

Virtual, April 14-17
Sponsorship: Publication of this supplement was funded by the Clinical Immunology Society. All content was reviewed and approved by the Program Committee, which held full responsibility for the abstract selections.
All contributors have provided original material as submitted for publication in the Journal of Clinical Immunology.
01 Oral Presentations
(1) A new manufacturing process to remove thrombogenic factors (II, VII, IX, X, and XI) from intravenous immunoglobulin gamma preparations
Alan Huber, PharmD MBA1, Dong Hwarn Park, PhD2, Gil Bu Kang, PhD3, Dae Eun Kang, MSc4, Ki Yong Kim, PhD4, Jeung Woon Hong, PhD5, Min Gyu Lee, PhD6, Jeung Whan Han, PhD7
1Director of Medical Affairs US/GC Mogam Inc
2Head, Research and Development/GC Corporation
3Deputy Director, Research and Development/GC Corporation
4Research and Development/GC Corporation
5Director of Bioassay/GC Corporation
6Faculty, School of Pharmacy/Sungkyunkwan University
7Faculty, School of Medicine/SungkyunKwan University
Coagulation factors (II, VII, IX, X, and particularly XIa) remaining in high concentrations in intravenous immunoglobulin (IVIG) preparations can form thrombi, causing thromboembolic events, and in serious cases, death. Therefore, manufacturers of biological products must investigate the ability of their production processes to remove procoagulant activities. Previously, we were able to remove coagulation factors II, VII, IX, and X from our IVIG preparation through ethanol precipitation, but factor XIa, which plays an important role in thrombosis, remained in the intermediate products. Therefore, our objective was to develop and test a process to remove factor XIa from IVIG.
The study samples were cleared cryo-poor plasma. A chromatographic process using a new cation-exchange (CEX) resin that binds with high capacity to IgG and removes procoagulant activities was added in a sequential step to the standard removal/inactivation process. Testing of the samples was performed using the standard process alone and then with sequential addition of the new CEX process. Procoagulant activity was tested using several standard methods, including, thrombin generation assay, chromogenic FXIa assay, non-activated partial thromboplastin time (NaPTT), and FXI/FXIa ELISA. We further spiked our samples with additional coagulation factor XIa, in amounts exceeding any variability that may be caused due to sample differences, and tested these samples for procoagulant activity using the same methods.
The procoagulant activities were reduced to low levels as determined by the thrombin generation assay: < 1.56 mIU/mL, chromogenic FXIa assay: < 0.16 mIU/mL, NaPTT: >250 s, FXI/FXIa ELISA: < 0.31 ng/mL. Even after spiking with FXIa at a concentration 32.5 times higher than the concentration in normal specimens, the procoagulant activities were below the detection limit ( < 0.31 ng/mL).
We successfully removed the coagulation factors FII, FVII, FIX, and FX through cold ethanol precipitation, and removed FXIa using chromatography. Using this novel technology can potentially reduce future thromboembolic events with IVIG since FXIa is virtually eliminated.
These results demonstrate the ability of our manufacturing process to remove procoagulant activities to below the detection limit (except by NaPTT), suggesting a reduced risk of thromboembolic events that may be potentially caused by our IVIG preparation.

Keywords: immunoglobulin, chromatography, Factor Xia
Disclosures: All authors indicated they had no financial relationships to disclose.
(2) CARMIL2 Deficiency And Various Clinical Phenotypes: Warning Signs For Early Diagnosis
Burcu Kocamis Kolukisa, MD1, Nurhan Kasap, MD1, Sevgi Bilgic Eltan, MD2, Dilek Baser, MSc2, Gamze Akgun,2, Asena Pınar Sefer, MD1, Yasemin Kendir Demirkol, MD3, Elif Karakoc Aydiner, MD4, Ekrem Unal, MD5, Ahmet Ozen, MD4, Safa Baris, MD4
1Clinical Fellow/Marmara University Hospital, Department of Pediatric Immunology and Allergy
2Marmara University Hospital, Department of Pediatric Immunology and Allergy
3Umraniye Research and Training Hospital, Department of Pediatric Genetic Diseases
4Professor of Pediatrics/Marmara University Hospital, Department of Pediatric Immunology and Allergy
5Erciyes University, Department of Pediatric Hematology and Oncology
CARMIL2(RLTPR) gene regulates CD28 co-signalization and cytoskeletal dynamics of immune cells. Immune deficiency caused by homozygous mutations in CARMIL2 has been linked to a broad range of manifestations, including allergies of the skin and respiratory tract; serious bacterial, fungal and viral infections such as disseminated warts and molluscum; EBV-related smooth muscle tumors; chronic diarrhea and growth retardation. We present a single center experience on CARMIL2 patients.
We studied seven patients (1 Male, 6 Females; current age: 16.7 years) from 4 independent families. Mean age at onset of symptoms was 48,8 months. P1 and P2 presented with chronic abdominal pain and bloody diarrhea. P3 and P4, sisters, had eczema, recurrent respiratory and skin infectins including warts and molluscum. P5 presented with early-onset IBD and wheezing. P6 and P7, cousins, had recurrent skin and airway infections, eczema and warts. Eosinophilia was observed in 3/6 patients. Serum immunoglobulins were normal in half, low IgG in two, high IgG, IgA, IgM in one patient. Protein antibody responses were poor in all patients. Flow cytometry revealed low NK-cells in 5 of 6 subjects; elevated naïve CD4+ T cells in 3 of 6, and reduced memory B cells in 2 of 6. Regulatory T-cells (Tregs) and induction of CTLA4 were reduced in all patients. Defective CD28 T-cell stimulation and cytokine production was confirmed in 2 patients (P3 and P7).
CARMIL2 deficiency may present with early onset-IBD, viral infections, eczema and malignancies. Increased naive T cells, observed in majority of patients suggests defective differentiation. Knowledge of diverse manifestations related to CARMIL2 deficiency should be envisaged for timely diagnosis.
This work was supported by the Scientific and Technological Research Council of Turkey (318S202).

Keywords: CARMIL2, combined immunodeficiency, Treg
Disclosures: All authors indicated they had no financial relationships to disclose.
(3) Excess IL-18 and perforin deficiency distinctly and synergistically promote pathologic CD8 T-cell activation and experimental Hemophagocytic Lymphohistiocytosis
Emily Landy, B.S.1, Scott Canna, MD2
1PhD Candidate/University of Pittsburgh
2Assistant Professor/University of Pittsburgh, UPMC Childrens Hospital
Hemophagocytic Lymphohistiocytosis (HLH) and Macrophage Activation Syndrome (MAS) are life-threatening hyperinflammatory cytokine storm syndromes. Familial HLH is associated with genetic impairment of cytotoxic function (e.g. perforin deficiency), and Prf1-/- mice succumb to typically-mild LCMV infection via CD8 T-cell/IFNγ-mediated immunopathology. MAS is clinically similar to HLH, but occurs in certain rheumatic/autoinflammatory diseases and has been associated with extraordinarily and chronically elevated serum IL-18. Improved understanding of their pathogenesis could significantly alter patient screening, diagnosis, and management.
Using mice with excess IL-18 (Il18tg) to model a state of susceptibility to MAS, we found some baseline abnormalities included decreased IL-18 receptor expression on NK cells and increased PD-1+ CD8 T-cells. Il18tg mice, but not Prf1-/- mice, developed more severe immunopathology in the TLR9-triggered model of MAS. Il18tg mice also developed MAS-like immunopathology upon infection with LCMV (Armstrong), which was largely mediated through CD8 T cells and IFNγ. As with LCMV-infected Prf1-/- mice, CD8 T cells from LCMV-infected Il18tg mice showed increased activation and cytokine production, but did not exhibit cytotoxic impairment or persistent antigen presentation. They retained KLRG1+ terminal effector differentiation and their transcriptional program was more comparable to WT than Prf1-/- mice in their absence of an exhaustion signature.
Mounting evidence suggests heterozygous mutations in cytotoxicity-related genes like PRF1 may promote hyperinflammatory responses in MAS patients, who also have highly elevated IL-18 levels. Though neither excess IL-18 nor perforin-deficiency individually cause immunopathology without inflammatory challenge, we observed lethal spontaneous hyperinflammation in Il18tg;Prf1-/- mice. We even observed subclinical MAS in Il18tg mice heterozygous for Prf1. These mice showed expansion of a splenic PD-1+, TIGIT+, and Tim-3+ CD8 T-cell population, yet show increased IFNg production. Additionally, spontaneous immunopathology was partially abrogated by CD8 depletion or IFNg neutralization. Together, these data suggest that IL-18 and cytotoxicity can independently and synergistically drive pathologic CD8 T-cell activation and life-threatening immunopathology in HLH and MAS.
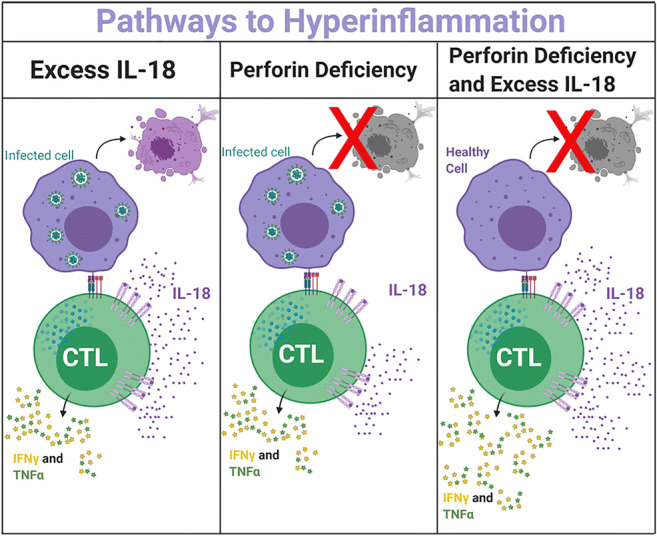
Figure 1: Dysregulation of the immune systems response to antigen/infection can lead to hyperinflammation. Excess free IL-18 (as seen in MAS) can hyperactivate CD8 T cells. Perforin deficiency (as seen in fHLH) can also lead to hyperactivated CD8 T cells along with lack of antigen removal. Together these factors can combine to cause spontaneous hyperinflammation in mice.
Keywords: HLH, MAS, IL-18, Perforin Deficiency, CD8 T cell, activation
Disclosures: Scott Canna received research grants from AB2Bio Ltd and IMMvention Therapeutix. Emily Landy had no financial relationships to disclose.
(4) Long-term Safety Data from Pregnant Women Treated with Facilitated Subcutaneous Immunoglobulin (fSCIG) in a Pregnancy Registry Study
Michael Borte, Prof. Dr. med.1, Stefan Raffac, MUDr.2, Martin Hrubisko, MD, PhD3, Karina Jahnz-Rozyk, MD, PhD4, Milada Cvackova, MD5, Enrique Garcia, MD, MBA6, Andras Nagy, MD7, Barbara McCoy, PhD8, Leman Yel, MD9
1Director, Clinic for Child and Adolescent Medicine/St. Georg Hospital
2Clinic of Clinical Immunology and Allergology/RAFMED s.r.o.
3Department Head, Clinical Immunology and Allergy/Oncology Institute of St. Elisabeth
4Head, Department of Internal Diseases, Pneumonology, Allergology & Clinical Immunology/Military Institute of Medicine
5Faculty Hospital Kralovske Vinohrad/3rd Medical Faculty of Charles University
6Senior Medical Director, GSL/Takeda Pharmaceutical Company Limited
7Associate Medical Director, Clinical Development/Baxalta Innovations GmbH, a Takeda company
8Senior Clinical Director/Baxalta Innovations GmbH, a Takeda Company
9Vice President, Head Clinical Medicine, Global Research & Development, PDT/Baxalta US Inc., a Takeda company, University of California
fSCIG is an immunoglobulin G (IgG) replacement therapy comprised of a dual-vial unit of IgG 10% and recombinant human hyaluronidase (rHuPH20). A postauthorization registry conducted in the United States and Europe assessed safety data on the courses/outcomes of pregnancy in women ever treated with fSCIG treatment and assessed the growth/development of their infants (NCT02556775; EUPAS5798).
Women pregnant during or after fSCIG exposure who provided informed consent participated in this non-interventional, 2-arm, prospective, uncontrolled, open-label, multicenter registry study. During pregnancy, women received alternative treatment (Arm 1) or continued fSCIG (Arm 2) as determined by their physician. Assessments were performed per standard of care. Data were obtained from medical records. Infants were followed for up to 2 years postdelivery.
The study included 9 mothers (Arm 1: n=2; Arm 2: n=7) and 7 infants between Dec 2015 and Dec 2019. Thirteen adverse events (AEs) occurred among 4 mothers (1 in Arm 1, 3 in Arm 2), including 2 serious AEs (SAEs; thrombocytopenia and preeclampsia) in Arm 2. None of these were considered treatment-related by the investigator, or led to fSCIG treatment changes. No local or immunologic AEs were recorded. No discontinuations of fSCIG occurred during pregnancy, and fSCIG was not associated with labor/delivery complications. All pregnancies with known outcomes (n=8) resulted in live births (mean gestational age [weight]: 38.7 weeks [3.1 kg]). Of the 7 infants enrolled, all had normal appearance, pulse, grimace, activity, and respiration (APGAR) scores. Seventeen AEs, all unrelated to the mothers’ treatment, occurred in 6 infants (1 in Arm 1, 5 in Arm 2). Among these, 2 were SAEs (cleft lip and talipes calcaneovalgus, both of mild severity) assessed as incidental findings in 2 infants in Arm 2 who had normal growth and development during the 2-year follow-up. All anti-rHuPH20 binding antibody results (from 4 mothers, 2 in each arm) were negative (titers < 160).
No AEs related to fSCIG were reported in this pregnancy registry in women ever treated with fSCIG treatment or their infants. All infants had normal APGAR scores.
Funding: Baxalta US Inc. (a Takeda company) funded this study and medical writing support.
Keywords: immunoglobulin replacement therapy, safety, pregnancy, infant, real-world data, primary immunodeficiency diseases, facilitated immunoglobulin
Disclosures: Steffan Raffac received speaker honoraria from Takeda. Karina Jahnz-Rozyk received speaker honoraria from Alyogen, AstraZeneka, Chiesi, CSL Behring, GSK, Novartis, Sanofi, and Takeda. Enrique Garcia, Barbara McCoy and Leman Yel are employees of Takeda. All other authors had no financial relationships to disclose.
(5) Long-term Outcomes after Gene Therapy for Adenosine Deaminase Severe Combined Immune Deficiency (ADA SCID)
Donald Kohn, M.D.1, Omar Habib, n/a2, Bryanna Reinhardt, n/a2, Kit Shaw, PhD3, Elizabeth Garabedian, MSN4, Dayna Terrazas, RN5, Beatriz Campo Fernandez, PhD6, Satiro De Oliveira, MD7, Theodore Moore, MD7, Alan Ikeda, MD7, Barbara Engel, MD,PhD8, Gregory Podsakoff, MD9, Roger Hollis, PhD6, Augustine Fernandes, PhD10, Connie Jackson, n/a10, Sally Shupien, n/a10, Suparna Mishra, PhD11, Alejandra Davila, MS11, Jack Mottahedeh, PhD11, Andrej Vitomirov, n/a11, John Everett, PhD12, Aoife Roche, n/a12, Pascha Hokama, n/a12, Shantan Reddy, n/a12, Xiaoyan Wang, PhD13, Kenneth Cornetta, MD14, Michael Hershfield, MD15, Robert Sokolic, MD16, Harry Malech, MD17, Frederick Bushman, PhD12, Fabio Candotti, MD18
1Distinguished Professor/University of California, Los Angeles
2Research Associate/University of California, Los Angeles
3Project Scientist/Dana Farber Cancer Center
4Research Nurse/National Genome Research Institute
5Research Nurse/University of California, Los Angeles
6Project Scientist/University of California, Los Angeles
7Clinical Investigator/University of California, Los Angeles
8Regulatory Administration/Children's Hospital of Philadelphia
9Regulatory Administration/Children's Hospital of Philadelphia
10Regulatory Administration/University of California, Los Angeles
11GMP Lab Scientist/University of California, Los Angeles
12Bioinformatician/University of Pennsylvania
13Biostatistican/University of California, Los Angeles
14Vector Manufacturing/Indiana University
15Investigator/Duke University School of Medicine
16Clinical Investigator/Brown University
17Clinical Investigator/National Institute of Allergy and Infectious Disease, NIH
18Clinical Investigator/University of Lausanne
Patients lacking functional adenosine deaminase activity suffer from severe combined immunodeficiency (ADA SCID), which can be treated with ADA enzyme replacement therapy (ERT), allogeneic hematopoietic stem cell transplantation (HSCT), or autologous HSCT with gene-corrected cells (gene therapy-GT). A cohort of 10 ADA SCID patients, aged 3 months to 15 years, underwent GT in a Phase II clinical trial between 2009 and 2012. Autologous bone marrow CD34+ cells were transduced ex vivo with the MND-ADA gamma-retroviral vector (gRV) and infused following busulfan reduced intensity conditioning. These patients were monitored in a long-term follow-up protocol over 8-11 years. Nine of ten patients have sufficient immune reconstitution to protect against serious infections, and have not needed to resume ERT or proceed to secondary allogeneic HSCT. ERT was restarted 6 months after GT in the oldest patient who had no evidence of benefit from GT. Four of nine evaluable patients with the highest gene marking and B cell numbers remain off immunoglobulin replacement therapy and responded to vaccines. There were broad ranges of responses, in terms of normalization of ADA enzyme activity and adenine metabolites in blood cells, and levels of cellular and humoral immune reconstitution. These outcome parameters were generally better in younger patients and those receiving higher doses of gene-marked CD34+ cells. No patient experienced a leukoproliferative event after GT, despite persisting prominent clones with vector integrations adjacent to proto-oncogenes. These long-term findings demonstrate enduring efficacy of GT for ADA SCID, but risks of genotoxicity with gRVs.
(Clinicaltrials.gov #NCT00794508)
Keywords: ADA SCID, Gene Therapy, Long-term Follow-up, Gamma-retroviral vector
Disclosures: Donald Kohn was a consultant for Leadiant Biosciences. Kit Shaw was a consultant for Orchard Therapeutics. Roger Hollis is an employee of ImmunoVec. All other authors had no financial relationships to disclose.
(6) Common Variable Immunodeficiency in a young male uncovers Nuclear Factor 훋B-1 (NFKB1) haploinsufficiency with variable phenotype in several relatives: the importance of pursuing a genetic diagnosis
Maria Chitty Lopez, MD1, Farnaz Tabatabaian, MD2, Hana Niebur, MD3, Erinn Kellner, MD4, Gulbu Uzel, MD5, Jennifer Leiding, MD6
1Physician Fellow/Division of Allergy and Immunology, Department of Pediatrics, University of South Florida, Tampa, FL, USA
2Assistant Professor/Division of Allergy & Immunology, Department of Internal Medicine, Morsani College of Medicine, University of South Florida, Tampa, Fla
3Assistant Professor/Division of Allergy and Immunology, Department of Pediatrics, University of Nebraska, Omaha, NE, USA
4Assistant Professor/Division of Allergy and Immunology, Department of Pediatrics, University of Cincinnati, Cincinnati, OH, USA
5Physician scientist/Laboratory of Clinical Immunology and Microbiology, National Institutes of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland, USA
6Associate Professor/Division of Allergy and Immunology, Department of Pediatrics, University of South Florida at Johns Hopkins-All Children’s Hospital, St Petersburg, FL
Common Variable Immunodeficiency (CVID) is a genetically heterogeneous disorder characterized by increased susceptibility to infections and humoral immunodeficiency. Monogenic CVID accounts for 10% of cases and is often associated with immunodysregulation. NFKB1 haploinsufficiency is a monogenic cause of CVID complicated by early-onset infection susceptibility, cytopenias, lymphoproliferative disease, autoimmunity, and malignancy. We present a young male with CVID, autoimmunity, and non-malignant lymphoproliferation who was found to have NFKB1 haploinsufficiency, leading to subsequent diagnosis in several previously undiagnosed family members.
Clinical details regarding proband and relatives were obtained. NFKB1 variant was identified by whole-exome sequencing (WES) and confirmed by Sanger sequencing in the proband and by variant-based Sanger sequencing in relatives.
A 14-year-old male with sinopulmonary infections, persistent cervical and abdominal lymphadenopathy, splenomegaly, Crohn's-like colitis, and idiopathic thrombocytopenic purpura (ITP) presented for evaluation. Family history showed father with CVID, ITP, splenomegaly post-splenectomy, and autoimmune hepatitis; paternal uncle with CVID deceased from lymphoma; paternal aunt with autoimmune hemolytic anemia (AIHA) and psoriatic arthritis; and paternal cousin with refractory thrombocytopenia post-splenectomy (Figure-1, Table-1).
Initial immunophenotyping revealed poor antibody responses to tetanus, diphtheria, and pneumococcus.; low IgM, normal IgG and IgA, T-cell lymphopenia, platelet-antibody-positive thrombocytopenia, and elevated double-negative-T-cells (DNTs), (Table-2). Bone marrow biopsy showed trilineage hematopoiesis; lymph node biopsy showed reactive hyperplasia. Immunoglobulin replacement therapy (IgRT) decreased the frequency of infections and improved platelet count. The use of rapamycin further controlled thrombocytopenia and lymphoproliferation. Infliximab improved his colitis. After several genetic evaluations, at age 21, WES identified a novel pathogenic heterozygous nonsense variant in NFKB1 (c.538C>T, p.Gln180Ter). The same pathogenic variant was found in the proband’s father, paternal aunt, cousin, and deceased paternal uncle. NFKB1 variant was also present in the proband’s asymptomatic sister and paternal uncle with a known daughter with ITP (Figure-1).
NFKB1 haploinsufficiency is a monogenic cause of CVID with a high incidence of immunodysregulatory features and variable penetrance leading to diverse immunophenotypes even amongst subjects of the same family. Clinicians should pursue genetic testing in the evaluation of CVID patients especially when symptoms of immune dysregulation or significant family history are present as this case series illustrates.

Table 1: Clinical Phenotype of Family Members

Figure-1. Variable clinical phenotype of NFKB1 haploinsufficiency in a single-family.
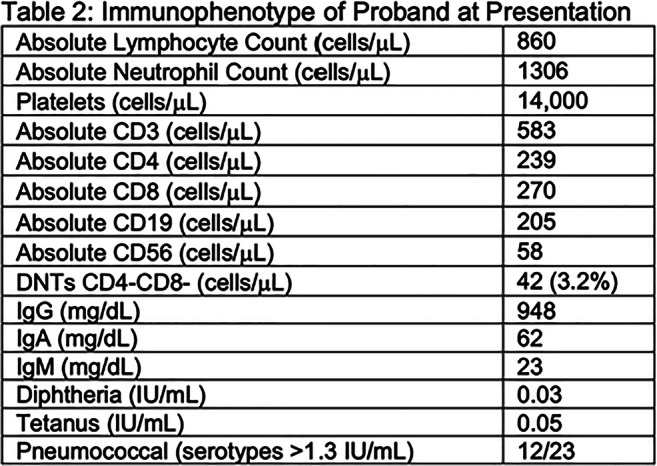
Table 2: Immunophenotype of Proband at Presentation
Keywords: Common Variable Immunodeficiency, NFKB1 loss of function, Monogenic CVID
Disclosures: Hana Niebur was a member of Horizon Therapeutics Advisory Board. Jennifer Leiding received speaker honoraria from CSL Behring and Horizon Therapeutics and was an advisory board member for Pharming. All other authors had no financial relationships to disclose.
(7) A novel primary atopic disorder associated with a homozygous missense variant in OSMR
Mehul Sharma, MSc1, Christina Michalski, BSc1, Kate Del Bel, MSc2, Henry Lu, PhD3, Ashish Sharma, PhD4, Maja Tarailo-Graovac, PhD5, Bhavi Modi, PhD3, Britt Drogemoller, PhD6, Géraldine Blanchard Rohner, MD7, Christof Senger, MS8, Wingfield Rehmus, MD MPH9, Julie Prendiville, MD10, Colin Ross, PhD11, Clara van Karnebeek, MD, PhD12, Wyeth Wasserman, PhD13, Pascal Lavoie, MD PhD11, Margaret McKinnon, MD8, Stuart Turvey, MBBS, DPhil13
1Trainee/University of British Columbia
2Research Coordinator/University of British Columbia
3Post Doctoral Research Fellow/University of British Columbia
4Instructor/Emory University
5Assistant Professor/University of Calgary
6Assistant Professor/University of Manitoba
7Physician/University Hospitals of Geneva
8Clinical Assistant Professor/University of British Columbia
9Clinical Associate Professor/University of British Columbia
10Physician/University of British Columbia
11Associate Professor/University of British Columbia
12Professor/University of Amsterdam
13Professor/University of British Columbia
Primary atopic disorders are monogenic disorders characterized by profound dysregulated allergic responses. Studying patients with these disorders has been instrumental in expanding our understanding of the pathogenesis of allergic inflammation with therapeutic implications for common polygenic versions of allergic disease.
Clinical findings: We describe a 9-year old boy who presented with severe eczema, high blood eosinophil counts (5.8x109 cells/L, normal range: 0-0.85x109 cells/L) and high serum IgE levels (2645υg/L, normal range: 0-500ug/L) since birth. After ruling out known allergic disorders and parasitic infections are ruled out, whole exome sequencing was performed on patient and his parents trio. The patient was found to have a homozygous variant in the evolutionarily conserved fibronectin III domain of the OSMR gene (c.1307T>A, p.V436D). OSMR encodes oncostatin M receptor-beta, a component of both the OSM type II and the IL31 receptor, and is important for keratinocyte cell proliferation, differentiation, apoptosis and inflammation. Variants in OSMR have been reported in association with familial primary localized cutaneous amyloidosis, however this condition was ruled out in this patient as skin biopsies were absent for amyloid deposits.
We transfected the c.1307T>A OSMR variant in both HEK293 cells and primary fibroblasts obtained from the patient, and observed a loss of expression of the mutated OSMR receptor on the cell surface. Signal transduction through phosphorylation of STAT1 and STAT5 was absent and phosphorylation of STAT3 was significantly reduced after stimulation with OSM in patient fibroblasts. These signaling defects were “rescued” upon lenti-viral transduction of the wild-type (WT) OSMR gene (Figure 1). RNAseq analysis confirmed that OSM mediated JAK-STAT and interferon signalling pathways were deficient in the patient fibroblasts and were rescued with WT OSMR. Furthermore, an enhanced atopic dermatitis gene expression signature was observed in patient fibroblasts at baseline which was also rescued upon lenti-viral transduction (Figure 1) or upon treatment with dexamethasone (by qPCR).
Our findings shed light on the disease mechanism of a novel primary atopic disorder, caused by a homozygous missense variant in OSMR.

Figure 1. Patient pedigree and clinical findings. (A) Patient family pedigree. (B) Patient displayed consistently high eosinophil and lgE levels since birth. (C) Impaired STAT1 and STAT5 signaling was observed in patient fibroblasts upon OSM stimulation, which was “rescued” with WT OSMR. (D) RNAseq showed high baseline expression of barrier disruption and inflammation genes in patient fibroblasts compared to healthy controls (HC1, HC2, HC3) and “rescued” fibroblasts.
Keywords: Primary Immunodeficiency, Atopic Dermatitis, Allergy, Eosinophilia, Primary Atopic Disorder
Disclosures: Wingfield Rehmus was an advisory board member of Leo Pharma Inc and Pfizer Canada. All other authors had no financial relationships to disclose.
(8) Mortality in Combined Immunodeficiency: Data From the USIDNET Registry
Jessica Durkee-Shock, MD1, Hua Liang, PhD2, Hannah Wright, MSPH3, Elizabeth Garabedian, MSN4, Rebecca Marsh, MD5, Kathleen Sullivan, MD, PhD6, Michael Keller, MD7
1Clinical Fellow in Allergy and Immunology/National Institute of Allergy and Infectious Diseases/ Children's National Medical Center
2Professor of Statistics/George Washington University Department of Statistics
3Research Data Analyst/The United States Immunodeficiency Network (USIDNET)
4Research Nurse/National Genome Research Institute
5Attending Physician/Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center
6Professor of Pediatrics, Chief of the Division of Allergy and Immunology/The Children’s Hospital of Philadelphia, Philadelphia, PA, USA
7Assistant Professor/Children's National Research Institute, Program for Cell Enhancement and Technologies for Immunotherapy
Combined Immunodeficiency (CID) is a broad category of inborn errors of immunity. Improved understanding of the determinants of early mortality may help to identify patients who will benefit from early definitive therapy.
The USIDNET database was queried for participants with a diagnosis of CID or a genetic diagnosis consistent with CID as defined by IUIS genetic classification. Participants with primary thymic defects, SCID, and Wiskott-Aldrich syndrome were excluded. Akaike Information Criterion (AIC) was used for variable selection for multivariate analysis of factors associated with mortality.
337 participants met the inclusion criteria, with a median age of symptom onset of 0.5 years (IQR 0.1years-4.0years), and a median age of diagnosis of 3.6 years (IQR 0.3years-15 years). Of this population, 38 of 332 individuals for whom data was available were deceased (11%), with a median age at death of 20.7 years. Lower mean values of absolute lymphocyte count, CD3+, CD4+, or CD8+ T cells were not statistically significantly associated with increased mortality. The most common genetic variants in the deceased participants regardless of transplant status were CD40L (17%), GATA2 (14%), DOCK8 (14%), NEMO (8%) and CTLA4 haploinsufficiency (8%). The most common genetic variants in those patients who were deceased and remained un-transplanted at time of death were CTLA4 haploinsufficiency, CD40L, ATM, STAT3, and RMRP. AIC model selection found statistically significantly decreased odds of survival for participants with variants in CTLA4 (OR 0.13), DOCK8 (OR=0.02), CD40L (OR 0.11) and ATM (OR 0.02); as well as participants with renal disease (OR 0.24), bone abnormalities (OR 0.19), invasive bacterial (OR 0.30), viral (OR 0.1) and fungal (OR 0.13) infections . Cognitive (OR 4.8), Mucocutaneous fungal (OR 14.4), and localized viral (OR 4.8) were statistically significantly associated with an increased odds of survival. Other variables selected for the AIC model included RMRP, NEMO, and neurologic comorbidities.
We identified multiple genetic variants, comorbidities, and infectious complications which appear to impact survival in CID. Determining patients at risk for increased mortality and poor outcome may influence the decision to proceed to early definitive therapy, such as hematopoietic stem cell transplant and thereby improve outcomes.
Keywords: Combined Immunodeficiency, USIDNET, Mortality
Disclosures: Kathleen Sullivan was a consultant of the Immune Deficiency Foundation. Michael Keller was an advisory board member of Enzyvant Sciences. All other authors had no financial relationships to disclose.
(9) Mosaic And Germline Gain-Of-Function Variants In TLR8 Leading To Immunodeficiency With Lymphoproliferation And Bone Marrow Failure
Jahnavi Aluri,1, Alicia Bach, MD2, Saara Kaviany, D.O.3, Luana Chiquetto Paracatu, PhD4, Maleewan Kitcharoensakkul, M.D., MSCI5, Magdalena Walkiewicz, PhD6, Christopher Putnam, Ph.D.7, Marwan Shinawi, MD8, Nermina Saucier, MS9, Elise Rizzi, BA10, Michael Harmon, BA11, Molly Keppel, MS12, Michelle Ritter, RN, BSN13, Morgan Similuk, MS14, Elaine Kulm, C.R.N.P.15, Michael Joyce, MD16, Adriana De Jesus, MD, PhD17, Raphaela Goldbach-Mansky, MD18, Yi-Shan Lee, MD,PhD19, Marina Cella, MD20, Peggy Kendall, MD21, Mary Dinauer, MD,PhD20, Jeffrey Bednarski, MD,PhD22, Christy Bemrich-Stolz, MD23, Scott Canna, MD24, Shirley Abraham, MD25, Matthew Demczko, MD26, Jonathan Powell, MD27, Stacie Jones, MD28, Amy Scurlock, MD29, Suk See De Ravin, MD,PhD30, Jack Bleesing, MD31, James Connelly, MD32, V. Koneti Rao, MD30, Laura Schuettpelz, MD,PhD22, Megan Cooper, MD,PhD33
1Postdoctoral Research Associate/Washington University in St. Louis
2Clinical Fellow/Department of Pediatrics, Division of Hematology/Oncology, Washington University School of Medicine, St. Louis, MO.
3Instructor/Pediatric Hematology Oncology, Vanderbilt University Medical Center, Nashville, TN Pediatric Hematology Oncology, Vanderbilt University Medical Center, Nashville, TN
4Postdoctoral Research Associate/Department of Pediatrics, Division of Hematology/Oncology, Washington University School of Medicine, St. Louis, MO
5Assistant Professor/Department of Pediatrics, Division of Rheumatology/Immunology and Allergy and Pulmonary Medicine, Washington University School of Medicine, St. Louis, MO,
6Certified Molecular Geneticist/Division of Intramural Research (DIR), NIAID, National Institutes of Health, Bethesda, MD
7Assistant Professor/Department of Medicine, University of California School of Medicine, San Diego, La Jolla, CA, and San Diego Branch, Ludwig Institute for Cancer Research, La Jolla, CA
8Professor/Department of Pediatrics, Division of Genetics and Genomic Medicine, Washington University School of Medicine, St. Louis, MO
9Research Technician/Department of Pediatrics, Division of Rheumatology/Immunology, Washington University School of Medicine, St. Louis, MO.
10Research Technician/Department of Medicine, Division of Allergy and Immunology, Washington University School of Medicine, St. Louis, MO
11Undergraduate Research assistant/Department of Pediatrics, Division of Rheumatology/Immunology, Washington University School of Medicine, St. Louis, MO
12Researcher/Department of Pediatrics, Division of Rheumatology/Immunology, Washington University School of Medicine, St. Louis, MO
13Research Nurse Coordinator/Department of Pediatrics, Division of Rheumatology/Immunology, Washington University School of Medicine, St. Louis, MO
14Genetic counselor/Centralized Sequencing Initiative and Division of Intramural Research (DIR), NIAID, National Institutes of Health, Bethesda, MD
15Pediatric Nurse Practitioner/Clinical Research Directorate, Frederick National Laboratory for Cancer Research sponsored by the National Cancer Institute,Frederick, MD
16Assistant professor/Nemours Children's Specialty Care, Jacksonville
17Staff Scientist/Translational Autoinflammatory Disease Section (TADS), Laboratory of Clinical Investigation and Microbiology (LCIM), NIAID, NIH
18Chief/Translational Autoinflammatory Diseases Section, NIAID, National Institutes of Health, Bethesda, MD,
19Assistant Professor/Department of Pathology and Immunology, Division of Anatomic and Molecular Pathology, Washington University School of Medicine, St. Louis, MO
20Professor/Department of Pediatrics, Division of Hematology/Oncology, Washington University School of Medicine, St. Louis, MO
21Associate Professor/Department of Medicine, Division of Allergy and Immunology and Department of Pathology and Immunology,Division of Immunology, Washington University School of Medicine, St. Louis, MO
22Assistant Professor/Department of Pediatrics, Division of Hematology/Oncology, Washington University School of Medicine, St. Louis, MO
23Assistant Professor/Department of Pediatrics, Division of Hematology and Oncology, University of Alabama School of Medicine, Birmingham, AL
24Assistant Professor/University of Pittsburgh, UPMC Childrens Hospital
25Assistant Professor/Department of Pediatrics, Division of Hematology and Oncology, University of New Mexico, Albuquerque, NM
26Physician/Department of Pediatrics, Division of Diagnostic Referral, Nemours Alfred I. DuPont Hospital for Children, Wilmington, DE, 19803
27Physician/Department of Pediatrics, Division of Pediatric Hematology/Oncology, Nemours Alfred I. DuPont Hospital for Children, Wilmington, DE, 19803
28Professor/Department of Pediatrics, Division of Allergy & Immunology, University of Arkansas for Medical Sciences and Arkansas Children’s Hospital, Little Rock, AR
29Associate Professor/Department of Pediatrics, Division of Allergy & Immunology, University of Arkansas for Medical Sciences and Arkansas Children’s Hospital, Little Rock, AR
30Staff Clinician/Laboratory of Clinical Immunology and Microbiology, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD
31Professor/Division of Bone Marrow Transplantation and Immunodeficiency, Cincinnati Children's Hospital Medical Center, Cincinnati, OH
32Assistant Professor Pediatric Hem/Onc/BMT/Vanderbilt University Medical Center, Nashville, TN
33Associate Professor/Department of Pediatrics, Division of Rheumatology/Immunology, Washington University School of Medicine, St. Louis, MO
Inborn errors of immunity (IEI) are a genetically heterogeneous group of disorders that affect the development and/or function of one or more components of the immune system. We describe a cohort of six unrelated male patients with a clinical and immunological presentation of recurrent infections, severe neutropenia, humoral defects, and lymphoproliferation with hepatosplenomegaly and lymphadenopathy. Multiple patients had a relatively poor response to therapeutic or high doses of GCSF. Anti-neutrophil antibody testing was performed in 3 patients and was positive in 2 cases. Three patients required hematopoietic stem cell transplantation. Exome sequencing identified novel missense variants in TLR8 gene. Five patients were mosaic for the variants, with four patients sharing the same variant. Patients with mosaic variants had less than 30% mosaicism, with similar allele frequencies in sorted immune cells, saliva and fibroblast lines. The sixth patient harbored a de-novo germline variant. All variants result in a gain-of-function (GOF) of the encoded protein in a TLR8-deficient NF-κB reporter cell line. Immune phenotyping revealed the presence of activated T cells, including the presence of T cell clones in some cases and large granular lymphocytic leukemia (T-LGL) in 1 patient. Multiple patients required IgG replacement therapy due to low B cells numbers and antibody defects. Analysis of serum cytokines demonstrated significantly increased levels of TNFα, IL-1β, IFNg, BAFF, IL-2Rα, IL-12/23 p40 and IL-18. The functional consequence of the GOF variants on primary cells was established using patient-derived iPSCs. Differentiation of myeloid cells from patient-derived iPSCs identified cells with the variant as having increased phosphorylation of NF-κB to low doses of TLR8 stimulation. Additionally, enhanced production of pro-inflammatory cytokines like IL-6, TNF-α, and IL-1β was also identified, supporting the presence of cytokine-driven mechanism of disease pathogenesis. Our finding of 3 novel variants in TLR8 gene in six unrelated patients suggests a novel monogenic TLR8-associated PID.
Keywords: Inborn errors of immunity, neutropenia, toll-like receptor
Disclosures All authors indicated they had no financial relationships to disclose.
(10) Autoantibodies against type I IFNs in Patients With Life-Threatening Disease Due To Yellow Fever Live Attenuated Vaccine
Paul Bastard, n/a 1
1MD-PhD Student/Laboratory of human genetics of infectious diseases, Imagine Institute
Yellow fever virus (YFV) live attenuated vaccine can rarely cause life-threatening disease, typically in patients with no history of severe viral illness. Autosomal recessive (AR) complete IFNAR1 deficiency was reported in one 14-year-old patient. Here, we studied eight other, previously healthy patients aged 13 to 68 years, with unexplained life-threatening YFV vaccine-associated disease. One 13-year-old patient had AR complete IFNAR2 deficiency. Three other patients vaccinated at the ages of 47, 62, and 64 years, had high titers of circulating auto-Abs against at least 14 of the 17 individual type I IFNs, a condition recently shown to underlie at least 10% of cases of life-threatening COVID-19 pneumonia. The auto-Abs were neutralizing in vitro, blocking the protective effect of IFN-a2 against YFV vaccine strains. AR IFNAR1 or IFNAR2 deficiency and neutralizing auto-Abs against type I IFNs thus account for more than half the cases of life-threatening YFV vaccine-associated disease studied here. Apparently healthy subjects could be tested for both deficiencies before vaccination against YFV.
Keywords: Yellow fever vaccine, Type I intereferons, autoantibodies
Disclosures: The author had no financial relationships to disclose.
(11) Patient, Parent, and Provider Perceived Barriers in Primary Immunodeficiency Transition of Care
Neha Agnihotri, MD1, Aisha Ahmed, MD2
1Assistant Professor of Medicine/University of Illinois at Chicago
2Attending Physician, Allergy and Immunology/Ann and Robert H Lurie Children's Hospital of Chicago
We explored the perspectives of primary immunodeficiency (PID) patients and their parents/guardians toward transition of care from pediatric to adult providers. We also compared how transition components were perceived by immunologists at our institution.
Using ICD-9/10 codes, patients with a PID were identified at Lurie Children’s Hospital (LCH) in Chicago, IL. Patients and their parents/guardians were sent separate surveys with questions derived from the validated ‘ATTITUDE’ and ‘QUARTT’ instruments for transition. Pediatric and adult immunologists at LCH and Northwestern Hospital completed respective surveys anonymously (www.surveymonkey.com). Respondents were asked to rate their level of agreement on a 5-point Likert scale, ranging from strongly disagree to strongly agree.
Overall, 17 patients, 18 parents, 9 pediatric immunologists and 11 adult immunologists participated. Regarding the current transition process, 71% of patients reported satisfaction (mean score 3.82, SD 1.33) compared to 31% of parents (mean score 2.94, SD 1.18). All parents, providers, and over 88% of patients agreed that during transition patients should be educated about medications, patient condition, and symptoms that require seeking health care. A transition coordinator was preferred by 94.1% of patients and 77.8% of parents. About 82.4% of patients and 72.2% of parents indicated a written individualized plan for transition should be provided; 70% of providers favored this. All parents and 94% of patients wanted an adult program to have phone access to a nurse. The majority of patients and parents preferred a joint appointment with pediatric and adult immunologists during transition; providers communicating directly without patient/parent present was strongly rejected. About 53% of patients and 56% of parents indicated that providers should prepare PID patients for transition around ages 15-17; in comparison, 47% of patients, 37.5% of parents, and 55% of total providers indicated transition preparation to start when 18 or older.
This study highlights key transition components. Families identified clear communication among all involved parties and specific elements (written transition plan, phone access) as being important. Interestingly, there were differences between current recommendations regarding age of transition preparation initiation and patient/parent perspectives. Further large-scale work is needed to build transition guidelines for the PID population.
Keywords: transition, primary immunodeficiency, adolescents, immunology provider
Disclosures: All authors indicated they had no financial relationships to disclose.
(12) COVID-19 in 3 patients with CLTA4 haploinsufficiency and absence of autoantibodies to type 1 interferons
Sebastian Ochoa, MD1, Lindsey Rosen, BS2, Michail Lionakis, MD, ScD3, Gulbu Uzel, MD4, Daniel Suez, MD5
1Clinical Fellow/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases/National Institutes of Health, Bethesda, MD, USA
2Investigator/Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD
3Chief, Fungal Pathogenesis Section/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases/National Institutes of Health, Bethesda, MD, USA
4Physician scientist/Laboratory of Clinical Immunology and Microbiology, National Institutes of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland, USA
5Past President of the Consortium of Independent Immunology Clinics/Allergy, Asthma & Immunology Clinic PA
Despite a worldwide increase in COVID-19 cases, clinical experience with SARS-COV2 in primary immunodeficiency diseases remains limited. Recent studies showed patients with defects in type 1 interferon (INF)-related pathways or those with auto-antibodies (auto-Abs) against type 1 interferons developed severe COVID-19. We report the clinical course of three patients with CTLA4 haploinsufficiency and COVID-19, and interrogated for autoantibodies to type 1 interferons at baseline.
Data was obtained via patient interview and chart review. Screening for anti-INF auto-Abs was carried out using a multiplex particle-based. Auto-Abs were tested for their neutralizing activity.
Patient characteristics and COVID-19 disease course are shown in table 1. Patients were adults (ages 20-34), had multiple autoimmune manifestations of CLTA4 haploinsufficiency, and were managed with mTOR inhibitors, IVIG replacement, and abatacept. All patients had a known close contact with COVID-19 and tested positive via nasopharyngeal rapid antigen or PCR. Most common symptoms were nasal congestion and anosmia. Patients 1 and 2 received monoclonal antibodies (mAb) to SARS CoV2 within four days of symptom onset and had mild disease course with no hypoxemia or need for hospitalization. Patient 3 received remdesivir and dexamethasone on day 7 (day 0 is defined as the first day of symptoms), was admitted due to bilateral pulmonary infiltrates and an a SaO2 of 92%. He was discharged after 3 days with delayed resolution of shortness of breath and fatigue after 4 weeks. Patients 1, 2 and 3 tested negative for SARS CoV2 at day 22, 30 and 19, respectively. Qualitative SARS CoV-19 IgG/IgM serologies obtained on day 103 (patient 1) and day 31 (patient 3) were negative. All patients were negative for autoantibodies to IFN-α, IFN-β and IFN-ω at baseline.
To our knowledge, this is the first report of COVID-19 in patients with CLTA4 haploinsufficiency. Unlike reported patients with autoantibodies to type 1 interferons at baseline, 2 of our patients (patients 1 and 2) who had an early therapeutic intervention with anti SARS-COV2 mAb had a benign disease course. Whether this is related to the early therapeutic intervention and/or absent autoantibodies to type 1 interferons remains to be elucidated.

Keywords: CTLA4, COVID19, Coronavirus, Interferon, SARS CoV2, CTLA4 haploinsufficiency, Primary immunodeficiency, Type 1 interferon, COVID-19, Autoantibodies to interferon
Disclosures: All authors indicated they had no financial relationships to disclose.
(13) Dupilumab Therapy in STAT3 Deficient Hyper IgE Syndrome
Pavan Nataraj, MD1, Jenna Bergerson, MD, MPH2, Shashi Kumar, MD3, Kimberly Risma, MD, PhD4, Michelle Sabo, MD, PhD5, Amanda Urban, CRNP6, Alexandra Freeman, MD7
1Resident Physician/George Washington University
2Staff Clinician/LCIM, NIAID, NIH
3Attending Physician/Alabama Asthma, Allergy & Immunology Center
4Associate Professor/Division of Allergy and Immunology, Cincinnati Children's Hospital Medical Center; Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH
5Assistant Professor of Medicine/University of Washington
6Nurse Practitioner/Clinical Research Directorate, Frederick National Laboratory for Cancer Research
7Director, Primary Immune Deficiency Clinic/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institute of Health
Dupilumab is a humanized monoclonal antibody blocking IL-4 and IL-13 signaling, approved to treat atopic dermatitis (AD), asthma, and nasal polyps from chronic sinusitis. Patients with dominant negative STAT3 mutations (LOF STAT3; Job’s syndrome) express increased IL-4R suggesting dupilumab may treat some clinical manifestations. We examined the clinical response and safety of dupilumab in LOF STAT3 patients treated for dermatitis, asthma, and/or allergic bronchopulmonary Aspergillus (ABPA).
We reviewed the charts of 10 LOF STAT3 patients treated with dupilumab. We reviewed age at initiation, length of therapy, indication, subjective clinical response, and IgE level, absolute eosinophil count (AEC) and FEV1 prior to initiation and on therapy.
Dupilumab was initiated in 10 patients (7 female), aged 10 to 41 years. Indications included AD (6 patients), asthma (1 patient), AD and asthma (1 patient), and ABPA (2 patients). Pre-dupilumab IgE (516 to 41,336 IU/ml) was increased compared to IgE on dupilumab (available for 6 patients; 458 to 12,233IU/mL); average percent decrease in IgE was 24.7% (n=6). Baseline AEC (40 to 3320 cells/uL) was lower than AEC on dupilumab (available for 6 patients; 70 to 5480 cells/uL); average percent change 14.5% increase (n=5). FEV1 was unchanged pre- and post-therapy for the one patient with asthma and available data. All patients treated at least 2 months endorsed significant improvement in dermatitis, with decreased or discontinuation of topical steroids. One patient had severe AD flare during a treatment interruption. One asthmatic patient endorsed significant improvement in chest tightness and need for rescue inhaler, while the other reported no symptom changes. One patient treated for ABPA had significant improvement in cough, decreased sputum production, and improved radiographic findings. The second ABPA patient developed difficulties with mental clarity and discontinued use. Dupilumab was otherwise well tolerated.
Dupilumab administration in LOF STAT3 appears clinically promising and safe for treatment of AD, with mixed results treating asthma and ABPA thus far. Preliminary studies showed a decrease in serum IgE, but an increase in eosinophilia. Further studies are needed to better evaluate the safety and efficacy in this population, as well as the mechanism behind the improvement.
Keywords: Dupilumab, STAT3, Job's Syndrome, IL-4 receptor α inhibition, asthma, atopic dermatitis, allergic bronchopulmonary aspergillus, ABPA, eczema, Hyper IgE Syndrome
Disclosures: All authors indicated they had no financial relationships to disclose.
(14) Investigating the Impact of Germline STAT3 Gain-of-Function on Regulatory T Cells
Erica Schmitt, M.D., Ph.D.1, Molly Keppel, MS2, Nermina Saucier, MS3, Kelsey Toth, B.A.4, Tiphanie Vogel, M.D, Ph.D.5, Peter Vogel, DVM, Ph.D.6, Megan Cooper, MD,PhD7
1Clinical Fellow Pediatric Rheumatology/Washington University School of Medicine in St. Louis
2Researcher/Department of Pediatrics, Division of Rheumatology/Immunology, Washington University School of Medicine, St. Louis, MO
3Research Technician/Department of Pediatrics, Division of Rheumatology/Immunology, Washington University School of Medicine, St. Louis, MO.
4Graduate Student/Washington University School of Medicine
5Assistant Professor, Division of Rheumatology, Department of Pediatrics & Medicine/Texas Children’s Hospital, Baylor College of Medicine
6Director, Veterinary Pathology Core Laboratory/St Jude Children’s Research Hospital
7Associate Professor/Department of Pediatrics, Division of Rheumatology/Immunology, Washington University School of Medicine, St. Louis, MO
Autosomal dominant germline gain-of-function variants in STAT3 result in immune dysregulation and a broad spectrum of clinical features. A decreased frequency of Foxp3+ regulatory T (Treg) cells has been observed in the peripheral blood of STAT3 GOF patients. STAT3 signaling is involved in both pro- and anti-inflammatory pathways and in the regulation and balance of the Treg/Th17 cell polarization axis. To investigate the pathogenesis of disease we developed a mouse model of STAT3 GOF with a protein variant discovered in patients, p.G421R. STAT3 GOF mice heterozygous for the G421R mutation have a normal or increased frequency of Treg cells, and similar expression of canonical Treg markers and Treg suppressive capacity in vitro. However, in vitro induction of iTreg cells is significantly impaired in these mice. With aging, STAT3 GOF mice develop progressive lymphoproliferation. There is an increased frequency of activated CD4+ T cells and increased IFN-gamma secretion from splenocytes re-stimulated ex-vivo. To further investigate the implications of STAT3 GOF in T cells we utilized the T cell transfer model of colitis. Naïve T cells were isolated from WT or STAT3 GOF mice and adoptively transferred into C57BL/6 Rag1-/- mice. Weight loss, survival, and intestinal pathology were similar. However, phenotypic analysis of the transferred T cells after 28 days demonstrated an increased frequency of IFN-gamma positive cells, but not IL-17A-producing cells, in the intestine lamina propria and mesenteric lymph nodes of mice receiving STAT3 GOF T cells. Mice with colitis induced by STAT3 GOF T cells had a significant reduction in the frequency of peripherally-induced Treg cells in the mesenteric lymph nodes and intestine lamina propria, suggesting decreased formation of iTreg cells in vivo. Interestingly though, preliminary data suggest that treatment of colitis mice with STAT3 GOF Treg cells is sufficient to allow for weight gain and survival, inferring that Treg function may not be significantly altered in STAT3 GOF. Collectively, these data may imply an altered function of effector T cells due to STAT3 GOF early in the disease course of mice with colitis, and in aged STAT3 GOF mice, with a pathogenic Th1 phenotype potentially dominating.
Keywords: immune dysregulation, STAT3, regulatory T cells
Disclosures: All authors indicated they had no financial relationships to disclose.
(15) Germline-encoded loss of RHOG specifically abrogates human lymphocyte cytotoxicity and cause HLH
Artem Kalinichenko1, Giovanna Casoni, n/a2, Loïc Dupré, PhD3, Luca Trotta, PhD4, Jakob Huemer, MSc5, Donatella Galgano, PhD6, Yolla German, MSc7, Ben Haladik, MSc5, Julia Pazmandi, MSc5, Marini Thian, MSc5, Özlem Petronczki, PhD8, Samuel Chiang, PhD6, Mervi Taskinen, MD, PhD9, Anne Hekkala, PhD10, Saila Kauppila, MD, PhD11, Outi Lindgren, MD, PhD11, Terhi Tapiainen, MD, PhD12, Michael Kraakman, PhD13, Kim Vettenranta, Prof, MD, PhD14, Alexis Lomakin, PhD15, Janna Saarela, MD, PhD16, Mikko Sepanenn, n/a17, Yenan Bryceson, PhD18, Kaan Boztug, MD, PhD19
1Postdoctoral fellow/St. Anna Children’s Cancer Research Institute / St. Anna Kinderkrebsforschung (CCRI)
2Pre-doctoral fellow/Center for Hematology and Regenerative Medicine, Department of Medicine Huddinge, Karolinska Institutet, Stockholm, Sweden
3Principal Investigator/Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases, Vienna, Austria
4Postdoctoral Fellow/6Institute for Molecular Medicine Finland, University of Helsinki, Finland
5Pre-doctoral fellow/St. Anna Children’s Cancer Research Institute, Vienna, Austria
6Postdoctoral Fellow/Center for Hematology and Regenerative Medicine, Department of Medicine Huddinge, Karolinska Institutet, Stockholm, Sweden
7Pre-doctoral fellow/Center for Pathophysiology of Toulouse Purpan, INSERM UMR1043, CNRS UMR5282, Paul Sabatier University, Toulouse, France
8Staff scientist/St. Anna Children’s Cancer Research Institute, Vienna, Austria
9Consultant/8Oulu University Hospital and University of Oulu, Oulu, Finland
10Postdoctoral Fellow/Oulu University Hospital and University of Oulu, Oulu, Finland
11Staff scientist/Oulu University Hospital and University of Oulu, Oulu, Finland
12Consultant/Oulu University Hospital and University of Oulu, Oulu, Finland
13Postdoctoral Fellow/St. Anna Children’s Cancer Research Institute, Vienna, Austria
14Consultant/Rare Disease and Pediatric Research Centers, University of Helsinki and Helsinki University Hospital, Helsinki, Finland
15Program Leader and Deputy Lab Head/St. Anna Children’s Cancer Research Institute, Vienna, Austria
16Director/Institute for Molecular Medicine Finland, University of Helsinki, Finland
17Rare Disease and Pediatric Research Centers, Hospital for Children and Adolescents, and Adult Immuno/Unit, Inflammation Center, University of Helsinki and HUS Helsinki University Hospital, Helsinki, Finland
18Principal Investigator/Center for Hematology and Regenerative Medicine, Department of Medicine Huddinge, Karolinska Institutet, Stockholm, Sweden
19Director/St. Anna Children’s Cancer Research Institute, Vienna, Austria
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening condition characterized by immune dysregulation and massive, aberrant hyperactivation of cytotoxic T cells and macrophages. The molecular pathologies underlying HLH are diverse. The monogenic or primary form of HLH is caused by mutations selectively disrupting perforin-mediated cytotoxicity in human lymphocytes.
We studied a patient with early-onset HLH who fulfilled all diagnostic criteria, including almost absent killing activity of NK cells. Targeted NGS-based panel sequencing did not reveal any germline mutations in established HLH-associated genes. Using exome sequencing and genome-wide SNP array, we identified biallelic germline mutations in RHOG. Genetic ablation of the RHOG gene in a model cell line and primary cytotoxic T lymphocytes (CTLs) from healthy individuals confirmed its crucial role in lymphocyte cytotoxicity. Notably, despite the severe defect in exocytosis, RhoG-deficient lymphocytes showed normal activation, proliferation, and cytokine production.
To decipher the molecular pathomechanism of the RhoG deficiency, we performed interaction proteomics analysis and defined the molecular partners of RhoG in human lymphocytes. In addition to the strong association with the cytoskeleton regulators, this analysis also revealed a direct link of RhoG with the exocytosis machinery. Hence, we discovered that RhoG interacts with the regulator of cytotoxic granule (CG) release Munc13-4 and regulates docking of Munc13-4-positive CGs to the plasma membrane. This step is required for subsequent fusion of the membranes to release cytolytic cargo toward target cells. Using molecular biology and biochemical approaches, we showed that RhoG is essential for the proper function of hematopoietic Munc13-4, assisting it in binding to the membrane phospholipids. We further confirmed that this requirement for the RhoG assistance is unique for Munc13-4, which lacks the C1 membrane-binding domain present in other Munc13 isoforms. Collectively, our work i) discovers a novel Mendelian disease affecting human immune function and homeostasis, potentially representing familial HLH type 6; ii) defines a molecular pathomechanism of the discovered disorder; iii) identifies a novel layer of exocytosis regulation unique for cytotoxic lymphocytes.
Keywords: Cytotoxic lymphocytes, Hemophagocytic lymphohistiocytosis, regulated exocytosis
Disclosures: All authors indicated they had no financial relationships to disclose.
(16) Immunometabolic Phenotyping in Father-Daughter Pair with GATA2 Haploinsufficiency
James Maiarana, MD1, Saara Kaviany, D.O.2, Todd Bartkowiak, Phd3, Yasmin Khan, MD4, Daniel Dulek, MD5, Sarah Neumann, RN6, Jeffrey Rathmell, PhD7, Jonathan Irish, PhD8, James Connelly, MD9
1Intern/Vanderbilt University Medical Center
2Instructor/Pediatric Hematology Oncology, Vanderbilt University Medical Center, Nashville, TNPediatric Hematology Oncology, Vanderbilt University Medical Center, Nashville, TN
3Postdoctoral Scholar/Vanderbilt University
4Assistant Professor Pediatrics AI/Vanderbilt University Medical Center
5Assistant Professor Pediatric ID/Vanderbilt University Medical Center
6Pediatric BMT Case Manager/Vanderbilt University Medical Center
7Associate Director - Vanderbilt Institute for Infection, Immunology and Inflammation, Professor PMI/Vanderbilt University
8Associate Professor of Cell and Developmental Biology/Vanderbilt University
9Assistant Professor Pediatric Hem/Onc/BMT/Vanderbilt University Medical Center, Nashville, TN
The transcription factor GATA2 is required for hematopoietic stem cell homeostasis and lymphangiogenesis (1,2,3). Heterozygous mutations in GATA2 result in haploinsufficiency leading to a spectrum of clinical phenotypes: susceptibility to viral and bacterial infections, cytopenias, pulmonary alveolar proteinosis, lymphedema, and myelodysplasia (2,4,5). We present a father-daughter pair with heterozygous GATA2 (c.890del, p.Asn297Thr*29) variants leading to haploinsufficiency and their distinct clinical phenotypes. Using mass cytometry (CyTOF), we performed comparative immune-profiling of peripheral blood, demonstrating immune phenotypic and metabolic characteristics.
The daughter presented at 15 years old with cytopenias and recurrent urinary tract infections. Bone marrow biopsies revealed hypocellularity, dyserythropoiesis, karryohexis, and erythrocyte and granulocyte atypia. An NGS myeloid panel revealed a c.890del variant in GATA2 with a 47% allelic frequency. She has recently developed recurrent panniculitis and lower extremity lymphedema.
The same variant was present in the father who presented with more severe clinical features, including HPV-associated squamous cell carcinoma, T-cell large granular lymphocytic leukemia, and recurrent viral and bacterial infections.
Patients were enrolled in the Human Immune Discovery Initiative (HIDI) at Vanderbilt University permitting immunophenotyping of human leukocytes using CyTOF. Cytometric evaluation revealed that the patients with GATA2 haploinsufficiency had systemically perturbed peripheral immunity compared to healthy controls including 1) increased frequencies of TEMRA, 2) 8-fold reduction in myeloid cells 3) inverted CD4:8 ratios, 4) increased expression of markers of activation (CD44, CD95, CXCR3, and CD57) and metabolism (ATP5a, CPT1a, GLUT1, GLUT3) in T- and B-cell subsets, and 5) increased expression of markers of glycolysis (GLUT1, GLUT3) in monocytes.
GATA2 haploinsufficiency leads to a spectrum of clinical illness with monocytopenia, and decreased frequencies of B- and NK-cells. Utilizing CyTOF, we have gained a more complete understanding of immunity in patients with known or suspected inborn errors of immunity (IEIs). Our findings are consistent with reports of known GATA2 haploinsufficiency (6,7); however, this is the first analysis, to our knowledge, of the immunometabolic profile of patients with GATA2 haploinsufficiency. Our data suggests an undescribed metabolic activation across lymphoid and myeloid lineages in our patients.
Figure 1:

Immunometabolic phenotyping of T cells in patients with Gata2 Haploinsufficiency compared to healthy control
Keywords: GATA2 Haploinsufficiency, Immunometabolism, Mass Cytometry
Disclosures: All authors indicated they had no financial relationships to disclose.
(17) Unusual presentation of subcutaneous panniculitis-like T cell lymphoma in patients with BENTA disease
Bradly Bauman, MS1, Stefania Pittaluga, MD, PhD2, Yu Zhang, PhD3, Sergio Rosenzweig, MD, PhD4, Ronald Anderson, MD5, Gregory Gulcher, MD6, Iwona Auer, MD7, Renee Perrier, MD8, Martin Campbell, MBSS Msc9, Magdalena Schelotto, MD10, Nicola Wright, MD, MSc, FRCPC5, Helen Su, MD PhD11, Andrew Snow, PhD12
1Graduate Student/Department of Pharmacology and Molecular Therapeutics, Uniformed Services University of the Health Sciences, Bethesda, MD, USA
2Senior Research Physician/Lab of Pathology, National Cancer Institute, Bethesda, MD, USA.
3Staff Scientist/Laboratory of Clinical Immunology & Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
4Senior Investigator/Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
5Clinical Associate Professor/Department of Pediatrics, Alberta Children's Hospital, University of Calgary, Calgary, AB, Canada
6Associate Professor/Department of Pediatrics, Alberta Children's Hospital, University of Calgary, Calgary, AB, Canada
7Clinical Associate Professor/Alberta Precision Laboratories, University of Calgary, Calgary, AB, Canada
8Clinical Associate Professor/Department of Medical Genetics, University of Calgary, Calgary, AB, Canada
9Consultant Oncologist/Children's Cancer Centre, Royal Children's Hospital, Melbourne, Australia
10Clinical Investigator/Department of Pediatric Hematology and Oncology, Fundación Pérez Scremini, Hospital Pereira Rossell, Montevideo, Uruguay
11Senior Investigator/Laboratory of Clinical Immunology & Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
12Associate Professor/Department of Pharmacology and Molecular Therapeutics, Uniformed Services University of the Health Sciences, Bethesda, MD
Subcutaneous panniculitis-like T cell lymphoma (SPTCL) is a rare form of non-Hodgkin’s lymphoma, comprised of neoplastic CD8+ TCRalpha/bet+ cytotoxic T cells that surround adipocytes in the panniculus. Primary SPTCL is frequently associated with hemophagocytic lymphohistiocytosis (HLH) and germline, biallelic loss-of-function mutations in HAVCR2, which encodes for the immunomodulatory receptor T cell immunoglobulin mucin 3 (TIM-3). In contrast, atypical lymphocytic lobular panniculitis (ALLP) or secondary SPTCL can occur in the context of infection or autoimmune disease, often accompanied by somatic mutations in infiltrating T cells, and can be difficult to distinguish from primary SPTCL. Here we describe two unrelated patients with confirmed B cell Expansion with NF-□B and T cell Anergy (BENTA) disease and a novel presentation of SPTCL/ALLP. Patient 1 presented early in life with recurrent infections and transitional B cell lymphocytosis, linked to a novel gain-of-function (GOF) mutation in the lymphocyte-specific scaffold protein caspase activation and recruitment domain 11 (CARD11, p.Lys238del), consistent with BENTA disease. By age 2, he developed SPTCL-like lesions and membranoproliferative glomerulonephritis / nephrotic syndrome. Following relapse after initial CHOP chemotherapy, the patient was successfully treated with cyclosporine A and has remained in remission since, with prophylactic TMP-SMX and no infections. Subsequent histopathological analyses of skin biopsies suggested lupus-associated ALLP with no clonality and normal TIM-3 expression. Patient 2 presented with splenomegaly, lymphadenopathy, elevated naïve B cells, and confirmed SPTCL at 18 months, with widespread cutaneous involvement and evidence of HLH. Genetic analysis revealed two de novo, in cis germline GOF mutations in the LATCH domain of CARD11 (p.Glu121Asp, p.Gly126Ser), confirming a diagnosis of BENTA disease. No HAVCR2 mutations were detected. After a poor response to multiple rounds of chemotherapy, she required autologous bone marrow transplant and has been in remission since, with no infectious complications. These cases illuminate an unusual pathological manifestation for BENTA disease, and further suggest that CARD11 GOF mutations can contribute to the development of cutaneous T cell dyscrasias and/or malignancies involving both CD4+ (Sézary syndrome) and CD8+ (SPTCL) lineages, or both (ALLP).
Keywords: CARD11, SPTCL, BENTA, lobular panniculitis, B cell lymphocytosis, ALLP
Disclosures: Gregory Gulcher received a research grant from Blue Bird Bio. All other authors had no financial relationships to disclose.
(18) Clinically Unsolvable Autoinflammatory Disorder Diagnosed by RNA and Long Read Sequencing
Jessica Nguyen, MD1, Marietta M. DeGuzman, MD2, Jessi Xu, n/a3, Stephen Wu, n/a3, Patricia Rosillo, MD2, Caridad Martinez, MD4, Judith Campbell, MD5, Sarah Nicholas, M.D.6, Jill Rosenfield, MS7, Fan Xia, PhD8, Lindsay Burrage, MD, PhD9, Medhat Mahmoud, n/a10, Fritz Sedlazeck, PhD11, Undiagnosed Disease Network, n/a12, Eric Hanson, MD13, M. Cecilia Poli, MD, PhD14, David Murdock, MD7, Tiphanie Vogel, M.D, Ph.D.15
1Resident/Baylor College of Medicine
2Division of Rheumatology, Department of Pediatrics Associate Professor/Baylor College of Medicine/Texas Children's Hospital
3Division of Rheumatology, Department of Pediatrics/Indiana University School of Medicine
4Division of Hematology and Oncology, Department of Pediatrics Associate Professor/Baylor College of Medicine/Texas Children's Hospital
5Division of Infectious Diseases, Department of Pediatrics Attending Physician/Baylor College of Medicine/Texas Children's Hospital
6Assistant Professor, Division of Immunology, Allergy and Retrovirology, Department of Pediatrics/Texas Children's Hospital, Baylor College of Medicine
7Department of Molecular and Human Genetics Assistant Professor/Baylor College of Medicine
8Department of Molecular and Human Genetics Associate Professor/Baylor College of Medicine
9Assistant Professor/Baylor College of Medicine
10Human Genome Sequencing Center Postdoctoral Associate/Baylor College of Medicine
11Human Genome Sequencing Center Assistant Professor/Baylor College of Medicine
12Undiagnosed Disease Network/Undiagnosed Disease Network
13Division of Rheumatology, Department of Pediatrics Associate Professor/Indiana University School of Medicine
14Director of the Immunogenetics and Translational Immunology Program; Assistant Professor/Universidad del Desarrollo; Baylor College of Medicine/ Texas Children's Hospital
15Assistant Professor, Division of Rheumatology, Department of Pediatrics & Medicine/Texas Children’s Hospital, Baylor College of Medicine
IKBKG on chromosome X encodes NEMO, a critical regulator of NF-κB and interferon signaling. Hypomorphic NEMO mutations cause anhidrotic ectodermal dysplasia with immunodeficiency in males. Recently, NEMO deleted exon 5 autoinflammatory syndrome (NEMO-NDAS) was reported in both males and females, characterized by infantile-onset fevers, panniculitis, and immunodysregulation. We report a case of NEMO-NDAS and the evaluation necessary for the diagnosis.
The patient presented at age 6 weeks with fevers and rash; inflammatory markers were elevated. Infectious etiologies were excluded and she responded to corticosteroids, but symptoms returned with tapers. Extensive clinical investigations pursuing an etiology were most notable for granuloma formation (in bone marrow, stomach, liver, and dura mater) and low B cells with hypogammaglobulinemia. Clinical trio exome sequencing (ES) was negative. Research analysis suggested dysregulated interferon responses in patient-derived cells. RNA-sequencing from fibroblasts revealed a deletion of IKBKG exon 5, present in 97% of transcripts, consistent with skewed X-inactivation. Targeted exome analysis of IKBKG was uninformative due to a nearby pseudogene. Nanopore long read sequencing was then done and identified a presumed de novo IKBKG splice site variant, c.519-2A>C, originating on the paternal allele. Fibroblast protein electrophoresis confirmed 95% of the patient’s NEMO protein was the shorter form.
Her predominate clinical features have been recurrent fever and wide-spread panniculitis. She has also developed multiple infections in the setting of chronic immunosuppression, requiring IVIG replacement and anti-microbial prophylaxis, including frequent upper respiratory and urinary infections, Pneumocystis pneumonia, HSV-1 and BK viremia, and disseminated candidiasis. She has trialed numerous immunomodulatory treatments (anti-TNF, IL-1, IL-6R, and IL-12/23 agents, costimulatory blockade, JAK inhibition). However, her disease is refractory to all steroid-sparing attempts and she has remained on corticosteroids since presentation. She exhibits multiple sequelae of chronic illness and steroid use including adrenal insufficiency, hypertension, short stature, osteoporosis, and papilledema. She is now 10 years old and undergoing evaluation for bone marrow transplant.
NEMO-NDAS is a new monogenic autoinflammatory disease characterized by an interferon signature. Thorough genetic evaluation for NEMO-NDAS should be considered in cases of early-onset fevers, severe immune dysregulation and panniculitis if clinical ES is unrevealing.
Keywords: IKBKG, NEMO, immune dysregulation, genetics, case report
Disclosures: Jill Rosenfield, Fan Xia, Lindsay Burrage, and David Murdock work for the Human Genetics Laboratory at Baylor College of Medicine which receives revenue from clinical genetic testing completed at Baylor Genetics Laboratory. All other authors had no financial relationships to disclose.
(19) Splice Site Variants in IKBKG Detected by a Customized Next-Generation Sequencing Analysis Cause an Early-onset Autoinflammatory Syndrome of Panniculitis and Cytopenias in Male and Female Patients
Adriana De Jesus, MD, PhD1, Bin Lin, PhD2, Eric Karlins, MSc3, Andrew Oler, PhD4, Sofia Torreggiani, MD5, Andre Rastegar, BS6, Dana Kahle, BS6, Jacob Mitchell, BS6, Eric Hanson, MD7, Magdalena Walkiewicz, PhD8, Sara Alehashemi, MD, MHS9, Kader Cetin-Gedik, MD9, Cassandra Holmes, MS, CRNP, FNP-C10, Kat Uss, RN, BSN11, Eveline Wu, MD12, Christiaan Scott, MD13, Timothy Leahy, MD, PhD14, Emma MacDermott, MD15, Orla Killeen, MD16, Thaschawee Arkachaisri, MD17, Scott Canna, MD18, Seza Ozen, MD19, Zoran Gucev, MD20, Kathryn Cook, DO21, Vafa Mammadova, MD, PhD22, Gulnara Nasrullayeva, MD22, Amer Khojah, MD23, Timothy Moran, MD, PhD24, Raphaela Goldbach-Mansky, MD25
1Staff Scientist/Translational Autoinflammatory Disease Section (TADS), Laboratory of Clinical Investigation and Microbiology (LCIM), NIAID, NIH
2Staff Scientist/Translational Autoinflammatory Disease Section, NIAID, NIH
3Bioinformatics Scientist/Bioinformatics and Computational Biosciences Branch, Office of Cyber Infrastructure and Computational Biology, NIAID, NIH
4Senior Bioinformatics Scientist/Bioinformatics and Computational Biosciences Branch, Office of Cyber Infrastructure and Computational Biology, NIAID, NIH
5Post-doctoral visiting fellow/Translational Autoinflammatory Disease Section (TADS), Laboratory of Clinical Investigation and Microbiology (LCIM), NIAID, NIH
6Post-baccalaureate fellow/Translational Autoinflammatory Disease Section (TADS), Laboratory of Clinical Investigation and Microbiology (LCIM), NIAID, NIH
7Division of Rheumatology, Department of Pediatrics Associate Professor/Indiana University School of Medicine
8Certified Molecular Geneticist/Division of Intramural Research (DIR), NIAID, National Institutes of Health, Bethesda, MD
9Clinical Fellow/Translational Autoinflammatory Disease Section (TADS), Laboratory of Clinical Investigation and Microbiology (LCIM), NIAID, NIH
10Clinical Trials Research Nurse Practitioner/Translational Autoinflammatory Disease Section (TADS), Laboratory of Clinical Investigation and Microbiology (LCIM), NIAID, NIH
11Research Nurse/Translational Autoinflammatory Disease Section (TADS), Laboratory of Clinical Investigation and Microbiology (LCIM), NIAID, NIH
12Associate Professor/University of North Carolina School of Medicine
13Associate Professor/University of Cape Town, Red Cross War Memorial Children's Hospital
14Consultant in Paediatric Immunology and Infectious Diseases/CHI at Crumlin
15Consultant Pediatric Rheumatologist/CHI at Crumlin
16Consultant Adolescent and Pediatric Rheumatologist/CHI at Crumlin
17Associate Professor/KK Women's and Children's Hospital
18Assistant Professor/University of Pittsburgh, UPMC Childrens Hospital
19Professor/Division of Rheumatology, Department of Pediatrics, Hacettepe University Faculty of Medicine
20Professor of Pediatrics/University Children's Hospital, Medical Faculty Skopje
21Pediatric Rheumatologist/Akron Children’s Hospital
22Pediatric Immunologist/Azerbaijan Medical University
23Attending Physician, Allergy, Immunology, and Rheumatology/Ann & Robert H. Lurie Children's Hospital of Chicago/ Northwestern University Feinberg School of Medicine
24Assitant Professor/University of North Carolina School of Medicine
25Chief/Translational Autoinflammatory Diseases Section, NIAID, National Institutes of Health, Bethesda, MD,
Loss-of-function mutations in the X-linked gene, IKBKG, encoding NEMO, cause immunodeficiency with ectodermal dysplasia in males and incontinentia pigmenti in females. Previously, we identified 4 patients with gain-of-function splice site variants in IKBKG using targeted sequencing. These variants caused in-frame splicing-out of exon 5 and a novel chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE)-like autoinflammatory disease (AID) called NEMO-deleted exon 5 autoinflammatory syndrome (NEMO-NDAS). Because an IKBKG pseudogene (IKBKGP1) complicates genetic diagnosis of NEMO-NDAS by standard methods, we sought to develop a customized bioinformatics approach to identify disease-causing splice site variants in IKBKG.
A bioinformatics pipeline to computationally mask the IKBKGP1 pseudogene was used in whole exome/genome sequencing (WES/WGS) from patients with undifferentiated AIDs enrolled in an IRB-approved protocol. Western blot, cDNA sequencing, RNA-seq and an interferon (IFN) response gene (IRG) score (Nanostring) were performed.
Out of 682 individuals, including patients with undiagnosed AIDs and their parents, 13 patients (9 females and 4 males) with 8 different de novo splice-site variants in IKBKG were identified. All patients had early-onset disease (2 days to 3 years old), panniculitis, systemic inflammation and elevated IFN signatures. Other manifestations (>70%) included failure to thrive, lipodystrophy, hepatosplenomegaly, thrombocytopenia and B-cell lymphopenia. All patients were steroid-dependent and partially responded to anti-TNF (n=8) or JAK-inhibition (n=5) therapies. One patient died from opportunistic infections and another from sudden cardiac arrest. cDNA sequencing showed exon 5 skipping in the 10 patients tested and Western blot confirmed the splice product in 5/5 patients. RNA-seq showed a high frequency of exon 5 skipping in the 6 patients tested. The IFN scores had higher expression of IRG’s harboring NF-kB binding sites (CXCL10, GBP1 and SOCS1) than IRG’s without. Screening of IKBKG exon 5 splice-site variants in internal and public WES/WGS databases with more than 2,000 subjects is ongoing.
We describe a novel CANDLE-like autoinflammatory disease caused by de novo splice-site variants in IKBKG. Our bioinformatics pipeline masking the pseudogene provides a diagnostic tool for the diagnosis of WES/WGS data and early recognition may allow for better outcomes.
Acknowledgements: This work was supported by the NIH-NIAID-IRP
Keywords: IKBKG, NEMO, autoinflammatory disease, NEMO-deleted exon 5 autoinflammatory syndrome, panniculitis, cytopenia, bioinformatics
Disclosures: All authors indicated they had no financial relationships to disclose.
(20) Thymus Hypoplasia Resulting from 22q11.2 Deletion Syndrome Corrected by Reconstitution with Normal Mesenchymal Cells
Pratibha Bhalla, PhD1, Igor Dozmorov, PhD2, Christian Wysocki, PhD, MD3, Ashwani Kumar, n/a4, Chao Xing, n/a5, Ondine Cleaver, PhD6, Mary Louise Markert, MD PhD7, M. Teresa De La Morena, MD8, Nicolai van Oers, PhD6
1Postdoctoral Researcher/UT Southwestern Medical Center
2Associate Professor/UT Southwestern Medical center
3Pediatric Immunologist. Assistant Professor/UT Southwestern Medical center
4Computational Biologist/Bioinformatics Lab , UT Southwestern Medical Center
5Director/Bioinformatics Lab , UT Southwestern Medical Center
6Professor/UT Southwestern Medical Center
7Professor/Department of Immunology, Duke University, Durham
8Allergist/Division of Immunology, Department of Pediatrics, University of Washington and Seattle Children’s Hospital
Thymus hypoplasia occurs in many clinical conditions including particular congenital disorders, 22q11.2 Deletion Syndrome (22q11.2DS), Down syndrome, and individuals with autosomal recessive FOXN1 mutations. 22q11.2 deletion syndrome (22q11.2del) is the most common human microdeletion disorder known, affecting ~1/4000. Thymus tissues that were obtained from 22q11.2DS patients are size-restricted. This results in reduced T cell output, measured as low TRECs. This is very similar for patients with autosomal recessive FOXN1 mutations. In both clinical settings, an allogenic thymus tissue graft is a preferred treatment option to resolve the stromal cell defects characteristic for 22q11.2DS and FOXN1 mutations. To determine the molecular mechanisms contributing to a small thymus size caused by 22q11.2del, we compared the development of the thymus in embryos from mouse models of 22q11.2del relative to those from normal controls and from Foxn1 mutant mice. Reaggregate fetal thymic organ culture assays established that mesenchymal cells were causal to the formation of a small thymus in the setting of 22q11.2. Experimentally, replacing mesenchymal cells from 22q11.2del hypoplastic lobes with equivalent numbers of normal ones restored thymus tissue growth and thymopoiesis. Normal thymic epithelial cells could not support this tissue regeneration. This is distinct from the Foxn1 mutant mice, wherein the thymic epithelial cells cause the thymus hypoplasia/aplasia. Single-cell RNA sequencing of normal and hypoplastic thymus lobes revealed a differential gene expression that primarily impacted 5 distinct mesenchymal cell subsets. Differential expression of transcripts involved in cell-cell interactions, collagen matrices, and growth factors were evident in the hypoplastic lobes. This contrasted the transcriptome differences using hypoplastic lobes from Foxn1 mutant mice, wherein TECs are primarily impacted. Taken together, these findings suggest novel approaches for restoring thymus function in various clinical settings.
Keywords: DiGeorge Syndrome, Thymus Hypoplasia, Low TRECs, 22q11.2 Syndrome, Chromosomal Microdeletion
Disclosures: All authors indicated they had no financial relationships to disclose.
(21) Constrained chromatin accessibility in PU.1-mutated agammaglobulinemia patients
Carole Le Coz, PhD1, David Nguyen, MD PhD2, Amy Stiegler, PhD3, Amy Rymaszewski, PhD4, Jennifer Puck, MD5, Kathleen Sullivan, MD, PhD6, John Routes, MD7, Joud Hajjar, MD8, Sarah Nicholas, M.D.9, Prescott Atkinson, MD, PhD10, James Verbsky, MD, PhD11, Ivan Chinn, MD12, Alexander Marson, MD PhD13, Neil Romberg, MD14
1Research Associate/Children's Hospital of Philadelphia
2Assistant Adjunct Professor/UCSF
3Research Associate & Scientist/Yale university
4Research Associate/Medical College of Wisconsin
5Attending Physician/Pediatric Allergy, Immunology, and Blood and Marrow Transplant Division, University of California San Francisco Benioff Children’s Hospital
6Professor of Pediatrics, Chief of the Division of Allergy and Immunology/The Children’s Hospital of Philadelphia, Philadelphia, PA, USA
7Professor of Pediatrics, Medicine, Microbiology and Immunology/Medical College of Wisconsin
8Assistant Professor of Immunology, Allergy, and Rheumatology/Baylor College of Medicine
9Assistant Professor, Division of Immunology, Allergy and Retrovirology, Department of Pediatrics/Texas Children's Hospital, Baylor College of Medicine
10Professor of Pediatrics/Division of Pediatric Allergy & Immunology University of Alabama at Birmingham
11Associate Professor of Pediatrics, Medicine, Microbiology and Immunology/Medical College of Wisconsin
12Assistant Professor/Baylor College of Medicine/ Texas Children's Hospital
13Associate Professor/UCSF, Gladstone Institutes
14Assistant Professor of Pediatrics, Perelman School of Medicine at the University of Pennsylvania/Children's Hospital of Philadelphia
PU.1 is a pioneer transcription factor that determines hematopoietic cell fate by decompacting stem cell heterochromatin, regulating expression of PU.1-controlled genes and recruiting non-pioneer transcription factors to otherwise inaccessible nucleotides. Although PU.1 deficiency fatally arrests lymphopoiesis and myelopoiesis in mice and PU.1 mutations are common in human myeloid leukemias, primary PU.1-mediated diseases have yet not been described. Here, we identify six unrelated, agammaglobulinemic subjects each harboring novel, non-synonymous heterozygous mutations (four de novo, two unphased) in SPI1, the gene encoding PU.1.
To determine if and how SPI1 mutations cause PU.1-mutated agammaglobulinemia (PU.MA).
PU.MA subject DNA, peripheral blood and bone marrow samples were analyzed with whole exome sequencing, Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-Seq), flow cytometry and immunohistochemistry. The transcriptional capacity, tertiary stability, nuclear localization and DNA binding strength of PU.1 mutants were assessed with reporter lines, confocal microscopy, electrophoretic mobility shift assays, circular dichroism spectroscopy and quantitative fluorescence polarization. Hematopoiesis was modelled in vitro using genetically edited RS4;11 pro-B cell lines and human hematopoietic stem cells (HSCs).
All PU.MA subjects lacked circulating B cells and were deficient of PU.1-high expressing conventional dendritic cells. CITE-seq analysis of PU.MA marrow displayed developmental arrest between the pro and pre-B cell stages when there was a rising demand for PU.1 and PU.1-dependent gene products like BTK, FCRLA and IGLL5. In vitro HSCs genetically edited to carrying patient-similar SPI1 mutations, failed to differentiate into B cells. When overexpressed in HEK293 lines, three of six mutant, patient SPI1 alleles failed to produce detectable protein. The three detectable PU.1 mutants failed to drive transcription in reporter cells but also did not interfere with wild type PU.1’s ability to do so. Of these, one PU.1 mutant failed to localize to the nucleus and two mutants localized but could not bind target DNA. In SPI1-excised RS4;11 lines, PU.1 loss constrained chromatin access for non-pioneer TFs critical for B-cell development.
Our findings identify and molecularly describe a novel form of autosomal dominant agammaglobulinemia. PU.MA agammaglobulinemia underscores the essential, dose-dependent role of PU.1 in human B-cell lymphopoiesis.
Keywords: agammaglobulinemia, hematopoiesis, Pioneer Transcriptional Factor, PU.1, B cells
Disclosures: John Routes had contracted research at CSL Behring and Evolve Biologics. Joud Hajjar received research grants from Amplimmune, Arcus Biosciences, ARMO BioSciences, Atterocor/Millendo, Baxalta, BMS, Calithera Biosciences, Chao Physician-Scientist Foundation, CytomX Therapeutics, Eli Lilly, EMD Serono, Healios Onc, Immune Deficiency Foundation, ImmuneOncia, Incyte, Jeffrey Modell Foundation, Karyopharm, Kymab, MedImmune, Merck, National Cancer Institute, NeoImmuneTech, Neon Therapeutics, Novartis, OncoSec KEYNOTE-695, Pfizer, PsiOxus, Regeneron, Surface Oncology, The Texams Medical Center Digestive Diseases Center and Top Alliance; she was an Advisory Board member of Alfaisal University, Genome & Company, Horizon, and Pharming. All other authors had no financial relationships to disclose.
(22) Results of 105 patients who received cultured thymus tissue implants
Mary Markert, MD, PhD1, Stephanie Gupton, CPNP2, Elizabeth McCarthy, RN, MSN3
1Professor of Pediatrics/Department of Pediatrics and Department of Immunology, Duke University Medical Center, Durham, North Carolina, USA
2Nurse Practitioner/Duke University Medical Center
3Research Program Leader, Senior/Duke University Medical Center
Congenital athymia results in high early mortality due to infection and immune dysregulation. One hundred and three children with congenital athymia and 2 with severe combined immune deficiency received cultured thymus tissue implantation (CTTI) over 27 years at a single center.
Objectives: Objectives of the study were to assess survival, T cell numbers and function, adverse events, and infections.
The 105 patients received CTTI under one of 10 IRB approved protocols. Ninety-five patients with athymia met the eligibility criteria for complete DiGeorge anomaly or FOXN1 and had naïve T cells < 50/mm3. Ten additional patients were enrolled in an expanded access protocol and were evaluated separately. Ninety-five patients were included in the Efficacy Analysis Set (EAS); 10 additional patients were included in the Full Analysis Set (FAS).
The EAS shows Kaplan-Meier estimated survival rates at year 1 and year 2 post-implantation of 77% and 76% respectively. The median follow-up time for the EAS is 7.6 years and ranged from 0 to 25.5 years. The median CD3 T cell counts in the EAS were >600/mm3 2 years post implantation and remained below the 10th percentile for age. After developing naïve T cells, the patients were able to clear disseminated infections such as mycobacteria and parainfluenza III virus. The proliferative response to phytohemagglutinin was normal. In the first year of life, there were adverse events such as autoimmune disease, fever, and respiratory, gastrointestinal, and central line infections. Infections were responsible for 12 deaths, all during the first year post implantation. Of the 10 additional patients in the FAS, there were 3 deaths with a survival similar to the EAS.
The use of CTTI in athymic infants led to an excellent survival based on the Kaplan Meier Curve of the EAS analysis set. Of 95 patients, there are 70 survivors whose median age is 12.7 years post CTTI. All deaths related to poor immunity occurred in the first 2 years post CTTI. The development of T cell immunity enabled the patients to fight off infection. In summary, CTTI is a life-saving investigational therapy for congenital athymia.

