

Abstract
Angelman syndrome (AS) is a severe neurodevelopmental disorder caused by mutation or deletion of the maternally-inherited UBE3A allele. In neurons, the paternally-inherited UBE3A allele (patUBE3A) is silenced in cis by a long non-coding RNA called UBE3A-ATS. As part of a systematic screen, we found that Cas9 can be used to unsilence paternal Ube3a in cultured mouse and human neurons when targeted to Snord115 genes, small nucleolar RNAs (snoRNAs) that are clustered in the 3´ region of Ube3a-ATS. A short Cas9 variant and guide (gRNA) targeting ~75 Snord115 genes was packaged into an adeno-associated virus (AAV) and administered to embryonic and early postnatal AS mice, when the therapeutic benefit of restoring Ube3a is predicted to be greatest1,2. This early treatment unsilenced patUbe3a throughout the brain for at least seventeen months and rescued anatomical and behavioral phenotypes in AS mice. Genomic integration of the AAV vector into Cas9 target sites caused premature termination of Ube3a-ATS at the vector-derived polyA cassette, or when integrated in the reverse orientation, by transcriptional collision with the vector-derived Cas9 transcript. Our study shows that targeted genomic integration of a gene therapy vector can restore patUBE3A function throughout life, providing a path towards a disease-modifying treatment for a syndromic neurodevelopmental disorder.
Drugs and antisense oligonucleotides have been identified that unsilence paternal UBE3A by downregulating UBE3A-ATS3,4. However, these potential therapeutics have a short half-life and require repeated invasive injections, which is not desirable for a pediatric onset disorder that lasts a lifetime. CRISPR/Cas9 has the potential to permanently reduce expression of non-coding RNAs via mutagenesis5. To determine if Cas9 could be used to reduce Ube3a-ATS and activate (“unsilence”) paternal UBE3A, we designed 260 Staphylococcus pyogenes (Sp)Cas9 gRNAs that target putative regulatory regions and genes in or near Ube3a-ATS (Fig. 1a, Supplementary Table 1). Each gRNA was cloned into an expression plasmid containing SpCas9 and was transiently transfected into cultured cortical neurons from Ube3am+/pYFP mice, which harbor a paternal Ube3a-YFP knock-in allele6. Several gRNAs unsilenced paternal UBE3A-YFP as effectively as topotecan, the positive control3, while gRNAs targeting Ube3a decreased paternal UBE3A-YFP below its low baseline level (Fig. 1b,c, Supplementary Table 1). Guide RNAs that were most effective at unsilencing paternal UBE3A-YFP were located in or near Snord115 and Snord116 genes (also known as H/MBII-52 and H/MBII-85, respectively), two clusters of C/D box snoRNAs that are processed from introns of Ube3a-ATS7. Since deletion of SNORD116 genes causes Prader-Willi Syndrome8–10, we did not pursue gRNAs that targeted Snord116 genes or upstream regions of Ube3a-ATS.
Figure 1. Screen to identify SpCas9 gRNAs that unsilence paternal Ube3a.
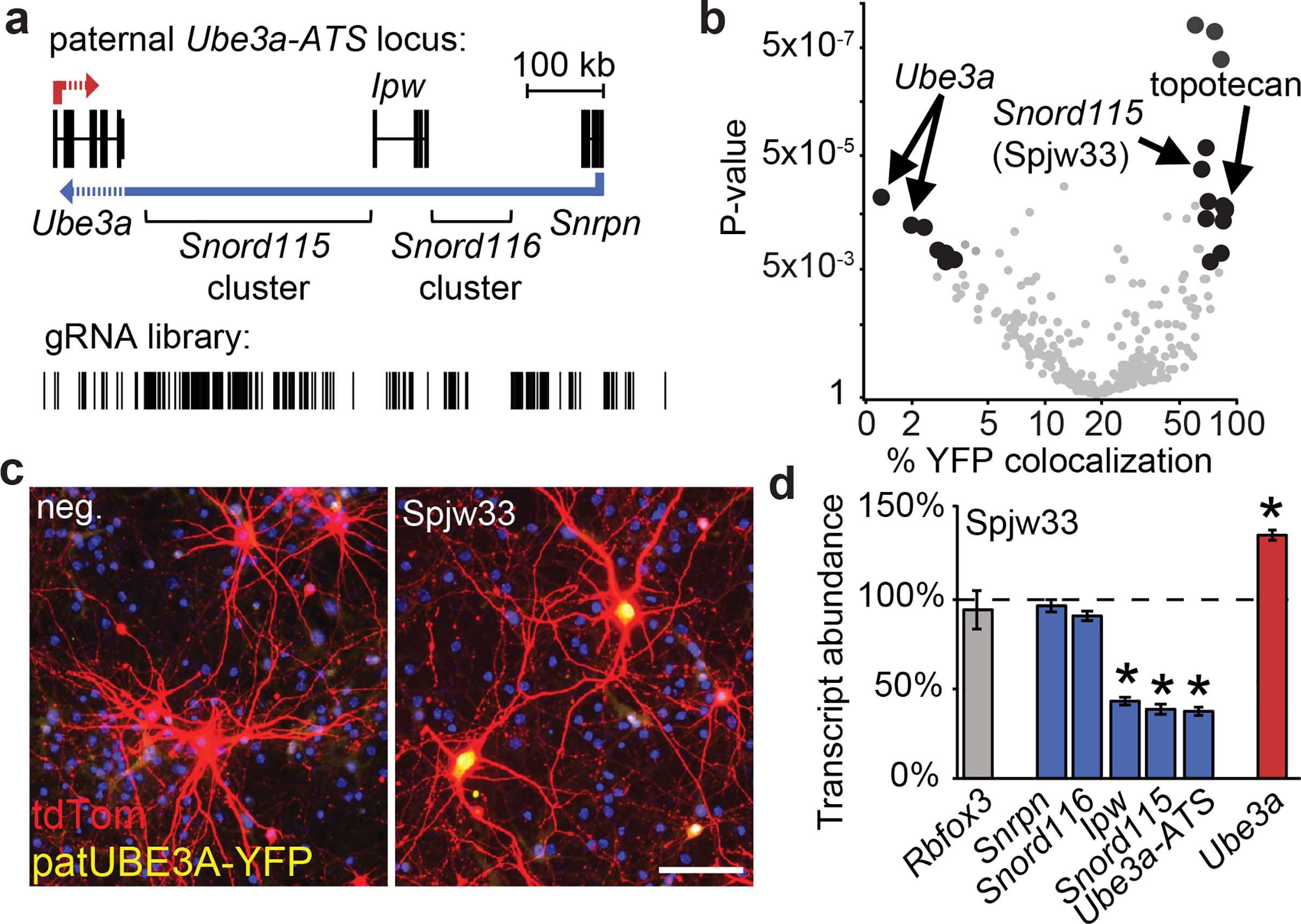
a. Paternal Ube3a-ATS locus showing the location of genes, transcription collision region (dashed lines), and gRNA library target sites.
b. Percentage of Ube3am+/pYFP neurons co-stained for UBE3A-YFP and tdTomato; co-transfected with CamKIIα promoter:tdTomato, SpCas9 and one gRNA (circles) or treated with 300 nM topotecan for 72 h. Black circles, P<0.005 and fold change >4.
c. Ube3am+/pYFP neurons co-transfected with CamKIIα promoter:tdTomato, SpCas9 and negative control gRNA (neg.) or Spjw33 gRNA. Nuclei, blue (DAPI). Scale bar, 100 μm.
d. Expression of the indicated genes seven days after transducing Ube3am+/pYFP cortical neurons with lentiviral SpCas9+Spjw33, relative to SpCas9+neg. control gRNA (dashed line). qPCR, normalized to Eif4a2, n=3. Error bars, standard error of the mean (S.E.M.), * P<0.01.
Snord115 genes are located 3´ of the Prader-Willi Syndrome critical region, in the distal portion of Ube3a-ATS that is only expressed in neurons11. Snord115 genes are highly similar, and in some cases identical, at the sequence level. One of the most effective gRNAs (Spjw33) is predicted to target 76 sites in the Snord115 cluster (Extended Data Fig. 1a,b, Supplementary Table 1), and was far more effective than gRNA pairs that were predicted to delete defined regions of Ube3a-ATS (Extended Data Fig. 1c).
To evaluate how targeting multiple Snord115 genes affected expression across the Ube3a-ATS locus, we transduced Ube3am+/pYFP cortical neurons with a lentivirus carrying SpCas9 and Spjw33 and quantified gene expression via reverse transcriptase-polymerase chain reaction (qRT-PCR). We found that expression of the paternal Ube3a-YFP allele was increased in neurons transduced with Spjw33 (Extended Data Fig. 1d–f). Furthermore, Spjw33 reduced expression of Snord115 as well as regions of Ube3a-ATS that are immediately upstream (Ipw) and downstream (Ube3a-ATS 3´) of the Spjw33 target sites (Fig. 1d). This contrasted with topotecan, which decreased expression of all transcripts derived from Ube3a-ATS (Extended Data Fig. 1g,h). A gRNA targeting human SNORD115 (negative control) had no effect in mouse neurons (Extended Data Fig. 1i). One or more base mismatches in the gRNA eliminated unsilencing activity (Extended Data Fig. 1j). We detected indel mutations at a low frequency at the Spjw33 target site (Extended Data Fig. 1k, Supplementary Table 2), but not at imperfectly matched (by 1–4 base pairs) sites (Extended Data Fig. 1k, Supplementary Table 2). These low frequency indels are unlikely to disrupt the Ube3a-ATS non-coding RNA.
Since double strand breaks are resolved in ~24 h, the likelihood that a break will be present at any point in time and physically impede transcription is expected to increase as a function of the number of target sites. We thus tested an additional 66 gRNAs that target different numbers of repetitive sites in Ube3a-ATS (Supplemental Table 1). We observed a correlation between the number of genomic target sites and patUbe3a unsilencing efficiency (Extended Data Fig. 1l, Supplemental Table 1). These data suggest that transcription through Ube3a-ATS is terminated at transient double strand breaks in these short-term (7 day) cell culture experiments.
We next evaluated the extent to which Cas9-directed targeting of SNORD115 genes can unsilence paternal UBE3A in primary human neural progenitor-derived (phNPC) neurons12. We used a single nucleotide variant (SNV) in UBE3A exon 5 to quantify expression of the presumed maternal and paternal UBE3A alleles (Extended Data Fig. 2a,b). As expected, the presumed maternal UBE3A allele was predominantly expressed in differentiated neurons, coincident with the emergence of paternal UBE3A-ATS expression (Extended Data Fig. 2c). We then transduced phNPC-derived neurons with lentiviral SpCas9:mCherry and AAV2 EGFP driven by the human Synapsin 1 (hSyn1) neuron-specific promoter, and evaluated three SpCas9 gRNAs that target a different number of sites in the SNORD115 cluster (Extended Data Fig. 2d,e). To quantify gene expression in different cell types, we FACS isolated 1) hSyn1:EGFP− progenitors, 2) SpCas9−/hSyn1:EGFP+ neurons (internal negative control), and 3) SpCas9+/hSyn1:EGFP+ neurons (Extended Data Fig. 2e–h). We confirmed that SpCas9 transcripts were expressed in SpCas9+ sorted cells, and that genes indicative of neuronal differentiation, specifically RBFOX3, SNORD115, and UBE3A-ATS 3´ region, were upregulated in hSyn1:EGFP+ sorted neurons (Extended Data Fig. 2g–n). We used SNV specific qRT-PCR probes to quantify allelic expression of UBE3A. As expected, UBE3A was expressed biallelically in progenitors and, in SpCas9− neurons, the maternal UBE3A allele was expressed nine-fold higher than the paternal allele (Extended Data Fig. 2o,p). In SpCas9+/hsa#3+ neurons, expression of the paternal UBE3A allele was increased to near maternal UBE3A levels (relative to neurons transduced with the negative control gRNA, Extended Data Fig. 2o,p). Furthermore, there was a correlation between the number of gRNA target sites and the magnitude of paternal UBE3A unsilencing (Extended Data Fig. 2p).
Total UBE3A levels increased in human neurons transduced with SpCas9 and hsa#3, consistent with increased expression of the paternal allele, while SNORD115 and UBE3A-ATS 3´ expression were significantly decreased (Extended Data Fig. 2q). IPW levels did not change in human neurons, which contrasted with results in mouse neurons (Fig. 1d), possibly because hsa#3 target sites are ~50 kb 3´ of human IPW whereas Spjw33 target sites are immediately downstream of mouse Ipw (compare Extended Data Figs. 1a, 2d).
The restrictive packaging limit of AAV prevented us from incorporating SpCas9 and a gRNA into a single AAV vector. Staphylococcus aureus (Sa)Cas9 is 1 kb smaller and can be incorporated into a single AAV vector with a gRNA13–15. The protospacer adjacent motif sites for SpCas9 and SaCas9 differ, so we designed and tested several SaCas9 gRNAs, settling on one (Sajw33) that was nearly identical to Spjw33 at the sequence level and that effectively unsilenced paternal Ube3a in cultured neurons (Extended Data Fig. 3a,b).
