

SUMMARY
Oligodendrocytes (OLs) are important for myelination and shuttling energy metabolites lactate and pyruvate toward axons through their expression of monocarboxylate transporter 1 (MCT1). Recent studies suggest that loss of OL MCT1 causes axonal degeneration. However, it is unknown how widespread and chronic loss of MCT1 in OLs specifically affects neuronal energy homeostasis with aging. To answer this, MCT1 conditional null mice were generated that allow for OL-specific MCT1 ablation. We observe that MCT1 loss from OL lineage cells is dispensable for normal myelination and axonal energy homeostasis early in life. By contrast, loss of OL lineage MCT1 expression with aging leads to significant axonal degeneration with concomitant hypomyelination. These data support the hypothesis that MCT1 is important for neuronal energy homeostasis in the aging central nervous system (CNS). The reduction in OL MCT1 that occurs with aging may enhance the risk for axonal degeneration and atrophy in neurodegenerative diseases.
Graphical Abstract
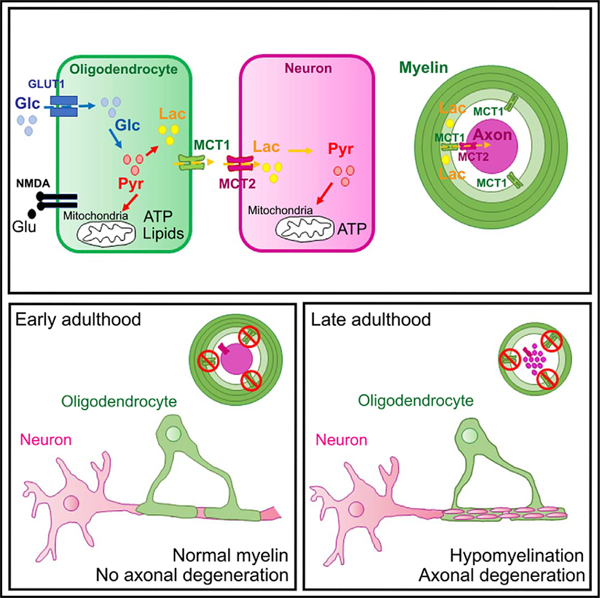
In Brief
Using conditional cell-specific deletion of MCT1, Philips et al. learn that oligodendrocyte lineage cells are actually dispensable for normal myelination and axonal energy homeostasis during early life but that the oligodendroglial lactate/MCT1-based support is critical for the aging of the nervous system.
INTRODUCTION
In the central nervous system (CNS), the capacity of neurons to provide for their own energy homeostasis is insufficient and highly dependent on metabolic and trophic support from glial cells (Saab and Nave, 2017; Magistretti and Allaman, 2018; Alberini et al., 2018; Argente-Arizón et al., 2017). Oligodendrocytes (OLs), which are myelin-generating cells, play a key role in glial-axonal signaling and metabolic support to neurons (Griffiths et al., 1998; Lappe-Siefke et al., 2003; Brady et al., 1999; Yin et al., 1998). Among the metabolites that mediate this OL energy support function are the monocarboxylates lactate and pyruvate (Lee et al., 2012; Fünfschilling et al., 2012). OLs appear to generate lactate and pyruvate in an activity-dependent manner: glutamate release from neurons activates NMDA receptors on OLs, increasing glucose uptake and lactate production through glycolysis (Saab et al., 2016). In order to prevent lactate accumulation, acidosis, and cell death, OLs shuttle lactate into the extracellular space through monocarboxylate transporters. Along with other glial and endothelial cells, OLs express monocarboxylate transporter 1 (MCT1, Slc16a1) that passively transports lactate and hydrogen toward the intra- or extracellular space, depending on the substrate concentration gradient across the plasma membrane (Lee et al., 2012; Rinholm et al., 2011). From the extracellular space, neurons can take up lactate through MCT1 and monocarboxylate transporter 2 (MCT2) and use lactate to fuel energy demands (Pierre et al., 2000; Rinholm et al., 2011). Prior studies have highlighted the importance of MCT1 in providing neurons with trophic support (Lee et al., 2012). Mice with only one genetic copy of Mct1 develop axonal degeneration by 8 months of age. Moreover, focal lentiviral vector-mediated knockdown of Mct1 specifically in OLs causes axonal injury, suggesting that the phenotype observed in Mct1 heterozygous (het) mice is, in part, mediated by a reduction in MCT1 expression by OLs. As loss of OL MCT1 expression has also been observed in human neurodegenerative disease and animal models of these diseases (Lee et al., 2012; Philips et al., 2013; Tang et al., 2019; Andres Benito et al., 2018), it could severely affect neuronal function and contribute to neuronal degeneration.
In this study, we test the prevailing hypothesis that OL MCT1 is required for axonal survival. In addition, the contribution of aging, in which neuronal energy homeostasis is altered (Mattson and Magnus, 2006), to the maintenance of OL-dependent neuronal function through OL MCT1 is evaluated. Our previous study focused on the pathological consequences of either ubiquitous knockdown of MCT1 in Slc16a1 het mice or the effects of focal knockdown of OL MCT1 on the surrounding axons, but essential questions still remain unanswered (Lee et al., 2012). First, MCT1 is expressed by multiple cells types including astrocytes and endothelial cells; therefore, the specific contribution of OL MCT1 metabolic support is impossible to extrapolate from the studies using Mct1 het mice. Second, focal knockdown of OL MCT1 by injection of lentivirus-encoding small hairpin RNAs (shRNAs) targeting MCT1 causes local tissue damage and inflammation that may impact axon degeneration. And third, the past lentiviral studies do not provide information on the long-term consequences of OL MCT1 loss throughout the entire CNS. To address these issues, we generated a conditional MCT1 mouse line (Jha et al., 2020) that we used to knock out MCT1 either specifically in mature OLs or across the entire OL lineage. These two approaches allow us to examine the consequences of MCT1 loss at various stages of OL development, including OL precursor cells (OPCs), differentiating OLs, and mature OLs, on both OL myelination and neuronal metabolic support throughout the animal’s lifespan.
RESULTS
OL MCT1 Expression Is Significantly Reduced with Aging
We first evaluated whether OL MCT1 expression undergoes dynamic changes with aging that could impact neuronal energy demands. To test Mct1 mRNA expression in OLs, we used Mct1-tdTomato mice, which express tdTomato (tdT) under control of the Mct1 promoter (Lee et al., 2012). To assess promoter activity in OLs over time, immunofluorescence co-localization studies were performed for tdT and the mature OL marker adenomatous polyposis coli clone (CC1) in lumbar gray matter spinal cord, at ages ranging from post-natal day 60 (P60) to P550. Aging had no effect on the percentage of tdT+ OLs in the spinal cord of Mct1-tdTomato mice (Figure 1A). Subsequently, MCT1 promoter activity during OL maturation was evaluated using Mct1-tdTomato mice crossed with Pdgfrα-CreER and RosaYFP reporter mice. These triple-transgenic animals were injected intraperitoneally with tamoxifen at different ages (P60, P200, P360, and P500) to induce yellow fluorescent protein (YFP) labeling of OPCs. Thirty days after the first tamoxifen injection, YFP-labeled OPCs that differentiated into tdT+CC1+-expressing OLs were quantified in the lumbar spinal cord gray matter. Despite the significant decrease in oligodendrogenesis within the YFP-labeled cell pool with aging (fraction CC1+YFP+/YFP+ cells: 54% by P230 and 80% by P530), the ability of newly generated CC1+YFP+ OLs to express tdT was unaffected (fraction CC1+YFP+MCT1tdT+/CC1+YFP+; Figures 1B–1D), suggesting that these newly generated OLs maintain Mct1 promoter activity. Dynamic changes in OL MCT1 protein with aging were evaluated in purified myelin protein extracts derived from the spinal cord of aged animals. Interestingly, unlike for the Mct1 promoter activity, spinal cord myelin MCT1 protein expression had declined by 35% by the age of P360, when mice are considered middle age, corresponding to age 38–47 years in humans (Flurkey et al., 2007) (Figure 1E). These data suggest that OLs gradually lose their ability to provide monocarboxylates to neurons. These observations are in line with earlier reported changes in neuronal integrity, energy homeostasis, and autophagic flux that occur with normal aging in mouse and human CNS (Stavoe and Holzbaur, 2019; Mattson and Magnus, 2006; Salvadores et al., 2017).
Figure 1. Loss of Oligodendrocyte (OL) MCT1 Protein Expression with Aging.
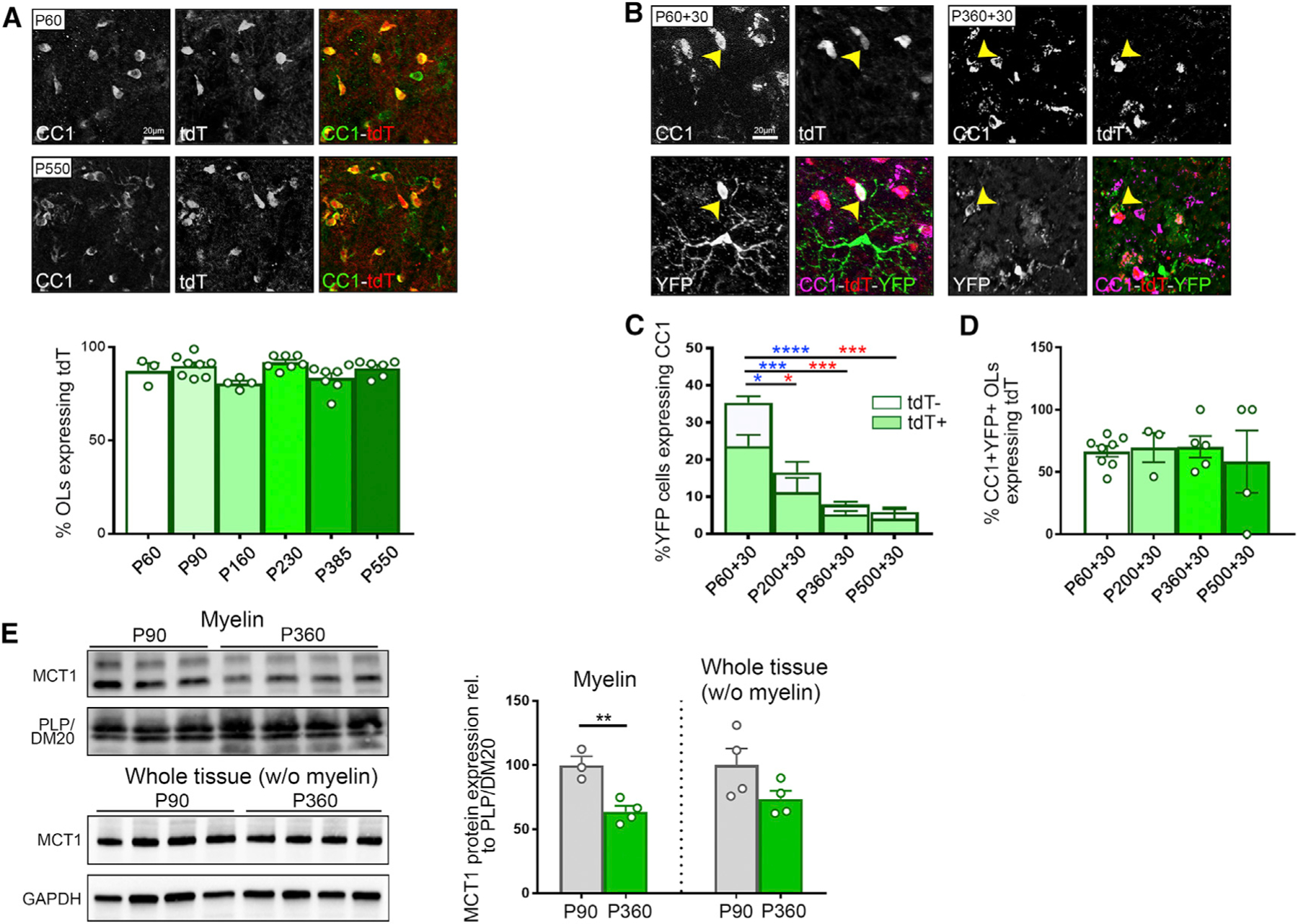
(A) Immunostaining and quantification of CC1+ OLs expressing MCT1-tdTomato (tdT) between the ages of P60 and P550 (n = 3–8). Data are represented as mean ± SEM.
(B) Evaluation of the ability of newly generated OLs to express tdT. P60-, P200-, P360-, and P550-old Pdgfrα-CreER-RosaYFP-MCT1td-Tomato mice were injected with tamoxifen to induce YFP labeling of OPCs and were euthanized 30 days later. Immunostaining highlights differentiated CC1+ OLs expressing tdT and YFP at the ages of P60+30 and P360+30. Images are representative of n = 5–8.
(C and D) Quantification of the fraction of newly generated OLs (# CC1+YFP+/# YFP+, full bars, statistics in blue) and the fraction of newly generated OLs expressing tdT (# CC1+YFP+MCT1tdTomato+/# YFP+, light green bars, statistics in red) reveals a significant decrease with aging (C) (n = 4–8, *p < 0.05, ***p < 0.001, ****p < 0.0001, one-way ANOVA with Tukey’s multiple comparison test). The fraction of newly differentiated OLs that turn on tdT expression (# CC1+YFP+MCT1tdTomato+/# CC1+YFP+) is unaffected by aging (D) (n = 3–8). Data are represented as mean ± SEM.
