanese ATLL cases from a different cohort by another group. 40 This finding suggests that CD28 fusions play a role in ATLL as driver alterations. Another study identified a super‐enhancer region at CD28, 41 and other alterations of CD28 have been identified in ATLL. 10 As such, the CD28 pathway is considered to be important in the pathophysiology of ATLL. We performed a functional analysis to understand CTLA4‐CD28 and found that this fusion gene acts in a ligand‐dependent manner (Figure 3) and confirms the importance of the CD28 domain in CTLA4‐CD28. CTLA4 inhibits TCR signaling, whereas CD28 and ICOS activate TCR signaling through interactions with antigen‐presenting cells. 42 Although both CTLA4 and CD28 share the CD80 and CD86 ligands, their affinity to ligands is higher for CTLA4 than for CD28 (Figure 3). 43 In ATLL, we found that ligands for CD80 and CD86 were expressed on tumor cells and nontumor cells, respectively. 12 Collectively, CTLA4‐CD28 can be activated by cell‐nonautonomous interactions. Because CTLA4 is expressed on the cell surface as a homodimer, 44 further studies aiming to identify the binding partner of CTLA4‐CD28 may help better understand the fusion gene. Our in vitro study found that the nonautonomous interactions between CTLA4‐CD28 and the ligands were targetable with anti‐CTLA4 antibody. Indeed, a previous case report of a patient with PTCL and CTLA4‐CD28 fusion showed a deep but transient response to the anti‐CTLA4 antibody. 45 To characterize the response in detail, further studies assessing whether the response is resulted from blocking the interaction or activation of immune response would be needed. We also confirmed that CD28 signaling driven by CTLA4‐CD28 caused PI3K activation. From the perspective of therapeutic options, PI3K inhibitors may be active against ATLL with CTLA4‐CD28, 46 , 47 although suitable clinical trials including in vivo models will be required in the future.
FIGURE 3.
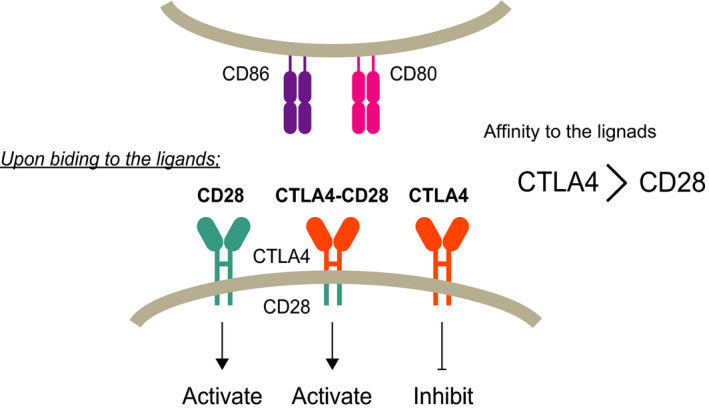
Schematic model of CD28, CTLA4, and CTLA4‐CD28. CD28 and CTLA4 interact with the shared ligands CD80 and CD86. Upon binding to the ligand, CD28 activates T‐cell receptor (TCR) signaling, whereas CTLA4 inhibits signaling. Because ligand affinity is greater in CTLA4 than CD28, normal T cells are controlled to prevent overactivation by CTLA4. In CTLA4‐CD28, the affinity of this fusion is suspected to be identical to that of CTLA4, and the fusion gene activates downstream
6. HETEROGENEITY OF HTLV‐1 CARRIERS
Most HTLV‐1 carriers do not develop any related diseases, although approximately 5% develop ATLL, and 0.3% of carriers are assumed to be affected by HTLV‐1–associated myelopathy (Figure 4). Only a small proportion of carriers develop these diseases, and several studies have identified the risk factors that cause ATLL, including high proviral load, advanced age, family history of ATLL, first opportunity for HTLV‐1 screening during treatment for other diseases, and types of HLA alleles. 48 , 49 Individuals infected in adulthood rarely, if ever, develop ATLL, 26 suggesting that age at the time of HTLV‐1 infection is extremely important. ATLL is characterized by the monoclonal proliferation of tumor cells with CD4 and CD25. Therefore, it was speculated that ATLL developed following a progression from oligoclonal expansion of HTLV‐1–infected cells. However, high‐throughput sequencing of HTLV‐1 integration sites of genomic DNA revealed that there are many HTLV‐1–infected T‐cell clones underlying the largest, putatively malignant clone in ATLL, and that one or more HTLV‐1–infected clones are abundant in some ATLL cases. 50 The analyses also found that the malignant clone can suddenly develop from a clone with low abundance before diagnosis. 51 A recent genomic analysis of HTLV‐1 carriers who developed ATLL later identified somatic mutations in peripheral blood samples before ATLL diagnosis. 52 The identified mutations were those found in ATLL including CCR4, NOTCH1, PLCG1, and PRKCB, and such mutations were not identified in individuals who did not develop ATLL. Thus, a “pre‐ATLL” status may exist before diagnosis of ATLL (Figure 4). The status is considered to be equivalent to monoclonal gammopathy of undetermined significance for myeloma, monoclonal B‐cell lymphocytosis for chronic lymphocytic leukemia, and clonal hematopoiesis of indeterminate potential for hematological malignancies. 53 , 54 , 55 , 56 Detection of these mutations may lead to the early identification of and intervention in ATLL in the future. In addition, the frequency of pre‐ATLL status among HTLV‐1 carriers and the risk for ATLL development are important in understanding the development of ATLL in HTLV‐1 carriers.