We packaged hSyn1 promoter-SaCas9 and U6 promoter-Sajw33 into AAV9 (Extended Data Fig. 3c), and injected this vector intracerebroventricularly (i.c.v., bilaterally) into embryonic day (E)15.5 or postnatal day (P)1 Ube3am+/pYFP mice. When examined histologically at P30, animals injected at E15.5 showed biased unsilencing of paternal UBE3A-YFP in lower layer cortical neurons, while animals injected at P1 showed biased unsilencing in upper layer cortical neurons (Extended Data Fig. 3d). We therefore opted to use a dual E15.5+P1 injection strategy to maximize the number of transduced neurons. Following dual injections in AS mice (Ube3am-/p+), paternal UBE3A was unsilenced throughout the P30 and P90 brain, including all layers of the cerebral cortex, all regions of the hippocampus, some cerebellar neurons, and the spinal cord (Fig. 2a–b; Extended Data Fig. 4a–g). Based on western blot analyses, paternal UBE3A protein was restored to ~37%, ~38%, and ~40% of wild-type levels in the cerebral cortex, hippocampus, and spinal cord of AS model mice at P90 (Fig. 2c,d), but showed no significant change in cerebellum. Moreover, paternal UBE3A was unsilenced in ~58% of all NEUN+ cortical neurons at P90, with median intensity levels 63% of maternal UBE3A (based on staining intensity in >9,000 cortical neurons), and was primarily localized to the nucleus, indicative of proper isoform expression16. We did not observe evidence of neuroinflammation or loss of hippocampal neural progenitors (Extended Data Fig. 4h–o)17. We also did not detect changes in expression or splicing patterns of known Snord115 target genes in AS mice treated with Sajw33 (Extended data Fig. 5), suggesting that transcriptional downregulation and/or mutagenesis of some Snord115 genes does not compromise putative Snord115 functions18,19. Remarkably, paternal UBE3A was unsilenced 17 months after a single E15.5 viral injection into AS mice (Extended Data Fig. 6a–c), revealing an extremely long duration of action.
Figure 2. AAV delivery of SaCas9 and Sajw33 unsilences paternal Ube3a throughout the nervous system.
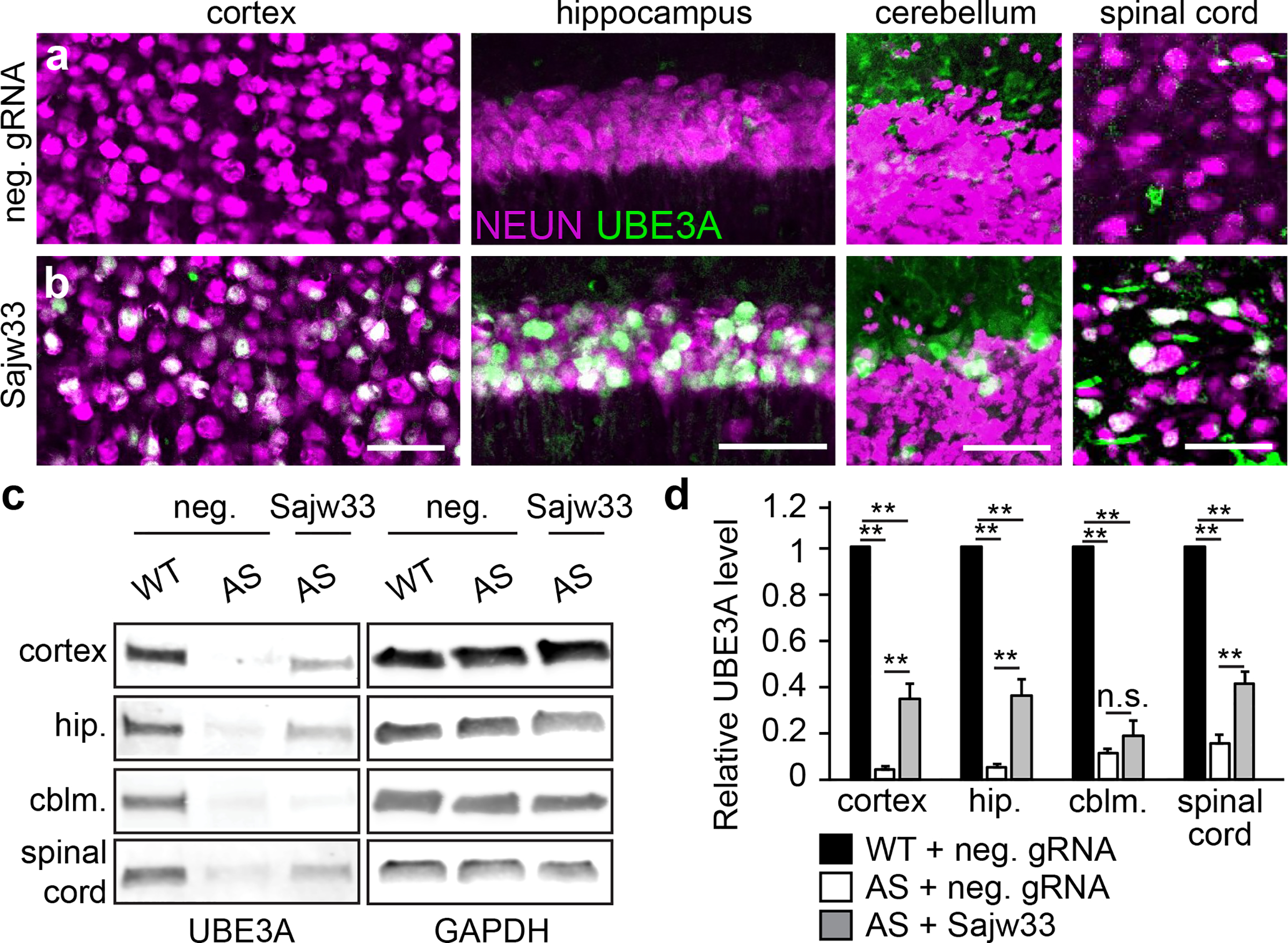
a-b. Ube3am-/p+ (AS model) mice were injected i.c.v. bilaterally at E15.5+P1 with 1.5 × 1010 AAV particles per ventricle (6 × 1010 AAV particles per mouse) containing SaCas9 and either the negative control gRNA (a) or Sajw33 (b) and immunostained for UBE3A and NEUN at P90. Lower magnification images shown in Extended Data Fig. 4f,g. Scale bar, 50 μm.
c-d. Western blot and quantification. UBE3A levels in the cerebral cortex, hippocampus (hip.), cerebellum (cblm.), and spinal cord of WT and AS (Ube3am-/p+) mice treated with control gRNA (neg.) or Sajw33. n=5 per group. ** P<0.001, not significant (n.s.). For confocal image and gel source data, see Supplementary Figures 1 and 2.
It is currently unknown if AAV can pass from treated fetuses to the mother, which is a potential safety concern for in utero gene therapies. To address this issue, and to evaluate tissue distribution of the SaCas9 vector more broadly, we quantified viral DNA (vDNA) in the cerebral cortex and liver of dams and their P60 offspring (all dual injected). In the treated offspring, vDNA levels were higher in the cerebral cortex relative to liver (Extended Data Fig. 7a). No vDNA was detected in the cerebral cortex or liver of the dams (Extended Data Fig. 7a), indicating that AAV did not transfer to the dams during pregnancy. Furthermore, SaCas9 RNA expression was largely restricted to the brain of treated offspring (Extended Data Fig. 7b), consistent with neuron-specific expression of the hSyn1 promoter. Lastly, organ morphology was normal and no tumors were detected in 10 month old treated mice (Supplemental Table 3).
Restoring Ube3a function during the embryonic and early postnatal period is predicted to be more efficacious at treating AS than restoring Ube3a in adulthood1,2. Thus, we next evaluated the extent to which dual E15.5+P1 i.c.v. injection of the SaCas9 AAV vector rescued anatomical and behavioral phenotypes in AS model mice that are reproducible20,21, and that interrogate core symptoms of AS, including microcephaly and deficits in proprioception and motor function (Fig. 3a, raw data presented in Supplementary Table 4). AS mice also become obese as they age4. Body weight returned to WT levels in female, but not male, AS mice that were treated with the SaCas9+Sajw33 vector (Fig. 3b, Extended Data Fig. 8a). Brain weight at 10 months of age was also increased in AS mice treated with the SaCas9+Sajw33 vector (Fig. 3c), suggesting that microcephaly can be partially rescued. AS mice treated with the SaCas9+Sajw33 vector also demonstrated behavioral improvements in hindlimb clasping and center time in the open field, but showed no rescue in distance travelled in the open field or marble burying (Fig. 3d–g, Extended Data Fig. 8b,c). Improvements in the rotarod test were evident at two months of age and endured at seven months of age (Fig. 3h,i, Extended Data 8d,e). In our hands, AS mice showed no deficits in contextual and cue based learning and memory tasks (Extended Data Fig 8f,g), consistent with several other labs (reviewed in Ref. 20), so we were unable to evaluate rescue of cognitive deficits. In sum, our data suggest that in utero reactivation of patUbe3a can enduringly treat many symptoms of AS.
Figure 3. SaCas9 gene therapy vector rescues phenotypes in AS mouse model.
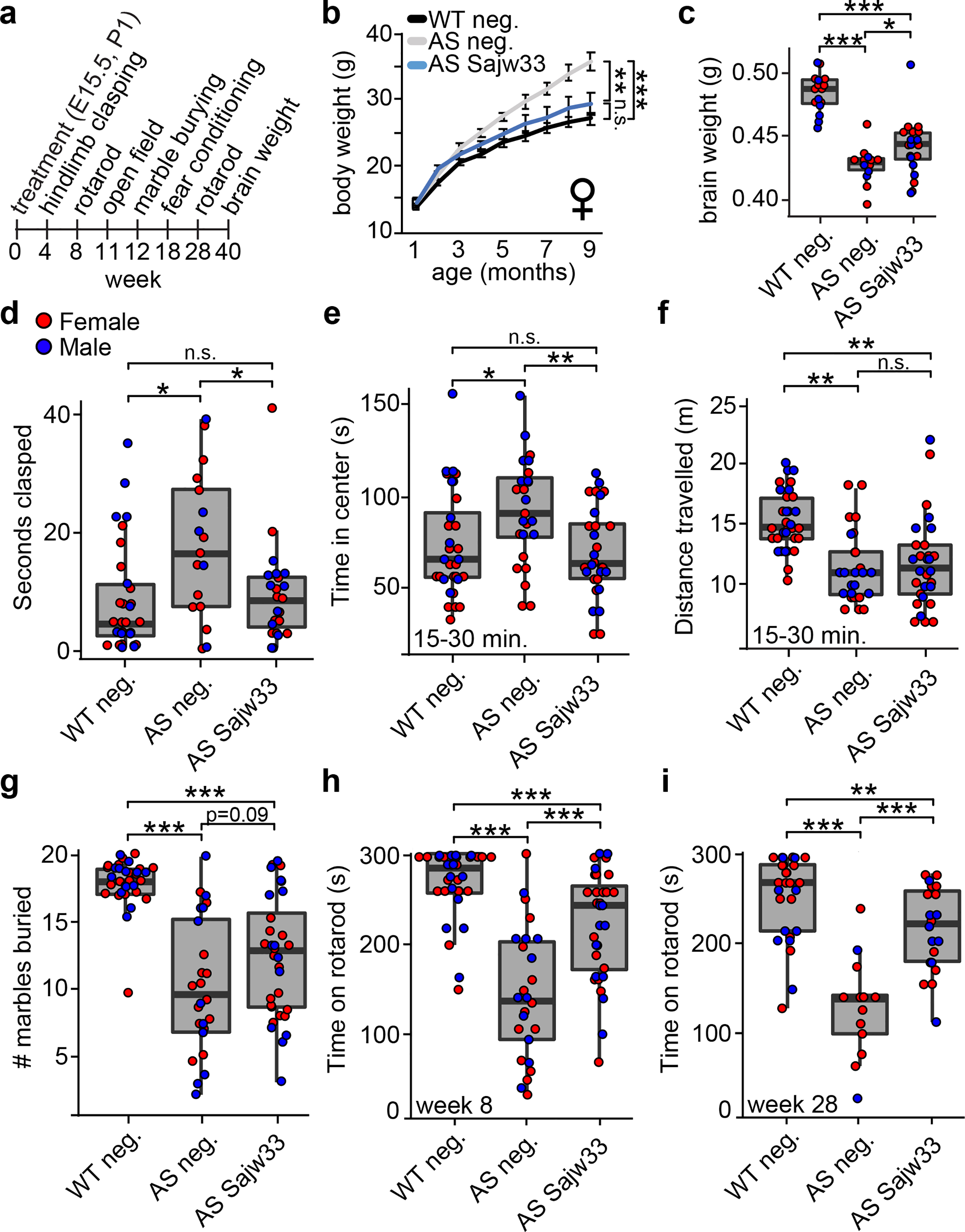
a. Timeline of assays performed on WT and AS mice injected i.c.v. bilaterally at E15.5+P1 with 1.5 × 1010 AAV particles per ventricle (6 × 1010 AAV particles per mouse) containing SaCas9 and the negative control gRNA (neg.) or Sajw33. P<0.05, ** P<0.01, *** P<0.001.
b. Body weight of female mice measured monthly over nine months.
c. Brain weight measured at 40 weeks of age.
d. Hindlimb clasping assay in four week old mice.
e-f. Open field data for minutes 15–30 in 11 week old mice. Time spent in center (e), distance travelled (f).
g. Marble burying task in 12 week old mice, 30 minute trial.
h-i. Rotarod testing data (average of two trials) at 8 weeks of age (h) and 28 weeks of age (i).
We next sought to determine how the gene therapy vector disrupted Ube3a-ATS in vivo by focusing on brain samples from 10 month old AS mice and controls. Using high-throughput DNA sequencing, we detected indels and substitutions in 0.3% of the Sajw33 target site amplicons (Extended Data Fig. 9a,b), and no evidence for mutations at predicted off target sites (Supplementary Table 5, see Methods). Moreover, Snord115 genomic copy number was not increased or decreased (Extended Data Fig. 9c). Thus, indels and deletions/duplications were unlikely to contribute to widespread unsilencing of patUbe3a in mice.
