

Abstract
Hypervirulent Klebsiella pneumoniae (hvKP) has been increasingly reported over the past three decades and causes severe infections. To increase our understanding of hvKP at the genome level, genome sequencing and comparative genome analysis were performed on 6 hvKPs. The whole genome DNA from 6 hvKPs with different capsular serotypes isolated in China was extracted. The genome sequencing and assembly results showed the genome size of the six hvKPs and GC content. Comparative analyses of the genomes revealed the gene homology and genome rearrangement in the 6 hvKPs compared with Klebsiella pneumonia NTUH-K2044. The phylogenetic tree based on full-genome SNPs of the 7 hvKPs showed that NTUH-K2044 formed a single clade, showing distant evolutionary distances with the other six strains, and the non-K1 hvKP strains had a relatively closer phylogenetic relationship. BLAST comparison analysis found that some selected virulence genes had different degrees of deletion in the non-K1 hvKPs. SNP-based virulence gene mutation analysis showed that some virulence genes had different degrees of SNP mutations. The whole-genome sequencing and comparative genome analysis of six hvKP strains with NTUH-K2044 provide us with a basic understanding of the genome composition, genetic polymorphism, evolution and virulence genes of hvKP and a basis for further research on these genes and the pathogenesis of hvKP.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00203-021-02263-0.
Keywords: Hypervirulent Klebsiella pneumoniae (hvKP), Whole genome sequencing, Comparative genome genomics, Virulence genes
Introduction
Klebsiella pneumoniae (K. pneumoniae) is a Gram-negative opportunistic pathogenic Enterobacteriaceae that is commonly the cause of nosocomial and community infections, such as septicemia, pneumonia, urinary tract infections, surgical site infections and catheter-related infections (Paczosa and Mecsas 2016).
Over the past three decades, hypervirulent Klebsiella pneumoniae (hvKP) has been increasingly reported worldwide, particularly in Asia (Hu et al. 2019; Lee et al. 2017; Siu et al. 2011; Zhang et al. 2016). In contrast to the classic K. pneumoniae strains, hvKP can cause serious community-acquired infections, and it has the potential for metastatic spread in healthy individuals, such as pyogenic liver abscesses, endophthalmitis and meningitis (Shon et al. 2013). hvKP is associated with a significant mortality rate, ranging from 3 to 42% (Shon et al. 2013). The capsule is an important virulence factor of K. pneumoniae, and there are at least 78 capsular polysaccharide serotypes existing to date (Pan et al. 2008). Eight capsular serotypes, K1, K2, K5, K16, K20, K54, K57, and KN1, are recognized as hypervirulent variants of K. pneumonia (Cheng et al. 2012; Pan et al. 2008, 2019). Multiple factors are required for the virulence of hvKP, such as capsular serotypes for the capsular polysaccharide, rmpA for extracapsular polysaccharide synthesis, kfu, ybtPQXS, ybtA-irp2-irp1-ybtUTE-fyuA (encoding yersiniabactin), iucABCDiutA (encoding aerobactin) for iron uptake and allS for allantoin-utilization (Li et al. 2014). There are some studies of the clinical and phenotypic characteristics and epidemiology of hvKP (Catalán-Nájera et al. 2017; Pomakova et al. 2012; Zhang et al. 2016), but data based on genetic backgrounds are limited. Genomics studies are promising for providing us with a deeper understanding of K. pneumoniae–host interactions from a genome-scale view (Li et al. 2014).
In this study, 6 hypervirulent Klebsiella pneumoniae strains isolated in mainland China were subjected to whole-genome sequencing to analyse the genetic characteristics of the strains and to increase our understanding of hypervirulent Klebsiella pneumoniae virulence. Comparative analyses of the genomes with the genome sequence of the hvKP strain NTUH-K2044 were performed to show the difference and evolutionary relationship between the sequencing samples and the reference sequence. This study includes structural variation (synteny), gene family, unique genes, single nucleotide polymorphisms (SNPs), small insertions and deletions (InDels) and phylogenetic analyses. In addition, the genomes were annotated and screened for virulence genes and further assessed for single-nucleotide polymorphism (SNP)-based differentiation of K. pneumoniae virulence genes.
Materials and methods
Bacterial strains, growth conditions and DNA extraction
The 6 Klebsiella pneumoniae strains used in this study are listed in Table 1. These strains belong to different capsular serotypes and were isolated from different specimens, mainly from patients in hospitals located in three geographical regions of China. The hypermucoviscosity (HV) phenotype was semi-quantitatively defined by a positive “string test”. The string test is positive when a bacteriology inoculation loop or needle is able to generate a viscous string > 5 mm in length by stretching bacterial colonies on an agar plate (Shon et al. 2013). Strains were grown at 37 °C in Luria–Bertani (LB) medium for at least 24 h. Genomic DNA of each strain was isolated using a traditional DNA extraction method(Xiao et al. 2011). Briefly, the bacteria culture was collected and washed using SE solution (0.15 M NaCl, 0.1 M EDTA, pH8.0). Then, SDS dry powder (final concentration 2%) was added to the tube with 65 °C water bath to lyse bacteria. Protein was removed from lysate solution with Tris-saturated phenol and chloroform. The nucleic acids were subjected to RNA digestion with 100ug/ml RNase. DNA was precipitated using absolute ethanol. Finally, the DNA was dissolved in TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH8.0). The quality and quantity of genomic DNA were evaluated by ultraviolet spectrophotometry and agarose gel electrophoresis.
