

Abstract
Oncolytic virotherapy has been tested in numerous early phase clinical studies. However, the antitumor activity of oncolytic viruses thus far has been limited. Numerous strategies are being explored to enhance their antitumor activity by activating the adaptive arm of the immune system. We reasoned that it might also be possible to engineer oncolytic viruses to redirect tumor-associated macrophages to tumor cells for therapeutic benefit. We engineered an oncolytic vaccinia virus (VV) to disrupt the CD47/SIRPα interaction by expressing a chimeric molecule that consists of the ectodomain of SIRPα and the Fc domain of IgG4 (SIRPα-Fc-VV). SIRPα-Fc-VV readily replicated in tumor cells and redirected M1 as well as M2 macrophages to tumor cells in vitro. In contrast, control VVs that either encoded YFP (YFP-VV) or SIRPα (SIRPα-VV) did not. In vivo, SIRPα-Fc-VV had greater antitumor activity than YFP-VV and SIRPα-VV in an immune competent osteosarcoma model resulting in a significant survival advantage. Pretreatment with cytoxan further augmented the antitumor activity of SIRPα-Fc-VV. Thus, arming oncolytic viruses with SIRPα-Fc may present a promising strategy to enhance their antitumor activity for the virotherapy of solid tumors.
Keywords: Vaccinia virus, CD47, SIRPα, osteosarcoma, pediatric cancer, immunotherapy
INTRODUCTION
Oncolytic viruses are actively being explored for a broad range of refractory and/or recurrent solid tumors.1 For example, adenoviruses (Adv), herpes simplex viruses (HSV) and vaccinia viruses (VV) have been genetically engineered to preferentially replicate in tumor cells, causing tumor cell lysis and immunogenic cell death.2,3 Numerous early phase clinical studies with oncolytic viruses have been conducted, and while their administration has been safe, antitumor activity was limited.4–9 Lack of antitumor activity is most likely due to limited viral spread through the tumor and the immunosuppressive tumor microenvironment. To overcome the latter, several strategies are actively being explored including genetically engineering oncolytic viruses to secrete cytokines, chemokines, and/or bispecific antibodies, or combining oncolytic viruses with checkpoint blockade.10–12 For example, arming VVs or HSVs with granulocyte macrophage colony stimulating factor (GM-CSF) has shown promising results in early Phase clinical studies, leading to FDA approval of the GM-CSF encoding HSV (talimogene laherparepvec) for melanoma.5,7
While the majority of approaches to enhance the antitumor activity have focused on activating the adaptive arm of the immune system, many solid tumors are infiltrated with tumor associated macrophages (TAMs).13–15 To protect themselves from macrophage-mediated killing, solid tumors express the ‘don’t eat me’ signal CD47.16 Blocking the interaction between CD47 on cancer cells and signal regulatory protein alpha (SIRPα), which is expressed on myeloid cells and TAMs, is actively being explored as a cancer therapeutic.17–19 For example, monoclonal antibodies (MAbs) targeting CD47 and chimeric molecules, consisting of the high affinity ectodomain of SIRPα fused to the Fc region of IgG (SIRPα-Fc), have shown potent antitumor activity in preclinical models.20–22 Early phase clinical testing of CD47 blockade as monotherapy has demonstrated limited efficacy,19 however combining it with the CD20 MAb rituximab resulted in high complete response rates in patients with Non Hodgkin Lymphoma.23
Here we report the generation of an oncolytic VV that is genetically engineered to secrete SIRPα-Fc (SIRPα-Fc-VV) for the virotherapy of osteosarcoma, a solid tumor for which outcomes remain poor for recurrent/refractory cases, with five-year survival rates of only 20%.24 SIRPα-Fc-VV replicated similarly to control VV in tumor cells, redirected macrophages to tumor cells, and had greater antitumor activity than control VV in an immune competent osteosarcoma model.
MATERIALS AND METHODS
Cell Lines and Culture Conditions
The African green monkey kidney fibroblast cell line CV-1, human cervical adenocarcinoma cell line HeLa, and human osteosarcoma cell lines SAOS2, G292, HOS, and U20S were purchased from ATCC (American Type Culture Collection, Manassas, VA). The murine adenocarcinoma cell line MC38 was provided by Dr. Serge Y. Fuchs (University of Pennsylvania, Philadelphia, PA) in 2014.25 The metastatic osteosarcoma cell line LM7 was provided by Dr. Eugenie Kleinerman (MD Anderson Cancer Center, Houston, TX) in 2011.26 The generation of LM7.eGFP.ffluc was previously described.27 Briefly, LM7 were genetically modified with a retroviral vector encoding a fusion protein consisting of enhanced green fluorescent protein and firefly luciferase (eGFP.ffluc). Human ovarian cancer cell lines OV10 (CD47-) and OV10–315 (CD47+) were provided by Dr. William Fraizer (Washington University, St Louis, MO) in 2014.28 The murine osteosarcoma cell lines F420 and CTC were derived fromsingly floxed p53+/F-Col2.3 transgenic mice.29 Cells were grown in Dulbecco’s Modified Essential Medium (GE Healthcare Life Sciences Hyclone Labatories, Logan, UT) supplemented with 10% fetal bovine serum (FBS, Hyclone) and 2 mmol/L GlutaMAX-1 (Thermo Fisher Scientific, Waltham, MA) and cultured at 37°C in 5% CO2. Cell lines were verified mycoplasma free with MycoAlert Mycoplasma Detection kit (Lonza, Switzerland). Human cell lines were validated at the Characterized Cell Line Core Facility (MD Anderson, Houston TX).
Generation of Macrophages
Healthy donor leukapheresis products (Gulf Coast Regional Blood Center, Houston, TX) were processed by Lymphoprep (Abbott Laboratories, Chicago, IL) gradient centrifugation. Monocytes were isolated from the resulting peripheral blood mononuclear cells (PBMCs) by CD14 MACS bead selection using LS Column and MidiMACS Separator (Miltenyi Biotec, Germany). Monocytes were cultured in Roswell Park Memorial Institute medium (RPMI, Hyclone) supplemented with 10% FBS, 2 mmol/L GlutaMAX-1, and 100ng/ml M-CSF (Peprotech, Rocky Hill, NJ) for five days and polarized to M1 macrophages with 100ng/ml LPS (MilliporeSigma, St Louis, MO) and 20ng/ml IFN-γ (Peprotech) or to M2 macrophages with 20ng/ml IL-4 (R&D Systems Inc, Minneapolis, MN) for 48 hrs.30
Construction of Recombinant VV
The minigenes containing SIRPα or SIRPα-Fc were synthesized by Thermo Fisher Scientific (Grand Island, NY) and subcloned into the pCMS1 shuttle plasmid downstream of the F17R late promoter (provided by Dr. David Bartlett, University of Pittsburgh, PA).31 Double-deleted VVs (vvDD, Western Reserve strain) expressing YFP, SIRPα, or SIRPα-Fc were generated by recombination of pCMS1 shuttle plasmids encoding YFP alone, SIRPα and YFP, or SIRPα-Fc and YFP into the TK locus of the VSC20 strain of Western Reserve VV.32,33 Viral stocks were prepared by infecting HeLa cells with 200 pfu (~0.0005 MOI) of YFP-VV, SIRPα-VV, or SIRPα-Fc-VV in DMEM-2.5% FBS for 2 hours at 37°C. DMEM, supplemented with 10%FBS and 2 mmol/L GlutaMAX-1, was added and cells were incubated until harvesting at 48–72 hours post-infection. Virus was quantified by plaque titering on CV-1 cells as previously reported.32
Flow Cytometric Analysis
10,000 cells per sample were collected on a FACSCalibur instrument (BD Biosciences, San Jose, CA) using Cell Quest Software (BD Bioscience) or a BD Canto II Instrument (BD Biosciences) using FACSDIVA software (BD Biosciences) and analyzed using FlowJo v10 (FlowJo LLC, Ashland, OR). For phenotyping macrophages, the following monoclonal antibodies (MAbs) were used: CD14-PerCP (BD), CD33-PE (BD), CD80-FITC (BD Biosciences), and CD163-AlexaFluor647 (Biolegend, San Diego, CA). For detection of VV transgenes, the following MAbs were used: AffiniPure F(ab’)2 Fragment Goat Anti-Human IgG: AlexaFluor 647 (Jackson ImmunoResearch Laboratories, West Grove, PA), human CD47-APC (eBioscience), murine CD47-APC (eBioscience). For murine immune infiltrate studies, the following MAbs were used: CD3-APC (BD Biosciences), CD11b-AlexaFluor 488 (BD Biosciences), CD11c-PerCP-Cy5 (BD Biosciences), CD206-PECy7 (eBioscience), F4/80-PE (Biolegend), Ly6G-AlexaFluor 700 (BD). Isotype-matched controls (BD) were used for each fluorophore.