FIGURE 4.
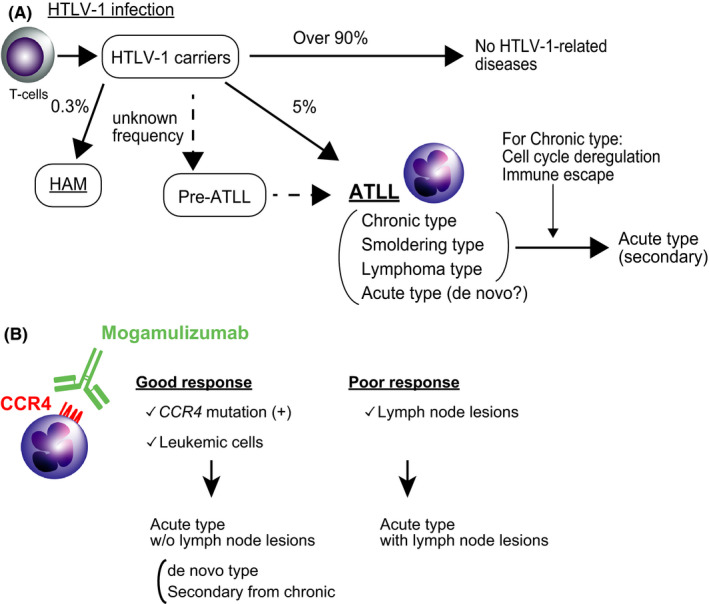
Progression patterns of HTLV‐1–related diseases. A, HTLV‐1 infection is considered to be an initial disease “hit.” The majority of HTLV‐1 carriers do not develop any related diseases. Individuals at pre‐ATLL stage do not experience any symptoms, but ATLL‐related mutations exist. Although it is known that approximately 5% of carriers develop ATLL, it is unclear what percentages of the carries with pre‐ATLL develop ATLL. Some HTLV‐1 carriers develop acute‐type ATLL with intervention from other subtypes of ATLL (secondary). They may also directly develop acute‐type ATLL (de novo). HTLV‐1, human T‐cell lymphotropic virus type 1; ATLL, adult T‐cell leukemia/lymphoma; HAM, HTLV‐1–associated myelopathy. B, Possible differences of response to mogamulizumab in ATLL cases. Presence of CCR4 mutation and the location of tumor cells have been currently identified as markers for response to mogamulizumab. Collectively, among acute‐type ATLL, the cases having CCR4 mutation without lymph node lesions are suspected to have good response to mogamulizumab. The cases with lymph node lesions may have poor response to mogamulizumab
7. HETEROGENEITY OF ATLL SUBTYPES
Based on laboratory and imaging findings, ATLL has been classified into four subtypes: acute, lymphoma, chronic, and smoldering. 57 According to a recent, nationwide, hospital‐based study, the acute subtype was the most common, followed by the lymphoma subtype, which was also frequently observed. 29 Among the subtypes, chronic and smoldering are regarded to be indolent ATLL, whereas the lymphoma and acute subtypes are considered to be aggressive ATLL. The chronic subtype is further divided into unfavorable or favorable groups based on laboratory findings. Because intensive chemotherapies including allogenic stem cell transplantation are recommended for aggressive ATLL and the unfavorable chronic type, 58 , 59 precise classification is required. In terms of genomic alterations, indolent and aggressive types exhibit different genomic landscapes. 9 , 60 From an epidemiological perspective, one‐half of indolent ATLL cases progress only to the acute, not the lymphoma subtype, approximately 1.5 years from diagnosis. 61 Clinically, soluble interleukin‐2 receptor levels have been reported as a predictive marker in indolent ATLL for transformation. 62 We found that cell cycle deregulation and immune escape mechanisms were involved in the progression from the chronic to the acute subtype. 9 Kataoka et al also reported that IRF4 mutation, CDKN2A loss, and PD‐L1 amplification were associated with poor prognosis among the unfavorable chronic type. 60 These indicate heterogeneity, even in the defined subtype.
