

Conspectus
Electrochemical measurements conducted in confined volumes provide a powerful and direct means to address scientific questions at the nexus of nanoscience, biotechnology, and chemical analysis. How are electron transfer and ion transport coupled in confined volumes and how does understanding them require moving beyond macroscopic theories? Also, how do these coupled processes impact electrochemical detection and processing? We address these questions by studying a special type of confined-volume architecture - the nanopore electrode array, or NEA, which is designed to be commensurate in size with physical scaling lengths, such as the Debye length, a concordance that offers performance characteristics not available in larger scale structures.
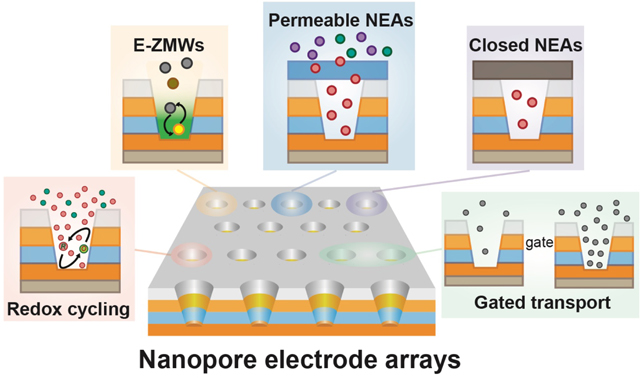
The experiments described here depend critically on carefully constructed nanoscale architectures that can usefully control molecular transport and electrochemical reactivity. We begin by considering the experimental constraints that guide the design and fabrication of zero-dimensional nanopore arrays with multiple embedded electrodes. These zero-dimensional structures are nearly ideal for exploring how permselectivity and unscreened ion migration can be combined to amplify signals and improve selectivity by enabling highly efficient redox cycling. Our studies also highlight the benefits of arrays, in that molecules escaping from a single nanopore are efficiently captured by neighboring pores and returned to the population of active redox species being measured - benefits that arise from coupling ion accumulation and migration. These tools for manipulating redox species are well-positioned to explore single molecule and single particle electron transfer events through spectroelectrochemistry, studies which are enabled by the electrochemical zero-mode waveguide (ZMW) - a special hybrid nanophotonic/nanoelectronic architecture in which the lower ring electrode of an NEA nanopore functions both as a working electrode to initiate electron transfer reactions and as the optical cladding layer of a ZMW. While the work described here is largely exploratory and fundamental, we believe that the development of NEAs will enable important applications that emerge directly from the unique coupled transport and electron-transfer capabilities of NEAs - including in situ molecular separation and detection with external stimuli, redox-based electrochemical rectification in individually encapsulated nanopores, and coupled sorters and analyzers for nanoparticles.
1. Introduction
Single entity electrochemistry (SEE) is a fast-growing field that addresses emerging questions at the boundaries of electrochemical measurement and processing. Since the term was popularized in 2016,1, 2 there has been mounting interest in exploiting the electrochemical behavior of single entities in electrochemical sensing and processing.3–5 SEE covers a range of topics from single redox molecules and single enzymes with redox-active centers, to single composite species, like nanoparticles or cells6–12. Unlike conventional bulk electrochemical measurements, SEE can characterize stochastic processes and heterogeneous properties of entities, where discrete events dominate dynamics.13, 14
SEE measurements are enabled by instrumentation that can capture small signals at acceptable signal-to-noise ratio, with fast temporal and high spatial resolution, and at reasonable cost. Shrinking the size of an electrode to the nanoscale reduces the cross-sectional area, realizing advantages, including enhanced mass transport, fast reaction kinetics, negligible ohmic drop, and permselectivity at low ionic strength.15–17 However, along with enhanced performance comes the need for better understanding, since ion transport and electron transfer from individual entities can deviate from predictions of conventional theories.13, 18
Nanopore electrodes constitute a subset of nanoelectrodes, in which annular ring or disk electrodes are inset within a cylindrical or conical nanopore with typical volumes ~ 10−18 L which confers special capabilities for executing electrochemical measurements and processing.19, 20 However, to realize the full potential of these architectures it is necessary to understand how electron transfer and ion transport are coupled in confined environments, understanding that can illuminate phenomena ranging from the biological sciences to energy conversion and storage.
Early work from our laboratory concentrated on embedding metallic electrodes in one-dimensional nanofluidic structures so that the same small potential (≤1 V) could be used both to drive electrokinetic flow and mediate faradaic electron transfer, allowing for simultaneous coupling of electrokinetic injection and electrochemical processing.21–23 Since then, our focus has moved toward electron transfer reactions in structures where the nanostructure size is commensurate with physical scaling lengths, such as the Debye length - a concordance that can lead to enhanced performance characteristics not available in larger structures. In the last five years, we have described a number of unusual effects enabled by working in confined volumes: nanopore-confined redox cycling24, 25, closed bipolar coupling of nanopore electrochemistry to electrofluorogenic reactions26, electrochemistry in the absence of supporting electrolyte,27–29 single electron transfer events in electrochemical zero-mode waveguides (E-ZMW),30, 31 nanopore-based electrochemical rectification32 and nanoparticle sorting.33
In this Account, we highlight nanopore electrode arrays (NEAs) - nanoscale electrode architectures that integrate all the above advantages. We begin by discussing the design of NEAs, especially the critical role of nanopore geometry, materials selection, device fabrication, and fabrication strategies. Next, we discuss how significant signal amplification can be obtained from redox cycling and the sensitivity and selectivity enhancements that arise from careful control of ion accumulation and ion migration. Subsequently, we consider spectroelectrochemical approaches to the study of single electron transfer events and single particle collisions - an approach to real-time observation of electron transfer at the single-entity level. Lastly, we hypothesize that the development of NEAs will enable important applications, such as in situ molecular separations, redox-based electrochemical rectification in encapsulated nanopores, and coupled sorters and analyzers for nanoparticles, all of which connect to emergent nanoscale behaviors and provide motivation for extending NEA-based work.