Table 1.
6 hvKP strains used in this study
| Strain ID | Location | Specimen | Serotype | |
|---|---|---|---|---|
| K038 | Chongqing | Pyogenic fluid | K2 | |
| K090 | Chongqing | Sputum | K20 | |
| K095 | Beijing | Ascitic fluid | K57 | |
| K323 | Shenzhen | Ascitic fluid | K5 | |
| K396 | Shenzhen | Blood | K54 | |
| K406 | Beijing | Ascitic fluid | K1 |
Genome sequencing, assembly, and annotation
After DNA extraction, the genomes of the 6 hvKP strains were sequenced using an Illumina HiSeq 4000 sequencing platform (Illumina, San Diego, CA, USA) at the Beijing Genomics Institute (Shenzhen, China). Genomic DNA was sheared randomly to construct three read libraries with lengths of less than or equal to 800 bp by a Bioruptor ultrasonicator (Diagenode, Denville, NJ, USA) and physico-chemical methods. The paired-end fragment libraries were sequenced according to the Illumina HiSeq 4000 system protocol. Raw reads of low quality from paired-end sequencing (those with consecutive bases covered by fewer than five reads) were discarded. After filtering, the sequenced reads were assembled using SOAPdenovo software (Version 2.04) (Li et al. 2008). Gene prediction from the assembled results was performed using Glimmer software (Version: 3.02) (Delcher et al. 2007) with Hidden Markov models. The N50 statistic was calculated by sorting the contigs in decreasing order and taking the contig length for which all contigs of that length or longer contained at least 50% of the summed length of all contigs. Gene annotation was performed on the Rapid Annotation System Technology (RAST, http://rast.nmpdr.org/) server (Overbeek et al. 2014).
Comparative genomics and phylogenetic analysis
Comparative genomics analysis was performed using hypervirulent K. pneumoniae NTUH-K2044 (GenBank accession no. AP006725) as a reference. This reference strain, belonging to capsular serotype K1, was originally isolated from a patient with a liver abscess and meningitis and has high virulence and hypermucoviscosity (Wu et al. 2009). The six strains are identified as hypervirulent K. pneumomiae using the string test. NTUH-K2044 is a representative strain of K1 serotype and is also recognized as hypervirulent strain (Russo and Marr 2019; Shon et al. 2013). The complete genome sequence for NTUH-K2044 is available from the NCBI (http://www.ncbi.nlm.nih.gov) and DDBJ (http://www.ddbj.nig.ac.jp/index-e.html) public websites (Wu et al. 2009). Many studies on NTUH-K2044 have been carried out from various aspects, such as virulent factor (Hsieh et al. 2019), transcriptional regulation (Luo et al. 2017; Peng et al. 2018). We can get a comprehensive understanding to the 6 hvKP strains while comparing with NTUH-K2044. We used the progressive Mauve program in Mauve2.3.1 software (Darling et al. 2010) to compare the genomes of the seven strains to obtain a synteny relationship between the genomes. The program used the default parameters.
The gene family was constructed by the genes of NTUH-K2044 and 6 hvKPs, integrating multiple software as follows: protein sequence alignment in BLAST, redundancy elimination by solar and gene family-clustering treatment for the alignment results using Hcluster_sg software and Muscle software (version: 3.8.31) (Edgar 2004) for multiple sequence alignment of the clustered gene families. The protein alignment results were converted into amino acid multiple sequence alignments in the coding sequence (CDS) regions.
SNPs were detected based on the alignment between the assembly result and reference using the alignment software MUMmer (version: 3.22). Then, 100 bp sequences on both sides of the SNP were extracted from the reference sequence and aligned with the assembly results to verify SNP sites using BLAST. If the length of the aligned sequence was shorter than 101 bp, this SNP was considered improbable and was removed. If the extracted sequence could be aligned with the assembly results several times, this SNP was considered located in a repeat region and was also be removed. The credible SNP could be obtained by filtering SNPs located in repeat regions. The InDel results were preliminarily obtained with LASTZ software (version: 1.01.50) (Chiaromonte et al. 2002), and the reference sequence and query sequence were aligned to obtain the alignment results. The alignment results were verified with Burrows–Wheeler Aligner (BWA) (Li and Durbin 2009) and SAMtools (Li et al. 2009). Detected SNPs and InDels were annotated.