Heterogeneity in the subtype may also be present among the acute type. The presence of tumor cells in peripheral blood can distinguish between the acute and the lymphoma subtypes, although both are considered to be aggressive. 57 It is, therefore, speculated that the chronic subtype hardly progresses to the lymphoma subtype because the chronic subtype has already had peripheral blood involvement in tumor cells. Umino et al compared the genomic alterations in tumors from peripheral blood and lymph nodes in identical acute subtypes. 63 The analysis found that genomic alterations were identified in more lymph node lesions than in peripheral blood. This analysis identified unique alterations in tumor cells in the peripheral blood, which were not detected in the lymph nodes. It is, therefore, believed that multiple subclones develop in the lymph nodes, and some are involved in the peripheral blood. This finding was also supported by studies investigating HTLV‐1 clonality in ATLL. 50 This may be one of the mechanisms of progression from the lymphoma to the acute subtype. Collectively, the acute subtype of ATLL may be classified into de novo acute type and secondary acute type from other subtypes (Figure 4A), although the de novo type ATLL may have other subtypes before diagnosis.
8. HETEROGENOUS RESPONSE OF ATLL TO MOGAMULIZUMAB
We speculate that heterogeneity of the acute subtype is related to the response of ATLL to anti‐CCR4 antibody (mogamulizumab), which is a humanized CCR4 monoclonal antibody that stimulates antibody‐dependent cellular toxicity. 64 More than 90% of ATLL cases express CCR4, 65 and it is now recognized as a marker for ATLL. Administration of mogamulizumab remarkably improved prognosis, though the overall response rate was 50%, 66 , 67 suggesting the existence of factors to stratify the sensitivity. CCR4 mutation is common in ATLL, with two types of mutations: nonsense (NS) and frameshift mutations (FS). 7 We previously found that NS mutation of CCR4 caused strong expression of CCR4 and that FS mutation of CCR4 was a poor prognostic factor in ATLL without mogamulizumab treatment. 68 A previous investigation reported that CCR4 mutations determine sensitivity to mogamulizumab treatment in ATLL. 69 In the study, the 5‐year overall survival rate from the day of first administration of mogamulizumab in aggressive ATLL cases with CCR4 mutation was 80.0%, while the cases without the mutation was 24.7%. However, in our analysis of another cohort, the CCR4 mutation was not necessarily involved in the responsiveness to mogamulizumab. 64 Actually, the overall response rate was lower in CCR4 mutated cases than wild type cases. In fact, several cases with CCR4 mutations exhibited response to mogamulizumab in peripheral blood tumors, but not to tumor cells in the lymph nodes. In agreement with this aspect, other studies also reported that mogamulizumab was more effective against the leukemic type than the lymphoma type. 67 , 70 This point was also identified in combination therapy using zidovudine and interferon‐alfa. 71 In other words, acute‐type ATLL with lymph node lesions and acute type without lymph node lesions could be distinguished according to treatment responsiveness (Figure 4B). The acute type with lymph node lesions may progress from the lymphoma type, whereas those without may be from the chronic type. Analyses that take this point into consideration may be required to identify biomarkers for therapeutic response in the future. In addition, further analysis from multiple aspects including the mutated clone size will be also needed. Of note, a study by Rowan et al found that somatic mutations associated with the development of ATLL were identified in the peripheral blood before patients were diagnosed with lymphoma‐type ATLL. 52 Analysis of how these clones in the peripheral blood are involved in the development of lymphoma type is considered to be one direction of research investigating the development of ATLL.