2. NEA fabrication and processing
NEAs contain electrodes that exhibit the intrinsic properties of single nanopores, e.g. confined environment, surface charge-induced ion transport, and the capability to enhance signal levels, but in a massively parallel format. Figure 1 summarizes the various structural motifs, fabrication strategies, and materials and geometry choices that dictate the family of NEA architectures described here.
Figure 1.

Schematic diagram illustrating the characteristics, fabrication methods and post-processing of NEAs. (A) Single- (top) and dual-electrode embedded NEAs, including dual-ring (center) and ring-disk (bottom) geometries. (B) Exploded layer view of multilayered NEAs, the nanopore array being formed where the Au layers overlap. (C) Parallel processing using nanosphere lithography (NSL) with subsequent reactive ion etching (RIE). (D) Direct writing method using focused ion beam (FIB) milling. (E) Orthogonal chemical functionalization of electrodes and dielectrics to tune the surface properties of NEAs. (F) Two variants of NEAs that provide transport control (semi-permeable NEAs) and confinement (closed NEAs), respectively.
NEA characteristics.
The most important geometric choices for NEA design are the nanopore diameter, and spacing of adjacent nanopores. Focused ion beam (FIB) milling30, 34 results in conical frustum pores with bottom diameters as small as 30 nm, while nanotemplating results in larger pores.24, 25 Nanopore spacing can vary, with large pitch (≥ 2 μm) arrays being ideal for optical observations30, while tightly-packed arrays fabricated by nanosphere lithography (NSL) and graphoepitaxy of block copolymers - down to ~ 35 nm spacing - are good candidates for signal enhancement and high-flux analysis35. The nanopore volume and shape affect mass transport and local concentration of redox species as well as the surface charge distribution. The nanopore volume can be extended from ~10−16 L down to ~10−20 L, leading to a single entity occupancy at concentrations from nM to μM.
Fabrication methods.
As shown in Figure 1A, NEAs used in our work are fabricated as either single- or dual-electrode embedded nanopore arrays, the latter including both ring-disk and dual-ring geometries. Single electrode NEAs can be used for single enzyme immobilization36 and nanoparticle collision experiments37, however the faradaic currents are limited, and they are less amenable to tuning electrochemical and optical responses. To circumvent these limitations, dual-electrode arrays are realized by placing an insulating SiNx layer between conducting layers. Typically, Au electrodes are lithographically patterned as overlapping vertically separated fingers, (100 × 100 μm2, typ.) before nanoscale array patterning, Figure 1B. Nanopores are then fabricated using either a combination of NSL and reactive ion etching (RIE), Figure 1C, or FIB milling, Figure 1D. FIB milling is a direct-write method capable of producing highly uniform nanopore arrays with variable structural parameters, e.g. nanopore diameter, depth, pitch, and array area, but NSL followed by RIE, a parallel method, is more cost-effective for the production of densely packed nanopores over large areas.
Surface-modified NEAs.
Fabrication, as shown in Figures 1A-D, produces NEAs composed of electrodes and insulating spacers in multilayered nanopore structures. Importantly, the materials, e.g. Au and SiO2, can be chosen to be chemically orthogonal, thus facilitating the post-fabrication derivatization, Figure 1E. Using this strategy, we covalently immobilized single copies of flavin adenine dinucleotide (FAD), a redox-active enzyme cofactor, onto the Au layer of NEAs starting with self-assembly of aminoethanethiol. Upon immobilization, single molecule fluorescence dynamics were observed to be controlled by applied potential, indicating the possibility that redox states could be controlled at the single molecule level.36 Dielectric materials, such as SiO2 or SiNx, can be used to control surface charge density, and various functional groups can be introduced through silanization, without cross-reacting to the electrode surface. We demonstrated this type of chemical orthogonality in preparing negatively-charged, cation-permselective silica surfaces. Then the charge selective gating was reversed by switching the outer surface positive with protonated 3-aminopropyltriethoxysilane35.
NEA add-ons.
The nanopore electrode architectures can also be extended by attaching layers that integrate other functions to the top NEA surface, Figure 1F. Adding supplemental layers on top of the NEAs can control access of species into NEAs (semi-permeable NEA) or trap (closed NEA) species within individual nanopores. Semi-permeable NEAs can be realized through the addition of permselective, e.g. Nafion, or stimulus-responsive polymeric coatings, providing directional control of ion flow and additional charge selectivity.32 Closed-pore NEAs are typically made by the addition of a thin elastomer, which seals the opening of each nanopore, thus preventing escape of redox species from NEAs.38
3. Pushing the detection limit
NEAs can enhance detection performance, e.g. sensitivity and selectivity, through optimized dual-electrode-embedded arrays.24 We developed a robust approach to fabricate NEAs over large areas by NSL/RIE, Figures 2A(i) and (ii). These structures enabled independent control of top and bottom nanopore electrodes, thus establishing the ability for generator-collector (GC) redox cycling, Figure 2B. The signal amplification from redox cycling in dual-electrode embedded nanopores (55x) is larger than thin layer cell, interdigitated electrode arrays, rotating ring disk electrodes, etc.39–41 and is derived from the high yield-cycling reaction that converts oxidized (reduced) species at the bottom (generator) electrode, into the complementary form at the top collector electrode. Defining the amplification factor, AF, as the ratio of the limiting currents, AF = ilim,GC/ilim,non-GC, allows a quantitative comparison of device performance, Figure 2B. By tuning the nanopore diameter, pitch and the gap distance between generator and collector electrode, AF was increased to >100x.42 Finite element simulation results confirmed that the large signal amplification results from the coupling of transport and accumulation within each nanopore, as well as the capture of escaped species by adjacent nanopores.24, 25 To investigate how the nanopore geometry affects redox cycling, we fabricated NEAs with a wide range of nanopore size, pitch, and nanogap25 and found that reaction efficiency is strongly correlated with geometry. Furthermore, ultra-small capacitive currents were observed from the collector electrode at fast scan rates, indicating the possibility of exploring transient electrochemical phenomena associated with short-lived species, such as radicals and reaction intermediates, viz. Figure 2C.