All SNPs were connected with the same order, and sequences with the same length were obtained as input files in fasta format. Then, the full-genome SNP-based phylogenetic tree was constructed by TreeBeST (Nandi et al. 2010) using the maximum-likelihood method, with 1000 bootstraps.
Comparative analysis of virulence genes
Approximate virulence-related genes in the genome of six K. pneumoniae strains were screened based on the core dataset in the VFDB (Virulence Factors of Pathogenic Bacteria, http://www.mgc.ac.cn/VFs/) database (Chen et al. 2016). At the same time, the virulence genes of K. pneumoniae were queried in the literature, by searching the NCBI nucleic acid database to identify the reference sequence of virulence genes. A local BLAST analysis was used to reveal the distribution of virulence-related genes in the genome sequence of six hvKP strains (the filtering parameters for comparison results were set to identify > 80, reference coverage > 0.5, evalue > 1e-5). Then, the SNPs of these virulence genes were found in the genome SNP file based on the location.
Results
General genome features
The genomes of 6 hvKPs were sequenced and de novo assembled. The genome size ranged from 5.34 to 5.58 Mb, represented by 41–73 contigs with a max. contig length ranging from 540,905 to 1,254,093 bp and a min. contig length ranging from 235 to 552 bp. The GC percentage of the K. pneumoniae strains sequenced in this study ranged from 57.22 to 57.46%. The genome sizes of the 6 hvKPs (K038, K090, K095, K323, K396, K406) were larger than that of the reference strain NTUH-K2044 (approximately 5.24 Mbp), and the GC% of the K. pneumoniae strains was nearly similar to that of the reference strain (57.68%). A total of 5061–5304 protein coding genes were predicted, with an average length of 912–915 bp. Coding region sizes ranged from 4.67 to 4.88 Mb. The overall features of the completely sequenced genomes of the 6 hvKP strains are shown in Table 2.
Table 2.
General features of the draft genomes of the 6 hypervirulent Klebsiella pneumoniae strains examined in this study
| Strain | K038 | K090 | K095 | K323 | K396 | K406 |
|---|---|---|---|---|---|---|
| Size (bp) | 5,445,525 | 5,479,618 | 5,342,398 | 5,448,453 | 5,513,846 | 5,580,814 |
| GC content of genome (%) | 57.27 | 57.32 | 57.32 | 57.46 | 57.22 | 57.26 |
| Contig number (> 500 bp) | 45 | 73 | 55 | 41 | 49 | 43 |
| Max/min contig length (bp) | 645,719/372 | 540,905/534 | 605,833/533 | 1,254,093/235 | 1,026,693/515 | 1,181,813/552 |
| Contig N50 (bp) | 356,027 | 293,449 | 267,799 | 369,888 | 325,095 | 307,460 |
| Scaffold number | 43 | 72 | 53 | 36 | 47 | 41 |
| Gene number | 5213 | 5207 | 5061 | 5191 | 5267 | 5304 |
| Coding regions size (bp) | 4,754,298 | 4,794,747 | 4,669,965 | 4,800,993 | 4,834,782 | 4,884,834 |
| Gene average length (bp) | 912 | 921 | 923 | 925 | 918 | 921 |
| Coding regions/genome length (%) | 87.31 | 87.50 | 87.41 | 88.12 | 87.68 | 87.53 |
| Intergenic region size (bp): | 691,227 | 684,871 | 672,433 | 647,460 | 679,064 | 695,980 |
| Ratio of intergenic region (%) | 12.69 | 12.50 | 12.59 | 11.88 | 12.32 | 12.47 |
Gene annotation
The assembled scaffolds for each strain were annotated by RAST; open reading frames (ORFs) were identified and subsequently classified in functional subsystems, which are the sets of proteins that perform related functional roles (Table 3). According to the annotation results, approximately 20% of the predicted CDSs encode hypothetical proteins. Approximately 86% of the CDSs, between 4352 and 4523 genes per strain, could be assigned to functional subsystems. The genes with the highest proportion for all seven strains were metabolic-related genes, such as carbohydrates, amino acids and derivatives, DNA and protein metabolism. There were also many genes related to virulence.
Table 3.
