

Abstract
Objectives
Efforts to develop and deploy effective vaccines against severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) continue at pace. Here, we describe rational antigen design through to manufacturability and vaccine efficacy of a prefusion‐stabilised spike (S) protein, Sclamp, in combination with the licensed adjuvant MF59 ‘MF59C.1’ (Seqirus, Parkville, Australia).
Methods
A panel recombinant Sclamp proteins were produced in Chinese hamster ovary and screened in vitro to select a lead vaccine candidate. The structure of this antigen was determined by cryo‐electron microscopy and assessed in mouse immunogenicity studies, hamster challenge studies and safety and toxicology studies in rat.
Results
In mice, the Sclamp vaccine elicits high levels of neutralising antibodies, as well as broadly reactive and polyfunctional S‐specific CD4+ and cytotoxic CD8+ T cells in vivo. In the Syrian hamster challenge model (n = 70), vaccination results in reduced viral load within the lung, protection from pulmonary disease and decreased viral shedding in daily throat swabs which correlated strongly with the neutralising antibody level.
Conclusion
The SARS‐CoV‐2 Sclamp vaccine candidate is compatible with large‐scale commercial manufacture, stable at 2–8°C. When formulated with MF59 adjuvant, it elicits neutralising antibodies and T‐cell responses and provides protection in animal challenge models.
Keywords: Molecular Clamp, neutralising antibodies, polyfunctional T cells, rapid response, SARS‐CoV‐2, subunit vaccine
We describe preclinical development of a prefusion stabilized spike protein subunit vaccine for SARS‐CoV‐2. Results include structural resolution by cryo‐electron microscopy, mouse immunogenicity and a hamster protection study.

Introduction
The scientific community, including critical industry and academic partnerships, has embarked on an unprecedented race to develop and produce effective COVID‐19 vaccine(s) for global use. 1 As of February 2021, there are 74 individual vaccine candidates in clinical trials and many more in preclinical development. 2 At the beginning of 2020, our vaccine platform, termed the Molecular Clamp, was at an early stage of development as a rapid response pipeline, supported through a 2018 call for platform technologies to rapidly respond to Disease X by the Coalition for Epidemic Preparedness Innovations (CEPI). Acting in response to the outbreak, and with support from CEPI, we immediately refocused our efforts towards the rapid development of a molecular clamp stabilised vaccine against severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), the causative agent of COVID‐19. This manuscript describes the entire preclinical development of our vaccine programme (timeline provided in Supplementary figure 1), which is now advancing in partnership with CSL for further manufacturing and pivotal stage clinical development.
Vaccine candidates in clinical trials in development include inactivated viruses, viral vectored approaches, nucleic acid‐based vaccines and subunit vaccines. As each platform has intrinsic strengths and weaknesses around aspects of manufacturability, ease of transport, safety and efficacy, it is crucial that multiple programmes are taken forward to allow selection of the most favorable approaches to meet the global challenge of the COVID‐19 pandemic. Adjuvanted protein‐based, subunit vaccines represent an important class of vaccine and its development has somewhat been lagged compared with the other platforms owing primarily to the need to tailor the manufacturing process to each antigen. However, protein vaccines paired with clinically validated adjuvants remain an attractive option to counter a global pandemic, as such pairings have already been proven to confer a tailored immune response to result in efficacy with an excellent safety profile, and can therefore be utilised with existing global manufacturing capacity to enable rapid production scale‐up to the levels required to counter a pandemic threat.
To date, the most advanced subunit programme described is the Novavax candidate produced via insect cells as a detergent‐stabilised nanoparticle in combination with a saponin‐based adjuvant. 3 Our candidate vaccine, SARS‐CoV‐2 Sclamp, consists of the recombinant viral S glycoprotein stabilised in its prefusion trimeric form, through the incorporation of the ‘Molecular Clamp’ stabilisation domain, and produced in mammalian cells (Chinese hamster ovary). 4 Stabilisation of the prefusion antigen is a major goal of next‐generation vaccines that target enveloped viruses. This approach ensures that vaccines elicit the production of specific antibodies that recognise a wider array of epitopes, including conformational epitopes, that are displayed on the virion surface. 5 , 6 Prefusion‐stabilised spike protein from the related Middle East respiratory syndrome coronavirus (MERS‐CoV) has previously been shown to elicit a higher level of neutralising antibodies compared with the native MERS‐CoV S. 7
Severe acute respiratory syndrome coronavirus 2 Sclamp is one of seven candidate subunit vaccines comprising the entire ectodomain of spike that is currently being evaluated in clinical trials 2 . Similar subunit vaccines being evaluated include alternate stabilisation methods including site‐directed mutagenesis of two proline residues, 3 , 8 , 9 , 10 or the incorporation of an alternate trimerisation domain based on human collagen. 11 The Molecular Clamp trimerisation domain we have employed comprises heptad repeat regions 1 and 2 (HR1/2) of glycoprotein 41 (gp41) (amino acids 540–576 and 619–656) from human immunodeficiency virus 1 (HIV‐1). These sequences encode a common fold found in many viral families (Supplementary figure 2) and assemble into an extremely stable six‐helix bundle that in the native context confers stability to the post‐fusion form of class I viral proteins. We have repurposed this feature and have reengineered it as a modular tag capable of stabilising the prefusion form of a wide range of trimeric viral antigens. We have validated the platform against established human pathogens including respiratory syncytial virus (RSV) and influenza, where it is able to stabilise the prefusion conformation without the need for additional site‐specific mutations common to other stabilisation strategies (Supplementary figure 2d). 12 , 13 Furthermore, we utilise an antibody to the molecular clamp to enable immunoaffinity purification, thereby exploiting a consistent methodology to purify novel antigens as a rapid response platform to pandemic threats. 14
In the ongoing phase I clinical trial investigating SARS‐CoV‐2 Sclamp, initial findings have demonstrated favorable safety and the induction of a strong neutralising immune response. 15 However, the HIV sequences included within the Molecular Clamp were also found to stimulate an immune response that could cross‐react with some HIV diagnostic tests. To avoid the issue of HIV diagnostic interference, we are in the process of re‐engineering the sequences used in the Molecular Clamp trimerisation; however, herein we describe the rapid development of the Sclamp vaccine, including antigen design and characterisation, production, and animal immunogenicity, safety and efficacy studies which permitted the transition to a human Phase I trial. These results serve as an important proof‐of‐principle demonstration of the underlying platform technology.
Results
Selection of the prefusion conformed Sclamp lead candidate
The virus genomic sequence first became available on 12 January 2020, 16 and we immediately commenced development of a candidate subunit vaccine utilising the Molecular Clamp platform (a timeline of research and development and an overview of the Molecular Clamp platform, including two proof‐of‐concept examples are included in Supplementary figures 1 and 2). Without a high‐resolution structure available, the candidate screen focussed on three main features of the molecule: (1) the signal peptide, (2) modification of the furin cleavage site and (3) fusion of the molecular clamp sequence. Within 34 days, we had expressed and screened > 200 antigens incorporating truncations and modifications at these key sites to identify a lead candidate that demonstrated suitable levels of trimer expression while maintaining affinity to the S‐specific monoclonal antibody (mAb), CR3022 17 (Figure 1a–c and Supplementary table 1). Our screen revealed that the most critical element for expression yield was modification of the furin‐like cleavage site. Without modification of this site, no spike protein could be recovered from transfected culture supernatants (Figure 1a). Given that early spike comparative sequence analysis against related SARS‐like viruses revealed an unusual extended and poly‐basic furin 1 site, we examined both site‐targeted mutations and larger deletions of the whole site. While most of the constructs showed similar expression to the single P1 site R685A mutation, two constructs that utilised a deletion of aa680–690 and either a GSG or GGSGG flexible linker were observed to express significantly higher yields. We examined a second generation of these constructs, screening through both the entire 80 aa membrane proximal region and testing alternative signal sequences, but determined that the original length and native signal sequence were optimal. The selected lead construct incorporated the S protein native signal peptide, replacement of aa680–690 with a glycine linker (GSG), truncation at aa1204 and followed by the Molecular Clamp coding sequence as an extension of the carboxy‐terminal sequence. This construct was originally termed M1GSG and then subsequently referred to as ‘Sclamp’ (Supplementary figure 3 and Supplementary table 1).
Figure 1.
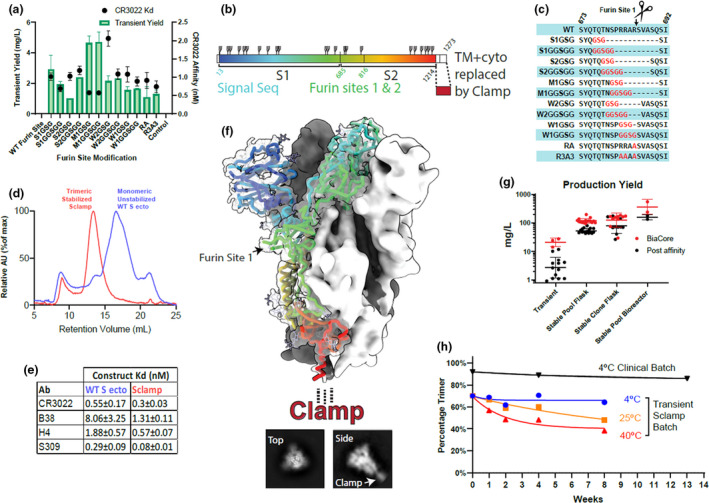
Antigen design and analysis. (a) In vitro screening of S1/S2 linker modifications for yield and CR3022 affinity (K D), untransfected cell supernatant included as control. (b) Linear representation of recombinant spike antigen. (c) Sequence details for the furin site‐modified constructs assayed in a. (d) Size‐exclusion chromatography analysis of non‐stabilised wild‐type spike and Sclamp. (e) Antibody binding affinities (K D) for unstabilised vs Sclamp. (f) Cryo‐TEM reconstruction of antigen structure, fitted with prefusion spike structure PDB:6VXX 10 shown in rainbow cartoon for one S monomer. Representative top and side‐on 2D classes shown below, with a density for clamp indicated. (g) Production yield from CHO cell culture transient expression, stable pools and clones in flasks or bioreactors. Red—antigen concentration in cell culture supernatant estimated by BiaCore. Black—protein recovery following immunoaffinity purification as measured by absorbance at 280 nm. (h) Percentage trimer analysis of stability assessed using SE‐HPLC for the development batch and clinical trial batch of Sclamp following incubation of the protein at 4, 25 or 40°C for up to 13 weeks.
We utilised several, patient‐derived, SARS‐CoV‐2 cross‐reactive mAbs (CR3022, 17 309, 18 H4 and B38 19 ), to demonstrate high‐affinity binding to the Sclamp protein as an indicator of the required conformational integrity (CR3022 kD = 0.3 nm; S309 kD = 0.08 nm; H4 kD = 0.6 nm; B38 kD = 1.3 nm). Without stabilisation via the Molecular Clamp, or an alternate methodology such as the insertion of structure stabilising mutations, 8 the spike protein dissociates into monomers and the binding affinity of spike conformation‐specific monoclonal antibodies to the monomers is reduced (Figure 1d and e). Sclamp was also shown to bind to soluble recombinant angiotensin‐converting enzyme 2 (ACE2) receptor (kD = 56 nm ± 5).