9. PTCL‐NOS: A HETEROGENEOUS GROUP OF T‐CELL LYMPHOMAS
At the end of this review, we will briefly introduce heterogeneity in PTCL‐NOS. Among the PTCL subtypes, the fact that PTCL‐NOS is one of the most common types reflects its heterogeneity. Recently, PTCL‐NOS has been classified in several aspects (Figure 5). First, PTCL‐NOS can be classified into two groups based on the expression of two transcriptional factors: GATA3 and TBX21. 72 This is a tumor phenotype‐based approach. Second, Watatani et al showed that genomic alteration patterns can be used to classify PTCL‐NOS. 73 Third, our group used signatures of cells in the tumor microenvironment to stratify prognosis in PTCL‐NOS. 74 Another approach for disease recognition is based on clinicopathological findings, especially the organs of the original tumor sites. In fact, a part of the recent World Health Organization classification of PTCL is based on this finding (eg, cutaneous T‐cell lymphoma and hepatosplenic T‐cell lymphoma). 1 From this perspective, we found that PTCL‐NOS that develops in the thyroid is characteristic in terms of clinical, pathological, and genomic findings; therefore, primary thyroid T‐cell lymphoma (PTTL) has been introduced as a new disease entity. 75
FIGURE 5.
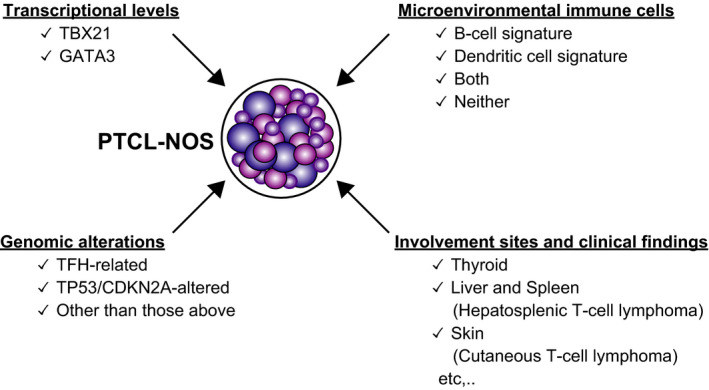
Peripheral T‐cell lymphoma, not otherwise specified (PTCL‐NOS) classification from multiple aspects. Two master transcriptional factors, GATA3 and TBX21, divide PTCL‐NOS into two groups with a different clinical course. Genetically, PTCL‐NOS is classified into three groups. T follicular helper (TFH)‐related genes represent TET2, RHOA, IDH2, and DNMT3A. Signatures of microenvironmental immune cells stratify PTCL‐NOS prognosis. From a clinicopathological aspect, PTCL‐NOS can be further divided, and the group may have characteristic genomic findings
The most common type of malignant lymphoma that develops in the thyroid is diffuse large B‐cell lymphoma, followed by follicular lymphoma, and extranodal marginal zone lymphoma of mucosa‐associated lymphoid tissue (MALT lymphoma). 76 PTCL that develops in the thyroid gland is extremely rare, and only a few case reports have been published. It is very interesting that most were from East Asia, including Japan, and were from otolaryngology and surgical disciplines rather than hematology.
From the perspective of clinical findings, we found that PTTL was associated with autoimmune thyroiditis as an underlying disease with indolent behavior. Of note, some PTTL patients exhibited spontaneous regression without any therapy. Pathologically, the tumor cells were small to medium in size, few had atypia, and were difficult to distinguish from MALT lymphoma using only histopathological techniques. Immunohistochemical analysis revealed that tumor cells expressed CD3, CD4, and CXCR3 antigens, suggesting that the tumor cells were from type 1T helper cells (Th1). It is known that Th1 plays a key role in the development of autoimmune thyroiditis, and we speculate that the autoreactive cells originated from PTTL. We found that the genomic copy number alteration pattern of PTTL differed from that of PTCL‐NOS, and we identified STX11 as a tumor suppressor gene in T‐cell malignancy. 75 , 77 Although PTTL generally has an indolent clinical course, we encountered one PTTL patient who experienced an aggressive clinical course. In this case, the size of the tumor cells was large, and the patient had a CDKN2A deletion. Because the alterations have been reported to be associated with aggressiveness of the chronic subtype of ATLL, 9 , 60 we believe that the deletion also plays an important role in the aggressive behavior of PTTL. Collectively, the series of studies investigating PTTL clearly reflect the heterogeneity of PTCL. In addition, it is possible to identify genomic alterations related to disease pathophysiology by focusing on diseases that exhibit specific clinicopathological characteristics and/or features.