Keywords: Thymus Transplantation, congenital athymia, athymia, complete DiGeorge, CHARGE, 22q11.2 deletion
Disclosures: Mary Markert received a research grant from Enzyvant Therapeutics, GmbH. All other authors had no financial relationships to disclose.
(23) Somatic reversion of pathogenic DOCK8 variants alters lymphocyte differentiation and function to effectively cure DOCK8 deficiency
Stuart Tangye, PhD1
1Theme Leader/Garvan Institute of Medical Research
Inborn errors of immunity cause monogenic immune dysregulatory conditions such as severe and recurrent pathogen infection, inflammation, allergy and malignancy. Somatic reversion refers to the spontaneous repair of a pathogenic germline genetic variant and has been reported to occur in a number of inborn errors of immunity with a range of impacts on clinical outcomes of these conditions. DOCK8 deficiency due to bi-allelic inactivating mutations in DOCK8 causes a combined immunodeficiency characterised by severe bacterial, viral and fungal infections, as well as allergic disease and some cancers. Here, we describe the clinical, genetic and cellular features of three patients with bi-allelic DOCK8 variants who, following somatic reversion in multiple lymphocyte subsets, exhibited improved clinical features, including complete resolution of infection and allergic disease, cure over time. Acquisition of DOCK8 expression restored defective lymphocyte signalling, survival and proliferation, as well as CD8+ T cell cytotoxicity, CD4+ T cell cytokine production, and memory B cell generation compared to typical DOCK8-deficient patients. Our temporal analysis of DOCK8-revertant and DOCK8-deficient cells within the same individual established mechanisms of clinical improvement in these patients following somatic reversion and revealed further non-redundant functions of DOCK8 in human lymphocyte biology. Lastly, our findings have significant implications for future therapeutic options for the treatment of DOCK8 deficiency.
Keywords: DOCK8, Somatic reversion, lymphocyte effector function, inborn errors of immunity
Disclosures: The author had no financial relationships to disclose.
(24) LRBA facilitates autophagy through binding to PIK3R4
Laura Gamez-Diaz, PhD1, Bodo Grimbacher, Prof. Dr. med.2
1Postdoc/Center for Chronic Immunodeficiency, Faculty of Medicine, Medical Center of the University Hospital, University of Freiburg, Freiburg, Germany
2Head of the laboratory/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
Patients with lipopolysaccharide responsive beige-like anchor protein (LRBA) deficiency present with a plethora of immune related defects including low numbers of switched memory B cells and plasma cells, as well as an impaired production of antibodies, leading to recurrent infections. However, the molecular mechanisms behind the defective B cell response remain unknown. To gain better insights into the possible roles of LRBA in B cell physiology, we screened for LRBA-interacting proteins using in silico analysis. Following validation by co-IP and PLA, we identified that endogenous LRBA interacts through its WD40 domain to the phosphoinositide 3-kinase regulatory subunit 4 (PIK3R4) in B cells. PIK3R4 (aka VPS15) is the regulatory subunit of VPS34, the catalytic subunit of the PI3K-III complex, which acts as a positive regulator of autophagy by producing phosphatidyl inositol-3 phosphate (PI(3)P). In fact, we observed that reduced LRBA impaired the production of PI(3)P upon autophagy induction leading to a blockade of the autophagosome-lysosome fusion, and resulting in a reduced mobility, abnormal accumulation and increased size of autophaghosomes, accompanied by an atypical lysosomal positioning, altogether causing an overall impaired autophagy flux and a decreased degradation of cargo material. Interestingly, LRBA-deficient cells exhibited enhanced activity of mTORC1 signaling, a key suppressor of autophagy whose activation possibly contributes to defective autophagy. Taken together, B lymphocytes lacking LRBA can form autophagosomes but they fail to fuse with lysosomes. Thus, we propose a role of LRBA at late stages of autophagy through the binding to PIK3R4, which is essential for plasma cell differentiation.
Keywords: LRBA, autophagy, B cells, primary immunodeficiency, mTOR, PIK3R4
Disclosures: Bodo Grimbacher received a research grant from Baxalta. The other author had no financial relationship to disclose.
02 Poster Presentations
(25) The origins of IL18 in models of Macrophage Activation Syndrome
Vinh Dang, B.S.1, Lauren Van Der Kraak, PhD2, Corinne Schneider, BS3, Timothy Hand, Phd4, Scott Canna, MD4, Emily Landy, B.S.5
1Research Technician/University of Pittsburgh
2Post Doctoral/University of Pittsburgh
3Lab Manager/University of Pittsburgh
4Principal Investigator/University of Pittsburgh
5PhD Candidate/University of Pittsburgh
Macrophage Activation Syndrome (MAS) is a life threatening hyperinflammatory complication of systemic juvenile idiopathic arthritis (sJIA) and other rheumatic diseases. IL-18 is an inflammasome-activated cytokine associated with augmenting the production of Th1 cytokines like IFNg and promoting cellular cytotoxicity. IL-18 is naturally and potently inhibited by IL-18 Binding Protein (IL-18BP). Serum IL-18 levels correlate strongly and specifically with MAS, particularly in the context of activating mutations in the NLRC4 inflammasome. What induces IL18 production and where it comes from in MAS remain unknown. Mice with excess IL-18 signaling (IL18BP KO) develop more severe TLR9 driven MAS. However, in a model of murine NLRC4 inflammasome hyperactivation, excess serum IL-18 comes from intestinal epithelial cells but, is not sufficient to exacerbate the TLR9 driven model of MAS. By contrast, the intestinal microbiota are critical for the development of TLR9-MAS in WT mice. We treated WT and IL-18BP KO mice with broad spectrum antibiotics prior to TLR9 stimulation with CpG and found that antibiotics were minimally protected in IL-18BP KO mice, suggesting that the microbiota may not be necessary for pathogenic IL-18 induction. Indeed, mice colonized with immunostimulatory microbes (Segmented Filamentous bacteria or Tritrichomonous) were not more susceptible to TLR9-MAS. Furthermore, increased IL-18 in TLR9-MAS did not derive from intestinal microbiota, suggesting it may derive from myeloid cells. We queried publicly available transcriptional data and found that IL18 transcription was frequently stimulated by Interferon signaling and strongly correlated with PPARg transcription in macrophages. Overall, these data suggest that although the intestine is a rich source of IL-18, gut-derived IL-18 may not be efficient at promoting MAS. Understanding the signals that drive IL-18, potentially including IFN and PPARg may help us to understand susceptibility to MAS.
Keywords: Macrophage Activation Syndrome, Hyperinflammation, IL-18, Cytokine, PPARg, Interferon, Microbiome, Antibiotics
Disclosures: All authors indicated they had no financial relationships to disclose.
(26) Recurrent inflammatory episodes, urticaria, and severe T-cell lymphopenia due to a novel heterozygous CD48 mutation
Laura Ann Wang, MD, MPH1, Vijaya Knight, MD, PhD2, Kaylee Dollerschell, MS, CGC3, Jordan Abbott, MD, MA4, Cullen Dutmer, MD4
1Allergy & Immunology Fellow/Children's Hospital Colorado, University of Colorado School of Medicine, Aurora, CO, USA
2Associate Professor of Pediatrics/Section of Allergy & Immunology, Children's Hospital Colorado, University of Colorado School of Medicine, Aurora, CO, USA
3Instructor of Pediatrics/Section of Genetics & Metabolism, Children's Hospital Colorado, University of Colorado School of Medicine, Aurora, CO, USA
4Assistant Professor of Pediatrics/Section of Allergy & Immunology, Children's Hospital Colorado, University of Colorado School of Medicine, Aurora, CO, USA
CD48 is a glycosylphosphatidylinositol (GPI)-anchored protein of the signaling lymphocytic activation molecule (SLAM) family that is expressed on the surface of hematopoietic cells and plays an important role in lymphocyte activation and differentiation. Volkmer et al recently described a 24-year-old man with a history of recurrent inflammatory disease in which a de novo S220Y mutation in the GPI-anchor region of CD48 was reported as causative.1 Herein, we describe a young man with a novel CD48 mutation affecting the same codon (S220F), resulting in a similar clinical phenotype.
A 17-year-old male presented with a 14-year history of recurrent fever, fatigue, and atypical urticaria. Episodes lasted 4-7 days. The episodes occurred as frequently as monthly but became less frequent. Two severe episodes required hospitalization, where lymphopenia and thrombocytopenia were observed. His infection history included 2 episodes of norovirus gastroenteritis, 1 episode of rotavirus gastroenteritis, and 1 influenza A (2009-H1N1) infection, all of which triggered inflammatory episodes.
His inter-episode diagnostic evaluation revealed severe T-cell lymphopenia (214 cells/mcL), high sCD25 (1233 pg/mL), high triglycerides (171 mg/dL), low IgM (19 mg/dL), and Streptococcus pneumoniae antibody deficiency. T-cell proliferation to phytohemagglutinin, NK-cell cytotoxicity (chromium release), ESR, CRP, and ferritin were normal. Trio whole-exome sequencing identified a de novo heterozygous variant in CD48 (c.659C>T, p.S220F) and flow cytometric analyses demonstrated reduced T-cell, B-cell, and monocyte surface CD48 expression.
We report a novel, de novo, heterozygous variant in CD48 associated with recurrent inflammatory episodes and severe T-cell lymphopenia that recapitulates the clinical phenotype induced by another aromatic amino acid substitution at codon 220.1 Serine 220 is the site of covalent GPI anchor attachment, and loss of that attachment point is proposed to result in reduced surface membrane CD48. The precise mechanism of pathogenicity remains to be determined.
References: 1. Volkmer B, Planas R, Gossweiler E, et al. Recurrent inflammatory disease caused by a heterozygous mutation in CD48. J Allergy Clin Immunol. 2019;144(5):1441-1445.e17. doi:10.1016/j.jaci.2019.07.038
Keywords: CD48, T-cell lymphopenia, Recurrent fever
Disclosures: All authors indicated they had no financial relationships to disclose.
(27) BTK inhibitors for Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): A Systematic Review
Michael Stack, BS1, Keith Sacco, MD2, Alicia Livinski, MPH, MLS3, Luigi Notarangelo, MD4, Michail Lionakis, MD, ScD5
1Postbaccalaureate Fellow/Laboratory of Clinical Immunology and Microbiology, National Institute for Allergy and Infectious Diseases, NIH, Maryland, USA
2Clinical Fellow/Lab of Clinical Immunology and Microbiology, Immune Deficiency Genetics Section, NIAID, NIH, Bethesda, MD, USA
3Biomedical Librarian/Informationist/National Institutes of Health Library, Division of Library Services, Office of Research Services
4Chief/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
5Chief, Fungal Pathogenesis Section/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases/National Institutes of Health, Bethesda, MD, USA
Bruton tyrosine kinase (BTK) regulates B cell and macrophage signaling, and activation. Blood monocytes from patients with severe COVID-19 showed increase in BTK activation and production of interleukin-6 correlating with systemic inflammation. Inhibiting BTK has been hypothesized to prevent lung injury in COVID-19 patients, alike to its protective effect in mice for lethal influenza-induced acute lung injury. Our aim was to evaluate BTK inhibitor (BTKi) use during acute COVID-19 infection and assess whether BTK inhibitors are associated with specific clinical outcomes.
Our primary aim was to describe clinical outcomes following BTKi use in acute SARS-CoV-2. We searched PubMed, Embase, Web of Science, MedRxiv, BioRxiv and 5 clinical trials registries on 30 December, 2020. Clinical studies with at least 5 patients were included. Case reports and reviews were excluded.
126 unique articles were identified, of which 6 met our inclusion criteria. Sample size ranged from 6 to 126. Patient populations observed included subjects hospitalized with COVID-19 (6/6) and admitted to ICU (5/6). Patient age range was between 35 and 98 years among the pooled 6 studies. 4 studies followed patient cohorts that were on BTKi for treatment of their lymphoproliferative disease -1 cohort of Waldenstrom’s macroglobulinemia and 3 studies of leukemia patients (Table 1). The most common clinical outcomes measured were oxygen requirements (4/6) and hospitalization (3/6), secondary clinical outcomes included length of intubation and most aggressive method of oxygenation. Standard of care reflected not only the date of study, but also the pre-existing conditions of the cohort. Studies used full doses of acalabrutinib (3/6) and/or ibrutinib (2/6). Three studies found favorable hospitalization rate/duration in those on full-dose BTK inhibitors. One study did not find any significant difference in survival for those on BTKi.
BTK inhibition shows promise for ameliorating hyper-inflammation in patients with severe COVID-19. Randomized Clinical trials assessing three BTK inhibitors (ibrutinib, acalabrutinib, zanubrutinib) in patients with severe SARS-CoV-2 are underway.
Acknowledgment: This research was supported by the Intramural Research Program of the NIH.

Table 1. Summary of studies evaluating clinical outcomes following BTK inhibitor use in acute SARS-CoV-2.
Keywords: COVID-19, Bruton's tyrosine Kinase (BTK), X-linked agammaglobulinemia (XLA), Systematic Review, Clinical Outcomes, BTK inhibitors
Disclosures: All authors indicated they had no financial relationships to disclose.
(28) A case of Spondyloenchondrodysplasia with immune dysregulation presenting as Systemic Lupus Erythematous
Juanita Valdes Camacho, MD1, Deepika Singh, MD2, Rosa Bacchetta, MD3, Kenneth Weinberg, MD4, Alma-Martina Cepika, MD, PhD5, Mansi Narula, MS6, David B Lewis, MD7, Yael Gernez, MD, PhD8, Katja Weinacht, MD, PhD9
1Allergy and Immunology Fellow/Stanford University HealthCare
2Pediatric Rheumatologist/Valley Children's Hospital Madera
3associate Professor (Research) of Pediatrics (Stem Cell Transplantation)/Stanford
4Professor of Pediatrics - Stem Cell Transplantation/Stanford
5Instructor - Pediatric Stem Cell Transplantation/Stanford
6Life Science Research Professional/Stanford
7Chief, Department of Pediatrics, Division of Allergy, Immunology and Rheumatology/Stanford
8Assistant Professor of Pediatric Allergy and Immunology/Stanford University
9Assistant Professor of Pediatric Stem Cell Transplantation/Stanford University
Monogenic lupus is a form of systemic lupus erythematosus that occurs in patients with a single gene defect. This rare variant of lupus generally presents with early onset severe disease, often with recalcitrant autoimmune cytopenias, renal or neurologic disease. Spondyloenchondrodysplasia with immune dysregulation (SPENCDI) is a rare type I interferonopathy, caused by biallelic mutations in the ACP5 gene. Clinically, SPENCDI is characterized by the triad of skeletal dysplasia, autoimmunity and variable neurologic findings, ranging from asymptomatic brain calcifications to severe developmental delay with spasticity. SPENCDI has been implicated in monogenic lupus, however, therapeutic considerations targeting the underlying mechanism have never been described.
A 7 year old girl with history of short stature and developmental delay, was diagnosed in 2017 with SLE characterized by petechial and purpuric rash, hematologic involvement with anemia, thrombocytopenia and splenomegaly, with elevated inflammatory markers (ESR 78mm/h, CR 1.2), hypergammaglobulinemia with secondary complement deficiency (CH50 = 0 mg/dl mg/dl, reduced C2 function, C3 41 mg/dl, C4 < 2.9 mg/dl), positive high titer ANA >1:1280, elevated dsDNA >300 IU/ml, elevated MPO antibody, without evidence of renal involvement. Initially, she had a favorable response to steroids, however, cytopenias immediately flare once steroids were withdrawn. The patient received rituximab and IVIG with transient effect, and was started on Mycophenolate to achieve disease control.
Extended immune function testing was not suggestive of a primary immunodeficiency. Work up for genetic causes of refractory Evans syndrome revealed biallelic mutations in the ACP5 gene, including one previously reported pathogenic variant and one variant of undetermined significance. Hands and spine radiographs demonstrated the characteristic radiolucent metaphyseal and vertebral lesions of spondyloepimetaphyseal dysplasia establishing the diagnosis of SPENCDI.
SPENCDI is a type I interferonopathy. Based on the favorable therapeutic response of patients with other type I interferonopathies Janus kinase (JAK) inhibitors, a therapeutic trial with the JAK inhibitor ruxolinitib has been initiated.
SPENDCI is a monogenic disease which can present with refractory cytopenias and other clinical features of SLE. Although SLE is a clinical diagnosis, vigilance for underlying genetic defects is warranted and may have important implications on management
Keywords: ACP5, SPENCDI, Monogenic Lupus, Interferonopathy, JAK inhibitor
Disclosures: All authors indicated they had no financial relationships to disclose.
(29) Severe scabies In Two Patients with Gain-of-function mutations in Signal transducer and activator of transcription 1
Sara Van Meerbeke, MD1, Robin Gehris, MD2, Brian Campfield, MD3, Hey Chong, MD, PhD4
1Allergy & Immunology Fellow/UPMC Children's Hospital of Pittsburgh
2Clinical Associate Professor of Dermatology and Pediatrics/UPMC Children's Hospital of Pittsburgh
3Assistant Professor of Pediatrics, Division of Infectious Diseases/UPMC Children's Hospital of Pittsburgh
4Associate Professor of Pediatrics, Division of Allergy & Immunology/UPMC Children's Hospital of Pittsburgh
Gain-of-function mutations in STAT1 are the most common syndromic cause of chronic mucocutaneous candidiasis and can lead to a wide spectrum of infections and autoimmune conditions as well as predispose to cerebral aneurysms and malignancy. Norwegian scabies, or crusted scabies, is characterized by infestation with millions of Sarcoptes scabei mites and thick, hyperkeratotic crusted skin and linked with immunosuppression most notably HIV although not classically STAT1 mutation.
Patient 1 is a 26-year-old female who presented at 22 years of age with recurrent oral candidiasis, hypothyroidism, and persistent hyperkeratotic skin rash. She later developed pulmonary Cryptococcal infection. Her immune evaluation revealed the following results: Serum immunoglobulins with elevated IgG but normal IgM and IgA. Lymphocyte subset showed CD3+, CD4+, and NK lymphocytopenia (691, 322 and 33 respectively). Lymphocyte proliferative response normal to PHA and PWM. Pt underwent punch biopsy which was consistent with Norwegian scabies treated with oral ivermectin and topical permethrin. Family genetic testing revealed STAT1 pathogenic variant c.820C>T (p.Arg274Trp).
Patient 2 is an 11-year-old male with chronic mucocutaneous candidiasis, who presented at 7 years of age with several week history of erythematous, pruritic rash across trunk and legs. His immune evaluated revealed elevated IgG at 1320 but normal IgA and IgM. Lymphocyte subset showed CD3+, CD4+, CD8+, B and NK lymphocytopenia (960, 446, 379, 103 and 24 respectively). WES revealed STAT1 c.1256C>G, p.T419R and c.1874(-8)C>T mutations. Microscopic exam of a skin scraping showed evidence of mites consistent with Norwegian scabies treated with oral ivermectin and topical permethrin.
While Norwegian scabies can be associated with immunosuppression, there is no clear association with defects in STAT1 pathway. We report two patients with STAT1 GOF who developed persistent hyperkeratotic skin crusts and ultimately diagnosed with Norwegian scabies. Literature review also reveals a case report by Sampaio et al. of patient with STAT1 GOF mutation who developed disseminated Rhodococcus infection, chronic mucocutaneous candidiasis, hypothyroidism and Norwegian scabies. These three cases suggest that in patients with STAT1 GOF, Norwegian scabies should be considered as etiology of skin rash. Further studies are needed to further elucidate underlying susceptibility to scabies infection.
Keywords: STAT1 GOF, Norwegian scabies, Skin rash
Disclosures: All authors indicated they had no financial relationships to disclose.
(30) The effect of critical illness on flow cytometric testing in identifying primary immune deficiency
Ali Hemyari, MD1, Liyun Zhang, MS2, Jenny Andres, MBA, BSN, RN3, John Routes, MD4, Ke Yan, PhD2, James Verbsky, MD, PhD5
1Pediatric Critical Care Fellow/Medical College of Wisconsin
2Biostatistician/Medical College of Wisconsin
3Quality and Outcomes Manager/Children's Wisconsin
4Professor of Pediatrics, Medicine, Microbiology and Immunology/Medical College of Wisconsin
5Associate Professor of Pediatrics, Medicine, Microbiology and Immunology/Medical College of Wisconsin
Flow Cytometry is used often in the diagnosis of primary immune deficiencies. Often these tests are sent in patients in varying states of health while in outpatient and inpatient settings, and it is unknown how clinical status can affect flow cytometric results.
We retrospectively analyzed flow cytometry results from the Clinical Immunology Research Laboratory at the Medical College of Wisconsin, a CLIA approved reference laboratory, and included any subject with two or more flow cytometric tests. Each test measured the absolute counts of up to four lymphocyte subsets: CD3+CD4+, CD3+CD8+, CD19+, and/or CD56+16+. Tests results were designated a percentage of the measurements that were normal. We then identified which clinical division ordered the test and the final diagnosis. A generalized linear mixed model with Maximum Likelihood Estimation was used to compare the tests ordered by clinical areas after considering the repeated measures within patients. Statistical significance was set at p < 0.05.
Of a total of 6,462 tests from 1,515 patients, 11% of all tests had measurements which were 100% normal, and only 42 patients (2.7% of total patients) had every test with 100% normal measurements. Of the 1,370 patients who had tests with < 100% normal measurements (an abnormal test), 224 patients had testing that “normalized” in future testing. When the mean percent normal measurements were ranked by the eight clinical divisions (after excluding tests from the bone marrow transplant, oncology, solid organ transplant, and HIV divisions), tests ordered by the Allergy division had the highest percent normal measurements and tests ordered in the ICU had the lowest percent normal measurements. Both were statistically significant when compared with other clinical divisions (Table 1).
These results confirm that flow cytometry can be greatly affected by the clinical status of the patient, as supported by the high rate of abnormal tests in the ICU, while outpatient testing had higher rates of normal tests. These data would argue that flow cytometric testing to identify primary immune deficiency during critical illness may result in a high false positive rate, and that this testing should be delayed until the clinical status has improved.

Table 1. Percent normal measurements for each ordering clinical area
Keywords: Primary Immunodeficiency testing, Flow Cytometric Testing, Critical Illness, Lymphocyte Subset Testing
Disclosures: John Routes had contracted research with CSL Behring and Evolve Biologics. All other authors had no financial relationships to disclose.
(31) Child with hyperinflammatory syndrome and aplastic anemia with CD247 pathogenic variant
Lourdes Ramirez, MD1, Carmen Ballestas, MD2, Maria Pilar Gutierrez, MD3, Luigi Notarangelo, MD4, Hanadys Ale, MD5
1Pediatric Resident/Joe DiMaggio Children's Hospital/Memorial Healthcare System
2Attending Physician/Joe DiMaggio Children's Hospital, Division of Pediatric Hematology and Oncology
3Attending Physician/Joe DiMaggio Children's Hospital, Division of Pediatric Infectious Diseases
4Chief/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
5Attending Physician/Joe DiMaggio Children's Hospital, Division of Pediatric Immunology and Allergy
Severe combined immunodeficiency (SCID) as a result of mutations in the invariant homodimer CD247 (CD3-zeta) lead to mild T-cell lymphopenia, in contrast to more severe forms of SCID. CD247 is also expressed in NK cells, and mutations in CD247 have been linked to NK cell hypo-responsiveness, even in heterozygous carriers. The full clinical manifestations of these patients however, have not been clearly described. Here we present a case of a patient with a pathogenic variant in CD247 who developed hyperinflammatory syndrome and refractory aplastic anemia.
A 12-year-old female with Noonan syndrome with multiple lentigines, associated prolonged QT syndrome, unrepaired VSD, recurrent sinopulmonary infections and hypogammaglobulinemia of unknown etiology, presented with a 4-day history of jaundice, abdominal pain and emesis. Evaluation demonstrated leukopenia, thrombocytopenia, transaminitis and direct hyperbilirubinemia. She developed fevers with hyperferritinemia, hypertriglyceridemia, elevated lactate dehydrogenase and worsening transaminitis. Soluble IL-2 receptor-α was significantly elevated at 12,340 pg/mL at its peak. Immunologic profiling revealed low IgG, significant T-cell lymphopenia, and borderline low NK cells with depressed NK-cell function. Invitae primary immunodeficiency panel revealed a heterozygous pathogenic variant in CD247 (c.301C>T p.Gln101*). Bone marrow (BM) biopsy initially showed normal cellularity without evidence of malignancy. She was treated with IVIG and high dose steroids with clinical improvement. Upon weaning steroids, she developed worsening cytopenias with a follow-up BM biopsy demonstrating aplastic anemia. Rituximab was started without improvement of cell counts. She was transferred to an outside institution where she received immunosuppressive therapy for aplastic anemia with Eltrombopag, cyclosporine and ATG. That hospital course was complicated by neutropenic fevers, multiple deep-seated infections and respiratory failure. Since failing immunosuppressive therapy, she is undergoing palliative management while awaiting BM transplantation.
Heterozygous carriers of CD247 pathologic variants have not been reported to exhibit CD247 disease. This report, however, suggests that perhaps the carrier status may contribute to this patients’ underlying immunodeficiency characterized by hypogammaglobulinemia, low NK cells, and absent NK function and T cell lymphopenias; ultimately leading to a hyperinflammatory state and aplastic anemia. These patients may require BM transplantation as definitive treatment early in their presentation.
Keywords: Primary immunodeficiency, aplastic anemia, hyperinflammatory syndrome, CD247
Disclosures: All authors indicated they had no financial relationships to disclose.
(32) Cortico-resistant Seronegative Autoimmune hepatitis
Eunice Sandoval-Ramirez1, Nery Eduardo Solis-Perales, MD2, Juan Manuel Alcántar-Fierros, MD3, Luis-Humberto Cruz-Contreras, MD4, Ignacio Camacho-Meza, MD1
1Pediatric Allergy/Hospital de Especialidades Pediatrico Leon
2Pediatric Gastroenterology/Hospital de Especialidades Pediatrico Leon
3Pediatric Surgery/Hospital de Especialidades Pediatrico Leon
4Pathologist/Hospital Materno Infantil Irapuato
Autoimmune hepatitis is characterized by chronic, progressive inflammation of liver with the presence of autoantibodies, hypergammaglobulinemia and interface hepatitis in the histological study, it presents several phenotypes, we focus on the seronegative; patients present a characteristic clinical picture, compatible laboratory and histological findings without the presence of "standard" antibodies, with a frequency of presentation between 1 and 5% of all reported cases, the association with thrombocytopenia is rare (1.4%).
The case of an 11-year-old patient is described, who begins suffering jaundice, hepatic pain, elevated liver enzymes and direct bilirubin. She was admitted to our hospital with ascites, hepatomegaly, anemia, and thrombocytopenia (52 thousand / μL platelets). No dilation of the bile duct or presence of stones was found. Negative viral serology, anti-KLM 18.6 antibodies, anti-smooth muscle, anti-mitochondrial, rheumatoid factor, Anti DNA, antinuclear and VDRL antibodies were negative, serum IgG 2780mg / dL, with a biopsy report of severe chronic active hepatitis and 60% necrosis, severe activity 3 of 4, mild fibrosis stage EFE1 DF4.
After excluding viral, toxic or hereditary causes of hepatitis, in the presence of another autoimmune disease, the most likely cause is a hepatic autoimmune process, despite not finding standard immunological markers but meeting criteria of the international group of autoimmune hepatitis. management begins with steroid and azathioprine without complete remission of the biochemical markers of hepatitis, so alternative treatment with mycophenolate mofetil at a dose of 1gr / day and increase in the dose of Azathioprine to 150 mg / day and prednisone 0.75mg / kg / day.
Upon evaluation 6 weeks later, we found a decrease in the values of liver enzymes and platelets 103 thousand / μL. Currently, it continues with the alternative management established with mycophenolate mofetil (MMF) associated with standard treatment of azatiprine and steroid with adequate response at the hepatic and hematological level.
Figure 1. Nejib trichrome (40x) Panoramic view showing viable parenchyma areas marked with (*) alternating with large areas of necrosis.
Figure 2. Nejib and PAS trichrome (100x). Interface hepatitis is shown with a predominance of lymphocytic infiltrate with severe inflammatory activity and necrosis, rosette formation (arrows) is observed. We can also observe moderate intracellular and canalicular cholestasis.
Keywords: Autoimmune, Hepatitis, Seronegative, Cortico-resistan
Disclosures: All authors indicated they had no financial relationships to disclose.
(33) X-linked agammaglobulinemia incidentally identified during pre-surgical ABO blood cross-matching: case report and review of the literature
Jennifer Blase, MD, PhD1, Kelly Walkovich, MD2, Thomas Michniacki, MD3, Mark Hannibal, MD, PhD4
1Fellow Physician, Pediatric Hematology & Oncology/University of Michigan
2Associate Professor, Pediatric Hematology & Oncology/University of Michigan
3Assistant Professor, Pediatric Hematology & Oncology/University of Michigan
4Associate Professor, Genetics, Metabolism & Genomic Medicine/University of Michigan
Mismatch between forward and reverse blood group typing, i.e. ABO typing discrepancy, is most commonly caused by cold agglutinins or weak/missing antibodies due to age or use of immunosuppressive agents. However, inborn errors of immunity with humoral defects can also result in ABO typing discrepancy. While more commonly diagnosed following frequent or unusual infections, we describe a case of X-linked agammaglobulinemia (XLA) where ABO typing discrepancy served as the primary trigger for diagnosis as well as a related literature review.
The medical record of the patient case was reviewed. A literature review was conducted using the keywords “immunodeficiency” and either “ABO typing” or “isoagglutinin” in Pubmed, with review of English articles published between 1958 and 2020.
We describe a 6-year-old male patient with a history of left knee septic arthritis and sinopulmonary infections who presented with fever, cough, rhinorrhea, left knee arthralgia and edema. He was found to have leukocytosis, severe thrombocytopenia, elevated inflammatory markers, and left knee effusion, consistent with recurrent septic arthritis. He was initially transfused with O negative platelets urgently for epistaxis. However, subsequent type and screen performed prior to left knee irrigation and debridement revealed type B blood group with forward typing but no evidence of anti-A antibodies during reverse typing. This prompted an immunodeficiency workup, revealing hypogammaglobulinemia and B cell deficiency. Bruton tyrosine kinase (BTK) protein expression was decreased, and a hemizygous pathogenic variant in BTK gene, c.1775C>A (p.Ser592Tyr), diagnostic of XLA, was found. IVIG supplementation was initiated.
A literature search, as above, yielded 202 articles, seven of which detailed immunodeficiency cases with abnormal blood bank testing. This included case reports of common variable immunodeficiency (CVID), XLA, selective IgM immunodeficiency (SIgMID), Good’s syndrome, and Wiskott-Aldrich syndrome (WAS). This revealed only a small number of published cases detailing ABO typing discrepancies in patients with primary immunodeficiency syndromes (Table 1).
This case and subsequent literature review demonstrate the utility of inter-specialty communication and highlight the need for a high index of clinical suspicion for primary immunodeficiencies in cases of ABO typing discrepancy.

Keywords: X-linked agammaglobulinemia, ABO typing discrepancy, Immunodeficiency
Disclosures: Kelly Walkovich is a consultant for Sobi Pharmaceuticals. All other authors had no financial relationships to disclose.
(34) G6PD deficiency with recurrent infections in a pediatric case
Tyrone Coyle, MD1, Sherry Farzan, MD2, Artemio Jongco, MPH, MD, PhD, FACP, FACAAI, FAAAAI2
1Allergy/Immunology fellow/Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
2Assistant Professor of Medicine and Pediatrics/Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
Glucose-6-phosphate dehydrogenase (G6PD) is a critical enzyme for erythrocytes and neutrophils. G6PD reduces NADP+ to NADPH which protects erythrocytes from oxidative injury and is a key substrate for neutrophil respiratory burst. G6PD deficiency is a congenital X-linked disorder. Clinical manifestations are often absent but various triggers can precipitate episodes of acute hemolytic anemia. Usually, diminished G6PD levels will not cause neutrophil dysfunction unless enzyme levels are below 5% of normal.
A 6-year-old Turkish male with history of asthma and chronic neutropenia was seen in an outpatient setting. He had a prolonged history of frequent infections characterized by fever, cough, diarrhea and myalgias. He was regularly treated with courses of antibiotics but no known bacterial infections were reported. Family history was significant for a brother, aunt and cousin with cystic fibrosis. Prior chloride sweat test and CFTR genetic testing were normal.
Initial immunodeficiency workup showed normal levels of immunoglobulins, T and B cells, CH50, MBL, mitogen proliferation, myeloperoxidase stain, DHR assay and negative anti-granulocyte antibodies. Vaccination titers were protective to all tested vaccines except varicella. Abnormal labs included CBC with neutropenia (range 1100 – 1700cells/μL), mildly decreased AH50 (40) and undetectable G6PD level. A commercial targeted primary immunodeficiency panel showed a hemizygous pathogenic c.563C>T (p.Ser188Phe) G6PD variant, confirming G6PD deficiency. He was also heterozygous for variants of uncertain significance in C8B, C9, CIITA, COL7A1, FANCA, PRKDC, RNASEH2B, and SPINK5.
The diagnosis of G6PD deficiency was initially surprising given the normal DHR assay and no prior clinical history suggestive of hemolysis. Despite the normal DHR assay, G6PD deficiency in conjunction with neutropenia is the suspected etiology of recurrent infections for our patient. The DHR assay has proven less sensitive than other modalities such as nitroblue tetrazolium (NBT) for assessing oxidative burst in G6PD. Severe G6PD deficiency can closely mimic chronic granulomatous disease (CGD) with recurrent bacterial and fungal infections, due to impaired reactive oxygen species dependent neutrophil extracellular trap (NET) formation, which occurs in the absence of NADPH. Given the similar pathogenesis, he may benefit from prophylactic antimicrobials which has proven effective in reducing morbidity in CGD.
Keywords: G6PD deficiency, recurrent infections, neutropenia, DHR assay, Pediatrics, NADPH, chronic granulomatous disease, neutrophil extracellular trap
Disclosures: All authors indicated they had no financial relationships to disclose.
(35) Clinical and Treatment History of Patients with Partial DiGeorge Syndrome and Autoimmune Cytopenia at a Tertiary Center
Priya Patel1, Emma Emma Westermann-Clark, MD2, Sonia Joychan, MD3, Ahmad Shaker, MD4, Boglarka Ujhazi, BS5, Cristina Meehan, BS6, Maryssa Ellison, n/a7, Krisztian Csomos, PhD8, Sumai Gordon, B.S.9, Daime Nieves, APRN10, Anthony Nguyen, MSPH11, Irmel Ayala, MD12, Jolan Walter, MD, PhD13, Panida Sriaroon, MD14
1Resident/Johns Hopkins All Children's Hospital
2Assistant Professor/Division of Allergy and Immunology, University of South Florida Morsani College of Medicine
3Physician/Dearborn Allergy & Asthma Clinic, P.C.
4Physician/Baycare Medical Group
5Senior Biological Scientist/University of South Florida
6Medical student/University of Alabama
7Department of Pediatrics/University of South Florida
8PhD/Massachusetts General Hospital
9B.S./University of South Florida
10APRN/University of South Florida
11Biostatistician/Johns Hopkins All Children's Hospital
12Physician/Johns Hopkins All Children's Hospital
13Division Chief and associate professor/Johns Hopkins All Children’s Hospital; Department of Pediatrics, Division of Allergy and Immunology, University of South Florida Morsani College of Medicine
14Medical Director and Associate Professor/Johns Hopkins All Children’s Hospital; Department of Pediatrics, Division of Allergy and Immunology, University of South Florida Morsani College of Medicine
Partial DiGeorge Syndrome (pDGS) presents with a wide phenotypic heterogeneity. Patients with pDGS can present with immune dysregulation, most commonly autoimmune cytopenia (AIC). The clinical spectrum, treatment outcome, and biomarkers for AIC in patients with pDGS are not well understood, and a standardized treatment pathway is needed.
The aim of this project is to characterize the natural history and immune phenotype of the heterogeneous population of pDGS with AIC, ranging from manageable to severe, and biomarkers predicting refractory AIC in pDGS.
Data on clinical presentation, disease severity, immunological phenotype, treatment selection and response for patients with pDGS with AIC were collected via retrospective chart review.
Of 69 pDGS patients, 28 (40%) had a history of cytopenia. 10 of 28 patients with cytopenia had AIC (14% of cohort) at our tertiary academic center. Seven patients had multi-lineage AIC (Evans syndrome (ES)) and 3 had immune thrombocytopenia alone. Five of the 7 pDGS patients with ES had refractory cytopenias requiring second line therapy beyond steroids and high dose immunoglobulin. Six of the 7 patients with ES were considered refractory for repeated hospitalizations and requiring multiple different combinations of treatments for stabilization. These 6 refractory patients had lower average nadir of hemoglobin (6.35 g/dL), white blood cell count (2.92x103/μL), and platelet (14x103/μL) count during acute AIC episodes compared to the 4 patients who were not refractory to initial therapy (11.9 g/dL, 9.15 x103/μL, and 77 x103/μL respectively). Those 6 patients with refractory AIC also had both evidence of antibody deficiency syndrome (ADS) and lower naïve T cell counts, compared to pDGS patients with mild AIC and the general population. Three of the 6 refractory patients received rituximab; other agents included mycophenolate mofetil, cyclophosphamide, and azathioprine. One patient is stabilized on sirolimus and high dose subcutaneous immunoglobulin.
AIC is common in pDGS and often treatment-refractory. The findings of this study suggest that lower naïve T cell count and ADS with need for high dose immunoglobulin replacement therapy distinguish pDGS patients with severe refractory AIC. Long term T cell therapy may improve AIC for these patients after initial treatment with rituximab.

Table 1 and Figure 1
Keywords: partial DiGeorge Syndrome, Autoimmune, Cytopenia
Disclosures: All authors indicated they had no financial relationships to disclose.
(36) Novel Use of Dupilumab in the Treatment of Recurrent Bacterial Scalp Infections and Axillary Lymphadenitis in a Patient with Hyper-IgE Syndrome
Jason Moraczewski, MD1, Katherine Tison, MD2, Lisa Kobrynski, MD, MPH3
1Pediatrics Resident/Emory University and Children's Healthcare of Atlanta
2Fellow/Emory University and Children's Healthcare of Atlanta
3Professor of Pediatrics, Marcus Professor of Immunology, Section Chief Allergy-Immunology/Emory University and Children's Healthcare of Atlanta
Hyper-IgE syndrome (HIES) is a primary immunodeficiency associated with elevated serum immunoglobulin E (IgE), bacterial pneumonias complicated by pneumatoceles, recurrent skin abscesses, and early-onset allergic manifestations, including an eczematous, impetiginized, pruritic rash similar to atopic dermatitis. Here we describe the successful treatment of bacterial skin infections and secondary axillary lymphadenitis with dupilumab in a 13-year-old African American female with HIES. By 2 years of age, she had recurrent staphylococcal infections of her scalp requiring incision and drainage procedures, otitis media requiring myringotomy tube placement, and recurrent pneumonia requiring hospitalization. Serum IgE levels were markedly elevated at 14,000 kU/mL, and she was subsequently diagnosed with HIES based on clinical features and the NIH scoring system. Further genetic testing revealed a STAT3 loss-of-function mutation. At 13 years of age, the patient presented with suppurative infections of her scalp, sinusitis, and left axillary lymphadenitis despite multiple courses of antimicrobial therapy and adequate intravenous immunoglobulin replacement. After 3-months of doxycycline therapy followed by a course of culture-directed antimicrobial and antifungal therapy status post axillary lymph node incision and drainage, she continued to have progression of her scalp involvement and worsening left axillary lymphadenitis. Due to refractory symptoms despite appropriate antibiotic treatment, the decision was made to initiate dupilumab. Dupilumab is an IgG4 human monoclonal antibody that inhibits IL-4 and IL-13 signaling by binding to the IL-4Rα subunit, thus preventing the release of proinflammatory cytokines, chemokines, nitric oxide, and IgE. She received the pediatric weight-based dosing for children ≥6 years for moderate to severe atopic dermatitis with initial 600mg dose followed by a maintenance dose of 300mg every other week. At follow-up 3 weeks after starting dupilumab, she had significant clearing of her scalp lesions and a decrease in her reactive left axillary lymphadenitis. While dupilumab has been used to treat severe atopic dermatitis in patients with HIES, this is a novel case where its use resulted in clinical improvement in skin infections and secondary lymphadenitis. This case suggests that dupilumab may be an effective treatment for the secondary bacterial infections that develop due to skin pathology in patients with HIES.
Keywords: HIES, Hyper-IgE syndrome, Dupilumab, Biologic therapy, Recurrent infections, STAT3 mutations, Atopic dermatitis
Disclosures: All authors indicated they had no financial relationships to disclose.
(37) TNFAIP3: Evolving clinical picture from Autoinflammatory syndrome to Autoimmune lymphoproliferative syndrome
Esosa Adah, MD1, Mary Nguyen, MD2, Nikita Raje, MD3, Voytek Slowik, MD4, Julia Harris, MD5
1Pediatric Resident/Children's Mercy Hosp. Kansas-City
2Allergy & Immunology Fellow/Children's Mercy Hosp. Kansas-City
3Allergy & Immunology Attending/Children's Mercy Hosp. Kansas-City
4Gastroenterology Attending/Children's Mercy Hosp. Kansas-City
5Rheumatology Atttending/Children's Mercy Hosp. Kansas-City
Autoimmune lymphoproliferative syndrome (ALPS) is a rare genetic disorder of the immune system that affects children and adults. It can present with lymphoproliferation to lymphoid organs with the development of neutropenia, lymphopenia, and thrombocytopenia. Most cases are caused by either a germline or somatic mutation in the FAS, FASLG, or CASP10 genes that are involved in cell apoptosis. Tumor Necrosis Factor Alpha, Induced Protein 3 (TNFAIP3) is a protein coding gene associated with A20 haploinsufficiency.
A 3-year-old female presented at 4-months old to the intensive care unit in shock with a vesiculopustular skin rash & indurated plaques on her extremities. Upon presentation, she was febrile and in respiratory failure requiring intubation. Infectious workup was unrevealing. Following discharge, genetic evaluation revealed heterozygous TNFAIP3 c.475del, p.Tyr159Metfs*57. Brain MRI/MRA/MRV were normal, EGD/colonoscopy showed chronic gastritis with normal biopsy, and eye exam showed no ocular inflammation. Immunology evaluation revealed T cell lymphopenia: CD3 1576 mm^3, CD4 1182 mm^3, CD8 368 mm^3, CD4−CD8− TCRαβ+ T cells (1.6 % T cells) with normal CD 19, NK cells, immunoglobulins, hib/tetanus titers, oxidative burst and lymphocyte proliferation to mitogen. At the age of 2, patient developed elevated AST/ALT (~300s-700 units/L) & pancytopenia 8 months later with physical findings of recurrent oral ulcers. Abdominal ultrasound remarkable for splenomegaly. Liver biopsy showed acute & chronic hepatitis with lymphocyte predominance, bile duct injury and bridging fibrosis. Bone marrow biopsy was negative for malignancy. Flow cytometry on peripheral blood had elevated CD4−CD8− TCRαβ+ (4.7% T cells ), suggestive of ALPS. Fas mediated assay, sFASL, IL-10, IL-18 were obtained & pending.
TNFAIP3 is known to present with autoinflammatory symptoms. Our case shows that patients with TNFAIP3 variants can have an evolving clinical picture that can include a spectrum of autoimmune lymphoproliferative syndrome -like disease. A hallmark finding of ALPS is a high proportion of CD4−CD8− TCRαβ+ T cells, called double-negative T cells. A 2017 case report revealed a 1 year old male with findings of bilateral cervical lymphadenopathy and hepatosplenomegaly. TNFAIP3, A20 was identified in this patient showing the ALPS-like phenotype with elevated double-negative T cells (5.1% T cells). (https://doi.org.10.1016/i.iaci.2016.09.038)
Keywords: TNFAIP3, ALPS, autoinflammatory, lymphoproliferation, autoimmune
Disclosures: All authors indicated they had had no financial relationships to disclose.
(38) Post-operative absolute lymphocyte counts in pediatric patients with congenital heart disease
Katie Kennedy, MD1, Kathryn Dodds, MSN, CRNP2, Jack Rychik, MD1, Jennifer Heimall, MD3
1Attending Physician/The Children's Hospital of Philadelphia
2Advanced nurse practitioner/The Children's Hospital of Philadelphia
3Assistant Professor/Division of Allergy and Immunology, Children’s Hospital of Philadelphia
Previous studies regarding T-cell excision circle (TREC) screening in newborns report a high proportion of false positive findings in infants with congenital heart disease (CHD). These observations are postulated to be secondary to removal of thymus tissue during routine cardiac surgery. We aimed to describe the absolute lymphocyte counts (ALC) trend over time of infants born at our institution with CHD requiring surgical intervention and subsequent thymectomy.
We performed a retrospective chart review of 441 infants born with CHD at our institution from September 2012 through August 2015. Absolute lymphocyte counts over time were analyzed in addition to cardiac defects, genetic diagnoses and effect of surgical intervention. Exclusion criteria included known chylous loss or post-natal administration of immune modulating medications such as high dose corticosteroids or chemotherapy.
The majority of subjects were male (57%) and born full term (85%). Genetic diagnoses were noted in 13%, most commonly 22q11.2 deletion syndrome (3.7%), Trisomy 21 (3.5%) and Turner Syndrome (1.6%). The most common cardiac defects included hypoplastic left heart syndrome (15.6%), transposition of the great arteries (15.3) and tetralogy of fallot (13.5%). No significant difference was found between average ALC at birth in infants with CHD who later underwent thymectomy and those that did not (p>.9999). Similar findings were present when comparing average ALC 6 months post-operatively and 15 months post-operatively (p>.9999, p>.9999 respectively). Of note, all newborn screen studies for severe combined immunodeficiency sent in Pennsylvania following thymectomy were normal.
In this large retrospective cohort, removal of thymus tissue did not affect ALC up to 15 months post-operatively. These findings suggest that the cause of lymphopenia in these patients is not due to routine thymectomy during cardiac surgery. Further study is necessary to elucidate infection risk in these patients as well as late follow up of ALC.
Keywords: lymphopenia, trec, newborn screen, congenital heart disease
Disclosures: All authors indicated they had no financial relationships to disclose.
(39) Yield of Genetic Testing in a Large Autoinflammatory Disease Cohort
Amanda Salih, MD, MPH1, Bo Yuan, PhD, FACMG2, Anaid Reyes, BA3, Justin Branch, BA4, Gina Cahill, MPH3, W. Blaine Lapin, MD5, Jennifer Rammel, MD, MPH6, Martha Curry, MS, RN, C-PNP7, Lisa Forbes-Satter, MD8, Joud Hajjar, MD, MS8, Nicholas Rider, DO8, M. Cecilia Poli, MD, PhD9, Zeynep Coban Akdemir, PhD10, James Lupski, M.D., Ph.D., D.Sc.11, Ivan Chinn, MD8, Tiphanie Vogel, M.D, Ph.D.12
1Fellow/Baylor College of Medicine/ Texas Children's Hospital
2Assistant Director/Seattle Children's
3Research Coordinator/Baylor College of Medicine/ Texas Children's Hospital
4Research Technician/Baylor College of Medicine/ Texas Children's Hospital
5Assistant Professor/Connecticut Children's
6Assistant Professor/UF Jacksonville
7Instructor/Baylor College of Medicine/ Texas Children's Hospital
8Assistant Professor/Baylor College of Medicine/ Texas Children's Hospital
9Director of the Immunogenetics and Translational Immunology Program; Assistant Professor/Universidad del Desarrollo; Baylor College of Medicine/ Texas Children's Hospital
10Postdoctoral Associate/Baylor College of Medicine/ Texas Children's Hospital;
11Professor/Baylor College of Medicine/ Texas Children's Hospital
12Assistant Professor, Division of Rheumatology, Department of Pediatrics & Medicine/Texas Children’s Hospital, Baylor College of Medicine
Systemic autoinflammatory diseases (SAIDs) represent a heterogenous group of disorders characterized by dysregulated activation of the innate immune system and a pathogenic inflammatory response. Genetic advances have identified a rapidly expanding number of monogenic SAIDs, and many can be diagnosed using commercial gene panels. Although most patients with autoinflammation do not have monogenic disease, identifying those who do is important to inform treatment, and is particularly advantageous in atypical presentations. We aimed to investigate genetic etiologies in a large cohort of patients with autoinflammation.
We identified 74 patients with autoinflammation and clinical genetic sequencing. Clinical sequencing was diagnostic for 30 patients (41%). The remaining 44 patients with non-diagnostic clinical sequencing included 16 exomes and 28 targeted panels, containing a range of 7-207 genes. These patients were further investigated using research exome sequencing (ES) and bioinformatic evaluation with a candidate gene approach.
Our cohort is 52% male and 67% Caucasian; 20% of the patients are Hispanic. The median age of onset is 2.2 years old. Fever episodes ranged from 1-10 days with an average periodicity of 3-6 weeks. Predominant clinical features include fatigue (84%), rash (69%), lymphadenopathy (61%), abdominal pain (60%), and arthritis (51%). Elevations in C-reactive protein (74%) and erythrocyte sedimentation rate (59%) were noted during flares, but no patients demonstrated elevated inflammatory markers outside of flares. A majority of patients (76%) demonstrated symptom improvement with steroids. Most patients (96%) required a controller medication, either colchicine (24%) or a biologic agent (72%). Research ES yielded no additional cases of known monogenic autoinflammatory diseases. Variants of uncertain significance (VUSes) in known or suspected primary immunodeficiency (PID) genes were identified in 46% of patients. Interestingly, 47% of patients with a VUS in a PID-associated gene carried heterozygous NLRP12 VUSes.
Identification of genetic causes of SAIDs is of critical importance for rapid disease diagnosis, targeted therapeutic selection, and family counseling for recurrence risk. Use of clinically available expanded genetic panels was sufficient to diagnose known diseases in 40% of our autoinflammatory cohort. Research investigations are ongoing into the VUSes identified in our patients.
Keywords: periodic fever syndromes, recurrent fevers, autoinflammation, genetics
Disclosures: Bo Yuan works for the Department of Molecular and Human Genetics at Baylor College of Medicine which receives revenue form clinical genetic testing completed at Baylor Genetics Lab. Lisa Forbes-Satter is a Consultant at ADMA and Grifols, and is an Advisory Board member at CSL Behring, Horizon, and Takeda. Joud Hajjar received research grants from Amplimmune, Arcus Biosciences, ARMO BioSciences, Atterocor/Millendo, Baxalta, BMS, Calithera Biosciences, Chao Physician-Scientist Foundation, CytomX Therapeutics, Eli Lilly, EMD Serono, Healios Onc, Immune Deficiency Foundation, ImmuneOncia, Incyte, Jeffrey Modell Foundation, Karyopharm, Kymab, MedImmune, Merck, National Cancer Institute, NeoImmuneTech, Neon Therapeutics, Novartis, OncoSec KEYNOTE-695, Pfizer, PsiOxus, Regeneron, Surface Oncology, The Texams Medical Center Digestive Diseases Center and Top Alliance; she was an Advisory Board member of Alfaisal University, Genome & Company, Horizon, and Pharming. Nicholas Rider is an Advisory Board member at CSL Behring, Pharming, Takeda; received a research grant from The Jeffrey Modell Foundation; Royalties from UpToDate and Wolters Kulwer Publishing. James Lupski has ownership interested in 23andMe and Regeneron and is an employee of the Department of Molecular and Human Genetics at Baylor College of Medicine All other authors had no financial relationships to disclose.
(40) Gain-of-function mutations in JAK3 in CVID and Interstitial Lung Disease
Michael Nordness, B.S.1, Amy Rymaszewski, PhD2, Jeffrey Woodliff, PhD3, Shaoying Chen, MD2, John Routes, MD4, James Verbsky, MD, PhD5
1Medical Student/Medical College of Wisconsin
2Research Associate/Medical College of Wisconsin
3Clinical Immunology Research Laboratory Manager/Medical College of Wisconsin
4Professor of Pediatrics, Medicine, Microbiology and Immunology/Medical College of Wisconsin
5Associate Professor of Pediatrics, Medicine, Microbiology and Immunology/Medical College of Wisconsin
Common variable immune deficiency (CVID) is a primary immune deficiency characterized by hypogammaglobulinemia, poor specific antibody responses, and infectious and non-infectious complications.
We evaluated a 64-year-old male with CVID, recurrent sinopulmonary infections, and progressive interstitial lung disease (ILD). He exhibited a chronic lymphocytosis (~13K cells/mm3), and flow cytometry demonstrated increased numbers of T cells (CD4: 3820 cells/mm3, CD8: 7163 cells/mm3) and NK cells (3104 cells/mm3), but an absence of B cells. The percentage of activated (HLA-DR+) CD4 and CD8 T cells was markedly increased, and there was a decreased percentage of naïve T cells. Open lung biopsy demonstrated lymphocytic interstitial pneumonitis (LIP) (CD4+>CD8+ T cells), an absence of B cells or granulomas, and severe pulmonary fibrosis. HIV-1 RNA was negative. Despite immunoglobulin replacement, he experienced repeated episodes of pneumonia requiring IV antibiotics and chronic CMV viremia.
Whole exome sequencing (WES) was conducted. JAK3 plasmids underwent site-directed mutagenesis and were transfected into an IL-2/IL-15 responsive HEK reporter line (Quanti-Blue). Cells were simulated with IL-2 in the presence or absence of a JAK3 inhibitor (tofacitinib). Peripheral blood mononuclear cells (PBMCs) and HEK cells were stimulated with IL-15, and phospho-STAT5 was assessed with flow cytometry.
WES demonstrated two rare, damaging variants in JAK3: S835C and R925S. No other known CVID-causing variants were detected. JAK3 cDNA cloning demonstrated cis orientation of these variants. To evaluate variant function, wild type (WT) JAK3, S835C/R925S, and a known gain-of-function (GOF) variant A573V were transfected into an IL-2/IL-15 responsive reporter line and stimulated with IL-15. WT JAK3 increased phospho-STAT5 levels and transcriptional activity versus controls, while S835C/R925S further increased phospho-STAT5 activity comparable to A573V. IL-15 stimulation of the patient’s PBMCs demonstrated delayed STAT5 dephosphorylation.
The dual presence of S835C and R925S variants in JAK3 caused STAT5 activation in reporter cell lines and delayed STAT5 dephosphorylation consistent with a GOF. We speculate that the GOF S835C and R925S mutations in JAK3 led to a chronic T-cell lymphocytosis and T cell predominant LIP and CVID. This is the first report of GOF mutations in JAK3 in a patient with CVID and an unusual form of ILD.
Keywords: CVID, JAK3 GOF, Interstitial Lung Disease
Disclosures: John Routes has contracted research with CSL Behring and Evolve Biologics. All other authors had no financial relationships to disclose.
(41) Multicentric Castleman Disease revealing STAT1 deficiency
Camille Beaufils, M.D.1, Marie-Paule Morin, M.D.. Ph.D.2, Jean Jacques De Bruycker, MD3, Lorie Marchitto, M.Sc.4, Isabel Fernandes, PhD5, Sonia Cellot, M.D. Ph.D.6, Philippe Ovetchkine, M.D. Ph.D.7, Jean-François Soucy, M.D.8, Luc Oligny, M.D. Ph.D.9, Elie Haddad, MD, Phd2, Fabien Touzot, M.D. Ph.D.2
1Clinical Fellow/Immunology-Rheumatology Division, Department of Pediatrics, University of Montreal
2Attending physician/Immunology-Rheumatology Division, Department of Pediatrics, University of Montreal
3Assistant clinical professor, division of Pediatric Immunology and Rhumatology/CHU Sainte-Justine
4Ph.D student/CHU Sainte-Justine Research Center, University of Montreal
5Professor/Immunology-Rheumatology Division, Department of Pediatrics, University of Montreal
6Attending Physician/Hematology Oncology Division, Department of Pediatrics, University of Montreal
7Attending Physician/Infectious Disease Division, Department of Pediatrics, University of Montreal
8Attending Physician/Genetic Division, Department of Pediatrics, University of Montreal
9Attending Physician/Pathology Division, Department of Pediatrics, University of Montreal
Castleman disease (CD), first described in 1956, is a rare lymphoproliferative disorder characterized by enlargement of lymphoid tissue with follicular hyperplasia, vascular proliferation, and plasmocytosis. Multicentric Castleman Disease (MCD) is an infrequent pathology in the pediatric population and is often reported in regions where HHV-8 is endemic. We report an 11-month-old girl who presented with 6 criteria of hemophagocytic lymphohistiocytosis (HLH): (i) prolonged fever > 38.5° C, (ii) splenomegaly, (iii) bicytopenia with severe thrombocytopenia and anemia, (iv) increased triglycerides and decreased fibrinogen, (v) hyperferritinemia (vi) increased circulating activated HLA-DR+ CD8 T. Additional remarkable features were a tumoral syndrome (multiple diffuse adenopathies, hepatomegaly), jaundice secondary to mild hepatitis with cholestasis, eosinophilia and an extensive rash. All the infectious workup was negative. A PET-scan revealed diffuse hypermetabolic adenopathies, hepatomegaly and splenomegaly). Bone marrow biopsy also excluded malignant hematological disease. An axillary lymph node biopsy was consistent with the diagnosis of MCD in its hyaline-vascular form. Immunohistochemistry for HHV-8 was negative. Perforin expression and NK degranulation were normal, excluding familial HLH. The patient was treated with 2 mg/kg/day prednisolone, 4 infusions of rituximab (375 mg/m2 per dose), and sirolimus (for trough level between 10-15 ng/mL). Despite an initial response to therapy, the patient relapse during steroid tapering (at 1 mg/kg/day) with recurrence of fever, skin rash, and eosinophilia. Meanwhile, the whole genome sequencing results were obtained and evidenced a maternal unidisomy of chromosome 1, resulting in a homozygous deletion encompassing exon 5 to 9 of STAT1. STAT1deficiency was confirmed in our patient through western blot and functional testing of STAT1 phosphorylation by flow cytometry. We decided to introduce subcutaneous Ig replacement and substitute sirolimus with ruxolitinib, given the initial HLH presentation and reports showing imbalanced STAT3 pathway activation in STAT1 deficiency. Modification of therapy allowed to maintain a sustainable remission of the disease and a complete withdrawal of steroids. While in complete remission, our patient underwent hematopoietic stem cell transplantation with a match unrelated donor.
Keywords: Primary Immunodeficiency, STAT1 deficiency, Multicentric Castleman Disease, Hemophagocytic Lymphohistiocytosis, ruxolitinib
Disclosures: Elie Haddad is a consultant for Jasper Therapeutics and Rocket Pharma. All other authors had no financial relationships to disclose.
(42) Newborn Screening for Severe Combined Immunodeficiency in New Jersey from 2014 to 2017
Amandeep Sandhu, MD1, Jennifer Heimall, MD2, Miriam Schachter, PhD3
1Allergy and Immunology Fellow/Children’s Hospital of Philadelphia
2Attending Physician/Children's Hospital of Philadelphia
3Research Scientist/New Jersey Department of Health
New Jersey department of health (NJ DOH) initiated newborn screening (NBS) for severe combined immunodeficiency (SCID) in July 2014. The NBS uses a T-cell receptor excision circle (TREC) assay reported as cycle threshold (CT) to evaluate for SCID but also identifies other causes of T-cell lymphopenia. The objective of this study was to report outcomes of abnormal NBS for SCID in NJ from its initiation in 2014 through December 2017.
Retrospective chart review of infants with abnormal NBS for SCID were identified by New Jersey Department of Health (NJ-DOH). The data reviewed included demographics, TREC CT, lymphocyte enumeration, lymphocyte proliferation to mitogens, quantitative immunoglobulins, genetic analysis, family history and clinical history. The TREC CT parameters for a presumptive positive screen is 35.8, while the test is repeated if the CT is between 34.5 and 35.7.
Two hundred and thirteen infants were referred for flow cytometry and 146 of these subsequently had a normal evaluation. Fifty-three patients expired prior to further evaluation for SCID. Fourteen patients (6.6%) were diagnosed with either SCID or T-cell lymphopenia: 4 patients (1.9%) with SCID, 2 patients (0.9%) with 22q11.2 deletion syndrome, and 8 patients (3.8%) with idiopathic T-cell lymphopenia. Of the 4 patients with SCID, a genetic mutation was identified in all and included the following genotypes: ADA (n=2), RAG1 (n=1), MSN (n=1). Demographic information, TREC cycle threshold values and lymphocyte enumeration data are summarized in tables 1 and 2.
NBS for SCID in NJ was one of the first in the US to use TREC-CT rather than TREC count as a reporting strategy. Most SCID cases detected in NJ were autosomal recessive genotypes, rather than the x-linked IL2RG genotype most commonly reported to cause SCID. In addition, female and non-Caucasian infants comprised half the population identified via NJ-DOH NBS to have significant lymphopenia. These data further support the value of population based NBS for SCID and significant T cell lymphopenia utilizing TREC CT reporting to detect these conditions in populations previously less likely to be detected prior to symptom onset.

Table 1: Demographic information for infants with SCID or T-cell lymphopenia identified on NJ NBS
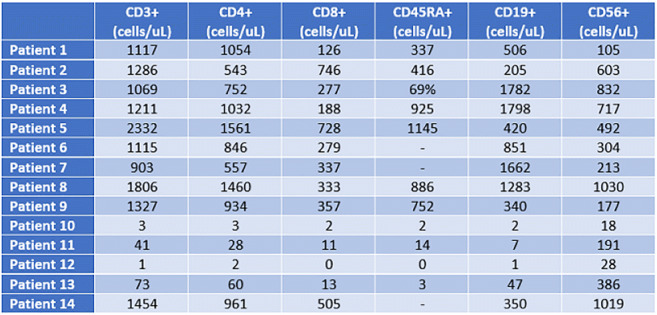
Table 2: Initial lymphocyte enumeration for infants with SCID or T-cell lymphopenia identified on NJ NBS
Keywords: Newborn screening, Severe combined immunodeficiency (SCID), T-cell Lymphopenia, TREC, DiGeorge, Cycle threshold, 22q11.2 deletion syndrome
Disclosures: Jennifer Heimall is an advisory board member of CIRM; received research grants from Regeneron and CSL Behring; and received speaker honoraria from Horizon. All other authors had no financial relationships to disclose.
(43) IRAK 4 deficiency diagnosed in a 14-year-old male presenting with bacterial meningitis
Paulina Tran, DO1, Juhee Lee, MD2
1Fellow Physician/Childrens Hospital of Philadelphia
2Attending Physician/Childrens Hospital of Philadelphia Department of Pediatrics, Allergy Immunology Division, Children’s Hospital of Philadelphia
IRAK4 (Interleukin-1 receptor-associated kinase 4) deficiency is an autosomal recessive innate immune defect involving the toll-like receptor pathway. The majority of patients present in infancy with pyogenic bacterial infections due to Staphylococcus aureus, Streptococcus pneumoniae, Pseudomonas aeruginosa, or less commonly Neisseria Meningitidis. (1) Common clinical manifestations include meningitis, osteomyelitis, skin abscesses, lymphadenitis, sinopulmonary infections, and bacteremia. Patients tend to have milder disease later in life. In one series, no patients beyond age 14 years developed invasive bacterial infection or death.(1) This improvement with age is a unique feature of IRAK4 deficiency and thought to be due to a maturing adaptive immune system. We report the first case of IRAK4 deficiency diagnosed in an adolescent male with meningococcal meningitis.
A 15-year-old male presented with sudden onset of severe headache with photophobia, nausea, vomiting, and low-grade fever. Cerebral spinal fluid culture gram stain and culture confirmed Neisseria meningitidis infection. Prior infectious history included bilateral lymphadenitis with lymph node removal at 7 years of age and recurrent sinus infections that improved after sinus surgery and allergen immunotherapy. Immune work up revealed an abnormal TLR assay with a decreased response in TLR2-TLR1, TLR3, TLR5, TLR7-TLR8, TLR4 and normal responses to TLR2-TLR6. Complement studies showed a reduced alternative complement response (AH50 = 59 units/mL) and normal classical pathway testing. Pneumococcal titers were 60% protective and meningococcal titers were protective to 3/4 serotypes. His immunoglobulin levels were normal. Whole exome sequencing was pursued and revealed IRAK4 deficiency with a pathogenic variant that results in a premature stop codon and loss of protein expression. The patient fully recovered from his infection and has not any further infectious issues. He has since completed the MCV4 and Serogroup B vaccination series and continues on daily amoxicillin prophylaxis.
To our knowledge this is the first case of IRAK4 deficiency that developed an invasive bacterial infection after the age of 14. Although rare, it is important to consider innate immune defects in older patients presenting with invasive pyogenic disease.
Reference: 1.) PMID: 21057262
Keywords: IRAK 4 deficiency, Invasive bacterial disease, Neisseria meningitidis, Adolescence, Innate Immunity, Toll Like Receptor Pathway
Disclosures: All authors indicated they had no financial relationships to disclose.
(44) Transcriptional dysregulation of CVID patients harboring C104R TNFRSF13B mutation
Neftali Ramirez, MSc1, Niko Langer, MTA2, Nils Klammer, graduate degree3, Alla Bulashevska, PhD4, Bärbel Keller, PhD4, Claudia Bossen, PhD5, Bodo Grimbacher, Prof. Dr. med.6
1PhD Sstudent/University Clinic Freiburg, CCI
2MTA/University Clinic Freiburg, CCI
3MD student/University Clinic Freiburg, CCI
4Postdoc/University Clinic Freiburg, CCI
5Group leader/University Clinic Freiburg, CCI
6Head of the laboratory/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
To determine additional factors contributing to the development of common variable immunodeficiency (CVID), we investigated the perturbations of transcription factor binding and transcriptome profiles in CVID patients harboring the C104R mutation in TNFRSF13B. The C104R mutation in TNFRSF13B however, has a prevalence of approx. 1% in the healthy population.
Assay for transposase accessible chromatin-sequencing (ATAC-seq) was performed on naïve and class switched memory B cells of six CVID patients heterozygous for the C104R mutation, six of their healthy relatives carrying the same heterozygous mutation, and eight healthy wild-type donors. In addition, ATAC-seq was performed on naïve CD4+ T cells of two C104R-heterozygous CVID patients, their unaffected relatives, and three healthy wild-type donors. RNA-sequencing was performed on three C104R-heterozygous CVID patients, their unaffected relatives, and three healthy donors. For functional validation, intra-cellular staining was performed by flow cytometry.
Our analysis revealed 25 % less accessible chromatin in class-switched memory B cells from TNFRSF13B-mutant carriers compared to healthy donors. 1.356 and 1.069 differential accessible regions were detected in naïve and class-switched memory B cells from mutation carriers compared to healthy donors, respectively. The most enriched transcription factor binding motif was for NF-kB in both B cell subsets, but not observed for the T cell population. IkBa expression (downstream of NF-kB) was significantly increased in patients, as determined by flow cytometry. RNA-seq analysis revealed 687 dysregulated genes in naïve B cells and 617 in class-switched B cells from CVID patients, respectively. Gene ontology analysis highlighted the NF-kB signaling and systemic lupus erythematosus pathway in naïve- and class-switched memory B cells, respectively. Integration of ATAC- and RNA-seq data pointed towards a dysregulation MAP3K8. Flow cytometry analysis of pERK (downstream of MAP3K8) confirmed this trend.
Here we showed the transcriptional dysregulation of naïve- and class-switched memory B cells of individuals with the C104R mutation in TNFRSF13B. The most enriched transcription factor binding motif was for NF-kB, confirmed by transcriptome analysis, and the increase of IkBa expression. NF-kB is essential for regulating immune response to infections. Thus, its changes may lead to variety of disease-causing differences. Furthermore, the data suggest NF-kB dysregulation is B cell intrinsic.
Keywords: CVID, TNFRSF13B, epigenetics, NF-kB
Disclosures: Bodo Grimbacher received a research grant from Baxalta. All other authors had no financial relationships to disclose.
(45) Using switchable NF-κB signaling to uncover the molecular defects of pathogenic NFKB1 and NFKB2 variants
Manfred Fliegauf 1, Bodo Grimbacher, Prof. Dr. med.2
1Researcher/Institute for Immunodeficiency, Center for Chronic Immunodeficiency, Medical Center, Faculty of Medicine, Albert-Ludwigs-University of Freiburg, Germany; CIBSS – Centre for Integrative Biological Signalling Studies, Albert-Ludwigs University, Freiburg, Germany
2Head of the Laboratory/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
Common variable immunodeficiency (CVID) represents the most prevalent symptomatic primary antibody deficiency with heterozygous NFKB1 and NFKB2 mutations collectively accounting for up to 20% of the monogenetic forms. NFKB1 encodes the transcription factor precursor p105, which is processed to p50 (canonical NF-κB signaling pathway), while NFKB2 encodes p100/p52 (non-canonical pathway). Known pathogenic variants either cause (haplo)insufficiency, precursor-skipping or non-processable p100, while most of the identified missense changes have yet unknown effects.
The disease-causing mechanisms presumably originate from imbalanced p105/p50 and p100/p52 ratios and altered NF-κB signaling dynamics. To assess the pathogenic relevance of NFKB1 and NFKB2 variants, we first analyze the localization, expression and processing, DNA-binding activity and reporter gene activation of mutant NF-κB proteins in transfected HEK293T cells. We then test their functional capabilities following experimentally induced precursor-processing, IκB-mediated cytoplasmic retention, nuclear translocation and DNA-binding competition, respectively. To accomplish NF-κB manipulation, we use switchable models based on co-expression of the NF-κB proteins together with diverse upstream kinases, natural and synthetic IκBs, Rel interaction partners, competitors and the combination thereof. With these tools, we aim at exploring the NF-κB signaling defects associated with diseases of the immune system.
Keywords: NFKB1, NFKB2, NF-kappaB, CVID
Disclosures: Bodo Grimbacher received a research grant from Baxalta. The other author had no financial relationships to disclose
(46) Effects of Aging on Immune parameters In Patients with STAT3 Hyper IgE Syndrome
Jennifer Heimall, MD1, Amanda Urban, CRNP2, Hastings Williamson, n/a3, Jean Ulrick, CRNP4, Alexandra Freeman, MD5
1Assistant Professor/Division of Allergy and Immunology, Children’s Hospital of Philadelphia
2Nurse Practitioner/Clinical Research Directorate, Frederick National Laboratory for Cancer Research
3Research Assistant/NIAID
4CRNP/NIAID
5Director, Primary Immune Deficiency Clinic/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institute of Health
Patients with Hyper IgE Syndrome (HIES) due to dominant negative STAT3 mutations (STAT3 HIES) have recurrent skin and lung infections, eczematous dermatitis, and connective tissue changes. Laboratory abnormalities include elevated serum IgE, elevated absolute eosinophil count (AEC), lower memory T lymphocytes, and lower switched memory B lymphocytes. We sought to describe the change in immune function parameters with age in this population.
METHODS: We retrospectively reviewed 157 patients with STAT3 HIES, with a focus on clinically obtained immune parameters. These included cells counts (AEC, CD19, CD20/CD27/IgM+, Cd20/CD27/IgM-, CD3-/CD16+ and/or CD56+, CD3, CD3/CD4, CD3/CD8, CD4/CD62L+/ CD45RA+/CD3+, CD3/CD4/CD62L+/CD45RA-, CD3/CD4/CD62L-/CD45RA-, CD8/CD62L+/ CD45RA+/CD3+, CD3/CD8/CD62L+/CD45RA-, CD3/CD8/CD62L-/CD45RA-) and levels of IgG, IgA, IgM, IgE. Patients were clustered by age groups of 0-5 years of age, 6-10 years of age, 11-15 years of age, 16-30 years of age, 31-45 years of age, and >45 years of age. We assessed median level of each parameter in each age cluster. Most patients only were represented in one cluster.
RESULTS: Results are summarized in Table 1. As expected, median IgE was elevated, but was higher in children, adolescents and young adults as compared to toddlers and older adults. Median IgG, IgA and IgM were normal. Surprisingly, median AEC was at the high end of normal and stable across age groups. Overall, CD19, CD3, and CD3-/CD16+ and/or CD56+ cell counts were slightly lower than expected for age across all categories. Naïve B cell populations did decrease with age, but there was not a commensurate increase in switched or unswitched memory B cell populations over time leading to progressive total B cell number decline. T cell populations demonstrated an appropriate 2:1 ratio of CD4:CD8 cells that was maintained over time; increased DNT were not seen. Naïve T cell populations did decrease with age, but there was not a commensurate increase in memory T cell populations over time leading to decreasing total T lymphocytes with age.
Further study is needed to correlate clinical outcomes with variations from these median immunologic parameters in patients with STAT3 HIES.
Table 1: Median Values of immune parameters by age
Keywords: Primary Immunodeficiency, Aging, Hyper IgE Syndromes, Immunophenotyping
Disclosures: All authors indicated they had no financial relationships to disclose.
(47) Chronic Granulomatous Disease presenting with Early Onset Chronic Recurrent Multifocal Osteomyelitis
Christopher Costin, MD1, Amer Khojah, MD2, Michael Miller, MD3
1Pediatric Rheumatology Fellow/Ann & Robert H. Lurie Children's Hospital of Chicago/ Northwestern University Feinberg School of Medicine
2Attending Physician, Allergy, Immunology, and Rheumatology/Ann & Robert H. Lurie Children's Hospital of Chicago/ Northwestern University Feinberg School of Medicine
3Clinical Practice Director, Division of Rheumatology/Ann & Robert H. Lurie Children’s Hospital of Chicago
Chronic Recurrent Multifocal Osteomyelitis (CRMO) is a rare autoinflammatory bone disease presenting with pain and noninfectious osteomyelitis. The pathophysiology of CRMO remains unclear; however, bone metabolism, IL-10 and IL-1 pathways have been linked in its pathogenesis. CRMO may present as distinct clinical entity or may be associated with other disease: such as Crohn’s disease, HLA B27 associated diseases. Early onset CRMO is typically linked to monogenic syndromes such as Majeed syndrome or Deficiency of the interleukin-1 receptor antagonist (DIRA). Rarely chronic granulomatosis disease (CGD) has been associated with CRMO. Here we present a case of CGD presenting with early onset CRMO.
A 3-year-old boy presented to the ED for left sided facial swelling. An MRI of the face showed left sided hemimaxilla enhancement. A whole-body MRI showed additional hyperintensity of the left ulna and radius concerning for CRMO. A bone biopsy of hemimaxilla showed fibroproliferative lesion with chronic inflammation consistent with CRMO. Infectious studies were negative. He was discharged home on scheduled NSAIDs with eventual resolution of his swelling and pain.
From ages 5-8 he then developed recurrent facial sores and skin infections that resolved with clindamycin or mupirocin. He developed a gluteal abscess requiring oral antibiotics. He was seen by dermatology and diagnosed with a Serratia facial infection. Oxidative burst testing was obtained and resulted low at 2.4. He was diagnosed with chronic granulomatous disease.
CRMO is a relatively rare disease. It typically presents in females ages 7-12. This case highlights the importance of considering monogenic autoinflammatory diseases in patients with early onset CRMO such as CGD, Majeed Syndrome and DIRA.
Keywords: CRMO, CGD, osteomyelitis, Chronic, granulomatous, Disease
Disclosures: All authors indicated they had no financial relationships to disclose.
(48) A pediatric case of trisomy 8 mosaicism with periodic fevers
Tyrone Coyle, MD1, Sujatha Rajan,, MD2, Artemio Jongco, MPH, MD, PhD, FACP, FACAAI, FAAAAI3
1Allergy/Immunology fellow/Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
2Pediatrics Infectious Disease/Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
3Assistant Professor of Medicine and Pediatrics/Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
Trisomy is the most common aneuploidy or defect with an abnormal number of chromosomes. Full trisomies are often lethal in utero. Trisomy mosaicism increases survivability but also leads to high variability in phenotypes. Trisomy 8 Mosaicism (T8M), or Warkany syndrome, occurs in 1:25,000 to 1:50,000 of live births, with 3:1 male predominance.
An 8-year-old Asian male was seen in an outpatient setting for evaluation of recurrent high grade fevers (to 103°F) for 3 years. A fever log showed episodes every 24-28 days, lasting for 3 days and then spontaneously resolving. Episodes were associated with abdominal pain, vomiting, diarrhea and chills. Initially, fevers were treated with antibiotics and antipyretics which were ineffective. A thorough infectious disease evaluation detected no infectious etiology. The synchronicity of fevers were highly suspicious for a periodic fever syndrome.
Preliminary workup revealed neutropenia (1200cells/μL), mild eosinophilia (520 cells/μL), thrombocytosis (458K/μL) and mild macrocytosis (MCV 100.2). Otherwise, screening of cellular immunity, complement, humoral immunity and for periodic fevers with IgD level and periodic fever syndromes panel (ELANE, LPIN2, MEFV, MVK, NLRP3, PSTPIP1, TNRFSF1A) were unremarkable.
A commercial targeted primary immunodeficiency panel showed copy number variants (CVNs) of uncertain significance (VUS) in ANGPT1, ASAH1, CHD7, DGAT1, EXTL3, GTF2E2, IKBKB, LYN, MCM4, NBN, PRKDC, TONSL, VPS13B, and heterozygous VUS in AP3B1, CTC1, FANCI, FCHO1, IL17RC, LRBA, PEPD, POLD1, SLC39A7, SPINK5, TTC37. All CVNs were on chromosome 8. A chromosomal microarray confirmed diagnosis of trisomy 8.
T8M can present with an array of clinical findings. Several articles from predominantly Asian adult cohorts show an association amongst T8M, myelodysplastic syndrome (MDS) and Behçet disease (BD)-like symptoms, and one Japanese review proposes a designation of “trisomy 8 syndrome” (T8S) for such cases. Several Japanese adults with T8S were reported to have periodic fevers that are reminiscent to the clinical presentation seen in our pediatric case. While our patient has not shown clinical signs of BD or MDS, prior Japanese reports of T8S were exclusively in adults. Periodic fevers could be the initial insult, prior to developing clinical signs of T8S.
Keywords: Trisomy 8, periodic fevers, Mosaicism, Warkany syndrome, pediatric, neutropenia, Behçet disease, myelodysplastic syndrome
Disclosures: All authors indicated they had no financial relationships to disclose.
(49) Suspected Lymphocytic Variant Hypereosinophilic Syndrome with Concomitant Immune Thrombocytopenia
Susamita Kesh, MD1, Ian Slack, MD2, Juan Abonia, MD3, Kimberly Risma, MD, PhD3, Stephanie Ward, MD4
1Allergy/immunology fellow/Cincinnati Children's Hospital Medical Center
2Clinical Fellow/Division of Allergy and Immunology, Cincinnati Children's Hospital Medical Center; Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH
3Associate Professor/Division of Allergy and Immunology, Cincinnati Children's Hospital Medical Center; Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH
4Assistant Professor/Division of Allergy and Immunology, Cincinnati Children's Hospital Medical Center; Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH
Hypereosinophilic syndrome (HES) is a rare condition which can lead to irreversible end organ damage with various manifestations. Here we describe a unique case of suspected lymphocytic-variant hypereosinophilic syndrome (L-HES) with concomitant immune thrombocytopenia (ITP).
A previously healthy 19-year-old Asian male who presented with a six-week history of bilateral lower extremity rash, angioedema, weight loss, and fatigue was admitted with thrombocytopenia (13,000 platelets/mcL) and leukocytosis (44,800 cells/mcL) with severe eosinophilia of 33,700 cells/mcL. Additional disease sequelae included decreased ejection fraction (50%), acute kidney injury, transaminitis, and hepatic vein thrombosis. Skin biopsy demonstrated eosinophilic panniculitis; rheumatologic and infectious workups were unrevealing. Bone marrow biopsy demonstrated eosinophilic hyperplasia (70-80%) with normal eosinophil morphology and unremarkable flow cytometry. Genetic workup for myeloproliferative variants including chronic eosinophilic leukemia were unremarkable including FISH (negative BCR/ABL1, JAK2, PDGFRA, PDGFRB, CHIC2, FGFR1), normal karyotype, microarray SNP, and no genetic alterations on FoundationOne heme next-generation sequencing. Peripheral flow cytometry was significant for an inverted CD4:CD8 ratio with increased CD3+/CD4+ γ/δ cells (21%) and minimal elevations of IL5 and IL6. Additional evaluation for a lymphocytic clonal process revealed no evidence of T or B cell clonality. Workup for his thrombocytopenia identified GpIIb/IIIa antibodies. Without any clear evidence for malignancy or immunodeficiency, he was started on systemic steroids which were escalated for recalcitrant thrombocytopenia with a nadir platelet count that was undetectable despite initial eosinophil normalization. He subsequently started mepolizumab 300 mg subcutaneously every 4 weeks allowing a slow taper of systemic steroids
Although cytogenetic testing can categorize patients into myeloproliferative HES, the diagnosis of L-HES, as in this case, is often challenging as a subset of patients can have clones which are difficult to identify. We suspect L-HES based on the increased γ/δ T cells, evidence of additional adaptive immune dysfunction (ITP), and lack of other findings to suggest a myeloproliferative variant. To our knowledge, this is the only reported case of HES with GpIIb/IIIa positive antibody ITP. Our patient is undergoing serial laboratory monitoring for end organ damage. Repeat bone marrow biopsy is planned for oncologic surveillance as well as risk of HES recurrence.
Keywords: hypereosinophilic syndrome, immune thrombocytopenia, malignancy
Disclosures: All authors indicated they had no financial relationships to disclose.
(50) Pneumococcal Antibody(PA) Levels Are Decreased in Subjects with Allergic Sensitization
Charles Song, M.D.1, Diana Chernikova, MD, Phd2, Dennys Estevez, MSc3
1Chief of pediatric allergy and immunology/Harbor-UCLA
2Resident/Harbor-UCLA
3Statistician/Harbor-UCLA
In the previous study of 313 subjects with recurrent respiratory symptoms aged 6-70 years, we reported allergic skin sensitization was significantly increased in those with inadequate baseline pneumococcal antibody levels. 1 We extended the study to include the 57 children under 6 years of age to further investigate the relationship between allergy and pneumococcal antibody (PA) levels.
We investigated 313 subjects aged 6 to 70 years and 57 subjects aged 2 to 5 years with recurrent respiratory symptoms for atopy and pneumococcal antibody; Inclusion criteria were: persistent cough >4 weeks, persistent rhinitis>4 weeks, otitis media > 4/year, sinusitis > 1x/year, chronic sinusitis ≥12 weeks. Exclusion criteria were the history of incomplete vaccination, primary immune deficiency, autoimmune disorders, and malignancy. Diagnostic studies included allergy skin test, IgE, IgG, IgA, IgM, and pneumococcal antibody levels. Pneumococcal antibodies were considered protective (adequate) if ≥50% of the tested serotypes were in a protective range (≥1.3 mg/dL) for ages 2 to 5 and ≥70% for ages 6 to 70.
The prevalence of adequate baseline PA among 194 patients aged 6-70 years with positive allergic skin sensitization was significantly lower compared to the 119 patients with negative skin sensitization (15% vs. 24%, P < 0.05). The prevalence of adequate baseline PA among 35 patients aged 2-5 years with positive allergic skin sensitization also showed lower trend compared to the 22 patients with negative skin sensitization (20% vs. 36%, P =0.29) (Figure 1).
Our findings demonstrated a strong association between the baseline PA and allergic sensitization. This raises the hypothesis that allergic propensity (Th2 pathway) may have an initial suppressive effect on the immunological response (IgM/G) to polysaccharide antigens (Th1 pathway). Patients with recurrent respiratory symptoms need to be checked both for allergic sensitization and PA levels.
Reference:
1. Song CH, Estevez D, Chernikova D, et al. Low baseline pneumococcal antibody titers predict specific antibody deficiency, increased upper respiratory infections, and allergy sensitization. Allergy and Rhinology . 2020; 11;1-10