Using size‐exclusion high‐performance liquid chromatography (SE‐HPLC), we demonstrated that the expressed antigen separates into two peaks both of which have sizes corresponding to trimers with slightly different hydrodynamic radiuses and that exposure to varying pH or temperature can drive transition between the two forms (Supplementary figure 4). The structure of the smaller trimer peak was determined to 5 Å resolution (Fourier shell correlation 0.143) by cryo‐electron microscopy (cryo‐EM) and found to be consistent with the previously defined closed trimeric prefusion conformation of SARS‐CoV‐2 spike (Figure 1f and Supplementary figure 5). 8 No clamp density was resolved in the final structure, likely due to flexibility in the long C‐terminal spike stalk that is disordered in published structures, 10 , 20 and has been shown to be highly flexible in native virions. 21 However, the clamp could be readily identified in 2D classification images of the cryo‐EM particle set (Figure 1f, inset). The structure of the larger trimer peak was unable to be resolved by cryo‐EM, but it is consistent with an open prefusion conformation previously defined, 8 and reports that SARS‐CoV‐2 spike exists in an equilibrium between these two conformations. 22
Transient gene expression in CHO cells was initially utilised to allow rapid screening of antigen design panels with CHO stable cell lines being established following lead candidate selection. Transitioning to CHO stable pools and using a fed‐batch bioprocess resulted in increased expression yields in shaker flasks of 100–150 mg L–1 and in bioreactors up to 500 mg L–1 (Figure 1g). This level of mammalian cell‐based protein expression has the potential to generate many millions of doses per bioreactor run using industry standard mammalian cell bioprocessing facilities. Subsequent studies have shown that Sclamp expressed in the CHO stable pools is decorated with additional sialylation, likely contributing to maximising the trimer form (data not shown). Furthermore, a downstream refined bioprocess was able to achieve batch‐to‐batch consistency (data not shown). We also confirmed that the trimeric conformation of Sclamp is unaffected by storage at 4°C for 3 months and is well retained even after prolonged exposure to heat stress (Figure 1h and Supplementary figure 6).
MF59‐adjuvanted Sclamp vaccination elicited neutralising antibodies against SARS‐CoV‐2
Purified protein‐based vaccines require adjuvant co‐administration to enhance immunogenicity and to reduce the total amount of antigen required to provide protective immunity. 23 The squalene‐oil‐in‐water adjuvant MF59® (Seqirus, Parkville, Australia) was selected for use with our vaccine because of its well‐established safety record, use in existing licensed vaccines and its ability to stimulate a balanced T helper (Th1/Th2) cell response. 24 Alhydrogel (Croda) was included as a comparator adjuvant. BALB/c mice received intramuscular (IM) injections of PBS (placebo) or two doses of Sclamp with or without Alhydrogel or MF59 (Figure 2a). Vaccinated mice developed a robust antigen‐specific IgG response after a single dose with either adjuvant, which was boosted following the second dose (Figure 2b).
Figure 2.
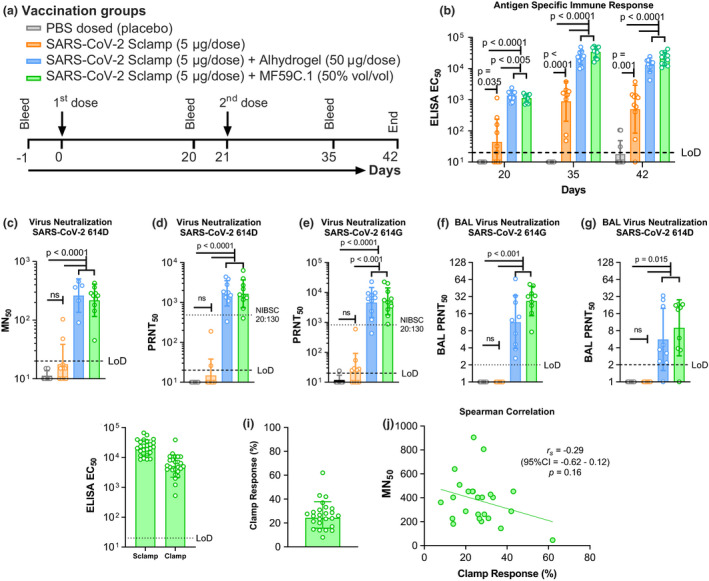
The antibody response following SARS‐CoV‐2 Sclamp vaccination in BALB/c mice (data from two of five repeat experiments shown). (a) Prime/boost vaccination and bleed schedule for the study. (b) SARS‐CoV‐2 Sclamp‐specific IgG EC50 titre (reciprocal EC50) in vaccinated mice (n = 10) 20, 35 and 42 days following IM injection of the first dose. (c) Day 42 serum MN titre against infectious SARS‐CoV‐2 614D as evaluated by MN50 assay (n = 10). (d) Day 42 serum MN titre against SARS‐CoV‐2 614D as evaluated by PRNT50 assay (n = 10). (e) Day 42 serum MN titre against SARS‐CoV‐2 614G as evaluated by PRNT50 assay (n = 10). (f) Day 42 BAL MN titre against SARS‐CoV‐2 614D as evaluated by PRNT50 assay (n = 10). (g) Day 42 BAL MN titre against SARS‐CoV‐2 614G as evaluated by PRNT50 assay. (h) Day 42 serum total Sclamp‐specific IgG EC50 titre and clamp‐specific IgG EC50 titre (n = 25). (i) Relative percentage of total IgG response directed to the clamp domain, and spike epitopes, calculated based on EC50 titres in h. (j) Spearman correlation between relative percentage clamp‐specific IgG and MN50 titre. P‐values were calculated following transformation of the neutralisation titres to log10 using (1) one‐way ANOVA with Tukey's multiple comparison post hoc test for normally distributed and homoscedastic data, (2) Welch's ANOVA with Games–Howell post hoc analysis for all heteroscedastic data and (3) the Kruskal–Wallis H‐test for non‐normally distributed and homoscedastic data sets.
Analysis of serum samples on day 42 showed that vaccination with two doses of Sclamp with either Alhydrogel or MF59 produced a strong serum neutralising antibody response against the matched strain of SARS‐CoV‐2 as assessed by a traditional microneutralisation (MN) assay (Figure 2c). 25 Further analysis of the sera and bronchoalveolar lavage (BAL) collected on day 42 from Alhydrogel‐ or MF59‐adjuvanted Sclamp‐vaccinated mice using plaque reduction neutralisation test (PRNT50) analysis showed that both 614D and the now dominant 614G SARS‐CoV‐2 variants 26 were similarly neutralised (Figure 2d–g). Serum PRNT50 analysis also included a WHO‐recommended reference serum (NIBSC code 20:130) 27 to allow benchmarking of the neutralising titre to other platforms. In this assay, sera from mice vaccinated with MF59‐adjuvanted Sclamp neutralised SARS‐CoV‐2 with a ~ 2‐fold higher geometric mean titre (GMT) compared with the reference serum (Figure 2d and e).
Clamp‐specific immune response was assessed at day 42 for 25 mice each receiving two doses of Sclamp with MF59 adjuvant (Figure 2h). Based on comparison of EC50 values in ELISA, we produced an estimate of the relative immune response directed against the spike ectodomain and the molecular clamp domain for each of 25 individual mice (Figure 2i). The geometric mean of these values suggests that 76% of IgG directed against the spike ectodomain (ranging between 38 and 92% for individual mice) and 24% of IgG to the molecular clamp domain. Interestingly, while the relative percentage of the immune response to the clamp itself varied between individual animals, this non‐spike specific response was not associated with a statistically significant correlation with MN50 titres (Figures 2j). In summary, the level of clamp‐specific response had no impact on the serum neutralisation response.
MF59‐adjuvanted Sclamp vaccination elicited robust S‐specific polyfunctional/cytotoxic CD4+ and CD8+ T‐cell immunity
We evaluated SARS‐CoV‐2 S‐specific CD4+ and CD8+ T‐cell responses in vivo using a fluorescent target array (FTA) analysis and complementary intracellular cytokine staining (ICS) analysis to determine type 1 vs type 2 immunity. 28 , 29 For the FTA analysis, the targets in the FTA were prepared following serial labelling with titrated concentrations of cell proliferation dye efluor™ 670 (CPD) and cell trace violet (CTV) that allowed the labelled cells to retain a unique, easily distinguishable fluorescent bar‐code following flow cytometry analysis (Figure 3a and b). This allowed us to accurately pulse each fluorescent bar‐coded cell population with 7 peptide pools that collectively span the SARS‐CoV‐2 S1–1226 sequence, S16–554 (S1), S673–1218 (S2), S1–1226 pool (Total) and the Peptivator array that comprise in silico mapped human immunodominant peptides (S304–338, S421–475, S492–519, S683–707, S741–770, S785–802 and S885–1273; Figure 3a and b). Following intravenous challenge of mice with the FTA, up‐regulation of CD69 on peptide‐pulsed B220+ FTA cells recovered from challenged mice is dependent on antigen‐specific CD4+ Th cell responses and the killing of cognate peptide‐pulsed targets is a result of cytotoxic T lymphocyte (CTL) responses in vivo. 30 , 31 Our findings in the current study revealed that Alhydrogel‐ and MF59‐adjuvanted regimens were more effective in eliciting S‐specific Th cell and CTL responses compared with Sclamp (Ag) only and placebo control groups with the MF59‐adjuvanted regimen being superior in eliciting the highest magnitude and the broadest S‐specific Th cell and CTL responses in vivo (Figure 3c and Supplementary tables 2 and 3). The superior immunogenicity of the MF59‐adjuvanted regimen was evident for Th cell responses to S185–362 (P2), S353–530 (P3), S521–698 (P4), S689–886 (P5), S857–1070 (P6), S1041–1226 (P7), S1, S2, total pool and Peptivator, but not for S1–194 (P1). CTL immunity followed a similar trend, but not with responses to P6 and P7.
Figure 3.