10. CONCLUSION
Genomic analysis of PTCL has yielded characteristic molecular findings, and at the same time, the heterogeneity of this disease has become clearer. In ATLL, recognition of pre‐ATLL status is a major step forward because evaluation may predict the development of ATLL, provide guidance for early therapeutic intervention, and prevent disease development. Classification of PTCL‐NOS using several approaches can precisely identify distinct subgroups with characteristic pathophysiology. This classification may reveal novel therapeutic targets for PTCL. Genome analysis is considered to be a deductive method based on the premise that cancers have genomic alterations. In contrast, it is considered to be an inductive method to establish new disease entities among heterogeneous groups by focusing on characteristic molecular and clinicopathological features.
The integration of these types of analyses and abduction analysis will be required in the future to better understand PTCL.
CONFLICT OF INTEREST
The authors declare no relevant conflict of interest.
ACKNOWLEDGMENTS
This work was supported, in part, by a Grant‐in‐Aid for Early‐Career Scientists (19K16574). We would like to thank Editage (www.editage.com) for English language editing.
Yoshida N, Yamada K, Ohshima K. Comprehensive genomic analysis identifying heterogeneity in peripheral T‐cell lymphoma. Cancer Sci. 2021;112:1339–1347. 10.1111/cas.14849
REFERENCES
- 1. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375‐2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roberti A, Dobay MP, Bisig B, et al. Type II enteropathy‐associated T‐cell lymphoma features a unique genomic profile with highly recurrent SETD2 alterations. Nat Commun. 2016;7:12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McKinney M, Moffitt AB, Gaulard P, et al. The genetic basis of hepatosplenic T‐cell lymphoma. Cancer Discov. 2017;7:369‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chihara D, Ito H, Matsuda T, et al. Differences in incidence and trends of haematological malignancies in Japan and the United States. Br J Haematol. 2014;164:536‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muto R, Miyoshi H, Sato K, et al. Epidemiology and secular trends of malignant lymphoma in Japan: Analysis of 9426 cases according to the World Health Organization classification. Cancer Med. 2018;7(11):5843‐5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Edlich RF, Arnette JA, Williams FM. Global epidemic of human T‐cell lymphotropic virus type‐I (HTLV‐I). J Emerg Med. 2000;18:109‐119. [DOI] [PubMed] [Google Scholar]
- 7. Nakagawa M, Schmitz R, Xiao W, et al. Gain‐of‐function CCR4 mutations in adult T cell leukemia/lymphoma. J Exp Med. 2014;211:2497‐2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakahata S, Ichikawa T, Maneesaay P, et al. Loss of NDRG2 expression activates PI3K‐AKT signalling via PTEN phosphorylation in ATLL and other cancers. Nat Commun. 2014;5:3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoshida N, Karube K, Utsunomiya A, et al. Molecular characterization of chronic‐type adult T‐cell leukemia/lymphoma. Can Res. 2014;74:6129‐6138. [DOI] [PubMed] [Google Scholar]
- 10. Kataoka K, Nagata Y, Kitanaka A, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47:1304‐1315. [DOI] [PubMed] [Google Scholar]
- 11. Shah UA, Chung EY, Giricz O, et al. North American ATLL has a distinct mutational and transcriptional profile and responds to epigenetic therapies. Blood. 2018;132:1507‐1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoshida N, Shigemori K, Donaldson N, et al. Genomic landscape of young ATLL patients identifies frequent targetable CD28 fusions. Blood. 2020;135:1467‐1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kataoka K, Shiraishi Y, Takeda Y, et al. Aberrant PD‐L1 expression through 3'‐UTR disruption in multiple cancers. Nature. 2016;534:402‐406. [DOI] [PubMed] [Google Scholar]
- 14. Matsuoka M, Jeang KT. Human T‐cell leukemia virus type 1 (HTLV‐1) and leukemic transformation: viral infectivity, Tax, HBZ and therapy. Oncogene. 2011;30:1379‐1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mesri EA, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host Microbe. 2014;15:266‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jacobson S, Shida H, McFarlin DE, Fauci AS, Koenig S. Circulating CD8+ cytotoxic T lymphocytes specific for HTLV‐I pX in patients with HTLV‐I associated neurological disease. Nature. 1990;348:245‐248. [DOI] [PubMed] [Google Scholar]