Annotation System Technology (RAST) server annotation functional subsystems classification of 6 hypervirulent Klebsiella pneumoniae and the Klebsiella pneumoniae reference strain NTUH-K2044
| Subsystem | Number of genes | ||||||
|---|---|---|---|---|---|---|---|
| K038 | K090 | K095 | K323 | K396 | K406 | NTUH-K2044 | |
| Cofactors, vitamins, prosthetic groups, pigments | 368 | 400 | 365 | 370 | 371 | 370 | 364 |
| Cell wall and capsule | 219 | 220 | 218 | 213 | 226 | 222 | 217 |
| Capsular and extracellular polysacchrides | 43 | 44 | 41 | 37 | 50 | 46 | 41 |
| Gram-negative cell wall components (90) | 90 | 90 | 91 | 90 | 90 | 90 | 90 |
| Cell wall and capsule—no subcategory | 86 | 86 | 86 | 86 | 86 | 86 | 86 |
| Virulence, disease and defence | 148 | 140 | 133 | 128 | 138 | 145 | 128 |
| Adhesion | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| Bacteriocins, ribosomally synthesized antibacterial peptides | 12 | 12 | 12 | 12 | 12 | 12 | 12 |
| Resistance to antibiotics and toxic compounds | 115 | 113 | 100 | 95 | 105 | 112 | 95 |
| Invasion and intracellular resistance | 14 | 8 | 14 | 14 | 14 | 14 | 14 |
| Potassium metabolism | 34 | 35 | 33 | 33 | 33 | 34 | 33 |
| Miscellaneous | 66 | 65 | 66 | 67 | 68 | 69 | 68 |
| Phages, prophages, transposable elements, plasmids | 14 | 53 | 32 | 22 | 18 | 29 | 4 |
| Membrane transport | 218 | 223 | 231 | 208 | 267 | 223 | 213 |
| Iron acquisition and metabolism | 105 | 113 | 72 | 75 | 65 | 109 | 76 |
| Siderophores | 52 | 60 | 24 | 23 | 17 | 54 | 24 |
| Iron acquisition and metabolism—no subcategory | 53 | 53 | 48 | 52 | 48 | 55 | 52 |
| RNA metabolism | 251 | 253 | 251 | 249 | 249 | 255 | 250 |
| Nucleosides and nucleotides | 138 | 138 | 138 | 141 | 142 | 142 | 140 |
| Protein metabolism | 320 | 288 | 312 | 304 | 304 | 315 | 306 |
| Cell division and cell cycle | 43 | 40 | 42 | 41 | 42 | 43 | 38 |
| Motility and chemotaxis | 9 | 9 | 9 | 10 | 11 | 11 | 11 |
| Regulation and cell signaling | 180 | 184 | 176 | 171 | 167 | 179 | 164 |
| Secondary metabolism | 5 | 5 | 5 | 5 | 17 | 5 | 5 |
| DNA metabolism | 130 | 118 | 121 | 132 | 138 | 132 | 131 |
| Fatty acids, lipids, and isoprenoids | 140 | 144 | 139 | 139 | 134 | 145 | 138 |
| Nitrogen metabolism | 49 | 47 | 47 | 46 | 47 | 63 | 62 |
| Dormancy and sporulation | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Respiration | 180 | 183 | 182 | 191 | 187 | 184 | 180 |
| Stress response | 180 | 181 | 180 | 182 | 183 | 183 | 180 |
| Metabolism of aromatic compounds | 79 | 79 | 79 | 82 | 78 | 85 | 85 |
| Amino acids and derivatives | 572 | 550 | 548 | 544 | 545 | 546 | 536 |
| Sulfur metabolism | 79 | 80 | 79 | 80 | 79 | 82 | 81 |
| Phosphorus metabolism | 69 | 67 | 67 | 68 | 69 | 69 | 67 |
| Carbohydrates | 863 | 896 | 849 | 846 | 875 | 878 | 872 |
| Total | 4464 | 4516 | 4379 | 4352 | 4458 | 4523 | 4354 |
Genomic synteny analysis
In this experiment, the synteny between NTUH-K2044 and the whole-genome alignments of the sample strains were conducted using Mauve software. The results are shown in Fig. 1. Because the 6 hvKP genome sequences were not complete, their contig sequences were used for the alignment; therefore, the synteny alignment of the genome was more cluttered, but overall, the genomic synteny of the seven strains was relatively high. There were many local collinear blocks (LCBs) between the genomes. In addition, there were insertions and deletions between the strain genomes and many genome rearrangement events, such as translocations and inversions, as shown in the reference strain NTUH-K2044 and six hvKPs.
Fig. 1.

Global alignment of 7 hypervirulent Klebsiella pneumoniae genomes. The same colour modules connected by the lines represent the collinear portions between the genomes, and there was no genome rearrangement inside. Areas outside the same colour region indicate that no homology was detected between the input genomes. The completely white area inside the fragment indicates that there was no alignment between the genomes, which may contain specific components or mutations
Gene family and phylogenetic analysis
In this study, the gene families of six strains and NTUH-K2044 were analysed and compared, and the results are shown in Table 4. The K038, K090, K095, K323, K396 and NTUH-K2044 strains had 2793, 2832, 2748, 2831, 2801, 2904 and 2899 gene families, each consisting of 3954, 4038, 3888, 3956, 3949, 4083 and 4397 genes, respectively. A total of 2252 gene families were shared by the seven strains, and 2, 2, 5, 7, 4, 1, and 12 gene families were unique to K038, K090, K095, K323, K396 and NTUH-K2044, respectively. The genes contained in the gene families unique to the seven strains of bacteria were annotated according to the Cluster of Orthologous Groups (COG) functional annotation. The detailed annotation information of the unique family and genes contained therein is shown in Table S1. Compared to the six samples, NTUH-K2044 had more unique genes, which were distributed to various functional categories. K090 had two unique genes grouped into the category “replication, recombination and repair; mobilome: prophages, transposons”. The unique genes of K095 were distributed in two categories, lipid transport and metabolism and signal transduction mechanisms. K323 had two unique genes categorized as cell cycle control, cell division, and chromosome partitioning, and K396 had two unique genes classified into two categories, general functional prediction and defence mechanisms.
