

Summary:
Gut microbiota metabolites may be important for host health yet few studies investigate the correlation between human gut microbiome and production of fecal metabolites and their impact on the plasma metabolome. Since gut microbiota metabolites are influenced by diet, we performed a longitudinal analysis of the impact of three divergent diets, vegan, omnivore, and a synthetic enteral nutrition (EEN) diet lacking fiber, on the human gut microbiome and its metabolome, including after a microbiota depletion intervention. Omnivore and vegan, but not EEN, diets altered fecal amino acid levels by supporting the growth of Firmicutes capable of amino acid metabolism. This correlated with relative abundance of a sizable number of fecal amino acid metabolites, some not previously associated with the gut microbiota. The effect on the plasma metabolome, in contrast, were modest. The impact of diet, particularly fiber, on the human microbiome influences broad classes of metabolites that may modify health.
Keywords: microbiome, metabolome, enteral nutrition, dietary fiber
Graphical Abstract
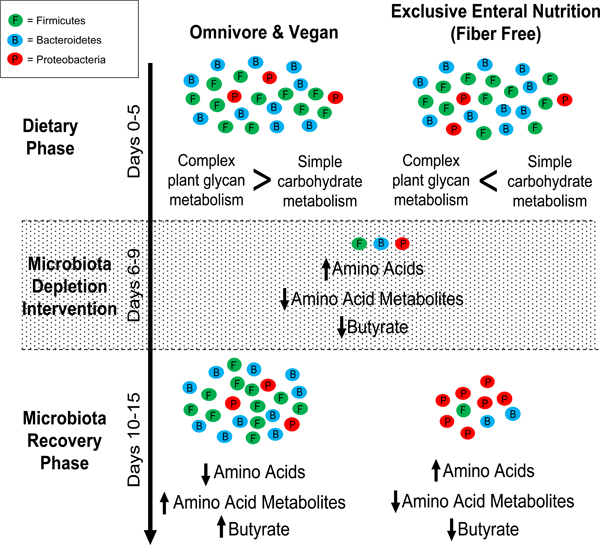
eTOC blurb
Tanes et al. show that consumption of a fiber free diet shifts the human gut microbiome towards metabolism of simple carbohydrates. Following an ecological stress, omnivore and vegan diets, but not the fiber-free diet, support growth of Firmicutes capable of carbohydrate and amino acid metabolism, altering fecal amino acids level.
Introduction
The gut microbiota produces bioactive small molecule metabolites (Donia and Fischbach, 2015). Examples include carbohydrate derivatives such as short chain fatty acids, lipids such as N-acyl amides (Cohen et al., 2017), amino acid metabolites (Dodd et al., 2017; Wlodarska et al., 2017), and the modification of bile acids (Fiorucci and Distrutti, 2015). Many of these metabolites influence mammalian physiology as ligands for G-protein coupled receptors and nuclear hormone receptors (Chen et al., 2019; Colosimo et al., 2019; Venkatesh et al., 2014) that could be targets for small molecule drugs (Brown and Hazen, 2017) to treat and/or prevent diseases such as coronary vascular disease (Wang et al., 2011), diabetes (Koh et al., 2018), inflammatory bowel disease (Furusawa et al., 2013; Venkatesh et al., 2014), and autism (Hsiao et al., 2013). The production and functionality of these molecules have been demonstrated primarily in animal model systems such as gnotobiotic mice; the relevance to human physiology remains to be determined (Walter et al., 2020).
Interventions directed at the composition or function of the gut microbiome often result in larger effects in murine models than in humans where inter-subject variability in microbiome composition is greater. For example, the impact of diet on the composition of the gut microbiota is larger in mice than in humans (Baxter et al., 2019; Johnson et al., 2019; Wu et al., 2011). The inter-subject variability of the gut microbiota’s response to diet is likely the result of complex community interactions. Better understanding these interactions can inform the design of precision diets that lead to a predictable human response (Johnson et al., 2019; Zeevi et al., 2015).
It is believed that interactions of diet and the microbiome influence the fecal and plasma metabolome, but this has not been well studied in humans. In some settings, diet appears to have a strong impact on the plasma metabolome independently of the gut microbiota. For example, the plasma metabolome differs among humans consuming an omnivore or vegan diet, with only few metabolites being produced primarily by the gut microbiota (Wu et al., 2016). In contrast, with extreme dietary changes such as elimination of fruits and vegetables from a diet composed of either whole foods or dietary formulas, there are relatively large changes in the composition of the gut microbiota (David et al., 2013; Lewis et al., 2015). To what extent these taxonomic changes lead to alteration in the fecal and plasma metabolome remains to be determined.
Dietary fiber from fruits and vegetables has large effects on the structure and function of the murine microbiota, with some effects being reproducible in humans (David et al., 2014; Kovatcheva-Datchary et al., 2015). Dietary fiber influences the composition of the murine gut microbiome, helping to maintain the diversity of the community (Sonnenburg et al., 2016) and leading to the production of SCFAs via fermentation. In part, this is due to the high representation of genes encoding glycan degrading enzymes in gut bacteria, such as glycoside hydrolases, where the bacterial contribution of these genes exceeds the mammalian host contribution by many-fold (Cantarel et al., 2012; El Kaoutari et al., 2013). The ability of dietary fiber to stimulate production of SCFAs by the gut microbiota may be greater and more consistent in murine models than in humans (Baxter et al., 2019; Wu et al., 2016), which may be due in part to the loss of bacterial taxa well adapted to degrade complex plant polysaccharides during the process of industrialization (Smits et al., 2017). The typical American diet is relatively low in fiber with less than 10% of Americans consuming the recommended daily intake of fiber (NHANES). In contrast, most, but not all, vegetarians and vegans consume a greater amount of dietary fiber. Based on this, we hypothesized that comparing the microbiome and metabolome of adults following divergent diets including a vegan diet that is relatively high in fiber, a typical American diet that contains intermediate amounts of fiber, and a formula-based diet that is devoid of fiber we could learn about the influence of diet on the gut microbiota composition and the fecal and plasma metabolome. Moreover, we hypothesized that by transiently reducing the gut bacterial load, we would be able to isolate the relationship between different bacterial taxa, diet and metabolite production. In the Food And Resulting Microbial Metabolites (FARMM) study described herein, we determined the effect of diet on the composition and metabolic function of the human gut microbiome using a controlled feeding experiment with three divergent diets - vegan, typical American (hereafter referred to as omnivore), and an exclusive enteral nutrition diet (EEN) devoid of dietary fiber. We included an antibiotic and polyethylene glycol (Abx/PEG) intervention to transiently reduce the concentration of bacteria within the gut, thereby allowing for an assessment of the effect of diet on the recovery of the gut microbiota and its associated fecal and plasma metabolome. The results of this study demonstrate the importance of diet on the human gut microbiota composition and the production of both carbohydrate dependent bacterial metabolites as well as those generated from amino acids, a number of which have not been previously associated with the gut microbiota.
Results
Analyzing the effects of exclusive enteral nutrition (EEN), vegan, or omnivore diets
We performed a controlled feeding study comparing the effect of three different diets: vegan, EEN (a liquid diet with no fiber), and omnivore. The microbiota and the metabolome were compared during three phases: the dietary phase (days 1–5), the Abx/PEG gut microbiota purge designed to reduce bacterial load (days 6–8), and the recovery phase (days 9–15). The purge involved treatment with oral antibiotics (vancomycin and neomycin) and polyethylene glycol (Abx/PEG) (Figure 1A). The fecal and plasma samples collected are summarized in Figure S1A. The analysis of fecal and plasma samples before and after the Abx/PEG intervention allowed us to interrogate the effects of these three diets on the gut microbiome, the fecal metabolome, and the plasma metabolome. The time points after the Abx/PEG intervention characterized the dietary effects on recovery of the gut microbiome.
Figure 1.
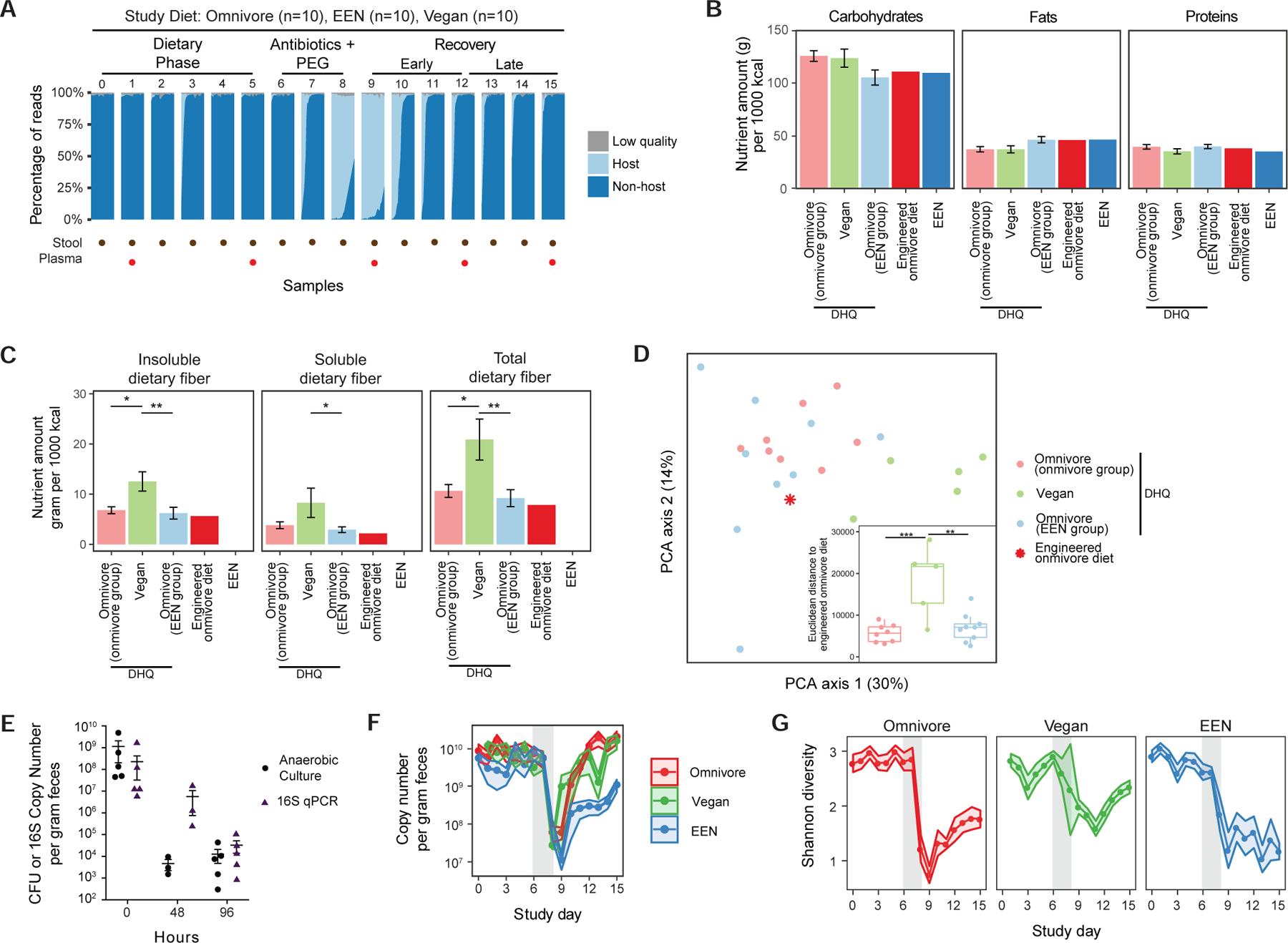
General response of the human gut microbiota during the three phases of the Food and Resulting Microbiota and Metabolite (FARMM) study. A) Overall study design is annotated at the top of the figure. The dots at the bottom of the figure represent the study days where either the fecal samples or plasma samples were collected. The plot shows the percentage of reads that were filtered for having low quality and for matching to the human genome (host) for each sample grouped by study day. The remaining non-host reads were further annotated with bacterial taxonomy and gene databases. B) Macronutrient compositions obtained from the NDSR analysis of the diet history questionnaires (DHQ) collected before the start of the study as well as the compositions of the study diets (N=17 for omnivores, N=5 for vegans). The omnivores were randomized to either receive the engineered omnivore diet or the EEN diet. C) Amounts of insoluble, soluble and total fiber consumed at baseline by all the diet groups as well as the levels in the engineered omnivore diet and EEN. Baseline diets were compared using linear models as shown (*q<0.05, **q<0.01, ***q<0.001). Ages of the subjects were added as a covariate. D) A Principal Component Analysis of the nutrient compositions from DHQ data with respect to the engineered omnivore diet. The sub plot shows the Euclidean distance between the DHQ data points and the engineered omnivore diet (*p<0.05, **p<0.01, ***p<0.001). Statistical testing was not performed for engineered omnivore diet and EEN diet since their composition does not vary from subject to subject (standard deviation is 0). E) The colony forming units (CFU) and 16S qPCR data in response to Abx/PEG intervention from a previous study (Ni et al., 2017). Data are represented as mean +/− standard error of the mean (SEM). F) qPCR results showing copy number of 16S genes per gram feces for each day in the study. The confidence intervals represent the SEM. The gray shaded area represents the antibiotic/PEG phase of the study. Linear mixed effects model was used to assess differences in copy number for each diet per study phase. G) Shannon diversity of the samples throughout the study. The confidence intervals represent the SEM. Linear mixed effects model was used to assess differences in diversity for each diet per study phase.