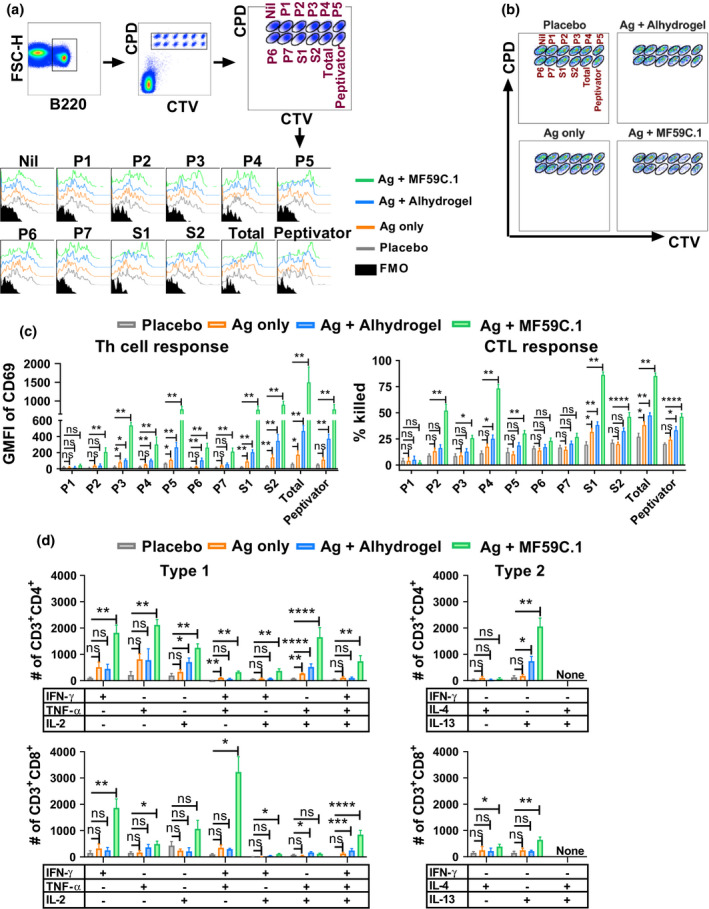
MF59‐adjuvanted SARS‐CoV‐2 Sclamp vaccination elicits potent T‐cell responses in mice (data from two separate experiments shown). (a) Representative flow cytometry plots show the gating strategy for analysis of S‐specific Th cell responses based on the up‐regulation of CD69 on peptide‐pulsed B220+ single cells in the FTA. BALB/c mice vaccinated as in Figure 2a were challenged 19 days after boost with the FTA comprising S peptide‐pulsed, CPD and CTV‐labelled targets. FMO—fluorescent minus one control. (b) Dot plots showing all targets recovered from a representative FTA‐challenged mouse which were used to analyse the S‐specific CTL responses as described in the Methods. (c) Bar graphs show the mean (n = 5) and SEM of the geometric mean fluorescent intensity (GMFI) of CD69 on gated B220+ cells and the percentage of peptide‐pulsed targets that were killed in the FTA. (d) Mean (n = 5) and SEM of the number of the indicated cytokine‐producing S1–1226‐specific CD4+ or CD8+ T cells. For the ICS analysis, splenocytes recovered 24 days following boost from mice prime/boost vaccinated at 2‐weekly intervals were stimulated for 16 h with the total pool. P‐values for all the comparisons are shown in Supplementary tables 2–5 and the P‐values shown indicate significance relative to placebo (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
The ICS analysis showed that the mice vaccinated with MF59‐adjuvanted Sclamp developed the highest number of S‐specific CD4+ or CD8+ T cells that expressed mono‐ or polyfunctional interferon (IFN)‐γ, tumor necrosis factor (TNF)‐α and/or interleukin (IL)‐2 (Figure 3d, Supplementary figure 7 and Supplementary tables 4 and 5). MF59‐ and Alhydrogel‐adjuvanted Sclamp‐vaccinated mice developed higher numbers of S‐specific CD4+ T cells that produced IL‐13 and a more subtle elevation in the numbers of S‐specific CD8+ T cells that produced IL‐4 or IL‐13 was also observed in the MF59‐adjuvanted Sclamp regimen compared with the placebo and/or Sclamp only (Figure 3d, and Supplementary figure 7 and Supplementary tables 4 and 5). Overall, using 2 independent T‐cell assays, we confirmed that MF59‐adjuvanted Sclamp vaccination elicited S‐specific CD4+ and CD8+ T‐cell responses with the highest magnitude and breadth of recognition encompassing type 1 (IFN‐γ, TNF‐α and IL‐2) and type 2 (IL‐4 and IL‐13) responses, albeit with the type 2 responses being less prominent compared with type 1.
Sclamp vaccination induced protection against pathogenic SARS‐CoV‐2 challenge
Given that vaccination with Sclamp adjuvanted with MF59 elicited the most robust antibody and T‐cell immunity, we further evaluated this regimen relative to parameters deemed important for safety evaluation (i.e. vaccine‐mediated enhancement of disease) and protection in the established hamster model of SARS‐CoV‐2 infection. 32 To investigate the possibility of vaccine‐enhanced respiratory disease, we completed two studies in which hamsters were challenged with intranasal (IN) inoculation of SARS‐CoV‐2 following either a single‐dose (Figure 4a) or a prime/boost vaccination regimen (Figure 5a). The challenge dose selected was based on the ability to induce disease and to be of relevance to natural exposure, rather than an artificially high dose as has been used in a range of non‐human primate (NHP) studies. 33 , 34 , 35 , 36 Both studies included a formalin‐inactivated virus combined with Alhydrogel (Croda) as a comparator based on the initial concern that vaccines that drive a Th2 response and/or a poorly neutralising response may result in immunopathology as has previously been observed for MERS‐CoV and SARS‐CoV. 37 , 38 Of note is that more recent studies have now shown that there is no compelling evidence for this undesired effect or antibody‐dependent enhancement of infection for inactivated SARS‐CoV‐2 vaccines adjuvanted with aluminium‐based adjuvants. 39 To further validate protection data following vaccination, we also included an additional group in the prime/boost study whereby hamsters were infected (intranasally) with SARS‐CoV‐2 and then allowed to recover over a 7‐week period prior to a second challenge.
Figure 4.
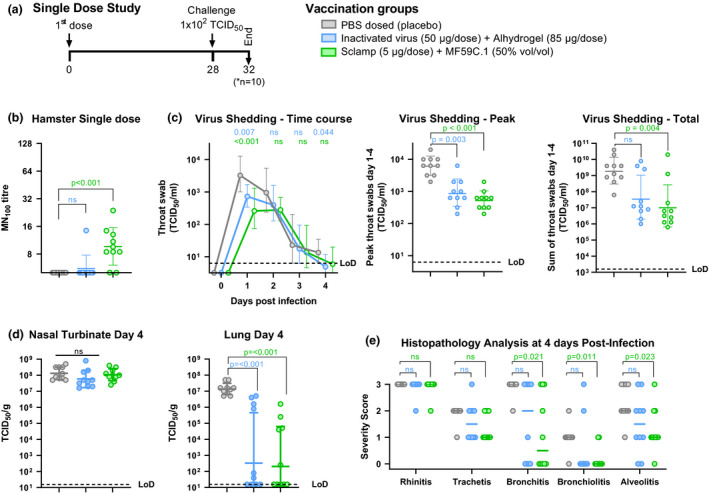
Protection of hamsters following a single dose of MF59‐adjuvanted Sclamp. (data from a single experiment shown). (a) Single‐dose study schedule. Syrian hamsters (n = 10/group) were vaccinated with a single dose of either a placebo, 50 μg inactivated virus with Alhydrogel or 5 μg SARS‐CoV‐2 Sclamp with MF59 prior to IN challenge with 100 TCID50 of SARS‐CoV‐2. (b) Average SARS‐CoV‐2 MN100 titre measured using hamster serum collected at day 21 and 28. (c) Kinetics of infectious viral load in daily throat swabs on days 1–4 post‐challenge (left panel), the peak virus titre from the kinetics analysis (middle panel), and the sum of virus titre for swabs collected on days 1–4 post‐challenge (right panel). (d) Viral loads in nasal turbinates and lung tissue at day 4 post‐challenge. (e) Severity score of inflammatory response based on histology at day 4 post‐challenge: 0 = no inflammatory cells, 1 = few inflammatory cells, 2 = moderate number of inflammatory cells and 3 = many inflammatory cells. P‐values were calculated using a Kruskal–Wallis ANOVA with Dunn's multiple comparison tests or a two‐way ANOVA with Dunnett's multiple comparison tests. Error bars show SD.
Figure 5.
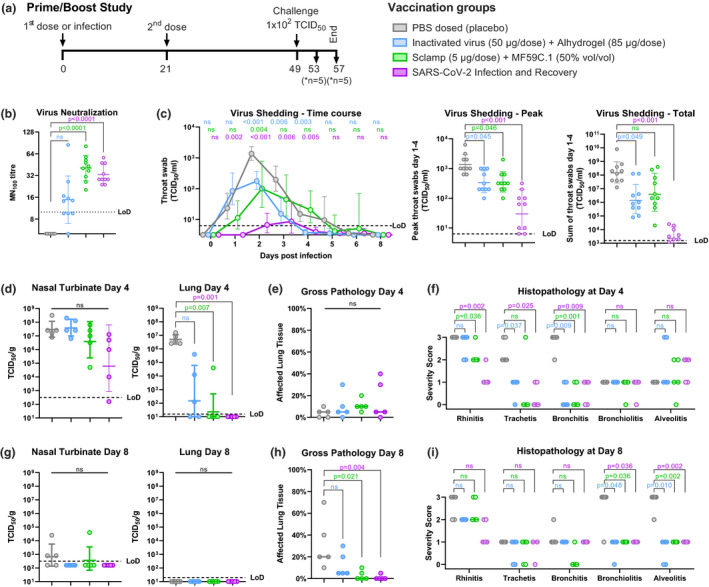
Protective outcomes following prime/boost vaccination (data from a single experiment shown). (a) Prime/boost study schedule. Syrian hamsters (n = 10/group) were vaccinated with two doses of either a placebo, 50 μg inactivated virus with Alhydrogel or 5 μg of SARS‐CoV‐2 Sclamp with MF59, or were infected with 102 TCID50 SARS‐CoV‐2 via IN administration and allowed to recover prior to challenge. (b) Average SARS‐CoV‐2 MN100 titre measured using serum collected at day 42 and 49. (c) Kinetics of infectious viral load in daily throat swabs on days 1–8 post‐challenge (left panel), the peak virus titre from the kinetics analysis (middle panel) and the sum of virus titre for swabs collected on days 1–4 post‐challenge (right panel). (d, g) Viral loads in nasal turbinate and lung tissue at day 4 and day 8 post‐challenge. (e, h) Extent of affected lung tissue damage as assessed by gross pathology at day 4 and day 8 post‐challenge. (f, i) Severity score of inflammatory response based on histology at day 4 and day 8 post‐challenge: 0 = no inflammatory cells, 1 = few inflammatory cells, 2 = moderate number of inflammatory cells and 3 = many inflammatory cells. P‐values were calculated using a Kruskal–Wallis ANOVA with Dunn's multiple comparison tests or a two‐way ANOVA with Dunnett's multiple comparison tests. Error bars show SD.
A single vaccination in hamsters with MF59‐adjuvanted Sclamp resulted in modest but statistically significant induction of neutralising antibodies (MN100 GMT = 9.6) while the inactivated virus did not (Figure 4b). Two doses of either vaccine elicited a more potent neutralising antibody response (MN100 GMT = 40.6), as did prior infection and recovery from SARS‐CoV‐2 infection (MN100 GMT = 33.1; Figure 5b). Note that the NIBSC convalescent human serum control (20:130) in this MN100 assay format has a GMT = 30 (results not shown).
The highly permissive nature of the hamster model was initially observed in a study that evaluated viral replication in daily throat swabs collected following IN challenge up to 4 days following a single dose and 8 days following a prime‐boost regimen (Figures 4c and 5c). These studies showed that infectious virus titre, as measured by TCID50, peaked at 1–2 days post‐challenge and then gradually decreased to undetectable levels between 5 and 8 days post‐challenge (Figures 4c and 5c). Vaccination with either one or two doses of MF59‐adjuvanted Sclamp or inactivated virus with Alhydrogel resulted in a decrease in both the peak virus titre detected in throat swabs and total virus shedding as measured by summation of virus detected in daily throat swabs collected between days 1 and 4 (Figures 4c and 5c). Prior exposure to SARS‐CoV‐2 infection reduced peak and total virus shedding by a greater level than either vaccination regimen (Figure 5c). This was expected given that immunity in the upper respiratory tract is more efficiently elicited during natural, IN exposure of the virus as opposed to IM vaccination. These findings further validate the hamster model in recapitulating infection outcomes in humans.
