

Abstract
We report the preparation of enantioenriched secondary alkylcarbastannatranes via a stereoinvertive SN2 reaction of enantioenriched alkyl mesylates and carbastannatranyl anion equivalents. Using this process, enantioenriched secondary alcohols may be converted into highly enantioenriched alkylcarbastannatranes, which are useful in stereospecific cross-coupling reactions.
Graphical Abstract
The use of configurationally stable enantioenriched organometallic nucleophiles in stereospecific transformations constitutes a powerful approach to the preparation of complex, nonracemic molecules.1,2 Accordingly, enantioenriched organoboron and organotin compounds have been extensively used in metal-catalyzed cross-coupling reactions. These transformations rely on stereospecific mechanisms that effectively translate chiral information from the starting materials to final products. Thus, it is vital that such precursors can be prepared with high enantiopurity in a straightforward manner.
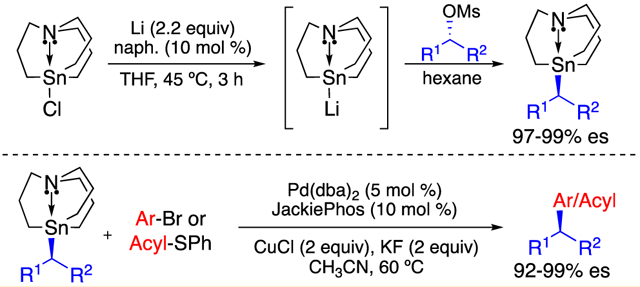
Building upon the pioneering work of Jurkschat3 and Vedejs,4 in 2013, our laboratory developed a Stille cross-coupling reaction of enantioenriched secondary alkylcarbastannatrane nucleophiles and aryl electrophiles (Figure 1).5-7
Figure 1.

Stereospecific Stille cross-coupling reaction using enantioenriched secondary alkylcarbastannatranes.
This method afforded arylation products in high yields and with high enantiospecificity. The unique reactivity of alkylcarbastannatranes is attributed to the selective labilization of its apical alkyl substituent as a consequence of the dative N–Sn interaction in the “atrane” tin backbone.4 This approach enabled the first example of a stereospecific cross-coupling reaction using an unactivated secondary organotin species.8 Subsequently, we extended this chemistry to stereospecific acylation reactions, as well as stereospecific fluorination reactions.9,10
The lack of an efficient route to prepare enantioenriched carbastannatrane nucleophiles has been a major bottleneck to their broader synthetic application. Because unactivated secondary alkylstannanes typically exhibit low reactivity, there has previously been limited value in devising synthetic strategies to prepare enantioenriched variants. As a result, our laboratory has relied heavily on stereospecific lithiation/stannylation processes11 and chiral preparatory HPLC separation of racemates to obtain enantioenriched alkylcarbastannatrane nucleophiles. We set out to address this limitation by developing a more general approach to the preparation of enantioenriched alkylcarbastannatranes. Herein, we report a study of carbastannatranyl anion equivalents in stereoinvertive SN2 reactions with enantioenriched alkyl mesylates. We have found that carbastannatranyllithium (2) readily undergoes substitution reactions with secondary alkyl mesylates. This strategy enables the preparation of highly enantiopure secondary alkylcarbastannatranes from commercially available single-enantiomer alcohols. With access to more diverse alkylcarbastannatrane nucleophiles, the scope of stereospecific Stille cross-coupling reactions is significantly broadened.
In 2015, Wang and Uchiyama reported the highly efficient lithiation of trialkyltin chlorides using in situ generated naphthalide radical anions.12 Using this method, clean substitution of methyl and primary allylic/benzylic halides was observed, which enabled preparation of the corresponding organostannanes in excellent yield. We hypothesized that carbastannatranyllithium (2) could be produced under similar reductive conditions and might be employed in SN2 reactions with enantioenriched secondary alkyl electrophiles to afford enantioenriched secondary alkylcarbastannatranes via stereoinversion. To circumvent potential racemization via a background radical pathway, we opted to investigate the use of secondary alkyl sulfonates, which have a lower propensity to form alkyl radicals than do alkyl halides. We quickly found that sec-butyl mesylate underwent efficient substitution with tributylstannyllithium prepared by the method of Wang and Uchiyama. However, an analogous approach using carbastannatranyl chloride failed to generate the corresponding secondary alkylcarbastannatrane. Ensuing studies revealed that carbastannatranyllithium (2) could be generated through sonication of the reaction mixture at 45 °C (see the Supporting Information).