AAV integrates into SaCas9-generated double strand break sites at a relatively high frequency22,23. To identify AAV integration events in Snord115 genes, we performed PCR from cortical genomic DNA (gDNA) using primers that bind in the Snord115 locus and in the AAV vector (Extended Data Fig. 9d). We detected forward and reverse integration events in Sajw33-treated animals, but not in animals treated with the negative control gRNA vector (Extended Data Fig. 9e, a primer). Using primers that anneal farther within the AAV vector (Extended Data Fig. 9e, b primer), we detected additional faint bands that corresponded to truncated AAV vectors; which we validated by Sanger sequencing and high-throughput amplicon sequencing (Extended Data Fig. 9f,g). No AAV integration events were detected at predicted off-target sites (Extended Data Fig. 9h). Using qPCR, we detected approximately one AAV integration event per diploid genome (Extended Data Fig. 9i). Assuming 50% of all diploid cells in the brain are neurons24, and since ~58% of all cortical neurons showed patUbe3a unsilencing, our data suggests there are ~4 integrated viral vectors per patUbe3a unsilenced neuron.
Additionally, we performed RT-PCR with primers that amplify across AAV integration sites. We detected fusion RNAs between Ube3a-ATS and the AAV vector only in Sajw33 treated animals (Extended Data Fig. 9j). In the forward orientation, fusion transcripts contained the polyadenylation sequence element from the 3ÚTR of SaCas9, resulting in premature transcription termination of Ube3a-ATS (Fig. 4a). Thus, AAV vector integration is functionally analogous to a gene trap4,25. In the reverse orientation, fusion transcripts were not detected beyond SaCas9 using a primer walking strategy (Fig. 4b), suggesting that convergent RNA Polymerase II transcription blocks Ube3a-ATS, analogous to the paternal Ube3a/Ube3a-ATS collision model4. Together, these data suggest AAV integration can disrupt Ube3a-ATS (Extended Data Fig. 10) and occurs frequently enough in vivo to account for the long term, likely permanent, unsilencing of paternal Ube3a.
Figure 4. AAV integration traps Ube3a-ATS.
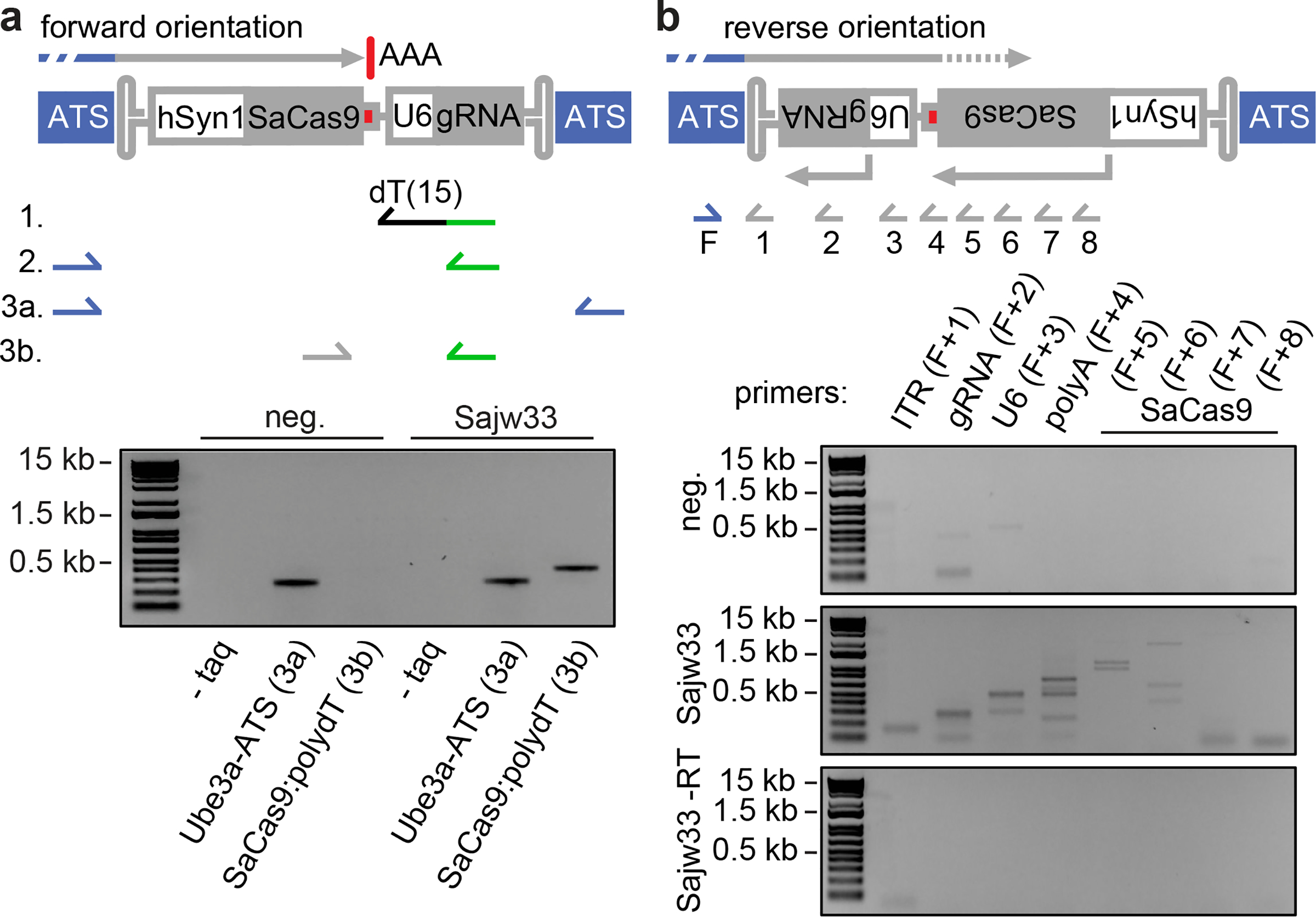
a. Identification of polyadenylated Ube3a-ATS/AAV fusion transcripts. 1. Total RNA was extracted from cortexes of 10 mo. old mice that were injected at E15.5+P1 with AAV containing SaCas9 and the negative control gRNA or Sajw33. cDNA was synthesized using a polydT primer with a 3´ adaptor sequence (green line). 2. RT-PCR with forward primer which anneals in Ube3a-ATS upstream of Cas9 target site, and reverse primer annealing in polydT 3´ adaptor. PCR resulted in a smear (not shown) due to the repetitive binding site of the forward primer and complex splicing patterns over the Snord115 locus, as previously described36. 3. Product from #2 was purified and used as template in a second PCR with primers to detect polyadenylated Ube3a-ATS (3a), and polyadenylated Ube3a-ATS/AAV fusion RNA (3b). Minus (–) taq polymerase with Ube3a-ATS primers (negative control).
b. Primer walking strategy to detect Ube3a-ATS/AAV fusion mRNAs. One forward primer (F) was used in combination with reverse primers that anneal progressively further into the AAV genome. The largest band in each lane is the correct predicted size based on integration in the ITR, smaller bands reflect integrations of truncated AAV genomes.
Methods
Animals
C57BL/6 mice, Ube3am-/p+ mice and Ube3am-/pYFP mice on the C57Bl/6 background and genotyping procedures were previously described3. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill, and in accordance with NIH guidelines.
Guide RNA cloning and Cas9 expression plasmids
To identify possible gRNA target sites, we analyzed RNA-seq data from cortical neuron cultures26, and publicly available datasets such as conservation (PhyloP), CTCF binding sites, DNase hypersensitivity sites, CHiP-seq, polyadenylation sites, and predicted RNA secondary structure. All gRNAs were cloned using Golden Gate assembly. Experiments using SpCas9 described in Fig. 1 and Extended Data Fig. 1 used v2 lentiCRISPR27. Experiments using SaCas9 used pX601-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA;U6::BsaI-sgRNA13, in which the CMV promoter was replaced with the hSyn1 promoter using XbaI and AgeI restriction sites. Experiments using mCherry tagged SpCas9 described in Extended Data Fig. 2 used lentiCRISPRv2-mCherry (Addgene #99154). The pLenti-CamKIIα-tdTomato was previously described28.
Guide RNA library screen
Primary mouse neuronal cultures were prepared as described3,26,29. Briefly, cortical neurons were dissected from E15.5 Ube3am+/pYFP embryos and plated in 384 well poly-D lysine coated plates. On days in vitro (DIV)3, each well was transfected with 50 ng CamKIIα:tdTomato and 50 ng of lentiCRISPR:gRNA plasmid using Lipofectamine 2000 (ThermoFisher). Each gRNA was transfected in quadruplicate. On DIV10, cells were fixed with 4% phosphate-buffered paraformaldehyde (PFA), and stained with primary rabbit anti-GFP antibody (Novus NB600–308), secondary donkey anti-rabbit IgG alexa 647 (ThermoFisher A31573) and 4′,6-diamidino-2-phenylindole (DAPI). Images were acquired using the GE IN CELL Analyzer 2200 high-content imager. YFP expression was quantified in tdTomato+ nuclei using a custom Cell Profiler pipeline. The screen was performed in triplicate, presented as the average of the three replicates. All subsequent experiments using mouse primary cortical neuron cultures followed the same culture protocol and timeline.
RNA extractions, qPCR, and RT-PCR
All RNA and DNA extractions were performed using Trizol (ThermoFisher). Total RNA was treated with DNase (New England Biolabs; NEB) for all qPCR experiments. cDNA synthesis for experiments involving standard qPCR and RT-PCR were performed using SuperScript IV VILO (ThermoFisher). All standard qPCR experiments were performed using SsoAdvanced Universal SYBR Green Supermix (NEB) on the QuantStudio3 or Quantistudio5 (Applied Biosystems). RT-PCR experiments from P60 mouse cortical RNA (Extended Data Fig. 5) were performed with Platinum Taq Polymerase (Invitrogen) and primers from Kishore et al.30. Primers used in all experiments are listed in Supplemental Table 6.
Spjw33 target site mutation analysis
E15.5 WT primary mouse neuron cultures were treated on DIV3 with lentivirus carrying SpCas9 and either Spjw33 or a scrambled gRNA. Genomic DNA was extracted at DIV12, and the Spjw33 target site was amplified using nine primer sets designed to amplify nearly all copies of Snord115: Forward primers: Snord115 1–3 F, Reverse: Snord115 1–3 R. Amplicons were cloned via TopoTA cloning (Invitrogen), and individual colonies were analyzed for mutations by Sanger sequencing.
Use of gRNA pairs to delete defined regions of Ube3a-ATS
Guide RNAs were selected based on target location in the Ube3a-ATS locus and lack of efficacy when tested alone in the primary screen (Supplementary Table 1). 50 ng of each gRNA was transfected either alone (−) or in combination with one other gRNA in E15.5 Ube3am+/pYFP embryos as described above. Unsilencing of paternal UBE3A-YFP was determined as previously described, with the only exception being the replacement of CamK2a:td:tomato reporter plasmid with immunofluorescent labelling of SpCas9+ cells with an SpCas9 antibody (Biolegend, clone 7A9).
Analysis of UBE3A expression in differentiated phNPCs
Human fetal brain tissue was obtained from the UCLA Gene and Cell Therapy Core following IRB regulations. Primary human (ph)NPCs were grown and differentiated as previously described12. Cells were mycoplasma tested and confirmed to be mycoplasma free. Briefly, cells were thawed and plated in 10 cm plates with proliferation media (Neurobasal A supplemented with primocin, BIT9500, glutamax, heparin, EGF, FGF, LIF, PDGF) in a humid incubator at 37°C with 5% (vol/vol) CO2. After two passages cells were transferred into 6 well plates, 4 × 105 cells per well, and changed to differentiation media (Neurobasal A, primocin, B27+, glutamax, NT3, BDNF) 24 hours post plating. A 50% media change was performed every 2–3 days. On day 14, 1.6 × 1010 AAV2 particles carrying hSyn1:eGFP (pAAV-hSyn1-EGFP, UNC Vector Core) were added to each well. On day 42, 4 × 108 lentivirus particles carrying SpCas9-mCherry and gRNAs were added to cultures, six wells per gRNA. hsa#1: 5´-CCTCTCTTCAGAACAATATA, hsa#2: 5´-TGGTCTCCTGCACTGAGCTG, hsa#3: 5´-TGCTCAATAGGATTACGCTG. On day 56 cells were lifted, six wells for each gRNA were pooled, stained with DAPI for live/dead discrimination, and sorted using the FACSAria II cell sorter. Cells were collected and total RNA was extracted using Trizol. RNA was treated with DNase I (NEB). For allele specific qPCR, cDNA was generated from 100 ng total RNA using SuperScript III (ThermoFisher) and 2 pM of UBE3A specific primer (5´-TCCTTTAGATCATACATCATTGG). Allele specific expression levels were determined using TaqMan Universal Master Mix II (ThermoFisher), and TaqMan genotyping probes for rsID:61734190 (Applied Biosystems). Quantification of allele specific expression was calculated by assessing the distance between Ct values for each allele within each replicate at ΔRn=0.5 (n=4). For qPCR of genes in the UBE3A-ATS locus, cDNA was synthesized from 100 ng total RNA using the VILO SSIV reverse transcriptase (ThermoFisher). Quantification was performed using the ΔΔCt method, normalized to EIF4A2. Presumption of the parental allele was based on stranded RNA-seq reads, with the paternal allele assigned to the SNV (A) that was observed from the UBE3A-ATS strand (which was observed only in neurons), and the maternal allele assigned to the alternate SNV (T).
Brain injections
Ube3am-/p+ dams were anesthetized under 2% isoflurane throughout the procedure. Stock virus was diluted in PBS and a final concentration of 0.05% fast green were added immediately before the procedure. 1.5 × 1010 particles/ventricle of virus was delivered into each embryo via bilateral intracerebroventricular (i.c.v.) injection at E15.5. Standard protocol for in utero injection was followed with no electroporation step31. The dams were then individually housed and delivered the pups naturally. At P1, neonatal pups were placed on ice to induce hypothermia anesthesia and bilaterally injected with virus at 1.5 × 1010 particles/ventricle following previously published protocol32. Injected pups were allowed to recover on heated pad before returning to their dam.