Throughout the study, vegan participants continued their diet as outpatients. Participants who had been consuming an omnivore diet prior to the study were randomized 1:1 to consume either an omnivore or an EEN diet under direct supervision in an inpatient research unit. The omnivore diet was designed to have similar carbohydrate, protein, and fat composition as the EEN diet, however there were differences in the constituents of the macronutrients such as the lack of fiber and a lower ratio of unsaturated to saturated fats in the EEN diet (Figure 1B and C). Diet history questionnaires (DHQs) of baseline diets showed that the subjects consumed comparable levels of total carbohydrates, fats, and proteins (Figure 1B, linear model p>0.05), however the vegan diet consisted of significantly fewer total calories, higher total fiber to sugar ratio, higher unsaturated to saturated fat ratio, and higher levels of both soluble and total dietary fiber compared to the omnivores (Figure S1B, linear model p<0.001 and Figure 1C). Baseline nutrient composition was not significantly different among omnivores assigned to consume either an omnivore diet or EEN (Figures 1B and C, linear model p>0.05). This is supported by the principal component plot of the nutrients normalized to amount per 1000 kcal consumed where the vegan diets are significantly different than the omnivores (Figure 1D, PERMANOVA p=0.04 and Figure S1C). The engineered omnivore diet was closer to the omnivore groups than the vegan group (Figure 1D, linear model p=0.001 and p<0.001 for EEN and omnivore groups, respectively).
As previously reported, based on counting colony forming units (CFU), the Abx/PEG intervention reduced viable bacteria by approximately 5 logs (Figure 1E) (Ni et al., 2017). The Abx/PEG intervention led to a reversible reduction in microbial biomass as documented by the proportion of shotgun reads annotated “host” vs. “non-host” (Figure 1A) and quantitative 16S copy number (Figure 1F). The percent of host reads were not different between diets during the three phases of the study (linear mixed effects model). The bacterial load did not change during the diet phase amongst any group (linear mixed effects model, p>0.05). However, recovery of bacterial load in the EEN diet group was slower than in the omnivore diet group (p<0.001). Shannon diversity was also not significantly altered during the dietary phase of the study (p>0.05) but recovery was lower in the EEN group relative to the other two diets (Figures 1G, linear mixed effects model p<0.001). A longer dietary phase would be needed to determine if this is due to a delayed recovery of the microbiome following Abx/PEG or the natural trajectory of the microbiome composition over a longer period of time on an EEN diet in the absence of the Abx/PEG intervention. In addition, the vegan microbiota was more resilient to the effects of the Abx/PEG intervention with a smaller decrease and greater recovery of diversity; this may have resulted from the vegan diet or due to vegans participating in the study as outpatients and as such being exposed to a different environmental reservoir (Ng et al., 2013) (Figure 1G, p=0.003).
EEN led to a significant change in the microbiota composition within 3 days of the dietary phase relative to the vegan and omnivore groups (Figure 2A, PERMANOVA test on Bray-Curtis distances p=0.03 and p=0.01 respectively). The vegan and omnivore groups were not significantly different from each other until day 7, which marks the introduction of PEG (Figure 2A, PERMANOVA test on Bray-Curtis distances). During the dietary phase of the study (days 1–5), two Rumincoccus genera belonging to Clostridia clade XIVa, R. gnavus and R. torques, increased in relative abundance on the EEN diet, and other taxa decreased, some also belonging to the same Clostridia clade (Figure 2B, linear mixed effects model).
Figure 2.
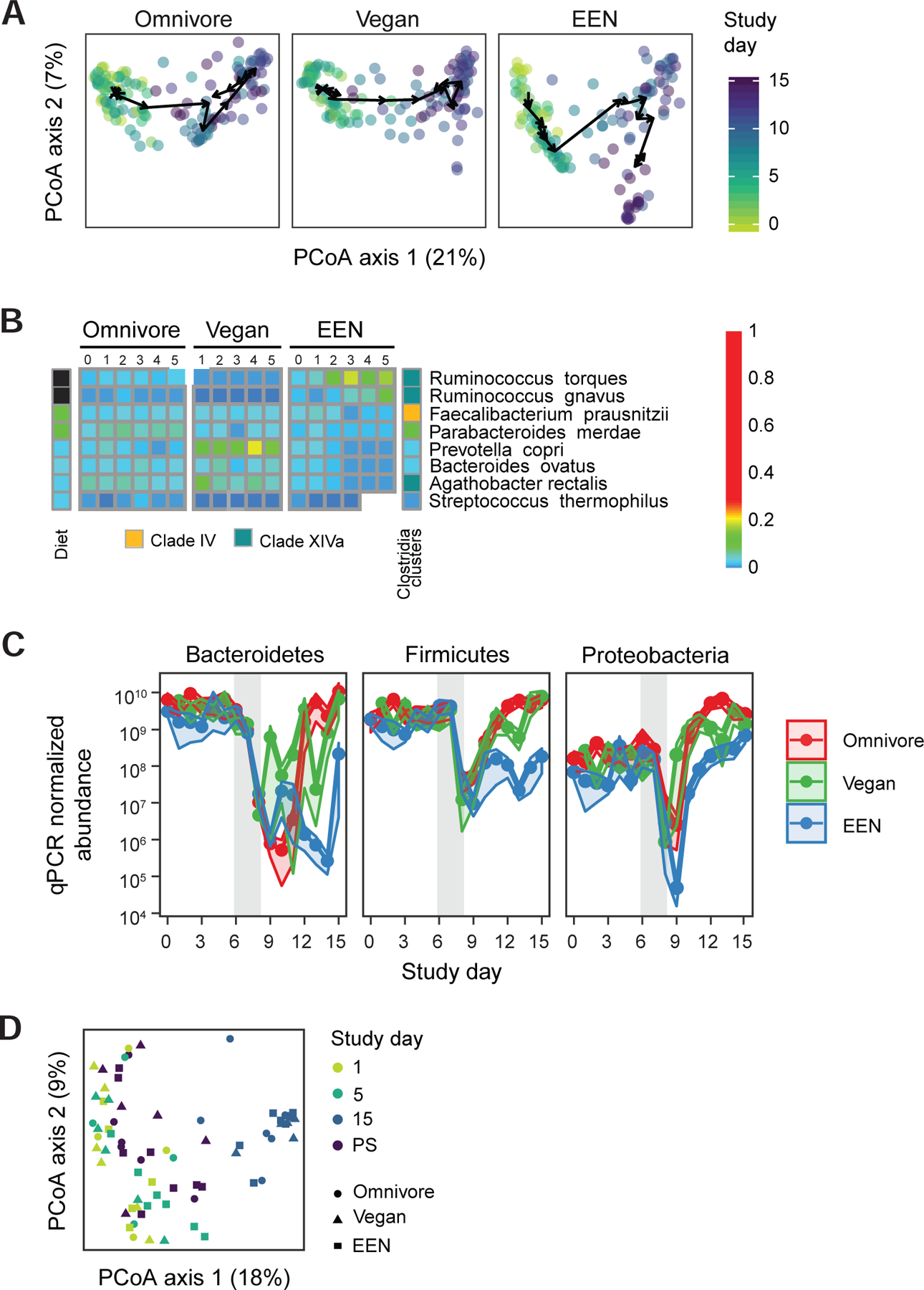
Taxonomic alterations of the human gut microbiome throughout the course of the FARMM study. A) Principal coordinate analysis of Bray-Curtis distances. All three facets share the same axes and can be overlaid. The axes are labeled with the percent variance explained. The arrows connect the centroids of consecutive time points for each diet. PERMANOVA test on Bray-Curtis distances was used to assess if the microbiome communities of the diet groups are different for each day. B) The taxa that are significantly different in EEN diet compared to the omnivore diet during the diet phase based on linear mixed effects models (q<0.05). The taxa that increase during the diet phase with the EEN diet are annotated in black and the taxa that decrease in abundance are annotated with white squares. Taxa are further annotated with the Clostridia clade to which they belong. C) The qPCR corrected relative abundance of three major phyla in three diets studied. The confidence intervals represent the SEM. The gray shaded areas represent the antibiotic/PEG phase of the study. Linear mixed effects model was used to assess differences in copy number corrected relative abundances for each diet per study phase. D) Principal coordinate analysis of Bray-Curtis distances of shotgun metagenomics data representing the samples collected 14–28 months after the Abx/PEG intervention (PS) from the subjects that participated in the original study as well as the samples collected on day 1 (pre Abx/PEG intervention), 5 (end of the diet phase), and 15 (end of study) of the original study.