Next, we investigated virus replication and associated pathology in the nasal turbinates and at more distal sites (i.e. the lungs) as these parameters can be indicative of a productive infection that can impact disease progression. For this purpose, all animals were sacrificed at 4 days post‐challenge (n = 10) in the single‐dose study, and in the prime/boost study, half were sacrificed at day 4 (n = 5) and half at day 8 (n = 5) to better evaluate resolution of infection. Placebo‐vaccinated animals sacrificed at 4 days post‐challenge had high levels of virus present in both lung and nasal turbinate tissue (Figures 4d and 5d). This was completely cleared from the lungs of control animals by day 8 and 3/5 hamsters had low levels of detectable virus that remained in the nasal turbinate tissue (Figure 5g).
A single dose of either the inactivated virus with Alhydrogel or MF59‐adjuvanted Sclamp resulted in a significant reduction in virus load in the lungs at day 4 post‐challenge (P < 0.001) but no reduction in viral titre within the nasal turbinate tissue (Figure 4d). In the prime/boost study, administration of two doses of MF59‐adjuvanted Sclamp resulted in complete absence of detectable virus in the lungs for all but one animal at 4 days post‐challenge (Figure 5d). The two‐dose MF59‐adjuvanted Sclamp regimen reduced viral geometric mean TCID50 titres in the nasal turbinates from 2.9 × 107 to 3.8 × 106, although this was not statistically significant (Figure 5d). Two doses of inactivated virus with Alhydrogel reduced viral titre in the lungs but not within the nasal turbinate (Figure 5d). Prior infection and recovery from SARS‐CoV‐2 infection resulted in the lowest viral titres in both lung and nasal turbinate tissue although the effect was not statistically significant in the nasal turbinate (P = 0.053 relative to placebo) (Figure 5c).
Histopathological assessment of placebo‐vaccinated hamsters at day 4 and at day 8 provided an opportunity to follow disease progression. At day 4 post‐challenge, inflammation was primarily confined to the upper respiratory tract and larger airways (trachea and bronchus); however, by day 8 severe inflammation was present in the smaller airways (bronchioles and alveoli) (Figure 5f and i, and representative histopathology images in Figure 6). At day 8 post‐challenge, the overall percentage of lung tissue affected was increased (Figure 5e and h) and there was also evidence of alveolar oedema and haemorrhage (Supplementary table 6). Of note, these findings of increased inflammation and associated pathologies were evident despite the apparent clearance of virus from the lung by day 8 (Figure 5g).
Figure 6.
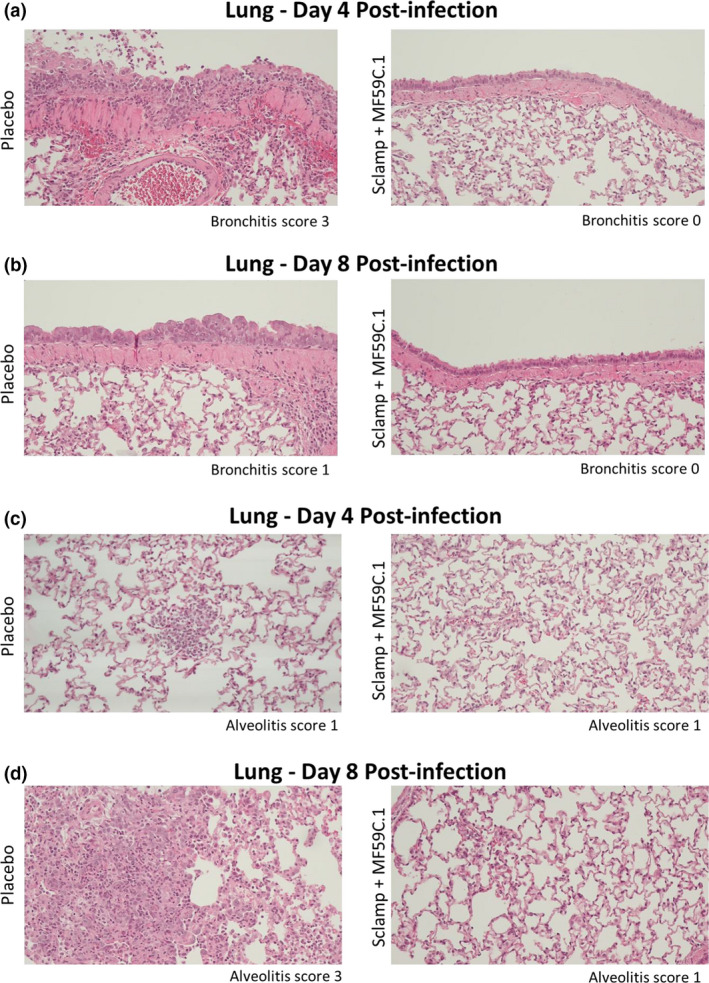
Representative histopathology images from the lungs of placebo and Sclamp + MF59‐vaccinated hamsters in the prime/boost study (10× objective; data from a single experiment are shown). (a, b) Representative images used for bronchitis scoring on day 4 and day 8 post‐challenge. (c, d) Representative images used for alveolitis scoring on day 4 and day 8 post‐challenge.
A single dose of MF59‐adjuvanted Sclamp was shown to provide a significant level of protection against bronchitis, bronchiolitis and alveolitis (Figure 4e). While a single dose of inactivated virus with Alhydrogel did not provide significant protection against disease pathology, neither did it result in increased inflammation despite the failure to elicit any detectable level of neutralising antibody (Figure 4b and e).
Two doses of MF59‐adjuvanted Sclamp or inactivated virus with Alhydrogel, or prior infection and recovery, were each able to provide a high level of protection against rhinitis, tracheitis and bronchitis at day 4, as well as bronchiolitis and alveolitis at day 8 (Figure 5f and i, and representative histopathology images in Figure 6). Pre‐existing immunity from vaccination either with MF59‐adjuvanted Sclamp, inactivated virus with Alhydrogel, or from prior infection, reduced the overall percentage of affected lung at day 8 (Figure 5h) and reduced the findings of peribronchial and perivascular cuffing, alveolar oedema and alveolar haemorrhage (Supplementary table 6).
Consistent with preclinical trials of all other vaccine candidates, sterilising protection in the upper respiratory tract was not observed; however, reduction in viral shedding was seen. In order to assess correlates of protection in the hamster model, we compared viral MN100 titre prechallenge with total virus shedding post‐challenge (sum of throat swabs collected on days 1–4) on a combined dataset including all hamsters in the single and prime/boost studies (n = 70) (Figure 7a) and with virus lung titre at day 4 post‐challenge on a combined dataset including hamsters in the single and prime/boost studies (n = 50; Figure 7b). There was a clear correlation between neutralising antibody level and a reduction in virus shedding (r s = −0.64; 95% CI = −0.76 to −0.46; P < 0.0001) and lower virus lung titre at day 4 (r s = −0.71; 95% CI = −0.83 to −0.53; P < 0.0001). Interestingly, all hamsters within the group that were previously infected and recovered from SARS‐CoV‐2 infection showed substantially lower levels of virus shedding than that expected based on linear regression of the entire data set.
Figure 7.
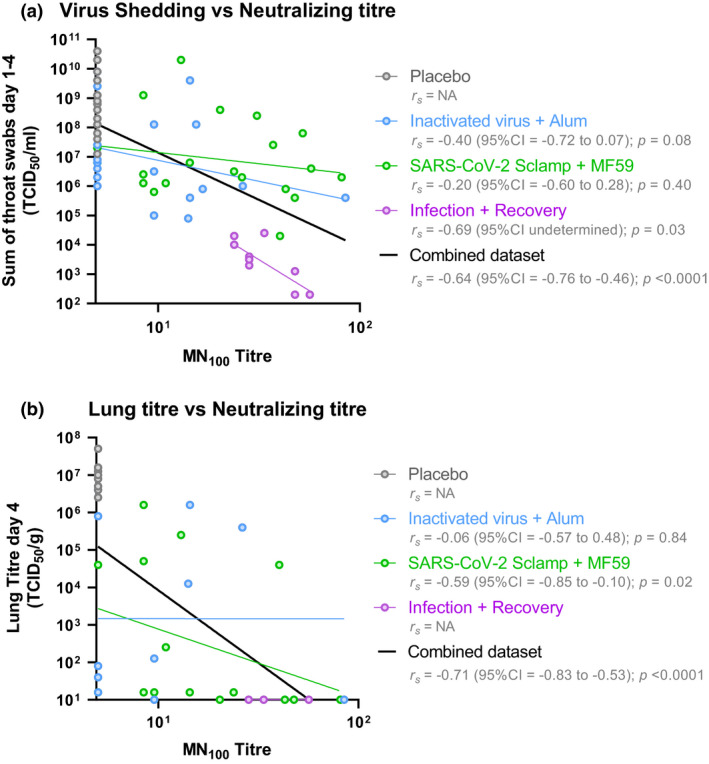
Correlation between neutralising antibodies and protection in hamster (data from two experiments shown). (a) Log‐transformed MN100 titre was compared against the log‐transformed measure of total virus shedding (sum of throat swabs day 1–4 TCID50 mL−1) for all 70 animals included in the single‐dose and prime/boost challenge studies. (b) Log‐transformed MN100 titre was compared against the log‐transformed measure of virus in lung tissue at day 4 post‐challenge for 50 animals sacrificed at day 4 included in the single‐dose and prime/boost challenge studies. Spearman non‐parametric correlation and associated values were calculated for each treatment and the combined dataset using GraphPad Prism 8.4.3.
Discussion
Vaccine development in response to the COVID‐19 pandemic has been unprecedented in terms of both speed and the diversity of the underlying technology platforms. These approaches have included inactivated viruses, nucleic acid‐based vaccines, viral vectors and protein subunits. 2 While each of these platforms comes with its own strengths and limitations, this diversity should also increase the likelihood of success and allow for the selection of the optimal vaccine(s) to progress into widespread global use based on key parameters, including safety and efficacy. 40 The utility of the SARS‐CoV‐2 spike protein as the target for an effective vaccine has also been strengthened through the release of efficacy data for multiple vaccines in phase 3 studies; however, data around longevity and type of protection will take longer to determine.
Driven by the rationale that any improvement in protein yield directly translates into a future increase in dose availability, we dedicated considerable effort to the selection of an optimal candidate and development of a high‐yielding and scalable bioprocess. The ability to manufacture at large scale, ensuring consistent product quality, together with supply chain stability, will be a critical differentiator in achieving global vaccine supply. 40 Furthermore, we exploit an antibody to the clamp domain for immunoaffinity purification that allows high‐level purification using a predefined and standard protocol and reagents that are independent of antigen, therefore speeding vaccine development. Selective engineering of an antigen for high expression can increase production, while selection of a homogeneous and stable antigen reduces product loss during the purification process. Our streamlined process for screening of clamp‐stabilised candidate antigens, 14 facilitated the development of a manufacturing process that can yield 100s of milligrams per litre of culture, which potentially translates to 10s of millions of doses per production run using industry standard, mammalian cell bioproduction platforms.