Figure 1 : Percentage of adequate (protective) pneumococcal antibody levels among allergic and non-allergic subjects.
Keywords: Pneumococcal antibody deficiency, allergy, recurrent respiratory symptoms
Disclosures: All authors indicated they had no financial relationships to disclose.
(51) Worsening Cough in Patient with CVID and GILD - what we least expected
Ana Luiza Graneiro, MD1, Yatyng Chang, MD2, Vivian Hernandez-Trujillo, MD3
1Pediatric Resident/Nicklaus Children’s Hospital
2Allergy and Immunology Fellow/Nicklaus Children's Hospital
3Allergy Immunology Attending/Nicklaus Children's Hospital
Antidegradation agents used to enhance rubber products include paraphenylenediamine (PPD), among others, known collectively as black rubber mix. This can also be found in some hair products. The rationale for their use stems from the fact that when oxidized, PPD turns hair a darker color. The substance can also cause allergic reactions. Here, we present an adult with primary immunodeficiency and chronic cough with acute exacerbation likely due to hair dye.
To describe a case of allergic reaction manifesting as worsening cough in a patient with CVID and GILD due to exposure to black rubber mix in hair dye.
This patient had a history of CVID and GILD on subcutaneous gamma globulin replacement, granulomatous interstitial lung disease, congenital heart disease, allergic rhinitis, chronic sinusitis and contact dermatitis, who presented with a dry cough for months. During this time, she was followed by multiple specialties including pulmonology for her history of stable basilar pulmonary fibrosis, who suspected her cough was upper respiratory in nature. A CT scan of the sinus was normal. Over the prior months, she also developed skin rashes to tape, jewelry and leads from EKG. Patch testing showed positive results to nickel, gold, Cl+Me-Isothiazolinone and black rubber mix. Skin prick testing was positive only to feathers. She was treated with cefdinir for three weeks for suspected sinus infection. Despite the treatment with allergy medications, her cough persisted. One day, patient went to the hair salon to have her hair done and had an acute worsening of her symptoms. She had not dyed her hair in over one year.
In patients with primary immunodeficiency and chronic lung disease presenting with acute exacerbation of cough, uncommon causes should be considered. For patients with persistent cough, who have eliminated pertinent environmental allergens and are on appropriate treatment, clinicians should consider infection versus products that may contain contact allergens such as PPD/black rubber mix as potential causes. We recommend consideration of testing for contact allergens. Patients should read labels carefully, eliminate hair products with black rubber mix/PDD or opt for natural hair products without these chemicals.
Keywords: Paraphenylenediamine, Black rubber mix, CVID, GILD
Disclosures: Vivian Hernandez-Trujillo is an advisory board member of Covis, CSL Behring, DBV, Kaleo, Takeda, and US WORLD MEDS. All other authors had no financial relationships to disclose.
(52) Hematopoietic Stem Cell Transplant Outcomes in Adolescent and Young Adults with Immune Disorders
Andrea Bauchat, DO1, Laura Lucas, NP2, Debbie Leavenworth, RN3, Suhag Parikh, MD4, Shanmuganathan Chandrakasan, MD5
1Bone Marrow Transplant Fellow/Aflac Cancer and Blood Disorders Center, Children's Healthcare of Atlanta
2Nurse Practitioner/Children's Healthcare of Atlanta
3RN/Children's Healthcare of Atlanta
4Attending Physician/Division of Bone Marrow Transplant, Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta, Emory University School of Medicine
5Pediatric Hematologist/Oncologist/Children's Healthcare of Atlanta
Hematopoietic stem cell transplantation (HSCT) is an effective therapy for immune dysregulations and immunodeficiencies. The adolescent and young adult (AYA) patients in this diagnostic group pose unique challenges as their pre-HSCT course may be characterized by recurrent infections, inflammatory manifestations, atopy, and autoimmunity, increasing post-HSCT complication risks.
We performed a chart review at a single institution for HSCT outcomes of AYA patients with immune disorders.
We identified 22 patients aged 11 to 20 years (median 14 years) who received HSCT between 2005 and 2020. Indications for HSCT include Hemophagocytic Lymphohistiocytosis(n=6), Combined Immune Deficiency with autoimmunity(n=5), Common Variable Immune Deficiency(n=2), chronic granulomatous disease(n=3), GATA2 haploinsufficiency(n=2), SCID for second HSCT(n=1), CD40 Ligand deficiency(n=1), IPEX(n=1), and CTLA4 haploinsufficiency(n=1). Conditioning regimens included Busulfan with Fludarabine (45%), Busulfan with Cytoxan (14%), and low-dose TBI (23%). Donors were matched siblings (36%), matched unrelated (27%) mismatched unrelated (14%) or haploidentical (23%). Graft sources included bone marrow (77%), peripheral blood (14%), and cord blood (5%).
Overall survival is 86% with 14% transplant related mortality. All patients achieved engraftment. Post-HSCT median follow-up is 3 years (range 1-10years). At last follow-up, 45% had complete donor chimerism in T cell and myeloid lineages. 32% had mixed T cell chimerism (31-91% donor). 9% had mixed myeloid chimerism (50-92% donor). A CGD patient had combined mixed chimerism of 83% donor T cell and 1% donor myeloid but remains clinically well. Acute graft versus host disease (GVHD) occurred in 50% (18% grade III-IV) and chronic GVHD in 22% (8.7% severe). Causes of death include a CID patient from intracranial hemorrhage due to autoimmune vasculitis (day+40) and 2 HLH patients from septic shock with respiratory failure (day+62) and respiratory failure with ADV colitis, CMV reactivation and EBV viremia (day+133) both without evidence of HLH recurrence. Among survivors, 90% are off immunoglobulin replacement and 95% have normal or significantly improved lymphocyte subsets.
This study documents HSCT outcomes in a complex cohort of patients with a high risk of developing therapy complications. Increased reporting of HSCT outcomes in AYA patients with immune disorders is critical to understanding the challenging areas for clinical improvement.
Keywords: Hematopoietic Stem Cell Transplant, Immune disorders, Immune dysregulation, Bone marrow transplant outcomes, Adolescent and young adult
Disclosures: All authors indicated they had no financial relationships to disclose.
(53) Type I interferon signature in RALD mimicking inflammatory CVID
Riccardo Papa, MD1, Marta Rusmini, PhD2, Sara Signa, MD1, Maria Cristina Coccia, MD3, Roberta Caorsi, MD1, Francesca Schena, PhD4, Giovanni Spirito, PhD5, Remo Sanges, PhD5, Diego Vozzi, PhD6, Andrea Cavalli, PhD6, Stefano Gustincich, PhD6, Elisabetta Traggiai, PhD7, Isabella Ceccherini, PhD8, Marco Gattorno, MD9, Stefano Volpi, MD, PhD1
1Pediatrician/Autoinflammatory Diseases and Immunodeficiencies Centre, IRCCS Istituto Giannina Gaslini, Genoa, Italy
2Geneticist/UOC Medical Genetics, IRCCS Istituto Giannina Gaslini, Genoa, Italy
3Pathologist/UO Pathology, IRCCS Istituto Giannina Gaslini, Genoa, Italy
4Biologist/Autoinflammatory Diseases and Immunodeficiencies Centre, IRCCS Istituto Giannina Gaslini, Genoa, Italy
5Geneticist/Area of Neurobiology, SISSA, Trieste
6Geneticist/RNA Central lab, IIT, Genova
7Biologist/Novartis Institutes for Biomedical Research, Basel 4056, Switzerland
8Head, Geneticist/UOC Medical Genetics, IRCCS Istituto Giannina Gaslini, Genoa, Italy
9Head, Pediatrician/Autoinflammatory Diseases and Immunodeficiencies Centre, IRCCS Istituto Giannina Gaslini, Genoa, Italy
RAS-associated autoimmune leukoproliferative disease (RALD) is a rare immune dysregulation syndrome caused by somatic gain-of-function mutations of either NRAS or KRAS gene in a subset of hematopoietic cells. We describe a patient with atypical RALD displaying hypogammaglobulinemia, multi-organ autoimmunity and liver failure.
Immune and genetic analyses were conducted as part of the patient's clinical workup and in the contest of a research project on pediatric autoimmune diseases approved by our hospital institutional ethics committee.
The patient is a 27-year-old boy who presented at 5 months of age with EBV infection, followed by episodes of recurrent otitis media and lymphadenopathy. During childhood, he developed polyarthritis, nephrotic syndrome with minimal change disease, growth and pubertal delay, pulmonary fibrosis, autoimmune hypothyroidism, alopecia, celiac disease, anemia and hypogammaglobulinemia. Immunological studies showed normal lymphocyte subpopulations including CD4/CD8 double negative T cells and Fas-mediated apoptotosis. Peripheral blood type I interferon signature was positive. Partial control of the disease was obtained during the years with oral steroids, hydroxichloroquine, and mofetil mycophenolate, together with IVIG prophylaxis. Since 18 years of age, regenerative nodular hyperplasia of the liver was detected, causing hepatopulmonary syndrome Whole exome sequencing analysis revealed the heterozygous p.Gly13Asp missense mutation in the NRAS gene. As reported, the mutation was demonstrated to be somatic, being absent at Sanger sequencing of oral swab, urine sediment and hair follicle DNA.
Our report highlights the possibility of detecting somatic NRAS gene variants in patients with inflammatory CVID. NRAS-dependent type I interferon pathways activation may be involved in the multi-organ autoimmunity of RALD. Life-threatening complications, as end-stage liver failure, can occur in case of a prolonged diagnostic delay.
Keywords: CVID, Interferon, NRAS, SLE, RALD
Disclosures: All authors indicated they had no financial relationships to disclose.
(54) Heterozygous transcription factor 3 gene (TCF3) mutation is associated with absent B cells, normal immunoglobulin levels and function and severe thrombocytopenia, which responded well to Rapamycin
Manar Abdalgani, MBBS1, Tam Pozos, MD PhD2, Diana Vilkama, APRN CNP3, Yoav Messinger, MD4
1Clinical Immunologist/Children's Hospitals and Clinics of Minnesota
2Medical Director of Immunology/Children's Hospitals and Clinics of Minnesota
3Immunology Nurse Practitioner/Children's Hospitals and Clinics of Minnesota
4Medical Director-Hematology Oncology/Children's Hospitals and Clinics of Minnesota
We present the case of 17 month old male with a novel heterozygous mutation in TCF3 and previously unreported phenotype: absent circulating CD19+ B cells and significant thrombocytopenia that improved with immunosuppression. There is also a family history of two half-sisters who died during early infancy with similar phenotype.
The male index case was born with respiratory failure and generalized rash. At birth, he had thrombocytopenia (24k/uL) and lymphopenia (465/uL). Initial absolute CD3 T cell count was low (442/uL), yet he had normal thymic output. CD19+ B cells were < 5/uL and bone marrow biopsy revealed decreased hematagones, yet he had initially preserved humoral function. He later developed low IgG levels requiring replacement.
Rapid genome sequencing revealed a heterozygous, predicted deleterious VOUS in TCF3, in the second transactivation domain (c.1138 C>T, p Pro380Ser). His deceased half-sisters and the father showed the same mutation: all 3 children had different mothers, suggesting an autosomal dominant inheritance pattern. An infant sister was born 5 months ago to the same parents as the index case, with a similar immunophenotype yet her TCF3 targeted gene sequencing is normal.
In the months after birth, the index case continued to require frequent platelet transfusions. He demonstrated increased T cell activation with elevated levels of soluble IL-2R (2510 pg/ml) and increased percentage of T cells expressing HLA-DR, CD95, CD25, CD71, and CD69 activation markers. A 10-day trial of prednisone resulted in significant improvement of his thrombocytopenia. He was switched to rapamycin, and his platelet count has remained normal since then. His B cell counts and lymphopenia almost normalized on rapamycin. His 5-month-old sister’s thrombocytopenia also resolved on rapamycin.
In this abstract, we present a case of an infant with absent B cells as well as severe thrombocytopenia responsive to rapamycin. The lack of TCF3 mutation in the 5-month-old sister calls into question the relevance of this mutation to the index case and his older half-sisters despite the established role of TCF3 in early B cell development. In collaboration with colleagues at NIH, studies are underway to evaluate if there is a contribution of TCF3 to this family’s phenotype.
Keywords: Thrombocytopenia, TCF3, Absent B cells
Disclosures: Tamara Pozos is employed by Smiths Medical. All other authors had no financial relationships to disclose.
(55) Skin disease related to NFKB1 abnormality improve by anti IL1 treatment
Elma Nievas, Hospital Pediátrico A Fleming1, Leonardo Mannino, n/a2, Natalia Tamburri, n/a3, Diego Cristal, n/a4, Mikko Sepanenn, n/a5, Silvina Denita, n/a6
1Pediatric Inmunologist/Hospital Pediátrico A Fleming
2Pediatric Infetology/Hospital Pediatrico A Fleming
3Pediatric Surgery/Hospital Pediátrico A Fleming
4Peditric Nursery/Hospital Alexander Fleming
5Rare Disease and Pediatric Research Centers, Hospital for Children and Adolescents, and Adult Immuno/Unit, Inflammation Center, University of Helsinki and HUS Helsinki University Hospital, Helsinki, Finland
6Molecular Biologist/Biogenotec Lab
14 years old male, born from non consanguineous couple. Past history of deep necrotizing ocular cellulitis at 2 years old associated to fever, neutrophilia and increased inflammatory markers. Lesion’s microscopy: inflammatory infiltration, abscess areas, mononuclear infiltration associated with medium vessels vasculitis, necrobiosis on epidermis. Panniculitis with vasculitis assumed and managed as pyoderma gangrenosum
He was followed up for dermatologist for 8 years, and received several oral steroids treatment after minimal traumatism because of pathergy phenomena.
On April 2019 was admitted on our Hospital because of prolonged fever of unknown etiology. He presented abdominal pain, severe leukocytosis and increase CRP. Pneumonia was diagnosed after chest-X-ray. After 3 days of intravenous antibiotics pleural effusion was observed and thoracentesis was made without any result but presented complications: diaphragm damage and secondary stomach ulcer by puncture. After gastroraphy, he developed dehiscence of the abdominal skin incision as post-surgery gangrenosum pyoderma
CT chest scan was made after surgical complications: nodules, consolidation, bilateral pleural effusion and ground glass opacity were the main findings. Laboratory assay: ANA, ANCA and antiphospholipid antibody were negative. Normal level of Immunoglobulins (IgG 1193, IgA 171, IgM213, IgE 40,8) Normal response to vaccines. Total Lymph: 3840 CD3:77% CD4:45% CD8: 29% CD19-20:12%. CD16-56: 11%. CD4/RA: 61% CD4/RO :39% CD19+ CD27 +: 9,6%. CD19+ C27+ IgD+: 5,3% CD19+CD27+IgD-: 4,3% . Normal DHR Test.Elevated serum amyloid
Corticosteroid was indicated and continued after 6 weeks to high dose (2mg/kg/day) with good results. In order to tapper steroids and look for long term mainstay treatment anti Il 1 (canakinumab 4 mg / kg / dose each 4 weeks) was indicated suspecting autoinflammatory disease.
Abdominal postsurgical incision pyoderma demonstrated improvement and the wound healed after high dose of steroids, local care and canakinumab. Nowadays he is on anti - IL 1 treatment and diaphragm damage resolved and stomach fistula healed. Pulmonary abnormalities improved too. Inflammatory parameters normalized. Genetic test by NGS was made and an NFKB1 p.(Arg156Pro) pathogenic variant was found, confirmed by sanger sequencing. Genetic test on parents are ongoing.
Keywords: NFKB1, ANTI IL1, PYODERMA GANGRENOSUM
Disclosures: All authors indicated they had no financial relationships to disclose.
(56) Risk of TNF-alpha Inhibitor Use in Patients with Undiagnosed Mendelian Susceptibility to Mycobacterial Disease
Jacqueline Squire, MD1, Jared Nelson, MD2, Lisa Brumble, MD1, Claudia Libertin, MD1, Federico Laham, MD3, Jennifer Leiding, MD4
1Physician/Mayo Clinic
2Infectious Disease Fellow/Mayo Clinic
3Physician/Orlando Health Arnold Palmer Hospital for Children
4Associate Professor/Division of Allergy and Immunology, Department of Pediatrics, University of South Florida at Johns Hopkins-All Children’s Hospital, St Petersburg, FL
Tumor necrosis factor-alpha (TNF-alpha) is a proinflammatory cytokine that has become a target for therapeutic blockade in the treatment of rheumatic and inflammatory conditions. Infection, particularly reactivation of latent tuberculosis (TB) or nontuberculous mycobacterium (NTM), is the most notable risk. We present two patients with undiagnosed Mendelian susceptibility to mycobacterial disease (MSMD) that developed disseminated mycobacterial infection following use of TNF-alpha inhibitors.
Case 1: A 4-year-old African American female presented with persistent fever and progressive cervical lymphadenopathy. She was diagnosed with juvenile idiopathic arthritis a year prior and was being treated with methotrexate and infliximab. On admission, she also had a scalp abscess with osteolytic skull lesion. Skull biopsy showed acute osteomyelitis, lymph node biopsies demonstrated non-caseating granulomas. All cultures grew Mycobacterium avium complex (MAC). Genetic testing revealed a heterozygous pathogenic IFNGR1, c.819_822del (p.Asn274Hisfs*7) mutation consistent with autosomal dominant IFNGR1 deficiency. Treatment with anti-mycobacterial agents and IFN-gamma has led to substantial improvement.
Case 2: A 50-year-old Caucasian female presented with retroperitoneal lymphadenopathy, splenomegaly, and right infrahilar pulmonary mass during evaluation for 50-pound weight loss. Lymph node and infrahilar mass biopsy demonstrated necrotizing granulomas, cultures negative. She was treated with prednisone and then infliximab for idiopathic granulomatous disease. After starting infliximab, she developed panniculitis and worsening right lower lobe pulmonary opacity, BAL positive for MAC. Immune evaluation demonstrated panlymphopenia with markedly reduced NK cells 0-1 cells/mcL (ref:101-678 cells/mcL), B-cells 6-10 cells/mcL (ref:99-527 cells/mcL), and monocytes .02x109/L (ref:0.26-0.81x109/L). Genetic testing revealed heterozygous pathogenic mutation in GATA2, c.1192C>T (p.Arg398Trp). Now improving on anti-mycobacterial treatment and undergoing evaluation for HCT.
Blockade of TNF-alpha is effective in treating inflammation associated with autoimmune conditions. While generally well tolerated, there are known risks with use, most notably TB or NTM infection. Complicating the diagnosis, disseminated mycobacterial infections can mimic systemic inflammatory syndromes which can delay diagnosis and appropriate treatment. Evaluation for underlying or latent infection prior to starting therapy with TNF-alpha inhibitors is indicated. Underlying immune deficiency, particularly MSMD, should be suspected in the right clinical context prior to initiating treatment with TNF-alpha inhibitors due to the risks of disseminated, life-threatening mycobacterial disease.
Keywords: Mendelian susceptibility to mycobacterial disease, TNF- alpha inhibitor, Disseminated mycobacterial disease, GATA2, IFNGR1 deficiency
Disclosures: Federico Laham is an advisory board member of Tyme, Inc. Jennifer Leiding is an advisory board member and received speaker honoraria from CSL Behring; an advisory board member of Phamring; and received speaker honoriaria and a research grant from Horizon Therapeutics. All other authors had no financial relationships to disclose.
(57) X-linked Hyper IgM Syndrome (HIGM): A novel pathogenic variant causing decreased function of CD40L
Arjola Cosper, DO1, Rachel Eisenberg, MD2, Arye Rubinstein, MD PhD3
1Fellow In training/Albert Einstein College of Medicine/Children's Hospital at Montefiore
2Attending in Allergy and Immunology/Montefiore Medical Center
3Professor of Pediatrics, Microbiology and Immunology Attending in Medicine Chief Division of Aller/Albert Einstein College of Medicine
X linked HIGM due to hemizygous pathogenic variants in CD40L present in the first two years of life with recurrent upper and lower respiratory infections. Typical lab findings include neutropenia and elevated IgM in the setting of low IgA and IgG consistent with a defect in class switching.
An 18-month-old male presented with recurrent acute otitis media, sinusitis, and pneumonia. Initial work up revealed an IgG of 333mg/dL, IgA of 5mg/dL and IgM of 122mg/dL with no protective titers to measles, mumps, or rubella. B-cell phenotyping was notable for increased naive B-cells, low class switched memory B-cells and decreased memory B- cells. His infection burden and immunoglobulin levels did not improve over time and replacement immunoglobulin was initiated at 4 years of age. A 207 gene primary immunodeficiency panel (Invitae) was notable for a maternally inherited hemizygous variant of uncertain significance (VUS) in CD40L c.373 C>T (p. His125Tyr). While this missense substitution has not been observed in X-linked HIGM, in silico analysis all suggested this variant to likely be disruptive. Functional validation showed decreased expression of CD40L on 32% of CD4 stimulated T cells (normal range 86-98%). Additionally, CD40-Ig FP stimulated, measuring the binding capability of CD40L protein, was decreased to 29% (normal range 73-98%). Our patient’s milder phenotype can possibly be explained by the finding of reduced instead of aberrant expression of CD40L. In addition, typically CD40-Ig FP will not be able to bind mutated CD40L protein, however the variant in our patient allowed for some function of CD40L. The variant was soon after reclassified as pathogenic by Invitae.
HIGM due to CD40L pathogenic variants should be considered in any male patient presenting with low IgG and normal or elevated IgM with recurrent infections. These patients need to be followed for possible complications of abdominal tumors, PJP risk during infancy and pure red cell aplasia when infected with parvovirus. In some cases, HSCT (hematopoietic stem cell transplant) is performed however the risk/benefit assessment may not be in favor in cases with milder phenotype of CD40L deficiency like our patient.
Keywords: Hyper IgM, CD40 Ligand, novel variant
Disclosures: All authors indicated they had no financial relationships to disclose.
(58) Post COVID-19 multisystem inflammatory syndrome in adults; a case series
Sibel Buabeng Atmaca, s.atmaca@erasmusmc.nl1, Jasper Schuurbiers, MD2, C.A. Den Uil, MD3, V.A.S.H. Dalm, MD,Phd4, P.M. van Hagen, Proffesor4, K Caliskan, MD, Phd5, S.M. Rombach, MD6
1Resident internal medicine/Erasmus Medical Centre
2resident intensive care medicine/Erasmus Medical Centre
3Intensivist-Cardiologist/Erasmus Medical Centre
4internist immunologist/Erasmus Medical Centre
5Cardiologist/Erasmus Medical Center
6Internist- immunologist/Erasmus Medical Center
Since the start of the COVID-19 pandemic multiple reports have been published about a new disease entity called multisystem inflammatory syndrome in children (MIS-C). The World Health Organization published the case definition of MIS-C as children and adolescents younger than 19 years old, presenting with fever > 38°C for > 3 days, multi organ (>2) ) life-threatening inflammation following SARS-CoV2 infection. Clinical data about MIS in adults is rare. We present a descriptive study of five adult patients with life-threatening cardiac insufficiency after COVID-19, meeting the criteria for MIS-C turning the diagnosis into MIS-in adults (MIS-A).
We included 5 adults admitted at the intensive care unit of the Erasmus Medical Center, 25-58 years of age who otherwise met the case definition of MIS-C. Prior and during treatment we performed routine blood test to monitor the course of cardiac damage and systemic inflammation. These include, CRP, ferritin, sIL-2R, interleukin 6, NT-Pro-BNP and Trop T. Cardiac function was monitored by ultrasound.
All patients were treated with intravenous immunoglobulins (IVIG) 2 g/kg bodyweight and methylprednisolone 2 mg/kg bodyweight in addition to medication for hemodynamic and cardiac support. In case of a good response we tapered MPS to zero within three weeks. We also performed whole exome sequencing in order to identify genetic determinants which may predict the development of MIS-A.
Within one week after start of treatment, routine inflammatory markers of all patients decreased with 87-96% and cardiac function improved significantly on cardiac ultrasound. Three patients could be discharged from the hospital after 7, 10 and 12 days after diagnosis. They recovered completely without significant residual disease. One patient had a prolonged hospital stay because of delayed recovery because of muscle weakness and the other patient because of neurological complications after coronary artery angiography.
The adult variant of MIS warrants early recognition and the start of high dose IVIG and methylprednisolone treatment. This resulted in our series in rapid improvement of cardiac function and systemic inflammatory parameters. WES results are pending, hopefully these contribute to more insights in the genetic determinants of MIS-A.
Keywords: Case series, MIS-A, Sars-CoV-2, kawasaki like disease, intravenous immunoglobulins (IVIG), methylprednisolone, cardiogenic shock, myocardial biopsy, COVID 19
Disclosures: V.A.S.H. Dalm has contracted research with CSL Behring and Takeda. P.M. van Hagen has contracted research with CSL Behring and Takeda. All other authors had no financial relationships to disclose.
(59) Fulminant Renal Failure in a Patient with Autosomal Dominant Hyper IgE Syndrome
Jason Moraczewski, MD1, Safia Nawaz, MD2, Tricia Lee, MD3
1Resident/Emory University School of Medicine/Children's Healthcare of Atlanta
2Fellow/Emory University School of Medicine/Children’s Healthcare of Atlanta
3Assistant Professor/Emory University School of Medicine/Children's Healthcare of Atlanta
Hyper IgE syndrome (HIES) is a primary immunodeficiency characterized by elevated serum IgE, recurrent bacterial pulmonary and cutaneous infections, eczema, and various connective tissue and craniofacial anomalies. The autosomal dominant variant of HIES is due to a loss of function (LOF) mutation in STAT3. Here we describe a 12-year-old male with autosomal dominant HIES who presented with acute renal failure requiring dialysis.
The patient was diagnosed in early childhood following multiple abscesses requiring drainage and recurring episodes of pneumonia resulting in partial right upper lobectomy. At age 8, he was diagnosed with cutaneous Fusarium solani and received intermittent amphotericin B infusions for 6 months. This resulted in acute kidney injury with improvement in renal function following completion of therapy. Two months prior to presentation the patient was restarted on Bactrim prophylaxis, which he had previously tolerated. At age 12, he presented to the ED with emesis, dizziness, and blood pressure of 151/105. Laboratory workup revealed pancytopenia, hyperkalemia, hypocalcemia, hyperphosphatemia, hyperparathyroidism, elevated BUN, and markedly elevated creatine at 16.8 mg/dL, prompting concern for chronic kidney disease. Renal ultrasound revealed increased echogenicity bilaterally with no evidence of obstruction. Renal biopsy revealed immune complex deposition and immunofluorescence was positive for deposits of IgM, IgG, C1q, and C3c. Autoimmune workup yielded positive ANA (1:80, speckled), low C3, and negative dsDNA IgG, while elevated LDH, decreased haptoglobin, anemia, and absence of schistocytes suggested autoimmune hemolytic anemia. Malignancy was ruled out with no masses visualized on chest x-ray or abdominal ultrasound. The patient was stabilized following initiation of dialysis for his end-stage renal disease.
The rare presentation of fulminant renal failure in a patient with HIES suggests immune dysregulation as a primary contributing factor, but the correlation between STAT3 LOF and said dysregulation has only sparsely been alluded to in previous literature. A small portion of patients with STAT3 LOF have been seen to present with a lupus-like picture with predominant renal involvement. There may be a role for screening these patients for lupus-like disease in an attempt to mitigate disease burden and allow for targeted therapies.
Keywords: Hyper IgE, Renal failure, STAT3 LOF, Immune dysregulation
Disclosures: All authors indicated they had no financial relationships to disclose.
(60) Mass Cytometry Reveals Immune Cellular-Signaling Defects in Common Variable Immunodeficiency with Granulomatous Lymphocytic Interstitial Lung Disease
Victor Lui1, Ryan Baxter, MSc2, Amy Rymaszewski, PhD3, Shaoying Chen, MD3, Tusharkanti Ghosh, PhD4, James Verbsky, MD, PhD5, John Routes, MD6, Elena Hsieh, MD7
1Student/University of Colorado Anschutz Medical Center
2Senior Professional Research Assistant/University of Colorado Anschutz Medical Center
3Research Associate/Medical College of Wisconsin
4Postdoctoral Fellow/University of Colorado Anschutz Medical Center
5Associate Professor of Pediatrics, Medicine, Microbiology and Immunology/Medical College of Wisconsin
6Professor of Pediatrics, Medicine, Microbiology and Immunology/Medical College of Wisconsin
7Assistant Professor/Department of Pediatrics, Section of Allergy and Immunology, University of Colorado, Anschutz School of Medicine; Children’s Hospital Colorado; Department of Immunology and Microbiology, University of Colorado, Anschutz School of Medicine, Aurora, CO
Common Variable Immunodeficiency (CVID) is a clinical, immunological, and genetically heterogenous primary immunodeficiency (PID) characterized by hypogammaglobulinemia, failure to make specific antibodies, and recurrent infections. However, due to the immune-dysregulation in CVID, non-infectious complications such as B cell lymphomas, GI tract disorders, autoimmune cytopenias and granulomatous lymphocytic interstitial lung disease (GLILD) are the most common causes of morbidity and mortality in this disorder. The immunopathogenesis of GLILD, the pulmonary component of multisystemic granulomatous and lymphoproliferative disorder, is poorly understood. To further investigate the underlying immunological mechanisms leading to the development of GLILD within CVID, we utilized mass cytometry to identify unique immune cellular and signaling signatures from patients’ peripheral blood cells. We report that patients with CVID (n=25) or CVID with GLILD (n=20) had increased frequency of HLADR+ CD4+ T cells, CD57+ CD8+ T cells, and CD21lo B cells in the periphery compared to healthy controls (n=25). Within these cellular populations in CVID patients with GLILD, engagement of T and B cell receptors (respectively) results in an asynchronous downstream signaling response when compared to CVID patients without GLILD. In CVID patients with GLILD, CD21lo-B cells show perturbed protein kinase B (Akt) and extracellular signal-regulated kinase (ERK) activation, while HLADR+ CD4+ T cells and CD57+ CD8+ T cells display disrupted activation of kinases most proximal to the antigen receptor. These findings likely represent altered T/B receptor signaling responses in exhausted lymphocyte subsets in these patient groups, and may contribute to our understanding of the mechanisms of immune dysregulation in CVID with GLILD.
Keywords: Common Variable Immunodeficiency, Granulomatous Lymphocytic Interstitial Lung Disease, Mass Cytometry
Disclosures: John Routes has contracted research with CSL Behring and Evolve Biologics. Elena Hsieh is a consultant for Enzyvant. All other authors had no financial relationships to disclose.
(61) A Case Report of Hyper-IgM syndrome with Normal Serum IgA Level
Ameera Bukhari1, Amer Khojah, MD2
1PhD Student/Loyola University Chicago
2Attending Physician, Allergy, Immunology, and Rheumatology/Ann & Robert H. Lurie Children's Hospital of Chicago/ Northwestern University Feinberg School of Medicine
CD40L deficiency is an X-linked primary immunodeficiency disorder representing the most common form of hyper-IgM syndromes. CD40 and CD40L interaction is critical for the initiation of T cell-dependent B cell class switching. Therefore, patients with CD40L deficiency typically have no or low IgA and IgG. We present a case of CD40L defect who had normal IgA.
A 20-month-old boy with a history of congenital subglottic stenosis who presented with failure to thrive, developmental delay, recurrent fever, and oral ulcers. He had a low-grade fever (100.4-101) almost every two weeks for the last few months with recurrent mouth ulcers. He had four bilateral ear infections in the past six months but no recent pneumonia or sinus infections. He also had intermittent watery, non-bloody diarrhea lasting 2-3 days per episode with normal stools in between for many months. He also has delayed motor function (although he was sitting at the age of 8 months, he cannot walk yet) and speech delay. His laboratory evaluation was notable for intermittent elevated CRP (0.3-21 mg/dl), intermittent neutropenia (ANC 190 /uL), anemia and negative stool cryptosporidium Ag. His Immunoglobulin levels showed elevated IgM (397 mg/dl), normal IgA (78 mg/dl) and low IgG (51 mg/dl). He has undetectable antibody response Hib, tetanus, measles, and all streptococcus pneumonia serotypes. Whole exome testing showed likely pathological hemizygotes variant in the CD40LG (c.674 T>C, P.L225S) inherited from his mother. This variant has not been observed in large cohorts such as Gnomad or ExAC. CD40L expression is markedly decreased on the stimulated T cell ( 5%, and the reference range is more than 85%) with intact ICOS expression.
Although hyper-IgM syndromes classically present with absent serum IgA and IgG due to class switch defect, the diagnosis should not be excluded just because the patient has a normal serum IgA level. Of note, T cell-independent B cells IgA class switch has been reported in the intestinal mucosa after APRIL stimulation.
Keywords: Hyper-IgM syndrome, CD40L deficiency, class switching
Disclosures: All authors indicated they had no financial relationships to disclose.
(62) Increased detection of genetic mutations in patients with Common Variable Immunodeficiency (CVID) and associated pulmonary involvement
Emily Haltigan, MD1, Zerka Wadood, MD2, Divya Patel, DO3, Diana Gomez-Manjarres, MD4, Lyda Cuervo-Pardo, MD5
1Resident Physician/Department of Medicine, University of Florida
2Fellow/Division of Pulmonary, Critical Care and Sleep Medicine; University of Florida
3Clinical Assistant Professor/Division of Pulmonary, Critical Care and Sleep Medicine; University of Florida
4Assistant Professor of Medicine/Division of Pulmonary, Critical Care and Sleep Medicine; University of Florida
5Assistant Professor of Medicine/Division of Rheumatology, Allergy, and Clinical Immunology, University of Florida
Common Variable Immunodeficiency (CVID) is the most common symptomatic immunodeficiency. It is characterized by both B and T-cell abnormalities, predisposition to infections, and noninfectious complications in in nearly 50% of patients. Pulmonary involvement has been documented in up to 46% of cases. Even though etiology is still unknown, CVID is considered a polygenic disorder, with multiple genes implicated in its pathogenesis. Genetic testing is advocated in the evaluation of patients, as results may have implications on diagnosis, prognosis, and treatment. The objective of this study is to characterize common pulmonary manifestations and genetic mutations in patients with CVID.
A retrospective chart review of CVID patients evaluated at the University of Florida between 2012 and 2019 was performed. Patients with diagnosis of primary CVID by consensus from our immunology and pulmonary departments were included. We collected demographic, clinical, physiological, radiographic, pathologic and genetic testing results.
Fifty patients were included in the analysis. Genetic testing data was available for twenty patients. Pathogenic mutations were identified in 55%. Variants of unknown significance were present in 70%. The most common genetic mutation identified was TNFRSF13B (TACI mutation) in 20%. Lung parenchymal involvement on chest imaging was present in 78% of patients. The most common CT findings were: nodules (38%), bronchiectasis (32%), and ground glass opacities (22%). Eight patients had a lung biopsy. Pathology showed lymphoid aggregates in most patients (78%). Granulomas and bronchiolitis were also seen (44%).
We described characteristics of genetic mutations and pulmonary manifestations among CVID patients at our institution. Prevalence of lung disease in this cohort emphasizes the importance of screening asymptomatic patients with chest imaging for early detection. Even though most cases of CVID have no clear associated genetic defect, and monogenic causes are thought to account only for 10% of cases, our data documented pathogenic mutations in 55% of cases. Genetic testing, especially in patients with pulmonary involvement should be advocated, which could result in a higher yield. Efforts to characterize disease phenotype may lead to early detection of organ involvement and potentially allow for earlier intervention and improved outcomes.
Keywords: CVID, genetics, lung disease, immunodeficiency, PID, genetic testing
Disclosures: All authors indicated they had no financial relationships to disclose.
(63) Hyper IgM Syndrome in Toddler with 18p Deletion
Erin Dennis, MD1, Jason Caldwell, DO2
1Fellow PGY4/Wake Forest Baptist Medical Center
2Fellowship Director/Wake Forest Baptist Medical Center
An 18 month old male with 18p deletion presented following low immunoglobulin levels in setting of history of previous chylothorax, worsening bronchiectasis, and recurrent infections concerning for primary immunodeficiency. His immunoglobulin levels revealed an IgG of < 75 , IgA of < 10, and IgM of 55 at 3 months of age in the setting of chylothorax. Repeat immunoglobulins at 18 months old continued to demonstrate low IgG, undetectable IgA and IgE, and normal IgM. Although the chylothorax resolved, he continued to show abnormal antibody levels, as well as experience infections requiring hospitalization.
Given his picture, primary immunodeficiency is being considered, particularly Hyper IgM Syndrome. Initial evaluation showed an absence of switched memory B cells. There are relatively normal T, B, and NK cell populations. He did have protective titers to tetanus and diphtheria, although maternal antibodies may still be present. His streptococcal titers showed adequate response. The lymphocyte response to mitogens was normal, but he had markedly reduced lymphocyte responses to both tetanus and Candida. An assay to evaluate the presence of CD40L was normal. A literature search revealed that none of the known genes associated with Hyper IgM Syndromes are located on chromosome 18 . (1)
While 18p deletion is associated with Selective IgA deficiency (2), this patient’s immune pattern is concerning for a more significant immunodeficiency. It is likely he has an unrelated primary immunodeficiency based on known gene associations with Hyper IgM Syndrome. Phenotypically the patient has an immune evaluation that meets the criteria for Hyper IgM Syndrome. Absent switched memory B cells supports this diagnosis. His normal CD40L study suggests a mutation outside the CD40 or CD40L genes. Genetic tests will help confirm this diagnosis as well as better classify his phenotype for a more tailored treatment approach.
References: 1. Yazdani R, Fekrvand S, Shahkarami S, Azizi G, Moazzami B, Abolhassani H, Aghamohammadi A. The hyper IgM syndromes: Epidemiology, pathogenesis, clinical manifestations, diagnosis and management. Clin Immunol. 2019 Jan;198:19-30. doi: 10.1016/j.clim.2018.11.007. Epub 2018 Nov 13.
2. Browning MJ. Specific polysaccharide antibody deficiency in chromosome 18p deletion syndrome and immunoglobulin A deficiency. J Investig Allergol Clin Immunol. 2010;20(3):263-6.
Keywords: Hyper IgM Syndrome, 18 p deletion, chromosome abnormality, primary immunodeficiency, chylothorax
Disclosures: All authors indicated they had no financial relationships to disclose.
(64) Efficacy and safety of administration of cutaquig (16.5% IGSC) in pediatric patients with primary immunodeficiencies (PI): Data from two phase 3 studies
Roger Kobayashi, MD1, Jose Fernando Mandujano, MD2, Syed Rehman, MD3, Ai Lan Kobayashi, MD4, Bob Geng, MD5, Elisabeth Clodi, PhD6, Eva Turpel-Kantor, MD7, Sudhir Gupta, MD, PhD8
1Allergist Immunologist/Allergy, Asthma, and Immunology Associates PC
2Pediatric Pulmonologist/Pediatric Pulmonary Associates of North Texas, P.A.
3Allergist Immunologist/Asthma and Allergy Center
4Allergist Immunologist/Midlands Pediatrics PC
5Allergist Immunologist/Rady's Children's Hospital San Diego
6Senior Global Medical Advisor/Octapharma PPG
7Clinical Project Manager/Octapharma Pharm. ProduduktionsgesmbH
8Professor of Medicine, Pathology & Laboratory Medicine, and Microbiology & Molecular Genetics/University of California, Irvine
Cutaquig is a 16.5% immunoglobulin solution for subcutaneous administration, currently approved for adult subjects with PI.
Two prospective, open-label, non-controlled, non-randomized, multicenter trials including pediatric patients with PI have recently been completed.
The pivotal study (65 weeks long, NCT01888484) assessed the pharmacokinetics (PK), efficacy and safety of Cutaquig in pediatric and adult patients with PI and was followed by an extension study (NCT03907241) assessing the medium-to-long-term safety and efficacy (max. treatment 168 weeks).
The pivotal study included 38 pediatric patients (76% male) in 3 subgroups: 12 young children (aged 2 to 5yrs), 14 older children (6 to 11yrs) and 12 adolescents (12 to 16yrs). Ten pediatric subjects continued in the extension study (2 young, 4 older children, 4 adolescents). Pediatric patients received a total of 3,283 cutaquig infusions.
PK data from 19 pediatric subjects were collected. Weekly cutaquig administration resulted in flat PK profiles with lower fluctuations at steady state after cutaquig administration than after IGIV. No patient had IgG trough levels below 5 g/L.
Clinical efficacy of cutaquig was confirmed by zero SBI (serious bacterial infection) reported in pediatric patients in either study. Overall rate of infections was similar across the age groups at between 2 and 4 infections/person-year in the pivotal study with the highest rate in young children (4.2). In the 10 pediatric patients in the extension study the rate of infections/person year was highest in the adolescent group (2.9). Most common infections were respiratory infections. The number of days absent from school/person-year was 8.5 in the youngest, 3 in older children and 4 days in adolescents in the pivotal study. Similar results were seen in the extension study.
Cutaquig administration was well tolerated with no related serious adverse reactions reported in pediatric patients in either study and 5 related adverse reactions in both studies (none in the youngest subjects). Infusion site reactions were reported in 25 (66%) pediatric patients during the pivotal study and in 7 (70%) in the extension study (all except one of mild/moderate intensity).
Data from both studies demonstrate similar efficacy and safety of cutaquig administration in pediatric patients as in adults.
Keywords: IgG, SCIG, Efficacy, subcutaneous, primary immune deficiency, pediatric patient, safety, PI, immunoglobulin
Disclosures: Roger Kobayashi received speaker honoraria from IgNS and a research grant and speaker honoraria from Octapharma. Syed Rehman received contracted research and speaker honoraria from Astra Zeneca, Genentech, and Takeda; contracted research with Kedrion, Novartis, Sagent, Sanofi, Takeda, and TEVA; speaker honoraria from GSK. Ai Lan Kobayashi received a research grant from Octapharma. Bob Geng was an advisory board member for Biocryst and received speaker honoraria from Grifols, GSK, Horizon, Kedrion, Octapharma, Optinose, Regeneron, Sanofi, and Takeda; he was a consultant for RMS. Elizabeth Clodi, Eva Turpel-Kantor are employees of Octapharma PPG. Sudhir Gupta received research grants from Octapharma and Takeda. All other authors had no financial relationships to disclose.
(65) Increased susceptibility to allergic and autoimmune diseases in patients with activated phosphoinositide 3-kinase (PI3K) delta syndrome (APDS)
Stephanie Kubala, MD1, Marjohn Rasooly, MSN, CRNP2, Gulbu Uzel, MD3, Pamela Frischmeyer-Guerrerio, MD, PhD4
1Clinical Allergy/Immunology Fellow/Laboratory of Allergic Diseases, National Institute of Allergy and Infectious Diseases, NIH
2Family Nurse Practitioner/Laboratory of Allergic Diseases, National Institute of Allergy and Infectious Diseases, NIH
3Physician scientist/Laboratory of Clinical Immunology and Microbiology, National Institutes of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland, USA
4Chief/Laboratory of Allergic Diseases, National Institute of Allergy and Infectious Diseases, NIH
Activated phosphoinositide 3-kinase (PI3K) delta syndrome (APDS) is caused by gain-of-function mutations (GOF) in the genes PIK3CD and PIK3R1, encoding for the p110δ and p85 subunits of phosphoinositide 3-kinase δ (PI3Kδ). Hyperactivation of the PI3K/AKT/mTOR/S6K pathway leads to immune dysregulation, lymphoproliferation, and immunodeficiency. Additionally, PI3K is a central regulator of mast cell, T follicular helper cell and Th2 cell function. Therefore, we hypothesize that patients with APDS would be predisposed to allergic, as well as autoimmune disease.
Thirty-seven patients (18 male; median age 17 years, range 6-54) with confirmed PI3K activating mutations were evaluated in a prospective natural history study. Twenty-eight patients had peripheral blood obtained to evaluate hematologic variables, immunoglobulins, total and allergen specific IgE, and tryptase.
Thirty-two of the thirty-seven patients had a positive allergic history; 23 had allergic rhinitis, 15 atopic dermatitis, 5 chronic urticaria, 1 chronic angioedema, and 16 physician-diagnosed asthma. Five patients had IgE-mediated food allergies with avoidance encompassing peanuts, tree nuts, eggs, and milk. Five had biopsy-proven eosinophil-associated gastrointestinal disorders. All patients denied contact dermatitis. Eleven patients reported autoimmune disorders including granulomatosis with polyangiitis, ulcerative colitis, hashimoto's, sjogren's, lupus, autoimmune hemolytic anemia, immune thrombocytopenic purpura, and other immune cytopenias. White blood cell counts were within normal limits (median 5.58 K/uL, range 2.12-15.05 K/uL) with absolute lymphocyte counts trending low (median 1.28 K/uL, range 0.45-3.07 K/uL). Median total IgE and eosinophil counts were within normal limits (20.3 IU/mL; range < 1–54276 IU/mL and 0.14 K/uL; range 0-1.84 K/uL, respectively) but varied widely. All other immunoglobulins were within normal limits. Of the 16 patients who had vaccine titers drawn, 4 had decreased responses. Median baseline serum tryptase level was 5 ng/ml (range 1.5-14.3 ng/ml). Seven out of twenty-eight patients had detectable specific IgE to common food allergens. Patients were most likely to be sensitive to milk (6 patients), egg (4 patients), peanut (3 patients), and wheat (3 patients). No patients were sensitized to finned fish or shellfish.
Patients with PI3K activating mutations exhibit an increased prevalence of atopic and autoimmune disorders, demonstrating an important role for PI3K in tolerance development in humans.
Keywords: PI3K, PIK3CD, APDS, allergy, autoimmunity
Disclosures: All authors indicated they had no financial relationships to disclose.
(66) Use of emapalumab and cyclophosphamide in a patient with X-linked lymphoproliferative disease and EBV-associated hemophagocytic lymphohistiocytosis complicated by retinal vasculitis
Sebastian Ochoa, MD1, Sameeya Ahmed-Winston, CPNP-PC2, Michael Keller, MD3, Blachy Blachy Dávila Saldaña, MD4
1Clinical Fellow/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases/National Institutes of Health, Bethesda, MD, USA
2Pediatric Nurse Practitioner/Division of Blood and Marrow Transplantation, Children's National Health System, Washington, DC, USA
3Pediatric Immunologist/Division of Allergy and Immunology, Children's National Health System, Washington, DC, USA
4Pediatric Hematologist/Oncologist/Division of Blood and Marrow Transplantation, Children's National Health System, Washington, DC, USA
A 15-year-old boy was admitted due to worsening fever, adenopathy, and splenomegaly after diagnosis of infectious mononucleosis. Diagnostic workup showed cytopenias (absolute neutrophil count of 0.3k/μL, platelets 83k/μL), AST 277 units/L, ALT 416 units/L, ferritin 1832.6ng/mL, fibrinogen 144mg/dL, soluble CD25 1606 (NR <=1033pg/mL), and borderline decreased NK cell activity. Bone marrow biopsy showed decreased marrow cellularity and hemophagocytosis, thus meeting criteria for hemophagocytic lymphohistiocytosis (HLH). Quantitative EBV polymerase chain reaction (PCR) analysis demonstrated 103,409 copies/mL. Cerebrospinal fluid (CSF) studies showed 25 WBCs/mm3 (86% lymphocytes, 14% monocytes) and CSF EBV PCR 11,137 copies/mL. Immunologic testing was notable for normal IgG (1280mg/dL), elevated IgA (442mg/dL) and IgM (317mg/dL); elevated CD3 (2958, NR 820-2035 cells/μL) and CD8 (2589, NR 262-864 cells/μL) and low CD4 (319, NR 527-1380 cells/μL) and NK cells (123, NR 160-390 cells/μL). Genetic analysis revealed a pathogenic variant in chromosome X SH2DIA (c.23A>C, p.His8pro) and NK and CD8 SAP expression by flow cytometry was undetectable, uncovering the diagnosis of X-linked lymphoproliferative disease type 1 (XLP-1). He was treated for HLH with CNS disease with I.V. methylprednisolone and etoposide, as well as intrathecal (IT) methotrexate (MTX) and methylprednisolone, in addition to ganciclovir and rituximab. A search for a potential stem cell transplant donor was initiated. Due to refractory disease and elevated CXCL9 (671 pg/mL, NR <=121pg/mL), he was started on emapalumab 150mg (2.718 mg/kg) once weekly. Following 4 doses of IT therapy and 5 doses of rituximab, he was continued on Decadron and emapalumab. He had onset of retinal vasculitis prompting resumption of IT chemotherapy, addition of cyclophosphamide, and an increase in emapalumab to twice weekly dosing, resulting in clinical improvement, with minimal residual visual field loss. He underwent successful hematopoietic stem cell transplant from a matched unrelated donor 6 months after initial HLH diagnosis, which was complicated by EBV-PTLD, treated with rituximab and third-party EBV-specific T-cell therapy. Retinal vasculitis is an uncommon manifestation of XLP-1, and there are only a few cases reported in the literature. We report the use of emapalumab and cyclophosphamide in a patient with XLP-1 and EBV-associated HLH complicated by retinal vasculitis, resulting in clinical improvement.
Keywords: XLP-1, XLP, Duncan syndrome, SAP deficiency, Retinitis, Vasculitis, Emapalumab, Cyclophosphamide, HLH, hemophagocytic lymphohistiocytosis
Disclosures: Sameeya Ahmed-Winston received speaker honoraria from Jazz Pharmaceuticals. Michaek Keller is an advisory board member of Enzyvant. All other authors had no financial relationships to disclose.
(67) Rituximab-induced Hypogammaglobulinemia and Infection Risk in Pediatric Patients
Roxane Labrosse, MD, MMSc1, Sara Barmettler, MD2, Beata Derfalvi, MD., PhD3, Annaliesse Blincoe, MBChB4, Guilhem Cros, MD5, Jonathan Lacombe-Barrios, MD1, Julie Barsalou, MD1, Nancy Yang, n/a6, Nora Alrumayyan, MD7, Jan Sinclair, MBChB4, Mei-Sing Ong, PhD8, Carlos Camargo, MD/DrPH2, Jolan Walter, MD, PhD9, Elie Haddad, MD, Phd10
1Physician/CHU Sainte-Justine
2Physician/Massachusetts General Hospital
3Associate Professor/Dalhousie University/IWK Health Centre
4Physician/Starship Children’s Hospital
5Physician/Centre Hospitalier de l'Université de Montréal
6Clinical Research Coordinator II/Massachusetts General Hospital
7clinical fellow/Division of Immunology, Department of Paediatrics, Dalhousie University, IWK Health Centre
8Assistant Professor/Harvard Medical School and Harvard Pilgrim Health Care Institute
9Division chief and associate professor/Johns Hopkins All Children’s Hospital; Department of Pediatrics, Division of Allergy and Immunology, University of South Florida Morsani College of Medicine
10Attending Physician/Immunology-Rheumatology Division, Department of Pediatrics, University of Montreal
Rituximab (RTX) is a B-cell depleting agent used in B-cell malignancies and autoimmune diseases. A subset of adult patients may develop prolonged and symptomatic hypogammaglobulinemia following RTX. However, this phenomenon has not been well delineated in the pediatric population.
Our objective was to determine the prevalence, risk factors and clinical significance of hypogammaglobulinemia following RTX therapy in children.
We conducted a multi-center, retrospective cohort study and extracted clinical and immunological data from pediatric patients who received RTX. Patients were excluded if they had a prior diagnosis of primary immune deficiency (PID) or if they received RTX after having undergone hematopoietic stem cell transplant (HSCT).
The cohort was comprised of 207 patients (median age 12.0 years). Compared to baseline values, there was a significant increase in reported hypogammaglobulinemia post-RTX, with an increase in prevalence of hypo-IgG (28.7% to 42.6%, p=0.009), hypo-IgA (11.1% to 20.4%, p=0.02) and hypo-IgM (20.0% to 62.0%, p < 0.0001). Additionally, low IgG levels at any time post-RTX were associated with a higher risk of serious infections (34.4% vs 18.9%; OR 2.3, 95% CI 1.1-4.8, p=0.03). Persistent IgG hypogammaglobulinemia (PH-IgG) was observed in 27 (26.7%) of 101 evaluable patients in whom IgG levels were measured beyond a year after RTX initiation. Significant risk factors for PH-IgG included low IgG and IgA levels pre-RTX and co-administration of prednisone. Nine patients (4.3%) within the study were subsequently diagnosed with a PID, seven of whom received RTX for autoimmune cytopenia. Genetic confirmation was present in 4 of the 9 patients (44.4%).
In the largest pediatric cohort described thus far, we found that hypogammaglobulinemia post-RTX was frequently diagnosed, and that low IgG levels were associated with a significant increase in serious infections. Our results highlight the importance of immunologic monitoring both before and after RTX therapy, and suggest that immunoglobulin replacement therapy should be considered in these patients. Furthermore, underlying PIDs were relatively common in children receiving RTX, particularly in those with autoimmune cytopenia. In patients with a suspected PID, genetic testing may help distinguish between iatrogenic (secondary) and primary immunodeficiency.
Keywords: rituximab, B-cell depleting therapy, hypogammaglobulinemia, infection, children, primary immunodeficiency, secondary immunodeficiency, IgG, IgA, IgM
Disclosure:. Elie Haddad was a consultant for Jasper Therapeutics and Rocket Pharma All other authors had no financial relationships to disclose
(68) Use Of Rituximab And Brentuximab Vedotin To Treat EBV-Associated Lymphoproliferative Disease Resembling Classical Hodgkin Lymphoma In A Patient With Ataxia Telangiectasia
Joshua Goldman, MD1, Amer Heider, MD2, Thomas Michniacki, MD3
1Pediatric Hematology/Oncology Fellow/University of Michigan
2Associate Professor of Pathology/University of Michigan
3Clinical Assistant Professor/University of Michigan
Patients with DNA repair disorders, such as ataxia telangiectasia (AT), both are at an increased risk for malignancy and experience low survival with standard cytotoxic chemotherapy given increased treatment toxicity. Median survival for AT patients with Hodgkin disease is approximately three months. We report novel use of rituximab and brentuximab vedotin (BV) to treat an EBV-associated lymphoma resembling classical Hodgkin disease in a patient with AT that resulted in prolonged overall survival compared to standard therapy while maintaining a good quality of life.
A 14-year old with AT presented with fever, night sweats, and hypoxia. MR chest/abdomen revealed pulmonary nodules, paratracheal/mediastinal adenopathy, and hepatic lesions. Liver biopsy showed an EBV-associated lymphoproliferative disorder resembling classical Hodgkin lymphoma with CD30+/CD20- cells (previously given rituximab dose for EBV viremia). The patient’s family declined cytotoxic chemotherapy, so treatment was initiated with rituximab 375 mg/m2 days 1/8/15/22 and BV 1.2 mg/kg days 1/8/15 mirroring a protocol for immunocompromised adults with EBV+ and/or CD30+ lymphomas. A partial response was noted following induction. Four additional weekly doses of 375 mg/m2 rituximab were completed with BV therapy moved to a more traditional dosing regimen of 1.8 mg/kg every 3 weeks. Given occurrence of peripheral neuropathy and exacerbation of the patient’s baseline tremor, therapy was transitioned to maintenance cycles of dose-reduced BV 1.2 mg/kg q3weeks and rituximab 375 mg/m2 q6weeks. Therapy was additionally augmented with prednisone 20 mg/m2 BID during Week 1 of Cycles 3-5. No convincing evidence of lymphoma was noted following Cycle 7. Therapy was well tolerated other than neuropathic pain managed with gabapentin and an episode of osteomyelitis. Unfortunately, the patient then experienced disease progression and rising EBV titers after 9 months of therapy and is attempting salvage therapy with additional chemotherapy-sparing agents.
Combination rituximab and brentuximab therapy should be considered for EBV+/CD30+ lymphomas in immunodeficient patients who poorly tolerate cytotoxic chemotherapy.
Table 1. Summary of administered therapy
Keywords: Primary immune deficiency, lymphoma, ataxia telangiectasia, rituximab, brentuximab, EBV
Disclosure: All authors had no financial relationships to disclose
(69) Combined Immunodeficiency With Autoimmunity Due Rag1 Mutation In A Preschooler With Systemic Lupus Erythematosus, Systemic Sclerosis, Antiphospholipid Syndrome, Vitiligo And Cytomegalovirus Infection
Oscar Moreno-Laflor, MD1, Lyuva Ramirez-Devars, MD2, Omar Saucedo-Ramirez, MD3, Blanca Del Rio-Navarro, MD, MSc4
1Allergy and Immunology fellow-in-training/Hospital Infantil de México
2Dermatology fellow in-training/Hospital Infantil de México
3Pediatric Allergy and Immunology/Hospital Infantil de México
4Pediatric Allergy and Immunology Training Program Director/Hospital Infantil de México
Immune dysregulation is emerging as a key feature of patients with human inborn error of immunity. RAG mutations with higher residual recombination activity are more likely to result in immune dysregulation and they have been identified in patients presenting later in childhood or in young adulthood with combined immunodeficiency with granulomas and/or autoimmunity (CID-G/AI).
METHODS : A 4 years and 5 months female infant with history of Vitiligo at 1 year 3 months old, Systemic Sclerosis (SSc) at 2 years 8 months old, Systemic Lupus Erythematosus (Overlap syndrome SLE/SSc) at 3 years 7 months, Antiphospholipid Syndrome at 4 years 1 month old and asymptomatic cytomegalovirus infection. The mother has a history about threatened miscarriage during pregnacy. The patient has not history about recurrent or severe infections. Laboratory evaluation for human inborn errors of immunity was made, lymphocyte subsets showed decreased T and B cells and normal count of NK cells: CD3+ 374.66mm3, CD8+ 146.1mm3, CD4+ 214.12mm3, CD16/56 110.8mm3, CD19+ 82mm3, CD4+CD45RA 105.3mm3, CD4+CD45RO 108.34mm3, CD8+CD45RA 103.6mm3, CD8+CD45RO 42.37mm3, CD4:CD8 1.36. Serum immunoglobins with decreased IgA: IgA 24 mg/dl, IgM 140 mg/dl, IgG 1210 mg/dl, IgE 42.9 mg/dl. It was started monthly intravenous gammaglobulin (IVIG) therapy. She is currently in treatment with mycophenolate mofetil, hydroxychloroquine, tocilizumab, acetylsalicylic acid and antibiotic prophylaxis.
Whole exome secuencing revealed a missense variant in RAG1 gene: c.1822G>A, p.(Val608Met). Exon 2/2. The tools that predict the effect of missense variants consider the change observed in this patient is deleterious. The variant found in this patient is located in the RNase H domain. The variant found in the patient is close to the active site, alters the function of the RAG1-RAG2 complex, reducing its activity and, therefore, the efficiency of the V(D)J recombination process.
RAG mutations show a diversity of clinical and immunological phenotypes. A broad range of serum autoantibodies has been demonstrated in patients with CID-G/AI, which is consistent with the diverse spectrum of clinical manifestations of autoimmunity. Our patient is currently in the program for hematopoietic stem-cell transplantation. This is the first case report about patient with four autoimmune diseases and primary inmunodeficiency.
Table 1: Lymphocytes subsets
Keywords: combined immunodeficiency, RAG mutation, Immune dysregulation, Vitiligo, autoimmune disease
Disclosure: All authors had no financial relationships to disclose
(70) The clinical coarse and outcome of SARS-COVID 19 in patients with inborn error of immunity , does it relate to the type of the immune defects
Roya Sherkat, M.D , Suspecialist1, Paria Bolourinejad, MD2, Negin Salemi, MD2, Farhoodeh Rahmati, PHd3, Hamid Aria, PHd3, Mahdieh Azizi, PHd4, Mahbubeh Jafari, MSc2, Somayeh Najafi, MSc2
1Associate Prof/Acquired Immunodeficiency Research Center, Isfahan University of Medical Sciences , Isfahan , Iran
2Research Fellow/Acquired Immunodeficiency Research Center, Isfahan University of Medical Sciences , Isfahan , Iran
3Research Fellow/Acquired Immunodeficiency Research Center, Immunology Department , Isfahan University of Medical Sciences , Isfahan , Iran
4Research Fellow/Acquired Immunodeficiency Research Center, Immunology Department ,Isfahan University of Medical Sciences , Isfahan , Iran
There are still enigmas about the impact of SARS-CoV-2 infection in individuals with inborn errors of immunity (IEI) and potential genetic effects on host immune response that may leading to increased susceptibility to SARS-CoV-2 infection. Understanding the pathways would help not only in unraveling disease pathogenesis, but also in suggesting targets for therapy and prophylaxis. It seems risk factors predisposing to severe disease and mortality in patients with IEI are related to the type of immune defects.
During a prospective study among our registry of IEI’s at Isfahan Immunodeficiency Research Center,14 cases with COVID19 declared their primary symptoms through tele-visit and clinical and immunological diagnosis made according to the PCR and laboratory tests. The clinical courses, treatment, and outcome of SARS-CoV-2 infection were followed in relation with the immunodeficiency disorders.
Among 14 patients, 7 (50%) were female, five patients (35.7%) suffer from Bruton Disease, three (21.4%) Common Variable Immunodeficiency, two (14.3%) Combined Immunodeficiency (CID), one (7.1%) Autosomal Recessive Hypogammaglobinemia, one (7.1%) CID with syndromic features, one (7.1%) IgM deficient and one (7.1%) Severe Congenital Neutropenia and the median age was 27 years. 10 (71.4%) patients were controlled and treated as outpatients via remote management, 3 (21.4%) required ward hospitalization and oxygen administration and one CID case (7.1%) admitted to the ICU without intensive ventilation.
This study showed IEI’s does not predispose all patients to severe COVID19 or related mortality. Among enrolled patients 9 (64.2%) of patients experienced moderate infection with satisfactory outcomes and 5(35.7%) suffered from severe COVID19 infection but were handled well. It seems that the course of infection and appearance of real cytokine storm is not the same as normal population but to find out about the best management and therapy in IEI’s more study has to be planned.

Table 1: Summary of patients Manifestations
Keywords: inborn error in immunity, SARS - covid19, clinical presentations
Disclosure: All authors had no financial relationships to disclose
(71) The evaluation of radiosensitivity in patients with STAT3 deficiency
Sukru Cekic, n/a1, Huzeyfe Huriyet, n/a2, Melika Hortoglu, n/a2, Safa Baris, MD3, Ayşe Metin, MD4, Ahmet Ozen, MD3, Elif Karakoç Aydıner, MD5, Candan Abakay, n/a6, Tolga Cavas, n/a7, Sara Kilic, n/a8
1Dr./Uludag University Faculty of Medicine, Pediatric Immunology
2Research Asistant/Faculty of Sciences and Letters, Biology Department, Uludag University, Bursa, Turkey.
3Professor of Pediatrics/Marmara University Hospital, Department of Pediatric Immunology and Allergy
4Prof. Dr./Ankara City Hospital
5professor/Marmara University Pendik Research and Training Hospital
6Assoc. Prof./Department of Radiation Oncology, Uludag University Faculty of Medicine, Bursa, Turkey.
7Prof./Faculty of Sciences and Letters, Biology Department, Uludag University, Bursa, Turkey.
8Prof./Uludag University Faculty of Medicine, Pediatric Immunology
The aberrant activity of STAT3, a powerful oncogene, leads to persistent activation of upstream regulators, such as cytokine and growth factor receptors. Therefore, a number of oncogenic pathways is up-regulated to promote aggressive tumor growth and migration. A relationship between abnormal STAT3 expression and increased radiosensitivity has been reported.
The aim was to investigate the radiation sensitivity in patients with STAT3 deficiency.
Twelve patients diagnosed with STAT3 LOF and age/sex-matched 11 healthy control were included in the study. Radiation sensitivity was evaluated by the Comet test. In the Comet test; the extents of spontaneous (0Gy), initial (immediately after 2Gy irradiation), and residual (2h after irradiation) DNA damage was evaluated by quantifying the Olive Tail Moment under an epifluorescence microscope using a computer-based image analysis system (Kameram Komet Module, Micro System Ltd. Turkey). Experiments were performed in triplicate and the obtained data is presented as the mean ± standard error of the mean.
The female/male ratio in patients STAT3 LOF is 1.4 (7/5). The age of patients diagnosed with STAT3 LOF is between 3.6 and 40.6 years old. There is no patient who did not develop cancer. The lymphocyte subgroups and immunoglobulin levels were normal in all patients. However, 10 of 12 patients with STAT3 LOF were treated with IVIG because of recurrent skin and pulmonary infections. The extent of initial DNA damage induced by 2Gy radiation was significantly higher in the patient’s lymphocytes in comparison with the control. On the other hand, after 2h of recovery, the level of DNA damage was low in control cells, whereas the extent of DNA damage increased in the patient’s lymphocytes, compared to initial values (table 1, and figure 1).
DNA repair mechanisms were impaired in all patients compared to controls. Impaired tumor immunosurveillance contributes to the high rate of malignancies in PID. Genetic defects that lead to impairment in DNA repair mechanisms can result in cancer. In this study, we showed an impairment in the DNA repair mechanisms in patients with LOF-STAT3 defects. This is the first study to disclose the increase sensitivity to radiation in these patient groups.

Table 1. The differences between Olive Tail Moments of patients with STAT3 LOF and healthy controls

Figure 1. The Olive Tail Moment of STAT3 LOF patients and healthy controls
Keywords: STAT3 LOF, Cancer, Radiosensitivity, DNA impairment
Disclosure: All authors had no financial relationships to disclose
(72) The phenomenon of high cytotoxic activity of the T-cell component of the immune system in patients with X-linked agammaglobulinemia
Lyudmila Sizyakina, PhD1, Irina Andreeva, PhD2
1Head. Dep. Clinical Immunology and Allergology/Rostov State Medical University
2professor of the Department of Clinical Immunology and Allergology/Rostov State Medical University
Primary immunodeficiency is an ideal natural model for studying the possibilities of functioning of the immune system with a defect of one of its links. In this regard, the primary immunodeficiency of the humoral type,which allows to evaluate the potential of the cell-based system in the whole is very informative. The analysis of the examination results of patients with X-linked agammaglobulinemia (XLA) was carried out to reveal the features of the functioning of the T-cell component of the adaptive immune response in the conditions of genetic antibody disorder.
12 men at the age of 10-25 years were under the supervision. The disease debuted in all patients in the first year of life and bacterial infections of the respiratory tract became a clinical manifestation. The diagnosis was confirmed by the detection of a genetic defect of BTK and/or a characteristic family history. The results of immunological testing after the detection of XLA before the replacement therapy were analyzed.
Expression of surface receptors and intracellular lymphocyte proteins were studied by flow cytometry. Healthy blood donors were examined as a comparison group. In patients with XLA an increase in the number of mature T-lymphocytes was revealed (91,40±2,5%, in the control 68.88±0.38%, p=0.03). The increase in the total pool of T- cells was mediated by increase of CD8+effectors (40.80±3.44%, in the control of 21.88 ±0.33%, p=0.001) with the strengthening of their cytolytic resources (CD8+Gr+ 32.60±3.84, in control of 9.38 ±2.21% , p < 0.0001). Changes in CD4+T-cells are associated with a decrease in the peripheral circulation of part of naive CD4+CD45RA+lymphocytes (12.6±5.3%, in the control 29.2±6.1%) and CD4+CD25+FoxP3+Treg (0.40±0.01%, in the control 1.3±0.3%, p=0.001).
Thus, the genetically determined absence of mature B-cells and complete antibody genesis contributes to the increase of quantitative and functional parameters of T-effectors of adaptive immunity with the involvement of immunoregulatory mechanisms which support their high potential.
Keywords: X-linked agammaglobulinemia, T-cells, immunoregulatory mechanisms
Disclosure: All authors had no financial relationships to disclose
(73) Second tier test options in newborn screening for inborn errors of immunity
Maartje Blom, MD1, Ingrid Pico-Knijnenburg, n/a2, Sandra Imholz, n/a3, Janika Schulze, Phd4, Jeannette Werner, Phd5, Mirjam van der Burg, PhD6
1Clinical Researcher/Leiden University Medical Center (LUMC)
2Research technician/Leiden University Medical Center (LUMC)
3Research technician/National Institute for Public Health and the Environment (RIVM)
4Senior Scientist/Epiume GmbH
5Scientist/Epiume GmbH
6Associate Professor Primary Immunodeficiencies/Leiden University Medical Center (LUMC)
Newborn screening (NBS) for severe combined immunodeficiency (SCID) and X-linked agammaglobulinemia (XLA) are based on the detection of indirect markers for newly formed T-lymphocytes i.e. T-cell receptor excision circles (TRECs) and B-lymphocytes i.e. kappa-deleting recombination excision circles (KRECS). There is a range of neonatal disorders and conditions that lead to low T- and/or B-lymphocytes around birth, causing NBS for SCID and XLA to be accompanied by secondary findings and false positive referrals.
The main objective was to explore different second tier options after TREC/KREC analysis in order to reduce the number of secondary findings, increase the positive predictive value for NBS for SCID/XLA and reduce the emotional impact for parents that is associated with a referral procedure.
NBS cards of newborns with low TRECs (N=56) and KRECs (N=110) measured with the SPOT-it kit (ImmunoIVD) were analyzed with a second TREC/KREC assay (NeoMDx PerkinElmer). Epigenetic immune cell counting (Epimune GmbH) based on unique DNA methylation markers was used for relative quantification of CD3+ T-cells and B-cells in NBS cards of healthy controls (N=311), newborns with low TRECs (N=58) and low KRECs (N=103). DNA was isolated for TREC region sequencing from NBS cards of healthy controls (N=12), idiopathic lymphocytopenia cases (N=4) and false positive referrals (N=8).
When analyzed with the second TREC/KREC assay, 25/56 newborns with low TRECs and 16/110 newborns with low KRECs had TREC/KREC levels above cut-off. With epigenetic qPCR, 15/58 newborns with low TRECs and 18/103 newborns with low KRECs had relative T-cell/B-cell counts within the 99.9% CI ellipse of healthy controls. A SNP was found in the TREC region of four false positive cases (14-22475276-G-T (GRCh38)) leading to a primer/probe mismatch and no TREC amplification with the SPOT-it kit.
All second tier options have the potential to reduce the number of referrals and secondary findings in NBS for SCID and XLA. Second tier testing with NGS with a SCID gene panel or BTK gene analysis are currently being explored as well. Sensitivity, specificity, costs, feasibility for the screening laboratories and throughput time should be further evaluated before second tier options can be implemented.
Keywords: second tier test, newborn screening, inborn errors of immunity, SCID, XLA, TRECs, KRECs, epigenetic immune cell counting, sequencing
Disclosure: All authors had no financial relationships to disclose
(74) A Heterozygous Pathogenic Mutation in FOXN1 in a patient with CVID
Michell Lozano Chinga, MD1, John Bohnsack, MD2, Angelica Putnam, MD3
1Pediatric Hematology-Oncology Fellow/University of Utah
2Professor and Chief - Division of Allergy and Immunology/University of Utah
3Associate Professor - Pathology/Department of Pathology, University of Utah, Salt Lake City, UT, USA
FOXN1 heterozygous mutations have been associated to thymic hypoplasia and T-cell lymphopenia. We report a case of a female teenager with CVID and mild T-cell lymphopenia found to have a pathogenic heterozygous FOXN1 mutation.
A 16-year-old female presented with an enlarging left neck mass and shortness of breath with exertion. Her medical history was remarkable for one episode of warm AIHA that resolved with prednisone. PE showed splenomegaly in addition to cervical and supraclavicular lymphadenopathy. Her CBC exhibited mild leukopenia with lymphopenia. CT of the neck and chest showed cervical, supraclavicular and mediastinal lymphadenopathy, and multiple pulmonary nodules. Biopsy of the cervical lymphadenopathy showed non-caseating granulomas. Cultures were negative, as was PCR for bacterial, fungal, and mycobacterial RNA. PFTs were consistent with mild persistent asthma.
Additional workup included low CD3, low normal CD4 and CD8 cells, low CD45RA, absent class-switched memory and increased transitional B-cells, with associated diffuse hypogammaglobulinemia. All IgG subclasses were low except for IgG3. Serum antibodies to pneumococcal polysaccharide, diphtheria and tetanus toxoids, and rubeola were below protective levels. The absolute number of peripheral blood T-regs were normal. ANA and ANCA were negative, and serum ACE was normal. Fluid obtained by BAL was negative for malignancy, Aspergillus species and pneumocystis by PCR, and culture was negative for routine pathogens, Legionella, Nocardia and AFB.
The InVitae 207 gene Immunodeficiency panel showed a pathogenic heterozygous mutation in FOXN1 gene, exon 4, c.823del (p.Ser275Profs*27) and heterozygous VUS in LYST and VPS13B.
The patient was diagnosed with CVID with lymphocytic interstitial lung disease and granulomatous lymphadenopathy. She is being treated with scIG replacement, inhaled albuterol and budesonide.
CVID is characterized by impaired B cell function with secondary hypogammaglobulinemia and variable defects in T cell function. Homozygous LOF FOXN1 mutations cause SCID with alopecia, while heterozygous LOF FOXN1 is associated with thymic hypoplasia, nail dystrophy and T-cell lymphopenia. There are no reports describing CVID in patients with FOXN1 mutation and only one patient has been described to have autoimmunity. Our patient has immunologic features of CVID. We suspect that her heteroxygous LOF in FOXN1 contributes to her immunologic phenotype.
Keywords: common variable immunodeficiency, FOXN1 mutation, lymphopenia
Disclosure: John Bohnsack has ownership interst in General Electric. All other authors had no financial relationships to disclose.
(75) Hypomorphic phenotype of XLA a Case Report
Cristina Vo, MD1, Stephen McGeady, MD2
1First year Allergy/Immunology fellow/Thomas Jefferson University and Nemours Children's Hospital
2Attending physician/Thomas Jefferson University and Nemours Children's Hospital
X-linked agammaglobulinemia (XLA) occurs 1 in 200,000 individuals (1), and results from mutations of Bruton’s tyrosine kinase (BTK) (1). The defect prevents precursor B cells in the bone marrow from forming mature B-lymphocytes, capable of differentiating into antibody producing plasma cells resulting in greatly diminished levels of all immunoglobulin isotypes (2). Individuals with XLA are susceptible to invasive encapsulated bacteria (2).
A 5 year old male presented with 2 episodes of bacterial pneumonia, one complicated by streptococcus pneumococcal bacteremia, skin abscess requiring drainage, and recurrent viral infections. Immune evaluation at 6 years of age revealed decreased CD19+ cells with an absolute count of 4 cells/mcL (normal 310 – 1120) and a CD19 percentage of 0% (normal 14-33). At age 6 years there was no detectable antibody to diphtheria, tetanus, or pneumococcal antigens but normal quantitative immunoglobulins. At 7 years old, he had normal IgG, elevated IgA 756mg/dL (normal 89-559), and low IgM 12 mg/dL (normal 24-98) and persistently low absolute CD19 count of 3 cells/mcL (normal 270-860). Following vaccine boosters for tetanus, diphtheria, and 23 pneumococcal serotypes, there was a normal recall response to tetanus, but no response to diphtheria and protective antibody titers to 6/23 pneumococcal serotypes. Genetic testing revealed a c.-31+5G>A mutation, in the BTK gene which was reported to be of unknown clinical significance. Flow cytometry for BTK expression was not possible in B cells due to the low numbers, but showed diminished monocyte expression (MFI =1.5 vs. normal control MFI= 5.05). He was also found to be heterozygous for UNC93B. The BTK mutation, greatly diminished B cell numbers, weak antibody responses and clinical history strongly suggestive of antibody deficiency, are felt likely to be due to the mutation of BTK resulting in a hypomorphic phenotype of XLA. Normal levels of IgG and IgA, with low IgM have been described in patients with hypomorphic mutations of BTK (3). The association of mutations in BTK with a UNC93B mutation, is previously unreported to our knowledge, but might suggest unusual susceptibility to herpesvirus meningoencephalitis. The BTK mutation in this patient has previously been reported in Korean patients with XLA(4).
Keywords: Hypomorphic phenotype of XLA, Bruton’s tyrosine kinase, UNC93B mutation, antibody deficiency
Disclosure: All authors had no financial relationships to disclose
(76) X-linked CGD Presenting with Candida lusitaniae Fungemia and Hemophagocytic Lymphohistiocytosis
Nivedita Muralidhar, MD1, Mia Chandler, MD, MPH2, Alicia Johnston, MD3
1Clincial Fellow/Boston Children's Hospital
2Clinical Fellow/Boston Children's Hospital
3Infectious Disease Attending Physician/Boston Children's Hospital
Chronic granulomatous disease (CGD) results from defective neutrophil and monocyte bacterial and fungal killing associated with mutations in NADPH oxidase including CYBB that results in X-linked CGD. Excessive cytokine release may lead to hemophagocytic lymphohistiocytosis (HLH) and this can be associated with infection. We report two infants with X-linked CGD presenting with Candida lusitaniae fungemia and HLH.
Case 1: A 5 wo term male presented with fever, diarrhea and rash. Labs revealed elevated inflammatory markers and transaminases. Blood cultures grew C. lusitaniae. The patient developed pancytopenia, adenopathy, hepatosplenomegaly, hyperferritinemia, and elevated soluble IL2 consistent with HLH. Dihydro-rhodamine assay showed no neutrophil oxidative burst. CYBB sequencing revealed a hemizygous mutation: c.253-8A>G or IVS3-8A>G. He was treated with liposomal amphotericin, caspofungin, decadron and IVIG with clearance of candidemia and resolution of HLH. The patient underwent a single allele HLA-B mismatched unrelated donor HSCT at 10 mo with peripheral blood chimerism demonstrating 97% donor engraftment and neutrophil oxidative index (NOI) of 275 (nl >100) in all neutrophils.
Case 2: A 6 wo term male presented with fever, bloody diarrhea, mesenteric adenopathy and splenomegaly. Pancytopenia, hyperferritinemia, elevated inflammatory markers, transaminases, and soluble IL2, CXCL9, and IL18 led to a diagnosis of HLH. IVIG and anakinra were initiated. Up-trending ferritin prompted the addition of decadron and emapalumab. Blood cultures grew C. lusitaniae. Emapalumab was held and liposomal amphotericin, micafungin and fluconazole were initiated. WES revealed a variant in CYBB: c.469C>T, p.R157X resulting in a nonsense mutation and the NOI was 1. Fungemia resolved after daily granulocyte infusions, however HLH persisted. Treatment with ruxolitinib led to clinical improvement and HSCT is planned.
C. lusitaniae represents 1-2% of all candida infections. While C. lusitaniae sepsis has been reported in very premature infants; it is rare in term infants without risk factors. Five cases of CGD and C. lusitaniae have been reported; two were fatal. Fourteen patients with CGD and HLH have been reported; including one infant with C. lusitaniae fungemia and HLH. We report two additional cases. C. lusitaniae fungemia and/or HLH in an infant without known risk factors should prompt an evaluation for CGD.
Keywords: Chronic Granulomatous Disease, Candida Lusitaniae, Primary Immunodeficiency, Fungemia, Hemophagocytic lymphohistiocytosis
Disclosure: All authors had no financial relationships to disclose
(77) Prolidase deficiency, a rare inborn error of immunity, presents with different phenotypes, immunological features, and proposed treatment modalities in twins
Beata Derfalvi, MD., PhD1, Nora Alrumayyan, MD2, Sarah M McAlpine, PhD3, Thomas Issekutz, MD4, Drew Slauenwhite, PhD5, Adam M Huber, MD4, Zaiping Liu, MD6
1associate Professor/Dalhousie University/IWK Health Centre
2clinical Fellow/Division Of Immunology, Department Of Paediatrics, Dalhousie University, IWK Health Centre
3research Associate/Division Of Immunology, Department Of Paediatrics, Dalhousie University, IWK Health Centre
4professor/Division Of Immunology, Department Of Paediatrics, Dalhousie University, IWK Health Centre
5postdoctoral Fellow/Department Of Paediatrics, Dalhousie University, IWK Health Centre
6associate Professor/Division Of Clinical Biochemistry & Maritime Newborn Screening, Department Of Pathology And Laboratory Medicine, Dalhousie University, IWK Health Centre
Prolidase deficiency (PD) is an autosomal recessive disorder of defective collagen synthesis caused by mutations in the PEPD gene encoding the enzyme prolidase D. PD typically presents with recurrent infections, intellectual disabilities, dysmorphic features, splenomegaly, and increased proline metabolites in urine. PD was recently classified under the immune dysregulation subgroup within inborn errors of immunity, however, detailed immunological assessments have not been reported. PD is also characterized as a “lupus-mimic” with intractable skin ulceration and systemic autoimmunity; thrombocytopenia, hypocomplementemia, and hypergammaglobulinemia are frequent laboratory findings. Standard therapies for PD have not yet been described.
We report teenage twin females of non-consanguineous asymptomatic carrier parents exhibiting variable severity of the phenotype caused by novel compound heterozygous mutations (c.977G>A, c.550C>T; premature stop codons) in the PEPD gene discovered using PID panel NGS. Clinically, twin A presented with more severe disease phenotype, including elevated inflammatory parameters, thrombocytopenia, autoimmune thyroiditis, and “livedoid vasculopathy”, characterized by debilitating skin ulceration that prevented ambulation for a year. Twin B has atopic symptoms of allergic rhinitis, eczema and asthma, and recently painful skin ulceration. Both patients have slightly dysmorphic facial features, learning difficulties, recurrent upper respiratory tract infections (URTI) and massive imidodipeptiduria. Immunological assessment revealed low memory B cell counts with IgG2 subclass deficiency and normal antibody responses to vaccines. Lupus specific autoantibodies were not present. In twin A, regulatory T (Treg) cell counts were low, possibly contributing to the more severe inflammatory symptoms. In both patients, lymphocyte proliferation was normal, whereas NK cell cytotoxicity was low in twin B. Both patients have MBL complement deficiency, possibly contributing to the frequent URTI. Both twins had normal serum amyloid A, but highly increased plasma IL-18 levels. A new therapeutic regimen included immunomodulatory IVIG, microcirculation-improving low-molecular-weight heparin, red cell filterability increasing pentoxifylline, and collagen synthesis cofactor vitamin C combined with topical proline and tacrolimus, leading to resolution of twin A’s skin ulcers, allowing her to walk again.
PD may feature defects in memory B cells and Tregs. Profoundly high IL-18 plasma levels suggest underlying autoinflammatory processes. Ulcer improvement has been seen with the current regimen.
Keywords: prolidase deficiency, inborn error of immunity, immune dysregulation, ulcer
Disclosure: Beata Derfalvi has contracted research with CSL Behring and is an advisory board member of SOBI. All other authors had no financial relationships to disclose
(78) Dupilumab successfully controls severe eczema in a child with hyper-IgE syndrome: a case report
Tejas Joshi, B.S.1, Sara Anvari, MD2, Meera Gupta, MD3, Carla Davis, MD4, Joud Hajjar, MD5
1Medical Student/Baylor College of Medicine
2Assistant Professor/Baylor College of Medicine
3Associate Professor/Baylor College of Medicine
4Professor/Baylor College of Medicine
5Assistant Professor of Immunology, Allergy, and Rheumatology/Baylor College of Medicine
Managing the severe atopic manifestations in Hyper-IgE syndromes (HIES) is challenging. Although dupilumab has been approved to treat severe asthma and atopic dermatitis, its effectiveness in HIES is unknown, especially in children. We present the case of a child with HIES who achieved remarkable control of his eczema and recurrent skin infections with dupilumab.
Our patient is a 12-year-old Caucasian male. He developed eczema in infancy, which became severe at 8-years, with a scoring atopic dermatitis (SCORAD) score of 91.7/103. Despite excellent skincare, he developed multiple Staphylococcus aureus skin infections, requiring systemic antibiotics and hospitalizations. He also developed herpes simplex skin infections and recurrent generalized lymphadenopathy. Furthermore, he experienced vernal keratoconjunctivitis of both eyes requiring amniotic membrane transplantation. Labs showed elevated IgE (>50,000 kU/L), normal IgG/A/M levels, and normal responses to protein/polysaccharide-based vaccines. He had occasional lymphocytosis with normal lymphocyte subsets, and low-normal intracellular IL-17 in CD4+T-cells upon stimulation. Chromosomal Microarray Analysis showed a gain of 9p24.3 involving the dedicator of cytokinesis 8 (DOCK8) gene, of unclear significance. DOCK8 protein expression on peripheral blood mononuclear cells was normal. Trio whole exome sequencing revealed a heterozygous c.7339C>T (p.Arg2447Ter) change in the Filaggrin gene inherited from father. Although genetic workup was not definitive, our patient’s clinical phenotype is that of HIES and not simply severe eczema and atopy.
Treatment regimen included cyclosporine 2-3 mg/kg, trimethoprim/sulfamethoxazole, and omalizumab 300 mg. The latter provided skin improvement, but our patient developed anaphylaxis. He was also started on intravenous immunoglobulin 1g/kg, which helped decrease infections and recurrent lymphadenitis. At 12-years, he was started on dupilumab 300 mg subcutaneously every 2 weeks, which led to considerable improvement in his skin infections and eczema (SCORAD score of 23.95/103). IgE levels decreased to 5,820 IU/mL, and CD4+T-cell induction of intracellular IL-17 normalized (Table 1). Moreover, he no longer needed systemic steroids, and his growth chart improved significantly.
Our case suggests that dupilumab might be used to treat the severe atopic symptoms seen in HIES. The steroids sparing effect led to improved patient’s symptoms, quality of life, and growth.

Table 1: Humoral immunity panel and CD4 T cell induction of intracellular IL-17 both before and after treatment: patient had elevated IgE levels but normal IgG/M/A and normal responses to protein and polysaccharide-based vaccines prior to treatment. Patient showed markedly reduced IgE levels following treatment. Also, treatment led to normalization of CD4 T cell induction of intracellular IL-17 following stimulation. Abbreviations: IgE, immunoglobulin E; IgG, immunoglobulin G; IgM, immunoglobulin M; IgA, immunoglobulin A
Keywords: Hyper IgE syndrome (HIES), dupilumab, eczema, primary immunodeficiency
Disclosure: Meera Gupta is an advisory board member of Sanofi Genzyme. Joud Hajjar received research grants from Amplimmune, Arcus Biosciences, ARMO BioSciences, Atterocor/Millendo, Baxalta, BMS, Calithera Biosciences, Chao Physician-Scientist Foundation, CytomX Therapeutics, Eli Lilly, EMD Serono, Healios Onc, Immune Deficiency Foundation, ImmuneOncia, Incyte, Jeffrey Modell Foundation, Karyopharm, Kymab, MedImmune, Merck, National Cancer Institute, NeoImmuneTech, Neon Therapeutics, Novartis, OncoSec KEYNOTE-695, Pfizer, PsiOxus, Regeneron, Surface Oncology, The Texams Medical Center Digestive Diseases Center and Top Alliance; she was an Advisory Board member of Alfaisal University, Genome & Company, Horizon, and Pharming. All other authors had no financial relationships to disclose
(79) Global T-cell lymphopenia and hypogammaglobulinemia in a 15-month-old patient with Takenouchi-Kosaki syndrome (CDC42 mutation)
Lydia Zhang, MD1, Jean Jacques De Bruycker, MD2, Yves Pastore, MD3, Jean-François Soucy, M.D.4, Marie-Paule Morin, M.D.. Ph.D.5, Elie Haddad, MD, PhD5
1Fellow-in-Training/McGill University
2Assistant clinical professor, division of Pediatric Immunology and Rhumatology/CHU Sainte-Justine
3Associate Professor in Pediatrics, department of pediatrics, hematology-oncology division/Centre hospitalier universitaire Sainte-Justine
4Attending Physician/Genetic Division, Department of Pediatrics, University of Montreal
5Attending physician/Immunology-Rheumatology Division, Department of Pediatrics, University of Montreal
Human CDC42, an intracellular member of the Rho-family GTPases, is a small GTPase that is highly conserved among eukaryotes and plays a crucial role for cell cycle development and actin cytoskeleton formation as it acts as a regulator of cell polarity. While there is a growing evidence of genotype-phenotype correlation, more data are needed to better characterize the role of CDC42 mutations on immune system dysfunction.
Here, we describe a case of Takenouchi-Kosaki syndrome presenting with transposition of the great arteries, microcephaly and club feet. Arterial switch post-operative course was complicated by a sternal wound dehiscence but no infectious complications. Additionally, neutropenia and global T-cell lymphopenia with decreased numbers of recent thymic migrant cells and hypogammaglobulinemia were present without thrombocytopenia. Whole genome sequencing was pursued after a normal CGH array and revealed a de novo mutation (NM_001791.3: c.68 A>G p.Tyr23Cys) in CDC42 affecting the N-terminal α-helix, corresponding to Takenouchi-Kosaki Syndrome. This mutation is known to cause an amino acid substitution at the highly conserved Tyr23 position, which is part of a cavity on the CDC42 surface that accommodates the CDC42/RAC-interacting binding motif that allows binding to effector proteins such as TAK1, WASP and FMNL2. PAK1 is responsible for cell motility and contraction, WASP is important for cell migration and cell cycle progression, whereas FMNL2 has a role in cell division, cell polarity and migration. This mutation can therefore explain the various clinical manifestations of our patient, including the absence of thrombocytopenia. However, the associated T-cell lymphopenia is not typical of the described amino acid substitution Tyr23 in the literature. In our patient, global T-cell lymphopenia with decreased numbers of recent thymic migrant cells could have been worsened by a partial thymectomy during cardiac surgery.
This report suggests that even with a better knowledge of phenotype-genotype correlation, close follow up in immunology clinic is warranted to address indication of pneumocystis pneumonia prophylaxis and immune globulin replacement treatment.
Keywords: CDC42, Takenouchi-Kosaki Syndrome, Congenital cardiac defect, Microcephaly, T-cell lymphopenia, Hypogammaglobulinemia
Disclosure: Yves Pastore has contracted research with Novartis and Principia, and is an advisory board member of Pfizer. Elie Haddad is a consultant with Jasper Therapeutics and Rocket Pharma. All other authors had no financial relationships to disclose
(80) Severe Congenital Neutropenia due to G6PC3 Deficiency with Autoinflammatory Complications
Monica Bray, MD1, MaiLan Nguyen, MD2
1Fellow Physician/Baylor College of Medicine
2Assistant Professor/Baylor College of Medicine
Severe congenital neutropenia (SCN) due to pathogenic variants in glucose-6-phosphatase catalytic subunit 3 (G6PC3) have been associated with the extra-hematopoietic features of cardiac abnormalities, urogenital malformation, and venous angiectasis. Patients typically present in infancy with failure to thrive and severe infections. A French cohort of 14 patients revealed 3 with diagnoses of inflammatory bowel disease (IBD) and 2 patients with polyarthritis. We aim to add to the existing body of knowledge of autoinflammatory conditions that can be associated with pathogenic variants in G6PC3.
The patient is a 4-year-old Hispanic male with a history of prematurity (born at 34 weeks gestation) and atrial septal defect. He required hospitalization at birth for respiratory distress and at 5 months of age for failure to thrive. He was admitted 3 times at 6 months of age for recurrent infectious myositis. On third admission it was noted that his absolute neutrophil count (ANC) had been low since birth and he was diagnosed with SCN. He was started on G-CSF with normalization of his ANC. Genetic testing confirmed inherited compound heterozygous pathogenic variants in G6PC3: c.210delC (p.F71fs) and c.778G>C (p.G260R). His weight improved from < 0.01 percentile to the 50th percentile by 1 year of age. From age 3 to 4, his weight decreased from the 50th percentile to the 10th percentile. At 4 years of age, he developed fatigue, abdominal pain, anorexia, oral ulcers, and splenomegaly. Bone marrow biopsy was negative for malignancy. He was referred to gastroenterology for the poor weight gain and intermittent abdominal pain. Biopsies from colonoscopy revealed surface erosion with reactive lymphonodular hyperplasia and were not consistent with IBD. He was also noted to have a persistent bilateral ankle swelling and limited range of motion, which was confirmed by rheumatology to be consistent with inflammatory arthritis. The bilateral ankle arthritis did not resolve with intra-articular steroid injections, so the decision was made to start adalimumab and methotrexate. His care team is also hopeful this treatment regimen may benefit his non-specific colonic inflammation. This case report demonstrates increasing recognition that autoinflammatory conditions can complicate primary immunodeficiency.
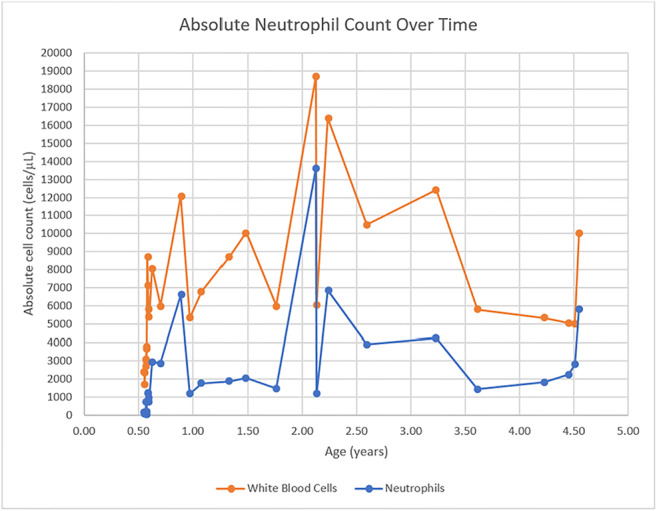
Figure 1. Absolute Neutrophil Count Over Time
Keywords: autoinflammatory, severe congenital neutropenia, immune dysregulation, rheumatology, arthritis
Disclosure: All authors indicated they had no financial relationships to disclose.
(81) Atypical presentation of T-/B+/NK+ SCID with Omenn-like phenotype attributable to a hemizygous variant in IL2RG
Abigail Lang, MD1, Nashmia Qamar, DO2, Aaruni Khanolkar, MBBS, PhD3, Kai Lee Yap, PhD3, Aisha Ahmed, MD4, Amer Khojah, MD5
1Fellow Physician/Ann and Robert H. Lurie Children's Hospital of Chicago
2Attending Physician/Ann and Robert H. Lurie Children's Hospital of Chicago
3Assistant Professor/Ann and Robert H. Lurie Children's Hospital of Chicago
4Attending Physician, Allergy and Immunology/Ann and Robert H Lurie Children's Hospital of Chicago
5Attending Physician, Allergy, Immunology, and Rheumatology/Ann & Robert H. Lurie Children's Hospital of Chicago/ Northwestern University Feinberg School of Medicine
Defects in interleukin-2 receptor gamma (IL2RG) lead to X-linked severe combined immunodeficiency (SCID). The common gamma chain is utilized by multiple interleukin receptors important for T and NK cell survival, and defects typically lead to T-/B+/NK- SCID phenotype. We present a case of T-/B+/NK+ SCID with Omenn-like phenotype attributable to a hemizygous variant in IL2RG.
Case Report: A full-term Caucasian male born to non-consanguineous parents was evaluated after Illinois newborn screen was positive for SCID with 0 TRECs/μL. The infant was transferred to the NICU for poor feeding and diffuse erythematous desquamating rash.
The initial complete blood count demonstrated white blood cell count of 21.7 103/μL [reference range (RR): 9.4-34.4 103/μL], eosinophil count of 9.76 103/μL (RR: 0-2.04 103/μL), and lymphocyte count of 1.085 103/μL (RR: 3.38-15.64 103/μL). Flow cytometry demonstrated T cell lymphopenia (CD3 count: 763/mm3, RR: 1375-7129/mm3), elevated B cells (CD19 count: 1687/mm3, RR: 104-1448/mm3) and elevated NK cells (CD3-CD16/CD56 count: 642/mm3, RR: 60-434/mm3). T cell subsets demonstrated a predominance of memory CD4 T cells with absent naïve T cells and recent thymic emigrants. Lymphocyte proliferation to PHA was sub-optimal, and NK cell function was reduced by lytic assay. TCR V-beta repertoire analysis revealed polyclonal T cells, and chimerism studies ruled out maternal engraftment.
Expedited genetic testing for a gene subset (CD247, CD3D, CD3E, CORO1A, IL7R, PTPRC) associated with T-/B+/NK+ SCID was negative. An expanded panel revealed a likely pathologic hemizygous variant in IL2RG (NM_000206.2, p.Tyr125Cys, c.374A>G). Flow cytometry demonstrated normal expression of CD127 (IL-7R) and CD132 (common gamma chain). Functional assays showed reduced STAT-5 phosphorylation after stimulation with IL-2, 7, and 15 consistent with common gamma chain defect.
Treatment with corticosteroids and cyclosporine resulted in normalization of eosinophilia and rash. The infant underwent a matched unrelated hematopoietic stem cell transplantation without major complications.
We present a case of atypical T-/B+/NK+ SCID with Omenn-like phenotype attributable to a hemizygous missense variant in IL2RG. The hypomorphic variant likely contributed to normal NK cell number with abnormal function. This case highlights the need for rapid genetic diagnostics followed by functional testing to confirm variant pathogenicity.
Keywords: Severe Combined Immunodeficiency, SCID, Omenn's syndrome, IL-2 receptor, Common gamma chain
Disclosure: All authors indicated they had no financial relationships to disclose.
(82) First reported complete C6 deficiency in Chinese patients
Valerie Chiang, MBBS1, WILLIAM WONG, PHD2, EVELYN LEUNG, MSc3, ELAINE AU, MBBS; MRCP; FHKCP; FHKAM; FRCPA; FHKCPath; FHKAM; D (ABMLI)4, PHILIP LI, MBBS, MRes(Med), PDipID, MRCP, FHKCP, FHKAM5
1Resident/Queen Mary Hospital, Hospital Authority
2Scientific Officer/Queen Mary Hospital, Hospital Authority
3Medical Technologist/Queen Mary Hospital, Hospital Authority
4Consultant/Queen Mary Hospital, Hospital Authority
5Clinical Assistant Professor/The University Of Hong Kong
Complete C6 deficiency (C6Q0) is a rare primary immunodeficiency leading to increased susceptibility to recurrent Neisseria infections. Patients with C6Q0 have mostly been reported in individuals of African ancestry previously, but never in Chinese. We identify the first Chinese patients with C6Q0 through family screening of an index case presenting with recurrent Neisseria meningitis with septicaemia.
Three C6Q0 patients had near‐absent C6 levels, and absent CH50/AH50 activity. Genetic testing identified them to be compound heterozygous for two nonsense mutations in the C6 gene: NM_000065.4:c.1786C>T (p.Arg596Ter) and NM_000065.4:c.1816C>T (p.Arg606Ter). Neither mutations have been reported to be pathogenic previously. Two other family members who were heterozygous for either only p.Arg596Ter or p.Arg606Ter had intermediate C6 levels but preserved CH50/AH50 activity. These two loss‐of‐function mutations showed a strong genotype–phenotype correlation in C6 levels; both nonsense mutations likely lead to abolishment of C6 protein level and function.
We demonstrate that heterozygous family members with subtotal C6 levels had preserved complement haemolytic function and demonstrate a threshold effect of C6 protein level. In this study, even about half the level of normal C6 levels was sufficient to maintain normal haemolytic activity as demonstrated by normal CH50/AH50 assays (i.e. a threshold effect of C6 protein level). These single heterozygotes develop C6SD without the classical predisposition to recurrent meningococcal disease seen in C6Q0.
A high prevalence of C6 deficiency has been reported among African Americans. In South Africa, where C6Q0 seems most prevalent, four common frameshift defects in the C6 gene account for the vast majority of cases. To the best of our knowledge, this is the first pedigree with C6Q0 deficiency reported in Chinese and the first TCC deficiency reported in Hong Kong. It is likely that C6Q0 patients of different ethnic and geographic backgrounds carry distinct mutations, hence the significance in detecting these two mutations in our reported pedigree. It is likely that the genetic causes of C6Q0 (and other PIDs) in Chinese may vary significantly compared with other populations and highlights the importance of future immunology studies dedicated to specific ethnicities.
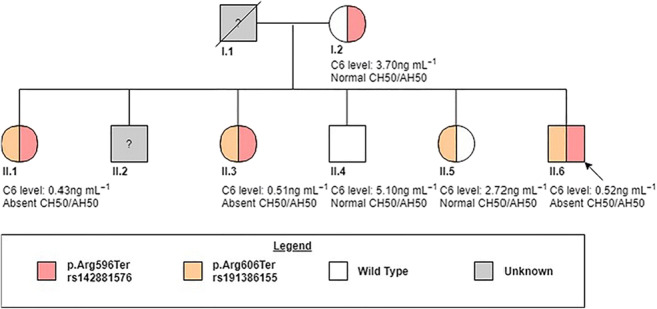
Figure 1: Family tree and summary of C6 genotypes
Keywords: c6, complement, deficiency, neisseria, immunodeficiency, chinese, pedigree
Disclosure: All authors indicated they had no financial relationships to disclose.
(83) Clinical Outcomes of SARS-CoV2 infection in STAT3 Deficiency
Muhammad Khalid, MD1, Amanda Urban, CRNP2, Dirk Darnell, RN3, Alexandra Freeman, MD4
1Clinical Fellow/National Institute of Allergy and Infectious Diseases, National Institute of Health
2Nurse Practitioner/Clinical Research Directorate, Frederick National Laboratory for Cancer Research
3Senior Clinical Research Nurse/National Institute of Allergy and Infectious Diseases, National Institute of Health
4Director, Primary Immune Deficiency Clinic/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institute of Health
With the continued global spread of SARS-CoV2, we continue to learn immunological responses and clinical outcomes in immunocompetent hosts. Our understanding is far more limited in patients with an underlying immune dysregulation.
The objective is to describe clinical outcomes of SARS-CoV2 infection in patients with HyperIgE immunophenotype due to STAT3 loss of function (LOF) mutations.
We tele-interviewed five patients with confirmed or clinically suspected SARS-CoV2 infection and assessed for nature, duration, and severity of symptoms.
All patients had confirmed STAT3 LOF and were on antimicrobial prophylaxis during the period of SARS-CoV2 infection. Two patients had SARS-CoV2 PCR confirmed infection, and three had suspected disease with a PCR-positive household family contact and compatible symptoms. Four of these five patients suffered from mild disease and fully recovered. They ranged in age from 14-45 years and included three males. Two had underlying mild parenchymal lung disease. Symptoms included fever, myalgias, fatigue, congestion, cough, dyspnea on exertion, diarrhea, anosmia, and ageusia. The duration of symptoms ranged from 6 to 20 days, except one patient had persistent fatigue for additional 21 days. One patient received additional antibiotics due to clinical worsening with resultant improvement.
One of the 5 patients suffered from fatal disease. This patient was a 41-year-old male who also had history of severe bronchiectasis with chronic Pseudomonas colonization, smoking history, coronary artery aneurysm with focal area of cardiac hypokinesis, hypertension, prolonged corticosteroid therapy, and class-1 obesity. He was admitted to the hospital after 2 days of fever and shortness of breath and was found to be SARS-CoV2 PCR-positive. He subsequently developed hypoxic respiratory failure with Pseudomonas pulmonary infection, requiring mechanical ventilation and blood pressure support, further complicated by kidney failure. Despite treatment with broad-spectrum antibiotics and corticosteroids, he died after 26 days of illness.
While four of five patients with STAT3 LOF completely recovered, one patient with additional co-morbidities and bacterial co-infection succumbed to COVID-19. This underscores the need for research to understand differences in immunological responses when compared to the healthy population.