- 17. Kurihara K, Harashima N, Hanabuchi S, et al. Potential immunogenicity of adult T cell leukemia cells in vivo. Int J Cancer. 2005;114:257‐267. [DOI] [PubMed] [Google Scholar]
- 18. Yamada K, Miyoshi H, Yoshida N, et al. Human T‐cell lymphotropic virus HBZ and tax mRNA expression are associated with specific clinicopathological features in adult T‐cell leukemia/lymphoma. Mod Pathol. 2021;34(2):314‐326. [DOI] [PubMed] [Google Scholar]
- 19. Ahn E, Araki K, Hashimoto M, et al. Role of PD‐1 during effector CD8 T cell differentiation. Proc Natl Acad Sci USA. 2018;115:4749‐4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Satou Y, Yasunaga J, Yoshida M, Matsuoka M. HTLV‐I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci USA. 2006;103:720‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Satou Y, Yasunaga J‐I, Zhao T, et al. HTLV‐1 bZIP factor induces T‐cell lymphoma and systemic inflammation in vivo. PLoS Pathog. 2011;7:e1001274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kasugai Y, Yoshida N, Ohshima K, Matsuo K, Seto M, Tsuzuki S. New mouse model of acute adult T‐cell leukemia generated by transplantation of AKT, BCLxL, and HBZ‐transduced T cells. Cancer Sci. 2016;107:1072‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakagawa M, Shaffer AL, Ceribelli M, et al. Targeting the HTLV‐I‐Regulated BATF3/IRF4 Transcriptional Network in Adult T Cell Leukemia/Lymphoma. Cancer Cell. 2018;34(2):286‐297.e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tagaya Y, Matsuoka M, Gallo R. 40 years of the human T‐cell leukemia virus: past, present, and future. F1000Research. 2019;8:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoshida N, Chihara D. Incidence of adult T‐cell leukemia/lymphoma in nonendemic areas. Curr Treat Options Oncol. 2015;16:321. [DOI] [PubMed] [Google Scholar]
- 26. Iwanaga M, Watanabe T, Yamaguchi K. Adult T‐cell leukemia: a review of epidemiological evidence. Front Microbiol. 2012;3:322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood. 2016;127:42‐52. [DOI] [PubMed] [Google Scholar]
- 28. Watanabe T, Yamashita S, Ureshino H, et al. Targeting aberrant DNA hypermethylation as a driver of ATL leukemogenesis by using the new oral demethylating agent OR‐2100. Blood. 2020;136:871‐884. [DOI] [PubMed] [Google Scholar]
- 29. Imaizumi Y, Iwanaga M, Nosaka K, et al. Prognosis of patients with adult T‐cell leukemia/lymphoma in Japan: a Nationwide Hospital‐Based Study. Cancer Sci. 2020;111(12):4567‐4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Verdonck K, Gonzalez E, Van Dooren S, Vandamme AM, Vanham G, Gotuzzo E. Human T‐lymphotropic virus 1: recent knowledge about an ancient infection. Lancet Infect Dis. 2007;7:266‐281. [DOI] [PubMed] [Google Scholar]
- 31. Rosadas C, Taylor GP. Mother‐to‐child HTLV‐1 transmission: unmet research needs. Front Microbiol. 2019;10:999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ichimaru M, Ikeda S, Kinoshita K, Hino S, Tsuji Y. Mother‐to‐child transmission of HTLV‐1. Cancer Detect Prev. 1991;15:177‐181. [PubMed] [Google Scholar]
- 33. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4‐ALK fusion gene in non‐small‐cell lung cancer. Nature. 2007;448:561‐566. [DOI] [PubMed] [Google Scholar]
- 34. Bernt KM, Hunger SP. Current concepts in pediatric philadelphia chromosome‐positive acute lymphoblastic leukemia. Front Oncol. 2014;4:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tarlock K, Zhong S, He Y, et al. Distinct age‐associated molecular profiles in acute myeloid leukemia defined by comprehensive clinical genomic profiling. Oncotarget. 2018;9:26417‐26430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sacher AG, Dahlberg SE, Heng J, Mach S, Janne PA, Oxnard GR. Association between younger age and targetable genomic alterations and prognosis in non‐small‐cell lung cancer. JAMA Oncol. 2016;2:313‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iacobucci I, Wen JI, Meggendorfer M, et al. Genomic subtyping and therapeutic targeting of acute erythroleukemia. Nat Genet. 