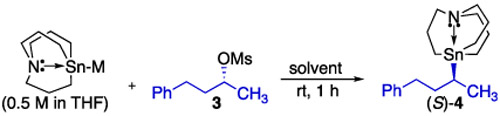
Having developed effective conditions for the in situ generation of tin lithium species 2, we investigated the enantiospecificity13 of the substitution reaction using enantioenriched alkyl mesylate 5 (Table 1). The reaction of 2 with 3 predissolved in THF resulted in eroded enantiopurity in the substitution product. However, we found that excellent enantiospecificity could be achieved when TMEDA was added to 2 or when ether or hexane was employed as the solvent. Access to carbastannatranyllithium (2) not only was found to be useful in forming tin–carbon bonds directly but could also be used as a precursor to other metalated tin nucleophiles.14 Using 5 as a model substrate for analysis, we examined the reactivity of carbastannatranyl anion equivalents 5–7 in this substitution reaction. Reactions involving 5 and 7 showed excellent enantiospecificity. Though none of these nucleophiles proved superior to the use of 2 in hexane or ether, the ability of attenuated nucleophiles such as 5 and 7 to undergo stereoinvertive substitution with high enantiospecificity suggests that alkyl mesylates bearing functional groups not compatible with tin lithium reagents will be still viable electrophiles in this process.
Table 1.
Optimization of Enantiospecificity in the Reaction of Carbastannatranyl Anion Equivalents and Alkyl Mesylate 5
 | ||||
|---|---|---|---|---|
| Entry | Tin Entry Nucleophile |
Solvent | Yield (%)a | % esb |
| 1 | 2 | THF | 74 | 74 |
| 2 | 2 (with TMEDA) | THF | 65 | 98 |
| 3 | 2 | ether | 63 | 99 |
| 4 | 2 | hexane | 68 | 99 |
| 5 | 5 | hexane | 30 | 99 |
| 6 | 6 | hexane | <5 | -- |
| 7 | 7 | hexane | 65 | 98 |
 | ||||
Calibrated 1H NMR yields.
Determined by chiral HPLC.
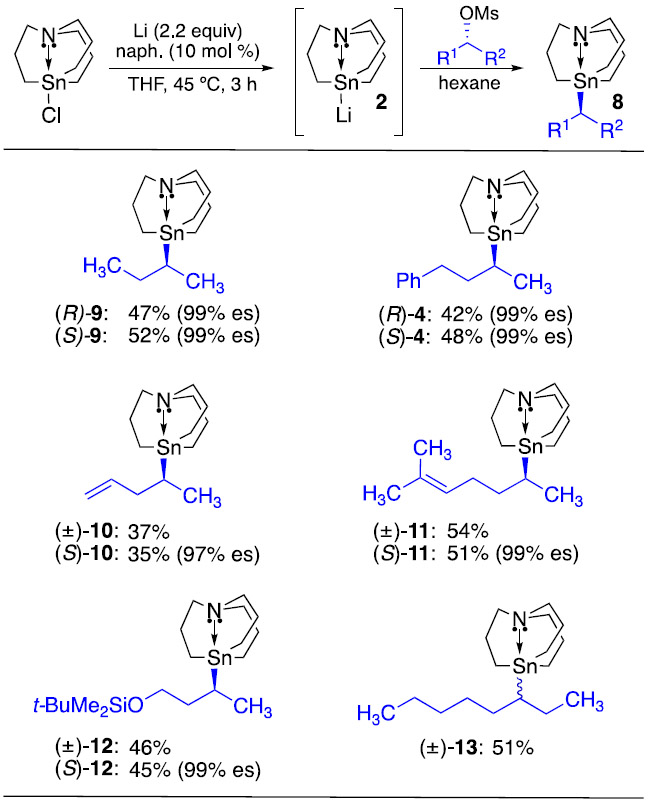
Using the conditions developed in Table 1, a series of secondary alkylcarbastannatrane nucleophiles was prepared from their corresponding mesylates (Table 2). Yields of ca. 50% were typically obtained alongside high levels of enantioenrichment, with separable elimination products accounting for the remaining mass balance. These unactivated enantioenriched secondary alkylcarbastannatranes (8) are completely tolerant of air and water. They are also inert to neutral, basic, and reductive reaction conditions, which enables synthetic modifications to be performed following installation of the carbastannatrane unit. For instance, desilylation of 12 was readily achieved by treatment with TBAF. Previously, we have