Taxonomic alterations were more extreme during the recovery phase for all three diets (Figure 2A and Figure S2). Even though all three phyla showed an increase based on qPCR corrected relative abundances (linear mixed effects models q<0.001 for all three), the level of Proteobacteria during late recovery period was greater than the diet phase (linear mixed effects model q<0.001). However, only in the EEN group was the proportion of Proteobacteria greater relative to either Bacteroidetes or Firmicutes upon recovery (Figure 2C). This was due to a dominance of Klebsiella pneumonia and Enterobacter cloacea (Figure S2). At a Phylum level, this reflects the delayed return of Bacteroidetes and Firmicutes in the EEN group during the recovery phase since 16S rRNA gene copy number is much slower to recover in this group (Figure 2C). Best visualized in the PCoA analysis (Figure 2A), these results show that diet can be used to modify the human gut microbiota into alternative configurations upon recovery following a profound ecological community disturbance. The complete separation of the EEN group from vegans and omnivores with a predominance of Proteobacteria emphasizes the potential importance of diet in recovery of Bacteroidetes and Firmicutes in the human gut microbiota. However, these alterations were transient since assessment of the post study (PS) composition of the microbiome 14 to 28 months after discontinuation of the study diets demonstrated a similar composition to baseline (Figure 2D, PERMANOVA test on Bray-Curtis distances p>0.05 for all study groups).
Glycoside hydrolase gene representation is associated with diet-dependent alterations in the composition of the gut microbiota and the production of short chain fatty acids during both dietary and recovery phases
The largest differences in composition of the gut microbiota were evident: 1) at the end of the diet phase with the EEN group changing relative to baseline and the other two groups and 2) during the recovery from the Abx/PEG intervention where the microbiome of the EEN group lagged behind the other two groups. We hypothesized that the absence of dietary fiber within the EEN formula could explain these related observations. Thus, we next examined representations of genes involved in the degradation of fiber and production of SCFAs (El Kaoutari et al., 2013; Louis and Flint, 2017). To provide evidence that fiber is a major dietary component responsible for the diet-associated compositional alterations in the gut microbiota described in Figures 1 and 2, we performed an analysis of reads that aligned to glycoside hydrolase genes as well as genomic pathways important in the production of the short chain fatty acid, butyrate (Figures 3 and 4).
Figure 3.
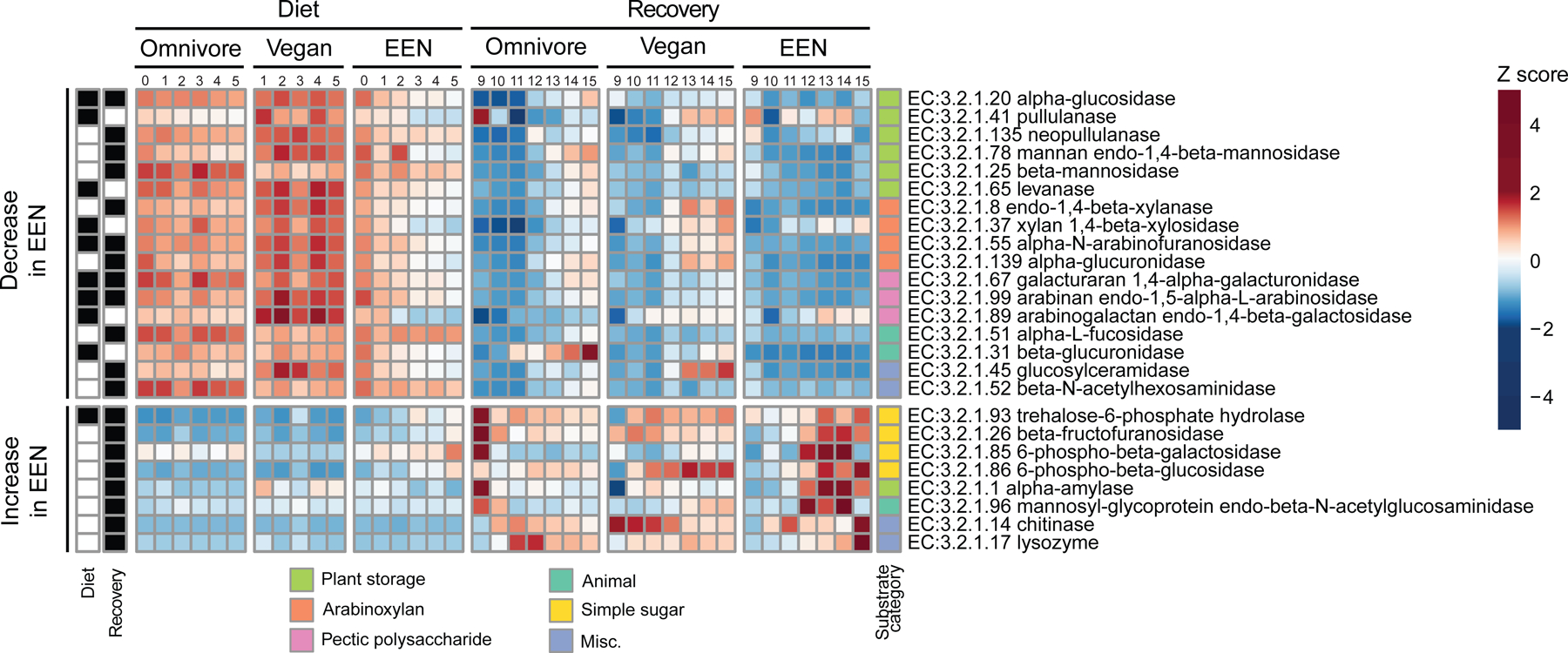
Genomic representation of glycoside hydrolase genes throughout the duration of the FARMM study. The glycoside hydrolase genes that have significantly different progression profiles in the EEN diet compared to the omnivore diet using linear mixed effects models on log transformed relative abundance of enzymes (q<0.05) are shown. The values represent the Z-scores of study day averages calculated per enzyme. The enzymes are grouped by their substrate category of plant based (storage, arabinoxylan, pectic polysaccharide), animal based, simple sugars or miscellaneous. The upper half of the plot shows any enzyme that shows a decreasing trend in EEN diet and the lower half of the plot shows an increasing trend in EEN diet compared to Omnivore diet. The black boxes denote if the statistically significant change was observed during the diet, recovery, or both phases. No enzymes had a statistically significant increase in one period and decrease in the other period.
Figure 4.
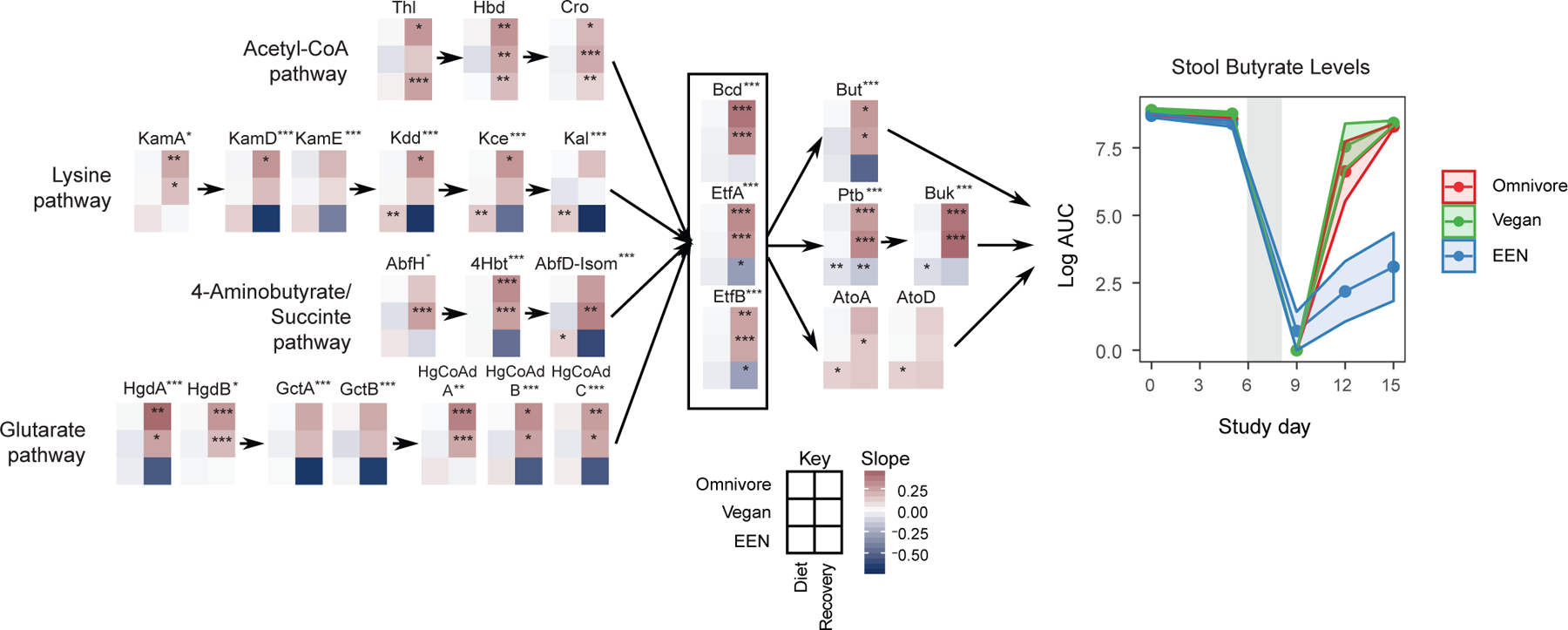
The human gut microbiome and butyrate production. Left Panel: The change in relative abundance of genes in four pathways responsible for butyrate production as curated by Vital et al.(Vital et al., 2014). Linear mixed effects models were fit to logit transformed gene relative abundance levels for each diet and study phase (diet or recovery) separately. Each 6 grid heatmap represents the slopes obtained from these linear mixed effects models where the rows represent the diet slopes and the columns represent the study phase. The stars within the heatmap boxes represent if the slope is significantly different than 0 based on linear mixed effects models. The p values were corrected for false discovery rate using Benjamini-Hochberg method (q<0.05). A separate linear mixed effects model was built to find the genes that show a different slope profile in EEN group compared to the omnivore group during the recovery phase. The stars next to the gene names represent if this progression profile of gene abundance is significantly different (*q<0.05, **q<0.01, ***q<0.001). Right Panel: Fecal butyrate levels throughout the duration of the FARMM study from untargeted metabolomics results. The confidence intervals represent the SEM. The gray shaded areas represent the antibiotic/PEG phase of the study.
Inferred protein sequences from metagenomic reads were aligned with the KEGG protein database focusing on the enzyme category 3.2.1, glycoside hydrolases. Relative to either omnivore or vegan diets, the EEN diet led to a reduction in the glycoside hydrolases responsible for degrading structural plant carbohydrates such as arabinoxylans (xylan 1,4 beta-xylanase, alpha-N-arabinofuranosidase) and pectic polysaccharides (galacturan 1,4-alpha-galacturonidase, arabinan endo-1,5-alpha-L-arabinosidase) and an increase in an enzyme, trehalose-6-phosphate hydrolase, as well as a trend for three others, involved in the digestion of sucrose or short fructooligosaccharides (Figure 3). This pattern is consistent with the abundance of simpler carbohydrates such as sucrose in EEN and absence of fermentable fiber. Similarly, during the recovery phase, subjects on the EEN diet showed a slower recovery of enzymes focused on plant-based glycans with a very robust increase in those involved in the digestion of more simple carbohydrates (Figure 3) consistent with the predominance of taxa belonging to the Proteobacteria phylum relative to either Firmicutes or Bacteroidetes phyla (Figure 2C) known to have a lower representation of glycoside hydrolases (El Kaoutari et al., 2013). The responsiveness of the human gut microbiome to dietary fiber is consistent with the transcriptomic signatures for the reduction in the mRNA expression of glycoside hydrolases in Bacteroides thetaiotomicron monoassociated gnotobiotic mice fed a fiber-free diet (Sonnenburg et al., 2005). We did not observe a robust genomic signal for an increase in animal glycan-degrading glycoside hydrolases (Figure 3), contrary to studies in mice where a fiber-free diet led to an increase in expression of genes for animal glycan degrading enzymes (Desai et al., 2016; Sonnenburg et al., 2005).