Table 4.
Gene family statistics
| Sample ID | Gene number | Clustered gene | UnClustered gene | Family num | Unique family |
|---|---|---|---|---|---|
| K038 | 5213 | 3954 | 1259 | 2793 | 2 |
| K090 | 5207 | 4038 | 1169 | 2832 | 2 |
| K095 | 5061 | 3888 | 1173 | 2748 | 5 |
| K323 | 5191 | 3956 | 1235 | 2831 | 7 |
| K396 | 5267 | 3949 | 1318 | 2801 | 4 |
| K406 | 5304 | 4083 | 1221 | 2904 | 1 |
| NTUH-K2044 | 5129 | 4397 | 732 | 2899 | 12 |
Gene number gene number of each strain Clustered gene genes that can be clustered into a gene family, UnClustered Gene genes not clustered into any gene family, Family Num number of gene family, Unique family number of unique gene family
The results of the statistical analysis of homologous genes in the seven strains are shown in Fig. 2. Gene families that were shared among the seven strains had similar single copy homologous genes, and NTUH-K2044 had more multicopy homologous genes.
Fig. 2.
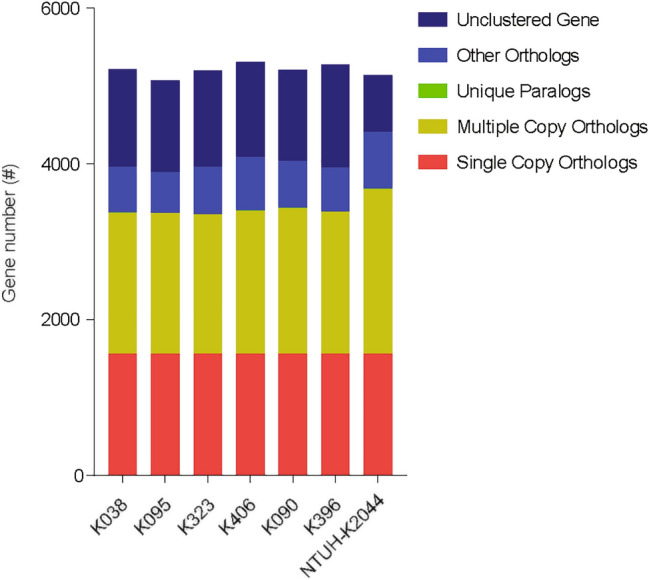
Statistical graph of homologous gene number. Single-copy orthologs, single copy homologous genes in the gene families shared among species; multiple-copy orthologs, multicopy homologous genes in gene families shared among species; unique paralogs, genes of the strain unique to the family; other orthologs, all other genes; unclustered genes, genes not clustered into any family
SNPs and InDels analysis
The analysis of SNPs and InDels was performed by mapping all individual reads onto the Klebsiella pneumoniae NTUH-K2044 reference genome. We found very abundant SNP and InDel polymorphisms in 6 the hvKPs compared with NTUH-K2044 (see Table S2, S3, S7 in the supplemental material). The results showed that except for K406, the other five non-K1 strains displayed between 27,574 and 29,308 nucleotide variations, including SNPs and InDels, compared with NTUH-K2044. In each of the five strains, approximately 88% of SNPs occurred in the CDS region; approximately 71% of these were synonymous mutations; whereas, the remaining 17% were non-synonymous mutations. The K1 serotype strain K406 had a high similarity to strain NTUH-K2044, with only 364 nucleotide variations. Approximately 83% of SNPs occurred in the CDS region, and 51% of these were non-synonymous mutations. Most of the InDel mutations of the six strains were located in the middle of the CDS. For the mutation types caused by InDels, except for mutations caused by other changes, there was no significant influence on the open reading frame with InDels, and most mutation types were frame shift mutations. Meaningful mutation sites, such as SNP mutations that caused non-synonymous mutations and InDel mutations causing frameshift mutations on the CDS region, may cause a change of the traits of an organism and will provide a basis for our understanding of the differences in virulence among the hvKPs.
Phylogenetic analysis
A phylogenetic tree was constructed, and the SNP analysis results are shown in Fig. 3. The results showed that NTUH-K2044 formed a single clade and showed a distant evolutionary distance from the other 6 strains and that the five non-K1 hvKP strains had a relatively closer phylogenetic relationship.
Fig. 3.