In mice, vaccination with MF59‐adjuvanted Sclamp was found to elicit a robust humoral immune response that efficiently neutralised both the original SARS‐CoV‐2 strain and the D614G variant which has since become the dominant global circulating strain. A major issue in comparing results between vaccine platforms is that there is no standardised method for assessing virus neutralisation. To address this issue, WHO has recommended the use of a convalescent patient reference serum (NIBSC code 20:130) until an international standard can be made available at the end of 2020. 27 Serum from mice that received two doses of MF59‐adjuvanted Sclamp was found to neutralise SARS‐CoV‐2 at twice the level of this reference serum. To our knowledge, no other programmes have reported neutralising titres relative to this reference serum; however, as these data provide a useful cross‐platform comparator for vaccine developers and regulatory authorities, we would encourage others to utilise this resource and openly present the data.
To assess whether an anticipated immune response to the clamp itself would detract from the protective immune response to the spike ectodomain, we quantified the relative response to each domain in mice. The data showed that on average roughly 76% of antigen‐specific IgG were specific to spike. We also assessed whether serum from mice with higher antibody responses to the clamp domain was less capable of neutralising SARS‐CoV‐2 virus but found no significant correlation. The preclinical studies showed that the molecular clamp in the Sclamp antigen is immunogenic and while cross‐reactivity of the clamp response to HIV‐1 GP41 present in some diagnostic reagents was recognised as a possibility, this was considered to likely have minimum impact due to the exclusion of the immunodominant region (aa577–618). 34 For this reason and the urgency to develop a vaccine for the COVID‐19 pandemic, progression to the phase I clinical trial was deemed warranted and approved by an independent ethics committee. 15
In addition to the humoral immune response, type 1 biased S‐specific T‐cell immunity is likely to contribute to protection in convalescent COVID‐19 patients. 41 Broadly reactive and/or type 1 cytokine‐producing T‐cell responses are also characteristic of DNA, adenovirus serotype 26 and chimpanzee adenovirus vaccines that protected non‐human primates against SARS‐CoV‐2 challenge and are in clinical development. 33 , 35 , 36 , 42 In order to assess the T‐cell response in detail, we evaluated the magnitude and breadth of the T‐cell response in vivo to complement the in vitro T‐cell stimulation and cytokine production analysis. Although such analyses are expected to be beneficial when targeting mutable RNA viruses, SARS‐CoV‐2 vaccines tested in preclinical models previously have relied on in vitro analysis with limited indications about the breadth of the T‐cell response. Using the in vivo FTA and ICS analysis, our study for the first time demonstrates that an MF59‐adjuvanted subunit vaccine, Sclamp, can elicit a robust, broad and predominantly type 1 cytokine‐producing polyfunctional T‐cell response. We also identified several novel immunodominant regions following in vivo functional analysis of Th cell and CTL responses to benefit the future development of SARS‐CoV‐2 vaccines.
MF59‐adjuvanted Sclamp vaccination elicited some type 2 cytokine (i.e. IL‐4 and mostly IL‐13)‐producing T cells as has also been shown in mice vaccinated with a SARS‐CoV‐2‐stabilised spike mRNA construct. 35 The type 2 T‐cell immunity elicited following MF59‐adjuvanted Sclamp vaccination is likely to be beneficial for promoting a neutralising antibody response, but not for triggering immune‐mediated pathology for the following reasons: (1) There was no evidence of vaccine‐enhanced disease in the hamster model and the hamsters were protected following vaccination; (2) pathological/excessive production of type 2 cytokines is characteristic of poorly cytolytic and low‐quality T‐cell responses, 28 , 43 which was not the case in the current study, and (3) there is no evidence to suggest that exposure to type 2 cytokines during SARS‐CoV‐2 infection is detrimental.
As a result of their genetic similarity to humans, NHPs, particularly Rhesus macaque, are thought to be a relevant model for SARS‐CoV‐2 vaccine development. However, infection kinetics are poorly correlated and NHPs experience only mild disease. 33 , 34 , 35 , 36 Multiple studies, 32 , 44 , 45 including our own, suggest that the Syrian hamster provides a more effective animal model to evaluate SARS‐CoV‐2 vaccine efficacy for the following reasons: (1) low‐dose (i.e. 102–103 TCID50) intranasal administration of SARS‐CoV‐2 more accurately reflects likely natural transmission dynamics and results in replication of infectious virus to high titres in the respiratory tract (lungs and nasal turbinates), (2) hamsters are permissive to communicable transmission, 32 (3) hamsters can develop severe pneumonia and lung injury similar to COVID‐19 patients, 45 and (4) protection can be reproducibly evaluated based on infectious viral load rather than viral genomic copy levels. 32 , 44 , 45 In the light of the extensive safety record for MF59C.1 and subunit vaccines and the ethics of NHP experiments that do not provide more informative data, we considered the immunogenicity data from mice, the protection data from Syrian hamsters and toxicology data from Sprague Dawley rats sufficient to support moving this product to the clinic without further testing in NHPs.
A single dose of MF59‐adjuvanted Sclamp or inactivated virus with Alhydrogel afforded a level of protection to hamsters from SARS‐CoV‐2 infection, with a significant decrease in peak viral shedding detected in throat swabs and in virus detected within lung tissue upon sacrifice at 4 days post‐infection. A single dose of MF59‐adjuvanted Sclamp showed significant protection against disease severity as measured by the level of inflammation within the bronchus, bronchioles and alveoli. While a single dose of the inactivated virus with Alhydrogel adjuvant did not provide a significant level of protection against disease severity, there was a trend towards reduced pathology. The absence of an increased inflammatory response as well as decreased virus titres following administration of inactivated virus with Alhydrogel, despite the absence of detectable virus neutralising antibodies, adds further weight to the mounting evidence against antibody‐dependent enhancement (ADE) or vaccine‐associated enhanced respiratory disease (VAERD) being of significant concern for SARS‐CoV‐2. 46 , 47
Administration of two doses of either MF59‐adjuvanted Sclamp or inactivated virus with Alhydrogel in the prime/boost study further reduced viral titres within the lung at day 4 post‐infection compared with the single dose. In the case of MF59‐adjuvanted Sclamp, there was also an apparent reduction in viral load within nasal turbinate tissue, suggesting that protection may extend to the upper respiratory tract; however, this trend did not reach statistical significance. Both vaccines decreased virus shedding collected in throat swabs; however, neither was to the same level as the group exposed to prior SARS‐CoV‐2 infection. This was expected as prior intranasal infection can elicit robust, localised immunity (e.g. IgA) to rapidly prevent re‐infection resulting from IN exposure of SARS‐CoV‐2. 48
Histopathological assessment of lungs from naïve (placebo‐vaccinated) animals at day 8 post‐infection revealed severe inflammation affecting a high percentage of total lung tissue and evidence of alveolar oedema and haemorrhage. Both vaccines, as well as prior SARS‐CoV‐2 infection, were able to significantly reduce disease severity assessed at both 4 and 8 days post‐infection. In particular, the observation of severe lung inflammation at day 8 has important parallels to the pathogenic proinflammatory cytokine storm observed in patients suffering the most severe consequences of infection. 49 We propose that the protection against severe lung pathology at this time point in the hamster model could be highly relevant to support vaccine‐mediated protection against the severe consequences of SARS‐CoV‐2 infection and also vaccine safety against the potential of VAERD.
Total virus shedding as measured by summation of the TCID50 SARS‐CoV‐2 titres from daily throat swabs collected between 1 and 4 days post‐infection provided a quantitative measure of productive viral replication. There was a high degree of correlation between higher virus neutralisation titre prior to challenge and reduction in total virus shedding. Neutralising antibodies are the primary goal of most SARS‐CoV‐2 vaccination strategies; however, to our knowledge this is the first reported evidence that neutralising antibody level is correlated with reduced virus shedding in a relevant animal model. Importantly, our findings also suggest that neutralising antibodies are not the only correlate of protection as animals previously exposed to SARS‐CoV‐2 infection and reinfected showed substantially lower virus shedding than predicted by neutralising antibody levels alone. It will be useful to understand additional mediators of protection, which could potentially target other SAR‐CoV‐2 proteins, to inform second‐generation vaccine approaches. This will be particularly relevant should the leading vaccine candidates already in clinical stages fail to elicit sufficient immunity to break the transmission cycle and end the COVID‐19 pandemic.
Overall, this work demonstrates the ability of MF59‐adjuvanted Sclamp to provide protection against SARS‐CoV‐2 infection and supports continued development and progression through human clinical trials. Subsequently to completion of this work, we have shown that the immune response to the HIV‐1 sequences presents within the clamp trimerisation domain. This finding has prevented progression into later‐stage clinical trials until re‐engineering of the molecular clamp trimerisation domain is completed to alleviate HIV diagnostic interference. Despite this issue, this candidate vaccine has significant advantages that demonstrate the suitability of the underlying platform technology to meet the global response to the ongoing COVID‐19 pandemic as well as novel viruses that may emerge in the future. Most notably, these advantages include accelerated progression from sequence to product in the absence of structural information and compatibility of the manufacturing process with industry standard mammalian cell‐based bioprocess facilities and favorable liquid stability for future global distribution.
Methods
Constructs and plasmids
To express the prefusion S ectodomain, codon‐optimised SARS‐CoV‐2 S (GenBank accession number: MN908947) gene with variations including (1) substitution at the furin cleavage site, (2) substitution at signal peptide, (3) truncation at C‐terminal domain was generated with primers containing overlapping sequence by PCR mutagenesis using Phusion polymerase (New England Biolabs, Ipswich, MA, USA). These amplicons were introduced upstream of the clamp trimerisation motif. Variable domains of heavy and light chain of CR3022, 17 S309, 18 B38, 18 H4, 50 CB6, 18 G4 (anti‐MERS S) 51 or anti‐clamp HIV1281 52 were cloned into the mammalian expression vector, pNBF‐Hv or pNBF‐Lv in‐frame with IgK signal peptide.
Recombinant protein expression
The ExpiCHO‐S expression system (ThermoFisher Scientific, Waltham, MA USA) was used for transient spike protein and antibody expression. CHO cells were cultured in ExpiCHO‐S Expression Medium (Gibco™, ThermoFisher Scientific) and transfection was conducted following the manufacturer's protocols for 5 or 7 days prior to harvest of the culture supernatant and protein purification. Stable cell lines were generated using the Lonza GS Xceed® System (Lonza, Basel, Switzerland). CHOK1SV GS‐KO® cells were transfected via electroporation with linearised GS expression vector encoding SARS‐CoV‐2 Sclamp as per manufacturer's instructions (GS Xceed® manual, version 06 2019). Approximately 24 h later, enriched pools were selected using 50 µM l‐methionine sulphoximine (MSX) over a period of 3–4 weeks. Stable pool shaker flask expression was assessed over 12 days via Lonza's abridged fed‐batch shake flask screen (v8.10), and clone selection was performed using the Beacon Optofluidic platform (Berkeley Lights, Emeryville, CA, USA). Stable pools were loaded onto the OptoSelect™ 1750b Chip (Berkeley Lights) as single cells. Cells were cultured on a chip for 3–5 days before pens were analysed for the secretion of SARS‐CoV‐2 Sclamp using fluorescently tagged anti‐clamp IgG. Selected pens were then exported into 96‐well plates and scaled up into shaker flasks. Clones were further assessed via Lonza's abridged fed‐batch shake flask screen (v8.10).