Butyrate is produced in the gut mainly from breakdown of complex polysaccharides by resident gut microbes (Louis and Flint, 2017). Our mass spectrometry platform allows for quantification of butyrate levels, but not acetate or propionate. Fecal butyrate levels were comparable during the diet phase among the groups (linear mixed effects model, p>0.05), declined significantly and comparably among groups during the Abx/PEG intervention phase (p<0.001), and recovered to a greater degree in the vegan and omnivore groups than in the EEN group during the recovery phase (p<0.001) (Figure 4). Consistent with previous reports (Baxter et al., 2019), there was a significant level of inter-subject variability in fecal butyrate levels in all three dietary groups, including omnivores where there was no decrease in variability over time despite all subjects consuming the same diet (Table S1 and Figure S3A). To see the changes in butyrate producing potential of the microbiome, we aligned inferred protein sequences from metagenomic reads against the amino acid sequences of proteins responsible for butyrate production categorized into four pathways, the major one based on carbohydrate metabolism via acetyl-CoA and three involving amino acid metabolism-lysine, aminobutyrate/succinate, and glutarate (Vital et al., 2014) (Figures 4 and S3B). There were no differences between diets in the relative abundance of the genes during the dietary phase of the study (Figure 4). During the recovery phase, the recovery of the butyryl-CoA dehydrogenase (Bcd) electron transfer flavoprotein (EtfA) complex (q<0.001), which is a keystone gene to produce butyrate regardless of the starting substrate, was slower to recover in the EEN group compared to the omnivore group (Figure 4). Two of the terminal genes to produce butyrate, butyryl-CoA:acetate CoA transferase (But) (q<0.001) and butyryl-CoA:4-hydroxybutyrate CoA transferase (4Hbt) (q<0.001), decreased in the EEN group during the recovery phase in contrast to the other two diets which showed increases. Several genes in pathways that start with amino acids as their major substrates, including KamD, KamE, Kdd, and Kce, showed similar significant trends with negative slopes. GctA, GctB, and HgCoAd C in the glutarate pathway and 4Hbt and Afbt-Isom in the 4-aminobutyrate pathway also showed reductions but these pathways were lower in abundance (Figure S3B). These results suggest that the lack of dietary fiber in EEN effects both carbohydrate- and amino acid-based production of butyrate.
Diet is a determinant of amino acid metabolism by the gut microbiome
Given that the analysis of butyrate production pathways revealed that both carbohydrate- and amino acid-based pathways were reduced in the EEN group upon the recovery of the microbiome (Figure 4), we sought to determine if there was a link between the resulting microbiome with the bacterial metabolite production dependent on non-carbohydrate substrates like amino acids. One example is the bacterial production of metabolites of aromatic amino acids such as tryptophan, tyrosine, and phenylalanine (Dodd et al., 2017). After production by the gut microbiota, some of these metabolites circulate systemically and can have physiologic effects on the host such as the enhancement of barrier (Venkatesh et al., 2014) and immune function (Zhang and Davies, 2016). We focused on indoleproprionic acid (IPA), a bacterial metabolite produced by several Clostridium spp. through a series of reductive aromatic amino acid metabolism steps. These steps have been defined specifically in Clostridium sporogenes by phenylactate dehydratase (FldABC) and etfA, which encodes electron transport flavoprotein subunit A (Dodd et al., 2017). Proportional gene representation for all four of these genes were significantly reduced in the microbiome of the EEN group during the recovery phase of the study (q<0.005 for all comparisons; Figure 5A). The gene acdA was most strongly associated with IPA fecal and plasma levels using a generalized linear mixed effect model (Figure 5B). Fecal metabolomic analysis revealed similar levels of tryptophan, the precursor aromatic amino acid needed for IPA generation, across the diet groups during the dietary phase of the study. Likewise, fecal and plasma levels of IPA were similar among groups during the dietary phase of the study (Figure 5A, p>0.05). Immediately after the Abx/PEG intervention on day 9, non-host reads and 16S rRNA gene copy number were at their nadir (Figures 1A and F, respectively), plasma levels of tryptophan were similar among the three diet groups and fecal levels were increased (linear mixed effect model p<0.001), yet IPA levels were undetectable in fecal and plasma samples (Figure 5). This confirms that the gut microbiome is the predominant source for the production of IPA. During the recovery phase, fecal and plasma IPA levels rose in the omnivore and vegan groups (p<0.001), but not in the EEN group (p=0.002 and p<0.001 for fecal and plasma levels respectively), consistent with the lower abundance of phenylactate dehydratase cluster gene representation in the EEN group during these same time points (Figure 5A). Fecal levels of tryptophan were elevated during the recovery phase in the EEN group relative to the other two diets (p=0.004) suggesting that, at least in part, this may be due to lack of conversion to IPA. Thus, diet can shape the functionality of the gut microbiome by altering both carbohydrate- and amino acid-based metabolism.
Figure 5.
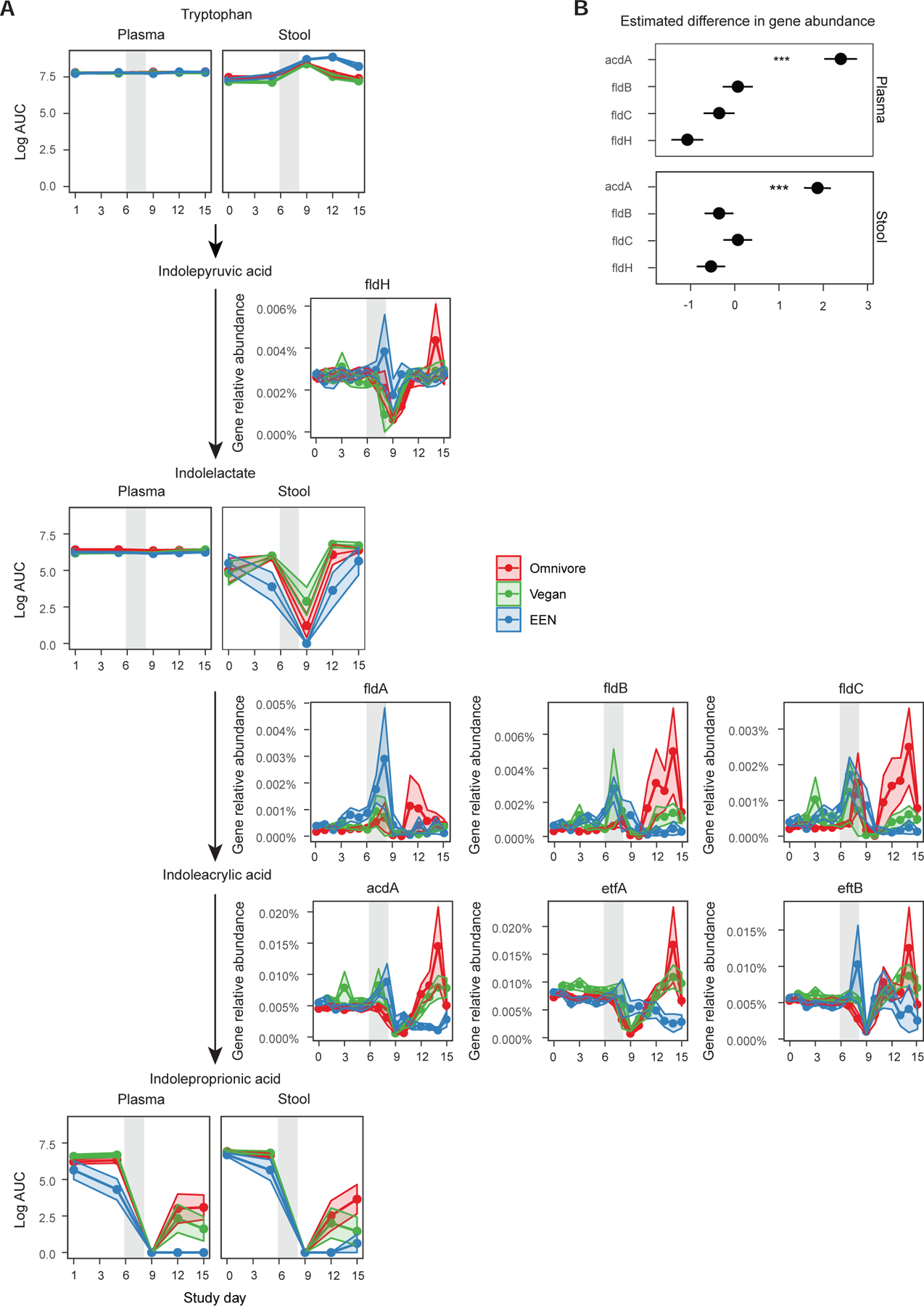
The reduction pathway of tryptophan to indole propionic acid. A) The intermediate metabolite levels in plasma and stool are shown for each diet group connected by the arrows to denote the progression of enzymatic reactions. The relative abundance of genes and their co-activators responsible for a reaction are denoted next to the arrows. The confidence intervals represent the SEM and the gray shaded areas represent the antibiotic/PEG phase of the study. Linear mixed effects model was used to assess differences in gene relative abundances or metabolite log area under the curve (AUC) values for each diet per study phase. B) Results of the generalized linear mixed effects model to determine which enzyme is a better predictor of indolepropionic acid levels in the plasma or the stool. The outcome was the presence or absence of indole propionic acid in stool or plasma (binary outcome transformed with binomial link) and the predictors were limited to the main enzymes and did not include any co-activators or cofactors to avoid collinearity.
Modeling of the metabolome reveals new associations between the gut microbiota, amino acids, and their metabolites
The robust reduction in bacterial biomass induced by the Abx/PEG intervention (Figure 1) allowed us to characterize the production of bacterial metabolites dependent upon carbohydrate and nitrogen metabolism via substrates delivered by diet. An untargeted analysis of known fecal metabolites at five time points across the sampling period showed that the trajectory of metabolite alteration was different for the EEN group on day 5 and onwards relative to the vegans and omnivores as visualized by a Principal Component Analysis (PCA) plot (Figure 6A, PERMANOVA fdr<0.01 for each time point). The fecal metabolomics profile of all three groups were the same at baseline (Figure 6A, PERMANOVA p>0.05). We developed a statistical nomenclature to classify longitudinal alterations in the abundance of fecal metabolites to specify metabolic functions contributed by the gut microbiome. We classified responses as an increase (coded as “1”), decrease (coded as a “3”), or no change (coded as a “2”) in the level of each fecal metabolite at each time interval based on the log ratio between each of the five sampled times points (four intervals) and a paired t-test corrected for multiple comparisons with q<0.1 (Figure 6B). In this manner, the pattern of each metabolite was assigned a 4-digit code (Table S2). The overall distribution of metabolites based on diet group classified by this code is shown in Figure 6C as relative proportions. The histogram to the right (log10 scale) shows the absolute numbers (Figure 6D). Among the 669 known metabolites identified in fecal samples, 333, 246, and 325 (50%, 37%, and 49%) were coded “2222” for the EEN, omnivore, and vegan groups, respectively (i.e., the concentration of these fecal metabolites appears to be independent of both the gut microbiome and diet). All metabolites in the vegan group have the code pattern “2XXX” indicating that there was no significant alteration during the dietary phase, which was expected since all 10 vegans remained outpatients and did not alter their diets during the study. Another common pattern was “2XX2” which indicates that the metabolites were mostly perturbed during the Abx/PEG intervention and early recovery.