Figure 1: Duration and outcomes of COVID-19 illness in patients with underlying HyperIgE Syndrome due to STAT 3 Loss of function.
Keywords: STAT3 Loss of Function, SARS-CoV2, HyperIgE Syndrome, COVID-19
Disclosure: All authors indicated they had no financial relationships to disclose.
(84) Infodemiological study on primary immunodeficiency disorders: A global report
Umair Ahmed Bargir, MBBS,DCP1, Manisha Madkaikar, MD2
1Clinical Scientist/ICMR-National Institute of Immunohaematology
2Director/ICMR National Institute of Immunohaematology
Primary immunodeficiency disorder (PID) now known as inborn errors of immunity is a group of more than 430 monogenic defects presenting with a wide phenotype from recurrent infections to autoimmune, autoinflammatory, and other immune dysregulatory features. Infodemiological studies apply user-generated data from various search engines and social media to analyze and explore the trend of different diseases and to speculate human behaviour towards distinct health topics. We investigated a Google Trends (GT) based approach to monitor selected PID globally. Relative search volume for ‘Severe Combined Immunodeficiency Disorder’ (SCID), ‘Ataxia-Telangiectasia’ (AT), ‘Digeorge Syndrome’ (DGS), ‘Hyper IgE Syndrome’ (HIGE), ‘Common Variable Immunodeficiency Disorder’ (CVID), ‘Chronic Granulomatous Disease’ (CGD), ‘Hemophagocytic Lymphohistiocytosis’ (HLH), ‘Familial Mediterranean Fever’ (FMF), ‘Hyper IgD syndrome’ (HIGD) and ‘PFAPA Syndrome’ disorders was extracted from Google Trends since its inception till 30 June 2020 to study the trend and across different countries. We also analysed the number of publications published per year using Web of Science. Our analysis shows a decreasing trend for SCID, AT, HIGE, CGD, and HIGD and an increasing trend for DGS, HLH, and PFAPA syndrome. No trend was observed for CVID. The peaks observed for seasonality signify the importance of awareness programs and educational conferences. The geographical distribution of different PID matches with the search interest. We report for the first time an infodemiological study on PIDs with a global perspective. GT acts as a valuable tool to understand the trend and seasonality and geographical distribution based on the search interest of different PIDs. This may help in tracing the impact of awareness programs and advocacy measures to promote newborn screening.

Figure 1: Search interest expressed as relative search volumes. A Trend for SCID. B, Trend for AT. C, Trend for DGS. D, Trend for HIGE (X=RSV, Y=Year)
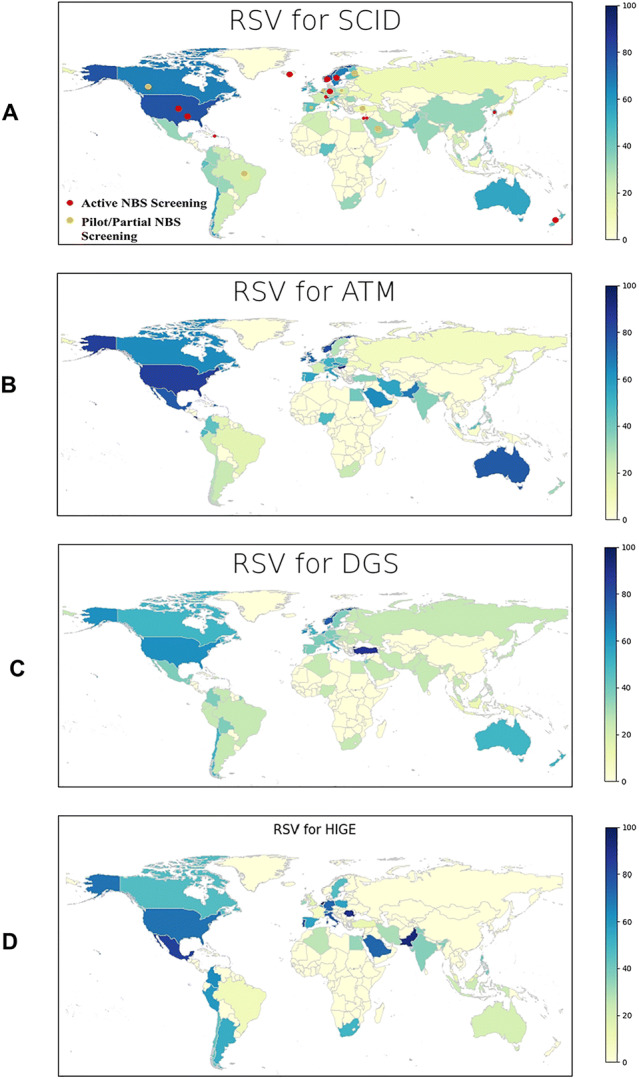
Figure 2: Choropleth maps for relative search volumes of A, SCID. B, AT. C, DGS. D, HIGE

Figure 3: No of publications per year for A-SCID, B-AT, C-DGS, D- HIGE
Keywords: Infodemiology, Google Trends, SCID, Primary immunodeficiency disorders, Time series analysis, Forecast
Disclosure: All authors indicated they had no financial relationships to disclose.
(85) Effects of HDAC activity modulation on disease pathogenesis in mice infected with Influenza – implications for rheumatic disease interventions
Milena Leseva, PhD1, Lora Simeonova, PhD2, Kalin Stoyanov, MD1, Petya Dimitrova, PhD3
1Researcher/The Stephan Angeloff Institute of Microbiology, Laboratory of Experimental Immunotherapy
2Assistant Professor/The Stephan Angeloff Institute of Microbiology, Laboratory of Experimental chemotherapy of Influenza
3Associate Professor/The Stephan Angeloff Institute of Microbiology, Laboratory of Experimental chemotherapy of Influenza
HDACs are epigenetic regulators with a role in infection, inflammation and autoimmunity. Their inhibition or activation can alter the host’s response to infection, but the specific effects on flu pathogenesis remain largely unexplored. Sirtuins, classified as class III HDACs, target a remarkably large set of proteins and are involved in numerous cellular processes with an impact on immune cell activity and function: from transcriptional and metabolic regulation, to cell differentiation. We are interested in new therapeutic approaches for rheumatoid arthritis (RA). Because patients with RA are susceptible to infections, we are focused on RA complicated by the flu. We have observed that modulation of Sirt1 activity alleviates joint swelling and inflammation in mouse models of RA in part through ligand-CXCR2 pathways, mobilization of neutrophils from the bone marrow and reduced cartilage destruction. Based on our data and on reports that sirtuins have a conserved anti-viral activity, we hypothesized that SRT2183 - an activator specific for Sirt1 – can be used for treating both RA and infections with Influenza. SRT2183 was administered subcutaneously at different doses, selected for their protective effects against RA-mediated inflammation and joint tissue damage. We used an adapted IAV H3N2 virus in two different experimental settings: high- and low-multiplicity of infection. Treatment with SRT2183 in the context of a severe infection did not alleviate the disease; it did not increase survival time, nor did it decrease the lung viral titers. On the contrary, lung tissue damage was exacerbated in mice treated with SRT2183 compared to controls treated with oseltamivir, a clinically relevant anti-viral. This effect was dose-dependent. Moreover, we observed a massive infiltration of mature neutrophils in the bronchoalveolar lavage of SRT2183 – treated infected mice. However, during a mild infection low-dose SRT2183 treatment exhibited some protective effect by extending the mean survival time by 1.2 days compared to controls.
Activation of Sirt1 using natural or synthetic compounds can be beneficial for RA complicated with the flu. However, it should be done with caution, especially during the flu season, since the effects depend on the severity of the infection.
Project funding from Bulgarian National Science Fund (DN11/5_2017).
Keywords: Rheumatoid arthritis, Influenza, autoimmunity, innate immunity, infection, sirtuins, Sirt1
Disclosure: All authors indicated they had no financial relationships to disclose.
(86) The other side of the coin: immunodysregulation in primary immunodeficiency. Analysis of the United States Immunodeficiency Network (USIDNET) database
Maria Chitty Lopez, MD1, Rahul Mhaskar, MPH, PhD2, Hannah Wright, MSPH3, Rebecca Marsh, MD4, Elizabeth Garabedian, RN, MSLS5, Ramsay Fuleihan, MD6, Kathleen Sullivan, MD, PhD7, Jennifer Leiding, MD8
1Physician Fellow/Division of Allergy and Immunology, Department of Pediatrics, University of South Florida, Tampa, FL, USA
2Director of College of Medicine Office of Research/Center for Evidence Based Medicine and Health Outcomes Research, USF Morsani College of Medicine, Tampa, Florida
3Research Data Analyst/The United States Immunodeficiency Network (USIDNET)
4Attending Physician/Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center
5Research Nurse/Office of the Clinical Director, National Human Genome Research Institute, NIH, Bethesda, MD
6Professor of Pediatrics/Division of Allergy and Immunology, Department of Pediatrics, Columbia University, New York, NY, USA
7Professor of Pediatrics, Chief of the Division of Allergy and Immunology/The Children’s Hospital of Philadelphia, Philadelphia, PA, USA
8Associate Professor/Division of Allergy and Immunology, Department of Pediatrics, University of South Florida at Johns Hopkins-All Children’s Hospital, St Petersburg, FL
Substantial effort has been devoted to characterizing infectious susceptibility in patients with primary immunodeficiencies diseases (PIDD). Immunodysregulation, autoimmunity, and auto-inflammation are increasingly identified as major PIDD features. We aim to describe the frequency of immunodysregulatory conditions in a large US PIDD cohort.
The USIDNET is a national research consortium with 43 contributing academic and private centers (Figure1). Data from PIDD patients is collected in a registry after obtaining informed consent. Patients were grouped by their diagnosis according to the IUIS 2019 classification (Table1). Immunodysregulatory conditions were categorized by affected organ system by investigators (Table2). The frequency of immunodysregulatory conditions and organ systems affected were determined within each IUIS category. Association between immunodysregulatory conditions and survival was investigated using Chi-square or Fisher’s exact test.
4,182 subjects with a diagnosis of PIDD were identified. Median-age-of diagnosis and death (n=259) were 8 and 17years respectively. Male:female ratio was 1.2:1. Of surviving patients (n=3.672), 57.8% had ≥1 organ systems affected; 9.4% had ≥3 affected. Within IUIS categories, the most common systems affected were respiratory (30.6%), integumentary (19.3%), gastrointestinal (18.4%), immune (13.9%), and hematopoietic (11.5%). The most frequent IUIS categories to have immunodysregulatory conditions associated were Diseases of Immune Dysregulation (76.4%), and Defects of the Innate Immune System (73.0%). Specific conditions segregated within IUIS categories: hematologic dyscrasias (34.3%) were most common within Diseases of Immune Dysregulation, gastrointestinal conditions (32.5%) within Congenital Defects of Phagocytes, and respiratory conditions (38.1%) within Predominantly Antibody Deficiency. Immunodysregulatory conditions affecting cardiovascular (p=0.014), endocrine (p=0.046), gastrointestinal (p < 0.0001), hematopoietic (p < 0.0001), and immune systems (p < 0.0001), were associated with death. Within each organ system affected, specific immunodysregulatory conditions were associated with worse survival (Table3).
More than half of patients within the USIDNET PIDD cohort had immunodysregulatory conditions associated with their diagnosis. Hematologic, GI, and respiratory conditions had the most negative effect on survival. This large cohort of patients was predominantly contributed by academic centers which may have influenced the observations, towards more severe phenotypes. Given the high frequency of immunodysregulatory features, recognition of autoimmunity and auto-inflammatory symptoms should be used to guide surveillance strategies for recognition and diagnosis of PIDD.


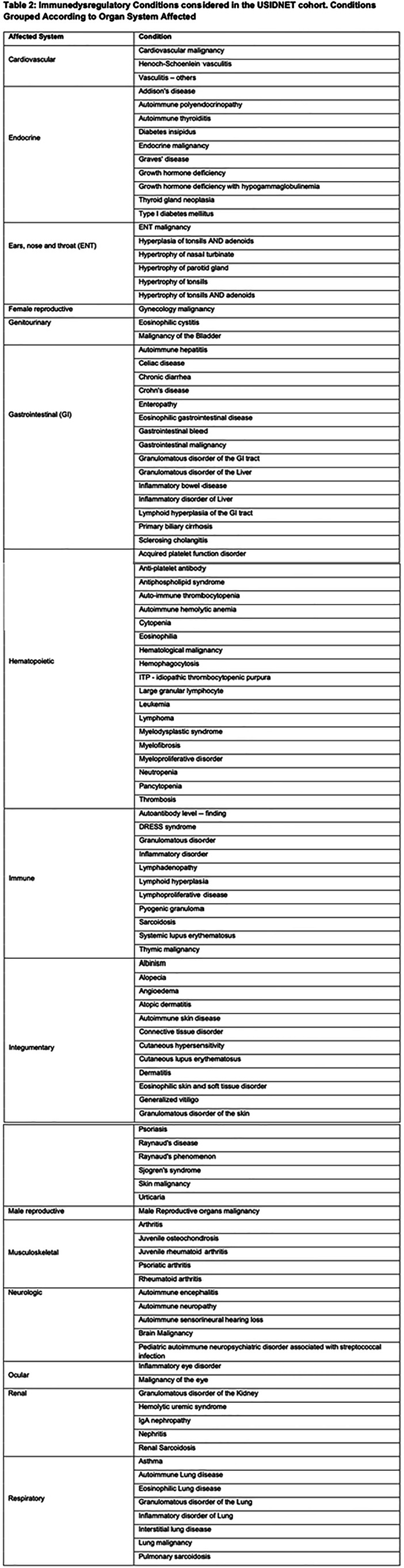

Keywords: primary immunodeficiencies diseases, immunodysregulation, PIDD, autoimmunity, auto-inflammation, USIDNET
Disclosure: Jennifer Leiding is an advisory board member and received speaker honoraria from CSL Behring; an advisory board member of Phamring; and received speaker honoriaria and a research grant from Horizon Therapeutics.
(87) Damaging Biallelic Variants in OSBPL5 are associated with T Cell Immunodeficiency in Humans
Amy O'Connell, MD, PhD1, Monica Wojcik, MD, PhD2, Casie Genetti, MS, CGC3, Jill Madden, PhD, MSc, CGC3, David Breault, MD, PhD4, Pankaj Agrawal, MBBS, MMSC4
1Assistant Professor/Boston Children's Hospital/Harvard Medical School
2Instructor/Boston Children's Hopsital/Harvard Medical School
3Genetic Counselor/Boston Children's Hospital
4Associate Professor/Boston Children's Hospital/Harvard Medical School
A male infant born at 24 5/7 weeks’ gestation developed fulminant necrotizing enterocolitis with multiple intestinal perforations at 1 month of age. The patient initially had normal TREC levels. He had a prolonged recovery complicated by Candida parapsilosis septicemia. At 4 months the patient died from Klebsiella sepsis. Immunologic testing at 41 weeks postmenstrual age demonstrated CD4 and CD8 T cell lymphopenia despite a normal absolute lymphocyte count with diminished naïve CD4 T cells, a paucity of switched memory B cells, and an abnormal T cell response to stimulation with anti-CD3 and Candida antigen. The family was enrolled in a research study and trio exome sequencing was performed. Bialellic missense variants in OSBPL5 were identified in a conserved region. The maternal variant, (GRCh38) chr11:3129108 C>T; c.41G>A, p.Cys14Tyr (NM_020896.4) is absent from gnomAD. The paternal variant (GRCh38) chr11:3129114 G>T; c.35C>A, p.Ser12Tyr (NM_020896.4) had a gnomAD frequency of 0.000019 (one heterozygote in gnomAD exomes, none in genomes). Both variants were assessed by in silico predictors as probably damaging (Polyphen), damaging (SIFT), and disease causing (Mutation Taster). OSBPL5 encodes ORP5, which is known to be involved in membrane trafficking of phospholipids and has been sown to be important for cell proliferation via mTOR signaling. Highest tissue mRNA expression of ORP5 is found in the liver and spleen, and CD4 T cells have the highest cellular mRNA expression. Shortly after we began investigating this result, a mechanistic study (Xu et al. J Immunol 2020) showed that ORP5 is responsible for calcium trafficking in T cells by facilitating PIP2 hydrolysis and potentiates T cell activation, confirming our suspicion about the genotype-phenotype correlation. This case report suggests ORP5 deficiency as a novel cause of inborn error of immunity in humans; ongoing functional evaluation in the laboratory seeks to further understand the impacts of ORP5 deficiency. The case also highlights that TREC screening alone does not identify all T cell functional disorders and emphasizes the clinical point that immunologic evaluation is challenging in premature infants. Further, premature infants with clinical symptoms of abnormal immunity deserve prompt genetic evaluation.
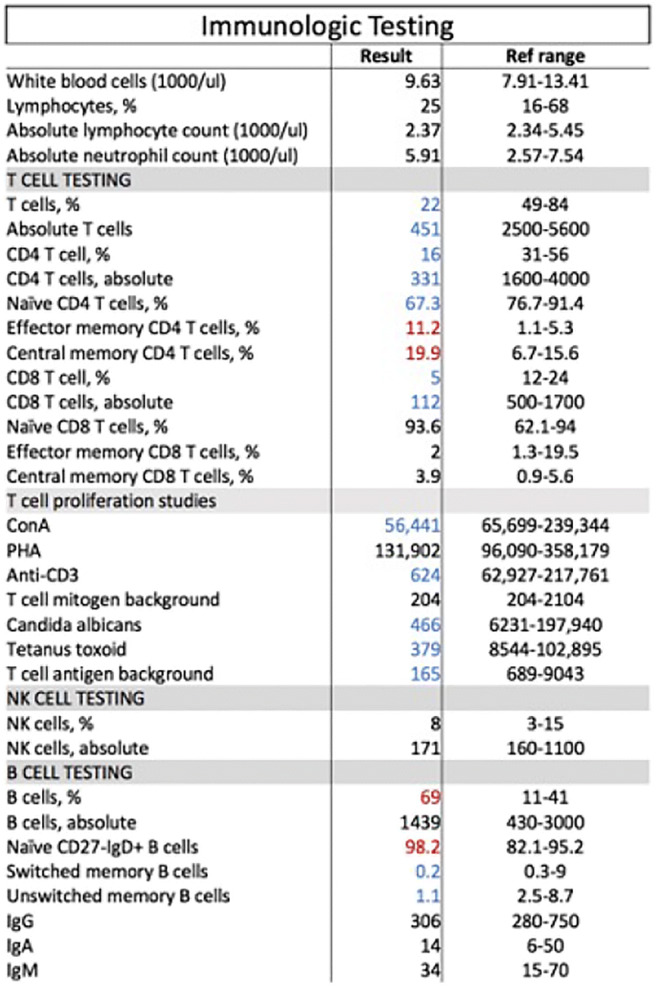
Table 1: Immunologic testing at 4 months of age (41 weeks postmenstrual age).

Keywords: Immunodeficiency, T cell, OSBPL5, Inborn error of immunity, lipid transfer, prematurity, proliferation
Disclosure: All authors indicated they had no financial relationships to disclose.
(88) Validation of APECED Expanded Diagnostic Criteria in Independent Patient Cohorts
Elise Ferre, PA-C, MPH1, Monica Schmitt, CRNP2, Michail Lionakis, MD, ScD3
1Physician Assistant/Fungal Pathogenesis Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases/National Institutes of Health, Bethesda, MD, USA
2Nurse Practitioner/Fungal Pathogenesis Section, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
3Senior Investigator/Fungal Pathogenesis Section, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
Autoimmune-Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED), caused by AIRE mutations, is typically diagnosed with any dyad among the classic triad manifestations of chronic mucocutaneous candidiasis, hypoparathyroidism, and adrenal insufficiency. We previously evaluated 35 American APECED patients (Ferré et al., JCI Insight, 2016), and reported APECED’s clinical spectrum is far broader, and identified an adjunct triad of APECED rash, intestinal dysfunction, and enamel hypoplasia, which upon incorporation within the classic criteria, could accelerate clinical diagnosis.
To validate the expanded diagnostic criteria in independent APECED cohorts from the Americas and Europe.
American (n=57) and European (n=12) APECED patients were enrolled in an IRB-approved prospective natural history protocol (11-I-0187) at the NIH Clinical Center and underwent uniform comprehensive clinical evaluation.
American patients reached a clinical APECED diagnosis 4.2 years earlier with the expanded diagnostic criteria (i.e., reaching any dyad among the six classic and adjunct manifestations) compared to the classic diagnostic criteria (mean age at diagnosis, 4.6 versus 8.8 years, respectively). Similarly, European patients reached a clinical APECED diagnosis 4 years earlier with the expanded diagnostic criteria compared to the classic diagnostic criteria (mean age at diagnosis, 3.8 versus 7.8 years, respectively). A total of 10 genetically-confirmed American and European APECED patients who have not yet reached a diagnosis based on the classic diagnostic criteria meet a clinical diagnosis by the expanded diagnostic criteria. APECED rash, intestinal dysfunction, and enamel hypoplasia were the three most common early-onset manifestations occurring before reaching diagnosis by the classic diagnostic criteria. Expanded diagnostic criteria-based diagnosis could have prevented acute life-threatening endocrine failure complications (i.e., adrenal crisis, hypocalcemic seizures) as presenting manifestations of unsuspected APECED in a total of 28 (41%) American and European patients.
Incorporation of APECED rash, intestinal dysfunction, and enamel hypoplasia into the classic diagnostic criteria can accelerate APECED diagnosis in both American and European patients. Earlier diagnosis based on utilization of expanded diagnostic criteria can a) facilitate early screening for endocrinopathies that would ameliorate acute life-threatening endocrine failure complications and b) promote early recognition and treatment for non-endocrine manifestations such as pneumonitis, hepatitis, and keratoconjunctivitis.
Keywords: APECED, APS-1, Expanded Diagnostic Criteria
Disclosure: All authors indicated they had no financial relationships to disclose.
(89) Mycophenolate-induced colitis in APECED patients: A case series
Monica Schmitt, CRNP1, Michail Lionakis, MD, ScD2, Elise Ferre, PA-C, MPH3, Theo Heller, MD4
1Nurse Practitioner/Fungal Pathogenesis Section, Laboratory of Clinical Immunology and Microbiology, NIAID, NIH, Bethesda, MD, USA
2Chief, Fungal Pathogenesis Section/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases/National Institutes of Health, Bethesda, MD, USA
3Physician Assistant/Fungal Pathogenesis Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases/National Institutes of Health, Bethesda, MD, USA
4Senior Investigator/Translational Hepatology Section, Liver Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NIH, Bethesda, MD 20892, USA
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) is an autoimmune disorder that often requires immunomodulation to prevent end-organ damage. Mycophenolic acid (MPA) is the active metabolite of mycophenolate mofetil (MMF) and mycophenolate sodium (MPS), which have been used to treat autoimmune manifestations in APECED. Mycophenolate can cause colitis in 2-9% of treated patients.
To describe the clinical and histological features of biopsy-proven mycophenolate-induced colitis in 3 APECED patients.
Patients were enrolled in an NIH IRB-approved protocol (11-I-0187) and were evaluated with history, physical examination, laboratory testing, and colonoscopy with biopsies and pathology evaluation.
Patient 1 is a 19-year-old woman with tubulointerstitial nephritis treated with MPS (540 mg twice daily). Seven months after MPS initiation, she developed persistent watery diarrhea (4-5 times daily), periumbilical cramping, and foul-smelling flatulence with significant nocturnal symptoms. Colonoscopy revealed diffuse macroscopic ulcerations and histological evidence of chronic colitis with crypt destruction and an increased number of eosinophils in the lamina propria. Patient 2 is a 14-year-old girl with autoimmune hepatitis and pneumonitis who was treated with MMF. Two weeks upon increase of MMF dosing to 1000 mg twice a day, she experienced 5-6 episodes of diarrhea per day. Colonoscopy demonstrated chronic active colitis with focal cryptitis, apoptotic bodies, and a lymphoplasmacytic infiltrate within the lamina propria. Patient 3 is a 10-year-old girl with tubulointerstitial nephritis who was treated with MMF (500 mg twice daily). Eight months later, she developed vomiting, abdominal pain and weight loss. Colonoscopy revealed increased apoptosis in the ileum and transverse colon. Abdominal symptoms resolved in all 3 patients within 1-3 weeks of MPS or MMF discontinuation, and colitis resolution was confirmed by colonoscopy in one of the patients.
Among 104 evaluated APECED patients, mycophenolate was used for autoimmune end-organ involvement in 10 patients. Three (30%) of them developed biopsy-proven mycophenolate-induced colitis, which was reversible upon drug discontinuation. Mycophenolate-induced colitis appears to be a common complication affecting the autoimmune syndrome APECED and clinicians should maintain a high index of suspicion in recognizing the development of this complication in mycophenolate-treated APECED patients.
Keywords: Mycophenolate, colitis, APECED
Disclosure: All authors indicated they had no financial relationships to disclose.
(90) Case Series of Pediatric Patients with Kabuki Syndrome
Elisa Ochfeld, MD1, Aisha Ahmed, MD2, A. Kyle Mack, MD3, Robert Liem, MD3, Allison Remiker, MD3, Amer Khojah, MD4
1Fellow/Northwestern University/ Lurie Childrens Hospital
2Attending Physician, Allergy and Immunology/Ann and Robert H Lurie Children's Hospital of Chicago
3Attending, Hematology/ Oncology/Lurie Children's Hospital of Chicago
4Attending Physician, Allergy, Immunology, and Rheumatology/Ann & Robert H. Lurie Children's Hospital of Chicago/ Northwestern University Feinberg School of Medicine
Introduction: Kabuki syndrome (KS) is a rare multi-organ system disorder that can present with distinct facial features, developmental delay/ intellectual disability, short stature, congenital heart disease, skeletal anomalies, and recurrent infections. KS is associated with genetic mutations in KMT2D (autosomal dominant inheritance) and KDM6A (X-linked inheritance), which encode histone-modifying proteins. The phenotype of Kabuki syndrome is heterogenous and can also involve endocrine dysfunction, autoimmune cytopenias, neurosensory abnormalities and immunodeficiency. Immunodeficiency associated with KS is variable but can resemble CVID, with hypogammaglobulinemia, low IgA and/ or impaired specific antibody responses. Methods: This is a retrospective case series from one pediatric academic institution including eight patients with Kabuki syndrome. Results: 5/8 patients were male, 3/8 female. All patients had characteristic facial features of KS. Age at genetic diagnosis of KS had a bimodal distribution, either occurring prior to age 3 (n=4) or from age 9-13 (n=4). All patients displayed developmental delay, 5/8 had hearing impairment, 3/8 had endocrine dysfunction, 3/8 had congenital cardiac abnormalities, 3/8 had autoimmune cytopenia. All associated variants were in KMT2D, and no patients had relevant family histories. Those with parental genetic testing showed de-novo variants. Recurrent infections were most commonly frequent AOMs in childhood which improved as the patient aged or after tympanostomy tube placement. None have required IgG replacement or prophylactic antibiotics for hypogammaglobulinemia with recurrent infections. Of patients with complete evaluations, 6/6 displayed robust vaccine response to protein and polysaccharide antigens, with no evidence of specific antibody dysfunction. Mild hypogammaglobulinemia was present in 3/8 patients, and 7/8 display low IgA (though detectable). Interestingly, 2/8 displayed T cell lymphopenia (though one of these patients had a significant cardiac history), and 3/8 had low NK cell levels. Conclusion: Kabuki syndrome is a highly heterogenous multi-system disorder with a variable immunologic phenotype. While humoral immunodeficiency is most commonly reported in KS, patients may display other immunologic abnormalities including T or NK cell lymphopenia, and should undergo a broad immunologic evaluation. Poor specific antibody response was not a hallmark of our population. Recurrent infections were typically mild and none of these patients have required IVIG replacement or prophylactic antibiotics.
Keywords: Kabuki Syndrome, Case Series, Immunologic phenotypes, Recurrent infections
Disclosure: All authors indicated they had no financial relationships to disclose.
(91) Neurologic Manifestations reported in Autoinflammatory Disorders and Immune Dysregulatory Diseases from the USIDNET Registry
Elizabeth Wagman1, Dana O’Toole, MD2, Hannah Wright, MSPH3, Elizabeth Garabedian, MSN4, Ramsay Fuleihan, MD5, Devorah Segal, MD6, Elizabeth Feuille, MD7
1Student/Emory University
2Assistant Professor of Clinical Pediatrics/Weill Cornell Medicine
3Research Data Analyst/The United States Immunodeficiency Network (USIDNET)
4Research Nurse/National Genome Research Institute
5Professor of Pediatrics/Division of Allergy and Immunology, Department of Pediatrics, Columbia University, New York, NY, USA
6Assistant Professor of Clinical Pediatrics/NYU Langone Health
7Assistant Professor of Pediatrics/Weill Cornell Medicine
Autoinflammatory and immune dysregulatory conditions may be associated with various neurologic manifestations including seizures, behavioral and developmental psychiatric conditions, fatigue, and headache. Here we aim to identify the types and frequencies of neurologic manifestations among patients in the USIDNET (United States Immunodeficiency Network) Registry with immune dysregulatory and autoinflammatory conditions.
Demographic and clinical data were obtained and described for patients in the USIDNET Registry with neurologic manifestations who had a genetic diagnosis of an autoinflammatory or immune dysregulatory disorder as defined by the International Union of Immunological Societies.
Among those patients with an immune dysregulatory disorder who reported neurologic symptoms (N=101), the most commonly reported was seizures (32%), followed by behavioral/psychiatric (23%), fatigue/pain (22%), headache (19%), and developmental conditions (18%). Among those patients (N=17) with an autoinflammatory disorder who reported neurologic symptoms, the most commonly reported were behavioral/psychiatric (30%), followed by fatigue/pain (24%), ophthalmic/visual (24%), auditory complications (24%), and developmental conditions (18%). Genetic mutations most commonly associated with neurologic conditions are listed in Table 1.
Various neurologic conditions may arise in patients with immune dysregulatory and autoinflammatory conditions, either as a direct manifestation of disease, as a consequence of infections, or as a complication of treatments. Knowledge of neurologic conditions associated with immune dysregulatory and autoinflammatory conditions may aid practitioners in suspecting and making a diagnosis and in anticipating neurologic complications that may arise as a consequence of these conditions.

Table 1. Genetic mutations associated with neurologic manifestations of patients in the USIDNET Registry

Table 2. Neurological manifestations of patients with autoinflammatory disorders and diseases of immune dysregulation in the USIDNET registry
Keywords: Autoinflammatory disorders, Diseases of immune dysregulation, Nervous system
Disclosure: All authors indicated they had no financial relationships to disclose.
(92) Treatment Of COVID-19 With Convalescent Plasma in a Patient With X-linked Agammaglobulinemia
Yashu Dhamija, MD1, Susan Xie, MD2, Simin Zhang, MD1, Bridget Wilson, MD3, Paul Spearman, MD4, Grant Paulsen, MD4, Lara Danziger-Isakov, MD4, Amal Assa'ad, MD5
1Fellow/University of Cincinnati
2Fellow/Cincinnati Children's Hospital Medical Center
3Resident/Cincinnati Children's Hospital Medical Center
4Attending Physician/Cincinnati Children's Hospital Medical Center
5Attending Phsyician/Cincinnati Children's Hospital Medical Center
X-linked agammaglobulinemia (XLA) is a primary immunodeficiency consisting of humoral deficiency and subsequent respiratory and gastrointestinal infections. Convalescent plasma contains immunoglobulins of patients who recovered from a specific infection. The US Food and Drug Administration has approved emergency use of convalescent plasma during the COVID-19 pandemic.
A 24-year-old white man with XLA adherent to intravenous immunoglobulin (IVIG) every 3 weeks presented with 1 week of nasal congestion and sore throat, followed by 3 days of fever, night sweats, productive cough, chest pain, and diarrhea. On presentation to the emergency department (ED), his COVID-19 PCR was positive. Chest x-ray showed right upper lobe pneumonia, and he was started empirically on IV ceftriaxone. IgG was 1,480 mg/dL (3 days after IVIG). He received convalescent plasma on the day of admission. His symptoms resolved over the next 2 days, and he remained afebrile and normoxic throughout admission, and he was discharged on a 10-day course of amoxicillin for pneumonia. SARS-CoV-2 IgG was 1:800 on day of discharge, indeterminate 8 days after discharge, and negative 14 days after discharge. The patient returned to the ED one month after admission with nasal congestion and cough. Chest X-ray showed worsening right upper lobe pneumonia; PCR testing was negative for COVID-19 but positive for rhinovirus. He was admitted overnight and discharged in stable condition on a course of amoxicillin/clavulanate and azithromycin for recrudescent community acquired pneumonia.
In patients with humoral immunodeficiencies, use of convalescent plasma provides passive immunity. In this case report, a patient with XLA was treated with early convalescent plasma for an acute COVID-19 infection with good response and maintenance of quality of life. Recurrence of this patient’s symptoms of fevers and cough suggest bacterial pneumonia may have been the preeminent infection. While there are no data on the half-life of COVID-19 convalescent plasma products, the half-life of IgG is thought to be 21 days. This patient displayed rapid loss of passive immunity. Antibody age in plasma products cannot be guaranteed. Research on use of early convalescent plasma in immunodeficient patients will provide further clarity on outcome measures.
Keywords: COVID-19, X-linked agammaglobulinemia, primary immunodeficiency, convalescent plasma, pneumonia
Disclosure: Lara Danziger-Isakov has contracted research with Ansun BioPharma, Astellas, Merck, Takeda and Viracor. All other authors had no financial relationship to disclose.
(93) 22q11.2 deletion and biomarkers of autoimmunity
Terrence Blaine Crowley, BA1, Daniel McGinn, BA2, Emily Liebling, MD3, Michele P Lambert, MD MSTR4, Katz Lorraine, MD5, Donna McDonald McGinn, MS LCGC6, Kathleen Sullivan, MD, PhD7
1Analyst/Children's Hospital of Philadelphia
2Research Assistant/Children's Hospital of Philadelphia
3Assistant Professor/Children's Hospital of Philadelphia
4Associate Professor/Children's Hospital of Philadelphia
5Professor of Pediatrics/Children's Hospital of Philadelphia
6Director of the 22q and You Center/Children's Hospital of Philadelphia
7Professor of Pediatrics, Chief of the Division of Allergy and Immunology/The Children’s Hospital of Philadelphia, Philadelphia, PA, USA
Chromosome 22q11.2 deletion syndrome is strongly associated with autoimmunity with a frequency that increases over time. There are recent reports suggesting that greater compromise in thymic production is associated with at least some specific types of autoimmune diseases in 22q11.2 deletion syndrome. Yet, there are other studies that suggest the autoimmunity is seen predominantly in patients with an altered B cell compartment. We undertook this retrospective analysis of the trajectory of laboratory features in our carefully phenotyped cohort of >1500 patients with 22q11.2 deletion syndrome to understand possible associations with autoimmunity. Three subcohorts with autoimmunity were identified after manual chart review to validate the diagnosis initially extracted by ICD codes. 25 patients with autoimmune thyroid disease with 80 available immunologic studies, 5 patients with idiopathic thrombocytopenia purpura (ITP) who had 24 immunologic evaluations and 5 patients with JIA with 13 immunologic evaluations were included in the study along with 668 patients with no autoimmune diagnosis who had 1207 immunologic evaluations. We found that ITP was associated with significantly lower CD4 counts. This same finding was not observed in the patients with autoimmune thyroid disease or JIA, suggesting a different mechanism for solid organ autoimmunity. We further looked at CD4 CD45RA as a common clinical marker for naïve T cells and found the lowest levels at all ages in the subcohort with ITP. The subcohort with ITP had on average CD4CD45RA counts of 327 cells/mm3 less than the non-autoimmune comparator group (p=0.0034). These findings support the hypothesis that the patients with the greatest compromise in thymic T cell production are at the highest risk of ITP. These results pave the way for emerging risk stratification for patients with 22q11.2 deletion syndrome.
Keywords: 22q11.2 deletion syndrome, DiGeorge syndrome, T cells
Disclosure: Michele P Lambert received research grants from Novartis, Octapharma, Principia, and Rigel; is a consultant for Dova; and Advisory board member and consultant for Argenx. Kathleen Sullivan is a consultant for Enzyvant and an advisory board member of the Immune Deficiency Foundation. All other authors had no financial relationship to disclose.
(94) A challenging case of recurrent idiopathic hemophagocytic lymphohistiocytosis (HLH) initially presenting in an infant with Pneumocystis jirovecii pneumonia
Benjamin Solomon, MD, PhD1, Jay Balagtas, MD2, May Chien, MD3, Rajdeep Pooni, MD, MS4, Imelda Balboni, MD, PhD5, Katja Weinacht, MD, PhD6, Yael Gernez, MD, PhD7
1Pediatric Resident Physician/Stanford University
2Associate Professor of Pediatric Hematology-Oncology/Stanford University
3Assistant Professor of Pediatric Hematology-Oncology/Stanford University
4Pediatric Rheumatology Instructor/Stanford University
5Associate Professor of Pediatric Rheumatology/Stanford University
6Assistant Professor of Pediatric Stem Cell Transplantation/Stanford University
7Assistant Professor of Pediatric Allergy and Immunology/Stanford University
Here, we report the case of a one-year-old girl with HLH who initially presented at 3 months of age with Pneumocystis jirovecii pneumonia (PJP) requiring intubation for hypoxemic respiratory failure. She developed anemia, neutropenia, and splenomegaly, which improved with treatment of her infection. She had hypogammaglobulinemia, but no evidence of malignancy or lymphocyte abnormalities and her newborn screen was normal. After discharge, a genetic panel identified a single copy variant of unknown significance (VUS) in STXBP2.
While the patient had no further infections, she had recurrent splenomegaly with severe cytopenias requiring repeat admissions. At 6 months of age, she met 7/8 of the HLH-2004 criteria (maximum ferritin of 1,189 ng/mL) and improved with methylprednisone (1 mg/kg). While on a prolonged steroid taper, she experienced another HLH flare at 9 months of age (maximum ferritin of 6,862 ng/mL) which ultimately required high-dose steroids in combination with anakinra (4mg/kg) to induce remission. The patient was discharged on anakinra and a steroid taper and remains well at 10 months of age.
Despite her unusual presentation, our patient’s immune evaluation has been relatively normal. She had persistent hypogammaglobulinemia with low isohemagglutinin and hepatitis B titers and was started on IVIG. She had moderately low CD27+IgM+ B-cells (1.2 cells/uL), though the rest of her lymphocyte phenotyping was normal, as was her T-cell proliferation to cytokines, TCR spectratyping, and CD40 genetic testing. Targeted genetic and whole exome sequencing did not identify an underlying genetic cause.
This patient posed a diagnostic and therapeutic challenge due to the absence of a clear HLH etiology. Her initial episode of PJP was suspicious for an immunodeficiency, though only a mild humoral impairment was found. Though her young age, history of a severe opportunistic infection, and two HLH flares suggest an underlying genetic predisposition to immune dysregulation, her single copy STXBP2 VUS alone does not account for her condition. She is currently being evaluated for possible allogenic hematopoietic stem cell transplantation. The inability to categorize our patient as primary vs. secondary HLH complicated coordination of her care and highlights the importance of interdisciplinary teams in management of HLH.
Keywords: Hemophagocytic lymphohistiocytosis (HLH), Macrophage activation syndrome (MAS), Primary immune deficiency (PID), Pneumocystis jirovecii pneumonia (PJP), Anakinra
Disclosure: All authors indicated they had no financial relationships to disclose.
(95) Diagnostic Utility of Genomic Testing in Disorders of the Immune System
Amber Begtrup, PhD, FACMG1, Ushna Ahmad, MS2, Rashmi Chikarmane, MsC, CGC3, Jane Juusola, PhD, FACMG4
Inborn errors of the immune system (IEI) are caused by monogenic germline variants that lead to disruption of immune function. Rapid gene discovery over the past decade has resulted in over 400 different genes now recognized to cause an IEI. Due to the large degree of genetic heterogeneity, and overlapping clinical features of many IEI, genomic sequencing, defined as exome or genome sequencing, is a cost-effective diagnostic tool to assist with diagnosis and management of individuals with IEI. To better understand the utility of genomic sequencing, we performed a retrospective review of our experience with individuals with IEI in our clinical laboratory. Our cohort included 2,624 patients, with trio (62%), duo (10%), singleton (23%), or other (5%) clinical exome testing strategies. Our cohort had an age range from 5 days to 76 years, with 74% of patients under 18 years. Overall, we found 14% of cases had a diagnostic finding, 47% with a nondiagnostic finding, and 38% with no findings reported. Patients < 2 years of age had the highest diagnostic rate (25%) and diagnostic yield decreased with increasing age of patients (6% in >40 years). Including family members in the testing strategy increased diagnostic rates and reduced the number of nondiagnostic variants reported in both the pediatric and adult patients. We identified 378 patients with diagnostic results, 176 (47%) with a diagnostic gene consistent with an IEI, 164 (42%) with a non-IEI diagnostic finding, and 38 cases with a diagnostic multigene CNV (10%). The most frequently observed diagnostic IEI genes were STAT1 (10), KMT2D (8), and PIK3CD (7). Our diagnostic cases included 97 distinct IEI genes and 133 non-immune gene findings, highlighting the genetic heterogeneity of this cohort. Additionally, over 300 distinct candidate genes with emerging associations to disease were reported in this cohort, suggesting that the rapid pace of identification of new genetic causes of IEI is likely to continue with the use of clinical genomic sequencing.
Keywords: genomics, gene discovery, diagnostics
Disclosure: All authors are employed by GeneDx.
(96) Developing threshold parameters to administer live vaccines in cellular immunodeficiency: A survey of practicing clinical immunologists
Justine Ade, MD1, Avni Joshi, MD, MS2
1Allergy Immunology Fellow/Mayo Clinic
2Allergist-Immunologist/Mayo Clinic
Live vaccines are contraindicated in patients with severe cellular immunodeficiencies. Guidelines regarding the administration of live vaccines in patients with more mild disease, however, are ill-defined. We sought to decipher different parameters used by practicing immunologists for the administration of live vaccines in cellular immunodeficiency patients.
A 27-question survey assessing clinical and laboratory threshold parameters used in the administration of live vaccines to immunodeficient patients was distributed amongst members of the Clinical Immunology Society (CIS) listserv after approval. Variables including CD3, CD4, CD8 counts and T-cell functional studies were used to inquire about thresholds for the administration of three live vaccines - measles, mumps and rubella (MMR), varicella, and rotavirus vaccines.
There were 83 survey respondents, 65% identified as female, and 71% were based in the United States. Allergy / Immunology and Immunodeficiency were the most common identified specialties, accounting for 84% of respondents. Most clinicians did administer live vaccines to patients with humoral (54/67; 80.6%), cellular (41/67; 61.2%), and combined diseases (37/67; 55.2%). Most clinicians who administer live vaccines to patients with immune deficiencies considered a threshold CD4 count of □ 400 cells/mm3 (MMR 48/60 [80%], Varicella 42/53 [79%], Rotavirus 40/45 [88.89%]), a CD8 count of □ 250 cells/mm3 (MMR 30/39 [76.92%], Varicella 29/37 [78.34%], Rotavirus 27/34 [79.41%]), and normal mitogen function (MMR 44/53 [83.02%], Varicella 40/48 [83.33%], Rotavirus 37/40 [92.5%]).
More than half of clinicians who care for immune deficient patients included in this survey do administer live vaccines to patients with cellular immunodeficiency. The most commonly used parameters used in clinical decision making include numeric and functional assessments of the T cell compartment. Using these results, we propose a treatment threshold of using CD4 count of □ 400 cells/mm3, a CD8 count of □ 250 cells/mm3 and normal mitogen function testing. Future studies are needed to determine clinical efficacy and safety using these thresholds.
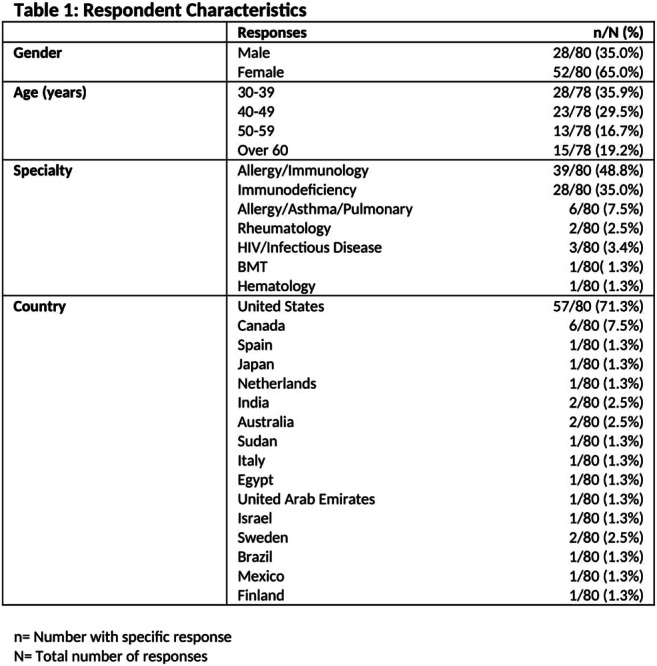


Keywords: live vaccine, primary immunodeficiency, cellular immunodeficiency
Disclosure: All authors indicated they had no financial relationships to disclose.
(97) Common variable immunodeficiency revised: expanding the spectrum of IRF2BP2 deficiency
Julia Körholz, MD1, Henrike Sczakiel, MD2, Livia Schulze, n/a3, Timm Amendt, Phd4, Anastasia Gabrielyan, Phd5, Matin Laass, MD6, Daniela Aust, Prof.7, Joachim Roesler, PD6, Axel Roers, Prof.8, Catharina Schuetz, Prof.6
1Resident/Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
2resident/Institute of Medical Genetics and Human Genetics, Charité - Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Berlin 13353, Germany.
3Technician/Institute of Immunology, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
4Researcher/Institute of Immunology, University Hospital Ulm, Ulm, Germany
5Postdoc/Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany.
6Chief resident/Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany.
7Assistant head of institute/Institute of Pathology, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany.
8Head of institute/Institute of Immunology, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany.
Interferon regulatory factor-2 binding protein 2 (IRF2BP2) is a nuclear protein that has been described as important transcriptional cofactor. It interacts with interferon regulatory factor 2 (IRF2) as transcriptional repressor and has recently been described as positive and negative regulator of gene expression. It impacts on different cellular functions influencing macrophage regulation, lymphocyte activation and angiogenesis especially in cancer development. In humans dominant IRF2BP2 deficiency is associated with an inborn error of immunity (IEI). Although IRF2BP2-deficiency is mentioned in almost every classification of monogenic CVID there has only been one family described to date with a CVID phenotype associated with a variant in IRF2BP2. The phenotypic spectrum was characterized by hypogammaglobulinaemia and infections but also psoriasis, colitis and type 1 diabetes.
We present a 32-year old male with a history of recurrent infections since childhood including sinusitis, bronchitis, pneumonia and otitis media attributed to hypogammaglobulinaemia. Immunoglobulin substitution significantly reduced susceptibility to infections although IgG remained low. Colonoscopy revealed inflammation of the intestinalgut mucosa with chronic lymphocytic and focal granulocytic infiltration similar to inflammatory bowel disease but with remarkableabscence of plasma cells. This may point to the cause underlying low Ig levels. At age 25, the patient developed a rheumatoid factor negative chronic active and erosive polyarthritis, resulting in progressive stiffness and severely restricted mobility within 2-3 years despite treatment with methotrexate.
Immunophenotyping revealed decreased memory B cells with an impairment of class-switch and a lack of plasmablasts. T-cell numbers, subpopulations and function were normal. Whole exome sequencing (and Sanger confirmation) revealed a de novo nonsense mutation in IRF2BP2,c.1618C>T, p.(Q540*) (ACMG class IV). This mutation may result in expression of a truncated IRF2BP2 protein lacking some highly conserved amino acids and leading to the inability to properly form the RING domain. This domain was also affected in the family described by Keller et al.
This report adds to the described spectrum of human IRF2BP2 deficiency expanding the phenotype of this rare monogenic CVID. Further functional investigations and patient cohort studies will broaden the understanding of this deficiency, its role for B-cell development and support tailored therapeutic management.
Keywords: IRF2BP2, CVID, Erosive polyarthritis, Hypogammaglobulinaemia
Disclosure: Axel Roers has contracted research with Celgene and Roche. All other authors had no financial relationship to disclose
(98) Reliability of shipped dried blood spot samples for IgG trough level monitoring in patients with primary immunodeficiency disease
Hanna Haberstroh, MSc1, Ulrich Salzer, MD2, Bodo Grimbacher, Prof. Dr. med.3, Sigune Goldacker, MD4, Aleksandra Hirsch, MD5, Arianna Troilo, MD, PhD6, Monika Erler, n/a7, Martina Eigenbrod-Damerau, n/a7, Norbert Zessack, n/a8, Klaus Warnatz, MD9
1technician/Center for Chronic Immunodeficiency, MedicalCenter, University of Freiburg, Germany
2Research group leader/University Clinic Freiburg, CCI
3Head of the laboratory/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
4Physician for internal medicine and rheumatology/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
5Physician/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
6Resident in Rheumatology/Department of Rheumatology and Clinical Immunology, Medical Center - University of Freiburg. Faculty of Medicine, University of Freiburg, Germany
7Study assistants/Department of Rheumatology and Clinical Immunology, Medical Center - University of Freiburg. Faculty of Medicine, University of Freiburg, Germany
8Associate Director, Country Medical Lead Immunology/Shire Deutschland GmbH
9Head of Division/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
Primary immunodeficiency disease (PID) and concomitant antibody deficiencies are effectively treated with immunoglobulin G (IgG) administered either intravenously (IVIG) at a medical facility or subcutaneously as home self-therapy (SCIG). One drawback of home-based SCIG is the lack of easy options for monitoring IgG levels. We and others have previously shown that IgG levels can be analyzed from dried blood spots (DBS) using a standard nephelometric assay. The aim of this study was to test the reliability of this method under real life conditions comparing sampling on-site and from shipped DBS cards with standard serum measurement.
IgG levels in DBS eluates from shipped and on-site samples and serum from 100 prospectively enrolled patients on IVIG or SCIG therapy were tested by nephelometry (BN Prospec, Siemens, Germany). All patients provided informed consent (IRB University of Freiburg #514/18) to the study (DRKS-ID: DRKS00020522)
We found a highly significant correlation of both on-site and shipped DBS samples with serum samples (n=100, Figure 1A and B; p < 0.0001) as well as within non-shipped and shipped DBS samples (n=100, Figure 1C; p < 0.0001). However, mean IgG levels determined from DBS samples were consistently slightly lower than those from serum samples for both non-shipped and shipped DBS samples (mean IgGserum: 10.07 g/l versus mean IgGon-site DBS 8.768 g/L, mean %CVon-site DBS vs serum: 10.54; or versus mean IgGshipped DBS 8.754 g/L, mean %CVshipped DBS vs serum: 10.77), whereas there was no significant difference between non-shipped and shipped DBS samples (mean %CVon-site DBS vs shipped: 4.552; Wilcoxon teston-site DBS vs shipped DBS: p= 0.6983).
Our data show that patient-friendly, home-based monitoring of IgG levels from DBS is reliable and the results not influenced significantly by shipment of DBS cards, confirming the applicability of the method in routine clinical practice. Slightly lower IgG levels were consistently found in DBS samples when compared with serum. It is currently unclear if this is related to the DBS sampling procedure or different properties of capillary blood itself. Therefore, reference ranges specific for IgG derived from capillary DBS samples might be necessary.

Figure 1. Correlations between on-site and shipped DBS sample and serum sample IgG Levels
Keywords: immunoglobulin, assay, primary immunodeficiency disease, dried blood spot
Disclosure: Hanna Haberstroh received a travel grant from Baxalta/Takeda. Ulrich Salzer is an advisory board member of Baxalta. Bodo Grimbacher received a research grant from Baxalta. Norbert Zessack is employed by Takeda. Klaus Warnatz received speaker honoraria from Baxalta/Takeda. All other authors had no financial relationship to disclose.
(99) Recurrent pneumonia and sinusitis: an interplay of alcoholism and selective IgM deficiency
Minh Nguyen, DO1, Taylor Cable, MD1, Ricardo de Castro, MD1, Ali Shah, MBBS1, Mark Loehrke, MD2
1Resident Physician/Western Michigan University School of Medicine
2Attending Physician/Western Michigan University School of Medicine
Selective immunoglobulin M deficiency (sIgMD) is a rare immune disorder, defined as IgM levels below two standard deviations of the mean with normal IgG and IgA and exclusion of other causes of immune deficiencies. Most patients with sIgMD present with frequent infections. Here, we detail a patient with recurrent pneumonia (PNA) and sinusitis; and a workup consistent with sIgMD. Further, his course is complicated by alcoholism. Chronic alcohol use is linked with changes in the immune system, leading to higher risk of infections. This case represents a clinical conundrum involving alcoholism and sIgMD.
A 42-year-old man with a history of homelessness, severe alcoholism, and recurrent PNA presented with worsening dyspnea and cough. He required oxygen supplementation on arrival. Chest XR revealed left basilar PNA. His CBC and CMP were unremarkable. Chart review revealed seven admissions related to PNA during the last 12 months. Imaging studies demonstrated opacities/infiltrates involving varying lobes: right lower, middle, lingula and upper lobes, and left lower lobe (figure 1). He reported multiple hospitalizations as a child for PNA. Since 2017, multiple brain CT scans showed sinus disease (figure 2). The patient confirmed recurrent sinusitis. Immunologic workup revealed IgM of 29 mg/dL (ref range 53-334) on two separate checks, IgE of 51 kU/L (ref range < 41), NK cells of 38 cells/uL (ref range 59-513) and normal IgG, IgA, IgG subclass, total complement, CD3, CD4, CD8 and CD19 cells. HIV antibodies were negative. ANA titers were less than 1:40. Vaccine titers were not obtained, as the patient left against medical advice.
The patient’s recurrent sinusitis and PNA involving various lobes suggested an underlying immunological condition. His workup is consistent with sIgMD. Concurrent alcohol abuse plays a role in his ongoing infections, as chronic alcohol use has profound effects on immune response. It is arguable that his recurrent PNA is partly due to inadequate treatment in the setting of noncompliance, although this is less likely with PNA occurring in varying lobes and recurrent PNA in childhood. Overall, the interplay of his alcoholism and an underlying immunological disorder, most likely sIgMD, contributed to a prolonged course of PNA and sinusitis.
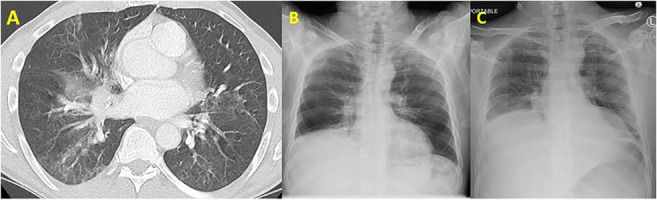
Figure 1: A) Computed tomography of chest taken in 1/2020 showed multifocal patchy groundglass opacities involving the lingula. B) Chest X-ray (CXR) on 4/2020 demonstrated right lung base consolidation. C) CXR on 12/2020 revealed left basilar infiltrates.

Figure 1: A) Computed tomography (CT) of brain in 8/2017 showed moderate amount of mucosal thickening involving the paranasal sinuses (red arrow). B) Brain CT in 8/2020 again demonstrated moderate opacification of the paranasal sinuses (red arrows).
Keywords: Selective IgM Deficiency, Alcoholism, Recurrent pneumonia, Recurrent Sinusitis
Disclosure: All authors indicated they had no financial relationships to disclose.
(100) Real-world healthcare resource utilization in untreated congenital athymia: A medical chart audit study
Megan Cooper, MD,PhD1, Cathleen Collins, MD, PhD2, Julie Kim-Chang, MD3, Shruti Nambiar, PhD4, Matthew O'Hara, MBA5, Sarah Kulke, MD6, Elena Hsieh, MD7
1Associate Professor/Department of Pediatrics, Division of Rheumatology/Immunology, Washington University School of Medicine, St. Louis, MO
2Assistant Professor/Department of Pediatrics, Division of Allergy Immunology, University of California San Diego; Rady Children’s Hospital, San Diego, CA
3Division of Allergy, Immunology, and Pulmonary Medicine, Department of Pediatrics, Duke University School of Medicine, Durham, NC
4Senior Consultant/Trinity Life Sciences, Waltham, MA
5Managing Director/Trinity Life Sciences, Waltham, MA
6Vice President, Medical Affairs/Enzyvant, Cambridge, MA
7Assistant Professor/Department of Pediatrics, Section of Allergy and Immunology, University of Colorado, Anschutz School of Medicine; Children’s Hospital Colorado; Department of Immunology and Microbiology, University of Colorado, Anschutz School of Medicine, Aurora, CO
Congenital athymia is an ultra-rare disorder characterized by absence of a thymus at birth. These patients lack naïve T-cells critical to immune regulation and defense against infections, and typically die within two to three years of life if they do not receive treatment to establish T-cell immunity.
To estimate the healthcare resource utilization (HCRU) associated with congenital athymia for patients receiving supportive care prior to cultured thymic tissue implantation (CTTI; if received) using medical charts.
Board-certified physicians participated in an IRB-approved online survey abstracting HCRU data from medical charts for living or deceased patients with congenital athymia (defined by naïve T-cell count < 100 cells/mm^3 recorded at birth or diagnosis). Data was collected for at least 12 continuous months starting from birth to three years of age, until either CTTI or death. Descriptive statistics were conducted to estimate the average annual HCRU in the first three years of life for future application as inputs in a model to estimate cost of care for congenital athymia from birth to age three.
Interim results from the ongoing survey (n=5 charts) are reported here; complete results will be reported in the presentation. Two patients were born to diabetic mothers, one had DiGeorge syndrome, one had CHARGE syndrome and one had no known associated conditions. Before age three, three patients received CTTI, and two patients died. Annual mean length of inpatient stay was 120.4 days, with maximum stay as high as 365 days. All patients required immunoglobulin replacement therapy on a weekly or monthly basis. All patients received prophylactic antibiotics and antifungals. Patients required multiple procedures such as feeding tube placement (100%), central line placement (80%), bronchoscopy (60%), skin biopsy (60%), and ventilators (60%). All patient charts reported abnormal T-cells counts, and 80% reported more than one infection type. Sepsis was reported in 60% of patients.
Congenital athymia is associated with high HCRU in the initial three years of life prior to CTTI, placing a significant burden on healthcare systems, providers, patients and their families.
Keywords: thymic aplasia, congenital athymia, real-world evidence, healthcare resource utilization, cultured thymic tissue implantation
Disclosure: Megan Cooper, Cathleeen Collins, Shruti Nambiar, Matthew O’Hara and Elena Hsieh are all consultants for Enzyvant. Sarah Kulke is employed by Enzyvant.
(101) Age isn’t just a number when it comes to genetic testing: diagnostic yields across age groups for patients with PIDDs
Hui Yu, PhD1, Jennifer Holle, MS, CGC2, Rebecca Truty, PhD3, Shiloh Martin, MD, PhD1, Jessica Connor, MS, LCGC1, Lauren Bryl, MS, CGC1, Olga Sarmento, PhD1, Britt Johnson, PhD, FACMG, HCLD(ABB)4
1Clinical Genomics Scientist/Invitae
2Genetic Counselor/Invitae
3Bioinformatic Analyst/Invitae
4Medical Director, Metabolic Genetics and Immunology,Medical Affairs Director, Biopharma Partnerships/Invitae Corporation, San Francisco, CA
The clinical presentations of individuals with inborn errors of immunity may be complicated by developmental stages and environmental exposures during life. Those with early presentations are typically considered more likely to test positive than older patients, while some primary immunodeficiencies and immune dysregulatory diseases (PIDDs) may be more commonly diagnosed in patients of different age groups. To better understand the genetic contribution to clinical presentations, we examined the diagnostic yield of genetic testing panels across PIDD patients of different age groups at a commercial laboratory over a 2.5 year period. Patients were divided into 3 cohorts based on testing age: < 3 years; 3-17 years; ≥18 years.
During the studied period 10,864 unrelated patients were tested with immunology panels with an overall positive rate of 6.22% (n=676). Among them, 2,003 patients were infants younger than 3 years, 4,876 were age 3-17 years, and 3,985 were adult patients ≥ 18 years. The < 3 age group had the highest positive rate (12.13%). The 3-17 group had a 5.46% positive rate. Adults had the lowest diagnostic yield: 4.19%. Similar trends were seen when results were separated by type of PID panel. The severe/combined immunodeficiencies (SCID/CID) panels were most frequently ordered (n=137) in the < 3 age group with a positive rate of 40.15%, compared to a 5% positive rate in the 3-17 group (n=20), and a 4.76% positive rate in adults (n=21). The large 207-gene PID panel accounted for over half of the tests ordered throughout all age groups, and its diagnostic yield correlated with the overall positive rates: 11.77%, 6.54%, and 4.32%, respectively.
Several factors are likely contributing to the different diagnostic yields among age groups. In very young patients, PIDD presentations tend to be classic, severe and less likely to escape diagnosis than in older individuals. Clinical manifestations in adult patients are frequently mild or moderate, involving nonspecific features. Adult patients with complex histories are more likely to undergo genetic testing for the purpose of “ruling out” primary immunodeficiency. Understanding the expected positive rate for a given patient in a specific age group is valuable for setting appropriate expectations for clinicians and families.
Keywords: genetic testing, diagnostic yields, primary immunodeficiencies and immune dysregulatory diseases
Disclosure: All authors are employed by Invitae.
(102) Diagnostic efficacy of next-generation sequencing panel testing in the diagnosis of inherited bone marrow failure syndromes
Alicia Scocchia, MS, LCGC1, Päivi Kokkonen, PhD2, Elina Hirvonen, PhD2, Emma Mårtensson, PhD2, Hatice Duzkale, MD, MPH, PhD, FACMG, CGMB3, Kim Gall, MSc, CGC4, Joe Jacher, MS, CGC5, Inka Saarinen, MSc6, Johanna Sistonen, PhD7, Juha Koskenvuo, MD, PhD8, Lotta Koskinen, PhD9, Tero-Pekka Alastalo, MD, PhD10
1Clinical Liaison, Licensed and Certified Genetic Counselor/Blueprint Genetics
2Geneticist/Blueprint Genetics
3Senior Geneticist/Blueprint Genetics
4Clinical Liaison, Certified Genetic Counselor/Blueprint Genetics
5Certified Genetic Counselor and Marketing Manager/Blueprint Genetics
6Data Scientist/Blueprint Genetics
7Clinical Development Manager/Blueprint Genetics
8Executive Director, Medical & Lab Director/Blueprint Genetics
9Clinical Interpretation Manager/Blueprint Genetics
10Executive Director, Medical/Blueprint Genetics
Inherited bone marrow failure syndromes (IBMFS), recently classified as inborn errors of immunity, represent a genetically heterogenous group of conditions characterized by aplastic anemia, congenital malformations, and increased risk to develop malignancies. Identifying the molecular etiology of IBMFS can allow for personalized management, surveillance, and risk estimation for the patient and their family members. Comprehensive next-generation sequencing (NGS) panel testing can be a useful molecular diagnostic tool, where broad inclusion of genes associated with IBMFS along with robust analysis of both multigenic and intragenic copy number variation (CNV) are expected to significantly contribute to diagnostic yield. To determine the diagnostic efficacy of a broad panel test including robust CNV analysis, we conducted a retrospective review of test results from patients with suspected IBMFS.
We reviewed clinical reports from consecutive patients with an indication of suspected IBMFS who underwent panel testing at Blueprint Genetics (a CLIA-certified diagnostic laboratory). The Bone Marrow Failure Syndrome panel was utilized for testing, which contains 135 genes and includes sequence variant, CNV, and targeted noncoding variant analysis. CNV analysis was performed bioinformatically from NGS data using two variant calling algorithms, including a proprietary method specific for small, intragenic, exon-level CNVs. Variant interpretation was performed according to ACMG guidelines. A molecular diagnosis was defined as identification of a pathogenic or likely pathogenic variant consistent with the patient’s reported phenotype and with known associated disease inheritance.
The median age of patients at testing was 14 years (ranging from fetal to 90 years). A molecular diagnosis was established in 82 of 442 tested patients (18.6%). CNVs ranged from 241 bp to 2.2 Mb and contributed to the diagnosis of 17.1% (14/82) patients; 64.3% (9/14) of these CNVs were intragenic. Diagnostic variants were identified in 34 genes, with nearly half of these genes (15, 44.1%) identified in a single patient in this cohort. The most commonly reported genes in diagnostic reports include FANCA (n=17, 20.7%) associated with Fanconi anemia and SBDS (n=12, 14.6%) associated with Shwachman-Diamond syndrome.
This study demonstrates that next-generation sequencing panel testing that includes robust CNV detection can contribute to the diagnostic yield among patients with IBMFS.
Keywords: Bone marrow failure, Next-generation sequencing, Genetic testing, Molecular diagnostics, Copy number variant
Disclosure: All authors are employeed by Blueprint Genetics.
(103) Trends in pediatric primary immunodeficiency: incidence, healthcare utilization, hematopoietic stem cell transplantation, and mortality
Taylor Eddens, MD/PhD1, Molly Mack, MD2, Meghan McCormick, MD3, Ramasubramanian Kalpatthi, MD4
1Allergy/Immunology Fellow/UPMC Children’s Hospital of Pittsburgh
2Pediatrics Resident/UPMC Children’s Hospital of Pittsburgh
3Assistant Professor/Pediatric Hematology/Oncology, UPMC Children's Hospital of Pittsburgh
4Associate Professor/UPMC Children’s Hospital of Pittsburgh, Hematology/Oncology
The term ‘primary immunodeficiency’ (PID) encompasses a myriad of diseases caused by inherited defects within the immune system. As the number of identified genetic defects associated with PID increases, understanding the incidence and outcomes of patients with PID becomes imperative.
The objective is to characterize the frequency of new diagnoses (i.e. incidence), patterns of healthcare utilization, rates of hematopoietic stem cell transplantation, and mortality over time in pediatric patients with PID.
A retrospective cohort analysis of pediatric inpatients from 2004-2018 with an ICD9/ICD10 diagnosis code associated with primary immunodeficiency using the Pediatric Health Information System (PHIS) database. Causes of secondary immunodeficiency were excluded. Incidence was calculated using first-time hospitalization.
There were a total of 17,234 patients hospitalized with a primary immunodeficiency from 2004-2018. There were 2.8 new PID diagnoses and 6.3 PID hospitalizations per 1,000 discharges; these metrics were unchanged during the study period. The number of new diagnoses for B cell and antibody defects significantly increased over time. The number of new PID diagnoses significantly increased in adolescents/adults and decreased in infants. T cell disorders had the highest number of ICU admissions. There were 747 patients that received hematopoietic stem cell transplantation (HSCT); complications of HSCT significantly decreased over time. Mortality significantly decreased in all PID patients and in the subgroup of patients receiving HSCT.
The total hospitalization rate and incidence of PID within the hospitalized pediatric population was stable. There were significant changes in the class of PID diagnosed, the age at diagnosis, and healthcare utilization metrics. Mortality significantly decreased over time within the PID cohort.
Keywords: Primary immunodeficiency, Hematopoietic stem cell transplantation, Pediatrics, incidence, Mortality
Disclosure: All authors indicated they had no financial relationships to disclose.
(105) Endocrine-Specific Autoimmunity in Primary Immunodeficiencies: a Report from the USIDNET Registry
Teresa Pelletier, MD1, Hannah Wright, MSPH2, Elizabeth Garabedian, MSN3, Ramsay Fuleihan, MD4, Elizabeth Feuille, MD5
1Resident/Weill Cornell Medicine- New York Presbyterian
2Research Data Analyst/The United States Immunodeficiency Network (USIDNET)
3Research Nurse/National Genome Research Institute
4Professor of Pediatrics/Division of Allergy and Immunology, Department of Pediatrics, Columbia University, New York, NY, USA
5Assistant Professor of Pediatrics/Weill Cornell Medicine
Primary Immunodeficiencies (PIDs) are typically thought of as presenting with susceptibility to infection, but may also manifest with other immune dysregulatory processes including autoimmunity which may be systemic or organ-specific. Subspecialists who evaluate these patients and may be the first point-of-contact with such patients may not be aware of the breadth and types of autoimmunity associated with PID. Here we report the numbers and types of endocrine-organ associated autoimmune conditions found in the USIDNET Registry, as well as the PIDs most often associated with endocrine-organ associated autoimmunity.
Investigators obtained clinical data for the patients within the USIDNET Registry with endocrine-organ autoimmunity. Frequency of endocrine-organ associated autoimmunity among various immune defects were reported. Of note, due to high probability of having iatrogenic cause, those with adrenal insufficiency that were not specifically reported as Addison’s Disease were excluded. In addition, patient’s with DiGeorge Syndrome and unspecific hypoparathyroidism were also excluded.
Among the 5486 patients in the USIDNET Registry with PIDs, 521 patients (9.5%) were found to have a condition associated with endocrine-organ autoimmunity. The most commonly reported endocrine-specific conditions among patients in the USIDNET Registry are thyroid specific, found in 356 patients (6.5% of registry patients); this is followed by pancreatic (N=105, 1.9%), parathyroid (N=82, 1.5%), ovarian (N=19, 0.3%, where 6 of these patients had undergone stem cell transplant), Addison’s (N=16, 0.3%), polyglandular (N=4, 0.1%), and testicular (N=1, 0.02%). The PID entities associated with more endocrine-organ associated autoimmunity were Mucocutaneous Candidiasis (affecting 60.9% of the 69 patients with Mucocutaneous Candidiasis) and Autoimmune Lymphoproliferative Syndrome (49.2% of 59, with majority affected by parathyroid disease), followed by Interferonopathy (23.1% of 13), Combined Immune Deficiency (14.2% of 106), and Common Variable Immune Deficiency (12.8% of 1820).
Endocrine-specific autoimmune conditions are common among patients with PIDs. Further study is needed to determine the impact of therapeutics on the development of endocrine-associated autoimmunity. This information may help guide clinicians to suspect PID in certain patients with endocrine-organ associated autoimmunity and/or to monitor for endocrine-organ autoimmunity in patients with certain PIDs.
Keywords: Primary Immunodeficiencies, Autoimmunity, Endocrine-Specific Autoimmunity
Disclosure: All authors indicated they had no financial relationships to disclose.
(106) Quality of Life in Adult and Pediatric Patients with Chronic Granulomatous Disease
Grace Gleason, BS, BA1, Samantha Kreuzburg, RN, BA2, Jennifer Treat, PA-C, MSHS, MPH3, Ericka Garrett, RN4, Dawn Shaw, MBA, MN, RN5, Betty Marciano, MD6, Christa Zerbe, MD7
1Post-Baccalaureate IRTA/National Institutes of Health
2Research Nurse Specialist/National Institutes of Health
3Physician Assistant/National Institutes of Health
4Nurse/National Institutes of Health
5Nurse Coordinator II/Leidos Biomedical Research, Inc.
6Staff Scientist/National Institutes of Health
7Director, Clinical Patient Services/National Institutes of Health
Chronic Granulomatous Disease (CGD) is an inherited primary immunodeficiency (PID) which results in both inflammatory response dysregulation and an increase in susceptibility to certain bacterial and fungal infections. Chronic disease can involve physical, psychological and social effects on a patient’s quality of life. In order to expand our understanding of CGD’s effects on quality of life, a six-month study of CGD patients began October 2019. The study was concluded in March 2020 due to concerns over the potential significant effect of the COVID-19 pandemic on patients’ quality of life.
All patients (inpatient/outpatient) were consented and administered either the WHO QOL-BREF instrument (26 years and older) or the PEDS-QL instrument (2-25 years old) enrolled on NIH protocol 93-I-0119 (NCT00404560). The WHO QOL-BREF is a validated questionnaire comprised of 26 items, encompassing: physical health, psychological health, social relationships and environment. The PEDS-QL is a validated questionnaire comprised of 23 items encompassing: physical, emotional, social, and school functioning. The PEDS-QL includes a module to assess the impact of CGD on the family for patients under 18. Each item in the WHO QOL-BREF and PEDS-QL is rated on a 5-point Likert scale. The surveys were interview-administered to 32 adult patients (16 males, 16 females) and 21 pediatric patients (17 males, 4 females) with genetically confirmed CGD. The age range was 4 - 62 years old (mean 31.4 years) with a distribution of 31% x-linked CGD, 22% CGD-carrier, and 47% autosomal recessive CGD among adult patients and a distribution of 76% x-linked CGD, 10% CGD-carrier, and 14% autosomal recessive CGD among pediatric patients. The WHO QOL-BREF domain scores were as follows: physical =14.14, psychological =14.38, social =16.63, environmental =16.69. The PEDS-QL domain scores were physical =74.63, emotional =67.32, social =78.93, school =38.22. Overall, adult domains showed an unexpected high score compared to the US general population; while pediatric domains showed a lower score, especially in school functioning. Perhaps being pulled out of school, frequent hospitalizations and illness impact the quality of life in pediatrics for this population more than it affects the quality of life in adults.
Keywords: Chronic Granulomatous Disease, Quality of Life, WHO QOL-BREF, PEDS-QL, Psychosocial, Inherited Primary Immunodeficiency
Keywords: Primary antibody deficiency, Screening algorithm, Diagnostic delay, Primary care
Disclosure: Mariannae A. Messelink, Roos-Marjin Berbers and Helen L. Leavis received research grants from Takeda. Joris van Montfrans is an advisory board member for Takeda. Giovanna Devercelli has ownership interest in Amgen and Takeda and is employed by Takeda. All other authors had no financial relationship to disclose.
(109) Efficacy and Safety of PlasmaCap IG, A New 10% Intravenous Immunoglobulin Manufactured Using an Innovative Chromatography Process, in Adults and Children with Primary Immunodeficiency Disorders
Richard Wasserman, MD, PhD1, Jennifer Leiding, MD2, William Lumry, MD3, Mark Scarupa, MD4, Daniel Suez, MD5, Sudhir Gupta, MD, PhD6, John Routes, MD7, Tina Zecca, MD8
1Medical Director, Pediatric Allergy / Immunology/Medical City Children's Hospital
2Associate Professor/Division of Allergy and Immunology, Department of Pediatrics, University of South Florida at Johns Hopkins-All Children’s Hospital, St Petersburg, FL
3Clinical Professor/University of Texas Southwestern Medical School
4Clinical Faculty/Johns Hopkins Asthma & Allergy Center
5Past President of the Consortium of Independent Immunology Clinics/Allergy, Asthma & Immunology Clinic PA
6Professor of Medicine, Pathology & Laboratory Medicine, and Microbiology & Molecular Genetics/University of California, Irvine
7Professor of Pediatrics, Medicine, Microbiology and Immunology/Medical College of Wisconsin
8Clinical Director/Allergy & Asthma Associates of Monmouth County
Primary immunodeficiency disorder (PIDD) patients require life-long immunoglobulin (IG) replacement therapy to minimize recurrent infections, especially serious bacterial infections (SBIs). Ongoing shortages of IG have exposed the need for increased manufacturing efficiency to meet growing global demand. PlasmaCap IG, a new 10% intravenous immunoglobulin (IGIV), is prepared from > 1000 donor plasma pools through an innovative chromatography process that can enable higher yields of certain plasma proteins, including IG, compared to traditional cold ethanol plasma fractionation. A clinical study evaluated the safety and efficacy of PlasmaCap IG in PIDD patients.
The primary efficacy outcome was the number of SBIs. Secondary endpoints included (among others) tolerability and safety, number of days missed at work or school, number of days hospitalized due to infections or with fever, and days of antibiotic therapy.
A Phase 3, prospective, open-label, multicenter study was conducted at 12 sites in the United States and Canada. Participants received 300-900 mg/kg of PlasmaCap IG either every 21 or 28 days for a period of one year.
A total of 57 of 63 participants (90.5%) completed the study and received 839 infusions. Six adults discontinued prior to study completion due to withdrawal of informed consent (4 subjects [8.3%]) and adverse events (2 subjects [4.3%]). Adults, age 20-70 years (median 51.0 years) and children age 2-16 years (median 9.0 years) were enrolled. No SBIs occurred in either group. Quality of life measures comprising days absent from work or school due to an infection (mean 6.5 days/patient/year), hospitalization due to infection (mean 0.2 days/patient/year), or febrile days > 38.5ºC, (mean 0.9 days/patient/year), were all low. Antibiotic use was also low. The most common treatment-related adverse events were headache, fatigue, and nausea; > 97% of all treatment-related adverse events reported were mild or moderate. No subject experienced a treatment-related serious adverse event (SAE), hemolysis, thromboembolism, or renal failure.
This study demonstrated that PlasmaCap IG is safe and effective in the treatment of adult and pediatric patients with PIDD.
Keywords: Intravenous Immunoglobulin, Primary Immunodeficiency Disorder, PlasmaCap IG, Chromatography, IGIV, PIDD
Disclosure: Richard Wasserman received research grants from CSL Behring, Evolve Biologics, Grifols, Kedrion, Korean Green Cross, and Takeda. Jennifer Leiding received speaker honoraria from CSL Behring; research grant and speaker honoraria from Horizon Therapeutics; and is an advisory board member for Pharming Therapeutics. William Lumry received a research grant from ADMA, ALK, BioCryst, Grifols, Ionis, Kedrion, Shire/Takeda, and Therapure; speaker honoraria from Astra Zeneca, CSL Behring, Grifols, GSK, Pharming, Sanofi/Regeneron; and is a consultant for Biomarin, Fresenius Kabi, Intellia, Kalyista, Phavarus and US Hereditary Angioedema Assocation. Mark Scarupa received a research grant from ALK, AstraZeneca, BioCryst, CSL Behring, Genentech, Grifols, Novartis, Octapharma, Pfizer, Regeneron and Takeda. John Routes has contracted research with CSL Behring and Evolve Biologics. All other authors had no financial relationship to disclose.
(110) Cross-sectional, caregiver-assessed burden of congenital athymia in patients without treatment in the first years of life
Cathleen Collins, MD, PhD1, Julie Kim-Chang, MD2, Bhagyashree Oak, PhD3, Matthew O'Hara, MBA4, Sarah Kulke, MD5, Elena Hsieh, MD6
1Assistant Professor/Department of Pediatrics, Division of Allergy Immunology, University of California San Diego; Rady Children’s Hospital, San Diego, CA
2Division of Allergy, Immunology, and Pulmonary Medicine, Department of Pediatrics, Duke University School of Medicine, Durham, NC
3Senior Consultant/Trinity Life Sciences, Waltham, MA
4Managing Director/Trinity Life Sciences, Waltham, MA
5Vice President, Medical Affairs/Enzyvant, Cambridge, MA
6Assistant Professor/Department of Pediatrics, Section of Allergy and Immunology, University of Colorado, Anschutz School of Medicine; Children’s Hospital Colorado; Department of Immunology and Microbiology, University of Colorado, Anschutz School of Medicine, Aurora, CO
Congenital athymia is an ultra-rare disorder characterized by the absence of a thymus in utero (naïve T-cell count < 100 cells/mm^3 at diagnosis); without treatment patients typically die within two to three years of life due to recurring infections and sequelae of immune dysregulation. Congenital athymia is associated with genetic/syndromic disorders, including DiGeorge Syndrome, 22q11.2 deletion, CHARGE syndrome, FOXN1 deficiency, diabetic embryopathy, and maternal alcohol exposure.
The objective was to characterize the economic, humanistic, and clinical burden of illness of congenital athymia on patients receiving supportive care prior to cultured thymic tissue implantation (CTTI; if received), and caregivers/families.
This cross-sectional study enrolled adult caregivers of patients with congenital athymia. Caregivers of patients who have not received a CTTI responded to questions about congenital athymia in the patient’s most recent 12 months (or patient’s lifetime, if < 12 months). Additionally, the caregiver proxy version of the PedsQL Generic instrument was administered. For caregivers of patients who had received CTTI at the time of participation, questions were asked to consider only the time prior to treatment.
Interim quantitative results from the survey (n=18) are reported here; insights from qualitative interviews will be reported in the poster. The sample included caregivers of seven patients who never received CTTI [mean age (SD): 1.7 (0.8) years] and eleven patients that received CTTI [8.8 (6.2) years]. The impact of congenital athymia on patients and caregivers was significant across many characteristics including the need to live in isolation (100% of respondents), financial hardship (78%), inability to meet family/friends (72%), high burden of medical care, and impact on siblings/families (67% each). Caregivers reported the impact on their own lives on a scale from 1-7, 7 being “very high impact” for a list of potential dimensions. Responses were rated highly, with all nearly > 6; “Ability to plan for the future” was reported with the highest average burden [mean: 6.9]. Caregiver-reported (n=7) PedsQL summary scores indicated low quality of life across domains.
Conclusion: Caregivers of patients with congenital athymia reported high emotional, social, financial, and clinical burden on patients and their families as a result of the disease.
Keywords: congenital athymia, burden of illness, real-world evidence, cultured thymic tissue implantation
Disclosure: Cathleen Collins, Bhagyashree Oak, Matthew O’Hara and Elena Hsieh are consultants for Enzyvant. Sarah Kulke is employed by Enzyvant.
(111) WES 102: Whole-exome sequencing analysis and diagnostic yield in 100 patients with recurrent infections and immune alterations
Saul Lugo Reyes, MD, MS1, Lina M Castano-Jaramillo, MD2, Luis Moises Silva Goytia, MD3, Edgar A Medina Torres, PhD4, Marco Antonio Yamazaki-Nakashimada, MD5, Aide Tamara Staines Boone, MD6, Selma C Scheffler Mendoza, MD, MSc.7, Ana Eunice Fregoso Zuñiga, MD8, Francisco Rivas Larrauri, MD9, Alonso Gutierrez Hernandez, MD9, Nideshda Ramirez Uribe, MD10, Juan Carlos Bustamante Ogando, MD, MSc11, Maria Edith Gonzalez Serrano, MD, MSc11, Laura Berron Ruiz, PhD11, Hector Gomez Tello, MD12, David Muzquiz Zermeño, MD13, Maria del Mar Saez de Ocariz Gutierrez, MD, MSc14, Melissa Espinosa Navarro, MD15, Leticia Hernandez Nieto, MD16, Rosa Maria Cortes Grimaldo, MD17, Ana Paola Macias Robles, MD18, Diana G Ramirez Vasquez, MD19, Kannelva M Gomez Castillo, MD20, Sandra Rajme Lopez, MD21, Joel Barroso Santos, MD22, Rosa Martha Covarrubias Carrillo, MD23, Aristoteles Alvarez Cardona, MD24, Beatriz A Llamas Guillen, MD25, Edna Venegas Montoya, MD26, Ana Isabel Jimenez Romero, MD27, Carmen Zarate Hernandez, MD28, Mario Ernesto Cruz Muñoz, PhD29, Chiharu Murata, MSc30, Sara Elva Espinosa Padilla, MD, PhD31
1Researcher/National Institute of Pediatrics
2Fellow in training/National Institute of Pediatrics
3General practitioner/Immune deficiencies lab, National Institute of Pediatrics
4Senior researcher/Immune deficiencies Lab, National Institute of Pediatrics
5Researcher/Clinical Immunology department, National Institute of Pediatrics
6Researcher/Hospital de Especialidades UMAE 25 IMSS Monterrey
7Attending/Clinical Immunology department, National Institute of Pediatrics, Mexico City
8Attending physician/Immunology department, Hospital Angeles Morelia, Michoacan
9Attending physician/Clinical Immunology Department, National Institute of Pediatrics
10Attending physician/Transplantation Department, National Institute of Pediatrics
11Researcher/Immune deficiencies lab, National Institute of Pediatrics
12Attending physician/Clinical Immunology Service, Hospital del Niño Poblano
13Attending physician/Clinical Immunology Department, Hospital de Especialidades, UMAE 25 IMSS Monterrey, NL
14Researcher/Dermatology Service, National Institute of Pediatrics
15Consulting physician/Immunology Department, Escuela Nacional de Ciencias Biologicas, Instituto Politecnico Nacional
16Attending physician/Allergy and Immunology, Secretaria de Salud, Hospital Juarez de Mexico
17Head/Clinical Immunology Service, Hospital de Especialidades, Centro Medico Nacional de Occidente, Guadalajara, Jalisco
18Head/Immunology Service, Hospital de Pediatria, Centro Medico Nacional de Occidente IMSS, Guadalajara, Jalisco
19Head of Pediatrics/Allergy and Immunology service, Hospital de la Niñez Oaxaqueña, Oaxaca
20Attending physician/Allergy and Immunology service, Hospital de Especialidades El Mirador
21Resident/Instituto Nacional de Nutricion y Ciencias Medicas
22Attending physician/Allergy and Immunology, Hospital del Niño DIF Pachuca, Hidalgo
23Physician/Immunology Department, Hospital General de Zacatecas
24Attending physician/Allergy and Immunology, Universidad Autonoma de Aguascalientes
25Attending/Immunology Department, Hospital del Niño Morelense
26Attending physician/Clinical Immunology Service, Hospital de Especialidades UMAE 25 IMSS Monterrey
27Head/Immunology Department, Hospital Angeles Leon
28Attending/Immunology Department, Hospital Universitario de Monterrey
29Head/Molecular Immunology Lab, Faculty of Medicine, Universidad Autonoma del Estado de Morelos
30Researcher/Research Methodology Department, National Institute of Pediatrics
31Head/Immune deficiencies lab, National Institute of Pediatrics, Mexico City
At a national referral center we discuss and pursue the molecular and genetic diagnoses of patients with suspected inborn errors of immunity (IEI) from all around the country. Since two years ago, we process and analyze our own whole-exome sequencing (WES) results at the lab. Assessing our diagnostic yield as a measure of performance and quality control might help optimize processes, improve patient selection and resource allocation.
We aimed to characterize our patients with suspected inborn errors of immunity and WES, to compare our diagnostic yield, and to identify attributes that might predict a positive diagnosis.
We extracted gDNA from whole blood of patients with suspected IEI in Mexico. Exome was sequenced in New Jersey using Illumina HiSeq with a 90% coverage and 50-100x raw depth of the IDT Xgen library, human genome version 38. We obtained two FASTQ files per patient to complete the bioinformatic workflow using Galaxy on the cloud for quality control, mapping & alignment of the reads, as well as variant detection, annotation and filtering; Ensembl Variant Effect Predictor to analyze said variants; Broad Institute IGV and UCSC Genome Browser to visualize them. We defined diagnostic yield as the proportion of patients with a genetic diagnosis after WES results analysis. Through multivariate logistic regression and tree partitioning algorithm we explored differences between diagnosed and undiagnosed cases.
We found a rare pathogenic variant in at least one gene known to cause IEI, and matching the patient phenotype in 42 of 100 exomes, and of other monogenic causes in 5, for a total yield of 47%. Statistical explorations suggested some predictingn features: family history, dysgammaglobulinemia, hemophagocytosis, etc.
We describe our experience analyzing our first 100 exomes at home. Our results compare well with those from other centers. A recent systematic review found 14 NGS studies for IEI, and for mixed groups like ours the dx yield was 15-46%, a median of 25%. Genetic diagnoses impact management, counseling, classification and epidemiology of rare diseases, including IEI. WES is a powerful and cost-effective diagnostic tool.
Keywords: whole-exome sequencing, inborn errors of immunity, diagnostic yield
Disclosure: All authors indicated they had no financial relationships to disclose.
(112) Chronic Granulomatous Disease with Associated IgG4-Related Disease Masquerading as Pulmonary Pseudotumor
Mahnaz Jamee, MD1, Mazdak Fallahi, MD2, Javad Enayat, MD2, Zahra Chavoshzadeh, MD3
1Medical Doctor, Research Assistant/Pediatric Nephrology Research Center, Research Institute for Children's Health, Shahid Beheshti University of Medical Sciences, Tehran, Iran
2Medical Doctor, Fellowship/Immunology and Allergy Department, Mofid Children's Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran
3Medical Doctor, Professor/Immunology and Allergy Department, Mofid Children's Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran
The association between IgG4-related disease (IgG4RD) and inborn errors of immunity (IEI) is barely understood. Herein, we presented the first report of pediatric IgG4RD and chronic granulomatous disease (CGD).
An 8.5-year-old male presented with complaints of dyspnea. He was the third child of non-consanguineous parents and the family history was unremarkable.
He was hospitalized two years ago with exertional dyspnea, mild cough, chest pain, and nocturnal sweating and was found to have a tumor-like mass in the right lung. In the chest MRI obtained at the last admission, the mass had a diameter of 52*15*61 millimeters with invasion to atria, completely obstructing right upper lobe bronchus and both right pulmonary veins. The histopathologic findings were consistent with non-necrotizing granulomatous inflammation, central neutrophilic micro-abscess, and extensive peripheral fibrosis without any evidence for acid-fast bacilli or fungal elements.
Treatment with prednisolone resulted in considerable symptom resolution. After 15 months, following the discontinuation of prednisolone by the patient, symptoms recurred, gradually exacerbated, and he developed anorexia and weight loss.
On the physical examination, respiratory distress, absent sound on auscultation and dullness on percussion of the right lung were detected. The chest spiral CT scan showed a large mass in the right lung, right lung collapse, and mediastinal metastasis. The abdominal ultrasound and CT scan were normal.
In laboratory evaluation, the complete blood count, leukocyte differentiation, lactate dehydrogenase, and liver function tests were normal, however, inflammatory markers (ESR: 112, CRP:108) were high. In immunologic workup, lymphocyte subsets were normal and serum immunoglobulin levels included IgG: 2530, IgM: 178, IgA: 151, IgG1: 1885, IgG2: 561, IgG3: 32, and IgG4: 311 (normal range: 2.3-189). The NBT test was zero in two consecutive evaluations. In virtue of high IgG4 level, the immunostaining of lung specimen was performed which was inconclusive for IgG4+ cells, and staining for CD138 was not available.
He was eventually diagnosed with concurrent CGD and IgG4RD, but progressed to respiratory failure and died despite the reinstitution of steroid therapy.
Further studies to investigate IgG subsets among IEI patients with pulmonary manifestations can help elucidate clinicopathological correlations between IgG4RD and IEIs.
Keywords: Inborn Error of Immunity, IgG4-related Disease, Chronic Granulomatous Disease, Pulmonary manifestations
Disclosure: All authors indicated they had no financial relationships to disclose.
(113) An easily available score to detect patients at risk of death after COVID19 infection
Javier Carbone, MD, PhD1, Jimena Gomez-Perez, MD2, Elizabeth Sarmiento, MD, PhD3
1Consultant Clinical Immunologist/Clinical Immunology Department. Hospital General Universitario Gregorio Marañón
2Immunology registrar/Clinical Immunology Department. Hospital General Universitario Gregorio Marañon
3Consultant Clinical Immunologist/Laboratory Medicine Department. Hospital Universitario La Paz
Background. COVID19 severe disease is associated with high mortality. There are no widely accepted proved therapeutic interventions that decrease mortality rates. So, all the efforts to detect patients at higher risk of developing severe disease are of utmost importance because this could aid to select patients for intervention. Taking into account that COVID19 is a pandemic infection biomarkers should be easily available all around the world including developing countries.
Objectives. We aimed to evaluate an easily available laboratory profile of progression to death at time of admission for COVID19 pneumonia that could be easily introduced into clinical practice.
Methods. The proposed basic profile to assess the risk of progression to COVID19 related death was the assessment at hospital admission of neutrophil count, lymphocyte count, total proteins and albumin concentration. 100 adult COVID19 patients with positive PCR to SARS-CoV-2 were prospectively evaluated at the time of admission at the Gregorio Marañón Hospital in Madrid, Spain, during the first wave (March-May 2020). During follow-up 14 patients (14%) died after admission.
Results. Patients who died were found to have higher levels of neutrophils and neutrophil to lymphocyte ratio as compared with survivors (6469±4522 vs 4302±2965 cells/ul, p=0.026 and 8.29±6.41 vs 5.00±4.34, p=0.020, respectively). Using ROC curves we identified cut-offs. Patients with neutrophil to lymphocyte ratio >4 and albumin < 3.3 g/dL were at higher risk of death (odds ratio, OR 4.16; 95%CI 1.06-16.27, p=0.04 and OR 7.49, 95%CI 1.22-45.94, p=0.029, respectively). Patients with both factors were at even higher risk of death (OR 12.99, 2.06-81.90, p=0.0063). Calculated globulin (difference between total protein and albumin results) at admission < 1.9 g/dL was not associated with risk of death.
Conclusions. An easily available laboratory profile (neutrophil to lymphocyte ratio >4 and albumin < 3.3 g/dL) was useful to assess the risk of death in COIVID19 patients. Multivariate analysis in greater number of patients is the next stept to further evaluate the independence of this potential predictor of death in hospitalized COVID19 patients. This profile has real potential to be introduced in the routine for better selection of patients for early therapeutical interventions.
Keywords: COVID19, Risk factors of death, Prediction
Disclosure: All authors indicated they had no financial relationships to disclose.
(114) High Dose Intravenous Immunoglobulin. The next step in therapy for severe COVID19 infection?
Maria Alejandra Mejia, MD1, Marisa di Natale, MD2, Hector Balastegui, MD3, Jimena Gomez-Perez, MD1, Elena Seoane, MD4, Javier Carbone, MD, PhD2
1Immunology Registrar/Clinical Immunology Department. Hospital General Universitario Gregorio Marañon
2Consultant Clinical Immunologist/Clinical Immunology Department. Hospital General Universitario Gregorio Marañón
3Immunology Registrar/Clinical Immunology Department
4Consultant Immunologist/Allegology Department, Hospital General Universitario Gregorio Marañon
COVID-19 disease is associated with high mortality among high-risk patients. Up today the only widely accepted interventions for hospitalized patients are enoxaparin and dexamethasone. All new information in this field is important. Distintc immunebased therapies are under investigation in clinical trials. We report a case serie of 5 patients admitted in our hospital with COVID-19 pneumonia who were treated with high-dose intravenous immunoglobulin (IVIG). Severely ill COVID-19 patients were treated with high-dose IVIG. Results. Patient 1 with previous history of lymphoma, secondary IgG hypogammaglobulinemia and persistence of SARS-Cov2 viremia (more than 3 months), received high-dose IVIG, 0.5 g/kg for 5 days (total 150 grams). Previous therapy included corticosteroids, tocilizumab, anakinra, lopinavir/ritonavir, remdesivir and 2 sesions of convalescent plasma. Low and transitory maintenance of SARS-Cov2 IgG antibodies was observed after plasma infusions. Patient 2 was treated with 0.4 g/kg for 3 days (total 150 grams). Obesity (120 kg), hypertension and diabetes were risk factors. Previous therapy included lopinavir/ritonavir. Due to severe bacterial infection corticosteroids and tocilizumab were not added as therapy. Patient 3 with severe pneumonia received therapy with remdesivir. High dose IVIG was added at a dose of 2 g/kg. Patient 4 was a CVID patient that demostrated bad radiological evolution soon after admission in hospital and received therapy with 1 g/kg of IVIG. Patient 5 was a 17 years-old women with previous history of myeloid leukemia. Previous therapy included tocilizumab, remdesivir and convalescent plasma. High dose IVIG was added at a dose of 2 g/kg (1gr/kg during 2 days). Biomarkers associated with bad prognosis (including high IL6) were observed in all cases. High-dose IVIG prevented deterioration of clinical symptoms and progression to mechanical ventilation in 4 cases and coincided with extubation a few days after addition of IVIG in patient 5. After high-dose IVIG was completed 4 patients were discharged from the hospital with a stable clinical condition in a few days and 1 patient was discharged from ICU. Infusions were tolerated well. DVT was observed in patient 2. Conclusion. High-dose IVIG can improve clinical condition and prevent progression to ICU usage in COVID-19 patients with severe symptoms.
Keywords: COVID19, Therapy, High dose intravenous immunoglobulin
Disclosure: All authors indicated they had no financial relationships to disclose.
(115) A Self-Limited Qualitative Complement Deficiency in Infancy
Ian Slack, MD1, Stephanie Ward, MD2, Kimberly Risma, MD, PhD3
1Clinical Fellow/Division of Allergy and Immunology, Cincinnati Children's Hospital Medical Center; Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH
2Assistant Professor/Division of Allergy and Immunology, Cincinnati Children's Hospital Medical Center; Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH
3Associate Professor/Division of Allergy and Immunology, Cincinnati Children's Hospital Medical Center; Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH
Complement deficiencies are a rare cause of innate immune defects with variable phenotypes depending on the particular component deficiency. While these deficiencies may be inherited or acquired, patients presenting in young childhood typically have a genetic etiology. Here we present a case of a young infant with initial near-absent complement activity who recovered function after 6 months with no underlying genetic difference identified.
A previously healthy 3-month old boy was hospitalized for prolonged fever and respiratory symptoms and diagnosed with necrotizing pneumonia. He responded to antibiotic therapy and surgical drainage. Immunology was consulted due to the severity of infection. His workup revealed normal lymphocyte subsets apart from expansion of terminally differentiated CD8 T cells suggesting recent systemic viral infection; HIV testing was negative. Additionally, he demonstrated normal neutrophil oxidative burst and normal IgM/IgA levels. However, his CH50 returned decreased at 5 CH50 units (normal range 101-300 units). On recheck one month later, his CH50 remained depressed at 2 CH50 units. Alternative pathway functional assay and lectin pathway functional assay at that time showed undetectable activity ( < 10% and < 4%, respectively). Based on these results, a defect in C3 or terminal complement was suspected (figure 1). Complement component levels did not identify quantitative complement defects. Workup for qualitative complement defects included normal C3 and C5 functional assays, as well as a normal next-generation sequencing panel encompassing 80 genes and C5 Sanger sequencing. At 9 months of age (6 months after his initial complement screen), both CH50 and AH50 recovered to normal levels. The patient was placed on amoxicillin prophylaxis upon initial diagnosis of complement deficiency and has not had additional serious infections.
This case highlights an unusual recovery of complement function in infancy. Although the inciting etiology for decreased complement activity was not identified in our patient, his course suggests that some infants without identified genetic differences in the complement system may regain function.