Recombinant protein purification
Severe acute respiratory syndrome coronavirus 2‐stabilised spike protein was purified using immunoaffinity chromatography on an ÄKTA pure protein purification system (Cytiva, Marlborough, MA, USA). This was achieved using an in‐house made immunoaffinity chromatography column—the anti‐clamp mAb HIV1281 coupled to 1‐ or 5‐mL HiTrap‐NHS‐activated HP columns (Cytiva). CHO expression cultures were centrifuged at 4000 g for 10 min at 4°C and resultant supernatant was filtered through a filter unit (0.22 µm pore size). Supernatant was added to an anti‐clamp protein affinity column to purify clamp‐tagged proteins or protein A HP column (Cytvia) to purify antibodies. Eluates following purification from culture supernatant were neutralised immediately and buffer‐exchanged into PBS using Merck Amicon Ultra‐4 or Ultra‐15 centrifugal filter units. Protein concentration was determined using the NanoDrop One (ThermoFisher Scientific) or via the Pierce BCA protein assay kit (ThermoFisher Scientific). Homogeneity of purified proteins was analysed using SDS/PAGE and visualised following staining with Coomassie blue.
SE‐HPLC analysis of Sclamp
High‐resolution assessment of the oligomeric state of Sclamp was conducted using an Agilent 1200 HPLC. Duplicate 95‐µL samples were injected onto a Waters X‐Bridge 300‐mm column precalibrated with PBS mobile phase and using a flow rate of 0.8 mL min–1.
Negative staining electron microscopy of prefusion Sclamp protein
Severe acute respiratory syndrome coronavirus 2 proteins were diluted at ~ 10 µg mL−1 in PBS. Diluted proteins (4 μL) were adsorbed onto carbon‐coated grids (ProSciTech, Thuringowa, QLD, Australia) for 2 min and glow‐discharged for 5 s in 25 mA. The grids were blotted and washed three times in water and stained twice with 1% uranyl acetate with blotting in between. The grids were air‐dried and imaged using a Hitachi HT7700 microscope operated at 120 Kv.
Cryo‐EM analysis of Sclamp
Severe acute respiratory syndrome coronavirus 2 proteins were diluted to 0.5 mg mL−1 in PBS. Diluted proteins (4 μL) were adsorbed onto glow‐discharged quantifoil grids (Q2/1) and plunge frozen using an EMGP2 system (Leica, Mount Waverley, VIC, Australia). Grids were imaged on a CRYO ARM 300 (JEOL, Tokyo, Japan) equipped with a K3 detector (Gatan, Pleasanton, CA, USA). 50 frame, 5‐s movies were collected using JADAS software (JEOL) at a magnification of 50 000×, corresponding to a pixel size of 0.48 Å per pixel and a dose rate of 9e pix−1 s−1. Movies were binned 2× during motion correction using MotionCor2 (v1.1.0), 53 giving a final pixel size of 0.96 Å. The contrast transfer function (CTF) parameters of each image were determined using CTFFIND (v. 4.1). 54 Initial 2D references were generated using particles manually picked in RELION 3.1. 55 A 60 Å filtered map from EMD‐21452 was used as an initial model for 3D classification using C1 symmetry, and the most ordered class was further refined using the RELION 3D auto‐refine procedure with C3 symmetry. CTF refinement and particle polishing were performed using RELION. The final half‐maps were masked with a soft, extended mask and Fourier shell correlation calculated using the gold standard Fourier shell correlation cut‐off of 0.143.
Thermal stability testing
Purified antigen was sterile‐filtered and diluted in PBS to a final concentration of 0.18 mg mL−1. 250‐µL aliquots were added to 1.5‐mL sterile microcentrifuge tubes which were then stored at either 4, 25 or 40°C for 1, 2, 4 or 8 weeks. At each designated time point, samples were removed from incubation and assessed by ELISA and SE‐HPLC.
Mouse vaccinations and immune analysis
Female, 5‐ to 7‐week‐old BALB/c mice were purchased from the Australian Resource Centre, Perth, and housed in individually ventilated HEPA‐filtered cages at the University of Queensland Biological Resources facility, The Australian Institute for Bioengineering and Nanotechnology. Mice were acclimatised for at least 5 days prior to vaccination via the IM route into the hind leg muscle with 50 µL of PBS (placebo) or 5 µg per mouse of SARS‐CoV‐2 Sclamp with or without Alhydrogel (50 µg per mouse; Croda, Snaith, UK) or MF59 (50% vol:vol of SARS‐CoV‐2 Sclamp:MF59 as recommended by the manufacturer) under anaesthesia.
Blood was collected via the tail vein prior to each vaccination and by cardiac puncture at the study end point. After overnight incubation of blood samples at 4°C, serum was collected via centrifugation for 10 min at 10 000 g, heat‐inactivated at 56°C for 30 min and stored at −80°C prior to analysis.
Bronchoalveolar lavage collection was performed by inserting a catheter into the trachea and flushing the lungs with 400 µL of PBS. After centrifugation at 300 g for 7 min at 4°C, clarified BAL supernatants were frozen at −80°C prior to analysis.
To assess T‐cell responses in mice at the study end point, splenocyte suspensions depleted of red blood cells (RBC) were analysed using ICS or the FTA as described below.
ELISA
A capture ELISA was used to screen ExpiCHO‐S supernatants and purified proteins for expression of Sclamp vaccine candidates. Nunc MaxiSorp ELISA plates were coated with 2 µg mL–1 of the anti‐clamp mAb HIV1281 in PBS overnight at 4°C. Plates were then blocked with 150 µL per well of 5% KPL milk diluent/blocking solution concentrate (SeraCare, Milford, MA, USA) in PBS with 0.05% Tween‐20 for 1 h at room temperature (RT). Blocking buffer was removed and plates were incubated with serial dilutions of harvested ExpiCHO‐S supernatant for 1 h at 37°C. Plates were washed three times with water before incubation with 5 µg mL−1 of an in‐house produced recombinant CR3022 mAb 17 with a mouse IgG1 backbone for 1 h at 37°C. Following another wash step, plates were incubated with a 1:2000 dilution of a horseradish peroxidase (HRP)‐conjugated goat anti‐mouse secondary antibody (ThermoFisher Scientific) for 1 h at 37°C. After a final wash, plates were developed for 5 min using TMB single solution chromogen/substrate (ThermoFisher Scientific) before the reaction was stopped by addition of 2 N H2SO4. Absorbance at 450 nm was then read on a Spectramax 190 Microplate reader (Molecular Devices, San Jose, CA, USA).
For ELISA analysis of mouse serum from placebo‐ or Sclamp‐immunised mice, Nunc Maxisorp ELISA plates were coated with 2 µg mL−1 of Sclamp antigen or an alternate clamp‐stabilised viral glycoprotein (influenza virus haemagglutinin (HA)clamp) and blocked as above. The blocked plates were incubated with serial dilutions of each mouse serum at 37°C for 1 h. The plates were then washed, developed and read as described above. EC50 values were calculated by three‐parameter curve fitting using nonlinear regression in GraphPad Prism (version 8.3.1) (GraphPad, San Diego, CA, USA). The limit of detection (LoD) was defined as the reciprocal of the highest concentration of sera tested and any values falling below the LoD were reported as ½ LoD.
MN assay
Neutralising activity against infectious SARS‐CoV‐2 was assessed by a traditional MN assay as described for SARS‐CoV. 25 Briefly, SARS‐CoV‐2 isolate CoV/Australia/VIC01/2020 56 was passaged in Vero cells and stored at −80°C. Serum samples were heat‐inactivated at 56°C for 30 min and serially diluted 1:20 to 1:10 240 before addition of 100 TCID50 of SARS‐CoV‐2 in MEM/0.5% BSA and incubation at RT for 1 h. Residual virus infectivity in the plasma/virus mixtures was assessed in quadruplicate wells of Vero cells incubated in serum‐free media containing 1 μg mL−1 of TPCK trypsin at 37°C/5% CO2; viral cytopathic effect was read on day 5. The neutralising antibody titre was calculated using the Reed/Muench method as previously described. 25 LoD was defined as the reciprocal of the highest concentration of sera tested and any values falling below the LoD were reported as ½ LoD.
PRNT50 analysis
Two SARS‐CoV‐2 isolates generated from infected patients in Queensland, Australia, were used for the PRNT assay, QLD02/2020—30/1/2020 (GISAID accession EPI_ISL_407896) and QLDID935/2020—25/03/2020 (GISAID accession EPI_ISL_436097) and were referred here as 614D and 614G, respectively. Both isolates were passaged three times in Vero E6 cells. Viral titration and PRNT were performed on Vero E6 cells by an immuno‐plaque assay (iPA) as recently described. 57 Briefly, serial dilutions of mouse sera and BAL were incubated with ~ 6000 focus‐forming units mL−1 of SARS‐CoV‐2 for 1 h at 37°C. Serum virus mixtures were added to Vero E6 cell monolayers (preseeded at 40 000 cells per well in 96‐well plates and incubated overnight) and incubated at 37°C for 30 min. Subsequently, cells were overlaid with 1% (w/v) medium viscosity carboxymethyl cellulose in M199 (ThermoFisher Scientific) supplemented with 2% heat‐inactivated foetal bovine serum supplemented with 1% penicillin–streptomycin (Sigma‐Aldrich, St. Louis, MO, USA) and incubated at 37°C in 5% CO2 for 14 h. After incubation, overlay was removed and cells fixed with 80% cold‐acetone in PBS for 30 min at −20°C. Plates were then dried, blocked with blocking buffer (1xKPL in 0.1% PBS–Tween‐20) for 1 h and then incubated with 1 µg mL−1 of CR3022 anti‐S mAb and 0.2 µg mL−1 IR‐Dye800‐conjugated goat anti‐human IgG (LI‐COR Biosciences, Lincoln, NE, USA) in blocking buffer. Plates were washed 3 times after antibody incubations by submerging in PBS with 0.1% Tween‐20. Plates were then dried prior to visualising using Odyssey (LI‐COR Biosciences). Immunoplaques were manually counted in blinded fashion.
FTA analysis
Fluorescent target array analysis was performed using a well‐established method as previously described. 30 , 31 In brief, naïve autologous BALB/c splenocytes were evenly split and serially labelled with 0.12, 0.46, 1.7, 6.2, 23 or 85 µm of CTV (Invitrogen) and 10 or 39 µm of CPD (Invitrogen) for 5 min at RT. Each of the dye‐labelled populations was pulsed with DMSO (nil) or 10 µg mL−1 peptide of the indicated peptide pools composed of 15–18 aa peptides (10–11 aa overlap between adjacent peptides) for 4 h at 37°C with 5% CO2. Overlapping peptides spanning the SARS‐CoV‐2 S1–1226 and the Peptivator array used for peptide pulsing were purchased from Shanghai RoyoBiotech (Shanghai, China) and Miltenyi Biotec (Bergisch Gladbach, Germany), respectively. The FTA was then injected intravenously into placebo or vaccinated mice such that each mouse received 24 × 106 cells (2 × 106 cells from each fluorescent bar‐coded target cell population) in 200 µL of PBS.