(E) Western blot analysis of MCT1 protein in purified myelin fractions reveals a 35% reduction at P360 compared with P90 (n = 3–4, **p < 0.01, Student’s t test), whereas MCT1 protein expression in the ‘‘whole tissue (without [w/o] myelin)’’ fraction is unchanged. Data are represented as mean ± SEM.
Conditional Loss of MCT1 in the OL Lineage Cells Significantly Affects MCT1 Expression and Transporter Activity
Mct1 conditional null mice were generated in our laboratory by transgenic insertion of loxP sites flanking exon 2 of the Mct1 gene (Jha et al., 2020). To target Mct1 specifically in OL lineage cells, two approaches were followed. The conditional Mct1 null mice (MCT1lox) were crossed with either the mature OL-specific Cre line MogCre (MogCre) or with the pan-OL lineage Cre mouse line Sox10Cre (Sox10Cre) to generate either Sox10Cre-MCT1lox or MogCre-MCT1lox mice, respectively, hemizygous for Cre and homozygous for MCT1lox. Homozygous age-matched MCT1lox mice were used as littermate controls throughout all experiments. Sox10Cre mice were used to evaluate the importance of MCT1 during differentiation and myelination in addition to its function in mature OLs. MogCre mice were used to determine the role of MCT1 in mature OLs specifically.
Efficacy of MCT1 conditional ablation was measured at RNA and protein level in whole-tissue CNS lysates prepared from either P45 MogCre-MCT1lox or SoxCre-MCT1lox mice and their MCT1lox littermate controls. In spinal cord and cortex, modest reductions were observed for MCT1 protein (Figures 2A and 2B), which likely reflect MCT1 expression in other cell types such as astrocytes and endothelial cells as we confirmed by immunostaining (Figure S1B). Whereas Mct1 mRNA was only modestly affected in the MogCre-MCT1lox mice (Figure S1A), Sox10Cre-MCT1lox mice had a 51% reduction of Mct1 mRNA expression in the spinal cord and a 38% reduction in the cortex compared with MCT1lox controls (Figure 2C). This is a far greater effect compared with loss of Mct1 mRNA in spinal cord (24%) and cortex (12%) in the astrocyte conditional MCT1 null mice (unpublished data). In addition, O4+ OL lineage cells isolated from the cortex of P150 Sox10Cre-MCT1lox mice have 76% reduction in Mct1 mRNA compared with MCT1lox controls (Figure 2C). To enrich for OL-specific MCT1 protein, myelin fractions were biochemically fractionated and purified from cortex and spinal cord. In P30 MogCre-MCT1lox mice, MCT1 expression in myelin fractions derived from spinal cord was reduced by 81%, whereas cortical myelin MCT1 expression was reduced by 40% at P30 and 80% at P180. (Figure 2D; Figure S1C). In myelin fractions derived from P30 Sox10Cre-MCT1lox mice, MCT1 protein expression was reduced by 84% in spinal cord and 81% in cortex (Figure 2E). Lastly, MCT1 transporter activity was measured in primary cultures of OPCs/OLs generated from cortices of P7-old Sox10Cre-MCT1lox and MCT1lox mice. Radioactive [14C]lactate uptake in cultures obtained from Sox10Cre-MCT1lox mice was reduced by 75% compared with MCT1lox controls, comparable to the uptake obtained when treating the MCT1lox OL lineage cultures with 1 µM MCT1 inhibitor AR-C155858 (Figure 2F). The uptake of radioactive [14C]pyruvate revealed a 68% reduction in [14C]pyruvate uptake in the Sox10Cre-MCT1lox cultures compared with MCT1lox controls (Figure 2F). In summary, these data indicate that our conditional knockout approach results in loss of MCT1 function in the OL lineage.
Figure 2. Loss of MCT1 Protein and Transporter Activity in the MogCre-MCT1lox and Sox10Cre-MCT1lox Conditional Null Mice.
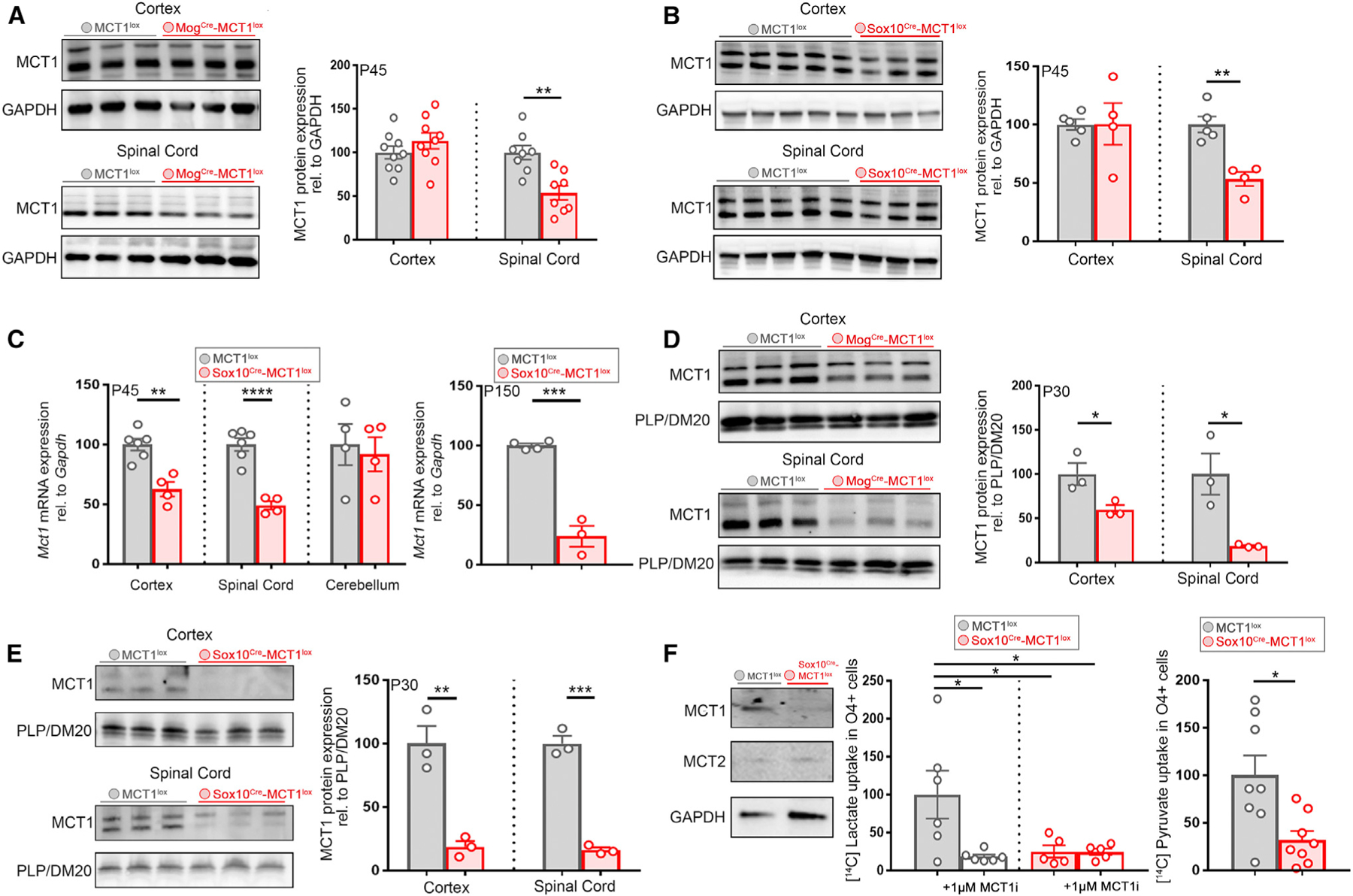
(A) MCT1 expression in whole spinal cord tissue lysates from P45 MogCre-MCT1lox mice is reduced by 46% compared with MCT1lox controls, whereas the cortical MCT1 expression is unchanged (n = 8–9, **p < 0.01, Student’s t test). Data are represented as mean ± SEM.
(B) MCT1 protein expression in whole-tissue lysates from spinal cord derived from P45 Sox10Cre-MCT1lox and MCT1lox controls is reduced by 47% in the conditional nulls, whereas cortical MCT1 protein expression is unchanged (n = 4–5, **p < 0.01, Student’s t test). Data are represented as mean ± SEM.
(C) Left: Mct1 exon 2 mRNA expression analysis in P45 Sox10Cre-MCT1lox and MCT1 lox mice whole CNS tissue RNA extracts is reduced by 51% in spinal cord and 37% in the cortex (Ctx) of the conditional nulls, whereas cerebellar Mct1 mRNA expression is unchanged (n = 4–6, **p < 0.01 and ****p < 0.0001, Student’s t test). Right: Mct1 mRNA expression in cortical O4+ progenitors isolated from P150 MCT1lox and Sox10Cre-MCT1lox mice is reduced by 76% in the conditional nulls (n = 3–4, ***p < 0.001, Student’s t test). Data are represented as mean ± SEM.
(D) MCT1 protein in myelin fractions derived from Ctx and spinal cord of P30 MogCre-MCT1lox mice and MCT1lox mice is reduced by 40% in the Ctx and 81% in the spinal cord of the conditional nulls (n = 3, *p < 0.05, Student’s t test). Data are represented as mean ± SEM.
(E) Quantification of MCT1 protein expression in myelin fractions derived from spinal cord and Ctx from P30 Sox10Cre-MCT1lox and MCT1 lox mice reveals an 84% reduction in the spinal cord and an 81% reduction in the Ctx of the conditional null mice (n = 3, ***p < 0.001, **p < 0.01, Student’s t test). Data are represented as mean ± SEM.
(F) MCT protein expression in OL lineage cultures derived from Sox10Cre-MCT1lox and MCT1lox P3 pups. MCT1 expression is absent from O4+ progenitor cell cultures derived from Sox10Cre-MCT1lox pups and is not compensated by changes in MCT2 expression. Quantification of [14C]lactate uptake in OL lineage cultures reveals a 75% reduction in the Sox10Cre-MCT1lox-derived cultures compared with the MCT1lox-cultured cells. Treatment with 1 µM MCT1 inhibitor AR-C155858 (MCT1i) affects [14C]lactate uptake in MCT1lox cultures by 82% (n = 5–6, *p < 0.05, one-way ANOVA with Tukey’s multiple comparison test). Quantification of [14C]pyruvate uptake between Sox10Cre-MCT1lox and MCT1lox OL lineage cultures reveals a 68% reduction in [14C]pyruvate uptake in the conditional nulls (n = 8, *p < 0.05, Student’s t test). Data are represented as mean ± SEM.
Conditional Loss of MCT1 in OLs Has Subtle Effects on the mRNA, but Not Protein, Expression of Other MCTs and Connexin Hemichannels
Apart from MCTs, glial cells express glucose transporters and connexin hemichannels that allow for the transport of energy metabolites across plasma membranes. Loss of MCT1 expression in OL lineage cells could be compensated by increased expression of MCTs MCT2 and MCT4, glucose transporter 1 (GLUT1) and GLUT3, or connexin hemichannels (OL: Gjc2 (Cx47); astrocytes: Gja1 (Cx43) and Gjb6 (Cx30)). Spinal cord mRNA and protein extracts isolated from P45-old MogCre-MCT1lox mice and MCT1lox controls were evaluated for changes in transporter or connexin hemichannel expression. A 1.3- and 1.2-fold increase in the expression of Mct2 and Mct4 mRNA was observed in the spinal cord of MogCre-MCT1lox mice and a 1.3-fold increase in astrocyte connexin Cx43 mRNA and OL connexin Cx47 mRNA (Figures 3A and 3B). Protein analysis did not reveal significant changes in MCT2 or Cx43 protein, and MCT4 expression was undetectable by western blotting (Figures 3D and 3E; data not shown). There were no changes in GLUT1 and GLUT3 expression at either mRNA or protein level (Figures 3C and 3F). In addition, no significant changes were observed in Glut, connexin, and Mct2/4 mRNA expression in spinal cord tissue derived from Sox10Cre-MCT1lox and MCT1lox mice (Figures 3G–3I). In summary, these data suggest that neither Sox10Cre-nor Mog-Cre-mediated depletion of MCT1 has a strong impact on the expression of other metabolic transporters. We did observe a subtle upregulation in Cx43 and Cx47 hemichannel and Mct2/4 mRNA expression in the MogCre conditional nulls, but not in the Sox10Cre conditional nulls, which could underlie a metabolic compensation for the loss of MCT1.
Figure 3. Conditional Loss of MCT1 in OLs Has Subtle Effects on the mRNA, but Not Protein, Expression of Other MCTs and Connexin Hemichannels.