Figure 6.
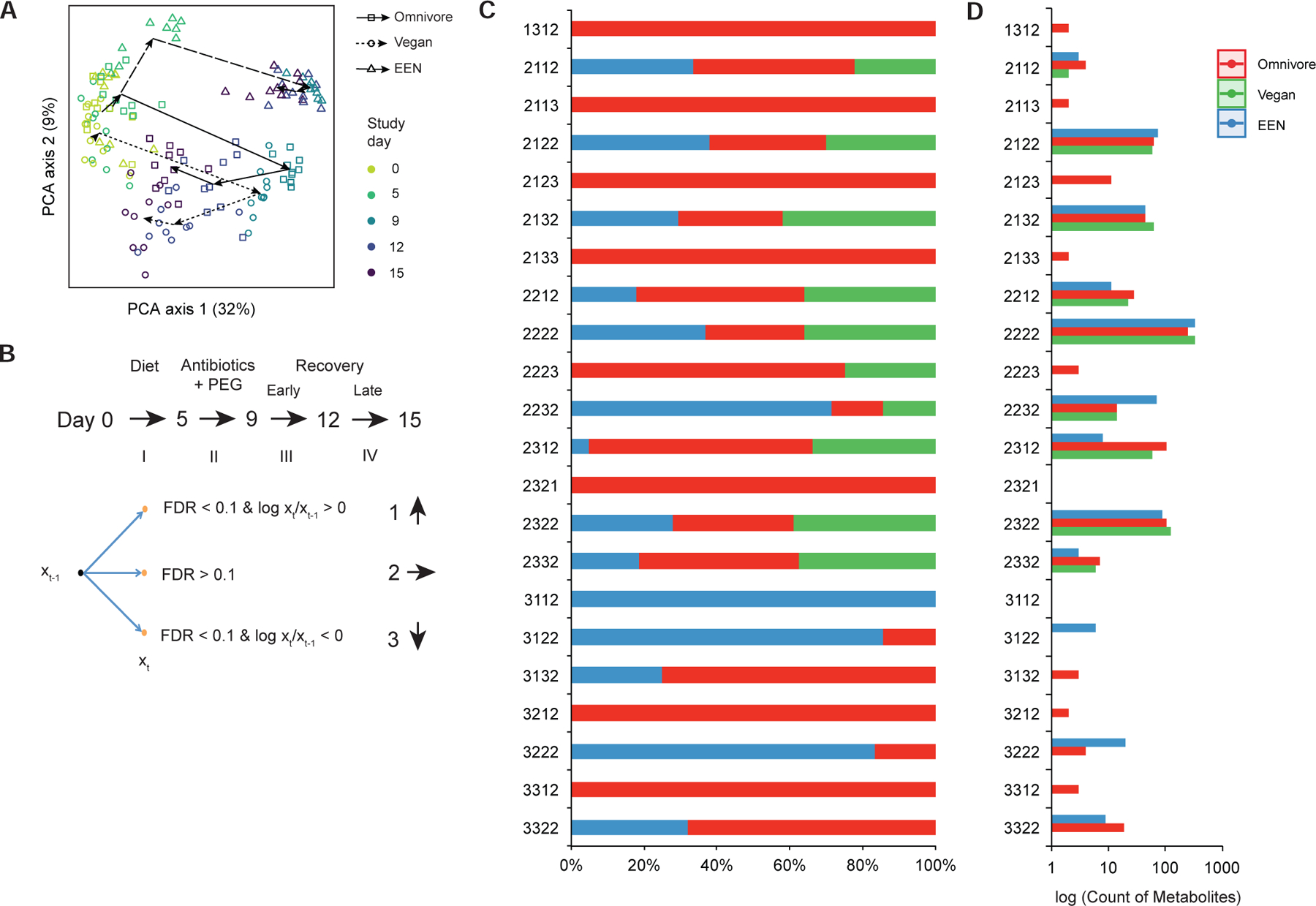
A statistical model to describe alterations in fecal metabolite levels across the four time intervals in the FARMM study. A) A Principal component analysis of fecal metabolites across the three diets (symbols) with colors representing each day of the study as indicated. The arrows connect the centroids of consecutive time points for each diet. B) Statistical model to provide a four-digit code per diet showing statistically-significant alterations of fecal metabolites across each of four time intervals using paired t-tests. Metabolite level changes in each interval were coded as an increase, no change or decrease (coded as a 1, 2, and 3, respectively) based on the criteria shown. C) Percent of fecal metabolites assigned to each of the four-digit codes that showed statistically-significant changes color coded by diet. D) Count of metabolites assigned to each four-digit code, log scale.
Based on the robust impact of the Abx/PEG intervention on bacterial load (Figure 1) and the effect of the EEN diet on the subsequent recovery of the gut microbiome and its impact on amino acid metabolism and IPA production (Figure 5), we performed an analysis focused on the effect of the Abx/PEG intervention (days 6–8) and the early recovery of the gut microbiome (days 9–12) on fecal and plasma amino acid levels and their derivatives. There was a consistent increase in fecal amino acids associated with the Abx/PEG intervention that was independent of diet (Figure 7A). This was likely a consequence of the purge of dietary nutrients from the gut before they can be absorbed in the small intestine, the colon, or metabolized by the gut microbiome given its low biomass immediately after the Abx/PEG intervention (Figures 1A and F). Most of these same amino acids decreased in the vegan and omnivore groups during the early recovery phase of the study (Figure 7A). It is likely that some of these amino acids, like tryptophan, tyrosine, and phenylalanine, are substrates for the production of amino acid metabolites as previously described (Dodd et al., 2017) (Figure 7B, asterisks). However, most of these same amino acids did not decrease in the EEN group during the early recovery phase, possibly due to the lack of bacterial consumption because bacterial biomass remained low (Figures 7A and 1F). We observed the opposite relationship for amino acid derivatives and other nitrogen-based metabolites. These generally decreased during the Abx/PEG intervention phase in all three dietary group and increased during the early recovery phase in both the vegan and omnivore groups, but did not increase in the EEN group (Figure 7B). This is also consistent with the impaired recovery of the gut microbiota on the EEN diet (Figure 1F). Despite the nearly three dozen fecal amino acid and nitrogen-based metabolites whose relative levels were significantly altered during either the Abx/PEG intervention and/or early recovery phases of the study, only a small fraction of these exhibit concurrent alterations in plasma levels (Figure 7). Interestingly, imidazole propionate, a product of bacterial histidine metabolism shown to be elevated in patients with type 2 diabetes and induces insulin resistance in mice (Koh et al., 2018), show an opposite change in plasma concentration relative to stool in the omnivore group during the purge phase (Figure 7B). Quantification of this metabolite in all subjects reveals that alterations in stool levels throughout the different intervals in FARMM (Figure S4A) do not reflect those observed in plasma levels (Figure S4B). There were significant group, time, and group by time effects in stool whereas plasma levels are generally consistent throughout the study in all three groups. In total, these results indicate that the impact of the gut microbiota on the human plasma metabolome may be modest. It is possible that species-specific pharmacokinetics of specific metabolites as well as alterations in intestinal absorption, which is reduced during and immediately after PEG consumption due to short transit time, may also be factors.
Figure 7.
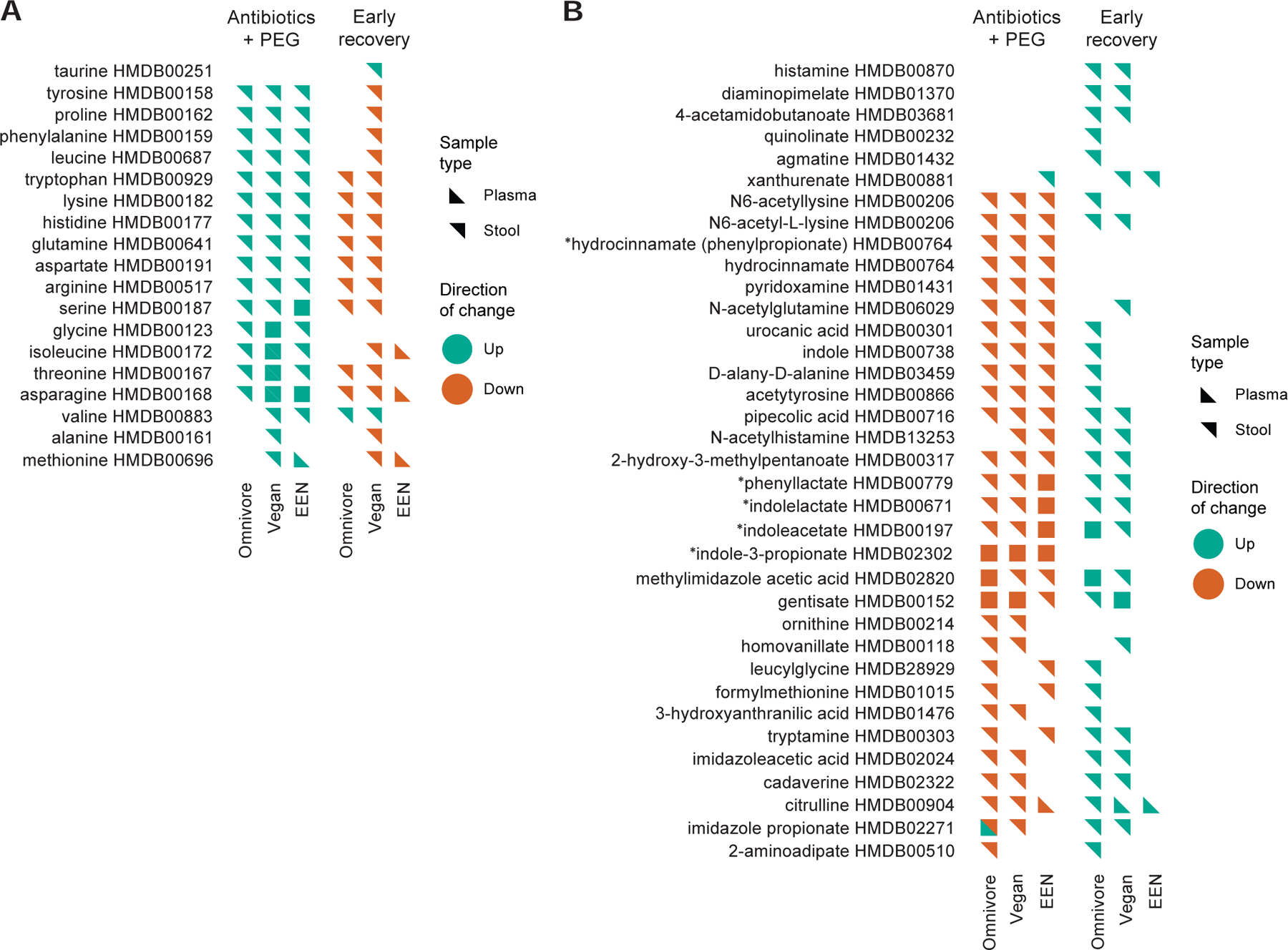
Lists of fecal metabolites, annotated as “amino acids” by the Human Metabolome Database (HMDB) with statistically-significant interval changes in both the plasma and stool using the paradigm described in Figure 6B. Interval changes during both the “Abx/PEG intervention” and “Early Recovery” phases of the FARMM study for each of the three diets are shown. A) Amino acids, many purged from the gut and likely to be consumed by the gut microbiota upon early recovery. B) Amino acid and other nitrogen-based metabolites likely to be produced by the gut microbiota.*=Bacterially-produced amino acid metabolites described in(Dodd et al., 2017). Green=Interval increase in relative abundance. Orange=Interval decrease relative abundance. Orientation of the triangle symbol indicates whether the interval alteration in the metabolite was observed in the plasma or stool. HMBD annotations are provided for each metabolite.