Co-culture Assay
Conditioned media was generated by infecting OV10 cells (CD47-) with YPP-VV, SIRPα-VV, or SIRPα-Fc-VV at a MOI of 0.1. After 48 hours media was collected and filtered through a 0.45 micron filter (Pall Corporation, Port Washington, NY). M1 or M2 macrophage were co-cultured with OV10–315 cells with either media alone or supernatant from infected cells at a 5:1 effector to target ratio. Cells were harvested for FACS analysis 48 hours later. Cells were stained with anti-CD33 PE (BD Biosciences) to distinguish macrophages from tumor cells, and 7AAD (BD Biosciences) to exclude dead cells. The absolute number of macrophage and tumor cells were quantified by CountBright Absolute Counting Beads (Thermo Fisher Scientific) using flow cytometry as previously described, except 20,000 beads was added to each sample and 2,000 beads per sample were collected.34
In Vivo Experiments
All animal experiments were performed on a protocol approved by the Baylor College of Medicine Institutional Animal Care and Use Committee in accordance with the American Association for Laboratory Animal Science.
LM7 xenograft model:
8–10 week-old female SCID Beige (CB17.Cg-PrkdcscidLystbg-J/Crl, Charles River, Wilmington, MA) were injected i.p. with 4×106 LM7.eGFP.ffluc cells. Mice were treated with 1×106 PFU of YFP-VV, SIRPα-Fc-VV on day 7. PBS (vehicle control) injected mice served as controls. Tumors were monitored weekly by bioluminescent imaging on an IVIS System (Xenogen, Alameda, CA) as previously described.35
F420 immune-competent model:
7–9 week-old male C57BL/6 mice (C57BL/6J, The Jackson Laboratory, Bar Harbor, ME) were injected s.c. into the right flank with 5×105 F420 cells. After 11–12 days, when tumors were approximately 50–100 mm3 in volume, 1×107 PFU of YFP-VV or SIRPα-Fc-VV was injected i.t. PBS injected mice served as controls. Tumors were monitored by caliper measurements biweekly. For the experiment with cytoxan, the same experimental outline was used; only mice received 250 mg/kg cytoxan (Patterson Veterinary Supply, Greeley CO) i.p. 48 hours before the VV or PBS injection. For analysis of tumor-infiltrating immune cells, 1×106 F420 cells were injected s.c. into the right flank. After 15 days, when tumors were ~300 mm3, 1×108 PFU of YFP-VV or SIRPα-Fc-VV were injected i.t. PBS injected mice served as controls. After 48 hours, mice were sacrificed, and tumor and spleen were removed. A gentleMACS Dissociator (Miltenyi) was used to obtain a single cell suspension for FACS analysis.
Euthanasia criteria:
Mice were euthanized when tumor radiance was greater than 5×109 RLU, or when tumor was greater than 1.5 cm in diameter, or when they met other defined euthanasia criteria (weight loss, signs of distress) in accordance with the Center for Comparative Medicine at Baylor College of Medicine.
Statistical Analysis
Continuous data were summarized as mean ± standard deviation. Two-sample t-test was used for comparing the means between two groups. One-way analysis of variance followed by t-test with Holm method for multiple comparison adjustment was used for comparing the means among three or more groups. A tumor volume greater or equal to 600 mm3 was defined as tumor progression. Time to tumor progression was used to compare the differences in tumor growth. Time to tumor progression and survival were analyzed by the Kaplan-Meier method and comparisons were made using the Wilcoxon test with Holm method for multiple comparison adjustment. Graphpad Prism 6 (Graphpad Software Inc, San Diego, CA) and SAS 9.4 were used for statistical analysis. P values <0.05 were considered statistically significant
RESULTS
Generation of SIRPα-Fc-VV, SIRPα-VV and YFP-VV
We generated double-deleted VVs (vvDD) expressing SIRPα-Fc (SIRPα-Fc-VV), SIRPα (SIRPα-VV) or YFP (YFP-VV) by homologous recombination of the parental VSC20 virus (Western Reserve strain) with a shuttle plasmid encoding our transgene(s) (Figure 1A). To determine if the VVs expressed the desired transgene, we first screened a panel of human and murine osteosarcoma cells for CD47 expression using OV10 as negative and OV10–315 and MC38 as positive controls.28,36 All murine and human osteosarcoma cells expressed CD47 (Figure S1). OV10, OV10–315, MC38, LM7, or F420 cells were infected with YFP-VV, SIRPα-VV, or SIRPα-Fc-VV at a multiplicity of infection (MOI) of 0.1. Non-infected cells served as controls. After 24–48 hours, cells were harvested and FACS analysis was performed to detect CD47 and Fc expression. While the majority of control and YFP-VV-infected cells were CD47 positive (Figure 1B), CD47 expression could not be detected on SIRPα-Fc-VV-infected tumor cells. SIRPα-Fc-VV-infected cells however stained positive for Fc (Figure 1C), indicating that SIRPα-Fc bound to CD47 on the surface of cells, and interfered with CD47 detection. The ability of SIRPα to interfere with CD47 detection was inconsistent, and SIRPα-VV infected tumor cells were negative for Fc. To provide direct evidence that SIRPα-Fc is secreted and binds to neighboring cells, we infected LM7 and F420 cells with decreasing MOIs of SIRPα-Fc-VV, and after 48 hours performed FACS analysis for VV-infected cells (YFP-positive) and Fc-positive cells. Even when only 2–6% of cells were YFP positive, all cells stained positive for Fc, indicating secretion and binding of SIRPα-Fc to neighboring non-infected cells (Figure S2).
Figure 1: Generation of SIRPα-Fc-VV.
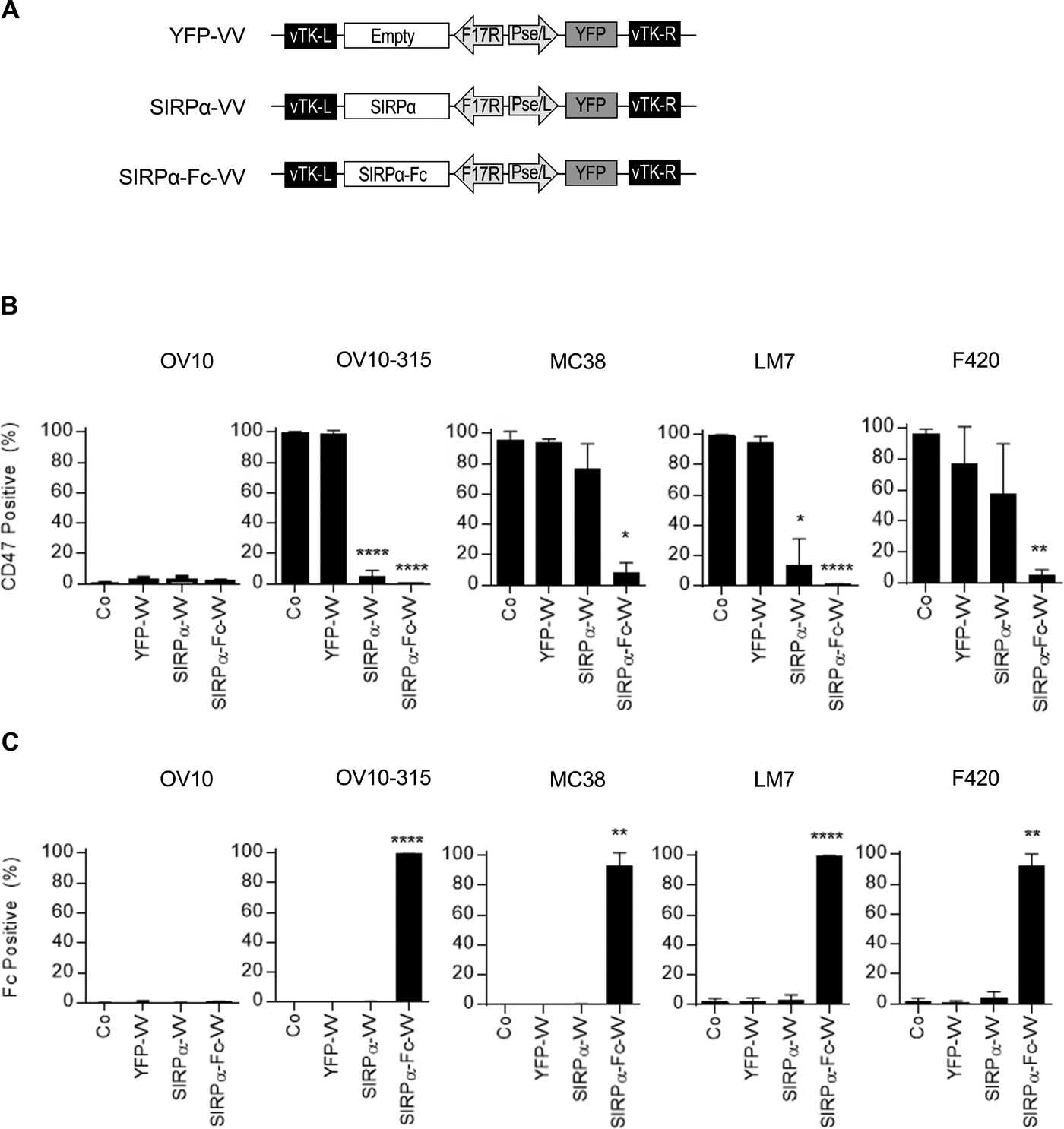
(A) Scheme of shuttle plasmids for generation of recombinant VV. SIRPαor SIRPα-Fc expression is controlled by the late F17R promoter, and yellow fluorescent protein (YFP) expression by the early/late Pse/I promoter. (B-C) OV10, OV10–315, MC38, LM7, or F420 were infected at a MOI of 0.1 with YFP-VV, SIRPα-VV, or SIRPα-Fc-VV. 24–48 hours post-infection expression of (B) CD47 or (C) Fc was determined by FACS analysis (n=3; mean percent expression +/−SD; ANOVA; *p<0.05, **p<0.01; ****p<0.0001).