Phylogenetic tree based on the full-genome SNPs of the 7 K. pneumoniae strains using maximum-likelihood analysis
Virulence gene detection and SNP analysis
Local BLAST comparison was performed between the virulence genes and the whole-genome sequence of each strain, and a SNP-based differentiation analysis of the virulence genes was conducted. Statistics of the BLAST result and virulence genes SNP analysis are shown in Table S4. The detailed information of the BLAST result is shown in Table S5, and the detailed information of the virulence gene SNP analysis is shown in Table S6.
In this study, the virulence gene BLAST analysis with the full-genome sequence of the six strains revealed that K406, the K1 strain, carried all the selected virulence genes; however, some virulence genes, which are mainly related to iron uptake and allantoin metabolism, had different degrees of deletion in the other five different capsular serotypes strains. In addition, we found some non-synonymous mutations in the virulence genes in the SNP analysis results. For example, compared with the reference strain, the five non-K1 strains K038(K2), K090(K20), K095(K57), K323(K5), K396(K54) showed a single nucleotide non-synonymous substitution in mrkD, a gene encoding the type 3 fimbriae adhesion protein related to biofilm formation (Jagnow and Clegg 2003). This nucleotide substitution resulted in an amino acid change from glutamic acid in strain NTUH-K2044 to glutamine in strains K090, K095, and K396 and from threonine in strain NTUH-K2044 to serine in strain K323.
Discussion
Compared with classic Klebsiella pneumoniae strains, hvKP has unique phenotypic and genotypic characteristics, such as hypermucoviscosity colonies, unique serotypes, and virulence genes closely related to high pathogenicity (Shon et al. 2013). More studies on K1 and K2 serotypes are needed since they are common clinical serotypes among the hvKP-related capsular serotypes (K1, K2, K5, K16, K20, K54, K57 and KN1) (Liu et al. 2014). To understand the characteristics of hvKP at the whole-genome level more comprehensively, the 6 hvKPs belonging to the K1, K2, K5, K20, K54, and K57 serotypes were screened from 310 K. pneumoniae clinical isolates, which were isolated and identified from Chongqing, Shenzhen and Beijing. The genomes of the 6 hvKP strains were sequenced and used in a comparative genome analysis.
The NTUH-K2044 genome was selected as a reference sequence for comparative genomics because NTUH-K2044 is a clinical isolate of type K1 hvKP, and the genome sequencing of NTUH-K2044 was completed. However, the phylogenetic analysis shows that the 6 hvKPs and NTUH-K2044 are in two phylogenetic branches. This may be due to the different areas of separation. NTUH-K2044 was isolated in Taiwan, and the 6 hvKPs in this experiment were isolated from mainland Chinese. Due to the lack of communication between these two areas for many years, the hvKP of Taiwan and the mainland evolved in different directions. Of course, the number of hvKP strains used to construct the phylogenetic tree in this study was small, and follow-up work can analyse the phylogenetic relationship with additional hvKP genome sequence based on hvKP genome sequences in NCBI. Furthermore, the isolation background of the strains can be combined to clarify the phylogenetic relationship of hvKP.
It has been reported that a few or even a single-nucleotide mutation may influence the virulence of bacteria. SNPs in porA were found to be responsible for the hypervirulence in Campylobacter jejuni by using a “directed genome evolution” strategy, which was developed to identify SNPs (Wu et al. 2016). Astrid’s work found that a SNP upstream of the orf2 promoter could change orf2 expression and then affect the virulence of Streptococcus suis (de Greeff et al. 2014). Bailey’s study also suggested that SNPs identified in four regulatory genes were associated with enhancing virulence in Vibrio cholerae clinical isolates (Carignan et al. 2016). In this study, through the comparison of sequence loci, we identified meaningful mutation sites and then performed a functional verification of mutant loci at the cell and animal level, which can help analyse the pathogenicity of bacteria. To assess the genetic diversity of the 6 isolates, the genome sequences were subjected to a variation analysis with NTUH-K2044. SNP/InDel analysis revealed a large number of variations between the six strains and NTUH-K2044; this may be because these strains were isolated from different geographical origins, and there was a lower homology between the bacteria and the reference. SNP and InDel analysis showed that the K1 strain K406 had fewer SNPs and InDels compared with the other five non-K1 strain, possibly because it has the same capsular serotypes as NTUH-K2044.
Many studies of the virulence genes (rmpA, magA, kfu, alls and siderophore) related to hvKP pathogenicity have been performed. In this study, we found that the distribution of these virulence genes in the genome of 6 serotypes strains was different. The kfu gene encoding iron transport system was only detected in K406 and K323, suggesting the difference of iron uptake capacity in 6 hvKP strains. The rmpA gene is responsible for the hypermucoviscous phenotype, and we found rmpA present in five strains (K038, K090, K095, K323, K406), but not in K54 strain (K396). Although rmpA is present in the vast majority of isolates with a hypermucoviscosity (HV) phenotype, a small proportion of HV isolates do not possess this gene (Lee et al. 2010). The hypermucoviscous phenotype was due to the overproduction of capsular polysaccharides and may be regulated by many factors (Hsu et al. 2011).