Figure 1. The complement cascade can be triggered by 1 of 3 pathways: lectin, classical, or alternative. These pathways converge with the assembly of a C3 convertase and ultimately the formation of the membrane attack complex. Complement deficiencies can be interrogated by measuring function of the 3 distinct pathways. The patient had deficient function in all 3 pathways, suggesting a C3 or terminal complement deficiency. Complement cascade schematic adapted from: Murphy, Kenneth, and Casey Weaver. Janeway's Immunobiology. Garland science, 2016
Keywords: Innate Immunity, Complement Deficiency, Complement System
Disclosure: All authors indicated they had no financial relationships to disclose.
(116) Missing in Plain Sight: Chromosomal Microdeletion and GATA2 Deficiency
Catherine Freeman, MD1, Cindy Bauer, MD2, Holly Miller, DO3, Benjamin Wright, MD4
1Allergy and Immunology Fellow/Mayo Clinic Arizona
2Section Chief, Allergy and Immunology/Phoenix Children's Hospital
3Divison of Hematology and Oncology/Phoenix Children's Hospital
4Division of Allergy, Asthma and Clinical Immunology/Mayo Clinic Arizona
The relationship between chromosomal deletion and immunodeficiency is best established in 22q11.2 deletion syndrome. Widespread availability of genetic testing has resulted in a greatly increased number of recognized chromosomal deletion and microdeletion syndromes. Deletions, especially on the X chromosome, have been ascribed to immunodeficiency states including X-linked chronic granulomatous disease, Wiskott-Aldrich syndrome and X-linked SCID. However, the association between chromosomal deletions and immunodeficiency remains incompletely characterized.
A six-year-old girl was evaluated for recurrent sinopulmonary and urinary tract infections. Medical history was notable for developmental delay with a de novo 3q13.33-q21.3 deletion and vesicoureteral reflux. Surgical history was remarkable for adenotonsillectomy age three. Physical examination demonstrated dysmorphic facies, microcephaly and low set ears. Laboratory evaluation revealed B and T-cell lymphopenia (CD4+ cells 351/mm3, CD8+ cells 312/mm3, CD19+ cells 247/mm3). Absolute monocyte count was normal. IgG was low for age at 381mg/dL but with normal IgM and IgA. Oxidative burst, total complement and vaccine titers were protective. The patient underwent ureteral reimplantation in conjunction with trimethoprim/sulfamethoxazole prophylaxis and was asymptomatic thereafter. Following discontinuation of antibiotic prophylaxis, her course was complicated by viral gastroenteritis and recurrent pneumonia. Genetic sequencing revealed a heterogeneous pathogenic mutation in GATA2, with the entire coding sequence missing. Bone marrow biopsy showed no evidence of dysplasia and computerized tomography of the chest was without findings suggestive of bronchiectasis or pulmonary alveolar proteinosis. The patient is currently managed on immunoglobulin replacement therapy and daily azithromycin prophylaxis, with improvement in infection frequency and severity.
GATA2 is a zinc finger transcription factor essential for hematopoiesis and lymphatic angiogenesis. GATA2 deficiency is caused by mutations in the GATA2 gene and follows an autosomal dominant pattern of inheritance. Patients are susceptible to infection (mycobacterial, viral and fungal), marrow failure and myeloid leukemias, lymphedema and pulmonary alveolar proteinosis. Disease onset, clinical features and outcomes are heterogenous. The GATA2 gene is located at 3q21.3 and has not previously been reported in microdeletion syndromes affecting chromosome 3. Meticulous review of the genes encompassed in a chromosomal microdeletion is critical in immunodeficiency evaluation as it can allow for earlier detection of primary immunodeficiency.
Keywords: GATA2 Deficiency, chromosomal deletions, primary immunodeficiency, microdeletions, gene deletions
Disclosure: All authors indicated they had no financial relationships to disclose.
(117) PATH4WARD: A Genetic Testing Program for Primary Immunodeficiencies With Neutropenia Including WHIM Syndrome
Britt Johnson, PhD, FACMG, HCLD(ABB)1, James Connelly, MD2, Sarah Cohen, MD3, Lukas Dillinger, PhD4, Adriana Badarau, MSc, PhD5, Swaroop Aradhya, Ph.D., FACMG6, Ana Morales, MS, CGC7, Peter Newburger, MD8
1Medical Director, Metabolic Genetics and Immunology,Medical Affairs Director, Biopharma Partnerships/Invitae Corporation, San Francisco, CA
2Assistant Professor Pediatric Hem/Onc/BMT/Vanderbilt University Medical Center, Nashville, TN
3Medical Director, Rare Disease/X4 Pharmaceuticals Inc, Boston, MA
4Director, Translational Sciences & Preclinical QA/X4 Pharmaceuticals Inc, Boston, MA
5Discovery Director/X4 Pharmaceuticals Inc, Boston, MA
6Medical Director/Invitae, San Francisco, CA
7Medical Affairs Liaison/Invitae Corporation, San Francisco, CA
8Physician/University of Massachusetts Medical School, Worcester, MA
PATH4WARD is a no-charge, sponsored genetic testing program utilizing a next-generation targeted sequencing panel to provide early and accurate diagnosis for patients with suspected congenital neutropenia (CN) including WHIM (Warts, Hypogammaglobulinemia, Infections, Myelokathexis) syndrome, a condition with a therapeutic option currently under investigation in clinical trials. Patients with WHIM present with heterogeneous symptoms that include neutropenia, increased susceptibility to infections, and persistent warts. Early diagnosis of CN is key to reduce progression of long-term morbidities and improve quality of life. We report methods and demographics for initial utilization of the PATH4WARD program from July 2019–September 2020.
At the time of data acquisition, PATH4WARD utilized a 23-gene CN panel, with reflex to a 207-gene primary immunodeficiency (PID) panel. Patients with a clinical presentation compatible with congenital or chronic idiopathic neutropenia and history of chronic severe neutropenia (absolute neutrophil count [ANC] ≤500/μL) were eligible. Analysis was performed at Invitae using multiple algorithms to identify single nucleotide variants, small/large indels, structural variants, and exon-level copy number variants. Sequencing was performed at an average depth of 350X (minimum 50X). Variants were classified per Sherloc, Invitae’s score-based refinement of recent guidelines.[1]
PATH4WARD was utilized by 156 unique physicians, most frequently by specialists in pediatric hematology/oncology (52.5%), followed by adult hematology (14.6%), clinical genetics (12.0%), other pediatric (9.5%), allergy/immunology (8.2%), and medical oncology (3.2%). The median age of patients tested was 5 years (range, 0–81).
The PATH4WARD program, sponsored by X4 Pharmaceuticals and Invitae, is a valuable tool for facilitating early genetic evaluation of patients with suspected PIDs with neutropenia, including WHIM. Pediatric hematology/oncology physicians most commonly referred patients, and a pediatric focus was highlighted by the age those enrolled. Owing to the success of PATH4WARD and the greater opportunity to help additional patients with CN, inclusion criteria were broadened in September 2020 to include patients with ANC ≤750 cells/μL, and the panel was extended, without need for reflex, to analyze 407 genes associated with PIDs including many associated with CN or neutropenia acquired in the context of an inborn error of immunity.[2]
References:
1. Nykamp K et al. Genet Med. 2017;19(10):1105-1117.
2. PATH4WARD Program. https://www.invitae.com/en/path4ward/
Keywords: WHIM Syndrome, Primary Immunodeficiency, PATH4WARD
Disclosure: Britt Johnson, Swaroop Aradhava, and Ana Morales is an employee of Invitate. Sarah Cohen received financial and material support from Clinical and Laboratory Standards institute and is employed by X4 Pharmaceuticals. Lukas Dillinger is employed by X4 Pharmaceuticals and UCB Pharma GmbH. Adriana Badarau is employed by X4 Pharmaceuticals. All authors indicated they had no financial relationships to disclose. Peter Newburger is an advisory board member of X4 Pharmaceuticals.
(118) Immune Dysregulation With Mucocutaneous Ulceration in a Large Cohort Due to A Novel Pathogenic RELA Variant.
Kelsey Lecerf, MD1, Hyesun Kuehn, PhD2, Jennifer Yonkof, MD3, Daniel Koboldt, MS4, Theresa Mihalic Mosher, MS, LGC5, Mari Mori, MD6, Scott Hickey, MD7, Shoghik Akoghlanian, MD8, Vidya Sivaraman, MD8, Sergio Rosenzweig, MD, PhD9, Roshini Abraham, PhD10
1Allergy and Immunology Fellow/The Ohio State University Wexner Medical Center/Nationwide Children's Hospital, Columbus, OH
2Staff Scientist/Immunology Service, Department of Laboratory Medicine, NIH Clinical Service, Bethesda, MD, USA
3Allergy and Immunology Physician/ProMedica Physicians Allergy and Immunology, Toledo, OH
4Principal Investigator, Institute for Genomic Medicine/Nationwide Children's Hospital, Columbus, OH
5Licensed Genetic Counselor/Institute for Genomic Medicine, Nationwide Children's Hospital, Columbus, OH
6Clinical and Biochemical Geneticist, Division of Genetic & Genomic Medicine/Nationwide Children's Hospital, Columbus, OH
7Clinical Geneticist, Division of Genetic & Genomic Medicine/Nationwide Children's Hospital, Columbus, OH
8Pediatric Rheumatologist/Nationwide Children's Hospital, Columbus, OH
9Senior Investigator/Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
10Director of the Diagnostic Immunology Laboratory/Department of Pathology and Laboratory Medicine, Nationwide Children’s Hospital, Columbus, OH
NFκB includes a family of inducible transcription factors important for immune regulation. One of the key components of the canonical pathway of NFκB is p65 (RELA gene). Here, we report four patients, from a large family kindred, with chronic mucocutaneous disease with a novel frameshift variant in RELA (c.1044dupC, p.Tyr349LeufsTer13).
The objective was to assess the functional impact of this RELA variant on the canonical NFκB pathway.
Immunoblotting for the canonical NFκB pathway components was performed after LPS stimulation of cells PBMCs from four affected individuals and healthy controls. Immunophenotyping of T and B cell subsets was performed.
WES on P3 and P4, which revealed the previously mentioned novel variant, which segregated with disease. This RELA variant results in premature truncation of the protein before the serine (S) 536 residue, which is a key phosphorylation site, which could result in enhanced degradation of the protein. Immunoblotting revealed significantly decreased phosphorylated (p) p65 with increased levels of native IKBα and decreased pERK. The levels of pIKKα and pIKKβ (and native IKKα and IKKβ), along with pIKBα were comparable to controls. The decrease in phosphorylated p65 in the patients suggests reduced heterodimer formation between p50/p65 [NFκB1/RelA]. Extensive T cell immunophenotyping revealed decreased CD4+ and CD8+ CD45RA+ T cells with increased memory CD4+ T cells in one of the 4 patients (P1). This patient also had increased CD4+25+ activated T cells, and senescent CD57+CD4+ and CD57+CD8+ T cells. B cell phenotyping was unremarkable. Cellular apoptosis and death was evaluated at baseline, 4 hours and 24 hours after stimulation with 100 ng LPS and showed no evidence of increased apoptosis or death in lymphocytes in any of the patients after stimulation.
Family members with this novel RELA variant have a clinical phenotype similar to other reported RELA cases with predominant chronic mucocutaneous ulceration, however, the clinical phenotype is expanded to include Behçet’s disease and inflammatory bowel disease (IBD, Crohn’s). There is impairment of the canonical pathway of NFκB. These findings will improve our understanding of the pathogenesis of early-onset or familial Behçet’s, and IBD, and may direct therapeutic/management decisions for these patients.

Figure 1. RELA Variant Pedigree

Table 1. Demographics and Clinical Features of Affected Patients
Keywords: Immune Dysregulation, Primary immunodeficiency, RELA, NFkB, Mucocutaneous ulceration
Disclosure: All authors indicated they had no financial relationships to disclose.
(119) A novel pathogenic variant in FANCA uncovers a previously undiagnosed case of Fanconi Anemia (FA) in a 22-year-old woman
Hannie Zomer, MD1, Arye Rubinstein, MD, PhD2, Rachel Eisenberg, MD3, Robert Marion, MD4, Monisha Sebastin, MS, CGC5
1Fellow/Montefiore Medical Center
2Chief of the Pediatrics Division of the Department of Allergy & Immunology/Montefiore Medical Center
3Attending in Allergy and Immunology/Montefiore Medical Center
4Chief of the Divisions of Genetics and of Development Medicine/Montefiore Medical Center
5Certified Genetic Counselor/Montefiore Medical Center
FA is an autosomal recessively inherited condition characterized by physical features, bone marrow failure and increased risk of malignancy. We present a 22-year-old female with multiple medical issues in whom a Primary Immunodeficiency (PID) panel led to an unexpected diagnosis of FA.
TC was referred because of recurrent upper respiratory infections with fevers, abnormal liver enzymes since childhood, and chronic fatigue. Past history revealed megaloblastic anemia non-responsive to vitamin B12 and growth failure unresponsive to growth hormone. Immune evaluation revealed normal serum immunoglobulins, protective titers to measles, mumps, rubella, and absent response to streptococcus pneumoniae (14/14 titers were < 0.5mcg/mL). Pan-activation was noted on a cytokine panel along with elevated double positive T-cells of unknown significance (23% CD3+CD4+CD8+ T – cells). A PID panel (Invitae) demonstrated a heterozygous deletion of one FANCA allele and two variants (c.3539T>A and c.4199G>A) noted by the lab to be of unknown significance in the second allele. Based on in-silico analysis, the c.4199G>A variant was suspected to likely be pathogenic. Diepoxybutane assay demonstrated chromosome breakage in TC that was 3-10 times greater than control, confirming the diagnosis of FA. Further proband and maternal genetic studies, including microarray, revealed maternal inheritance of a 16q24.3 deletion encompassing FANCA. Further studies are needed to confirm that the second variant (c.4199G>A) is paternally inherited. Bone marrow biopsy was within normal limits. A multi-gene molecular profiling assay covering genes implicated in the pathogenesis of solid and/or hematological malignancies was within normal limits. The patient is currently undergoing evaluation for allogenic-hematopoietic stem cell transplantation.
We describe a 22-year-old woman with FA in whom a variant in FANCA (c.4199g>A) previously described as a VUS was determined to be pathogenic. In retrospect, TC had multiple findings seen in FA, including macrocytosis, growth failure, horseshoe kidney, multiple café-au-lait macules, and abnormal left thumb. Individuals with FA often exhibit immune activation early in life leading to neoplastic disease. In TC, diagnosis was delayed until the third decade because of the mild nature of her symptoms. We suspect this is because the c.4199G>A variant produced partial protein function. We are closely following her course.
Keywords: Fanconi Anemia, Immune activation, Pathogenic variant
Disclosure: All authors indicated they had no financial relationships to disclose.
(120) Early T-cell Development in Twins with Heterozygous FOXN1 Mutations
Saara Kaviany, DO1, Todd Bartkowiak, PhD2, Kelsey Voss, PhD3, Yasmin Khan, MD4, Daniel Dulek, MD5, Sarah Neumann, RN6, Jeffrey Rathmell, PhD7, Jonathan Irish, PhD8, James Connelly, MD9
1Pediatric Hematology/Oncology/BMT Instructor/Vanderbilt University Medical Center
2Research Fellow, Department of CDB/Vanderbilt University
3Research Fellow, Department of PMI/Vanderbilt University
4Assistant Professor Pediatrics AI/Vanderbilt University Medical Center
5Assistant Professor Pediatric ID/Vanderbilt University Medical Center
6Pediatric BMT Case Manager/Vanderbilt University Medical Center
7Associate Director - Vanderbilt Institute for Infection, Immunology and Inflammation, Professor PMI/Vanderbilt University
8Associate Professor of Cell and Developmental Biology/Vanderbilt University
9Assistant Professor Pediatric Hem/Onc/BMT/Vanderbilt University Medical Center, Nashville, TN
The transcription factor FOXN1 plays an essential role in thymic epithelial development, mediating selection of maturing thymocytes1. Heterozygous loss-of-function FOXN1 variants are associated with low T-cell receptor excision circles (TRECs) and T-cell lymphopenia at birth1. CD4+ T-cell reconstitution is not completely understood, but a lower proportion of naïve T-cells in adult patients has suggested a role for homeostatic proliferation1. We present fraternal twins with low TRECs at birth, found to have a pathogenic heterozygous variant in FOXN1. Using 40+ plex single-cell mass cytometry (CyTOF) we performed extensive immune-profiling to understand T-cell development in newborn patients with lymphopenia. Increased understanding of the immune milieu will guide potential need for therapeutics, and risk for infection or autoimmunity over time.
The twins presented at birth with severely low TRECs by newborn screen and T-cell lymphopenia with quantitatively normal B- and NK- cells.
Genetics- Targeted primary immunodeficiency testing revealed two heterozygous variants of unknown significance: in DOCK8 (C.277G>T, p.Val93Leu), and FOXN1 (c.1205del, p.Pro402Leufs*148). The FOXN1 variant found shares a similar frameshift mutation to reported patients2, affecting the transactivation domain necessary for increasing DNA affinity in a DNA binding assay3.
Immunophenotype- The patients were enrolled in the Human Immune Disease Initiative (HIDI) at Vanderbilt University, combining multidimensional cytometry and genomics to gain deeper insight into inborn errors of immunity. Key findings (depicted in Figure 1) include an expansion of terminally differentiated CD4+ TEMRA cells and contraction of the CD8+ naïve (CD45RA+ CCR7+) T-cells in peripheral blood of the patients with FOXN1 mutations relative to healthy controls. Both patients had increased CD38 and ICOS expression, suggesting a T-cell activation state.
Through deeper immunophenotyping of this variant, our findings suggest that defective FOXN1 expression may impact peripheral T-cell differentiation, potentially through impaired thymic selection and subsequent early peripheral expansion and maturation in a lymphopenic environment. We present HIDI as a platform capable of high-dimensional immune phenotyping to provide further understanding of pediatric inborn errors of immunity. Our findings support a future focus exploring T-cell development and the role of CD4+ TEMRA cells in patients with severe lymphopenia, such as the FOXN1 heterozygous variants.


Figure 1: Deep phenotyping of CD45+ CD3+ T-cells from FOXN1 heterozygous twins and a healthy donor using mass cytometry and t-SNE: t-SNE plots from mass cytometric data demonstrating greater frequencies of CD4+ TEMRA cells (red) and fewer CD8+ naïve T cells (green) observed in the T cell compartment of the FOXN1 twins than in the healthy reference.
Keywords: lymphopenia, mass cytometry, immunophenotyping, FOXN1, CD4+ TEMRA
Disclosure: Jeffrey Rathmell received research grants from Incyte, Kadmon, and Tempest; advisory board member of Caribou Biosciences, Nirogy, Sitryx; and is a consultant for Mitobridge. All other authors had no financial relationship to disclose.
(121) The lockdown in 2020 has opened a new path in the genetic diagnosis of patients with hereditary angioedema.
Irina Guryanova, Phd student1, Ekaterina Polyakova, PhD student2, Iryna Ausianik3, Andrei Salivonchik4, Valeria Pugacheva1, Yulia Zharankova, Phd student5, Mikhail Belevtsev, PhD6
1Bench scientist/Belarusian research center for pediatric oncology, hematology and immunology
2PhD student,Research in the field of genetics of PIDS, determination of TREC /KREC/Belarusian Research Center for Pediatric Oncology, Hematology and Immunology,clinical research laboratory of the scientific department
3President of HAE Belarus/Belarussian National Public Organization "HAE Patients Care"
4Physician/Republican Scientific and Practical Center of Radiation Medicine and Human Ecology
5Physician/Belarusian research center for pediatric oncology, hematology and immunology
6PhD/Belarusian Research Center for Pediatric Oncology, Hematology and Immunology, Deputy Director for Research
Hereditary angioedema (HAE) is a rare, but potentially life-threatening genetic disorder, characterized by acute, recurring, and self-limiting edematous episodes of the face, extremities, trunk, genitals, upper airways, or the gastrointestinal tract. HAE due to C1-inhibitor deficiency (Type I) or dysfunction (Type II) occurs in about 1 in 10,000 to 1 in 50,000 people without racial or gender differences.
In 2020 due to the COVID-19, we started using the Dry Blood Spot (DBS) method for the genetic diagnostic for 33 blood relatives of previously diagnosed HAE patients (8 were symptomatic and 25 were asymptomatic), who were not able to come to the Center. Patients sent their DBS using postal envelopes. Genetic analysis was performed by Sanger sequencing.
In all symptomatic patients, we identified family mutations. 20 from 25 asymptomatic blood relatives were wild type for the family mutations and 5 patients were diagnosed before their first attacks. Thus, there are now 85 genetically confirmed HAE patients in Belarus (50 females): 64 are type I (75.3%) and 21 - type II (24.7%). Based on these results, the estimated minimal prevalence of HAE in Belarus is 1: 110 700 (0.90 in 100 000).
Since HAE attacks can start without any reason or can be caused by the trigger (stress, trauma, etc.) patients are afraid of moving within the country for diagnostic purposes. Thanks to the DBS method, we diagnosed 13 patients, some of whom could not come to the Belarusian research center for pediatric oncology, hematology and immunology for more than 5 years. We are confident that the DBS method has received high evaluation and response among HAE patients, primarily due to its simplicity and convenience.
Keywords: Hereditary angioedema (HAE), c1 inhibitor deficiency, genetic diagnosis, Dry Blood Spot, Belarus
Disclosure: All authors indicated they had no financial relationships to disclose.
(122) A novel semi-quantitative ELISA for measuring circulating SARS-CoV-2 neutralizing antibody levels in human plasma or serum that correlates with a plaque reduction cell-based neutralization assay
Andrew Gibson1, Wm Pat Leinert2, Dima Decker, PhD3, Gene Wetzstein, PharmD, BCOP4, James Mond, MD/PhD5
1Manager Assay Development/Leinco Technologies, Inc
2President and CEO/Leinco Technologies, Inc
3Associate Director of Medical Affairs/ADMA Biologics
4Executive Director & Head of Scientific Engagement/ADMA Biologics
5Chief Scientific Officer/Chief Medical Officer/ADMA Biologics
The protective quality of convalescent plasma and vaccine efficacy is reflected in a large part by the level of neutralizing antibodies (NAbs). The gold standard for detecting NAbs against viruses is the plaque reduction cell-based neutralization assay (PRNT) that requires specialized facilities, is costly and takes several days. To overcome these challenges, we have developed a rapid, semi-quantitative ELISA that measures COVID-19 NAbs, correlates closely with the live virus plaque reduction assay and can be used for high throughput screening of samples.
Results obtained from the ELISA were compared to the results from PRNT for positive percent agreement (PPA) and negative percent agreement (NPA) with 115 samples (95% confidence interval [CI] was calculated using the Wilson Method). Cross-reactivity to antibodies against 11 viruses including the common coronaviruses NL63, 2293, OC43 and HKU1 was analyzed with this ELISA.
When comparing the ELISA and PRNT PPA for the 115 samples, there was a very close correlation between the extent of neutralization in these two assays. ELISA specificity was demonstrated, as there was no cross-reactivity with the 55 potentially cross reactive samples tested. In addition, of the 531 negative plasma samples collected from healthy donors prior to the COVID-19 outbreak, 527 samples were negative for NAbs resulting in 99.3% specificity. Furthermore, when screening 100 convalescent plasma donor samples, only 61% of the samples contained COVID-19 NAbs. Eighty percent of these exhibited low to moderate neutralization and only 20% contained high neutralization activity.
We have developed a rapid high throughput ELISA assay that measures COVID-19 NAbs and has demonstrated a significant correlation to PRNT. This assay can be used to: 1) screen donor plasma units for NAbs prior to their inclusion in the manufacture of IG to achieve a plasma pool with the highest level of NAbs, 2) test vaccinated subjects to ensure that they have mounted a protective NAb titer to COVID-19 and 3) continue to monitor the vaccinated population for sustained levels of protective NAbs.
Keywords: ELISA, COVID-19, SARS-CoV-2, convalescent plasma
Disclosure: Andrew Gibson and Wm Pat Leinert are employed by Leinco Technologies, Inc. Dima Decker, Gene Wetzstein and James Mond are employed by ADMA Biologics.
(123) A Case of Zhu‐Tokita‐Takenouchi‐Kim (ZTTK) Syndrome in a Child With Significant Hypogammaglobulinemia and Poor Vaccine Response
Lauren Gunderman, MD1, Aisha Ahmed, MD2, Amer Khojah, MD3
1Allergy and Immunology Fellow/Northwestern McGaw and Ann and Robert H Lurie's Hospital
2Attending Physician, Allergy and Immunology/Ann and Robert H Lurie Children's Hospital of Chicago
3Attending Physician, Allergy, Immunology, and Rheumatology/Ann & Robert H. Lurie Children's Hospital of Chicago/ Northwestern University Feinberg School of Medicine
Zhu‐Tokita‐Takenouchi‐Kim (ZTTK) Syndrome is a rare, congenital anomaly syndrome caused by de novo variants in the SON gene located on 22q22.11. Given the heterogeneity of variants, there is no clinical diagnostic criteria, but patients often have intellectual disability, brain malformation, facial dysmorphia, musculoskeletal abnormalities, and visceral malformations. We present a case of ZTTK Syndrome with significant hypogammaglobulinemia and need for immunoglobulin replacement.
A 2-year-old girl with known ZTTK syndrome, diagnosed on whole exome sequencing (WES), complicated by subglottic stenosis, hypothyroidism, hypoglycemia, developmental delay, dysphagia with GJ tube dependence, and left ventricular dysfunction, presented to the emergency department (ED) in respiratory failure. The patient was intubated with concern for aspiration and found to be rhinovirus/enterovirus positive. She was admitted to the pediatric intensive care unit (PICU), and required prolonged intubation for five days. Immunology was consulted given a past medical history significant for 4 episodes of acute otitis media, multiple viral infections (rhinovirus/enterovirus, norovirus and rotavirus), hypovolemic shock secondary to E. faecalis urosepsis and recurrent fevers without a clear infectious trigger. Pertinent Immunology labs included a severely low IgG of 150 mg/dL, low IgM of 29 mg/dl, normal IgA and poor antibody response to both Streptococcus pneumonaie(only 1 out 23 serotype was >1.3) and Tetanus ( < 0.1IU/ml). T and B cell lymphocyte enumeration was within normal limits. In the setting of hypogammaglobulinemia, concern for poor vaccine responses and recurrent infections, the child received Intravenous Immunoglobulin (IVIG) with an appropriate increase of her IgG. The patient was discharged home with plans for continued IVIG, unfortunately she continued to have ED visits and admissions for emesis and died a month later after an episode of emesis, choking and exhaustion of Cardiopulmonary Resuscitation (CPR).
ZTTK Syndrome is a rare congenital anomaly syndrome characterized by multiple congenital anomalies, including facial dysmorphia, musculoskeletal abnormalities and visceral abnormalities. Our patient’s presentation was further complicated by having a humoral immunodeficiency and the need for replacement Immunoglobulin. In their approach to children with ZTTK Syndrome and recurrent infections, clinicians should consider investigating for immunodeficiency.
Keywords: Zhu‐Tokita‐Takenouchi‐Kim (ZTTK), hypogammaglobulinemia, CVID
Disclosure: All authors indicated they had no financial relationships to disclose.
(124) The interrelationship of CVID gastrointestinal manifestations, autoimmunity, splenomegaly and lymphoproliferative disease.
Hannah Wright, MSPH1, Ramsay Fuleihan, MD2, Charlotte Cunningham Rundles, MD3, Kathleen Sullivan, MD, PhD4, Edith Schussler, MD5
1Research Data Analyst/The United States Immunodeficiency Network (USIDNET)
2Professor of Pediatrics/Division of Allergy and Immunology, Department of Pediatrics, Columbia University, New York, NY, USA
3David S. Gottesman Professor of Immunology/Mount Sinai School of Medicine
4Professor of Pediatrics, Chief of the Division of Allergy and Immunology/The Children’s Hospital of Philadelphia, Philadelphia, PA, USA
5Assistant Professor/Weill Cornell Medicine
The GI tract is the largest lymphoid organ and gastrointestinal (GI) complaints are frequent in CVID, leading in some to increased morbidity and higher mortality. Previous studies have shown associations between autoimmunity(AI), lymphoproliferation (LP), Splenomegaly(SM) and CVID enteropathy, however results have been varied. We suggest this variation may be due to the variable definitions of enteropathy used in these evaluations. We investigated the relationship of GI status as a whole as well as the most common GI complaints to AI, LP and SM in CVID patients in the USIDNET registry.
Data on 939 patients with a diagnosis of CVID in the United States Immunodeficiency Network (USIDNET) Patient Registry were analyzed. GI+ was defined as any condition affecting the GI tract from mouth to anus excluding the spleen. Evaluation of the most common GI complaints was used to further delineate which manifestations are most associated with AI and LP, and SM.
Results: CVID GI disease showed significant associations with autoimmunity (OR 2.04, 95% CI:1.53-2.71, p < 0.001), lymphoproliferation (OR 2.44, 95% CI:1.67-3.54, p < 0.0001) and splenomegaly (OR 2.26, 95% CI:1.441-3.528, p < 0.0004). When looking at the most common GI manifestations the only significant association was between aphthous ulcers and AI (p=0.03), although chronic diarrhea approached statistical significance (p=0.06). Chronic diarrhea was significantly associated with lymphoproliferation (OR 1.55, 95% CI:1.03-2.34, p=0.04), and splenomegaly (OR 1.94, 95% CI:1.22-3.09, p=0.005), while infectious diarrhea was not. Abdominal pain was significantly associated with lymphoproliferation (OR 1.7, 95% CI:1.15-2.5, p=0.007), but not lymphadenopathy or splenomegaly. Non-IBD colitis was significantly associated with lymphoproliferation (OR 1.86, 95% CI:1.20-2.90, p=0.006), lymphadenopathy (OR 2.01, 95% CI:1.22-3.29, p=0.006), but not splenomegaly. Infectious colitis was not associated with LP, LA or SM.
Conclusions: GI disease is common in CVID affecting 65% of patients in our cohort. GI+ CVID patients have a higher frequency of AI manifestations, LP and SM than those without GI complaints. We found that specific GI complaints were very specifically associated with autoimmunity, lymphoproliferation and/or splenomegaly. These results suggest that specific GI symptoms/manifestations rather than groupings may be useful in the evaluation of CVID.
Keywords: CVID, Gastrointestinal, Autoimmune, Lymphoproliferative, Splenomegaly
Disclosure: Kathleen Sullivan is a consultant for the Immune Deficiency Foundation. Edith Schussler is an advisory board member for Horizon Therapeutics. All other authors had no financial relationship to disclose.
(125) Chronic Epstein-Barr virus infection and atypical sarcoidosis vs. granulocytic/lymphocytic interstitial lung disease in a child hemizygous for a pathogenic variant on SKIV2L
Arthur Chang, M.D.1, Danielle Goetz, M.D.2, Teresa Hennon, M.D.3, Rafal Kozielski, M.D.4, Gail Deutsch, M.D.5, Karl O.A. Yu, M.D., Ph.D., F.A.A.P.3
1Fellow/University at Buffalo / Oishei Children's Hospital
2Clinical Associate Professor of Pediatrics/University at Buffalo / Oishei Children's Hospital
3Clinical Assistant Professor of Pediatrics/University at Buffalo / Oishei Children's Hospital
4Clinical Assistant Professor of Pathology/University at Buffalo
5Professor of Pathology/University of Washington / Seattle Children's Hospital
A 9-year-old female with asthma and allergic rhinitis was admitted to the intensive care unit for 3 weeks of cough, intermittent fever and malaise. Her exam was remarkable for diffuse lymphadenopathy, hepatosplenomegaly and clubbing. She was hypoxic when febrile, intermittently requiring oxygen by nasal cannula. Symptoms slowly improved without targeted therapy. After discharge, she had slow improvement in lymphadenopathy and splenomegaly, but continued to have dyspnea on exertion and mild intermittent cough. Approximately 4 months after discharge, she developed fever, dyspnea and cough. This responded well to corticosteroid therapy.
Laboratories showed pancytopenia, mildly elevated inflammatory markers, and Epstein Barr viremia. Imaging showed bilateral pulmonary infiltrates, diffuse adenopathy and hepatosplenomegaly. On biopsy, bone marrow was mildly hypocellular with intact trilineage hematopoiesis. An inguinal lymph node had non-necrotizing epithelioid granulomas. A lung needle biopsy showed lymphocytic and lymphohistiocytic infiltrates with few multinucleated giant cells but no well-formed granulomas. A pathologic diagnosis of granulocytic/lymphocytic interstitial lung disease (GLILD) versus a clinical diagnosis of atypical sarcoidosis are considered.
The patient’s viremia is persistent (now > 6 months), with negative IgM but positive IgG against viral capsid antigen, and seropositivity against early and nuclear antigens. This suggests an aberration on the cellular immunologic capacity for herpesvirus control. She had intact antibody responses to common vaccine antigens. Flow cytometry showed an exaggerated CD4:CD8 ratio and a mild B lymphocytopenia.
Targeted panel sequencing uncovered a heterozygous pathogenic mutation in SKIV2L, p.Arg374*. SKIV2L encodes a helicase component of the RNA exosome complex, and is itself a negative regulator of Rig-1 like receptors and their associated antiviral/interferon response. Homozygous deficiency of SKIV2L causes trichohepatoenteric syndrome (syndromic diarrhea, Stankler syndrome), a rare condition characterized by intractable diarrhea in infancy, syndromic facies, and skin, hair, liver, intellectual and immunologic abnormalities.
Unpublished anecdotal reports on individuals hemizygous for SKIV2L pathogenic variants have identified them as either having trichohepatoenteric syndrome (presumed unidentified second defect) or being phenotypically normal (these patients’ parents). As GLILD is well-described in several primary immunodeficiencies, this report suggests that study is needed to determine if there may be a haploinsufficiency phenotype for this genetic defect.
Keywords: interstitial lung disease, Epstein Barr infection, RNA exosome
Disclosure: All authors indicated they had no financial relationships to disclose.
(126) Newly diagnosed chronic granulomatous disease in a 52 year-old male presenting with fulminant pulmonary aspergillosis
Louis Marois, MD, PhD1, David Drouin, MD2, Isabel Fernandes, PhD3, Charles Leduc, MD4, Hélène Manganas, MD4, Hugo Chapdelaine, MD4, Emilia Liana Falcone, MD, PhD4, Guilhem Cros, MD4
1Postgraduate Resident Year 6/Montreal University
2Postgraduate Resident Year 3/Montreal University
3Professor/Immunology-Rheumatology Division, Department of Pediatrics, University of Montreal
4Professor/Montreal University
Background: Chronic granulomatous disease (CGD) is caused by inherited NADPH (nicotinamide adenine dinucleotide phosphate) oxidase 2 defects resulting in severely reduced phagocyte-derived superoxide production that manifest clinically as recurrent infections and autoinflammation. Although the majority of patients with CGD are diagnosed before 10 years of age, only a handful of patients with autosomal recessive CGD have been diagnosed after the fourth decade of life. We present a 52 year-old male diagnosed with autosomal recessive (p47phox-deficient) CGD after an episode of fulminant pulmonary aspergillosis (a.k.a. mulch pneumonitis).
Case presentation: 52-year-old male excavator with no significant past medical history was admitted with fever, nausea, and headaches. Blood tests revealed a c-reactive protein (CRP) of 238 mg/L and a WBC count at 13 x109/L, while cerebral spinal fluid (CSF) analyses were within normal range. A chest X-Ray showed small multicentric consolidation and empirical treatment for community acquired pneumonia was begun along with corticosteroids. Despite initial improvement, the patient's respiratory condition rapidly deteriorated along with an increase in inflammatory parameters (CRP 299 mg/L, WBC 24 x109/L) leading to his transfer to the intensive care unit on day 3 of hospitalization. A lung biopsy revealed an extensive granulomatous inflammation, along with a massive presence of Aspergillus spp, confirmed by culture.. He was empirically started on voriconazole and caspofungin. Despite antifungal treatment, the patient developed refractory acute respiratory distress syndrome requiring extracorporeal membrane oxygenation (ECMO). A dihydrorhodamine 123 (DHR) assay was performed and demonstrated a marked reduction in neutrophil superoxide production. The patient was promptly started on anakinra (IL-1 receptor antagonist). However, the patient succumbed within 28 days of hospitalization. Genetic analysis showed a homozygous NCF1 (p47phox) deletion, confirming the diagnosis of CGD in this previously asymptomatic man.
Conclusion: Autosomal recessive CGD can present beyond the fourth decade of life with fulminant pulmonary aspergillosis in a previously completely asymptomatic individual. The severity and rapidly progressive clinical course in this seemingly healthy older individual demonstrates the importance of maintaining a high index of suspicion for primary immune defects in adults with atypical clinical presentation or evolution of infections.
Keywords: pulmonary aspergillosis, chronic granulomatosis disease, late-onset primary immunodeficiency
Disclosure: All authors indicated they had no financial relationships to disclose.
(127) Opportunistic Infections in non-HIV associated CD4+ T-cell Lymphopenia
Sara Barmettler, MD1, Nancy Yang, BS2, Jocelyn Farmer, MD/PhD1, Jolan Walter, MD/PhD3, Carlos Camargo, MD/DrPH1, Mei-Sing Ong, PhD4
1Physician/Massachusetts General Hospital
2Clinical Research Coordinator/Massachusetts General Hospital
3Department of Pediatrics/University of South Florida at Johns Hopkins All Children's Hospita
4Assistant Professor/Harvard Medical School and Harvard Pilgrim Health Care Institute
The most common clinical manifestations of CD4+ T-cell lymphopenia are opportunistic infections (OIs). Published literature on human immunodeficiency virus (HIV) shows that OIs are associated with significant morbidity and mortality, but data are lacking from patients with non-HIV associated CD4+ lymphopenia. Clinicians currently extrapolate from HIV guidelines to manage patients with non-HIV associated CD4+ lymphopenia.
We performed a retrospective review of patients with CD4+ lymphopenia
in our healthcare system. We evaluated demographics, severity of CD4+ lymphopenia, and incidence of OIs. CD4+ lymphopenia was defined by an absolute CD4+ T-cell count of < 300 cells/microL or < 20% of total T cells on more than one occasion, without evidence of HIV infection. We further stratified CD4+ lymphopenia as mild (CD4+ T-cells 201 to < 300 cells/microL), moderate (CD4+ T-cells 101 to ≤200 cells/microL), severe (CD4+ T-cells 51 to ≤100 cells/microL), and very severe (CD4+ T-cells ≤50 cells/microL). OIs of interest included pneumocystis jirovecii, toxoplasma gondii, mycobacterium avium complex, cryptococcus neoformans, coccidioidomycosis and histoplasmosis. We examined the association between severity of lymphopenia and risk of OI, and used logistic regression to identify risk factors for OIs.
We identified 366 patients with non-HIV associated CD4+ lymphopenia. The mean age was 44 years; there were 256 (70%) men. 38 (10%) patients had mild CD4+ lymphopenia, 22 (6%) moderate, 7 (2%) severe, and 299 (82%) very severe lymphopenia. 55/366 (15%) patients had one or more OIs, including pneumocystis jirovecii (n=31), toxoplasma gondii (n=14), mycobacterial infection (n=10), cryptococcus neoformans (n=10), and disseminated mycobacterium avium-intracellulare complex (n=5). Almost all cases of OIs occurred in those with very severe disease (n=51/55=93%). Very severe CD4+ lymphopenia was associated with a higher risk of OIs (OR 1.14; 95%CI, 1.04-1.25). Age and sex were not associated with OIs. Most infections (n=44/55=80%) required inpatient treatment.
15% of patients with CD4+ lymphopenia developed OIs. Severity of CD4+ lymphopenia was associated with a higher risk of OIs. Future studies on non-HIV associated CD4+ lymphopenia are needed to better understand risk factors for OIs, as well as the safety and efficacy of antibiotic prophylaxis.
Keywords: T-cell lymphopenia, opportunistic infections, immune deficiency
Disclosure: Jocelyn Farmer received a research grant from Bristol Myers Squibb and X4 Pharmaceuticals. All other authors had no financial relationship to disclose.
(128) Immunomodulatory Therapy with Doxycycline and Hydroxychloroquine Allows Excisional Debridement of Cutaneous Granulomas in a Patient with Ataxia Telangiectasia
Luis Murguia-Favela, MD, FRCPC1, Michele Ramien, MDCM, MSc, FRCPC2, Alan Harrop, MD, MSc (Exp Surg), MSc (Epi), FRCSC, FACS3
1Clinical Assistant Professor, Pediatric Immunology, Section of Hematology/Immunology/Department of Pediatrics, Alberta Children's Hospital, University of Calgary
2Clinical Associate Professor, Pediatric and Adult Dermatology/Departments of Pediatrics and Medicine, Alberta Children's Hospital, University of Calgary
3Clinical Professor, Section Head of Plastic Surgery, Department of Surgery/Alberta Children's Hospital, University of Calgary
Rubella virus vaccine strain-induced cutaneous granulomas are now a well-recognized pathologic feature in patients with inborn errors of immunity, including ataxia telangiectasia and other combined immunodeficiencies. These lesions can become persistent with high risk of tissue loss and infections.
There are no specific therapies and topical and systemic corticosteroids, nitazoxanide, anti-tumor necrosis factor agents, and interferon-alfa have been used with inconsistent results. In addition, patients usually require multiple courses of antibiotics for bacterial superinfections.
Immunomodulatory properties of doxycycline have been studied. These include inhibition of matrix metalloproteinases, downregulation of pro-inflammatory cytokines, inhibition of leukocyte chemotaxis, and inhibition of granuloma formation. Likewise, hydroxychloroquine has been shown to have anti-granulomatous effects, namely reduced toll-like receptor signaling resulting in decreased activation of dendritic cells and production of pro-inflammatory cytokines.
We present a 10-year-old boy with Ataxia Telangiectasia with skin granulomas since he was 3 years old. Biopsy materials from the two main lesions allowed for RT-PCR detection and sequencing analysis confirming the presence of the Wistar RA27/3 vaccine strain rubella virus.
Over the years, these lesions had a pattern of acute ulceration during the winter and slow healing over 8-12 weeks. The granulomatous inflammation would persist and each year the ulcerations would be larger and extend deeper into the subcutaneous tissues. Many courses of oral and intravenous antibiotics for bacterial secondary infections were required. Failed therapies included topical steroids, tacrolimus, and many types of dressings. Access to nitazoxanide therapy was not possible.
During the most recent episode of ulceration, combination therapy with doxycycline (4mg/Kg/day divided twice daily) and hydroxychloroquine (4mg/Kg/day) was trialed. The ulcerations fully closed within 4 weeks, without visible residual inflammation and no side effects. Three months later, both residual lesions were excised, and the wounds healed completely.
The immunomodulatory effects of doxycycline and hydroxychloroquine, a combination therapy repurposed from treatment of inflammatory granulomas, contributed to a faster healing of ulcers in rubella vaccine-strain-induced cutaneous granulomas in a patient with ataxia telangiectasia. This allowed for a complete surgical excision and may prevent recurrences. More studies are needed to fully evaluate the efficacy of this therapeutic approach.
Keywords: immunomodulation, cutaneous granulomas, ataxia telangiectasia, doxycycline, hydroxychloroquine
Disclosure: Michele Ramien is an advisory board member for Abbvie and Eli Lilly, a consultant for Pfixer, and received a research grant from Sanofi-Genzyme. All other authors had no financial relationship to disclose.
(129) Enzymatic Activity of Neutrophil Elastase (NE) and Myeloperoxidase (MPO); Putting it into Clinical Practice
Devora Eisenberg, MS41, Ronit Elhasid, MD2, Silvia Baron, Phd3
1Fourth year medical student/Tel Aviv University- Sackler School of Medicine
2Director of Pediatric Hemato-oncology/Dana-Dwek Children’s Hospital
3Senior Scientist/Pediatric Hemato-Oncology Research Laboratory, Tel Aviv Sourasky Medical Center affiliated to the Sackler Faculty of Medicine
Introduction: Neutrophil extracellular trap formation (NETosis), a potent method for killing extracellular pathogens, plays an important role in many inflammatory diseases such as juvenile idiopathic arthritis (JIA) and even cancer. However, the evaluation of NETosis is based on microscopy, a labor-intensive and subjective technique. In the previous study of our group, the activity of key enzymes of NETs formation, neutrophil elastase (NE) and myeloperoxidase (MPO), showed linear correlation to NETosis(1). Therefore, we suggested that the easy and fast measurements of NE activity could serve as a surrogate marker for NETosis. The aim of this study was to determine the reference range of NE and MPO activity to enable clinicians to use the activity of these enzymes to easily estimate NETosis in different pathological conditions.
Methods: 93 healthy volunteers between the ages of 18-62 (median age: 30) were recruited. Neutrophils were isolated by immunomagnetic negative selection kit from StemCell Technologies. Subsequently, cells were lysed by Triton X-100 and then incubated with chromogenic peptide MeOSuc-Ala-Ala-Pro-Val-pNA for 90 min at 37 °C for NE activity or with o-phenylenediamine and hydrogen-peroxide for 20 min at room temperature for MPO activity measurements. Enzymatic activity was measured with an iMark Microplate Absorbance Reader at 415 nm or at 450 nm respectively for NE and MPO. Calibration curves were set up using varying amounts of purified NE and MPO respectively.
Results: Enzyme activity level for NE ranged between 1.7 and 10.0 mU (mean 4.5 ± 1.9; N=89) and for MPO ranged between 0.22 and 2.53 mU (mean 0.91 ± 0.46; N=93). Both NE and MPO enzymatic activity was mildly higher in females than males, but there was no significant difference for NE or MPO values respectively PNE=0.77 and PMPO=0.82.
Conclusions: In this study, as opposed to our previous study, we used immunomagnetic negative selection for isolation of neutrophils, which contains milder treatments causing less activation of the cells resulting in a more reliable reference range. Future studies will involve data collection from elderly (over 60 years old) and children (below 18 years old), as well as the application of this assay in disease states.
Keywords: Neutrophil, Neutrophil extracellular trap, Neutrophil elastase, Myeloperoxidase
Disclosure: All authors indicated they had no financial relationships to disclose.
(130 The associations of T cell subsets, serum cytokines (IL-23, IL-17, TNF-α) and IL-23R and IL-12B gene polymorphisms with disease activity in patients with psoriasis
Shirin Tarafder, MBBS, MPhil1, Sabia Shahin Sultana, MBBS, MD2, Ismet Nigar, MBBS, FCPS3, ATM Asaduzzaman, MBBS, FCPS4
1Professor/Microbiology and Immunology Department, Bangabandhu Sheikh Mujib Medical University, Dhaka, Bangladesh
2Lecturer/Microbiology Department, Shaheed Suhrawardy Medical College, Dhaka, Bangladesh
3Assistant Professor/Microbiology and Immunology Department, Bangabandhu Sheikh Mujib Medical University, Dhaka, Bangladesh
4Associate Professor/Dermatology and Venereology Department, Bangabandhu Sheikh Mujib Medical University, Dhaka, Bangladesh
Psoriasis is a genetically regulated, T lymphocyte mediated autoimmune skin disease. Monoclonal antibodies targeting p19 subunit inhibits IL-23, a “master regulator” of Th17 cell development, offer potential disease control for long term disease management.
Objective: This study aims to demonstrate the T cell subsets, cytokines (IL-17, IL-23 and TNF-α) and polymorphisms in IL-12B and IL-23R genes in psoriasis patients to investigate their possible relationship with susceptibility to psoriasis.
Blood samples from 35 clinically diagnosed psoriatic patients and 35 healthy controls analysed at our laboratory between March 2019 and February 2020. T cell subsets were detected by flow cytometric immunophenotyping using monoclonal antibodies, serum cytokines levels by ELISA and gene polymorphism by Real-Time PCR.
The mean percentage of Th1, Th17, T-reg and pT-reg cells were significantly more in psoriatics than in controls (P < 0.05). In contrast mean percentage of tT-reg cells were decreased significantly in psoriatics (P < 0.0001). A significant difference was seen between psoriatics and healthy controls, for IL-17 (150 Vs 84.10, P=0.002), IL-23 (231.70 Vs 13.97, P < 0.0001) and TNF-α (534.68. Vs 84.26, P=0.002). IL-17 and IL-23 level showed difference among groups according to disease severity (IL-17, P= 0.03; IL-23, P=0.42). The allelic frequency of the major variant C of IL23R rs11805303 was 27 (77.10%) in patients and 18 (51.4%) in controls (P=0.02). The frequency of homozygous genotype AA of IL23R rs2201841 was 45.70% and 22.90% in patients and healthy controls respectively (P= 0.043). Predominant presence of AA genotype of rs10889677 and TT genotype of rs11805303 of IL-23R were statistically significant, suggesting a risk of developing psoriasis with these genotypes. Of 35 psoriasis patients, 23 (65.71%) had AA genotype of IL23R rs10889677 gene, along with increased number of all T cell subsets and increase serum cytokine level which corresponds with disease severity (PASI>10) and duration (>10 years).
This is the first study in Bangladesh showing association of mean IL-23 level with AA genotype of IL23R rs10889677 gene polymorphism provides evidence that IL-23 pathway can be an appropriate target for intervention in psoriasis as higher concentration of IL-23 are associated with severity of disease and duration.
Keywords: Psoriasis, T cell subsets, cytokines, gene polymorphisms, IL-23, IL-23R, IL-12B
Disclosure: All authors indicated they had no financial relationships to disclose.
(131) New homozygous activating RAC2 mutation causing combined immune deficiency with pulmonary, skin and neoplastic manifestations
Louis Marois, MD, PhD1, Agnes Donkò, PhD2, Géraldine Gosse, MSc3, Amy P Hsu, MD, PhD4, Thomas L Leto, PhD5, Hugo Chapdelaine, MD6, Emilia Liana Falcone, MD, PhD6, Guilhem Cros, MD6
1Postgraduate Resident Year 6/Montreal University
2Post-doctoral Fellow/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD.
3Genetic consellor/Institut de recherches cliniques de Montréal
4Professor/3Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD.
5Senior Investigator/3Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD.
6Professor/Montreal University
Ras-related C3 botulinum toxin substrate 2 (RAC2) is a member of the rho family of GTPases that is strictly expressed in hematopoietic cells, and involved in superoxide (O-2) production and actin remodeling. Dominant negative RAC2 mutations result in phagocyte defects, whereas heterozygous dominant activating mutations manifest as combined immune deficiencies (CID). We present a 44-year old woman diagnosed with a novel homozygous activating mutation in RAC2, and functionally evaluate the mutation in vitro.
A 44-year-old woman was evaluated following the resection of an anal epidermoid cancer. The patient was from a consanguineous French-Canadian family and her past medical history was significant for recurrent pneumonias and severe asthma leading to bronchiectasis at 9 years of age, as well as recurrent tonsilitis resulting in tonsillectomy. As a teenager, she suffered from recurrent cold sores and persistent warts on her hands and genitals which have persisted into adulthood. Her immune tests were significant for moderate hypogammaglobulinemia and lymphopenia. She was treated with intravenous immunoglobulins for presumed common variable immunodeficiency (CVID). She underwent a hysterectomy at age 40 for high grade cervical and vaginal intraepithelial neoplasia, which was followed by the resection of a perianal epidermoid cancer at age 44. At this time, her lymphocyte phenotyping demonstrated moderate T and B lymphopenia with marked reduction in naive T lymphocytes (Table 1). A homozygous mutation in RAC2 (c.202C>T, p.R68W) was identified using a gene panel. The patient’s family history was only significant for recurrent pneumonias in her mother and HPV mediated oropharyngeal cancer in her father.
Stimulated HEK cells transfected with plasmids carrying the R68W mutation showed a greater than 3-fold increase in O-2 production compared to controls. Interestingly, COS-7 cells transfected with the mutant plasmid showed decreased (presumably unstable) RAC2 protein expression associated with increased phosphorylated AKT (pAKT, marker of RAC2 activity) by Western blot analysis (Figure 1).
We present a novel homozygous RAC2 gain-of-function mutation resulting in a relatively moderate phenotype and family history compared to reported patients with heterozygous RAC2 dominant activating mutations likely due to RAC2 protein instability associated with our patient’s mutation.
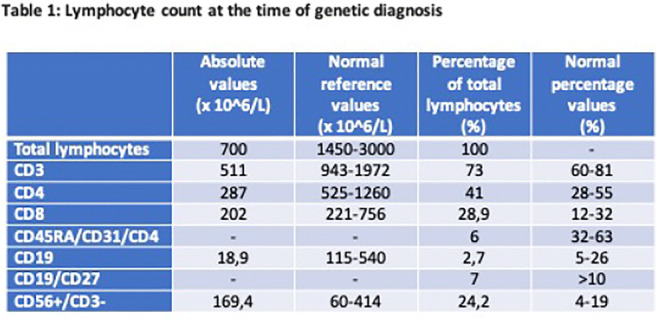

Keywords: RAC2, Reactive oxygen species, Combined immunodeficiency
Disclosure: All authors indicated they had no financial relationships to disclose.
(132) Novel coronavirus immunoglobulin G in a four-month-old infant
Melissa Mendoza Suyo, MD1, Yatyng Chang, MD2, Jose Calderon, MD3, Vivian Hernandez-Trujillo, MD3
1Pediatric Resident/Nicklaus Children's Hospital
2Allergy Immunology Fellow/Nicklaus Children's Hospital
3Allergy Immunology Attending/Nicklaus Children's Hospital
The COVID-19 pandemic has been a challenge for public health. There is scarce literature regarding the immune response to SARS-CoV-2 in the pediatric population. We present the case of a 4-month-old infant with positive IgG anti-SARS-CoV-2 antibodies.
4-month-old male with positive IgG anti-SARS-CoV-2 antibodies. When he was 2 months of age both parents were diagnosed with COVID-19, after they presented with anosmia and cough, and positive for SARS-CoV-2 PCR. He became symptomatic one week after his parents were diagnosed with COVID-19, with cough and rhinorrhea lasting for 2 weeks with an uncomplicated course. He was born full term via cesarean section. Mother was asymptomatic around the time of delivery. He has been breastfed since birth.
We present a 4-month-old infant with positive IgG anti-SARS-CoV-2 antibodies, who may be one of the youngest patients with positive antibodies reported in the literature. Although the positive antibodies could represent an adaptive immune response to SARS-CoV-2, the interpretation of this positive serology presents a diagnostic challenge for a variety of reasons. First, cross-reactivity between antibodies to SARS-CoV-2 and alpha (NL63 and 229E) and beta (OC43 and HKU1) coronaviruses has been reported in the literature. In fact, the adult population can have a prevalence of positive antibodies as high as 90% which might represent a high false positive rate. Second, there is the possibility of mother-to-fetus transplacental IgG passage of antibodies to common human coronaviruses. The duration of circulating maternal antibodies in the infant cannot be firmly established. Despite the lack of symptoms in his mother prior to delivery, there is a possibility of perinatal transmission of antibodies to SARS-CoV-2 or common human coronaviruses. Lastly, the role of breastfeeding should be considered. Animal studies have suggested the breast milk IgG can cross the gut barrier, although SARS-CoV-2 RNA has not been found in breast milk, and transmission through human breast milk has not been described.
In conclusion, this case describes a young infant with positive IgG anti-SARS-CoV-2 antibodies and the caveats to consider when interpreting this test in patients of this age group.
Keywords: Novel coronavirus, SARS-CoV-2 antibodies, Infancy
Disclosure: Vivian Hernandez-Trujillo is an advisory board member of Covis, CSL Behring, DBV, Kaleo, Takeda, and US WORLD MEDS. All other authors had no financial relationships to disclose.
(133) Novel STAT3-variant found in a family with presumed Hyper-IgE syndrome in four generations of women.
Camilla Drabe, Dr.1, Tania Masmas, MD, Phd2, Hanne Marquart, MD, Phd3, Jens Lundgren, MD, Phd4, Isabel Forss, Phd5, Line Borgwardt, MD, Phd6, Terese Katzenstein, MD, Phd, MSc7
1Clinical research assistant/Dept. of Infectious Diseases, Rigshospitalet, Copenhagen University hospital, Copenhagen, Denmark
2Consultant/Pediatric hematopoietic stem cell transplantation and immunodeficiency, The Child and Adolescent Clinic, Rigshospitalet, Copenhagen University Hospital
3Cunsultant/Dept. of Clinical Immunology, Rigshospitalet, Copenhagen University Hospital
4Professor, Consultant/Department of Infectious Diseases, PERSIMUNE, Centre of Excellence for Personalised Medicine of Infectious Complications in Immune Deficiency, Rigshospitalet, Copenhagen University Hospital, Denmark
5Molecular biologist/Center for Genomic Medicine, University Hospital of Copenhagen, Rigshospitalet, Denmark
6Consultant/Center for Genomic Medicine, University Hospital of Copenhagen, Rigshospitalet, Denmark
7Consultant/Department of Infectious Diseases, Rigshospitalet, Copenhagen University Hospital, Denmark
Variants in the signal transducer and activator of transcription 3 (STAT3) gene cause autosomal dominant hyper-IgE syndrome (AD-HIES) also known as Job’s syndrome. Here we present a family of four generations of women with a medical history compatible with AD-HIES, where we identified a novel variant in STAT3 in the two youngest generations.
A four-year-old girl was referred for evaluation of primary immunodeficiency due to recurring severe staphylococcus abscesses in the last two years. Her mother also suffered recurring abscesses whereas her grandmother had eczema, allergy, and lung-disease, and died at an early age. The great-grandmother had a similar phenotype and died young too. Reportedly the mother and grand-mother had been clinically diagnosed with Job’s syndrome elsewhere in the past, but for unknown reasons the mother was not followed for her immunodeficiency. No connective tissue, skeletal or dental abnormalities were found or reported. Both the girl and her mother initiated Co-Trimoxazole prophylaxis with a markedly reduction of abscesses.
The girl and her mother had significantly elevated levels of IgE. By whole-genome sequencing a heterozygote missense variant in the DNA-binding domain of STAT3 (c.1250G>C, p.(Arg417Thr)) was found in the child and mother. The variant was confirmed by Sanger sequence.
The identified STAT3 variant has not been previously reported in the literature in conjunction with AD-HIES or in population databases. It resides in the DNA-binding domain of STAT3, where other pathogenic missense variants have previously been reported in AD-HIES patients. The family history and phenotype are highly suggestive for AD-HIES. As the two oldest generations are deceased, it was not possible to further illuminate co-segregation in family members. The functional impact of the variant remains to be determined. We suspect that the identified variant is disease-causing, however according to the American College of Medical Genetics and Genomics (ACMG) classifications, we classify the variant as of uncertain significance (class 3).
Keywords: Hyper-IgE, HIES, Jobs syndrome, Immunodeficiency, STAT3, Whole Genome Sequencing, WGS, Genetics, Diagnostics
Disclosure: Camila Drabe received speaker honoraria from Takeda. Terese Katzenstein received speaker honoraria from CSL Behring, Takeda, Baxalta, Janssen, Shire; a research grant from Gilead; and is an advisory board member of ViiV/GSK, MSD, Chiesi. All other authors had no financial relationships to disclose.
(134) Analysis of soluble mediators of promotion and suppression of the immune response in patients with the 22q11.2 deletion syndrome
Diogo Soares, MD, PhD1, Chong Kim, MD, PhD2, Leuridan Torres, PhD3, Anelisa Dantas, PhD4, Natalia Nunes, MSc5, Maria Isabel Melaragno, PhD6, Magda Carneiro-Sampaio, MD, PhD7
1Researcher/Faculdade de Medicina da Universidade de São Paulo
2Associate Professor/Faculdade de Medicina da Universidade de São Paulo
3Head of Research Department/Instituto de Medicina Integral Prof. Fernando Figueira
4Researcher/Universidade Federal de São Paulo
5PhD Student/Universidade Federal de São Paulo
6Full Professor/Universidade Federal de São Paulo
7Full Professor/Faculdade de Medicina da Universidade de São Paulo
22q11.2 deletion syndrome (22q11.2DS) is the most common human microdeletion syndrome, with an estimated prevalence of 1:4000 live births. It has a wide phenotypic variability, with more than 180 different related manifestations. Among the clinical findings, immune abnormalities are present in approximately 75% of the cases and includes recurrent infections, atopy and/or autoimmune diseases, suggesting that it is a disease of dysregulation of the immune system. OBJECTIVE: to evaluate soluble mediators of promotion and suppression of the immune response in 22q11.2DS patients. Fifteen patients with a confirmed diagnosis of 22q11.2DS and 15 healthy patients without a family history of 22q11.2DS, major congenital malformations or a clinical history suggestive of immunodeficiencies were enrolled in the study. Through the sample of peripheral blood, analyzes of mediators related to immune regulation were performed, using techniques of Cytometric Bead Array (CBA) and enzyme-linked immunosorbent assay (ELISA). The study comprised a total of 15 22q11.2DS patients (15.5 years ± 8.4; 4 women and 11 men) and 15 healthy individuals (17.5 years ± 7.2; 7 men and 8 women). When comparing 22q11.2DS patients with healthy individuals, the patients presented higher levels of TREM1, MCP1, IL8 and reduced levels of sOX40 and RANTES. When comparing 22q11.2DS patients with and without autoimmune disease, the group with autoimmune disease has increased levels of sTREM1, s4-1BB, IP10, RANTES, sPD1 and reduced levels of sOX40 and sPDL1. 22q11.2DS patients have defects in the production of soluble mediators involved in the promotion and suppression of the inflammatory response, being essential to maintain the physiological balance of the immune system, regardless of exposure to external factors that may trigger the activation of the immune response. The mediators evaluated can be used as biomarkers to predict autoimmunity in patients with 22q11.2DS.
Keywords: 22q11.2 deletion syndrome, DiGeorge syndrome, autoimmunity
Disclosure: All authors indicated they had no financial relationships to disclose.
(135) Spectrum of COVID-19 in patients with secondary immunodeficiency
Jessica Galant-Swafford, MD1, Lori Broderick, MD, PhD2, Marc Riedl, MD, MS3
1Clinical Fellow, Allergy/Immunology/University of California San Diego/Division of Rheumatology, Allergy & Immunology
2Assistant Professor of Pediatrics/University of California San Diego/Division of Rheumatology, Allergy & Immunology
3Professor of Medicine/University of California San Diego/Division of Rheumatology, Allergy & Immunology
Little is known about the spectrum of COVID-19 in patients with secondary/acquired immunodeficiency diseases, such as those arising from immunosuppressive medications and/or hematologic malignancies. We hypothesized that those with hypogammaglobulinemia may be at particular risk due to a decreased ability to produce protective antibodies.
Methods: In this retrospective analysis, patients were identified who had been diagnosed with hypogammaglobulinemia via ICD-10 code and had a positive SARS-COV-2 PCR or antibody test. Descriptive information about each patient’s clinical history, course of SARS-COV-2 infection, and relevant laboratory data were obtained.
A total of 11 patients with secondary immunodeficiency were identified as COVID-19 positive: 27.2% female, median age 55 years (25-80). Seven had a history of malignancy, 1 with autoimmune disease, 2 with history of transplant, and 1 with hypogammaglobulinemia of unknown etiology. All had received chemotherapy or immunosuppressive therapy. Less than half were prescribed routine immunoglobulin (Ig) replacement. Median IgG level at COVID diagnosis was 607 mg/dL (446-862). The most common subjective symptom was fever (87.5%), followed by cough/shortness of breath (75%), however less than one-third of these had objective temperature >100.4°C. Approximately 73% of patients were hospitalized, 45% received supplemental oxygen, 18% were intubated, and 27.2% died. Five patients received remdesivir, 2 received dexamethasone, and 1 received additional Ig replacement. Lymphopenia and decrease in absolute lymphocyte count from baseline, but not baseline IgG, were predictors for severe disease. The mortality rate among those on routine Ig replacement was 25%, compared to 28.6% in those without routine replacement. Similar rates of hospitalization, oxygen administration, and ICU admission were observed in the two groups. Of the 3 deceased patients, two had multiple myeloma and one had acute myeloid leukemia.
Particular subtypes of secondary immunodeficiency may be more predisposed to severe disease from COVID-19, specifically those arising from hematologic malignancy. A larger sample size is needed to determine wither routine Ig replacement has a protective effect on COVID-19-associated mortality in this group. Examining the course of COVID-19 in patients with immunodeficiency can promote greater understanding of the pathogenesis of SARS-COV-2 infection, protective and deleterious immune responses, and protective strategies for susceptible patients.

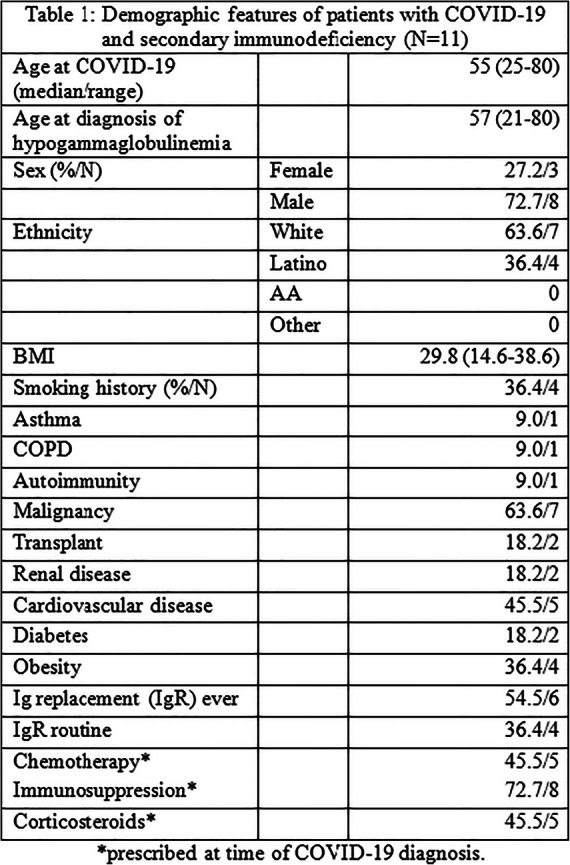

Keywords: COVID-19, Immunodeficiency, Hypogammaglobulinemia, Secondary immunodeficiency
Disclosure: Lori Broderick received financial or material support from IFM Pharmaceuticals. Marc Redl is a consultant for Adverum, Attune, Biocryst, Biomarin, CSL Behring, Ionis, Kalvista, Pfizer, Pharming, Pharvaris, RegenexBio, Shire/Takeda; received a research grant from Biocryst, CSL Behring, Ionis, and Shire/Takeda; and speaker honoraria from CSL Behring, Pharming, and Shire/Takeda. All other authors had no financial relationship to disclose.
(136) A case of disseminated Mycobacterium flavescens in a child with a novel combination of compound heterozygous variants in IFNGR1 leading to complete IFNGR1 deficiency
Elizabeth Christian, MD1, Brenna LaBere, MD2, Alicia Johnston, MD3
1Infectious Disease Fellow/Boston Children's Hospital and Beth Israel Deaconess Medical Center
2Allergy and Immunology fellow/Boston Children's Hospital
3Infectious Disease Attending Physician/Boston Children's Hospital
Mendelian susceptibility to mycobacterial diseases (MSMD) is a rare condition characterized by predisposition to infection with weakly virulent mycobacteria, salmonella, candida, and other intracellular organisms. Pathogenic variants have been described in nine genes involved in IFNγ-dependent immunity: IFNGR1, IFNGR2, STAT1, IL12B, IL12RB1, ISG15, IRF8, NEMO, and CYBB. Autosomal recessive (AR) complete IFNGR1 deficient patients are at risk for severe disseminated mycobacterial disease in early childhood, with fewer than 20% surviving beyond 12 years. Hematopoietic stem cell transplant (HSCT) may be curative. To date, there have been fewer than 40 cases described worldwide.
The patient presented at 16 months of age with fever, leukocytosis and elevated inflammatory markers. CT revealed diffuse lymphadenopathy concerning for lymphoma. Bone marrow biopsy revealed normocellular marrow, while cervical lymph node biopsy showed a reactive lymph node without granulomas. Flow cytometry from both samples was negative for a lymphoproliferative process. AFB stains were negative, however lymph node culture grew an AFB positive organism, later identified as Mycobacterium flavescens by 16s rRNA sequencing. Isolation of a low virulence organism prompted an immune evaluation. The patient’s immunologic profile was generally reassuring, though with significant elevation in IFNγ. Prevention Genetics PID panel revealed two heterozygous variants in IFNGR1: C523del, p.Tyr175Metfs*2 and C.200+1G>A. Both have been shown to be pathogenic as homozygous variants. Parental sequencing revealed that each parent was heterozygous for one variant. Parents are non-consanguineous. Functional studies revealed no expression of CD119/IFNγR1 on monocytes and no activation of pSTAT1 in monocytes following IFNγ stimulation, supporting the diagnosis of AR complete IFNGR1 deficiency. The patient received linezolid, isoniazid, ciprofloxacin and azithromycin with resolution of fever and lymphadenopathy, and normalization of inflammatory markers and IFNγ levels. Parents are considering HSCT with 9/10 A or B mismatched unrelated donors versus HSCT following IVF with preimplantation genetic diagnosis.
We report a novel combination of heterozygous variants in IFNGR1 leading to complete IFNGR1 deficiency, identified after presentation with disseminated M. flavescens, a rarely pathogenic scotochromogenic mycobacterium. To our knowledge this is the first report of M. flavescens infection in a patient with MSMD.
Keywords: Mendelian susceptibility to mycobacterial disease, Primary immunodeficiency, Non-tuberculous mycobacterium, IFNGR1 deficiency
Disclosure: All authors indicated they had no financial relationships to disclose.
(137) Retroprotenial fibrosis is arare immunlogical disease that cause heamaturia in young pateints
Awatif Alobeid1, Mawia Hassan, MD2, Hani Aldien, MD2
1Dr/Clinical immunology
2Urologist/Sudanese urlogy association
Retroperitoneal fibrosis is characterized by development of extensive fibrosis throughout the retroperitonium .its thought to be apart of IGg related autoimmune diseas,and its responding to steroid and immunesupressive therapy.This is a case of 16 years old male patient who was presented with haematuria,CTKUB showed retroperitoneal mass.
Keywords: Retroperitoneal fibrosis, Heamaturia, IGg related disease, Autoimmune
Disclosure: All authors indicated they had no financial relationships to disclose.
(138) Utility of expanded molecular testing for primary immunodeficiencies: disease-specific panels may miss diagnoses
Jessica Connor, MS, LCGC1, Rebecca Truty, PhD2, Jennifer Holle, MS, CGC3, Shiloh Martin, MD, PhD1, Hui Yu, PhD1, Olga Sarmento, PhD4, Britt Johnson, PhD, FACMG, HCLD(ABB)5
1Clinical Genomics Scientist/Invitae
2Bioinformatic Analyst/Invitae
3Genetic Counselor/Invitae
4Clinical Genomics Scientist/Inviate
5Medical Director, Metabolic Genetics and Immunology,Medical Affairs Director, Biopharma Partnerships/Invitae Corporation, San Francisco, CA
Primary immunodeficiency disorders (PIDs) exhibit “phenotype expansion” in which genes previously thought to be associated with specific phenotypes have an increasing circle of clinical presentations. Traditionally, genetic testing for PIDs is phenotype-specific, guided by genes associated with a given clinical presentation. A disease-specific panel may not include the actual causative gene in a patient with a suspected PID, making a phenotype-agnostic panel a more powerful test.
This study compared the diagnostic value of a 207 gene PID panel to smaller, disease-focused panels. Patient phenotypes were compared to keywords and ICD.10 codes corresponding to each focused panel and each requisition was assigned a virtual focused panel. Positive diagnoses on the expanded panel and the focused panel were compared for each requisition to determine the frequency of missed diagnoses if only the focused panel had been ordered.
Our commercial diagnostic laboratory performed 6,257 expanded PID panels between April 2017 and October 2019. A virtual focused panel could be assigned based on clinical indication for 3,384 unique orders. Of the 2,573 cases in which a focused panel could not be assigned, 623 (18%) had no clinical information provided and 1,950 had information not useful for analysis. Of the 3,384 unique orders, 105 had positive results. Positive results would have been missed in 24.8% (26/105) of cases had the expanded panel not been ordered. The Combined T/B Cell Deficiencies panel missed three positive results (14%) while the Antibody Deficiencies panel missed 17 positive results (42.5%). The Autoinflammatory Syndromes panel missed four results (25%) and for orders that indicated both the Antibody Deficiencies panel and Autoinflammatory Syndromes panel, 2 positive results were missed (16%). For example, an infant presented with failure to thrive and perirectal abscess. Two of her older siblings are deceased, one of whom had a genetic diagnosis of hemophagocytic lymphohistiocytosis, an autoinflammatory syndrome. The expanded PID panel identified a homozygous MALT1 truncation, which is consistent with a diagnosis of combined immunodeficiency. These results indicate the importance of ordering an expanded panel or having the option to re-requisition to an expanded panel for conditions with overlapping symptoms.
Keywords: primary immunodeficiency, genetic testing, diagnostic yield
Disclosure: All authors are employed by Invitae.
(139) Isolated CNS HLH during adolescence as the initial manifestation of Griscelli Syndrome type 2 due to a novel homozygous mutation in RAB27A
Miriah Gillispie-Taylor, MD1, Philip Roehrs, MD2, Sharat Chandra, MD3
1Assistant Professor/Atrium Health/Levine Children's Hospital
2Associate Professor/Atrium Health/Levine Children's Hospital
3Associate Professor/Cincinnati Children's Hospital
Primary/familial HLH is an inherited syndrome of immune dysregulation caused by impairment of the cytotoxic function of NK and T cells leading to uncontrolled immune cell activation, and multi-organ immunopathology. While CNS involvement is common in primary HLH, presentation with isolated CNS HLH without any systemic manifestations is rare. We report isolated CNS HLH as the initial manifestation of Griscelli Syndrome type 2 due to a novel homozygous mutation in RAB27A.
A 17-year-old Pakistani-American female was admitted for evaluation of headache, fever, and left sided weakness. Her medical history was significant for altered mental status and status epilepticus at the age of 13 years. Exam then was significant for hypopigmented skin lesions. She is the product of a consanguineous marriage. CSF studies revealed pleocytosis and elevated protein, as well as anti-NMDAr antibodies in serum that led to the diagnosis of anti-NMDAr encephalitis and treatment followed BrainWorks protocol. Recurrence of CNS symptoms at age 17 prompted repeat admission. CSF studies showed elevated protein and pleocytosis. Spine MRI showed a demyelinating lesion at the level of C6/7. She developed status epilepticus and was treated with high dose steroids, rituximab and IVIG for a presumed autoimmune neuroinflammatory process, but repeat serum and CSF encephalitis panels were negative. She developed respiratory distress and chest CT showed parenchymal opacities. Due to worsening clinical status she underwent brain and lung biopsies. Brain pathology shows leptomeningeal infiltration of T cells and lymphohistiocytes and lung pathology showed organizing/fibrinous pneumonia with histiocytic infiltration. Further evaluation revealed minimal elevation in soluble IL-2 and ferritin, but impaired T cell degranulation. She was diagnosed with HLH and started on HLH therapy which led to clinical response. HLH genetic panel revealed a novel homozygous variant in RAB27A (p. Trp113Arg), predicted to be pathogenic. She underwent allogeneic HCT and successfully engrafted with 100% donor chimerism. She is currently 3 months post BMT and doing well.
Our report suggests that Griscelli syndrome type 2 due to homozygous mutation in RAB27A can present with isolated CNS HLH. This case highlights the importance of evaluating for isolated CNS-HLH in any patient with neuroinflammation of unclear etiology.
Keywords: HLH, CNS, seizure
Disclosure: Philip Roehrs received speaker honoraria from Sobi Pharmaceuticals and is a consultant for Spark Therapeutics. All other authors had no financial relationship to disclose.
(140) ICF Type 2: A Novel Mutation
Priyanka Seshadri, MD1, Hilary Gordon, MD2, Stephen McGeady, MD2
1Fellow in Training/Thomas Jefferson University Hospital/ Nemours A.I. DuPont Hospital for Children
2Attending/Thomas Jefferson University Hospital/ Nemours A.I. DuPont Hospital for Children
Immunodeficiency, centromeric instability, and facial dysmorphism Type 2 (ICF Type 2) is a rare primary immunodeficiency resulting from abnormalities in the ZBTB24 gene involved in DNA methylation . Patients often present with global developmental delays and either humoral or combined immunodeficiencies that can be progressive. The mortality rate is high with a majority of patients succumbing to opportunistic infections. Therapies that have been used include immune globulin replacement and hematopoietic stem cell transplant. We describe a novel genetic mutation leading to ICF Type 2 and address the ethics of declining treatment when no standard of care has been established.
A 3 month old female presented with severe hypotonia and dysmorphic facial features. Family history is significant for consanguinity in parents (1st cousins) and a sister deceased due to sepsis at 2 years of age. Whole Exome Sequencing revealed 2 mutations in the ZBTB24 exome, one previously unreported (p.Arg398Ter: c.1192C>T) and one in which functional studies demonstrate moderate to no damaging effects (p.Gly486Asp:c.1457). Immune evaluation at age 4 months revealed hypogammaglobulinemia (IgG 128mg/dL normal 228-636, IgA 3mg/dL normal 27-72, IgM 7mg/dL normal 26-60) and reduced isotype-switched memory B cells on flow cytometry (normal is 5-30%). Lymphocyte subsets and lymphocyte mitogen proliferation were normal. At age 6 months, the patient had no infections nor required any anti-microbials however, she remained hypogammaglobulinemic (IgG 134mg/dL normal 228-636mg/dL, IgA 2mg/dL normal 27-72mg/dL, IgM 9mg/dL normal 25-60mg/dL). Immunoglobulin replacement was recommended but the family declined treatment due to concerns about inflicting pain with IV placement and the religious belief that it would be wrong to prolong life if the quality would be poor. An ethics committee was consulted due to the child’s significant hypogammaglobulinemia and posed the question of whether immune globulin replacement as a life sustaining therapy could ethically be refused. The committee concluded it would be unethical to refuse immune globulin replacement if the immunology department considered IVIG life-sustaining. The family ultimately consented to immune globulin replacement.
This case highlights a novel mutation in a rare primary immunodeficiency and addresses the ethics of declining treatment when no standard of care has been established.
Keywords: ICF Type 2, IVIG, Ethics
Disclosure: All authors indicated they had no financial relationships to disclose
(141) Moderate to severe CARD11 Loss of Function-associated atopic dermatitis treated with biologic modifiers
Natalie Diaz-Cabrera, MD1, Bradly Bauman, MS2, Mildred Iro, MD, PhD3, Gina Dabbah, BS4, Vered Molho-Pessach, MD5, Abraham Zlotogorski, MD6, Oded Shamriz, MD7, Yael Dinur-Schejter, MD8, Tatyana Dubnikov Sharon, PhD9, Polina Stepensky, MD10, Yuval Tal, MD, PhD11, Eli Eisenstein, MD12, Joshua Milner, MD13, Anthony Williams, PhD14, Gil Armoni-Weiss, MD15, Andrew Snow, PhD16, Jennifer Leiding, MD17
1Fellow Physician/Division of Allergy and Immunology, Department of Internal Medicine, University of South Florida College of Medicine, Tampa, FL
2Graduate Student/Department of Pharmacology and Molecular Therapeutics, Uniformed Services University of the Health Sciences, Bethesda, MD, USA
3NIHR Academic Clinical Lecturer/Faculty of Medicine and Institute for Life Sciences, University of Southampton, Southampton, United Kingdom
4Graduate Student/Department of Pharmacology & Molecular Therapeutics, Uniformed Services University of the Health Sciences, Bethesda, MD
5Senior Lecturer/Pediatric Dermatology Service, Department of Dermatology and Venereology, The Faculty of Medicine, Hadassah Medical Center, Hebrew University of Jerusalem, Israel
6Associate Professor/Department of Dermatology and Venereology, Hadassah Medical Center, Jerusalem, Israel
7Fellow Clinical Immunologist/Clinical Immunology and Allergy Unit, Hadassah-Hebrew University Medical Center; The Lautenberg Center for Immunology and Cancer Research, Institute of Medical Research Israel-Canada, Hebrew University-Hadassah Medical School, Jerusalem, Israel
8Staff Clinical Immunologist/Department of Bone Marrow Transplantation and Cancer Immunotherapy, Hadassah University Medical Center, Jerusalem, Israel
9Head of Immunodiagnostics lab/Department of Bone Marrow Transplantation and Cancer Immunotherapy, Hadassah University Medical Center, Jerusalem, Israel
10Professor, Director of Bone Marrow Transplantation Cancer Immunotherapy & Immunobiology Research Cen/Department of Bone Marrow Transplantation and Cancer Immunotherapy, Hadassah University Medical Center, Jerusalem, Israel
11Associate Professor/Clinical Immunology and Allergy Unit, Hadassah-Hebrew University Medical Center, Jerusalem, Israel
12Pediatric Rheumatologist/Department of Pediatrics, Hadassah-Hebrew University Medical Center,Mount Scopus, Jerusalem, Israel
13Professor/Laboratory of Allergic Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA
14Professor/Faculty of Medicine and Institute of Life Sciences, University of Southampton, Southampton, United Kingdom
15Dermatologist/Department of Dermatology and Venereology, Hadassah Medical Center, Jerusalem, Israel
16Associate Professor/Department of Pharmacology and Molecular Therapeutics, Uniformed Services University of the Health Sciences, Bethesda, MD
17Associate Professor/Division of Allergy and Immunology, Department of Pediatrics, University of South Florida at Johns Hopkins-All Children’s Hospital, St Petersburg, FL
The caspase activation and recruitment domain 11 (CARD11) gene encodes a scaffold protein required for lymphocyte antigen receptor signaling. Dominant negative, loss-of-function (LOF) mutations in CARD11 are associated with common variable immune deficiency, cutaneous and respiratory infections, neutropenia, and atopy. Nearly 90% of patients display severe atopic disease such as atopic eczema, allergic rhinitis, food allergies, and eosinophilic esophagitis. Omalizumab and dupilumab are two humanized monoclonal antibodies directed against free circulating IgE and the interleukin-4 receptor, respectively, successfully used in the treatment of refractory allergic asthma, idiopathic urticaria, and atopic dermatitis. Data is limited regarding their safety and efficacy in patients with CARD11 LOF.
We assessed the response to dupilumab in two patients and omalizumab in one patient with severe atopic disease. CARD11 mutations were validated for pathogenic potential using a T cell transfection assay to assess impact on activation-induced signaling to NF-kB, JNK, and mTORC1.
This series included 3 unrelated females with a mean age of 20 years; all had novel heterozygous LOF variants in CARD11 that were shown to disrupt wild-type CARD11 signaling (Table 1). All 3 presented with atopic disease in infancy and susceptibility to infections. Patients 1 and 2 were treated with dupilumab for treatment-refractory atopic dermatitis and patient 3 was treated with omalizumab for treatment-refractory chronic urticaria. Patients 1 and 2 experienced substantial improvement in atopic dermatitis following therapy with dupilumab. SCORing Atopic Dermatitis (SCORAD) for patient 1 reduced from 84 to 40 and Eczema Area and Severity Index (EASI) score reduced from 21.75 to 4.0 in patient 2, both within a few months of therapy. Chronic urticaria improved substantially in patient 3. Additional atopic conditions improved as well: asthma, allergic rhinitis, and food sensitivities. There were no complications or adverse effects from use of dupilumab or omalizumab.
Treatment with dupilumab and omalizumab for severe, refractory atopic disease in patients with CARD11-associated atopy with dominant interference of NF-kB signaling (CADINS) disease appears to be effective and well tolerated in our cohort. Further follow-up and larger sample sizes are required to determine additional efficacy and safety data.
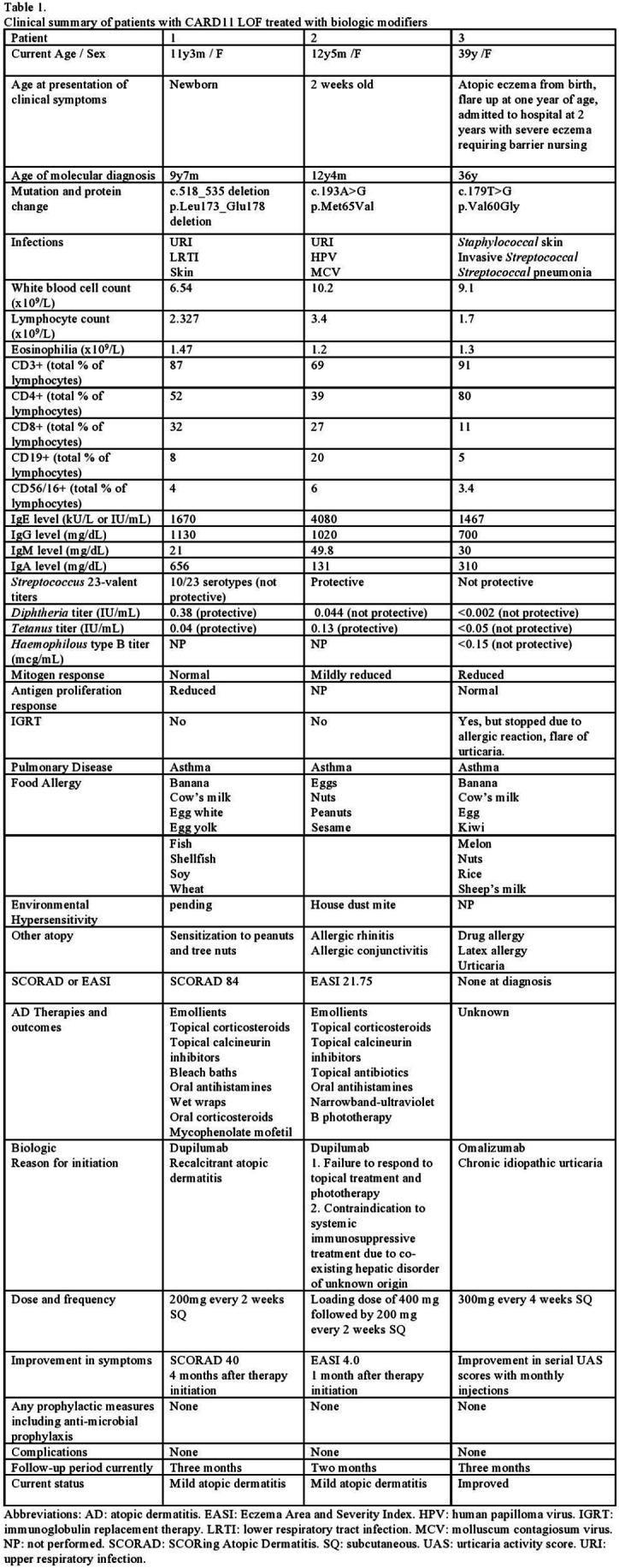
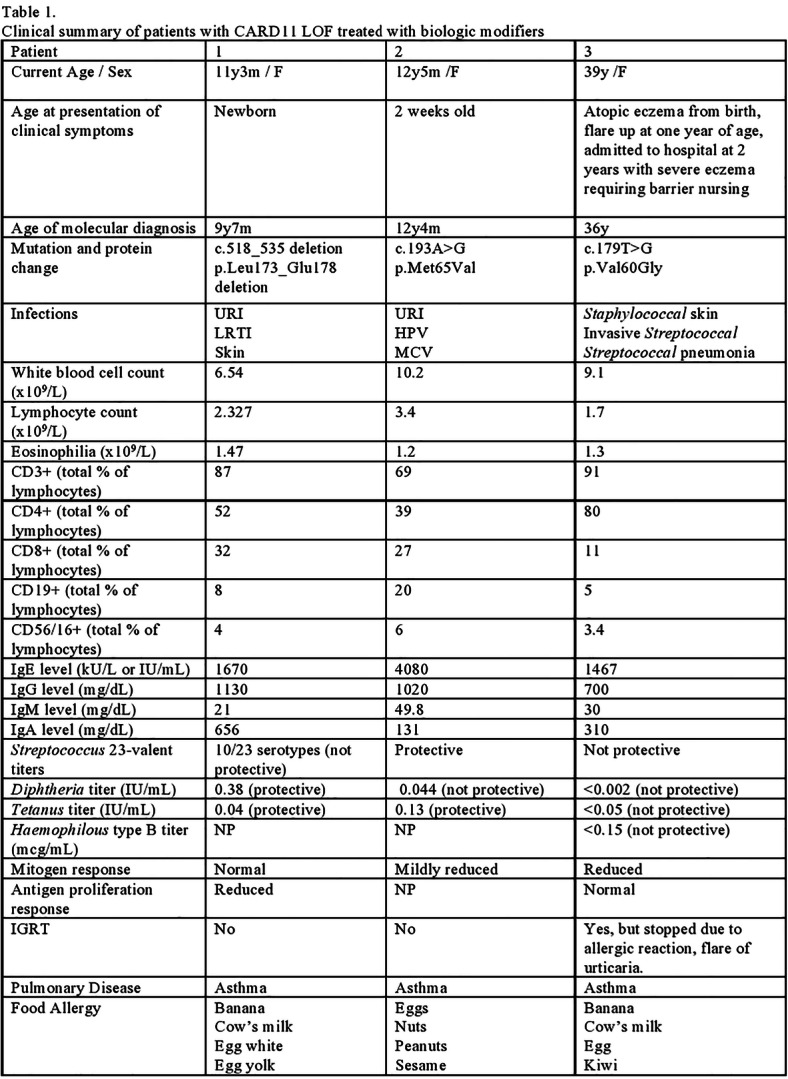