SIRPα-Fc expression does not impair the oncolytic activity of VV and redirects macrophages to non-infected tumor cells
Since transgene expression can potentially interfere with the oncolytic activity of VV, we first compared the ability of YFP-VV, SIRPα-VV, or SIRPα-Fc-VV to induce tumor cell lysis in the absence of macrophages. OV10, OV10–315, MC38, LM7, or F420 cells were infected with VV at MOI 0.1 or 0.01 and the percentage of dead cells (7AAD-positive cells) was determined by FACS analysis at 24, 48, and 72 hours post infection (Figure 2A). There was no significant difference in the oncolytic activity of YFP-VV, SIRPα-VV, and SIRPα-Fc-VV at both MOIs for all five evaluated cell lines (50–60% 7AAD-positive cells 48–72 hours post infection). These results demonstrate that the oncolytic activity of the VVs are not affected by expression of SIRPα or SIRPα-Fc.
Figure 2: SIRPα-Fc-VV kills tumor cells through direct killing and redirecting macrophages.
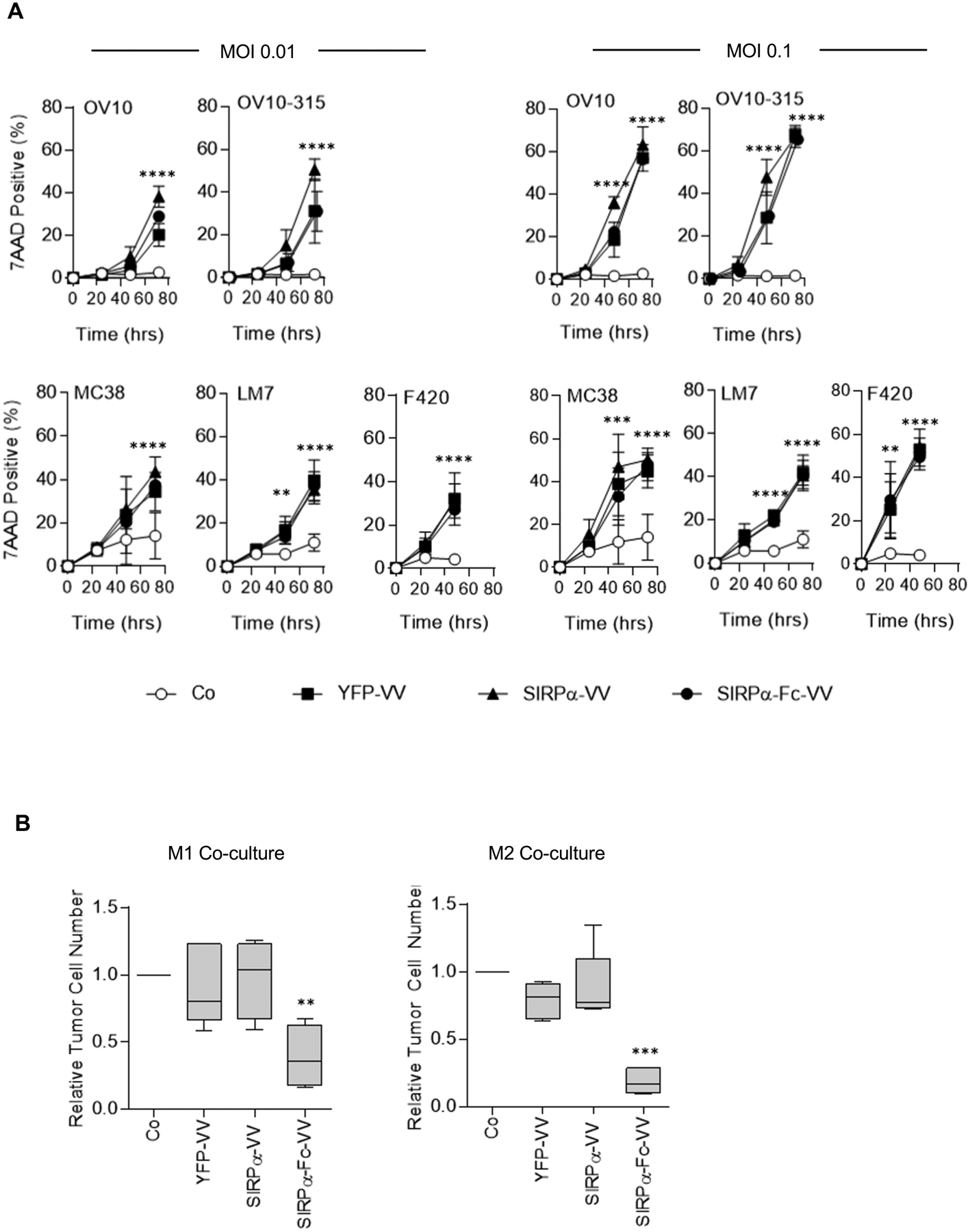
(A) OV10, OV10–315, MC38, LM7, or F420 cells were infected at a MOI of 0.01 or 0.1 with YFP-VV, SIRPα-VV, or SIRPα-Fc-VV. The percentage of dead (7AAD-positive) cells was determined 24, 48, and 72 post infection by FACS analysis. Non-infected cells served as controls (Co), (n=6; mean percent 7AAD+ expression +/−SD; ANOVA; Co versus YFP-VV, SIRPα-VV, or SIRPα-Fc-VV: **p<0.01; ***p<0.001; ****p<0.0001). (B) M1 or M2 macrophages were co-cultured with OV10–315 cells with either media alone or conditioned media from YFP-VV, SIRPα-VV, or SIRPα-Fc-VV-infected cells at an effector to target ratio of 5:1. After 48 hours, the absolute number of viable (7AAD-), CD33-negative OV10–315 cells was enumerated by FACS analysis (n=4–5; Tukey box and whiskers plot of relative tumor cell number (VV media tumor cell number/media alone tumor cell number; ANOVA; **p<0.01; ***p<0.001).
Next, we investigated whether SIRPα-VV or SIRPα-Fc-VVs could induce macrophages to kill tumor cells. Macrophages were generated and polarized to M1 or M2 phenotype (Figure S3). Since the oncolytic activity of VVs would confound the analysis of macrophages-mediated killing if VV-infected tumor cells were used as targets, we performed coculture assays with filtered, conditioned media that was obtained from YFP-VV-, SIRPα-VV-, or SIRPα-Fc-VV-infected cells. OV10–315 cells were cocultured with M1 or M2 polarized macrophages at a 5:1 effector to target ratio in the presence of control or conditioned media, and after 48 hours viable tumor cells were enumerated by FACS analysis. Only supernatant from SIRPα-Fc-VV infected cells redirected M1 and M2 macrophages to kill tumor cells, resulting in a significant (p<0.01) decrease in the number of viable tumor cells (Figure 2B). While the mean tumor cell killing of M2 macrophages was greater than of M1 macrophages (81.0 vs 61.2%), this difference did not reach significance.