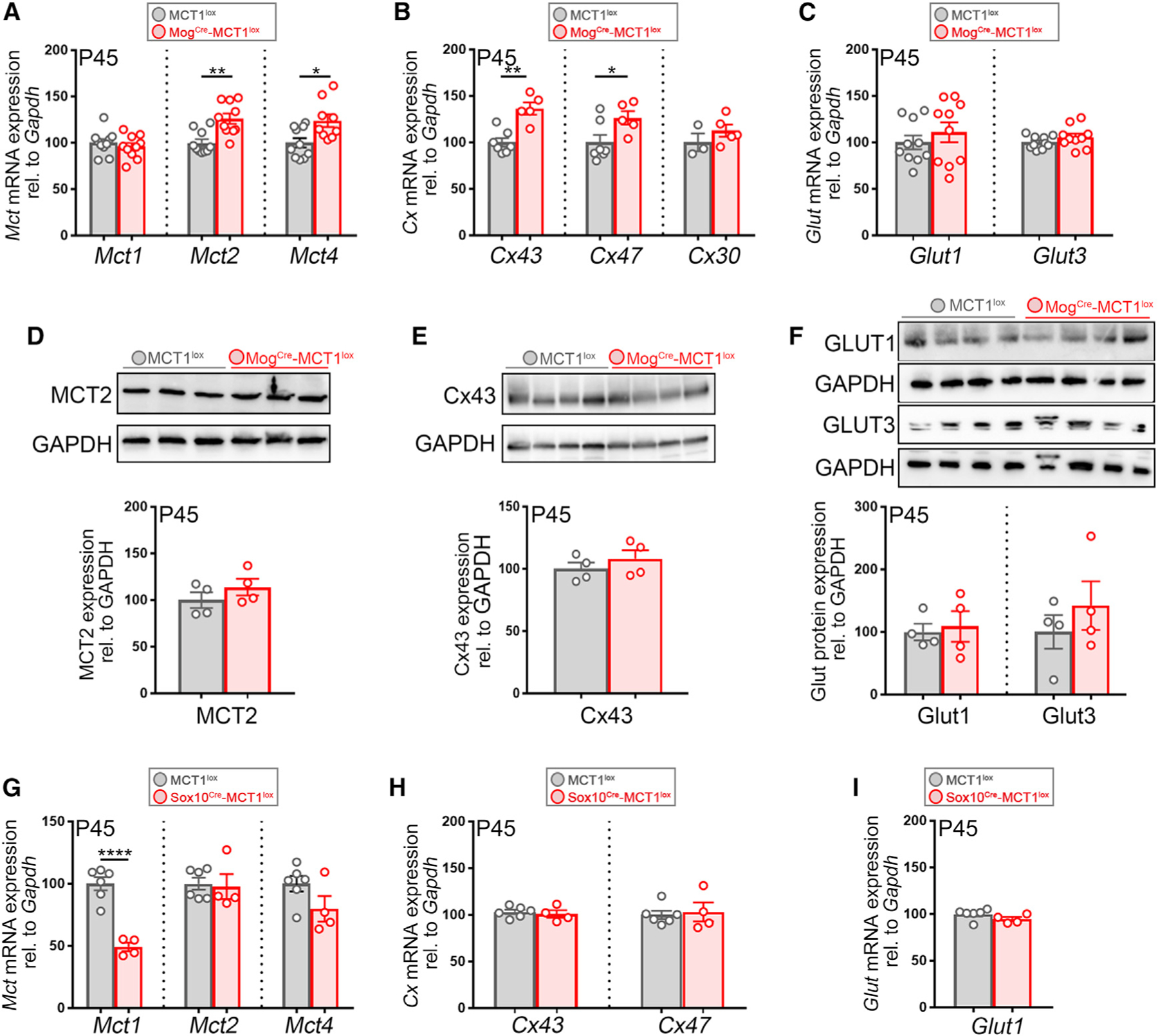
(A–C) Mct, connexin, and Glut mRNA expression in the spinal cord of P45 MogCre-MCT1lox and MCT1lox mice. Mct2 mRNA expression is elevated by 26% and Mct4 expression by 24% in the conditional nulls, whereas Mct1 mRNA expression is unchanged (n = 9–10, **p < 0.01, *p < 0.05, Student’s t test) (A). Cx43 hemichannel mRNA expression is increased by 27% and Cx47 is increased by 30% in the MogCre-MCT1lox mice compared with the MCT1lox controls (n = 5–7, **p < 0.01, *p < 0.05, Student’s t test) (B). There are no changes in expression for glucose transporter 1 (Glut1) and glucose transporter 3 (Glut3) (n = 10) (C). Data are represented as mean ± SEM.
(D–F) Western blot on whole spinal cord protein lysates from P45 MogCre-MCT1lox and MCT1lox controls does not reveal changes in the expression of MCT2 (D), Cx43 (E), and GLUT1 and GLUT3 (F) (n = 4). Data are represented as mean ± SEM.
(G–I) Mct, connexin, and Glut1 mRNA expression in whole spinal cord extracts from P45 Sox10Cre-MCT1lox and MCT1lox controls. There is a 51% reduction in Mct1 mRNA, but no changes in Mct2 or Mct4, in the conditional nulls (n = 4–6, ****p < 0.0001, Student’s t test) (G). There are no changes in mRNA expression of Cx43 and Cx47 (H) or Glut1 (I) (n = 4–6). Data are represented as mean ± SEM.
OL MCT1 Null Mice Develop Normally through Early Adulthood
If OL MCT1 is essential for providing metabolic support to neurons, we expect that loss of MCT1 would impact animal behavior, including locomotion and cognition. P90 Sox10Cre-MCT1lox mice and age-matched MCT1lox littermates were examined for behavioral changes related to general locomotor activity, anxiety, and long-term memory consolidation (summarized in Table S1). The motor performance and exploratory behavior of Sox10Cre-MCT1lox mice were indistinguishable from those of MCT1lox littermate controls (Figures S2A–2D). In addition, Y-maze testing memory consolidation showed no significant differences between Sox10Cre-MCT1lox animals and MCT1lox controls (Figure S2E). Sox10Cre-MCT1lox mice spent more time in the open arms of the elevated plus maze (Figure S2H), which is indicative of reduced anxiety levels; however, no difference in anxiety in the open field test was observed (Figure S2D). In addition, passive avoidance test or fear trace conditioning testing did not reveal significant differences in long-term memory consolidation between Sox10Cre-MCT1lox and MCT1lox controls (Figures S2G and S2I). In summary, behavior testing at the age of P90 in Sox10Cre-MCT1lox mice was mostly indistinguishable from their littermate MCT1lox transgenic controls. Given that targeting MCT1 or the flow of glycogen-derived lactate from glia to neurons causes memory retention deficits and alterations in dendritic spine density (Suzuki et al., 2011; Vezzoli et al., 2019), we performed spine density measurements in MogCre-MCT1lox and MCT1lox mice up to the age of P550 and found no differences in cortical or hippocampal spine density (Figure S2J). Therefore, our studies targeting MCT1 in OL lineage cells specifically do not support a role for OL MCT1 in memory retention.
We next examined multiple CNS areas of the Sox10Cre-MCT1lox mice for pathological changes indicative of either axonal degeneration or dysregulation of myelination. At P45, there were no signs of reactive astrogliosis or microglial activation in either cortex, spinal cord, or cerebellum (Figure 4A; data not shown). Transmission electron microscopy (TEM) analysis was performed on isolated optic nerves sections from both genotypes to examine more subtle signs of axonal degeneration. The optic nerve was chosen for our analysis as a CNS region with high density of small myelinated axons with active OL turnover (Tripathi et al., 2017). There were no significant differences in the number of actively degenerating or degenerated axons between P45-old Sox10Cre-MCT1lox mice and MCT1lox controls (Figure 4B). In addition, there were no differences in mitochondrial morphological parameters at either P18 or P45 (Figures S3A–S3C).
Figure 4. MCT1 Is Dispensable for Early Development and Myelination.
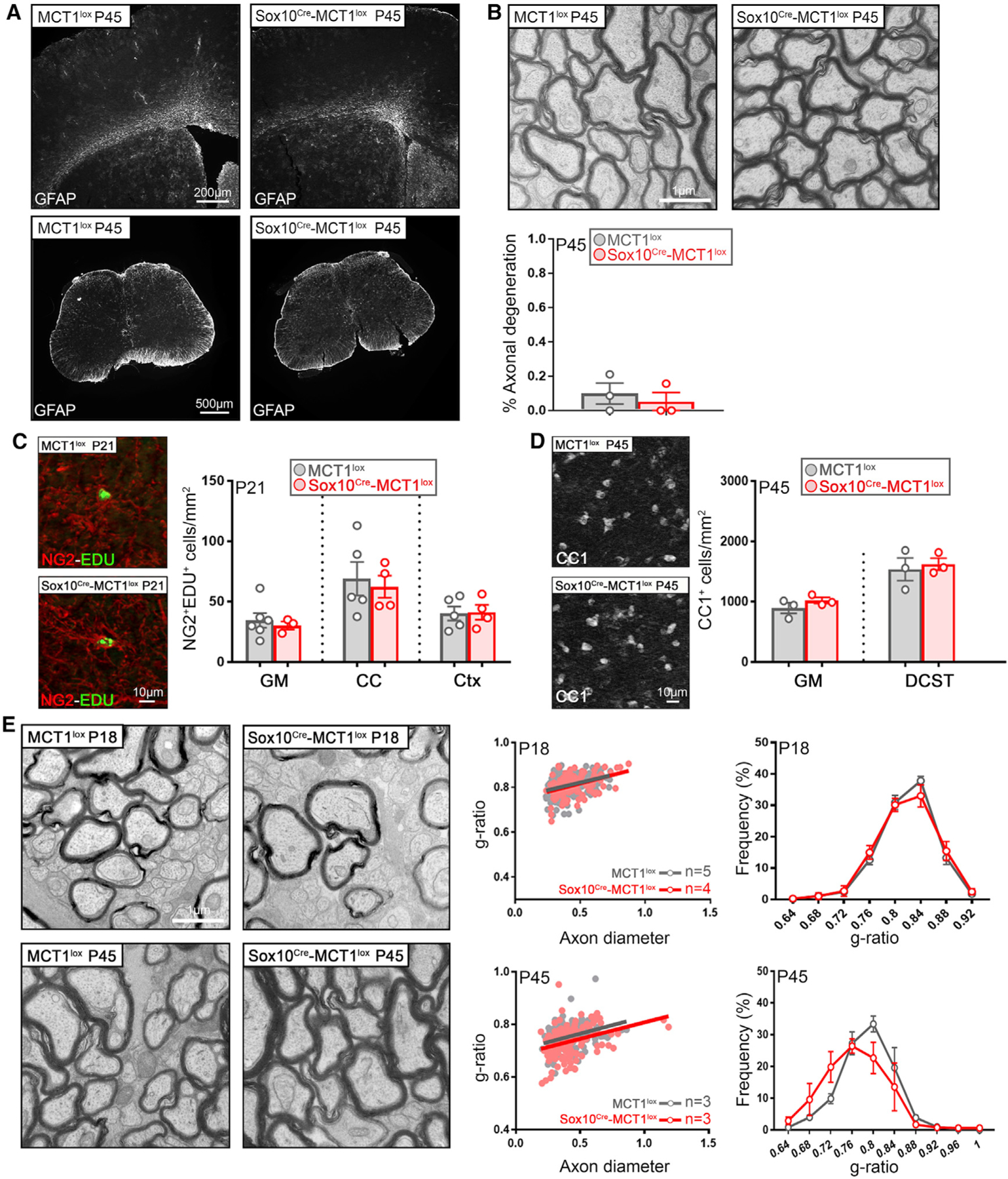
(A) GFAP immunolabeling in Ctx, subcortical white matter (WM), and lumbar spinal cord is unaffected in P45 Sox10Cre-MCT1lox and MCT1lox mice (representative of n = 3). Data are represented as mean ± SEM.
(B) Electron microscopic evaluation of axonal integrity in the optic nerves of P45 Sox10Cre-MCT1lox and MCT1lox mice does not reveal significant differences (n = 3). Data are represented as mean ± SEM.
(C) Quantification of proliferating OPCs (NG2+EdU+) in the gray matter (GM), corpus callosum (CC), and Ctx of P21 Sox10Cre-MCT1lox and MCT1lox mice does not reveal changes in OPC proliferation (n = 3–6). Data are represented as mean ± SEM.
(D) There are no differences in the number of CC1+ OLs in spinal cord GM and dorso-corticospinal tract (DCST) of P45 Sox10Cre-MCT1lox and MCT1lox mice (n = 3). Data are represented as mean ± SEM.
(E) Myelin integrity in P18- and P45-old Sox10Cre-MCT1lox is maintained compared with MCT1lox mice (n = 3–5). Data are represented as mean ± SEM.
As myelination is an energetically expensive process, it could exert a significant metabolic burden on OLs during rounds of de novo myelination. In order to understand the influence of MCT1 loss on oligodendrogenesis and myelination, P15 Sox10Cre-MCT1lox and MCT1lox mice were injected with 5-ethynyl-2′-deoxyuridine (EdU) for six consecutive days to label proliferating OPCs, which is a hallmark of ongoing oligodendrogenesis as well as a response to OL injury (Kang et al., 2013; Philips et al., 2013). We did not observe a significant difference in EdU+ OPCs in different CNS regions such as spinal cord, corpus callosum, and cortex from Sox10Cre-MCT1lox and MCT1lox mice (Figure 4C). In addition, CC1+ OL cell count in the gray and white matter lumbar spinal cord areas of P45-old Sox10Cre-MCT1lox and MCT1lox mice did not reveal significant differences (Figure 4D). Myelin thickness in optic nerve was comparable between Sox10Cre-MCT1lox and MCT1lox mice at both P18 and P45 (Figure 4E). No changes in major myelin protein levels were detected in myelin fractions prepared from cortices of P90 Sox10Cre-MCT1lox mice or spinal cords of P30 MogCre-MCT1lox mice (Figures S3D and S3E). In summary, these data suggest that monocarboxylates such as lactate and pyruvate are not essential nutrients during early myelination.