2019;51:694‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Serebriiskii IG, Connelly C, Frampton G, et al. Comprehensive characterization of RAS mutations in colon and rectal cancers in old and young patients. Nat Commun. 2019;10:3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vallois D, Dupuy A, Lemonnier F, et al. RNA fusions involving CD28 are rare in peripheral T‐cell lymphomas and concentrate mainly in those derived from follicular helper T cells. Haematologica. 2018;103:e360‐e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sakamoto Y, Ishida T, Masaki A, et al. Clinical significance of CD28 gene‐related activating alterations in adult T‐cell leukaemia/lymphoma. Br J Haematol. 2020;192(2):281‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wong RWJ, Ngoc PCT, Leong WZ, et al. Enhancer profiling identifies critical cancer genes and characterizes cell identity in adult T‐cell leukemia. Blood. 2017;130:2326‐2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen L, Flies DB. Molecular mechanisms of T cell co‐stimulation and co‐inhibition. Nat Rev Immunol. 2013;13:227‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walker LS, Sansom DM. The emerging role of CTLA4 as a cell‐extrinsic regulator of T cell responses. Nat Rev Immunol. 2011;11:852‐863. [DOI] [PubMed] [Google Scholar]
- 44. Brunet J‐F, Denizot F, Luciani M‐F, et al. A new member of the immunoglobulin superfamily–CTLA‐4. Nature. 1987;328:267‐270. [DOI] [PubMed] [Google Scholar]
- 45. Sekulic A, Liang WS, Tembe W, et al. Personalized treatment of Sezary syndrome by targeting a novel CTLA4:CD28 fusion. Mol Genet Genomic Med. 2015;3:130‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Horwitz SM, Koch R, Porcu P, et al. Activity of the PI3K‐delta, gamma inhibitor duvelisib in a phase 1 trial and preclinical models of T‐cell lymphoma. Blood. 2018;131:888‐898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Katsuya H, Cook LBM, Rowan AG, Satou Y, Taylor GP, Bangham CRM. Phosphatidylinositol 3‐kinase‐delta (PI3K‐delta) is a potential therapeutic target in adult T‐cell leukemia‐lymphoma. Biomark Res. 2018;6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Iwanaga M, Watanabe T, Utsunomiya A, et al. Human T‐cell leukemia virus type I (HTLV‐1) proviral load and disease progression in asymptomatic HTLV‐1 carriers: a nationwide prospective study in Japan. Blood. 2010;116:1211‐1219. [DOI] [PubMed] [Google Scholar]
- 49. Sonoda S, Li HC, Tajima K. Ethnoepidemiology of HTLV‐1 related diseases: ethnic determinants of HTLV‐1 susceptibility and its worldwide dispersal. Cancer Sci. 2011;102:295‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bangham CR, Cook LB, Melamed A. HTLV‐1 clonality in adult T‐cell leukaemia and non‐malignant HTLV‐1 infection. Semin Cancer Biol. 2014;26:89‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cook LB, Melamed A, Niederer H, et al. The role of HTLV‐1 clonality, proviral structure and genomic integration site in adult T cell leukemia/lymphoma. Blood. 2014;123(25):3925‐3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rowan AG, Dillon R, Witkover A, et al. Evolution of retrovirus‐infected premalignant T‐cell clones prior to adult T‐cell leukemia/lymphoma diagnosis. Blood. 2020;135:2023‐2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kyle RA, Therneau TM, Rajkumar SV, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354:1362‐1369. [DOI] [PubMed] [Google Scholar]
- 54. Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood‐cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477‐2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jaiswal S, Fontanillas P, Flannick J, et al. Age‐related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488‐2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shim YK, Rachel JM, Ghia P, et al. Monoclonal B‐cell lymphocytosis in healthy blood donors: an unexpectedly common finding. Blood. 2014;123:1319‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T‐cell leukaemia‐lymphoma. A report from the Lymphoma Study Group (1984–87). Br J Haematol. 1991;79:428‐437. [DOI] [PubMed] [Google Scholar]