To identify those metabolites that are likely produced by the gut microbiota, we considered those amino acid metabolites that were reduced in the intervention phase across all three group and increased in the early recovery phase only among the vegan and omnivore groups. The aromatic amino acid reduction pathway, which is critical for the production of IPA and was originally described for C. sporogenes, has also been identified in three strains of Clostridium cadaveris as well as in Peptostreptococcus anaerobius, where it can lead to the production of 12 metabolites-9 are known to accumulate in host plasma, at least 3 of which are exclusively produced by the gut microbiota (Dodd et al., 2017). Abx/PEG intervention greatly reduced the number of reads that aligned to the genomic region responsible for IPA production from these bacterial taxa in all three diet groups (Figures S5A and B, days 7 to 9). During the recovery phase, there was a robust increase in the abundance of these taxa in the omnivore and vegan groups, but not the EEN group (Figures S5A and B, day 9 to 15). Five of these 12 metabolites were reduced during the Abx/PEG intervention phase of the study independent of diet whereas three were increased during the recovery phase of the study in both the omnivore and vegan groups but not the EEN group (Figure 7B, asterisks). There are about two dozen fecal metabolites that have similar patterns (i.e., decreased levels in the Abx/PEG intervention phase across all three dietary groups followed by increased levels in only the omnivore and vegan groups during the recovery phase) (Figure 7B). Production of some of these metabolites may also be dependent upon the gut microbiome. For example, cadaverine is a natural polyamine with multiple biological activities that has been characterized as a bacterial metabolite (Miller-Fleming et al., 2015).
With the notion that these metabolite patterns closely track with the bacterial load results (Figure 1F), we looked further into the possibility that the metabolites that decrease in abundance during the purge when there is low bacterial load and increase as the bacterial load recovers might be produced by or associated with gut bacteria. We reviewed data in the Human Metabolome Database (HMDB) v4.0 for the 36 metabolites listed in Figure 7B (Wishart et al., 2018). Eight of these metabolites have been described to be of microbial origin and found in feces (cadaverine, diaminopimelate, formylmethionine, hydrocinnamate/phenylpropionate, indole-3-propionate, indole, indoleacetate, and phenyllactate) which, when added to others that have previously been shown to be generated from bacterial amino acid metabolism (indole lactate) (Dodd et al., 2017), leaves 27 metabolites that have not been previously annotated as having a microbial origin associated with a mammalian host. One of these, D-alany-D-alanine, is a constituent of peptidoglycan in bacterial cell walls. A manual literature search (PubMed and Google Scholar using a combination of the metabolite name/HMDB ID, “microbial production”, “microbial metabolism”, “bacterial production”, and “bacterial metabolism”) was conducted for the remaining 26 metabolites and identified primary source literature describing the involvement of the metabolite in a bacterial metabolic pathway for 24 of the 26 metabolites. Many of the studies identified are not in host-associated organisms or communities, and many were published prior to 2000 (Table S3). Thus, these represent known microbial metabolites that have not previously been linked to gut-associated communities.
The remaining two metabolites lack primary source literature linking them to bacterial metabolism. However, one (methylimidazole acetic acid) can be linked to bacterial processes via two-step pathways in the literature (e.g., the metabolite has been placed in a specific pathway based on literature and/or available pathway descriptions [e.g., KEGG], and that pathway is present in bacteria, but the individual metabolite has not been studied in bacteria) (Table S3). The remaining metabolite, acetyltyrosine, to the best of our knowledge, has not been previously linked to microbial metabolism. It is possible that this represents a previously unknown microbial metabolite that is linked to host-associated communities. Together, through the manipulation of the gut microbiota with Abx/PEG purge and three different diets, we have identified 27 novel metabolites that appear to be a product of human gut microbiota metabolism.
Discussion
There is great interest in the impact of dietary fiber on health and disease, in part, mediated via its effects on the composition of the gut microbiota and its metabolites. Despite a multitude of studies in rodent models showing that dietary fiber supplementation has a large effect on the composition of the gut microbiota and the production of metabolites such as SCFAs, data in humans is more limited. Most studies observed that the effects are relatively modest (Baxter et al., 2019; Johnson et al., 2019; O’Keefe et al., 2015; Wu et al., 2016). In this study, the use of a fiber-free EEN formulation had pronounced effects on the human gut microbiome that exceeds the difference between the effects of an omnivore vs. vegan diet. We also assessed the impact of the EEN diet on the recovery of the microbiome after an intervention that reduced bacterial load by about 5 logs. The short-term recovery of the gut microbiome was dramatically reduced on an EEN diet, and, unexpectedly, the resulting bacterial metabolites of both carbohydrate and amino acid origins were altered suggesting a broad impact of dietary fiber on bacterial metabolome. This may represent a conceptual advance over the current thinking that the gut microbiota metabolizes a single substrate class into a similar class of small molecules. Rather, depriving or supplying the human microbiome with one dietary component (i.e., fiber) can directly impact metabolites of an unrelated portion of the diet (i.e., amino acids) via the induction of specific gut bacterial taxa. Based on statistical modeling of the latter, we provide evidence for a number of amino acid metabolites that are likely to be of bacterial origin but have not been previously associated with the gut microbiome providing additional support for the importance of dietary fiber on a broad spectrum of bacterial metabolites.
During the dietary phase of our FARMM study, the composition of the gut microbiota was similar between omnivores and vegans, consistent with our previous research (Wu et al., 2016). In contrast, EEN consumption led to fecal microbiome composition and metabolites that were distinct from omnivores and vegans. These changes occurred within a few days and were notable for an induction of two bacterial taxa belonging to the Clostidium clade XIVa, Ruminococcus torques and gnavus. The lack of fiber in EEN may have led to a growth advantage of these taxa, which some strains are known to be mucinophilic (Crost et al., 2013; Henke et al., 2019; Png et al., 2010). Analysis of genomic representation of glycoside hydrolases showed that the fiber-free composition of the EEN diet led to a decrease in the ability to hydrolyze plant cell wall glycans with an increase in the ability to digest more simple carbohydrates found in the EEN formula. Consumption of EEN did not lead to a robust increase in glycoside hydrolases targeting animal glycans found in mucus, as has been observed in gnotobiotic mouse studies (Desai et al., 2016; Sonnenburg et al., 2005). Although this might be an indication of the differences between murine models and human biology, it is also possible that 5 days on an EEN diet was not sufficient to observe such an effect. Indeed, we did not observe a decrease in fecal butyrate levels within the first 5 days.
The combination of antibiotics, a gut purge, and a diet free of fermentable carbohydrates profoundly altered the human gut microbiome configuration leading to a high proportions of Proteobacteria. This was due to the reduced ability of both Bacteroidetes and Firmicutes to repopulate the gut during the recovery phase, demonstrating the phylum-wide dependence on dietary fiber to re-establish their intestinal niche. Similar findings have been observed in humanized gnotobiotic mice where a fiber-deficient diet exacerbated microbiota collapse upon antibiotic treatment and delayed recovery (Ng et al., 2019). The presumed mechanism is the predominance of glycan degrading enzymes encoded in the genome of taxa in both the Bacteroidetes and Firmicutes phyla relative to Proteobacteria (El Kaoutari et al., 2013). Indeed, during the recovery phase of those subjects on an EEN diet, there was a reduction of glycoside hydrolases, a reduced representation of butyrate producing pathways, and a reduction in fecal butyrate levels relative to the other two diets.
The reconfiguration of the gut microbiome into a reduced fermentative state through a combination of both antibiotics and a fiber-free diet has a number of potentially important implications for gut microbiome-dependent outcomes in patients. Since an increased relative proportion of Proteobacteria, particularly Enterobacteriaceae, with a reduction in Firmicutes is a predominant signature for dysbiosis in inflammatory bowel diseases (Nagalingam and Lynch, 2012), a configuration known to be deleterious in animal models of colitis (Sartor and Wu, 2017; Zhu et al., 2018), our results suggest that the combination of antibiotics with EEN may be less effective in patients with Crohn’s disease than EEN alone, and could be potentially harmful. Although antibiotics with a gut purge (PEG) and EEN diet is not a standard clinical care of patients with Crohn’s disease, the results of this study suggest that there may be merit in testing supplementation of EEN diets with fermentable fiber for the treatment of Crohn’s disease and/or ulcerative colitis; in the latter EEN is thought to be ineffective. Similarly, there might be value in exploring the impact of dietary fiber supplementation to reduce the risk for bacterial sepsis in seriously ill patients receiving antibiotics plus enteral nutrition by reducing Proteobacteria-dominance of the gut microbiota and preserving richness (McDonald et al., 2016). Given the robust effect of a fiber-free diet coupled with antibiotics and bowel lavage on the human gut microbiome in this study and in mouse models where dietary fiber has been depleted (Desai et al., 2016; Sonnenburg et al., 2016; Zou et al., 2018), we confirmed that EEN-induced alterations in the gut microbiota spontaneously resolved post-study.
The robust taxonomic and metabolomic alterations of the gut microbiome observed during the recovery phase of subjects consuming EEN, where there is a much lower abundance of Firmicutes relative to the other two diets (Figure 2C), led to an unexpected finding: not only does the lack of dietary fiber reduce the production of carbohydrate metabolites such as butyrate, but also a number of amino acid-based metabolites that have been shown to be produced by Clostridium spp. (Dodd et al., 2017; Wlodarska et al., 2017). As an example, we show that there is reduced production of indolepropionic acid from tryptophan upon the recovery phase in the EEN group (Figure 5) which corresponds to a lower abundance of Clostridium spp. known to express enzymes in the aromatic amino acid reductive pathway (Figure S5). Since four additional amino acid metabolites produced by similar pathways (Dodd et al., 2017) also do not increase in the EEN group during the recovery phase (see asterisks, Figure 7B), we believe that Firmicutes may play a role in a number of additional amino acid metabolites listed in Figure 7B and Table S3. The broad impact of dietary fiber on the microbiota-dependent generation of metabolites from both carbohydrate and amino acid components of diet suggests that the effects of fiber on host physiology has the capability to extend well beyond those only attributed to bacterial carbohydrate metabolism (Dodd et al., 2017; Zhang and Davies, 2016).