OL Lineage MCT1 Null Mice Develop Late-Onset Axonal Degeneration and Hypomyelination
Having established that OL MCT1 is not required for energy homeostasis during early myelination and early adulthood, we evaluated pathological abnormalities in the CNS regions of conditional null mice at a more advanced age. In the OL lineage null Sox10Cre-MCT1lox mice at P270, there were no changes observed in the expression of microglial ionized calcium binding adaptor molecule 1 (Iba1) or astrocytic glial fibrillary acidic protein (GFAP) in the cortex, spinal cord, or cerebellum or changes in the density of cortical neurons (Figures S4A and S4C). Similar results were obtained in the MogCre-MCT1lox mice (Figures S4B and S4D). Optic nerve myelin integrity in Sox10Cre-MCT1lox mice appeared normal up to the age of P180 compared with MCT1lox controls (Figure S5A). Lastly, quantification of aspartoacylase (ASPA)+ OL cell number in the gray matter spinal cord showed no significant differences between P270 MogCre-MCT1lox mice and MCT1lox controls (Figure S5A). These data suggest that despite the earlier reported axonal degenerative changes in the 8-month-old het Mct1 null mice, conditional loss of Mct1 using either Sox10Cre or MogCre does not lead to overt gliosis or axonal degeneration up to the age of P270.
Intriguingly, pathological changes were observed in multiple CNS areas in Sox10Cre-MCT1lox mice at P360 when mice are considered middle age, equivalent to a human age of 38–47 years (Flurkey et al., 2007). Patches of increased microglial reactivity suggestive of underlying injury were found throughout spinal cord, cerebellum, and brain stem of the Sox10Cre-MCT1lox mice compared with the age-matched MCT1lox controls (Figures 5A–5E). Clusters of Iba1+lysosomal associated membrane protein-2 (LAMP-2)+ microglia with amoeboid ‘‘reactive’’ morphology were often found scattered throughout white matter areas along the rostro-caudal axis but absent from the corpus callosum (Figures 5A–5E, insets). Quantification of these clustered reactive microglia showed a 2.7-fold increase in the lumbar spinal cord in P360 Sox10Cre-MCT1lox mice (Figure 5F). Cerebellum white matter had a 1.6-fold increase in the number of clustered Iba1+-reactive microglia (Figure 5F). Interestingly, changes in astroglial reactivity were not observed in these regions, indicative of the subtle nature of these pathological findings (Figures 5A–5E, insets). In addition, OL numbers were not affected in different CNS regions of P360 Sox10Cre-MCT1lox and MCT1lox mice (Figure 5G).
Figure 5. Increased Microglial Reactivity in the CNS WM of Aged Sox10Cre-MCT1lox Mice.
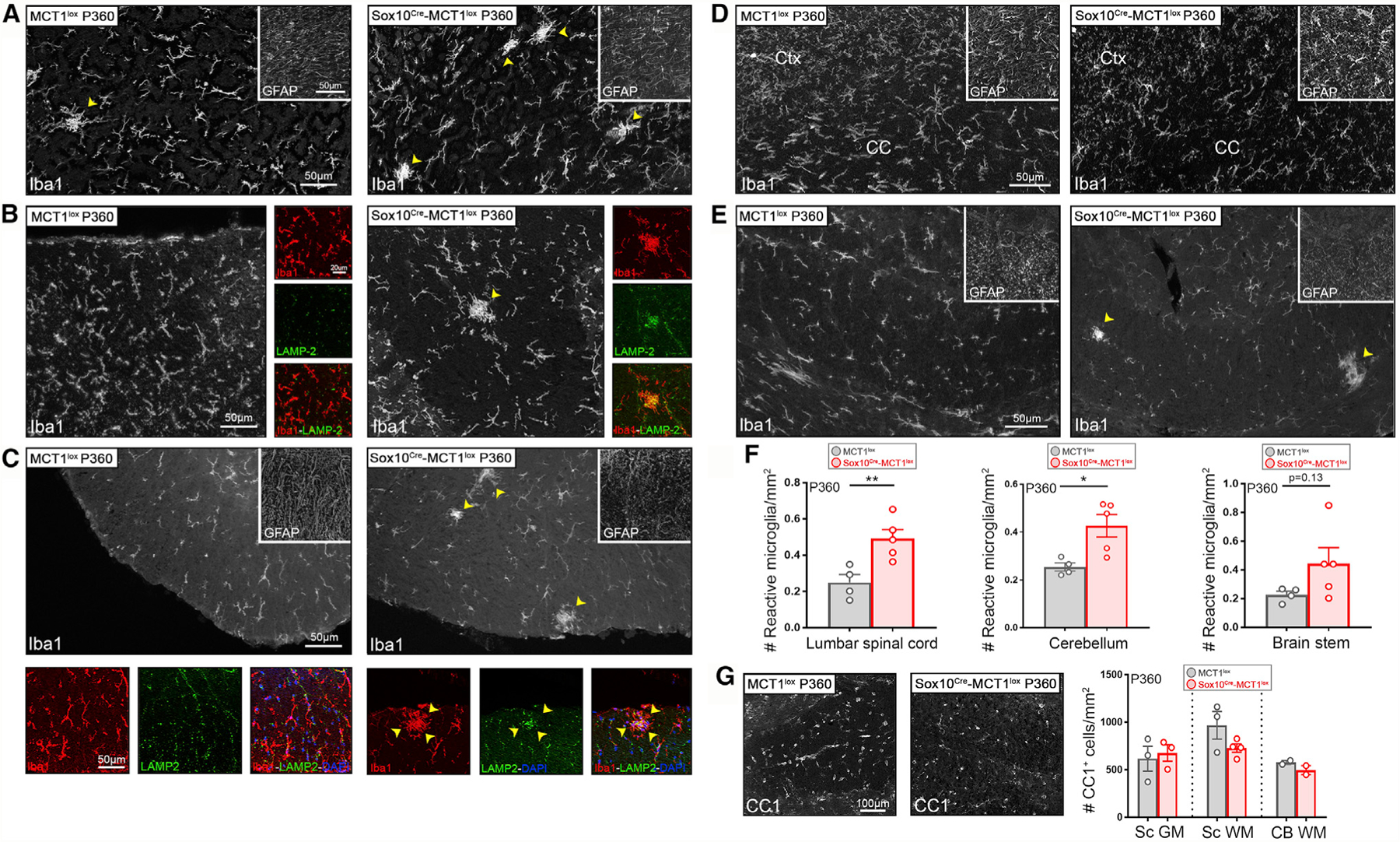
(A–E) Microglial Iba1+ reactivity is enhanced in the cerebellum (A), DCST (B), and ventral spinal cord WM (C) of P360 Sox10Cre-MCT1lox mice compared with MCT1lox controls (arrowheads). Iba1+ immunoreactivity co-localized with LAMP-2 (Mac 3), confirming that microglia acquire a more reactive phenotype (small panels taken from larger images in B and C). There was no change in GFAP immunoreactivity (insets). Microglial Iba1+ and astrocyte GFAP+ reactivity in the Ctx and CC of P360 Sox10Cre-MCT1lox is unaffected compared with MCT1lox controls (D). Images are representative of n = 4–5.
(F) Quantification of changes in microglial Iba1+ reactivity reveals a 2-fold increase in the spinal cord and a 1.7-fold increase in the cerebellum of P360 Sox10Cre-MCT1lox mice compared with MCT1lox controls (spinal cord: **p < 0.01; cerebellum: *p < 0.05; n = 4–5, Student’s t test). No change in the number of reactive microglia was observed in the brain stem (n = 4–5). Data are represented as mean ± SEM.
(G) There are no differences in the number of CC1+ OL cell number in the GM spinal cord (Sc GM), WM spinal cord (Sc WM), and WM cerebellum (CB WM) between P360 Sox10Cre-MCT1lox and MCT1lox mice (n = 2–4). Data are represented as mean ± SEM.
As axonal degeneration is observed in optic nerves isolated from 8-month-old Mct1 het mice (Lee et al., 2012), we addressed whether optic nerve axons in aged Sox10Cre-MCT1lox mice were degenerating as well. Optic nerves were isolated from P360 Sox10Cre-MCT1lox mice and MCT1lox age-matched controls and processed for TEM. Image analysis revealed the presence of optic nerve axons with accumulations of swollen, intracellular organelles, indicative of active axonal degeneration. Degenerated axons were also detected with accumulation of redundant myelin loops (myelin ovoids, Figure 6A). The prevalence of these actively degenerating and degenerated axons increased 3.8-fold by P360 in Sox10Cre-MCT1lox mice compared with MCT1lox controls (Figure 6B). In addition, some mitochondria were enlarged and had lost their typical cristae structure (Figure 6C), but this increase in mitochondrial size did not reach statistical significance (Figure 6B; Figure S5B). Furthermore, there were no changes in the number of mitochondria (Figure S5C). From P360 onward, the g-ratio (ratio between inner and outer diameter of a fiber) was increased in the Sox10Cre-MCT1lox optic nerves and there was a 3.2-fold increase in the proportion of hypomyelinated axons (Figure 6C). Nevertheless, the analysis of myelin isolated from the whole brain from P360-old Sox10Cre-MCT1lox mice revealed normal expression of abundantly expressed myelin proteins, except for a significant increase in the expression of myelin proteolipid protein (PLP) (Figure 6D).
Figure 6. Axonal Degeneration and Hypomyelination in Optic Nerves Isolated from P360 Sox10Cre-MCT1lox Mice.
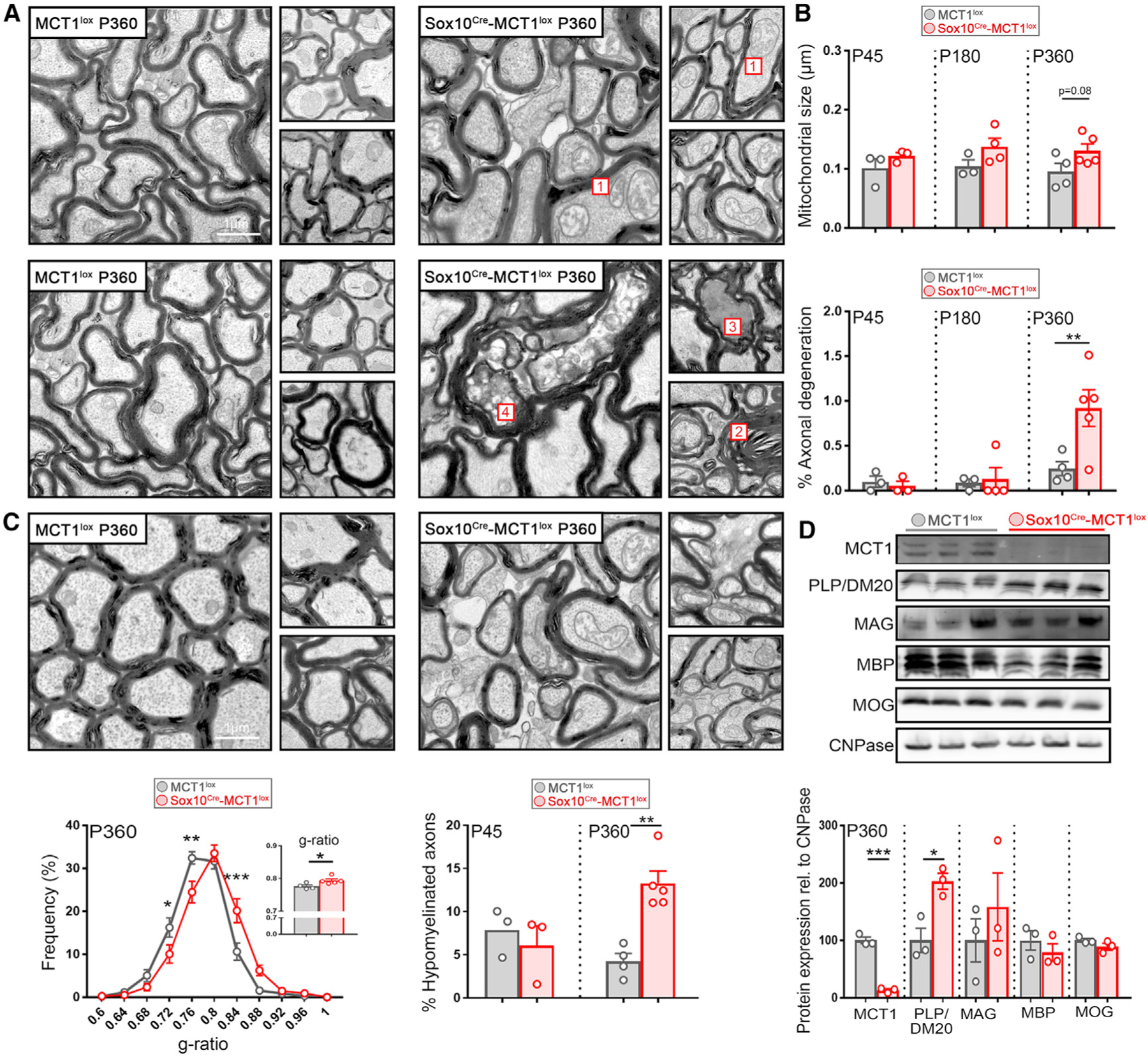
(A) Electron microscopy images from P360 optic nerves from Sox10Cre-MCT1lox reveals the presence of enlarged mitochondria with abnormalities in christae structure (1), accumulation of redundant myelin loops (2), and active degenerating (3) as well as degenerated (4) axons containing multivesicular structures.