- 58. Tsukasaki K, Hermine O, Bazarbachi A, et al. Definition, prognostic factors, treatment, and response criteria of adult T‐cell leukemia‐lymphoma: a proposal from an international consensus meeting. J Clin Oncol. 2009;27:453‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cook LB, Fuji S, Hermine O, et al. Revised adult T‐cell leukemia‐lymphoma international consensus meeting report. J Clin Oncol. 2019;37:677‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kataoka K, Iwanaga M, Yasunaga J‐I, et al. Prognostic relevance of integrated genetic profiling in adult T‐cell leukemia/lymphoma. Blood. 2018;131:215‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Takasaki Y, Iwanaga M, Imaizumi Y, et al. Long‐term study of indolent adult T‐cell leukemia‐lymphoma. Blood. 2010;115:4337‐4343. [DOI] [PubMed] [Google Scholar]
- 62. Katsuya H, Shimokawa M, Ishitsuka K, et al. Prognostic index for chronic‐ and smoldering‐type adult T‐cell leukemia‐lymphoma. Blood. 2017;130:39‐47. [DOI] [PubMed] [Google Scholar]
- 63. Umino A, Nakagawa M, Utsunomiya A, et al. Clonal evolution of adult T‐cell leukemia/lymphoma takes place in the lymph nodes. Blood. 2011;117:5473‐5478. [DOI] [PubMed] [Google Scholar]
- 64. Kawano N, Yoshida N, Kawano S, et al. Clinical features, pathological features, and treatment outcomes of 22 patients with aggressive adult T‐cell leukemia‐lymphoma treated with a humanized CCR4 antibody (Mogamulizumab) at a single institution during a 6‐year period (2012–2018). Intern Med. 2019;58:2159‐2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ishida T, Utsunomiya A, Iida S, et al. Clinical significance of CCR4 expression in adult T‐cell leukemia/lymphoma: its close association with skin involvement and unfavorable outcome. Clin Cancer Res. 2003;9:3625‐3634. [PubMed] [Google Scholar]
- 66. Phillips AA, Fields PA, Hermine O, et al. Mogamulizumab versus investigator's choice of chemotherapy regimen in relapsed/refractory adult T‐cell leukemia/lymphoma. Haematologica. 2019;104:993‐1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ishida T, Joh T, Uike N, et al. Defucosylated anti‐CCR4 monoclonal antibody (KW‐0761) for relapsed adult T‐cell leukemia‐lymphoma: a Multicenter Phase II Study. J Clin Oncol. 2012;30:837‐842. [DOI] [PubMed] [Google Scholar]
- 68. Yoshida N, Miyoshi H, Kato T, et al. CCR4 frameshift mutation identifies a distinct group of adult T‐cell leukemia/lymphoma with poor prognosis. J Pathol. 2016;238(5):621‐626. [DOI] [PubMed] [Google Scholar]
- 69. Sakamoto Y, Ishida T, Masaki A, et al. CCR4 mutations associated with superior outcome of adult T‐cell leukemia/lymphoma under mogamulizumab treatment. Blood. 2018;132:758‐761. [DOI] [PubMed] [Google Scholar]
- 70. Cook LBM, Demontis MA, Sagawe S, et al. Molecular remissions are observed in chronic adult T‐cell leukemia/lymphoma in patients treated with mogamulizumab. Haematologica. 2019;104:e566‐e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bazarbachi A, Plumelle Y, Carlos Ramos J, et al. Meta‐analysis on the use of zidovudine and interferon‐alfa in adult T‐cell leukemia/lymphoma showing improved survival in the leukemic subtypes. J Clin Oncol. 2010;28:4177‐4183. [DOI] [PubMed] [Google Scholar]
- 72. Heavican TB, Bouska A, Yu J, et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T‐cell lymphoma. Blood. 2019;133:1664‐1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Watatani Y, Sato Y, Miyoshi H, et al. Molecular heterogeneity in peripheral T‐cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia. 2019;33:2867‐2883. [DOI] [PubMed] [Google Scholar]
- 74. Sugio T, Miyawaki K, Kato K, et al. Microenvironmental immune cell signatures dictate clinical outcomes for PTCL‐NOS. Blood Adv. 2018;2:2242‐2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yoshida N, Nishikori M, Izumi T, et al. Primary peripheral T‐cell lymphoma, not otherwise specified of the thyroid with autoimmune thyroiditis. Br J Haematol. 2013;161:214‐223. [DOI] [PubMed] [Google Scholar]
- 76. Graff‐Baker A, Roman SA, Thomas DC, Udelsman R, Sosa JA. Prognosis of primary thyroid lymphoma: demographic, clinical, and pathologic predictors of survival in 1,408 cases. Surgery. 2009;146:1105‐1115. [DOI] [PubMed] [Google Scholar]
- 77. Yoshida N, Tsuzuki S, Karube K, et al. STX11 functions as a novel tumor suppressor gene in peripheral T‐cell lymphomas. Cancer Sci. 2015;106:1455‐1462. [DOI] [PMC free article] [PubMed] [Google Scholar]