Fifteen hours after the injection, RBC‐depleted splenocytes from FTA‐challenged mice were stained with PE‐Cy7‐conjugated anti‐mouse CD69 (clone H1.2F3; BD Biosciences, San Jose, CA, USA) and BUV395‐conjugated anti‐mouse B220 (clone RA3‐6B2; BD Biosciences) and fixed using 0.5% paraformaldehyde. Subsequently, stained samples were acquired using the BD LSRII and analysed using the FlowJo software (version 8.8.7) (BD, Franklin Lakes, NJ, USA). GMFI of CD69 plotted was calculated using the formula: B220+ peptide‐pulsed target value (GMFI of CD69) ‐ B220+ nil target value (GMFI of CD69). The following formula was used to calculate the % killed data: [(nil target value % − peptide‐pulsed target value %)/nil target value %] × 100.
ICS analysis
2 x 106 RBC‐depleted splenocytes from vaccinated or placebo control mice were seeded into wells of a 96‐well round‐bottom plate and stimulated with 5 µg mL−1 of S1–1226 at 37°C with 5% CO2 for 12 h. DMSO and PMA/ionomycin (PMA at 25 ng mL–1 and ionomycin at 1 µg mL–1) stimulations were included as negative and positive controls, respectively. Following the 12‐h incubation, 1 µg mL–1 of brefeldin A (BioLegend, San Jose, CA, USA) was added to each well and incubated for further 4 h prior to staining the cells with fluorochrome‐conjugated monoclonal antibodies. The stimulated cells were stained for cell‐surface markers, fixed and permeabilised using IC Fix/Perm buffer (BioLegend) prior to the intracellular stain to analyse cytokine expression. The following fluorochrome‐conjugated monoclonal antibodies were used to stain the cells: CD3 (clone: 17A2; BioLegend), CD4 (clone: GK1.5; BioLegend), CD8 (clone: 53‐6.7; BioLegend), IFN‐γ (clone: XMG1.2; BioLegend), TNF‐α (clone: MP6‐XT22; BioLegend), IL‐2 (clone: JES6‐5H4; BioLegend), IL‐4 (clone: 11B11; BioLegend) and IL‐13 (clone: eBio13A; eBioscience, San Jose, CA, USA). Stained cells were acquired using the BD LSRII flow cytometer and analysed using the FlowJo software (version 10.8).
Preparation of formalin‐inactivated virus
To prepare formalin‐inactivated virus, SARS‐CoV‐2 was cultured in Vero E6 cells for 3 days before harvesting supernatant and clarification via centrifugation. Formalin was added to the virus stock at 1:4000 and incubated at 37°C for 3 days. The formalin‐treated virus stock was centrifuged using Millipore Amicon filters (Merk, Darmstadt, Germany) to concentrate the virus prior to removal of formalin and buffer‐exchange using PBS. Virus was passaged on Vero E6 cells to confirm loss of infectivity. Total protein concentration was quantified by BCA analysis.
Hamster challenge study
Male, 10–12‐week‐old Syrian hamsters (n = 10 per group) were vaccinated with either a single dose or two doses as shown in Figures 4a and 5b, respectively. Vaccination with PBS alone was used as a placebo and vaccination with 50 µg of formalin‐inactivated SARS‐CoV‐2 virus formulated with Alhydrogel was included as a comparator vaccination. All vaccines were delivered by IM immunisation. For formalin‐inactivated SARS‐CoV‐2 + Alhydrogel vaccination, 100 µL total volume was delivered per dose, while all other vaccines comprised a total volume of 50 µL per dose. For an infection and recovery comparator group, hamsters were housed separately in a BSL‐3 containment isolator and infected with 102 TCID50 of SARS‐CoV‐2 (BetaCoV/Munich/BavPat1/2020) diluted in PBS to a dose volume of 100 µL delivered via IN administration.
At 4 weeks after the single‐dose and the boost vaccination in the two dose, or 7 weeks after infection in the infection and recovery group, hamsters were challenged with 102 TCID50 of SARS‐CoV‐2 (BetaCoV/Munich/BavPat1/2020) diluted in PBS to a dose volume of 100 µL delivered via IN administration. Following challenge, animal weight loss was recorded daily, and throat swabs were taken for virus quantification by TCID50 and qPCR. At day 4 or day 8 of infection, animals were sacrificed, and tissue samples collected from the lung lobes and nasal turbinate to measure virus titre by TCID50. Samples were also fixed and sectioned to enable the assessment of lesions by gross pathology as well as histopathological assessment for evidence of congestion, emphysema, presence of foreign body, alveolar haemorrhage, bronchioloalveolar hyperplasia and inflammation and oedema.
Statistical analysis
Statistical analysis of the data was performed using the IBM SPSS (Armonk, NY, USA) Statistics Software (version 25) (Armonk, NY, USA) or GraphPad Prism (version 8.3.1). P‐values are reported for statistically significant comparisons with P < 0.05. Non‐significant data with P > 0.05 are denoted ‘ns’.
Conflict of interest
KJC, DW and PRY are inventors of the ‘Molecular Clamp’ patent, US 2020/0040042.
Author Contributions
Dan Watterson: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Project administration; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Danushka Wijesundara: Data curation; Formal analysis; Investigation; Methodology; Validation; Writing‐original draft; Writing‐review & editing. Naphak Modhiran: Data curation; Formal analysis; Investigation; Methodology; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Francesca Mordant: Data curation; Formal analysis; Investigation; Methodology; Writing‐review & editing. Zheyi Li: Data curation; Formal analysis; Investigation; Methodology; Writing‐review & editing. Michael S Avumegah: Formal analysis; Investigation; Writing‐review & editing. Christopher McMillan: Investigation; Methodology; Writing‐review & editing. Julia Lackenby: Investigation; Methodology; Writing‐review & editing. Kate Guilfoyle: Data curation; Investigation; Methodology; Writing‐review & editing. Geert van Amerongen: Investigation; Methodology; Writing‐review & editing. Koert Stittelaar: Methodology; Writing‐review & editing. Stacey Cheung : Investigation; Writing‐review & editing. Summa Bibby: Investigation; Writing‐review & editing. Mallory Daleris: Investigation; Writing‐review & editing. Kym Hoger: Investigation; Writing‐review & editing. Marianne Gillard: Investigation; Methodology; Writing‐review & editing. Eve Radunz: Methodology; Writing‐review & editing. Martina L Jones: Methodology; Writing‐review & editing. Karen Hughes: Methodology; Writing‐review & editing. Ben Hughes: Methodology; Writing‐review & editing. Justin Goh: Investigation; Writing‐review & editing. David Edwards: Investigation; Writing‐review & editing. Judith Scoble: Methodology; Writing‐review & editing. Lesley Pearce: Investigation; Writing‐review & editing. Lukasz Kowalczyk: Investigation; Writing‐review & editing. Tram Phan: Methodology; Writing‐review & editing. Mylinh La: Methodology; Writing‐review & editing. Louis Lu: Methodology; Writing‐review & editing. Tam Pham: Investigation; Writing‐review & editing. Qi Zhou: Investigation; Writing‐review & editing. David A Brockman: Investigation; Writing‐review & editing. Sherry J Morgan: Investigation; Writing‐review & editing. Cora Lau: Investigation; Writing‐review & editing. Mai H Tran: Investigation; Writing‐review & editing. Peter Tapley: Investigation; Writing‐review & editing. Fernando Villalón‐Letelier: Investigation; Writing‐review & editing. James Barnes: Investigation; Writing‐review & editing. Andrew Young: Investigation; Writing‐review & editing. Noushin Jaberolansar: Investigation; Writing‐review & editing. Connor AP Scott: Investigation; Writing‐review & editing. Ariel Isaacs: Investigation; Writing‐review & editing. Alberto Amarilla Ortiz: Writing‐review & editing. Alexander Khromykh: Investigation; Writing‐review & editing. Patrick Reading: Funding acquisition; Investigation; Methodology; Writing‐review & editing. Charani Ranasinghe: Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Writing‐original draft; Writing‐review & editing. Kanta Subbarao: Formal analysis; Investigation; Methodology; Writing‐review & editing. Trent P Munro: Data curation; Investigation; Methodology; Project administration; Writing‐review & editing. Paul Young: Conceptualization; Funding acquisition; Writing‐review & editing. Keith Chappell: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Validation; Writing‐original draft; Writing‐review & editing.
Supporting information
Acknowledgments
This work was funded by CEPI. We thank Mike Whelan, Raafat Fahim and the rest of the project management team from CEPI for all their expert advice and guidance. MF59 adjuvant was provided by Seqirus, and we thank CSL and Seqirus for the assistance with manufacturing process development. We thank Cytiva for their development of the custom immunoaffinity resin, Berkeley lights for assistance optimising clonal cell line selection and Lonza and Thermofisher for the provision of CHO cell lines. We thank Christina Henderson for administrative coordination and The University of Queensland Legal, Finance and Communications teams. We thank Virginia Nink and Nadia De Jager at the Queensland Brain Institute for their help with the flow cytometry. Antigen production was supported by the National Biologics Facility and Therapeutic Innovation Australia. We thank the facilities, and the scientific and technical assistance, of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy and Microanalysis and the Research Compute Centre, the University of Queensland, in particular the assistance of Drs Roger Wepf, Lou Brillault and Matthias Floetenmeyer. We thank Nvidia for the supply of a DGX workstation to aid computational analysis of cryo‐EM data and Michael Landsberg for assistance. We thank NIBSC for the provision of the reference serum. We also thank Jake Carrol and the Research Computing Center at UQ for technical assistance. KS is supported by NHMRC investigator grant 1177174 and generous support of the Jack Ma Foundation and the a2 Milk Company. The Melbourne WHO Collaborating Centre for Reference and Research on Influenza is supported by the Australian Government Department of Health. SARS‐CoV‐2 isolates QLD02 and QLD935 were provided by Queensland Health Forensic & Scientific Services, Queensland Department of Health. Patricia Pilling, Laura Castelli, Luisa Pontes‐Braz assisted with assay development. We also thank Jonneke de Rijck, Guido van der Net, Stephane Nooijen and the rest of the Viroclinics Xplore team for their work on the hamster challenge studies as well as Judith van den Brand of Utrecht University who performed the gross and histopathological examinations and analysis.
Contributor Information
Daniel Watterson, Email: d.watterson@uq.edu.au.
Paul R Young, Email: p.young@uq.edu.au.
Keith J Chappell, Email: k.chappell@uq.edu.au.