(B) Quantification of mitochondrial size in optic nerves of P45, P180, and P360 Sox10Cre-MCT1lox mice does not reveal differences compared with MCT1lox littermate controls. Quantification of axonal degeneration revealed a 3.8-fold increase in the number of degenerating axons at P360 in the Sox10Cre-MCT1lox mice (n = 3–5, **p < 0.01, two-way ANOVA with Šidák’s multiple comparison test). Data are represented as mean ± SEM.
(C) Analysis of myelin integrity in P360 Sox10Cre-MCT1lox and MCT1lox mice reveals an increase in the abundance of thinly myelinated axons in Sox10Cre-MCT1lox compared with MCT1lox controls (n = 4–5, *p < 0.05, **p < 0.01, ***p < 0.001, two-way ANOVA with Šidák’s multiple comparison test). The g-ratio was increased in the Sox10Cre-MCT1lox mice (0.776 ± 0.004 in MCT1lox versus 0.794 ± 0.005, n = 4–5, *p < 0.05, Student’s t test). At P360, there was a 3.2-fold increase in the percentage of hypomyelinated axons (g-ratio > 0.85) in the Sox10Cre-MCT1lox mice compared with the MCT1lox controls (n = 3–5, **p < 0.01, two-way ANOVA with Šidák’s multiple comparison test). Data are represented as mean ± SEM.
(D)Western blot of myelin proteins in total brain derived myelin extracts from P360 Sox10Cre-MCT1lox and MCT1lox mice. MCT1 expression was lowered by 87% in the Sox10Cre-MCT1lox mice compared with controls (n = 3, ***p < 0.001, Student’s t test). PLP/DM20 expression was increased by 2-fold in the Sox10Cre-MCT1lox group compared with controls (n = 3, *p < 0.05, Student’s t test). Expression is normalized to 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase). Data are represented as mean ± SEM.
In order to explore the physiologic consequences of MCT1 loss in the OL lineage of P360-old Sox10Cre-MCT1lox mice, compound action potential (CAP) recordings were performed on acutely isolated optic nerves from P360 Sox10Cre-MCT1lox and MCT1lox mice. Optic nerves were subject to high-frequency (100-Hz) stimulation (HFS) for 30 s after an initial baseline recording period (0.1 Hz), followed by a 5-min recovery period (0.1 Hz). CAP waveforms recorded at baseline and during recovery appeared very similar between P360 MCT1lox and Sox10Cre-MCT1lox control nerves (Figure S6A). A trend toward a delayed recovery in normalized amplitude was observed in the Sox10Cre-MCT1lox nerves compared with MCT1lox controls, but this did not reach statistical significance (Figures S6B and S6C). In addition, despite the accumulation of hypomyelinated axons as observed with TEM, there was only a trend in reduced peak conduction velocity in the Sox10Cre-MCT1lox compared with the MCT1lox controls (Figure S6D). Interestingly, all optic nerves with slowest amplitude recovery and conduction velocity values were Sox10Cre-MCT1lox conditional nulls, suggestive of a bimodal distribution for both parameters in the conditional null cohort, in correspondence with the findings from the EM analysis. In summary, loss of MCT1 expression in the OL lineage causes subtle axonal degeneration and hypomyelination by P360 that could impact axons already at metabolic risk during aging.
Loss of MCT1 in Mature OLs Causes Severe Late-Onset Axonal Degeneration
In order to understand the contribution of MCT1 in mature OLs to provide metabolic support to neurons, we performed EM on MogCre-MCT1lox mice at the ages of P90, P360, and P750. Only at the age of P750 did we detect profound axonal degeneration in optic nerves in the MogCre-MCT1lox conditional null mice compared with MCT1lox controls (Figures 7A–7D). These pathological findings included ‘‘dark axons,’’ axons containing multi-vesicular bodies, and redundant myelin loops scattered throughout the optic nerve, all reflective of active axonal degeneration (Figure 7A). The percentage of degenerated axons increased by 2.6-fold by P750 in the MogCre-MCT1lox mice (Figure 7D). This did not lead to differences in the axonal diameter distribution, indicating that axons were lost irrespective of caliber (Figure S7B). Overall, ~5% of all optic nerve axons were undergoing active neurodegeneration at the time of sacrifice (Figure 7D). The mitochondrial average size was increased 1.6-fold in the MogCre-MCT1lox mice, and individual mitochondria often appeared swollen, with loss of normal cristae structures, all indicative of mitochondrial metabolic disturbances (Figures 7A and 7B; Figure S7A). In addition, clusters containing swollen mitochondria increased 2.2-fold in the P750 MogCre-MCT1lox mice compared with controls (Figure 7C). Interestingly, axonal degeneration was not observed in the MogCre-MCT1lox conditional nulls at the earlier time points, either at P360 or P90, suggesting that this is a very late-onset phenomenon (Figures 7A–7D). There was no significant difference in myelin thickness between MogCre-MCT1lox and MCT1lox mice at P90 or P750 (Figure 7E), suggesting that the axonal degeneration observed in the MogCre-MCT1lox mice at P750 is not due to hypomyelination. In summary, these data highlight the essential role for OLs in providing neurons with metabolic support at advanced age and that loss of MCT1 at this age is an additional risk factor for developing axonal degeneration.
Figure 7. Age-Dependent Axonal Degeneration in Optic Nerves Isolated from MogCre-MCT1lox Mice.
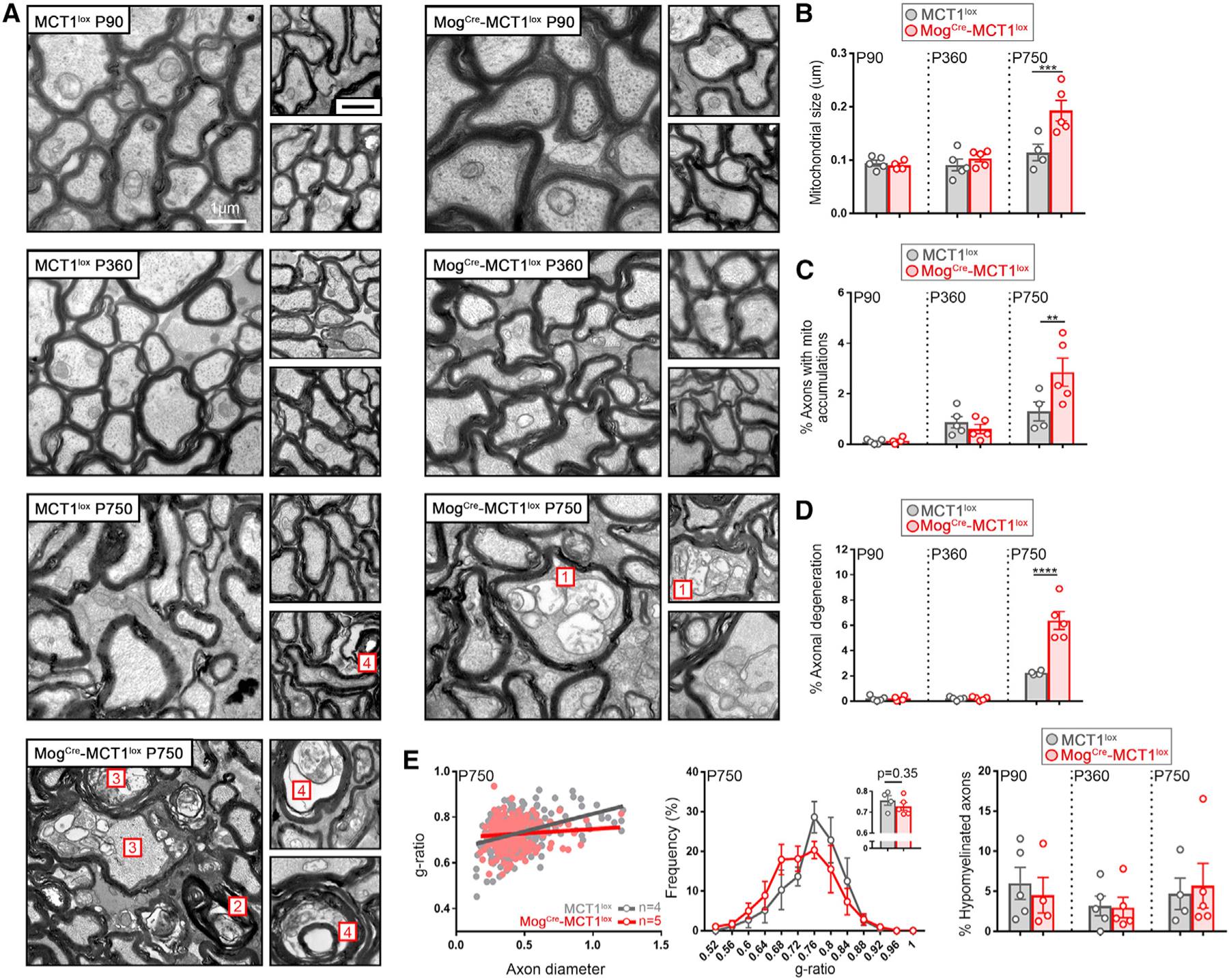
(A) Electron microscopy images from P750 optic nerves from MogCre-MCT1lox reveal the presence of swollen mitochondria (1), accumulation of redundant myelin loops (2), and active degenerating (3) as well as degenerated (4) axons containing multivesicular structures. These morphological changes were only rarely observed in the age-matched MCT1lox controls.
(B–D) At P750, mitochondrial size is 1.7-fold increased in the MogCre-MCT1lox mice compared with the MCT1lox controls (B), the accumulation of intra-axonal mitochondria is 2.2-fold increased in the MogCre-MCT1lox mice compared with the MCT1lox controls (C), and the extent of axonal degeneration is 2.9-fold increased in the MogCre-MCT1lox mice compared with the MCT1lox controls (D) (n = 4–5, **p < 0.01, ***p < 0.01, ****p < 0.0001, two-way ANOVA with Šidák’s multiple comparison test). Data are represented as mean ± SEM.
(E) Evaluation of myelin thickness in P90- and P750-old MogCre-MCT1lox mice does not reveal changes in g-ratio, g-ratio distribution, or percentage of hypomyelinated axons (g-ratio > 0.85 [n = 4–5]). Data are represented as mean ± SEM.
DISCUSSION
MCT1 Importance for Myelination
This study focused on the importance of OL lineage MCT1 in providing metabolic support to OL and neurons during aging. A key observation from this study is that MCT1 expression from OL lineage cells is not required for myelination and neuronal energy homeostasis during development and the mature adult stage (3–6 months). However, when mice are middle age (10–14 months old), OL lineage MCT1 loss causes hypomyelination and axonal degeneration, which dramatically worsens in older age (18–24 months) (Flurkey et al., 2007). Unexpectedly, hypomyelination was not observed when MCT1 is ablated specifically in mature OLs, suggesting that expression of MCT1 in maturing OL progenitor cells is critical for myelination in aging brain. Earlier studies suggested an important role for the MCT1 substrate lactate during OL myelination, and lactate metabolism contributes to lipid biosynthesis in OLs (Ichihara et al., 2017; Rinholm et al., 2011; Sánchez-Abarca et al., 2001). Loss of MCT1 would limit lactate/pyruvate uptake and deprive OLs from the energy necessary for proper myelination. However, direct in vivo evidence for the role of OL MCT1 in OL myelination had not been obtained to date. Our in vivo observations clearly indicate that when either OLs or their progenitor cells lack MCT1, developmental myelination is unaffected during the early postnatal stages and into early adulthood, possibly due to the availability of other energy intermediates such as glucose. By contrast, the loss of MCT1 in maturing OL progenitors causes hypomyelination, potentially in concert with other age-related factors that affect the ability of OPCs to fully mature (Kang et al., 2013, Philips et al., 2013). It would be of interest to test this hypothesis in animal models of demyelination and/or limited energy intermediates to determine the consequences of MCT1 loss in the OL lineage for remyelination at either young or advanced age. With the hypomyelination observed in our OL lineage conditional nulls, there was a concomitant increase in expression of myelin proteolipid protein PLP. It remains to be explored whether the hypomyelination observed in the aged Sox10Cre-MCT1lox animals is due to loss of metabolites essential for myelination at this age, changes in PLP expression, or a different, yet unknown mechanism.
Interestingly, our recent study using the same conditional MCT1 null approach indicates that P0 Cre-mediated Schwann cell MCT1 depletion leads to adult-onset hypomyelination in sensory dorsal root and sural peripheral nerves, but not in motor peripheral nerves (Jha et al., 2020). The hypomyelination occurred much earlier than in OL lineage MCT1 knockout (4 months versus 1 year of age) and leads to deficits in mechanical sensitivity by 1 year of age. Unlike OL lineage MCT1 null, hypomyelination in the Schwann cell MCT1 conditional nulls was not concomitant with axonal degeneration with aging, at least up to 1 year. A second study using an independently generated MCT1 conditional null line, also induced Schwann cell depletion of MCT1 using myelin protein zero-Cre mice (Boucanova et al., 2020), which lead to changes in motor neuron endplate innervation and altered motor neuron transcriptome. Similar to what is observed with OL lineage nulls, the phenotypes in both of these transgenic mice were relatively mild, suggesting that other metabolic transporters could compensate for the loss of MCT1 in the myelinating cells.