Immunohistochemistry
After behavioral experiments, mice were anesthetized with sodium pentobarbital (60 mg/kg). Each brain was cut sagittally through the midline. One half was flash frozen on dry ice for use in western blotting analysis while the other half was immersion fixed in 4% PFA in PBS, pH 7.4 before cryoprotecting by incubation in 30% sucrose in PBS (for 48 h at 4°C) and sectioning them to a thickness of 60 μm (P30 and P90 samples) or 100 μm (17 month old samples) on a freezing microtome (ThermoFisher Scientific). For immunostaining, we permeabilized sections with 0.01 M PBST (PBS containing 0.2% Triton X-100) for 30 minutes, and then blocked with 5% donkey serum in PBST for 1 h at room temperature. We incubated blocked sections with mouse IgG2A anti-UBE3A (1:1000; clone 3E5, SAB1404508, Sigma-Aldrich) and guinea pig anti-NEUN (1:1000; ABN90P, EMD Millipore) overnight at 4°C with gentle shaking. For AAV toxicity study we used Goat anti-GFAP (abcam ab53554, 1:1000), Rabbit anti-Iba1 (Wako 019–19741, 1:300), rat anti-Tbr2 (eBioscience 14–4875-82, 1:300), goat anti-DCX (Santa Cruz sc-8066, 1:500). The next day, we washed sections several times with PBST, and incubated with AlexaFluor 488-conjugated anti-mouse IgG2A (1:500; A21131, Invitrogen), AlexaFluor 568-conjugated anti-rabbit (1:500; A10042, Invitrogen), AlexaFluor 568-conjugated anti-rat (1:500; A21208, Invitrogen), AlexaFluor 568-conjugated anti-goat (1:500; A11057, Invitrogen), AlexaFluor 647-conjugated anti-guinea pig (1:500; A21124, Invitrogen), and DAPI (7 mg/mL, D1306, Invitrogen) during the secondary antibody incubation. We imaged sections via laser-scanning confocal microscopy (Zeiss LSM 710 and 780). Images were quantified with Fiji image processing software and Cell Profiler.
Western blotting
Brain regions were dissected from one hemisphere and protein lysates were prepared in RIPA Lysis and Extraction buffer (Thermo Scientific) containing protease inhibitor (Sigma, P8340) and phosphatase inhibitor (78426, ThermoFisher). Total proteins (60 μg) were fractionated by SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were blotted with rabbit anti-UBE3A (1:500, 10344–1-AP, Proteintech) and rabbit anti-GAPDH (1:2,000, 2118, Cell Signaling) in Intercept™ Blocking Buffer (927–70001, LI-COR) and secondary anti-rabbit antibody IRDye®680RD (1:10,000, C60813–05, LI-COR). Membranes were imaged with the ODYSSEY CLx Infrared Imaging System (LI-COR). Relative UBE3A protein level is measured based on loading control (GAPDH) with Fiji image processing software.
Behavioral assays
All behavioral assays were carried out blind to genotype and treatment of animals.
Hindlimb Clasping Assay.
At P30 each mouse was held by the tail at least 10 inches away from a hood bench (measured from the tip of the tail) for 30 s. Each mouse received two trials, with 5 minutes inter-trial intervals. Trials were recorded with a Samsung HMX-F80 camera and scored offline. Clasping behavior was defined by movement of the hindlimbs (either one or both) curling inward toward the belly of the animal. Data presented as the total time spent clasping across two trials.
Accelerating Rotarod.
Accelerating rotarod (3–30 rpm over 5 min; model 47600, Ugo Basile Biological Research Apparatus, Varese, Italy). Mice were given three trials on the first day with a 10 minute inter-trial interval (training session) and re-tested on day three with two trials (testing session). For each day, the average time spent on the rotarod was calculated, or the time until the mouse made three consecutive wrapping/passive rotations on the rotarod (latency in seconds). Maximum duration of a trial was 5 minutes.
Open Field.
Mice were individually placed in a 45 × 45 cm square open field and allowed to explore for 30 minutes. The total distance moved by each mouse in the open arena was recorded by camera (Noldus® Wageningen, NL) connected to the EthoVision® software (Noldus® Wageningen, NL). Center zone defined as 35 × 35 cm square field in the middle of the arena.
Marble Burying.
Open polycarbonate cages (50 × 26 × 18 cm) were filled with 3 L of bedding material (1/8” Corn Cob Bedding, irradiated, CC8-IRR, Lab Supply). On top of the bedding material, 20 black glass marbles were arranged in an equidistant 5 × 4 grid and the animals were given access to the marbles for 30 minutes. After the test, the mice were gently removed from the cage. Marbles covered by more than 50% by bedding were scored as buried.
Fear Conditioning:
All mice were brought into experiment room at least 20 min. before testing. Day 1 conditioning: each mouse was placed in individual chamber with house light on and given three presentations of a 30 s tone, paired with a 2 s foot shock of 0.4 mA after two minutes of acclimation, with 80 seconds between the first and second pairing and then 120 seconds between the second and third pairing. Day 2 Context: each mouse was put back into the chamber from Day 1 with house light on and filmed for 5 min. Day 3 Cue: each mouse was put in a chamber with new floor panel, a new black triangular insert. 0.1 mL of vanilla extract was also placed in the chamber. The house light is turned off, and tone is presented for the last 3 min. after 2 minutes of acclimation. All activities were videotaped, number and length of freeze were called using the Near-Infrared image tracking system (MED Associates, Burlington, VT).
Viral DNA qPCR
Organs from treated mice or dams were extracted at P30. Tissue (0.5–0.8 g) was isolated from each organ and DNA/RNA was extracted using Trizol. 100 ng of DNase (NEB) treated total RNA was used in cDNA synthesis using the VILO SSIV reverse transcriptase mix (ThermoFisher). SaCas9 levels were quantified in genomic DNA and cDNA samples using TaqMan probes and normalized to EIF4A2 or ACTB, respectively.
In silico off-target prediction
Cas-OFFinder33 was used to identify Sajw33 off-target binding sites in the mm10 reference genome with the following parameters: Pam type = SaCas9 (5´-NNGRRT-3´), target genome = Mus musculus (mm10), seq = 5´-CTGAGGCCCAACCAGGGCGA, mismatch number = 6, DNA bulge size = 0, RNA bulge size = 0. Primers were designed to amplify the loci flanking the top ten matches.
Next-generation amplicon sequencing
PCR and Library Preparation:
We performed PCR from 5 ng genomic DNA isolated (Trizol) from the cortex of 10 month old mice dual injected with either negative gRNA (n=3), or Sajw33 (n=3). Multiple primer sets were designed to amplify all Sajw33 target sites across the Ube3a-ATS locus, and AAV integration events (Snord115 1 F, Snord115 4 F, Snord115 5 F, Snord115 1 R, Snord115 2 R, Snord115 3 R, ITR C R, ITR 2 R, AAV hSyn R). Off target primers are listed in Supplementary Table 6. PCR reactions were performed on the QuantStudio5 qPCR machine and halted once all samples reached exponential growth phase (cycle #26) in order to avoid over-amplification bias. Each PCR reaction was performed in triplicate to reduce random PCR sampling bias (360 PCRs total). PCRs from individual mice were pooled, PCR purified (Qiagen), end repaired and A-tailed (KAPA HyperPlus kit), and adaptors were ligated using KAPA dual-indexed adaptors ligation kit (KAPA Biosystems, Wilmington, MA). Libraries were purified using AMPure XP beads (Beckman Coulter, Brea, CA), and sequenced using the Illumina Miniseq system with Miniseq Mid Output kit (Illumina) (300 cycles), 150 bp paired end reads. Reads were filtered for adaptor contamination using cutadapt34 and filtered such that at least 90% of bases of each read had a quality score > 20.
Off-target:
Reads from top 10 predicted off target sites were analyzed for mutations using the following approach: 1. Reads were aligned to mm10, retaining reads that did not align perfectly. 2. Reads were removed that were present in negative samples, retaining putative off-target mutations that contain at least one read in each of the three Sajw33 replicates. Remaining reads were manually inspected for mismatches to mm10 reference within 10 bp of PAM site. No reads survived these criteria (966,058 total reads from Sajw33 treated animals analyzed).
AAV Integration PCR
Genomic DNA PCR:
Genomic DNA was isolated from total cortical lysate stored in RIPA buffer using ethanol precipitation. 5 ng gDNA was used in all PCR reactions. Primers in Fig. 5a,b: A: 5F, Z :1R, a: ITR 1 C Rev, b: AAVhSyn Rev, SaCas9: SaCas9 1 For/Rev. Primers in Fig. 5c: Ube3a-ATS 3´ 1 F/R, priwalk F/ITR 2 R. AAV integration events in Fig. 5c were normalized to a region of Ube3a-ATS that exists only once in each allele, representing signal obtained for one diploid genome. PCR reactions were performed using Platinum Taq Polymerase (Invitrogen). qPCR reactions were performed using SsoAdvanced Universal SYBR Green Supermix (NEB).
RT-PCR:
RNA was extracted from the cortex of 10 month old E15.5/P1 AAV treated mice using Trizol. RNA was DNaseI treated (NEB) and extracted using Trizol a second time. To identify Ube3a-ATS/AAV fusion transcripts (Extended Data Fig. 9j) 500 ng total was used to synthesize cDNA using SuperScriptIV VILO (ThermoFisher). 0.05 uL of the cDNA was loaded into a PCR using primers to amplify Ube3a-ATS (Ube3a-ATS 3´ 1 F/R), SaCas9 (SaCas9 1 F/R), and Ube3a-ATS/AAV fusion transcripts (priwalk F, ITR 1 R).
To identify polyA trapped Ube3a-ATS/AAV fusion transcripts (Fig. 4a) cDNA was synthesized from 500 ng treated RNA using SuperScript IV (Invitrogen) and 3ŔACE poly-dT primer35. 0.05 uL of cDNA was loaded into a PCR using primers to amplify polyadenylated Ube3a-ATS transcripts (For: Snord115 HG, Rev: 3ŔACE anchor). The resulting PCR product was PCR purified (PCR Purification Kit, Qiagen). 1 pg of purified PCR was used as template with primers to amplify Ube3a-ATS (positive control, primers Ube3a-ATS 3´ 1 For/Rev), and Ube3a-ATS:AAV:pA fusion transcripts (For: SaCas9 2 F, Rev: 3ŔACE anchor). PCR products were purified and Sanger sequenced to confirm identity.
For the primer walking strategy (Fig. 4b) cDNA was synthesized from 500 ng total RNA using SuperScriptIV VILO (ThermoFisher). RT-PCR primers are listed in Supplementary Table 6. RT-PCR performed using Platinum Taq Polymerase (Invitrogen), 5.5’ extension time, 3% KB extender, 40 cycles, and touchdown PCR annealing temperatures (67˚C-60˚C for 15 cycles, −0.5˚C per cycle) to ensure optimum specificity of primer annealing.
Statistical Analyses
All results from analyses are presented as the mean ± standard error of the mean (s.e.m.) and differences were considered significant when P < 0.05. Two-tailed unpaired Student’s t-test was used for comparisons between two normally distributed groups. For behavioral data consisting of more than two groups, varying in a single factor, one-way analysis of variance (ANOVA) and Kruskall–Wallis were used. Comparison of non-normally distributed behavioral data were performed using a nonparametric Kruskall–Wallis test.
Extended Data
Extended Data Figure 1. Genomic map of Spjw33 targets and functional outcomes on Ube3a and Snord115 locus.
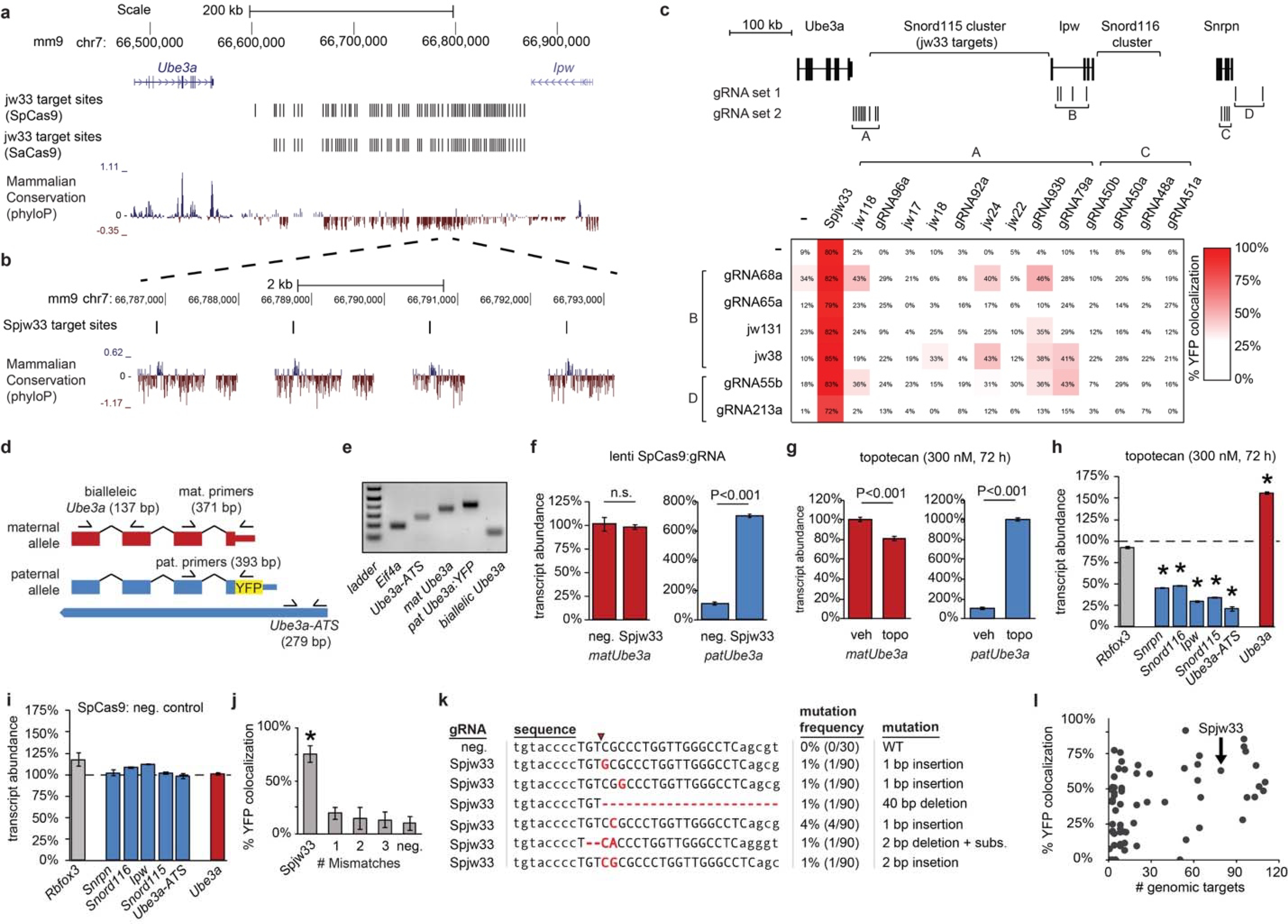
a. Mouse genome browser view.