The hvKP strain can cause health care-associated and community-acquired infections. Vaccine development could be a method to prevent these infections. Whole-genome analysis could be used to predict and screen out potential antigenic sites to accelerate vaccine development (de Alwis et al. 2021). Meanwhile, the complete genome sequence analysis of many pathogenic microbes could provide information for potential drug targets (Gomez-Simmonds and Uhlemann 2017; Wyres et al. 2020).
Conclusion
In summary, the differences in genomic structure and functional genes of 7 hvKP strains were revealed through comparative genomic analysis, which provided a molecular basis for further study of the genetic evolution, physiological metabolism and pathogenic mechanism of hvKP. It may also provide a useful genetic basis for effective vaccine development and therapy approaches to prevent and control hvKP infection.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31071093, 31170129 and 31200064).
Declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval
The study was approved by the Ethics Committee of the Chongqing Medical University Approval. And patients provided written informed consent for publication.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ling Du and Jiaxue Zhang contributed equally to this work and should be considered co-first author.
Contributor Information
Yingli Li, Email: 1150563687@qq.com.
Jingfu Qiu, Email: jfqiu@126.com.
References
- Carignan BM, Brumfield KD, Son MS. Single nucleotide polymorphisms in regulator-encoding genes have an additive effect on virulence gene expression in a Vibrio cholerae clinical isolate. mSphere. 2016;1:e00253–e316. doi: 10.1128/mSphere.00253-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalán-Nájera JC, Garza-Ramos U, Barrios-Camacho H. Hypervirulence and hypermucoviscosity: two different but complementary Klebsiella spp. phenotypes? Virulence. 2017;8:1111–1123. doi: 10.1080/21505594.2017.1317412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zheng D, Liu B, Yang J, Jin Q. VFDB 2016: hierarchical and refined dataset for big data analysis–10 years on. Nucleic Acids Res. 2016;44:D694–697. doi: 10.1093/nar/gkv1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng NC, Yu YC, Tai HC, et al. Recent trend of necrotizing fasciitis in Taiwan: focus on monomicrobial Klebsiella pneumoniae necrotizing fasciitis. Clin Infect Dis. 2012;55:930–939. doi: 10.1093/cid/cis565. [DOI] [PubMed] [Google Scholar]
- Chiaromonte F, Yap VB, Miller W. Scoring pairwise genomic sequence alignments. Pac Symp Biocomput. 2002 doi: 10.1142/9789812799623_0012. [DOI] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alwis R, Liang L, Taghavian O, et al. The identification of novel immunogenic antigens as potential Shigella vaccine components. Genome Med. 2021;13:8. doi: 10.1186/s13073-020-00824-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Greeff A, Buys H, Wells JM, Smith HE. A naturally occurring nucleotide polymorphism in the orf2/folc promoter is associated with Streptococcus suis virulence. BMC Microbiol. 2014;14:264. doi: 10.1186/s12866-014-0264-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Simmonds A, Uhlemann AC. Clinical implications of genomic adaptation and evolution of carbapenem-resistant Klebsiella pneumoniae. J Infect Dis. 2017;215:S18–s27. doi: 10.1093/infdis/jiw378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh PF, Lu YR, Lin TL, Lai LY, Wang JT. Klebsiella pneumoniae Type VI secretion system contributes to bacterial competition, cell invasion, type-1 fimbriae expression, and in vivo colonization. J Infect Dis. 2019;219:637–647. doi: 10.1093/infdis/jiy534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CR, Lin TL, Chen YC, Chou HC, Wang JT. The role of Klebsiella pneumoniae rmpA in capsular polysaccharide synthesis and virulence revisited. Microbiology (Reading) 2011;157:3446–3457. doi: 10.1099/mic.0.050336-0. [DOI] [PubMed] [Google Scholar]
- Hu F, Guo Y, Yang Y, Zheng Y, Wu S, Jiang X, Zhu D. Resistance reported from China antimicrobial surveillance network (CHINET) in 2018. Eur J Clin Microbiol Infect Dis. 2019;38:2275–2281. doi: 10.1007/s10096-019-03673-1. [DOI] [PubMed] [Google Scholar]
- Jagnow J, Clegg S. Klebsiella pneumoniae MrkD-mediated biofilm formation on extracellular matrix- and collagen-coated surfaces. Microbiology (Reading) 2003;149:2397–2405. doi: 10.1099/mic.0.26434-0. [DOI] [PubMed] [Google Scholar]
- Lee CH, Liu JW, Su LH, Chien CC, Li CC, Yang KD. Hypermucoviscosity associated with Klebsiella pneumoniae-mediated invasive syndrome: a prospective cross-sectional study in Taiwan. Int J Infect Dis. 2010;14:e688–692. doi: 10.1016/j.ijid.2010.01.007. [DOI] [PubMed] [Google Scholar]