MCT1 and Late-Onset Axonal Degeneration
In addition to the importance of MCT1 in myelination with advanced age, our results indicate that the loss of MCT1 expression in OL lineage cells causes late-onset axonal degeneration, beyond the age of P360. During development and early postnatal life, the conditional null mice do not develop axonal degeneration, gliosis, or gross behavioral deficits and are indistinguishable from their littermate controls. We did observe significant differences when performing elevated plus maze, a test commonly used to test for anxiety, suggesting that the MCT1 conditional nulls are less anxious than their littermate controls. However, this finding was not confirmed by measuring anxiety in the open field test, where no differences were observed in time spent in the central versus peripheral area and therefore is not indicative of potential anxiety differences. Therefore, the potential impact of oligodendroglial MCT1 on anxiety remains unresolved. A previous study suggested that loss of MCT1 in hippocampal neurons of MCT1 het null mice leads to learning deficits in a specific long-term memory task (Tadi et al., 2015). In addition, rats that had MCT1 antisense oligonucleotides injected into the hippocampus developed deficits in memory retention (Suzuki et al., 2011). In our study, we did not observe learning deficits in our Sox10Cre-MCT1lox conditional null line when tested with passive avoidance testing, Y-maze, or fear trace conditioning testing. Regardless of differences in species and strain backgrounds used in the different studies, our data would suggest that OL lineage MCT1 is not important for memory retention compared with MCT1 expression in other cells such as astrocytes in which glycogenolysis is critical in the molecular and synaptic changes underlying memory retention (Suzuki et al., 2011; Vezzoli et al., 2019). These glycogen stores are most strongly associated with astrocytes, not OLs, and astrocyte-derived glycogen when converted to glucose and lactate could bypass the need for OL-derived lactate in providing the metabolic support needed during memory retention. Alternatively, knockdown of MCT1 in multiple cell types (OLs, astrocytes, and endothelial cells), as shown in previously published reports, may work in concert to regulate memory retention in this particular task. Studies selectively knocking out MCT1 in other cell types such as astrocytes and endothelial cells are critical to elucidate each individual contribution to memory formation through MCT1-mediated lactate release.
Of note is the delayed degeneration we observed in the MogCre-MCT1lox conditional null compared with the Sox10Cre-MCT1lox conditional null mice. Axonal degeneration was only observed after ~2 years of age in the MogCre-MCT1lox animals, whereas the Sox10Cre-MCT1lox animals developed axonal degeneration already by P360. Multiple hypotheses could explain this difference: hypomyelination, which is only observed in the Sox10Cre-MCT1lox line, could accelerate axonal degeneration in this line compared with the MogCre transgenic line. Alternatively, changes in expression in connexin hemichannels or other MCTs could metabolically compensate for the loss of MCT1, but this appears less likely as only subtle changes at the mRNA level were observed in the MogCreMCT1lox mice, but not with the Sox10Cre-MCT1lox mice.
Axonal Degeneration in the Conditional Nulls: Comparison with Prior Findings
The onset of axonal degeneration in our conditional nulls as assessed with EM was developed at middle age (10–14 months). Although the electrophysiological CAP recording data did not reach statistical significance, the bimodal distribution of the amplitude recovery data in the conditional nulls, but not the controls, clearly indicates that degenerative changes have developed. These data could be considered confusing in light of our earlier observations in Mct1 het null mice and mice with targeted delivery of lentiviral vectors targeting Mct1 (Lee et al., 2012) (Table S2). In the Mct1 het null mice, axonal degeneration was observed from the age of 8 months onward. The earlier phenotype observed in these mice could be explained by the fact that MCT1 protein is expressed in multiple cell types as clearly demonstrated by our immunostainings with a new polyclonal antibody directed at the C-terminal end of MCT1. The contribution of OL MCT1 to the phenotype observed in these Mct1 het null mice is therefore uncertain. On the other hand, in the lentiviral injection experiments, where MCT1 is targeted with shRNA expressed under either a ubiquitous or OL-specific promoter, onset of degeneration is acute and very profound (Lee et al., 2012). Of note, the injection techniques in those prior experiments produced distinct degeneration in the control injected optic nerves and spinal cord, suggesting that a pre-injury condition might propel axonal degeneration when MCT1 expression is downregulated. Additionally, these viral injections cause local inflammation that could also alter the OL metabolic demands. This is consistent with the far milder phenotype without prior CNS damage, as occurs in the Mct1 het null mice as well our conditional MCT1 null experiments (Lee et al., 2012). In addition, the conditional null mice experiments allow us to assess pathology at advanced age (e.g., up to P750 in the MogCre-MCT1lox mice) and throughout the entire CNS, as opposed to just locally near the site of lentivirus injection. It is only with advanced age that OL MCT1 becomes essential for providing neuronal trophic support, potentially due to other risk factors for degeneration that become more prominent with aging, such as abnormalities in neuronal mitochondrial energy homeostasis and autophagic flux (Salvadores et al., 2017; Mattson and Magnus, 2006). Interesting, in this light, MCT1 expression is reduced in normal aging and may enhance the risk of developing neurodegeneration. Aging is, in fact, the number one risk factor in the development of neurodegenerative diseases. The death and degeneration of OLs that we and others have reported in these age-dependent diseases, as well as in their animal models, could consequently severely affect MCT1 expression levels and contribute to developing neuronal degeneration (Kang et al., 2013; Philips et al., 2013; Patel and Balabanov, 2012; Ettle et al., 2016).
In summary, we have found that OL MCT1 expression is not required for normal energy homeostasis during early development, while in adulthood, from middle-age onward, loss of OL MCT1 leads to axonopathy and hypomyelination. These data indicate that monocarboxylates such as lactate and pyruvate have an essential role in OL and neuronal energy homeostasis with advanced age and that loss of transporter expression, as observed in the normal-aging CNS and models of neurodegenerative diseases, could contribute to severe axonal injury and degeneration.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeffrey D. Rothstein (jrothstein@jhmi.edu).
Materials Availability
The MCT1 conditional null mice generated in this study will be made available upon request. We will require a Material Transfer Agreement to be completed prior to sharing.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Non-transgenic B6SJLF1/J mice were purchased from the Jackson Laboratory (Stock No: 100012). Mct1-tdTomato mice were generated in our laboratory and described in our earlier study (Lee et al., 2012). B6N.Cg-Tg(Pdgfra-cre/ERT)467Dbe/J ((Pdgfrα-CreER) mice were obtained from Dr. Dwight Bergles at Johns Hopkins University. B6.129X1Gt(ROSA)26Sortm1(EYFP)Cos/J (RosaYFP) reporter mice were obtained from Jackson Laboratories (Stock No: 006148). Mct1 conditional null mice (MCT1lox) were recently generated in our laboratory (Jha et al., 2020). Mogtm1(Cre)Gkl (MogCre) mice were generated by Dr. Ari Waisman from University of Mainz, Germany and kindly shared with our laboratory (Buch et al., 2005). B6;CBA-Tg(Sox10-cre)1Wdr/J mice (Sox10Cre mice) were obtained from Jackson Laboratories (Stock No: 025807). Both male and female littermates were used in this study and randomly allocated to each of the experimental groups at ages varying from P21 to P750. All mice are kept in 14 hours light/10 hours dark cycle. All animal experiments were carried out in compliance with animal protocols approved by the Animal Care and Use Committee at the Johns Hopkins University School of Medicine.
Primary oligodendrocyte cultures
OL lineage cell cultures were prepared from P7 mouse cortices from Sox10Cre-MCT1lox and MCT1lox mice. Both male and female pups were allocated to each experimental group. Cortices were dissociated using the adult mouse brain dissociation kit according to the manufacturer’s protocol (Miltenyi). In short, meninges were removed and cortices underwent papain digestion for 30 min. using the adult brain dissociator (Miltenyi). After passage of cells through a 70µm cell strainer, myelin was removed with ‘myelin debris removal solution’ and single cells were pelleted. Red blood cells were removed using ‘Red Blood cell Removal Solution’ and cells were incubated with anti-O4 magnetic microbeads for 15 minutes on ice. O4+ oligodendrocyte progenitors were then magnetically sorted and collected in oligodendrocyte cell medium. O4+ cells derived from the same genotype and from either male or/ female pup were pooled in order to increase cell plating density. Cells were plated at 400K per well in a 6-well plate and maintained in the cell incubator at 37.5°C with 0.5% CO2. The oligodendrocyte medium used contains DMEM-F12 with HEPES and Glutamax, N2 supplement, B27 supplement, 20ng/µl basic fibroblast growth factor (bFGF) and 20ng/µl platelet derived growth factor α (PDGFα). Cells were plated on 6-well plates that were coated overnight with Matrigel at 37.5°C and 0.5% CO2.
METHOD DETAILS
Crossing of transgenic mouse lines
The Pdgfrα-CreER mice were crossed with RosaYFP and Mct1-tdTomato mice to generate triple transgenic animals, hemizygous for either transgene.
The MCT1lox conditional null mice were crossed with hemizygous MogCre or hemizygous Sox10Cre mice to obtain double transgenic MogCre-MCT1lox and Sox10Cre-MCT1lox mice, hemizygous for Cre and homozygous for the MCT1 conditional allele. All mice used in this study were hemizygous for Cre and homozygous for the MCT1lox allele.
Animal behavior and treatment procedures
All mouse behavior experiments were performed at the Johns Hopkins animal behavior core.
Accelerated Rotarod
Motor learning and coordination were examined by the rotarod test (Rotamex-5 System with a spindle dimensions of 3.0 cm x 9.5 cm; Columbus Instruments). Testing was conducted over a 3 day period, where each mouse was given three trials per day and latency to fall off the rotarod was measured as it accelerated from 4 to 99 rpm over a 5 min. period. Before the start of testing on the first day, each mouse was given a habituation trial by being placed on the rotarod, which was rotating at a constant speed of 4 rpm for 10 min. The mean latencies were calculated for each animal.
Open field testing
Spontaneous locomotion was assessed over a 30 min. period using activity chambers (16’’ (W) x 16’’ (D) x 15’’ (H)) with Photobeam Activity System (PAS) open field system equipped with 16 × 16 infrared beams (San Diego Instruments, San Diego, CA, USA) during the dark period from 10:00am to 06:00pm (reversed light cycle). Total locomotor activity was automatically measured as the number of beams breaks. The center zone was defined as the surface area covered by central 14 × 14 beams of the box (14 × 14 inches) with the remaining peripheral area being designated as the peripheral zone. The amount of time mice spend along the walls was used as a measure of anxiety.
Y-maze
Spatial working memory assessed by spontaneous alternation and spatial recognition memory were assessed in the y-maze test as previously described (Shevelkin et al., 2017).
Passive avoidance test
Passive avoidance was assessed using GEMINI Active and Passive Avoidance System (Stoelting Co., Wood Dale, IL). Briefly, a mouse was placed in the light compartment of the shuttle box for 30 s. to acclimate. Following the habituation, the door between two compartments was opened, and the latency to enter the dark compartment was measured. Entering the dark compartment led to closing the door. Three seconds after entering the dark compartment, the mouse was exposed to an inescapable foot shock delivered for at the intensity of 0.5mA. Twenty-four hours following the training session, the mouse was returned to the light compartment, the door between the compartments was opened and the latency to enter the dark compartment was measured. The cut-off time was 5 min.
Elevated Maze Plus
Anxiety was evaluated in the plus-maze (San Diego Instruments Inc., San Diego, CA, USA) as previously described (Pletnikov et al., 2008).
Trace fear trace conditioning test
Trace fear conditioning (TFC) was assessed as previously described (Jouroukhin et al., 2018; Terrillion et al., 2017). Briefly, TFC is a 3 day test consisting of the habituation day, training day, and test day. On day 1, a mouse was habituated to the shock box (Coulbourn, Holliston, MA) for 10 min. On Day 2, the mouse was placed in the shock box, and a 20 s. 90dB tone was delivered. Twenty seconds following the termination of the tone, a scrambled 2 s. 0.5 mA shock was delivered. This tone-shock pairing was repeated three times. On Day 3, the mice were placed in the shock box for 3 min and the freezing behavior was assessed as a measure of contextual memory. Following the context test, the cued test was performed. The mice were placed in a different testing box, and the tone presented during the training session was presented three times. The freezing behavior in response to the tones was assessed as a measure of cue-dependent fear memory. Freezing was automatically detecting using the Freeze Scan software (Clever Sys Inc.).
Tamoxifen injections
Tamoxifen (MilliporeSigma) was dissolved in ethanol, vortexed for 10 s. and dissolved in sunflower seed oil (ratio ethanol-oil 1:10). PDGFRαCreER mice crossed with RosaYFP and Mct1-tdTomato mice were injected intraperitoneally with 4 doses of tamoxifen (200mg/kg) for 4 consecutive days. Four different cohorts were injected: P60-P150-P300 and P550. Mice were sacrificed 30 days after the first injection.
EDU proliferation assay
P14 old mice were injected with 5mg/kg EDU for 5 consecutive days. Mice were sacrificed one week after the first injection at P21. EDU immunostaining in different CNS areas was performed according to the manufacturer’s protocol (Thermo Scientific).
Assays
Immunohistochemistry
Mice were deeply anesthetized by I.P. injections of anesthetic cocktail acepromazine maleate (3mg/kg; MWI Veterinary Supply), ketamine (100mg/kg; MWI Veterinary Supply), and xylazine (20mg/kg; MWI Veterinary Supply). Mice were transcardially perfused with ice-cold PBS followed by ice-cold PBS containing 4% paraformaldehyde (PFA). Spinal cord, cerebellum, optic nerve and forebrains were dissected and post-fixed in 4% PFA in PBS for 4 hours. Samples were stored overnight in 30% sucrose solution in PBS at 4°C and subsequently embedded in OCT compound (Sakura Finetek). 20 µm spinal cord and 40 µm coronal brain and cerebellar floating sections were cut on a cryostat (Thermo Scientific), washed in PBS and blocked for 1 hour at room temperature in PBS solution containing 0.1% Triton X-100 (PBST) and 10% normal goat serum (Vector). Sections were then incubated overnight at 4°C with primary antibodies diluted in PBST: chicken ant-MCT1 (our lab), rabbit anti-Iba1 (WAKO), rabbit anti-NG2 (MilliporeSigma), chicken anti-GFAP (MilliporeSigma), rabbit anti-GFP (Rockland), mouse anti-CC1 (Abcam), rat anti-LAMP-2 (Biolegend), mouse anti-CD31 (BD), rabbit anti-GLT-1 (our lab) and mouse anti-NeuN (MilliporeSigma)(for more details see Key Resources Table). Sections were washed in PBST and incubated with fluorescent labeled secondary antibodies (Thermo Scientific) for 1 hour at room temperature. Sections were subsequently washed in PBST and mounted on glass slides in Prolong Gold antifade reagent containing DAPI (Thermo Scientific). Section were imaged on a Zeiss Axioimager Z1 equipped with Apotome 2 or on a Zeiss 800 confocal microscope (Zeiss).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse monoclonal anti-APC antibody [CC-1] | Abcam | Cat#ab16794; RRID:AB_443473 |
| mouse monoclonal anti-CNPase antibody clone 11–5B | MilliporeSigma | Cat#MAB326; RRID:AB_2082608 |
| rabbit polyclonal anti-Connexin-43 antibody | MilliporeSigma | Cat#C6219; RRID:AB_476857 |
| mouse monoclonal anti-GAPDH (6C5) | Thermo Fisher Scientific | Cat#AM4300; RRID:AB_437392 |
| chicken polyclonal anti-GFAP | MilliporeSigma | Cat#AB5541; RRID:AB_10579068 |
| rabbit polyclonal anti-GFP | Rockland Immunochemicals | Cat#600–401-215; RRID:AB_828167 |
| rabbit polyclonal anti-Glut1 | Abcam | Cat#Ab652; RRID:AB_305540 |
| rabbit monoclonal anti-Glut3 | Abcam | Cat#Ab191071; RRID:AB_2736916 |
| rabbit polyclonal anti-Iba1 | WAKO | Cat#019–19741; RRID:AB_839504 |
| rat monoclonal anti-CD107b (LAMP-2) | Biolegend | Cat#816101; RRID:AB_2564796 |
| mouse monoclonal anti-MAG | MilliporeSigma | Cat#MAB1567; RRID:AB_2137847 |
| mouse monoclonal anti-MBP | Biolegend | Cat#808403; RRID:AB_2628849 |
| chicken polyclonal anti-MCT1 | Dr. Jeffrey Rothstein (Jha et al., 2020) | N/A |
| rabbit polyclonal anti-MCT2 | MilliporeSigma | Cat#AB3542; RRID:AB_177409 |
| mouse monoclonal anti-MOG | MilliporeSigma | Cat#MAB5680; RRID:AB_1587278 |
| rabbit polyclonal anti-NG2 | MilliporeSigma | Cat#AB5320; RRID:AB_11213678 |
| mouse monoclonal anti-Myelin proteolipid protein clone PLPC1 | MilliporeSigma | Cat#MAB388; RRID:AB_177623 |
| rabbit anti-GLT-1 | Dr. Jeffrey Rothstein (Rothstein et al., 1994) | N/A |
| mouse anti-CD31 | BD | Cat#550389; RRID:AB_2252087 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AR-C155858 | Tocris | Cat#4960 |
| Lactate acid, sodium salt. L-14C | PerkinElmer | Cat#NEC599050UC |
| Pyruvic acid, sodium salt, 1-14C | PerkinElmer | Cat#NEC255050UC |
| Tamoxifen | MilliporeSigma | Cat#T5648 |
| Critical Commercial Assays | ||
| Taqman assay: Slc16a1 (MCT1) | Thermo Scientific | Cat#4331182; Mm01306378_m1 |
| Taqman assay: Slc16a7 (MCT2) | Thermo Scientific | Cat#4331182; Mm00445115_m1 |
| Taqman assay: mouse Slc16a3 (MCT4) | Thermo Scientific | Cat#4331182; Mm00446102_m1 |
| Taqman assay: mouse Slc2a1 (GLUT1) | Thermo Scientific | Cat#4331182; Mm00441480_m1 |
| Taqman assay: mouse Slc2a3 (GLUT3) | Thermo Scientific | Cat#4331182; Mm00441483_m1 |
| Taqman assay: mouse Gjb6 (Cx30) | Thermo Scientific | Cat#4331182; Mm00433661_s1 |
| Taqman assay: mouse Gjc2 (Cx47) | Thermo Scientific | Cat#4331182; Mm00519131_s1 |
| Taqman assay: mouse Gja1 (Cx43) | Thermo Scientific | Cat#4331182; Mm00439105_m1 |
| Taqman assay: mouse GAPD | Thermo Scientific | Cat#4352339E |
| O4 microbeads | Miltenyi | Cat#130–094-543 |
| Adult brain dissociation kit | Miltenyi | Cat#130–107-677 |
| Click-iT EdU Cell Proliferation Kit for Imaging, Alexa Fluor 488 dye | Thermo Scientific | Cat#C10337 |
| Experimental Models: Cell Lines | ||
| Mouse primary oligodendrocytes (Sox10Cre-MCT1lox) | This paper | N/A |
| Mouse primary oligodendrocytes (MCT1lox) | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6SJLF1/J | Jackson Laboratory | Cat#100012 |
| Mouse: MCT1tdTomato | Dr. Jeffrey Rothstein (Lee et al., 2012) | N/A |
| Mouse: B6N.Cg-Tg(Pdgfra-cre/ERT) 467Dbe/J | Dr. Dwight Bergles /Jackson Laboratory | Cat#018280 |
| Mouse: B6.129X1Gt(ROSA)26Sortm1(EYFP)Cos/J | Jackson Laboratory | Cat#006148 |
| Mouse: Mogtm1(Cre)Gkl | Dr. Ari Waisman (Buch et al., 2005) | http://www.informatics.jax.org/allele/MGI:3689957 |
| Mouse: B6;CBA-Tg(Sox10-cre)1Wdr/J | Jackson laboratory | Cat#025807 |
| Mouse: MCT1lox | Dr. Jeffrey Rothstein (Jha et al., 2020) | N/A |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| GraphPad Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Zen 2.3 Lite | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen-lite.html |
| MATLAB (R2014b) | Mathworks | https://www.mathworks.com/ |
| Clampfit 11 | Molecular Devices | https://mdc.custhelp.com/app/answers/detail/a_id/20260/~/axon™-pclamp™-11-electrophysiology-data-acquisition-%26-analysis-software |
| Origin 2020 | OriginLab | https://www.originlab.com/2020 |
Myelin preparation
Whole brain and spinal cord were dissected and homogenized in 0.32M sucrose using an ultra-turrax homogenizer. The homogenate was carefully layered over a 0.85M sucrose solution and centrifuged at 75000 g for 30 min. at 4°C in Beckman ultracentrifuge (Beckman). After centrifugation, the myelin containing interface was collected, washed with deionized water and centrifuged at 75000 g for 15 min at 4°C. The pelleted fractions then underwent two rounds of osmotic shock by incubation in deionized water for 10 min. on ice followed by ultracentrifugation at 12000 g for 15 min. at 4°C. Pellets were then dissolved in 0.32M sucrose, layered over 0.85M sucrose and myelin containing interface was collected after centrifugation at 75000 g for 30 min. at 4°C. After another wash in deionized water, myelin was pelleted by centrifugation at 75000 g for 15 min at 4°C. Myelin pellets were dissolved in RIPA buffer (Thermo Scientific), complemented with protease and phosphatase inhibitors and stored at −80°C.
Western Blot
The protein concentration of each sample was measured using the Lowry method and the colorimetric reaction was measured on a SpectraMax M3 plate reader (Molecular Devices). 10–40µg of total protein was loaded on mini Protean TGX Precast gels (Biorad). After electrophoresis, proteins were blotted on a PVDF membrane (Biorad) and blocked in 5% Blotting Grade Blotter (Biorad) diluted in Tris buffered saline (TBS) containing 0.1% Tween 20 (TBST). Blots were then incubated with either of the following antibodies: chicken anti-human MCT1 antibody, mouse anti-PLP (MilliporeSigma), mouse anti CNPase (MilliporeSigma), rabbit anti-MCT2 (MilliporeSigma), rabbit anti-GLUT1 (abcam), rabbit anti-GLUT3 (abcam), mouse anti-MBP (Biolegend), mouse anti-MOG (MilliporeSigma), rabbit anti-Connexin-43 (MilliporeSigma), mouse anti-GAPDH (Thermo Scientific), and mouse anti-MAG (MilliporeSigma)(for more details see Key Resources Table). Membranes were overnight incubated with primary antibodies at 4°C. Membranes were washed with TBST and incubated with HRP labeled secondary antibodies for 1 hour at room temperature. After two washes in TBST and one wash in TBS, membranes were exposed to ECL reagent for 2 min. (Thermo Scientific) and chemiluminescent signal was detected with the LAS Imager 4000 (GE Healthcare).
Isolation of adult oligodendrocyte progenitor cells
P150 Sox10Cre-MCT1lox and MCT1lox mice were deeply anesthetized by I.P. injections of anesthetic cocktail acepromazine maleate (3mg/kg; MWI Veterinary Supply), ketamine (100mg/kg; MWI Veterinary Supply), and xylazine (20mg/kg; MWI Veterinary Supply). Mice were transcardially perfused with ice-cold PBS. Brain cortices were dissected and placed in PBS with calcium/magnesium. O4+ oligodendrocyte progenitors were isolated according to the adult brain dissociation protocol (Miltenyi) as outlined under ‘primary oligodendrocyte cultures’ (see above). After magnetic sorting, cells were centrifuged for 5 min. at 300 g at 4°C. Pellets were dissolved in Tri Reagent (Zymo Research) and RNA was extracted using the Direct-zol RNA MicroPrep kit (Zymo Research).
Reverse transcriptase-q-PCR
Mice were deeply anesthetized by I.P. injections of anesthetic cocktail acepromazine maleate (3mg/kg; MWI Veterinary Supply), ketamine (100mg/kg; MWI Veterinary Supply), and xylazine (20mg/kg; MWI Veterinary Supply). Mice were transcardially perfused with ice cold PBS and tissue was dissected and submerged in Tri Reagent (Zymo Research). Alternatively, oligodendrocyte lineage cells were sorted from whole tissue as described above. RNA was isolated using either the Direct-zol RNA MicroPrep kit (Zymo Research) (for cells) or by conventional chloroform-isopropanol-ethanol extraction methods (for tissue). For the latter, tissue in Tri Reagent is diluted in chloroform in a 5:1 Tri Reagent:chloroform ratio. After centrifugation at 12000rpm, the clear upper phase is collected and ice-cold isopropanol is added. After 12 minutes centrifugation at 12000rpm, 75% ethanol is added to the pellet. After subsequent centrifugation (12000rpm, 10 minutes), the RNA pellet is dried and dissolved in purified water. RNA concentration was measured by nanodrop (ThermoScientific). RNA then underwent reverse transcription into cDNA using the High Capacity cDNA reverse transcription kit (Thermo Scientific). To detect relative gene expression, inventoried assay Taqman probes targeting Mct1, Mct2, Mct4, Cx43, Cx30, Cx47, Glut1, Glut3 and Gapdh (endogenous control) were used throughout the manuscript (for details see Key Resources Table).
Electron microscopy
Mice were deeply anesthetized by I.P. injections of anesthetic cocktail acepromazine maleate (3mg/kg; MWI Veterinary Supply), ketamine (100mg/kg; MWI Veterinary Supply), and xylazine (20mg/kg; MWI Veterinary Supply). Mice were subsequently transcardially perfused with 0.1M Sorensen’s phosphate buffer to flush out the blood and subsequently perfused with 4% paraformaldehyde/2.5% glutaraldehyde in 0.1M phosphate buffer. Optic nerves were dissected and post-fixed with 2% osmium for 2 hours. The tissue was subsequently dehydrated in graded ethanol and embedded in Embed 812 resin (EMS). Thin sections (70–80nm) were cut on a Reichert Jung Ultracut E microtome and placed on formvar coated 100 mesh copper grids. The sections were stained with uranyl acetate followed by lead citrate. All imaging was performed on a Zeiss Libra 120 with a Veleta (Olympus) camera at an accelerating voltage of 120Kv. For light microscopy, 0.5µm sections were cut and stained with 1% toluidine blue (EMS).