b. Zoom in showing four Spjw33 target sites. Snord115 genes (blue peaks) are conserved (phyloP track) relative to other regions of Ube3a-ATS.
c. Systematic testing of pairs of gRNAs in Ube3am+/pYFP primary cortical neurons. Top panel: locations of gRNAs positioned to selectively target the Snord115 cluster (gRNA sets A and B), the Snord116 cluster (gRNA sets B and C), or the Ube3a-ATS promoter (gRNA sets A/C and D). Bottom panel: Percentage patYFP+/Cas9+ neurons for each pair of gRNAs relative to the positive control (SpCas9+Spjw33 alone).
d. Location of primers used to quantify transcript expression and discriminate between maternal Ube3a and paternal Ube3a-YFP alleles. Expected band size indicated.
e. Agarose gel showing RT-PCR products amplified from Ube3am+/pYFP cortical neuron cultures. All bands are of the expected size.
f-g. Expression (qPCR) of maternal or paternal Ube3a alleles in cortical neuron cultures transduced with lentivirus carrying SpCas9 and a gRNA targeting human SNORD115 (neg. control), Spjw33 (f), or treated with topotecan (g). Data normalized to Eif4a2, n=3.
h-i. Expression of the indicated genes in wild-type cortical neuron cultures treated with topotecan (h), or lentivirally transduced with SpCas9 and a gRNA targeting human SNORD115 (neg. control) (i). Dashed line marks vehicle (h) or neg. control gRNA (i) expression levels. Normalized to Eif4a2, n=3. * P<0.01.
j. Ube3am+/pYFP neurons transfected with gRNAs containing the indicated number of base mismatches relative to Spjw33. n=4, * P<0.05.
k. Table summarizing mutations identified at the Spjw33 target site. Primary cortical neurons were lentivirally transduced with SpCas9 and either a neg. control gRNA or Spjw33. Genomic DNA spanning the Spjw33 target site was PCR amplified and individual clones were analyzed by Sanger sequencing. Red arrow marks SpCas9 cleavage site. Red letters denote mutations that were introduced by SpCas9, based on the observation that these mutations were not found in neurons treated with the neg. control gRNA nor were they found in other Snord115 genes (mm9 genome build).
l. Percentage YFP colocalization in neurons transfected with SpCas9 and gRNAs targeting the indicated number of genomic sites in the Snord115 locus.
Extended Data Figure 2. Targeting SNORD115 unsilences paternal UBE3A in human neurons.
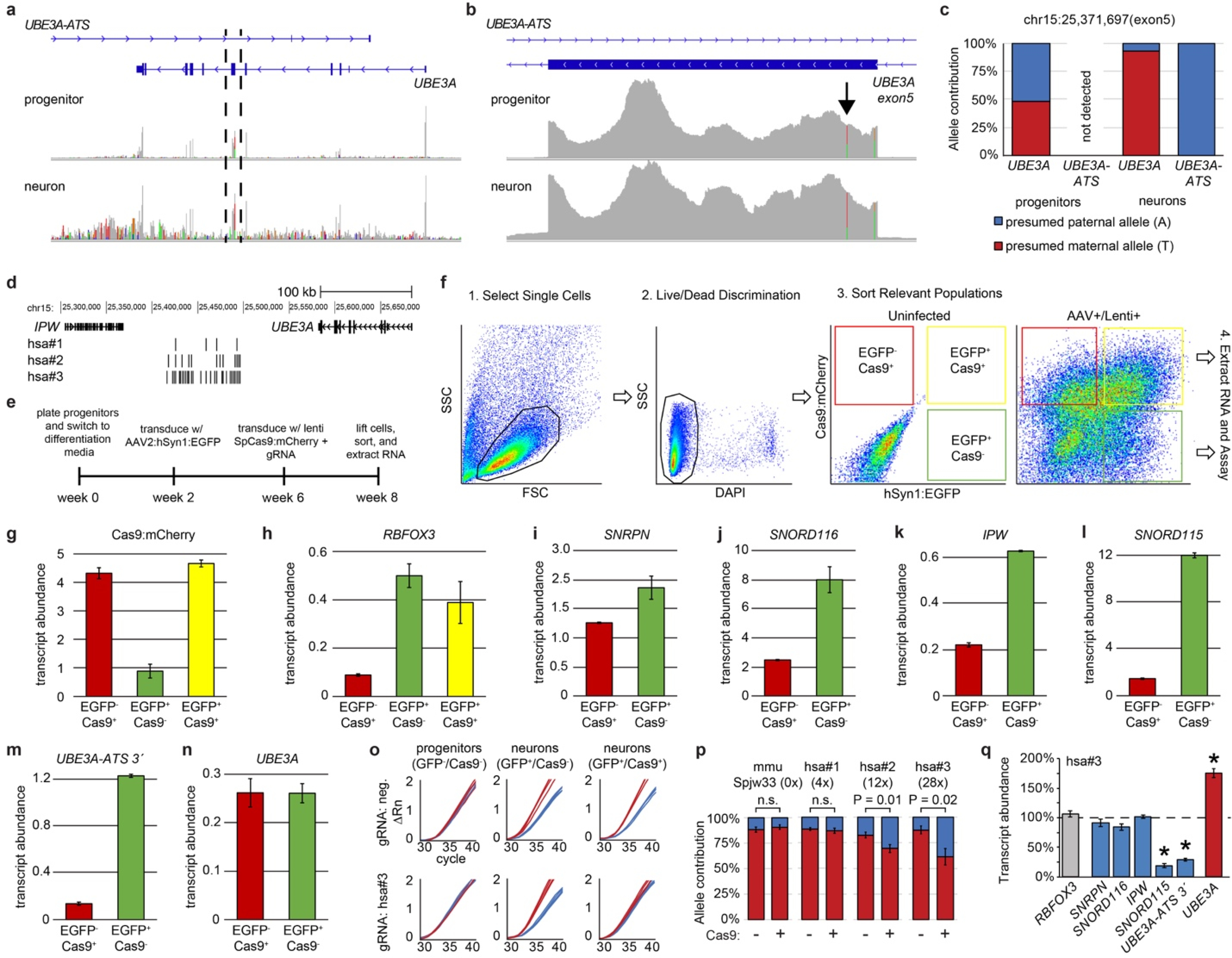
a. Alignment of RNA-seq reads from human neural progenitors (top panel) and 8 week differentiated neurons (bottom panel). Colored lines mark polymorphisms not present in the reference genome.
b. Zoom in of the region between dashed lines in a). Arrow denotes SNV (rsID:61734190) used to quantify allele specific expression in Extended Data Figs. 2c,o,p.
c. Allelic expression of UBE3A and UBE3A-ATS before and after differentiation of phNPCs into neurons, calculated as the percentage of stranded RNA-seq reads containing a SNV at chr15:25,371,697 (rsID:61734190).
d. Guide RNA locations in the human SNORD115 cluster.
e. Experimental design for transducing differentiated human neurons with lentiviruses containing SpCas9 and gRNAs.
f. Sorting strategy to isolate differentiated neuron populations from non-neuronal cells. Neurons were labeled and identified using AAV2:hSyn1:EGFP (green), cells lentivirally transduced with Cas9 were identified based on mCherry (red=non-neuronal cells, yellow=neurons). Side scatter counts (SSC), forward scatter counts (FSC).
g-n. Expression of the indicated genes in different cell populations using qPCR and SYBR green, normalized to EIF4A2. Values on Y-axis are presented as ΔCt values (not normalized to a specific sample), so expression values can be directly compared across multiple genes. n=3 for each qPCR reaction.
o. Raw qPCR curves from populations of progenitors and neurons containing the indicated fluorescent markers. Spjw33 negative control (mouse-specific). Presumed maternal (T; red line) and paternal (A; blue line) allele. ΔRn is change in reporter dye intensity per cycle, normalized to passive reference dye. One line per replicate, n=4.
p. Allele specific expression after lentiviral transduction of human neurons with SpCas9 and the indicated gRNAs, relative to neurons from the same cultures that were not transduced with Cas9. Number in parentheses refers to the number of SNORD115 target sites.
q. Expression of the indicated genes from Cas9+ neurons transduced with gRNA:hsa#3 versus Cas9− human neurons (dashed line). qPCR, normalized to EIF4A2, n=3. * P<0.01.
Extended Data Figure 3. SaCas9 vector development and testing.
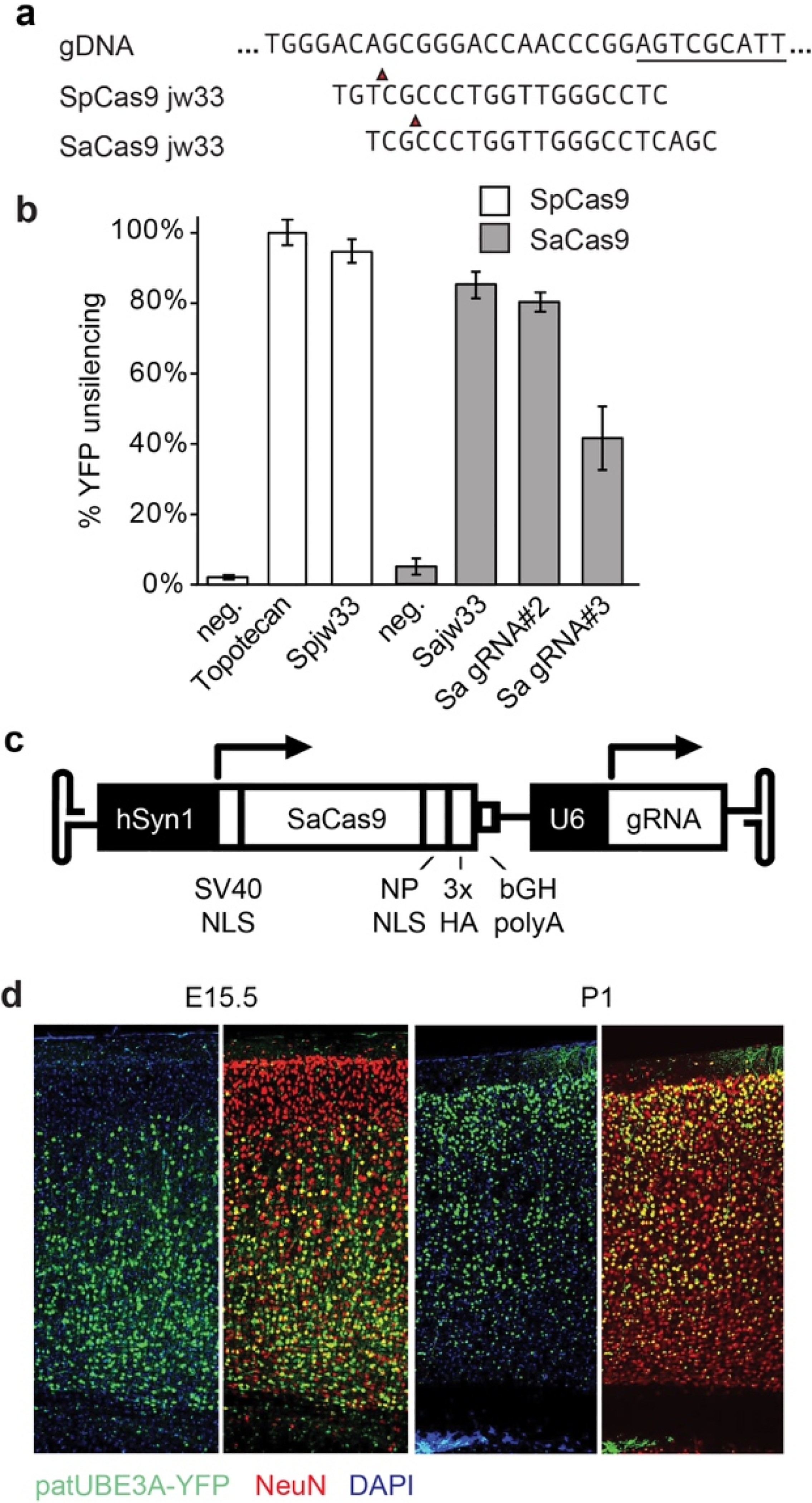
a. Sequence alignment between Spjw33 and Sajw33 gRNA and genomic target site. Underlined sequence marks a portion of Snord115. Red arrow marks Cas9 cleavage site.
b. Percentage of UBE3A-YFP+ neurons from Ube3am+/pYFP cortical neurons cultures transiently transfected with SpCas9 or SaCas9 and their respective gRNAs; relative to neg. control gRNAs and relative to topotecan treatment (300 nm, 72 h).
c. Map showing SaCas9 expression cassette in AAV backbone (not to scale). Human synapsin-1 promoter (hSyn1), simian virus 40 (SV40) nuclear localization sequence (NLS), nucleoplasmin (NP), hemagglutinin tag (HA), bovine growth hormone polyadenylation sequence element (bGH polyA), U6 promoter.
d. Immunofluorescence for indicated proteins in the cerebral cortex of P30 mice injected i.c.v. with AAV9 SaCas9:Sajw33 at E15.5 or P1. Note bias for deeper layer neurons when AAV was injected at E15.5 and bias for upper layer neurons when AAV was injected at P1.