Keywords: CARD11, atopic dermatitis, dupilumab, omalizumab, urticaria
Disclosure: Jennifer Leiding received speaker honoraria from CSL Behring and Horizon Therapeutics and was an advisory board member for Pharming. All other authors had no financial relationships to disclose.
(142) MHC Class II deficiency: up-and-down road from the diagnosis to the follow-up
Claudia Rita1, Ignacio Iturrieta-Zuazo, PhD2, Paloma Lapuente Suanzes, MD2, Ana García-Soidan, MD2, Ana De Andrés, MD3
1Immunology resident/Hospital Universitario Ramón y Cajal
2Immunology resident/Ramón y Cajal University Hospital
3Immunologist/Ramón y Cajal University Hospital
MHC II deficiency is caused by a mutation in the genes encoding transcription factors of MHC II. Most cases have been reported from consanguineous marriages.
Herein, we report a twenty-three-year-old Spanish boy from a consanguineous family. He was referred to our centre for a highly inverted CD4/CD8 ratio, chronic diarrhoea and early onset of recurrent and severe respiratory tract infections. Since the first year of life, he presented with frequent upper and lower respiratory tract infections. More than 20 hospital admission for bacterial pneumonia and gastroenteritis were recorded. Besides he had history if septic arthritis of the knee complicated with osteomyelitis, two episodes of immune thrombocytopenic purpura and at the age of 15 years, bronchiectasis was identified needing oxygen therapy. In family history, one brother and one sister have demised during the first year of life for septicaemia. One sister presented with recurrent otitis media and respiratory tract infections being diagnosed with IgA deficiency.
Laboratory findings revealed IgG2 deficiency and IgG hypogammaglobulinemia with normal levels of IgA and IgM. T-cell compartment showed a T CD4+ lymphopenia (224 cell/μL) and inverted CD4/CD8 ratio (0,12),. T-cell proliferation to antigens (Candida albicans) was decreased but normal to mitogens (OKT3 and PHA), delayed-type hypersensitivity was negative for purified protein derivative (PPD) and candidin. Flow cytometry revealed no expression of HLA-DR antigen on resting monocytes and B-cells and absence of inducible expression of MHC class II molecules after interferon-gamma stimuli on B-cells, T-cells or monocytes. Genetic analysis revealed a homozygous mutation in RFXANK gene. During the study of the family, one sister showed the same homozygous mutation, while six siblings showed heterozygous mutation and one had no mutation.
According to the high risk of transplant-related mortality and significant comorbidities hematopoietic cell transplantation was not accepted by the family. He was treated for 22 years with prophylactic cotrimoxazole and intravenous immunoglobulin. During the following years, the frequency and severity of infectious episodes diminished until he develop nephrotic syndrome requiring to stop immunoglobulins and dying for sepsis.
Unknown factors may influence the residual innate or CD8+ T cell–mediated immunity, which potentially contributes to this more favourable outcome.
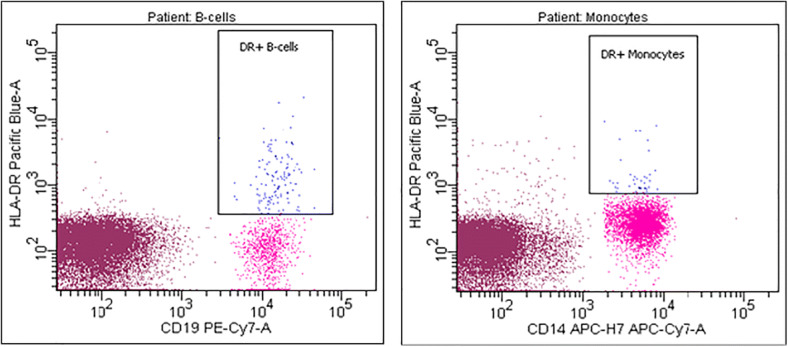
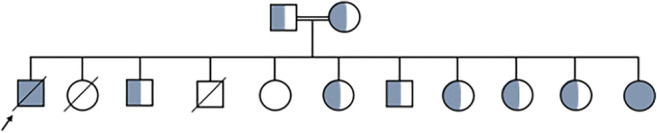
Figure 2: Pedigree of the family. The index patient is pointed by an arrow.
Keywords: MHC class II deficiency, bafe lymphocyte syndrome, primary immunodeficiency
Disclosure: All authors indicated they had no financial relationships to disclose.
(143) Novel Compound Heterozygous variants in IKBKB associated with Autoimmunity and Autoinflammation
Nouf Alsaati, MD1, Keith Sacco, MD2, Lauren Smith, MD3, Sameeya Ahmed-Winston, CPNP-PC4, Blachy Blachy Dávila Saldaña, MD5, Hyesun Kuehn, PhD6, Sergio Rosenzweig, MD, PhD7, Michael Keller, MD8
1Resident Physician/Division of Allergy and Immunology, Children's National Health System, Washington, DC, USA
2Clinical Fellow/Lab of Clinical Immunology and Microbiology, Immune Deficiency Genetics Section, NIAID, NIH, Bethesda, MD, USA
3Assistant Professor of Pediatrics/Children's Hospital of the King's Daughters, Eastern Virginia Medical School, Norfolk, VA, USA
4Pediatric Nurse Practitioner/Division of Blood and Marrow Transplantation, Children's National Health System, Washington, DC, USA
5Pediatric Hematologist/Oncologist/Division of Blood and Marrow Transplantation, Children's National Health System, Washington, DC, USA
6Staff Scientist/Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
7Senior Investigator/Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
8Pediatric Immunologist/Division of Allergy and Immunology, Children's National Health System, Washington, DC, USA
Homozygous null mutations of IKBKB encoding IKK2 are known to cause severe combined immunodeficiency (SCID) with normal peripheral T cell and B cell counts, and hypogammaglobulinemia. Heterozygous missense mutations in the same gene were described to cause an autosomal dominant combined immunodeficiency. To our knowledge, a phenotype of autoinflammation with organ-specific autoimmunity has not been described.
A 6-week-old ex-full term male developed a self-limiting episode of rash, fever, and autoimmune hemolytic anemia (AIHA) – hemoglobin (hgb) 10.4 g/dL, platelets 407k/mcL, and absolute neutrophil count (ANC) 3321/mcL at presentation. Symptoms recurred at 7 weeks (hgb 6.5 g/dL, platelets 702k/mcL, ANC 8474/mcL) with serological testing being positive for anti-D antibodies and anti-neutrophil antibodies, requiring packed red cell transfusion. His rash was clinically described as erythematous papules coalescing into plaques scattered on the arms, legs, trunk, and diagnosed by biopsy as Sweet syndrome. Serum immunoglobulins were not decreased (IgG 493 mg/dL, IgM 278 mg/dL and IgA 55.2 mg/dL). The patient developed a fever after receiving scheduled vaccinations at 2 months which led to a third autoimmune crisis. He had received the second dose of Hepatitis B, and first doses of Diphtheria, Tetanus, Pertussis, Haemophilus influenzae type b, inactivated Polio vaccine, pneumococcal conjugate vaccine (PCV13), and Rotavirus. A targeted primary immunodeficiency gene panel identified two IKBKB variants in trans (IKBKB: c.1552G>A, p.D518K; c.1676 C>T, p.T559M). Variant c.1676 C>T, p.T559M had a low minor allele frequency of 5.24e-5 (Gnomad®) while c.1552G>A, p.D518K was absent from major gene databases. Functional assays showed decrease in IkBα degradation in patient’s PBMCs following TNFα stimulation, as well as elevation of baseline phospho-AKT.
Our case is the first to demonstrate a clinical phenotype of autoinflammation with organ-specific autoimmunity due to novel compound heterozygous variants in IKBKB. Though the specific consequences of these individual IKBKB variants remains unknown, the phenotype could suggest dysfunctional signaling of the canonical NF-κB pathway affecting its interaction with NLRP3, TNFα, and IL-1β. These findings suggest that a IKBKB mutations should be considered in the setting of disease manifesting with features of autoinflammation or autoimmune dysregulation.
Keywords: IKBKB, NfKB signaling, Immune dysregulation, Combined immunodeficiency, Autoimmunity, Autoinflammation
Disclosure: Sameeya Ahmed-Winston received speaker honoraria from Jazz Pharmaceuticals. Michael Keller is an advisory board member of Enzyvant. All other authors had no financial relationship to disclose.
(144) Improvement of SLC29A3 spectrum disorder-related sensorineural hearing loss after initiation of IL-6 inhibitor
Hallie Carol, MD1, Amer Khojah, MD2, Nurcicek Padem, MD3, Taher Valika, MD4
1Resident Physician/McGaw Medical Center of Northwestern University/ Ann & Robert H. Lurie Children's Hospital
2Attending Physician, Allergy, Immunology, and Rheumatology/Ann & Robert H. Lurie Children's Hospital of Chicago/ Northwestern University Feinberg School of Medicine
3Attending Physician, Allergy and Immunology/Riley Children's Hospital
4Attending Physician, Otorhinolaryngology-Head & Neck Surgery/Ann & Robert H. Lurie Children's Hospital of Chicago/ Northwestern University Feinberg School of Medicine
SLC29A3 spectrum disorder is an autosomal recessive, autoinflammatory syndrome, that is characterized by mutations in the SLC29A3 gene, which encodes the human equilibrative nucleoside transporter 3 (hENT3). This defect precipitates as a multisystem autoinflammatory disorder with a wide spectrum of severity and presentations, commonly including hyperpigmentation, hypertrichosis, hepatosplenomegaly, heart anomalies, hearing loss, hypogonadism, low height, and hyperglycemia in the setting of immune mediated diabetes. Interleukin 6 (IL-6) is a cytokine that plays an important role in the regulation of the immune response and inflammation, and over production has been linked to the etiologyof inflammatory diseases such as rheumatoid arthritis.. Therefore, IL-6 inhibitors havebeen used in the treatment of autoinflammatory diseases with great success.
A 15 month old boy initially presented to immunology clinic with increased inflammatory markers and sensorineural hearing loss in the setting of a SLC29A3 mutation (homozygous pathogenic variant c.1309 G>A p.G437R). Of note, this variant has been reported in the published literature to cause SLC29A3 related disorders. Family history was significant for parental consanguinity and a maternal aunt with hearing loss secondary to Rosai Dorfman syndrome. His inflammatory markers were elevated (ESR 31-43 mm/hr and CRP 2-10.4 mg/dl).The patient’s sensorineural hearing loss was progressive and presumed secondary to his underlying autoinflammation. In the hopes of slowing the progression of his hearing loss, he was started on an IL-6 inhibitor (tocilizumab 162mg subcutaneously, every 21 days) at age 25 months. Continued follow up has shown improvement in his hearing and overall down-trending inflammatory markers (ESR 1mm/hr CRP < 0.1).
This case provides insight into the use of Il-6 inhibitors to slow the progression and potential reversal of autoinflammatory related hearing loss associated with SLC29A3 spectrum disorders.
Keywords: SLC29A3, L-6 inhibitor, sensorineural hearing loss
Disclosure: Taher Valika has ownership interest in Save My Scope, Inc. All other authors had no financial relationship to disclose.
(145) A Compare and Contrast of COVID-19 Disease Progression in Two Siblings with APECED in Relation to the Timing of Treatment Initiation
Alyssa James, MD1, Monica Schmitt, CRNP2, Sebastian Ochoa, MD1, Elise Ferre, PA-C, MPH3, Thomas Dimaggio, RN4, Michail Lionakis, MD, ScD5
1Allergy/Immunology Clinical Fellow/National Institute of Allergy and Infectious Diseases/National Institutes of Health, Bethesda, MD, USA
2Certified Registered Nurse Practitioner/Fungal Pathogenesis Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases/National Institutes of Health, Bethesda, MD, USA
3Physician Assistant/Fungal Pathogenesis Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases/National Institutes of Health, Bethesda, MD, USA
4Nurse/Fungal Pathogenesis Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases/National Institutes of Health, Bethesda, MD, USA
5Chief, Fungal Pathogenesis Section/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases/National Institutes of Health, Bethesda, MD, USA
Early treatment of COVID-19 in patients with high-risk primary immunodeficiencies (PIDs) is of critical importance. Here we discuss two siblings with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) who developed distinct severities of COVID-19 associated with differential timing of treatment initiation.
Methods: Two COVID-19 positive siblings with APECED provided consent and were studied using an NIH IRB approved protocol. Their charts were reviewed.
Siblings are Caucasian with APECED caused by compound heterozygous deletions in AIRE [c.967_979del13 (p.L323SfsX51) and c.190_226del37 (p.S64TfsX71)] with positive autoantibodies to IFN-α and IFN-ω, which are associated with high risk COVID-19 disease. Sibling 1 is a 48 year old woman with chronic mucocutaneous candidiasis (CMC), hypoparathyroidism, adrenal insufficiency, hypothyroidism, primary ovarian failure, and Sjogren’s syndrome. Sibling 2 is a 45 year old man with CMC, hypoparathyroidism, adrenal insufficiency, hypothyroidism, autoimmune gastritis, intestinal malabsorption, alopecia, vitiligo, B12 deficiency, and end stage renal disease secondary to hypertensive nephropathy. Sibling 1 tested positive for COVID-19 by SARS-CoV-2 PCR in March 2020 after developing fever, cough and shortness of breath. She was treated with hydroxychloroquine, but developed respiratory failure requiring intubation for nine days. She was treated with high dose corticosteroids, extubated and made a full recovery. Sibling 2 tested positive for COVID-19 by SARS-CoV-2 PCR in October 2020 after developing fever, congestion and cough. He was admitted and treated with remdesivir and high dose corticosteroids given CT chest findings of bilateral ground glass opacities and mild hypoxemia (95% on room air). A bronchoalveolar lavage was positive for SARS-CoV-2 PCR with negative bacterial and fungal cultures. He developed acute pulmonary emboli involving the distal pulmonary arterial branches to the right lower lobe and was treated with apixaban. Throughout his 2-week hospitalization, he remained on room air.
We describe siblings with APECED with the same genetic profile who experienced distinct clinical courses of COVID-19. Sibling 2 was treated in the beginning of his course with corticosteroids and remdesivir which may have prevented the development of respiratory failure like in his sibling. This report highlights the importance of prompt therapeutic intervention in patients with certain PIDs that confer high risk for severe COVID-19.
Keywords: APECED, COVID-19, Primary Immunodeficiency, Early Treatment
Disclosure: All authors indicated they had no financial relationships to disclose.
(146) case of activated phosphoinositide 3-kinase (PI3K) delta syndrome associated with growth hormone deficiency, aseptic arthritis, nephromegaly, and asthma
Megan Craig, MD1, Mateja Cernelc-Kohan, MD2, Kristen Wigby, MD3, Susan Phillips, MD4, Bob Geng, MD5
1Resident Physician/Department of Pediatrics, University of California San Diego, Rady Children’s Hospital
2Assistant Professor/Department of Respiratory Medicine, University of California San Diego, Rady Children's Hospital
3Clinical Geneticist, Assistant Clinical Professor of Pediatrics/Division of Genetics, Department of Pediatrics, University of California San Diego, Rady Children's Hospital
4Assistant Professor/Department of Endocrinology, University of California San Diego, Rady Children's Hospital
5Assistant Professor of Pediatrics and Medicine/Divisions of Adult and Pediatric Allergy & Immunology, University of California San Diego, Rady Children's Hospital
We report the case of a 9-year-old boy with a heterozygous de novo variant in PIK3CD (p.E1021K) resulting in activated PI3K delta syndrome (APDS), a rare form of primary immunodeficiency. Our patient exhibits classic findings of this syndrome, including recurrent sinusitis and bronchitis, bronchiectasis, mild developmental delay (DD), splenomegaly, and lymphadenopathy. His laboratory abnormalities include immune cytopenias and specific antibody deficiency requiring subcutaneous immunoglobulin replacement therapy.
In addition to the previously described features, he is also affected by growth hormone (GH) deficiency, aseptic arthritis, asthma, and nephromegaly. He showed marked growth deceleration by age 5 (short stature is seen in APDS2). GH provocative testing with arginine-clonidine revealed low peak GH consistent with deficiency. Subsequent pituitary MRI demonstrated mildly hypoplastic adenohypophysis with thinning and hypoplasia of the infundibular stalk. He started GH therapy with an excellent growth response.
He underwent evaluation of chronic cough and asthma at age 7. Despite appropriate therapy and normalization of PFTs, chronic wet cough persisted, and further work-up was notable only for bronchiectasis. Taken together with his other clinical symptoms, there was increasing suspicion for a syndrome that would provide a unifying diagnosis, and he was referred to genetics. Whole exome sequencing (WES) revealed APDS.
Since GH signals through the PI3K pathway, his diagnosis prompted concern that GH could increase risk of malignancy. However, he was allowed to continue as the pathway was considered constitutively active at baseline and he had been tolerating GH for over three years prior to diagnosis.
Shortly after diagnosis, he was admitted for acute on chronic left hip pain. Laboratory studies revealed mild leukocytosis with normal inflammatory markers. MRI showed moderate left hip effusion with synovial enhancement, moderately prominent inguinal and iliac lymph nodes, and no avascular necrosis. He underwent arthrocentesis and was diagnosed with aseptic arthritis. Symptoms resolved spontaneously soon after the procedure.
This patient’s unique findings add to the clinical information available for this rare form of primary immunodeficiency and illustrate the importance of a multidisciplinary collaborative approach in managing this complex syndrome. WES was instrumental in obtaining a timely diagnosis, especially as targeted therapies are being developed.
Keywords: activated phosphoinositide 3-kinase (PI3K) delta syndrome, whole exome sequencing, growth hormone deficiency, pituitary abnormality, aseptic arthritis, asthma, nephromegaly
Disclosure: Bob Geng was an advisory board member for Biocryst and received speaker honoraria from Grifols, GSK, Horizon, Kedrion, Octapharma, Optinose, Regeneron, Sanofi, and Takeda; he was a consultant for RMS. All other authors had no financial relationship to disclose.
(147) Diagnosis and treatment of hepatopulmonary syndrome in an adolescent patient with partial DiGeorge syndrome
Minh Nguyen, DO1, Nicholas George, MD1, Katherine Scott, MD2, Roua Azmeh, MD3
1Resident Physician/Western Michigan University School of Medicine
2Attending Physician/Bronson Pediatric Oncology & Hematology Specialists (A Bronson Methodist Hospital facility)
3Attending Physician/Western Michigan University School of Medicine
Chromosome 22q11.2 deletion syndrome, or DiGeorge syndrome (DGS), is the most common chromosome deletion syndrome. Immunodeficiency is one of the key features. Hepatopulmonary syndrome (HPS) is characterized by hypoxemia caused by intrapulmonary vascular dilatation in patients with liver disease. Here, we present an adolescent with DGS and immune dysregulation who developed HPS.
The patient was a 13-year-old girl with partial DGS, immune dysregulation (low IgG, IgA, T, B, natural killer, helper and suppressor cells along with poor response to vaccinations), Evans’ syndrome, and hemodynamically insignificant cardiac anomalies. During an outpatient IVIG infusion, she was found to be hypoxemic on room air. A chest CT showed pulmonary hypertension, splenomegaly along with enlarged splenic and portal veins, concerning for portal hypertension (figure 1). An echocardiogram with bubble study suggested pulmonary arteriovenous malformations (pAVMs) (figure 2). A cardiac catherization identified pAVMs which were not amenable to intervention. Genetic testing for hereditary hemorrhagic telangiectasia was negative. A high-resolution chest CT reported no signs of parenchymal diseases. A liver biopsy showed epithelioid granulomas without evidence of fibrosis. A diagnosis of HPS was made. Her clinical course was notable for persistent hypoxemia and multiple intensive care unit admissions. Liver transplant was not feasible due to her comorbidities. Altogether, she enrolled in hospice care. In light of her immune dysregulation, rituximab was considered, but held due to side effects and the patient’s frail status. Instead, high dose prednisone was initiated and slowly tapered. After one year, she improved, and supplemental oxygen was discontinued. She was discharged from hospice.
Although HPS is typically associated with cirrhosis, her case was related to granulomatous liver disease secondary to DGS. Granulomas are inflammatory infiltrates occurring as a result of immune dysregulation and have been described in primary immunodeficiency disorders such as chronic granulomatous disease and common variable immunodeficiency. Her liver epithelioid granulomas caused poor clearance of angiogenic factors and resulted in the development of AVMs in the lungs, with subsequent shunting and hypoxemia.
To our knowledge, this is the first reported case of HPS in a DGS patient. The case also offers a potential treatment for HPS associated with immunodysregulation.

Figure 2: Echocardiogram with bubbly study, apical 4-chamber view. (A) demonstrates the atria and ventricles prior to the injection of saline. After the injection, (B) shows the presence of the saline in the right side of the heart (yellow arrow), which is a normal finding. After 4 cardiac cycles, (C) indicates the saline (red arrows) in the left heart. This finding is positive for intrapulmonary shunting, consistent with pulmonary arteriovenous malformations. RA: right atrium. RV: right ventricle. LA: left atrium. LV: left ventricle. R: right heart. L: left heart.
Keywords: DiGeorge Syndrome, Hepatopulmonary syndrome, Granulomatous disease
Disclosure: All authors indicated they had no financial relationships to disclose.
(148) Organizing pneumonia in a Down syndrome child with T cell lymphopenia
Minh Nguyen, DO1, Roua Azmeh, MD2, Mariam Ischander, MBChB2
1Resident Physician/Western Michigan University School of Medicine
2Attending Physician/Western Michigan University School of Medicine
Organizing pneumonia (OP) is a rare but distinct clinical and pathologic entity, characterized by the formation of organized buds of granulation tissue. These lead to obstruction of the alveolar lumen and bronchioles. This inflammatory process occurs in response to an injury, which can be primary or secondary to other causes. We describe a rare case of OP secondary to chronic aspiration exacerbated by rhinovirus infection in a pediatric patient.
A 5-year-old female with Down Syndrome (DS) and chronic aspiration presented with shortness of breath and was hypoxic on room air. A nasal swab tested positive for rhino/enterovirus. CXR revealed bibasilar pulmonary opacities, suspicious for a community acquired pneumonia. She was started on a course of amoxicillin. At 3-day follow-up, she was again hypoxic. Repeat CXR showed worsening bilateral opacities (figure 1). Chest CT showed bilateral patchy ground-glass opacities distributed along bronchovascular bundles in the lower lung zones, concerning for a secondary OP (figure 2). Lung biopsy was not pursued as the patient improved with a prolonged course of steroids. After she completed her steroid course, immunologic workup revealed low T lymphocyte counts (CD45, CD3, and CD4).
OP is rare in pediatric patients. OP typically manifests as dyspnea and cough. Biopsy is the gold standard for diagnosis. Histology is defined by buds of granulation issue located in the distal airspaces. The classic radiologic features of OP, as in this case, are areas of focal consolidation and ground glass opacities. The radiographic findings consistent with OP and our patient’s improvement with corticosteroids persuaded against a biopsy. Her chronic pulmonary aspiration is also a predisposing factor for development of OP. Further, patients with DS can have high rates of infections, particularly of the respiratory tract. Their disease courses are often complicated, attributed to T and B cell dysfunction and deficiencies, as found in our patient. This rare case of pediatric OP in the setting of DS, T cell lymphopenia and chronic pulmonary aspiration highlights the need for a high index of suspicion when evaluating respiratory symptoms in patients with DS or other conditions associated with immune deficiency.
Figure 1: Chest X-Ray (CXR) (A) from the first hospital admission showing bibasilar pulmonary opacities. Repeat CXR (B) five days later showing increasing bilateral lower lobe opacities.
Figure 2: Axial view of lung (A) computed tomography (CT) with patchy consolidation in lower lobes, right middle lobe, and the lingual. Coronal view of lung CT (B) with patchy consolidation in both lower lobes, right middle lobe, and the lingula.
Keywords: Organizing pneumonia, T cell lymphopenia, Down syndrome
Disclosure: All authors indicated they had no financial relationships to disclose.
(149) Comparing Hemophagocytic Lymphohistiocytosis Between Adult and Pediatric Patients: Perspectives from a Tertiary Care Institution
Minh Nguyen, DO1, Tahnee Spoden, MD1, Gisel Rivera, MD1, Melissa Baker, DO1, Roua Azmeh, MD2
1Resident Physician/Western Michigan University School of Medicine
2Attending Physician/Western Michigan University School of Medicine
Hemophagocytic lymphoshistiocytosis (HLH) describes a rare syndrome resulting from immune system overactivation. This retrospective observational study characterizes adult and pediatric patients diagnosed with HLH at our small tertiary care center in order to further our understanding of the condition.
A report was generated via the EMR of all patients with the ICD-10 code D76.1 for HLH and D89.42 for macrophage activation syndrome in their problem list or encounter diagnoses seen between March 2007 and May 2020.
15 patients fulfilled the HLH-2004 diagnostic criteria. Of these, 9 were adult patients with an average age of 44.6 years and a mean time to diagnosis of 6.7 days. Causes included 4 cases of viral-induced (EBV, HIV, and CMV), 1 case of T-cell lymphoma, 1 case of familial HLH, and 3 idiopathic cases. The survival rate in adult patients was 66.7%. For our pediatric population, 6 met diagnostic criteria, their average age being 4.7 years with a mean time to diagnosis of 11.5 days. Causes included 5 cases of viral-induced (HSV-2, HHV-6 and CMV), 1 case of malignancy (B-cell leukemia) and 2 cases of familial HLH. The survival rate was 83.3% in this population. Diagnostic criteria of prolonged fever were met in 14 patients. Elevated serum ferritin above 500 ng/mL was present in all patients. Time to diagnosis was shorter in adults by 5 days. However, one complicated case of HLH of the liver took 46 days to diagnose. Treatment with etoposide and dexamethasone was used in 9 patients. Bone marrow transplant was performed in two children and two adults. Two patients died while receiving treatment and one was transitioned to comfort care.
HLH remains rare as 15 cases were identified out of 875,690 patients. Viral causes were the most common etiology in both groups, in contrast to prior studies that found malignancy as the most common inciting factor in adults. Shorter time to diagnosis was seen in adults, along with a higher mortality rate. Overall, diagnosing HLH requires a high index of clinical suspicion, with treatment being dependent on the cause of immune overstimulation.
Keywords: Hemophagocytic Lymphohistiocytosis, Macrophage activation syndrome, Retrospective observational study
Disclosure: All authors indicated they had no financial relationships to disclose.
(150) Immune-mediated chronic macrothrombocytopenia, musculoskeletal abnormalities, and dysmorphic features in a patient with Takenouchi-Kosaki Syndrome
Leigh Stubbs, MD1, Amanda Grimes, MD2, Lisa Forbes, MD3, Lindsay Burrage, MD, PhD2, Tiphanie Vogel, MD, PhD2, Maria Pereira, MD2
1Rheumatology Fellow/Baylor College of Medicine
2Assistant Professor/Baylor College of Medicine
3Assisant Professor/Baylor College of Medicine
CDC42 is a Rho-family GTPase important for proper cell polarization and migration. A spectrum of human disease has been associated with heterozygous, autosomal dominant mutations in CDC42, ranging from neurodevelopmental to hyperinflammatory phenotypes. Takenouchi-Kosaki syndrome (TKS) is a rare, multisystemic disorder associated with CDC42 mutations and typically characterized by delayed psychomotor development, dysmorphic features, brain malformations, macrothrombocytopenia, and lymphedema. We describe a patient with TKS that presented with chronic macrothrombocytopenia responsive to corticosteroids.
A 17-year-old male presented for evaluation and management options for chronic macrothrombocytopenia prior to surgical release of a tethered spinal cord. He had a history of global developmental delay, short stature, asymptomatic external hydrocephalus, bilateral sensorineural hearing loss, recurrent inguinal hernias, obstructive sleep apnea and a left lower lobectomy in infancy for congenital lobar emphysema. Physical examination was significant for dysmorphic features including biparietal prominence, high forehead, upslanting palpebral fissures, low-set ears with thick helices, a long and smooth philtrum, a thin upper lip, midface hypoplasia, and mandibular prognathia. Musculoskeletal examination showed limited abduction of the shoulders, limited extension of the elbows, camptodactyly of the 5th digits, decreased rotation of the hips, severe thoracic dextroscoliosis, and a leg length discrepancy with no sign of inflammatory arthritis. His platelets ranged from 40-60 x 10^3/uL at baseline. Exome sequencing revealed a presumed de novo pathogenic mutation in CDC42 (c.203G>A, p.Arg68Gln). Subsequently, extensive immune phenotyping and a hyperinflammation evaluation were unremarkable. It is not clear if thrombocytopenia in TKS is associated with an increased risk of bleeding. However, due to his planned surgery a trial of 4 mg/kg/day of prednisone was given, to which his platelets promptly responded. He was managed peri-operatively with a short course of corticosteroids.
TKS should be considered in the differential for patients with neurodevelopmental disorders, dysmorphic features, and thrombocytopenia. The chronic macrothrombocytopenia in our TKS patient was rapidly responsive to glucocorticoids, suggesting an immune-mediated component which is consistent with the expanding immune phenotypes reported in patients with CDC42 mutations. As additional patients are described, it may allow clarification of genotype-phenotype associations between CDC42 variants and the role of immunomodulation for these patients.
Keywords: CDC42, Takenouchi-Kosaki syndrome, case report, thrombocytopenia
Disclosure: Lisa Forbes-Satter is a Consultant at ADMA and Grifols, and is an Advisory Board member at CSL Behring, Horizon, and Takeda. Lindsay Burrage works at the Baylor Genetics Laboratory which receives revenue from clinical genetic testing completed at Baylor Genetics Laboratory. All other authors had no financial relationship to disclose.
(151) Developing a Rag1-mutant Murine Model of Rubella vaccine-associated Granulomatous Inflammation
Keith Sacco, MD1, Marita Bosticardo, PhD2, Francesca Pala, PhD3, Stefania Pittaluga, MD, PhD4, Cristina Corsino, .5, LiJuan Hao, PhD6, Ludmila Perelygina, PhD7, Luigi Notarangelo, MD8
1Clinical Fellow/Lab of Clinical Immunology and Microbiology, Immune Deficiency Genetics Section, NIAID, NIH, Bethesda, MD, USA
2Staff Scientist/Lab of Clinical Immunology and Microbiology, Immune Deficiency Genetics Section, NIAID, NIH, Bethesda, MD, USA.
3Postdoctoral Fellow/Lab of Clinical Immunology and Microbiology, Immune Deficiency Genetics Section, NIAID, NIH, Bethesda, MD, USA.
4Senior Research Physician/Lab of Pathology, National Cancer Institute, Bethesda, MD, USA.
5Research Technician/Lab of Clinical Immunology and Microbiology, Immune Deficiency Genetics Section, NIAID, NIH, Bethesda, MD, USA.
6Biologist/Viral Vaccine Preventable Diseases Branch. National Center for Immunization & Respiratory Diseases, Centers for Disease Control & Prevention (CDC), Atlanta, GA, USA
7Senior Researcher/Viral Vaccine Preventable Diseases Branch. National Center for Immunization & Respiratory Diseases, Centers for Disease Control & Prevention (CDC), Atlanta, GA, USA
8Chief/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
Mutated RA27/3 viral sequences (termed Immunodeficiency-related vaccine derived rubella viruses [iVDRVs]) have been identified in granulomas of patients with combined immunodeficiencies. Mice carrying Rag1 hypomorphic mutations (R972Q and R972W) replicate the phenotype of combined immune deficiency with granulomas and/or autoimmunity (CID-G/AI) seen in patients. We used the R972Q and R972W mice to investigate the quality and kinetics of an inflammatory response to iVDRV recovered from skin granuloma.
The Rag1 R972Q mouse is the leakiest model of CID-G/AI. We intramuscularly (IM) injected 4-6-week-old R972Q and WT mice with 10^3 pfu/mouse iVDRV in 50uL followed by an IM injection at week +4 of iVDRV 10^7 pfu/mouse in 50uL. Age-and-sex matched R972Q and WT control mice were injected with equal volume of PBS. Serum was collected at baseline, week +4, +9 and +20, and titer of anti-rubella virus antibodies was measured. Serum cytokines were analyzed at week +20. Mice were euthanized at week +22, and tissue was sent for histology, RNA extraction, and splenic tissue immunophenotyping. In a second experiment, we injected WT and Rag1 R972W mice with 2 doses of 10^7 pfu/mouse iVDRV in 50uL 4 weeks apart. In parallel, R972W and WT control mice were injected with 50 uL of PBS as a control.
No significant weight changes were observed. Titers of anti-rubella virus antibodies at 4 weeks were negative in WT and R972Q mice, however they became positive at +9 weeks (following 2 doses). Mean titer OD values were higher for WT males (2.5) and WT females (2.74) versus R972Q males (0.77) and R972Q females (1.48). In the second experiment, titers were positive in WT mice after 1 dose of 10^7 pfu however remained absent in R972W after the second 10^7 pfu dose. No granulomas were observed on histology. iVDRV RNA was extracted from bone marrow and lymph nodes of WT mice but was weakly positive by PCR (Ct 34-36) in bone marrow and lymph nodes of R972Q mice.
Higher iVDRV doses may be required to produce an inflammatory phenotype. iVDRV in bone marrow suggests a role for development and persistence of immunological memory.
Keywords: Combined Immunodeficiency, Granuloma, Rubella vaccine, Chronic Rubella Infection
Disclosure: All authors indicated they had no financial relationships to disclose.
(152) Fluctuations in Quality of Life and Immune Responses During the Intravenous Immunoglobulin Infusion Cycles
Jordan Abbott, MD, MA1, Sanny Chan, MD, PhD2, Morgan Macbeth, BS3, James Crooks, PhD2, Cathy Hancock, RN4, Vijaya Knight, MD, PhD5, Erwin Gelfand, MD6
1Associate Professor/University of Colorado Aschutz Medical Campus, Children's Hospital colorado
2Assistant Professor/National Jewish Health
3Professional Research Assistant/University of Colorado School of Medicine
4Research Coordinator/National Jewish Health
5Associate Professor of Pediatrics/Section of Allergy & Immunology, Children's Hospital Colorado, University of Colorado School of Medicine, Aurora, CO, USA
6Distinguished Professor/National Jewish Health, University of Colorado School of Medicine
Despite adequate infection prophylaxis, decline in self-reported quality of life throughout the IVIG infusion cycle is a widely reported but infrequently studied phenomenon.
Objective: To better understand this phenomenon, 18 antibody-deficient subjects receiving replacement doses of IVIG were studied over 3 infusion cycles.
Questionnaire data from 6 time points spread over 3 IVIG infusions cycles (infusion day and 7 days after each infusion) assessing well-being were analyzed in conjunction with monitoring the number of regulatory T-cells (Treg) and levels of 40 secreted analytes: primarily cytokines, chemokines, and growth factors at 9 time points over the same time period (immediately before the infusion, 1 hour after infusion completion, and 7 days after the infusion for 3 cycles).
Across all subjects and all infusion cycles, a statistically significant increase in well-being (visual analog scale, VAS, p=0.001) between the day of infusion and 7 days following the infusion was shown. The percentage of CD4+ T-cells that had a Treg phenotype increased from before the infusion to 7 days following the infusion but did not correlate with VAS score improvement. Cytokine level changes were significant between the pre-infusion measurement and 1 hour after infusion and included increases in CCL4 (MIP-1b), CCL3 (MIP-1a), CCL2 (MCP-1), CCL20, TNF-alpha, granzyme B, IL-10, IL-1RA, IL-8, and IFN-gamma and decreases in IL-17E and EGF. These cytokine changes were more dramatic in subjects receiving IVIG for the first time (naïve), and additional increases in IL-6, GM-CSF, and decreases in EGF and CD40L were observed in this group. Non-naïve subjects experienced a decrease in IL-17E that was not seen in the IVIG-naïve group. The decrease in IL-17E was detectable 7 days following the infusion, indicating the impact of IVIG on this cytokine lasts beyond the week following infusion.
The data demonstrate that following IVIG infusion, patients experience significant immediate changes in multiple blood cytokine levels, particularly in those receiving IVIG for the first time. In addition, increases in quality of life and blood Tregs arose 1 week after infusion. The relationships between these observed perturbations remain to be elucidated in further studies.
Keywords: IVIG, Quality of Life, Cytokines, Tregs, IVIG-Naive, CVID, Hypogammaglobulinemia
Disclosure: Sanny Chan received a research grant from CSL Behring. All other authors had no financial relationship to disclose.
(153) Immune implications of SARS-CoV-2 in Nasopharynx
Jose Ordovas-Montanes, PhD1, Carly Ziegler, BS2, Haley Williams, MS3, George Abraham, MD4, Tanya Robinson, PhD5, Michal Senitko, MD5, Meredith Sloan, MD6, Anna Owings, MD6, Hannah Laird, BS7, Taylor Christian, BS7, Adam Parker, MD8, Yilianys Pride, MHA9, Andrew Navia, BS10, Vincent Miao, BS10, Ying Tang, PhD11, Bruce Horwitz, MD12, Alex Shalek, PhD13, Sarah Glover, DO14
1Assistant Professor of Pediatrics/Boston Children's Hospital
2Graduate Student/Massachusetts Institutes of Technology
3Research Specialist/University of Mississippi Medical Center
4Associate Professor/University of Mississippi Medical Center
5Assistant Professor/University of Mississippi Medical Center
6Chief Resident/University of Mississippi Medical Center
7Medical Student/University of Mississippi Medical Center
8Gastroenterology Fellow/University of Mississippi Medical Center
9Division Business Administrator/University of Mississippi Medical Center
10Graduate Student/Massachusetts Institute of Technology
11Post Doctorate Assistant/Boston Children's Hospital
12Associate Professor of Pediatrics/Boston Children's hospital
13Associate Professor/Massachusetts Institute of Technology
14Division Chief/University of Mississippi Medical Center
COVID-19 presents with respiratory symptoms including sore throat, cough and congestion, and can progress to multi-organ system involvement. Nasopharyngeal (NP) epithelial cells express the SARS-CoV-2 spike protein receptor, angiotensin-converting enzyme 2 (ACE2), and infection of the NP appears to be an obligate step in the development of COVID-19. Immune responses initiated in the NP are almost certainly crucial for directing viral clearance but may also play a role in the generation of inflammatory injury observed in a subset of patients infected with the virus. While several studies have identified infected cells within the respiratory epithelium of the lung, neither the target cells for SARS-CoV-2 infection nor the response of target and bystander cells within the nasal mucosa have been characterized in detail. We have previously presented preliminary results employing single-cell RNA sequencing to evaluate cellular composition and targets of viral infection using material isolated on nasal swabs obtained from a cohort of adults with severe COVID-19. Here we report on an extended cohort that includes patients with mild disease as well as those that have recovered from acute disease. We have identified cellular targets of infection including subsets of ciliated and secretory epithelial cells. Further, we have identified a potent type 1 interferon response within a subset of ciliated cells that appears to be a direct response to viral infection, as well secondary anti-viral responses in several diverse cell populations. Finally, we will present initial correlations between cellular targets of infection, cellular subset composition, and gene expression profiles with the severity of COVID-19. These studies provided a framework for understanding the cellular and transcriptional response to viral infection within both epithelial and immune cells of the nasal mucosa, and lay the groundwork for understanding protective cellular immune responses to this disease.
Keywords: COVID-19, SARS-CoV-2, single cell RNA sequencing
Disclosure: All authors indicated they had no financial relationships to disclose.
(154) Granulocyte Transfusions in Patients with Chronic Granulomatous Disease Undergoing Hematopoietic Cell Transplantation or Gene Therapy
Danielle Arnold, MD1, Deepak Chellapandian, MD2, Suhag Parikh, MD3, Kanwaldeep Mallhi, MD4, Rebecca Marsh, MD5, Jennifer Heimall, MD6, Debra Grossman, BS7, Maria Chitty Lopez, MD8, Luis Murguia-Favela, MD9, Andrew Gennery, MBChB, MD10, Farid Boulad, MD11, Erin Arbuckle, MS12, Morton Cowan, MD13, Christopher Dvorak, MD13, Linda Griffith, MD, MHS, PhD14, Elie Haddad, MD, Phd15, Luigi Notarangelo, MD16, Sung-Yun Pai, MD17, Jennifer Puck, MD13, Michael Pulsipher, MD18, Troy Torgerson, MD, PhD19, Elizabeth Kang, MD20, Harry Malech, MD20, Jennifer Leiding, MD21
1Assistant Research Physician/National Cancer Institute
2Attending Physician/Center for Cell and Gene Therapy for Non-Malignant Conditions, Blood and Marrow Transplant Program, John Hopkins All Children’s Hospital
3Attending Physician/Division of Bone Marrow Transplant, Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta, Emory University School of Medicine
4Attending Physician/Fred Hutchinson Cancer Research Center, Seattle Children’s Hospital, The University of Washington School of Medicine
5Attending Physician/Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center
6Assistant Professor/Division of Allergy and Immunology, Children’s Hospital of Philadelphia
7Contract Worker/Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
8Fellow Physician/Division of Allergy and Immunology, Department of Pediatrics, John Hopkins All Children’s Hospital, University of South Florida
9Attending Physician/Section of Hematology/Immunology, Department of Pediatrics, Alberta Children's Hospital, Cumming School of Medicine, University of Calgary
10Attending Physician/Translational and Clinical Research Institute, Newcastle University and Paediatric Immunology and Haematopoietic Stem Cell Transplantation, Great North Children's Hospital
11Attending Physician/Department of Pediatrics, BMT Service, Memorial Sloan Kettering Cancer Center
12Clinical Research Coordinator/Department of Pediatrics, Duke University
13Attending Physician/Pediatric Allergy, Immunology, and Blood and Marrow Transplant Division, University of California San Francisco Benioff Children’s Hospital
14Attending Physician/Division of Allergy, Immunology and Transplantation, National Institute of Allergy and Infectious Diseases, National Institutes of Health
15Attending Physician/Immunology-Rheumatology Division, Department of Pediatrics, University of Montreal
16Chief/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
17Attending Physician/Immune Deficiency-Cellular Therapy Program, National Cancer Institute, National Institutes of Health
18Attending Physician/Blood and Marrow Transplant Program, Division of Hematology, Oncology and Blood and Marrow Transplantation, Children's Hospital Los Angeles, Keck School of Medicine, University of Southern California
19Attending Physician/Experimental Immunology, Allen Institute
20Attending Physician/Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
21Associate Professor/Division of Allergy and Immunology, Department of Pediatrics, University of South Florida at Johns Hopkins-All Children’s Hospital, St Petersburg, FL
Granulocyte transfusions are sometimes used as an adjunctive therapy for the treatment of infection in patients with chronic granulomatous disease (CGD). However, granulocyte transfusions can be associated with alloimmunization, and their role in CGD patients undergoing allogeneic hematopoietic cell transplantation (HCT) and gene therapy (GT) is unknown.
We conducted a retrospective survey of 27 CGD patients who received granulocyte transfusions as a bridge to (within 3 months of) HCT or GT and/or post-HCT using a spreadsheet questionnaire and the Primary Immune Deficiency Treatment Consortium (PIDTC) database.
Median age at HCT or GT was 12 (range 2-25) years. Twelve patients received granulocyte transfusions pre- +/- post-HCT or GT, and 15 patients received granulocyte transfusions post-HCT only (Table 1). Patients received a median of 11 (range 4-75) granulocyte transfusions. Overall response rate was 50% in patients who received granulocytes as a bridge to HCT or GT. Six of 10 (60%) patients for whom testing was performed developed anti-HLA antibodies, and 3 (25%) patients had autoimmune cytopenia within the first 100 days post-HCT or GT. HLA antibodies were not checked for any of the 15 patients who received granulocyte transfusions post-HCT only, but there were no cases of early autoimmune cytopenia. HCT and GT characteristics are shown in Table 2. Five of 25 (20%) patients who underwent HCT experienced primary graft failure. Three of the patients with primary graft failure received granulocyte transfusions pre-HCT. One of the three patients received a graft from a 5/10 HLA-matched related donor and was found to have donor-specific antibodies. The other two patients had high anti-HLA antibody levels but did not have donor-specific antibodies identified. Overall and event-free survival were 36% (CI 11-63%) and 33% (CI 10-59%) in patients who received granulocytes pre- +/- post-HCT or GT versus 100% (p < 0.001) and 87% (CI 56-97%, p=0.005) in patients who received them post-HCT only (Figure 1).
Granulocyte transfusions pre-HCT or GT are associated with a high rate of alloimmunization and may increase the risk of primary graft failure and early severe autoimmune cytopenia post-HCT or GT. Granulocyte transfusions post-HCT appear to be safe.

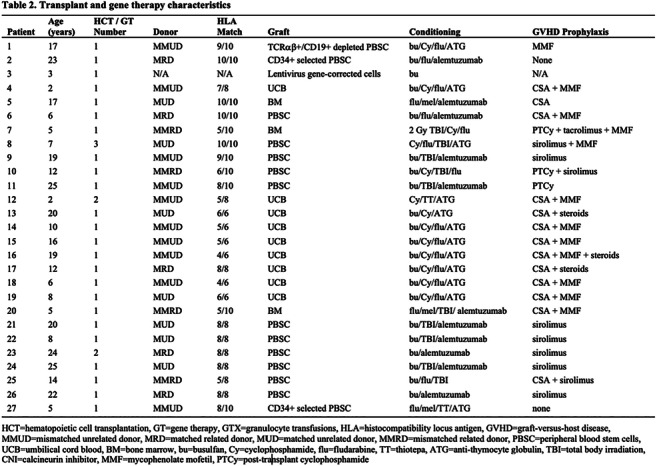

Keywords: granulocyte transfusions, chronic granulomatous disease, alloimmunization, hematopoietic cell transplantation, graft failure
Disclosure: Morton Cowan is an advisory board member of Bluebird Bio, Leadiant, Rocket Pharma; ownership interest in Homology Medicine; and other financial material support from UpToDate. Christopher Dvorak is a consultant for Alexion Inc., Omeros Corp. Elie Haddad is a consultant for Jasper Therapeutics and Rocket Pharma. Michael Pulsipher received other financial material support from Adaptive and Miltenvi; is an advisory board member of Mesoblast; and received speaker honoraria from Novartis. Troy Torgerson is a consultant for CSL Behring and Grifols; received speaker honoraria from Takeda and X4 Pharmaceuticals; and is an advisory board member of Enzyvant. Jennifer Leiding received speaker honoraria from CSL Behring and Horizon Therapeutics and was an advisory board member for Pharming. All other authors had no financial relationship to disclose.
(155) A Case of Undetectable T-cell Receptor Excision Circle (TREC) and Multiple Congenital Anomalies
B. Paige DePriest, MD1, Mamatha Mandava, MD1, Michelle Hudspeth, MD2, Mary Markert, MD, PhD3, Elie Haddad, MD, Phd4, Kelli Williams, MD, MPH2
1Pediatric Hematology & Oncology Fellow/Department of Pediatrics, Medical University of South Carolina, Charleston, South Carolina, USA
2Associate Professor of Pediatrics/Department of Pediatrics, Medical University of South Carolina, Charleston, South Carolina, USA
3Professor of Pediatrics/Department of Pediatrics and Department of Immunology, Duke University Medical Center, Durham, North Carolina, USA
4Attending Physician/Immunology-Rheumatology Division, Department of Pediatrics, University of Montreal
A Caucasian girl was born at 32 weeks to a G3P3 mother with type II diabetes. Prenatal ultrasound revealed ventriculomegaly. Postnatally she had ventriculomegaly, PDA, left renal agenesis, rib anomalies, lumbar scoliosis, hypoparathyroidism with associated hypocalcemia. Initial NBS had inconsistent TREC and repeat was undetectable. Lymphocyte phenotyping revealed T-B+NK+ with CD3+ 26cells/mcl (1484-5327), CD4 15 cells/mcl (733-3181), CD8 6 cells/mcl (370-2555), CD56 681 cells/mcl, CD19 >1000 cells/mcl. CD4 recent thymic emigrants revealed 1% naïve and 99% memory CD4 cells. Mitogen proliferation to phytohemagglutinin was decreased, whereas proliferation to pokeweed mitogen was essentially normal. Serum immunoglobulins were IgG 563, IgA 49, IgM 179, and IgE 25.4 mg/dL. Genetics identified a normal karyotype and microarray. Next generation sequencing primary immunodeficiency panel (Invitae) and subsequently whole exome sequencing failed to identify any pathogenic mutation. At 4 weeks, the patient developed a diffuse maculopapular rash. Skin biopsy was consistent with maternal engraftment, confirmed by chimerism assay (16% maternal cells). By 8 weeks old, her T cells increased (CD3 104, CD4 91, CD8 undetectable). At 4 months, she developed recurrence of her diffuse maculopapular rash, marked diffuse lymphadenopathy, tachypnea, thrombocytopenia and eosinophilia. IgE was now >10,000 IU/mL and T cells increased (CD3 1897, CD4 1740, CD8 undetectable). TCR Vb repertoire identified 25 oligoclonal populations. Lymph node biopsy showed predominant histiocytes with small follicles. Clinically, presentation was most consistent with atypical complete DiGeorge Syndrome associated with maternal diabetes. Research evaluation confirmed normal in vitro differentiation of CD34+ hematopoietic stem cells into T cells on 3D organoid, consistent with functional athymia. The patient clinically decompensated and had immune dysregulation, so she received systemic steroids followed by alemtuzumab with improvement. At 11 months, the patient’s IgE increased to >10,000 IU/mL with intermittent exfoliative rash. A second treatment with alemtuzumab has been ordered. She is currently awaiting cultured thymus tissue implantation and continues on prophylaxis with intravenous immunoglobulin, azithromycin, trimethoprim-sulfamethoxazole, and voriconazole.
This case highlights the impact maternal diabetes can have on the immune system and its association with complete DiGeorge Syndrome. Additionally it further expands the differential diagnoses to consider with an abnormal TREC on NBS.
Keywords: T-cell receptor excision circles (TRECs), Newborn screen (NBS), Congenital athymia
Disclosure: Michelle Hudspeth is an advisory board member of Mesoblast. Mary Markert received a research grant and has patent/intellectual property with Ezyvant Therapeutics. Elie Haddad is a consultant for Jasper Therapeutics and Rocket Pharma. Kelli Williams is an advisory board member of Horizon Therapeutics and Kenota Health and received a research grant from Regeneron Pharmaceuticals. All other authors had no financial relationships to disclose.
(156) Anti-TNF-α therapy leads to recurrent and invasive S. aureus infections in patient with compound heterozygous NOD2 variants
Nissim Stolberg, DO1, James Verbsky, MD, PhD2, John Routes, MD3, Amy Rymaszewski, PHd4, Shaoying Chen, MD5, Jeffrey Woodliff, PhD6, Adrian Miranda, MD7
1Rheumatology Fellow/Medical Collège of Wisconsin
2Associate Professor of Pediatrics, Medicine, Microbiology and Immunology/Medical College of Wisconsin
3Professor of Pediatrics, Medicine, Microbiology and Immunology/Medical College of Wisconsin
4Research Associate II/Medical College of Wisconsin
5Research Associate/Medical College of Wisconsin
6Clinical Immunology Research Laboratory Manager/Medical College of Wisconsin
7Professor/Medical College of Wisconsin
NOD2 gain of function (GOF) variants result in Blau syndrome and loss of function (LOF) variants result in Crohn’s disease. NOD2 facilitates the detection of S. aureus and generation of inflammatory cytokines (i.e. IL-1β, IL-6 and TNF-α) which promotes recruitment of neutrophils and other immune cells required to resolve infection. Herein we describe a case of severe recurrent S. aureus infection following TNF-α inhibition in a patient with compound heterozygous NOD2 variants.
Exome sequencing was performed with Sanger confirmation of subject and parents. NOD2 expression plasmid were modified by site-directed mutagenesis, transfected into HEK293T cells, stimulated with IL-1β and MDP, and IL-8 production measured. PBMCs were stimulated with MDP and IL-8 and TNF-α detected.
A 10-year-old Caucasian male diagnosed with Crohn’s colitis was started on infliximab and within 2 weeks developed recurrent cellulitis, lymphadenitis (12 episodes), and abscesses involving his neck, psoas muscle, axilla, and perianal region due to S. aureus. Psoriasiform skin lesions developed and resolved upon transition to adalimumab, however S. aureus infections continued. Transition to Ustekinumab resolved his colitis and infections.
Exome sequencing revealed two NOD2 variants: Leu1007Profs*2, a known LOF variant associated with Crohn’s disease; and Lys421Asn, a novel variant in NOD2 located in the nucleotide binding domain usually associated with Blau syndrome. Sanger sequencing confirmed the variants, and sequencing of parents demonstrating compound heterozygous inheritance. Transfection studies confirmed LOF of the Leu1007Profs*2 variant, but the Lys421Asn variant was functional. However, stimulating patients PBMCs with MDP resulted in complete loss of IL-8 and TNF-α production, demonstrating loss of function of NOD2.
This case illustrates the important role of NOD2 in defense against S. aureus, as complete loss of NOD2 function, combined with TNF-α blockade, resulted in recurrent and severe S. aureus infections. It remains unclear how the Lys421 variant, which is in the nucleotide binding domain and close to other GOF variants, results in NOD2 unresponsiveness. We propose that TNF-α blockade inhibited redundant inflammatory pathways that are critical in sensing S. aureus in the setting of NOD2 deficiency. These results may have implications in the treatment of Crohn’s disease.
Keywords: NOD2, CARD15, Crohn's, Anti-TNF-α, Staphylococcus aureus
Disclosure: John Routes has contracted research with CSL Behring and Evolve Biologics. All other authors had no financial relationship to disclose.
(157) Facilitated Subcutaneous Immunoglobulin (fSCIG) Usage in Children and Adolescents with Primary or Secondary Immunodeficiency Diseases: A Retrospective Chart Study (RAHPP)
Ulrich Baumann, Prof. Dr. med.1, Maria Fasshauer, Dr. Med2, Christine Pausch, Dr. Med3, Corinna Hermann, Dr. Med4, David Pittrow, Prof. Dr. med.5, Michael Borte, Prof. Dr. med.6
1Department of Paediatric Pulmonology, Allergy and Neonatology/Hannover Medical School
2Senior Physician, Paediatrics and Adolescent medicine/St. Georg Hospital
3Pharmacoepidemiology/GWT-TUD GmbH
4Global Medical Team Lead Immunology IG/Baxalta Innovations GmbH, a Takeda company
5Clinical Research/Pharmacoepidemiology/Technical University of Dresden
6Director, Clinic for Child and Adolescent Medicine/St. Georg Hospital
Pediatric patients ( < 18 years) included in a phase 3 trial of fSCIG in primary immunodeficiency diseases (PID; NCT00814320) showed low rates of infection and systemic reactions with fSCIG treatment. The RAHPP study (DRKS-ID: DRKS00015436) assessed real-world fSCIG utilization in pediatric patients with PID or secondary immunodeficiency diseases (SID) in Germany.
This multicenter retrospective chart review study enrolled pediatric patients ( < 18 years) with PID or SID on fSCIG for ≥6 months from 3 German centers. Informed consent was obtained for all patients. Patient characteristics, immunoglobulin treatment history, and reason for initiating fSCIG were collected at fSCIG initiation. fSCIG administration and experience parameters and adverse drug reaction (ADR) reporting were collected at fSCIG initiation and 6 months. Data were analyzed for the overall population and by age subgroup (children: 0– < 12 years; adolescents: 12– < 18 years).
Thirty patients (16 male/14 female; mean [SD] age: 11.1 [4.9] years) with PID (n=26) or SID (n=4) were enrolled. While 90.0% of patients received their first fSCIG infusion at a hospital/doctor’s office, by 6 months all received infusions at home, with >95.0% of patients infusing every 3–4 weeks. The median dose (volume) was 10.0 g (100.0 mL) at initiation and 15.0 g (150.0 mL) at 6 months. At both timepoints, patients used a median of 1 infusion site. Four local ADRs occurred at initiation, and 6 local and 2 systemic ADRs occurred at 6 months. No serious ADRs or technical infusion issues were reported. The median monthly dose at 6 months was 0.4 g/kg in both children (n=14; mean age: 6.6 years) and adolescents (n=16; mean age: 15.1 years). Most patients in both age groups received their first infusion from a nurse (84.6% and 87.5%, respectively), while at 6 months, children primarily (85.7%) received treatment via caregiver, and adolescents primarily (75.0%) self-administered treatment. This real-world study confirms the feasibility and tolerability of administering fSCIG at home every 3–4 weeks to pediatric patients with PID or SID, with administration parameters similar to clinical trials.
Funding: Takeda Pharmaceuticals International AG funded this study. Baxalta US Inc. (a Takeda company) funded writing support.
Keywords: hypogammaglobulinemia, immunoglobulin, pediatric, real-world study, primary immunodeficiency diseases, secondary immunodeficiency diseases
Disclosure: Ulrich Baumann received speaker Honoria from Takeda, CSL Behring and Octapharma. Corinna Hermann is employed by Baxalta Innovations GmbH – part of Takeda. David Pittrow is a consultant for Actelion Amgen, Baxalta, Biogen and Daiichi Sankyo. All other authors had no financial relationship to disclose.Michael Borte: No financial relationships or conflicts of interest
(158) Persistently Positive SARS-CoV-2 RT-PCR Testing in a Pediatric Patient with X-Linked Agammaglobulinemia
Aaron Westreich, MD1
1Fellow/National Jewish Health
Limited information is available regarding SARS-CoV-2 infection in patients with primary immunodeficiency and the effect this has on disease course and duration. Recommendations for self-isolation after SARS-CoV-2 infection have been informed by retrospective studies that examined the duration of viral shedding and infectivity in patients. Though immunosuppressed patients were included in these studies, patients with primary immunodeficiency were not specifically studied. I present a case of a pediatric patient with X-linked agammaglobulinemia (XLA) who had persistently positive SARS-CoV-2 RT-PCR testing as well as prolonged symptoms.
Our patient was a 12-year-old male with XLA with known hemizygous A523V mutation in the BTK gene. His infection history included recurrent acute otitis media, chronic conjunctivitis and possible sinus disease. Patient was diagnosed with SARS-CoV-2 infection by RT-PCR 3 days after he began developing symptoms including sore throat, nausea, vomiting, headache, dyspnea, non-productive cough and chest pain. He also developed a fever which was not typical of his previous infections. During the first week of illness, he was diagnosed with acute otitis media and conjunctivitis for which he was treated with standard courses of oral and ophthalmic antibiotics, respectively. On day 25 of illness, patient was admitted for persistent symptoms and dehydration and discharged the following morning. He received an IVIG infusion while admitted as his routine monthly infusion had been delayed by 3 weeks. He continued to report persistent symptoms over the next several weeks, particularly fatigue and nausea. His workup during this time included normal inflammatory and cardiac biomarkers, ECG, echocardiogram and chest x-ray. A chest CT revealed diffuse mild bronchiectasis and an 8mm ground-glass nodule in his left upper lobe of unclear etiology. Repeat SARS-CoV-2 RT-PCR testing revealed persistently positive results until 46 days after symptom onset with negative results after 53 days. He was discharged from isolation on day 57 from symptom onset after his second negative PCR test. A significant improvement in symptoms was noted approximately 58 days from symptom onset.
This case highlights the limitations of applying generalized isolation protocols to patients with primary immunodeficiency where the significance of prolonged viral shedding is unknown.

Fig. 1. Cycle threshold (Ct) values of N gene (a nucleocapsid protein) on SARS-CoV-2 real-time reverse transcription polymerase chain reaction are shown in blue. The horizontal dashed red line represents the cutoff for positivity at 40, with undetectable values presented below this line.
Keywords: Immunodeficiency, COVID-19, SARS-CoV-2, Coronavirus, Viral shedding, Immunosuppression, X-linked agammaglobulinemia
Disclosure: The author had no financial relationship to disclose.
(159) Immunoglobulin Initiation Among Patients with Primary Immunodeficiency Diseases (PIDD): A Retrospective Claims-based US Cohort Study
Colin Anderson-Smits, MPH1, Jordan Orange, MD, PhD2, Michelle Park, PharmD3, Zhongwen Huang, PhD4, J. Bradley Layton, PhD5, Mary E. Ritchey, PhD6
1Director Outcomes Research & Epidemiology/Shire US Inc., A Takeda company
2Vagelos College of Physicians and Surgeons/Columbia University Irving Medical Center
3Director, Global Medical Affairs, Rare Immunology/Shire US Inc., A Takeda company
4Sr. Principal Statistician/Takeda Pharmaceuticals International Co.
5Senior Research Epidemiologist/RTI Health Solutions
6RTI Health Solutions/Med Tech Epi, LLC
Immunoglobulin (IG) replacement therapy, administered intravenously (IVIG) or subcutaneously (SCIG), is standard first-line treatment for most forms of PIDD with defective antibody production. SCIG allows for home administration of highly concentrated 20% IG or facilitated 10% IG delivered with recombinant human hyaluronidase. Limited data exist regarding characteristics of patients with PIDD initiating IVIG and SCIG.
To identify and describe demographic, clinical, and treatment characteristics of patients with PIDD in the United States initiating IVIG and SCIG.
This claims-based cohort study identified patients initiating IVIG or SCIG from 2012 to 2018, via diagnosis codes, in IBM® MarketScan® Research Databases. Clinical and demographic characteristics were documented and described for IVIG and SCIG initiators. Risk Vital Sign (RVS), a claims-based weighted algorithm for PIDD risk, was used to assess disease severity.
The study population comprised 15,327 IVIG (57% female, median age 54 years) and 2604 SCIG initiators (69% female, median age 48 years). Compared with IVIG initiators, SCIG initiators were more likely to have asthma (59% vs 38%), chronic obstructive pulmonary disease (71% vs 58%), fibromyalgia (25% vs 20%), and inflammatory bowel disease (12% vs 8%), but less likely to have cancer (11% vs 22%), coronary artery disease/hypertension (45% vs 58%), and peripheral vascular disease (11% vs 16%). Compared with IVIG initiators, SCIG initiators had greater use of high-potency oral antibiotics (37% vs 31%) but lower use of IV antibiotics (23% vs 29%), systemic high-dose corticosteroids (53% vs 67%), antifungals (25% vs 29%), and growth factors (1% vs 10%). Markers of disease severity differed: A greater proportion of IVIG initiators had RVS predicted risks in the medium/high range (46% vs 37%) and pneumonia (28% vs 23%), while recurrent sinusitis was less common compared with SCIG initiators (38% vs 59%).
This exploratory analysis suggests differences between characteristics of patients with PIDD who initiated SCIG versus IVIG with respect to comorbidities, markers of PIDD severity, and previous PIDD treatments. Misclassification of PIDD status was possible, which warrants further research.
Funding: Baxalta US Inc. (a Takeda company) funded this study and writing support.
Keywords: subcutaneous, intravenous, immunoglobulin, primary immunodeficiency
Disclosure: Colin Anderson-Smits, Michelle Park and Zhongwen Huang are employed by Takeda. Joran Orange is a consultant for CSL, Enzyvant, Grifols, Takeda, and an advisory board member of ADMA. J. Bradley Layton is employed by RTI Health Solutions. Mary E. Ritchey has contracted research with AbbVie, Boehringer-Ingelheim, Gilead, Pasage Bio, Regeneron, Takeda and Urovant; consultant of Johnson and Johnson and UCB; and ownership interest in Procter & Gamble.
(160) Model-Based Simulation of Varied Subcutaneous Immune Globulin (Human), 20% (Ig20Gly) Loading and Maintenance Dosing Regimens in Immunoglobulin-Naïve Patients with Primary Immunodeficiency Diseases
Zhaoyang Li, PhD1, Todd Dumas, PharmD2, Barbara McCoy, PhD3, N. Seth Berry, PharmD4, Leman Yel, MD5
1Senior Director, Clinical Pharmacology, Clinical Medicine, PDT/Shire US Inc., a Takeda company
2Director, Research Pharmacometrics, Decision Sciences/IQVIA
3Senior Clinical Director/Baxalta Innovations GmbH, a Takeda Company
4Senior Director, Clinical PK-PD Modeling and Simulation/IQVIA
5Vice President, Head Clinical Medicine, Global Research & Development, PDT/Baxalta US Inc., a Takeda company, University of California
While the pharmacokinetic (PK) profile of Ig20Gly is well characterized in immunoglobulin (IG)-experienced patients with primary immunodeficiency diseases (PIDD), information in IG-naïve patients is limited. This study used population PK modeling to simulate serum IgG profiles and trough levels of Ig20Gly in IG-naïve patients with different dosing regimens for treatment initiation and maintenance in PIDD.
Simulations of Ig20Gly PK profiles in IG-naïve patients were performed using a validated population PK model developed from two pivotal phase 2/3 trials of weekly Ig20Gly in PIDD (NCT01412385, NCT01218438). Two scenario sets were simulated: 1) a 400-mg/kg loading dose and 100-mg/kg weekly maintenance dose, and 2) an 800-mg/kg loading dose and 200-mg/kg weekly maintenance dose. Endogenous IgG baseline levels of 1.5, 2.0, 4.0, and 6.0 g/L for IG-naïve patients were evaluated. Dosing regimens were simulated to determine the time to achieve the therapeutic target IgG trough level (7 g/L).
Across various loading and maintenance regimens, irrespective of endogenous baseline IgG levels, serum IgG approached steady state around Week 12. The time to achieve the target median IgG trough level was dependent on endogenous baseline IgG level and Ig20Gly loading scheme. Achievement of target IgG trough levels was faster when the loading dose was dispensed over 1 week vs 2 weeks. Patients with endogenous IgG < 4 g/L may require the 800-mg/kg loading dose given within 1 week to achieve target IgG levels within 1 to 2 weeks. Both maintenance regimens sustained serum IgG levels above the target level.
These simulations suggest that attainment of serum IgG levels above a protective target threshold (assumed 7 g/L in this study) in IG-naïve patients with PIDD can be achieved within 1 to 2 weeks using appropriate loading-dose regimens and that, after the loading phase, a 400- or 800-mg/kg/month maintenance regimen is adequate to maintain stable IgG levels. Regular serum IgG trough-level monitoring should guide subsequent dose adjustments and dosing intervals.
Funding: Baxalta US Inc. (a Takeda company) funded this study and writing support.
Keywords: subcutaneous immunoglobulin G, pharmacokinetic, primary immunodeficiency diseases, treatment-naïve, Immune Globulin Subcutaneous (Human) 20% Solution
Disclosure: Barbara McCoy and Leman Yel are employed by Takeda. N. Seth Berry is employed by IQVIA. The other authors had no financial relationship to disclose.
(161) The assessment of brain evoked potentials in patients with CAPS
Sukru Cekic1, Aylin Bican Demir2, Sara Kilic3
1Dr./Uludag University Faculty of Medicine, Pediatric Immunology
2Dr./Uludag University Faculty of Medicine, Neurology
3Prof./Uludag University Faculty of Medicine, Pediatric Immunology
CAP syndromes are extremely rare, affecting about one person in every one million people. Sensorinoral complications of the disease reduce the quality of life of patients. Evoked potentials such as ABR and VEP are non-invasive methods used to measure the electrophysiological response of the central nervous system. They have been used in the diagnosis and monitoring of many immune and inflammatory diseases that affect central nervous system. We aimed to assess the neurological involvement and outcome of nine patients with CAPS.
Material and methods: We enrolled nine patients with CAPS from the same family. Visual evoked potentials (VEP) and auditory brainstem responses (ABR) were performed in all patients using Neuropack S1 MEB 9402, Nihon Kohden, Tokyo, Japan.
Results: The female to male ratio was 1.25. The median age at the diagnosis was 25 years (9–65), and the median duration of delay in diagnosis was 24 years (2–58). Six patients were treated with canakinumab and three patients with anakinra. In ABR tests, three cases (6 ears) considered acoustic nerve damage because no brainstem activity occurred at 90 and 95 dB. The prolongation of P100 latency in VEP was detected in five cases (bilateral in two cases,).
Conclusion: There was no previously reported data on the nervous system evoked potentials such as ABR and VEP in patients with CAPS. Supporting audiometry with ABR is more convenient than audiometry alone in recognizing hearing loss. Besides, VEP is also a useful method to monitor the visual acuity of the patients. Subsequent studies are needed to clarify this issue.
Keywords: CAPS, hearing loss, brain evoked potentials
Disclosure: The authors had no financial relationship to disclose.
(162) Which triggers could help to enhance timely identification of primary antibody deficiency (PAD) – a qualitative study from the perspective of the patients
Lisanne M.A. Janssen, MD, MSc1, Kim van den Akker, MD, MSc2, Mohamed A. Boussihmad, BSc3, Esther de Vries, MD, PhD4
1PhD student/Tilburg University
2Pediatric resident/Radboud UMC
3Master student/Radboud University
4Professor, Physician researcher in immunology/Tranzo, Tilburg School of Social and Behavioral Sciences, Tilburg University, Tilburg; Laboratory for Medical Microbiology and Immunology, Elisabeth Tweesteden Hospital, Tilburg, the Netherlands
Patients with predominantly (primary) antibody deficiencies (PADs) commonly develop recurrent respiratory infections which can lead to bronchiectasis, long-term morbidity and increased mortality. Recognizing symptoms and making a diagnosis is vital to enable timely treatment. Studies on disease presentation have mainly been conducted using medical files rather than direct contact with PAD patients. In order to ascertain how patients appraised their symptoms and which factors were involved in a decision to seek medical care, we conducted semi-structured interviews with 14 PAD-patients (11 women, median 44, range 16-68yrs) until saturation of key emergent themes was achieved.
Being always ill featured in all participant stories. Often from childhood onwards periods of illness were felt to be too numerous, too bad, too long-lasting, or antibiotics were always needed to get better. Recurrent or persistent respiratory infectious episodes were the main triggers for patients to seek care. All participants developed an extreme fatigue, described as a feeling of physical and mental exhaustion and thus an extreme burden on daily life that was not solved by taking rest. Despite this, participants tended to normalize their symptoms and carry on with usual activities. Non-immunologists, as well as patients, misattributed the presenting signs and symptoms to common, self-limiting illnesses or other ‘innocent’ explanations.
Participants in a way understand the long diagnostic delay. They know that the disease is rare and that doctors have to cover a broad medical area. But they are more critical about the way the doctors communicate with them. They feel that doctors often don’t listen very well to their patients. The participants’ symptoms as well as the interpretation of these symptoms by their social environment and doctors had a major emotional impact on the participants and a negative influence on their future perspectives.
In conclusion, to timely identify PAD, ‘pattern recognition’ should not only focus on the medical ‘red flags’, but also on less differentiating symptoms, such as ‘being always ill’ and ‘worn out’ and the way patients cope with these problems. And, most important, really listening to the patient, and remembering that things are not always what they seem remains the key.
Keywords: primary antibody deficiency, qualitative research, timely diagnosis, trigger
Disclosure: The authors had no financial relationship to disclose.
(163) Real-World Safety and Tolerability of Facilitated Subcutaneous Immunoglobulin in Pediatric Patients with Primary Immunodeficiency Diseases: Interim Analysis from a Post-authorization Study in Europe
Peter Ciznar, MD, PhD1, Marion Roderick, MD2, Helena Schneiderová, MUDr.3, Milos Jesenak, MD, PhD4, Gergely Kriván, MD, PhD5, Nicholas Brodszki, MD, PhD6, Stephen Jolles, MD, PhD7, Katharina Fielhauer, n/a8, Shumyla Saeed-Khawaja, MD9, Barbara McCoy, PhD10, Leman Yel, MD11
1Associated Professor of Pediatrics/Children University Hospital Bratislava, National Institute of Children’s Diseases
2Department of Paediatric Immunology/Bristol Royal Hospital for Children
3Department of Pediatrics/Faculty of Medicine Masaryk University
4Department of Pediatrics/Jessenius Faculty of Medicine, Comenius University in Bratislava, University Hospital in Martin
5Department of Pediatric Hematology & Stem Cell Transplantation/Szent László Hospital
6Department of Pediatric Oncology/Hematology/Immunology/Skåne University Hospital
7Clinical Lead – Immunodeficiency Centre for Wales/University Hospital of Wales
8Associate Clinical Director/Baxalta Innovations GmbH, a Takeda company
9Associate Clinical Director/Baxalta US Inc., a Takeda company
10Senior Clinical Director/Baxalta Innovations GmbH, a Takeda Company
11Vice President, Head Clinical Medicine, Global Research & Development, PDT/Baxalta US Inc., a Takeda company, University of California
In a phase 3 trial in primary immunodeficiency diseases (PIDD; NCT00814320), facilitated subcutaneous immunoglobulin (fSCIG), a dual-vial unit containing immunoglobulin G (IgG) 10% and recombinant human hyaluronidase (rHuPH20), was effective and bioequivalent to intravenous immunoglobulin with fewer systemic adverse events. This ongoing phase 4, prospective, European post-authorization study is evaluating fSCIG safety, tolerability, and immunogenicity in pediatric patients with PIDD (NCT03116347).
Patients aged 2 to < 18 y with PIDD receiving immunoglobulin therapy from 16 European centers enrolled with informed consent and received fSCIG for up to 3 years (Epoch 2). fSCIG-pretreated and fSCIG-naïve (new starters) patients were included. New starters initiated fSCIG with a ramp-up of up to 6 weeks (Epoch 1). fSCIG safety, immunogenicity, tolerability, use, and IgG trough-level data were collected approximately every 3 months. An interim analysis was preplanned for when 75% of patients completed 1 year in Epoch 2 (data cut-off: May 14, 2020).
Of 42 patients (81.0% male) enrolled, 23 were fSCIG-naïve and 19 were fSCIG-pretreated (mean age: 10.3 and 11.7 y, respectively). At interim analysis, patients received a mean of 12.5 infusions. A total of 42 treatment-related adverse events occurring after first fSCIG dose (AEs), excluding infections, were reported in 12 patients; most AEs were mild. Treatment-related AEs occurred more frequently in fSCIG-naïve (33 local, 3 systemic treatment-related AEs in 10 patients; 1.7 events/patient-year [PY]) than fSCIG-pretreated (2 local, 4 systemic-related AEs in 2 patients; 0.3 events/PY). No serious treatment-related AEs were reported. No patients developed positive anti-rHuPH20 antibody titers (≥1:160). Patients received a median of 1.2 infusions/month, with fSCIG-pretreated receiving higher median infusion volumes/site than fSCIG-naïve (150 vs 80 mL/site) and fewer interrupted/adjusted/stopped infusions (4.9% vs 9.3% of infusions). Mean IgG trough levels at enrolment and 12 months were 9.6 and 8.2 mg/dL, respectively.
This interim analysis supports the long-term safety of fSCIG in pediatric patients with PIDD, with a safety and tolerability profile consistent with previous clinical studies, and indicates that incidence of local AEs declines with fSCIG treatment duration.
Funding: Baxalta US Inc. (a Takeda company) funded this study and writing support.
Keywords: hypogammaglobulinemia, immunoglobulin, pediatric, safety, immunogenicity, primary immunodeficiency diseases
Disclosure: Peter Ciznar received speaker honoraria from Shire/Takeda. Stephen Jolles is an advisory board member of Biocryst, Biotest, Grifols, LFB, Pharming; received a research grant from CSL Behring and Takeda; and speaker honoraria from Octapharama, Takeda, UCB Pharma; and has ownerhip interest in Zarodex. Katharina Fielhauer, Shumyla Saeed-Khawai, Barbara McCoy and Leman Yel are employed by Takeda. The other authors had no financial relationship to disclose.
(164) A Case of Drug Allergy – Or is it?
Yatyng Chang, MD1, Stephania Lairet, MD2, Jose Calderon, MD3, Vivian Hernandez-Trujillo, MD3
1Allergy Immunology Fellow/Nicklaus Children's Hospital
2Pediatric Resident/Nicklaus Children's Hospital
3Allergy Immunology Attending/Nicklaus Children's Hospital
The Jarisch-Herxheimer reaction is a systemic inflammatory reaction secondary to the release of endotoxins. It results in high levels of TNF훂, IL6, and IL8. Symptoms are similar to anaphylaxis and occur immediately after administration of antibiotics, thereby often being confused as a drug allergy. Here we present a child with E. coli urosepsis and likely Jarisch-Herxheimer reaction.
To describe a case of systemic cytokine release and septic shock not responding to treatment for anaphylaxis.
This patient initially presented with 2 days of fever, back pain, and dysuria. She was found to have left pyelonephritis and treated with cefotaxime. Soon after antibiotic administration, she developed tachycardia, difficulty breathing, rash, and hypotension. She was given multiple doses of epinephrine and transferred to the PICU. Given concern for drug allergy, she was changed to meropenem and developed hypotension requiring epinephrine. She was subsequently labeled as allergic to penicillins, cephalosporins, and meropenem. When consulted, the allergy team diagnosed septic shock and suspected Jarisch-Herxheimer reaction, as the patient was not improving despite aggressive treatment for anaphylaxis. She improved clinically once treatment for septic shock was initiated.
In this age of growing antibiotic resistance, a misdiagnosis of drug allergy, especially in a child, can greatly affect and restrict patient care. It can turn simple outpatient treatments into long inpatient stays. We believe that in patients not responding to appropriate treatment for anaphylaxis, septic shock and Jarisch-Herxheimer reaction should be considered. The patient should be followed closely with consideration of antibiotic testing and/ or reintroduction.
Keywords: Jarisch-Herxeimer, Drug allergy, Drug reaction
Disclosure: Disclosures: Vivian Hernandez-Trujillo is an advisory board member of Covis, CSL Behring, DBV, Kaleo, Takeda, and US WORLD MEDS. All other authors had no financial relationships to disclose.
(165) A Novel Intronic Genetic Variant in an Adolescent with Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy Syndrome
Mohamed Ebrahim, MD1, Rachel Eisenberg, MD2, Melissa Wasserstein, MD3, Katie Gallagher, MS, C.G.C.4, Arye Rubinstein, MD, PhD5
1Allergy and Immunology fellow in Ttraining/Montefiore medical center
2Attending in Allergy and Immunology/Montefiore Medical Center
3Chief of the department of Pediatric Genetic Medicine/Montefiore medical center
4Certified Genetic Counselor/Montefiore medical center
5Chief of the Pediatrics Division of the Department of Allergy & Immunology/Montefiore Medical Center
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) is a rare autosomal recessive disease caused by bi-allelic pathogenic variants in the Autoimmune Regulator (AIRE) gene. AIRE is a transcriptional regulator, critical in the expression of tissue-specific antigens (TSAs). Thymocytes with high affinity to TSAs undergo apoptosis, thereby eliminating escape of self-reactive T cells to the periphery. Aberrations in this process lead to multiorgan autoimmunity, commonly in endocrine tissues and susceptibility to chronic mucocutaneous candidiasis. We describe a boy with classic features of APECED whose diagnosis was confirmed 16 years later by whole genome sequencing (WGS).
A 17-year-old male of mixed European, including Finnish, ancestry presented at 14 months, with a hypotensive crisis and severe hyponatremia and was diagnosed with adrenal insufficiency. Over the years, he developed esophageal candidiasis, hypothyroidism, hypoparathyroidism, vitiligo, enamel dysplasia, alopecia, uveitis, autoimmune hepatitis and poor bowel motility. His management included antifungal therapies, calcium supplementation, oral fludrocortisone and tacrolimus. At 11 years, he developed recurrent sinopulmonary infections. Chest imaging showed interstitial pneumonitis, bilateral nodular ground glass and tree-in-bud opacities, mild bilateral lower lobe bronchial wall thickening and eventual bronchiectasis. Immune work up revealed poor response to streptococcus pneumonia vaccines. His infection burden improved with gammaglobulin replacement. The bowel motility abnormality led to failure to thrive requiring gastro-jejunal tube insertion and a cecostomy.
Genetic testing as a young child demonstrated a heterozygous, maternally inherited, pathogenic variant (c.769C>T) in AIRE. Whole exome sequencing (WES) at age 16 years did not identify any additional pathogenic variants. At 17 years, he enrolled in NYCKidSeq, an NIH funded WGS, that identified a second, paternally inherited, intronic AIRE variant of uncertain significance (c.1504-818G>A). The Genome Aggregation Database (gnomAD) supports the rarity of this allele, and in silico tools predict conflicting evidence of pathogenicity. However, In the setting of bi-allelic variants in a patient with a consistent clinical diagnosis, it highly likely that they are causative of APECED.
In this era of evolving genetic testing, this case is a reminder of the limitations of genetic panels and WES. In patients with highly suspicious clinical findings, follow up with WGS may be of imperative significance.
Keywords: APECED, AIRE, Intronic, gene, Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy
Disclosures: Melissa Wasserstein has contracted research with BioMarin, National Niemann Pick Disease Foundation, Orchard Therapeutics, Retrophin, Sanofi Genzyme and Ultragenyx. All other authors had no financial relationships to disclose.
(166) Utilizing Newborn Screening to Facilitate Immunologic Diagnosis of 22q11.2 Deletion Syndrome
Erika Cherk, MD1, Victoria Dimitriades, MD, FAAAAI, FACAAI2
1Resident Physician/UC Davis Medical Center
2Clinical Professor of Pediatrics; Division Chief, Pediatric Allergy, Immunology & Rheumatology/UC Davis Medical Center
Historically, 22q11.2 deletion syndrome has been underdiagnosed. However, since initiation of Severe Combined Immune Deficiency newborn screening (SCID-NBS) in California in 2010, immunologists have become involved in earlier identification of cases of 22q11.2 deletion syndrome.
A clinically well-appearing 10 month-old, ex-28 WGA male diagnosed with 22q11.2 deletion syndrome after a positive SCID NBS prompted further immunological workup. He did not have any of the cardiac (congenital heart disease), facial (cleft palate, hypertelorism, posteriorly rotated ears), or endocrinologic abnormalities (hypoparathyroidism) commonly associated with 22q11.2 deletion syndrome.
SCID NBS (measuring numbers of T cell receptor excision circles [TRECs]), CBC, flow cytometry, tracking lymphocyte subpopulations (from birth until diagnosis), Primary Immunodeficiency panel genetic testing
On day 16 of life, the patient was found to have positive SCID newborn screening with low TREC numbers. Subsequent immunologic testing showed mild hypogammaglobulinemia (which resolved over time) and T cell lymphopenia (which persisted over time). Genetic testing identified a pathogenic deletion in TBX1, a gene associated with 22q11.2 deletion syndrome.
Without SCID newborn screening and immunologic follow-up, this case of 22q11.2 deletion syndrome without typical clinical features may not have been detected. Ultimately, earlier detection of 22q11.2 deletion syndrome will greatly improve health and quality of life for affected children, allowing them to receive appropriate preventative care (e.g. associated disease tracking, developmental assessments, infection prevention) prior to developing complications related to their genetic disorder.
Keywords: 22q11.2 deletion syndrome, primary immunodeficiency, SCID newborn screening, genetic testing
Disclosures: Victoria Dimitriades is received speaker honoraria from Grifols and is an advisory board member of Horizon Therapeutics. The other author had no financial relationships to disclose.
(167) Dysregulation of multiple cytokine signaling pathways underlies the pleiotropy of SOCS1 haploinsufficiency
Julia Körholz1, Anastasia Gabrielyan, PhD2, John Sowerby, PhD3, Felix Boschann, Dr. med.4, Lan-Sun Chen, PhD5, Diana Paul6, Karsten Conrad, PD. Dr. med7, Nadja Roeber8, Min Ae Lee-Kirsch, Prof. Dr. med.9, Ken Smith, Prof.10, Alexander Dalpke, Prof. Dr. med.11, Catharina Schütz, Prof. Dr. med.12, William Rae, MSc13
1Resident/Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
2PostDoc/Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
3Postdoctoral Research Associate/Cambridge Institute of Therapeutic Immunology and Infectious Disease, Jeffrey Cheah Biomedical Centre, Cambridge Biomedical Campus, Cambridge, UK. / Department of Medicine, University of Cambridge, Cambridge Biomedical Campus, Cambridge, UK.
4Physician/Institute of Medical Genetics and Human Genetics, Charité - Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health
5PostDoc/Institute of Medical Microbiology and Hygiene, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
6Lab Assistant/Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany.
7Head of the laboratory, scientific staff/Institute of Immunology, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
8Scientific Staff/Institute of Immunology, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden,
9Head of the clinical research group 249/Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany.
10Professor of Medicine/Cambridge Institute of Therapeutic Immunology and Infectious Disease, Jeffrey Cheah Biomedical Centre, Cambridge Biomedical Campus, University of Cambridge, Cambridge, UK / Department of Medicine, University of Cambridge, Cambridge Biomedical Campus, Cambridge, UK.
11Head of Infection & Immunity, Microbiome Studies/Institute of Medical Microbiology and Hygiene, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
12Professor at the Department of Pediatric immunology/Department of Perdiatrics, Uniklinikum Carl Gustav carus Dresden
13PhD Student/Cambridge Institute of Therapeutic Immunology and Infectious Disease, Jeffrey Cheah Biomedical Centre, Cambridge Biomedical Campus, Cambridge, UK. / Department of Medicine, University of Cambridge, Cambridge Biomedical Campus, Cambridge, UK.
Inborn errors of immunity (IEI) present diagnostic and therapeutic challenges, as they are characterized by highly diverse phenotypic spectrums, genetic heterogeneity and incomplete penetrance (1,2). Suppressor of cytokine signaling 1 (SOCS1) is a key negative regulator of cytokine signaling, limiting phosphorylation of different targets, including signal transducer and activator of transcription (STAT) 1 (3). SOCS proteins exert their action by interacting with Janus kinase (JAK) and non-receptor tyrosine kinase-2 (TYK2) kinases with varying affinities across different cell surface cytokine receptors3. SOCS1 haploinsufficiency was recently identified as an autosomal dominant monogenic IEI (4-6).
We assessed impacts of reduced SOCS1 expression across multiple immune cell pathways utilizing patient cells and CRISPR/Cas9 edited primary human T cells. We conducted molecular genetics analysis, measurements of interferon signature, immunoblotting and flow cytometric analysis.
SOCS1 haploinsufficiency phenotypes straddle across the International Union of Immunological Societies classification of IEI. Reduced SOCS1 expression leads to dysregulation of multiple intracellular pathways in immune cells. STAT1 phosphorylation is enhanced, comparably with STAT1 gain-of-function mutations, and STAT3 phosphorylation is similarly reduced with concurrent reduction of Th17 cells. Furthermore, nuclear factor 'kappa-light-chain-enhancer' of activated B-cells (NF-κB)1 activity appears increased in SOCS1 haploinsufficient cells. Reduced E3 ligase functions of SOCS1 lead to increased focal adhesion kinase (FAK) in immune cells resulting in increased AKT (protein kinase B) and p70 ribosomal protein S6 kinase phosphorylation. We also find increased Toll-like receptor responses in SOCS1 haploinsufficient patients.
SOCS1 haploinsufficiency is a pleiotropic monogenic IEI. Dysregulation of multiple immune cell pathways may explain the variable clinical phenotype associated with this new condition. Knowledge of these additional dysregulated immune pathways is important when considering the optimum management for SOCS1 haploinsufficient patients.
References:
1. Bousfiha A, et al. J Clin Immunol. 2017.
2. Gruber C, Bogunovic D. Human Genetics. 2020.
3. Yoshimura, A. et al. Front Immunol. 2012.
4. Thaventhiran J, et al. Nature. 2020.
5. Lee PY, et al. J Allergy Clin Immunol. 2020.
6. Hadjadj J, et al. Nat Commun. 2020.
Keywords: Inborn error of immunity, primary immunodeficiency, SOCS1
Disclosures: All authors indicated they had no financial relationships to disclose.
(168) CTLA-4 and CVID: Analysis of USIDNET Patients
Sara Ghannam, MD1, John McDonnell, MD, FAAP1, Seth Rotz, MD2, James Fernandez, MD, PhD3
1Allergy Immunology Fellow/Cleveland Clinic Foundation
2Staff, Pediatric Hematology Oncology and Blood and Bone Marrow Transplant/Cleveland Clinic Foundation
3Staff, Allergy and Clinical Immunology/Cleveland Clinic Foundation
Common variable immunodeficiency disorder (CVID) is common, affecting up to 1 in 25,000 patients. Improvements in medical science have led to the discovery of several monogenic defects of this clinical syndrome, which is broadly characterized by decreased antibody levels, decreased antibody function, and immune dysregulation. One recently described monogenic defect of CVID is a pathogenic mutation in cytotoxic T lymphocyte associated protein four (CTLA-4), a soluble factor produced by regulatory T cells.
To examine clinical characteristics of a cohort of subjects in the United States Immunodeficiency Network (USIDNET) registry with known CTLA-4 mutations.
USIDNET is a registry of data from multiple participating sites concerning patients with primary immune deficiencies in the US and Canada. Information was obtained on patients diagnosed with CTLA-4 mutations and entered into the registry through 2019.
There were a total of 22 subjects with diagnosed CTLA-4 mutations in the registry, all born between 1944 and 2018. Of these, 13 were male (59.1%). The vast majority were white (90.9%). The average age of onset of symptoms was 13.5 years, and the average time to molecular diagnosis was 11.4 years. 68.2% of patients had a family history of primary immunodeficiency and 95% of subjects had a single pathogenic mutation (CTLA-4). The most common infections reported were sinopulmonary (49.3%), cutaneous (25.4%), and diarrheal (9.4%). The top immunomodulators prescribed by category included m-tor inhibitors (33%), abatacept (24.7%), and corticosteroids (15.3%). Other immunomodulators used include rituximab, colony stimulators, mycophenolate, calcineurin inhibitors, phosphodiesterase-4 inhibitors and antimalarials. One subject underwent a stem-cell transplant, and 4 subjects died during the period of observation.
Data from the USIDNET registry summarizes the currently reported clinical characteristics of patients with CTLA-4, a monogenic cause of CVID.