We extended this notion beyond previously described bacterial amino acid metabolites. From approximately 700 known metabolites in this non-targeted analysis, we identified a dozen additional amino acid derivatives that follow the same pattern of expression and which are known to be produced by bacteria as well as approximately an equal number not previously known to be microbial metabolites. The latter might serve as a starting point to identify novel bacterial metabolic pathways that may link diet to the health of humans. Only a small number of these fecal metabolites show similar alterations in plasma, possibly due to the impact of intestinal epithelial metabolism and/or first pass metabolism by the liver. One metabolite, imidazole propionate, showed opposite trends in the stool and plasma of the omnivore group. Despite a decrease in omnivore stool levels, imidazole propionate increased in the plasma of omnivores during the purge phase whereas plasma levels remained unchanged while stool levels increased during the recovery phase. Reasons that might explain these differences include variation in the half-life of this metabolite in stool vs. plasma or the altered metabolism of the subjects during the purge phase that might have led to an increase. With respect to the latter, germ-free mice have quantifiable levels of plasma imidazole propionate (Koh et al., 2018).
A challenge in most feeding studies is the inability to isolate the effect of a single dietary component. By conducting this study in an inpatient setting, we had complete control of the diet for participants in the omnivore and EEN arms. We used random assignment to balance known and unknown confounders. We also matched the omnivore diet to the overall macronutrient composition of the EEN formula. Nonetheless, by design it was necessary for the composition of the two diets to differ. EEN lacks complex carbohydrates and replaces these calories with simple sugars. Other components of the diets differed slightly and could potentially have contributed to the observations. Indeed, we only had access to publicly available information about the overall composition of Modulen®. However, the absence of fiber as well as the consistent changes in both the taxonomy of the microbiome and its gene representations all point to fiber as the driving force in the observed microbiome and metabolome alterations observed in this study. A future study of fiber-free EEN versus EEN plus fiber could further isolate the effect of fiber on these physiologic processes.
In summary, by characterizing the effect of three different diets in a controlled feeding experiment, we show the importance of dietary fiber on both the composition and metabolite production of the human gut microbiome. Effects were context-dependent, the greatest impact being on taxa belonging to the Firmicutes phylum. Normally, the absence of dietary fiber leads to the expansion of specific mucinophilic Clostridium spp. However, upon depletion of the gut microbiota with antibiotics, fiber plays an important role in both preservation of microbial biomass and diversity and the recovery of the gut microbiome with an effect on a broad range of metabolites that are both carbon- and nitrogen-based. Furthermore, with this model, we have identified new metabolites that appear to be derived by the microbiota from dietary sources in humans. The lessons learned from the FARMM study may not only have direct and practical impact on the approach to patients consuming diets low in fermentable fiber in whom there has been a disruption of the gut microbiota, but also a more general impact focused on diet-dependent production of microbiota metabolites and their role in human health and disease.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact:
Further information and requests for resources and reagents should be directed to Gary D. Wu (gdwu@pennmedicine.upenn.edu).
Materials Availability:
This study did not generate new unique reagents.
Data and Code Availability:
Shotgun metagenomic sequence files are available at the Sequence Read Archive (SRA) under accession number PRJNA675301. Metabolomics data are available at the National Metabolomics Data Repository (www.metabolomicsworkbench.org) under accession number PR001024. The code generated during this study are available at Github at https://github.com/PennChopMicrobiomeProgram/FARMM.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects:
Thirty-one healthy volunteers between the ages of 18 and 60 were included in the study, however one withdrew before completing the protocol. As a result, 30 are included in the analysis, 10 in each group (for further characteristics of the participants, see Table S4). Self-reported vegans were required to have followed a vegan diet for a minimum of 6 months prior to enrollment. Key exclusion criteria included inflammatory bowel disease, celiac disease, or other chronic intestinal disorders; prior bowel resection surgery other than appendectomy; baseline bowel frequency less than every 2 days or greater than 3 times daily; creatinine concentration greater than the upper limit of normal; diabetes mellitus; currently smoking; body mass index (BMI) <18.5 or >35; and use of antibiotics or probiotics in the prior 6 months. The 10 vegans continued to follow their usual diet as outpatients. All participants completed the Diet History Questionnaire II (DHQ II), a food frequency questionnaire developed by the Risk Factor Monitoring and Methods Branch of the National Cancer Institute. The 10 vegans continued to follow their usual diet as outpatients. They also completed three 24-hour diet recalls with a dietitian in the week prior to starting antibiotics. We randomly assigned the 20 omnivores to receive an omnivore diet or EEN (Modulen® IBD) while residing in an inpatient research unit. The macronutrient composition of Modulen®, as per the manufacturer, is protein 36g, fat 47g, and carbohydrate 110g per 1000 Kcal (more detailed information about composition is not publicly available). The two omnivore diets were engineered to have a similar composition to EEN. All the inpatient subjects consumed the menu A on day 11 and menu B on days 4 and 14. The menus of the inpatient omnivores are listed in Table S1. A clear liquid diet was used on the day of the bowel purge for all participants. Diets for omnivores were constructed to provide the expected total calories required per day for the participant to maintain their current weight and were adjusted if there was weight gain or loss of more than 2.5 pounds. On days 6, 7, and 8, inpatient participants received vancomycin 500mg orally every 6 hours and neomycin 1000mg orally every 6 hours. On day 7, participants consumed 4L of polyethylene glycol (PEG) based bowel purgative (GoLytely®). Participants in the omnivore and EEN arms left the inpatient research unit only under the direct supervision of a research staff member. The vegan outpatients reported to the hospital twice on days 6, 7 and 8 to receive antibiotics and to consume PEG on day 7. The first stool sample of each day of the inpatient groups was collected, aliquoted and frozen immediately at −80°C. Blood was collected on days 1, 5, 9, 12 and 15 from which plasma aliquots were immediately isolated and frozen at −80°C. The outpatient participants following the vegan diet also had blood collected on days 1, 5, 9, 12, and 15 from which plasma aliquots were immediately isolated and frozen at −80°C. Among these participants, the first stool of the day was collected at home daily and kept on ice packs until it was brought to the research unit where it could be aliquoted and frozen. Samples from vegans were received for aliquoting within 24 hours and on average within 4 hours (Wu et al., 2010). Day 0 stool was not collected from vegans since their diet did not change. Follow-up samples were collected from participants 14–28 months after completion of the study at an outpatient visit. The aliquoted amounts for all samples ranged from 500 mg to 1 g. The aliquots were taken from different areas of the sample. Remaining sample was then collected into one residual 50 ml conical tube with a tongue depressor. Any remaining stool was discarded. The University of Pennsylvania Institutional Review Board (IRB) approved the research protocol and considered it exempt from clinical trial registration requirements based on the protocol’s stated objectives.
METHOD DETAILS
DNA purification, library preparation and sequencing:
DNA was extracted from approximately 200 mg of stool using the Qiagen DNeasy PowerSoil kit. Extracted DNA was quantified with the Quant-iT PicoGreen® dsDNA assay kit (ThermoFisher). Shotgun libraries were generated using the NexteraXT kit and sequenced on the Illumina HiSeq 2500 using 2x125 bp chemistry. Extraction blanks and DNA free water were included to empirically assess environmental and reagent contamination. Laboratory-generated mock communities consisting of DNA from Vibrio campbellii and Lambda phage were included as positive sequencing controls (Figure S6).
qPCR analysis:
A quantitative PCR (qPCR) assay targeting the 16S rRNA gene was used to estimate bacterial abundance. Primer sequences were 5’-AGAGTTTGATCCTGGCTCAG-3’ and 5’-CTGCTGCCTYCCGTA-3’. The probe sequence was 5’-TAACACATGCAAGTCGA-3’(Hill et al., 2010). Reactions were performed in triplicate with TaqMan™ Fast Universal PCR Master Mix (Thermo Fisher Scientific, Waltham, MA) with the conditions: 20 s at 95°C followed by 40 cycles of 3 s at 95°C and 30 s at 60°C. A plasmid containing the full length 16S rRNA gene from Streptococcus was used to generate the standard curve.
Microbial Culturing:
Freshly voided fecal samples were weighed and placed in an anaerobic chamber (2.5% H2, balance nitrogen; Coy Labs, Grass Lake, MI) within one hour of collection. The samples were homogenized in phosphate buffered saline with 0.1% cysteine, and serial dilutions were plated on gut microbiota medium (Goodman et al., 2011) and incubated at 37°C for 24 hours.
Bioinformatics processing:
Sequences from the Illumina HiSeq 2500 were demultiplexed (https://github.com/PennChopMicrobiomeProgram/dnabc). The average read depth of the samples were 10.6 ± 4.7 million read pairs. The adapters were trimmed with the Trimmomatic software using default parameters(Bolger et al., 2014). Host and phiX derived reads were removed from the samples by aligning the reads to the Human genome version hg38.v4 and the phiX genome obtained from NCBI (Li and Durbin, 2009). The abundance of bacteria was estimated using MetaPhlAn software(Segata et al., 2012). Sample similarity was assessed by Bray-Curtis distance and alpha diversity was assessed by Shannon diversity metric (https://github.com/PennChopMicrobiomeProgram/PathwayAbundanceFinder). Reads were mapped to the KEGG protein database (Ogata et al., 1999) to estimate the abundance of bacterial gene orthologs using RAPSearch2(Zhao et al., 2012). The abundance of orthologs were then mapped to Enzyme Commission (EC) numbers annotations already present in the KEGG database. The enzymes related to glycoside hydrolases were defined as all the enzymes that start with an EC number of EC:3.2.1 and analyzed further. The reads were also aligned to curated databases of proteins involved in butyrate production(Vital et al., 2014) and amino acid reduction into indole propionic acid (IPA) (Dodd et al., 2017) using DIAMOND search(Buchfink et al., 2015). Additionally, the reads were aligned against the genomic regions responsible for IPA production using bwa(Li and Durbin, 2009). Quality control, read filtering, and the number of reads to these databases are provided in Supplementary Table S5.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis:
Sample size and statistical power was estimated based on detecting a difference in metabolite concentration between two time periods using a paired t-test. With a sample size of 10 per group and 80% power, the detectable difference for an alpha error of 0.05 is 1 unit of standard deviation. Using a Bonferroni correction for 600 metabolites, the detectable difference is 2.42 units of standard deviation. Ultimately, we included 669 metabolites in the analyses. The differences between diet groups across time in qPCR levels and Shannon diversity were assessed using linear mixed effects models with the diet groups and study days as fixed effects and the subject IDs as random effects. The models were built for each arm of the study separately (diet, antibiotics, recovery). Community-level differences between sample groups were assessed using the PERMANOVA test. Bacterial species and gene abundance levels were tested using linear mixed effects models on logit transformed relative abundances. When multiple tests were performed, the p-values were corrected for false discovery rate using Benjamini-Hochberg method. Subjects’ BMI and age were added as covariates in all of the regression models.
Metabolomic analyses of fecal and plasma samples.