Extended Data Figure 4. AAV delivery of SaCas9 and Sajw33 unsilences paternal Ube3a at P30 with no detectable AAV mediated toxicity.
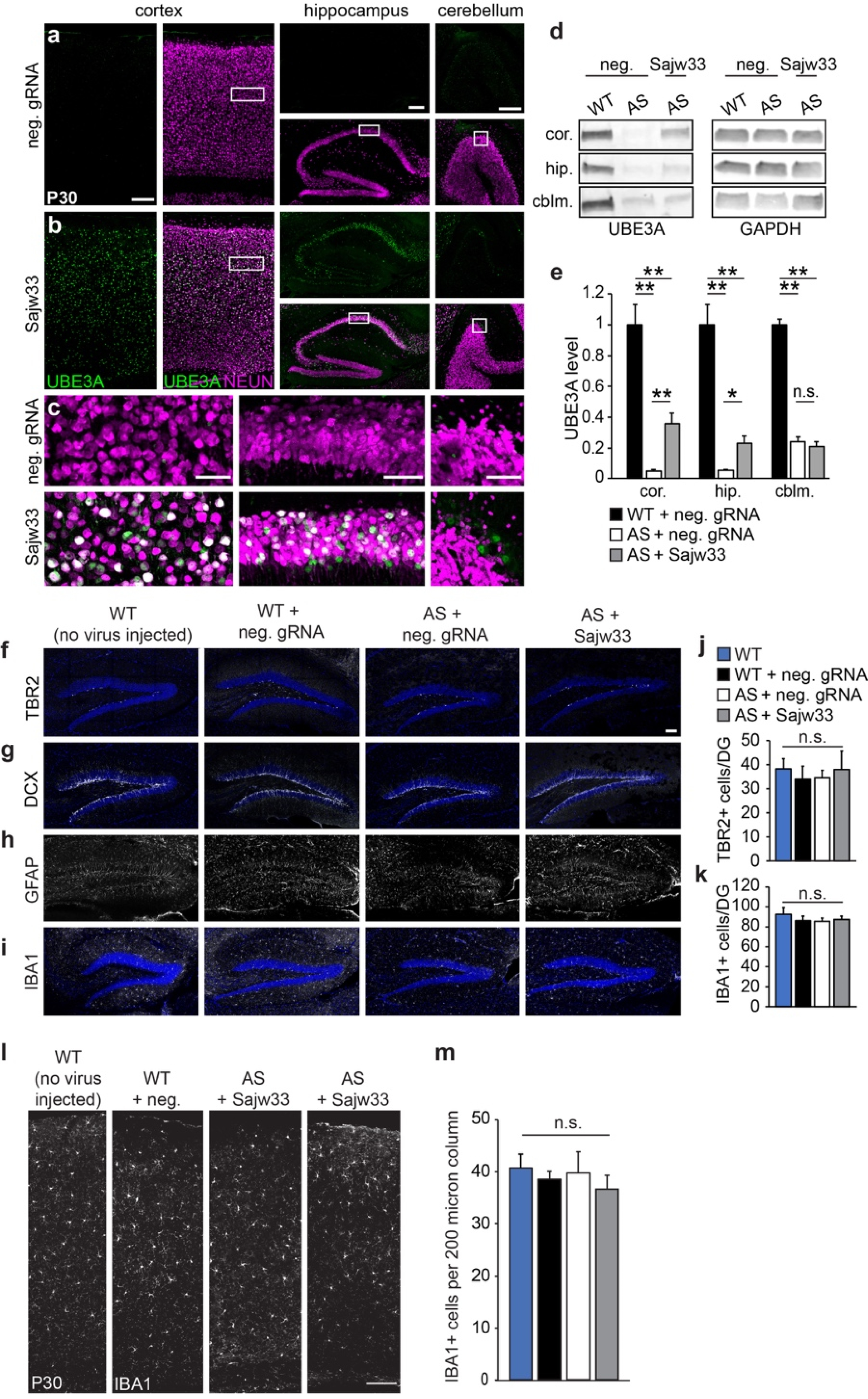
a-c. Histological staining for UBE3A and NEUN in the cortex, hippocampus, and cerebellum of P30 Ube3am-/p+ mice, injected at E15.5+P1 with AAV SaCas9 vector containing neg. control gRNA (a) or Sajw33 (b). c. Zoom-in view shows UBE3A protein in neurons (NEUN+). a and b, Cortex and hippocampus, scale bar, 200 μm. cerebellum, scale bar, 100 μm. c, scale bar, 50 μm.
d-e. Western blot quantification of UBE3A levels in the cortex (cor.), hippocampus (hip.), and cerebellum (cblm.) of P30 Ube3am-/p+ (AS) mice treated with neg. control gRNA or Sajw33 (n=3 per group; dual E15.5+P1 injections). WT=wild-type mice, age P30. * P<0.05, ** P<0.01.
f-i. Representative images of hippocampus from indicated genotypes and treatments at P30 from untreated or E15.5/P1 i.c.v. injected embryos. Immunofluorescence for progenitors (TBR2), immature neurons (DCX), astrocytes (GFAP), and microglia (IBA1).
j-k. Quantification of TBR2+ (j) and IBA1+ (k) cells showed no significant difference of cell numbers in dentate gyrus among indicated genotype and treatment groups.
l. Representative images of dorsal cortices stained for microglia (IBA1) of indicated genotypes and treatments at P30. Scale bar, 100 μm.
m. Quantification of IBA1+ cells in a 200 μm column of each dorsal cortex imaged. n=3 animals per genotype with treatment with 2 imaged sections each. n.s., not significant.
Extended Data Figure 5. Expression and alternative splicing of Snord115 target genes are not affected in the brain.
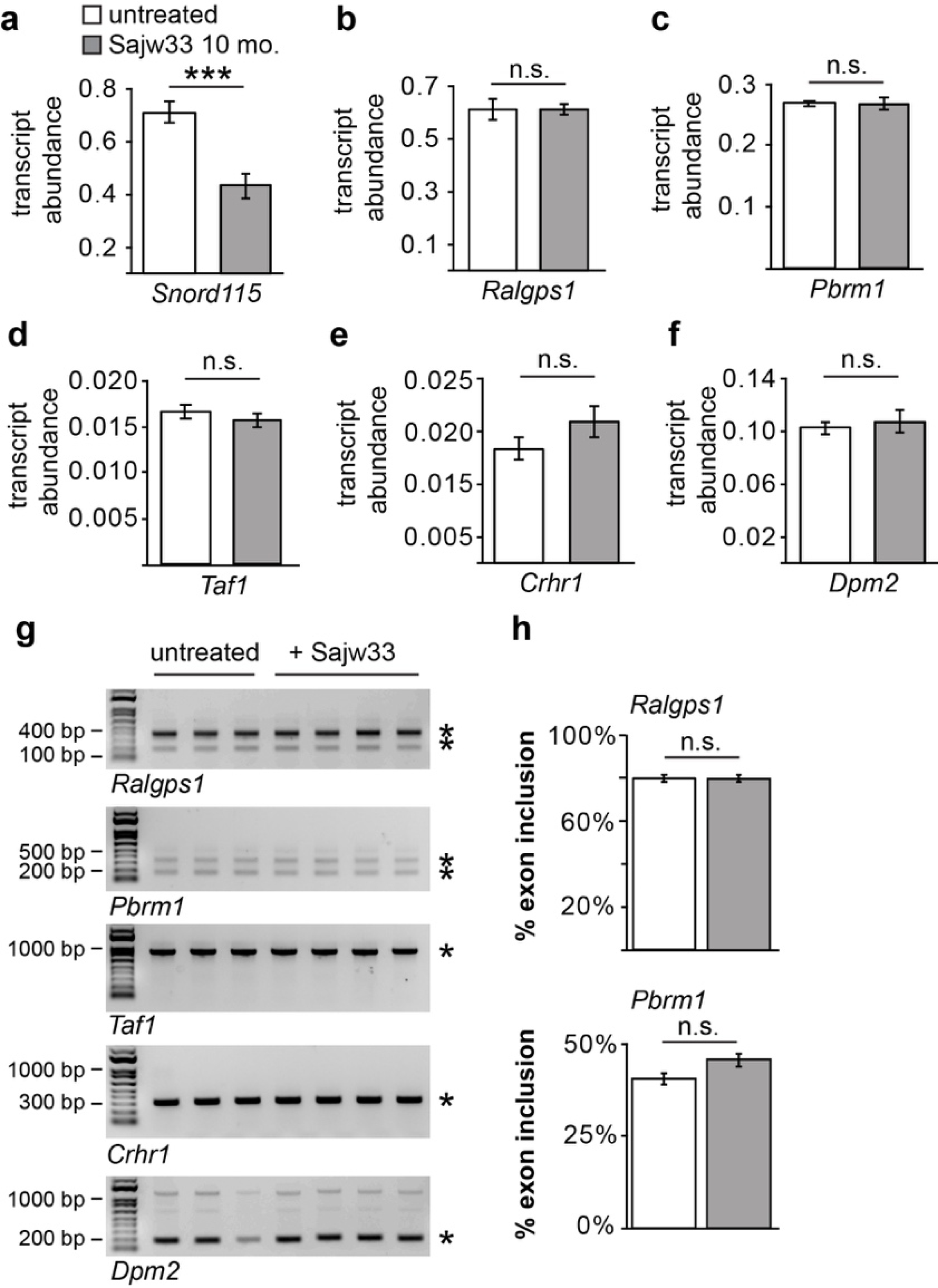
a. qPCR for Snord115 from cortex of mice either untreated or dual injected (i.c.v.) at E15.5+P1 with AAV carrying SaCas9 and Sajw33.
b.-f. Expression of Snord115 target genes in cortex of mice either untreated or dual injected (i.c.v.) at E15.5+P1 with AAV carrying SaCas9 and Sajw33.
g. Reverse transcription PCR for Snord115 target genes with published primers{Kishore, 2010 #54432}. Total RNA extracted from cortex of P60 mice dual injected at E15.5+P1 (i.c.v.) with AAV carrying SaCas9 and Sajw33.
h. Quantification of alternate splicing.
Extended Data Figure 6. Single injection of AAV containing SaCas9 and Sajw33 at P1 enduringly unsilences paternal UBE3A in 17 month old mice.
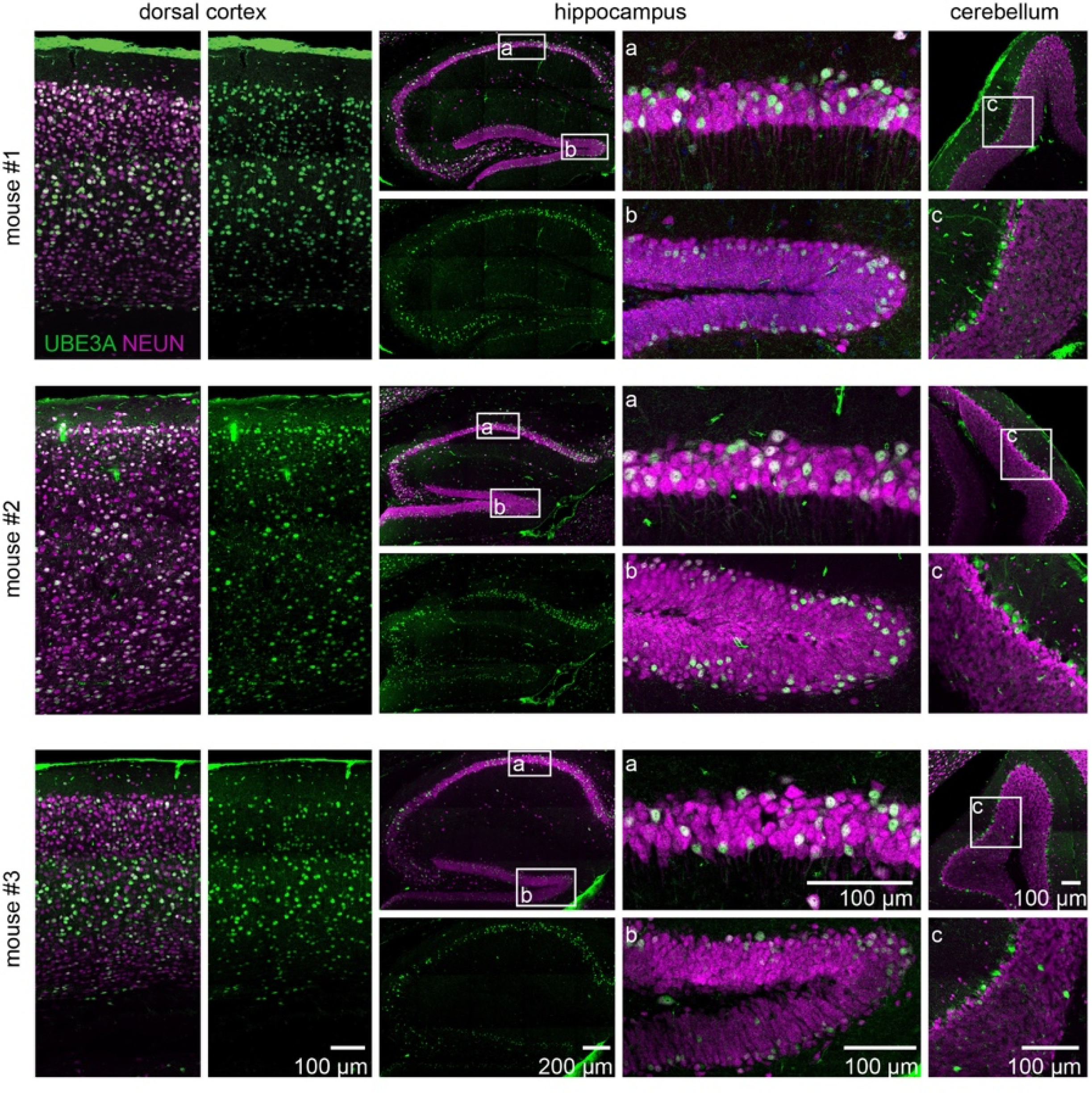
Representative images from three 17 month old Ube3am-/p+ animals treated with AAV9 carrying SaCas9 and Sajw33 at P1 through i.c.v. injection. Brains stained for UBE3A (green) showed extensive unsilencing in neurons (NEUN, magenta). Zoom in images of indicated region in hippocampus CA1 (a), dentate gyrus (b), and cerebellum (c).