SIRPα-Fc-VV has antitumor activity in immune competent osteosarcoma model
To investigate if SIRPα-Fc-VV has antitumor activity in vivo, we utilized the F420 immune competent murine osteosarcoma model. 5×105 F420 cells were injected subcutaneously (s.c.) into the right flank of C57BL/6 mice. On day 11–12, when the tumors were approximately 50–100 mm3 in volume, 1×107 plaque-forming units (PFU) of SIRPα-Fc-VV, SIRPα-VV, or YFP-VV were injected intra-tumorally (i.t.). PBS injected tumors served as controls. Tumor volume was monitored by caliper measurements (Figure 3A). The time to reach tumor progression, defined as a tumor volume of 600 mm3, was significantly delayed in mice receiving SIRPα-Fc-VV injections as compared with control (p<0.008; Table 1). This resulted in a significant survival advantage (p=0.005; Figure 3B). YFP-VV or SIRPα-VV injection did not significantly slow progression of the tumors in this aggressive osteosarcoma model. Based on the lack of efficacy of SIRPα-VV, we omitted it from further in vivo studies.
Figure 3: SIRPα-Fc-VV has superior antitumor activity in immune-competent osteosarcoma model.
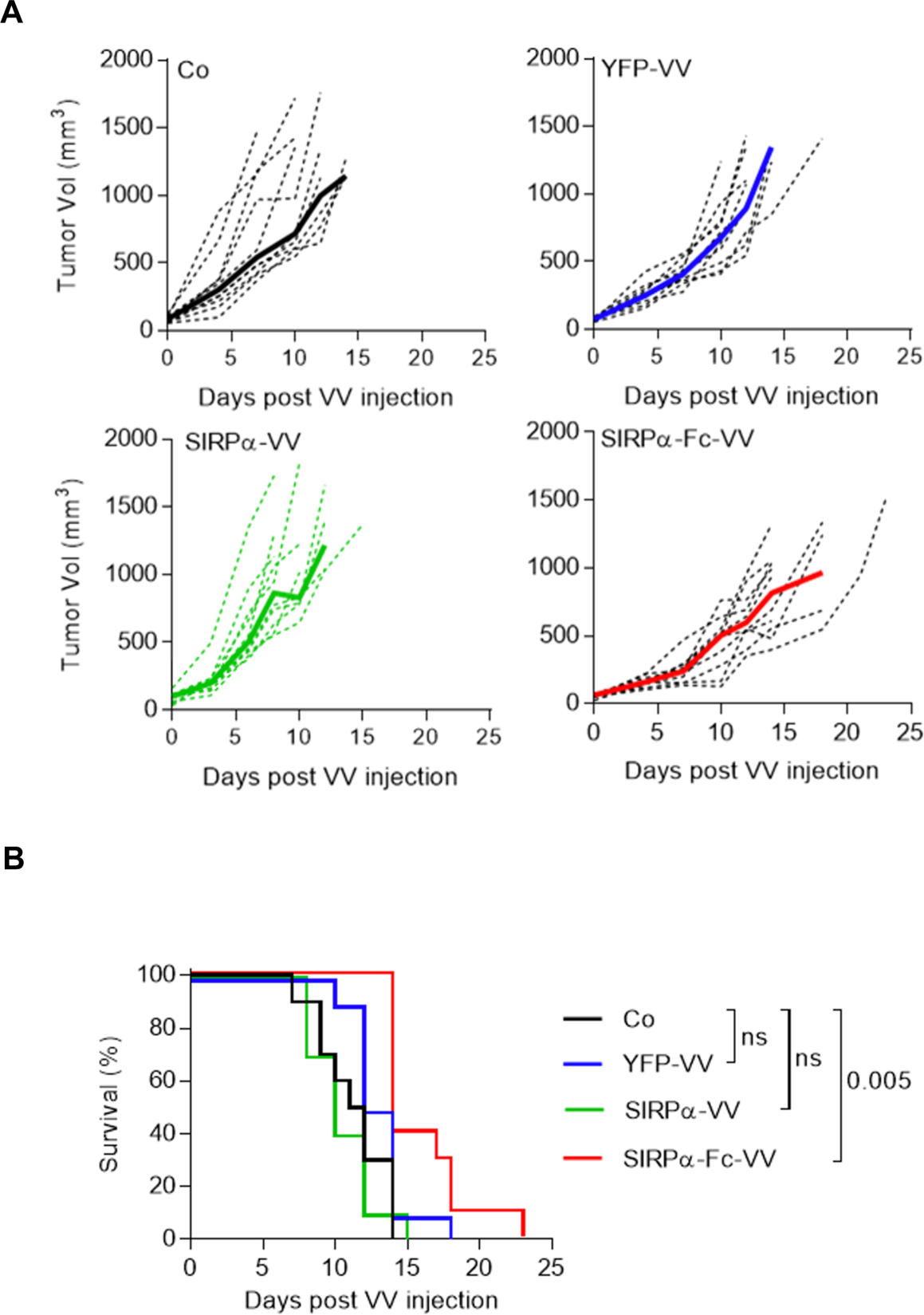
C57BL/6 mice were injected with 5×105 F420 cells s.c. into the right flank. Once tumors reached ~50–100mm3, mice (n=10 per group) were injected i.t with 1 ×107 PFU of SIRPα-Fc-VV, SIRPα-VV, YFP-VV or PBS (control; Co). Tumor size was monitored by caliper measurements (n=10 per group). (A) Tumor volume (dotted lines: individual mice; solid line: median). (B) Kaplan-Meier survival curves. The Wilcoxon test with Holm method for adjustment of multiple comparisons was used to determine significance.
Table 1: Time to tumor progression - SIRPα-Fc-VV.
| Comparisons | P┼ |
|---|---|
| Control vs. YFP-VV | 0.2759 |
| Control vs. SIRPα-VV | 0.3463 |
| Control vs. SIRPα-Fc-VV | 0.0080 |
The Wilcoxon test with Holm method for adjustment of multiple comparisons
SIRPα-Fc-VV and YFP-VV recruit macrophages and monocytes into tumors
Having observed either only modest (SIRPα-Fc-VV) or no (SIRPα-VV and YFP-VV) antitumor activity in the immune competent osteosarcoma model, we first wanted to confirm that SIRPα-Fc-VV and YFP-VV can replicate, and cause tumor cell destruction in vivo when not impeded by an intact immune system. 4×106 LM7 GFP.ffluc osteosarcoma cells were injected intraperitoneally (i.p.) into SCID-Bg mice and 7 days later, 1×106 PFU SIRPα-Fc-VV or YFP-VV or PBS were injected i.p. Tumor growth was monitored by bioluminescent imaging. Both YFP-VV and SIRPα-Fc-VV had significant antitumor activity as judged by a reduction in bioluminescent signal (Figure 4A, B). However, VV treated mice started to lose weight (Figure 4C), with YFP-VV treated mice having to be euthanized significantly earlier (p=0.0039) than SIRPα-Fc-VV treated mice. The weight loss was due to disseminated VV as determined by PCR for VV (data not shown).
Figure 4: SIRPα-Fc-VV has antitumor activity in immunodeficient osteosarcoma model.
