

Abstract
We tested the hypothesis that voluntary wheel running would complement microdystrophin gene therapy to improve muscle function in young mdx mice, a model of Duchenne muscular dystrophy. mdx mice injected with a single dose of AAV9-CK8-microdystrophin or vehicle at age 7 weeks were assigned to three groups: mdxRGT (run, gene therapy), mdxGT (no run, gene therapy), or mdx (no run, no gene therapy). Wild-type (WT) mice were assigned to WTR (run) and WT (no run) groups. WTR and mdxRGT performed voluntary wheel running for 21 weeks; remaining groups were cage active. Robust expression of microdystrophin occurred in heart and limb muscles of treated mice. mdxRGT versus mdxGT mice showed increased microdystrophin in quadriceps but decreased levels in diaphragm. mdx final treadmill fatigue time was depressed compared to all groups, improved in mdxGT, and highest in mdxRGT. Both weekly running distance (km) and final treadmill fatigue time for mdxRGT and WTR were similar. Remarkably, mdxRGT diaphragm power was only rescued to 60% of WT, suggesting a negative impact of running. However, potential changes in fiber type distribution in mdxRGT diaphragms could indicate an adaptation to trade power for endurance. Post-treatment in vivo maximal plantar flexor torque relative to baseline values was greater for mdxGT and mdxRGT versus all other groups. Mitochondrial respiration rates from red quadriceps fibers were significantly improved in mdxGT animals, but the greatest bioenergetic benefit was observed in the mdxRGT group. Additional assessments revealed partial to full functional restoration in mdxGT and mdxRGT muscles relative to WT. These data demonstrate that voluntary wheel running combined with microdystrophin gene therapy in young mdx mice improved whole-body performance, affected muscle function differentially, mitigated energetic deficits, but also revealed some detrimental effects of exercise. With microdystrophin gene therapy currently in clinical trials, these data may help us understand the potential impact of exercise in treated patients.
Keywords: duchenne muscular dystrophy, muscle physiology, muscle pathology, dystrophic grade, voluntary exercise, endurance, durability, muscle power, mitochondrial respiration, myosin heavy chain
Graphical abstract
We report that voluntary exercise complements microdystrophin gene therapy in the mdx mouse, a model of Duchenne muscular dystrophy (DMD), by increasing running endurance, muscle strength, and energy production. These and other findings may help us understand how exercise in DMD boys may influence microdystrophin gene therapy.
Introduction
Duchenne muscular dystrophy (DMD) is a degenerative neuromuscular disease affecting approximately 1:5,000 male births.1 Boys affected by this fatal disease experience progressive muscle weakness, respiratory stress, cardiomyopathy, and loss of ambulation.1,2 Muscles in DMD are deficient in the protein dystrophin due to various mutations in its gene that result in disrupted reading frames and premature stop codons.3 In normal muscle fibers, dystrophin links costameric actin to the sarcolemma via the dystrophin-glycoprotein complex (DGC).4,5 The DGC is thought to confer both structural and signaling functions integral to the health of the muscle fiber.6 In the absence of dystrophin, these functions are lost, and the muscles are more susceptible to injury and regenerate poorly.6
Treatment options for DMD have largely focused on modulating expression of the compensatory protein utrophin, decreasing activity of pathways associated with dystrophic pathology, and restoring dystrophin expression.7 Because the gene encoding dystrophin is the largest in the human genome at 2.2 Mb,8 over the past several decades it has been extensively engineered to reduce its size yet still maintain critical elements for proper function of the protein product in the muscle cell.8,9 These microdystrophin gene constructs (size < 4 kb) can be effectively delivered systemically when carried by adeno-associated virus (AAV) vectors (packaging size ∼5 kb) of serotype 6, 8, and 9.8,9 This approach has been demonstrated successfully in both mouse and dog models of DMD, yielding microdystrophin expression throughout muscles and corresponding improvements in muscle function.8,9
In addition to pre-clinical studies, microdystrophin gene therapy is currently being investigated in clinical trials for the treatment of DMD (ClinicalTrials.gov: NCT03368742, NCT03769116, and NCT03362502). With the potential clinical success of a microdystrophin gene therapy program on the horizon, additional questions related to real-world scenarios should be explored. For example, what are the effects of increased physical activity, such as voluntary exercise, on overall treatment efficacy and durability? While animal studies have supported the use of exercise as a potential therapeutic in DMD, studies in dystrophic boys to determine the beneficial or detrimental effects of exercise in the absence of a treatment have been limited for fear of exacerbating their condition.10, 11, 12, 13 Pre-clinical studies evaluating exercise combined with microdystrophin may help inform guidelines for physical activity for treated DMD boys.
The effects of exercise have been studied in the mdx mouse model of DMD across various modalities including voluntary wheel running,13, 14, 15 forced treadmill running,16,17 and swimming18,19 to better understand how dystrophic muscles respond. Whereas forced treadmill running to exacerbate the dystrophic phenotype in older mdx mice (6–12 months) was used to assess microdystrophin gene therapy,17 in the current study, voluntary wheel running in young mdx mice was used because the mice can get on and off the wheel as they choose and therefore may mimic the self-regulated physical activity of dystrophic boys if successfully treated with microdystrophin. Furthermore, although young mdx mice represent a mild dystrophic model of DMD that show functional and histopathological deficits relative to wild-type (WT) animals but do not recapitulate the severity of DMD disease progression,20 we selected the strategy to treat young mice based on (1) the juvenile age of DMD diagnosis, (2) the therapeutic goal to treat DMD boys as early as possible after diagnosis to prevent chronic accumulation of muscle damage, and (3) the demonstrated effectiveness of early treatments relative to age of diagnosis in other monogenic muscle diseases.21,22
Herein, we report the effects of 21 weeks of voluntary wheel-running exercise initiated in 7-week-old mdx mice after a single treatment with AAV9-CK8-microdystrophin. Our data demonstrate that young mdx mice treated with gene therapy combined with running-wheel exercise improved endurance capacity, exhibited partial to full restoration of muscle function across different assessments, mitigated mitochondrial energetic deficits, and revealed some potential negative effects of exercise compared to gene therapy treatment alone.
Results
Study strategy
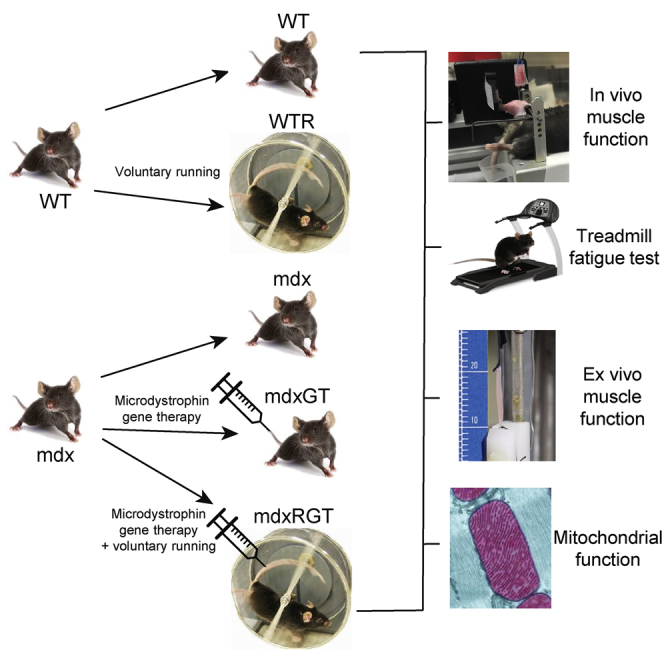
In this study, we wanted to know if voluntary running-wheel exercise would complement microdystrophin gene therapy to improve dystrophic muscle function in mdx mice. Treated animals were separated into gene therapy (mdxGT) and running wheel combined with gene therapy (mdxRGT) groups. Each group was administered a single intravenous dose of AAV9-creatine kinase 8 (CK8)-microdystrophin at approximately 7 weeks of age and followed for 22 weeks post-treatment. Control groups of mdx, WT, and WT animals undergoing running-wheel exercise (WTR) were assessed in parallel. Equivalent administration of gene therapy was confirmed by measurement of vector genome (vg) copies in blood samples collected 30 min post-dosing. Muscle vg levels, microdystrophin expression, and dystrophic histopathology were evaluated at the completion of the study to determine potential differences due to running-wheel exercise. Throughout the study, endurance capacity and muscle contractile function were evaluated using in vivo assays. At the end of the study, muscle contractile function was determined by ex vivo assays along with assessments of metabolic function (Figure 1). Although all study groups are discussed, the comparison between mdxGT and mdxRGT groups is highlighted.
Figure 1.

Project design
(A) Project timeline in weeks (−3 to 22). R, mice received at 4 weeks of age; T, 0 weeks, start of treatment: mdx mice received either AAV9-CK8-microdystrophin or excipient via tail-vein injection. Boxes indicate tests at specific time points. (B) Group identification and treatment descriptions.
Vector genome concentrations are similar in treated mdx mice irrespective of voluntary wheel running
An expression construct composed of two inverted terminal repeat (ITR) sequences, a muscle-specific CK8 promoter,23 a microdystrophin cDNA, and a poly(A) tail (pA) (Figure 2A) was packaged into an AAV9 vector. The AAV9-CK8 microdystrophin vector was injected via tail vein at 2e14 vg/kg into mdx mice aged 7 weeks to replace the absent full-length dystrophin (Figure 2B) with the microdystrophin protein product encoded by the microdystrophin gene construct (Figure 2C) (see Materials and methods for details). Following systemic delivery, AAV9-CK8-microdystrophin vg copies in the blood were similar between the mdxGT and mdxRGT groups 30 min after tail-vein injection at a value of ∼1e9 vg/μg genomic DNA (gDNA) and were present in the blood at 22 weeks (the end of the study) at ∼1e4 and ∼1e3 vg/μg gDNA for the mdxGT and mdxRGT groups, respectively (Figure 2D). Muscle vg concentrations for the mdxGT and mdxRGT groups were between 1e5 and 1e6 vg/μg gDNA for the heart, diaphragm, and three hindlimb skeletal muscles at 22 weeks. (Figure 2E).
Figure 2.

Vector strategy and biodistribution
(A) An expression construct composed of two inverted terminal repeat (ITR) sequences, a muscle-specific CK8 promoter,23 a microdystrophin cDNA, and a poly(A) tail (pA) (A) was packaged into an AAV9 vector. (B and C) The AAV9-CK8 microdystrophin vector was injected into 7-week-old mice via tail vein to replace (B) absent full-length dystrophin in mdx muscle fibers with (C) microdystrophin. (D) AAV9-microdystrophin blood concentration in treated mice 30 min after injection via tail vein and still present 22 weeks post-treatment. (E) AAV9-microdystrophin was present in heart, diaphragm, quadriceps, gastrocnemius, and EDL muscles at the conclusion of the study. There was no significant difference between groups. Dashed line indicates the lower limit of quantification in (D) and (E).