- Lee CR, Lee JH, Park KS, et al. Antimicrobial resistance of Hypervirulent Klebsiella pneumoniae: epidemiology, hypervirulence-associated determinants, and resistance mechanisms. Front Cell Infect Microbiol. 2017;7:483. doi: 10.3389/fcimb.2017.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Zhao Y, Liu C, Chen Z, Zhou D. Molecular pathogenesis of Klebsiella pneumoniae. Future Microbiol. 2014;9:1071–1081. doi: 10.2217/fmb.14.48. [DOI] [PubMed] [Google Scholar]
- Liu YM, Li BB, Zhang YY, Zhang W, Shen H, Li H, Cao B. Clinical and molecular characteristics of emerging hypervirulent Klebsiella pneumoniae bloodstream infections in mainland China. Antimicrob Agents Chemother. 2014;58:5379–5385. doi: 10.1128/AAC.02523-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M, Yang S, Li X, et al. The KP1_4563 gene is regulated by the cAMP receptor protein and controls type 3 fimbrial function in Klebsiella pneumoniae NTUH-K2044. PLoS ONE. 2017;12:e0180666. doi: 10.1371/journal.pone.0180666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi T, Ong C, Singh AP, et al. A genomic survey of positive selection in Burkholderia pseudomallei provides insights into the evolution of accidental virulence. PLoS Pathog. 2010;6:e1000845. doi: 10.1371/journal.ppat.1000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Olson R, Pusch GD, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST) Nucleic Acids Res. 2014;42:D206–214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paczosa MK, Mecsas J. Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol Mol Biol Rev. 2016;80:629–661. doi: 10.1128/MMBR.00078-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YJ, Fang HC, Yang HC, et al. Capsular polysaccharide synthesis regions in Klebsiella pneumoniae serotype K57 and a new capsular serotype. J Clin Microbiol. 2008;46:2231–2240. doi: 10.1128/JCM.01716-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YJ, Lin TL, Chen YY, et al. Identification of three podoviruses infecting Klebsiella encoding capsule depolymerases that digest specific capsular types. Microb Biotechnol. 2019;12(3):472–486. doi: 10.1111/1751-7915.13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D, Li X, Liu P, et al. Transcriptional regulation of galF by RcsAB affects capsular polysaccharide formation in Klebsiella pneumoniae NTUH-K2044. Microbiol Res. 2018;216:70–78. doi: 10.1016/j.micres.2018.08.010. [DOI] [PubMed] [Google Scholar]
- Pomakova DK, Hsiao C-B, Beanan JM, Olson R, MacDonald U, Keynan Y, Russo TA. Clinical and phenotypic differences between classic and hypervirulent Klebsiella pneumonia: an emerging and under-recognized pathogenic variant. Eur J Clin Microbiol Infect Dis. 2012;31(6):981–989. doi: 10.1007/s10096-011-1396-6. [DOI] [PubMed] [Google Scholar]
- Russo TA, Marr CM. Hypervirulent Klebsiella pneumoniae. Clin Microbiol Rev. 2019;32:e00001–19. doi: 10.1128/CMR.00001-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shon AS, Bajwa RP, Russo TA. Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed. Virulence. 2013;4:107–118. doi: 10.4161/viru.22718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu LK, Fung CP, Chang FY, Lee N, Yeh KM, Koh TH, Ip M. Molecular typing and virulence analysis of serotype K1 Klebsiella pneumoniae strains isolated from liver abscess patients and stool samples from noninfectious subjects in Hong Kong, Singapore, and Taiwan. J Clin Microbiol. 2011;49:3761–3765. doi: 10.1128/JCM.00977-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KM, Li LH, Yan JJ, et al. Genome sequencing and comparative analysis of Klebsiella pneumoniae NTUH-K2044, a strain causing liver abscess and meningitis. J Bacteriol. 2009;191:4492–4501. doi: 10.1128/JB.00315-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Periaswamy B, Sahin O, et al. Point mutations in the major outer membrane protein drive hypervirulence of a rapidly expanding clone of Campylobacter jejuni. Proc Natl Acad Sci U S A. 2016;113:10690–10695. doi: 10.1073/pnas.1605869113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyres KL, Lam MMC, Holt KE. Population genomics of Klebsiella pneumoniae. Nat Rev Microbiol. 2020;18:344–359. doi: 10.1038/s41579-019-0315-1. [DOI] [PubMed] [Google Scholar]
- Xiao X, Zhang JL, Zhang QY, et al. Two methods for extraction of high-purity genomic DNA from mucoid Gram-negative bacteria. Afr J Microbiol Res. 2011;5:4013–4018. doi: 10.5897/AJMR11.785. [DOI] [Google Scholar]
- Zhang Y, Zhao C, Wang Q, et al. High prevalence of Hypervirulent Klebsiella pneumoniae Infection in China: geographic distribution, clinical characteristics, and antimicrobial resistance. Antimicrob Agents Chemother. 2016;60:6115–6120. doi: 10.1128/AAC.01127-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.