demonstrated that diborylation of olefins and LiAlH4 reduction of esters do not affect the integrity of the carbastannatrane unit.10,15,16 The majority of the enantioenriched alkylcarbastannatranes prepared using this method contain a methyl group on the stereogenic carbon center. Unfortunately, analytical chiral HPLC conditions could not be found to separate enantiomers of carbastannatrane 13, in which an ethyl group replaces the methyl group. Though the enantiopurity of 13 could not be determined, we feel that the enantiospecificity would likely be very high from the enantioenriched alkyl mesylate, as was observed when other mesylates were employed alongside our standard conditions.
Table 2.
Preparation of Enantioenriched Secondary Alkylcarbastannatranes from Their Corresponding Mesylates
 |
Previously, we had only reported two examples of stereospecific arylation reactions using unactivated secondary alkylcarbastannatranes (prepared using preparatory chiral HPLC).5 Now having broader access to enantioenriched alkylcarbastannatranes nucleophiles, we conducted an investigation of their use in stereospecific Stille reactions. The cross-coupling products in Table 3 were all generated with high enantiospecificity. In contrast to our observations for stereospecific Suzuki reactions,2 the electronic properties of the electrophilic component did not influence the transfer of stereochemistry during the cross-coupling reaction. Both electron-rich and electron-deficient electrophiles underwent coupling with nearly perfect transfer of the initial stereochemistry. The presence of heteroatoms and acidic protons was well tolerated in these reactions. The free alcohol formed from desilylation of (S)-12 underwent arylation with high yield and excellent enantiospecificity. Use of (rac)-13 as a nucleophile resulted in the formation of arylation product 14h in high yield. Thus, transmetalation of a secondary alkylcarbastannatrane still occurs efficiently from the bulkier, non-methyl-containing alkylcarbastannatrane nucleophile. Finally, when (S)-11 was employed in a coupling reaction with the thioester of (S)-naproxen, acylation product 14j was obtained with completely reagent controlled diastereoselectivity.
Table 3.
Stereospecific Stille Cross-Coupling Reactions Using Enantioenriched Secondary Alkylcarbastannatranesa
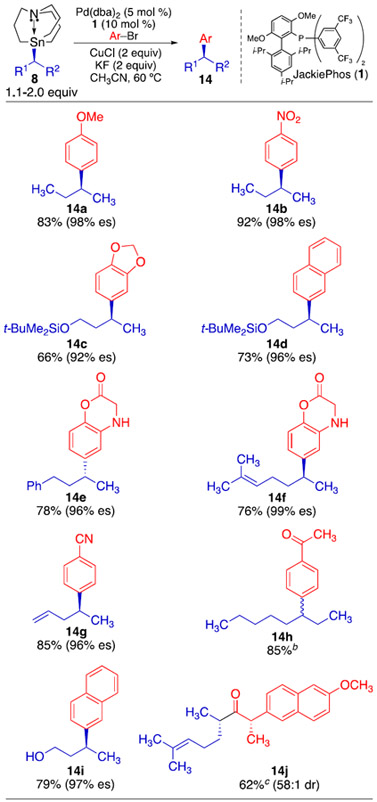 |
Isolated yields of duplicate runs.
Using racemic 13.
Using the thioester of naproxen as electrophile; no KF added.
In summary, we have developed a reliable synthetic method to access enantioenriched secondary alkylcarbastannatrane from their corresponding alkyl mesylates. Treatment of enantioenriched alkyl mesylates with stannatranyllithium (2) results in the formation of highly enantioenriched secondary alkylcarbastannatranes via a stereoinvertive SN2 pathway Other carbastannatranyl anion equivalents (e.g., 5 and 7 were also shown to undergo substitution reactions with high enantiospecificity. We subsequently demonstrated that these enantioenriched alkylcarbastannatrane nucleophiles readily undergo cross-coupling reactions with aryl bromides, with highly general preference for retention of stereochemistry. We expect that this method to access enantioenriched alkylcarbastannatranes will facilitate the future development of new stereospecific transformations of alkylcarbastannatrane compounds.
Supplementary Material
ACKNOWLEDGMENTS
We thank the City College of New York, the National Institutes of Health (R01GM131079), and the National Science Foundation (CHE-1665189) for support of this work.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.organomet.9b00467.
Synthetic procedures, spectral data (1H and 13C NMR), and HPLC data (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).For review articles on stereospecific cross-coupling reactions see:Rygus JPG; Crudden CM Enantiospecific and Iterative Suzuki-Miyaura Cross-Couplings. J. Am. Chem. Soc. 2017, 139 18124–18137.Sandford C; Aggarwal VK Stereospecific Functionalizations and Transformations of Secondary and Tertiary Boronic Esters. Chem. Commun. 2017, 53, 5481–5494.Cherney AH; Kadunce NT; Reisman SE Enantioselective an Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents to Construct C–C Bonds. Chem Rev. 2015, 115, 9587–9652.Wang CY; Derosa J; Biscoe MR Configurationally Stable, Enantioenriched Organometallic Nucleophiles in Stereospecific Pd-Catalyzed Cross-Coupling Reactions: An Alternative Approach to Asymmetric Synthesis. Chem. Sci. 2015, 6, 5105–5113.
- (2) (a).Zhao S; Gensch T; Murray B; Niemeyer ZL; Sigman MS; Biscoe MR W–C Bond Formation Enabled through Ligand Parameterization. Science 2018, 362, 670–674. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li L; Zhao S; Joshi-Pangu A; Diane M; Biscoe MR Stereospecific Pd-Catalyzed Cross-Coupling Reactions of Secondary Alkylboron Nucleophiles and Aryl Chlorides. J. Am. Chem. Soc 2014, 136, 14027–14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3) (a).Jurkschat K; Tzschach A; Meunier-Piret J; Van Meerssche M Crystal and Molecular Structure of 1-Aza-5-Stanna-5-Chlorotricyclo[3.3.3.01,5]undecane, a 2,8,9-Tricarbastannatrane. J. Organomet. Chem 1985, 290, 285–289. [Google Scholar]; (b) Jurkschat K; Tzschach A; Meunier-Piret J Crystal and Molecular Structure of 1-Aza-5-Stanna-5-Methyltricyclo[3.3.3.01,5]undecane. Evidence for a Transannular Donor-Acceptor Interaction in a Tetraorganotin Compound. J. Organomet. Chem 1986, 315, 45–49. [Google Scholar]
- (4) (a).Vedejs E; Haight AR; Moss WO Internal Coordination at Tin Promotes Selective Alkyl Transfer in the Stille Coupling Reaction. J. Am. Chem. Soc 1992, 114, 6556–6558. [Google Scholar]; (c) Theddu N; Vedejs E Stille Coupling of an Aziridinyl Stannatrane. J. Org. Chem 2013, 78, 5061–5066. [DOI] [PubMed] [Google Scholar]
- (5).Li L; Wang CY; Huang R; Biscoe MR Stereoretentive Pd-Catalysed Stille Cross-Coupling Reactions of Secondary Alkyl Azastannatranes and Aryl Halides. Nat. Chem 2013, 5, 607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).For other examples of carbastannatrane use, see:Jensen MS; Yang C; Hsiao Y; Rivera N; Wells KM; Chung JYL; Yasuda N; Hughes DL; Reider PJ Synthesis of an Anti-Methicillin-Resistant Staphylococcus Aureus (MRSA) Carbapenem via Stannatrane-Mediated Stille Coupling. Org. Lett 2000, 2, 1081–1084.Sebahar HL; Yoshida K; Hegedus LS Effect of Adjacent Chiral Tertiary and Quaternary Centers on the Metal-Catalyzed Allylic Substitution Reaction. J. Org. Chem 2002, 67, 3788–3795.Fillion E; Taylor NJ Cine-Subsitution in the Stille Coupling: Evidence for the Carbenoid Reactivity of sp3-gem-Organodimetallic Iodopalladio-Trialkylstannylalkane Intermediates. J. Am. Chem. Soc 2003, 125, 12700–12701.Kavoosi A; Fillion E Synthesis and Characterization of Tricarbastannatranes and Their Reactivity in B(C6F5)3-Promoted Conjugate Additions. Angew. Chem., Int. Ed 2015, 54, 5488–5492.Srivastav N; Singh R; Kaur V Carbastannatranes: Powerful Coupling Mediators in Stille Coupling. RSC Adv. 2015, 5, 62202–62213.Fillion E; Kavoosi A; Nguyen K; Ieritano C B.(C6F5)3-Catalyzed Transfer 1,4-Hydrostannylation of α,β,-Unsaturated Carbonyls Using iPr-tricarbastannatrane. Chem. Commun 2016, 52, 12813–12816.Simidzija P; Lecours MJ; Marta RA; Steinmetz V; McMahon TB; Fillion E; Hopkins WS Changes in Tricarbastannatrane Transannular N–Sn Bonding upon Complexation Reveals Lewis Base Donicities. Inorg. Chem 2016, 55, 9579–9585.
- (7).For carbagermanatrane use, see: Xu M-Y; Jiang W-T; Li Y; Xu Q-H; Zhou Q-L; Yang S; Xiao B Alkyl Carbagermatranes Enable Practical Palladium-Catalyzed sp2-sp3 Cross-Coupling. J. Am. Chem. Soc 2019, 141, 7582–7588. [DOI] [PubMed] [Google Scholar]
- (8).Activated organotin nucleophiles feature the presence of a C(sp2) α-carbon, heteroatomic α-carbon, and/or strongly coordinating β-carbonyl group. [Google Scholar]
- (9).Wang CY; Ralph G; Derosa J; Biscoe MR Stereospecific Palladium-Catalyzed Acylation of Enantioenriched Alkylcarbastannatranes: A General Alternative to Asymmetric Enolate Reactions. Angew. Chem., Int. Ed 2017, 56, 856–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ma X; Diane M; Ralph G; Chen C; Biscoe MR Stereospecific Electrophilic Fluorination of Alkylcarbastannatrane Reagents. Angew. Chem., Int. Ed 2017, 56, 12663–12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Beak P; Basu A; Gallagher DJ; Park YS; Thayumanavan S Regioselective, Diastereoselective, and Enantioselective Lithiation–Substitution Sequences: Reaction Pathways and Synthetic Applications. Acc. Chem. Res 1996, 29, 552–560. [Google Scholar]
- (12).Wang DY; Wang C; Uchiyama M Stannyl-Lithium: A Facile and Efficient Synthesis Facilitating Further Applications. J. Am. Chem. Soc 2015, 137, 10488–10491. [DOI] [PubMed] [Google Scholar]
- (13).Enantiospecificity (%es) = (%eeproduct/%eestarting material) × 100%.
- (14).Hibino J-I; Matsubara S; Morizawa Y; Oshima K; Nozaki H Regioselective Stannylmetalation of Acetylenes in the Presence of Transition-Metal Catalyst. Tetrahedron Lett. 1984, 25, 2151–2154. [Google Scholar]
- (15).We have found secondary alkylcarbastannatranes to be incompatible with oxidative conditions, though it is plausible that very mild oxidants may be tolerated.
- (16).Cyclohexyl mesylate could be efficiently converted to the corresponding cyclohexylcarbastannatrane. However, 4-tert-butylcyclohexyl mesylate failed to react under similar conditions. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.