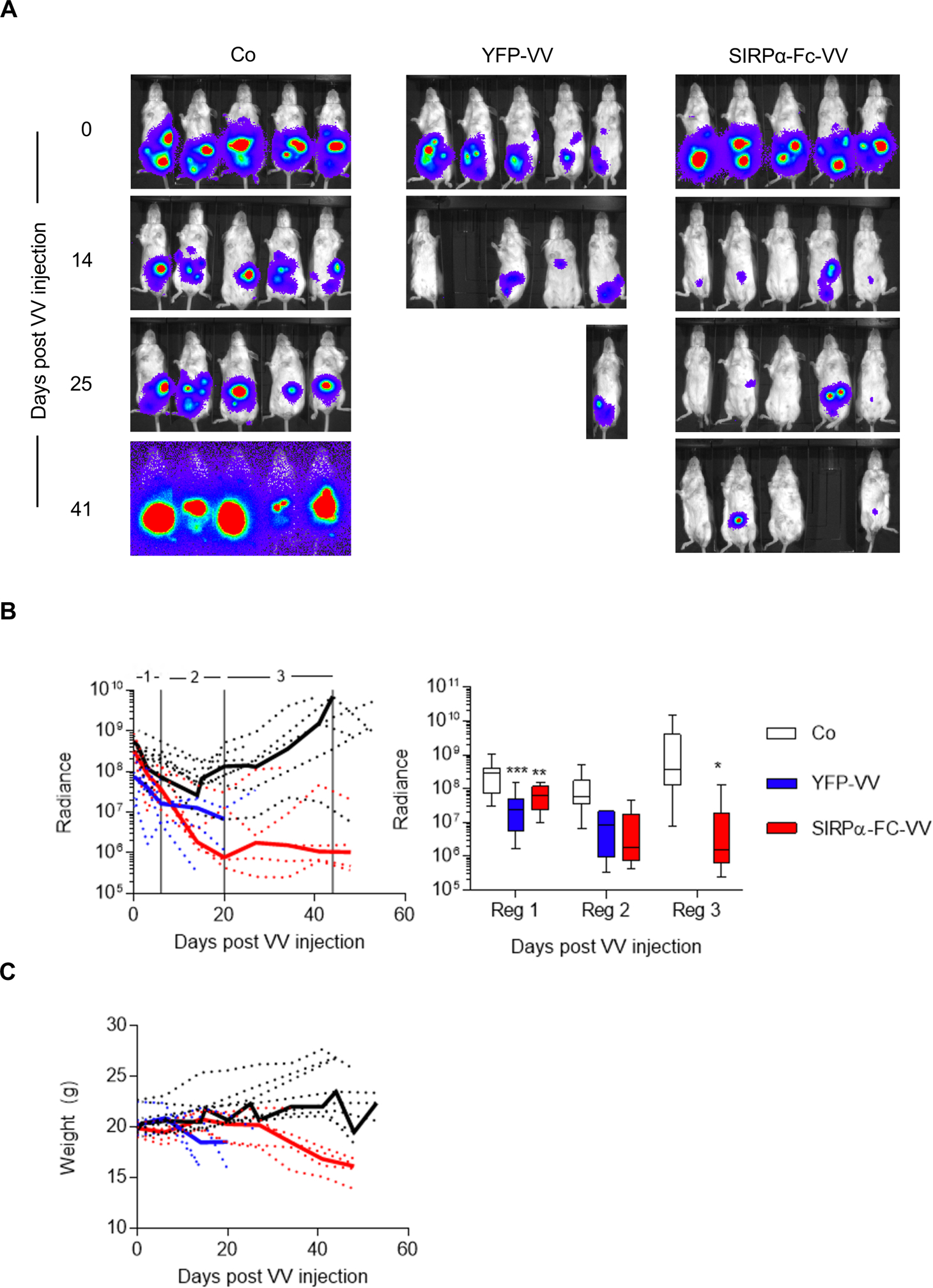
SCID Beige mice were injected with 4×106 LM7.GFP.ffluc cells i.p. After 7 days, mice (n=5 per group) were injected with 1×106 PFU of SIRPα-Fc-VV, YFP-VV or PBS (control; Co) i.p. Tumor growth was monitored by bioluminescent imaging. (A) Representative images of animals. (B) Left panel: Quantitative bioluminescent imaging results (dotted lines: individual mice; solid line: median; radiance = photons/s/cm2/sr). Right panel: Bioluminescence imaging results were divided into 3 regions to enable quantitative analysis (box and whiskers plot of median tumor luminescence +/− range; ANOVA; *p<0.05; **p<0.01; ***p<0.001; reg: region). (C) Weights of mice (dotted lines: individual mice; solid line: median).
Since the above results indicate that the immune system is critical for restricting VV replication in vivo, we next set out to determine the immune cell infiltrate in the immune competent F420 osteosarcoma model, and if VV injection changes its composition. 1×106 F420 cells were injected s.c. into the right flank of C57BL/6 mice. Fifteen days later, when the tumors were ~300 mm3 in volume, 1×108 PFU of SIRPα-Fc-VV or YFP-VV were injected i.t. PBS injected mice served as controls. Mice were sacrificed after 48 hours later, and the presence of T cells (CD3+), granulocytes (Ly6G+), M2 macrophages (F4/80+, CD206+), monocytes (CD11b+), and dendritic cells (CD11c+) in tumors and spleens was determined by FACS analysis.
Within control tumors, 6.6% (±9.9%) of cells were T cells, 0.5% (±0.4%) granulocytes, 8.2% M2 macrophages (±3.3%), 4.6% (±2.1%) monocytes, and 0.2% (±0.2%) dendritic cells (Figure 5A). SIRPα-Fc-VV or YFP-VV injection resulted in a significant (p<0.05) increase in the percentage of intratumoral immune cells. While the percentage of M2 macrophages and monocytes significantly (p<0.05) increased after YFP-VV or SIRPα-Fc-VV injection, the percentage of granulocytes only significantly (p<0.05) increased after YFP-VV injection (Figure 5A). No change in the percentage of T cells or dendritic cells was observed. Within spleens, SIRPα-Fc-VV or YFP-VV injection only resulted in a significant decrease in the mean percentage of T cells [Co: 27.9% (±2.6%); YFP-VV: 23.0% (±2.3%); SIRPα-Fc-VV: 22.2% (±1.9%)] (Figure 5B). Thus, our results indicate that VV injection increases the percentage of myeloid-derived cells within tumors.
Figure 5: Changes in myeloid cell immune infiltrate in F420 tumors after VV administration.
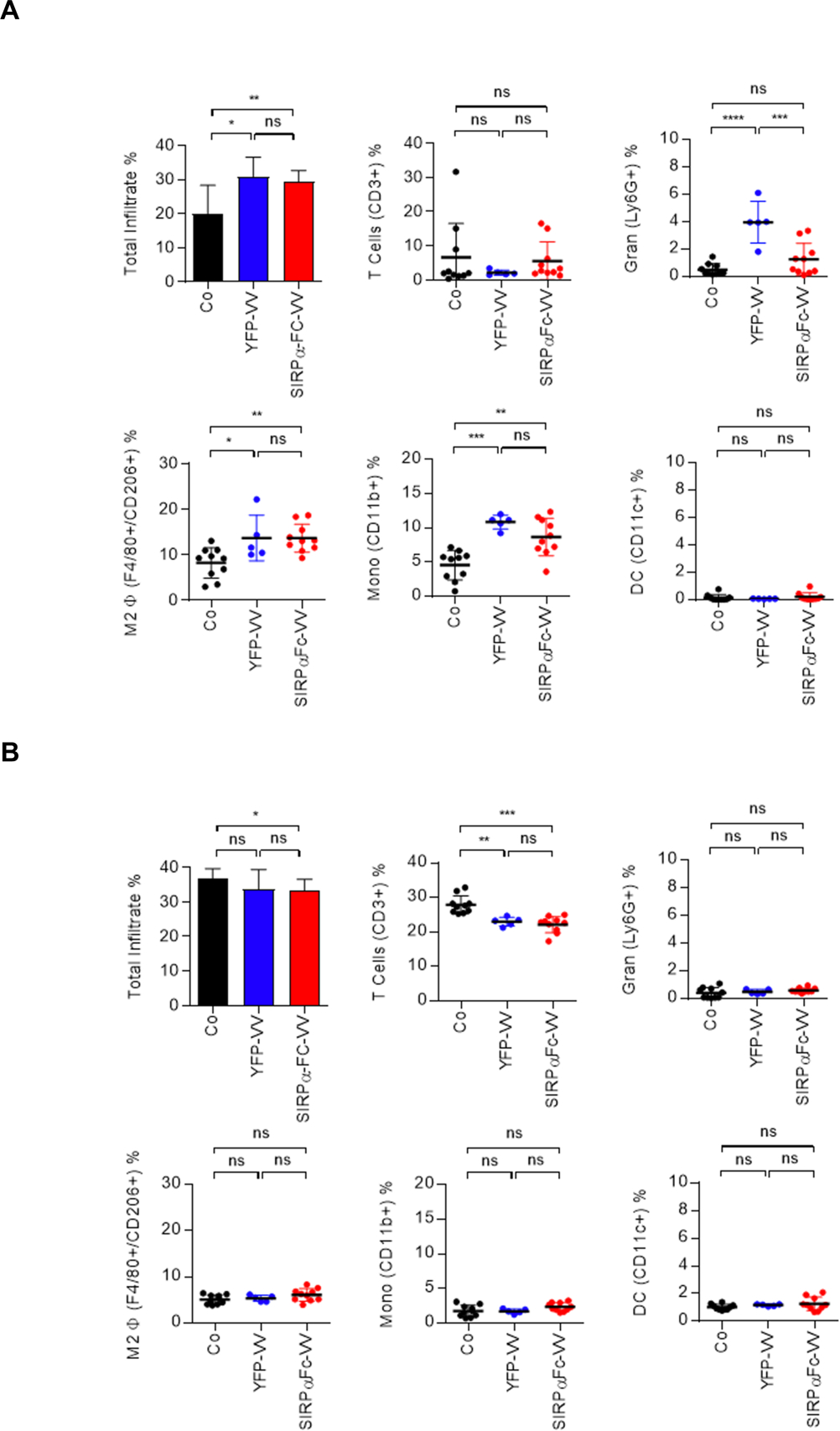
C57BL/6 mice were injected with 1×106 F420 cells s.c. into the right flank. Once the tumor was ~300mm3, 1×108 PFU of SIRPα-Fc-VV (n=10) or YFP-VV (n=5) were injected i.t. PBS injected mice (n=10) served as controls (Co). After 48 hours mice were euthanized, and single cell suspensions of (A) tumors and (B) spleens were prepared. The presence of T cells (CD3+), granulocytes (Ly6G+), M2 macrophages (F4/80+, CD206+), monocytes (CD11b+), and dendritic cells (CD11c+) was determined by FACS analysis (dot: individual mice; mean percent positive +/−SD; ANOVA; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001).
Combining SIRPα-Fc-VV with cytoxan enhances antitumor activity
Since F420 tumors at base line have an immune cell infiltrate that could potentially inhibit VV replication, we determined in the final set of experiments if depleting immune cells with cytoxan enhances antitumor effects. 5×105 F420 cells were injected s.c. into the right flank of C57BL/6 mice. On day 9, 250mg/kg cytoxan was injected i.p., followed by an i.t. injection of 1×107 PFU of SIRPα-Fc-VV or YFP-VV after 48 hours. PBS injected mice served as controls. Tumor volume was monitored by caliper measurements. Only SIRPα-Fc-VV injections significantly slowed tumor progression (p=0.026) (Table 2) and prolonged survival (p=0.002) (Figure 6B) as compared. In contrast, YFP-VV had no therapeutic benefit.
Table 2: Time to tumor progression - SIRPα-Fc-VV plus cytoxan.
| Comparisons | P┼ |
|---|---|
| Cy vs. YFP-VV +Cy | 0.912 |
| Cy vs. SIRPα-Fc-VV +Cy | 0.026 |
| YFP-VV +Cy vs. SIRPα-Fc-VV +Cy | 0.026 |
The Wilcoxon test with Holm method for adjustment of multiple comparisons
Figure 6: Cytoxan improves the antitumor activity of SIRPα-Fc-VV in immune-competent osteosarcoma model.
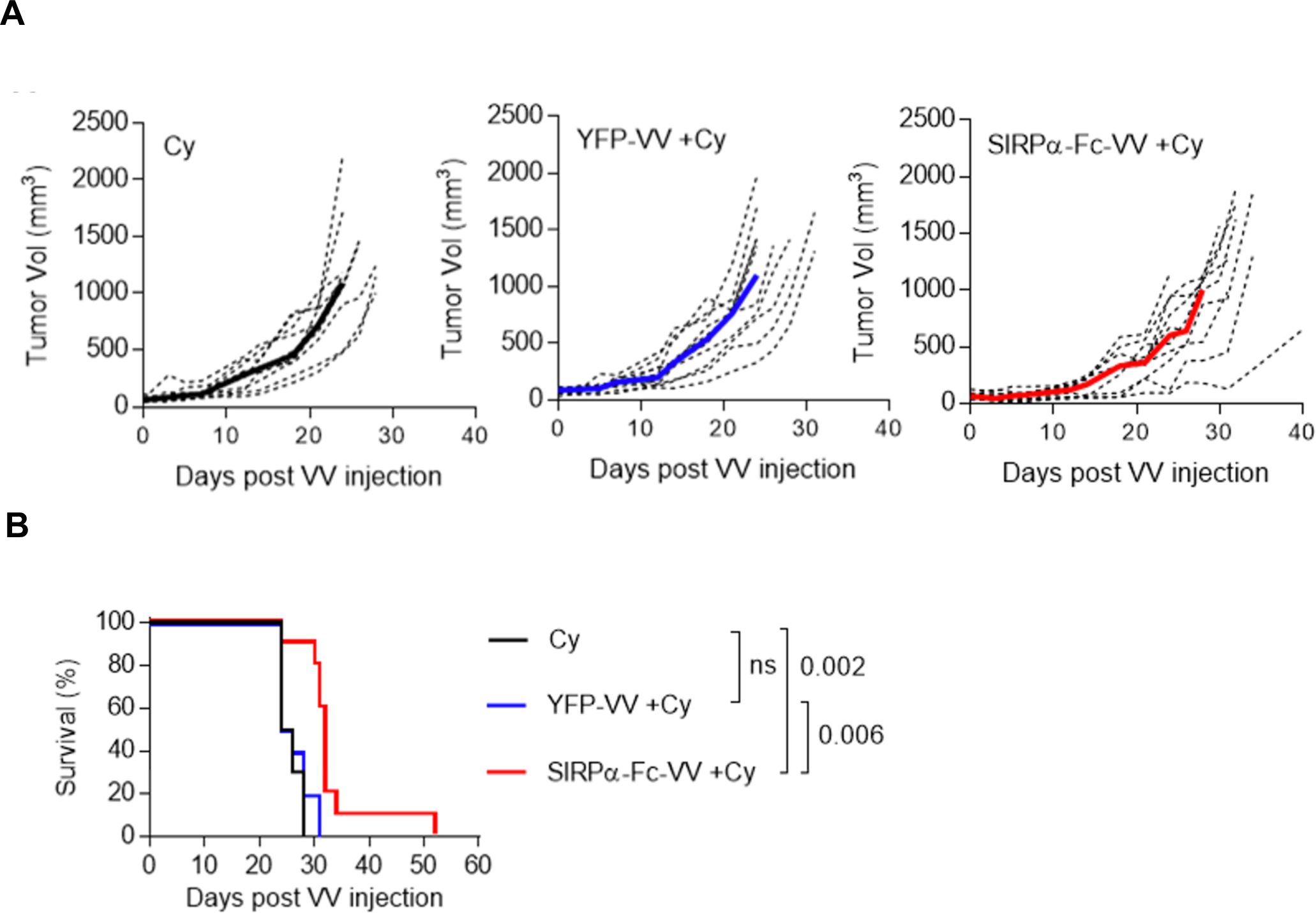
C57BL/6 mice were injected with 5×105 F420 cells s.c. into the right flank. After 9 days mice (n=10 per group) were injected i.p. with 250mg/kg cytoxan. 48 hours later, tumors were injected i.t. with 1×107 PFU of SIRPα-Fc-VV or YFP-VV. PBS-injected tumors served as controls (Co). Tumor size was monitored with caliper measurements. (A) Tumor volume (dotted lines: individual mice; solid line: median). (B) Kaplan-Meier survival curves. The Wilcoxon test with Holm method for adjustment of multiple comparisons was used to determine significance.
DISCUSSION
Here we describe the generation of an oncolytic VV genetically modified to express SIRPα-Fc, a chimeric molecule that blocks CD47 and opsonizes tumor cells. We demonstrate that expression of SIRPα-Fc does not interfere with the oncolytic activity of VV, and that SIRPα-Fc-VV redirects M1 and M2 macrophages to tumor cells. In vivo, SIRPα-Fc-VV had improved antitumor activity in comparison to YFP-VV and SIRPα-VV, which was further enhanced by administering cytoxan prior to VV injection.
Oncolytic viruses, including VV, HSV, and Adv are actively being explored as cancer therapeutics for solid tumors and brain tumors.1,3 Studies have highlighted that besides viral replication, activation of the immune system is critical for their antitumor activity.37,38 Oncolytic viruses have been genetically modified to express an array of immune stimulatory molecules including costimulatory molecules, cytokines, bispecific antibodies to activate T cells, and/or antibodies that block T-cell inhibitory molecules such as PD-1 or PD-L1.10–12,39,40 The benefit of arming oncolytic viruses with GM-CSF has been extensively evaluated in clinical studies, leading to the first in class FDA approval of a GM-CSF encoding HSV for melanoma.5,7 Here, we explored for the first time if genetically engineering oncolytic viruses to block CD47, an immune check point for macrophages, enhances their antitumor activity.
SIRPα and SIRPα-Fc encoded by VVs bound to CD47 as judged by interfering with CD47 detection by FACS analysis. However, SIRPα-Fc was more effective in blocking CD47 detection than SIRPα. This difference is most likely explained by the fact that SIRPα-Fc is larger than SIRPα (365 vs 147 amino acids); moreover, the Fc domain allows for dimer formation. Only SIRPα-Fc induced phagocytosis of tumor cells by macrophages, which confirms findings by others that not only blocking of CD47 but also opsonization is critical for macrophages to phagocytose tumor cells.22
M1 and M2 macrophages were both effective in killing SIRPα-Fc-positive tumor cells. In our in vitro studies we focused on OV10–315; clearly, additional studies are needed to expand our findings to other tumor cells. Other investigators have reported that M1 macrophages phagocytose glioblastoma cells, which had been incubated with a CD47-blocking MAb, more efficiently than M2 macrophages.41,42 Consistent with our findings, we found both M1 and M2 macrophages expressed Fc receptors (CD16, CD32, CD64; Figure S4). Differences between both studies is most likely due to the used macrophage generation/polarization protocol and/or target cells, and further studies are needed to reconcile these contradictory findings.
We evaluated the antitumor activity of YFP-VV, SIRPα-VV, and/or SIRPα-Fc-VV in xenograft and immune competent models similar to one previous report in which an oncolytic F35/F5 adenovirus was evaluated that was armed with a SIRPα-Fc fusion protein.43 In the SCID-Bg xenograft model, YFP-VV and SIRPα-Fc-VV had similar antitumor activity. However, mice died of disseminated VV infection. The same strain of VV (vvDD) has been evaluated by others in athymic nude mice xenograft models without the development of disseminated VV infection.32,44,45 The absence of functional NK cells in SCID-Bg mice in comparison to athymic nude mice is most likely responsible for our finding, since NK cells are critical for the control of poxvirus infections.46 YFP-VV-injected mice succumbed significantly earlier to disseminated VV infection than mice treated with SIRPα-Fc-VV. This difference might be due to the presence of functional macrophages in SCID-Bg mice.47 Foremost, our results suggests that preclinical studies with oncolytic viruses should, whenever possible, be conducted in immune competent animal models to model the interactions between tumor cells, virus, and the resident immune system.
In our immune competent model, intratumoral injection of YFP-VV or SIRPα-VV had no significant antitumor activity in contrast to SIRPα-Fc-VV. However, the antitumor activity of SIRPα-Fc-VV was transient. We therefore analyzed the immune cell infiltrate in control tumors, and post YFP-VV or SIRPα-Fc-VV injection. For these experiments we injected larger tumors (~300 mm3) versus 50–100 mm3 for the conducted efficacy experiments and therefore increased the VV dose (1×108 vs 1×107 PFU). T cells, M2 macrophages, granulocytes, and monocytes were present in control tumors. The percentage of M2 macrophages and monocytes increased significantly after VV injection with no difference between YFP-VV and SIRPα-Fc-VV. These results mirror the findings of others, indicating that VV increases myeloid cell infiltration.12 However, the increase in antitumor activity we observed was limited. To investigator potential mechanisms of therapeutic failure, we performed one additional experiment in which unmodified F420 cells or F420 cells genetically modified to express SIRPα or SIRPα-Fc were injected into C57BL/6 mice. While 10/10 mice injected with unmodified F420 cells developed tumors, only 2/10 with F420/SIRPα, and 0/10 with SIRPα-Fc (Figure S5). These results argue that VV spread through tumors or expression of the inserted transgene is most likely limiting its efficacy. Since we performed the analysis 2 days post VV injection, we did not observe an increase in the percentage of intratumoral T cells. We are planning to perform a more comprehensive time course to evaluate changes in the immune cell infiltrate post VV injection in the future.
The injection of cytoxan is a commonly used strategy to deplete inhibitory immune cells prior to the administration of biological-based therapies in patients and preclinical models.49–53 Cytoxan belongs to class of chemotherapeutic agents called alkylating agents, and has activity not only against immune cells but also cancer cells. Indeed, it is used in the clinic to treat patients with osteosarcoma.54,55 Thus, it was not unexpected that administration of cytoxan decreased tumor growth in our model. Combining SIRPα-Fc-VV with cytoxan also enhanced the antitumor activity in our model. However, cytoxan as a single agent also had antitumor activity. Thus, future studies are needed to determine if cytoxan also had an effect on immune cells, potentially increasing VV replication and production of SIRPα-Fc-VV. While our study demonstrated that arming VV with SIRPα-Fc enhances their antitumor activity, tumors eventually progressed. This is similar to results by other investigators that have armed VVs with single chain variable fragment (scFv) to block PD-1, the chemokine CXCL11, or 41BBL.11,39,56 Limited efficacy is most likely multifactorial and include limited intratumoral spread of oncolytic viruses including VV, and the immunosuppressive tumor microenvironment that inhibits immune cells that are activated by armed VVs.48 These barriers can most likely be addressed by engineering VVs to express high levels of transgenes and inserting multiple transgenes into the VV genome to activate the immune systems. Since VVs can hold large DNA fragments (up to 25kb), it should be feasible to generate VVs that are armed with several molecules to activate the immune system. In this regard, combining PD-1 blockade with SIRPα-Fc seems particular promising based on the recent finding that TAMs express high levels of PD-1.57 In addition, inserting cytokine genes such as IL-12 or IL-15 could change the immunosuppressive tumor microenvironment into a pro-inflammatory environment bolstering the effect of SIRPα-Fc.58,59
In conclusion, our results demonstrate that CD47 blockade with a VV encoding SIRPα-Fc is feasible, and redirects macrophages to tumor cells for therapeutic benefit. While we focused here on VV and osteosarcoma, our approach is applicable not only to other oncolytic viruses, but also to a broad range of tumors that express CD47.
Supplementary Material
CLINICAL IMPLICATIONS.
The outcome for patients with recurrent/refractory sarcoma remains poor. Here we demonstrate that arming oncolytic vaccinia virus with SIRPα-Fc enhances their antitumor activity warranting further active preclinical exploration of our modified approach to oncolytic virotherapy and future early phase clinical testing.
ACKNOWLEDGEMENTS
We thank the members of the Center for Cell and Gene Therapy at Baylor College of Medicine for discussion and assistance, and Drs. David Bartlett, Eugenie Kleinerman, Serge Fuchs, and William Frazier for providing cell lines or reagents detailed in the Material and Methods section. This work was supported by National Institute of Health (NIH) grants F30CA203270-02, T32DK060445, and P30CA125123, and the Alex Lemonade Stand Foundation. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
CONFLICTS OF INTEREST
FC, SG, and XTS have patent applications in the fields of immunotherapy, cancer gene therapy, and/or oncolytics for cancer. XTS is the Founding CEO of Icell Kealex Therapeutics.
ETHICS STATEMENT
The authors confirm that the ethical policies of the journal, as noted on the journal’s author guidelines page, have been adhered to. No ethical approval was required as this is a review article with no original research data.
REFERENCES
- 1.Russell SJ, Peng KW. Oncolytic Virotherapy: A Contest between Apples and Oranges. Molecular therapy : the journal of the American Society of Gene Therapy. 2017;25(5):1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nature reviews Cancer. 2009;9(1):64–71. [DOI] [PubMed] [Google Scholar]
- 3.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nature biotechnology. 2012;30(7):658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mell LK, Brumund KT, Daniels GA, et al. Phase I Trial of Intravenous Oncolytic Vaccinia Virus (GL-ONC1) with Cisplatin and Radiotherapy in Patients with Locoregionally Advanced Head and Neck Carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(19):5696–5702. [DOI] [PubMed] [Google Scholar]
- 5.Chesney J, Puzanov I, Collichio F, et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2017:JCO2017737379. [DOI] [PMC free article] [PubMed]
- 6.Downs-Canner S, Guo ZS, Ravindranathan R, et al. Phase 1 Study of Intravenous Oncolytic Poxvirus (vvDD) in Patients With Advanced Solid Cancers. Molecular therapy : the journal of the American Society of Gene Therapy. 2016;24(8):1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rehman H, Silk AW, Kane MP, Kaufman HL. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. Journal for immunotherapy of cancer. 2016;4:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cripe TP, Ngo MC, Geller JI, et al. Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Molecular therapy : the journal of the American Society of Gene Therapy. 2015;23(3):602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breitbach CJ, Moon A, Burke J, Hwang TH, Kirn DH. A Phase 2, Open-Label, Randomized Study of Pexa-Vec (JX-594) Administered by Intratumoral Injection in Patients with Unresectable Primary Hepatocellular Carcinoma. Methods in molecular biology. 2015;1317:343–357. [DOI] [PubMed] [Google Scholar]
- 10.Chard LS, Maniati E, Wang P, et al. A vaccinia virus armed with interleukin-10 is a promising therapeutic agent for treatment of murine pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(2):405–416. [DOI] [PubMed] [Google Scholar]
- 11.Liu Z, Ravindranathan R, Li J, Kalinski P, Guo ZS, Bartlett DL. CXCL11-Armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. Oncoimmunology. 2016;5(3):e1091554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Z, Ravindranathan R, Kalinski P, Guo ZS, Bartlett DL. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nature communications. 2017;8:14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Cao X. The origin and function of tumor-associated macrophages. Cellular & molecular immunology. 2015;12(1):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denton NL, Chen CY, Scott TR, Cripe TP. Tumor-Associated Macrophages in Oncolytic Virotherapy: Friend or Foe? Biomedicines. 2016;4(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu JF, Pan XH, Zhang SJ, et al. CD47 blockade inhibits tumor progression human osteosarcoma in xenograft models. Oncotarget. 2015;6(27):23662–23670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willingham SB, Volkmer JP, Gentles AJ, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(17):6662–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Logtenberg MEW, Scheeren FA, Schumacher TN. The CD47-SIRPalpha Immune Checkpoint. Immunity. 2020;52(5):742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jalil AR, Andrechak JC, Discher DE. Macrophage checkpoint blockade: results from initial clinical trials, binding analyses, and CD47-SIRPalpha structure-function. Antib Ther 2020;3(2):80–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J, Wang L, Zhao F, et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PloS one. 2015;10(9):e0137345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petrova PS, Viller NN, Wong M, et al. TTI-621 (SIRPalphaFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(4):1068–1079. [DOI] [PubMed] [Google Scholar]
- 22.Weiskopf K, Ring AM, Ho CC, et al. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341(6141):88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Advani R, Flinn I, Popplewell L, et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. The New England journal of medicine. 2018;379(18):1711–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Durfee RA, Mohammed M, Luu HH. Review of Osteosarcoma and Current Management. Rheumatology and therapy. 2016;3(2):221–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang KJ, Yin XF, Yang YQ, et al. A Potent In Vivo Antitumor Efficacy of Novel Recombinant Type I Interferon. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(8):2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jia SF, Worth LL, Kleinerman ES. A nude mouse model of human osteosarcoma lung metastases for evaluating new therapeutic strategies. Clinical & experimental metastasis. 1999;17(6):501–506. [DOI] [PubMed] [Google Scholar]
- 27.Ahmed N, Salsman VS, Yvon E, et al. Immunotherapy for osteosarcoma: genetic modification of T cells overcomes low levels of tumor antigen expression. MolTher. 2009;17(10):1779–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonald JF, Zheleznyak A, Frazier WA. Cholesterol-independent interactions with CD47 enhance alphavbeta3 avidity. The Journal of biological chemistry. 2004;279(17):17301–17311. [DOI] [PubMed] [Google Scholar]
- 29.Zhao S, Kurenbekova L, Gao Y, et al. NKD2, a negative regulator of Wnt signaling, suppresses tumor growth and metastasis in osteosarcoma. Oncogene. 2015;34(39):5069–5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Semeraro M, Rusakiewicz S, Minard-Colin V, et al. Clinical impact of the NKp30/B7-H6 axis in high-risk neuroblastoma patients. Science translational medicine. 2015;7(283):283ra255. [DOI] [PubMed] [Google Scholar]
- 31.Yang Z, Reynolds SE, Martens CA, Bruno DP, Porcella SF, Moss B. Expression profiling of the intermediate and late stages of poxvirus replication. Journal of virology. 2011;85(19):9899–9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCart JA, Ward JM, Lee J, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer research. 2001;61(24):8751–8757. [PubMed] [Google Scholar]
- 33.Rintoul JL, Wang J, Gammon DB, et al. A selectable and excisable marker system for the rapid creation of recombinant poxviruses. PloS one. 2011;6(9):e24643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szoor A, Vaidya A, Velasquez MP, et al. T Cell-Activating Mesenchymal Stem Cells as a Biotherapeutic for HCC. Molecular therapy oncolytics. 2017;6:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible activation of MyD88 and CD40 in CAR T-cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discovery 2017; 7(11):1306–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X, Pu Y, Cron K, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nature medicine. 2015;21(10):1209–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sampath P, Li J, Hou W, Chen H, Bartlett DL, Thorne SH. Crosstalk between immune cell and oncolytic vaccinia therapy enhances tumor trafficking and antitumor effects. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21(3):620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ottolino-Perry K, Diallo JS, Lichty BD, Bell JC, McCart JA. Intelligent design: combination therapy with oncolytic viruses. Molecular therapy : the journal of the American Society of Gene Therapy. 2010;18(2):251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kleinpeter P, Fend L, Thioudellet C, et al. Vectorization in an oncolytic vaccinia virus of an antibody, a Fab and a scFv against programmed cell death −1 (PD-1) allows their intratumoral delivery and an improved tumor-growth inhibition. Oncoimmunology. 2016;5(10):e1220467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu F, Wang X, Guo ZS, Bartlett DL, Gottschalk SM, Song XT. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Molecular therapy : the journal of the American Society of Gene Therapy. 2014;22(1):102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang M, Hutter G, Kahn SA, et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages In Vivo. PloS one. 2016;11(4):e0153550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gholamin S, Mitra SS, Feroze AH, et al. Disrupting the CD47-SIRPalpha anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Science translational medicine. 2017;9(381). [DOI] [PubMed] [Google Scholar]
- 43.Huang Y, Lv SQ, Liu PY, et al. A SIRPalpha-Fc fusion protein enhances the antitumor effect of oncolytic adenovirus against ovarian cancer. Mol Oncol. 2020;14(3):657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guse K, Sloniecka M, Diaconu I, et al. Antiangiogenic arming of an oncolytic vaccinia virus enhances antitumor efficacy in renal cell cancer models. Journal of virology. 2010;84(2):856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chalikonda S, Kivlen MH, O’Malley ME, et al. Oncolytic virotherapy for ovarian carcinomatosis using a replication-selective vaccinia virus armed with a yeast cytosine deaminase gene. Cancer gene therapy. 2008;15(2):115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez J, Huang X, Yang Y. Direct TLR2 signaling is critical for NK cell activation and function in response to vaccinia viral infection. PLoS pathogens. 2010;6(3):e1000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia Z, Taylor PR, Locklin RM, Gordon S, Cui Z, Triffitt JT. Innate immune response to human bone marrow fibroblastic cell implantation in CB17 scid/beige mice. Journal of cellular biochemistry. 2006;98(4):966–980. [DOI] [PubMed] [Google Scholar]
- 48.Zheng M, Huang J, Tong A, Yang H. Oncolytic Viruses for Cancer Therapy: Barriers and Recent Advances. Mol Ther Oncolytics. 2019;15:234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao J, Cao Y, Lei Z, Yang Z, Zhang B, Huang B. Selective depletion of CD4+CD25+Foxp3+ regulatory T cells by low-dose cyclophosphamide is explained by reduced intracellular ATP levels. Cancer research. 2010;70(12):4850–4858. [DOI] [PubMed] [Google Scholar]
- 50.Bracci L, Moschella F, Sestili P, et al. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(2 Pt 1):644–653. [DOI] [PubMed] [Google Scholar]
- 51.Li H, Zeng Z, Fu X, Zhang X. Coadministration of a herpes simplex virus-2 based oncolytic virus and cyclophosphamide produces a synergistic antitumor effect and enhances tumor-specific immune responses. Cancer research. 2007;67(16):7850–7855. [DOI] [PubMed] [Google Scholar]
- 52.Fulci G, Breymann L, Gianni D, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(34):12873–12878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng KW, Myers R, Greenslade A, et al. Using clinically approved cyclophosphamide regimens to control the humoral immune response to oncolytic viruses. Gene therapy. 2013;20(3):255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luetke A, Meyers PA, Lewis I, Juergens H. Osteosarcoma treatment - Where do we stand? A state of the art review. Cancer Treat Rev. 2014;40(4):523–32. [DOI] [PubMed] [Google Scholar]
- 55.Lamplot JD, Denduluri S, Qin J, et al. The Current and Future Therapies for Human Osteosarcoma. Curr Cancer Ther Rev. 2013;9(1):55–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim HS, Kim-Schulze S, Kim DW, Kaufman HL. Host lymphodepletion enhances the therapeutic activity of an oncolytic vaccinia virus expressing 4–1BB ligand. Cancer research. 2009;69(21):8516–8525. [DOI] [PubMed] [Google Scholar]
- 57.Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang B, Guo ZS, Bartlett DL, et al. Synergistic Combination of Oncolytic Virotherapy and Immunotherapy for Glioma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2020;26(9):2216–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ge Y, Wang H, Ren J, et al. Oncolytic vaccinia virus delivering tethered IL-12 enhances antitumor effects with improved safety. J Immunother Cancer. 2020;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.