Microdystrophin expression similarly improves dystrophic histopathology in animals performing voluntary exercise
Microdystrophin expression was evaluated at the protein level to assess distribution and relative content across muscles of treated groups. Immunofluorescence analysis was performed to evaluate both revertant fibers present in animals and microdystrophin-positive fibers following treatment. Using an antibody reactive to a sequence not found in microdystrophin, few revertant fibers were detected in any animals (Table S1). Using a separate antibody reactive to microdystrophin, robust, widespread expression was observed in the heart, quadriceps, and EDL muscles, with greater than 72% positive fibers in the mdxGT and mdxRGT groups (Figures 3A and 3C; Table S2). Semiquantitative western blots demonstrated similar microdystrophin protein content normalized to β-tubulin in heart muscles of the two groups but showed increased microdystrophin content in mdxRGT (mean ± SE; 0.85 ± 0.10) compared to mdxGT (0.52 ± 0.09) quadriceps muscles (p < 0.05) (Figures 3B and 3D). Interestingly, microdystrophin content was lower in mdxRGT (0.84 ± 0.06) compared to mdxGT (1.14 ± 0.13) diaphragm muscles (p < 0.05) (Figures 3B and 3D; Table S3). H&E staining (heart and quadriceps) and dystrophic grading (quadriceps; scale, 0 [normal] to 4 [severe]; mean ± SE) were performed to evaluate pathophysiological characteristics associated with active dystrophic disease (Figures 4A and 4B). mdx mice displayed the highest degree of dystrophic pathology (3.4 ± 0.2) across all groups, whereas WT (0.0 ± 0.0) and WTR (0.25 ± 0.1) muscles appeared relatively normal. Muscles from both groups administered microdystrophin gene therapy, mdxGT (1.9 ± 0.2) and mdxRGT (1.6 ± 0.2), showed significant decreases in dystrophic pathology compared to mdx (p < 0.05). No significant differences were observed in histopathological improvements between mdxGT and mdxRGT groups. These data suggest that overall benefits to muscle histopathology following microdystrophin gene therapy are retained in treated animals that also perform voluntary exercise.
Figure 3.

Microdystrophin content
(A) MANEX1011b (DSHB) to detect microdystrophin (red) in mdx, mdxGT, and mdxRGT quadriceps (quad, 200×; scale bar, 100 μm), heart (400×; scale bar, 100 μm), and EDL muscles (200×, scale bar, 100 μm). (B) Western blot to detect microdystrophin and β-tubulin protein in mdxGT and mdxRGT quad, heart, and diaphragm (dia) muscles. (C) Immunofluorescence quantification of microdystrophin-positive fibers in mdxGT and mdxRGT quad, heart, and EDL muscles. No difference between groups. (D) Western blot quantification of microdystrophin protein, normalized to β-tubulin in mdxGT and mdxRGT quad, heart, and dia muscles. ∗mdxGT < mdxRGT; ∗∗mdxRGT < mdxGT. All comparisons p < 0.05. Mean ± SE mdxGT, n = 8; mdxRGT, n = 8.
Figure 4.

Histopathology
(A) Representative images for hematoxylin and eosin in heart (H) and quadriceps (Q). Heart images, 400× (scale bar, 50 μm), and quadriceps, 200× (scale bar, 100 μm). (B) Dystrophic grade in quadriceps. y axis, dystrophic grade 0–5. ∗mdx > all groups; ∗∗mdxGT, mdxRGT > WT, WTR. All comparisons p < 0.05. Mean ± SE. All groups n = 4.
Microdystrophin gene therapy improved running ability and muscle strength
To assess endurance, a run-to-exhaustion assay was performed using a treadmill. To assess hindlimb muscle strength and power, anesthetized mice were analyzed using an in vivo contractile force measurement system. Treadmill and in vivo contractile assays were conducted before treatment (baseline) and at specific time points after treatment (n weeks) in the same mice. Responses between groups could therefore be compared longitudinally.
Each mouse was subjected to a treadmill fatigue test, which started at a low walking speed, then increased every few minutes to a slow run, and then to a fast run speed. Speed increased until the mice could no longer run (i.e., exhaustion) according to pre-specified criteria. The time from start to end was recorded as the time to fatigue. At baseline (i.e., before treatment), all groups showed shorter times to fatigue compared to the WTR group (mean ± SE; 48.8 ± 12.7 min; p < 0.05; Figure 5A; Table S5). At 21 weeks post-treatment, mdx time to fatigue (24.8 ± 3.6 min) in the final treadmill test was significantly less than all other groups (p < 0.05). mdxGT animals (70.9 ± 4.0 min) demonstrated times to fatigue ∼2.8 fold greater than untreated mdx mice, reaching times similar to WT animals (67.0 ± 6.6 min). Notably, mdxRGT animals (130.0 ± 0.9 min) yielded a mean time to fatigue ∼1.8-fold greater than the mdxGT (p < 0.05) and similar to that of the WTR group (126.2 ± 2.0 min; Figure 5B; Table S5). Expressed relative to baseline, the mdxGT group improved more than the mdx group (434% versus 156%, p < 0.05), while the mdxRGT group showed the greatest improvement of all groups (e.g., 898% versus 434% for mdxGT, 405% for WTR; and 204% for WT; all, p < 0.05) (Figure 5C; Table S5). These data demonstrate that wheel running combined with microdystrophin gene therapy improved endurance ∼2-fold more than the gene therapy alone.
Figure 5.

Running capacity
(A and B) Treadmill running time at (A) 2 weeks pre- and (B) 21 weeks post-treatment. (A) ∗WTR > mdx groups. (B) ∗WTR and mdxRGT > all other groups; ∗∗mdx < all other groups. (C) Treadmill data expressed as percent change from baseline. ∗mdxGT > mdx; ∗∗mdxRGT > all groups. (D) Wheel-running distance per week. No difference between groups. (E) Running-wheel activity expressed as percent change from first week of running. ∗mdxRGT > WTR. All comparisons p < 0.05. Mean ± SE. WTR, n = 7; WT, n = 8; mdx, n = 7; mdxGT, n = 8; mdxRGT, n = 8.
There was no difference in running-wheel performance between the WTR and mdxRGT groups over the 21 weeks (mean ± SE; 54.0 ± 1.9 versus 50.5 ± 1.6 km/week, respectively; Figure 5D). The reason for the decreased distance for the mdxRGT group at 9 weeks is unclear, but by 10 weeks, the distance run by the mdxRGT group was similar to the 8-week values, and thereafter closely tracked the WTR run distance (Figure 5D). Expressed relative to week 1 running distance, the mdxRGT group demonstrated greater improvement than the WTR group post-treatment (193.8% ± 4.3% versus 125.8% ± 4.6%, respectively; p < 0.05; Figure 5E).
An in vivo isometric plantarflexor torque-frequency assay, which measures muscle strength in response to increasing electrical frequencies of stimulation, was performed on all mice 1 week before treatment (baseline) and at 2, 4, 8, 12, 16, and 20 weeks post-treatment. Torque (mN × m) produced was normalized to body mass (g) for each mouse ([mN × m]/g). Throughout the study, the mdx group produced the weakest torque response compared to all other groups (Figures 6A–6C; p < 0.05). At baseline, all the mdx groups (mdx, mdxGT, mdxRGT) produced maximum torque that was ∼66% of the WT and WTR groups combined (Figure 6A; p < 0.05). At two weeks post-treatment, at a stimulation frequency of 120 Hz (representative of the plateau torque), the mdxGT (mean ± SE in [mN × m]/g, 0.34 ± 0.04) and mdxRGT (0.34 ± 0.03) torques were comparable to the WT (0.34 ± 0.04) and WTR (0.35 ± 0.04) values and were significantly greater than the mdx value (0.25 ± 0.02, p < 0.05; Figure 6B). At 20 weeks post-treatment, the mdxGT (0.31 ± 0.02) and mdxRGT (0.34 ± 0.02) 120-Hz torques were similar to WT (0.35 ± 0.02) and greater than the mdx value (0.26 ± 0.01; p < 0.05; Figure 6C). Notably, throughout the study, mdxRGT and mdxGT 120-Hz torque values remained similar (Figure S1A; Table S6) post-treatment, indicating that microdystrophin gene therapy alone improved plantarflexion torque independent of running. Compared to baseline 120-Hz torque values, post-treatment torques at 120 Hz for the mdxGT and mdxRGT groups showed significant improvements compared to WT, WTR, and mdx (Figure 6D; Table S6; p < 0.05).
Figure 6.

In vivo contractile properties
(A–C) Torque-frequency curves (A) 1 week pre-treatment,∗ mdx < WT; (B) 2 weeks post, ∗mdx < all groups; and (C) 20 weeks post-treatment, ∗mdx < all groups; ∗∗mdxGT < mdxRGT, WTR, WT; †WT, mdxRGT < WTR. (D) Torque at 120 Hz as percent of baseline over time, ∗mdxGT, mdxRGT > all groups. (E) Power curves 1 week pre-treatment, ∗mdx < WT. (F) Power 2 weeks post treatment, ∗mdx < all groups; ∗∗WTR > all groups. (G) Power 20 weeks post-treatment, ∗mdx < all groups; ∗∗WTR > all groups; †mdxGT < WT, mdxRGT. (H) Peak power as percent of baseline over time, ∗mdxRGT > all groups. All comparisons (p < 0.05). Mean ± SE. WTR, n = 7; WT, n = 8; mdx, n = 7; mdxGT, n = 8; mdxRGT, n = 8.
Power was measured at the same time points as torque frequency. Power describes the muscle’s ability to produce torque at some rate (i.e., power = torque × velocity). Power is based on torque-velocity measurements shown in Figure S2B. Power is expressed in milliwatts (mW) and normalized to body mass (i.e., mW/g; Figure S1B; Table S7). At baseline, all the mdx groups (mdx, mdxGT, mdxRGT) produced a power curve that was ∼51% of the WT and WTR groups combined (Figure 6E; p < 0.05). At 2 weeks post-treatment, peak power (determined at 40% maximum force or 0.4 fractional load)24 for the mdxGT (mean ± SE in mW/g, 0.9 ± 0.1) and mdxRGT (0.9 ± 0.1) groups were similar to the WT (0.9 ± 0.1) group. At this same time point, WTR animals produced power greater than all groups (1.2 ± 0.1 mW/g; p < 0.05), whereas mdx power output was lower than all groups (0.6 ± 0.1 mW/g; p < 0.05; Figure 6F; Table S7). At 20 weeks post-treatment, there was a clear hierarchy of peak power output among the groups in the following order (from greatest to least, in mW/g): WTR (1.4 ± 0.2), mdxRGT (1.1 ± 0.1) and WT (1.1 ± 0.1), mdxGT (0.8 ± 0.2), and mdx (0.5 ± 0.1) (Figure 6G; Table S7). From these values, peak power for mdx was less than all groups, WTR was greater than all groups, and mdxGT was less than WT (all p < 0.05). mdxRGT peak power was similar to WT and mdxGT power was less than mdxRGT and WT (p < 0.05; Figure 6G), demonstrating that running complemented the microdystrophin gene therapy to improve power similar to WT. Relative to baseline power values, across all time points, mdxRGT mice showed greater improvements in power compared to all other groups (p < 0.05), peaking at 16 weeks post-treatment (296.3% ± 62.9%; Figure 6H; Table S7).
Microdystrophin gene therapy improved ex vivo muscle strength and power differentially in diaphragm and EDL
After mice were euthanized, ex vivo muscle contractile assays were performed on isolated diaphragm strips and EDL muscles to determine if running complemented the microdystrophin gene therapy. The diaphragm was assayed because it is considered one of the most affected muscles in the mdx mouse.25, 26, 27 The EDL was assayed because it is a muscle composed of primarily fast fibers,28 and mdx fast fibers are considered more susceptible to damage compared to slow fibers.29 For both muscles, it was important to determine if their contractile function was improved by the running combined with the gene therapy or if gene therapy alone was beneficial.