Figure 1: Demographic data presented as mean (SD) for continuous measures, and percent (%) for categorical measures.

Figure 2: Infection types in CTLA-4 patients
Keywords: CTLA4, CVID, Primary Immunodeficiency, USIDNET
Disclosures: All authors indicated they had no financial relationships to disclose.
(169) Medium-to-Long-Term Safety, Efficacy, and Tolerability of Subcutaneous Human Immunoglobulin 16.5% in Patients with Primary Immunodeficiencies
Syed Rizvi, PharmD1, Sudhir Gupta, MD, PhD2, Roger Kobayashi, MD3, Ai Lan Kobayashi, MD4, Isaac Melamed, MD5, Jose Fernando Mandujano, MD6, Bob Geng, MD7, Syed Rehman, MD8, Bruce Ritchie, MD, FRCPC9, Shardae Young, PharmD10
1Associate Director Medical Affairs, Immunology/Octapharma
2Professor of Medicine, Pathology & Laboratory Medicine, and Microbiology & Molecular Genetics/University of California, Irvine
3Allergist Immunologist/Allergy, Asthma, and Immunology Associates PC
4Allergist Immunologist/Midlands Pediatrics PC
5Allergist Immunologist/IMMUNOe Research Centers
6Pediatric Pulmonologist/Pediatric Pulmonary Associates of North Texas, P.A.
7Allergist Immunologist/Rady's Children's Hospital San Diego
8Allergist Immunologist/Asthma and Allergy Center
9Hematologist/University of Alberta
10Medical Science Liaison, Immunology/Octapharma
Patients with primary immunodeficiencies (PID) require life-long replacement therapy with immunoglobulins (Ig) to prevent severe infections and irreversible complications. In this follow-up study, medium-to-long term safety, efficacy and tolerability of a subcutaneous human normal immunoglobulin 16.5% were evaluated in adult and pediatric PID patients.
Methods: A prospective, open-label, non-controlled, single-arm, multicenter phase 3 study involving 27 adult and pediatric patients with PID was conducted in North America to assess medium-to-long term safety, efficacy and tolerability of SCIg 16.5% as a follow-up extension study to the pivotal Phase 3 study. PID patients received weekly or bi-weekly doses of SCIg 16.5% (Cutaquig) with the option of increased infusion volume (up to 60 mL/site) and rate (up to 240 mL/hr/all sites) over a 42-month period.
Twenty-seven patients with a mean age of 39.26 years receiving a total of 2,777 SCIg infusions were included in the Full Analysis Set. The mean actual dose administered was 0.169 g/kg. One serious bacterial infection was recorded in 1 adult patient, for an overall rate of 0.018 SBIs per person-year. Among the 119 other infections observed, only 2 (1.7%) were graded as severe (pyrexia of unknown origin and E.coli bacteremia). All other infections were mild (61.3%) or moderate (37%) in intensity. Two patients had to be hospitalized on 3 occasions due to infection for an overall rate of hospitalization of 0.185 days. Of the 204 reported adverse events (excluding infusion site reactions and infections), only 7 (headache, nausea, chills, pyrexia) were assessed as being related to the study drug; all of which were non-serious. Overall, 44.4% of patients experienced infusion site reactions, with most being mild-to-moderate in intensity. In the majority of infusions (96.6%), there were no infusion site reactions.
: The efficacy of this SCIg 16.5% product was confirmed by an overall rate of 0.018 SBIs which is far below the FDA requested threshold of 1. The product was well tolerated, with 96.6% of all infusions having no infusion site reactions.
This study demonstrated that subcutaneous human normal immunoglobulin 16.5% is well-tolerated, safe and effective for long-term use in patients with primary immunodeficiency diseases.
Keywords: Primary immunodeficiency, Subcutaneous immunoglobulin, infections, immunoglobulins
Disclosures: Syed Rizvi and Shardae Young are employed by Octapharma. Sudhir Gupta and Isaac Melamed received contract research from Octapharma and Shire. Bob Geng was an advisory board member for Biocryst and received speaker honoraria from Grifols, GSK, Horizon, Kedrion, Octapharma, Optinose, Regeneron, Sanofi, and Takeda; he was a consultant for RMS. Roger Kobayashi received speaker honoraria from IgNS and a research grant and speaker honoraria from Octapharma. Ai Lan Kobayashi received a research grant from Octapharma. Bruce Ritchie is a consultant for Octapharma.
(170) Inflammatory Markers Correlate with Clinical Response to Immunomodulatory Agents in MIS-C: A Case Series
Sylwia Nowak, MD1, Andrew McGurn, MD2, Latania Logan, MD3, Colleen Nash, MD, MPH3, Julie Wohrley, MD3, Sindhura Bandi, MD3
1Allergy/Immunology Fellow/Rush University Medical Center
2Resident/Rush University Medical Center
3Attending/Rush University Medical Center
Multisystem Inflammatory Syndrome in Children (MIS-C) is a potentially severe disease with a varied clinical presentation, elevated markers of inflammation, and temporal relationship to a previous SARS-CoV-2 infection.
We report on 4 pediatric cases of multi-system inflammatory dysfunction in setting of SARS-CoV-2 positive IgG titers.
Patient 1 is a previously healthy 5-year-old who rapidly decompensated with cardiogenic shock and respiratory failure after presenting with a 2-day history of infectious gastroenteritis. He received IVIG and corticosteroids, with continued fever, organ involvement, and persistent elevation of inflammatory markers. Anakinra was administered with reduction in inflammatory markers and clinical improvement.
Patient 2 is an ex-24 week 18-month-old with history of IVH and BPD who presented with status epilepticus, hypotensive shock, and respiratory failure in the setting of 3-day history of fevers. She received IVIG and corticosteroids with clinical improvement and downtrending inflammatory markers thereafter.
Patient 3 is a previously healthy 7-year-old who presented with 5-day history of fevers, worsening abdominal and chest pain, altered mental status, and purpura requiring VA-ECMO and CRRT. After treatment with IVIG due to continued organ dysfunction, he was treated with anakinra and methylprednisolone that led to eventual improvement in inflammatory markers and clinical status.
Patient 4 is a previously healthy 12-year-old who presented with a 4-day history of fever, headache, and myalgias who developed persistent hypotension and troponin leak. She clinically improved after treatment with IVIG and methylprednisolone with normalization of her troponin and other inflammatory markers.
Patients with hyperinflammation and organ dysfunction due to MIS-C necessitate a guideline-driven multidisciplinary approach, which incorporates a stepwise approach to initiation of immunomodulatory agents specific to each patient’s clinical course, laboratory trends, and response to treatment.
Keywords: MIS-C, SARS-CoV-2, multidisciplinary, immunomodulatory
Disclosure: All authors indicated they had no financial relationships to disclose.
(171) Heterozygous AIRE Mutation in a Family with Multiple Autoimmune Organ Involvement
Asena Pınar Sefer, MD1, Burcu Kocamis Kolukisa, MD1, bengü akçam, pHd2, Yasemin Kendir Demirkol, MD3, Elif Karakoç Aydıner, MD4, ahmet Ozen, MD4, Safa Baris, MD5
1Clinical Fellow/Marmara University Hospital, Department of Pediatric Immunology and Allergy
2PhD/Marmara University Pendik Research and Training Hospital
3./Umraniye Research and Training Hospital, Department of Pediatric Genetic Diseases
4professor/Marmara University Pendik Research and Training Hospital
5Professor of Pediatrics/Marmara University Hospital, Department of Pediatric Immunology and Allergy
The AIRE (Autoimmune regulatory) gene plays an important role in controlling immune tolerance and preventing autoimmunity. APS -1 (Autoimmune polyendocrinopathy syndrome) is a syndrome that is caused by autosomal recessive mutations in the AIRE gene and presented with multiple autoimmune tissue and organ involvement. Recently, it is unveiled that autosomal dominant heterozygous mutations in the AIRE gene have also been cause autoimmune diseases like autoimmune thyroiditis, adrenal insufficiency, pernicious anemia and vitiligo like APS-1 but occurring at a older age.
A 16-year-old male patient who had symptoms of celiac disease, autoimmune hepatitis, vitiligo, and muscle weakness was immunologically evaluated due to his history of frequent fever and lymphopenia. The patient's immunoglobulin levels were normal and lymphocyte subgroup examination was ; CD3: 74% (absolute: 592-low), CD3CD4: 35% (absolute: 280-low), CD3CD8: 35% (absolute: 80-low), CD19: 12% (absolute: 96-low), CD16- 56: 10% (absolute: 80-low). Anti-parietal antibody positivity was detected in the autoimmune panel. The regulatory T cell ratios of the patient were lower than the control. Genetic analysis was done and C.926T> C, p.Ile309 * heterozygous change in the AIRE gene was detected. Sanger sequence analysis was performed to the patient's family. Similar heterozygous changes were detected in the AIRE gene of the mother who had no complaints, but lymphopenia and anti-thyroid autoantibody positivity were detected in her examinations.
AIRE gene mutations can cause multiple autoimmune organ involvement in addition to autoimmune polyendocrinopathy syndrome. In these patients, autosomal dominant transition, which is the rare, can be detected. This work was supported by the Scientific and Technological Research Council of Turkey to (318S202).
Keywords: AIRE, autoimmunity, heterozygous mutation, APDS
Disclosure: All authors indicated they had no financial relationships to disclose.
(172) Activated Phosphoinositide 3-Kinase Delta Syndrome 1: clinical and immunological data from an Italian cohort of eight patients
Giulio Tessarin, MD1, Manuela Baronio, PhD2, Stefano Rossi, MD1, Luisa Gazzurelli, PhD3, Michael Colpani, MD1, Fabio Cardinale, MD4, Baldassarre Martire, MD5, Letizia Brescia, MD6, Giorgio Costagliola, MD7, Laura Luti, MD8, Gabriella Casazza, MD8, Maria Cristina Menconi, MD8, Francesco Saettini, MD9, Raffaele Badolato, MD10, Marco Chiarini, MSc11, Daniele Moratto, MSc12, Antonella Meini, MD13, Maria Pia Bondioni, MD14, Silvia Giliani, MD15, Alessandro Plebani, MD, PhD16, Vassilios Lougaris, MD17
1Pediatric Resident/Pediatrics Clinic and “A. Nocivelli” Institute for Molecular Medicine, Department of Clinical and Experimental Sciences, University of Brescia, ASST Spedali Civili of Brescia, Brescia, Italy
2PhD/Pediatrics Clinic and “A. Nocivelli” Institute for Molecular Medicine, Department of Clinical and Experimental Sciences, University of Brescia, ASST Spedali Civili of Brescia
3PhD/Pediatrics Clinic and “A. Nocivelli” Institute for Molecular Medicine, Department of Clinical and Experimental Sciences, University of Brescia, ASST Spedali Civili of Brescia, Brescia, Italy
4Professor of Pediatrics/Allergy, Immunology and Pediatric Pulmonology Unit, “Policlinico-Giovanni XXII” Hospital, University of Bari, Bari, Italy;
5MD/Pediatrics Unit, Monsignor Dimiccoli Hospital, Barletta, Italy
6MD/Pediatric Oncohematology Unit, Ospedale Santissima Annunziata, Taranto, Italy
7Pediatric Resident/Division of Pediatrics, University of Pisa, Pisa, Italy
8MD/Pediatric Oncology and Hematology, University of Pisa, Pisa, Italy
9MD/Pediatric Hematology Oncology Unit, Department of Pediatrics, University of Milano Bicocca, MBBM Foundation, Monza, Italy
10Full Professor of Pediatrics/Pediatrics Clinic and “A. Nocivelli” Institute for Molecular Medicine, Department of Clinical and Experimental Sciences, University of Brescia, ASST Spedali Civili of Brescia, Brescia, Italy
11Msc/Clinical Chemical Analysis Central Laboratory, ASST Spedali Civili of Brescia, Brescia, Ital
12MSc/Clinical Chemical Analysis Central Laboratory, ASST Spedali Civili of Brescia, Brescia, Italy
13MD/Pediatrics Clinic, ASST-Spedali Civili of Brescia, Brescia, Italy
14MD/Pediatric Radiology, University of Brescia, ASST Spedali Civili di Brescia, Brescia, Italy;
15Assistant Professor/Cytogenetic and Medical Genetics Unit and “A. Nocivelli” Institute for Molecular Medicine, Department of Molecular and Translational Medicine, University of Brescia, ASST Spedali Civili of Brescia, 25123 Brescia,
16Full Professor of Pediatrics/ediatrics Clinic and “A. Nocivelli” Institute for Molecular Medicine, Department of Clinical and Experimental Sciences, University of Brescia, ASST Spedali Civili of Brescia, Brescia, Italy
17Assistant Professor of Pediatrics/ediatrics Clinic and “A. Nocivelli” Institute for Molecular Medicine, Department of Clinical and Experimental Sciences, University of Brescia, ASST Spedali Civili of Brescia, Brescia, Italy
Activated Phosphoinositide 3-Kinase Delta Syndrome 1 (APDS-1) is a recently described primary combined immunodeficiency caused by gain-of-function mutations in the PIK3CD gene.
The methods used were a review of medical records and laboratory data of APDS-1 patients regularly followed by three Italian Immunology Units.
We report on eight APDS-1 patients. All patients are alive. Ages at clinical onset and molecular diagnosis were variable (0.5 to 20.5; 2.2 to 43.2 years). Recurrent sinopulmonary infections were the most common initial feature of disease requiring medical evaluation. At onset or during follow-up, seven patients presented lymphoproliferative disease, one case complicated by HLH (Table 1). Immunohistochemical analysis of bioptic specimens showed reactive non-infective non-neoplastic lymphoproliferation. Only one patient presented an atypically mild clinical course: besides the B cell lymphopenia, she presented occasional respiratory infections, without signs of lymphoproliferation. Among our cohort, reduced CD4+ T-cells count with low Recent Thymic Emigrants, increased CD8+ effector-memory, raised CD8+CD57+ and hypogammaglobulinemia were common findings. Genetic analysis of PIK3CD gene revealed three novel variants: functional testing evaluating the phosphorylation pattern of S6K confirmed their activating nature (Table 2 and Figure 1). In the remaining five patients, the previously reported mutations p.E1021K (n=4) and p.E525A (n=1) were identified. Six patients were started on immunoglobulin replacement treatment (IgRT). In two patients, Sirolimus was used to control lymphoproliferation. One patient successfully underwent HSCT, with good chimerism and no GVHD at 16 months after-HSCT.
APDS-1 is a combined immunodeficiency with a wide variety of clinical manifestations and a complex immunological presentation. Besides IgRT, specific therapies targeting the PI3Kδ pathway will most likely become a valid aid for the amelioration of patients’ clinical management and their quality of life.


Table 2. Genetic analysis and phospo-S6K evaluation of eight APDS-1 patients

Figure 1. phospo-S6 kinase (pS6K) levels in peripheral T cells from Activated Phosphoinositide 3-Kinase Delta Syndrome-1 (APDS-1) patients (Pts). Summarized data for pS6K levels from patients harbouring the Y524D, P658L and R108L PIK3CD mutations are shown for CD4+ T cells (a) and CD8+ T cells (b) after anti-CD3 stimulation and/or CAL-101 treatment. (Data were summarized from n = 3 experiments from the index patients and four different healthy controls (HCs); statistical analysis was performed using the Student’s t-test (* p < 0.05)
Keywords: inborn errors of immunity, p110delta, lymphoproliferation, apds-1, PIK3CD, HSCT, Sirolimus, activated PI3K
Disclosure: All authors indicated they had no financial relationships to disclose.
(173) Reduction of Infusion Time Using a 10% Immunoglobulin Product During Staffing Shortages Due to COVID-19
Christine Miller, PharmD1, Timothy Walton, MHS, CCRP2, Barbara Prosser, RPh3, Craig Vollmer, BA4, David Luckey, MBA5, Drew Doyle, RPh6
1Manager, Health Economics and Outcomes Research/Soleo Health
2Director of Data Quality, Health Outcomes and Research/Soleo Health
3VP, Health Economics and Outcomes Research/Soleo Health
4Chief Commercial Officer/Soleo Health
5Director of Business Applications/Soleo Health
6Vice President, Sales & Market Development/Soleo Health
The recent healthcare crisis due to the novel Coronavirus disease 2019 (COVID-19) has created an increased demand for infusions administered within the home environment. Patients with immunodeficiencies who are receiving intravenous immune globulin (IVIg) are at greater risk for contracting infections, like COVID-19. Hospital and outpatient clinics providing scheduled infusions have referred patients to specialty pharmacy organizations in order to make availability for critically ill patients and reduce COVID-19 exposure risk to patients and employees. Due to increased demand for home infusion, it has become imperative to reduce nurse visit times to minimize COVID-19 exposure risk to nurses and patients while meeting these increased demands. The purpose of this study is to determine if a 10% immune globulin (Ig) product with a 15-minute titration protocol has a shorter total infusion time and could therefore reduce the amount of time spent in a patient’s home.
A retrospective review of patient medical records was conducted by a multidisciplinary team over a 30-month period. Customized reports identified patients receiving Gammaplex 10% after previously receiving other Ig products. This allowed for a comparison of infusion times for all Ig products pre and post transition to Gammaplex 10%.
A total of twenty-three patients met the inclusion criteria. Thirteen had an average infusion reduction time of 35 minutes per episode, with a maximum of 1 hour 15 minutes. Five patients had an average infusion increase time of 36 minutes per episode and five had no change per episode. 56% of patients experienced zero adverse events post-transition. The most frequent adverse events reported (>10% patients) included headache and fatigue. Fatigue may also have been attributed to premedication with diphenhydramine, which was taken by 83% of patients.
Conclusions: The home environment is an important site of care to help reduce the risk of exposure to COVID-19, especially for those with immunodeficiencies and receiving Ig infusions. Using a 10% Ig product with a 15-minute titration protocol with a shorter infusion time is one strategy to help reduce the amount of time a visiting nurse spends with a patient and allows greater flexibility with nursing scheduling.
Keywords: COVID, Immunoglobulin, Infusion Time, IVIG, Specialty Pharmacy, Staffing Shortage
Disclosure: All authors indicated they had no financial relationships to disclose.
(174) Exploring the Effects of BMI and Route of Administration on Efficacy of Immunoglobulin G Replacement Therapy - a Preliminary Report
Nikki Kimura, MD1, Miriam Samstein, MD, PhD2, Brianne Navetta-Modrov, MD3, David Rosenthal, DO, PhD4, Blanka Kaplan, MD4, Vincent Bonagura, MD5, Artemio Jongco, MPH, MD, PhD, FACP, FACAAI, FAAAAI4
1Resident Physician/Zucker School of Medicine at Hofstra/Northwell Residency Program in Internal Medicine at North Shore University Hospital and Long Island Jewish Medical Center (NS/LIJ)
2Assistant Professor of Clinical Pediatrics/Weill Cornell Medicine
3Assistant Professor of Medicine/Stony Brook University School of Medicine
4Assistant Professor of Medicine and Pediatrics/Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
5Chief, Division of Allergy/Immunology/Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
There is a paucity of information about whether and how BMI and administration route may affect infection rate in patients on IgG replacement therapy (IGRT). Our prior work suggested that mean infection rate did not differ by BMI when adjusting for administration route, but that IVIG patients have a lower infection rate compared to SCIG patients when adjusting for BMI (P < 0.01).
The overall study objective is to elucidate the relationship between BMI, dose, and route of administration in primary immunodeficiency patients receiving IGRT. Here, we build upon our prior work by expanding the original sample size and study time frame.
We performed a retrospective chart review at an outpatient, tertiary care, academic medical center allergy & immunology clinic from 1/1/2000 through 12/31/2019. Administration route, dose, infection history, IgG levels, height, weight, demographics, and diagnosis requiring IGRT were analyzed. BMI categories were based on height and weight upon study entry.
The study included a total of 177 subjects compared to 63 from the previous study. The median age was 41 years (range 11 months – 91 years), 45.1% (n=80) were male. Racial and ethnic demographics were as follows: 84.7% (n= 150) White, 6.2% (n=11) Hispanic or Latino, 6.2% (n=11) Other or Multiracial, 1.7% (n=3) Black or African American, and 1.1% (n=2) Asian. 5.1% (n=9) were underweight, 42.4% (n=75) had normal BMI, 24.3% (n=43) were overweight, and 28.2% (n=50) were obese. 59.3% (n=105) of subjects used SCIG. The diagnoses were distributed as follows: 54.2% (n=96) Common Variable Immunodeficiency, 28.8% (n=51) hypogammaglobulinemia, 6.2% (n=11) X-linked agammaglobulinemia, 2.8% (n=5) Specific Antibody Deficiency, 2.3% (n=4) Hyper IgM, 1.7% (n=3) Severe Combined Immunodeficiency, and 4.0% (n=7) other. At study start, median age (P=0.14), number of visits (P=0.35) or number of infections (P=0.37) did not differ among the four BMI cohorts.
The current study more than doubled the prior sample. Our cohort was mostly white middle-aged females, 52.5% of whom were overweight or obese, 54.2% of whom have CVID, with 59.3% on SCIG. Ongoing analyses with the larger cohort will help clarify the relationship between BMI, dose, and route of administration.
Keywords: Immunoglobulin Replacement Therapy, Infections in PIDs, Phenotypic and Molecular Spectrum of Primary Immune Deficiency
Disclosure: Nikki Kimura and Artemio Jongco received support from Takeda. All other authors indicated they had no financial relationships to disclose.
(175) Agranulocytosis and Severe Infection Secondary to Metimazole
Daniel Urschel, MD1, Vivian Hernandez-Trujillo, MD2, Jose Calderon, MD2
1Allergy/Immunology Fellow/Nicklaus Children's Hospital
2Allergy Immunology Attending/Nicklaus Children's Hospital
An eleven-month-old, previously healthy, vaccinated male traveled with family to Cuba. He developed fever and oral lesions after arrival. Patient was diagnosed with viral stomatitis and treated with an over the counter antipyretic. Symptoms worsened to include frequent drooling, decreased oral intake, cough, and intermittent stridor leading to multiple ER visits and admission after returning to the United States. Laboratory evaluation demonstrated neutropenia and leukopenia, with normal hemoglobin and platelet counts. Concerning imaging included unilateral infiltrate on chest x-ray and epiglottitis on CT of neck. Immunologic laboratory evaluation pertinent for borderline low IgG for age, normal IgM and IgA, significant T cell lymphopenia, normal B cell and NK cell lines, normal antibody/antitoxin titers to S.Pneumoniae, H.Influenzae B, Diphtheria and Tetanus.
Our patient was started on antibiotics, antifungals, steroids, IVIG and supplemental oxygen. Symptoms dramatically improved within one week of admission. Laboratory values, including complete blood count with differential, repeat lymphocyte subsets including T cell subsets, all normalized one week after admission. Blood cultures were negative. A skin lesion tested positive for Pseudomonas. Viral testing was positive for Rhinovirus/Enterovirus only. Newborn screen confirmed to be normal. Further history after admission revealed the antipyretic the patient had been treated with prior to severe symptom onset was Dipyrone (Metamizole). He remained well following discharge. Subsequent testing including complete blood counts with differentials and immunoglobulin levels were normal at follow up.
Dipyrone is a commonly used analgesic, antipyretic, antispasmodic, and anti-inflammatory throughout the world. The mechanism of agranulocytosis secondary to Dipyrone is not fully understood, although it is believed to be immune mediated via anti-neutrophil antibody production. Illness secondary to agranulocytosis can be severe and life threatening.
Concern in the case of an infant presenting with severe illness, leukopenia, T cell lymphopenia and neutropenia initially were for a primary immunodeficiency such as severe combined immunodeficiency. Identifying the underlying cause of these lab abnormalities in a severely ill infant without a significant medical history is challenging. Infections, primary immunodeficiencies, malignancies, idiosyncratic drug reactions, autoimmune and genetic conditions must be considered. This represents the importance of thorough history taking and awareness of secondary immunodeficiencies.
Keywords: Other Topics Related To Immune Deficiency and/or Dysregulation, Secondary Immunodeficiency, Agranulocytosis
Disclosure: Vivian Hernandez-Trujillo is an advisory board member of Covis, CSL Behring, DBV, Kaleo, Takeda, and US WORLD MEDS. All other authors had no financial relationships to disclose.
All other authors indicated they had no financial relationships to disclose.
(176) Psychosocial insights of parents of pediatric primary immune deficient (PID) patients who had genetic testing
Smriti Singh, BS1, Lauren Lichten, MS2, Nadia Ali, PhD2, Christopher Gray, MS3, Hong Li, MD, PhD4, Shanmuganathan Chandrakasan, MD5
1Graduate Student/Emory University Genetic Counseling Training Program, Atlanta, Georgia
2Department of Human Genetics/Emory University, Atlanta, Georgia
3Genetic Counselor/Roberts Individualized Medical Genetics Center and the Division of Human Genetics at Children's Hospital of Philadelphia
4Associate Professor and Medical Geneticist/Department of Human Genetics and Pediatrics, Emory University, Atlanta, Georgia
5Pediatric Hematologist/Oncologist/Children's Healthcare of Atlanta
Primary immunodeficiency syndromes (PIDs) are a group of immune conditions with variable severity and age of onset. Given PIDs include 400+ conditions, genetic testing utilizing next generation sequencing can shorten the diagnostic odyssey and can influence management. While psychosocial challenges of raising a child with a PID are significant, psychosocial challenges surrounding genetic testing for these patients are relatively unexplored.
We performed a qualitative study to evaluate experiences of 9 negative and 9 positive result families who had a child undergo panel (14) or exome (4) genetic testing for a PID at Children’s Healthcare of Atlanta’s Immune Dysregulation Clinic in the last three years. Eighteen qualitative semi-structured interviews were adopted from validated genomics outcome and psychosocial assessment scales and interpreted using thematic analysis.
Preliminary findings indicate genetic result disclosures left questions, concerns, or uncertainty in many parents regarding financial costs of testing, and adaption to understanding of results as related to patients’ long-term outcomes. Some disclosures took several months or were learned via a web portal/the advocacy of parents seeking results. Few participants were referred to genetic counseling, yet they demonstrated needs involving a) a variety of emotional reactions to results/long-term coping, b) support groups, c) planning next steps, and/or d) needing a central provider such as a genetic counselor to coordinate communication between all involved providers and the family. Approximately half of parents also desired help with a) follow-up testing regarding at-risk/symptomatic family members, b) realistically assessing genetic testing’s ability to end patients’ diagnostic odysseys or alter treatment/management plans, and c) anticipatory guidance for psychosocial challenges, including distress from uncertain results. Despite anticipated concerns being realized, a majority of parents viewed genetic testing for PIDs in a positive light, cognizant of benefits it provided for current/future care of their children.
For pediatric immunology providers offering genetic testing, a unified protocol or pre-/post-test counseling is suggested. A PID genetic testing protocol/counseling can be adapted using Rolland & Williams’ Family Systems Genetic Illness model (FSGI) to encompass comprehensive consent, timely/informed testing and result disclosure, and parent-friendly resources such as summary letters for patients’ multiple specialty providers.
Keywords: PID, Genetic, Testing, Psychosocial, Resources, Panel, Exome, Counseling, Coping, Pediatric
Disclosure: Nadia Ali received a research grant from Amicus Therapeutics, BioMarin, Pfizer, Sanofi Genzyme, Shire/Takeda and speaker honoraria from Vitaflo. All other authors indicated they had no financial relationships to disclose.
(177) Immunodeficiency of Kabuki Syndrome: A Case Series
Stefani Su, MD1, Alissa McInerney, MD2, Artemio Jongco, MPH, MD, PhD, FACP, FACAAI, FAAAAI3
1Allergy Immunology Fellow in Training/Donald and Barbara Zucker School of Medicine at Hofstra/ Northwell Health
2Allergy Immunology Fellow in Training/Donald and Barbara Zucker School of Medicine at Hofstra/Northwell Health
3Assistant Professor of Medicine and Pediatrics/Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
Kabuki syndrome is a rare syndrome characterized by dysmorphic facies, intellectual disability, anatomic malformations and recurrent infections. We present two cases of Kabuki syndrome.
(Patient 1): A 22 month old female with developmental delay, ex-35 week gestation, hypocalcemia at birth and multiple refractory complex febrile seizures presented for immune evaluation. Hematology evaluation showed anemia and thrombocytopenia, requiring multiple transfusions and oral steroids. Bone marrow biopsy was consistent with Autoimmune Myelofibrosis. She had one episode each of urinary tract infection and otitis media.
Physical exam demonstrated long palpebral fissures with eversion of lower eyelid, prominent ears and frontal bossing, flat nasal tip, prominent fingertip pads, café-au-lait spots, and expressive language delay.
Whole Exome Sequencing revealed heterozygous pathogenic variant c. 1234 dupCpL412PfsX4 in KMT2D, consistent with Kabuki Syndrome. She was also heterozygous for likely pathogenic variant c.1511 C>A p.T504N in PCDH19 so she was diagnosed with PCDH19-Related Epilepsy Disorder. Initial testing showed normal numbers of CD4+ and CD8+ T-cells and CD19+ B-cells. Subsequent testing showed CD8+ T-cell lymphopenia as well as B-cell lymphopenia likely due to Rituximab administration.
(Patient 2): A 4 year old male with history of hypocalcemia at birth, microcephaly, severe developmental delay, and eczema presented for immune evaluation. He had multiple pneumonias requiring hospitalization and chronic ear infections. He was diagnosed with Evans syndrome at age two due to thrombocytopenia, which improved with Rituximab.
Physical exam demonstrated elongated palpebral fissures, arched and broad eyebrows, and prominent ears.
He was diagnosed with Kabuki Syndrome at age four after genetic testing revealed a mutation in KMT2D. Immune labs done prior to Rituximab showed normal immunophenotype as well as normal immunoglobulins. Labs 1 year post-Rituximab showed continued normal numbers of T-cells, B-cell lymphopenia, as well as profound hypogammaglobinemia. He started on subcutaneous immunoglobulin replacement therapy with decreased frequency and severity of sinopulmonary infections.
The immunodeficiency associated with Kabuki syndrome is variable and can include humoral and cellular defects as well as autoimmunity. The most common infections seen in patients is otitis media. Further information on the immunologic manifestations of the syndrome is needed.
Keywords: Kabuki syndrome, Immunodeficiency, KMT2D
Disclosure:. All authors indicated they had no financial relationships to disclose.
(178) Common Variable Immune Deficiency in a patient with a heterozygous mutation in IRF2BP2
Alissa McInerney, MD1, Stefani Su, MD2, Artemio Jongco, MPH, MD, PhD, FACP, FACAAI, FAAAAI3
1Allergy Immunology Fellow in Training/Donald and Barbara Zucker School of Medicine at Hofstra/Northwell Health
2Allergy Immunology Fellow in Training/Donald and Barbara Zucker School of Medicine at Hofstra/ Northwell Health
3Assistant Professor of Medicine and Pediatrics/Donald and Barbara Zucker School of Medicine at Hofstra/Northwell
Common Variable Immune Deficiency (CVID) is a common primary immune deficiency, although a genetic cause is found in only a small minority of patients. We present a case with a heterozygous variant of uncertain significance in IRF2BP2, a gene that has been associated with autosomal dominant CVID in one family.
A 60-year-old woman with frequent sinopulmonary infections, chronic diarrhea, and fatigue presented for immune evaluation. She had a history of IgA deficiency and a family history of malignancy and autoimmune disease.
Laboratory evaluation revealed low IgG, IgM, IgA with elevated IgA antibodies. Non protective titers to measles, mumps, rubella, varicella, tetanus, and diphtheria were found. Immunophenotype revealed increased number of B cells and decreased switched memory B cells (CD27+IgM-IgD-). Commercially available immune deficiency genetic panel revealed a heterozygous mutation IRF2BP2 c.1688C>T(p. Pro563Leu), classified as a VUS.
The heterozygous variant of IRF2BP2 c.1654G>A(p. Ser551Asn), located in the carboxyl terminal ring domain, was reported by Keller et al., 2016 in 3 members of a single family and shown to be likely a cause of autosomal dominant CVID. In vitro, this mutation impacted B cell maturation and differentiation of plasmablasts. Our patient’s mutation is just downstream and within the ring domain, which interacts with nuclear factor of activated T-cells 1 (NFAT1), a transcription factor important in immune responses. This interaction strongly inhibits transcriptional activity of cytokine genes preventing differentiation of B cells in animal models. Based on the similar clinical presentation and the available information we suggest that this mutation is pathologic in our patient. Functional validation studies are underway to further characterize this patient's variant.
Keywords: Common Variable Immune Deficiency, IRF2BP2, CVID
Disclosure:. All authors indicated they had no financial relationships to disclose
(179) A patient with X-linked agammaglobulinemia and COVID-19 infection treated with remdesivir and convalescent plasma
Aled Iaboni, MD1, Stephen Betschel, MD2
1Resident/Allergy/Immunology
2Staff Physician/Allergy/Immunology
This 28 year-old male with X-linked agammaglobulinemia (XLA) presented to hospital with a one-week history of fevers, hyposmia and cough. He had received a throat swab prior to presentation that was positive for COVID-19. On presentation, he was tachycardic, tachypnic and mildly hypoxic. A chest X-ray showed bilateral airspace opacities suggestive of pneumonia. He had hyponatremia, leukopenia, thrombocytopenia, transaminitis and elevated inflammatory markers (Table 1).
The patient was admitted and treated with dexamethasone 6mg daily. By day 4, his hypoxia had worsened and he began a 5-day course of Remdesivir. On day 5, he was transferred to the intensive care unit and received 500mL of convalescent plasma (CP). By day 9, he began to improve clinically and had less oxygen requirements. By day 11, he had been completely weaned off of oxygen. He was was discharged on day 13 with near complete symptom resolution.
Patients with primary immunodeficiencies are suspected to be at elevated risk for more severe infections due to COVID-19. Two early studies reported 4 patients with agammaglobulinemia and 5 patients with common variable immunodeficiency (CVID) that had COVID-19 infection. Compared with CVID, patients with agammaglobulinemia had very mild courses. This led to a hypothesis that B-lymphocytes may be involved in COVID-19 related inflammation. However, a more recent study described 3 XLA patients who required more protracted hospital courses. Similar to our patient, these patients rapidly recovered after receiving CP. These cases suggest that XLA patients remain at risk of severe complications from COVID-19.
The rapid recoveries seen in XLA patients following administration of CP suggests antibodies are important for viral neutralization. However, a recent randomized trial of 334 adult patients with severe COVID-19 pneumonia showed administration of CP compared to placebo resulted in no difference in clinical outcomes or mortality. Whether CP has a unique mechanism of effect in patients with absence of B-lymphocytes remains unknown. The rapid response to CP in our patient suggests that humoral immunity is an important factor in recovery from COVID-19. The observed response to CP may be unique to patients who lack B-lymphocytes and this requires further study.
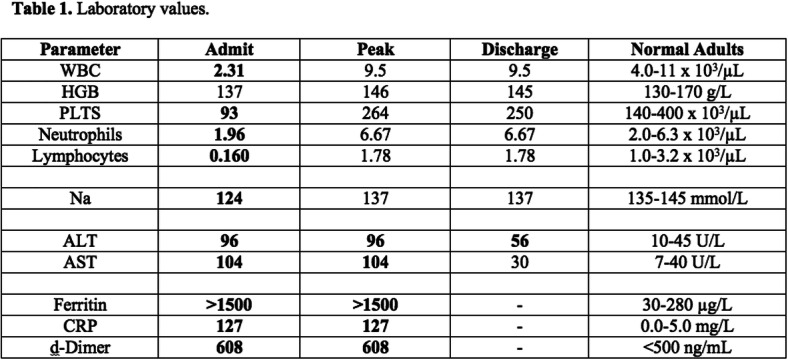
Keywords: PID, XLA, COVID-19, remdesivir, convalescent plasma, agammaglobulinemia
Disclosure:. All authors indicated they had no financial relationships to disclose.
(180) Confirmed SARS-COV-2 Infection In A Cohort Of Children And Young Adults With Moderate Or Severe Primary Immunodeficiencies
Angela Deyà-Martínez, MD, PhD1, Anna P. García-García, MD2, Alexandru Vlagea, MD, PhD3, Yolanda Jordan, MD, PhD4, Victoria Fumado, MD, PhD5, Claudia Fortuny, MD, PhD5, Marta Español, MD6, Azucena Gonzalez, MD,PhD6, anna Esteve-Solé, MD, PhD7, Manel Juan, MD, PhD8, Mariona Pascal, MD, PhD9, Laia Alsina, MD, PhD10
1Attending/Clinical Immunology and Primary Immunodeficiencies Unit. Pediatric Allergy and Clinical Immunology Department. Hospital Sant Joan de Déu, Barcelona, Spain. Institut de Recerca Sant Joan de Déu, Barcelona, Spain.4. Clinical Immunology Unit Hospital Sant Joan de Déu-Hospital Clínic Barcelona
2Attending/Clinical Immunology and Primary Immunodeficiencies Unit. Pediatric Allergy and Clinical Immunology Department. Hospital Sant Joan de Déu, Barcelona, Spain
3Attending/Clinical Immunology Unit Hospital Sant Joan de Déu-Hospital Clínic Barcelona, Barcelona, Spain. Department of Immunology-CDB, Hospital Clínic-IDIBAPS, Universitat de Barcelona, Barcelona, Spain.
4Attending/Hospital Sant Joan de Deu, Pediatric Intensive Care, esplugues llobregat, Spain
5Attending/Hospital Sant Joan de Deu, Pediatric Infectious Disease Unit, Esplugues llobregat, Spain
6Attending/Department of Immunology-CDB, Hospital Clínic-IDIBAPS, Universitat de Barcelona, Barcelona, Spain
7Post-Doctoral researcher/Clinical Immunology and Primary Immunodeficiencies Unit. Pediatric Allergy and Clinical Immunology Department. Hospital Sant Joan de Déu, Barcelona, Spain. Institut de Recerca Sant Joan de Déu, Barcelona, Spain.
8Head of the Unit/Universitat de Barcelona, Spain. Clinical Immunology Unit Hospital Sant Joan de Déu-Hospital Clínic Barcelona, Barcelona, Spain. Department of Immunology-CDB, Hospital Clínic-IDIBAPS, Universitat de Barcelona, Barcelona, Spain. Immunotherapy Platform, Hospital Sant Joan de Déu-Hospital Clínic, Universitat de Barcelona, Barcelona, Spain.
9Head of the Unit/Universitat de Barcelona, Spain; Clinical Immunology Unit Hospital Sant Joan de Déu-Hospital Clínic Barcelona, Barcelona, Spain; Department of Immunology-CDB, Hospital Clínic-IDIBAPS, Universitat de Barcelona, Barcelona, Spain. Immunotherapy Platform, Hospital Sant Joan de Déu-Hospital Clínic, Universitat de Barcelona, Barcelona, Spain.
10Head of the Unit/Clinical Immunology and Primary Immunodeficiencies Unit. Pediatric Allergy and Clinical Immunology Department. Hospital Sant Joan de Déu, Barcelona, Spain Institut de Recerca Sant Joan de Déu, Barcelona, Spain. Universitat de Barcelona, Spain. Clinical Immunology Unit Hospital Sant Joan de Déu-Hospital Clínic Barcelona, Barcelona, Spain
In December of 2019, an outbreak caused by SARS-CoV-2 started in Wuhan causing a worldwide pandemic. Evidence indicates lower incidence and milder clinical course in children, but there is a lack of information about primary immunodeficiency (PID) as a possible risk factor. The objective of the study is to determine de prevalence of SARS-CoV-2 infection in a cohort of children and young adults with a moderate or severe primary immunodeficiency using different immune assays and describe their clinical expression.
Cross-sectional study developed in a single-centre between May 2020- June 2020. Children and young adults, from 0-21 years old, with moderate-severe PID and with signed informed consent, were included. Patients' information was collected from the medical chart, focusing into presence of past COVID 19-related symptoms (from patients and household contacts) during epidemic period (January 2020-June 2020). In order to confirm the infection the following assays were performed: SARS-CoV-2 PCR, specific IgA/IgM/IgG serology and specific spike protein SARS-CoV-2 ELISpot* (* only in those patients with positive household contacts).
A total of 65 patients have been included (median age 12.3 (2-21.1); 58.5% males) from 7 different PID groups (IUIS classification). 18% of the patients had confirmed/highly suspected household contact. Five patients had confirmed infection (4/5 positive serology; 1/5 positive PCR), only 1/5 with symptoms (mild cough). Twelve patients had positive household contacts: 3/12 positive serology; 9/12 negative serology or PCR neither positive results in ELISpot assay.
The prevalence of COVID19 in the studied paediatric cohort of moderate-severe PID patients, was 7.7% (5/65), similar to that observed in Catalonia at that time (6.1%; 5.2-7.2). Of these, only 1/5 was symptomatic (mild symptoms; Jacobsen Syndrome + CVID-like phenotype). In the subgroup with higher SARS-CoV-2 exposure and negative PCR or serology, SARS-CoV-2 protein S ELISpot did not improve infection diagnosis.
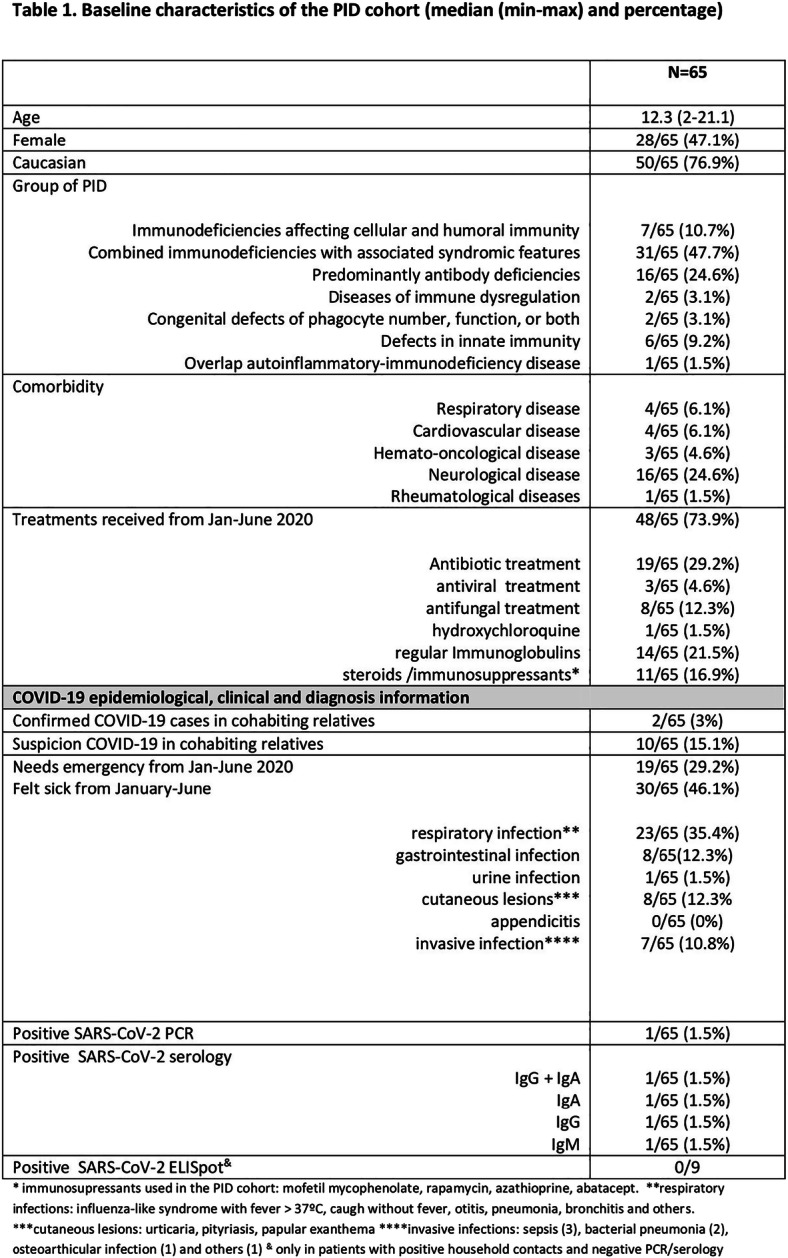
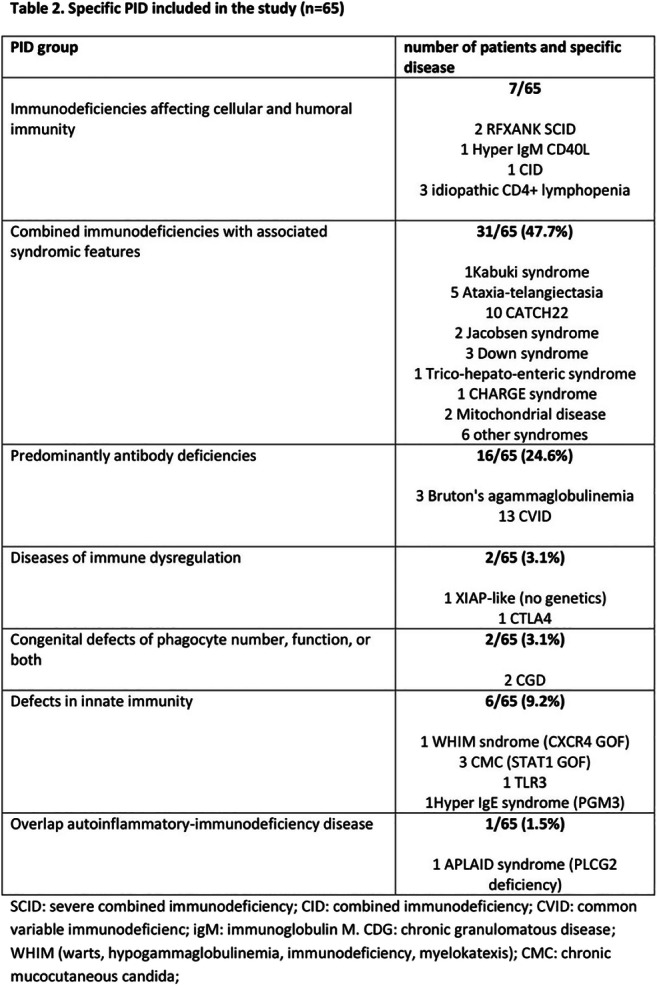

Keywords: Primary immunodeficiencies, Paediatric, SARS-CoV-2 infection, Diagnosis, COVID19
Disclosure:. All authors indicated they had no financial relationships to disclose.
(181) Comparative cost-effectiveness of hematopoietic stem cell transplantation versus lifetime immunoglobulin replacement for congenital agammaglobulinemia
Di Sun, MD1, Nancy Bunin, MD2, Jennifer Heimall, MD3, Neil Romberg, MD4
1Allergy/Immunology Fellow/Children's Hospital of Philadelphia
2Professor of Pediatrics, Perelman School of Medicine at the University of Pennsylvania/Children's Hospital of Philadelphia
3Attending Physician/Children's Hospital of Philadelphia
4Assistant Professor of Pediatrics, Perelman School of Medicine at the University of Pennsylvania/Children's Hospital of Philadelphia
To prevent severe infections, the mainstay of treatment for congenital agammaglobulinemia is lifelong immunoglobulin (Ig) replacement. Although hematopoietic stem cell transplantation (HSCT) is associated with significant morbidity and mortality, this therapy offers a one-time cost when compared to lifelong intravenous or subcutaneous Ig replacement. This study evaluates the cost-effectiveness of HSCT compared to Ig replacement for treatment of agammaglobulinemia in the United States (US).
The base-case scenario of a 12 month-old child with agammaglobulinemia receiving Ig replacement therapy compared with either matched sibling donor (MSD) or matched unrelated donor (MUD) HSCT was represented using a Markov model from the healthcare perspective over an 80-year time horizon. The model was populated through extensive analysis of published literature. Cohort analysis and microsimulations were used to determine the incremental cost-effectiveness ratio (ICER) expressed in 2020 US dollars (USD) per quality-adjusted life year (QALY) gained. One-way sensitivity and probabilistic uncertainty analyses were performed across plausible ranges.
For treatment of agammaglobulinemia, MSD and MUD HSCT yield a lower cost of $1,322,563 and $1,417,499 respectively when compared to Ig replacement ($3,743,107), but Ig replacement generates more QALYs (48.34) compared to MSD (36.12) and MUD (36.07) HSCT. Overall, Ig replacement results in an ICER of $198,056 and $189,532 per QALY gained when compared to MSD and MUD HSCT respectively, which is higher than the willingness-to-pay (WTP) threshold of $100,000 (per capita GDP of the US). In microsimulation analysis (n=10,000), compared to HSCT, Ig replacement costs less and is more effective only 5.98% of the time. The cost-effectiveness of Ig replacement therapy is highly dependent on the annual cost of Ig replacement; in fact, when annual cost of Ig replacement therapy is lower than $30,474, this strategy falls below the WTP threshold of $100,000.
Under current conditions, HSCT is more cost-effective than Ig replacement therapy but Ig replacement is more effective (generating greater QALYs). Importantly, when the annual cost of Ig replacement is reduced, this therapy falls below the WTP threshold of $100,000. In this reduced cost scenario, Ig therapy would be the preferred long-term treatment strategy for agammaglobulinemia in the US.


Keywords: Agammaglobulinemia, Cost-effectiveness analysis, Hematopoietic stem cell transplantation, Health economics
Disclosure:. ennifer Heimall is an advisory board member of CIRM; received research grants from CSL Behring and Regeneron; and speaker honoraria from Horizon. All other authors indicated they had no financial relationships to disclose.
(182) Monitoring of post-transplant naïve lymphocytes reconstitution in PID patients using TREC and KREC quantification
Ekaterina Polyakova, PhD student1, Maria Stegantseva, PhD2, Mikhail Belevtsev, PhD3
1PhD student,Research in the field of genetics of PIDS, determination of TREC /KREC/Belarusian Research Center for Pediatric Oncology, Hematology and Immunology,clinical research laboratory of the scientific department
2PhD/Belarusian Research Center for Pediatric Oncology, Hematology and Immunology, laboratory of genetic biotechnology, scientific department
3PhD/Belarusian Research Center for Pediatric Oncology, Hematology and Immunology, Deputy Director for Research
In this study the dynamics of TRECs and KRECs positive cells in PID patients after hematopoietic stem cell transplantation was evaluated.
Quantity of TREC and KREC were determined using RQ-PCR in 11 patients from 3 months to 12.3 years old (median age – 1.95 years). Following diagnoses were genetically confirmed: SCID-4, WAS-4, CGD-1, HLH-1, HLA-DR II deficiency-1). The number of CD3+ and CD19+ cells were determined in the routine immunogram after HSCT.
It was found that 2 SCID patients reconstituted the number of TREC-positive cells to day 180, when others keep undetectable TREC during the all follow-up. All of them reached normal KREC count closer to day 45 - 60, except one (day 180). Among the WAS patients three restored TREC level between 100-245 days and KREC level to day 60. Only one had both negative results during all monitoring period. HLH patient had the longest time of recovery: TREC –day 245, KREC – day 145. Patients with CGD and HLA-DR II deficiency had similar results: TREC reconstituted to day 180 and KREC between 30-45 days. The dynamics of TREC- and KREC molecules correlated with the CD3+CD45RA+ T-lymphocytes and differed from total population of CD3+ and CD19+ lymphocytes. The high quantity of TREC on the 15-30 days is caused by the presence of naïve T-cells in the transplant and is not related to thymopoiesis reconstitution.
Quantity of TREC and KREC molecules can be useful markers to assess the reconstitution of naïve lymphocytes after HSCT
Keywords: TRECs KRECs, RQ-PCR, Hematopoietic stem cell transplantation, PIDs, Reconstitution of naïve lymphocytes
Disclosure:. All authors indicated they had no financial relationships to disclose.
(183) Hypogammaglobulinemia, macrocytosis, and pancytopenia in a patient with LIG1 deficiency
Angela Chun, M.D.1, Juan C. Bernini, M.D.2, Tiphanie Vogel, M.D, Ph.D.3, Sarah Nicholas, M.D.4
1Clinical Fellow, Division of Rheumatology, Department of Pediatrics/Texas Children’s Hospital, Baylor College of Medicine
2Professor, Division of Hematology-Oncology, Department of Pediatrics/Vannie E. Cook Jr. Children's Cancer and Hematology Clinic, Baylor College of Medicine
3Assistant Professor, Division of Rheumatology, Department of Pediatrics & Medicine/Texas Children’s Hospital, Baylor College of Medicine
4Assistant Professor, Division of Immunology, Allergy and Retrovirology, Department of Pediatrics/Texas Children's Hospital, Baylor College of Medicine
DNA ligase 1 (LIG1) is critical for proper DNA replication and repair, and LIG1-deficient cells are prone to chemical and radiation-induced DNA damage. Deleterious biallelic mutations in LIG1 have been reported in only rare patients with primary immunodeficiency, varying from mild antibody deficiency to severe combined immunodeficiency. These patients exhibited variable growth retardation and hematologic abnormalities, including hypogammaglobulinemia, erythroid macrocytosis, and lymphopenia. We describe a patient with compound heterozygous variants in LIG1, who presented with short stature, hypogammaglobulinemia, macrocytosis, and pancytopenia.
A 15-year-old neurodevelopmentally normal male presented with chronic fatigue and a history of recurrent fevers responsive to empiric antibiotics. He was found to have growth retardation ( < 1st percentile). Laboratory evaluation revealed pancytopenia (nadir WBC 2.02 x 103/□L, ANC 0.33 x 103/□L, hemoglobin 8.2 g/dL, platelets 22 x 103/□L), macrocytosis (peak MCV 120 fL), and marked hypogammaglobulinemia (IgG < 7 mg/dL, IgA < 6 mg/dL, IgM 72 mg/dL). Retrospective review noted the patient was first documented to have macrocytosis at 5-years-old, without associated anemia. He was subsequently lost to follow up and experienced frequent infections (pneumonia, acute otitis media, gingivitis) during childhood, twice requiring hospitalization for pneumonia. At the time of evaluation, lymphadenopathy and marked hepatosplenomegaly was noted. Liver biopsy demonstrated diffuse lymphohistiocytic inflammation and multifocal hepatocyte necrosis and lymph node biopsy showed paracortical T-cell expansion and diminished B-cell follicles without germinal center formation. He had normocellular bone marrow, with moderate erythroid hyperplasia and dysplastic features. Peripheral evaluation revealed increased CD3+ T-cells with a low CD4:CD8 ratio, and a relative decrease in mature B- and NK-cells. His lymphocytes demonstrated impaired responses to mitogens. Telomere length was markedly shortened in T- and NK-cell populations, but normal in granulocytes, indicating replicative stress. Exome sequencing revealed novel compound heterozygous variants (c.1049G>T, p.G350V; c.2636G>A, p.R879H) in LIG1. Functional validation was not performed, but he was diagnosed with LIG1-deficiency based on the remarkable phenotypic match. He has been treated successfully with immunoglobulin replacement.
Defects in LIG1 can result in variable clinical phenotypes. Hallmark features include hypogammaglobulinemia and macrocytosis. Our patient is unique in his hepatosplenomegaly, neutropenia, and thrombocytopenia. Bone marrow transplant and splenectomy are being explored.
Keywords: LIG1 deficiency, Pediatric, Primary immunodeficiency, Macrocytosis, Hypogammaglobulinemia
Disclosure:. All authors indicated they had no financial relationships to disclose.
(184) Management Dilemmas as Newborn Screening for Severe Combined Immunodeficiency also Detects Newborns with T cell Lymphopenia: A Review of University of California, Los Angeles 2019-2020
Lisa KLohn, MD, PhD1, Alexis Stephens, BS2, Manish Butte, MD, PhD3
1Fellow/UCLA
2Research Coordinator/UCLA
3Associate Professor/Division of Allergy, Immunology, and Rheumatology, University of California Los Angeles
Since 2008, newborn screening (NBS) measuring T cell receptor excision circles (TRECs) has been used to detect patients with severe combined immunodeficiency (SCID) prior to the onset of infections or other complications. Published reviews demonstrate that in addition to detecting newborns with SCID, TREC screening also identifies infants with other potential immunodeficiencies.
From January 2019 through December 2020, UCLA treated 24 infants who were referred for low TRECs ( < 18 TREC/μL), all of whom underwent further diagnosis by flow cytometry to assess CD3+ T cells/μL, % of CD4+ that are CD45RA+ T cells (naive), CD19+ B cell/μL, and CD56+ NK/μL.
Of the 24 infants, six met criteria for typical SCID, with TRECs 0-2/μL and CD3+ T cells/μL < 300 (at initial or subsequent evaluation). Genetic testing identified variants in genes associated with SCID (IL2RG, JAK3, DCLRE1C, or RAG1) or cartilage hair hypoplasia with profound lymphopenia (RMRP). All patients have either undergone or are awaiting hematopoietic stem cell transplant or gene therapy, and the cohort has 100% survival at short-term follow up.
Of the remaining 18 patients detected on TREC screening without typical SCID, four exhibit syndromic immunodeficiency (partial DiGeorge, trisomy 22); three had T cell loss (chylothorax); two had lymphopenia secondary to other issues (extreme prematurity, maternal medication); one patient’s work-up remains pending; and eight infants had T cell lymphopenia (300-1500/μL).
Of the eight lymphopenic patients, three had low B cells (1500/μL) at birth and none had low NK cells. None have known pathogenic variations in immune-associated genes, but all patients had at least one variation of unknown significance (VUS). One patient has variation in RASGRP1, a gene associated with both lymphopenia and EBV driven lymphoproliferation and is undergoing evaluation for possible HSCT. Live vaccines were held in all patients, until 12-month pending re-evaluation of T cells and antibody response to non-live vaccines. Thus far, two of the patients have resolved T cell lymphopenia (>1500/μL).
This new cohort of T lymphopenic infants of unclear underlying diagnosis pose a management dilemma, requiring long-term follow-up and additional genetic investigations in order for us to provide well-informed prognoses and potential interventions.
Uploaded File(s)

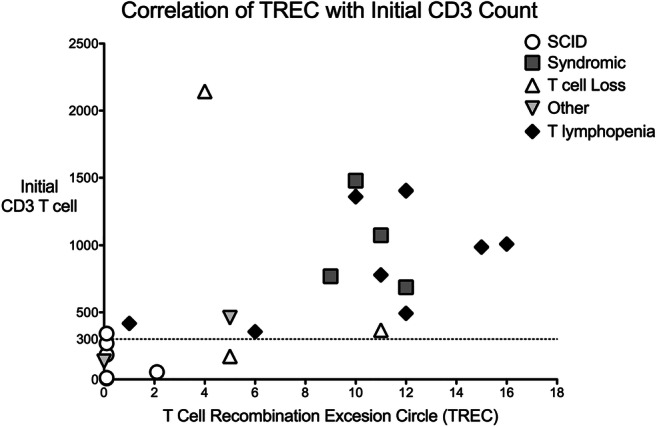
Figure 1: Correlation of TREC level with initial CD3+ T cell count, grouped by underlying diagnosis
Keywords: Severe Combined Immunodeficiency, T cell Lymphopenia, Newborn Screen, T cell Recombination Excision Circle (TREC)
Disclosure:. Manish Butte received research grant from Horizon, Regeneron and Takeda and speaker honoraria from CSL. All other authors indicated they had no financial relationships to disclose.
(185) A Proposed Targeted Treatment for Omenn Syndrome – A Case Report
Terrie Ahn, MD1, Gloria Sheng, MD, PhD1, Maria Garcia-Lloret, MD2, Manish Butte, MD, PhD2
1Allergy & Immunology Fellow/Division of Allergy, Immunology, and Rheumatology, University of California Los Angeles
2Associate Professor/Division of Allergy, Immunology, and Rheumatology, University of California Los Angeles
Omenn Syndrome (OS) is a type of severe combined immunodeficiency (SCID) most commonly caused by hypomorphic mutations in recombination-activating gene 1 (RAG1) or RAG2. It is characterized by presentation in infancy with cellular immunodeficiency, scaling erythroderma, diarrhea, lymphadenopathy and hepatosplenomegaly resulting from autoreactive, oligoclonal T cells. Prognosis in OS has improved with hematopoietic stem cell transplant (HSCT), but remains suboptimal often due to poor clinical status pre-transplant. Herein, we describe a patient with OS and comment on a possible targeted treatment.
A two-month-old male born to consanguineous parents presented with a positive SCID newborn screen. Immunophenotyping showed a T-B-NK-low profile with poor mitogen proliferation. We found a homozygous missense mutation in RAG1 (c.2867T>C, p.Ile956Thr) previously associated with OS.
At one month of age, he developed scaling erythroderma with concerning lymphocyte count (ALC 4,200 cells/μL), eosinophilia (AEC 4,800 cells/μL), and elevated IgE (9,421 mg/dL). There was no evidence of maternal T cell engraftment. T-helper cell profiling showed a strong Th2-skewing compared to control (Figure 1) consistent with previous reports.
We considered blocking Th2 cytokines to treat OS. In vitro, the patient’s CD4+ helper T cells showed modest decrease in IL-4 and IL-5 expression when cultured with dupilumab (IL-4 receptor-alpha blocker, IL-4Rɑ), whereas the reduction was more robust after IL-12 stimulation (Figure 2).
He received topical tacrolimus and systemic steroids with rapid resolution of his cutaneous disease, and currently awaits HSCT.
In OS, systemic immunosuppression against autoreactive T lymphocytes provides benefit, but often has associated adverse effects. Tailored therapies downregulating type 2 immunity may provide a novel approach to OS management. Interestingly, CD4+ helper T cells showed a weaker response to dupilumab in terms of IL-4 reduction as compared to control, but notably showed a robust decrease in both IL-4 and IL-5 expression with IL-12 stimulation. This could suggest that activated T cells in OS may not require IL-4 signaling for ongoing production of Th2 cytokines. Further studies of therapeutics directed against the Th2 pathway (such as IL-4 or IL-5 blockade) in conjunction with strategies for IL-12 augmentation are needed to fully understand their potential in OS.


Keywords: Omenn Syndrome, Immunodeficiency, Type 2 immunity, Dupilumab, Hypomorphic RAG1
Disclosure:. Manish Butte received research grant from Horizon, Regeneron and Takeda and speaker honoraria from CSL. All other authors indicated they had no financial relationships to disclose.
(186) X-linked Agammaglobulinemia and Massive Thoracic Lipoma: Not so Benign?
Aaron Chin, MD1, Nicholas Rider, DO2
1Resident Physician/Baylor College of Medicine
2Associate Professor/Texas Children's Hospital and the Baylor College of Medicine
X-linked agammaglobulinemia (XLA) is primary immunodeficiency resulting from mutations in the Bruton tyrosine kinase (BTK) gene which disrupt the development of humoral immunity. Although infectious complications of XLA are well characterized, risk of malignancies and tumors may also be a concern. We present a case of a 20 year old, stable XLA patient presenting with a massive thoracic lipoma.
The patient was initially diagnosed after suffering multiple serious bacterial infections within the first 2 years of life prompting evaluation. The patient, his brother, and maternal grandfather were ultimately diagnosed via sequencing of BTK and all have absent B-cells. He was started promptly on weekly subcutaneous immunoglobulin therapy will minimal recurrent infections and stable normal IgG levels since. During a hospitalization for pericarditis and 25-pound weight loss a right axillary mass was detected measuring 16 x 7 x 6 cm. Initial core needle biopsy revealed monomorphic mature fat with no MDM2 amplification consistent with lipoma. The patient underwent resection requiring radical axillary dissection to extricate the tumor from his brachial plexus. Notably, the patient also had multiple subcutaneous nodules above his patella the in years preceding this presentation which were self-revolved and also found to be benign lipomas.
To our knowledge this is the first case of a patient with XLA presenting with multiple lipomas including a massive thoracic lipoma. The association between XLA and malignant neoplasms has been well-documented; however, this case raises the possibility of unusual benign neoplasms as an additional complication of XLA. The understood relationship between XLA and benign neoplasms is limited but may still have important clinical implications. For example, thymolipomas, are reported in patients with hypogammaglobulinemia and suggest a potential relationship between adipose tissue and humoral deficiency. An association with colorectal and gastric polyps is also observed in patients with XLA with increased risk of later undergoing malignant transformation. Generally, patients with humoral deficiencies have a high incidence of benign lymphoproliferative hyperplastic growths that may increase susceptibility of developing a lymphoid malignancy. Our case highlights the need for further characterization of “benign” neoplasms in patients with XLA so they may be appropriately distinguished from malignancies.
Keywords: X-linked agammaglobulinemia, Lipoma, benign neoplasm, primary immunodeficiency, XLA
Disclosure:. Nicholas Rider is an advisory board member for CSL Behring, Horizon Therapeutics and Takeda. All other authors indicated they had no financial relationships to disclose.
(187) Molecular Diagnosis Of A Chinese Boy With X-Linked Agammaglobulinaemia
Jenny Ho, PhD1, Stephen Cheung, PhD1, Pamela Lee, MD2, Janette Kwok, MD PhD3
1Scientific Officer/Queen Mary Hospital
2Associate Professor/Department of Paediatrics and Adolescent Medicine,The University of Hong Kong
3Consultant/Queen Mary Hospital
X-linked agammaglobulinaemia (XLA) is characterised by recurrent bacterial infections in affected males in the first few years of life. Mutations in the Bruton Tyrosine Kinase (BTK) gene have been found to be associated with the clinical presentation of XLA. We investigated a Hong Kong Chinese 6-year old boy presenting with severe E. coli sepsis, pleural effusion and purulent pericardial effusion. Laboratory investigations found absence of IgG/A/M and lymphocyte subset confirmed absence of B-cells which were in concordance with the absence of KREC in the peripheral blood with the KREC assay by quantitative PCR. Nucleotide substitution c.3G>T of exon 2 in the BTK gene has revealed a start-loss variant p.Met1Ile of the BTK protein. Carrier status in the mother and the two maternal aunts has been confirmed. Family study has found the mutation was inherited from one of the biggest hindred of the Hong Kong Chinese XLA family reported in the 90s with different nomenclature as 135G>T. The family has only been tested with basic serology tests e.g. IgG/A/M levels as screening while molecular diagnosis was not widely available at the time. Retrospective or early molecular diagnosis is warranted to review previous similar family screening and hence reduce related complication and allow prompt medical intervention.

Keywords: BTK, XLA, molecular diagnosis, KREC
Disclosure:. All authors indicated they had no financial relationships to disclose.
(188) Systems biology reveals biomarkers and pathways involved in autoimmunity in 22q11.2 deletion syndrome
Carolyn Baloh, MD1, Guglielmo Venturi, Phd2, Bernard Fischer, DMV, Phd3, John Sleasman, MD4
1Fellow/Post-doc/Duke
2Staff Scientist/Duke
3Associate Professor/Duke
4Professor of Pediatrics, Chief of Division of Allergy, Immunology, and Pulmonary Medicine/Duke
Autoimmunity is common in 22q11.2 deletion syndrome (22q11.2DS) but biomarkers associated with its risks have not been identified. Typically, autoimmunity develops 8 years after 22q11.2DS diagnosis with up to 25% experiencing at least 1 autoimmune complication. We sought to identify novel biomarkers of autoimmunity and determine if they shed light on the mechanism of autoimmunity in 22q11.2DS.
Single site, retrospective chart review of clinical outcomes of 71 children with 22q11.2 deletion followed at Duke University Immunology Clinic from 2008 to 2020. Additional prospective assessment of immune biomarkers included flow cytometry of naive T regulatory cells, Th17 T cells, and total T and B cell subsets done on selected participants. Multiplex ELISA and RNA-sequencing were also performed. Results between 22q11.2 deletion participants and age balanced healthy controls were compared using Kruskal Wallis and Benjamini Hochberg correction. The study was approved by the Duke University IRB.
Results: Seventy-one patients with 22q11.2DS were identified with 19 of 71 (26.8%) having physician diagnosis of at least one autoimmune disease. The most common autoimmune diagnoses were hematologic cytopenias. Autoimmunity occurred only in children with 22q11.2DS who had absolute lymphocyte counts < 2680 cells/ul. In a subpopulation of 21 participants with 22q11 DS (7/24 had autoimmunity) and 4 healthy controls, those with 22q11.2DS and autoimmunity had significantly lower percentage CD3+ CD31+ naive T cells, a lower percentage CD4+CD25+CD127loCD45RA+ Naive T regulatory cells, and a shift of T regulatory cells away from Th17 pathways (CD4+CD25+CD127loCXCR3-CCR6-), when compared to 22q11.2 without autoimmunity and healthy controls. Analysis of plasma samples from 24 participants with 22q11.2DS (17/24 had autoimmunity) and 12 healthy controls revealed 3 cytokines (ST2, BAFF, and IFN-beta) that differed significantly based on whether or not the participants had 22q11.2DS. An additional 3 cytokines (MMP3, APRIL, and haptoglobin) differentiated those with autoimmunity. STRING analysis of these 6 cytokines converged on type 1 interferon pathways. Exploratory RNA-seq of a smaller subset identified ST2 as an upregulated gene among children with 22q11.2DS and autoimmunity.
Conclusion: Children with 22q11.2DS and autoimmunity have characteristic immune profiles involving Treg polarization, upregulation of ST2 signaling, and trends toward Type 1 interferon pathways.
Keywords: 22q11.2 deletion syndrome (22q11.2DS), DiGeorge Syndrome, Autoimmunity
Disclosure:. All authors indicated they had no financial relationships to disclose.
(189) Juvenile Sjögren’s Syndrome With Extraglandular Manifestations Mimicking Immunodeficiency Diseases
Akaluck Thatayatikom, MD1, Sthorn Thatayatikom, MD, MSc2, Silvana Carr, MD2, Dima Ezmigna, MD2, Luciana Paim-Marquest, MD, PhD3, Indraneel Bhattacharyya, DDS, MSD4, Seunghee Cha, DDS, PhD4
1Associate Professor/Department of Pediatrics, University of Florida
2Assistant Professor/Department of Pediatrics, University of Florida
3Fellow-in-training/Department of Pediatrics, University of Florida
4Professor/College of Dentistry, University of Florida
Juvenile Sjögren’s syndrome (jSS) is a rare and poorly defined systemic autoimmune disorder affecting children and adolescents. jSS is characterized by chronic lymphocytic infiltrates in the exocrine glands and positive autoantibodies. The clinical phenotype varies from benign glandular disease to aggressive systemic extraglandular manifestations. In the absence of jSS-specific diagnostic criteria, timely diagnosis of jSS is challenging. We present a single case report of a young boy who was evaluated for possible immunodeficiency but later diagnosed with jSS.
A nine-year-old boy was hospitalized for failure to thrive, oral thrush, fungal skin infections, chronic cough, abdominal distension with chronic diarrhea, and hyperpigmented rash of two years duration. Physical examinations revealed Tinea faceii, Tinea capitis, generalized erythematous gingiva, desquamative oral mucosa, angular cheilitis, cervical lymphadenopathy, fine crackles in both lungs, abdominal distension and generalized well-demarcated papules. While EBV on PCR, ANA (1:2560), SSA-52, and enterocyte antibodies were positive, normal CBC, immunoglobulins, lymphocyte subsets, lymphocyte mitogen stimulation, NK cell function, HIV PCR, XIAP, FoxP3, IL-18, and CF variants were found. Chest CT revealed bronchiectasis with diffuse peribronchial thickening and left lower lobe consolidation. A labial minor salivary gland biopsy was positive with a focus score >2. A cytologic smear and skin biopsies indicated oral candidiasis and lichen planus, respectively. Esophageal candidiasis and autoimmune enteropathy with decreased pancreatic enzymes were uncovered. Chronic hepatitis by a liver biopsy with negative CMV and EBV in situ hybridization tests and early lymphocytic interstitial pneumonia with subpleural and interstitial T and B lymphocytic infiltrates by a thoracoscopic lung biopsy were detected. A heterozygous variant of an uncertain significance in IL17RA (c.2428 G>A, p.E810K) was discovered by whole exome sequencing. The patient responded well to pulse methylprednisolone, rituximab, mycophenolate, antifungals, and pancreatic enzyme replacement.
We report the perplexing jSS with overlapping features of primary immunodeficiency including EBV infection, persistent oral and esophageal candidiasis and aggressive extraglandular manifestations, such as autoimmune enteropathy, exocrine pancreatic insufficiency, pulmonary and hepatic involvement and lichen planus. Future studies should investigate any potential association between IL17RA variant with jSS and establish jSS-specific diagnostic criteria to avoid a delay in jSS intervention.
Keywords: Juvenile Sjögren’s syndrome, Immunodeficiency, Candidiasis, ANA, autoimmune enteropathy, Bronchiectasis, Pancreatic insufficiency, Anti-SSA autoantibodies, Epstein-Barr virus, IL17RA
Disclosure:. All authors indicated they had no financial relationships to disclose.
(190) Lower IgG trough and CD4 count may lead to hospitalization for COVID-19 infection in patients on IgG maintenance therapy.
John Kuster, MD1, Serhan Unlu, MD2, Michael Simonov, MD3, Ryan Steele, DO4, Ida Hsu, MD5, Christina Price, MD6, Insoo Kang, MD7, Junghee Shin, MD8
1Clinical Fellow/Division of Allergy and Immunology, Section of Rheumatology, Allergy and Immunology, Department of Internal Medicine, Yale University School of Medicine
2Postdoctoral Research Fellow/Division of Allergy and Immunology, Section of Rheumatology, Allergy and Immunology, Department of Internal Medicine, Yale University School of Medicine
3Instructor; Director of Informatics, Clinical and Translational Research Accelerator (CTRA)/Department of Internal Medicine, Yale University School of Medicine
4Assistant Professor; Program Director, Contact Dermatitis Program/Division of Allergy and Immunology, Section of Rheumatology, Allergy and Immunology, Department of Internal Medicine, Yale University School of Medicine
5Assistant Professor of Clinical Medicine; Training Program Director: Allergy & Immunology Fellowship/Division of Allergy and Immunology, Section of Rheumatology, Allergy and Immunology, Department of Internal Medicine, Yale University School of Medicine
6Assistant Professor of Medicine; VA Medical Center Section Chief Allergy and Clinical Immunology/Division of Allergy and Immunology, Section of Rheumatology, Allergy and Immunology, Department of Internal Medicine, Yale University School of Medicine
7Professor; Director of Allergy & Immunology, Internal Medicine/Division of Allergy and Immunology, Section of Rheumatology, Allergy and Immunology, Department of Internal Medicine, Yale University School of Medicine
8Instructor/Division of Allergy and Immunology, Section of Rheumatology, Allergy and Immunology, Department of Internal Medicine, Yale University School
Immunoglobulin G (IgG) therapy is a pooled donor IgG preparation used in the treatment of antibody deficiency, autoimmune disease, rheumatologic disorders, and chronic infection. IgG therapy has also been utilized in COVID-19 infection to modulate inflammatory response with some benefit, however, studies on the effect of IgG maintenance therapy for IgG replacement or immune-modulation on COVID-19 infection is limited. Here, we hypothesized that IgG maintenance therapy implemented a vital role in protecting immune deficient and dysregulated patients from poor outcomes of COVID-19 infection.
To test this hypothesis, we performed a retrospective chart review of COVID-19 positive patients in the Yale-New Haven Health System from March 2020 to October 2020. We collected their demographics, Elixhauser comorbidities, home medications including IgG maintenance therapy and immunosuppressive therapy, laboratory data, and clinical outcome of COVID-19 infection.
We identified total of 14,359 patients who were COVID-19 positive in Yale-New Haven Health System. From this cohort, fourteen patients were found to be on IgG maintenance therapy. Of these, three and six patients met the criteria for Common Variable Immune deficiency (CVID) and IgG deficiency, respectively. Five patients had diagnosed autoimmune disorders. The outcome of COVID-19 infection of the fourteen patients showed that eight were hospitalized and there were no deaths. The IgG trough level (743.40 +/- 192.00mg/dL vs 1054.33 +/- 79.84mg/dL, p=0.02) and baseline CD4 counts (344.75 +/- 181.92cell/ul vs 733.85 +/- 94.54cell/ul, p=0.009 ) were found to be significantly lower in the hospitalized patients compared to those who were treated as outpatients, respectively. Their diagnosis for IgG maintenance therapy, 48 Elixhauser comorbidities, use of immunosuppressive therapies, CD8, CD19 and NK cell counts were similar between patients who were hospitalized vs. monitored as outpatients.
Our findings suggest that targeting higher IgG trough level (such as 1000 mg/dL) during the COVID-19 pandemic period may reduce the need for hospitalization of COVID-19 infection for patients on IgG maintenance therapy. Monitoring T cell subsets may serve as screening for high risk patients for hospitalization with COVID-19 infection in immune-deficient and -dysregulated patients.
Keywords: IVIG, COVID-19, immunoglobulin G maintenance therapy, autoimmune disease, IgG deficiency
Disclosure:. All authors indicated they had no financial relationships to disclose.
(191) Neurological Manifestations as a Sole Presentation of a Novel Gain-Of-Function Mutation in NLRC4
Sthorn Thatayatikom, MD, MSc1, Akaluck Thatayatikom, MD2
1Assistant Professor/Department of Pediatrics, University of Florida
2Associate Professor/Department of Pediatrics, University of Florida
Inflammasomes are cytosolic multiprotein complexes that sense microbial infections or host cell damage, triggering pro-inflammatory cytokine production including IL-1beta and IL1-8, and leading to cell death. A gain-of-function mutation (GOF) in the gene encoding NLRC4 leads to over activation of NLRC4 inflammasome and autoinflammatory syndrome characterized by persistent elevated IL-18. A recent study demonstrated a potential role of NLRC4 inflammasome activation related to neuroinflammation and neuropathology in animals and in human brain with multiple sclerosis.
A sixteen-year-old Hispanic female presented with intermittent severe throbbing frontal headache twice a week for several months. Her headache typically lasts for 30 minutes but may continue throughout the day without auras, hypersensitivity to light or sounds, nausea or vomiting, diarrhea, fever, skin rash, joint symptoms or signs of infections. She was previously asymptomatic and found with a novel gain-of-function mutation in NLRC4, c.1475G>A (p.Arg492Gln) without clinical symptoms and signs of inflammatory disease. The genetic test was obtained since the mutation was found in three family members including the father, 3-year-old brother and 12-year-old sister who were diagnosed with NLRC4 associated autoinflammatory disease. While abnormal laboratory tests including persistent highly elevated IL-18 in the range of 2234-2297 pg/ml, positive thyroid autoantibodies, and ANA (1:160) were detected, normal CBC, ESR, CRP, T4, TSH and cytokines studies including IL-1beta, IL-2, IL-6, IL-8, IL12, IFN-gamma, TNF-alpha, were uncovered. A brain MRI revealed mild to moderate generalized atrophy. Since an IL-18 inhibitor has not been approved by FDA, a trial of canakinumab, an IL-1 inhibitor was considered.
We report a teenager female with gain-of-function mutation in NLRC4 who developed severe headache with imaging evidence of brain pathology. This case highlights that neurological symptoms can be the only clinical manifestation of NLRC4 associated autoinflammatory disease and the deleterious effect of NLRC4 inflammasome activation in the brain development. Future evaluation of the role of NLRC4 inflammasome and the development of central nervous system in children is warranted.
Keywords: NLRC4, Autoinflammatory syndrome, IL-18, Headache, inflammasome
Disclosure:. All authors indicated they had no financial relationships to disclose.
(192) ARGINASE + MYELOID-DERIVED SUPPRESSOR CELLS ARE IMPORTANT IN THE PATHOPHYSIOLOGY OF SEVERE COVID-19.
Augusto Ochoa, MD1, matthew Dean, PhD2, Juan Ochoa, MD3, Maria Sanchez-Pino, PhD4, zabaleta Jovanny, PhD5, Jone Garai, PhD6, Luis Del Valle, MD, PhD7, Dorota Wyczechowska, PhD8, Lyndsey Buckner, PhD9, Phaethon Philbrook, MD, PhD Candidate10, Rinku Majumder, PhD11, Richard Vander Heide, MD12, Logan Dunkenberger, student13, Ramesh Thyllur, PhD6, Bobby Nossaman, MD14, W. Mark Roberts, MD15, Andrew Chapple, PhD4, Jack Collins, PhD16, Brian Luke, PhD17, Randall Johnson, PhD17, Claudia Morris, MD18, Julia Garcia Diaz, MD19
1Professor Pediatrics, Director LSU Cancer Center/Louisiana State University Health - New Orleans, Louisiana
2Postdoctoral Fellow/LSU Health
3Director, Surgical Intensive Care Unit/Ochsner Medical Center
4Assistant Professor/LSU Health
5Associate Professor, Director Genomics Core/LSU Cancer Center
6Postdoctoral Fellow/LSU Cancer Center
7Associate Professor/LSU Cancer Center
8Instructor/LSU Cancer Center
9Supervisor, Biorepository/Ochsner Medical Center
10Student/LSU Cancer Center
11Professor Biochemistry/LSU Health
12Professor, Pathology/LSU Health
13Research Associate/LSU Cancer Center
14Anesthesiologist - ICU/Ochsner Health
15Dean of Research/Ochsner Health
16Director, Advanced Biomedical Computational Science/Frederick National Laboratory for Cancer Research
17Investigator/Frederick National Laboratory for Cancer Research
18Professor of Pediatrics and Emergency Medicine/Emory University School of Medicine
19Director, COVIS-19 Clinics/Ochsner health
Approximately 35% of COVID-19 patients are asymptomatic, while 20% develop severe respiratory and coagulation complications. The mechanisms differentiating these presentations is unclear. Severe COVID-19 is associated with uncontrolled inflammation including a cytokine storm, increased granulocytes and decreased T cells. Genomic studies suggested the importance of granulocytes and two reports showed the presence of myeloid-derived suppressor cells (MDSC). MDSC produce arginase and deplete arginine which impairs T cell and endothelial cell function, and increases reactive oxygen species (ROS).
We compared the type of MDSC among patients with different stages of COVID-19, tested arginase expression and its effect on arginine levels, T cells and endothelial cells. We also aimed to determine if MDSC might play a role in respiratory complications of severe COVID-19. Transcriptomic signatures of MDSC between COVID-19 patients at different stages were compared.
A prospective observational study was conducted with 24 severe, 5 asymptomatic, and 26 convalescent COVID-19 patients, and 15 normal controls. Autopsy samples from 5 patients who perished from severe COVID-19 were also tested. All samples were tested for MDSC subpopulations, T cells and NK cells. Plasma was tested for arginine, nitrates, cytokines and markers for endothelial cell dysfunction. Transcriptomes of purified MDSC were compared using RNAseq.
Severe COVID-19 patients had a highly significant increases in circulating Arginase+ granulocytic-MDSC (Arg+G-MDSC), compared to other groups. High Arg+G-MDSC resulted in decreased plasma arginine, decreased numbers of T cells and NK cells and increased markers of endothelial cell dysfunction. Large accumulations of Arg+G-MDSC expressing NOX1 and NOX2, were found infiltrating lungs of patients who died from severe COVID. Transcriptome comparison showed that Arg+G-MDSC from asymptomatic patients had increased expression of Type I IFN genes and associated downstream pathways, while severe COVID-19 patients had increased expression of granulocyte functions. Statistical analysis suggests that G-MDSC may identify patients at risk for developing severe COVID-19.
Arg+G-MDSC play an important role in the pathophysiology of severe COVID-19. Arginine depletion can cause T cell and endothelial cell dysfunction, while infiltration of lungs by Arg+G-MDSC expressing NOX-1 and NOX-2 may produce ROS resulting in extensive pulmonary inflammation and damage and respiratory distress.
Keywords: Myeloid-derived suppressor cells (MDSC), Arginase 1, T cell receptor z chain, COVID-19
Disclosure:. Claudia Morris is a consultant for Pfizere. All other authors indicated they had no financial relationships to disclose.
(193) Immunodeficiency Identified In Organ Donor
Eszter Lazar-Molnar, PhD1, Archana Agarwal, MD2, Attila Kumanovics, MD3
1Assistant Professor/University of Utah
2Associate Professor/University of Utah
3Clinical Consultant/Mayo Clinic
Offer for an import kidney was received for a sensitized, 99% cPRA re-transplant patient. Prospective crossmatch performed using donor lymphocytes from whole blood revealed a compatible T cell crossmatch, however, the B cell crossmatch could not be interpreted due to very low number of B cells (130 B cells, 0.4% of enriched lymphocytes). Follow-up on the 34y old male donor’s history revealed that the cause of death was bacterial meningitis and pneumonia, laboratory results indicated increased white cell count, and cultures were positive for Streptococcus pneumoniae. The donor chart indicated a history of IgM deficiency, however, the presence of encapsulated bacteria in the context of the low B cells suggested the possibility of an immunodeficiency affecting IgG levels. Indeed, serum immunoglobulin test showed decreased levels of both IgM and IgG, and very low levels of Streptococcus pneumoniae antibodies (11 out of 23 serotypes above 1ug/ml, 4 over 1.3ug/ml).
Histological analysis of the residual spleen tissue indicated that the spleen was severely depleted of B cells, with markedly attenuated follicles and expansion of red pulp.
Whole exome sequencing of residual donor DNA from the donor identified a pathogenic R288Q variant of Bruton’s tyrosine kinase (Btk) enzyme, which was also confirmed by Sanger sequencing. Pathogenic variants of Btk are known to cause X-linked agammaglobulinemia.
Identification of a pathogenic Btk variant in this deceased donor explained the observed dramatically decreased B cell numbers in whole blood and spleen, and provided explanation for the fatal sensitivity to infection with encapsulated bacteria. The results of the findings were communicated to the originating Organ Procurement Organization to be used for informing and counseling the donor family.
Keywords: agammaglobulinemia, B cell, Bruton's tyrosine kinase
Disclosure:. All authors indicated they had no financial relationships to disclose.
(194) Combination Treatment with Cromolyn Sodium and Masitinib Displays Cell-Protective Effect and Shows Additive Anti-Oxidative Actions on an in vitro Neurodegenerative Model
A. Yasemin Göksu Erol, MD1, Fatma Gonca Kocanci, Phd2, Devrim Demir-Dora, Phd3, Hilmi Uysal, MD4
1Assoc. Prof. Dr./1- Akdeniz University, Department of Gene and Cell Therapy / 2- Faculty of Medicive, Department of Histology and Embryology
2Asst. Prof./Alaaddin Keykubat University, Division of Medical Techniques, Department of Medical Laboratory Techniques
3Asst. Prof. Dr./Akdeniz University, Faculty of Medicine, Department of Medical Pharmacology
4Prof. Dr./Akdeniz University, Faculty of Medicine, Department of Neurology
Neuroinflammation plays a key role in occurrence and development of neurodegenerative diseases. Microglia, the resident immune cells in the brain, have been recognized to contribute to neuroinflammation. Cromolyn sodium, which is an FDA-approved mast cell-stabilizer drug used in the treatment of asthma and known to prevent mast cells from triggering the immune response, has a neuroprotective and anti-inflammatory effect on microglial cells, as well. Masitinib [1] is an orally administered selective tyrosine kinase inhibitor which is developed for neurological (i.e. amyotrophic lateral sclerosis), inflammatory and oncological diseases. It modulates the activity of mast cells and macrophages for immunity, and can exert a neuroprotective effect in some neurodegenerative diseases by its activity on mast cells and microglia.
In our previous studies, we found that masitinib inhibits neuronal cell death on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) degenerated- differentiated (d)-SH-SY5Y human neuroblastoma cell model. In this study we tested the synergistic neuroprotective effects of cromolyn sodium and masitinib when applied together on the same model.
First, (d)-SH-SY5Y cells, with or without MPTP, were exposed to different doses of cromolyn sodium. MTT assay was used to detect cell survival rates. Spectrophotometric measurements of total oxidant capacity (TOC)/total antioxidant capacity (TAC) ratios in CM of (d)-SH-SY5Y cells were performed.
When (d)-SH-SY5Y cells were exposed to MPTP, a dose-dependent decrease in cell viability rates was observed. High doses of cromolyn ameliorated decreased viability rates (p < 0.05) (Fig 1). When cromolyn (6.25 μM) and masitinib (0.5 μM) were applied as combination treatment on d-SH-SY5Y cells, with or without MPTP (d)-SH-SY5Y cell viability was found to be increased (Fig 2), and TOC/TAC ratios in CM of d-SH-SY5Y cells were measured to be significantly decreased (p < 0.05) (Fig 3). These agents reduced TOC/TAC ratios more effectively when applied together, compared to their mono-treatments.
Combined therapy with cromolyn sodium and masitinib showed an additive effect on decreasing oxidative stress, and protecting cells against neurotoxin applications. Beneficial effects of this combined treatment should be considered in future investigations.
References
1- Dubreuil P, Letard S, Ciufolini M, Gros L, Humbert M, et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PloS One 2009;4(9):e7258