Extended Data Figure 7. Evaluation of viral transduction in tissues from injected mice and their dams.
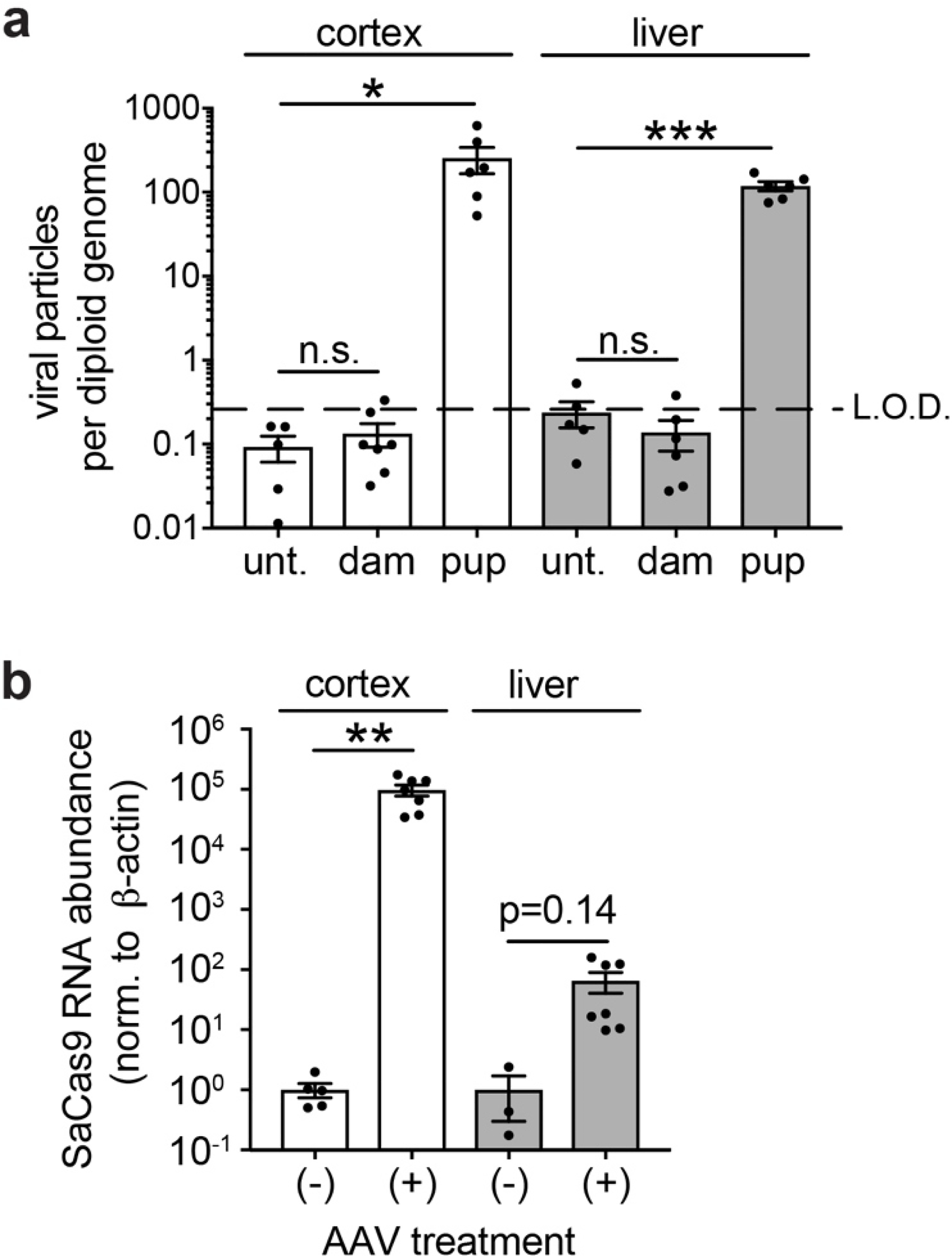
a. qPCR quantification of viral DNA (SaCas9 TaqMan probes) in cortex and liver of age matched untreated animals, P60 dams whose pups were injected i.c.v. at E15.5, and P60 mice that were dual injected i.c.v. at E15.5+P1. Data normalized to Eif4a2 (representing a gene with 2 copies per diploid genome). Limit of detection (LOD) determined by performing serial dilutions with known quantities of AAV particles spiked into gDNA samples from untreated mice. * P<0.05, ** P<0.01, *** P<0.001.
b. Expression of SaCas9 mRNA in cortex and liver from same animals as (a). ΔΔCt method, normalized to ß-actin.
Extended Data Figure 8. Additional Behavioral Assays.
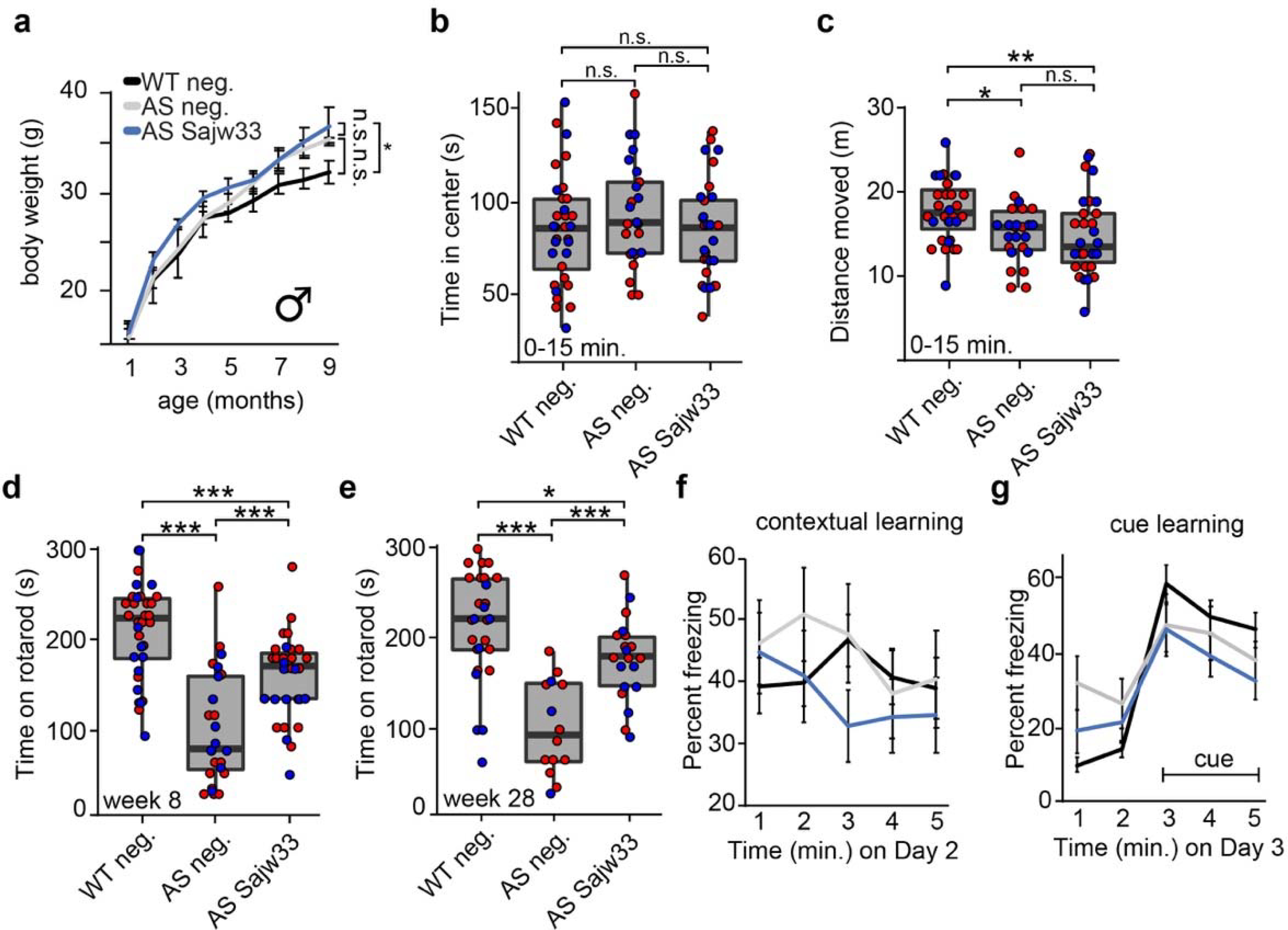
a. Body weight of male mice measured monthly over nine months.
b-c. Open field data at 11 weeks of age, first 0–15 minutes of experiment. Time spent in center in seconds (b), distance travelled in meters (c). * P<0.05, ** P<0.01, *** P<0.001.
d-e. Rotarod training data (average of three trials) at 8 weeks of age (d) and 28 weeks of age (e).
f-g. Contextual (f) and cue based (g) learning at 18 weeks of age. No statistically significant phenotypes were observed.
Extended Data Figure 9. Analysis of mutations and AAV integration in 10 month old mice.
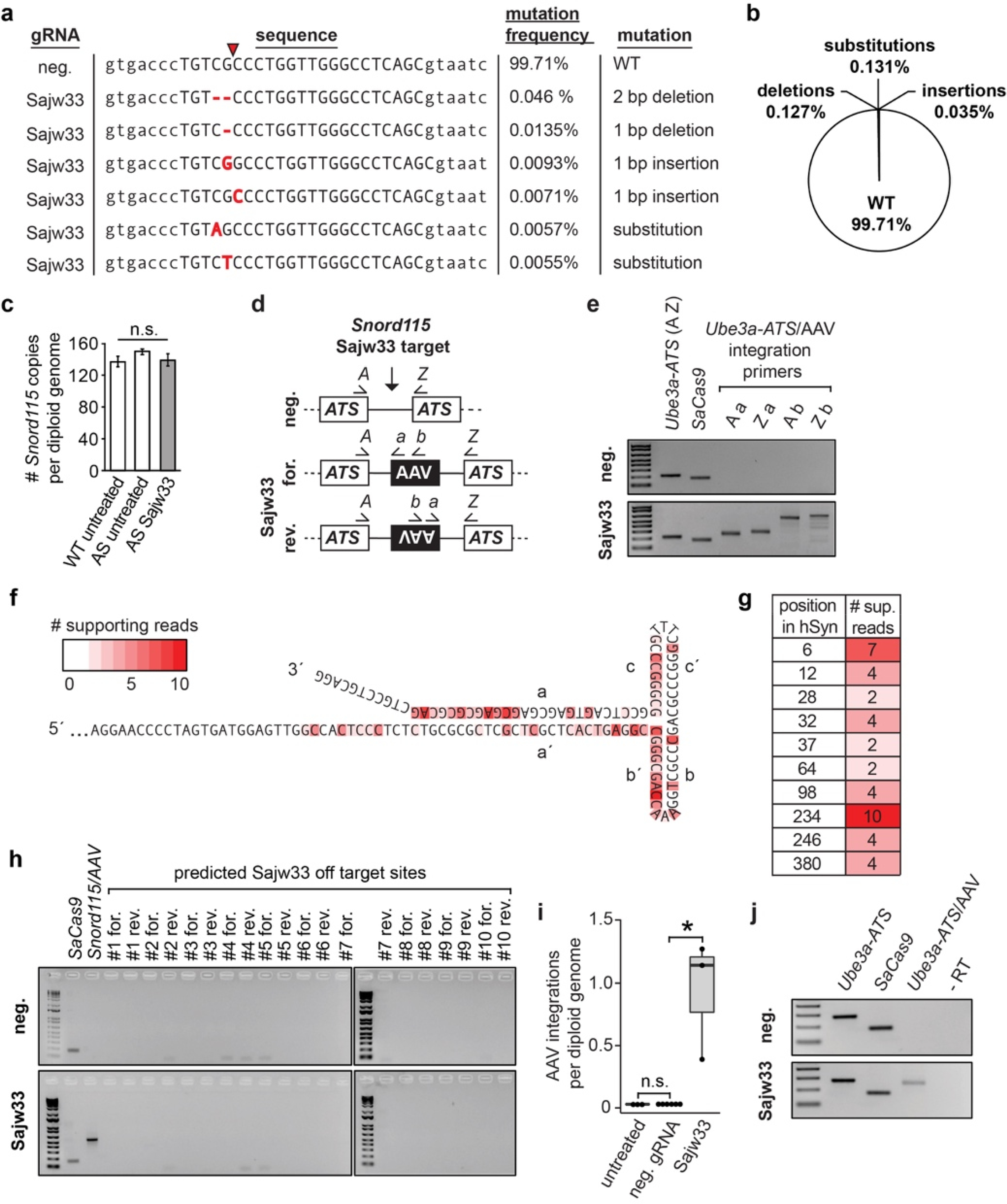
a. Analysis of mutations found in Sajw33 treated mice by high-throughput gDNA amplicon sequencing (no mutations found in controls). Capital letters denote Sajw33 target site, red arrow refers to SaCas9 cleavage site. Red nucleotides represent nucleotides not present in AS animals treated with neg. gRNA nor in the reference genome (mm9).
b. Percentage of specific mutation types identified in 10 month old Sajw33 treated mice (n=3).
c. qPCR from gDNA isolated from 10 month old animals of the indicated genotypes. Data normalized to a region of Ube3a-ATS which contains two copies per diploid genome by ΔΔCt method.
d. PCR strategy to detect AAV integration events.
e-j. All DNA/RNA extractions performed from cortexes of 10 mo. old mice, injected at E15.5+P1 with AAV containing SaCas9 and the negative control gRNA or Sajw33.
e. PCR of genomic DNA with the indicated primers.
f-g. DNA amplicon sequencing of AAV integration events, detailing the position in the AAV ITR or hSyn1 promoter that was immediately adjacent to endogenous gDNA at the Sajw33 target site.
h. gDNA PCR at predicted Sajw33 off target sites using primers to interrogate AAV integration in both orientations. SaCas9 and Snord115/AAV integrations are positive controls. PCR was performed for 40 cycles. The absence of bands suggests no AAV integration at the top 10 predicted off-target sites.
i. qPCR of genomic DNA. Forward primer anneals in Snord115 forward orientation, reverse primer anneals to both viral inverted terminal repeats (ITRs) to quantify the number of AAV integration events in the genome irrespective of orientation. * P<0.05.
j. RT-PCR with primers specific for the indicated genes/gene fusion. cDNA synthesized with random hexamers, no reverse transcriptase control (-RT) with Ube3a-ATS/AAV fusion primer pair.