Figure 2.

Pushing the limit of detection in NEAs. (A) Ring-disk NEAs schematically shown (i), with its cross-sectional SEM image, scale bar 200 nm (ii), and a device photo with 8 NEAs on glass (iii). (B) Redox cycling effect illustrated by comparing GC mode (blue) and non-GC mode (black, inset) operation, indicating 55x signal enhancement. (C) Capacitive current from collector and generator electrodes, indicating the possibility of transient electrochemistry for short-lived species. (D) Signal enhancement in the absence of supporting electrolyte for Ru(NH3)63+. (E) Migration-induced ion accumulation as a result of decreasing ionic strength for Ru(NH3)63+. (F) Increasing selectivity for dopamine in the presence of interfering ascorbic acid during electrochemical detection at decreasing ionic strength. Panels A and F are adapted with permission, ref 29. Copyright 2017 The Royal Society of Chemistry. Panels B and C are adapted with permission, ref 25. Copyright 2016 The Royal Society of Chemistry. Panel D is adapted with permission, ref 27. Copyright 2014 American Chemical Society. Panel E is adapted with permission, ref 28. Copyright 2016 American Chemical Society.
Besides boosting the current and achieving small non-Faradic signals, the combination of ionic strength and applied potential alters the surface charge distribution along the nanopore.27–29 Typically, the thickness of the electrical double layer (EDL), which is correlated with the Debye length, is on the scale of a few nm, increasing to 10s of nm when the electrolyte concentration decreases to the μM range. Thus, NEA nanopore dimensions are comparable to the EDL at low ionic strength, a relation captured by the dimensionless group κd, where κ = inverse Debye length, and d = pore diameter. When κd ~ 1, surface charge controls ion transport and electron transfer of redox species inside the nanopore. We exploited this phenomenon to show ~100× signal enhancement in the absence of electrolyte, as shown by the data at 1 μM in Figure 2D,27 compared to much smaller signal enhancement/suppression with supporting electrolyte at macroscopic electrodes.43–45 We attributed this observation to a combination of ion migration, accelerating the migration of charged species beyond diffusive transport, and permselectivity, accumulating redox species in the confined volume of the nanopore. Surprisingly, permselectivity is dominant, with the negatively charged surface passivation layer, i.e. SiO2, preferentially admitting cations into the nanopore. We systematically investigated the factors that contribute to this additional enhancement, including EDL effects, like: (1) ionic strength (low μM to 100 mM); (2) redox-species charge, |z| ≤ 3; (3) solvent polarity; (4) nanopore geometry (diameter, 30 nm - 3 μm, and spacing, 50 nm - 5 μm); and (5) nanopore surface charge and found that these all interact to determine the magnitude of enhancement, Figure 2E.28 White and coworkers obtained similar results from a nanoscale thin layer cell, their results confirming the migration-accumulation explanation for the behavior of the dual-electrode system at low ionic strength.46, 47 Later, we observed current amplifications as large as ~500× upon removal of supporting electrolyte using smaller nanopore diameters and gap distances, and thicker passivation layers.29 Combining the signal amplification from redox cycling alone (AFRC = 55) with operation in the absence of supporting electrolyte (AFASE = 500), produces a total amplification, AFtot ~27,500, similar to that observed by Lemay and coworkers in a parallel plate nanogap.48 In addition to these enormous enhancements in sensitivity, we found that selectivity could similarly be improved. For example, when detecting dopamine in the presence of an excess of ascorbic acid, the charge reversal of these species at physiological pH, as well differences in the reversibility of redox cycling reaction, produce a 3000-fold selectivity ratio, as shown in Figure 2F.29
Having achieved large signal enhancement and increased selectivity, it was natural to ask whether NEAs can support electrochemistry at the level of one, or a few, molecules. Two elegant approaches to conduct single molecule electrochemistry have been reported. The first, using a recessed nanopore, was described by Fan and Bard49 and later improved by Sun and Mirkin50. The astonishing redox cycling effect inside this highly confined volume amplified the signal from a single molecule to the pA level (AFtotal = ~107). Lemay and coworkers introduced individual molecules into a nanoscale dual-electrode thin layer cell,51 producing 10–100 fA signals and enabling frequency-domain and anode-cathode cross-correlation studies. As a comparison, the current produced from a single molecule in a single nanopore of a typical NEA under redox cycling conditions is ~1.8 fA, corresponding to ~104 electrons s−1,25 the measurement of which is difficult, but achievable with the demonstrated noise floor of state-of-the-art instrumentation. Furthermore, at low occupancy levels, 〈npore〉 ~ 0.001 single molecule population fluctuations, Δ〈narray〉 = ±1, on the array can be observed. The noise floor further sets limits on the NEA geometries that can realistically be studied, since the nanoelectrode spacing determines how fast diffusive transport can couple anode and cathode in redox cycling. Rather than continue to push the limits of the very demanding electrical detection of single electron transfer events, an alternative approach explores the coupling of electron transfer to changes in single molecule fluorescence.
4. Single entity spectroelectrochemistry
In contrast to single molecule electrochemistry, optical detection of single molecules has become commonplace. Detection of electron transfer events is Johnson noise-limited, due to the required current-to-voltage conversion in a transimpedance amplifier. In contrast, single molecule fluorescence under typical conditions can produce shot-noise limited signals. Thus, there is significant motivation to pursue the spectroelectrochemistry of electrofluorogenic molecules by combining optical and electrochemical measurements in NEAs for single entity detection.30, 31, 36, 37
Previously, Craighead, Webb, and their coworkers et. al. developed a nanophotonic structure, the zero-mode waveguide (ZMW), consisting of a small aperture in a metal layer. With proper choice of the refractive index and thickness-to-diameter ratio of the aperture, the ZMW admits no far-field propagating modes, thus effectively trapping optical radiation in the zeptoliter-scale volume inside the aperture.52 Similar to the evanescent wave of total internal reflection, the trapped radiation can interact with molecules by scattering (Rayleigh or Raman) or by producing excited states which can subsequently relax by fluorescence emission. The combination of small volume and efficient optical excitation make it possible to achieve single molecule sensitivity at μM concentrations. Building on this concept, we developed a bifunctional structure, the electrochemical ZMW (E-ZMWs) - a new kind of hybrid nanophotonic-nanoelectronic architecture in which the lower ring electrode of an NEA nanopore functions both as a working electrode to initiate electron transfer reactions and as the optical cladding layer of the ZMW, as shown in Figures 3A(i) and (ii).30, 31 Our approach to fabricating E-ZMWs starts with a thin-film stack, and a parallel array of nanopores is precisely FIB-milled into the structure, as shown in Figure 3B(iii).34 This approach allows control over electrode composition and spacing at the design level.
Figure 3.

Single molecule and single particle spectroelectrochemistry in NEAs. (A) E-ZMWs shown schematically (i) and in a cross-sectional SEM image (ii). E-ZMWs exhibiting fluorescence emission within the nanopore array, scale bar 2 μm (iii). The redox behavior of individual flavin mononucleotide (FMN), a voltage-sensitive dye is fluorescent in the oxidized state, but dark in the reduced state. An unusual intermediate state is tentatively assigned to a stabilized semiquinone species (iv). (B) Schematic illustration of a dual-ring E-ZMW operating in either non-GC (i) or GC mode (ii), along with cross-sectional image (iii). The light is strongly trapped within the bottom ring region of the nanopore, as indicated by the simulation (iv). The electrochemical signals from the anode and cathode are strongly correlated with each other and with the fluorescence signals from the interrogated sub-array. Transients are assigned to population fluctuations arising from capturing or losing individual FMN molecules (v). (C) Schematic illustration of families of collision dynamics represent either Type A: aggregation-induced collisions, (i) or Type B: multiple single particle collisions, (ii), of AgNPs in the confined environment. Top-view SEM image of a single ring NEA (iii) and simulated particle collisions inside nanopores (iv). Raman spectra of bis-MSB tagged nanoparticles are evident in SERS spectra displaying two distinct types of temporal dynamics (v). Panel A is adapted with permission, ref 30. Copyright 2015 The Royal Society of Chemistry. Panel B is adapted with permission, ref 31. Copyright 2017 The Royal Society of Chemistry. Panel C is adapted with permission, ref 37. Copyright 2019 American Chemical Society.
The central characteristic of E-ZMWs is that both single molecule spectroscopic and electrochemical data can be acquired simultaneously. In addition, the conical cross-section of the nanopores allows the electrochemically-active and optically-active volumes to be addressed individually, as illustrated in Figure 3B(iv). Furthermore, the composition of the internal surfaces can be altered by chemical derivatization or electrodeposition, and because Au and SiO2 are chemically orthogonal, judicious choice of linker functionality can direct the derivatizing agent to the desired surface. The design principles mean that the electrodes in each nanopore are held at the same potential, so that the electrochemical behavior is integrated over the entire array. In contrast, the spectroscopic response can be examined over a user-defined sub-array (by limiting the area of excitation)30, 31 or over the entire array (using wide-field imaging)36, as shown in Figure 3A(iii).
Initial studies from our laboratory established the functional utility of E-ZMWs for coupled electrochemical and spectroscopic investigations.53 First, we demonstrated that the E-ZMW can couple electrochemical potential control to spectroscopic behavior by immobilizing the redox-active cofactor flavin adenine dinucleotide (FAD) at the gold cladding layer of an NEA.36 We established the combined spectroscopic and electrochemical capabilities of the NEA by observing classic single molecule behavior, e.g. dispersion (static heterogeneity) in single molecule potentials for the 2e−/2H+ reduction of fluorescent FAD to non-florescent FADH2. Potential-dependent blinking in single FAD molecules was characterized, reflecting the potential dependence of electron transfer probabilities for single FAD molecules. To explore this approach with freely diffusing molecules, we switched to a confocal geometry to observe the spectroscopic response from a single electrochemically active nanopore under conditions where the average pore occupancy was 〈npore〉 ~ 1 molecule.30 Among the unprecedented observations were surprisingly long-lived semiquinone states of flavin mononucleotide (FMN) stabilized in the zeptoliter-volume E-ZMWs, Figure 3A (iv). The instrument was sufficiently stable to monitor single molecule fluctuations in the electrochemical current generated on the array and to cross-correlate these fluctuations with temporal changes in the single molecule florescence signal. E-ZMWs with dual-ring electrodes were then used to implement efficient redox cycling of electrofluorogenic species, demonstrating the ability to use electron transfer to trigger single molecule fluorescence switching, Figure 3B. Currents at top and bottom ring electrodes in the E-ZMWs were temporally correlated at ultralow molecular loadings, 〈npore〉 ~ 0.001 enabling the cross-correlation of single molecule electron transfer and single molecule fluorescence events arising from single molecule population fluctuations, Figure 3B(v).
Subsequently, we extended the NEA studies of single molecule spectroelectrochemistry to single nanoparticles, employing simultaneous amperometry and surface enhanced Raman scattering (SERS), as shown in Figure 3C.37 Since the seminal work by Bard and coworkers,54 nanoparticle collisions on ultramicroelectrode has drawn intense interest.7, 55 Previous studies reported both direct particle-electrode electron transfer as well as electrocatalytic reactions when nanoparticles impinge on the surface of a flat electrode. However, very few reports have addressed the interactions of nanoparticles with comparably-sized electrodes in confined environments.10 Collision experiments in an NEA are interesting, because once individual nanoparticles are captured in the nanopore, they are likely to undergo multiple electrode collisions before diffusing out of the pore. We coupled observations of enhanced Raman scattering with amperometry to map out particle dynamics inside NEA nanopores. Silver nanoparticles (AgNPs) were functionalized with the electrochemically stable Raman reporter, 1,4-bis(2-methylstyryl) benzene (bis-MSB), and then trapped inside the nanopore. Oxidation current transients from AgNPs and the appearance and dynamics of Raman peaks from bis-MSB were compared to indicate the collision frequency and lifetime of nanoparticles in the confined nanopore environment. These studies not only open up a new avenues for nanopore spectroelectrochemistry, they provide a view into confined particle dynamics that would be difficult to obtained by other means.
5. Gated transport in confined nanopores
The studies described above all employed immobilized or freely-diffusing redox species but did not use the individually addressable dual electrodes in NEAs to manipulate transport. In order to explore this capability of NEAs, the top electrode was employed to gate the transport of silver nanoparticles (AgNPs) entering NEA nanopores, while the oxidation of AgNPs colliding at the bottom disk electrode was used to assess access of the AgNPs to the nanopore, Figure 4A(i)-(iii). 33 First, we acquired quantitative collision rates and magnitudes from AgNPs colliding with the bottom electrode as a function of applied potential (EBE), while the top electrode, ETE, was kept at open circuit. These measurements showed incomplete oxidation of AgNPs beginning at EBE = 0.2 V, becoming increasingly more complete as EBE was increased from 0.2 to 0.6 V, a potential at which AgNPs were completely oxidized. Complete oxidation was confirmed by the calculated charge of the current transients for 50 nm particle (~0.5 pC) matching the experimental result (~0.55 pC). Several groups have reported that AgNPs undergoing incomplete oxidation can be associated with multiple sequential collisions,56–58 an observation that is consistent with our measurements. Furthermore, particle transport inside nanopores can be voltage-gated. Figure 4A(iv) shows a representative amperometric time trace of ~80 nm AgNPs impinging on the bottom electrode at EBE = 0.3 V while ramping ETE from 0.1 up to 0.7 V. While only sporadic collisions were detected at 0.1 V ≤ ETE ≤ 0.5 V, stepping ETE to 0.6 or 0.7 V resulted in a discontinuous increase in collision rate, indicating that the top electrode regulated the transport of AgNPs into the interior of the NEA nanopores, as indicated schematically in Figures 4A(i) and (ii).
Figure 4.

Gated ion and particle transport in confined nanopores. (A) Ring and disk electrodes of NEAs act as gate and sensing electrodes, respectively, to control the transport and collisions of single nanoparticles. The gate electrode is closed at low voltage (i), but open above a voltage threshold (ii). This behavior is confirmed by both simulations (iii) and experimental results (iv). (B) Nafion coated NEAs act as molecular gates to control the transport of ions into the NEAs as shown schematically (i) and in a cross-sectional SEM image (ii). The Nafion@NEA structure is permselective, as shown by experimental (iii) and simulated (iv) results comparing redox behavior of Ru(NH3)62/3+ and Fe(CN)63/4−. (C) PDMS sealed NEAs create a fully closed environment to trap redox species inside nanopores. Operating in two-electrode mode produces depletion (i) and accumulation (ii) when the bottom electrode is biased positive and negative, respectively, relative to the top electrode, resulting in both signal enhancement and electrochemical rectification (iii). Panel A is adapted with permission, ref 33. Copyright 2018 Wiley VCH. Panel B is adapted with permission, ref 32. Copyright 2018 American Chemical Society. Panel C is adapted with permission, ref 38. Copyright 2018 American Chemical Society.
Inspired by the voltage-controlled transport of nanoparticles, we placed an ion-exchange membrane (IEM) on top of attoliter-volume NEAs to extend the control paradigm for mass transport of redox species by combining two gating effects, i.e. voltage-gating by the top electrode and permselective gating by the IEM, Figures 4B (i) and (ii).32 When electrochemical measurements were performed on an uncovered NEA with 1 mM Ru(NH3)63+ or 1 mM Fe(CN)63− in 1 M KCl, no noticeable difference in current behavior was observed. However, after the NEA was capped with a Nafion cation-exchange membrane, an amplified current was achieved from Ru(NH3)63+, with nearly no current from Fe(CN)63−, indicating highly selective on/off charge-gating by the Nafion, Figure 4B(iii). Simulated concentration profiles also support that the selective gating response was driven by the Nafion membrane, Figure 4B(iv). We then examined how the voltage-driven gating characteristics of the top electrode were affected by the Nafion. When no Nafion was present, the voltammetric response from 1 mM Ru(NH3)63+ in 1 M KCl exhibited almost the same limiting current, regardless of ETE switched in the range +0.1 to −0.5 V vs. Ag/AgCl reference. However, significantly different current responses were obtained after the NEA was capped with Nafion, with the limiting current from Ru(NH3)63+ being ~ 6 times higher at ETE = −0.5 V than at +0.1 V, clearly signaling the voltage-responsive gating behavior of the top electrode.
Next, we asked whether completely closed NEAs, in which individual nanopores are isolated entirely from the bulk solution, would allow us to manipulate the populations of redox-active molecules. While high bulk concentrations (>1 mM) of redox species generally produce ensemble-averaged (redox cycling) currents, the current signals become digitized when the bulk concentration is lowered into the few μM range, because the nanopore populations reach single molecule occupancies, i.e. 〈npore〉 ~ 1. Thus, it is possible to remove or add digitized currents from the open nanopore arrays during single molecule measurements. To address this issue, we investigated electron-transfer and mass transport of molecules in individually encapsulated nanopores, generated by a PDMS cladding layer, Figures 4C(i) and (ii).38 Once an NEA was sealed by a PDMS layer, the redox cycling current increased with time, reaching enhancements as large as 250-fold compared to the current in open nanopore arrays. This current enhancement was derived from efficient trapping of redox species and from enhanced mass transport due to non-uniform solution distributions, i.e. evaporation-driven meniscus evolution. We also demonstrated that lowering ionic strength led to ion migration effects, enhancing redox cycling currents and producing strong rectification, mimicking diode functions, Figure 4C(iii).
6. Conclusions and Outlook
Compared to mature single molecule techniques used in optical imaging and spectroscopy, SEE is still in its infancy and still must develop to realize its full potential to impact studies of single molecules, nanoparticles, and cells. To further flourish, key areas for advancement include: (1) Pushing the limit of detection of purely electrochemical experiments to even lower concentration levels through judicious combination of signal enhancement strategies, e.g. redox cycling and migration-induced ion accumulation. (2) Developing hybrid techniques for single entity detection, including correlation approaches and high sensitivity spectroelectrochemical techniques, both fluorescence and Raman-based, for detection of single redox molecules and single reaction events, such as single enzyme turnovers. (3) Developing new molecular-scale tools for precise control of ion/particle transport and molecular trapping inside confined nanopore environments, especially those that utilize principles of active transport control. The advances described here are based on synthetic nanopore systems, which were inspired by the electrochemical and control behavior of biological pores and channels and mimic some, but certainly not all, of the properties of biological pores. Although the synthetic nanopores NEAs extend high-performance electrochemical measurement capabilities, including the capacity to manipulate and measure single molecules and particles, there remains much to be done. A key goal is the development of full electrochemical mapping of single entities and their activity across the whole nanopore array, such that each nanopore can addressed as an “electrochemical pixel” in analogy to optical image sensors. How powerful it would be to have high-resolution electrochemical sensors to capture the electrochemical equivalent of photographs in the world of redox reactions!
Acknowledgments
K.F. was supported by an ACS Division of Analytical Chemistry fellowship, sponsored by Eastman Chemical Company. D.H. was supported by a National Research Foundation of Korea (NRF) grant (MSIT, 2018R1C1B5085888). Work described in this article has been supported by NSF, DOE, and NIH, most recently through grants NSF 1904196, DE FG02 07ER15851, and 1R21GM126246.
Biographies
Kaiyu Fu is a postdoctoral associate at Stanford University. He received his bachelor’s and master’s degree from Sichuan University and Fudan University, respectively and his Ph.D. in Chemistry from the University of Notre Dame. His research focuses on the advancement of electroanalytical methods, using micro/nanoscale electrodes for lab-on-a-chip and the application of electrochemical biosensors for point-of-care diagnostics.
Seung-Ryong Kwon is currently a postdoctoral associate at the University of Notre Dame. He received a B.S. and M.S. from Incheon National University and a Ph.D. from Seoul National University under the supervision of Professor Taek Dong Chung. His research interests include ion/molecular transport in confined nanoporous structures.
Donghoon Han is an assistant professor in the Department of Chemistry at The Catholic University of Korea. He received his Ph.D. in chemistry from Seoul National University and worked as postdoctoral research associate at the University of Notre Dame. His research interests lie at the intersection of electrochemistry, nanoscience, and the biological interface to develop new (bio)analytical methods to probe chemical and biological systems at the micro/nanoscale.
Paul W. Bohn is the Arthur J. Schmitt Professor of Chemical and Biomolecular Engineering and Professor of Chemistry and Biochemistry at the University of Notre Dame. He received the B.S. in Chemistry from Notre Dame and the Ph.D. from the University of Wisconsin-Madison. In 1983 he joined the faculty at the University of Illinois at Urbana-Champaign (UIUC), where he remained until moving to Notre Dame in 2006. His research interests include: molecular nanotechnology, integrated micro-nanofluidic chemical measurements, and correlated chemical imaging.
Footnotes
The authors declare no competing financial interest.
References
- 1.Wang Y; Shan X; Tao N, Emerging tools for studying single entity electrochemistry. Faraday Discuss. 2016, 193, 9–39. [DOI] [PubMed] [Google Scholar]
- 2.Crooks RM, Concluding remarks: single entity electrochemistry one step at a time. Faraday Discuss. 2016, 193, 533–547. [DOI] [PubMed] [Google Scholar]
- 3.Gooding JJ, Single Entity Electrochemistry Progresses to Cell Counting. Angew. Chem. Int. Ed 2016, 55, 12956–12958. [DOI] [PubMed] [Google Scholar]
- 4.Baker LA, Perspective and Prospectus on Single-Entity Electrochemistry. J. Am. Chem. Soc 2018, 140, 15549–15559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edwards MA; Robinson DA; Ren H; Cheyne CG; Tan CS; White HS, Nanoscale electrochemical kinetics & dynamics: the challenges and opportunities of single-entity measurements. Faraday Discuss. 2018, 210, 9–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lemay SG; Kang S; Mathwig K; Singh PS, Single-Molecule Electrochemistry: Present Status and Outlook. Acc. Chem. Res 2013, 46, 369–377. [DOI] [PubMed] [Google Scholar]
- 7.Anderson TJ; Zhang B, Single-Nanoparticle Electrochemistry through Immobilization and Collision. Acc. Chem. Res 2016, 49, 2625–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brasiliense V; Berto P; Combellas C; Tessier G; Kanoufi F, Electrochemistry of Single Nanodomains Revealed by Three-Dimensional Holographic Microscopy. Acc. Chem. Res 2016, 49, 2049–2057. [DOI] [PubMed] [Google Scholar]
- 9.Fang Y; Wang H; Yu H; Liu X; Wang W; Chen H-Y; Tao NJ, Plasmonic Imaging of Electrochemical Reactions of Single Nanoparticles. Acc. Chem. Res 2016, 49, 2614–2624. [DOI] [PubMed] [Google Scholar]
- 10.Mirkin MV; Sun T; Yu Y; Zhou M, Electrochemistry at One Nanoparticle. Acc. Chem. Res 2016, 49, 2328–2335. [DOI] [PubMed] [Google Scholar]
- 11.Li X; Dunevall J; Ewing AG, Quantitative Chemical Measurements of Vesicular Transmitters with Electrochemical Cytometry. Acc. Chem. Res 2016, 49, 2347–2354. [DOI] [PubMed] [Google Scholar]
- 12.Patrice FT; Qiu K; Ying Y-L; Long Y-T, Single Nanoparticle Electrochemistry. Annu. Rev. Anal. Chem 2019, 12, 347–370. [DOI] [PubMed] [Google Scholar]
- 13.Chen S; Liu Y; Chen J, Heterogeneous electron transfer at nanoscopic electrodes: importance of electronic structures and electric double layers. Chem. Soc. Rev 2014, 43, 5372–5386. [DOI] [PubMed] [Google Scholar]
- 14.Singh PS; Lemay SG, Stochastic Processes in Electrochemistry. Anal. Chem 2016, 88, 5017–5027. [DOI] [PubMed] [Google Scholar]
- 15.Arrigan DWM, Nanoelectrodes, nanoelectrode arrays and their applications. Analyst 2004, 129, 1157–1165. [DOI] [PubMed] [Google Scholar]
- 16.Oja SM; Wood M; Zhang B, Nanoscale Electrochemistry. Anal. Chem 2013, 85, 473–486. [DOI] [PubMed] [Google Scholar]
- 17.Mathwig K; Albrecht T; Goluch ED; Rassaei L, Challenges of Biomolecular Detection at the Nanoscale: Nanopores and Microelectrodes. Anal. Chem 2015, 87, 5470–5475. [DOI] [PubMed] [Google Scholar]
- 18.Chen S; Liu Y, Electrochemistry at nanometer-sized electrodes. Phys. Chem. Chem. Phys 2014, 16, 635–652. [DOI] [PubMed] [Google Scholar]
- 19.Martin CR, Nanomaterials: a membrane-based synthetic approach. Science 1994, 266, 1961–1966. [DOI] [PubMed] [Google Scholar]
- 20.Fu K; Bohn PW, Nanopore Electrochemistry: A Nexus for Molecular Control of Electron Transfer Reactions. ACS Cent. Sci 2018, 4, 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piruska A; Branagan SP; Minnis AB; Wang Z; Cropek DM; Sweedler JV; Bohn PW, Electrokinetic control of fluid transport in gold-coated nanocapillary array membranes in hybrid nanofluidic–microfluidic devices. Lab Chip 2010, 10, 1237–1244. [DOI] [PubMed] [Google Scholar]
- 22.Branagan SP; Contento NM; Bohn PW, Enhanced Mass Transport of Electroactive Species to Annular Nanoband Electrodes Embedded in Nanocapillary Array Membranes. J. Am. Chem. Soc 2012, 134, 8617–8624. [DOI] [PubMed] [Google Scholar]
- 23.Gibson LR; Branagan SP; Bohn PW, Convective Delivery of Electroactive Species to Annular Nanoband Electrodes Embedded in Nanocapillary-Array Membranes. Small 2013, 9, 90–97. [DOI] [PubMed] [Google Scholar]
- 24.Ma C; Contento NM; Gibson LR; Bohn PW, Redox Cycling in Nanoscale-Recessed Ring-Disk Electrode Arrays for Enhanced Electrochemical Sensitivity. ACS Nano 2013, 7, 5483–5490. [DOI] [PubMed] [Google Scholar]
- 25.Fu K; Han D; Ma C; Bohn PW, Electrochemistry at single molecule occupancy in nanopore-confined recessed ring-disk electrode arrays. Faraday Discuss. 2016, 193, 51–64. [DOI] [PubMed] [Google Scholar]
- 26.Ma C; Zaino LP; Bohn PW, Self-induced redox cycling coupled luminescence on nanopore recessed disk-multiscale bipolar electrodes. Chem. Sci 2015, 6, 3173–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma C; Contento NM; Bohn PW, Redox Cycling on Recessed Ring-Disk Nanoelectrode Arrays in the Absence of Supporting Electrolyte. J. Am. Chem. Soc 2014, 136, 7225–7228. [DOI] [PubMed] [Google Scholar]
- 28.Ma C; Xu W; Wichert WRA; Bohn PW, Ion Accumulation and Migration Effects on Redox Cycling in Nanopore Electrode Arrays at Low Ionic Strength. ACS Nano 2016, 10, 3658–3664. [DOI] [PubMed] [Google Scholar]
- 29.Fu K; Han D; Ma C; Bohn PW, Ion selective redox cycling in zero-dimensional nanopore electrode arrays at low ionic strength. Nanoscale 2017, 9, 5164–5171. [DOI] [PubMed] [Google Scholar]
- 30.Zaino LP; Grismer DA; Han D; Crouch GM; Bohn PW, Single occupancy spectroelectrochemistry of freely diffusing flavin mononucleotide in zero-dimensional nanophotonic structures. Faraday Discuss. 2015, 184, 101–115. [DOI] [PubMed] [Google Scholar]
- 31.Han DH; Crouch GM; Fu K; Zaino LP; Bohn PW, Single-molecule spectroelectrochemical cross-correlation during redox cycling in recessed dual ring electrode zero-mode waveguides. Chem. Sci 2017, 8, 5345–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu K; Han DH; Kwon SR; Bohn PW, Asymmetric Nafion-Coated Nanopore Electrode Arrays as Redox-Cycling-Based Electrochemical Diodes. ACS Nano 2018, 12, 9177–9185. [DOI] [PubMed] [Google Scholar]
- 33.Fu K; Han DH; Crouch GM; Kwon SR; Bohn PW, Voltage-Gated Nanoparticle Transport and Collisions in Attoliter-Volume Nanopore Electrode Arrays. Small 2018, 14, 1703248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han D; Zaino LP; Fu K; Bohn PW, Redox Cycling in Nanopore-Confined Recessed Dual-Ring Electrode Arrays. J. Phys. Chem. C 2016, 120, 20634–20641. [Google Scholar]
- 35.Fu K; Bohn PW, Nanochannel Arrays for Molecular Sieving and Electrochemical Analysis by Nanosphere Lithography Templated Graphoepitaxy of Block Copolymers. ACS Appl. Mater. Interfaces 2017, 9, 24908–24916. [DOI] [PubMed] [Google Scholar]
- 36.Zhao J; Zaino LP; Bohn PW, Potential-dependent single molecule blinking dynamics for flavin adenine dinucleotide covalently immobilized in zero-mode waveguide array of working electrodes. Faraday Discuss. 2013, 164, 57–69. [DOI] [PubMed] [Google Scholar]
- 37.Kim JY; Han D; Crouch GM; Kwon SR; Bohn PW, Capture of Single Silver Nanoparticles in Nanopore Arrays Detected by Simultaneous Amperometry and Surface-Enhanced Raman Scattering. Anal. Chem 2019, 91, 4568–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwon SR; Fu K; Han D; Bohn PW, Redox Cycling in Individually Encapsulated Attoliter-Volume Nanopores. ACS Nano 2018, 12, 12923–12931. [DOI] [PubMed] [Google Scholar]
- 39.Anderson LB; Reilley CN, Thin-layer electrochemistry: steady-state methods of studying rate processes. J. Eletroanal. Chem 1965, 10, 295–305. [Google Scholar]
- 40.Barnes EO; Lewis GEM; Dale SEC; Marken F; Compton RG, Generator-collector double electrode systems: A review. Analyst 2012, 137, 1068–1081. [DOI] [PubMed] [Google Scholar]
- 41.White HS; McKelvey K, Redox cycling in nanogap electrochemical cells. Curr. Opin. Electrochem 2018, 7, 48–53. [Google Scholar]
- 42.Hüske M; Stockmann R; Offenhäusser A; Wolfrum B, Redox cycling in nanoporous electrochemical devices. Nanoscale 2014, 6, 589–598. [DOI] [PubMed] [Google Scholar]
- 43.Pendley BD; Abruna HD; Norton JD; Benson WE; White HS, Analysis of voltammetric half-wave potentials in low ionic strength solutions and voltammetric measurement of ion impurity concentrations. Anal. Chem 1991, 63, 2766–2771. [Google Scholar]
- 44.Ciszkowska M; Stojek Z, Voltammetric and Amperometric Detection without Added Electrolyte. Anal. Chem 2000, 72, 754A–760A. [DOI] [PubMed] [Google Scholar]
- 45.Belding SR; Compton RG, Cyclic voltammetry in the absence of excess supporting electrolyte: The effect of analyte charge. J. Electroanal. Chem 2012, 683, 1–13. [Google Scholar]
- 46.Xiong J; Chen Q; Edwards MA; White HS, Ion Transport within High Electric Fields in Nanogap Electrochemical Cells. ACS Nano 2015, 9, 8520–8529. [DOI] [PubMed] [Google Scholar]
- 47.Chen Q; McKelvey K; Edwards MA; White HS, Redox Cycling in Nanogap Electrochemical Cells. The Role of Electrostatics in Determining the Cell Response. J. Phys. Chem. C 2016, 120, 17251–17260. [Google Scholar]
- 48.Zevenbergen MAG; Wolfrum BL; Goluch ED; Singh PS; Lemay SG, Fast Electron-Transfer Kinetics Probed in Nanofluidic Channels. J. Am. Chem. Soc 2009, 131, 11471–11477. [DOI] [PubMed] [Google Scholar]
- 49.Fan F-RF; Bard AJ, Electrochemical Detection of Single Molecules. Science 1995, 267, 871–874. [DOI] [PubMed] [Google Scholar]
- 50.Sun P; Mirkin MV, Electrochemistry of Individual Molecules in Zeptoliter Volumes. J. Am. Chem. Soc 2008, 130, 8241–8250. [DOI] [PubMed] [Google Scholar]
- 51.Zevenbergen MAG; Singh PS; Goluch ED; Wolfrum BL; Lemay SG, Stochastic Sensing of Single Molecules in a Nanofluidic Electrochemical Device. Nano Lett. 2011, 11, 2881–2886. [DOI] [PubMed] [Google Scholar]
- 52.Levene MJ; Korlach J; Turner SW; Foquet M; Craighead HG; Webb WW, Zero-Mode Waveguides for Single-Molecule Analysis at High Concentrations. Science 2003, 299, 682–686. [DOI] [PubMed] [Google Scholar]
- 53.Crouch GM; Han D; Bohn PW, Zero-mode waveguide nanophotonic structures for single molecule characterization. J. Phys. D Appl. Phys 2018, 51, 193001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao X; Bard AJ, Observing Single Nanoparticle Collisions at an Ultramicroelectrode by Electrocatalytic Amplification. J. Am. Chem. Soc 2007, 129, 9610–9612. [DOI] [PubMed] [Google Scholar]
- 55.Kim J; Dick JE; Bard AJ, Advanced Electrochemistry of Individual Metal Clusters Electrodeposited Atom by Atom to Nanometer by Nanometer. Acc. Chem. Res 2016, 49, 2587–2595. [DOI] [PubMed] [Google Scholar]
- 56.Ma W; Ma H; Chen J-F; Peng Y-Y; Yang Z-Y; Wang H-F; Ying Y-L; Tian H; Long Y-T, Tracking motion trajectories of individual nanoparticles using timeresolved current traces. Chem. Sci 2017, 8, 1854–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oja SM; Robinson DA; Vitti NJ; Edwards MA; Liu Y; White HS; Zhang B, Observation of Multipeak Collision Behavior during the Electro-Oxidation of Single Ag Nanoparticles. J. Am. Chem. Soc 2017, 139, 708–718. [DOI] [PubMed] [Google Scholar]
- 58.Ustarroz J; Kang M; Bullions E; Unwin PR, Impact and oxidation of single silver nanoparticles at electrode surfaces: one shot versus multiple events. Chem. Sci 2017, 8, 1841–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]