

Abstract
In a substantial fraction of cancers TERT promoter (TERTp) mutations drive expression of the catalytic subunit of telomerase, contributing to their proliferative immortality. We conducted a pan-cancer analysis of cell lines and find a TERTp mutation expression signature dominated by epithelial-to-mesenchymal transition (EMT) and MAPK signaling. These data indicate that TERTp mutants are likely to generate distinctive tumor microenvironments and intercellular interactions. Analysis of high throughput screening tests of 546 small molecules on cell line growth indicated that TERTp mutants displayed heightened sensitivity to specific drugs, including RAS pathway inhibitors, and we found that inhibition of MEK1 and 2, key RAS/MAPK-pathway effectors, inhibited TERT mRNA expression. Consistent with an enrichment of mesenchymal states in TERTp mutants, cell lines and some patient tumors displayed low expression of the central adherens junction protein E-cadherin, and we provide evidence that its expression in these cells is regulated by MEK1/2. Several mesenchymal transcription factors displayed elevated expression in TERTp mutants including ZEB1 and 2, TWIST1 and 2 and SNAI1. Of note, the developmental transcription factor SNAI2/SLUG was conspicuously elevated in a significant majority of TERTp mutant cell lines, and knockdown experiments suggest that it promotes TERT expression.
Introduction
Stratification of cancer patient tumors according to genetic alterations in a tissue-agnostic manner is emerging as a valuable tool for directing therapies (1). Many cancers harbor heterozygous C>T transitions in the proximal promoter for telomerase reverse transcriptase (TERT) (2–4). The human TERT gene encodes the catalytic subunit of telomerase (5) which maintains telomere length in stem cells and most cancer cells (6–9). TERT promoter (TERTp) mutations drive allele-specific expression (10,11) and are especially prevalent in glioblastomas, melanomas, myxoid liposarcomas, liver cancers, thyroid cancers, and bladder cancers (4,12,13). For unknown reasons TERTp mutations frequently associate with poorer patient survival (14–17).
Mutant TERT promoters display distinct regulatory features (18,19) but the mechanistic details regulating transcription from mutant TERT promoters are incompletely understood. The mutation creates a de novo consensus binding sequence for E-twenty-six (ETS) transcription factors, a large family with more than 26 members. In glioblastoma, liver cancer, and bladder cancer cell lines, the housekeeping ETS transcription factors GABPα & GABPβ1 are recruited to the mutant TERT sequence (11,18). In glioblastoma, ETS1, and in thyroid cancer, ETV5, appear to play roles in these specific cancers (20,21). These specific ETS factors have not been found at wild-type TERT promoters (11,18), and published data suggest that such promoters generally may be reliant on a different subset of factors (13,22,23). Importantly, the specific isoform GABPβ1L was shown in mice with TERTp mutant xenografts of glioblastoma to regulate survival in a TERT-dependent manner (24) demonstrating that these factors drive aggressiveness in this established model of brain cancer. In addition to ETS factors, initial studies indicate that some TERTp mutant cancer types rely on RAS pathway signaling to maintain TERT mRNA expression (3,21,25–28), but the generality of this observation has not been established.
Our current study was motivated to understand (1) the high frequency of TERT promoter mutations in certain cancer types and their complete absence in other cancer types (4,13,22), and (2) the poorer patient outcomes that often associate with these mutations. It has been observed that TERTp mutations are common in cancers that arise from more slowly proliferating cell types and may provide a proliferative advantage (29). To more fully explore the underlying biological associations with TERTp mutations across cancer types, we employed a pan-cancer analysis including hundreds of tumor-derived cell lines from 19 tissue types. We hypothesized that TERTp mutations may provide a selective advantage in, or be necessitated by, specific cellular programs. We considered that such programs, if functionally linked with TERTp mutations, could be revealed by analysis of gene and protein expression profiles across multiple cancer types. Our expectation from these studies was to determine whether common cellular mechanisms operate in TERTp mutants that distinguish them from cells that lack these mutations, both biologically and clinically.
Methods
Gene Set Enrichment Analysis (GSEA) and Matching Gene Sets to Mutation Status Phenotypes
Our approach incorporated three interrelated types of analysis: i) using gene set enrichment analysis to analyze the differences between TERTp mutant vs. wild type cell lines, ii) generation of transcriptional signatures of TERTp mutations, including a TERT signature, and comparing these signatures vs. an independent signature of TERT mRNA activation, and iii) experimental validation of several findings focusing on specific EMT gene markers.
Onco-GPS Analysis (Figs 2 and S1)
Figure 2. TERTp mutations and the Onco-GPS map of cellular states.

(A) The EMT hallmark gene set delineates the EMT states of TERTp mutants on the Cancer Cell Line Encyclopedia (CCLE) map. (B) Map showing the location of the most frequent TERTp mutations; the black circles are TERTp mutant while the grey circles are wild-type cell lines (wt), and the very faint circles are unclassified CCLE lines. (C) Transcriptional signature of TERTp mutations used to define their expression profile; (D) Transcriptional signature of TERT overexpression. A, C, D: circles represent CCLE lines, color coded to how well they match the indicated signature (red, best match). pg, promoter genotype.
The map of cellular states (Onco-GPS map) displayed in Figs 2 and S1 was obtained following the Onco-GPS methodology (30). The Onco-GPS is a PanCancer map very similar to the one shown in Fig S1 of our previous study (30). The samples on the map are color coded to indicate the mutation status of TERT (Fig 2B) or the single-sample GSEA enrichment score of TERT transcriptional signatures (Fig 2C–D, Fig S1B-C) or Hallmark gene sets (Fig 2A, Fig S1A).
CCLE Gene Expression Analysis
RNAseq, Affymetrix, and reverse phase protein array data were analyzed from the Cancer Cell Line Encyclopedia and TERT promoter mutation genotype calls were annotated as previously described (10,31). For Fig 3 503 lines and for Fig S3 278 lines were analyzed for which the TERT promoter mutation status and RNAseq data were determined. For GABPβ1 isoform analysis, transcript level expression (02-Jan-2019 version) were downloaded from the CCLE data portal (https://portals.broadinstitute.org/ccle/data). Comparisons were performed using a Wilcoxon rank sum test.
Fig 3. TERTp mutant cell lines are enriched for mesenchymal drivers and markers.
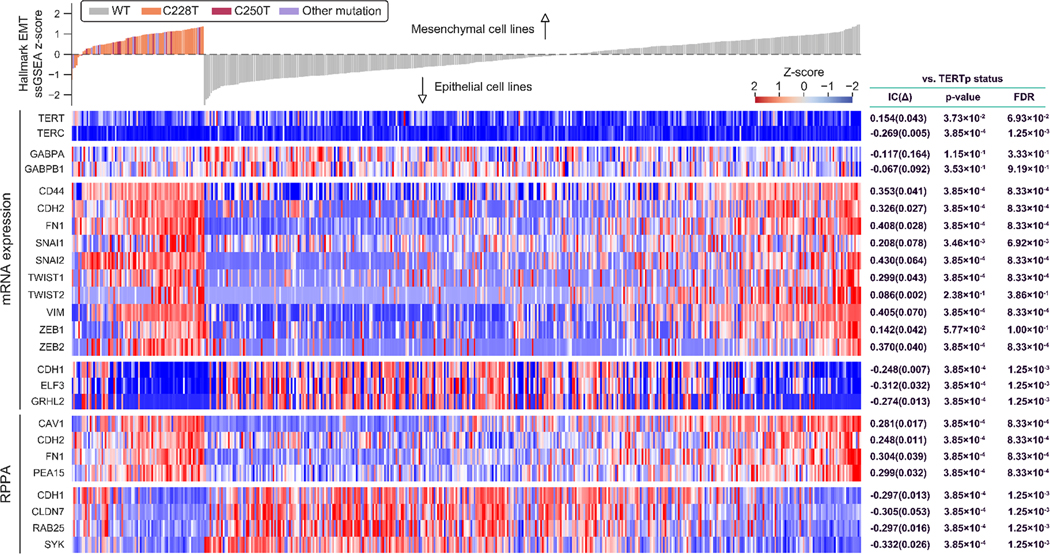
Gene expression (mRNA) and protein levels (RPPA) for selected genes illustrate the enrichment in TERTp mutants for key drivers and markers of the mesenchymal traits. In the displayed data, individual cell lines are represented by vertical bars. RPPA, reverse phase protein array; IC, information coefficient. FDR, false discovery rate. Box at top, WT = wild-type; C228T are C250T are, respectively, TERT promoter mutations at −124 and −146 base pairs from the TERT ATG. Expression levels are displayed as Z-scores (high expression, red; low expression, blue) which are normalized to a zero value against all CCLE lines. At the top, cell lines are classified as mesenchymal or epithelial based on their full expression profile compared with Hallmark EMT signature in ssGSEA (35).
CTRP analysis
Data for cell line drug response (EC50) were downloaded from the Cancer Therapeutics Response Portal (CTRP v2: ftp://caftpd.nci.nih.gov/pub/OCG-DCC/CTD2/Broad/CTRPv2.0_2015_ctd2_ExpandedDataset/). Cell lines were stratified into TERT promoter mutant and wild type. Cell lines with missing EC50 values were excluded from comparisons on a drug-by-drug basis. Comparisons were performed using a Wilcoxon rank sum test.
Cell culture and lentiviral infection
SNU-423, SNU-475, SNU-398, HEK293T, and DAOY were obtained from the American Type Culture Collection. U87MG, SCaBER, and MDA-MB-231 were obtained from the University of Colorado, Anschutz, Tissue Culture Shared Resource. HaCaT cells were a gift from X. Liu. Mel3249, Mel3616, and Mel1692 were gifts from K. Couts at the University of Colorado, Anschutz Medical Campus (32,33). Cancer cell lines and HaCaT cells were cultured in DMEM (VWR Scientific) with 2 mM GlutaminePlus (Atlanta Biologicals), 10% FBS (Thermo Fisher), 2 mM GlutaMAX-I (Gibco), 100 units/ml penicillin and 100 mg/ml streptomycin (Gibco) and 1 mM sodium pyruvate (Gibco). Cell lines were tested for mycoplasma contamination by the supplier or by PCR (MD Biosciences) at the University of Colorado Tissue Culture Facility and were used for experiments within 10 weeks of resuscitation. Cell lines from ATCC and the University of Colorado Tissue Culture Resource were authenticated by STR analysis by the supplier or at the Heflin Center for Genomic Sciences at University of Alabama. HEK293T cells were grown to 60% confluency in a 10 cm plate and transfected with 6 μg of siSNAI2#3-shRNA-pLKO.1 plasmid (Addgene #10905(34)), 3 μg of pDelta-8.9 plasmid containing gag, pol and rev genes, and 0.6 μg of a plasmid encoding VsVg (Functional Genomics Facility, University of Colorado, Anschutz Medical Campus). Cells were transfected with Lipofectamine 2000 Transfection Reagent (Life Technologies/Invitrogen 11668–019). Media was changed after 12 hours, followed by incubation for 48 hours in 20 mL. Media was harvested and filtered with 0.2 μm filters and 8 mLs were added to a 10 cm plate of cell lines for 24 hours, followed by fresh media for 48 hours at which point the cells were selected with puromycin until colonies emerged. Puromycin concentrations per mL used were 1 μg for DAOY, 1.2 μg for U87, 0.8 μg for MEL3616 and MEL3429 and 1.5 μg for MDA-MB-231.
RNA Extraction and cDNA Preparation
Following RNA extraction with Trizol (Life Technologies), reverse transcription was performed by treating 10 μg of RNA with 5 units of RQ1 DNase (Promega) according to the manufacturer’s protocol, followed by phenol extraction (pH 6.7, Amresco #0883), then chloroform: isoamyl alcohol extraction (VWR #X205), and then 70% ethanol precipitation. The cDNA was then generated from 2 μg of RNA synthesized using random hexamers, oligo (dT) 20-mer, and SuperScript III (Life Technologies). Following treatment with RNase H (New England Biolabs) quantitative PCR was performed with either SybrSelect (Thermo Fisher) or iQ SYBR Green (Bio-Rad) PCR mix using a Roche LightCycler 480 with the program 10 min at 98°C, 30 sec at 95°C, 30 sec at 60°C, 30 sec at 72°C, and 5 min at 72°C, followed by quantification using the Roche LightCycler 480 software. Melt curve analyses were examined to ensure the uniformity of relevant PCR amplicons and all PCR amplicons were sequenced at least once to confirm the product identity. Primers for TERT cDNA exon 2 were forward 5’-CGTGGTTTCTGTGTGGTGTC-3’, reverse 5’-CCTTGTCGCCTGAGGAGTAG-3’; and TERT cDNA exon 14 were those previously described (17). Primers for FOS cDNA were forward 5’-AGAATCCGAAGGGAAAGGAA-3’, reverse 5’-CTTCTCCTTCAGCAGGTTGG-3’; for GABPβ1L cDNA (24) were forward 5’-ATTGAAAACCGGGTGGAATC-3’, reverse 5’-CTGTAGGCCTCTGCTTCCTG-3’; for CDH1 cDNA were reverse 5’-GAACGCATTGCCACATACAC-3’, reverse 5’-ATTCGGGCTTGTTGTCATTC-3’; for GAPDH cDNA were forward 5’-CTGCACCACCAACTGCTTAG-3’, reverse 5’-GTCTTCTGGGTGGCAGTGAT-3’.
Cell lysis and immunoblots
After removing media from cells in a 6-well plate, 1.5 mL of ice-cold PBS was added and the cells were scraped into a 1.7 mL tube. Cells were collected by centrifugation at 500 x g for 4 min, PBS was removed and cells placed on ice for lysis, or snap-frozen in LN2 and stored at −80°C. Cell pellets were lysed in 10 mM Tris-Cl (pH 8.0), 150mM sodium chloride, 1% Triton X-100, 1 mM EDTA at 750,000 cells per 15 μL. Lysis buffer contained per 100 μL, 3 μL of Complete Protease Inhibitor (Sigma #P8340) and 3 μL benzonase (SIGMA-Aldrich E1014). Samples were incubated on ice for 10 min. Samples were made up to 1 x LDS loading buffer and 5% 2-mercaptoethanol (βME), incubated at 95°C for 7 min before loading 15 μL onto NuPAGE™ Novex™ 4–12% Bis-Tris polyacrylamide gels (Thermo Fisher Scientific NP0321). Gels were run in 1 x MES buffer for 35 min at 200 volts. Gels were transferred to GE Healthcare Amersham™ Hybond™ -N+ Membranes (Fisher Scientific 45–000-927) for 1 hour at 4°C at 0.5 amps. Membranes were blocked in 5% non-fat dry milk in PBS-T (PBS+tween 20, 0.05%) or Starting Block Blocking buffer (Fisher # 37539) with orbital shaking for 1 hour at RT or several hours at 4°C. Membranes were then cut into strips for appropriate sizes depending on the antibody. Antibodies were incubated with blots for one to eight hours at 4°C with orbital shaking, followed by antibody removal and rinsing of the membranes twice with 5 ml PBS-T followed by washing in 10 mL of PBS-T three times for 15 min at RT with orbital shaking. Primary antibodies were detected by addition of species-specific horseradish-peroxidase (HRP) conjugated-secondary antibody in blocking buffer, incubated with orbital shaking for 30–60 min followed by washing as for primary antibodies. After removing the last wash buffer, chemiluminescent visualization solution was added. For chemiluminescent detection, SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific Catalog number 34096) or SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific Catalog number 34577) was used. Membranes were visualized with Alpha Imager.
Antibodies used for immunoblots were ERK1/2 (p44/42 MAPK (Erk1/2) Antibody Cell Signaling Technology catalog number 9102S), phospho-ERK1/2 (pERK; Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E XP (R) Rabbit mAb Cell Signaling Technology 4370S), and SNAI2 (C19G7 Rabbit mAb Cell Signaling Technology 9585S). Dilutions for ERK, pERK, and SNAI2/SLUG were 1:1000 for both primary and secondary antibodies. For housekeeping genes, dilutions for both primary and secondary antibodies for GAPDH (D16H11 XP® Rabbit mAb Cell Signaling Technology 5174S) and histone 3 (Abcam ab1791) were 1:5000.
Chromatin Immunoprecipitation
ChIP was performed as previously described (11). For immunoprecipitation, 10 μg of solubilized chromatin was used with 5 μl of α-SNAI2/SLUG antibody (35)(C19G7 Rabbit mAb Cell Signaling Technology 9585S) or an equivalent mass of non-specific IgG control (12–370, EMD Millipore), and nutated overnight at 4°C. The antibody for SNAI2/SLUG ChIP has been validated by a previous study (35). Protein G/Protein A agarose beads (IP05–1.5 mL, EMD Millipore Corporation) were added for three hours and then treated as previously described (11). PCR analysis was performed for the TERT promoter as previously described (11) with the forward primer 5′-GTCCTGCCCCTTCACCTT-3′ and reverse primer 5′-AGCGCTGCCTGAAACTCG-3′, and for TERT intron 1/exon 2 boundary using primers forward 5’-GCAGGTGTCCTGCCTGAA-3’ and reverse 5’-GAAGGCCAGCACGTTCTTC-3’, or exon 2 with the forward primer 5′- CTACTCCTCAGGCGACAAGG-3′ and reverse primer 5′- TGGAACCCAGAAAGATGGTC -3′.
Results
Pan-cancer integrative omics analysis of TERTp mutant cancers reveals BRAF/MAPK activation and mesenchymal/EMT cellular states
We compared the expression profiles of a subset of cancer cell lines from the Cancer Cell Line Encyclopedia (31,36) harboring TERTp mutations (83 samples in 13 tissue types) against wild types (419 samples in 16 tissue types) in order to identify differentially expressed genes. Then we used single-sample gene set enrichment analysis (ssGSEA) (37) and gene sets from the Molecular Signatures Database (MSigDB.org)(38). to identify gene expression patterns in the TERTp cell lines. For our analysis we considered the set of distinct TERTp mutations shown in Table S1.
The analysis strongly suggested that the specific transcriptional profile of TERTp mutant cell lines is that of a mesenchymal/EMT cell type. This can be seen, for example, in Figure 1A which shows the top-scoring gene sets from the hallmark collection of the Molecular Signatures Database (MSigDB.org). Each hallmark in this collection of 52 gene sets consists of a “refined” gene set, derived from multiple “founder” sets, that conveys a specific biological process and displays coherent expression (38). The top hit is the hallmark for the epithelial-mesenchymal transition (EMT). We then matched a collection of gene sets representing major oncogenic pathway components that we previously introduced (30) and observed that the top two hits were transcriptional components C6, representing BRAF/MAPK activation, and C4 representing core EMT signaling (Fig 1B).
Figure 1. TERTp mutants from divergent tissues share specific expression profiles.
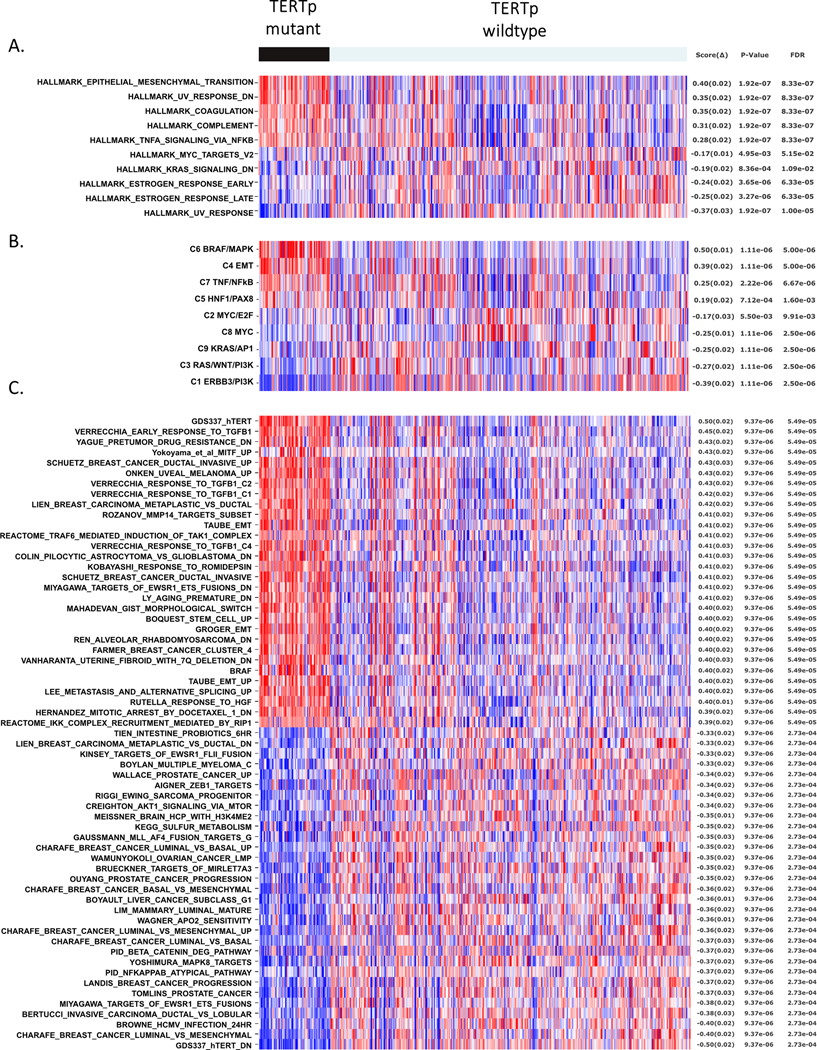
(A) Matching the single-sample GSEA (ssGSEA) profiles of MSigDB Hallmark gene sets stratified by TERTp status in the CCLE cancer cell lines. The figure shows the five most correlated and anticorrelated gene sets. The top hit is the hallmark that represents the epithelial-mesenchymal transition (EMT). (B) Matching the ssGSEA profiles of gene sets representing major oncogenic pathway components (9 gene sets) against the TERTp status on the CCLE cancer cell lines. The top two hits are the component C6 that represents BRAF/MAPK activation, and C4 that represents EMT. (C) Matching the ssGSEA profiles of a general collection of gene sets from the C2 subset of MSigDB, plus additional gene sets from the literature (5,334 gene sets), against the TERTp status on the CCLE cancer cell lines. The top hits include gene sets representing EMT, BRAF and MITF activation signatures, response to TGFβ1, invasion, stemness, metastasis and TERT overexpression. All the featured gene sets in Figs 1A–C displayed significance p ≤ 0.001.
We performed a second analysis using a broader range of gene sets from the C2 subcollection of MSigDB plus additional gene sets from the literature (5,334 gene sets). Among the top 30 hits are two independent gene sets representing EMT, one gene set representing BRAF and MITF activation signatures, one gene set representing the response to TGFβ1, and several representing invasive and metaplastic breast cancers, stemness and metastasis (Fig 1C). Interestingly, the top hit is gene set GDS337_hTERT derived from a TERT-rescue of late passage mammary epithelial cells (HMEC) (39).
In order to gain a more global and contextual perspective of the association between TERTp, mesenchymal characteristics, and BRAF/MAPK activation, we projected the TERTp mutation status onto a map of cancer cell line cellular states (30). The results are displayed in Figure 2 where we can see that the TERTp mutation-positive cell lines (Fig 1A–C) lie on top of the general area of EMT states.
Fig 2A shows the enrichment of the EMT hallmark signature (38) on the map and helps to delineate the EMT states at the bottom of the map. In Fig 2B cancer cell lines with TERTp mutations appear to fall across the full span of EMT cancers, including the core EMT state (C4), the partial EMT states (C2, C5, C7 and C9) and the BRAF/MAPK state (C6). Fig 2C shows the enrichment profile of a TERTp signature (‘TERTness’) and how closely it resembles the EMT hallmark signature (Fig 2A). A similar association was apparent when we projected the profiles of the individual mRNA signatures for the two most frequent TERTp mutations C228T and C250T on the same map (Fig S1). Interestingly, a mutation-specific signature for TERT_pg_5.1295250.G.A mutations (see Table S1) is more narrowly concentrated in the BRAF/MAPK component (C6, Fig S1B).
The signature also strongly resembles the gene expression profile generated from comparing passaged BJ fibroblasts overexpressing TERT plus the SV40 T antigen vs SV40-BJ cells lacking TERT expression (Fig 2D) (40). This TERT-driven rescue profoundly impacted gene expression in these slower-growing mesenchymal cells ((40), see methods) indicating that either telomere maintenance by TERT, or another TERT function, has dramatic consequences for the cells.
The association between TERTp mutations and the composite TERT transcriptional signature is quite high (Fig 2C and D). This is more clearly illustrated in the heatmap in Fig S2 showing correspondence between the different TERT signatures. The TERTp 250 signature appears to have a narrower scope centered on BRAF/MAPK activation (Fig S1b), while all the others have a broader context that encompasses the full spectrum of EMT cancers (Fig S1C, 2C-D).
TERTp mutant cells exhibit several canonical markers of EMT
To further examine the mesenchymal state in conjunction with TERTp status, we directly compared TERTp status with the gene expression patterns of several epithelial and mesenchymal markers across different tissue types and TERTp mutations. Canonical mesenchymal markers such as SNAI1, SNAI2 (SLUG), ZEB1, ZEB2, TWIST1, TWIST2, N-cadherin (CDH2), and VIMENTIN (VIM) showed statistically significant upregulation in TERTp mutants as opposed to TERTp wild-type cell lines (Fig. 3, S3A–C). Conversely, mRNA for epithelial markers such as E-cadherin (CDH1) and grainyhead-like transcription factor 2 (GRHL2)(41) showed significant downregulation in TERTp mutant lines (Fig. 3, S3A–C). Importantly, we observed these EMT traits in cells bearing different TERTp mutations and across most tissue types with these mutations (Fig. 3, S3A–C), indicating they are a frequently observed characteristic of TERTp mutant cancers. The relative prominence of these associations suggests TERTp mutants represent a distinct set of functional states.
Analysis of EMT markers on a tissue-specific basis reveal significant associations with TERTp mutations (Fig S3B,C). Loss of E-cadherin is associated with a mesenchymal cellular state and metastasis in cancer (42,43) and significantly alters outcomes in preclinical models(44). To determine if E-cadherin and other proteins associated with cell-cell attachment and cellular identity were altered in TERTp mutant cancers, we analyzed Reverse Phase Protein Array (RPPA) data for CCLE lines. These results revealed a strong correlation between E-cadherin mRNA and protein expression (Fig S4A) and that TERTp mutant cancer types have significantly reduced E-cadherin protein levels (Fig S3C, S4B). We also observe a strong correlation between E-cadherin expression and GRHL2, an epithelial marker that positively regulates E-cadherin expression (45,46) (Fig S4B). Claudin 7, which is an essential component of tight junctions and focal adhesions (47) and another epithelial marker protein, was also lower in TERTp mutant lines (Figure 3, S3B,C). In contrast, marker proteins for mesenchymal cells, fibronectin 1 (FN1) and N-cadherin (CDH2), were elevated (Fig 3, S3B,C). Tissue-specific analyses revealed that TERTp mutant bladder cancers frequently retain epithelial markers such as GRHL2 and fail to display key mesenchymal traits (Figure S3B,C). Collectively, these results for TERTp mutants indicate that their adhesive state and surface protein-mediated signaling may be distinct from most cancer types that typically lack these mutations.
To determine if patient tumor samples also displayed significantly reduced E-cadherin gene expression, we analyzed data from The Cancer Genome Atlas (TCGA). Both melanomas and liver cancers with TERTp mutations displayed significantly lower levels of E-cadherin gene expression in clinical samples (Figure S3D).
GABPβ1 is not elevated in most TERTp mutant cell lines
The GABPα/GABPβ1 heterodimer is an important driver of mutant TERT promoters in multiple tumor-derived cell types and is essential for GBM tumor formation in mouse xenografts (11,18,24). Previous studies identified elevated levels of GAPBβ1 in a subset of cell lines with TERTp mutations. To determine if TERTp mutations selectively occurred in cells with elevated GABPβ1 levels in most tumors, we analyzed a total of 19 cancer types from the CCLE (31) to assess GABPα/β1 levels. Unexpectedly, the results indicated that across a broad range of tissue types total GABPβ1 transcript levels were not consistently elevated in TERTp mutant cancers (Fig 3, 4, S5A). Previous work in glioblastoma identified that specifically the long isoform, GABPβ1L, regulates TERT (24). We therefore examined individual splice variants of GABPβ1. Short isoforms of GABPβ1 lack exon 9 and instead terminate at a stop codon downstream of exon 8 leading to a C-terminus containing an additional 15 amino acids. Long isoforms of GABPβ1 include exon 9 which encodes an additional 50 amino acids of distinct sequence from GABPβ1S. We analyzed CCLE lines for their levels of the main annotated isoforms of GABPβ1L that contain exon 9 (ENST00000380877.3, ENST00000220429.8, ENST00000543881.1) and those that do not (ENST00000359031.4, ENST00000396464.3, ENST00000429662.2). GABPβ1L isoforms were not elevated in TERTp mutant lines, although ENST00000380877.7 was nearly significant and two forms of GABPβ1S (ENST00000429662.2 and ENST00000359031.4) were significantly elevated (Fig 4, S5B). These results suggest that although GABPβ1L is critical to drive mutant TERT promoters in GBM, higher levels of GABPβ1L isoforms in cancer cell lines do not appear to broadly explain the selective incidence of TERTp mutations across these 19 tumor types.
Fig 4. GABPβ1 isoform mRNA expression levels in cell lines.

Three isoforms of GABPβ1L (long) and three isoforms of GABPβ1S (short) were assessed in TERTp mutants vs TERTp wild-type cancer cell lines in the CCLE. Mut, TERTp mutants, WT, TERTp wild type.
TERTp mutant cancers display selective drug sensitivities including to RAS/MAPK pathway inhibitors
Having established that TERTp mutations mark a subset of cancers that share a distinct expression profile, we considered the potential utility of this mark for re-deploying anti-cancer therapies specifically in this context. To address this possibility, we used The Cancer Therapeutics Response Portal (CTRP) (48,49) to assess whether the growth of TERTp mutants was selectively inhibited by specific compounds compared to TERTp wt cells. The most selective compound was a BRAF inhibitor, dabrafenib (Fig 5A). Dabrafenib inhibits the enzymatic activity of V600E/K-mutant BRAF and is FDA approved for use in metastatic melanoma. TERTp mutants are significantly enriched for BRAF mutations, suggesting the overall sensitivity of TERTp mutant lines may be due to the presence of these mutations. We therefore re-assessed the EC50 values with respect to BRAF mutations. This analysis indicated that even in the absence of mutant BRAF, >75% of the TERTp mutant lines displayed reduced growth at relatively low concentrations (Fig S6).
Fig 5. TERTp mutant cell lines exhibit enhanced sensitivity to some cancer therapeutics.

(A) TERTp lines treated with mutant-BRAF inhibitor dabrafenib selectively reduced growth of TERTp mutant cell lines (Wilcoxon, p = 3.3×10-5, T-test, p = 4.6×10−6). (B) Cells treated with a 4:1 combination of selumetinib + BRDA02303741 selectively reduced growth of TERTp mutant cell lines (Wilcoxon, p = 0.00082, T-test, p = 0.00011).
We also observed that a 4:1 combination of MEK1/2 inhibitor + DOT1L inhibitor (selumetinib+BRDA02303741) was significantly more selective against TERTp mutants (p = 0.00082; Fig 5B); interestingly, the combination of these two drugs was much more effective than either drug on their own. Two other selumetinib combinations were also moderately effective, with PLX-4032 (8:1, Wilcoxon, p = 0.00082, T-test, p = 0.00011) and with JQ-1 (4:1, Wilcoxon, p = 0.00082, T-test, p = 0.00011) (data not shown). These results from the CTRP confirm the functionality of the shared expression profiles of TERTp mutants, particularly a reliance on BRAF/MEK signaling, and suggest that the shared underlying biology of TERTp mutants may be clinically relevant.
MEK1/2 signaling regulates TERT expression in telomerase-positive cells
The preceding analysis indicated that BRAF/MAPK activation is a major feature of TERTp mutant cancers. Previous studies of melanoma showed that TERT expression in TERTp mutants relies on this signaling axis. MEK1 and MEK2 (MEK1/2) are effectors of the RAS-RAF signaling axis and they primarily target ERK1 and ERK2, which are major downstream kinases that localize to chromatin (50). To test the broad reliance of TERTp mutant cancers on MEK1/2 signaling to drive TERT expression, we inhibited MEK1 and MEK2 in cell lines derived from glioblastoma, liver cancer, bladder cancer, and melanoma. Treatments with low doses of trametinib for 24 hours (51) significantly decreased phosphorylation of ERK1 and ERK2 (Fig S7), and in each of the TERTp mutant cell lines that we tested, TERT mRNA was also substantially decreased (Fig 6A). Specifically, these data revealed that inhibition of MEK1/2 downregulates TERT mRNA expression in a broad range of TERTp mutant cells (Fig 6A; two-tailed T-test assuming heteroscedasticity, p = 4.4 × 10−4, n = 4 cancer types). These results are consistent with expectations from studies in melanoma and thyroid cancer (25–28) and extend the finding to other TERTp mutant cancer types. To test whether this effect of MEK1/2 inhibition was specific to mutant TERT promoters, we subjected two telomerase-positive, non-cancerous cell types to trametinib treatment. We used induced pluripotent stem cells (iPSCs) generated from human fibroblasts and spontaneously immortalized keratinocyte cells (HaCaT). These experiments suggested that the observed effect of MEK1/2 inhibition is not specific to mutant TERT promoter alleles, as both iPSC and HaCaT cells displayed significantly reduced TERT mRNA expression after treatment with trametinib (Fig 6B).
Fig 6. Regulation of TERT expression, but not GABPβ1L, by MEK1/2.

(A) Inhibition of MEK1 and MEK2 with 250 nM trametinib for 24 h significantly reduced TERT mRNA in cell lines from liver cancers (n = 5), brain cancers (medulloblastoma, n = 1, glioblastoma, n = 1), melanoma (n =3) or bladder cancer (n = 1). (B) Inhibition of MEK1/2 reduced TERT mRNA expression in telomerase positive, non-cancer cells lacking TERT promoter mutations, 100 nM p = 4.5×10−5, n = 3; 250 nM p = 0.01, n = 3. (C) Selective, short-term inhibition of MEK1/2 activity reduced TERT mRNA expression in cell lines (p = 0.002, n = 5) and FOS mRNA expression (p = 0.01, n = 4) but did not affect GABPβ1L mRNA expression (p = 0.79, n = 5). Data are mean + SEM. nd, not determined.
Consistent with a previous report (21), in multiple cell lines we observed the expected loss of FOS mRNA following MEK1/2 inhibition, but not GABPβ1L (Fig 6C). This finding indicates that additional regulatory mechanisms beyond GABPβ1L transcriptional regulation operate in response to MEK1/2 signaling to drive TERT mRNA expression.
MEK1/2 signaling contributes to E-cadherin gene repression in TERTp mutant cancer cell lines.
Diverse mechanisms regulating E-cadherin in cancer frequently involve elevated RAS signaling and transcription factors such as ZEB1, CTBP and SLUG. We found that treatment of TERTp mutants with MEK1/2 inhibition alleviated CDH1 mRNA repression in several cancer types (Fig S8A,B). A time course of CDH1 derepression in the TERTp mutant bladder cancer cell line SCaBER indicated that this derepression may occur as soon as six hours after drug treatment, in contrast to changes in TERT expression, which were not observed until 24 hours (Fig S8C). These results suggest that MAPK signaling may contribute to shaping the adhesive properties of TERTp mutant cancers.
SNAI2/SLUG supports TERT expression and localizes to TERT in some cells.
The developmental transcription factor SNAI2/SLUG was elevated in a significant majority of TERTp mutant cell lines. SLUG is a major regulator of mesenchymal cell gene expression impacted by RAS signaling (52). Although SLUG has not previously been reported to regulate telomerase expression, the nearly uniformly elevated levels of SLUG in TERTp mutant cell lines (p = 3.6 × 10−23) suggested a potential role in TERT expression. To test the importance of SLUG for TERT expression, we generated multiple cell lines deficient for SLUG using lentivirus that encodes a previously validated shRNA targeted to exon 3 (53) and selected for stable expression and SLUG knockdown. We successfully generated five lines from four tissue types (U87MG, DAOY, Mel3429, Mel3616, and MDA-MB-231). At the earliest time point assessable for each line, SLUG protein levels were found to be reduced by between 40–90% while TERT mRNA was reduced by 40–60% (Fig 7A–C). These results suggest that SLUG levels positively influence TERT mRNA expression in several TERTp mutant cell lines.
Fig 7. SLUG supports TERT expression and localizes to TERT in some cells.

(A) SLUG gene expression according to tissue type of origin for CCLE cell lines. (B, C) TERT expression in cell lines with stable knockdown of SLUG protein. U87 (n = 3), DAOY and Mel3429 (n = 2), Mel3616 and MDA-MB-231 (n = 1). Data are mean + SEM, p = 0.002, n = 5. (D) Schematic of the TERT promoter and intron 1 indicating TERTp mutation at −124, and potential consensus sequence binding sites for slug. (E) SLUG ChIP at TERT intron 1 (+317, see panel D) in induced pluripotent stem cells (iPSC), bone marrow derived mesenchymal progenitor cells (MPC) and osteocytes (Ost). (F) SLUG ChIP in a melanoma cancer cell line (Mel3429) with and without MEK1/2 inhibition at TERT intron 1 (+317). Data are mean + SEM.
We next tested whether this influence of SLUG on TERT expression may result from directly localizing to the TERT locus. The TERT gene has a SLUG consensus binding sequence CAGGTG (54) in the proximal promoter and also one each at the 5’ and 3’ ends of intron 1 (Fig 7D). To test if SLUG is recruited to these motifs we performed ChIP in mesenchymal progenitor cells (MPC), where SLUG is predicted to be highly expressed and active. We also tested induced pluripotent stem cells (iPSC) for SLUG binding to TERT. No SLUG binding was observed in iPSC at TERT. However, MPC displayed strong SLUG recruitment at intron 1 (p = 5.8 × 10−5), but not at the TERT promoter (Fig 7E). This intronic localization is consistent with the reported binding profile for SLUG in mice where it displays a 10-fold preference for introns (approximately 45% of all loci bound) over proximal promoters (35). To assess if SLUG occupancy at TERT in MPC persisted upon differentiation, osteocytes were generated from these MPC cells and subjected to SLUG ChIP. Osteocytes displayed little SLUG occupancy at TERT indicating that this recruitment may be a feature of bone marrow-derived MPCs.
We next tested a broad range of TERTp mutant tumor-derived cell lines for SLUG binding to the TERT locus. We did not detect binding to the TERT promoter in any cells, and most cells displayed no binding to intron 1 (data not shown), suggesting recruitment to this locus was not a common mechanism by which SLUG influenced TERT expression in cancer cells. However, the melanoma line Mel3429 consistently displayed strong SLUG recruitment near intron 1 (Fig 7F), but not at other nearby positions in the TERT gene or proximal promoter (data not shown). SLUG has been reported to be a direct target of ERK1/2 (55). Consistent with this, SLUG occupancy at TERT in Mel3429 depended on MEK1/2 pathway signaling, as inhibition with 250 nM trametinib for 24 hours followed by SLUG ChIP (Fig 7F) showed a significant reduction of SLUG occupancy at TERT (p = 0.04, n = 2). These results are consistent with the hypothesis that SLUG may mediate the effect of MEK1/2 on TERT mRNA expression through direct interactions at the TERT locus in a subset of cell lines.
Discussion
To achieve telomere maintenance, certain tumor lineages (e.g., glioblastoma, hepatocellular cancer, melanoma) rely on TERTp mutations while other tumor types (e.g., AML, prostate, colon) predominantly or exclusively activate telomerase without these mutations. Our study tested the hypothesis that this selective occurrence of TERTp mutations associates with a particular biological signature. To this end, we derived a TERTp signature by comparing gene and protein expression of TERTp mutant vs TERTp wild-type cell lines. Our results from GSEA analysis of CCLE lines revealed that TERTp mutants do indeed share a set of distinct expression profiles that are likely to impact their pathogenicity. Our data suggest that this signature is not a reflection of TERT overexpression but encompasses a cancer cell state that is marked by promoter mutations and is shared across tissue types. Moreover, these features are likely to generate distinct adhesion and tumor microenvironmental characteristics (e.g. CD44, E-cadherin, N-cadherin, Claudin 7).
TERTp mutations represent an alternative mechanism by which telomerase is activated in a majority of certain tumor types and has been found to co-occur with other mutations (31). We considered that in addition to tumors harboring TERTp mutations, a larger class enriched with this same transcriptional signature reflects a cancer cellular state for which we propose the term “TERTness”. The utility of this concept is the observation that although 90% of cancers express telomerase, this does not make them equivalent in terms of a TERTness signature. TERTp mutant cancers may be viewed as harboring more TERTness and can be uniquely identified, in a similar way that the concept of BRCAness has been useful to describe the set of tumors without BRCA1/2 mutations (56) with aberrant homologous recombination (HR). Our findings represent an initial report of common features that characterize TERTp mutants as a class and complement previous studies which identified important differences among TERTp mutants (11,18,20,21). Further efforts aimed at integrating these observations with our present study to delineate biologically and clinically relevant TERTp mutant variables will be informative.
TERTp mutants display a strong association with mesenchymal gene expression. Many tumors of epithelial origin acquire a subset of mesenchymal traits during oncogenesis. Other cancers, including soft tissue tumors, originate from mesenchymal cell types. That TERTp mutants display this association indicates that they undergo EMT or arise from mesenchymal cellular states. A mesenchymal state has major implications for tumors as it has been linked to persistence of therapy-tolerant cells (57–60). We speculate that the mesenchymal nature of TERTp mutant cancers may provide some biological plasticity for the cells (61). Under selective pressure from therapeutics, this mesenchymal phenotype may facilitate the emergence of altered cellular states that are therapy tolerant creating opportunities for tumor relapse. Thus, whether the TERTp mutant signature may predispose these cancers to enhanced therapy resistance is of interest.
Some TERTp wild-type cells displayed overlapping phenotypes with TERTp mutants. One possible interpretation is that for cells in mesenchymal or BRAF-driven cellular states, TERTp mutations provide a selective advantage (thus enriching for their occurrence) but they do not represent the only path to immortalization.
The prominent MAPK/RAS pathway signaling in TERTp mutants suggests that it may cooperate with (62) or drive (63,64) components of this EMT-like state. Importantly, we observed some mutation-specific characteristics to these signatures (Fig 2, S1, S2), suggesting the existence of multiple, related driver mechanisms. Previous work in melanoma (65) suggests that examining the correlation between these mutation-specific signatures with patient outcomes may be informative. Specifically, there is a differential distribution of TERT promoter mutations in primary cutaneous melanomas, which are enriched for C250T/−146 mutations, and these tumors frequently co-occur with BRAF and NRAS mutations (22). RAS pathway inhibition has proven effective in the treatment of some cancers, leading us to speculate that TERTp mutations may serve to stratify patients for treatment with MEK1/2 inhibitors. Supporting this rationale, TERTp mutants, regardless of their KRAS or BRAF mutation status, displayed enhanced sensitivity to dabrafenib as well as to a combination MEK+DOT1L inhibitor. Dabrafenib is FDA-approved only for BRAF-mutant melanomas. The basis for this selective approval is that in some cell lines with wild-type BRAF, paradoxical activation of the downstream effectors of RAF, MEK1/2 and ERK1/2, has been observed. This paradoxical activation observed in model systems has guided these restrictions in the clinic. However, to our knowledge, those cell types displaying paradoxical activation in these model systems have not been identified as carrying TERT promoter mutations (66–70). In our dataset, why the growth of most cell lines with wild-type BRAF were sensitive to dabrafenib remains unknown. Of note, 3 of the 14 TERTp mutant cell lines with wild type BRAF showed little growth inhibition, consistent with a previous report for a glioma line of similar genotype (BTL2176) (21); this heterogeneity among wtBRAF/TERTp mutant cell lines indicates that undiscovered factors can modulate the response to dabrafenib. Interestingly, it was shown that in the TERTp mutant breast cancer cell line MDA-MB-231, small molecule-mediated dimerization between BRAF and CRAF (a proposed mechanism of this paradoxical activation) was insufficient to recapitulate the activation (69)These studies, and our analysis of CTRP data, suggest that paradoxical activation mechanisms may be less relevant in a subset of tumors, including TERTp mutants.
A key initial report of TERTp mutations in melanoma speculated that TERTp mutations may be responsive to RAS pathway signaling (3) and subsequent reports have identified such sensitivities (25–28). We observed that TERT expression does rely on MEK1/2, but that this was not unique to the mutation status of the TERT promoter since non-tumor derived HaCaT cells and iPSCs displayed similar sensitivities. Our previous work identified that mutant TERT promoters exhibit chromatin features more similar to stem cell TERT promoters than to TERTp wild-type promoters. Thus, our results are consistent with our previous findings (19) and support the relevance of those observations to TERT regulation.
GABPβ1L is critical for TERTp mutant gene expression, but we found no evidence that GABPβ1L mRNA expression levels in cell lines broadly explains the distribution of TERTp mutations in different cancer types. This does not exclude the possibility that during early oncogenesis GABPβ1L may positively select for retention of TERTp mutations. However, in contrast to GABPβ1L, the majority of TERTp mutant cell lines displayed elevated SNAI2/SLUG expression. SLUG is a transcriptional repressor that recruits LSD1 and nuclear receptor corepressor (NCoR) complex and the DNA-binding scaffold protein CtBP1 in a phosphorylation-dependent manner (54,71,72), and may also act as a transcriptional activator. SLUG activity is supported by ERK1/2 signaling in breast cancer through phosphorylation (52,55). In our study, we observed that SLUG bound to the TERT locus in one melanoma cell line where its occupancy was sensitive to MEK1/2 inhibition, suggesting that SLUG can directly influence TERT expression in a MEK1/2 dependent fashion. SLUG also localized to the TERT locus in mesenchymal progenitor cells, but not in osteocytes terminally differentiated from these cells, suggesting a specific association at TERT for primary cells in a mesenchymal state. In contrast, we did not see SLUG at intron 1 of TERT or in the TERT promoter in most cancer cell lines tested, suggesting that in most cases the mechanism by which it influences TERT expression maybe be indirect. Such two-step regulation has been observed whereby ERK1/2-dependent activation of SLUG promotes vimentin mRNA expression in the absence of binding to the vimentin promoter (55), indicating the involvement of additional factors. SLUG genome occupancy has been mapped in mouse cells (35) and in human keratinocytes (73) but has not been examined in cancer cells with TERTp mutations. Thus, such experiments in TERTp mutant cancers may identify the SLUG circuitry impacting TERT transcription. Future experiments will be required to determine the effect of SLUG on TERT expression in cancers with wild type TERT promoters. In ALT cells, SNAI1 promotes telomere maintenance that involves telomeric (TERRA) transcription and TERRA was found to influence expression of mesenchymal genes (74). Future studies examining whether in TERTp mutants the elevated levels of SNAI1 or SNAI2 impact telomeric transcription and mesenchymal gene expression may also be informative.
The link between the signature derived from TERTp mutant cancers and TERT-overexpressing BJ fibroblasts and HMECs is not clear (Fig 1C, 2D, S2). It is not explained by total TERT mRNA expression levels in TERTp mutant cancers (Fig 3) because TERTp wild-type cancers displayed similar levels. One possible explanation is that the impact of telomerase on global gene expression is context dependent. If true, this would imply that gene expression differences between such aged cells rescued with telomerase may resemble gene expression differences between wild-type cancers vs TERTp mutants.
About 10–15% of cancers maintain telomeres though a non-telomerase, homologous recombination-based mechanism called Alternative Lengthening of Telomeres (ALT) (recently reviewed(75)). Interestingly, ALT cells also associate with a mesenchymal cellular state (76,77) suggesting that a mesenchymal state may necessitate alternative pathways to telomere maintenance. Importantly, it has been documented that cancer cells can switch telomere maintenance mechanisms between telomerase and ALT and that these two mechanisms in some cases co-exist(78). The TERT gene in ALT cancers is transcriptionally repressed and associates with H3K9me3, H3K27me3 and DNA methylation (79). These features are very similar to the transcriptionally silent TERT allele in cancers with heterozygous TERT promoter mutations (11,19) suggesting common mechanisms may operate at silent TERT promoters in these two cancer types.
Telomere maintenance is a critical requirement for the indefinite proliferation of cancer cells and underpins their ability to cause fatal disease. Activating mutations in the TERT promoter are the most common recurrent noncoding genetic alterations in cancer (31,80). Our study reports an unanticipated association between TERTp mutations and a biological state shared across multiple tumor types. Our results suggest that these mutations commonly arise in a specific cellular milieu and may capitalize on this state. This study represents the first global assessment of the biology of TERTp mutant cancers and suggests that a TERTp mutant ‘signature’, or “TERTness”, may exist with implications for a clinical understanding of cancers that share this common mutation.
Supplementary Material
Implications:
Cancers harboring TERT promoter mutations are often more lethal, but the basis for this higher mortality remains unknown. Our study identifies that TERTp mutants, as a class, associate with a distinct gene and protein expression signature likely to impact their biological and clinical behavior and provide new directions for investigating treatment approaches for these cancers.
Acknowledgments
We thank T. Rowland, K. Foster, and other Cech lab members, H. Ma, J. Silver, V. Rao, and K. Anseth (University of Colorado, Boulder), K. Couts (University of Colorado, Anschutz Medical Campus), B. Lasseigne (University of Alabama at Birmingham). JLS was supported by a postdoctoral fellowship 127621-PF-16-099-01-DMC from the American Cancer Society and a Mary Ann Harvard Young Investigator Grant from the O’Neal Comprehensive Cancer Center. FWH was supported by a Prostate Cancer Foundation Young Investigator Award (FWH). This project was supported by the following NIH grants: R01GM099705 (TRC), R01GM074024 (PT), U24CA194107 (PT), R01HG009285 (PT), U54CA209891 (PT), R01CA172513 (PT), U24CA220341 (PT), U01CA217885 (PT), 5P20CA233255 (FWH), and U19CA214253 (FWH). Support was received from the Biostatistics and Bioinformatics Shared Resource through the University of Colorado Cancer Center Support Grant (P30CA046934).
Footnotes
Disclosure of Potential Conflicts of Interest: T.R.C. is on the board of directors of Merck, Inc., and a scientific advisor to Storm Therapeutics. No other potential conflicts of interest to declare.
References
- 1.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell. 2018;173:291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang FW, Hodis E, Xu MJ, Kryukov G V., Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–61. [DOI] [PubMed] [Google Scholar]
- 4.Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110:6021–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. 1997;276:561–7. [DOI] [PubMed] [Google Scholar]
- 6.Greider CW, Blackburn EH. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature. 1989;337:331–7. [DOI] [PubMed] [Google Scholar]
- 7.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PLC, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–5. [DOI] [PubMed] [Google Scholar]
- 8.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer Part A. 1997;33:787–91. [DOI] [PubMed] [Google Scholar]
- 9.Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18:173–9. [DOI] [PubMed] [Google Scholar]
- 10.Huang FW, Bielski CM, Rinne ML, Hahn WC, Sellers WR, Stegmeier F, et al. TERT promoter mutations and monoallelic activation of TERT in cancer. Oncogenesis. 2015;4:e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stern JL, Theodorescu D, Vogelstein B, Papadopoulos N, Cech TR. Mutation of the TERT promoter, switch to active chromatin, and monoallelic TERT expression in multiple cancers. Genes Dev. 2015;29:2219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vinagre J, Pinto V, Celestino R, Reis M, Pópulo H, Boaventura P, et al. Telomerase promoter mutations in cancer: An emerging molecular biomarker? Virchows Arch. 2014. page 119–33. [DOI] [PubMed] [Google Scholar]
- 13.Ramlee MK, Wang J, Toh WX, Li S. Transcription regulation of the human telomerase reverse transcriptase (hTERT) gene. Genes (Basel). MDPI AG; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.George JR, Henderson YC, Williams MD, Roberts DB, Hei H, Lai SY, et al. Association of TERT promoter mutation, but not BRAF mutation, with increased mortality in PTC. J Clin Endocrinol Metab. Endocrine Society; 2015;100:E1550–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simon M, Hosen I, Gousias K, Rachakonda S, Heidenreich B, Gessi M, et al. TERT promoter mutations: A novel independent prognostic factor in primary glioblastomas. Neuro Oncol. 2015;17:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rachakonda PS, Hosen I, De Verdier PJ, Fallah M, Heidenreich B, Ryk C, et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc Natl Acad Sci U S A. 2013;110:17426–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borah S, Xi L, Zaug AJ, Powell NM, Dancik GM, Cohen SB, et al. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science. 2015;347:1006–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bell RJA, Rube HT, Kreig A, Mancini A, Fouse SD, Nagarajan RP, et al. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science. 2015;348:1036–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stern JL, Paucek RD, Huang FW, Ghandi M, Nwumeh R, Costello JC, et al. Allele-Specific DNA Methylation and Its Interplay with Repressive Histone Marks at Promoter-Mutant TERT Genes. Cell Rep. Elsevier B.V; 2017;21:3700–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bullock M, Lim G, Zhu Y, Åberg H, Kurdyukov S, Clifton-Bligh R. ETS Factor ETV5 Activates the Mutant Telomerase Reverse Transcriptase Promoter in Thyroid Cancer. Thyroid. 2019;29:1623–33. [DOI] [PubMed] [Google Scholar]
- 21.Gabler L, Lötsch D, Kirchhofer D, van Schoonhoven S, Schmidt HM, Mayr L, et al. TERT expression is susceptible to BRAF and ETS-factor inhibition in BRAFV600E/TERT promoter double-mutated glioma. Acta Neuropathol Commun. 2019;7:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heidenreich B, Kumar R. TERT promoter mutations in telomere biology. Mutat. Res. - Rev. Mutat. Res. Elsevier B.V; 2017. page 15–31. [DOI] [PubMed] [Google Scholar]
- 23.Yuan X, Larsson C, Xu D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: old actors and new players. Oncogene. 2019. page 6172–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mancini A, Xavier-Magalhães A, Woods WS, Nguyen K-T, Amen AM, Hayes JL, et al. Disruption of the β1L Isoform of GABP Reverses Glioblastoma Replicative Immortality in a TERT Promoter Mutation-Dependent Manner. Cancer Cell. 2018;34:513–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vallarelli AF, Rachakonda PS, André J, Heidenreich B, Riffaud L, Bensussan A, et al. TERT promoter mutations in melanoma render TERT expression dependent on MAPK pathway activation. Oncotarget. 2016;7:53127–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reyes-Uribe P, Adrianzen-Ruesta MP, Deng Z, Echevarria-Vargas I, Mender I, Saheb S, et al. Exploiting TERT dependency as a therapeutic strategy for NRAS-mutant melanoma. Oncogene. 2018;37:4058–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Cheng HS, Chng WJ, Tergaonkar V, Cleaver JE. Activation of mutant TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas. Proc Natl Acad Sci U S A. 2016;113:14402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu R, Zhang T, Zhu G, Xing M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat Commun. 2018;9:579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, Hockemeyer D. Cancer-associated tert promoter mutations abrogate telomerase silencing. Elife. 2015;4:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim JW, Abudayyeh OO, Yeerna H, Yeang CH, Stewart M, Jenkins RW, et al. Decomposing Oncogenic Transcriptional Signatures to Generate Maps of Divergent Cellular States. Cell Syst. 2017;5:105–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov G V., Lo CC, McDonald ER, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Couts KL, Bemis J, Turner JA, Bagby SM, Murphy D, Christiansen J, et al. ALK inhibitor response in melanomas expressing EML4-ALK fusions and alternate ALK isoforms. Mol Cancer Ther. 2018;17:222–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner JA, Bemis JGT, Bagby SM, Capasso A, Yacob BW, Chimed TS, et al. BRAF fusions identified in melanomas have variable treatment responses and phenotypes. Oncogene. 2019;38:1296–308. [DOI] [PubMed] [Google Scholar]
- 34.Gupta PB, Kuperwasser C, Brunet JP, Ramaswamy S, Kuo WL, Gray JW, et al. The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat Genet. 2005;37:1047–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 2015;525:256–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith LL, Coller HA, Roberts JM. Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat Cell Biol. 2003;5:474–9. [DOI] [PubMed] [Google Scholar]
- 40.Lovejoy CA, Li W, Reisenweber S, Thongthip S, Bruno J, de Lange T, et al. Loss of ATRX, Genome Instability, and an Altered DNA Damage Response Are Hallmarks of the Alternative Lengthening of Telomeres Pathway. Scott HS, editor. PLoS Genet. 2012;8:e1002772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Craene B, Berx G Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer. 2013. page 97–110. [DOI] [PubMed] [Google Scholar]
- 42.Aiello NM, Kang Y. Context-dependent EMT programs in cancer metastasis. J. Exp. Med. 2019. page 1016–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev Cell. 2018;45:681–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, et al. E-cadherin is required for metastasis in multiple models of breast cancer. Nature. Springer Science and Business Media LLC; 2019;573:439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pyrgaki C, Liu A, Niswander L. Grainyhead-like 2 regulates neural tube closure and adhesion molecule expression during neural fold fusion. Dev Biol. 2011;353:38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cieply B, Riley IV P, Pifer PM, Widmeyer J, Addison JB, Ivanov A V., et al. Suppression of the epithelial-mesenchymal transition by grainyhead-like-2. Cancer Res. 2012;72:2440–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hagen SJ. Non-canonical functions of claudin proteins: Beyond the regulation of cell-cell adhesions. Tissue Barriers. 2017. page e1327839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price E V., Gill S, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol. 2016;12:109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Basu A, Bodycombe NE, Cheah JH, Price E V., Liu K, Schaefer GI, et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 2013;154:1151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klein AM, Zaganjor E, Cobb MH. Chromatin-tethered MAPKs. Curr. Opin. Cell Biol. Elsevier Ltd; 2013. page 272–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. [DOI] [PubMed] [Google Scholar]
- 52.Chen H, Zhu G, Li Y, Padia RN, Dong Z, Pan ZK, et al. Extracellular signal-regulated kinase signaling pathway regulates breast cancer cell migration by maintaining slug expression. Cancer Res. 2009;69:9228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell. 2019. page 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shirley SH, Hudson LG, He J, Kusewitt DF. The skinny on slug. Mol Carcinog. 2010;49:851–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Virtakoivu R, Mai A, Mattila E, De Franceschi N, Imanishi SY, Corthals G, et al. Vimentin-ERK signaling uncouples slug gene regulatory function. Cancer Res. 2015;75:2349–62. [DOI] [PubMed] [Google Scholar]
- 56.Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16:110–20. [DOI] [PubMed] [Google Scholar]
- 57.Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK -rearranged lung cancer. Cancer Discov. 2016;6:1118–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raoof S, Mulford IJ, Frisco-Cabanos H, Nangia V, Timonina D, Labrot E, et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene. Springer Science and Business Media LLC; 2019;38:6399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Staalduinen J, Baker D, ten Dijke P, van Dam H. Epithelial–mesenchymal-transition-inducing transcription factors: new targets for tackling chemoresistance in cancer? Oncogene. 2018. page 6195–211. [DOI] [PubMed] [Google Scholar]
- 60.Yochum ZA, Cades J, Wang H, Chatterjee S, Simons BW, O’Brien JP, et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene. 2019;38:656–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Williams ED, Gao D, Redfern A, Thompson EW. Controversies around epithelial–mesenchymal plasticity in cancer metastasis. Nat Rev Cancer. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aguirre AJ, Hahn WC. Synthetic lethal vulnerabilities in kras-mutant cancers. Cold Spring Harb Perspect Med. 2018;8:a031518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ichikawa K, Kubota Y, Nakamura T, Weng JS, Tomida T, Saito H, et al. MCRIP1, an ERK Substrate, Mediates ERK-Induced Gene Silencing during Epithelial-Mesenchymal Transition by Regulating the Co-Repressor CtBP. Mol Cell. 2015;58:35–46. [DOI] [PubMed] [Google Scholar]
- 64.Sale MJ, Balmanno K, Saxena J, Ozono E, Wojdyla K, McIntyre RE, et al. MEK1/2 inhibitor withdrawal reverses acquired resistance driven by BRAF V600E amplification whereas KRAS G13D amplification promotes EMT-chemoresistance. Nat Commun. 2019;10:2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nagore E, Rachakonda S, Kumar R. TERT promoter mutations in melanoma survival. Oncotarget. 2019;10:1546–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–5. [DOI] [PubMed] [Google Scholar]
- 67.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell. Elsevier; 2010;140:209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carnahan J, Beltran PJ, Babij C, Le Q, Rose MJ, Vonderfecht S, et al. Selective and potent raf inhibitors paradoxically stimulate normal cell proliferation and tumor growth. Mol Cancer Ther. 2010;9:2399–410. [DOI] [PubMed] [Google Scholar]
- 69.Holderfield M, Merritt H, Chan J, Wallroth M, Tandeske L, Zhai H, et al. RAF Inhibitors Activate the MAPK Pathway by Relieving Inhibitory Autophosphorylation. Cancer Cell. 2013;23:594–602. [DOI] [PubMed] [Google Scholar]
- 70.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Phillips S, Prat A, Sedic M, Proia T, Wronski A, Mazumdar S, et al. Cell-state transitions regulated by SLUG are critical for tissue regeneration and tumor initiation. Stem Cell Reports. 2014;2:633–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferrari-Amorotti G, Fragliasso V, Esteki R, Prudente Z, Soliera AR, Cattelani S, et al. Inhibiting interactions of lysine demethylase LSD1 with snail/slug blocks cancer cell invasion. Cancer Res. 2013;73:235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mistry DS, Chen Y, Wang Y, Zhang K, Sen GL. SNAI2 controls the undifferentiated state of human epidermal progenitor cells. Stem Cells. Wiley-Blackwell; 2014;32:3209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mazzolini R, Gonzàlez N, Garcia-Garijo A, Millanes-Romero A, Peiró S, Smith S, et al. Snail1 transcription factor controls telomere transcription and integrity. Nucleic Acids Res. 2018;46:146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gaspar TB, Sá A, Lopes JM, Sobrinho-Simões M, Soares P, Vinagre J. Telomere Maintenance Mechanisms in Cancer. Genes (Basel). 2018;9:1–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cesare AJ, Reddel RR. Telomere uncapping and alternative lengthening of telomeres. Mech Ageing Dev. 2008;129:99–108. [DOI] [PubMed] [Google Scholar]
- 77.Dilley RL, Greenberg RA. ALTernative Telomere Maintenance and Cancer. Trends in Cancer. 2015. page 145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Vitis M, Berardinelli F, Sgura A. Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT). Int J Mol Sci. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zinn RL, Pruitt K, Eguchi S, Baylin SB, Herman JG. hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res. 2007;67:194–201. [DOI] [PubMed] [Google Scholar]
- 80.Rheinbay E, Nielsen MM, Abascal F, Wala JA, Shapira O, Tiao G, et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature. 2020;578:102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.