Extended Data Figure 10. AAV integration disrupts Ube3a-ATS via distinct mechanisms.
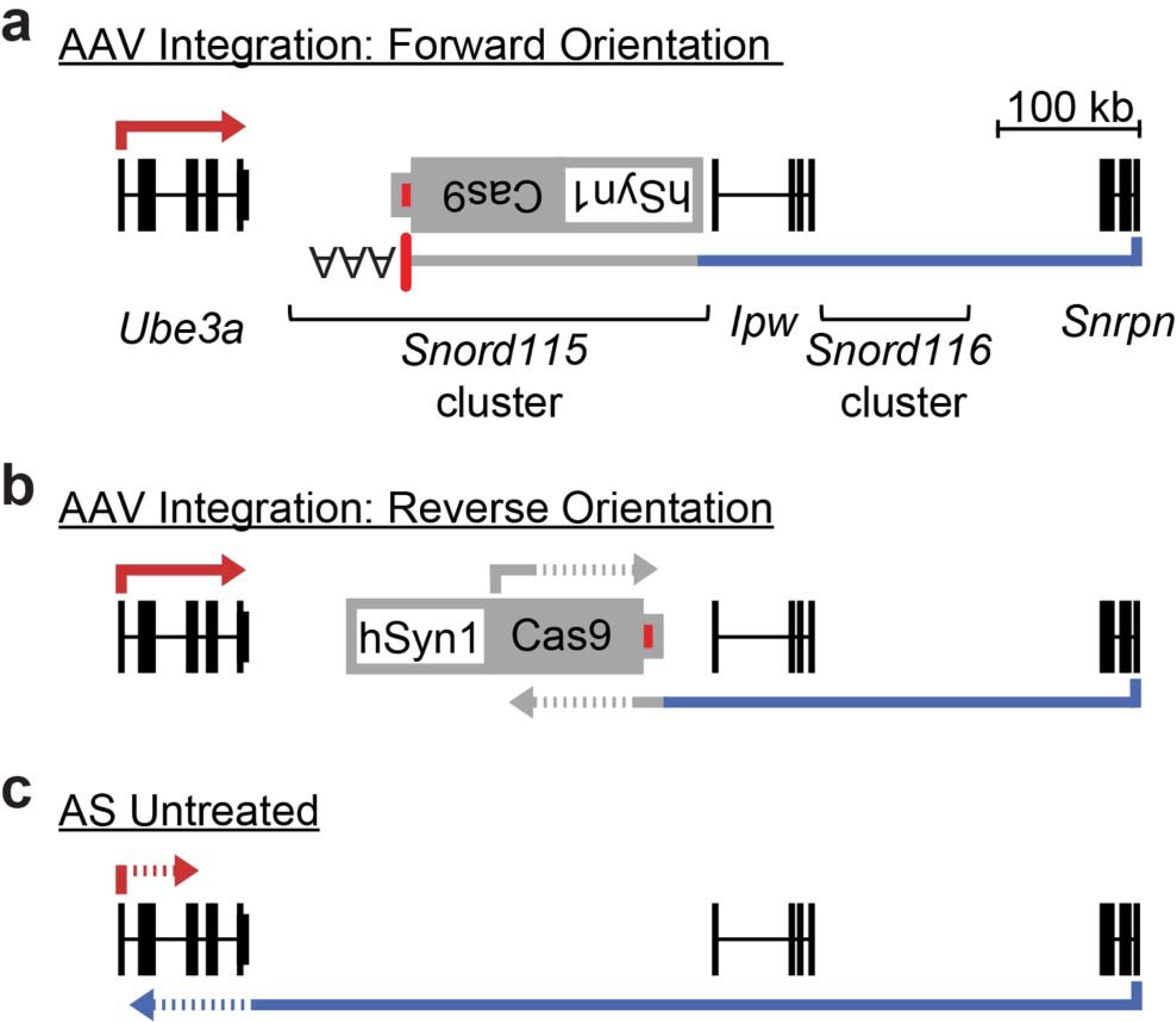
a. AAV integration in the forward orientation gene traps Ube3a-ATS, causing premature transcription termination at the AAV vector-derived polyadenylation sequence element (red box).
b. In the reverse orientation, convergent transcription with the AAV vector-derived Cas9 transcript disrupts Ube3a-ATS.
c. Paternal Ube3a (red line) is similarly disrupted (“silenced”) by convergent transcription.
Supplementary Material
Acknowledgments
We thank Eric McCoy, Gabriela Salazar, Erik Hopkins, Travis Ptacek, and Bonnie Taylor-Blake for technical assistance. We thank the UNC Catalyst for Rare Diseases for use of their high-throughput screening equipment. This work was supported by grants to M.J.Z. from The Angelman Syndrome Foundation, The Simons Foundation (SFARI, Award ID 631904), The National Institute of Neurological Disorders and Stroke (NINDS; 1R01NS109304-01A1), and The Eshelman Institute for Innovation. J.M.W. was supported by grants from the National Institute for Child Health and Human Development (NICHD T32HD040127) and a Pfizer-NCBiotech Distinguished Postdoctoral Fellowship in Gene Therapy. H.M. was supported by NICHD (T32HD040127). J.L.S. was supported by grants from the National Institute of Mental Health (R01MH118349, R00MH102357, R01MH120125). The microscopy core and J.M.S. in the bioinformatics core were supported by NICHD (U54HD079124) and NINDS (P30NS045892). The UNC Flow Cytometry Core Facility is supported in part by The National Cancer Institute (P30CA016086), awarded to the UNC Lineberger Comprehensive Cancer Center. The UNC Mouse Behavioral Phenotyping Core is supported by NICHD (U54HD079124).
Footnotes
Competing interest declaration
The authors declare the existence of a financial competing interest. M.J.Z. serves as a consultant to AskBio to which technologies evaluated in this paper have been licensed. J.M.W., H.M., G.F., J.M.S., M.J.Z. are inventors of the technology and could receive royalties. These relationships have been disclosed to and are under management by UNC-Chapel Hill. The remaining authors have no competing interests.
Additional Information
Contains six supplementary data tables, 10 extended data figures, and two Supplementary Figures containing raw data (gels, confocal images).
Data availability statement
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
Main References
- 1.Silva-Santos S et al. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. The Journal of clinical investigation 125, 2069–2076, doi: 10.1172/JCI80554 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zylka MJ Prenatal Treatment Path for Angelman Syndrome and Other Neurodevelopmental Disorders. Autism Res, doi: 10.1002/aur.2203 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang HS et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 481, 185–189, doi: 10.1038/nature10726 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meng L et al. Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS genetics 9, e1004039, doi: 10.1371/journal.pgen.1004039 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu S et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nature biotechnology 34, 1279–1286, doi: 10.1038/nbt.3715 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dindot SV, Antalffy BA, Bhattacharjee MB & Beaudet AL The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Human molecular genetics 17, 111–118, doi: 10.1093/hmg/ddm288 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Bortolin-Cavaille ML & Cavaille J The SNORD115 (H/MBII-52) and SNORD116 (H/MBII-85) gene clusters at the imprinted Prader-Willi locus generate canonical box C/D snoRNAs. Nucleic acids research 40, 6800–6807, doi: 10.1093/nar/gks321 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Smith AJ et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Human molecular genetics 18, 3257–3265, doi: 10.1093/hmg/ddp263 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bieth E et al. Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi Syndrome. Eur J Hum Genet 23, 252–255, doi: 10.1038/ejhg.2014.103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderlid BM, Lundin J, Malmgren H, Lehtihet M & Nordgren A Small mosaic deletion encompassing the snoRNAs and SNURF-SNRPN results in an atypical Prader-Willi syndrome phenotype. American journal of medical genetics. Part A 164A, 425–431, doi: 10.1002/ajmg.a.36307 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Hsiao JS et al. A bipartite boundary element restricts UBE3A imprinting to mature neurons. Proceedings of the National Academy of Sciences of the United States of America 116, 2181–2186, doi: 10.1073/pnas.1815279116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stein JL et al. A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron 83, 69–86, doi: 10.1016/j.neuron.2014.05.035 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ran FA et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191, doi: 10.1038/nature14299 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedland AE et al. Characterization of Staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome biology 16, 257, doi: 10.1186/s13059-015-0817-8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bengtsson NE et al. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nature communications 8, 14454, doi: 10.1038/ncomms14454 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Avagliano Trezza R et al. Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nature neuroscience 22, 1235–1247, doi: 10.1038/s41593-019-0425-0 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Johnston S et al. AAV Ablates Neurogenesis in the Adult Murine Hippocampus. bioRxiv, 2020.2001.2018.911362, doi: 10.1101/2020.01.18.911362 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kishore S & Stamm S The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science 311, 230–232, doi: 10.1126/science.1118265 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Doe CM et al. Loss of the imprinted snoRNA mbii-52 leads to increased 5htr2c pre-RNA editing and altered 5HT2CR-mediated behaviour. Human molecular genetics 18, 2140–2148, doi: 10.1093/hmg/ddp137 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sonzogni M et al. A behavioral test battery for mouse models of Angelman syndrome: a powerful tool for testing drugs and novel Ube3a mutants. Molecular autism 9, 47, doi: 10.1186/s13229-018-0231-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mandel-Brehm C, Salogiannis J, Dhamne SC, Rotenberg A & Greenberg ME Seizure-like activity in a juvenile Angelman syndrome mouse model is attenuated by reducing Arc expression. Proceedings of the National Academy of Sciences of the United States of America 112, 5129–5134, doi: 10.1073/pnas.1504809112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanlon KS et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nature communications 10, 4439, doi: 10.1038/s41467-019-12449-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nelson CE et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nature medicine 25, 427–432, doi: 10.1038/s41591-019-0344-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.von Bartheld CS, Bahney J & Herculano-Houzel S The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. The Journal of comparative neurology 524, 3865–3895, doi: 10.1002/cne.24040 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedel RH & Soriano P Gene trap mutagenesis in the mouse. Methods in enzymology 477, 243–269, doi: 10.1016/S0076-6879(10)77013-0 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 26.King IF et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature 501, 58–62, doi: 10.1038/nature12504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nature methods 11, 783–784, doi: 10.1038/nmeth.3047 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mabb AM et al. Topoisomerase 1 Regulates Gene Expression in Neurons through Cleavage Complex-Dependent and -Independent Mechanisms. PLoS One 11, e0156439, doi: 10.1371/journal.pone.0156439 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dickerson AS et al. Autism spectrum disorder prevalence and associations with air concentrations of lead, mercury, and arsenic. Environmental monitoring and assessment 188, 407, doi: 10.1007/s10661-016-5405-1 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Kishore S et al. The snoRNA MBII-52 (SNORD 115) is processed into smaller RNAs and regulates alternative splicing. Human molecular genetics 19, 1153–1164, doi: 10.1093/hmg/ddp585 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walantus W, Castaneda D, Elias L & Kriegstein A In utero intraventricular injection and electroporation of E15 mouse embryos. Journal of visualized experiments : JoVE, 239, doi: 10.3791/239 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim JY, Grunke SD, Levites Y, Golde TE & Jankowsky JL Intracerebroventricular viral injection of the neonatal mouse brain for persistent and widespread neuronal transduction. Journal of visualized experiments : JoVE, 51863, doi: 10.3791/51863 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bae S, Park J & Kim JS Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475, doi: 10.1093/bioinformatics/btu048 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin M CUTADAPT removes adapter sequences from high-throughput sequencing reads. EMBnet Journal 17, 10–12 (2011). [Google Scholar]
- 35.Blazie SM et al. Alternative Polyadenylation Directs Tissue-Specific miRNA Targeting in Caenorhabditis elegans Somatic Tissues. Genetics 206, 757–774, doi: 10.1534/genetics.116.196774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hogart A, Wu D, LaSalle JM & Schanen NC The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiology of disease 38, 181–191, doi: 10.1016/j.nbd.2008.08.011 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang D, Zhang F & Gao G CRISPR-Based Therapeutic Genome Editing: Strategies and In Vivo Delivery by AAV Vectors. Cell 181, 136–150, doi: 10.1016/j.cell.2020.03.023 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Landers M et al. Maternal disruption of Ube3a leads to increased expression of Ube3a-ATS in trans. Nucleic acids research 33, 3976–3984, doi: 10.1093/nar/gki705 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its supplementary information files).