An ex vivo isometric force-frequency (F-F) assay, which measures the strength of an isolated muscle, was assessed in all mice. Ex vivo F-F relationships for diaphragm demonstrated differences between groups as shown in Figure 7A. When compared at 150 Hz, which is representative of the plateau force the two WT groups were not different (mean ± SE; WT, 17.0 ± 1.0; WTR, 15.6 ± 1.1 mN/mg) but produced greater forces compared to all mdx groups (p < 0.05; Figure 7A; Table S8). Notably, the mdxGT group produced greater diaphragm force (in mN/mg, 12.8 ± 1.7; p < 0.05) than both the mdxRGT (10.5 ± 1.4) and mdx (9.4 ± 0.5) groups. (Figure 7A; Table S8). The ex vivo stress-frequency relationships for EDL demonstrated differences between groups, as shown in Figure 7D. When compared at 150 Hz, the two WT groups were not different (mean ± SE; WT, 312.8 ± 27.4; WTR, 319.2 ± 23.0 mN/mm2) but produced greater stresses compared to all mdx groups (p < 0.05; Figure 7D; Table S8). Although the mdxGT (266.4 ± 23.0) and mdxRGT (287.0 ± 17.2) EDL stresses were similar, only the mdxRGT group produced greater stresses than the mdx group (229.6 ± 16.9 mN/mm2; p < 0.05; Figure 7D; Table S8). Relative to WT, mdxGT and mdxRGT diaphragm forces were only 75% and 62%, respectively, while EDL stresses were 85% and 92% of WT values, respectively. In the diaphragm, running blunted isometric force production in the mdxRGT group, suggesting a negative effect of running on the gene therapy, whereas in the EDL, running positively influenced the gene therapy (Tables S8 and S9).
Figure 7.

Ex vivo contractile properties of diaphragm and EDL
(A–C) Diaphragm. (D–F) EDL. (A) Diaphragm force-frequency curves, ∗mdx, mdxRGT < WT, WTR, mdxGT; ∗∗mdxGT < WT, WTR. (B) Diaphragm power curves, ∗mdx < all groups; ∗∗mdxRGT < WT, WTR, mdxGT; ∗∗∗mdxGT < WT, WTR. (C) Diaphragm isometric force loss during eccentric contractions, ∗mdx < all groups; ∗∗mdxGT, mdxRGT < WT, WTR. (D) EDL stress-frequency curves, ∗mdx < WT, WTR, mdxRGT; ∗∗mdxGT, mdxRGT < WT, WTR. (E) EDL power curves, ∗mdx < all groups; ∗∗mdxGT, mdxRGT < WT. (F) EDL isometric force loss during eccentric contractions. No differences between groups. All comparisons p < 0.05. Mean ± SE. WTR, n = 7; WT, n = 8; mdx, n = 7; mdxGT, n = 8; mdxRGT, n = 8.
Ex vivo power curves and peak power were assessed as described for in vivo power. Ex vivo power curves for diaphragm demonstrated differences between groups, as shown in Figure 7B. Overall, the ex vivo diaphragm power curve was lowest for the mdx group compared to all other groups (p < 0.05; Figure 7B). Peak power for mdx (mean ± SE, in mW/mg; 0.07 ± 0.01) and mdxRGT (0.10 ± 0.02) were less than both WT groups (WT, 0.17 ± 0.02; WTR, 0.18 ± 0.02 mW/mg; p < 0.05; Figure 7B; Table S8). Peak power for the mdxRGT group was less than for mdxGT (0.13 ± 0.02 mW/mg; p < 0.05; Figure 7B). Gene therapy alone increased diaphragm peak power to ∼193% of the mdx value, but it was reduced to ∼151% when combined with running (p < 0.05). However, the mdxGT and mdxRGT groups only achieved ∼77% and ∼60% of WT peak power, respectively, demonstrating only partial rescue. These data suggest running negatively influenced diaphragm function when combined with the gene therapy, which seems incongruent with the mdxRGT mice, demonstrating a final treadmill-running time similar to WTR and ∼5 times greater than mdx mice.
Ex vivo power curves for EDL demonstrated differences between groups as shown in Figure 7E. Peak power was least for the mdx group compared to all other groups (mean ± SE; 1.46 ± 0.14 mW/mm2; p < 0.05; Figure 7E). Power for the mdxGT (1.69 ± 0.18) and mdxRGT (1.72 ± 0.11) groups were similar, and both produced less power compared to the WT groups (WT, 1.90 ± 0.18 mW/mg; WTR, 1.82 ± 0.16 mW/mm2; p < 0.05; Figure 7E; Table S8). Peak power for the two WT groups was not different. As an indication of functional restoration, EDL mdxGT and mdxRGT peak power values were ∼89% and ∼91% of WT peak power, respectively. Considered together, these data suggest that overall power curves and peak power for the diaphragm and EDL were differentially affected when running was combined with the microdystrophin gene therapy. Tables S8 and S9 summarize the mean relative levels of performance for all groups compared to WT or changes post-treatment relative to baseline for many of the variables discussed above.
An ex vivo eccentric or stretch-injury protocol can be used to discriminate dystrophic from normal muscles, because absence of dystrophin is thought to make the muscles more susceptible to stretch injury.30 To assess injury, force loss as a percent of initial isometric force output was measured between initial and subsequent stretches.30 The diaphragm eccentric-injury protocol31 revealed the two WT groups were similar, with no force loss over the 5 stretches (Figure 7C; p < 0.05). The mdx group demonstrated the greatest loss of initial isometric force to (mean ± SE) 87.1% ± 1.0% compared to all other groups at the fifth stretch (Figure 7C; p < 0.05). The mdxGT (93.0% ± 2.1%) and mdxRGT (95.1% ± 2.5%) groups were similar and demonstrated an intermediate but greater force loss compared to the two WT groups (Figure 7C; p < 0.05). These data suggest the running (mdxRGT) did not complement the gene therapy alone (mdxGT) because the force losses for both groups were similar. For the EDL, the isometric plateau stress over successive eccentric contractions was compared to the initial stress. Force loss was minimal and similar in all groups (gain/loss of force ± 2%–3%; Figure 7F).
Microdystrophin gene therapy and voluntary wheel running mitigated some energetic deficits
Quadriceps were separated into red muscle and white muscle portions to assess mitochondrial respiration. In red fibers from quadriceps, succinate-stimulated respiration was approximately 30%–40% lower in mdx than WTR (Figure 8A; p < 0.05). This respiration decrement was significantly improved with gene therapy in the mdxGT samples, but the mdxRGT complemented the gene therapy to provide the greatest bioenergetic benefit. Mitochondrial respiration in white versus red fibers was lower, with mdx, WTR, and WT demonstrating similar mitochondrial respiration across conditions. Surprisingly, succinate-stimulated mitochondrial respiration in white fibers was depressed in mdxGT versus mdx (p < 0.5; Figure 8B), indicating a negative effect of gene therapy alone, but the decrement was rescued in mdxRGT (Figure 8B). These data indicate that mitochondria in red and white quadriceps fibers demonstrated a greater increase in mitochondrial respiration compared to mdx when the gene therapy and running were combined.
Figure 8.

Mitochondrial function
(A and B) Oxygen consumption (O2 flux) in red (A) and white (B) fibers from quadriceps. (A) ∗mdx < WT, WTR, mdxRGT; †mdxGT < WTR; ‡WT < WTR; #mdx, mdxGT < WTR, mdxRGT. (B) ∗mdxGT < WTR, mdx, mdxRGT; †mdxGT < all groups. All comparisons p < 0.05. Mean ± SE. WTR, n = 7; WT, n = 8; mdx, n = 7; mdxGT, n = 8; mdxRGT, n = 8.
Metabolic enzyme activities were greater in red muscle (soleus combined the red portion of gastrocnemius) versus white quadriceps muscle (p < 0.05; Figure S3). In red muscle, mdxRGT mice had greater citrate synthase activity versus WT and greater cytochrome c oxidase activity versus WT and mdx mice. β-hydroxyacyl-CoA dehydrogenase (βHAD) was the only enzyme in the mdx group that demonstrated less activity versus WTR (p < 0.05; Figure S3). In white muscle, there were no differences in enzyme activities between any of the groups. These data suggest changes in enzyme activities were not influenced by gene therapy or running combined with gene therapy, with the exception of citrate synthase and cytochrome c oxidase.
Discussion
The purpose of this study was to determine if voluntary running-wheel exercise for 21 weeks would complement a single systemic treatment of AAV9-CK8-microdystrophin to improve dystrophic muscle function in 7-week-old mdx mice. A single dose of 2e14 vg/kg was used to treat all animals in mdxGT and mdxRGT groups to evaluate the relative effect of voluntary exercise on the level of microdystrophin expression and to assess the overall outcomes in the mdxGT animals. vg copies measured in blood samples 30 min post-administration were similar in mdxGT and mdxRGT animals and remained similar between groups in blood and muscle samples collected 22 weeks post-treatment, confirming that both groups received similar treatments. In both the mdxGT and mdxRGT groups, fibrosis and immune infiltration were reduced compared to untreated mdx mice. Importantly, mdxRGT animals showed similar improvements in dystrophic grade compared to those in the mdxGT group, indicating that voluntary exercise did not have a detrimental effect on muscle health. In addition, the hearts, quadriceps, and EDLs of both groups demonstrated similar robust distribution of microdystrophin. Microdystrophin content was modestly increased in the quadriceps but was decreased in diaphragm of mdxRGT compared to mdxGT mice, suggesting that running affected the gene therapy differentially depending on the muscle. The decreased mdxRGT diaphragm microdystrophin content could account for the significantly lower diaphragm power compared to mdxGT. Plantarflexor power and mitochondrial energetics from the red portion of quadriceps were improved in the mdxRGT compared to the mdxGT mice. Surprisingly, the mitochondrial respiration of white muscle was significantly depressed by the gene therapy alone but was rescued when combined with running. Functional restoration in mdxGT mice expressed relative to WT values was 75%–101%, indicating only partial restoration for many assessments. However, in mdxRGT mice, functional restoration was greater than 91% for all assessments with the exception of diaphragm normalized force and power. Despite the depressed mdxRGT diaphragm power, treadmill-running endurance was greatly improved for mdxRGT mice. Nevertheless, the partial rescue of diaphragm function with microdystrophin with and without running warrants additional investigation. However, the overall outcomes support the conclusion that voluntary exercise performed in dystrophic animals treated with microdystrophin gene therapy can provide an additive benefit to whole body and muscle function while importantly maintaining improvements in dystrophic histopathology.