Figure 1: Dose dependent effects of cromolyn sodium treatment on with or without MPTP (d)-SH-SY5Y cell viability.
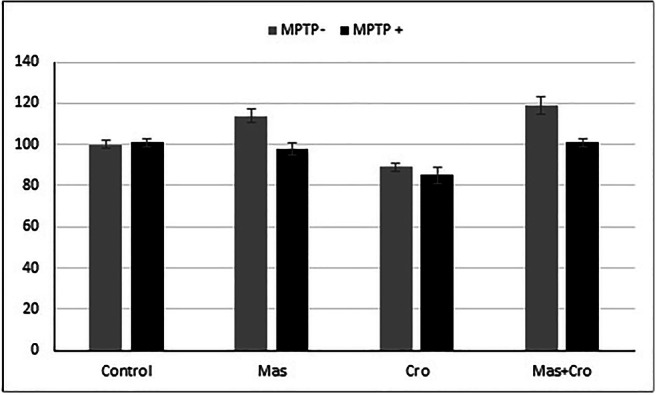
Figure 2. Effects of cromolyn sodium (6.25 μM) and masitinib (0.5 μM) treatment on with or without MPTP (d)-SH-SY5Y cell viability.
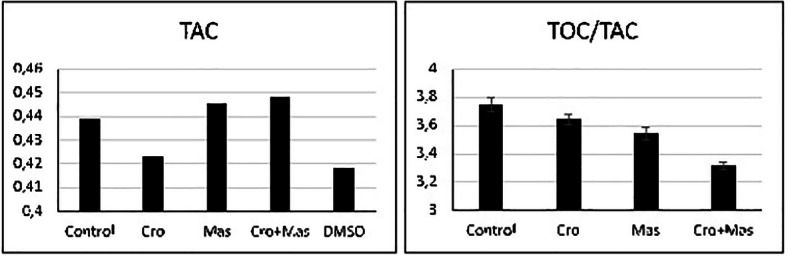
Keywords: Masitinib, Cromolyn sodium, Neurodegeneration, Neuroinflammation, Combined therapy, Cell culture, Oxidative stress, Total antioxidant capacity, SH-SY5Y cells, MPTP
Disclosure:. All authors indicated they had no financial relationships to disclose.
(195) A case of subcutaneous panniculitis-like T-cell lymphoma with hemophagocytic lymphohistiocytosis independent of germline variants in HAVCR2
Gloria Sheng, MD, PhD1, Maria Garcia-Lloret, MD2, Manish Butte, MD, PhD2
1Allergy & Immunology Fellow/Division of Allergy, Immunology, and Rheumatology, University of California Los Angeles
2Associate Professor/Division of Allergy, Immunology, and Rheumatology, University of California Los Angeles
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is an unusual type of non-Hodgkin skin lymphoma characterized by subcutaneous infiltration of pleomorphic alpha/beta T cells mimicking panniculitis. Patients have a good survival rate unless they develop hemophagocytic lymphohistiocytosis (HLH), which occurs in 20% of cases. A germline mutation in the hepatitis A virus-cellular receptor gene (HAVCR2) causing loss of function of T cell immunoglobulin mucin 3 (TIM-3) has been identified in 60-85% of SPTCL-HLH cases. Patients with wild-type HAVCR2 can develop SPTCL-HLH, albeit with a lower incidence and typically in the setting of underlying autoimmune disease. Herein we report a previously healthy 40 year old male who presented with a 4 year history of persistent fevers, lymphadenopathy, subcutaneous nodularities, seizures without apparent cause, found to have splenomegaly and pancytopenia. He underwent an extensive workup at an outside institution that was negative for infectious and autoimmune etiologies. He had numerous lymph and bone marrow biopsies, all of which were unrevealing. Upon transfer to our institution, patient met diagnostic criteria for HLH (including elevated ferritin, elevated soluble IL-2 receptor, hemophagocytosis on repeat bone marrow biopsy) with marginally elevated EBV titers. Skin biopsy of a lower extremity subcutaneous nodule was also consistent with SPTCL. Immune workup was notable for persistently low IgG, otherwise unremarkable. Cytokine panel was consistent with a dysregulated IFN-gamma-inducible inflammatory host response. Molecular assays were negative for chronic granulomatous disease and X-linked lymphoproliferative disease type 1/2. Whole exome sequencing did not identify a primary immunodeficiency associated with increased susceptibility to HLH (including no variants in HAVCR2). However, patient was found to have a heterozygous c.169G >A variant in the MCM4 gene, which may be associated with NK cell deficiency and EBV lymphoproliferative disease. Further work is needed to determine the significance of this variant and its possible relationship with SPTCL-HLH, as well as reanalysis of his exome to determine if he has an alternative variant which may affect TIM-3 expression or downstream pathways. While the patient has improved on etoposide and dexamethasone, he may require maintenance therapy with an immunomodulator to target this immune checkpoint inhibitor or its effectors.
Keywords: hemophagocytic lymphohistiocytosis, subcutaneous panniculitis-like T-cell lymphoma, dysgammaglobulinemia, immune overactivation, MCM4
Disclosure: Manish Butte received research grant from Horizon, Regeneron and Takeda and speaker honoraria from CSL. All other authors indicated they had no financial relationships to disclose.
(196) No association of viral infection with CTLA-4 insufficiency onset
Noriko Mitsuiki, MD, PhD1, Máté Krausz, MD2, Laura Gamez-Diaz, PhD3, Nadezhda Camacho, MD4, Hartmut Hengel, MD, PhD5, Bodo Grimbacher, Prof. Dr. med.6
1Physician/Center for Chronic Immunodeficiency, Faculty of Medicine, Medical Center of the University Hospital, University of Freiburg, Freiburg, Germany AND Department of Pediatrics, Tokyo Medical and Dental University, Tokyo, Japan.
2Physician scientist/Centre for Chronic Immunodeficiency
3Postdoc/Center for Chronic Immunodeficiency, Faculty of Medicine, Medical Center of the University Hospital, University of Freiburg, Freiburg, Germany
4PhD student/Center for Chronic Immunodeficiency, Faculty of Medicine, Medical Center of the University Hospital, University of Freiburg, Freiburg, Germany.
5Medical Director- Institute of Virology/Institute of Virology, Medical Center of the University Hospital, University of Freiburg, Freiburg, Germany.
6Head of the laboratory/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
Cytotoxic-T-lymphocyte-antigen-4 (CTLA-4) insufficiency is an immune deficiency and immune dysregulation syndrome caused by heterozygous mutations in CTLA4. Aiming to determine the influence of viral infections as triggers of the disease onset that explain the incomplete penetrance characteristically observed in CTLA-4 insufficiency patients, we evaluated the seroprevalence of antibodies against HSV (Herpes simplex virus), CMV (cytomegalovirus), EBV (Epstein Barr Virus), and human parvo virus B19.
We collected serum samples and corresponding clinical data from wild-type CTLA4 healthy individuals (n=12) and patients with CTLA-4 insufficiency, including affected CTLA4 mutation carries (n=5) and unaffected CTLA4 mutation carriers (n=7). The later showing normal immunoglobulin production or dysgammaglobulinemia. Following the seroprevalence evaluation of antibodies against HSV, CMV, EBV, and human parvo virus B19, we performed a Fc gamma receptor (FcγR) activation assay for EBV to measure the ability of the IgG against EBV to bind to viral antigens and to stimulate and activate FcγR. The FcγR activity was estimated by IL-2 levels produced by human FcrR-expressing BW5147 cells.
Affected CTLA4 mutation carriers did not present higher seroprevalence of any of the four pathogens evaluated, in comparison to unaffected CTLA4 mutation carriers and wild-type healthy individuals. In addition, the FcrR activation assay demonstrated that all of the unaffected CTLA4 mutation carriers showed normal FcγR activation, while two out of five affected CTLA4 mutation carriers showed low activity, suggesting that affected CTLA4 mutation carriers possess IgG with abnormal functionality. No history of severe EBV-associated diseases in these patients were reported.
Our data suggest that HSV, CMV, EBV and human parvo virus B19 do not trigger the disease onset in the context of CTLA-4 innsufficiency.
Keywords: CTLA4, Viral infections, Primary immunodeficiency, EBV, CMV, HSV
Disclosure: Bodo Grimbacher received research grant from Baxalta. All other authors indicated they had no financial relationships to disclose.
(197) Recurrent Infections and Profound Autoimmune Disease in a Patient with a Heterozygous TNFSF12 Variant
Daniel Cerrone, MD1, Manish Butte, MD, PhD2
1Clinical Fellow/Division of Allergy, Immunology, and Rheumatology, University of California Los Angeles
2Associate Professor/Division of Allergy, Immunology, and Rheumatology, University of California Los Angeles
TNFSF12 (TWEAK) has been implicated as a very rare, autosomal dominant cause of common variable immune deficiency (CVID). We present a case of a 63-year old female diagnosed with a TNFSF12 variant in the setting of recurrent infections and numerous comorbid autoimmune conditions.
Our patient first came to medical attention in childhood where she was noted to suffer from numerous episodes of otitis media, bacterial pharyngitis, and bacterial pneumonia. She developed bouts of oral thrush even aside from antibiotic use. At age 24 years she developed her first of two lifetime cases of optic neuritis. She was also diagnosed with hyperparathyroidism in setting of osteoporosis and numerous fractures. She had three children, with one diagnosed with type 1 diabetes and rheumatoid arthritis. Her infections persisted and she was referred to an immunologist at 57 years of age, who noted a mildly low IgG of 612 mg/dL and IgA of 75 mg/dL. Her response to the pneumococcal polysaccharide vaccine was inadequate and she would be diagnosed with CVID. Of note, both TPO and cardiolipin antibodies were elevated at time of diagnosis. She has received subcutaneous immunoglobulin since diagnosis, however, despite maintaining a treatment IgG level consistently above 1100 mg/dL, she has required hospitalization for treatment of Listeria bacteremia and treatment of submandibular cellulitis. She has also been diagnosed with granulomatous-lymphocytic interstitial lung disease.
At 63 years of age genetic testing revealed a single heterozygous missense variant in TNFSF12 (NM_003809:c.716T>C (p.Phe239Ser)). This variant is located in the conserved TNF homology domain. More recent immune testing revealed a B-cell count of 127 cells/μL but a relatively low switched memory B-cell count and percentage (6.8% of B cells, 8.6 cells/μL).
TNFSF12 encodes TWEAK, which promotes proliferation, fibrosis, and chronic inflammation in autoimmune diseases. Previous investigations have shown that R145C-variant TWEAK signals appropriately through its receptor, but forms aberrant oligomers with BAFF and perturbs B-cell signaling. Our case highlights not only the infections consequences of TNFSF12 mutations but also a profound autoimmune phenotype.
Keywords: TNFSF12, TWEAK, CVID
Disclosure: Manish Butte received research grant from Horizon, Regeneron and Takeda and speaker honoraria from CSL. All other authors indicated they had no financial relationships to disclose.
(198) HLA-typing analysis of patients with CTLA-4 insufficiency
Noriko Mitsuiki, MD, PhD1, Charlotte Schwab, MD, PhD2, Laura Gamez-Diaz, PhD3, Veronika Soetedjo, MSc4, Florian Emmerich, MD5, Bodo Grimbacher, MD6
1Physician/Center for Chronic Immunodeficiency, Faculty of Medicine, Medical Center of the University Hospital, University of Freiburg, Freiburg, Germany AND Department of Pediatrics, Tokyo Medical and Dental University, Tokyo, Japan.
2Resident/Center for Chronic Immunodeficiency, Faculty of Medicine, Medical Center of the University Hospital, University of Freiburg, Freiburg, Germany.
3Postdoc/Center for Chronic Immunodeficiency, Faculty of Medicine, Medical Center of the University Hospital, University of Freiburg, Freiburg, Germany
4PhD Student/Institute of Medical Biometry and Statistics, Freiburg Center for Data Analysis and Modeling (FDM), IMBI/ZKS, Freiburg, Germany.
5Principal Investigator- Laboratory for Immunogenetics/Institute for Transfusion Medicine and Gene Therapy, University Medical Center Freiburg, University of Freiburg, Freiburg, Germany.
6Principal Investigator -Genetic Basis of Immunodeficiency/Center for Chronic Immunodeficiency, Faculty of Medicine, Medical Center of the University Hospital, University of Freiburg, Freiburg, Germany.
Heterozygous mutations in Cytotoxic-T-lymphocyte-antigen-4 (CTLA4) causes an immune deficiency and immune dysregulation syndrome known as CTLA4 insufficiency, which is characterized by a heterogeneous clinical phenotype and an incomplete disease penetrance. Aiming to identify a disease modifier, we investigated the correlation of the HLA-type of CTLA4 patients with several clinical and baseline characteristics, as HLA type is known to be an underlying mechanism of autoimmune diseases and immune dysregulation.
We evaluated a total of seven families composed of 43 individuals carrying heterozygous mutations in CTLA4. Among them, 25 patients were clinically affected whereas 18 were unaffected. Genotype analyses at 4-digit-level of the HLA locus, HLA-A, -B, -C, -DRB1, and -DQB1 were performed in all patients. Statistical analysis using the method of generalized estimating equations (GEE) allowed us to estimate HLA-types with statistically significant odds ratio (OR).
Following multiple comparisons analyses of HLA-typing with different variables (including clinical features and age), we found that affected CTLA4 mutation carriers presented HLA-DRB1 *13:01 with a higher OR (OR 1.73; p value= 0.024) in comparison to unaffected CTLA4 mutation carriers. In addition, patients with CTLA-4 insufficiency who developed autoimmune diseases and lymphoproliferation had significantly more often HLA-DRB1 *15:01 (OR 3.71; p value= 0.013), and HLA-DRQ1 *06:02 (OR 4.82; p value=0.023). HLA-DRB1 *15:01 and -DRQ1 *06:02 are predicted to form a haplotype.
Our results showed that CTLA-4-insufficient patients with autoimmune diseases and lymphoproliferation who were categorized with a severe immune dysregulation phenotype, presented the haplotype HLA-DRB1 *15:01 and -DQB1 *06:02 with higher frequency. Further studies focused on the association of HLA-typing and CTLA-4 insufficiency onset should be performed.
Keywords: CTLA4, HLA type, primary immunodeficiency
Disclosure: All authors indicated they had no financial relationships to disclose.
(199) Two Unrelated Cases of Familial Hemophagocytic Lymphohistocytosis Presented as Severe Combined Immunodeficiency
Khaled Hazzazi, MD1, Bandar Al-Saud, MD2, Reem Mohammed, MD2
1Fellow in Pediatric Allergy/immunology/King Faisal Specialist Hospital and Research Centre
2Consultant Allergist/Immunologist/King Faisal Specialist Hospital & Research Centre
There is a growing data on primary immune deficient patients presenting with hemophagocytic lymphohistocytosis (HLH). On the other hand, It is less often reported for a patient with a known genetic mutation causing familial HLH to present clinically as severe combined immunodeficiency (SCID).
We present two unrelated Saudi patients with familial HLH presenting as SCID. The first child was a 6-month-old male, who presented with severe pneumonia at age of 4 months, requiring mechanical ventilation. His preliminary laboratory assessment showed severe lymphopenia and hypogammaglobulinemia. His lymphocytes markers confirmed a T-B-NK- SCID phenotype. At 8 months of age, he developed persistent fever and progressive cytopenia despite broad spectrum antimicrobials. His laboratory testing showed pancytopenia, hyperferritinemia, transaminitis and hypertriglyceridemia. A diagnosis of HLH was suspected and confirmed with bone marrow biopsy. Genetic testing revealed a homozygous missense mutation c.3145C>G p.(Pro1049Ala) in UNC13D gene.
The second case is a 2-year-old female presented at age of 9 months with recurrent skin abscesses and febrile neutropenia. Her initial lab showed neutropenia and severe lymphopenia. Immunological workup revealed severely depressed lymphocyte function and markedly low T and B lymphocytes markers consistent with T-B-NK+ SCID. Genetic testing showed a homozygous mutation variant c.1229G>C (p.Arg410Pro) in PRF1 gene.
This case series of familial HLH presenting with SCID phenotype adds to the spectrum of clinical presentation of familial HLH and highlights the importance of early genetic diagnosis in improving patients’ outcome.
Keywords: hemophagocytic lymphohistocytosis, FHL, combined immunodeficiency, mutation in UNC13D, PRF1
Disclosure: All authors indicated they had no financial relationships to disclose.
(200) Hereditary Angioedema Type 1, Pregnancy, and Acute SARS-CoV-2 Infection
Amanda Salih, MD, MPH1, Manisha Gandhi, MD2, Amir Shamshirsaz, MD3, Joud Hajjar, MD4
1Fellow/Baylor College of Medicine/ Texas Children's Hospital
2Associate Professor, Division of Maternal Fetal Medicine, Department of Obstetrics and Gynecology/Baylor College of Medicine
3Assistant Professor, Division of Maternal Fetal Medicine/Surgical Critical Care/Baylor College of Medicine
4Assistant Professor of Immunology, Allergy, and Rheumatology/Baylor College of Medicine
Hereditary angioedema (HAE) and coronavirus disease 2019 (Covid-19) have pathophysiologic overlap via the activation of complement, contact, and coagulation systems, and production of proinflammatory cytokines. Both HAE and Covid-19 can lead to an exaggerated inflammatory response. Common pathway blockade through C1 esterase inhibitor (C1-INH) may dampen inflammation and improve outcomes.
We report a case of a patient with HAE Type I, pregnant in her second trimester, who developed Covid-19.
SERPING1 sanger sequencing was performed by GeneDx. SARS-CoV-2 PCR was performed by Texas Children’s Hospital Laboratory.
A 20-year-old Caucasian female with known HAE type 1 (SERPING1 c.707 T>C) presented at 22-weeks gestation with abdominal pain and extremity swelling. She had been maintained on subcutaneous C1-INH (Haegarda®) at 60 IU/Kg with disease control prior to presentation.
Routine nasal swab surveillance screening in the emergency department for Covid-19 by PCR was positive. She experienced no fever, chills, nasal congestion, or dyspnea symptoms. Later, she reported loss of taste and smell. She was admitted to the hospital and received 2500 IU of intravenous human C1-INH. Following the abortive therapy, her swelling and abdominal pain resolved. No signs of fetal distress occurred despite flare. She was discharged 24 hours after admission. Her pregnancy progressed to 36 weeks and she delivered a healthy newborn. Repeat Covid-19 testing 2 days prior to delivery was negative.
Pregnancy is an independent risk factor in both Covid-19 and HAE. Pregnant women infected with Covid-19 are at increased risk for both intensive care unit admission and mechanical ventilation requirements. Elevated estrogen during pregnancy can trigger an angioedema flare. Despite having two independent risk factors for poor outcomes, our patient fared well. Her mild clinical course might be attributed to C1-INH therapy that blunted the inflammatory response associated with Covid-19.
While further studies are currently being conducted to investigate the effectiveness of C1-INH in decreasing severe inflammation associated with Covid-19, our case highlights the importance of maintaining adequate therapy in HAE patients, particularly during pregnancy.
Keywords: Hereditary Angioedema Type 1, SERPING1, Covid-19, SARS-CoV-2, Pregnancy, C1-INH, Angioedema
Disclosure: Joud Hajjar received research grants from Amplimmune, Arcus Biosciences, ARMO BioSciences, Atterocor/Millendo, Baxalta, BMS, Calithera Biosciences, Chao Physician-Scientist Foundation, CytomX Therapeutics, Eli Lilly, EMD Serono, Healios Onc, Immune Deficiency Foundation, ImmuneOncia, Incyte, Jeffrey Modell Foundation, Karyopharm, Kymab, MedImmune, Merck, National Cancer Institute, NeoImmuneTech, Neon Therapeutics, Novartis, OncoSec KEYNOTE-695, Pfizer, PsiOxus, Regeneron, Surface Oncology, The Texams Medical Center Digestive Diseases Center and Top Alliance; she was an Advisory Board member of Alfaisal University, Genome & Company, Horizon, and Pharming. All other authors indicated they had no financial relationships to disclose.
(201) At Both Ends of the Spectrum: A Case Report of an Infant with Anauxetic Dysplasia and Severe Combined Immunodeficiency
Lindsay Zayia1, Hana Niebur, MD2, Danita Velasco, MD3
1Medical Student/Creighton University School of Medicine, Omaha, NE
2Assistant Professor/Division of Allergy and Immunology, Department of Pediatrics, University of Nebraska, Omaha, NE, USA
3Assistant Professor/Division of Genetic Medicine, Munroe-Meyer Institute for Genetics and Rehabilitation, University of Nebraska Medical Center, Omaha, NE
Cartilage-Hair Hypoplasia-Anauxetic Dysplasia (CHH-AD) spectrum disorder includes a range of phenotypes related to biallelic variants in RMRP, the untranslated RNA component of mitochondrial RNA processing endoribonuclease. This spectrum includes severe short-limbed short stature and vertebral anomalies, with limited extraskeletal features in AD to combined immunodeficiency, sparse hair, anemia, gastrointestinal abnormalities, and metaphyseal dysplasia without vertebral involvement or even normal stature in CHH. While CHH is a rare cause of severe combined immunodeficiency (SCID), AD is not typically associated with SCID. Herein, we describe a case of an infant with skeletal features consistent with AD and SCID.
Next-generation sequencing was obtained with follow-up parental testing confirming biallelic inheritance.
A term female infant with prenatally detected micromelia and limb bowing suggestive of thanatophoric dysplasia presented with abnormal TREC assay on day of life 5. Prenatal genetic testing including FGFR3 was unrevealing. After birth, the infant had profound lymphopenia, and anemia, requiring a PRBC transfusion. While anemia improved, lymphopenia continued, and the infant developed moderate neutropenia. Flow cytometry demonstrated profound B- and T-cell lymphopenia with absent naïve T-cells. IgG, A, and M were low to absent. A skeletal survey showed characteristic features of AD including platyspondyly, hypoplastic iliac bones, and shortened, bowed femurs, humeri, radii, and ulnae. MRI demonstrated a small foramen magnum with an attenuated CSF signal at the cervicomedullary junction and upper cervical spinal cord. Genetic testing showed biallelic variants in RMRP with a known pathogenic variant in the promoter region (n.-19_-3dup) and a novel variant in the transcribed region (n.223C>T). The infant was referred for hematopoietic stem cell transplantation.
Conclusions: To our knowledge, this is the first reported case of an infant with both the severe skeletal findings of AD and extraskeletal features of CHH including SCID. This novel case demonstrates the importance of immunological screening in all individuals presenting with features consistent with a CHH-AD spectrum disorder, regardless of the severity of the skeletal dysplasia and expands the phenotype of CHH-AD spectrum disorders.
Keywords: Severe Combined Immunodeficiency, Cartilage Hair Hypoplasia, Anauxetic Dysplasia
Disclosure: All authors indicated they had no financial relationships to disclose.
(202) Seropositive IgG Responses to Coronaviruses in Children Pre-COVID-19 Provides Cross-Protection Against SARS-CoV-2
Nicolai van Oers, PhD1, Quan-Zhen Li, MD, Phd2, Fatma Coskun, BSc3, Prithvi Raj, Phd4, Patricia Pichilingue-Reto, MD, Phd5, Igor Dozmorov, PhD2, Jeffrey Kahn, MD, Phd1, Christian Wysocki, PhD, MD6
1Professor/UT Southwestern Medical Center
2Associate Professor/UT Southwestern Medical Center
3Graduate Student/UT Southwestern Medical Center
4Assistant Professor/UT Southwestern Medical Center
5Medical fellow/UT Southwestern Medical Center
6Pediatric Immunologist. Assistant Professor/UT Southwestern Medical center
The antibody repertoire forms in response to infections, the microbiome, vaccinations and environmental exposures. The specificity of such antibody responses was compared among a cohort of toddlers to identify associations between seropositive responses to SARS-CoV-2 relative to other infectious agents and vaccinations.
The serum IgG antibody reactivities in children and adults prior to and during the COVID-19 pandemic was compared with a microfluidic array containing 110 distinct antigens. Seropositivity to RNA and DNA viruses, including multiple Coronaviruses together with SARS-CoV-2 was compared. Neutralization assays were performed on individuals with seropositivity to SARS-CoV-2. A stratification was developed based on quantitative variations in the IgG responses. Clinical presentations were investigated in relation to IgG responses.
Approximately 5% of toddlers seen prior to the COVID-19 pandemic had seropositive antibody responses to the Spike protein from diverse Coronaviruses. Some of these IgG responses provided cross-protection against SARS-CoV-2 in a neutralization assay. This was in spite of limited IgG responses to the SARS-CoV-2 nucleocapsid protein. Current analyses are examining whether the SARS-CoV-2 responses are associated with enhanced IgG responses to different Coronaviruses along with diverse live-attenuated viral vaccines. Taken together, our findings suggest that prior Coronavirus infections in children may afford some protection against COVID-19. This may account for the less severe conditions seen in some the younger age group relative to adults, who may have diminished serospecific antibody responses.
Keywords: SARS-CoV-2, serum IgG profiling, Coronaviruses, vaccines, children antibody responses
Disclosure: All authors indicated they had no financial relationships to disclose.
(203) Heterozygous ATM Mutation Associated with Immunodeficiency and Recurrent Malignancy
Daniel Tcheurekdjian1, Shan Shan Wu, DO2, Devi Jhaveri, DO3, Robert Hostoffer, DO4, Haig Tcheurekdjian, MD4
1Visiting Student/Allergy/Immunology Associates, Inc.
2Attending Physician/University Hospitals of Cleveland
3Clinical Instructor/Case Western Reserve University
4Associate Clinical Professor/Case Western Reserve University
Ataxia-Telangiectasia is an early-onset autosomal recessive disorder caused by mutations in the Ataxia-Telangiectasia Mutated gene (ATM). It is characterized by immunodeficiency, predisposition to malignancy, and neurodegeneration. We present a patient with a heterozygous ATM mutation displaying adult-onset immunodeficiency and recurrent malignancies without neurodegeneration or telangiectasia.
A 51 year old male presented with a 10 year history of recurrent shingles, sinusitis, bronchitis and pneumonia. Physical examination was normal except for bilateral rhonchi and rales on pulmonary examination. Neurologic examination was normal, and there were no telangiectasia on the skin or ocular tissues. The complete blood count was normal with an absolute lymphocyte count of 1282 cells/μL (850-3900 cells/μL). The peripheral lymphocyte phenotyping demonstrated a low T-cell count of 764 cells/μL (840-3060 cells/μL), with absolute CD4 and CD8 counts of 443 cells/μL (490-1740 cells/μL) and 277 cells/μL(180-1170 cells/μL), respectively. The quantitative immunoglobulins demonstrated an IgG of 892 mg/dL (692-1618 mg/dL), IgA of 226 mg/dL (81-463 mg/dL), and IgM of 28 mg/dL (48-271 mg/dL). The pneumococcal titers were protective against 2/14 serotypes. Both unconjugated and conjugated polysaccharide pneumococcal immunization led to no change in titers. The patient was started on intravenous immunoglobulin therapy with a decrease in the number and severity of infections. He subsequently developed a diffuse large B-cell lymphoma in the retroperitoneum two years later, which was successfully treated with R-CHOP. He later developed a left occipital mass. An excisional biopsy revealed an ectopic salivary gland neoplasm. Excision was curative. Three years later there was a recurrence of a diffuse large B-cell lymphoma in the retroperitoneum and the liver which was successfully treated with an autologous stem cell transplantation. Whole exome sequencing was obtained because of the patient's history of unusual and recurrent malignancy in association with immunodeficiency identifying a heterozygous pathogenic mutation in ATM (c.2098 C>T, p.Q700X).
Heterozygous ATM mutations may cause an ATM-related disorder manifesting with immunodeficiency and predisposition to malignancy without neurodegeneration.
Keywords: Ataxia-Telangiectasia, ATM mutation, Immunodeficiency, Malignancy
Disclosure: All authors indicated they had no financial relationships to disclose.
(204) Serum proteomic analysis of inflammatory complications in common variable immunodeficiency
Hsi-en Ho, MD1, Lin Radigan, MD2, Charlotte Cunningham Rundles, MD3
1Assistant Professor/Mount Sinai School of Medicine
2Researcher/Icahn School of Medicine at Mount Sinai
3David S. Gottesman Professor of Immunology/Mount Sinai School of Medicine
Inflammatory complications in Common variable immunodeficiency (CVID) are heterogeneous, and together they lead to increased morbidity/mortality. The shared immune pathways underlying such heterogeneous manifestations are not fully identified.
We employed a proteomic-approach to identify the serum immune signatures that marked CVID subjects with autoimmune/inflammatory complications (“CVID+”) from those without.
\We performed Olink Protein Extension Assays, covering 92 inflammation-related proteins, in serums from 30 well-characterized CVID subjects (CVID+, n=20).
Among CVID+ subjects in this cohort, the clinical manifestations included interstitial lung disease (45%), granulomas (25%), nodular regenerative hyperplasia of liver (25%), enteropathy (50%), lymphoproliferations (90%), and a history of hematologic autoimmunity (65%). Our analysis identified five immune signatures, consist of (A) type 2 interferon and associated factors, (B) chemokines, (C) pro-inflammatory cytokines (IL-6, TNF-a/b), (D) immune-regulatory proteins (IL-10), and (E) hematopoietic growth factors (M-CSF), which were all significantly elevated in CVID+ compared to CVID subjects without inflammatory manifestations (FDR < 0.05). Signature A was notable for elevated circulating IFN-g, along with its associated cytokines, IL-12 and IL-18. Additionally, elevated IFN-g positively correlated with multiple circulating factors, including PD-L1, MMP-10, CDCP1, TNF-a/b, and IL-15RA (Spearman r=0.50-0.76, FDR < 0.05). Signature B included chemokines typically highly expressed in lymph nodes (CCL19), small intestines (CCL25), and lung/liver (CCL23). In addition, it included multiple chemokines that were significantly correlated with serum IFN-g levels in this cohort: CXCL9, CCL19, CXCL10 (Spearman r=0.54-0.73, FDR < 0.05).
Our serum proteomic analysis of CVID revealed common immune signatures that characterized those with inflammatory manifestations. Our data extend upon the IFN transcriptional signature previously noted in CVID+ subjects and identified dysregulated immune pathways and markers that may be of clinical and therapeutic importance.
Keywords: common variable immunodeficiency, proteomics, immune dysregulation
Disclosure: All authors indicated they had no financial relationships to disclose.
(205) Two types of independent malignancies in a patient with selective IgM deficiency
Yesim Demirdag, MD1, Linda Doan, MD2, Sudhir Gupta, MD3
1Director, Allergy and Immunology Fellowship Program Associate Professor of Clinical Medicine/Division of Basic and Clinical Immunology Department of Medicine University of California Irvine
2Assitant Clinidal Professor/University of California in Irvine
3Professor of Medicine/University of California in Irvine
Selective IgM deficiency (SIGMD), defined as IgM < 2 SD of age-adjusted levels, is a rare primary immunodeficiency. Although it is usually asymptomatic, individuals with selective IgM deficiency may present with recurrent respiratory tract infections and atopic tendency. Here we present a 76-year-old man with IgM deficiency associated with IgG2 subclass deficiency. In addition to infections, the patient developed melanoma as well as angiosarcoma, a rare and aggressive form of soft tissue malignancy with a poor prognosis. To our knowledge this is the first reported case of selective IgM deficiency who developed angiosarcoma, and, this is also the first case of selective IgM deficiency who developed two different types of malignancies.
Case: The patient was initially evaluated in our immunology clinic at age 73 because of recurrent shingles and a history of prolonged recovery from infections. His past medical history was significant for trauma-induced quadriplegia at age 29, neurogenic bladder causing recurrent urinary tract infections, and neurogenic colon requiring colostomy at age 64 which was complicated by perforation and sepsis. He also has type 2 diabetes, gout, and a history of melanoma on the right upper chest which was fully resected without any further therapy at age 72. Immunology evaluation revealed normal complete blood counts and differentials, normal IgG and IgA levels, but very low IgM levels (17 mg/dl, normal: 65-230 mg/dl). Lymphocyte subsets and IgG subclasses were normal and antibody response to pneumococcal polysaccharide vaccine was adequate. During follow-up, IgM levels stayed low and IgG2 level slightly decreased (227 mg/dl, normal: 242-700 mg/dl). In May 2019, he was diagnosed with aggressive angiosarcoma ( Figure 1) which was located on the right side of the umbilicus. It was resected and he was started on radiotherapy followed by chemotherapy.
Summary: While selective IgM deficiency is associated with a wide clinical spectrum from an asymptomatic state to recurrent infections and/or atopy, malignancies are rarely reported. We describe a patient with SIGMD with 2 different types of malignancies one of which is a rare and aggressive type.
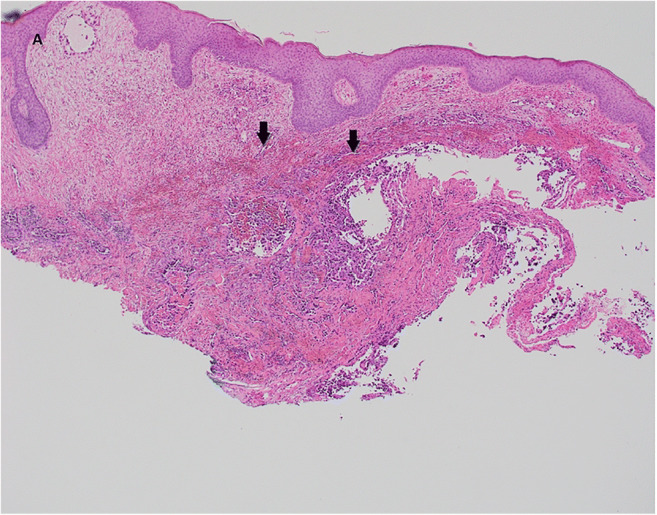
Figure 1: The skin biopsy shows extravasated blood in the dermis with inflammatory cells and the suggestion of vascular channels (black arrows).

Figure 2: The tumor cell nuclei show strong positive staining with ERG

Figure 3: The tumor cell nuclei show negative staining for Melanin
Keywords: Selective IgM deficiency, Angiosarcoma, Melanoma
Disclosure: All authors indicated they had no financial relationships to disclose.
(206) A Phase 1/2 Study of Lentiviral-mediated Ex-vivo Gene Therapy for Pediatric Patients with Severe Leukocyte Adhesion Deficiency-I (LAD-I): Interim Results
Donald Kohn, MD1, Gayatri Rao, MD, JD2, Elena Almarza, PhD3, Dayna Terrazas, RN4, Eileen Nicoletti, MD5, Augustine Fernandes, PhD6, Caroline Kuo, MD7, Satiro De Oliveira, MD8, Theodore Moore, MD9, Ken Law, PhD10, Brian Beard, PhD11, Julian Sevilla, MD, PhD12, Cristina Mesa-Nunez, PhD13, Claire Booth, MBBS, MSc, PhD14, Adrian Thrasher, MBBS, PhD15, Juan Bueren, PhD16, Jonathan Schwartz, MD17
1Distinguished Professor, Microbiology, Immunology & Molecular Genetics/University of California, Los Angeles
2Vice President, Global Program Head, LAD-1 Program/Rocket Pharmaceuticals, Inc.
3Senior Scientist/Rocket Pharmaceuticals, Inc.
4Study Coordinator/University of California, Los Angeles
5Medical Director/Rocket Pharmaceuticals, Inc.
6Administrative Director/University of California, Los Angeles
7Assistant Professor of Pediatrics/University of California, Los Angeles
8Clinical Instructor, Pediatrics/University of California, Los Angeles
9Professor of Pediatrics, Chief of Pediatric Hematology/Oncology/University of California, Los Angeles
10Associate Director, CMC/Rocket Pharmaceuticals, Inc.
11Associate Vice President, CMC/Rocket Pharmaceuticals, Inc.
12Hematologist/IB Hospital Infantil Universitario Niño Jesús, and Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER-ISCIII )
13Scientist/entro de Investigaciones Energéticas Medioambientales y Tecnológicas (CIEMAT) and Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER-ISCIII); IIS-FJD/UAM
14Senior Clinical Lecturer/UCL Great Ormond Street (GOS) Institute of Child Health
15Professor/UCL Great Ormond Street (GOS) Institute of Child Health
16Professor/entro de Investigaciones Energéticas Medioambientales y Tecnológicas (CIEMAT) and Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER-ISCIII); IIS-FJD/UAM
17Chief Medical Officer/Rocket Pharmaceuticals, Inc.
LAD-I is a rare disorder of leukocyte (primarily neutrophil) adhesion resulting from ITGB2 gene mutations encoding for the β2-integrin component, CD18. Severe LAD-I (i.e., CD18 expression on < 2% of PMNs) is characterized by severe infections, impaired wound healing, and childhood mortality. Although allogeneic hematopoietic stem cell transplant (alloHSCT) is potentially curative, utilization and efficacy are limited by donor availability and risk of graft-versus-host disease (GVHD). RP-L201-0318 (NCT03812263) is a phase 1/2 open-label clinical trial evaluating the safety and efficacy of RP-L201, consisting of autologous CD34+ cells transduced with a lentiviral vector (LV) carrying the ITGB2 gene encoding for CD18 (Chim-CD18-WPRE) in severe LAD-I.
Pediatric patients ≥ 3 months old with severe LAD-I are eligible. Peripheral blood (PB) HSCs are collected via apheresis after mobilization with granulocyte-colony stimulating factor (G-CSF) and Plerixafor and transduced with Chim-CD18-WPRE LV. Myeloablative busulfan conditioning is followed by RP-L201 infusion. Patients are followed for safety and efficacy (i.e., survival to age 2 and at least 1-year post-infusion, increase in PMN CD18 expression to at least 10%, PB vector copy number (VCN), decrease in infections/hospitalizations, and resolution of skin or periodontal abnormalities).
Four patients (ages 7 months to 9 years) have been treated with RP-L201 with follow-up ranging from 6 weeks to 12 months. RP-L201 cell doses ranged from 2.8x106 to 6.5x106 CD34+ cells/kg with VCNs from 1.83 to 3.8 copies/cell (liquid culture). No serious treatment-emergent adverse events were reported. Neutrophil engraftment was observed in ≤ 5 weeks. PB PMN CD18 expression in Patient 1 12-months post-treatment was 40% (sustained from 47% at 6-months, vs. < 1% at baseline), with PB VCN of 1.2. Skin lesions present at baseline resolved with no new lesions reported. Patient 2 PB PMN CD18 expression 6-months post-treatment was 23% with PB VCN at 0.75. Patients 3 and 4 PB PMN CD18 expression were 74% at 3-months and 59% at 6-weeks post-treatment, respectively. No new infections have been reported in patients post-infusion.
These results demonstrate that RP-L201 leads to durable neutrophil CD18 expression and improved clinical course. Additional patient treatment is planned for early 2021.
Keywords: Primary Immunodeficiency, Gene Therapy, Leukocyte Adhesion Deficiency Type 1, Rare Disease
Disclosure: Donald Kohn is an advisory board member of Allogene Therapeutics, MyoGene Bio and Orchard Therapeutics. Gayatri Rao, Elena Almarza, Eileen Nicoletti, Ken Law, Brian Beard are employees of Rocket Pharmaceuticals. Julian Sevilla has ownership interest in Rocket Pharmaceuticals, and is a consultant at Amgen, Miltenyi Biotec and Sobi. Claire Booth received educational honoraria from GSK, Orchard, Rocket, SOBI, and Takeda. Adrian Thrasher is a consultant at 4BioCapital, Generation Bio, Orchard, and Rocket Pharma. Juan Bueren and Jonathan Schwartz have ownership interest in Rocket Pharmaceuticals. All other authors indicated they had no financial relationships to disclose.
(207) Observed Treatment Adjustments and Complications in an Ovarian Cancer Patient with Inborn Error of Immunity
Jamila Mammadova1, Anna Redden2, Rachel Cruz1, Maryssa Ellison3, Tyra Gatewood, PharmD, BCOP4, Carla Duff, MSN3, Charurut Somboonwit, MD, FACP, FIDSA5, Chakrapol Sriaroon, MD5, Jolan Walter, MD/PhD6, Roohi Ismail-Khan, MD, MSc7
1Medical Student/University of South Florida
2Student/University of Southern California
3Department of Pediatrics/University of South Florida
4Clinical Pharmacist/H. Lee Moffitt Cancer Center and Research Institute
5Department of Medicine/University of South Florida
6Department of Pediatrics/University of South Florida at Johns Hopkins All Children's Hospita
7Co-Director, Cardio-Oncology Program, Moffitt/USF/H. Lee Moffitt Cancer Center and Research Institute
Inborn errors of immunity (IEI) prone patients to increased risk of developing cancer. However, there is sparse literature on optimizing their cancer treatment.
We present a patient with specific antibody deficiency (SAD), immune dysregulation (AI) and stage III high grade ovarian carcinoma, whose treatment regimen needed modification secondary to ongoing infections.
This is a retrospective chart review on the patient's cancer treatment course.
Our patient with SAD and AI (ANA+, polyarthralgia, autoimmune neutropenia) was diagnosed with stage III ovarian carcinoma at age 60. After robot-assisted laparoscopic primary debulking surgery she underwent 6 cycles of chemotherapy. Prior to cycle 1, routine immunoglobulin replacement therapy was adjusted from 40 mg of IVIG every two weeks to 80 mg of IVIG every 3 weeks (1.3 g/kg/3 weeks, wt 60 kg), administered 1 week prior to each chemo cycle. Chemo doses were lowered for all cycles: Carboplatin (area under the curve (AUC) 4 instead of standard 5), Paclitaxel (Taxol 150 mg/m^2 instead of standard 175 mg/m^2) and Avastin (15 mg/kg). Colony-stimulating factor pegfilgrastim throughout prevented febrile neutropenia. After cycle 1, the patient developed pulmonary mycobacterium avium-intracellulare (MAI) infection, hyperbilirubinemia, elevated liver enzymes, and urinary retention. Liver evaluation was reassuring. For post-cycle-1 adjustment, the IVIG dose was reduced to 60 mg (1g/kg/4 weeks, wt 50 kg). During cycle 2 and 3, the patient developed urinary tract infection (UTI) caused by Klebsiella pneumoniae, treated with Rocephin and Ciprofloxacin. Throughout cycles 4-6, UTI was prevented with prophylactic Bactrim and Azithromycin and received Myrbetriq for symptoms of urinary incontinence. The patient completed all 6 cycles. For MAI lung scarring and recurrent cough, she had repeat bronchoscopy that was unrevealing. Post-chemo CT scan revealed no evidence of cancer, therefore she will continue with 1-year maintenance therapy of Avastin (15 mg/kg). Cancer genetic analysis revealed no targetable markers, primary immunodeficiency gene panel (207 genes) was unrevealing.
Patients with IEI with cancer may require a modified treatment course and multidisciplinary team approach. Larger studies are needed on how to adjust therapy to decrease risk of infection but still treat cancer aggressively for optimal long-term outcomes.
Keywords: Primary immunodeficiency, Ovarian cancer, Inborn error of immunity, Treatment adjustment, Complications
Disclosure: All authors indicated they had no financial relationships to disclose.
(208) A rare case of Chronic Granulomatous Disease and Fanconi's Anemia
Candace Rypien, MD1, Luis Murguia-Favela, MD2, Tony Truong, MD3, Sneha Suresh, MD4, Nicola Wright, MD, MSc3
1Pediatrician/Alberta Children's Hospital
2Attending Physician/Section of Hematology/Immunology, Department of Pediatrics, Alberta Children's Hospital, Cumming School of Medicine, University of Calgary
3Associate Professor/Alberta Children's Hospital
4Clinical Assistant Professor/Stollery Children's Hospital
Chronic granulomatous disease (CGD) is a primary immunodeficiency leading to increased risk of life threatening infections with catalase positive bacteria and fungus, and inflammatory complications associated with poorly functioning neutrophils. In North America, the most common cause of CGD is mutations in CYBB (X-linked), however, CGD can be caused by mutations in any of the components of the NADPH oxidase complex (CYBB, NCF1, CYBA, NCF2, NCF4). In populations with higher rates of consanguinity, the recessive forms of CGD can be more common.
Regular use of prophylactic antimicrobials has decreased the rate of infections; however, patients continue to have significant morbidity in the form of inflammatory complications and ongoing risk of infections. Growing evidence supports the use of Hematopoietic Stem cell transplant (HSCT) in patients with CGD to prevent long term complications of infection and inflammation.
This patient presented at 3 months of age with chronic cough, congestion and failure to thrive since birth. Due to ongoing oxygen requirements, she had a chest CT which found multiple scattered pulmonary nodules leading to a bronchoalveolar lavage being performed which grew Aspergillus fumigatus. Immune investigations demonstrated an absence normal neutrophil oxidative burst on flow cytometry. A primary immunodeficiency genetic panel was sent which came back demonstrating homozygous stop codon variant in CYBA as well as a homozygous splice variant causing Fanconi’s anemia. She was started on voriconazole for treatment of pulmonary Aspergillosis and required admission to the PICU with respiratory failure requiring intubation.
Patients with a history of consanguinity are at increased risk of recessive monogenetic diseases. The identification of a second recessive disease, Fanconi’s Anemia, lead to significant changes to the conditioning regiment and bone marrow donor selection. Both parents were suboptimal bone marrow donors as they were carriers of Fanconi’s anemia. Given the unusual ethnic background and lack of sibling donor, a mismatched unrelated cord had to be utilized for transplant. The conditioning regiment had to be modified given the radio-sensitivity of the patient due to their underlying Fanconi’s anemia. This patient demonstrates a unique HSCT challenge and outlines the importance of genetic sequencing when preparing for bone marrow transplant.
Keywords: Chronic Granulomatous Disease, Fanconi's Anemia, Hematopoietic Stem Cell Transplant, Pulmonary Aspergillosis
Disclosure: Tony Truong is a consultant at Jazz Pharmaceutical. Sneha Suresh received speaker honoraria from Abbvie. Nicola Wright received speaker honoraria from Sobi Canada Inc. All other authors indicated they had no financial relationships to disclose.
(209) Post-COVID-19 Hemophagocytic Lymphohistiocytosis in an Infant with Trisomy 21
Taya Carpenter, DO1, Melissa Gans, MD2
1Resident Physician in Pediatrics/Westchester Medical Center
2Attending in Allergy and Immunology/Boston Children's Health Physicians
An 8 month-old ex-35 week baby girl with trisomy 21, atrioventricular canal defect status post repair, pulmonary hypertension, oxygen dependence, and nasogastric tube feed dependence was admitted to the pediatric intensive care unit for respiratory failure secondary to COVID-19 pneumonia. She was treated with remdesivir, COVID-19 convalescent plasma, and dexamethasone. Over the next several weeks, she had waxing and waning respiratory distress and was also diagnosed with a Klebsiella urinary tract infection, for which she completed treatment. She was afebrile for 5 consecutive days at the end of week 1 to beginning of week 2 of her treatment course, but soon her fevers returned accompanied by elevated inflammatory markers, anemia, thrombocytopenia, elevated ferritin, hypertriglyceridemia, low fibrinogen, and elevated liver function tests. Multisystem inflammatory syndrome in children was considered as a diagnosis and she was treated with immunoglobulin and dexamethasone with no improvement in symptoms. Further laboratory testing revealed elevated IL-2 receptor, low Natural Killer cell function, and normal perforin/granzyme B. Her clinical presentation was believed to be more consistent with hemophagocytic lymphohistiocytosis, even though her bone marrow aspiration did not show hemophagocytosis. The patient was started on HLH94 dexamethasone dosing regimen with a response to treatment evidenced by improvement in clinical status and some laboratory markers. Etoposide and other immunosuppressive agents were held due to clinical improvement on dexamethasone alone. Immune evaluation revealed lymphopenia (ALC 2679 lymphocytes per microliter) with normal distribution of lymphocyte subsets and normal IgA and IgM. Genetic analysis of 407 genes associated with primary immunodeficiency was unrevealing. Trisomy 21 is an underrecognized immune dysregulatory disorder and we believe that this underlying inborn error of immunity likely triggered her hemophagocytic lymphohistiocytosis after COVID-19. This case also demonstrates the diagnostic difficulty in discerning multisystem inflammatory syndrome in children from hemophagocytic lymphohistiocytosis. Bone marrow aspiration could distinguish the two disorders, but our patient had a negative aspiration, possibly due to partial treatment of her hemophagocytosis by her previous course of dexamethasone or bone marrow sampling error. Additionally, viruses, particularly Epstein-Barr virus, are well known triggers of hemophagocytic lymphohistiocytosis and this case demonstrates COVID-19 as a potential trigger as well.

Keywords: Trisomy 21, Hemophagocytic Lymphohistiocytosis, Immune Dysregulation, COVID-19
Disclosure: All authors indicated they had no financial relationships to disclose.
(210) Immune cell profiling and fitness in WHIM syndrome
Maryssa Ellison1, Ulrich Salzer, MD2, Christoph Geier, MD, MSc3, Mei-Sing Ong, PhD4, Rachel Cruz5, Sumai Gordon, B.S.6, Boglarka Ujhazi, BS7, Marton Keszei, PhD8, Svetlana Sharapova, PhD9, Morna Dorsey, MD10, Mica Muskat, Nurse Practitioner11, Amer Khojah, MD12, Eyal Grunebaum, MD, M.Sc. Paeds13, Oliver Wegehaupt, MD14, Klaus Warnatz, MD15, Krisztian Csomos, PhD16, Joao Pereira, PhD17, Jolan Walter, MD/PhD18
1Department of Pediatrics/University of South Florida
2Research group leader/University Clinic Freiburg, CCI
3MD, MSc/Immunology Outpatient Clinic, Vienna, Austria
4PhD/Department of Population Medicine, Harvard Medical School and Harvard Pilgrim Health Care Institute, Boston, MA, USA
5Medical student/University of South Florida
6B.S./University of South Florida
7Senior Biological Scientist/University of South Florida
8PhD/Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Sweden
9PhD/Belarusian Research Center for Pediatric Oncology and Hematology
10Director, Allergy/Immunology Program/University of California, San Francisco
11Ped-Immuno Nurse Practitioner, Pediatrics/University of California, San Francisco
12Attending Physician, Allergy, Immunology, and Rheumatology/Ann & Robert H. Lurie Children's Hospital of Chicago/ Northwestern University Feinberg School of Medicine
13Co-director Food Allergy and Anaphylaxis Program, Division of Immunology and Allergy/SickKids
14MD/Institute for Immunodeficiency, Center for Chronic Immunodeficiency, Medical Center University of Freiburg Faculty of Medicine, University of Freiburg, Freiburg, Germany
15Head of Division/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
16Research Associate/University of South Florida
17Associate Professor of Immunobiology/Yale
18Department of Pediatrics/University of South Florida at Johns Hopkins All Children's Hospita
CXCR4 gain-of function is the most common molecular basis for immunodeficiency in warts, hypogammaglobulinemia, infections, myelokathexis syndrome (WHIM) manifesting as both defect in innate immunity, but also variable levels of antibody deficiency. In our current retrospective international cohort of 59 WHIM patients, 37 (62%) had a diagnosis of hypogammaglobulinemia. There has been limited data on characterization of lymphocyte profile and fitness and its relationship to variability in antibody deficiency.
We aim to evaluate lymphocyte phenotype, function, and survival in a cohort of patients with WHIM syndrome.
Flow cytometry-based assay to assess extended immune phenotype, function, and fitness of peripheral lymphocytes (T, B and NK cell subsets).
We had seven patients available for immune studies from our international cohort of WHIM syndrome. The majority of patients have history of hypogammaglobulinemia. Immune phenotype was notable for decreased fraction of memory B, naïve CD4+ and CD8+ and central memory T cells in patients when compared to age matched healthy controls. In vitro differentiation of naïve B cells revealed early activation and decreased ability for expansion. BAFF levels were found to be increased (n=6). Regarding lymphocyte fitness, an increase in apoptosis was found in several lymphocyte subsets, but most prominently in total CD19+ B cell population post 24-hour incubation.
Our study has shown lymphocyte abnormalities in WHIM syndrome, specifically regarding development, function, and survival of B cells. Early activation may explain B cell apoptotic tendencies, but further investigation is warranted to understand the mechanisms and its link to variable phenotype of antibody deficiency in WHIM syndrome. Further investigation of the B cell immune phenotype, function and apoptosis could help with the development of biomarkers to predict the need for antibody replacement therapy in WHIM syndrome.
Keywords: WHIM Syndrome, lymphocyte, immune phenotype, apoptosis
Disclosure: Ulrich Salzeer is an advisory board member at Baxalta. Klaus Warnatz received speaker honoraria from Baxalta/Takeda. All other authors indicated they had no financial relationships to disclose.
(211) Neuroblastoma amplified sequence related disease manifesting with complex immune cytopenias
Dilawar Khokhar, MD1, Mark Hannibal, MD, PhD2, Lauren DeMeyer, MS3, Laura Howe, MD4, Beth Kurt, MD, MS5, Yelena Kier, DO, FACOI6, Kristle Habrichter, DO7, Kelly Walkovich, MD8
1Allergy and Immunology Fellow/University of Michigan
2Associate Professor, Genetics, Metabolism & Genomic Medicine/University of Michigan
3Genetic Counselor/University of Michigan
4Allergist/Bayside Allergy
5Section Chief, Pediatric Hematology and Oncology/Spectrum Health Helen DeVos Children’s Hospital
6Hematologist, Oncologist/Cowell Family Cancer Center
7Pathologist/Grand Traverse Pathology, PLLC
8Associate Professor, Pediatric Hematology & Oncology/University of Michigan
Neuroblastoma amplified sequence (NBAS)-associated disease is a multisystem disorder that may involve the liver, nervous system, immune system, retina, connective tissue and bone. The most commonly reported immunologic aberrations are hypogammaglobulinemia and NK cell deficiency. Hematologic features of isolated ITP or AIHA are rare, although the Pelger-Huet anomaly is frequently visualized on peripheral smear.
We present a 25-year-old male diagnosed with CVID requiring immunoglobulin replacement at age 8 years and Evans syndrome (ES) with ITP and warm AIHA at age 16 years. His ES required treatment with corticosteroids, vincristine, danazol, rituximab and splenectomy, with an ongoing need for chronic steroids to manage hemolysis. Additionally, he has legal blindness secondary progressive optic nerve atrophy, short stature, liver disease, type 1 diabetes mellitus and lymphopenia. He was referred for further evaluation due to serious recurrent infections despite adequate IgG supplementation (IgG > 1000 mg/dL) and refractory ES. Laboratory testing showed severe lymphopenia (CD3+ T cells 128 cells/mcL; CD4+ T cells 62 cells/mcL; CD8+ T cells 65 cells/mcL; CD19+ B cells 0 cells/mcL; CD16/56+ NK cells 39 cells/mcL) as well as undetectable IgA and IgM. His DAT showed 2+ IgG, negative C3; anti-platelet antibody screening was not obtained. His peripheral smear demonstrated easily appreciable neutrophils with the Pelger-Huet anomaly.
Genetic testing for immune disorders identified a pathogenic heterozygous variant in NBAS c.2827G>T, p.Glu943Ter, along with a heterozygous missense variant of uncertain significance in NBAS c.5740C>T, p.Arg1914Cys, consistent with the NBAS-associated disease. This missense variant occurs in the c-terminal region, at the same codon as the Yakut founder variant, NBAS c.5741G>A, p.1914His. C-terminal missense variants, in conjunction with null variants, define a phenotype-genotype group characterized by highly penetrant, pleiotropic, multisystem features.
Immune-mediated cytopenias, particularly in association with hypogammaglobulinemia, liver disease, optic atrophy and/or the presence of the Pelger-Huet anomaly, should prompt consideration for NBAS-associated disease as the underlying etiology. Additionally, this case underscores the value of genetic testing in adult patients with unexplained hypogammaglobulinemia as well as review of the peripheral blood smear as a diagnostic aid.
Keywords: NBAS, hypogammaglobulinemia, Evans Syndrome, Pelger Huet Anomaly
Disclosure: Kelly Walkovich is a consultant for Sobi Pharmaceuticals. All othre authors indicated they had no financial relationships to disclose.
(212) Improving Diagnosis of Immunodeficiency in the Premature Population
Lauren Frazer, MD, PhD1, Maureen Schnur, DNP, RN, CPN2, Rylee Kerper, MPH3, Vanessa Young, RN. BA2, Amy O'Connell, MD, PhD4
1Clinical Fellow/Boston Children's Hospital
2Clinical Research Nurse/Boston Children's Hospital
3Clinical Research Assistant/Boston Children's Hospital
4Assistant Professor/Boston Children's Hospital/Harvard Medical School
Newborn screening for severe combined immunodeficiency (SCID) leads to earlier detection and saves lives. Testing in premature neonates, however, is significantly complicated by the relative T cell lymphopenia common in very premature and extremely premature infants. Previous work by our group and others has shown the T cell receptor excision circle (TREC) screening is not highly specific in premature infants, and current diagnostic algorithms may be missing cases of primary immunodeficiency in premature infants. Meanwhile, a subset of extremely premature infants has lasting T cell lymphopenia even at term postmenstrual age, a phenomenon we are calling prematurity-related immune dysfunction (PRID). We are interested in improving detection and management of immunodeficiency in premature infants, both for primary immunodeficiency disorders and PRID. In the first phase of this project, we are using T cell receptor repertoire sequencing (RepSeq) to determine whether we can utilize RepSeq to enhance test specificity for premature infants with low TRECs. Using discard or small volume blood samples, we can generate diverse T cell repertoire sequencing outputs in premature infants and newborns. Patients with SCID and some combined immunodeficiencies are known to have abnormal diversity in the T cell repertoire, and repertoire diversity and skewing should be less affected by lymphopenia than flow cytometry, which is the current standard reflex test for low TRECs. We are currently testing whether RepSeq can be used as an effective second step test to improve specificity after an abnormal TREC screen in very and extremely premature infants. Our long-term project goal is to improve identification of SCID and other forms of PID in premature infants.
Keywords: TREC, SCID, Prematurity, Repertoire, Diagnosis, Newborn Screening, Immunodeficiency
Disclosure: All authors indicated they had no financial relationships to disclose.
(213) Deciphering The Genetic Diagnosis Of 325 Patients with Primary Antibody Deficiency By Targeted Next Generation Sequencing At The CCI In Freiburg.
Jessica Rojas-Restrepo, MSc1, Andrés Caballero-García de Oteyza, Phd2, Katrin Hübscher3, Hanna Haberstroh, MSc3, Michele Proietti, Dr. med.4, Bodo Grimbacher, Prof. Dr. med.5
1PhD student/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
2Postdoctoral scientist/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
3Research assistant/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
4Head of the laboratory/Department of Rheumatology and Immunology, Hannover Medical University, Hannover, Germany
5Head of the laboratory/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
Primary antibody deficiencies (PAD) are chronic disorders with an often markedly reduced quality of life and, if left untreated, also reduced life expectancy. A timely diagnosis has been shown to be associated with a significantly improved prognosis.
Here, we report on our experiences with targeted panel sequencing employing Agilent’s HaloPlex or SureSelect and Illumina’s MiSeq technologies in a group of 325 patients with common variable immunodeficiency, hypogammaglobulinemia and agammaglobulinaemia.
In total, we have detected over 50 relevant mutations in ADA, BTK, CECR1, CTLA4, CXCR, IL2RG, LRBA, NFKB1, NFKB2, STAT3, TNFRSF13B. Overall, a genetic diagnosis could be made in up to 21.8% leading to a definite or possible molecular diagnosis in 71 out of the 325 evaluated patients. Based on the type of variant we could identified 89 missense, 11 nonsense, 5 splice-site, 11 insertion or deletion and 3 frameshift. The most frequent mode of inheritance in our cohort was autosomal dominant (in 5 genes due to a loss of function or gain of function mutations), followed by autosomal recessive (in 4 genes) and in 2 genes were found X-linked mutations. Moreover, functional testing was performed in patients who had candidate variants for instance in CTLA4 and LRBA. This large genetic cohort analysis highlights once again that the primary antibody deficiencies have a diverse genetic background and genotype-phenotype correlation is often poor, although certain clinical features may hint towards a specific group of defects.
Employing targeted sequencing panels proves to be a very time-and cost-efficient, yet reliable, method for the establishment of a genetic diagnosis of PAD. In case of negative screening results by panel sequencing, further work-up including whole exome sequencing should be considered for patients with complex disease, a positive family history, or an early onset of disease.
Keywords: PAD, NGS, Targeted gene panel, Genetics, Sequencing
Disclosure: Bodo Grimbacher received a research grant from Baxalta. All other authors indicated they had no financial relationships to disclose.
(214) Epigenetic Immune Cell Quantification For Diagnosis and Monitoring of Patients with Inborn Errors of Immunity, Primary Immune Deficiencies, and Immune Regulatory Disorders
Neftali Ramirez, MSc1, Steffi Walter, Diploma2, Jeannette Werner, Phd3, Christoph Sachsenmaier, Phd4, Bodo Grimbacher, MD, Phd5, Ulrich Salzer, MD5, Janika Schulze, Phd6
1PhD student/University Clinic Freiburg, CCI
2Clinical Research Scientist/Epiume GmbH
3Scientist/Epiume GmbH
4Vice President Business Development/Epiume GmbH
5Research group leader/University Clinic Freiburg, CCI
6Senior Scientist/Epiume GmbH
Quantitative immune cell enumeration is important in all inborn errors of immunity, and all primary and secondary immune deficiencies. Within this study, the application of a new in vitro diagnostic test for the epigenetic quantification of CD3+, CD4+, and CD8+ T cells, B cells and NK cells from as little as 40 μl of fresh or frozen whole blood as well as dried blood spots is described. The method has been shown to yield identical results when compared to flow cytometry from fresh blood samples of a healthy donor cohort, with the advantage of being more sensitive with minimal sample requirements.
Using this in vitro diagnostic test, 247 whole blood samples of different patients with Primary Immune Deficiencies (PID) or Inborn Errors of Immunity (IEI) were analyzed and compared to flow cytometric data obtained in an independent diagnostic laboratory. Spearman correlation of > 0.9 was observed for all immune cell types analyzed except for NK cells. With regard to NK cells, the epigenetic analysis shows an overestimation of the cell percentages, but the correlation of 0.73 was still robust.
To demonstrate the utility of the epigenetic quantification method, we extended the immune cell panel to regulatory T cells (Treg), Th17 cells, Tfh cells, PD-1+ cells, memory B cells, monocytes and granulocytes. Data of these additional markers will be presented at the meeting.
This study underscores the suitability of epigenetic immune cell quantification for the accurate measure of multiple immune cell types from PID and IEI patients. We propose this method as uniquely suitable for novel molecular diagnostic applications in settings with limited fresh blood sample or limited cell number, at the point of care, as well as for newborn screening.
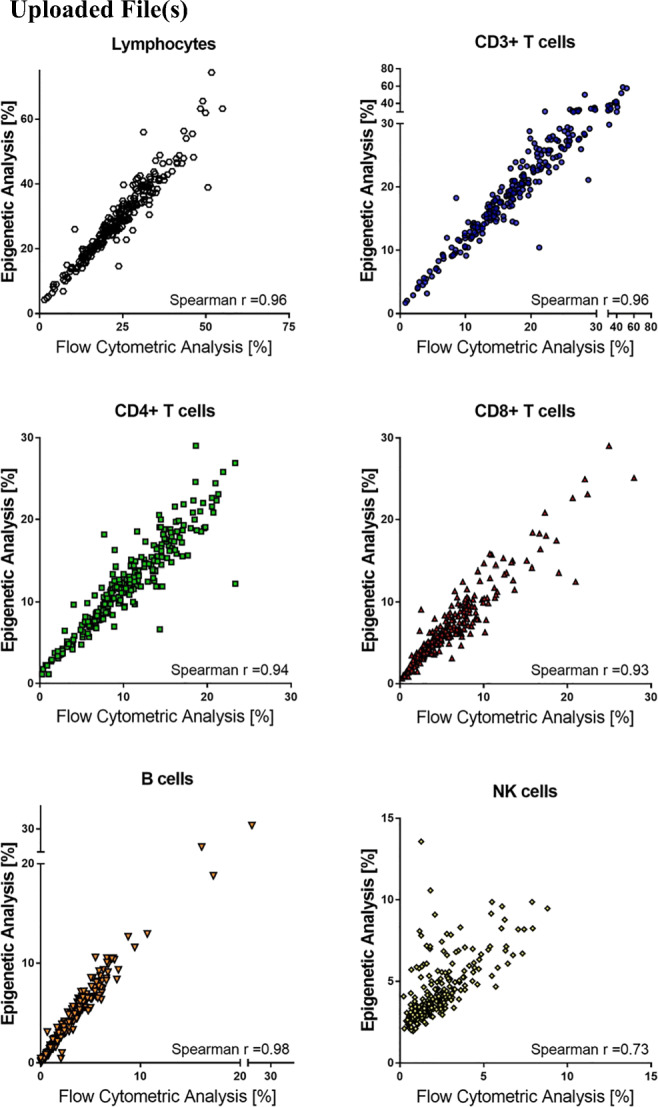
Figure 1: Comparison of epigenetic analysis and flow cytometric analysis in lymphocytes, CD3+, CD4+, and CD8+ T cells, B cells and NK cells from 247 patients with Primary Immune Deficiencies (PID) or Inborn Errors of Immunity (IEI). Spearman correlation for each cell type is indicated in the corresponding plot.
Keywords: epigenetics, diagnostic, lymphocyte subsets
Disclosure: Steffi Walter, Jeanette Werner, Christoph Sachsenmaier and Janika Schulze are all employed by Epimune GmbH. Bodo Grimbacher received a research grant from BMBF, Bristol Myers Squibb GmbH & Co. KgaA, CSL Behring, DFG, Merck KGaA, Novartis Pharma AG, Takeda Pharma Veeertrieb GmbH & Co. KG and is a consulstant at Atheneium Partners GmbH, Epimune GmbH, GigaGen Inc., Janssen-Cilag GmbH, Octapharma, UCB Pharma S.A. All other authors indicated they had no financial relationships to disclose.
(215) High-throughput CRISPR knock-in functional screening in primary human T cells to predict functionally pathologic mutations in the IL2RG gene
David Nguyen, MD PhD1, Peixin A. Chen, BS2, Charlotte Hui Wang, n/a3, Eric Shifrut, PhD4, Jennifer Puck, MD5, Alexander Marson, MD PhD6
1Assistant Adjunct Professor/UCSF
2Staff Research Associate/UCSF
3Undergraduate/Emory
4Postdoc/UCSF
5Attending Physician/Pediatric Allergy, Immunology, and Blood and Marrow Transplant Division, University of California San Francisco Benioff Children’s Hospital
6Associate Professor/UCSF, Gladstone Institutes
X-linked severe combined immunodeficiency (X-SCID) is a rare inborn error of immunity caused by loss of function (LOF) mutations in the IL2RG gene, encoding the cytokine receptor common gamma chain, □c. Newborn screening (NBS) for T cell excision circles (TRECs) in California identified 50 patients with SCID between 2010 and 2017 [Amatuni et al Pediatrics 2019]. IL2RG was most frequently mutated, affecting 14 patients, 5 of whom had mutations in Exon 5. NBS also missed one individual with a hypomorphic mutation in IL2RG exon 5 (Arg222Cys) who developed pneumonia at age 7 months. Hypomorphic mutations in IL2RG cause diminished, but not absent STAT5 phosphorylation and a “leaky” phenotype with susceptibility to infection, low but not absent T cells, and delayed clinical presentation.
While over 200 unique mutations have been observed in X-SCID across the IL2RG gene, additional characterization is required to understand the functional consequences of all possible variants; and patients with new variants continue to be found [Lebet et al, Genet in Med 2008]. We utilized high efficiency CRISPR knock-in editing to create every potential single nucleotide variant (SNV) across the 163 bases of Exon 5 in primary human T cells from healthy males. In a pooled screening format, we then assessed the impact of each SNV on cell proliferation, survival, and □c surface expression over 14 days of cell culture in vitro. Sequencing of cDNA from cells fluorescently-sorted for □c expression revealed preferential dropout of nonsense mutations (eg Ser201*) and other mutations (eg Arg226His/Cys) previously associated with X-SCID. We identified additional missense mutations (eg c.720G>A; Trp240*) with deleterious effects annotated with presumed LOF in the ClinVar database but never previously described in patients. We also noted a characteristic intolerance of missense mutations in the WSEWX motif (aa237-241) predicted to be critical for protein folding.. Further, certain variants had minimal effect, including p.Glu239Gln not listed in databases. More expansive functional annotation of IL2RG (and other genes associated with immunodeficiencies) could be leveraged to permit confident rapid interpretation of previously undescribed mutations. Further, these studies lay a foundation to overwrite pathogenic mutations with healthy code for corrective gene therapy.
Keywords: CRISPR, Human, IL2RG, High throughput screen, SCID
Disclosure: All authors indicated they had no financial relationships to disclose.
(216) Genetic analysis of a cohort of 275 patients with a hyper-IgE phenotype
Natalie Frede, MD1, Jessica Rojas-Restrepo, MSc2, Andrés Caballero-García de Oteyza, Phd3, Mary Buchta4, Katrin Hübscher4, Michele Proietti, Dr. med.5, Shiva Saghafi, MD6, Zahra Chavoshzadeh, MD7, Nermeen Galal, MD8, Zineb Jouhadi, MD, PhD9, Bodo Grimbacher, Prof. Dr. med.10
1Resident Doctor/University Medical Center Freiburg, Dept of Rheumatology & Clinical Immunology
2PhD student/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
3Postdoctoral scientist/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
4Technician/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Freiburg
5Head of the laboratory/Department of Rheumatology and Immunology, Hannover Medical University, Hannover, Germany
6Medical Doctor/Immunology Asthma and Allergy Research Institute Tehran, University of Medical Sciences Tehran, Iran
7Medical Doctor, Professor/Immunology and Allergy Department, Mofid Children's Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran
8Medical Doctor/Department of Pediatrics, Faculty of Medicine, Cairo University, Cairo, Egypt
9Medical Doctor/Department of pediatric infectious diseases, Children’s hospital CHU Ibn Rochd, University Hassan 2, Casablanca, Morocco
10Head of the laboratory/Center for Chronic Immunodeficiency, Medical Center, University of Freiburg, Germany
Hyper IgE syndromes (HIES) and chronic mucocutaneous candidiasis (CMC) constitute rare primary immunodeficiency syndromes with an overlapping clinical phenotype. During recent years a growing number of underlying genetic defects have been identified. To characterize the underlying genetic defects in a large international cohort of 275 patients, of whom 211 had been clinically diagnosed with hyper IgE syndrome and 64 with chronic mucocutaneous candidiasis, targeted panel sequencing was performed relying on Agilent HaloPlex and Illumina MiSeq technologies.
This approach allowed us to identify 86 mutations in 77 patients, which translates into a diagnostic hit rate of 28%. Specifically, mutations in DOCK8 (25 patients), STAT3 (22), STAT1 (15), CARD9 (6), AIRE (3), IL17RA (2), SPINK5 (3), ZNF341 (2), CARMIL2/RLTPR (1), IL12RB1 (1) and WAS (1) have been detected. Genetic data were correlated with clinical presentations. The most common clinical findings were elevated IgE (81.5%), eczema (71.7%) and eosinophilia (62.9%). Regarding infections, 54.7% of patients had a history of radiologically-proven pneumonia, while 28.3 % reportedly had other serious infection(s). 53% of patients had a history of fungal infection or chronic mucocutaneous candidiasis and 52.9% had at least one skin abscess. 46.2% of patients had skeletal or dental abnormalities with a characteristic face being the most commonly reported item (23.1%), followed by retained primary teeth reported in 18.9% of patients. The NIH HIES score was available for 122 patients with a clinical hyper IgE phenotype and averaged at 34.0 points, in contrast to on average 9.75 points in CMC patients. Individual clinical features proved to be largely unspecific and not predictive of detection of an underlying monogenic defect in general, likely due to clinical heterogeneity of hyper IgE phenotypes and associated genetic defects.
Targeted panel sequencing provides a cost-effective first-line genetic screening method. This approach has allowed us to identify mutations in patients with atypical clinical presentations, highlighting the importance of a genetic diagnosis, which has important implications for counseling the patient regarding treatment, prognosis and family planning.
Keywords: hyper IgE syndrome, chronic mucocutaneous candidiasis, targeted panel sequencing, genetics
Disclosure: Bodo Grimbacher received a research grant from Baxalta. All other authors indicated they had no financial relationships to disclose.
(217) Variant X-Linked Chronic Granulomatous Disease Associated with A409E Mutation in CYBB
S Deane, MD1, John Belko, MD2, Chioma Enweasor, MD3, Harry Hill, MD4
1Senior Physician; Assistant Clinical Professor of Medicine/The Permanente Medical Group; University of California, Davis, School of Medicine
2Senior Physician/The Permanente Medical Group
3Clinical Fellow/University of California, Davis, School of Medicine
4Professor of Pathology, Pediatrics and Medicine; Medical Director, ARUP Laboratories/University of Utah, School of Medicine
Variant X-linked chronic granulomatous disease (XLCGD) can present with atypical patterns on dihydrorhodamine (DHR) testing (PMID: 19483518). We present an adolescent male with serial DHR assays initially reported as normal, who was subsequently reclassified after a high index of suspicion led to further genetic testing and reanalysis of the DHR data. The case highlights the potential for variable expressivity of disease in patients with A409E missense mutations in CYBB and the potential for variant patterns that may be seen on DHR assays.
The patient is a 16-year-old male who developed his first serious infection, a lobar pneumonia and empyema requiring decortication, at approximately 34 months of age; cultures were unrevealing. He had episodic otitis media and respiratory infections starting at 6 months of age, and lymphadenitis at 15 months of age. Pneumonia requiring hospitalization reoccurred at age 4. At age 11, he developed recurrent lymphadenitis, rib, spine, and lung lesions, with both caseating and noncaseating granulomas found. Necrotizing granulomatous lung inflammation recurred at age 14, with granulomatous dermatitis and nodulocystic acne by age 16. Excepting hospitalizations for his two early pneumonias, he had largely done well with outpatient treatment. Multiple DHR assays were all initially reported as normal. However, CYBB sequencing revealed a A409E missense mutation. Subsequent review of his most recent DHR assay demonstrated abnormalities in the flow cytometry data, with a borderline stimulation index of 36, a population of cells that failed to demonstrate respiratory burst signal and a broad, irregular histogram peak in the population of cells that did, consistent with variant XLCGD.
DHR assays are commonly used to diagnose or exclude CGD in patients with compatible presentations. An A409E mutation causing CGD has been described previously (PMID: 27666509), with DHR findings and a clinical course typical of XLCGD. Our patient has had milder manifestations both clinically and in respiratory burst activity, resulting in serial DHR assays being initially reported as “normal.” This case highlights the importance of patient specific analysis of raw DHR flow cytometry data to identify variant XLCGD and the potential for variable expressivity of A409E mutations in CYBB.
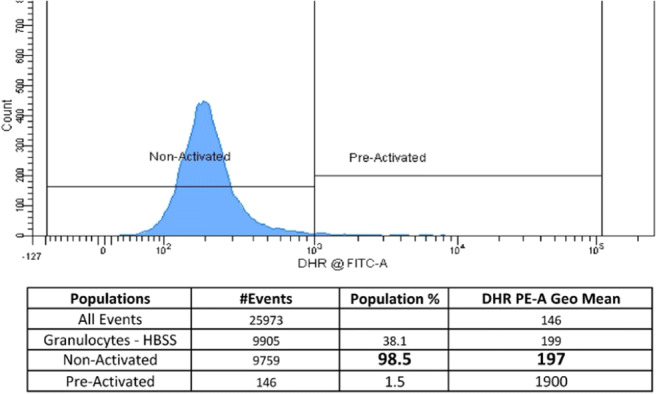
Figure 1: Control Unstimulated DHR
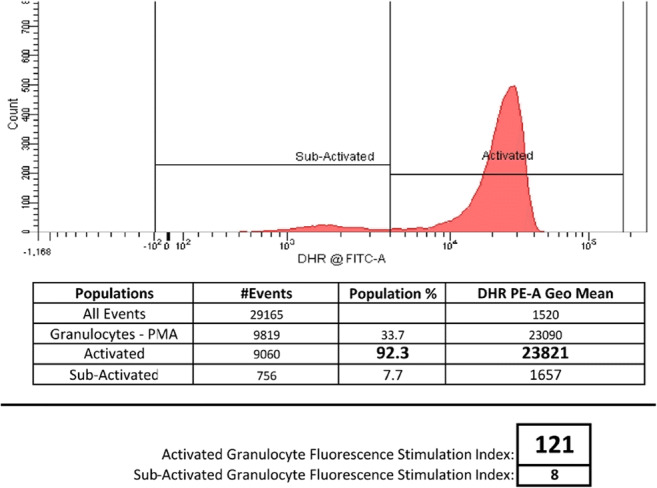
Figure 2: Control Stimulated DHR
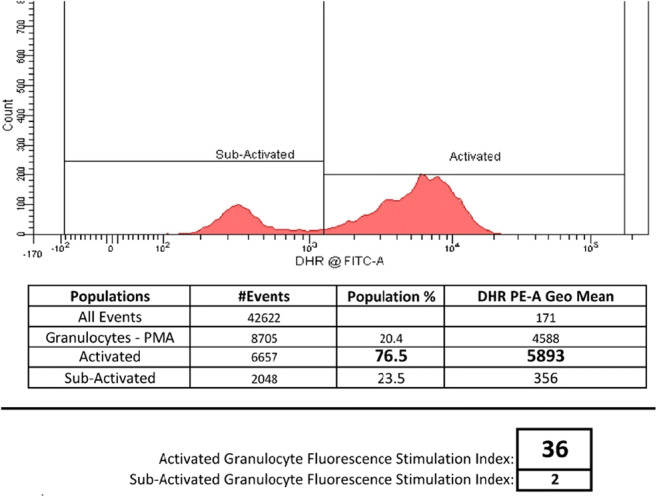
Figure 3: Patient Stimulated DHR
Keywords: Chronic Granulomatous Disease, CGD, CYBB, Variant CGD, DHR, Dihydrorhodamine, Necrotizing Lymphadenitis, Primary Immune Deficiency, Granulomatous Disease, A409E
Disclosure: Sean Deane is employed but Sutter Medical Group and The Permanente Medical Group. Harry Hill received speaker honoraria from Horizon Pharm. All other authors indicated they had no financial relationships to disclose.
(218) Nodular regenerative hyperplasia in X-linked agammaglobulinemia: an underestimated and severe complication
Cristiane Nunes dos Santos, MD1, Christopher Koh, MD, MHSc2, Anjali Rai, MD3, Keith Sacco, MD4, Betty Marciano, MD5, David Kleiner, MD, PhD6, Michael Stack, BS7, Jenna Bergerson, MD, MPH8, Gregory Constantine, MD9, Warren Strober, MD10, Gulbu Uzel, MD11, Ivan Fuss, MD12, Luigi Notarangelo, MD13, Steven Holland, MD14, Sergio Rosenzweig, MD, PhD15, Theo Heller, MD16
1Postdoctoral Visiting Fellow/Immunology Service, Department of Laboratory Medicine, National Institutes of Health (NIH) Clinical Center, Bethesda, MD, USA
2Director, Gastroenterology Fellowship Program, Digestive Diseases Branch; Staff Clinician/Liver Diseases Branch, National Institute of Diabetes & Digestive & Kidney Diseases, NIH, Bethesda, MD, USA
3Pediatric Gastroenterology Fellow/John's Hopkins Children's Center
4Clinical Fellow/Lab of Clinical Immunology and Microbiology, Immune Deficiency Genetics Section, NIAID, NIH, Bethesda, MD, USA
5Staff Scientist/National Institutes of Health
6Senior Research Physician/Laboratory of Pathology, National Cancer Institute, NIH, Bethesda, MD, USA
7Postbaccalaureate Fellow/Laboratory of Clinical Immunology and Microbiology, National Institute for Allergy and Infectious Diseases, NIH, Maryland, USA
8Staff Clinician/LCIM, NIAID, NIH
9Assistant Research Physician/National Institutes of Health
10Senior Investigator/Mucosal Immunity Section, Laboratory of Host Defenses, National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD, USA.
11Physician scientist/Laboratory of Clinical Immunology and Microbiology, National Institutes of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland, USA
12Staff Clinician/Mucosal Immunity Section, Laboratory of Host Defenses, National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD, USA
13Chief/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health
14Scientific Director – Division of Intramural Research/Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD, USA
15Senior Investigator/Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
16Senior Investigator/Translational Hepatology Section, Liver Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NIH, Bethesda, MD 20892, USA
Late-onset complications in X-linked agammaglobulinemia (XLA) are increasingly recognized. Nodular regenerative hyperplasia (NRH) has been reported in primary immunodeficiency but data in XLA are very limited. Objective: To assess and characterize NRH in patients with XLA. Methods: We retrospectively reviewed the medical records of all XLA patients referred to the NIH between 1994 and 2019. Liver biopsies were performed when clinically indicated. Patients were stratified into NRH+ or NRH- groups, according to their NRH biopsy status. Fisher’s exact test and Mann-Whitney test were used for statistical comparisons. Results: Twenty-one XLA patient records were reviewed, with a median age at start of follow-up (f/u) of 21y and a cumulative follow-up of 129 patient-years. Eight patients underwent at least one liver biopsy of whom 6 (29% of NIH XLA cohort) were NRH+. The median age at NRH diagnosis was 20y (17-31). Among patients who had liver biopsies, alanine aminotransferase (ALT) levels were mildly elevated in all, while alkaline phosphatase (ALP) levels were only increased in NRH+ patients (p=0.04). Both NRH+ and NRH- groups had similar aspartate aminotransferase (AST) levels at baseline but higher values were observed at the end of f/u in the NRH+ group (85 vs. 32 U/L, p=0.04). Persistently low platelet count ( < 100k/μL for more than 6 months), mildly to highly elevated hepatic venous pressure gradient (HVPG) and either hepatomegaly and/or splenomegaly were present in all NRH+ patients. In opposition, persistently low platelet counts were not seen in NRH- patients, and hepatosplenomegaly observed in one NRH- patient. HVPG was normal in the only NRH- patient tested. All-cause mortality was higher among NRH+ patients (5/6, 83%) than in the rest of the cohort (1/15, 7% among NRH- and Unknown patients, p=0.002). Conclusion: NRH is an underreported, frequent and severe late-onset complication in XLA, which is highly associated with increased mortality. Persistent thrombocytopenia, elevated ALP, elevated HVPG, hepato- and/or splenomegaly were common in liver biopsy-proven XLA/NRH+ patients and distinguished them from XLA/NRH- patients. Based on NRH prevalence, lack of specific treatment and poor outcome in XLA, immune-reconstitution should be considered early in this population in order to prevent fatal long term complications.
Keywords: X-linked agammaglobulinemia, Nodular regenerative hyperplasia, mortality, morbidity
Disclosure: All authors indicated they had no financial relationships to disclose.
(219) Hematopoietic stem cell transplantation using non-genotoxic anti-CD117 immunotoxin conditioning corrects immune deficiencies and restores immune function in ataxia telangiectasia mice
Athena Russell1, Chengyu Prince2, Deepak Kumar, PhD3, Kristopher Knight4, Sruti Rayaprolu, PhD3, Hailian Xiao2, Srikant Rangaraju, MBBS5, H. Trent Spencer, PhD6, Christopher Doering, PhD7, Shanmuganathan Chandrakasan, MD8
1Doctoral Candidate/Emory University
2Research Technologist/Emory University
3Postdoctoral Fellow/Emory University
4Graduate Student/Emory University
5Associate Professor/Emory University
6Director of Gene and Cell Therapy/Children's Healthcare of Atlanta
7Professor/Emory University
8Pediatric Hematologist/Oncologist/Children's Healthcare of Atlanta
Ataxia telangiectasia (AT) is a chromosomal instability syndrome caused by mutation in ATM kinase, a protein critical for DNA double strand break repair and cell cycle control. AT is characterized by cerebellar neurodegeneration, immune dysfunction and predisposition to malignancy. Like several other immune deficiency disorders with DNA repair defect, AT patients could benefit from HSCT and immune reconstitution with ATM-competent cells. Additionally, we hypothesized that correction of underlying immune defects could delay inflammation-enhanced neurodegeneration. However, increased sensitivity to DNA damaging agents and progressive cerebellar degeneration are major barriers precluding HSCT for AT. Conditioning with an antibody-based, non-genotoxic immunotoxin targeting CD117 on hematopoietic stem cells may offer a safer approach to allogeneic HSCT in this setting. We conditioned Atm-/- mice with CD117-saporin immunotoxin, then transplanted GFP+ Atm+/+ whole bone marrow or fetal liver cells. We analyzed multilineage donor chimerism, body weight, leukocyte counts, immunoglobulins, cytokine levels, and immunization responses in transplanted Atm-/- mice versus Atm+/+ and non-transplanted Atm-/- controls. CD117-saporin conditioning enabled high-level myeloid chimerism (97.5 +/- 0.8%) within six weeks that was sustained for 26 weeks. Chimerism reached 80.1 +/- 0.9% and 93.6 +/- 1.0% in T and B cell compartments, respectively. Atm-/- mice have a growth defect that was not corrected by HSCT. However, CD4+ and CD8+ T cells were increased in transplanted mice compared with age-matched Atm-/- controls and, following immunization with a T cell-dependent immunogen, mounted effective IgM and IgG immune responses after HSCT similar to Atm+/+ mice, whereas Atm-/- controls could not. IgA and IgG1 levels were low in Atm-/- controls but were significantly increased after transplant. Additionally, Atm-/- controls had low levels of cytokines known to be important for innate and adaptive immunity such as M-CSF, MIP-1a, and MIP-2, and these returned to Atm+/+ levels after HSCT. One mouse that received HSCT after CD117-saporin conditioning developed host-derived T cell leukemia. We are currently exploring non-genotoxic host lymphocyte depletion to complement CD117-saporin conditioning to prevent lymphoid malignancy caused by residual Atm-/- T cells. Additionally, the role of donor myeloid derived microglial-like cell reconstitution in brain and its effects on decreasing neuroinflammation and neurodegeneration are being evaluated.
Keywords: Ataxia telangiectasia, hematopoietic stem cell transplantation, non-genotoxic conditioning, immunotoxin, immune deficiency, neurodegeneration, neuroinflammation
Disclosure: H. Trent Spencer and Christopher Doering have ownership interest in Expression Therapeutics. All other authors indicated they had no financial relationships to disclose.
(3) Late-Breaking Oral Abstracts
(220) A germline AIOLOS variant is associated with abnormal T and B differentiation, pneumocystis pneumonia and increased risk for chronic lymphocytic leukemia
Hye Sun Kuehn, PhD1, Motoi Yamashita, MD, PhD2, Julie E. Niemela, MS3, Jennifer L. Stoddard, BS4, Ryan Baxter, MSc5, Elena Hsieh, MD6, Mary Garofalo, RN, BSN7, Thomas A. Fleisher, MD, PhD8, Tomohiro Morio, MD, PhD9, Cullen M. Dutmer, MD10, Sergio Rosenzweig, MD, PhD11
1Senior Associate Scientist, Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
2Scientist, Department of Pediatrics and Developmental Biology, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University
3Research Biologist, Immunology Service, Department of Laboratory Medicine, Clinical Center, NIH
4Molecular Biologist, Immunology Service, Department of Laboratory Medicine, Clinical Center, NIH
5Senior Professional Research Assistant, University of Colorado Anschutz Medical Center
6Assistant Professor, Department of Pediatrics, Section of Allergy and Immunology, University of Colorado, Anschutz School of Medicine; Children’s Hospital Colorado; Department of Immunology and Microbiology, University of Colorado, Anschutz School of Medicine, Aurora, CO
7Research Nurse, National Institutes of Health National Institute of Allergy and Infectious Diseases
8Executive Vice President, American Academy of Allergy, Asthma and Immunology
9Vice President / Deputy Director, Department of Pediatrics and Developmental Biology, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University
10Assistant Professor of Pediatrics, Section of Allergy & Immunology, Children's Hospital Colorado, University of Colorado School of Medicine, Aurora, CO, USA
11Senior Investigator, Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, USA
AIOLOS, encoded by IKZF3, is a member of the IKAROS family of proteins. Both Aiolos and Ikaros have been shown to be important regulators of lymphocyte development and differentiation. In Aiolos deficient mice, abnormal B cell differentiation and functions were observed including increased number of B cell precursors, breakdown in B cell tolerance, spontaneous autoantibody production, and the development of B cell lymphomas, suggesting the important role of Aiolos in murine B cell differentiation and development. Previously, germline IKAROS (IKZF1) mutations have been associated with common variable immunodeficiency (CVID) and combined immunodeficiency (CID). Here we describe a novel immunodeficiency due to an IKZF3 mutation in a family presenting with CID, Pneumocystis jirovecii pneumonia (PJP) and/or chronic lymphocytic leukemia (CLL). Individuals carrying IKZF3 c.479A>G p.N160S heterozygous variant displayed impaired humoral immune responses as well as abnormal B cell development with a high percentage of CD21low B cells. In addition, T cell development was also affected, with increased naïve T cells, abnormal T cell differentiation, and impaired CD40L upregulation following activation. Further in-vitro studies demonstrated that the mutant protein failed to bind to its DNA target sequence as well as to undergo pericentromeric localization. Mechanistically, the mutant was fully penetrant and had a dominant negative effect over AIOLOS wildtype function. Our findings demonstrate that AIOLOS plays a key role in T and B cell development in humans and also associates abnormal AIOLOS function with CID, PJP and potential development of CLL.
Keywords: AIOLOS, IKZF3, chronic lymphocytic leukemia, pneumocystis pneumonia, combined immunodeficiency
Disclosures: Elena Hsieh is a consultant for Enzyvant. All other authors had no financial relationships to disclose.
(221) Germline SAMD9L truncation variants trigger global translational repression
Eric J. Allenspach, MD, PhD1, Frank Soveg, BS2, Laura Finn, MD3, Lomon So, PhD4, Jacquelyn Gorman, PhD4, Aaron Rosen, BS5, Suzanne Skoda-Smith, MD6, Marsha Wheeler, PhD4, Jason Debley, MD, MPH6, Michael Bamshad, MD7, Deborah Nickerson, PhD7, Ram Savan, PhD7, Troy Torgerson, MD, PhD8, David Rawlings, MD7
1Assistant Professor of Pediatrics, University of Washington
2Graduate Student, University of Washington
3Professor, University of Pennsylvania
4Post-Doctoral Fellow, University of Washington
5Research Scientist, Seattle Children's Research Institute
6Associate Professor, University of Washington
7Professor, University of Washington
8Attending Physician, Experimental Immunology, Allen Institute for Immunology
SAMD9L is an interferon-induced tumor suppressor implicated in a spectrum of multi-system disorders including risk for myeloid malignancies and immune deficiency. We identified a heterozygous de novo frameshift variant in SAMD9L in an infant with B cell aplasia and clinical autoinflammatory features who died from respiratory failure with chronic rhinovirus infection. Autopsy demonstrated absent bone marrow and peripheral B cells as well as selective loss of Langerhans and Purkinje cells. The frameshift variant led to expression of a truncated protein with interferon treatment. This protein exhibited a gain-of-function phenotype resulting in interference in global protein synthesis via inhibition of translational elongation. Using a mutational scan, we identified a region within SAMD9L where stop-gain variants trigger a similar translational arrest. SAMD9L variants that globally suppress translation had no effect or increased mRNA transcription. The complex reported phenotype likely reflects lineage dominant sensitivities to this translation block. Taken together, our findings indicate that interferon- triggered SAMD9L gain-of-function variants globally suppress translation.
Keywords: SAMD9L, Translation, Autoinflammatory, B cell Aplasia
Disclosures: Jason Debley received a research grant from the NIH. Troy Torgerson is a consultant for CSL Behring and Grifols; received speaker honoraria from Takeda and X4 Pharmaceuticals; and is an advisory board member of Enzyvant. All other authors had no financial relationships to disclose.
(222) New machine learning approaches to estimate the functional consequence of mutations in diverse human populations
Yuval Itan, PhD1, Yiming Wu, PhD2, Cigdem Sevim Bayrak, PhD2
1Assistant Professor, Icahn School of Medicine at Mount Sinai
2Postdoctoral Researcher, Icahn School of Medicine at Mount Sinai
The genome of a patient with a genetic disease contains about 20,000 non-synonymous variations, of which only one (or a few) is disease-causing. Current computational methods cannot predict the functional consequence of a mutation: whether it results in gain-of-function (GOF) or loss-of-function (LOF). Moreover, computational predictions of mutation pathogenicity are still lacking specificity when analyzing diverse human genetic data. Here we present two novel approaches to address these shortcomings: (1) a machine learning study to computationally differentiate GOF from LOF mutations, using natural language processing (NLP) and feature selection to generate the first large-scale human inherited GOF and LOF mutation database; and (2) a deep learning neural network approach to classify mutations by the human phenotype ontology (HPO) disease group. We demonstrate the utility of our combining our state-of-the-art with gold standard methods in case-control studies across different diseases including susceptibility to severe COVID-19 and inflammatory bowel disease (IBD), where we discovered novel genetic etiologies.
Keywords: Mutation and gene predictions, Next generation sequencing, Computational methods, Disease genomics, Machine learning, Case-control associations, Phenome-wide associations, Polygenic risk score
Disclosure: All authors indicated they had no financial relationships to disclose.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.