Lactate and pyruvate transport assay
Lactate and pyruvate transport assays were performed on oligodendrocyte progenitor cell cultures. After culturing oligodendrocytes and plating, oligodendrocyte progenitors were grown for 3 days in oligodendrocyte before lactate/pyruvate transport in oligodendrocyte lineage cells was measured with the lactate/pyruvate transport assay.
For measuring lactate/pyruvate uptake, cells underwent two rounds of washing in HEBSS buffer (25mM HEPES, 150mM NaCl, 3mM KCl, 1.7mM KH2PO4, 0.6mM MgCl2 and 1mM CaCl2*2H2O). Cells were then exposed to 2X Hot Lactate/Pyruvate solution (0.5µCi/mL of [14C]-L-lactate/[14C] Pyruvate, PerkinElmer) for 1 min. on ice. The cells treated with MCT1 inhibitor were incubated with 1uM of AR-C1554858 (Tocris) in HEBSS buffer for 10 min. on ice. Cells were washed twice with HEBSS buffer containing 2mM 4-CIN. Cells were incubated in 0.1N NaOH. Plates were swirled to detach the cells from the bottom of the plate and cells were lysed by pipetting cells up and down for 30 times. 500µl of the supernatant is transferred to a scintillation vial and 3 mL of scintillation fluid is added to each sample. Samples are placed on a shaker overnight at room temperature. 200µl of each sample is transferred to a 96 well plate and radioactivity of the samples is measured with scintillation counting machine. The remaining supernatants obtained after cell lysis is used to measure protein concentration using the Bradford assay with the colorimetric reaction measured on a SpectraMax M3 plate reader (Molecular Devices).
Electrophysiological recording
Optic nerve recordings were performed on P360 Sox10Cre-MCT1lox mice and MCT1lox controls as described previously (Larson et al., 2018). Mice were anaesthetized with isoflurane and sacrificed by cervical dislocation. Optic nerves were then rapidly dissected and incubated at room temperature in oxygenated ACSF for ≥ 30 min. Nerves were then transferred to a recording chamber superfused with oxygenated ACSF at 37°C (temperature controller, Cell MicroControls TC2BIP 2/3Ch). Using gentle suction, each end of the nerve was drawn into the tip of a flared pipette electrode. The stimulating electrode (containing the retinal end of the nerve) was connected to a constant current isolated stimulator unit (Winston Electronics Co., St. Louis, MO) driven by pClamp9 software (Molecular Devices). CAPs were elicited by a 1 mA, 50 µs current pulse. The recording electrode (containing the chiasmatic end of the nerve) was connected to one input channel of a Multiclamp 700A amplifier (Axon Instruments). A second electrode, placed near the recording electrode but not in contact with the nerve, was connected to the second channel of the amplifier, and the two signals were subtracted on-line by routing through a differential amplifier (Model 440, Brownlee Precision), significantly reducing the stimulus artifact. Signals were filtered at 1 kHz, digitized at 100 kHz using a Digidata 1322A digitizer (Axon Instruments), and recorded to disk using pClamp9 software (Molecular Devices). Data were analyzed offline using MATLAB custom scripts.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image and data analysis
Images were analyzed using Image-J bundled with Java 1.8.0_112 and Zen 2.3 Lite software. For immunofluorescence, at least 2–15 sections per mouse were analyzed and quantified using Zen 2.3 Lite or Image-J. Cells were quantified within a defined area as # cells/µm2. On the electron microscopy images g-ratio’s and axonal degenerative phenotypes were measured on > 500 axons per sample from 3–10 images taken at 10000x magnification. Images were taken from a minimum of 5 randomly chosen areas of an optic nerve cross section. Inner and outer area of axons were measured with ImageJ from which the outer and inner axonal diameter was calculated respectively. Axons were scored as degenerated if they were either swollen, had accumulation of intracellular material (vesicles, swollen mitochondria) or dark cytoplasm, or if axons had disappeared altogether with accumulation of redundant myelin loops. Mitochondrial area was calculated with ImageJ bundled with Java 1.8.0_112. For the electrophysiological recordings, data were analyzed using Clampfit 11 (Molecular Devices) and MATLAB (Mathworks). Data collected from each optic nerve was considered a biological replicate.
Statistics
All data throughout the manuscript is represented as mean ± SEM as indicated in the figure legends. All statistical details of the experiments can be found in either the Figure legends or the Supplemental Figure legends including the value of n, the p value and statistical test being used. The p value was considered significant when p < 0.05. No subjects were excluded from the datasets in any of the experiments. All data met assumptions for the statistical approach, eg equal variance among groups. All statistical analysis has been performed using GraphPad Prism7.
Supplementary Material
Highlights.
Lack of MCT1-mediated oligodendrocyte metabolic support is well tolerated early on
Loss of oligodendrocyte MCT1-driven metabolic support causes axonal degeneration
Loss of oligodendrocyte MCT1-mediated metabolic support causes axonal hypomyelination
ACKNOWLEDGMENTS
This work was funded by the Muscular Dystrophy Association (MDA, development grant 381190) (T.P.), ALS Association (J.D.R.), Department of Defense (J.D.R.), and NIH (J.D.R. and B.M.M.). We thank Carol Cooke for her help with the EM at the Neurology-Peripheral Nerve Division at Johns Hopkins University School of Medicine. We thank members of J.D.R.’s lab at Johns Hopkins University School of Medicine for helpful discussions.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108610.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alberini CM, Cruz E, Descalzi G, Bessié res B, and Gao V (2018). Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 66, 1244–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres Benito P, Dominguez Gonzalez M, and Ferrer I (2018). Altered gene transcription linked to astrocytes and oligodendrocytes in frontal cortex in Creutzfeldt-Jakob disease. Prion 12, 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argente-Arizón P, Guerra-Cantera S, Garcia-Segura LM, Argente J, and Chowen JA (2017). Glial cells and energy balance. J. Mol. Endocrinol 58, R59–R71. [DOI] [PubMed] [Google Scholar]
- Boucanova F, Pollmeier G, Sandor K, Morado Urbina C, Nijssen J, Medard JJ, Bartesaghi L, Pellerin L, Svensson CI, Hedlund E, et al. (2020). Disrupted function of lactate transporter MCT1, but not MCT4, in Schwann cells affects the maintenance of motor end-plate innervation. Glia 69, 124–136. [DOI] [PubMed] [Google Scholar]
- Brady ST, Witt AS, Kirkpatrick LL, de Waegh SM, Readhead C, Tu PH, and Lee VM (1999). Formation of compact myelin is required for maturation of the axonal cytoskeleton. J. Neurosci 19, 7278–7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buch T, Heppner FL, Tertilt C, Heinen TJ, Kremer M, Wunderlich FT, Jung S, and Waisman A (2005). A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat. Methods 2, 419–426. [DOI] [PubMed] [Google Scholar]
- Ettle B, Schlachetzki JCM, and Winkler J (2016). Oligodendroglia and Myelin in Neurodegenerative Diseases: More Than Just Bystanders? Mol. Neurobiol 53, 3046–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flurkey KC, Currer JM, and Harrison DE (2007). Mouse models in aging research. In The Mouse in Biomedical Research, 2nd, Fox JG, Barthold SW, Davisson MT, Newcomer CE, Quimby FW, and Smith AL, eds. (Elsevier), pp. 637–672. [Google Scholar]
- Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Möbius W, et al. (2012). Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, and Nave KA (1998). Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 280, 1610–1613. [DOI] [PubMed] [Google Scholar]
- Ichihara Y, Doi T, Ryu Y, Nagao M, Sawada Y, and Ogata T (2017). Oligodendrocyte Progenitor Cells Directly Utilize Lactate for Promoting Cell Cycling and Differentiation. J. Cell. Physiol 232, 986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha MK, Lee Y, Russell KA, Yang F, Dastgheyb RM, Deme P, Ament XH, Chen W, Liu Y, Guan Y, et al. (2020). Monocarboxylate transporter 1 in Schwann cells contributes to maintenance of sensory nerve myelination during aging. Glia 68, 161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouroukhin Y, Kageyama Y, Misheneva V, Shevelkin A, Andrabi S, Prandovszky E, Yolken RH, Dawson VL, Dawson TM, Aja S, et al. (2018). DISC1 regulates lactate metabolism in astrocytes: implications for psychiatric disorders. Transl. Psychiatry 8, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, and Bergles DE (2013). Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci 16, 571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, and Nave KA (2003). Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet 33, 366–374. [DOI] [PubMed] [Google Scholar]
- Larson VA, Mironova Y, Vanderpool KG, Waisman A, Rash JE, Agarwal A, and Bergles DE (2018). Oligodendrocytes control potassium accumulation in white matter and seizure susceptibility. eLife 7, e34829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, and Allaman I (2018). Lactate in the brain: from metabolic end-product to signalling molecule. Nat. Rev. Neurosci 19, 235–249. [DOI] [PubMed] [Google Scholar]
- Mattson MP, and Magnus T (2006). Ageing and neuronal vulnerability. Nat. Rev. Neurosci 7, 278–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J, and Balabanov R (2012). Molecular mechanisms of oligodendrocyte injury in multiple sclerosis and experimental autoimmune encephalomyelitis. Int. J. Mol. Sci 13, 10647–10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips T, Bento-Abreu A, Nonneman A, Haeck W, Staats K, Geelen V, Hersmus N, Küsters B, Van Den Bosch L, Van Damme P, et al. (2013). Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 136, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre K, Pellerin L, Debernardi R, Riederer BM, and Magistretti PJ (2000). Cell-specific localization of monocarboxylate transporters, MCT1 and MCT2, in the adult mouse brain revealed by double immunohistochemical labeling and confocal microscopy. Neuroscience 100, 617–627. [DOI] [PubMed] [Google Scholar]
- Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, Mori S, Moran TH, and Ross CA (2008). Inducible expression of mutant human Dros. Inf. Serv.C1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol. Psychiatry 13, 173–186. [DOI] [PubMed] [Google Scholar]
- Rinholm JE, Hamilton NB, Kessaris N, Richardson WD, Bergersen LH, and Attwell D (2011). Regulation of oligodendrocyte development and myelination by glucose and lactate. J. Neurosci 31, 538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, and Kuncl RW (1994). Localization of neuronal and glial glutamate transporters. Neuron 13, 713–725. [DOI] [PubMed] [Google Scholar]
- Saab AS, and Nave KA (2017). Myelin dynamics: protecting and shaping neuronal functions. Curr. Opin. Neurobiol 47, 104–112. [DOI] [PubMed] [Google Scholar]
- Saab AS, Tzvetavona ID, Trevisiol A, Baltan S, Dibaj P, Kusch K, Möbius W, Goetze B, Jahn HM, Huang W, et al. (2016). Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 91, 119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvadores N, Sanhueza M, Manque P, and Court FA (2017). Axonal Degeneration during Aging and Its Functional Role in Neurodegenerative Disorders. Front. Neurosci 11, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Abarca LI, Tabernero A, and Medina JM (2001). Oligodendrocytes use lactate as a source of energy and as a precursor of lipids. Glia 36, 321–329. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevelkin AV, Terrillion CE, Abazyan BN, Kajstura TJ, Jouroukhin YA, Rudow GL, Troncoso JC, Linden DJ, and Pletnikov MV (2017). Expression of mutant DISC1 in Purkinje cells increases their spontaneous activity and impairs cognitive and social behaviors in mice. Neurobiol. Dis 103, 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavoe AKH, and Holzbaur ELF (2019). Neuronal autophagy declines substantially with age and is rescued by overexpression of WIPI2. Autophagy 16, 371–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, and Alberini CM (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadi M, Allaman I, Lengacher S, Grenningloh G, and Magistretti PJ (2015). Learning-Induced Gene Expression in the Hippocampus Reveals a Role of Neuron -Astrocyte Metabolic Coupling in Long Term Memory. PLoS ONE 10, e0141568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Li Z, Zhang W, and Yao Z (2019). Nitric oxide might be an inducing factor in cognitive impairment in Alzheimer’s disease via downregulating the monocarboxylate transporter 1. Nitric Oxide 91, 35–41. [DOI] [PubMed] [Google Scholar]
- Terrillion CE, Abazyan B, Yang Z, Crawford J, Shevelkin AV, Jouroukhin Y, Yoo KH, Cho CH, Roychaudhuri R, Snyder SH, et al. (2017). DISC1 in Astrocytes Influences Adult Neurogenesis and Hippocampus-Dependent Behaviors in Mice. Neuropsychopharmacology 42, 2242–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi RB, Jackiewicz M, McKenzie IA, Kougioumtzidou E, Grist M, and Richardson WD (2017). Remarkable Stability of Myelinating Oligodendrocytes in Mice. Cell Rep 21, 316–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzoli E, Cali C, De Roo M, Ponzoni L, Sogne E, Gagnon N, Francolini M, Braida D, Sala M, Muller D, et al. (2019). Ultrastructural Evidence for a Role of Astrocytes and Glycogen-Derived Lactate in Learning-Dependent Synaptic Stabilization. Cereb. Cortex 30, 2114–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Crawford TO, Griffin JW, Tu P.h., Lee VM, Li C, Roder J, and Trapp BD (1998). Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J. Neurosci 18, 1953–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.