References
- 1. Amanat F, Krammer F. SARS‐CoV‐2 vaccines: status report. Immunity 2020; 52: 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. WHO(WHO) . DRAFT landscape of COVID‐19 candidate vaccines; 2021. https://www.who.int/publications/m/item/draft‐landscape‐of‐covid‐19‐candidate‐vaccines2021
- 3. Keech C, Albert G, Cho I et al. Phase 1–2 trial of a SARS‐CoV‐2 recombinant spike protein nanoparticle vaccine. N Engl J Med 2020; 383: 2320–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chappell K, Watterson D, Young P. Chimeric Molecules and Uses Thereof US Patent 2020/0040042; 2018.
- 5. Ekiert DC, Kashyap AK, Steel J et al. Cross‐neutralization of influenza A viruses mediated by a single antibody loop. Nature 2012; 489: 526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang K, Incognito L, Cheng X, Ulbrandt ND, Wu H. Respiratory syncytial virus‐neutralizing monoclonal antibodies motavizumab and palivizumab inhibit fusion. J Virol 2010; 84: 8132–8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pallesen J, Wang N, Corbett KS et al. Immunogenicity and structures of a rationally designed prefusion MERS‐CoV spike antigen. Proc Natl Acad Sci USA 2017; 114: E7348–E7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hsieh CL, Goldsmith JA, Schaub JM et al. Structure‐based design of prefusion‐stabilized SARS‐CoV‐2 spikes. Science 2020; 369: 1501–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Amanat F, Strohmeier S, Rathnasinghe R et al. Introduction of two prolines and removal of the polybasic cleavage site lead to higher efficacy of a recombinant spike‐based SARS‐CoV‐2 vaccine in the mouse model. mBio 2021; 12: e02648‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell 2020; 181: 281–292.e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Richmond P, Hatchuel L, Dong M et al. Safety and immunogenicity of S‐Trimer (SCB‐2019), a protein subunit vaccine candidate for COVID‐19 in healthy adults: a phase 1, randomised, double‐blind, placebo‐controlled trial. Lancet 2021; 397: 682–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krarup A, Truan D, Furmanova‐Hollenstein P et al. A highly stable prefusion RSV F vaccine derived from structural analysis of the fusion mechanism. Nat Commun 2015; 6: 8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McLellan JS, Chen M, Joyce MG et al. Structure‐based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013; 342: 592–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wijesundara DK, Avumegah MS, Lackenby J et al. Rapid response subunit vaccine design in the absence of structural information. Front Immunol 2020; 11: 592370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chappell KJ, Mordant FL, Li Z et al. First Report of a Phase 1 Randomised Trial of Molecular Clamp‐Stabilised Spike Protein‐Based and MF59‐Adjuvanted Vaccine for SARS‐CoV‐2. Lancet ID 2021; 2021. Available at SSRN: https://ssrncom/abstract=3769210
- 16. (WHO) WHO . Novel Coronavirus (2019‐nCoV) Situation Report 1. https://www.who.int/docs/default‐source/coronaviruse/situation‐reports/20200121‐sitrep‐1‐2019‐ncov.pdf2020
- 17. ter Meulen J, van den Brink EN, Poon LL et al. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Medicine 2006; 3: e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pinto D, Park YJ, Beltramello M et al. Cross‐neutralization of SARS‐CoV‐2 by a human monoclonal SARS‐CoV antibody. Nature 2020; 583: 290–295. [DOI] [PubMed] [Google Scholar]
- 19. Wu Y, Wang F, Shen C et al. A noncompeting pair of human neutralizing antibodies block COVID‐19 virus binding to its receptor ACE2. Science 2020; 368: 1274–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wrapp D, Wang N, Corbett KS et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science 2020; 367: 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ke Z, Oton J, Qu K et al. Structures and distributions of SARS‐CoV‐2 spike proteins on intact virions. Nature 2020; 588: 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Edwards RJ, Mansouri K, Stalls V et al. Cold sensitivity of the SARS‐CoV‐2 spike ectodomain. Nat Struct Mol Biol 2021; 28: 128–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vogel FR. Improving vaccine performance with adjuvants. Clin Infect Dis 2000; 30(Suppl 3): S266–270. [DOI] [PubMed] [Google Scholar]
- 24. El Sahly H. MF59TM as a vaccine adjuvant: a review of safety and immunogenicity. Expert Rev Vaccines 2010; 9: 1135–1141. [DOI] [PubMed] [Google Scholar]
- 25. Subbarao K, McAuliffe J, Vogel L et al. Prior infection and passive transfer of neutralizing antibody prevent replication of severe acute respiratory syndrome coronavirus in the respiratory tract of mice. J Virol 2004; 78: 3572–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Korber B, Fischer WM, Gnanakaran S et al. Tracking changes in SARS‐CoV‐2 spike: evidence that D614G increases infectivity of the COVID‐19 virus. Cell 2020; 182: 812–827.e819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. (WHO) WHO . 1st WHO International Standard for anti‐SARS‐CoV‐2 antibodies. In: Mattiuzzo G, editor. https://www.who.int/biologicals/One_Pager_anti‐SARS_CoV‐2_antibodies.pdf?ua=12020 [Google Scholar]
- 28. Ranasinghe C, Trivedi S, Stambas J, Jackson RJ. Unique IL‐13Ralpha2‐based HIV‐1 vaccine strategy to enhance mucosal immunity, CD8+ T‐cell avidity and protective immunity. Mucosal Immunol 2013; 6: 1068–1080. [DOI] [PubMed] [Google Scholar]
- 29. Wijesundara DK, Ranasinghe C, Jackson RJ, Lidbury BA, Parish CR, Quah BJ. Use of an in vivo FTA assay to assess the magnitude, functional avidity and epitope variant cross‐reactivity of T cell responses following HIV‐1 recombinant poxvirus vaccination. PLoS One 2014; 9: e105366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mekonnen ZA, Grubor‐Bauk B, English K et al. Single‐dose vaccination with a hepatotropic adeno‐associated virus efficiently localizes T cell immunity in the liver with the potential to confer rapid protection against hepatitis C virus. J Virol 2019; 93: e00202‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grubor‐Bauk B, Wijesundara DK, Masavuli M et al. NS1 DNA vaccination protects against Zika infection through T cell‐mediated immunity in immunocompetent mice. Sci Adv 2019; 5: eaax2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sia SF, Yan LM, Chin AWH et al. Pathogenesis and transmission of SARS‐CoV‐2 in golden hamsters. Nature 2020; 583: 834–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Doremalen N, Lambe T, Spencer A et al. ChAdOx1 nCoV‐19 vaccine prevents SARS‐CoV‐2 pneumonia in rhesus macaques. Nature 2020; 586: 578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dorn J, Masciotra S, Yang C et al. Analysis of genetic variability within the immunodominant epitopes of envelope gp41 from human immunodeficiency virus type 1 (HIV‐1) Group M and its impact on HIV‐1 antibody detection. J Clin Microbiol 2000; 38: 773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Corbett KS, Flynn B, Foulds KE et al. Evaluation of the mRNA‐1273 vaccine against SARS‐CoV‐2 in nonhuman primates. N Engl J Med 2020; 383: 1544–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yu J, Tostanoski LH, Peter L et al. DNA vaccine protection against SARS‐CoV‐2 in rhesus macaques. Science 2020; 369: 806–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tseng CT, Sbrana E, Iwata‐Yoshikawa N et al. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS One 2012; 7: e35421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Agrawal AS, Tao X, Algaissi A et al. Immunization with inactivated middle east respiratory syndrome coronavirus vaccine leads to lung immunopathology on challenge with live virus. Hum Vaccin Immunother 2016; 12: 2351–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Poland GA, Ovsyannikova IG, Kennedy RB. SARS‐CoV‐2 immunity: review and applications to phase 3 vaccine candidates. Lancet 2020; 396: 1595–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Plotkin S, Robinson JM, Cunningham G, Iqbal R, Larsen S. The complexity and cost of vaccine manufacturing ‐ an overview. Vaccine 2017; 35: 4064–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weiskopf D, Schmitz KS, Raadsen MP et al. Phenotype and kinetics of SARS‐CoV‐2‐specific T cells in COVID‐19 patients with acute respiratory distress syndrome. Sci Immunol 2020; 5: eabd2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mercado NB, Zahn R, Wegmann F et al. Single‐shot Ad26 vaccine protects against SARS‐CoV‐2 in rhesus macaques. Nature 2020; 586: 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kienzle N, Olver S, Buttigieg K et al. Progressive differentiation and commitment of CD8+ T cells to a poorly cytolytic CD8low phenotype in the presence of IL‐4. J Immunol 2005; 174: 2021–2029. [DOI] [PubMed] [Google Scholar]
- 44. Chan JF, Zhang AJ, Yuan S et al. Simulation of the clinical and pathological manifestations of coronavirus disease 2019 (COVID‐19) in a golden Syrian hamster model: implications for disease pathogenesis and transmissibility. Clin Infect Dis 2020; 71: 2428–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Imai M, Iwatsuki‐Horimoto K, Hatta M et al. Syrian hamsters as a small animal model for SARS‐CoV‐2 infection and countermeasure development. Proc Natl Acad Sci USA 2020; 117: 16587–16595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arvin AM, Fink K, Schmid MA et al. A perspective on potential antibody‐dependent enhancement of SARS‐CoV‐2. Nature 2020; 584: 353–363. [DOI] [PubMed] [Google Scholar]
- 47. Lee WS, Wheatley AK, Kent SJ, DeKosky BJ. Antibody‐dependent enhancement and SARS‐CoV‐2 vaccines and therapies. Nat Microbiol 2020; 5: 1185–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Krammer F. SARS‐CoV‐2 vaccines in development. Nature 2020; 586: 516–527. [DOI] [PubMed] [Google Scholar]
- 49. Bohn MK, Hall A, Sepiashvili L, Jung B, Steele S, Adeli K. Pathophysiology of COVID‐19: mechanisms underlying disease severity and progression. Physiology (Bethesda) 2020; 35: 288–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huo J, Le Bas A, Ruza RR et al. Neutralizing nanobodies bind SARS‐CoV‐2 spike RBD and block interaction with ACE2. Nat Struct Mol Biol 2020; 27: 846–854. [DOI] [PubMed] [Google Scholar]
- 51. Wang L, Shi W, Joyce MG et al. Evaluation of candidate vaccine approaches for MERS‐CoV. Nat Commun 2015; 6: 7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Frey G, Chen J, Rits‐Volloch S, Freeman MM, Zolla‐Pazner S, Chen B. Distinct conformational states of HIV‐1 gp41 are recognized by neutralizing and non‐neutralizing antibodies. Nat Struct Mol Biol 2010; 17: 1486–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zheng SQ, Palovcak E, Armache J‐P, Verba KA, Cheng Y, Agard DA. MotionCor2: anisotropic correction of beam‐induced motion for improved cryo‐electron microscopy. Nat Methods 2017; 14: 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rohou A, Grigorieff N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J Struct Biol 2015; 192: 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Scheres SH. A Bayesian view on cryo‐EM structure determination. J Mol Biol 2012; 415: 406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Caly L, Druce J, Roberts J et al. Isolation and rapid sharing of the 2019 novel coronavirus (SARS‐CoV‐2) from the first patient diagnosed with COVID‐19 in Australia. Med J Aust 2020; 212: 459–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Amarilla AA, Modhiran N, Setoh YX et al. An optimized high‐throughput immuno‐plaque assay for SARS‐CoV‐2. Front Microbiol 2021; 12: 625136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials