

Abstract
DNA-dependent protein kinase catalytic subunit (DNA-PKcs) is a key member of the phosphatidylinositol-3 kinase-like (PIKK) family of protein kinases with critical roles in DNA-double strand break repair, transcription, metastasis, mitosis, RNA processing, and innate and adaptive immunity. The absence of DNA-PKcs from many model organisms has led to the assumption that DNA-PKcs is a vertebrate-specific PIKK. Here, we find that DNA-PKcs is widely distributed in invertebrates, fungi, plants, and protists, and that threonines 2609, 2638, and 2647 of the ABCDE cluster of phosphorylation sites are highly conserved amongst most Eukaryotes. Furthermore, we identify highly conserved amino acid sequence motifs and domains that are characteristic of DNA-PKcs relative to other PIKKs. These include residues in the Forehead domain and a novel motif we have termed YRPD, located in an α helix C-terminal to the ABCDE phosphorylation site loop. Combining sequence with biochemistry plus structural data on human DNA-PKcs unveils conserved sequence and conformational features with functional insights and implications. The defined generally progressive DNA-PKcs sequence diversification uncovers conserved functionality supported by Evolutionary Trace analysis, suggesting that for many organisms both functional sites and evolutionary pressures remain identical due to fundamental cell biology. The mining of cancer genomic data and germline mutations causing human inherited disease reveal that robust DNA-PKcs activity in tumors is detrimental to patient survival, whereas germline mutations compromising function are linked to severe immunodeficiency and neuronal degeneration. We anticipate that these collective results will enable ongoing DNA-PKcs functional analyses with biological and medical implications.
Keywords: Sequence analysis, Motifs, Kinase, DNA repair, DNA damage Response, Crystal structure, Cryo-electron microscopy
1. Introduction
1.1. The PIKK family of serine/threonine protein kinases
Human cells contain over 500 protein kinase genes, the majority of which belong to the canonical eukaryotic protein kinase family and have amino acid similarity in their kinase domains to cAMP-dependent protein kinase. However, the human kinome also contains a small group of “atypical” protein kinases, one of which is the phosphatidylinositol-3 kinase-like family of serine/threonine protein kinases, or PIKKs (Manning et al., 2002). The PIKK family is composed of six biologically important kinases: 1) the DNA-dependent protein kinase catalytic subunit (DNA-PKcs, gene names PRKDC and XRCC7); 2–3) Ataxia Telangiectasia Mutated (ATM) and ATM-and Rad3-related (ATR), which are involved in the cellular response to DNA double strand breaks (Blackford and Jackson, 2017); 4) mammalian/mechanistic target of rapamycin (mTOR), which has multiple roles in energy metabolism and protein synthesis (Liu and Sabatini, 2020); 5) suppressor of morphogenesis in genitalia 1 (SMG1), which is involved in non-sense mediated mRNA decay (Lloyd, 2018); and 6) the catalytically inactive pseudo kinase, transformation/transcription domain-associated protein (TRRAP), which is part of the Spt-Ada-Gcn5 acetyltransferase (SAGA) and TIP60 acetyltransferase complexes (Elias-Villalobos et al., 2019).
The human PIKKs are very large polypeptides that share a characteristic architecture that includes four major elements. One, a region consisting of multiple Huntingtin, Elongation Factor 3 A, Protein Phosphatase 2 A subunit, TOR (HEAT) repeats (Brewerton et al., 2004). Second, a conserved FRAP, ATM, and TRRAP (FAT) domain (Bosotti et al., 2000). Third, the highly conserved kinase domain (Hartley et al., 1995), and fourth, a short C-terminal FATC motif (Baretic et al., 2019; Bosotti et al., 2000; Elias-Villalobos et al., 2019) (see Fig. 1A).
Fig. 1. Structure of DNA-PKcs.
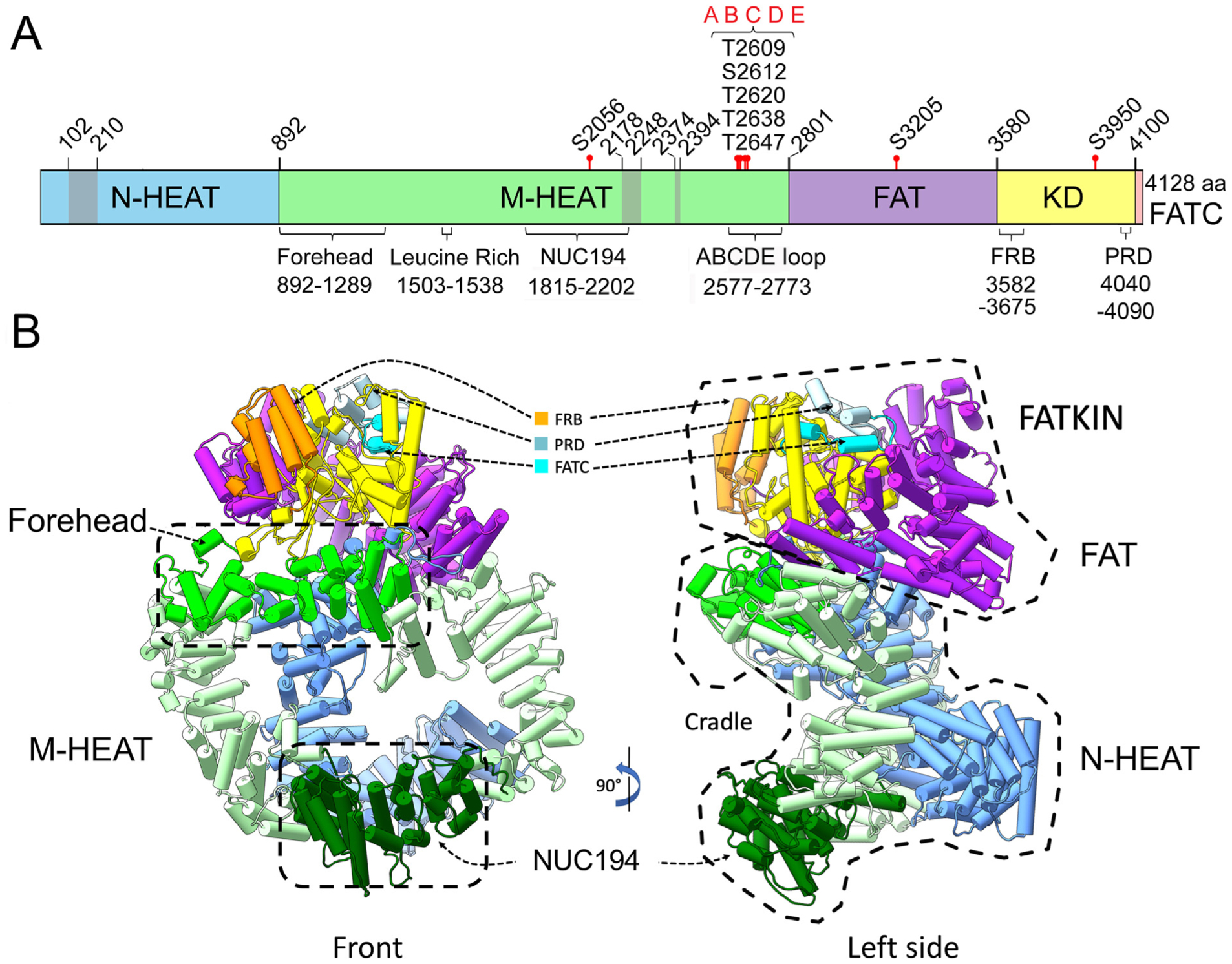
Panel A: Cartoon of DNA-PKcs showing the locations of the major domains, proposed Ku-interacting regions and autophosphorylation sites. DNA-PKcs is composed of a unique N-terminal HEAT domain (N-HEAT, residues 1–891, blue), a middle-HEAT domain, also called the circular cradle (M-HEAT, residues 892–2800, green), the FAT domain (residues 2801–3579, pink), the kinase domain (residues 3580–4099, yellow) and the FAT-C domain (residues 4100–4128, orange). Domain boundaries are from (Sibanda et al., 2017). Also shown are the locations of the putative Ku binding sites, residues 102–210, 2178–2248 and 2374–2394 (grey) from (Yin et al., 2017), the Forehead domain (residues 892–1289 (Sibanda et al., 2017)), a leucine rich region (residues 1503–1538) associated with DNA-binding activity (Gupta and Meek, 2005) and the NUC194 domain (residues 1815–2202 from (El-Gebali et al., 2019; Staub et al., 2004)). The location of the characterized phosphorylation sites at serine 2056 in the M-HEAT region, threonine 2609, serine 2612, threonine 2620, threonine 2638 and threonine 2647 (the ABCDE cluster), serine 3205 in the FAT domain (Neal et al., 2011) and threonine 3950 in the kinase domain (Douglas et al., 2007) are also shown. Residues 2577–2773 represent a flexible loop containing the ABCDE phosphorylation sites that was absent from available X-ray and cryoEM structures (Saltzberg et al., 2019). The location of regions of DNA-PKcs with similarity to the FKBP12erapamycin-binding domain of PI3K located at residues 3582–3675 (Sibanda et al., 2017; Yang et al., 2013), and a region reported to contain a PIKK Regulatory Domain (PRD) (Mordes et al., 2008) are also shown.
Panel B: Model of the DNA-PKcs-Ku-DNA structure. The structure of DNA-PKcs (PDB:5LUQ from (Sibanda et al., 2017)) is shown in pipes and planks representation. The N-HEAT domain is in blue, the M-HEAT/circular cradle is in green, the FAT domain is in purple and the kinase domain is in yellow. Also shown is the location of the Forehead domain (bright green with hatched box) and the NUC194 domain (dark green with hatched box). The FATC domain is shown in bright blue, the FRB motif in orange and the PRD motif in blue/grey.
Although the PIKK kinase domain sequence is more similar to that of the phosphatidylinositol-3 kinase (PI3K) catalytic subunit (gene PIK3CA) than canonical eukaryotic protein kinases (Hartley et al., 1995; Hunter, 1995), it is distinct from PI3K in that it only contains the lysine (K) of the valine-alanine-isoleucine-lysine (VAIK) ATP-binding site found in PI3K/PIK3CA (Elias-Villalobos et al., 2019). Like PI3K, the catalytic site of the PIKKs has a characteristic glycine-aspartic acid-arginine-histidine-X-X-asparagine (GDRHXXN) motif where D is the catalytic aspartic acid (DXXXXN) in all eukaryotic protein kinases (Taylor et al., 1992). The canonical aspartic acid-phenylalanine-glycine (DFG) Mg2+ binding site is conserved only in PI3K/PIK3CA, DNA-PKcs and mTOR, while ATR, ATM and SMG1 retain only the aspartic acid (Elias-Villalobos et al., 2019). Like PI3K, the PIKKs are inhibited by wortmannin. Unlike PI3K, the PIKKs are serine/threonine protein kinases rather than lipid kinases. DNA-PKcs, ATM and ATR prefer serines or threonines that are followed by a glutamine (SQ/TQ motifs) (Lees-Miller et al., 1992; O’Neill et al., 2000), although DNA-PKcs can also phosphorylate other substrates on serine or threonine followed by leucine, methionine or other aliphatic amino acids (Dobbs et al., 2010; Douglas et al., 2002).
1.2. DNA-PKcs function
At 4128 residues (approximately 469 kDa), the human DNA-PKcs polypeptide is the largest human PIKK family member. DNA-PK was originally discovered in human cells as a protein kinase that was stimulated by double stranded (ds) DNA (Carter et al., 1990; Lees-Miller et al., 1990; Walker et al., 1985). Alone, DNA-PKcs has weak protein kinase activity that is stimulated by interaction with the Ku70/80 heterodimer and ends of dsDNA (Chan et al., 1996; Gottlieb and Jackson, 1993). DNA-PKcs and Ku are required for the repair of ionizing radiation (IR)-induced DNA double strand breaks (DSBs) via the non-homologous end joining (NHEJ) pathway, and cells that lack either Ku or DNA-PKcs are sensitive to ionizing radiation and other DSB-inducing agents (Pannunzio et al., 2018; Wang and Lees-Miller, 2013). The DNA-PKcs nuclease partner Artemis (gene name DCLRE1C) is needed for processing some DNA end configurations at dsDNA breaks for NHEJ. The analogous nuclease partner MRE11 complex for the PIKK ATM is critical to license and then commit to homologous-recombination dsDNA break repair and without MRE11 the repair is instead completed by NHEJ (Syed and Tainer, 2018). Thus, better defining Artemis-DNA-PKcs functional interactions will likely be of substantial interest. DNA-PKcs and Artemis are also required for opening DNA hairpin ends during V(D)J recombination, a gene rearrangement process critical for antibody diversity in the adaptive immune system (Lieber, 2010). Accordingly, animals with inactivating mutations in DNA-PKcs, for example the severe combined immunodeficient (SCID) mouse, are both radiation sensitive due to defective NHEJ and immune-deficient due to defective V(D)J recombination (Lieber, 2010). In addition, DNA-PKcs has other key cellular functions, for example in regulation of androgen receptor-mediated transcription and metastasis in prostate cancer (Goodwin and Knudsen, 2014), mitosis (Douglas et al., 2020; Jette and Lees-Miller, 2015), detection of foreign DNA and innate immunity (Burleigh et al., 2020; Chu et al., 2000) and ribosomal RNA processing (Shao et al., 2020).
1.3. DNA-PKcs autophosphorylation
DNA-PKcs undergoes extensive autophosphorylation in vitro which, counterintuitively, leads to loss of its kinase activity (Chan and Lees-Miller, 1996). In vitro DNA-PKcs autophosphorylation sites include serines 2612 and 2624 plus threonines 2609, 2620, 2638 and 2647 (Douglas et al., 2002) as well as serine 3205 in the FAT domain (Douglas et al., 2002) and threonine 3950 in the kinase domain (Douglas et al., 2007) (see Fig. 1A). Serines 2612 and 2624 plus threonines 2609, 2620, 2638 and 2647 are frequently referred to as the ABCDE or T2609 cluster (Ding et al., 2003): they are located in a flexible loop extending from residues ~2577 to 2773 in human DNA-PKcs. In human cells, IR-induced phosphorylation of serines 2612 and 2624 and threonines 2609, 2620, 2638 and 2647 is inhibited by DNA-PK kinase inhibitor NU7441 but not by ATM kinase inhibitor KU55933, suggesting that phosphorylation of these sites in vivo is DNA-PKcs-dependent (Meek et al., 2007). In vitro autophosphorylation of DNA-PKcs results in its dissociation from Ku-bound DNA, while DNA-PKcs in which the ABCDE phosphorylation sites are mutated to alanine has reduced ability to dissociate from Ku-DNA (Hammel et al., 2010; Jette and Lees-Miller, 2015). This has led us to propose that DNA-PKcs is recruited by Ku to ends of dsDNA, autophosphorylates, undergoes a conformational change and then dissociates from the Ku-DNA complex (Dobbs et al., 2010). In support of this model, DNA-PKcs in which the ABCDE sites have been mutated to alanine, as well as kinase-dead DNA-PKcs are retained at sites of DNA damage in vivo, whereas wild type-DNA-PKcs is recruited to DNA damage sites but dissociates with a half-life of ~60 min (Uematsu et al., 2007). Other studies have reported that phosphorylation of the ABCDE cluster is ATM-dependent (Jiang et al., 2015), and can be ATR-dependent in response to UV radiation (Yajima et al., 2006). However, in our hands, T2609 phosphorylation is as robust in ATM-deficient cells as in wild type cells (Neal and Meek, 2019). Regardless, it seems clear that the ABCDE phosphorylation sites play an important role in DSB repair as their mutation to alanine results in extreme radiation sensitivity and defects in DSB repair and V(D)J recombination (Ding et al., 2003; Meek et al., 2008). Another well characterized auto-phosphorylation site is serine 2056 (Chen et al., 2005; Cui et al., 2005; Neal and Meek, 2019), which, with serines 2023, 2029, 2041 and 2053, is referred to as the PQR cluster (Cui et al., 2005). Mutational analysis reveals that phosphorylation of the PQR cluster inhibits access to DNA ends, suggesting that ABCDE and PQR phosphorylation clusters act in a reciprocal manner (Neal and Meek, 2011). Yet these autophosphoryation sites represent only a few of the total post-translational modifications on DNA-PKcs, with 88 phosphoserines, 34 phosphothreonines, 21 phosphotyrosines and almost 200 ubiquitination sites reported on Phosphosite Plus (Hornbeck et al., 2015) (see www.phosphosite.org).
1.4. DNA-PKcs structure
Elegant crystallographic and cryo-electron microscopy (cryo-EM) studies have determined the structure of DNA-PKcs alone (Sibanda et al., 2010), with a peptide from the Ku80 C-terminal region (Sibanda et al., 2017) and in the context of the DNA-PKcs-Ku70/80-dsDNA holoenzyme, DNA-PK (Sharif et al., 2017; Yin et al., 2017). The structures reveal that the N-terminal HEAT repeat-containing region of DNA-PKcs can be differentiated into an N-terminal unit (residues 1–892) that is absent from other PIKKs (Sibanda et al., 2017) (also called the N-HEAT region, shown in blue in Fig. 1) and a central cradle unit, also called the M-HEAT region (residues 893–2801, shown in green in Fig. 1). Within the M-HEAT domain is the Forehead region (residues 893–1289) which is located at the top of the ring and has a more irregular helical structure (Sibanda et al., 2017). The FAT domain occupies residues 2802–3564 and is followed by the kinase domain (residues 3565–4100) and the FATC domain, residues 4101–4128. Structural similarity led to the identification of a region with similarity to the FKBP12erapamycin-binding (FRB) domain of PI3K located at residues 3582–3675 in DNA-PKcs (Sibanda et al., 2017; Yang et al., 2013). DNA-PKcs also contains a PIKK regulatory domain (PRD) located at residues 4043–4090 (Mordes et al., 2008). A substantial conserved region, corresponding to “domain B” in the DNA-PKcs structure, called the NUC194 domain, was identified in a bioinformatics screen for nucleolar proteins (Staub et al., 2004). The NUC194 domain is defined in the Pfam database (El-Gebali et al., 2019), as corresponding to residues 1815–2202 of DNA-PKcs (http://pfam.xfam.org/family/Nuc194#tabview=tab9) and, like the N-HEAT domain, is unique to DNA-PKcs.
The HEAT repeat containing N-HEAT and M-HEAT domains form a super-helical α-solenoid structure, while the FAT-kinase-FAT-C domains form a “crown” at the top of the molecule (see Fig. 1B). The overall dimensions of DNA-PKcs are 180 Å × 130 Å × 105 Å. The Ku70/80 heterodimer nestles against the base and back of DNA-PKcs, aligning dsDNA to enter DNA-PKcs (Sharif et al., 2017; Yin et al., 2017). Potential Ku interaction sites have been proposed at amino acids 102–210, 2178–2248 and 2374–2394 (Yin et al., 2017) (marked in grey in Fig. 1A) and a region associated with DNA binding has been mapped to a leucine rich region between residues 1503–1538 (Gupta and Meek, 2005). The S2056 phosphorylation site is located at the base of the molecule, close to the Ku binding sites (dark green in Fig. 1B) (Sibanda et al., 2017). Absent from current X-ray and cryo-EM structures is a flexible loop beginning at residue 2577 and ending at residue 2733 containing the ABCDE phosphorylation sites (Neal et al., 2014; Saltzberg et al., 2019; Sheff et al., 2017) (see Fig. 1A). We suggest that autophosphorylation of the ABCDE sites leads to a conformational change in the synaptic complex that allows DNA-end processing enzymes, and ultimately DNA ligase IV, to access the extreme ends of the DSB (Dobbs et al., 2010). However, phosphorylation of the ABCDE sites alone is insufficient for DSB repair, suggesting that added phosphorylation events are required for efficient NHEJ (Meek et al., 2007).
1.5. DNA-PKcs phylogeny
Unlike its relatives ATM, ATR, mTOR and TRRAP, DNA-PKcs has not been detected in the Dikarya fungi Saccharomyces cerevisiae and Schizosaccharomyces pombe, the free-living nematode Caeno-rhabditis elegans, the Dipteran fly Drosophila melanogaster and the flowering plant Arabidopsis thaliana (Dore et al., 2004; Elias-Villalobos et al., 2019). These observations led the suggestion that DNA-PKcs arose later in evolution, at the invertebrate-vertebrate transition (Zhao et al., 2019). However, a putative DNA-PKcs orthologue was reported in the genome of the slime mould Dic-tostelium discoideum (Block and Lees-Miller, 2005; Eichinger et al., 2005) and indeed, DNA-PKcs is functionally important for NHEJ in D. discoideum (Hsu et al., 2006, 2011; Pears and Lakin, 2014). Moreover, potential DNA-PKcs orthologues have been reported in the mosquito Anopheles gambiae and the honey-bee Apis mellifera ligustica (Dore et al., 2004). DNA-PKcs has since been identified in the green algea Ostreococcus tauri (Hindle et al., 2014) and the radiation-resistant bdelloid rotifer Adineta vaga (an aquatic micro-invertebrate) (Hecox-Lea and Mark Welch, 2018). Interestingly, putative orthologues of Artemis have also been identified in A. vaga (Hecox-Lea and Mark Welch, 2018) and D. discoideum contains a metallo-beta-lactamase with similarity to Artemis that is required for NHEJ (Pears and Lakin, 2014). Potential orthologues of multiple DNA repair genes including Ku70, Ku80, DNA-PKcs and DNA ligase IV (but not Artemis) have also been reported in dinoflagellates, a group of phytoplankton (Li and Wong, 2019). Furthermore, sequence alignment of the kinase domains of the PIKK family members identified putative DNA-PKcs orthologues in a wide range of Metazoans, including sea urchins, sponges and hydra, as well as Choanoflagellates (free-living unicellular eukaryotes), amoeba, fungi of the Chytridiomycota and Muromycota phyla, plants of the Tracheophyte, Bryophyte and Chlorophyte phyla, Stramenopiles, Alveolates, Rhizaria, Euglenozoa, and Heterolobosea (Elias-Villalobos et al., 2019). Thus, although lost from some lineages, in particular many model organisms that are widely used in molecular biology, these data suggest that DNA-PKcs has a far more ancient emergence than previously supposed.
To more systematically explore the evolutionary conservation of DNA-PKcs, we screened NCBI databases for proteins with amino acid similarity to DNA-PKcs and identified a diverse array of additional organisms that contain DNA-PKcs, including a wide range of invertebrates, fungi, plants, and protists. Strikingly, phylogenetic analysis indicates that DNA-PKcs emerged over 1 billion years ago and has retained its core functions over time. In addition, we identify novel sequence motifs that are unique to DNA-PKcs that we propose as signature motifs that may aid in functional characterization and sequence-based searches. In the light of these combined evolutionary and structural analyses we consider DNA-PKcs mutations and their consequences.
2. Methodology
2.1. Protein sequence analysis
The NCBI protein sequence data base (Sayers et al., 2019) was searched using the FAT-Kinase-FAT-C domain of human DNA-PKcs (Accession number P78527.3). The NCBI Protein database was searched with the terms: DNA-activated protein kinase and NUC194. Given the large size of DNA-PKcs we expected reported sequences to be subject to gene prediction errors that can affect the analysis of sequence similarity across species (Banyai and Patthy, 2016). Indeed, obvious gene prediction errors were present in approximately 40% of the sequences that we used in this study. These included both fragmented and incomplete sequences. To correct these errors we first used Clustal Omega at EMBL (Madeira et al., 2019) to align the protein sequence in question with others that were as closely related as possible. Gene graphics in NCBI was used to identify the region containing the missing sequence. This region was translated using the translation map function in the free on-line Sequence Manipulation Suite (Stothard, 2000) available at (https://era.library.ualberta.ca/items/6852886f-f731-440c-b7a4-f05c3e60c4d5).
Potential coding sequences were sewn together using tBLASTn to identify overlapping RNAseq reads in the sequence read archive (NCBI) (Sayers et al., 2019). In many species, incomplete gene sequences resulted in gene prediction errors. In these cases, we scanned the Whole Genome Shotgun sequences in NCBI with tBLASTn (Sayers et al., 2019). using both surrounding sequences from the species in question and the predicted missing sequences from related species. As previously, the identified sequences were sewn together with RNAseq reads. Occasionally useful transcript information was also found in the Transcriptome and EST databases in NCBI (Sayers et al., 2019). It should be noted that we did not identify full-length transcripts for any of the sequences so some errors may still exist. Sequences were aligned using various multiple sequence alignment programs, including Clustal-Omega at EMBL (Madeira et al., 2019) and COBALT at NCBI (Sayers et al., 2019). All alignments were run using default settings (conservation setting 2 in COBALT). In Clustal-Omega * represents identical amino acids,: represents highly conserved amino acids and. similar amino acids. In COBALT, regions of highest amino acid similarity/highest conservation are represented by red, partial conservation in blue and non-conserved amino acids in grey.
2.2. Identification of the PRKDC gene in the extremophile Alvinella pompejana
We used a short read sequence library obtained from A. pompejana samples collected during past expeditions (2003–2004) in the East Pacific Rise (Shin et al., 2009) to build a partial genome sequence of the extremophile using SOAPdenovo2, and then conducted gene prediction analysis with the gene prediction tool AUGUSTUS (Stanke and Morgenstern, 2005) trained on a set of full-length ESTs previously reported (Holder et al., 2013). A putative full-length PRKDC gene was identified in a 124-kb scaffold (scaffold 113562); however, sequence alignment suggested that the protein product was missing ~20 amino acids between codons 3360 and 3380. We found a partial EST that spanned the gap and whose sequence was indeed present in scaffold 113562, implying that the missing 20 amino acids originated from incorrect splice detection by AUGUSTUS. Therefore, we used the additional EST sequence to reconstruct an ungapped PRKDC cDNA. The sequence of DNA-PKcs from A. pompejana has been deposited in GenBank with the accession number MT472577
2.3. Visualization of DNA-PK structures
Structures of DNA-PKcs [5LUQ (Sibanda et al., 2017) and DNA-PK holoenzyme structures 5Y3R (Yin et al., 2017), 5W1R (Sharif et al., 2017)] were downloaded from the Protein Data Bank and visualized using Pymol (Schrodinger) and CHIMERA (Pettersen et al., 2004), as indicated.
2.4. Modeling of a DNA-PK synaptic complex
Modeling of DNA-PKcs and DNA-PK synaptic complexes is described in Hammel et al., this issue (Hammel et al., 2020, this issue). Briefly, amino acid loops missing from the cryo-EM and X-ray structures, including the ~2576–2776 region that contains the ABCDE phosphorylation sites (Ding et al., 2003; Douglas et al., 2002) were built in to the X-ray crystal structure (Sibanda et al., 2017) using MODELLER (Sali and Blundell, 1993). A DNA-PKcs dimer was modelled by molecular docking two DNA-PKcs monomer-models using a rigid docking, geometric shape-matching algorithm PatchDock (Schneidman-Duhovny et al., 2005). The best docking model was selected for further analysis. Amino acids that were highly conserved (>95%) in representative examples of vertebrate and invertebrate DNA-PKcs were mapped onto the structure of human DNA-PKcs and the docking model of the human DNA-PKcs dimer using CHIMERA (Pettersen et al., 2004).
2.5. Multiple sequence alignment and phylogenetic reconstruction
Annotated protein sequences for all DNA-PKcs members were acquired as above. For the broadest-level analysis we used sequences ranging from metazoans to plants, fungi, unicellular protists and a putative Archaeon sequence using the related Drosophila TOR protein sequence as an outgroup. A total of 48 sequences were analyzed, while for the metazoan-specific analysis we used a total of 58 sequences from across Metazoa, with the fungus Glomus cerebriforme as an outgroup. DNA-PKcs sequences ranged from ~3500 to ~5000 amino acids in length, and were initially aligned using the Multiple Sequence Alignment by Log-Expectation (MUSCLE) program (Edgar, 2004). The optimal parameters for phylogenetic reconstruction analysis were taken from the best-fit evolutionary model selected using the Akaike Information Criterion (AIC) implemented in the PROTTEST3 software (Darriba et al., 2011), and were inferred to be the Le-Gascual (LG) model (Le and Gascuel, 2008), with gamma-distributed among-site rate variation and empirical state frequencies. Phylogenies were inferred from these alignments using RaXML v8.2.9 software (Stamatakis, 2014) and results were visualized using FigTree v1.4.4 (https://github.com/rambaut/figtree/releases).
2.6. Evolutionary trace analysis of DNA-PKcs function
Evolutionary Trace analysis (Lichtarge et al., 1996; Mihalek et al., 2004) was run using the “position-specific gap-reducing real-valued trace” option and as input the multiple sequence alignments shown in the supplemental figures or specific branches of them were used. PyMOL 2.3.4 was used to visualize the structures and the PyETV plugin (Lua and Lichtarge, 2010) to color the residues according to their ET importance. Residues with the lowest ET score were coloured red and those with the second lowest score in yellow.
2.7. Cancer and human inherited disease genome-wide analyses
RNA-seq gene expression data in cancer genomes were downloaded using the TCGA Assembler suite (Wei et al., 2018); comparisons between tumor and normal tissues were limited to those tumor types for which at least 10 normal control samples were available. Survival data related to gene expression levels were also retrieved from TCGA and analyzed with the R packages “survival” and “survminer”. PRKDC mutations and progression-free survival data were retrieved from cBioPortal (https://www.cbioportal.org) by choosing the “Curated set of non-redundant studies”. PRKDC missense mutations associated with inherited disease or predisposing to human disease were obtained from the HGMD Professional, version 2019.2.
2.8. Evolutionary Action analysis of tumor-associated PRKDC mutations
Evolutionary Action (EA) scores were generated for the NP_008835 human PRKDC sequence, using the multiple sequence alignment of Supplementary Fig. 9 and the methodology described previously (Katsonis and Lichtarge, 2014). PRKDC variants retrieved from cBioPortal (https://www.cbioportal.org), although they also referred to the NP_008835 sequence, had a residue number shift on and after the residue position 1245, apparently due to a residue addition in the reference sequence used by cBioPortal (this addition was found neither in the sequence databases nor in HGMD). We accounted for this discrepancy and assigned correctly the EA scores to the cBioPortal variants. The EA distributions were generated by binning the EA score range into deciles. The random nucleotide variants were collected by performing all three nucleotide changes to each nucleotide position of the PRKDC sequence (silent and nonsense variants were ignored). The difference between the observed and random distributions was quantified by the significance p-value of the Kolmogorov-Smirnov test (two-sample, right-tail).
3. Results
3.1. DNA-PKcs has a broad distribution across most eukaryotic phyla
Analysis of NCBI databases revealed the presence of DNA-PKcs orthologues in all major phyla including Metazoa (from human to sponge), fungi of the Chytridiomycota and Mucoromycota phyla and plants (Viridiplantae) of the Chlorophyte (green algae) and Streptyphyte phyla from Charophyte and Klebsormidiophyte (green algae), to Bryophytes (liverworts and mosses), Lycophytes (clubmoss) and Euphyllophytes of the class Acrogymnospermae (conifers, ferns, cycads and Ginkgo biloba). DNA-PKcs was also present in Stramenopiles, Ciliates (Paramecium, Stentor and Tetrahymena), Evocea (slime molds and social amoeba) and Choanoflagellates (free-living eukaryotes, considered to be the earliest living relatives of animals) as well a variety of protist phyla (Apusozoa, Heterolobosea, Haptista and Rhizaria) (Supplementary Table 1).
Supporting previous observations (Dore et al., 2004; Elias-Villalobos et al., 2019), we did not detect DNA-PKcs in D. melanogaster, a Dipteran fly of the of the Brachycera suborder. However, DNA-PKcs was present in Dipterans of the Nematocera suborder, such as the midge Clunio marinus and the mosquito Aedes aegypti, as well as in most other insect orders, with the possible exception of Lepidoptera (see below). Similarly, although DNA-PKcs was not present in nematodes of the class Chromadorea such as C. elegans, it was present in nematodes of the Enoplea class, including the parasites Trichinella pseudospiralis and Trichius muris. We did not detect DNA-PKcs in Dikarya fungi (yeast, mushrooms) or in Euphyllophyte plants of the class Magnoliopsida (flowering plants including model organisms Arabidopsis thaliana and Glycine max). DNA-PKcs was also absent from red algae of the phylum Rhodophyta, classes Bangiophyceae and Florideophyceae. Included within the Bangiophyceae are the extremophiles Galdiera sulphuraria and Cyanidioschyzon merolae that live under extreme conditions of temperature and pH (Qiu et al., 2013).
3.2. DNA-PKcs phylogeny reveals an ancient eukaryotic past
Phylogenetic analysis of DNA-PKcs sequences from a broad range of Eukaryotes largely conformed to current classification knowledge (Adl et al., 2019). The Diaphoretick clade including Alveolates, Stramenopiles, Viridiplantae, Rhizaria and Haptista grouped together as expected (Fig. 2A). Within the Amorphean clade, the Opisthokonts including Fungi, Choanoflagellates, and Metazoa grouped together. A small difference from the current classification was seen as the Amorphean clades Amoebozoa and Apuozoa formed a group with Heterolobosia that was slightly closer to the Diaphoreticks than the Amorpheans.
Fig. 2. Phylogenetic analysis of DNA-PKcs.
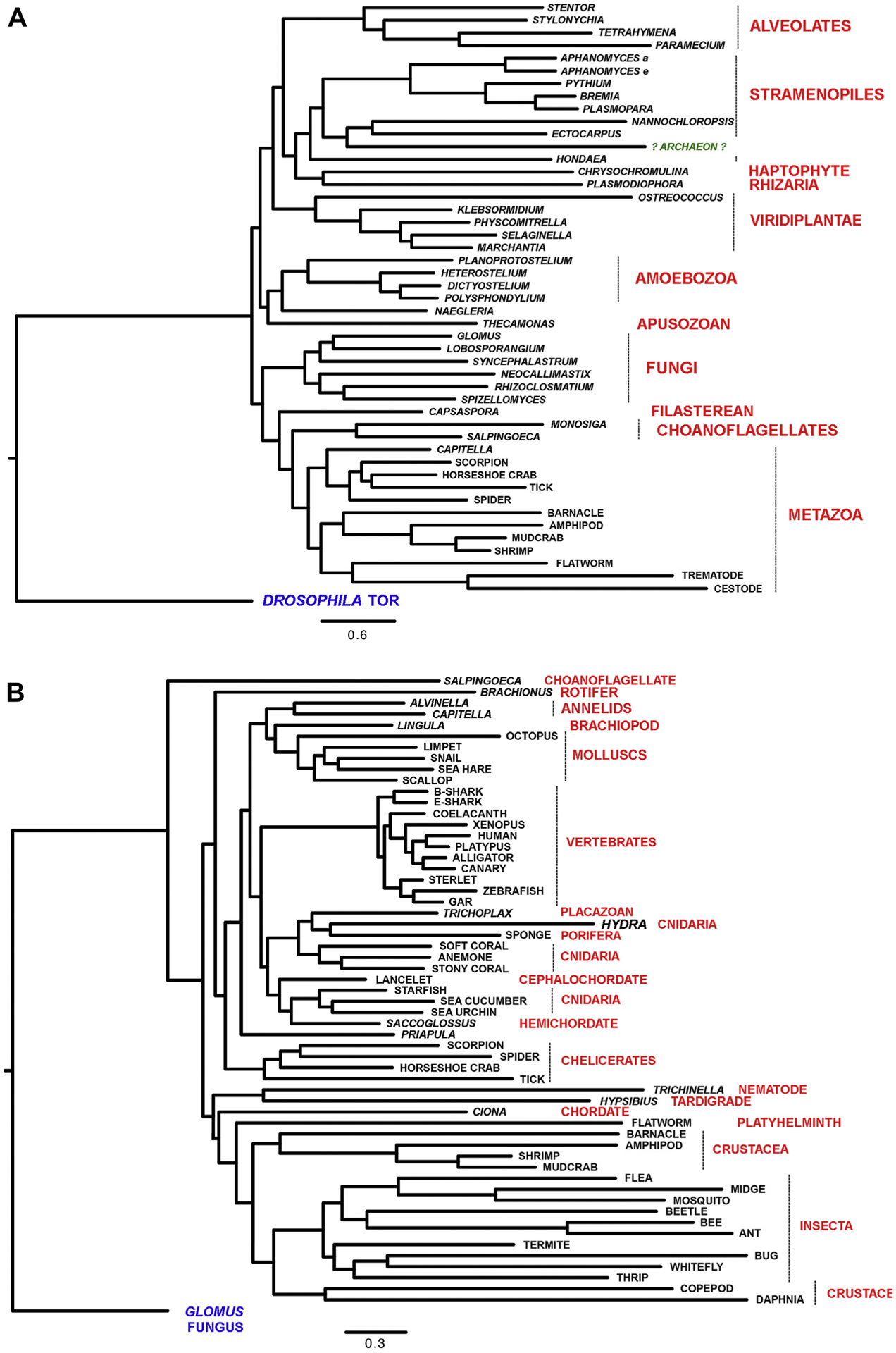
Panel A: DNA-PKcs sequences used were from Alveolates (Stentor coeruleus, Stylonychia lemnae, Tetrahymena thermophila and Paramecium tetraurelia); Stramenopiles [Aphanomyces astaci (Aphanomyces a), Aphanomyces euteiches (Aphanomyces e), Pythium oligandrum, Bremia latucae, Plasmopara halstedii, Nannochloropsis salina, Ectocarpus siliculosus and Hondaea fermentalgiana]; a Haptophyte (Chrysochromulina tobinii), a Rhizarium (Plasmodiophora brassicae); green plants (Viridiplantae) [Ostreococcus tauri, Klebsormidium nitens, Physcomitrella patens, Selaginella moellendorffii and Marchantia polymorpha]; Amoebazoa (Planoprotostelium fungivorum, Heterostelium album, Dictyostelium purpureum and Polysphondylium violaceum); a Heterolobosea (Naegleria gruberi); the Apuzosoan Thecamonas trahens; fungi (Glomus cerebriforme, Lobosporangium transversal, Syncephalastrum race-mosum, Neocallimastix californiae, Rhizclosmatium globosum and Spizellomyces sp. ‘palustris); the Filasterean Capsaspora owczarzaki; the Choanoglagellates Monosiga brevicollis and Salpingoeca rosetta as well as the metazoa Capitella teleta (Anellid worm), Scorpion (Centruroides sculpturatus), the horseshoe crab (Limulus polyphemus); tick (Ixodes scapularis); spider (Parasteatoda tepidariorum); barnacle (Amphibalanus amphitrite), amphipod (Hyalella azteca), mudcrab (Scylla olivacea), shrimp (Penaeus vannamei), the flatworm Macrostomum lignano, the trematode Opisthorchis viverrini and the cestode Hymenolepis microstoma. Also included was the “putative archaeon” sequence derived from sequences RYG70459.1, RYG70450.1, RYG70451.1 and RYG70452.1, see Supplementary Fig. 9 and text for details. Also see Supplementary Table 1 for taxonomy and additional details.
Panel B: Phylogenetic tree of DNA-PKcs in a variety of metazoan with fungal DNA-PKcs (Glomus cerebriforme) as the outgroup. DNA-PKcs sequences analyzed were from a Choanoflagellate (Salpingoeca rosetta), a rotifer (Brachionus plicatilis), the deep-sea annelid worm Alvinella pompejana, the annelid worm Capitella teleta, the brachiopod Lingula metazoa, the molluscs Octopus bimaculoides and Lottia gigantea (limpet), Pomacea canaliculate, (snail), Aplysia californica, Mizuhopecten yessoensis as well as sharks Chiloscyllium punctum, brown-banded bamboo shark (bshark) and elephant shark Callorhinchus milii (eshark), coelacanth (Latimeria chalumnae), xenopus (Xenopus laevis), platypus (Ornitho-rhynchus anatinus), alligator (Alligator sinensis), canary (Serinus canaria), sterlet (Acipenser ruthenus, fish), Zebrafish (Danio rerio), gar fish (Lepisosteus oculatus), trichoplax (Placazoa), hydra (Hydra vulgaris), sponge (Amphimedo queenslandia), softcoral (Dendronephthya gigantea), anemone (Nematostella vectensis), stonycoral (Stylophora pistillata), lancelet (Branchiostoma belcheri), starfish (Acanthaster planci), seacucumber (Apostichopus japonicus), seaurchin (Strongylocentrotus purpuratus), hemichordate (Saccoglossus kowalevskii, Acorn worm), priapula (Priapulus caudatus, worm), scorpion (Centruroides sculpturatus), spider (Parasteatoda tepidariorum), limulus (Limulus polyphemus, horseshoe crab), tick (Ixodes scapularis), trichinella (Trichinella pseudospiralis, parasitic roundworm), tardigrade (Hypsibius dujardini), ciona (Ciona intestinalis, tunicate), platyhelminth (Macrostomum lignano), barnacle (Amphibalanus amphitrite), amphipod (Hyalella aetazo), shrimp (Penaeus vannamei), mudcrab (Scylla olivacea), flea (Ctenocephalides felis), midge (Clunio marinus), mosquito Aedes aegypti, beetle (Tribolium castaneum), bee (Bombus impatiens), ant (Monomorium pharaonis), termite (Cryptotermes secundus), bug (Laodelphax striatellus), white fly (Bemisia tabaci), thrip (Frankliniella occidentalis), copepod (Eurytemora affinis), and daphia (Daphnia magna).
Unlike the Eukaryotic tree, the Metazoan tree bore little resemblance to the current classification (Telford et al., 2015) (Fig. 2B). While Sponges, Placozoa and Cnidaria are normally at the base of the tree, in the DNA-PKcs tree they occupy the apex and are replaced at the base by Ecdyzoan Protostomes, most prominently Insects and Crustaceans. The relationship of Sponges, Placozoa and Cnidaria to Deuterostomes (Echinoderms and Chordates) and Lophotrochozoan Protostomes (Molluscs and Annelids) is some-what more conventional. Also notable from Fig. 2B is the high level of conservation within vertebrates. Overall these findings indicate that DNA-PKcs appeared in early Eukaryotes, which evolved in the relatively stable ocean environment, and that it largely retained its core function(s) in Phyla that remained within this environment. However, in many organisms that adapted to life on land, DNA-PKcs was either lost or greatly diverged. The clear exception being vertebrates, where DNA-PKcs appears to have gained function.
3.3. DNA-PKcs sequence conservation in many major phyla including invertebrates and protists
We selected ~100 DNA-PKcs sequences from a broad representation of Eukaryotes and carried out four catagories of alignments: 1) jawed vertebrates, 2) Metazoa (vertebrates plus invertebrates), 3) selected Metazoa plus a fungus, a plant, an amoeba, a ciliate and an oomycete, and 4) 85 sequences representing the entire Eukaryotic spectrum. For each alignment except jawed vertebrates, we used both Clustal Omega and COBALT. At default settings, COBALT has low stringency for amino acid similarity while Clustal Omega has extremely high stringency, so that one mildly dissimilar or missing residue prevents any level of conservation from being indicated. For example, an alignment resulting in 19 glycines and 1 cysteine or 19 glycines and one gap will prevent any conservation being detected. Thus, many regions of high but not exclusive conservation may be missed using Clustal Omega alone. However, we posit that the combination of high stringency CLUSTAL and low stringency COBALT alignments provides a reasonable, overall view of amino acid conservation.
DNA-PKcs is required for V(D)J recombination, which is thought to have arisen abruptly in evolution by transposition of the RAG genes into jawed vertebrates (Market and Papavasiliou, 2003; Schluter et al., 1999). Therefore, we first aligned DNA-PKcs sequences from selected jawed vertebrates and analyzed their sequence similarity using Clustal Omega (Supplementary Table 1, Supplementary Fig. 1). The percent amino acid identity between full-length human and mouse DNA-PKcs was 79.3%, as determined by the Clustal Omega percent identity matrix function. Birds, reptiles and amphibians had ~67% amino acid identity to human while in sharks and fish the percent identity ranged from 58 to 62% (Supplementary Fig. 1A). Extensive regions of amino acid identity and similarity were observed throughout the sequence (Supplementary Fig. 1B). To visualize residue identity and similarity from the Clustal Omega alignment, we assigned identical amino acids (* in Clustal Omega) a score of 3, highly conserved amino acids (: in Clustal Omega), a score of 2 and similar amino acids (in Clustal Omega) a score of 1 and plotted these against residue number (Fig. 3A/B). These analyses reveal that DNA-PKcs is highly conserved across its entire length in vertebrates.
Fig. 3. Summary of identical and conserved amino acids from multiple sequence alignments.
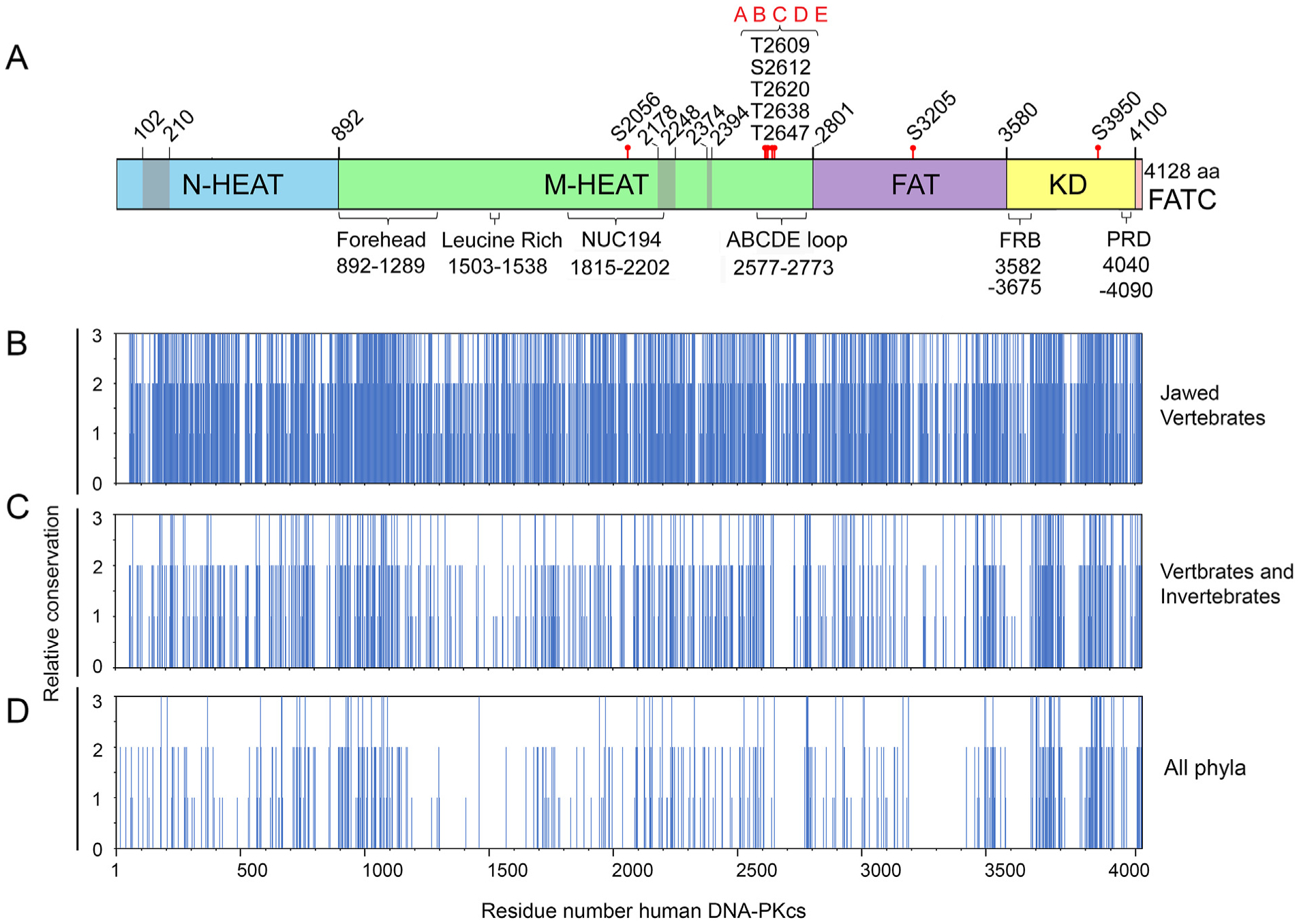
Panel A: Schematic of DNA-PKcs as in Fig. 1A showing location of the major domains and features. Panels B–D: Identical, conserved and semi-conserved amino acids from Clustal Omega alignments of (B) jawed vertebrates, (C) metazoa (vertebrates and invertebrates) and (D) representatives from all phyla. Complete alignments are shown in Suppl. Fig. 1, 2 and 4, respectively. The regions of amino acid identity and conservation from these alignments were plotted against amino acid number where 3 = amino acid identity (* in Clustal Omega alignments), 2 = amino acid conservation, (:) in Clustal Omega alignments and 3 = amino acid similarity (.) in Clustal Omega alignments.
In order to identify regions of DNA-PKcs that were conserved over a greater evolutionary distance, we carried out alignments on a greater diversity of species. For these alignments we included sequences from additional Metazoa including Chordates (Vertebrates, Tunicates and Lancelets), Annelids (worms), Cnidarians (sea anemones and corals), Echinoderms (starfish and sea urchins), Molluscs (snails and bivalves), Arthropods (Crustaceans, Insects and Chelicerates) and Porifera (Sponge). By Clustal Omega, the percentage amino acid identity in invertebrates (compared to human DNA-PKcs) was ~40% in Molluscs (oyster, Crassostrea virginica and snail Pomacea canaliculata), lancelet (Branchiostoma belcheri), starfish (Acanthaster planci), sea anemone (Nematostella vectensis) and corals (Stylophora pistillata and Dendronephthya gigantea), 35% in scorpion (Centruroides sculpturatus) and 30% in termite (Cryptotermes secundus) and shrimp (Penaeus vannamei) (Supplementary Fig. 2). Analysis by both Clustal Omega (Supplementary Fig. 2) and COBALT (Fig. 4A and B and Supplementary Fig. 3) again revealed multiple areas of amino acid conservation throughout the entire DNA-PKcs sequence, including the N-HEAT, Forehead, MHEAT, NUC194, FAT, kinase and FATC domains. However, some regions of DNA-PKcs were particularly prominent, such as the region in the Forehead domain corresponding to amino acids ~900–1200 in human DNA-PKcs that was highly conserved in all 33 sequences examined (Figs. 3C and 4A). However, there were also areas of high divergence, mainly within the M-HEAT and FAT domains (Figs. 3C and 4A and Supplementary Figs. 2 and 3).
Fig. 4. Amino acid conservation in DNA-PKcs from mammals to oomycetes.
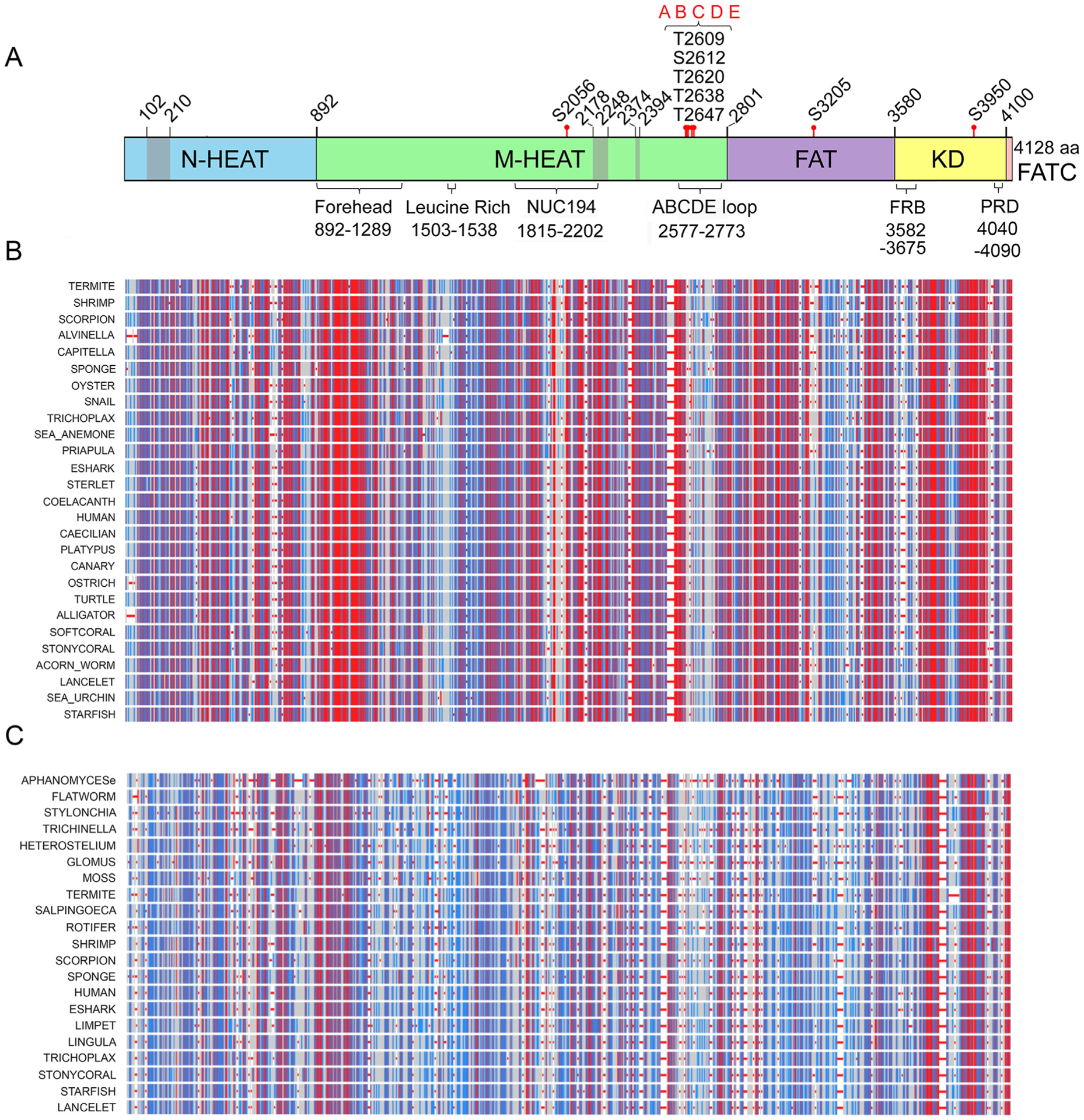
Panel A: Schematic of DNA-PKcs from Fig. 1A. Panel B: COBALT alignment of DNA-PKcs from a variety of vertebrate and invertebrate species, from human to sponge. Highly conserved amino acids are shown in red, moderately conserved in blue and non-conserved in grey. It should be noted that the location of conserved regions from the COBALT alignment relative to the schematic is only approximate as gaps may have been introduced into the COBALT alignments. The full alignment is shown in Supplementary Fig. 3.
Panel C: DNA-PKcs sequences from representative vertebrates, invertebrates, plants and fungi (Supplementary Fig. 5) were aligned using COBALT. Red, blue and grey coloration is as in panel B.
Closer examination of the alignments revealed that the sequence YR-x-G-(D/E)-(L/F)-PD-(I/V)-x-I-x5-I-x-P-x-Q beginning at tyrosine 2775 and ending at glutamine 2795 in the human sequence, was present in all 33 metazoans examined. In the sequence above, the residues in bold were identical in all 33 sequences examined, x = any amino acid and (D/E), (L/F) and (I/V) refer to conservative replacements (Supplementary Figs. 2 and 3). This motif, which we refer to as the YRPD motif, is located between the disordered loop (residues 2577 to 2773) that contains the ABCDE phosphorylation sites and is absent from the crystal and cryoEM structures, and the FAT domain (starting at residue 2801) (see Fig. 1A). There was also a second YR sequence at tyrosine 2772 and arginine 2773 which was conserved in all organisms except termite where the tyrosine was replaced by phenylalanine. Thus the YRPD motif could extend to Y2772 in some organisms, i.e. (Y/F)-R-x-YR-x-G-x-x-PD.
To further test our analysis on invertebrates, we examined and identified DNA-PKcs in the extremophile, Alvinella pompejana, a deep-sea polychaete worm found at hydrothermal vents in the Pacific Ocean (Shin et al., 2009). The amino acid sequence of A. pomejana DNA-PKcs aligned well with that of other metazoans and had 38% identity to human DNA-PKcs (ALVINELLA in Fig. 4A, Supplementary Figs. 2 and 3). Conservation in the Forehead domain and YRPD motif was also observed in A. pompejana (Supplementary Figs. 2 and 3).
We next extended our analysis to include representatives from more distantly related organisms including a brachiopod (Lingula antatina), rotifer (Brachionus plicatilis), roundworm (Trichinella pseudospiralis), flatworm (Macrostomum lignano), Choanoflagellate (Salpingoeca rosetta), fungus (Glomus cerebriforme), amoeba (Heterostelium album), ciliate (Stylonychia lemnae), oomycete (Aphanomyces euteiches), and the moss (Physcomitrella patens). Analyses by both COBALT and Clustal Omega showed that amino acids within the Forehead domain were again highly conserved, as were smaller regions throughout the HEAT and the kinase domain (Figs. 3D and 4C and Supplementary Figs. 4 and 5). In the Forehead domain, R924, A930, E932, G943, Q990, R1026, H1069, R1075 and R1090 were invariant from humans to oomycetes. At the YR-YRxGxxPD motif, the first Y (corresponding to Y2772 in humans) was not highly conserved (substitutions M, L, F or R). The R2773 was conserved in all except the parasitic roundworm, Trichinella. Remarkably, the second YR (equivalent to human Y2775/R2776), as well as the residues equivalent to human G2778, P2781 and D2782 were identical in all organisms in our alignment, from humans to amoeba and oomycete. Although the residues equivalent to D2779, I2780 and I2783 were not identical, they were replaced by highly similar amino acids supporting the notion that this entire region is important for DNA-PKcs structure and/or function (Fig. 5A and Supplementary Figs. 4 and 5).
Fig. 5. Amino acid conservation in the YRPD motif, ABCDE loop and kinase domain of DNA-PKcs.
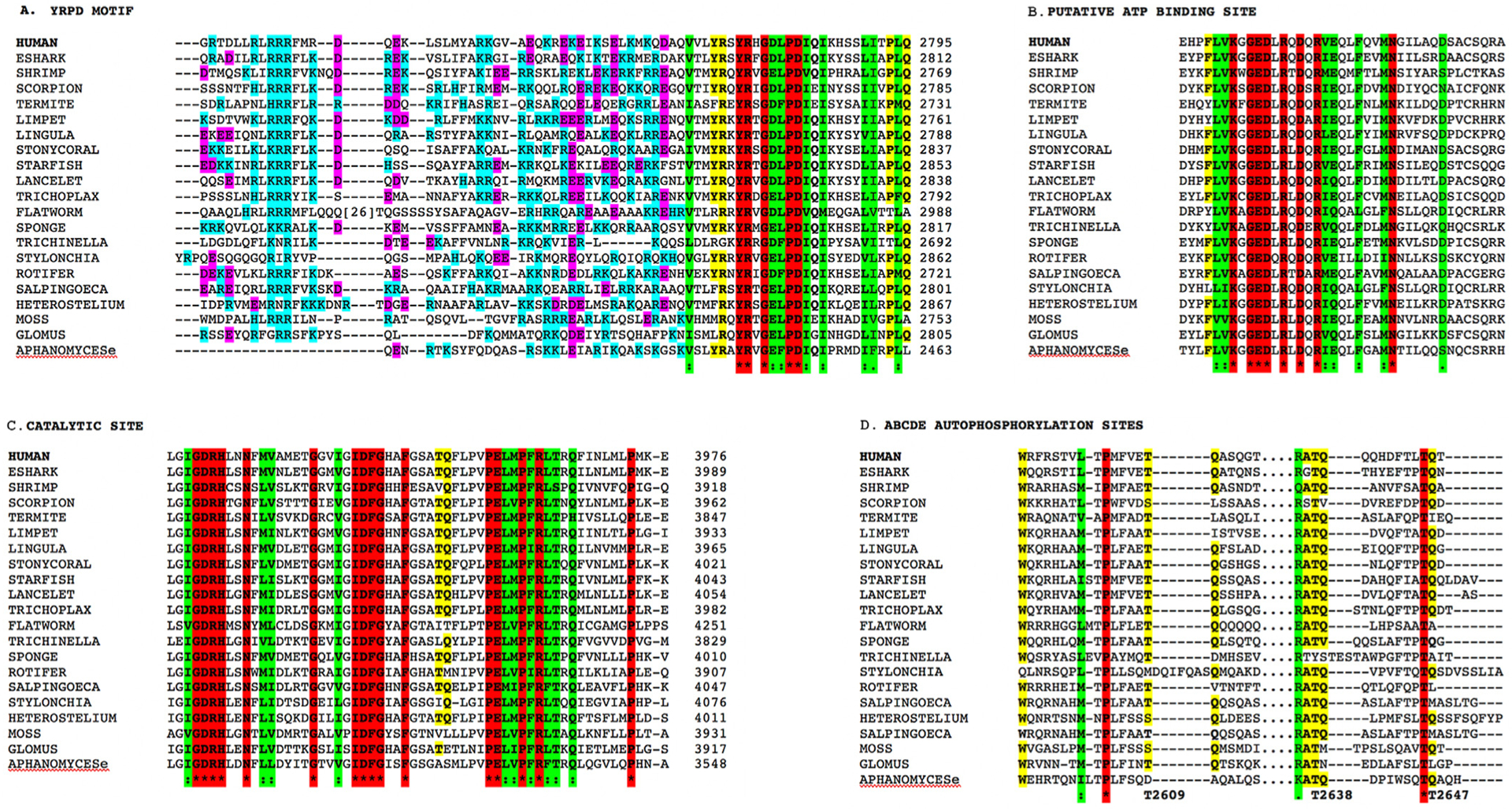
Panel A: Alignment of C-terminal portion of the ABCDE loop from representatives from all major phyla, showing the location of the basic patch and the YRPD motif. Basic amino acids (K, R and H) are shown in teal and acidic amino acids (D and E) in magenta. Identical amino acids are indicated by the * and are highlighted in red. Highly conserved amino acids/conservative substitutions are highlighted in green and partially conserved amino acids are highlight in yellow.
Panel B: Conservation of ATP binding site in representative sequences from all major phyla. Colours are as in panel A.
Panel C: Conservation of DXXXXN and DFG sequences in the catalytic site in representative sequences from all major phyla. Colours are as in panel A.
Panel D: Alignment of DNA-PKcs autophosphorylation sites 2609, 2638 and 2647 from all major phyla (Supplementary Fig. 4). Colours are as in panel A. The organisms shown are as follows: HUMAN; ESHARK = Callorhinchus milii, elephant shark; SHRIMP = Penaeus vannamei; SCORPION = Centruroides sculpturatus; TERMITE = Cryptotermes secundu; LIMPET = Lottia gigantea; LINGULA = Lingula anatina; STONYCORAL = Stylophora pistillata; STARFISH = Acanthaster planci; LANCELET = Branchiostoma belcheri; TRICHOLPAX = placazoa; FLATWORM = Macrostomum ligna; TRICHINELLA = Trichinella pseudospiralis, a parasitic roundworm; SPONGE Amphimedo queenslandia; ROTIFER = Brachionus plicatilis; SALPINGOECA = Salpingoeca rosetta, a choanoflagellate; STYLONCHIA = Stylonychia lemnae, a ciliate; HETEROSTELIUM = Heterostelium album, an amoeba; MOSS = Physcomitrella patens; GLOMUS = Glomus cerebriforme, a fungus; and APHANOMYCESe = Aphanomyces euteiches, an oomycte. See Supplementary Table 1 and Supplementary Fig. 4 for details.
Notably, the region immediately N-terminal to the YRPD motif contained a high percentage of charged amino acids, particularly positively charged residues R, K and H (Fig. 5A). There were also several invariant amino acids at the beginning of the ABCDE loop, including L2581 and P2604 (Supplementary Figs. 4 and 5). In the kinase domain, invariant amino acids included L3680, W3686, E3700, P3702, G3703, Y3705, P3711 and P3735. There was also a high degree of conservation in the ATP-binding site, beginning at K3753, at the catalytic site GDRHxxN, the metal binding site IDFG and at D4113, L4117, G4123 and W4124 in the FATC domain (Fig. 5B and C and Supplementary Figs. 4 and 5).
Even when the alignment was expanded to include 85 curated sequences across all major phyla, clear areas of amino acid conservation remained (Supplementary Figs. 6 and 7). In the Forehead domain, E932 and G943 were identical in all 85 organisms examined (Supplementary Figs. 6 and 7). Remarkably, the residues equivalent to Y2775, G2778, P2781 and D2782 of the YRPD motif were invariant in all 85 species examined including plants, amoeba and ciliates (Supplementary Figs. 6 and 7). W3008 in the FAT domain and P3702, P3745, K3753 (ATP-binding site), D3761, T3790, P3795, G3921, D3922, R3923, H3924, N3927 (DXXXXN), G3935, D3941 (DFG motif), F3946, E3957, R3962 and F4005 in the kinase domain as well as W4124 in the FATC domain were invariant in all 85 species.
Although several insects were included and shared a similar pattern of conservation to other organisms in our analysis, Lepidoptera seemed to display distinct characteristics. We aligned putative DNA-PKcs sequences from five Lepidopteran superfamilies, including 7 distinct families, with a diverse group of Metazoans (Supplementary Fig. 8). The only moderately conserved features were the N-HEAT, the M-HEAT from 1550 to 2650 (which includes the NUC194 domain) and the FAT-kinase-FATC domains. The kinase domain contained the essential residues predicted for activity, but overall the sequences were too divergent to be identified with confidence as DNA-PKcs. Notably absent from all Lepidopteran sequences examined were the Forehead domain and the YRPD (Supplementary Fig. 8).
3.4. Unique identifiers of DNA-PKcs
To identify regions that were unique to DNA-PKcs, we carried out a Blastp search comparing the various regions across a wide variety of Eukaryotic organisms. The regions included in the Blastp analysis were the N-HEAT (residues 1–892), the Forehead domain (residues 893–1200), two regions of the M-HEAT domain, residues 1200–1700 and residues 1700–2576 that contain the NUC194 domain, the ABCDE loop region (residues 2550–2820), the FAT domain (residues 2800–3580) and the kinase/FATC domains (residues 3580–4128). The FAT and especially the kinase/FATC domains were conserved in DNA-PKcs from all Eukaryotic phlya but also shared conservation with other PIKKs including TOR, SMG1, ATR, ATM and TRRAP (Supplementary Table 2). The N-HEAT, the Forehead, the region of the M-HEAT containing the NUC194 domain (1700–2576) and the ABCDE loop region all identified DNA-PKcs with a Blast score >30 but none of the other PIKKs were identified, indicating that these regions are unique identifiers for DNA-PKcs. Amongst these unique identifiers, the Forehead domain showed the greatest conservation over the broad range of Eukaryotes. The region of the ABCDE loop encompassing residues 2550–2820 had less overall conservation, but was still specific for DNA-PKcs compared to other PIKKs. The M-HEAT domain from 1200 to 1700 was not well conserved outside Metazoa. An exception to these observations was the Mormon butterfly, Papilio polytes. Its putative DNA-PKcs sequence showed similarity to only one of the unique DNA-PKcs identifiers - the M-HEAT region containing the NUC194 domain. Interestingly, its divergent kinase domain was more similar to TOR than DNA-PKcs, suggesting it could be a unique Lepidopteran PIKK of unknown evolutionary origin.
3.5. Evaluation of metagenomic project sequences
When searching the NCBI database for proteins containing the NUC194 domain, we found a scaffold that was binned as a putative archaeon (hypothetical protein EON64_00075 [archaeon]) within the poplar leaf phyllosphere (Crombie et al., 2018). The scaffold was predicted to contain 4 separate genes, RYG70452.1 which aligned with the N-terminal region of DNA-PKcs; RYG70451.1 with the Forehead domain; RYG70450.1 with the NUC194 domain and RYG70459.1 with the kinase domain. When compiled as one, the putative archaeon sequence retained over 20% amino acid identity to human DNA-PKcs (Supplementary Fig. 9) with 141 amino acids identical between it and human DNA-PKcs. The YRPD motif was also conserved in the reported sequence of this organism. By COBALT analysis, the putative archaeon DNA-PKcs had a similar pattern of conservation as other Metazoans, and phlyogenetic analysis grouped it with the Stramenopiles Nannochloropsis salina and Ectocarpus siliculosus, suggesting the possibility of a eukaryotic origin (Fig. 2A). We did not detect sequences similar to DNA-PKcs in the Archaeal taxid of the NCBI database, and, using the eukaryotic gene prediction program (AUGUSTUS), found that the four fragments could be spliced together, yielding a sequence with greater similarity to other orthologues, further supporting the possibility that these putative Archaeon sequences might actually be from a Eukaryote.
3.6. Conservation of phosphorylation sites
As described, DNA-PKcs autophosphorylation is critical for its function in DSB repair and V(D)J recombination. We therefore examined the amino acid alignments for conservation of ABCDE (T2609, T2638 and T2647), PQR (S2056), S3205 and T3950 phosphorylation sites. Serine 2056 and glutamine 2057 (SQ) were highly conserved in jawed vertebrates but less so in invertebrates and other phyla (Supplementary Figs. 2 and 6). Interestingly, an SQ site at 2041/2042 was identical in all vertebrates, as were several other serines and threonines in the vicinity, including S2046, T2047 and S2053, however, whether these residues are phosphorylated and/or functionally important in vivo remains to be determined. In contrast, T2609 and glutamine 2610 were conserved in most organisms, though in some, the threonine was replaced by serine. In several cases glutamine 2610 was substituted for leucine, but since DNA-PKcs is known to phosphorylate substrates at S/T followed by L, M and other aliphatic amino acids (Dobbs et al., 2010), these could still be DNA-PK-dependent phosphorylation sites. Similarly, T2638/Q2639 and T2647/Q2648 were highly conserved over a wide range of invertebrates, amoeba and oomycetes (Fig. 5 and Supplementary Figs. 2 and 6). SQ/TQ motifs at 2612, 2620 and 3950 were highly conserved in vertebrates and some invertebrates, while S3205 (SM) was not well represented outside vertebrates. Thus, in general, S2056 and S3205 were primarily present in vertebrates, while T2609, T3950 and, in particular T2638 and T2647 were more widely conserved, including in some insects, coral, fungi, plants and amoeba (Fig. 5D and Supplementary Figs. 4 and 6). The different level of conservation between the autophosphorylation sites suggests that these sites may have discrete functional consequences, with 2056 and 3205 having functions in vertebrates while T2609, T3950, T2638 and T2647 may have functions that are conserved throughout evolution.
3.7. Location of conserved residues within the structure of DNA-PKcs
We next mapped the regions of conservation between metazoa (vertebrates and invertebrates, Supplementary Fig. 3) and all phyla (Supplementary Fig. 6), onto the structure of human DNA-PKcs. Strikingly, in metazoa, there was a highly conserved patch of amino acids at the top of the solenoid, beneath the FAT/kinase domains corresponding to residues R924, A930, E932 and K983 from the Forehead domain and the YRPD motif, thus these two regions may form an extended patch of amino acid conservation (Fig. 6A). Also conserved was the NUC194 domain at the base of the ring and the kinase domain at the top (shown in red in Fig. 6A). Furthermore, rotation of the structure revealed that these regions of conservation were almost entirely located on the “front” face of DNA-PKcs, around the concave ring (Fig. 6A). These areas of conservation were observed even when sequences from all major phyla were examined (Fig. 6B). Again, regions of conservation on the front face of DNA-PKcs were particularly evident in the side view, with the YRPD and NUC194 domains particularly prominent (red in Fig. 6B).
Fig. 6. Location of highly conserved amino acids in DNA-PKcs from selected metazoans on the structure of human DNA-PKcs.
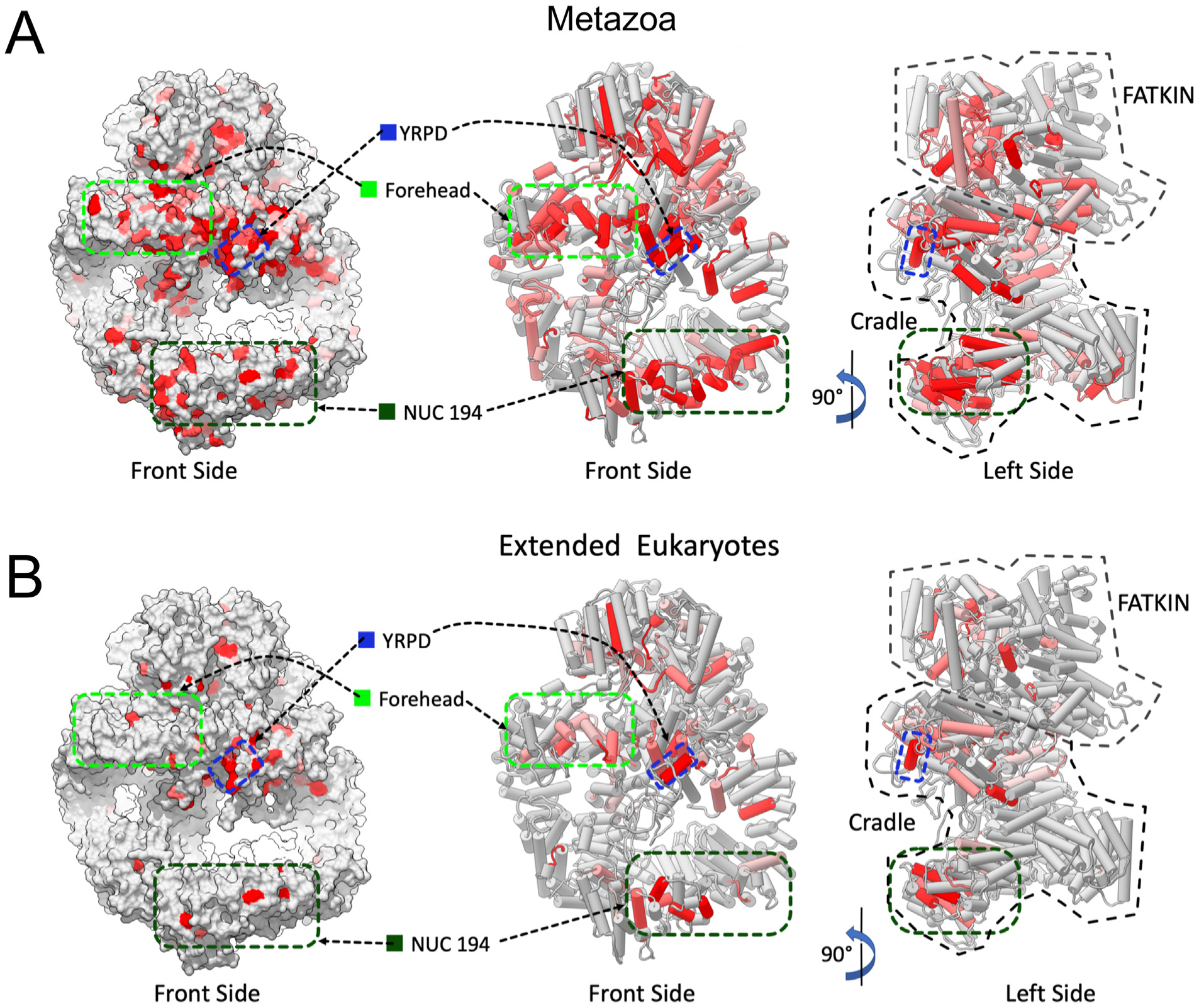
Panel A: Location of the highly conserved regions in DNA-PKcs from alignment of selected metazoans on the structure of human DNA-PKcs. Amino acids that were identical and/or highly conserved (>95%) in all vertebrate/invertebrates (Supplementary Fig. 2) were mapped onto the molecular surface (left panel) and two orthogonal views of the structure of human DNA-PKcs (PDB 5LUQ from (Sibanda et al., 2017)) are shown in pipes and planks representation (right panel). Regions containing identical or conserved amino acids are highlighted in red and light red respectively. The highly conserved region underneath the FAT-KINASE-FATC (Forehead) is indicated by the light green hatched box, the YRPD motif is indicated by the dark blue hatched box and the NUC194 domain by the dark green hatched box.
Panel B: Location of highly conserved regions in DNA-PKcs in vertebrates, invertebrates, plants and fungi (Supplementary Fig. 6) are shown as in panel A. The locations of the forehead domain, the YRPD helix and the NUC194 domain are shown by the light green, blue and dark green hatched boxes, respectively as in panel A.
Of particular interest was the conserved (YR)xYRxGxxPD motif. Residues Y2772 and R2773 in the first YR are part of the ABCDE loop (residues 2577–2773) and are not present in the cryo-EM or crystal structures. However, the conserved YRxGxxPD sequence beginning at Y2775 marked the beginning of a short alpha helix that protrudes from the front face of DNA-PKcs, at the top of the solenoid ring (dark blue in Fig. 7) and terminates with proline 2582. Thus, the YRPD motif connects the C-terminal end of the ABCDE loop to the M-HEAT domain, close to the start of the FAT domain: a position that could play a major role in regulation of the ABCDE loop and or allosteric interactions with the FAT-kinase-FATC domain (Fig. 7).
Fig. 7. Models of monomeric and dimeric DNA-PKcs showing the positions of the YRPD motif and the ABCDE loop.
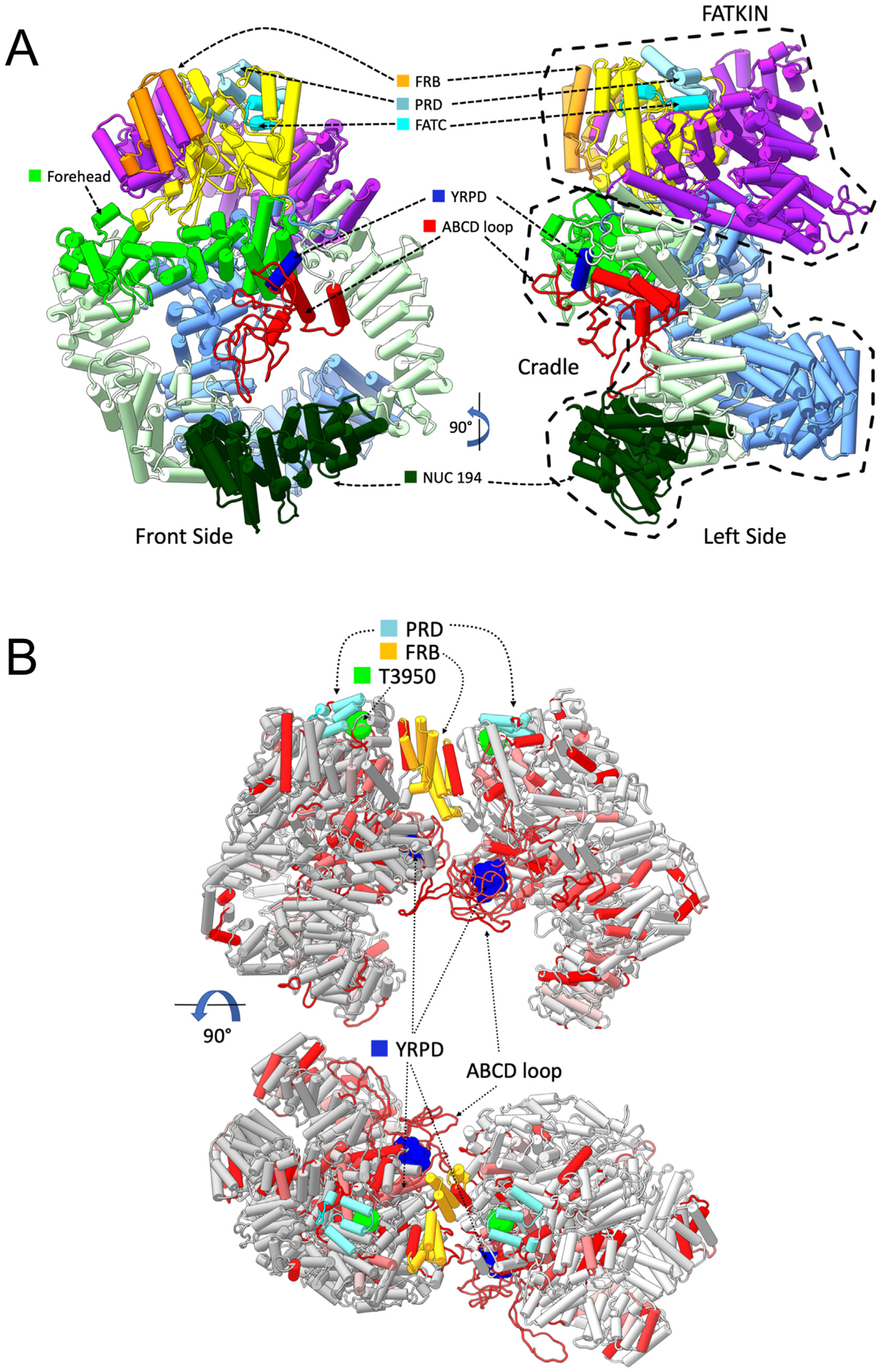
Panel A: DNA-PKcs model with added missing regions shown in pipes and planks representation. The model of DNA-PKcs was built based on the X-ray structure (PDB:5LUQ from (Sibanda et al., 2017)) by adding missing loops using MODELLER (Sali and Blundell, 1993), including the ABCDE loop region (red). Major domain in DNA-PKcs are coloured as highlighted, where the N-HEAT domain is shown in blue, the M-HEAT in green, the FAT in purple, the kinase in yellow and the FATC in bright light blue. Also shown is the location of the NUC194 domain (dark green) and the Forehead domain (light, bright green). The added disordered loop containing the ABCDE sites is shown in red. The conserved YRxGxxPD sequence beginning at Y2776 is shown in dark blue. Also shown are the FRB (orange) and PRD (grey/blue) motifs.
Panel B: The locations of highly conserved regions from selected metazoa were mapped onto the DNA-PKcs dimer model (see Methods and Hammel et al., submitted, this issue). Amino acids that were identical and/or highly conserved (>95%) in all vertebrate/invertebrates examined (Supplementary Fig. 2) were mapped onto two orthogonal views of dimer models shown in pipes and planks representation. Regions containing identical or conserved regions are coloured in red and light red, respectively. The highly conserved YRPD residues (dark blue) are shown in sphere representation. Phosphorylation site threonine 3950 is in bright green, the FRB in orange and the PRD in grey/blue. The position of the ABCDE loop is shown in red.
3.8. Modeling the position of the YRPD helix and ABCDE loop in a DNA-PKcs synaptic complex
In NHEJ, two DNA-PK holoenzymes assemble on either side of the DSB to form a synaptic complex (Wang et al., 2018). Biochemical studies suggest that Ku70/80 binds the DNA ends then translocates inward, allowing DNA-PKcs to interact with the DSB ends (Yoo and Dynan, 1999). In this scenario, two DNA-PKcs molecules would therefore be predicted to interact across the synapse. Although structures of NHEJ synaptic complexes have not been determined, DNA-PKcs can self-associate to form a dimer at high protein concentration (Hammel et al., 2010). In this special issue, Hammel and colleagues used small-angle-X-ray scattering (SAXS), atomistic modeling and X-ray structures of DNA-PKcs (Sibanda et al., 2017) to model the interactions within the self-associating DNA-PKcs dimer, and used this model to predict the structure of a DNA-PKcs-Ku-DSB synaptic complex (Hammel et al., 2020, this issue). By building on this SAXS model, we predicted the position of the YRPD motif and the ABCDE loop in the synaptic complex (Fig. 7B). This combined SAXS-EM model predicts that two DNA-PKcs molecules interact such that the kinase domain of one monomer faces the kinase domain of the opposite monomer. Within this complex, the Forehead domains face each other but are offset, predicting that the YRPD helices and ABCDE loops on DNA-PKcs molecules on opposite sites of the break could be in close proximity (Fig. 6D).
3.9. Defining the evolutionary importance of DNA-PKcs residues
To determine the evolutionary importance of the DNA-PKcs residues, identify functional sites, and observe how the evolutionary pressure differs in branches of the phylogenetic tree, we used the Evolutionary Trace (ET) algorithm (Lichtarge et al., 1996; Mihalek et al., 2004). ET measures how protein residues vary with respect to the phylogenetic tree and ranks each residue on a scale from 0 to 100 that indicates in that order an increasingly weaker relative functional sensitivity to mutations. As such, ET has been able to guide experimental studies to identify new functional sites in proteins (Adikesavan et al., 2011) or allosteric pathways (Rodriguez et al., 2010), to predict substrate specificity (Amin et al., 2013), to help interpret variant effects (Katsonis and Lichtarge, 2014, 2019; Neskey et al., 2015), and to associate genes to disease (Clarke et al., 2019). Using as input for the ET algorithm the Clustal alignments of homologous sequences from jawed vertebrates (Supplementary Fig. 1), selected vertebrate and invertebrates (human to sponge) (Supplementary Fig. 2), we obtained six sets of ET ranks that were strongly correlated to each other (the Pearson’s correlation coefficient ranged from 0.74 to 0.9996 with a media value of 0.89). We focused next on the ET result obtained for sequence alignment for Metazoa (human to sponge), including the putative archaeon sequence (Supplementary Fig. 9) because it provided deeper information of the DNA-PKcs evolution and overall it yielded the best correlation to the other ET results. From the three available pdb structures (PDB IDs: 5y3r, 5w1r, and 5lug), we decided to only use the 5y3r, because a structure alignment of all three showed a perfect overlap, except for some local flexibility when DNA and the XRCC6/XRCC5 (Ku70/80) proteins were bound (in structure pdb 5y3r), which may be biologically relevant.
To interpret the ET outcome in terms of structure and function, the ET ranks were mapped onto chain C of the 5y3r pdb structure, using a color-code palette ranging from red for the most important residues (ET rank = 0) to green for the least important residues (ET rank = 100), as shown in Fig. 8A. (The ABCDE loop is not shown in this analysis as it is not visible in the crystal or cryo-EM structures). Typically and as observed here, the most important sequence positions tend to cluster structurally, precisely at the location of functional sites on the surface, or at structurally, dynamically and allosterically important sites in the structure core, and the quality of an evolutionary trace analysis is correlated with the quality of this clustering (Madabushi et al., 2002; Mihalek et al., 2006). Here, the top 30% of the residues according to the ET ranks clustered with a z-score of 22.8, indicating a very reliable analysis.
Fig. 8. Evolutionary Trace analysis of the DNA-PKcs protein family.
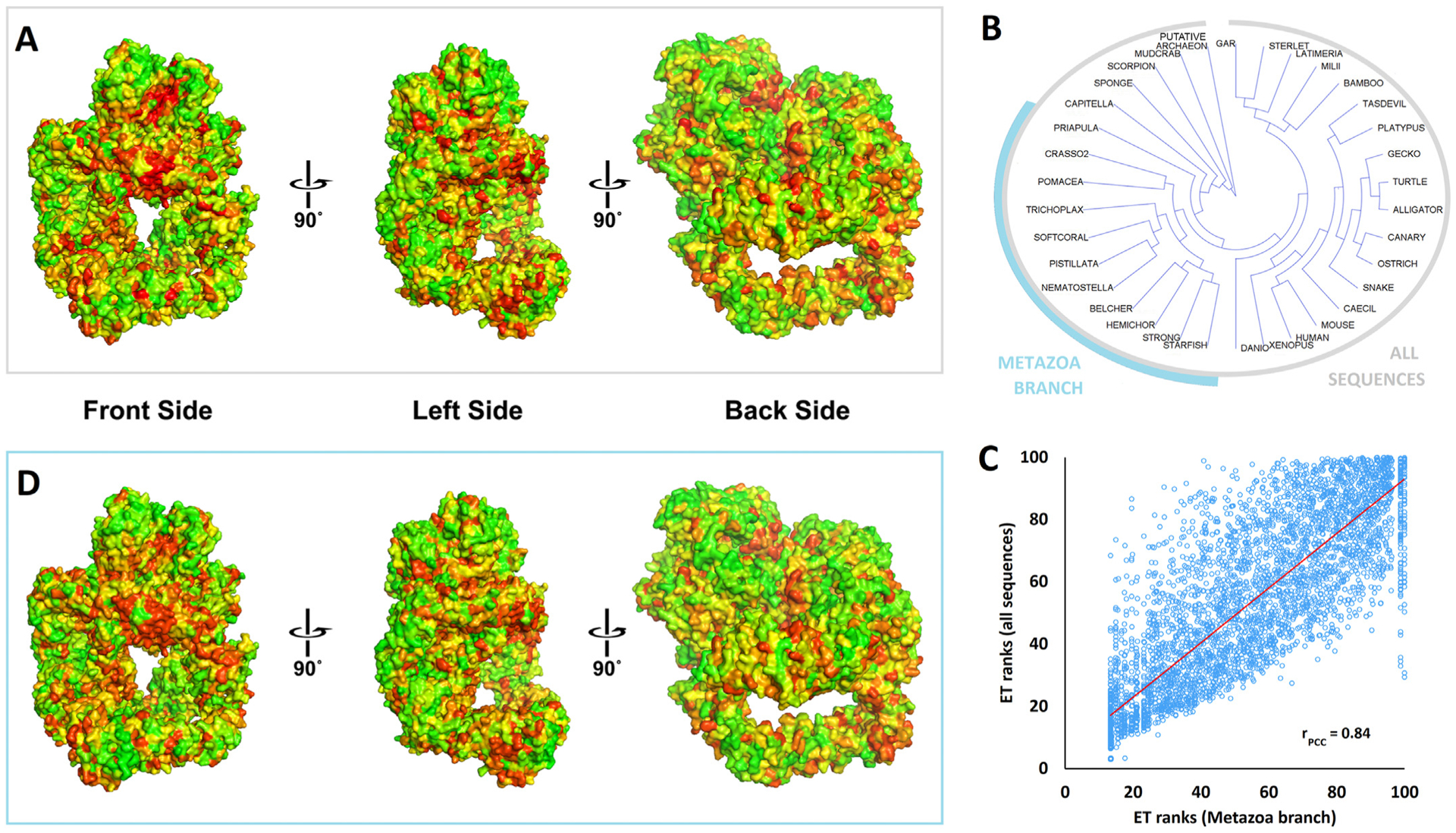
Panel A: The evolutionary importance of the DNA-PKcs residues was estimated by the Evolutionary Trace (ET) algorithm using as input the sequences alignment of Supplementary Fig. 9 (selected metazoan/vertebrates plus invertebrates and putative archaeon). The results are represented by a color scale from red (most important) to green (least important). The three viewpoints differ for about 90° and show the FAT-kinase-FATC domain (crown) on the top.
Panel B: The phylogenetic tree of the sequences found in Supplementary Fig. 9. A branch of closely related metazoan sequences was highlighted cyan. The phylogenetic tree was output of the ET algorithm and it was visualized using Archaeopteryx (version 0.9901).
Panel C: The correlation of ET ranks generated for all sequences shown in panel B versus the ET ranks generated for the metazoan branch highlighted in panel B. The Pearson’s correlation coefficient r was calculated to be 0.84.
Panel D: The ET ranks of the DNA-PKcs residues using as input the sequence alignment of the metazoan branch highlighted in panel B (same color scale and viewpoints as in panel A).
The figure panels A and D were generated by PyMol, using the PyETV plugin (Lua and Lichtarge, 2010) and chain C of the 5y3r pdb structure.
Strikingly, the color distribution and gradients showed only minor differences across all the different ET ranks maps, including ET rank maps that correspond to specific branches of the phylogenetic tree. E.g., focusing on the metazoan branch shown on Fig. 8B, the ET ranks strongly correlated to the ET ranks of the whole alignment with a Person’s correlation coefficient of 0.84 (Fig. 8C). The ET ranks map is very similar to the one obtained using all sequences, identifying the same 3D functional sites with some fluctuations on their intensity and boundaries (Fig. 8D). These data show the high quality of the analysis and moreover that the evolutionary important sites on the three-dimensional structure do not vary appreciably during DNA-PKcs evolution. In other words, the ET analysis of these alignments suggests that the underlying sequences all perform their functions at similar structural locations.
3.10. Alterations in the PRKDC gene impact cancer and human inherited disease
Tumorigenesis entails a profound reprogramming of gene expression and the acquisition of somatic mutations that in many cases alter protein function and enforces a substantial mutational burden (Bacolla et al., 2019; Hanahan and Weinberg, 2011). To assess the contribution of alterations in the PRKDC gene to cancer, we first compared its expression profile in the tumors relative to matched control tissues from the same patients from The Cancer Genome Atlas (TCGA), a vast repository of cancer genomic data from >11000 patients representing 33 tumor types. Strikingly, in 15 tumor types for which matched controls were available, PRKDC mRNA levels were upregulated in 10 of them (75%) and down-regulated in only 3 (20%) (Fig. 9A). Thus, PRKDC is often overex-pressed in tumors. The Kaplan-Meier estimator and Cox proportional regression (hazard ratio) are statistical methods that evaluate the impact of a variable on survival. We applied these analyses to inform whether patients that express PRKDC mRNA at higher levels in the tumor (above the mean for all patients for a given tumor type) would incur greater risk than those with lower mRNA levels (below the mean). In 8/33 (~25%) tumor types, higher PRKDC expression was a significant risk factor associated with shorter overall survival time (Fig. 9B and C).
Fig. 9. Alterations in the PRKDC gene impact cancer and human inherited disease.
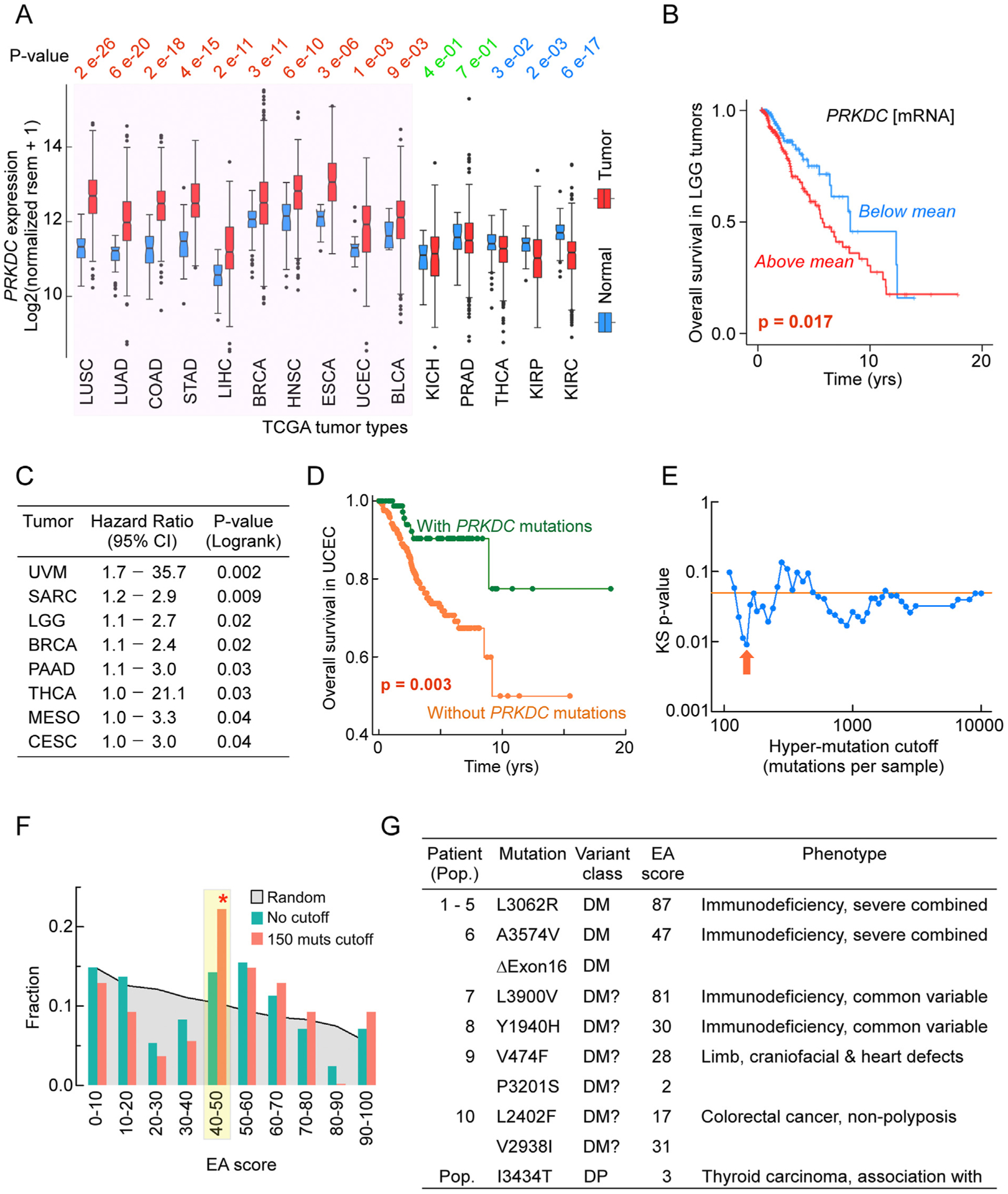
Panel A: PRKDC mRNA levels in tumor types and matched normal control tissues from TCGA. P-values from Wilcoxon tests; red, higher levels in the tumors than in controls; green, no difference between tumors and controls; blue, higher levels in controls than in tumors.
Panel B: Kaplan-Meier survival curve in LGG tumors for patients stratifies in two groups: group 1 with PRKDC mRNA levels above the mean (red) and group 2 with PRKDC mRNA levels below or at the mean (blue) in the tumor samples; p-value from log-rank test.
Panel C: Hazard ratios from Cox proportional hazards regression analysis for tumor types in which high (above the mean) PRKDC expression is associated with increased risk (decreased survival).
Panel D: Kaplan-Meier survival curve in uterine corpus endometrial carcinoma (UCEC) patients with (green) and without (orange) missense mutations in the PRKDC gene; p-value from log-rank test.
Panel E: KolmogoroveSmirnov test p-values for comparing the EA score distributions of PRKDC variants found in tumors to random nucleotide changes. This analysis used patients monitored for progression-free survival with one missense mutation in the PRKDC gene. P-values were obtained after limiting patients up to a varying total number of mutations (hyper-mutation cutoff). Arrow indicates the p-value for patients with ≤150 mutations.
Panel F: Fraction of occurrences in EA scores for PRKDC variants obtained by random nucleotide changes (grey shading) and in progression-free survival patients from Panel E without any applied hypermutation cutoff (dark green) or hypermutation cutoff at ≤150 (red arrow set, dark orange). Yellow highlight with red star, pronounced overrepresentation of intermediate EA scores.
Panel G: Germline PRKDC missense mutations (Mutation) reported to be associated with inherited disease in individual patients (Patient) or with susceptibility to cancer in the population (Pop). DM, Variant class and Phenotype, HGMD classifications; DM, disease-causing mutation; DM? possible pathologic mutation; DP, disease-causing polymorphism.
To address whether PRKDC missense mutations would also correlate with survival, we examined a total of 44,597 patients from 178 studies (https://www.cbioportal.org), 968 (2%) of which carried one or more mutations in the PRKDC gene. Overall survival was not impacted in the aggregate cohort (log-rank p-value 0.728); however, in endometrial carcinoma, subjects with PRKDC mutations exhibited a better outcome than those with wild-type PRKDC (Fig. 9D). A similar trend was observed for 6667 patients who were followed for disease-free survival (log-rank p-value 0.0182) and 11,085 patients who were monitored for progression-free survival (log-rank p-value 0.0235). Although the relationship between PRKDC alterations and patient survival is complex, these observations suggest that DNA-PKcs activity plays a role in tumor fitness and that DNA-PKcs inhibitors might offer useful therapeutic strategies in some cancers.
Next, we tested wether the PRKDC missense mutations found in these patients present any selection patterns, characteristic of cancer driver mutations. To do so, we calculated the Evolutionary Action (EA) scores for all possible single nucleotide changes that result in missense variants of PRKDC (random distribution) and compared them to the missense PRKDC variants found in patients (observed distribution). Including all observed variants yielded no different EA distribution than random variants (p-value of 0.91), suggesting that most of the observed changes are passenger mutations. To reduce the fraction of passenger PRKDC missense variants, we limited the observed variants to only those that came from tumors with a single PRKDC missense variant (since many appeared together with nonsense, frameshift, or more missense variants) and tumors with up to a maximum total number of variants (to avoid hypermutated tumors). This subset of PRKDC missense variants had significantly different distribution than random (p-value of 0.049), which became stronger (up to p-value of 0.009) as hypermutated tumors were left out (Fig. 9E). The difference between the observed and the random distributions was at the intermediate EA scores (40e70) and further increased for less conservative hypermutated tumor cutoffs (Fig. 9F). Similar over-representation of intermediate EA scores has been observed before (Clarke et al., 2019; Network, 2017) and indicates gain-of-function variants, suggesting PRKDC may act as oncogene in at least some of the cancer types.
The human gene mutation database (HGMD) is a collection of over 260,000 gene mutations reported to be associated with inherited human disease or predisposing to disease. Disease-causing mutations in PRKDC are rare (Fig. 9G), with only 10 patients reported to date. Of these, 5 cases of Turkish origin comprising two siblings shared a homozygous hypomorphic L3062R substitution in the DNA-PKcs FAT domain and presented with radiosensitive severe combined immunodeficiency (RS-SCID), which includes autoimmunity and granulomas (Esenboga et al., 2018; Mathieu et al., 2015; van der Burg et al., 2009). The mutation does not impair DNA-PKcs kinase activity, DNA-binding, autophosphorylation or interaction with Artemis, but it reduces end-joining activity and compromises the function of Artemis, which is also essential for V(D)J recombination.
Evolutionary Action (EA) is an effective computational method based on evolutionary tracing and mutation severity to score and identify point mutations that are likely to have negative impact (Neskey et al., 2015). Disease mutations with known negative impact generally score between 80 and 100, so any site mutation above 80 is likely to negatively impact the protein and its function(s). With a score of 87, our EA analysis confidently predicts that L3062R is a highly damaging substitution.
Patient 6 presented with SCID, complete absence of T and B cells, undetectable IgA and IgM levels, severe postnatal neuronal atrophies and poor developmental progress, hearing and visual impairment, and died at 31 months of age (Woodbine et al., 2013). Reduced DNA-PKcs protein level and no DNA-PK activity were caused by an inactivating PRKDC deletion on one allele and a hypomorphic A3574V substitution on the other allele, which our EA score suggests is moderately damaging. The clinical manifestations of patient 6 are revealing because they support a critical role for NHEJ factors in maintaining proper neuronal development, as seen in mice deficient of other NHEJ factors, including DNA Ligase 4 (Barnes et al., 1998) and Xrcc4 (Gao et al., 1998).
Patients 7 and 8 were identified in a study investigating the genetic landscape of common variable immunodeficiency disorders (CVID), a frequent defect in primary antibody production affecting 1 in 25,000 people (van Schouwenburg et al., 2015). Both patients were heterozygous for PRKDC variants, and our EA score suggests that both substitutions are at least partially damaging, raising the prospect that CVID may also ensue from DNA-PKcs haploinsufficiency. Patient 9 also was part of a broad cohort of cases, in this case sequenced for exome-wide variants after indications of fetal abnormalities following prenatal ultrasound (Carss et al., 2014). Post-mortem examination of the 22 weeks-old male fetus revealed extensive organ abnormalities of the heart, kidney, limbs, face and other organs while genotyping identified compound heterozygosity for PRKDC, HEPHL1, which regulates intracellular iron content and has been associated with rare developmental disorders, and ZNF44, a transcription factor. In addition, hemizygous variants were detected in BCORL1, a transcriptional corepressor linked to neurodevelopmental disorders, FAM47A, MAGEA6, ZCCHC12, a transcriptional coactivator, and a truncated KCNE5, a potassium channel ancillary subunit important for heart activity. Our EA score suggests a mild damaging effect for the two PRKDC variants, and thus the devastating phenotype may have been caused by the combined genetic variants.
Germline mutations in mismatch repair (MMR) genes have been linked to Lynch Syndrome, also known as hereditary nonpolyposis colorectal cancer (HNPCC), which is characterized by increased risk of developing colorectal (CRC), endometrial (EC) and other types of malignancies. In about half of HNPCC cases no germline mutations in MMR genes have been detected, and a comprehensive study has suggested that copy number variants of other non-MMR-associated genes may also predispose to HNPCC (Kayser et al., 2018). Two such cases involved PRKDC, one exhibiting a heterozygous deletion involving PRKDC and MCM4, the other carrying two missense variants with high combined annotation-dependent depletion (CADD) score and intermediate EA score (patient 10, Fig. 9G). Thus, it will be of interest to further explore the proposed link between germline-dependent DNA-PKcs dosage alterations and HNPCC susceptibility.
Other studies which focused on the contribution of PRKDC germline variants to susceptibility to cancer have concurred in attributing a high-risk factor for I3434T to a variety of cancer types, including papillary and follicular thyroid carcinomas (Rahimi et al., 2012) and colorectal cancer (Mehrzad et al., 2019) in the Iranian population. In summary, germline variants in PRKDC display a wide range of phenotypic manifestations in humans, from life-threatening to tolerated, and as additional studies are conducted it will be critical to relate the spectrum of PRKDC mutations to human disease, as this will inform on possible novel roles for this large and complex enzyme, thereby driving improved therapeutic interventions.
3.11. Location of disease causing mutations on the structure of the DNA-PK holoznzyme
Taking into account our new insights from multiple sequence alignment, ET and EA analysis, we next examined the location of the identified mutations and conserved features in the context of the DNA-PKcs-Ku-DNA holoenzyme (Yin et al., 2017) (Fig. 10, Supplemental Movies 1–3). In the cryoEM holoenzyme structure, Ku70/80 binds to the back face of DNA-PKcs and the Ku-bound DNA passes in between the N-HEAT (blue in Fig. 10A, Supplementary Movie 1) and the M-HEAT regions (green in Fig. 10A, Supplementary Movie 1) and emerges towards the front face of DNA-PKcs. The trajectory of the DNA goes along the bottom left of the largest DNA-PKcs cavity, underneath the YRPD helix, located at the top right of the cavity (Fig. 10A, Supplementary Movie 1). The YRPD helix (gold in Fig. 10A, Supplementary Movie 1) juts out from the regular helical topology of the HEAT repeats and interacts with residues distant in the primary sequence, visible when DNA-PKcs is rainbow coloured from the N- to C-terminus (Fig. 10A). We also mapped conserved amino acids (Supplementary Fig. 2) onto the structure of the DNA-PK holoenzyme where deep magenta is highly conserved, green partial conservation and grey not conserved (Fig. 10B). Notably, the YRPD signature sequence is part of the largest cluster of conserved residues on DNA-PKcs, even greater than the kinase domain where most conserved residues are located deep in the active site pocket or the Ku70/80 protein/protein interface (Fig. 10B, Supplemental Movie 2). The inset shows the high degree of conservation of Y2772, R2773, G2778, P2781 and D2782 (red) with S2774 in green (less well-conserved), as well as that of the underlying residues, suggesting that the exact position of the YRPD helix is important for DNA-PKcs function. The second largest cluster is directly below the YRPD, at the bottom of the largest cavity, corresponding to the NUC194 domain (red at base in Fig. 10B). Intriguingly, this second largest conserved cluster, on the side of six helices facing towards the cavity, would be positioned next to but not in the trajectory of the DNA. The juxtaposition of the two most conserved clusters is likely a clue to DNA-PKcs mechanisms. Third, mapping of the most and second lowest ET scores onto the structure indicates that the YRPD helix is as important as the kinase active site to DNA-PKcs function. These were the only two places that showed clustering of the lowest ET scoring residues. The YRPD surface cluster included Tyr2775, Arg2776, Gly2778, Pro2781, Asp2782, Ile2785 on the YRPD helix and nearby residues Gln924, Arg925, His935, Val979, Leu983, Glu930, Glu932 (corresponding to the Forehead domain), and Ser2569 (beginning of the ABCDE loop), and Ile2791, Pro2793, Gln2795 (immediately after the YRPD motif). In contrast, there was only one low scoring ET residue (Asn 2234) mapped in the DNA binding interface, and none to the Ku70/80 binding interface.
Fig. 10. Conservation of DNA-PKcs amino acids and motifs in the DNA-PK holoenzyme.
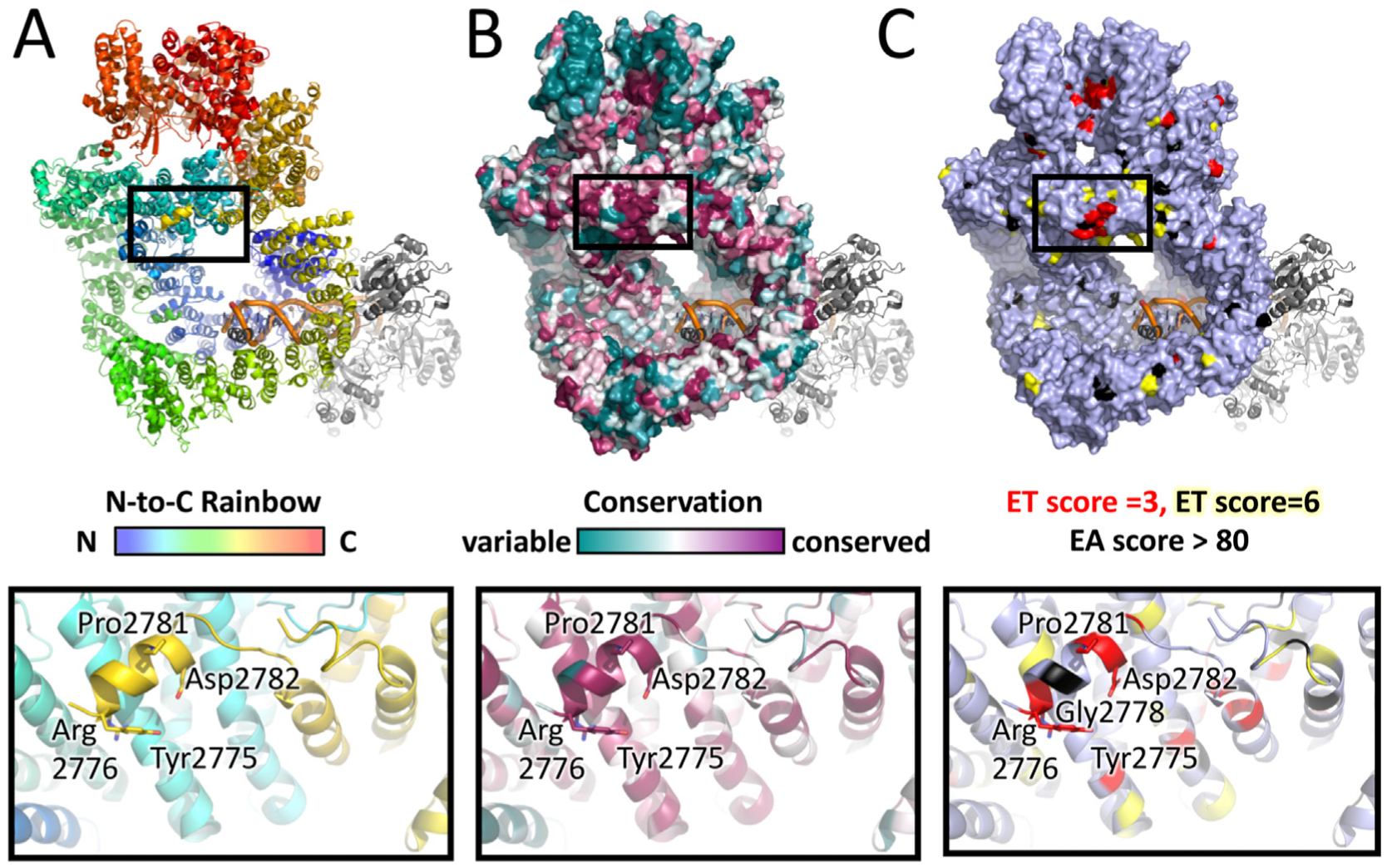
Panel A: Representation of the structure of the DNA-PKcs-Ku-dsDNA holoenzyme from cryo-EM (Yin et al., 2017) coloured in rainbow (a gradation of colours where the N-terminal region is in blue and the C terminal region in red). In the figure, this approximates to the N-HEAT in blue, the M-HEAT in green, the Forehead in cyan, the FAT in orange/bronze and the Kinase/FATC in red. The short helix containing conserved residues Y-R-H-G-D-L-P-D-I-Q (2775–2784) of the YRPD motif is shown in gold at the top of the central cavity (surrounded by the black box). Not shown are L2772, R2773 and S2774 and the ABCDE loop (residues 2577–2773) plus other small loops, which are missing from the structure. DNA is shown in bronze. Ku is in grey. See also Supplementary Movie 1. The panel below shows an expanded view of the YRPD helix from the boxed region above.
Panel B: Representation of conserved amino acids in vertebrates/invertebrates (Supplementary Fig. 2) mapped onto the structure of the DNA-PK holoenzyme where red = highly conserved, green = modestly conserved and white = not conserved. Grey = Ku70/80. See also Supplementary Movie 2. The expanded view below shows Y2772, R2773, G2778, P2781 and D2782 in red, and S2774 in green.
Panel C: Representation of the structure of the DNA-PKcs-Ku-DNA holoenzyme where DNA-PKcs residues with an ET score of 3 are shown in red and those with an ET score of 6 are in yellow. Mutations in cancer patients with an EA score >80 are shown in black. All other residues are in grey. See also Supplementary Movie 3. The expanded view below shows the YRPD helix with the highly conserved Gly2778, a tumor-associated mutation with an EA score >80 in black.
Mapping the tumor-associated mutations that scored above 80, a level suggesting severe impact, revealed one residue (Gly2778) in the YRPD helix and one (Glu2564) in the cluster outside the helix, corresponding to the beginning of ABCDE loop (Fig. 10C, Supplemental Movie 3). Unlike for ET scores, we observed a number of high EA scoring residues along the DNA binding path (Leu128, Pro447, Ser450, Asp2358) and near KU70/80 interfaces (Leu128, Leu201, Asp2358, Arg2377). Although both ET and EA analyses were consistent with a hypothetical critical role for the YRPD helix, the relative disproportionality between the YRPD and Ku70/80 or the DNA interfaces might suggest the opposite. However, in our experience with XPG and TFIIH disease mutations (Tsutakawa et al., 2020; Yan et al., 2019), we have found that mutations that disrupt essential processes are selected against and that some inconsistency between conservation and cancer mutation is not unexpected.
4. Discussion
The DNA damage response in vertebrate cells is controlled by DNA-PKcs along with the related protein kinases ATM and ATR (Blackford and Jackson, 2017). As DNA damage response signaling is the core element for the cellular response to genotoxic insults and the defense against neoplastic transformation, a detailed molecular understanding of DNA-PKcs is critical for cancer biology. In fact, DNA-PKcs inhibitors are actively being tested for potential in cancer therapy (Damia, 2020; Timme et al., 2018). Furthermore DNA-PKcs is generally active in stress responses for innate immunity (Burleigh et al., 2020; Ferguson et al., 2012; Meek, 2020) and even cellular aging (Park et al., 2017). For these reasons, we expect that the molecular understanding of DNA-PKcs sequence in terms of structure and evolution presented here will inform both cell biology and medicine.
More specifically, we have herein identified over 100 putative DNA-PKcs sequences from human, insects, amoeba, fungi, algae and plants. Multiple sequence analysis reveals that these sequences have a remarkable degree of amino acid conservation, not just in the FAT/kinase/FATC domains but in the N-terminal HEAT domain, the Forehead domain, and a novel region that we termed the YRPD motif, which is conserved in all organisms from single-celled Eukaryotes to humans. The YRPD is located in a small helix immediately after the flexible ABCDE loop and immediately before the start of the FAT domain, suggesting a critical role in regulating the function of the ABCDE phosphorylation loop and/or conformational changes between the ABCDE loop and FAT domain. In striking contrast to most other residues in the HEAT domains, the YRPD helix juts out from the regular helical topology of the HEAT repeats and interacts with residues distant in the primary sequence. Moreover, the YRPD motif appears to be part of a larger region of amino acids at the apex of the solenoid ring under the FAT/KIN domain and this entire region (YRPD plus Forehead region) was highly conserved in all metazoa examined and, by ET analysis, was functionally conserved through evolution. Although the function of the YRPD domain is not known, its broad conservation suggests that it is critical to DNA-PKcs function. Interestingly, Histone Parylation Factor 1 (HPF1) contains a sequence resembling the YRPD motif, namely Tyr238 and Arg239 which are followed by glutamic acid, leucine, proline and glutamic acid (i.e. YRxxPE in HPF1 compared to YRxGxxPD in DNA-PKcs). HPF1 residues Y238 and R239 are highly conserved through evolution and are required for interaction with poly-ADP ribose polymerase (PARP) 1 and 2, (Gibbs-Seymour et al., 2016; Suskiewicz et al., 2020). Since DNA-PK interacts with PARP1 (Spagnolo et al., 2012), this raises the tantalizing possibility that the YRPD motif in DNA-PKcs could be required for interaction with PARP1 and/or 2, proteins that play important roles in the DNA damage response and are targets for PARP inhibitors used in the treatment of DSB repair-deficient forms of breast, ovarian and prostate cancer (Jette et al., 2020; Lord and Ashworth, 2017; Slade, 2020). Indeed, inhibitors of the glycohydrolase that removes poly-ADP ribose and releases PARP1 are under active investigation preclinical cancer investigation (Houl et al., 2019), so defining the PARP interaction site in DNA-PKcs is of great interest.
We also noticed that the region N-terminal to the YRPD motif was highly charged with a net positive charge (Fig. 5). We propose that this positive charge could promote interaction with DNA, possibly linking autophosphorylation of the ABCDE loop with DNA binding. However, as shown in Fig. 9, in the available DNA-PK holoenzyme structures, the dsDNA is positioned at the base of the cradle, distant from the YRPD/Forehead patch. Therefore any interaction with dsDNA would require a mechanism to draw more duplex DNA into the central cavity and/or dramatic movement of the YRPR/Forehead region towards the base of DNA-PKcs. Another possibility is that this basic sequence interacts with the acidic C-terminus of Ku80, a 13 amino acid region that contains seven acidic amino acids and no basic residues, and is known to interact with DNA-PKcs (Gell and Jackson, 1999).
The relative low abundance of conserved residues in the Ku and DNA-binding regions of the holoenzyme was surprising. This could be due to the large size of these interfaces being able to accommodate variations. The kinase active site shows clustering of low ET scoring residues, as active sites require precise positioning of chemistry and therefore cannot withstand changes. However, if this is the case, what does this mean for the YRPD helix? It would argue against the YRPD being part of a protein/protein interface and toward perhaps a catalytic active site. Such an exposed active site is uncommon but the WSS1/SPARTN protein contains an exposed active site showing precedence (Neskey et al., 2015; Stingele et al., 2016; Yang et al., 2017).
Another unique signature found in DNA-PKcs was the NUC194 domain, which served as an excellent identifier of DNA-PKcs in many of the organisms examined. The NUC194 motif was originally identified in a screen for nucleolar proteins (Staub et al., 2004). Interesting, DNA-PKcs and its NHEJ partner protein Ku70/80 accumulate in the nucleolus in a ribosomal RNA-dependent manner, and are important for RNA processing (Shao et al., 2020), suggesting this may be an important and highly conserved function of DNA-PKcs.
Many of the previously described autophosphorylation sites were also highly conserved. E.g., threonines 2609, 2638 and 2647 were conserved in most invertebrates, protists, fungi and plants suggesting that the ABCDE loop is critical for DNA-PK function. In contrast, T3950 was only conserved in vertebrates and some invertebrates, and S2056 and S3205 were poorly conserved outside jawed vertebrates. Also, we did not see a high degree of amino acid conservation in the putative FRB and PRD domains.
Interestingly, the most degenerate DNA-PKcs sequences we detected were in Lepidopterans which lacked the Forehead, auto-phosphorylation loop and YRPD domain and had relatively weak conservation in the kinase domain. In contrast, the NUC194 domain was conserved in all Lepidopteran DNA-PKcs orthologues detected. Whether these sequences represent true orthologues of DNA-PKcs or a novel PIKK remain to be determined.
Although long thought to play an exclusive role in DSB repair, recent studies have revealed that DNA-PKcs is involved in many cellular processes, ranging from RNA processing, transcription and regulation of metastasis to innate immunity and mitosis. Our identification of evolutionarily conserved amino acids provides an important basis for mutational analysis to probe DNA-PKcs function. In addition, our identification of DNA-PKcs in the extremophile Alvinella pomejana could provide important clues to aid in expression of stable forms of DNA-PKcs for high quality structural studies, as shown for superoxide dismutase (Shin et al., 2009).
The relationship between human DNA-PKcs and DNA-PKcs of invertebrates, plants, fungi and protists revealed by phylogenetic analysis is best explained by a deep, ancient origin for the PRKDC family. The divergence of the Opisthokonta into Fungal and Metazoan kingdoms occurred over 1 billion years ago (Dohrmann and Worheide, 2017). The divergence of Opisthokonta from plants and protists is even more ancient. Therefore we can safely assume that the precursor to modern DNA-PKcs emerged well over a 1 billion years ago. Furthermore, the generally progressive diversification of protein sequences across extant PRKDCs suggests aspects of conserved functionality. Importantly, this functional conservation was supported by quantitative ET analysis, which reveals that the evolutionary important sites do not vary appreciably from one alignment to the other. This finding means that both the functional sites and the evolutionary pressures remain identical and fundamental to cell biology.
Notably, DNA-PKcs was absent in flowering plants, Chromadorean nematodes, Dikarya fungi and Rhodophytes (red algae), some of which are adapted to life under extreme geothermal conditions (Qiu et al., 2013). DNA-PKcs is also absent or highly divergent in insects and crustaceans. In general, these are organisms that left the ocean and diversified in a terrestrial environment. In contrast, DNA-PKcs is highly conserved in organisms that exist in the relatively stable ocean environment, for example some Molluscs, Sponges, Cnidarians and Echinoderms. While this may be due to random chance during the evolutionary process, it is possible that, given its role in DNA repair, loss of DNA-PKcs could correlate with increased genomic instability and therefore contribute to diversity allowing organisms to better adapt to conditions in an ever-changing environment, in contrast to life in the relatively constant conditions of the ocean. An extension to this idea is the high degree of amino acid conservation of DNA-PKcs in the vertebrates, which have highly stable genomes. We suggest that this conservation may be due to the role of DNA-PKcs in V(D)J recombination. Interestingly, DNA-PKcs is not the only NHEJ associated-gene to have a more ancient lineage than previously supposed. Recently, genes with similarity to the V(D)J recombination-specific RAG1/2 genes were reported in invertebrates including platyhelminths, nematodes, corals and tardigrades (Martin et al., 2020), organisms shown here to have putative PRKDC orthologues. In addition, putative orthologues of the V(D)J nuclease and DNA-PKcs partner Artemis have been identified in rotifers (Hecox-Lea and Mark Welch, 2018).
We find that while PRKDC is overexpressed in many tumor types, and over 15% of endometrial cancer patients have mutations in PRKDC (https://www.cbioportal.org). Furthermore, DNA-PKcs mutation correlates with better survival. Our analysis also informs on the predicted severity of PRKDC mutations in patients with severe combined immunodeficiency. These combined analyses provide a basis to examine disease-associated mutations and variants of unknown significance (VUS), which have not provided actionable information to date. The emerging unified sequence, evolution, and structural information suggests both possible markers and insights for ongoing cancer research and inhibitor trials.
Overall the analyses presented here provide a foundation for a unified understanding of DNA-PKcs sequence, phylogenetics, structure, and function by leveraging the wealth of the sequence databases with computational analyses and breakthrough X-ray and cryo-EM structures. The identified conserved features and signature sequences can be tested by ongoing structural and biochemical efforts by researchers everywhere. We therefore anticipate that these sequence level insights from structure and evolution will have biological relevance for better defining molecular roles of DNA-PKcs in biology as well as in disease susceptibility and potential advanced medical interventions.
Supplementary Material
Acknowledgements
We thank Grace Wang and Shyla Bhardia for help with preliminary analyses.
Funding
This work was funded by NIH/NCI program project grant P01 CA092584 (S.P.L.M. and J.A.T.), NIH grant R01 GM066099 (O.L. and P.K.), grant RGPIN-2019-04829 from the Natural Sciences and Engineering Council (Canada) (S.P.L.M.). J.A.T. is a CPRIT Scholar in Cancer Research and acknowledges support by a Robert A. Welch Chemistry Chair. This work used the Extreme Science and Engineering Discovery Environment (XSEDE) Bridges at the Pittsburgh Supercomputing Center through allocation MCB170053.
Footnotes
Declaration of competing interest
The authors declare no conflict of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.pbiomolbio.2020.09.010.
References
- Adikesavan AK, Katsonis P, Marciano DC, Lua R, Herman C, Lichtarge O, 2011. Separation of recombination and SOS response in Escherichia coli RecA suggests LexA interaction sites. PLoS Genet. 7, e1002244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adl SM, Bass D, Lane CE, Lukeš J, Schoch CL, Smirnov A, Agatha S, Berney C, Brown MW, Burki F, Cárdenas P, Čepička I, Chistyakova L, Del Campo J, Dunthorn M, Edvardsen B, Eglit Y, Guillou L, Hampl V, Heiss AA, Hoppenrath M, James TY, Karnkowska A, Karpov S, Kim E, Kolisko M, Kudryavtsev A, Lahr DJG, Lara E, Le Gall L, Lynn DH, Mann DG, Massana R, Mitchell EAD, Morrow C, Park JS, Pawlowski JW, Powell MJ, Richter DJ, Rueckert S, Shadwick L, Shimano S, Spiegel FW, Torruella G, Youssef N, Zlatogursky V, Zhang Q, 2019. Revisions to the classification, nomenclature, and diversity of eukaryotes. J. Eukaryot. Microbiol 66, 4–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin SR, Erdin S, Ward RM, Lua RC, Lichtarge O, 2013. Prediction and experimental validation of enzyme substrate specificity in protein structures. Proc. Natl. Acad. Sci. U. S. A 110, E4195–E4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A, Ye Z, Ahmed Z, Tainer JA, 2019. Cancer mutational burden is shaped by G4 DNA, replication stress and mitochondrial dysfunction. Prog. Biophys. Mol. Biol 147, 47–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banyai L, Patthy L, 2016. Putative extremely high rate of proteome innovation in lancelets might be explained by high rate of gene prediction errors. Sci. Rep 6, 30700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baretic D, Maia de Oliveira T, Niess M, Wan P, Pollard H, Johnson CM, Truman C, McCall E, Fisher D, Williams R, Phillips C, 2019. Structural insights into the critical DNA damage sensors DNA-PKcs, ATM and ATR. Prog. Biophys. Mol. Biol 147, 4–16. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T, 1998. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr. Biol 8, 1395–1398. [DOI] [PubMed] [Google Scholar]
- Blackford AN, Jackson SP, 2017. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell 66, 801–817. [DOI] [PubMed] [Google Scholar]
- Block WD, Lees-Miller SP, 2005. Putative homologues of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and other components of the non-homologous end joining machinery in Dictyostelium discoideum. DNA Repair 4, 1061–1065. [DOI] [PubMed] [Google Scholar]
- Bosotti R, Isacchi A, Sonnhammer EL, 2000. FAT: a novel domain in PIK-related kinases. Trends Biochem. Sci 25, 225–227. [DOI] [PubMed] [Google Scholar]
- Brewerton SC, Dore AS, Drake AC, Leuther KK, Blundell TL, 2004. Structural analysis of DNA-PKcs: modelling of the repeat units and insights into the detailed molecular architecture. J. Struct. Biol 145, 295–306. [DOI] [PubMed] [Google Scholar]
- Burleigh K, Maltbaek JH, Cambier S, Green R, Gale M Jr., James RC, Stetson DB, 2020. Human DNA-PK activates a STING-independent DNA sensing pathway. Sci Immunol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carss KJ, Hillman SC, Parthiban V, McMullan DJ, Maher ER, Kilby MD, Hurles ME, 2014. Exome sequencing improves genetic diagnosis of structural fetal abnormalities revealed by ultrasound. Hum. Mol. Genet 23, 3269–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter T, Vancurova I, Sun I, Lou W, DeLeon S, 1990. A DNA-activated protein kinase from HeLa cell nuclei. Mol. Cell Biol 10, 6460–6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DW, Lees-Miller SP, 1996. The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J. Biol. Chem 271, 8936–8941. [DOI] [PubMed] [Google Scholar]
- Chan DW, Mody CH, Ting NS, Lees-Miller SP, 1996. Purification and characterization of the double-stranded DNA-activated protein kinase, DNA-PK, from human placenta. Biochem. Cell. Biol 74, 67–73. [DOI] [PubMed] [Google Scholar]
- Chen BP, Chan DW, Kobayashi J, Burma S, Asaithamby A, Morotomi-Yano K, Botvinick E, Qin J, Chen DJ, 2005. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J. Biol. Chem 280, 14709–14715. [DOI] [PubMed] [Google Scholar]
- Chu W, Gong X, Li Z, Takabayashi K, Ouyang H, Chen Y, Lois A, Chen DJ, Li GC, Karin M, Raz E, 2000. DNA-PKcs is required for activation of innate immunity by immunostimulatory DNA. Cell 103, 909–918. [DOI] [PubMed] [Google Scholar]
- Clarke CN, Katsonis P, Hsu TK, Koire AM, Silva-Figueroa A, Christakis I, Williams MD, Kutahyalioglu M, Kwatampora L, Xi Y, Lee JE, Koptez ES, Busaidy NL, Perrier ND, Lichtarge O, 2019. Comprehensive genomic characterization of parathyroid cancer identifies novel candidate driver mutations and core pathways. J Endocr Soc 3, 544–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crombie AT, Larke-Mejia NL, Emery H, Dawson R, Pratscher J, Murphy GP, McGenity TJ, Murrell JC, 2018. Poplar phyllosphere harbors disparate isoprene-degrading bacteria. Proc. Natl. Acad. Sci. U. S. A 115, 13081–513086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Yu Y, Gupta S, Cho YM, Lees-Miller SP, Meek K, 2005. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol. Cell Biol 25, 10842–10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damia G, 2020. Targeting DNA-PK in cancer. Mutat. Res 821, 111692. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D, 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Reddy YV, Wang W, Woods T, Douglas P, Ramsden DA, Lees-Miller SP, Meek K, 2003. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol. Cell Biol 23, 5836–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs TA, Tainer JA, Lees-Miller SP, 2010. A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair 9, 1307–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohrmann M, Worheide G, 2017. Dating early animal evolution using phylogenomic data. Sci. Rep 7, 3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore AS, Drake AC, Brewerton SC, Blundell TL, 2004. Identification of DNA-PK in the arthropods. Evidence for the ancient ancestry of vertebrate non-homologous end-joining. DNA Repair 3, 33–41. [DOI] [PubMed] [Google Scholar]
- Douglas P, Cui X, Block WD, Yu Y, Gupta S, Ding Q, Ye R, Morrice N, Lees-Miller SP, Meek K, 2007. The DNA-dependent protein kinase catalytic subunit is phosphorylated in vivo on threonine 3950, a highly conserved amino acid in the protein kinase domain. Mol. Cell Biol 27, 1581–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas P, Sapkota GP, Morrice N, Yu Y, Goodarzi AA, Merkle D, Meek K, Alessi DR, Lees-Miller SP, 2002. Identification of in vitro and in vivo phosphorylation sites in the catalytic subunit of the DNA-dependent protein kinase. Biochem. J 368, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas P, Ye R, Radhamani S, Lees-Miller SP, 2020. June. Nocodazole-induced expression and phosphorylation of anillin and other mitotic proteins is decreased in DNA-dependent protein kinase catalytic subunit (DNA-PKcs)-deficient cells and rescued by inhibition of the Anaphase Promoting Complex/ Cyclosome (APC/C) with proTAME but not apcin. Mol. Cell Biol 40 (13) 10.1128/MCB.00191-19 e00191–e00119, Print 2020 Jun 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichinger L, Pachebat JA, Glockner G, Rajandream MA, Sucgang R, Berriman M, Song J, Olsen R, Szafranski K, Xu Q, authors, a.o., 2005. The genome of the social amoeba Dictyostelium discoideum. Nature 435, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, Sonnhammer ELL, Hirsh L, Paladin L, Piovesan D, Tosatto SCE, Finn RD, 2019. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–d432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias-Villalobos A, Fort P, Helmlinger D, 2019. December 20. New insights into the evolutionary conservation of the sole PIKK pseudokinase Tra1/TRRAP. Biochem. Soc. Trans 47 (6), 1597–1608. 10.1042/BST20180496. [DOI] [PubMed] [Google Scholar]
- Esenboga S, Akal C, Karaatmaca B, Erman B, Dogan S, Orhan D, Boztug K, Ayvaz D, Tezcan İ, 2018. Two siblings with PRKDC defect who presented with cutaneous granulomas and review of the literature. Clin. Immunol 197, 1–5. [DOI] [PubMed] [Google Scholar]
- Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL, 2012. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 1, e00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J, Bronson RT, Malynn BA, Bryans M, Zhu C, Chaudhuri J, Davidson L, Ferrini R, Stamato T, Orkin SH, Greenberg ME, Alt FW, 1998. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 95, 891–902. [DOI] [PubMed] [Google Scholar]
- Gell D, Jackson SP, 1999. Mapping of protein-protein interactions within the DNA-dependent protein kinase complex. Nucleic Acids Res. 27, 3494–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs-Seymour I, Fontana P, Rack JGM, Ahel I, 2016. HPF1/C4orf27 is a PARP-1-interacting protein that regulates PARP-1 ADP-ribosylation activity. Mol. Cell 62, 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin JF, Knudsen KE, 2014. Beyond DNA repair: DNA-PK function in cancer. Canc. Discov 4, 1126–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb TM, Jackson SP, 1993. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell 72, 131–142. [DOI] [PubMed] [Google Scholar]
- Gupta S, Meek K, 2005. The leucine rich region of DNA-PKcs contributes to its innate DNA affinity. Nucleic Acids Res. 33, 6972–6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M, Rosenberg D, Bierma J, Hura GL, Lees Miller SP and Tainer JA, submitted. Visualizing functional dynamicity in the DNA-dependent protein kinase holoenzyme DNA-PK complex by integrating SAXS with cryo-EM Progress in Biophysics and Molecular Biology. (Special Isse). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, Rambo RP, Hura GL, Pelikan M, So S, Abolfath RM, Chen DJ, Lees-Miller SP, Tainer JA, 2010. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J. Biol. Chem 285, 1414–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA, 2011. Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hartley KO, Gell D, Smith GC, Zhang H, Divecha N, Connelly MA, Admon A, Lees-Miller SP, Anderson CW, Jackson SP, 1995. DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell 82, 849–856. [DOI] [PubMed] [Google Scholar]
- Hecox-Lea BJ, Mark Welch DB, 2018. Evolutionary diversity and novelty of DNA repair genes in asexual Bdelloid rotifers. BMC Evol. Biol 18, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindle MM, Martin SF, Noordally ZB, van Ooijen G, Barrios-Llerena ME, Simpson TI, Le Bihan T, Millar AJ, 2014. The reduced kinome of Ostreococcus tauri: core eukaryotic signalling components in a tractable model species. BMC Genom. 15, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder T, Basquin C, Ebert J, Randel N, Jollivet D, Conti E, Jékely G, Bono F, 2013. Deep transcriptome-sequencing and proteome analysis of the hydrothermal vent annelid Alvinella pompejana identifies the CvP-bias as a robust measure of eukaryotic thermostability. Biol. Direct 8, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E, 2015. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houl JH, Ye Z, Brosey CA, Balapiti-Modarage LPF, Namjoshi S, Bacolla A, Laverty D, Walker BL, Pourfarjam Y, Warden LS, Babu Chinnam N, Moiani D, Stegeman RA, Chen MK, Hung MC, Nagel ZD, Ellenberger T, Kim IK, Jones DE, Ahmed Z, Tainer JA, 2019. Selective small molecule PARG inhibitor causes replication fork stalling and cancer cell death. Nat. Commun 10, 5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu DW, Gaudet P, Hudson JJ, Pears CJ, Lakin ND, 2006. DNA damage signaling and repair in Dictyostelium discoideum. Cell Cycle 5, 702–708. [DOI] [PubMed] [Google Scholar]
- Hsu DW, Kiely R, Couto CA, Wang HY, Hudson JJ, Borer C, Pears CJ, Lakin ND, 2011. DNA double-strand break repair pathway choice in Dictyostelium. J. Cell Sci 124, 1655–1663. [DOI] [PubMed] [Google Scholar]
- Hunter T, 1995. When is a lipid kinase not a lipid kinase? When it is a protein kinase. Cell 83, 1–4. [DOI] [PubMed] [Google Scholar]
- Jette N, Lees-Miller SP, 2015. The DNA-dependent protein kinase: a multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol 117, 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jette NR, Kumar M, Radhamani S, Arthur G, Goutam S, Yip S, Kolinsky M, Williams GJ, Bose P, Lees-Miller SP, 2020. ATM-deficient Cancers Provide New Opportunities for Precision Oncology. In: Cancers (Basel), vol. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Crowe JL, Liu X, Nakajima S, Wang Y, Li C, Lee BJ, Dubois RL, Liu C, Yu X, Lan L, Zha S, 2015. Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol. Cell 58, 172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsonis P, Lichtarge O, 2014. A formal perturbation equation between genotype and phenotype determines the Evolutionary Action of protein-coding variations on fitness. Genome Res. 24, 2050–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsonis P, Lichtarge O, 2019. CAGI5: objective performance assessments of predictions based on the Evolutionary Action equation. Hum. Mutat 40, 1436–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser K, Degenhardt F, Holzapfel S, Horpaopan S, Peters S, Spier I, Morak M, Vangala D, Rahner N, von Knebel-Doeberitz M, Schackert HK, Engel C, Büttner R, Wijnen J, Doerks T, Bork P, Moebus S, Herms S, Fischer S, Hoffmann P, Aretz S, Steinke-Lange V, 2018. Copy number variation analysis and targeted NGS in 77 families with suspected Lynch syndrome reveals novel potential causative genes. Int. J. Canc 143, 2800–2813. [DOI] [PubMed] [Google Scholar]
- Le SQ, Gascuel O, 2008. An improved general amino acid replacement matrix. Mol. Biol. Evol 25, 1307–1320. [DOI] [PubMed] [Google Scholar]
- Lees-Miller SP, Chen YR, Anderson CW, 1990. Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Mol. Cell Biol 10, 6472–6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller SP, Sakaguchi K, Ullrich SJ, Appella E, Anderson CW, 1992. Human DNA-activated protein kinase phosphorylates serines 15 and 37 in the amino-terminal transactivation domain of human p53. Mol. Cell Biol 12, 5041–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Wong JTY, 2019. DNA damage response pathways in dinoflagellates. Microorganisms 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtarge O, Bourne HR, Cohen FE, 1996. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol 257, 342–358. [DOI] [PubMed] [Google Scholar]
- Lieber MR, 2010. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem 79, 181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GY, Sabatini DM, 2020. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol 21, 183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd JPB, 2018. The evolution and diversity of the nonsense-mediated mRNA decay pathway. F1000Res 7, 1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A, 2017. PARP inhibitors: synthetic lethality in the clinic. Science 355, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lua RC, Lichtarge O, 2010. PyETV: a PyMOL evolutionary trace viewer to analyze functional site predictions in protein complexes. Bioinformatics 26, 2981–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabushi S, Yao H, Marsh M, Kristensen DM, Philippi A, Sowa ME, Lichtarge O, 2002. Structural clusters of evolutionary trace residues are statistically significant and common in proteins. J. Mol. Biol 316, 139–154. [DOI] [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, Lopez R, 2019. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–w641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S, 2002. The protein kinase complement of the human genome. Science 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- Market E, Papavasiliou FN, 2003. V(D)J recombination and the evolution of the adaptive immune system. PLoS Biol. 1, E16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EC, Vicari C, Tsakou-Ngouafo L, Pontarotti P, Petrescu AJ, Schatz DG, 2020. Identification of RAG-like transposons in protostomes suggests their ancient bilaterian origin. Mobile DNA 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu AL, Verronese E, Rice GI, Fouyssac F, Bertrand Y, Picard C, Chansel M, Walter JE, Notarangelo LD, Butte MJ, Nadeau KC, Csomos K, Chen DJ, Chen K, Delgado A, Rigal C, Bardin C, Schuetz C, Moshous D, Reumaux H, Plenat F, Phan A, Zabot MT, Balme B, Viel S, Bienvenu J, Cochat P, van der Burg M, Caux C, Kemp EH, Rouvet I, Malcus C, Meritet JF, Lim A, Crow YJ, Fabien N, Menetrier-Caux C, De Villartay JP, Walzer T, Belot A, 2015. PRKDC mutations associated with immunodeficiency, granuloma, and autoimmune regulator-dependent autoimmunity. J. Allergy Clin. Immunol 135, 1578–1588 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek K, 2020. An antiviral DNA response without the STING? Trends Immunol. 41, 362–364. [DOI] [PubMed] [Google Scholar]
- Meek K, Dang V, Lees-Miller SP, 2008. DNA-PK: the means to justify the ends? Adv. Immunol 99, 33–58. [DOI] [PubMed] [Google Scholar]
- Meek K, Douglas P, Cui X, Ding Q, Lees-Miller SP, 2007. Trans Autophosphorylation at DNA-dependent protein kinase’s two major autophosphorylation site clusters facilitates end processing but not end joining. Mol. Cell Biol 27, 3881–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrzad J, Dayyani M, Khorasani ME, 2019. Polymorphisms of XRCC3 and XRCC7 and colorectal cancer risk in khorasan razavi province, Iran. Asian Pac. J. Cancer Prev. APJCP 20, 2153–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalek I, Res I, Lichtarge O, 2004. A family of evolution-entropy hybrid methods for ranking protein residues by importance. J. Mol. Biol 336, 1265–1282. [DOI] [PubMed] [Google Scholar]
- Mihalek I, Res I, Lichtarge O, 2006. Evolutionary and structural feedback on selection of sequences for comparative analysis of proteins. Proteins 63, 87–99. [DOI] [PubMed] [Google Scholar]
- Mordes DA, Glick GG, Zhao R, Cortez D, 2008. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 22, 1478–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal JA, Dang V, Douglas P, Wold MS, Lees-Miller SP, Meek K, 2011. Inhibition of homologous recombination by DNA-dependent protein kinase requires kinase activity, is titratable, and is modulated by autophosphorylation. Mol. Cell Biol 31, 1719–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal JA, Meek K, 2011. Choosing the right path: does DNA-PK help make the decision? Mutat. Res 711, 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal JA, Meek K, 2019. Deciphering phenotypic variance in different models of DNA-PKcs deficiency. DNA Repair 73, 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal JA, Sugiman-Marangos S, VanderVere-Carozza P, Wagner M, Turchi J, Lees-Miller SP, Junop MS, Meek K, 2014. Unraveling the complexities of DNA-dependent protein kinase autophosphorylation. Mol. Cell Biol 34, 2162–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neskey DM, Osman AA, Ow TJ, Katsonis P, McDonald T, Hicks SC, Hsu TK, Pickering CR, Ward A, Patel A, Yordy JS, Skinner HD, Giri U, Sano D, Story MD, Beadle BM, El-Naggar AK, Kies MS, William WN, Caulin C, Frederick M, Kimmel M, Myers JN, Lichtarge O, 2015. Evolutionary action score of TP53 identifies high-risk mutations associated with decreased survival and increased distant metastases in head and neck cancer. Canc. Res 75, 1527–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network CGA, 2017. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 169, 1327–1341 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill T, Dwyer AJ, Ziv Y, Chan DW, Lees-Miller SP, Abraham RH, Lai JH, Hill D, Shiloh Y, Cantley LC, Rathbun GA, 2000. Utilization of oriented peptide libraries to identify substrate motifs selected by ATM. J. Biol. Chem 275, 22719–22727. [DOI] [PubMed] [Google Scholar]
- Pannunzio NR, Watanabe G, Lieber MR, 2018. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem 293, 10512–10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Gavrilova O, Brown AL, Soto JE, Bremner S, Kim J, Xu X, Yang S, Um JH, Koch LG, Britton SL, Lieber RL, Philp A, Baar K, Kohama SG, Abel ED, Kim MK, Chung JH, 2017. DNA-PK promotes the mitochondrial, metabolic, and physical decline that occurs during aging. Cell Metabol. 25, 1135–1146 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pears CJ, Lakin ND, 2014. Emerging models for DNA repair: Dictyostelium discoideum as a model for nonhomologous end-joining. DNA Repair 17, 121–131. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Qiu H, Price DC, Weber AP, Reeb V, Yang EC, Lee JM, Kim SY, Yoon HS, Bhattacharya D, 2013. Adaptation through horizontal gene transfer in the cryptoendolithic red alga Galdieria phlegrea. Curr. Biol 23, R865–R866. [DOI] [PubMed] [Google Scholar]
- Rahimi M, Fayaz S, Fard-Esfahani A, Modarressi MH, Akrami SM, Fard-Esfahani P, 2012. The role of Ile3434Thr XRCC7 gene polymorphism in differentiated thyroid cancer risk in an Iranian population. Iran. Biomed. J 16, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GJ, Yao R, Lichtarge O, Wensel TG, 2010. Evolution-guided discovery and recoding of allosteric pathway specificity determinants in psychoactive bioamine receptors. Proc. Natl. Acad. Sci. U. S. A 107, 7787–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL, 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Saltzberg DJ, Hepburn M, Pilla KB, Schriemer DC, Lees-Miller SP, Blundell TL, Sali A, 2019. SSEThread: integrative threading of the DNA-PKcs sequence based on data from chemical cross-linking and hydrogen deuterium exchange. Prog. Biophys. Mol. Biol 10.1016/j.pbiomolbio.2019.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Agarwala R, Bolton EE, Brister JR, Canese K, Clark K, Connor R, Fiorini N, Funk K, Hefferon T, Holmes JB, Kim S, Kimchi A, Kitts PA, Lathrop S, Lu Z, Madden TL, Marchler-Bauer A, Phan L, Schneider VA, Schoch CL, Pruitt KD, Ostell J, 2019. Database resources of the national center for biotechnology information. Nucleic Acids Res. 47, D23–D28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter SF, Bernstein RM, Bernstein H, Marchalonis JJ, 1999. ‘Big Bang’ emergence of the combinatorial immune system. Dev. Comp. Immunol 23, 107–111. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ, 2005. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 33, W363–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Flynn RA, Crowe JL, Zhu Y, Liang J, Jiang W, Aryan F, Aoude P, Bertozzi CR, Estes VM, Lee BJ, Bhagat G, Zha S, Calo E, 2020. DNA-PKcs has KU-dependent function in rRNA processing and haematopoiesis. Nature 579 (7798), 291–296. 10.1038/s41586-020-2041-2. Epub 2020 Feb 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif H, Li Y, Dong Y, Dong L, Wang WL, Mao Y, Wu H, 2017. Cryo-EM structure of the DNA-PK holoenzyme. Proc. Natl. Acad. Sci. U. S. A 114, 7367–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff JG, Hepburn M, Yu Y, Lees-Miller SP, Schriemer DC, 2017. Nanospray HX-MS configuration for structural interrogation of large protein systems. Analyst 142, 904–910. [DOI] [PubMed] [Google Scholar]
- Shin DS, Didonato M, Barondeau DP, Hura GL, Hitomi C, Berglund JA, Getzoff ED, Cary SC, Tainer JA, 2009. Superoxide dismutase from the eukaryotic thermophile Alvinella pompejana: structures, stability, mechanism, and insights into amyotrophic lateral sclerosis. J. Mol. Biol 385, 1534–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibanda BL, Chirgadze DY, Ascher DB, Blundell TL, 2017. DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science 355, 520–524. [DOI] [PubMed] [Google Scholar]
- Sibanda BL, Chirgadze DY, Blundell TL, 2010. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature 463, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade D, 2020. PARP and PARG inhibitors in cancer treatment. Genes Dev. 34, 360–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnolo L, Barbeau J, Curtin NJ, Morris EP, Pearl LH, 2012. Visualization of a DNA-PK/PARP1 complex. Nucleic Acids Res. 40, 4168–4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A, 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Morgenstern B, 2005. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub E, Fiziev P, Rosenthal A, Hinzmann B, 2004. Insights into the evolution of the nucleolus by an analysis of its protein domain repertoire. Bioessays 26, 567–581. [DOI] [PubMed] [Google Scholar]
- Stingele J, Bellelli R, Alte F, Hewitt G, Sarek G, Maslen SL, Tsutakawa SE, Borg A, Kjær S, Tainer JA, Skehel JM, Groll M, Boulton SJ, 2016. Mechanism and regulation of DNA-protein crosslink repair by the DNA-dependent metalloprotease SPRTN. Mol. Cell 64, 688–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stothard P, 2000. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28 (1102), 1104. [DOI] [PubMed] [Google Scholar]
- Suskiewicz MJ, Zobel F, Ogden TEH, Fontana P, Ariza A, Yang JC, Zhu K, Bracken L, Hawthorne WJ, Ahel D, Neuhaus D, Ahel I, 2020. HPF1 completes the PARP active site for DNA damage-induced ADP-ribosylation. Nature 579, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed A, Tainer JA, 2018. The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu. Rev. Biochem 87, 263–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Knighton DR, Zheng J, Ten Eyck LF, Sowadski JM, 1992. Structural framework for the protein kinase family. Annu. Rev. Cell Biol 8, 429–462. [DOI] [PubMed] [Google Scholar]
- Telford MJ, Budd GE, Philippe H, 2015. Phylogenomic insights into animal evolution. Curr. Biol 25, R876eR887. [DOI] [PubMed] [Google Scholar]
- Timme CR, Rath BH, O’Neill JW, Camphausen K, Tofilon PJ, 2018. The DNA-PK inhibitor VX-984 enhances the radiosensitivity of glioblastoma cells grown in vitro and as orthotopic xenografts. Mol. Canc. Therapeut 17, 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutakawa SE, Sarker AH, Ng C, Arvai AS, Shin DS, Shih B, Jiang S, Thwin AC, Tsai MS, Willcox A, Her MZ, Trego KS, Raetz AG, Rosenberg D, Bacolla A, Hammel M, Griffith JD, Cooper PK, Tainer JA, 2020. Human XPG nuclease structure, assembly, and activities with insights for neurodegeneration and cancer from pathogenic mutations. Proc. Natl. Acad. Sci. U. S. A 117, 14127–14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, Mari PO, van Gent DC, Chen BP, Chen DJ, 2007. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J. Cell Biol 177, 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, Mari PO, Tezcan I, Chen DJ, Zdzienicka MZ, van Dongen JJ, van Gent DC, 2009. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J. Clin. Invest 119, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schouwenburg PA, Davenport EE, Kienzler AK, Marwah I, Wright B, Lucas M, Malinauskas T, Martin HC, Lockstone HE, Cazier JB, Chapel HM, Knight JC, Patel SY, 2015. Application of whole genome and RNA sequencing to investigate the genomic landscape of common variable immunodeficiency disorders. Clin. Immunol 160, 301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AI, Hunt T, Jackson RJ, Anderson CW, 1985. Double-stranded DNA induces the phosphorylation of several proteins including the 90 000 mol. wt. heat-shock protein in animal cell extracts. EMBO J. 4, 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Lees-Miller SP, 2013. Detection and repair of ionizing radiation-induced DNA double strand breaks: new developments in nonhomologous end joining. Int. J. Radiat. Oncol. Biol. Phys 86, 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JL, Duboc C, Wu Q, Ochi T, Liang S, Tsutakawa SE, Lees-Miller SP, Nadal M, Tainer JA, Blundell TL, Strick TR, 2018. Dissection of DNA double-strand-break repair using novel single-molecule forceps. Nat. Struct. Mol. Biol 25, 482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Jin Z, Yang S, Xu Y, Zhu Y, Ji Y, 2018. TCGA-assembler 2: software pipeline for retrieval and processing of TCGA/CPTAC data. Bioinformatics 34, 1615–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbine L, Neal JA, Sasi NK, Shimada M, Deem K, Coleman H, Dobyns WB, Ogi T, Meek K, Davies EG, Jeggo PA, 2013. PRKDC mutations in a SCID patient with profound neurological abnormalities. J. Clin. Invest 123, 2969–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima H, Lee KJ, Chen BP, 2006. ATR-dependent phosphorylation of DNA-dependent protein kinase catalytic subunit in response to UV-induced replication stress. Mol. Cell Biol 26, 7520–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Dodd T, He Y, Tainer JA, Tsutakawa SE, Ivanov I, 2019. Transcription preinitiation complex structure and dynamics provide insight into genetic diseases. Nat. Struct. Mol. Biol 26, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP, 2013. mTOR kinase structure, mechanism and regulation. Nature 497, 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Li Y, Gao Z, Li Z, Xu J, Wang W, Dong Y, 2017. Structural analysis of Wss1 protein from saccharomyces cerevisiae. Sci. Rep 7, 8270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Liu M, Tian Y, Wang J, Xu Y, 2017. Cryo-EM structure of human DNA-PK holoenzyme. Cell Res. 27, 1341–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S, Dynan WS, 1999. Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res. 27, 4679–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Watanabe G, Morten MJ, Reid DA, Rothenberg E, Lieber MR, 2019. The essential elements for the noncovalent association of two DNA ends during NHEJ synapsis. Nat. Commun 10, 3588. 10.1016/j.pbiomolbio.2020.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.