Literature reports reveal a broad range of mdx mouse ages at initiation of voluntary running-wheel exercise (e.g., 3–4 weeks;13, 14, 15 6 months;32 15.5 months33) and training durations (3–52 weeks).4,14 One consistent outcome was mdx mice that started running at any age demonstrated impaired running performance compared to similarly aged wild-type controls.14,32, 33, 34, 35 We anticipated, therefore, that running distance on the wheel per week would improve in 7-week-old mdx mice when combined with the gene therapy. The closely matched run distances in km/week for the mdxRGT and WTR mice over the training period suggested that running plus the microdystrophin gene therapy were complementary. Expressed relative to week 1 running distance, mdxRGT running-wheel activity was greater than WTR at 2 weeks and remained elevated throughout the study, with a mean increase of ∼194% versus 126%, respectively. Furthermore, WTR and mdxRGT final treadmill run times in minutes were similar and greater than all other groups. When expressed relative to baseline run times, the mdxRGT group showed a greater percent improvement (∼898%) than all other groups and was ∼2-fold greater than mdxGT (434%). Increased microdystrophin content in the quadriceps and comparable content in the EDL may have contributed to the greater endurance in the mdxRGT versus the mdxGT mice, though decreased diaphragm content may have been a negative influence. These differential muscle effects suggest that the relationship between exercise and microdystrophin content may not be uniformly beneficial. The mechanisms behind these differences are unclear but may include differences in biodistribution of the virus, differences related to fiber type composition, or differences in blood flow, physical stress, or damage to the affected areas during exercise.
Mitochondria are dysregulated in DMD and in the dystrophic mdx and D2.mdx mouse models;36,37 therefore, correction of dystrophic mitochondrial function would be advantageous. We predicted microdystrophin gene therapy alone (mdxGT) would increase mitochondrial capacity and metabolic enzyme activities of both red and white muscle, but the gene therapy combined with running (mdxRGT) would provide a greater increase in both. The improvement in treadmill performance in the mdxRGT mice suggests the running plus gene therapy induced positive mitochondrial adaptations in both red and white muscle to meet the increased ATP demand consistent with regular endurance activity.38,39 Congruent with the improved endurance for the mdxRGT compared to the mdxGT mice, mitochondrial respiration (oxygen consumption) was improved in red fibers of mdxRGT quadriceps. This improvement suggests that defects evident in the red fibers of mdx quadriceps were mitigated by the gene therapy alone, with additional benefits when combined with running. Succinate-stimulated mitochondrial respiration was 2.5–3 times higher in mdxRGT red fibers than in white fibers, consistent with red muscle fibers having a greater mitochondrial volume density than white muscle fibers.40 Surprisingly, the gene therapy alone significantly attenuated the complex II respiration of mdxGT compared to mdx white fibers, but this attenuation was rescued by running in the mdxRGT mice. The depressed white muscle respiration in the mdxGT mice was an unexpected negative result not readily explained mechanistically. Functionally, the mdxGT final treadmill exhaustion times were significantly greater than mdx and similar to WT non-runners, suggesting the impact of depressed mitochondrial respiration in white muscle on whole-body movement was minimal. These data demonstrate that voluntary running exercise complemented the AAV9-CK8-microdystrophin by improving mitochondrial respiration in red and white muscles.
Remarkably, only modest changes in mitochondrial metabolic enzyme activities were observed, with no differences evident between the mdxRGT and mdxGT muscles nor between the WTR and WT muscles. When the red gastrocnemius (and soleus) and white quadriceps were assayed, all enzymes had activities greater in red than white muscles, independent of group. Gene therapy alone did not change metabolic enzyme activities in red or white muscle, but when combined with running, red muscles of mdxRGT mice had greater citrate synthase activity versus WT and greater cytochrome c oxidase activity versus WT and mdx mice. These data also indicate that running plus gene therapy drives positive adaptations in mitochondria of red muscle. The mitochondrial data in red muscles suggest that positive adaptations contributed to the improved mdxRGT endurance capacity. It is also possible that adaptations in the heart contributed in response to the running, the gene therapy, or both.
Both positive and negative effects of exercise on dystrophic hearts have been reported in untreated mdx mice. Decreased LV function, ejection fraction, and shortening fraction were reported for mdx mice that ran from 4 to 20 weeks of age41 and increased dystrophin-related cardiomyopathy and cardiac fibrosis for mdx mice that ran from 7 to 11 weeks of age.42 However, mdx mice that performed voluntary wheel running from 4 to 56 weeks of age showed increased LV function and end-diastolic and systolic volumes.15 Thus, starting age and duration of exercise could impact mdx heart function in the absence of gene therapy.
Improvements in heart pathophysiology and cardiac function43, 44, 45, 46 have been reported for mdx mice treated with various microdystrophin constructs. For example, electrocardiograph abnormalities in mdx mice were corrected with the AAV9-ΔR4/ΔC microdystrophin including normalization of heart rate, and PT and QT intervals.43 mdx mice treated with rAAV2/6-ΔR4-23/ΔCT microdystrophin45 significantly improved baseline end-diastolic volume compared to untreated mdx mice, but not systolic function (e.g., end-systolic pressure). However, this construct did prevent cardiac-pump failure in response to a dobutamine challenge. This outcome suggests that similar protection of the mdxGT and mdxRGT hearts was possible with microdystrophin treatment. Although heart function was improved, Townsend et al.45 commented it was less clear how vascular tone would be modulated in the absence of microdystrophin incorporation into vascular smooth muscle. Modulation of smooth muscle to regulate vascular tone would be critical to increasing blood flow to working muscles (i.e., active/functional hyperemia) during exercise.47 This aspect of potential microdystrophin rescue should be further elucidated.
To our knowledge, no studies have explored long-term exercise combined with microdystrophin to assess performance outcomes as described herein. Running-wheel performance was only assessed over 13 days for microdystrophin-treated and untreated older mdx mice (age not reported) with the cumulative running distance significantly greater for the treated mdx mice.44 In a recent study, 6-month-old mdx mice followed for 6 months after treatment with microdystrophin gene therapy were subjected to treadmill running in the final month.17 VO2 max was determined before and after 6 forced treadmill-training sessions intended to exacerbate the dystrophic phenotype. The microdystrophin countered the exercise-induced muscle damage from the forced treadmill running and preserved grip strength and VO2 max (pre-post), indicating that microdystrophin provided some protection, though values did not reach WT levels.17 In the current study, the robust microdystrophin content observed in mdxRGT hearts and the clear ability of mdxRGT mice to run voluntarily suggests hearts were functioning well enough to support the stress of running. The running performance also suggests the mdxRGT diaphragms could effectively cope; however, their reduced microdystrophin content could have compromised function.
The diaphragm is considered the most affected striated muscle in the mdx mouse, and that most closely mimics the pathophysiology of DMD muscles.25 One of the key outcomes for gene therapy, therefore, should be the rescue of diaphragm function, as respiratory failure often leads to death.25 In the absence of treatment, moderate treadmill exercise improves respiratory function in mdx-DBA/2J mice,48 while in contrast, impaired diaphragm function in mdx mice after 52 weeks of running yielded an ∼60% decline compared to sedentary controls.15 Treatment with microdystrophin in mdx mice at least partially rescued muscle strength and resistance to eccentric injury in both limb and diaphragm muscles.49, 50, 51, 52 In addition, dual minidystrophin delivery to 4-week-old mdx4cv mice was beneficial to muscle function when mice were challenged by treadmill exercise for 8 weeks.53 In the current study, consistent with several reports, the mdx diaphragm F-F profile was significantly depressed compared to the two WT groups.54, 55, 56, 57 Both mdxGT and mdxRGT diaphragm strips demonstrated increased resistance to eccentric injury. However, the mdxRGT group produced forces less than mdxGT and comparable to mdx. In addition, mdxRGT peak power was surprisingly less than the mdxGT value, indicating that running depressed power achieved with gene therapy alone.
Full-length dystrophin restored to 41% content in mdx diaphragms by exon skipping oligonucleotide treatment in non-exercised mice at age 3 weeks was reduced ∼59% to 17% content after 3 weeks of treadmill training.58 Specific force and protection from eccentric injury were significantly greater in the non-exercised treated versus untreated mdx mice. However, because comparable functional data for the exercised, treated mdx mice was not reported, it was not clear how the reduced dystrophin content affected function.58 In the current study, mdxRGT diaphragm microdystrophin content was decreased ∼26% versus mdxGT. Relative to WT, power for both mdxGT (∼77%) and mdxRGT (∼60%) was only partially restored. Nevertheless, the mdxRGT mice improved their final treadmill exhaustion time dramatically. Is the reduced power solely due to changes in microdystrophin content? One possibility is that adaptations to running shifted the fiber type distribution to reduce power but still support endurance. Transcriptome analysis (RNA sequencing [RNA-seq]; Figure S4) revealed that microdystrophin gene therapy alone rescues the fiber type transcript distribution close to WT and WTR (Figure S4) but when combined with running promotes a slow fiber phenotype in mdxRGT versus mdxGT. Notably, the biggest difference between the WT and mdx transcripts was for IIx (63% versus 22%, respectively), consistent with ∼2-fold decrease in mdx diaphragm IIx/IIb fibers versus WT,57 and which could explain the lowest power of all groups in the mdx diaphragms. Compared to mdx, mdxGT diaphragms demonstrated decreased type I and IIa and increased IIx transcripts, which tracks with increased power for this group. Finally, compared to mdxGT, mdxRGT had transcripts that were unchanged for type IIx, increased for type I ∼2.3-fold and IIa ∼1.3-fold, and decreased for IIb ∼5.8-fold (Figure S4). Although transcript expression cannot be directly compared to protein content, this shift in mdxRGT transcript distribution could explain the decreased mdxRGT diaphragm power. Given the diaphragm is one of the most affected muscles in DMD, future studies should investigate (1) the potential negative influence of running on microdystrophin content in the diaphragm, (2) the influence of microdystrophin with and without running on fiber type transcripts and proteins, and (3) the relationship between running, microdystrophin, and respiratory function (e.g., breathing frequency, tidal volume, etc.).
Boys with DMD suffer from a progressive decrease in functional muscle mass and increased infiltration of fat, which presents as pseudohypertrophy (e.g., calf).59 As a direct consequence, muscle strength progressively deteriorates.60 As strength is lost, shortening speed (velocity) is lost, and this yields decreased power for movement. Similar impairments in contractile properties are evident in mdx skeletal muscles ex vivo49,50,61 and in vivo.56 Plantarflexors, which include the medial and lateral gastrocnemius, soleus, and plantaris, among others,62 are high-load locomotory muscles used to “toe off” during gait. Voluntary exercise improves mdx plantarflexor strength. mdx mice that started at age ∼4 weeks improved running capacity and plantarflexor torque after 12 weeks of running-wheel activity.13 Similarly, ex vivo soleus-specific tetanic force was improved after 12 weeks63 and 16 weeks35 of wheel running. Physiological benefits, however, may be age dependent and/or training duration dependent, as mdx mice that ran on wheels starting at age 6 months showed no changes in soleus strength or fatigability after 4 weeks.32 In the current study, untreated mdx mice consistently demonstrated the lowest plantarflexor torque. At 2 weeks of treatment, both the mdxRGT and mdxGT groups produced torques similar to the WT groups. At 20 weeks, mdxRGT produced greater normalized torque than mdxGT. However, expressed relative to baseline torque, both mdxGT and mdxRGT values were significantly greater than all other groups throughout the study post-treatment, suggesting microdystrophin improved relative plantarflexor torque independent of running. Expressed relative to baseline peak power, only mdxRGT values were greater than all other groups throughout the study, indicating that running was complementary to microdystrophin for plantarflexor power.
The mdx EDL F-F curve was significantly less than that of all other groups, except mdxGT. mdxGT and mdxRGT force outputs were similar, indicating running combined with gene therapy did not increase EDL force production. EDL mdx peak power was significantly less than all other groups, in agreement with previous findings.64 mdxGT and mdxRGT peak power were similar and only significantly less than WTR values. These data indicate microdystrophin treatment in mdx mice improved EDL power, independent of running. The minor effects in EDL due to the gene therapy may be related to the function of the EDL as a low-load locomotory muscle.62 In untreated mdx mice subjected to running, type I and IIa fibers were increased and type IIb fibers decreased in EDL muscles of mdx runners versus sedentary,14,65 while no changes in type I and IIa but decreased type IIx fiber types occurred in soleus muscles.65 As noted above for the diaphragm, it is possible there was a shift to a slow fiber phenotype to support endurance activity in microdystrophin-treated limb skeletal muscles activated during running. Future studies should explore this possibility.
In summary, these data support the idea that voluntary running complements microdystrophin gene therapy as demonstrated by positive adaptations in running capacity, increased muscle power, protection against eccentric injury in the diaphragm, and enhanced mitochondrial bioenergetics in red fibers. Although running decreased microdystrophin content and power in the mdxRGT diaphragm, the fiber type transcriptome analysis suggested power was traded for endurance capacity. Studies at the transcriptomic and proteomic levels could provide important insights into the mechanisms of adaptation for all muscles.
Clinical trials with microdystrophin are currently ongoing. With their potential success comes a key question: can partially rescued microdystrophin-expressing muscles safely endure physical activity? Our data indicate several positive outcomes that may help guide complementary exercise prescriptions in the clinic if promising treatments for DMD, such as microdystrophin gene therapy, are successful. The primary caveat is that at present, it is not clear if the complementary outcomes of voluntary activity will be similar in young microdystrophin-treated mdx mice (a mild model of DMD) and microdystrophin-treated DMD boys. Specifically, if the DMD boys undertake activities such as slow walking or gentle running, the threshold below which these physical activities remain complementary to the gene therapy are not currently defined. Voet et al.66 concluded that current evidence to support strength and endurance training in individuals with neuromuscular diseases, including DMD, is still uncertain. The authors recommended more robust methodology and greater numbers of participants to obtain the needed evidence. It is clear this advice should be extended to clinical studies of DMD boys treated with microdystrophin combined with exercise. Greater insight into the mechanisms of adaptation in both pre-clinical (including older and/or physically challenged mdx mice, and other DMD mouse/rat models) and clinical studies will hopefully identify the types and amounts of activity that will safely complement the microdystrophin gene therapy.
Materials and methods
Animal studies
All animal experiments were approved by the Institutional Animal Care and Use Committee at Virginia Tech and in concordance with NIH guidelines. Four-week-old WT (C57BL-10ScSnJ/Jax strain #000476) and mdx (C57BL/10ScSn-DMDmdx/Jax strain #001801) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Mice were group-housed (3–4 mice/cage) in a temperature-controlled room (21.1°C,) with a 12-h light/12-h dark cycle and were given access to water and chow (Harlan-Teklad 2018) ad libitum. The mice were purchased in cohorts of 10 mice (age 4 weeks) per week for 4 weeks to stagger the necessary assays post-sacrifice. Of the original 40 mice, 2 mice died early in the study, 1 WTR and 1 mdx due to dehydration. Subsequently, in addition to the cage lixit system, all mice were provided hydrogel (ClearH2O, Portland, ME, USA) packs to ensure hydration.
One day after arrival of a given cohort (−3 weeks; Figure 1), the body mass of each mouse was determined. The mice were assigned to one of 5 treatment groups, so the average body mass was similar between the two WT groups and similar between the three mdx groups. The mice acclimated for an additional 5 days before the baseline treadmill test.
AAV construct and delivery
Recombinant AAV9 was produced at the University of Pennsylvania Vector Core (https://www.med.upenn.edu/gtp/vectorcore). The gene-of-interest (GOI) plasmid used for production coded for a four-repeat microdystrophin sequence, which does not contain the R16/R17 neuronal nitric oxide synthase (nNOS) binding-domain-specific repeats, was under the control of the CK8 skeletal and cardiac muscle-specific promoter. AAV9-CK8-microdystrophin at 2e14 vg/kg or an equivalent volume of vehicle (phosphate-buffered saline + 0.001% pluronic F-68) was injected via tail vein of each mouse in the mdxGT and mdxRGT groups at age 7 weeks (time 0 [T]; Figure 1) with a U100 28G × 1/2 insulin syringe (Becton Dickson, Franklin Lakes, NJ, USA).
Blood draws
Whole blood was collected from the submandibular vein 30 min following treatment administration and by cardiac puncture at sacrifice into 0.5 mL K2 EDTA Microtainer MAP tubes (Becton Dickson, Franklin Lakes, NJ, USA) and stored at −80°C for subsequent analysis of AAV9-microdystrophin vg copies.
Biodistribution
In addition to whole blood, heart, diaphragm, left quadriceps, left gastrocnemius, and left extensor digitorum longus were harvested at sacrifice in a manner that prevented cross-contamination, snap frozen in liquid nitrogen, and stored at −80°C until gDNA was extracted. gDNA was isolated using a DNeasy blood and tissue kit (QIAGEN, Valencia, CA, USA) according to the manufacturer’s instructions, and vector gDNA concentrations were determined using the NanoDrop One system (Thermo Fisher, Carlsbad, CA, USA). AAV vg copies present in gDNA were quantified by real-time PCR using the QuantStudio 3 Real-Time PCR system (Thermo Fisher, Carlsbad, CA, USA) according to the manufacturer’s instructions. Results were analyzed using the QuantStudio Design & Analysis v1.4.1 software with the threshold, start baseline, and stop baseline set automatically by the software. In brief, primers and probes designed to the human codon-optimized dystrophin (HuDys) sequence of the AAV vector were used. A standard curve was performed using plasmid DNA containing the same codon-optimized HuDys target sequence. PCR reactions contained a total volume of 50 μL and were run as follows: 50°C for 2 min, 95°C for 10 min, and 45 cycles of 95°C for 15 s and 60°C for 1 min. DNA samples were assayed in triplicate. To assess PCR inhibition, the third replicate was spiked with plasmid DNA at a ratio of 100 copies/μg gDNA. If this replicate was greater than 40 copies/μg gDNA, then the results were considered acceptable. If a sample contained greater than or equal to 100 copies/μg gDNA, it was considered positive for vgs. If a sample contained less than 100 copies/μg gDNA, it was considered negative for vgs. Vector copy numbers reported are normalized per μg gDNA. Assay controls included the following: no template control (NTC) with acceptability criteria < 15 copies and an established study-specific standard curve slope range (±0.3 of the study mean slope determined from three individual standard preparations and runs). Data were reported as AAV genome copies per μg total gDNA. Statistical analysis between groups was performed with an unpaired t test with significance set at p < 0.05.
Baseline and final treadmill fatigue tests
All mice were acclimated to a mouse treadmill (TSE Systems, Chesterfield, MO, USA) for 3 days before the study began (baseline test) and again shortly before sacrifice (final test). On the fourth day, the mice were run to exhaustion and total run time and distance recorded. Exhaustion, or failure to run, was defined as the inability of the mouse to keep running after aggressive physical nudges with a cotton-tipped swab. Each mouse was allowed three failures before it was considered fatigued. Time to fatigue was recorded. Relative treadmill time was calculated as final treadmill time/baseline treadmill time × 100%. The training and fatigue protocols are described in Table S4. This treadmill protocol was developed as an endurance run to fatigue. Another method to assess whole-animal performance is described in Rocco et al.67
Body mass
Body mass was obtained on a PT1200 balance (Sartorius, Bohemia, NY, USA) to the nearest 0.1 g.
Voluntary wheel running
At time 0 for each cohort, mice in the WTR and mdxRGT groups were single housed in cages with running wheels (15-cm diameter). The wheels were instrumented to count revolutions every 2 min, and the values were recorded by a laptop computer into an excel spreadsheet using our lab’s custom software. The software determined the distance per revolution (m) using the equation circumference = 2πr. Running data were collected continuously for 24 h each day for 7 days. At the end of the 7-day period, the resulting spreadsheet was downloaded for additional data manipulation (e.g., distance, km/week) and analysis, and the program was restarted to collect data for the next week. Mice in the mdx, mdxGT, and WT groups were single housed in cages with locked running wheels. Relative running-wheel data were calculated as each respective mouse value at each time point/that mouse’s week 1 running distance × 100%.
In vivo skeletal muscle contractile properties
In vivo isometric plantarflexor torque, velocity, and fatigue were assessed in all mice 1 week prior to, and at 2, 4, 8, 12, 16, and 20 weeks after, treatment (Figure 1). Body mass was recorded prior to each experiment.
Equipment and software
In vivo contractile properties were determined with Aurora Scientific (ASI, Aurora, ON, Canada) equipment and software including a 5N dual-mode servomotor (305C-FP-M) with a foot pedal displacement maximum of ∼40° (±20° from neutral position) and a 701C high-power follow stimulator set to constant current. Current was optimized for each mouse for each in vivo experiment (range, 45–65 mA). Dynamic Muscle Control (DMC) software controlled the timing and frequency of the stimulations and collection of force, torque, and foot pedal displacement. As noted below, the DMC units were set to either torque (mN × m) or force (mN) depending on the assay. The mouse platform was maintained at 37°C by an HTP-1500 heat-therapy pump (Adroit Medical Systems, Loudon, TN, USA). Dynamic Muscle Analysis (DMA) software was used to analyze the force, torque, and displacement data.
Preparation and recovery
Mice were anesthetized with isoflurane (VetOne Fluriso, Boise, ID, USA) delivered through an isoflurane vaporizer (VetEquip, Livermore, CA, USA) at 3% isoflurane, 1.0 L/min O2 in a sealed anesthesia box for 3 min. Mice were then placed on the temperature-controlled platform (37°C) of the contractile apparatus (ASI), and isoflurane was maintained via nosecone (1.5%–3% isoflurane, 1.0 O2 L/min). VaporGuard activated charcoal canisters (VetEquip, Livermore, CA, USA) were used to scavenge excess isoflurane. The eyes were treated with Puralube (Decra, Overland Park, KS, USA) to prevent drying. The right hindlimb was shaved, hair remover applied (Nair Hair Remover Lotion, Ewing, NJ, USA) for 30 s, cleaned with 2-inch × 2-inch gauze and tap water, and swabbed with providone-iodine (Betadine Solution Swabsticks, Stamford, CT, USA). The knee was clamped so the tibia was 90° to the femur. The foot at 90° to the tibia was secured with clear Transpore surgical tape (M3, St. Paul, MN, USA) to the foot pedal of the dual mode servomotor. The mouse tail was taped (M3, St. Paul, MN, USA) loosely to the platform to keep it clear of the foot pedal.
Following the experiment, mice recovered in a sealed chamber gassed with1.0 L/min O2. After mice were fully awake and mobile in the recovery chamber, they were placed in a clean cage with an active or locked running wheel as necessary and returned to the vivarium.
Electrodes and optimal current
Electrical stimulations were delivered from the 701C stimulator via F-E2 platinum-tipped needle electrodes (Natus Manufacturing, Gort, Ireland). The two electrodes were taped together with ∼3 mm between the tips. The paired electrodes were inserted ∼0.5–1.0 mm deep, ∼1.0 mm distal, lateral to the knee, and parallel to the tibia to depolarize the tibial nerve.
Isometric torque frequency
Torque is determined by the force (mN) applied to, and multiplied by the length of, the foot pedal (m). DMC units were therefore set to mN × m. Isometric torque was determined with the foot at 90° to the tibia (neutral position) over a series of stimulations at increasing frequencies: 1, 10, 30, 50, 65, 80, 100, 120, 150, 180, and 200 Hz. Data were plotted as torque normalized to mouse body mass (mN × m/g) versus frequency. Relative torque data were calculated as each respective mouse value at each time point/that mouse’s baseline torque × 100%.
Torque velocity and power
DMC units were set to mN. 2 min following the end of the torque-frequency assay, maximum isometric tetanic forces were recorded at 150 Hz with the foot pedal in dorsiflexion (+20°), in the neutral position (0°), and plantarflexion (−20°). The plantarflexor isometric tetanic force was greatest when the foot was dorsiflexed, in agreement with Ashton-Miller et al.,68 and was used to determine fractional loads. To maximize the range for displacement (full range from +20° to −20°, total 40°), the foot pedal was moved by software into the dorsiflexed position (+20°) prior to determining displacement at a given load.
Force velocity was determined with the tetanic afterload method at a stimulation frequency of 150 Hz.24,69 In this method, a load as a proportion of the maximum isometric tetanic force is set on the servomotor, and when the plantarflexor force during stimulation equals the load, the foot pedal is displaced. Displacement divided by time yields velocity. We determined velocity at fractional loads 0.30, 0.40, 0.75, and 0.90 of the maximal dorsiflexion tetanic force value to subsequently calculate maximum torque, which typically occurs at 0.40 fractional load.24 Because the time to full displacement became shorter as the fractional load decreased, we modified the 150 Hz stimulation duration at the various fractional loads: 0.2 s (0.90); 0.1 s (0.75, 0.50, 0.40); and 0.05 s (0.30). At fractional loads less than 0.30, there was insufficient time to capture the peak velocity before the pedal exceeded the displacement limit. Displacement in degrees was converted to mm, data were plotted as fractional load versus velocity in mm/s, and the Hill equation was used to fit a curve and generate an equation (GraphPad Prism, GraphPad Software, La Jolla, CA, USA).
Power = force × velocity. Power was determined as the product of a given load in mN and the associated velocity (mm/s) and then expressed in mW. A curve was fit to the resulting power values and the data plotted to compare power curves between the groups and to determine the peak power for each (GraphPad Prism). Relative power data were calculated as each respective mouse value at each time point/that mouse’s baseline power × 100%.
Tissue collection
Mice were deeply anesthetized intraperitoneally (i.p.) with ketamine/xylazine (200/100 mg/kg, respectively). Blood was obtained by cardiac puncture with a 1-mL insulin syringe fitted with a 26G × 5/8 needle (Becton Dickson, Franklin Lakes, NJ, USA). Immediately following, the mouse was euthanized by cervical dislocation and then skinned. The mouse spinal cord was transected just below the diaphragm. From the lower-body portion, tissues were harvested from the hindlimbs, while simultaneously, from the upper half, the diaphragm was dissected whole with ribs attached, and then the heart removed.
Mitochondrial respiration
Quadriceps white and red fiber portions
Visually, there is a distinct contrast between superficial white (e.g., vastus lateralis and rectus femoris, fast fibers) and deep red (vastus intermedius, ∼50% slow, ∼50% fast fibers) portions of quadriceps.70 Fiber type distribution of whole quadriceps was not different between mdxGT and mdxRGT groups (Figure S5), but because the white portion represents a greater proportion than the red, it likely biased the fiber type distribution analysis. We therefore separated left quadriceps into white and red portions as described to conduct mitochondrial respirometry studies.70 Permeabilized muscle fibers were prepared as described.71 Small portions of white and red quadriceps muscle were placed in ice cold BIOPS (pH 7.1) (7.23 mM K2EGTA, 2.77 mM Ca-K2EGTA, 20 mM imidazole, 20 mM taurine, 5.7 mM ATP, 14.3 mM PhosphoCreatine, 6.56 mM MgCl2∗6H2O, 50 mM MES). Small fiber bundles (∼2 × ∼5 mm for white muscle; ∼1 × ∼2 mm for red muscle; three separate fiber bundles per muscle sample) were separated along the longitudinal axis with fine-tipped 5/45 Dumont forceps (Roboz, Gaithersburg, MD, USA) under magnification with a Leica M80 stereo microscope (Leica Microsystems, Wetzlar, Germany).
Saponin treatment
Bundles were permeabilized by treatment with 30 μg/mL saponin (Sigma, St. Louis, MO, USA) for 30 min at 4°C on a rocking platform. Bundles were transferred to 8 mL wash buffer (105 mM MES [pH 7.1], 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2∗6H20, 0.5 mg/mL BSA, 1 μM EGTA) and incubated at 4°C for 15 min with rocking.
Assay procedures
High-resolution O2 consumption measurements were performed on the permeabilized fiber bundles in 2 mL buffer Z (105 mM K-MES, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2∗6H20, 0.5 mg/mL BSA, 1 μM EGTA, 20 mM creatine monohydrate (pH 7.1), and sterile filtered with a PVDF syringe filter (0.22 μm, 30 mm CELLTREAT Scientific Products, Pepperell, MA, USA) using an Oroboros Oxygraph-2k (Oroboros Instruments, Innsbruck, Austria) at 37°C with stirring at 750 rpm; measurements were acquired every 2 s. Blebbistatin (10 μM) was added to the chamber to prevent fiber contraction, and the chamber was hyperoxygenated to 400–450 μM O2. After closing the chamber of the O2k to maintain a known oxygen content, oxygen consumption rate (i.e., oxygen flux; JO2) was monitored until it reached a constant rate. The following were injected into the chamber sequentially once the oxygen flux returned to a constant rate after each addition: 2.5 mM malate, 5 mM pyruvate, 5 mM glutamate, and two injections of ADP at 2.5 mM to bring the final concentration to 5 mM ADP. Succinate was then added at 10 mM. Each substrate was used to test a different aspect of the mitochondrial respiratory chain (see Table S10). NADH (complex I)-supported respiration is the rate of oxygen consumption after addition of pyruvate, glutamate, and malate to the chamber. Contribution of complex II to respiration is assessed by calculating the difference between rates of respiration following ADP and succinate additions, respectively. The integrity of the outer mitochondrial membrane was assessed by injection of 10 μM cytochrome c, followed by addition of FCCP in 0.5-μM increments (data not shown). Each assay was completed before O2 concentration was depleted below 150 μM to avoid O2 diffusion limiting the reactions.71
Upon completion of the assay, each fiber bundle was removed from the chamber, rinsed in dH2O for 5 min, blotted dry, frozen at −80°C, and lyophilized overnight. Mass of the dried fiber bundles was measured with a Thermo Cahn C-35 microbalance (Thermo Scientific, Waltham, MA, USA) to the nearest 0.1 μg, and JO2 measurements normalized to dry bundle mass.
Western blot
Total protein was isolated from quadriceps and hearts of mdxGT and mdxRGT animals using RIPA buffer (Millipore, #20-188) containing protease inhibitors (Roche, #11836153001) (Roche, #05892970001). Proteins were transferred to nitrocellulose membranes and probed for microdystrophin using the NCL-DYSB primary antibody (Leica, 6052319, 1:50) and a horseradish peroxidase (HRP)-linked anti-mouse insulin growth factor (IgG) secondary antibody (Jackson Labs, #715-035-150, 1:4,000). Signal detection was performed using the Clarity Western ECL substrate (GE Life Sciences, #RPN2236) and measured by digital imaging using the ChemiDoc MP system (Bio-Rad). Quantification of band signal intensities normalized to β-tubulin (Abcam, Ab21058, 1:5,000) was performed with Image Lab Software (Bio-Rad). Relative protein levels were evaluated using a one-way ANOVA with Prism software (GraphPad Prism 8).
Immunofluorescence
Isopentane-frozen quadriceps muscle specimens were sectioned at 8-μm thickness and immunostained per standard techniques for dystrophin (Leica, NCL-DYS2), microdystrophin (Leica, NCL-DYSB), laminin (Sigma, L9393), myosin heavy-chain type 1 (Developmental Studies Hybridoma Bank [DSHB], BA-D5), or myosin heavy-chain type 2A (DSHB, SC-71). Evaluation of percent dystrophin- or microdystrophin-positive fibers was performed by a neuropathologist using a standard fluorescent microscope and estimated to the nearest 5%. For fiber type analysis, slides were scanned with an Olympus VS120 whole slide scanner, and scanned images were evaluated with VisioPharm software using 20× magnification. Immunostaining on the far-red channel highlighted laminin expression, which identified edges of myofibers. Type 1 myosin heavy-chain expression was visualized on the red channel, type 2A myosin heavy-chain expression was visualized on the green channel, and together these fibers were assessed as the oxidative fiber population.72 Fibers negative for type 1 and type 2A myosin heavy chain were assumed to be glycolytic and categorized separately. The Visiopharm software identified muscle fibers using laminin expression to determine cell edges and fiber size limits of between 5 and 110 μm, which was assessed by manual measurement of the smallest and largest fibers in samples from this dataset. Minferet diameter data was obtained for all muscle fibers and with respect to fibers that were positive for type 1, type 2A, or negative for both type 1 and 2A myosin heavy chain. Frequency histograms were constructed using Prism software (GraphPad Prism 8). Fiber size data and fiber proportion data were analyzed using a two-way ANOVA with Prism software (GraphPad Prism 8).
Determination of dystrophic grade
H&E-stained sections of frozen quadriceps-muscle tissue were assessed by a board-certified neuropathologist with respect to findings associated with active dystrophic disease, including myofiber degeneration, active myofiber regeneration (basophilic fibers), and inflammation. The goal of this approach was to capture histological features that were indicative of myofiber degeneration and regeneration in the post-treatment period, while excluding indications of longer-standing muscle damage that may have occurred prior to treatment. The area of each sample displaying these findings was visually estimated, and the following grading scheme was applied: grade 0 = normal, grade 1 = chronic regenerative changes only, grade 1.5 = very mild (<5% of muscle area with active dystrophic pathology), grade 2 = mild (6%–20% of muscle area with active dystrophic pathology), grade 2.5 = mild to moderate (21%–30% of muscle area with active dystrophic pathology), grade 3 = moderate (30%–50% of muscle area with active dystrophic pathology), grade 3.5 = moderate to severe (51%–60% of muscle area with active dystrophic pathology) and grade 4 = severe (>60% of muscle area with active dystrophic pathology). Areas indicative of chronic regeneration (internally nucleated fibers with appropriate eosinophilia, endomysial fibrosis, or fatty infiltration) were not integrated into the grading of dystrophic severity, as it is possible that the damage associated with these changes occurred before treatment. Dystrophic severity of each treatment group was analyzed with a repeated-measure one-way ANOVA using Prism software (GraphPad Prism 8).
Additional methods, including metabolic enzyme assays, ex vivo contractile properties, and transcriptome analysis are in the Supplemental materials and methods. Mouse morphology at sacrifice is presented in Table S11.
Acknowledgments
Funding was provided by Solid Biosciences, Inc. We are grateful for the contribution of A.J. Mansueto for data analysis.
Author contributions
Conceptualization, R.W.G., J.P.G., C.A.M., and M.W.L.; methodology, R.W.G., J.P.G., C.A.M., D.A.B., and M.W.L.; investigation, R.W.G., S.E.H., K.E.B, D.D.F., A.K.A., H.Z., R.P.M., K.E.C., M.J.P., M.A.V.A., and S.N.K.; formal analysis, R.W.G., S.E.H., K.E.B, D.D.F., A.K.A., H.Z., R.P.M., K.E.C., M.J.P., M.A.V.A., J.B.D., D.L.M., and S.N.K.; resources, R.W.G.; writing – original draft, R.W.G., S.E.H., D.D.F., K.E.B, and M.W.L.; writing – review & editing, R.W.G., S.E.H., J.P.G., M.W.L., J.B.P., J.B.D, and K.E.C.; visualization, R.W.G, S.E.H., M.W.L., M.A.V.A., K.E.C., and M.J.P.; supervision, R.W.G. and J.P.G.; project administration, R.W.G. and A.K.A.; funding acquisition, R.W.G. and J.P.G.
Declaration of interests
Research funding for R.W.G. was provided by Solid Biosciences, Inc.; R.W.G. is a consultant for Lion Therapeutics, Inc., Cytokinetics, Inc., Prothelia Inc., and Anagenesis Biotechnologies, Inc. Research funding for M.W.L. was provided by Solid Biosciences, Audentes Therapeutics, Taysha Therapeutics, and Prothelia. M.W.L. was a board member for Solid Biosciences and Audentes Therapeutics during the period when studies were performed. M.W.L. is a consultant for Audentes Therapeutics, AGADA Biosciences, Encoded Therapeutics, Kate Therapeutics, Lacerta Therapeutics, Affinia Therapeutics, Modis Therapeutics, Dynacure, and Biomarin. Research funding and D.L.M. was provided by Audentes Therapeutics and Cytokinetics. D.L.M. is a consultant for Audentes Therapeutics, Kate Therapeutics, and Affinia Therapeutics. Research funding for K.E.C. was provided by Solid Biosciences, Inc.; no other remuneration was received. J.P.G. and C.A.M. are employees of Solid Biosciences, Inc. All other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.02.024.
Supplemental information
References
- 1.Yiu E.M., Kornberg A.J. Duchenne muscular dystrophy. J. Paediatr. Child Health. 2015;51:759–764. doi: 10.1111/jpc.12868. [DOI] [PubMed] [Google Scholar]
- 2.Nigro G., Comi L.I., Politano L., Bain R.J.I. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990;26:271–277. doi: 10.1016/0167-5273(90)90082-g. [DOI] [PubMed] [Google Scholar]
- 3.Aartsma-Rus A., Van Deutekom J.C., Fokkema I.F., Van Ommen G.J., Den Dunnen J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34:135–144. doi: 10.1002/mus.20586. [DOI] [PubMed] [Google Scholar]
- 4.Rybakova I.N., Patel J.R., Ervasti J.M. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J. Cell Biol. 2000;150:1209–1214. doi: 10.1083/jcb.150.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao Q.Q., McNally E.M. The Dystrophin Complex: structure, function and implications for therapy. Compr. Physiol. 2015;5:1223–1239. doi: 10.1002/cphy.c140048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lapidos K.A., Kakkar R., McNally E.M. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ. Res. 2004;94:1023–1031. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- 7.Shieh P.B. Emerging Strategies in the Treatment of Duchenne Muscular Dystrophy. Neurotherapeutics. 2018;15:840–848. doi: 10.1007/s13311-018-00687-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chamberlain J.R., Chamberlain J.S. Progress toward Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2017;25:1125–1131. doi: 10.1016/j.ymthe.2017.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duan D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018;26:2337–2356. doi: 10.1016/j.ymthe.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grange R.W., Call J.A. Recommendations to define exercise prescription for Duchenne muscular dystrophy. Exerc. Sport Sci. Rev. 2007;35:12–17. doi: 10.1249/01.jes.0000240020.84630.9d. [DOI] [PubMed] [Google Scholar]
- 11.Markert C.D., Ambrosio F., Call J.A., Grange R.W. Exercise and Duchenne muscular dystrophy: toward evidence-based exercise prescription. Muscle Nerve. 2011;43:464–478. doi: 10.1002/mus.21987. [DOI] [PubMed] [Google Scholar]
- 12.Spaulding H.R., Selsby J.T. Is Exercise the Right Medicine for Dystrophic Muscle? Med. Sci. Sports Exerc. 2018;50:1723–1732. doi: 10.1249/MSS.0000000000001639. [DOI] [PubMed] [Google Scholar]
- 13.Baltgalvis K.A., Call J.A., Cochrane G.D., Laker R.C., Yan Z., Lowe D.A. Exercise training improves plantar flexor muscle function in mdx mice. Med. Sci. Sports Exerc. 2012;44:1671–1679. doi: 10.1249/MSS.0b013e31825703f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Call J.A., Voelker K.A., Wolff A.V., McMillan R.P., Evans N.P., Hulver M.W., Talmadge R.J., Grange R.W. Endurance capacity in maturing mdx mice is markedly enhanced by combined voluntary wheel running and green tea extract. J Appl Physiol (1985) 2008;105:923–932. doi: 10.1152/japplphysiol.00028.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selsby J.T., Acosta P., Sleeper M.M., Barton E.R., Sweeney H.L. Long-term wheel running compromises diaphragm function but improves cardiac and plantarflexor function in the mdx mouse. J Appl Physiol (1985) 2013;115:660–666. doi: 10.1152/japplphysiol.00252.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schill K.E., Altenberger A.R., Lowe J., Periasamy M., Villamena F.A., Rafael-Fortney J.A., Devor S.T. Muscle damage, metabolism, and oxidative stress in mdx mice: Impact of aerobic running. Muscle Nerve. 2016;54:110–117. doi: 10.1002/mus.25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodgers B.D., Bishaw Y., Kagel D., Ramos J.N., Maricelli J.W. Micro-dystrophin Gene Therapy Partially Enhances Exercise Capacity in Older Adult mdx Mice. Mol. Ther. Methods Clin. Dev. 2019;17:122–132. doi: 10.1016/j.omtm.2019.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayes A., Williams D.A. Contractile function and low-intensity exercise effects of old dystrophic (mdx) mice. Am. J. Physiol. 1998;274:C1138–C1144. doi: 10.1152/ajpcell.1998.274.4.C1138. [DOI] [PubMed] [Google Scholar]
- 19.Lynch G.S., Hayes A., Lam M.H., Williams D.A. The effects of endurance exercise on dystrophic mdx mice. II. Contractile properties of skinned muscle fibres. Proc. Biol. Sci. 1993;253:27–33. doi: 10.1098/rspb.1993.0078. [DOI] [PubMed] [Google Scholar]
- 20.McGreevy J.W., Hakim C.H., McIntosh M.A., Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis. Model. Mech. 2015;8:195–213. doi: 10.1242/dmm.018424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 22.Mack D.L., Poulard K., Goddard M.A., Latournerie V., Snyder J.M., Grange R.W., Elverman M.R., Denard J., Veron P., Buscara L. Systemic AAV8-Mediated Gene Therapy Drives Whole-Body Correction of Myotubular Myopathy in Dogs. Mol. Ther. 2017;25:839–854. doi: 10.1016/j.ymthe.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramos J.N., Hollinger K., Bengtsson N.E., Allen J.M., Hauschka S.D., Chamberlain J.S. Development of Novel Micro-dystrophins with Enhanced Functionality. Mol. Ther. 2019;27:623–635. doi: 10.1016/j.ymthe.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graber T.G., Kim J.-H., Grange R.W., McLoon L.K., Thompson L.V. C57BL/6 life span study: age-related declines in muscle power production and contractile velocity. Age (Dordr.) 2015;37:9773. doi: 10.1007/s11357-015-9773-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stedman H.H., Sweeney H.L., Shrager J.B., Maguire H.C., Panettieri R.A., Petrof B., Narusawa M., Leferovich J.M., Sladky J.T., Kelly A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 26.Coirault C., Pignol B., Cooper R.N., Butler-Browne G., Chabrier P.-E., Lecarpentier Y. Severe muscle dysfunction precedes collagen tissue proliferation in mdx mouse diaphragm. J Appl Physiol (1985) 2003;94:1744–1750. doi: 10.1152/japplphysiol.00989.2002. [DOI] [PubMed] [Google Scholar]
- 27.Burns D.P., Roy A., Lucking E.F., McDonald F.B., Gray S., Wilson R.J., Edge D., O’Halloran K.D. Sensorimotor control of breathing in the mdx mouse model of Duchenne muscular dystrophy. J. Physiol. 2017;595:6653–6672. doi: 10.1113/JP274792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burkholder T.J., Fingado B., Baron S., Lieber R.L. Relationship between muscle fiber types and sizes and muscle architectural properties in the mouse hindlimb. J. Morphol. 1994;221:177–190. doi: 10.1002/jmor.1052210207. [DOI] [PubMed] [Google Scholar]
- 29.Moens P., Baatsen P.H., Maréchal G. Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J. Muscle Res. Cell Motil. 1993;14:446–451. doi: 10.1007/BF00121296. [DOI] [PubMed] [Google Scholar]
- 30.Warren G.L., Lowe D.A., Armstrong R.B. Measurement tools used in the study of eccentric contraction-induced injury. Sports Med. 1999;27:43–59. doi: 10.2165/00007256-199927010-00004. [DOI] [PubMed] [Google Scholar]
- 31.Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carter G.T., Wineinger M.A., Walsh S.A., Horasek S.J., Abresch R.T., Fowler W.M., Jr. Effect of voluntary wheel-running exercise on muscles of the mdx mouse. Neuromuscul. Disord. 1995;5:323–332. doi: 10.1016/0960-8966(94)00063-f. [DOI] [PubMed] [Google Scholar]
- 33.Kogelman B., Putker K., Hulsker M., Tanganyika-de Winter C., van der Weerd L., Aartsma-Rus A., van Putten M. Voluntary exercise improves muscle function and does not exacerbate muscle and heart pathology in aged Duchenne muscular dystrophy mice. J. Mol. Cell. Cardiol. 2018;125:29–38. doi: 10.1016/j.yjmcc.2018.10.008. [DOI] [PubMed] [Google Scholar]
- 34.Wineinger M.A., Abresch R.T., Walsh S.A., Carter G.T. Effects of aging and voluntary exercise on the function of dystrophic muscle from mdx mice. Am. J. Phys. Med. Rehabil. 1998;77:20–27. doi: 10.1097/00002060-199801000-00004. [DOI] [PubMed] [Google Scholar]
- 35.Hayes A., Williams D.A. Beneficial effects of voluntary wheel running on the properties of dystrophic mouse muscle. J Appl Physiol (1985) 1996;80:670–679. doi: 10.1152/jappl.1996.80.2.670. [DOI] [PubMed] [Google Scholar]
- 36.Barker R.G., Wyckelsma V.L., Xu H., Murphy R.M. Mitochondrial content is preserved throughout disease progression in the mdx mouse model of Duchenne muscular dystrophy, regardless of taurine supplementation. Am. J. Physiol. Cell Physiol. 2018;314:C483–C491. doi: 10.1152/ajpcell.00046.2017. [DOI] [PubMed] [Google Scholar]
- 37.Hughes M.C., Ramos S.V., Turnbull P.C., Rebalka I.A., Cao A., Monaco C.M.F., Varah N.E., Edgett B.A., Huber J.S., Tadi P. Early myopathy in Duchenne muscular dystrophy is associated with elevated mitochondrial H2 O2 emission during impaired oxidative phosphorylation. J. Cachexia Sarcopenia Muscle. 2019;10:643–661. doi: 10.1002/jcsm.12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoppeler H., Vogt M., Weibel E.R., Flück M. Response of skeletal muscle mitochondria to hypoxia. Exp. Physiol. 2003;88:109–119. doi: 10.1113/eph8802513. [DOI] [PubMed] [Google Scholar]
- 39.Hood D.A. Invited Review: contractile activity-induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol (1985) 2001;90:1137–1157. doi: 10.1152/jappl.2001.90.3.1137. [DOI] [PubMed] [Google Scholar]
- 40.Picard M., Hepple R.T., Burelle Y. Mitochondrial functional specialization in glycolytic and oxidative muscle fibers: tailoring the organelle for optimal function. Am. J. Physiol. Cell Physiol. 2012;302:C629–C641. doi: 10.1152/ajpcell.00368.2011. [DOI] [PubMed] [Google Scholar]
- 41.Hourdé C., Joanne P., Medja F., Mougenot N., Jacquet A., Mouisel E., Pannerec A., Hatem S., Butler-Browne G., Agbulut O., Ferry A. Voluntary physical activity protects from susceptibility to skeletal muscle contraction-induced injury but worsens heart function in mdx mice. Am. J. Pathol. 2013;182:1509–1518. doi: 10.1016/j.ajpath.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 42.Costas J.M., Nye D.J., Henley J.B., Plochocki J.H. Voluntary exercise induces structural remodeling in the hearts of dystrophin-deficient mice. Muscle Nerve. 2010;42:881–885. doi: 10.1002/mus.21783. [DOI] [PubMed] [Google Scholar]
- 43.Bostick B., Yue Y., Lai Y., Long C., Li D., Duan D. Adeno-associated virus serotype-9 microdystrophin gene therapy ameliorates electrocardiographic abnormalities in mdx mice. Hum. Gene Ther. 2008;19:851–856. doi: 10.1089/hum.2008.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shin J.-H., Nitahara-Kasahara Y., Hayashita-Kinoh H., Ohshima-Hosoyama S., Kinoshita K., Chiyo T., Okada H., Okada T., Takeda S. Improvement of cardiac fibrosis in dystrophic mice by rAAV9-mediated microdystrophin transduction. Gene Ther. 2011;18:910–919. doi: 10.1038/gt.2011.36. [DOI] [PubMed] [Google Scholar]
- 45.Townsend D., Blankinship M.J., Allen J.M., Gregorevic P., Chamberlain J.S., Metzger J.M. Systemic administration of micro-dystrophin restores cardiac geometry and prevents dobutamine-induced cardiac pump failure. Mol. Ther. 2007;15:1086–1092. doi: 10.1038/sj.mt.6300144. [DOI] [PubMed] [Google Scholar]
- 46.Gregorevic P., Blankinship M.J., Allen J.M., Chamberlain J.S. Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol. Ther. 2008;16:657–664. doi: 10.1038/mt.2008.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Armstrong R.B., Delp M.D., Goljan E.F., Laughlin M.H. Distribution of blood flow in muscles of miniature swine during exercise. J Appl Physiol (1985) 1987;62:1285–1298. doi: 10.1152/jappl.1987.62.3.1285. [DOI] [PubMed] [Google Scholar]
- 48.Zelikovich A.S., Quattrocelli M., Salamone I.M., Kuntz N.L., McNally E.M. Moderate exercise improves function and increases adiponectin in the mdx mouse model of muscular dystrophy. Sci. Rep. 2019;9:5770. doi: 10.1038/s41598-019-42203-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nelson D.M., Lindsay A., Judge L.M., Duan D., Chamberlain J.S., Lowe D.A., Ervasti J.M. Variable rescue of microtubule and physiological phenotypes in mdx muscle expressing different miniaturized dystrophins. Hum. Mol. Genet. 2018;27:2090–2100. doi: 10.1093/hmg/ddy113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lynch G.S., Rafael J.A., Hinkle R.T., Cole N.M., Chamberlain J.S., Faulkner J.A. Contractile properties of diaphragm muscle segments from old mdx and old transgenic mdx mice. Am. J. Physiol. 1997;272:C2063–C2068. doi: 10.1152/ajpcell.1997.272.6.C2063. [DOI] [PubMed] [Google Scholar]
- 51.Deconinck N., Ragot T., Maréchal G., Perricaudet M., Gillis J.M. Functional protection of dystrophic mouse (mdx) muscles after adenovirus-mediated transfer of a dystrophin minigene. Proc. Natl. Acad. Sci. USA. 1996;93:3570–3574. doi: 10.1073/pnas.93.8.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Banks G.B., Judge L.M., Allen J.M., Chamberlain J.S. The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet. 2010;6:e1000958. doi: 10.1371/journal.pgen.1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y., Yue Y., Li L., Hakim C.H., Zhang K., Thomas G.D., Duan D. Dual AAV therapy ameliorates exercise-induced muscle injury and functional ischemia in murine models of Duchenne muscular dystrophy. Hum. Mol. Genet. 2013;22:3720–3729. doi: 10.1093/hmg/ddt224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Faulkner J.A., Ng R., Davis C.S., Li S., Chamberlain J.S. Diaphragm muscle strip preparation for evaluation of gene therapies in mdx mice. Clin. Exp. Pharmacol. Physiol. 2008;35:725–729. doi: 10.1111/j.1440-1681.2007.04865.x. [DOI] [PubMed] [Google Scholar]
- 55.Burns D.P., Murphy K.H., Lucking E.F., O’Halloran K.D. Inspiratory pressure-generating capacity is preserved during ventilatory and non-ventilatory behaviours in young dystrophic mdx mice despite profound diaphragm muscle weakness. J. Physiol. 2019;597:831–848. doi: 10.1113/JP277443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Capogrosso R.F., Mantuano P., Cozzoli A., Sanarica F., Massari A.M., Conte E., Fonzino A., Giustino A., Rolland J.-F., Quaranta A. Contractile efficiency of dystrophic mdx mouse muscle: in vivo and ex vivo assessment of adaptation to exercise of functional end points. J Appl Physiol (1985) 2017;122:828–843. doi: 10.1152/japplphysiol.00776.2015. [DOI] [PubMed] [Google Scholar]
- 57.Petrof B.J., Stedman H.H., Shrager J.B., Eby J., Sweeney H.L., Kelly A.M. Adaptations in myosin heavy chain expression and contractile function in dystrophic mouse diaphragm. Am. J. Physiol. 1993;265:C834–C841. doi: 10.1152/ajpcell.1993.265.3.C834. [DOI] [PubMed] [Google Scholar]
- 58.Betts C.A., Saleh A.F., Carr C.A., Muses S., Wells K.E., Hammond S.M., Godfrey C., McClorey G., Woffindale C., Clarke K. Implications for Cardiac Function Following Rescue of the Dystrophic Diaphragm in a Mouse Model of Duchenne Muscular Dystrophy. Sci. Rep. 2015;5:11632. doi: 10.1038/srep11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tyler K.L. Origins and early descriptions of “Duchenne muscular dystrophy”. Muscle Nerve. 2003;28:402–422. doi: 10.1002/mus.10435. [DOI] [PubMed] [Google Scholar]
- 60.Akima H., Lott D., Senesac C., Deol J., Germain S., Arpan I., Bendixen R., Lee Sweeney H., Walter G., Vandenborne K. Relationships of thigh muscle contractile and non-contractile tissue with function, strength, and age in boys with Duchenne muscular dystrophy. Neuromuscul. Disord. 2012;22:16–25. doi: 10.1016/j.nmd.2011.06.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dupont-Versteegden E.E., McCarter R.J. Differential expression of muscular dystrophy in diaphragm versus hindlimb muscles of mdx mice. Muscle Nerve. 1992;15:1105–1110. doi: 10.1002/mus.880151008. [DOI] [PubMed] [Google Scholar]
- 62.Charles J.P., Cappellari O., Hutchinson J.R. A Dynamic Simulation of Musculoskeletal Function in the Mouse Hindlimb During Trotting Locomotion. Front. Bioeng. Biotechnol. 2018;6:61. doi: 10.3389/fbioe.2018.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Call J.A., McKeehen J.N., Novotny S.A., Lowe D.A. Progressive resistance voluntary wheel running in the mdx mouse. Muscle Nerve. 2010;42:871–880. doi: 10.1002/mus.21764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lynch G.S., Hinkle R.T., Chamberlain J.S., Brooks S.V., Faulkner J.A. Force and power output of fast and slow skeletal muscles from mdx mice 6-28 months old. J. Physiol. 2001;535:591–600. doi: 10.1111/j.1469-7793.2001.00591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Landisch R.M., Kosir A.M., Nelson S.A., Baltgalvis K.A., Lowe D.A. Adaptive and nonadaptive responses to voluntary wheel running by mdx mice. Muscle Nerve. 2008;38:1290–1303. doi: 10.1002/mus.21141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Voet N.B., van der Kooi E.L., van Engelen B.G., Geurts A.C. Strength training and aerobic exercise training for muscle disease. Cochrane Database Syst. Rev. 2019;12:CD003907. doi: 10.1002/14651858.CD003907.pub5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rocco A.B., Levalley J.C., Eldridge J.A., Marsh S.A., Rodgers B.D. A novel protocol for assessing exercise performance and dystropathophysiology in the mdx mouse. Muscle Nerve. 2014;50:541–548. doi: 10.1002/mus.24184. [DOI] [PubMed] [Google Scholar]
- 68.Ashton-Miller J.A., He Y., Kadhiresan V.A., McCubbrey D.A., Faulkner J.A. An apparatus to measure in vivo biomechanical behavior of dorsi- and plantarflexors of mouse ankle. J Appl Physiol (1985) 1992;72:1205–1211. doi: 10.1152/jappl.1992.72.3.1205. [DOI] [PubMed] [Google Scholar]
- 69.Quiat D., Voelker K.A., Pei J., Grishin N.V., Grange R.W., Bassel-Duby R., Olson E.N. Concerted regulation of myofiber-specific gene expression and muscle performance by the transcriptional repressor Sox6. Proc. Natl. Acad. Sci. USA. 2011;108:10196–10201. doi: 10.1073/pnas.1107413108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bloemberg D., Quadrilatero J. Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PloS ONE. 2012;7:e35273. doi: 10.1371/journal.pone.0035273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Perry C.G.R., Kane D.A., Lin C.-T., Kozy R., Cathey B.L., Lark D.S., Kane C.L., Brophy P.M., Gavin T.P., Anderson E.J., Neufer P.D. Inhibiting myosin-ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem. J. 2011;437:215–222. doi: 10.1042/BJ20110366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tinklenberg J.A., Siebers E.M., Beatka M.J., Fickau B.A., Ayres S., Meng H., Yang L., Simpson P., Granzier H.L., Lawlor M.W. Myostatin Inhibition Using ActRIIB-mFc Does Not Produce Weight Gain or Strength in the Nebulin Conditional KO Mouse. J. Neuropathol. Exp. Neurol. 2019;78:130–139. doi: 10.1093/jnen/nly120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.