Stool samples were homogenized using a bead mill (TissueLyser II, QIAGEN), and the aqueous homogenates were aliquoted for metabolite profiling analyses. Four separate LC–tandem MS methods were used to measure polar metabolites and lipids in each sample. Methods 1, 2, and 3 were conducted using two LC-MS systems composed of Nexera X2 UHPLC systems (Shimadzu Scientific Instruments) and Q Exactive Hybrid Quadrupole-Orbitrap MSs (Thermo Fisher Scientific), and method 4 was conducted using a Nexera X2 UHPLC (Shimadzu Scientific Instruments) coupled to an Exactive Plus Orbitrap MS (Thermo Fisher Scientific). Raw data were processed using Progenesis QI software (NonLinear Dynamics) for feature alignment, nontargeted signal detection, and signal integration. Targeted processing of a subset of known metabolites was conducted using TraceFinder 3.3 software (Thermo Fisher Scientific).. Statistical analyses were conducted on log10 transformed area under the curve values, and metabolite values below the limit of detection were imputed as 1 to allow for log transformation. PERMANOVA on Euclidian distances was used to establish metabolomic composition differences between diets at each time point. Paired t-tests on individual metabolites over each of the 4 time intervals for each diet group was done (with Bonferroni multiple comparison adjustments). The results were then classified as an increase (coded as “1”), decrease (coded as a “3”) or no change (coded as a “2”). For each metabolite, to summarize the significance over 4 time intervals, we took the smallest of the Bonferonni adjusted p value and then performed FDR control over all the metabolites.
Method 1 – positive ion mode MS analyses of polar metabolites.
LC-MS samples were prepared from stool homogenates (10 μL) via protein precipitation with the addition of nine volumes of 74.9:24.9:0.2 v/v/v acetonitrile/methanol/formic acid containing stable isotope-labeled internal standards (valine-d8, Isotec; and phenylalanine-d8, Cambridge Isotope Laboratories; Andover, MA). The samples are centrifuged (10 min, 9,000 x g, 4°C), and the supernatants were injected directly onto a 150 x 2 mm Atlantis HILIC column (Waters; Milford, MA). The column was eluted isocratically at a flow rate of 250 μL/min with 5% mobile phase A (10 mM ammonium formate and 0.1% formic acid in water) for 1 minute followed by a linear gradient to 40% mobile phase B (acetonitrile with 0.1% formic acid) over 10 minutes. MS analyses were carried out using electrospray ionization in the positive ion mode using full scan analysis over m/z 70–800 at 70,000 resolution and 3 Hz data acquisition rate. Additional MS settings were: ion spray voltage, 3.5 kV; capillary temperature, 350°C; probe heater temperature, 300°C; sheath gas, 40; auxiliary gas, 15; and S-lens RF level 40.
Method 2 – negative ion mode MS analysis of polar metabolites.
LC-MS samples were prepared from stool homogenates (30 μL) via protein precipitation with the addition of four volumes of 80% methanol containing inosine-15N4, thymine-d4 and glycocholate-d4 internal standards (Cambridge Isotope Laboratories; Andover, MA). The samples were centrifuged (10 min, 9,000 x g, 4°C) and the supernatants were injected directly onto a 150 x 2.0 mm Luna NH2 column (Phenomenex; Torrance, CA). The column was eluted at a flow rate of 400 μL/min with initial conditions of 10% mobile phase A (20 mM ammonium acetate and 20 mM ammonium hydroxide in water) and 90% mobile phase B (10 mM ammonium hydroxide in 75:25 v/v acetonitrile/methanol) followed by a 10 min linear gradient to 100% mobile phase A. MS analyses were carried out using electrospray ionization in the negative ion mode using full scan analysis over m/z 60–750 at 70,000 resolution and 3 Hz data acquisition rate. Additional MS settings were: ion spray voltage, −3.0 kV; capillary temperature, 350°C; probe heater temperature, 325°C; sheath gas, 55; auxiliary gas, 10; and S-lens RF level 40.
Method 3 – negative ion mode analysis of metabolites of intermediate polarity (e.g. bile acids and free fatty acids).
Stool homogenates (30 μL) were extracted using 90 μL of methanol containing PGE2-d4 as an internal standard (Cayman Chemical Co.; Ann Arbor, MI) and centrifuged (10 min, 9,000 x g, 4°C). The supernatants (10 μL) were injected onto a 150 x 2 mm ACQUITY T3 column (Waters; Milford, MA). The column was eluted isocratically at a flow rate of 400 μL/min with 25% mobile phase A (0.1% formic acid in water) for 1 minute followed by a linear gradient to 100% mobile phase B (acetonitrile with 0.1% formic acid) over 11 minutes. MS analyses were carried out using electrospray ionization in the negative ion mode using full scan analysis over m/z 200–550 at 70,000 resolution and 3 Hz data acquisition rate. Additional MS settings were: ion spray voltage, −3.5 kV; capillary temperature, 320°C; probe heater temperature, 300°C; sheath gas, 45; auxiliary gas, 10; and S-lens RF level 60.
Method 4 – polar and nonpolar lipids.
Lipids were extracted from stool homogenates (10 μL) using 190 μL of isopropanol containing 1-dodecanoyl-2-tridecanoyl-sn-glycero-3-phosphocholine as an internal standard (Avanti Polar Lipids; Alabaster, AL). After centrifugation (10 min, 9,000 x g, ambient temperature), supernatants (10 μL) were injected directly onto a 100 x 2.1 mm ACQUITY BEH C8 column (1.7 μm; Waters; Milford, MA). The column was eluted at a flow rate of 450 μL/min isocratically for 1 minute at 80% mobile phase A (95:5:0.1 vol/vol/vol 10 mM ammonium acetate/methanol/acetic acid), followed by a linear gradient to 80% mobile-phase B (99.9:0.1 vol/vol methanol/acetic acid) over 2 minutes, a linear gradient to 100% mobile phase B over 7 minutes, and then 3 minutes at 100% mobile-phase B. MS analyses were carried out using electrospray ionization in the positive ion mode using full scan analysis over m/z 200–1100 at 70,000 resolution and 3 Hz data acquisition rate. Additional MS settings were: ion spray voltage, 3.0 kV; capillary temperature, 300°C; probe heater temperature, 300°C; sheath gas, 50; auxiliary gas, 15; and S-lens RF level 60. All raw data were processed using Progenesis QI software (NonLinear Dynamics) for feature alignment, nontargeted signal detection, and signal integration. Targeted processing of a subset of known metabolites was conducted using TraceFinder software (Thermo Fisher Scientific; Waltham, MA). Compound identities were confirmed using reference standards and reference samples.
Metabolite classification analysis.
Databases of metabolites with known biospecimen locations (i.e., blood, urine, saliva, cerebrospinal fluid, feces, sweat, breast milk, bile, amniotic fluid, or other) and origins (i.e., exogenous, endogenous, food, plant, microbial, toxin/pollutant, cosmetic, drug, or drug metabolite) were obtained from The Human Metabolome Database (HMDB) v4.0 (Wishart et al., 2018). There are 114,085 metabolites in the HMDB. Of these, 6,815 are classified as having been identified in fecal samples, and 172 are classified as being of microbial origin. There are 117 metabolites that are classified as identified in fecal samples and having a microbial origin. Manual literature searches were conducted on Pubmed and Google Scholar using a combination of the metabolite name/HMDB ID, ‘microbial production’, ‘microbial metabolism’, ‘bacterial production’, and ‘bacterial metabolism’).
Supplementary Material
Table S2: Patterns in stool metabolites across the four study phases (Diet, Antibiotics+PEG, Early Recovery, Late Recovery), Related to Figure 6. “1” indicates an increase in metabolite relative abundance, “2” indicates no change, and “3” indicates a decrease.
Table S5: The number of total reads per sample after quality control step (Trimmomatic) and removing the reads that align to the human genome, the number of reads that align to the KEGG orthology database, the number of reads that map to the enzyme category EC:3.2.1.- within the KEGG orthology alignments, as well as the number of reads that align to genes responsible for IPA and butyrate production. Related to STAR Methods (Bioinformatics processing).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Human fecal and plasma samples | This study | N/A |
| Critical Commercial Assays | ||
| DNeasy PowerSoil Kit | QIAGEN | 12888-100 |
| Nextera XT DNA Library Preparation Kit | Illumina | FC-131-1096 |
| Nextera XT Index Kit v2, Set A/B/C/D | Illumina | FC-131-2001/2/3/4 |
| Quant-iT™ PicoGreen™ dsDNA Assay Kit | ThermoFisher Scientific | P7589 |
| High Sensitivity NGS Fragment Analysis Kit (1-6000 bp), 500 Samples | Agilent Technologies, Inc. | DNF-474-0500 |
| HiSeq PE Cluster Kit v4 cBot | Illumina | PE-401-4001 |
| HiSeq SBS Kit V4 250 Cycle Kit | Illumina | FC-401-4003 |
| TaqMan™ Fast Universal PCR Master Mix (2X), no AmpErase™ UNG | ThermoFisher Scientific | CAT# 4352042 |
| Deposited Data | ||
| Metagenomics sequencing | This paper | SUB7782171 |
| Human genome | NCBI | GRCh38.p4 |
| Metabolomics data | This paper | http://dx.doi.org/10.17632/ykb5wh3gn3.1 |
| Stool metabolomics data: identified metabolites from HILIC-pos, C8-pos, HILIC-neg, and C18-neg methods | National Metabolomics Data Repository (www.metabolomicsworkbench.org) | Accession number: PR001024 |
| Stool metabolomics data: unknown peak data from HILIC-pos, C8-pos, HILIC-neg, and C18-neg methods | National Metabolomics Data Repository (www.metabolomicsworkbench.org) | Accession number: PR001024 |
| Plasma metabolomics data: identified metabolites from HILIC-pos, C8-pos, HILIC-neg, and C18-neg methods | National Metabolomics Data Repository (www.metabolomicsworkbench.org) | Accession number: PR001024 |
| Plasma metabolomics data: unknown peak data from HILIC-pos, C8-pos, HILIC-neg, and C18-neg methods | National Metabolomics Data Repository (www.metabolomicsworkbench.org) | Accession number: PR001024 |
| Oligonucleotides | ||
| BSF8, 16S qPCR forward primer: AGAGTTTGATCCTGGCTCAG | Hill et al, 2010 | N/A |
| BSR357, 16S qPCR reverse primer: CTGCTGCCTYCCGTA |
Hill et al., 2010 | N/A |
| 16S qPCR probe (+ indicates locked nucleic acid base): /56-FAM/TAA +CA+C ATG +CA+A GT+C GA/3BHQ_1/ |
Hill et al., 2010 | N/A |
| Software and Algorithms | ||
| Demultiplexing software | This paper | https://github.com/PennChopMicrobiomeProgram/dnabc |
| Trimmomatic | Bolger et al., 2014 |
https://github.com/PennChopMicrobiomeProgram/illqc, http://www.usadellab.org/cms/index.php?page=trimmomatic |
| Bwa2 | Li et al., 2009 | https://github.com/PennChopMicrobiomeProgram/decontam |
| MetaPhlAn2 | Truong et al., 2015 | https://huttenhower.sph.harvard.edu/metaphlan |
| Gene assignments | This paper | https://github.com/PennChopMicrobiomeProgram/PathwayAbundanceFinder |
| RAPSearch2 and DIAMOND search |
Zhao et al., 2012 Buchfink et al., 2015 |
https://omics.informatics.indiana.edu/mg/RAPSearch2/ http://www.diamondsearch.org/index.php |
| The Human Metabolome Database | Wishart et al., 2018 | www.hmdb.ca/ |
Highlights:
The gut microbiome on a fiber-free diet differs from that of an omnivore or vegan.
The lack of dietary fiber slows microbiome recovery after an ecological stress.
Dietary effects on Firmicutes alter carbohydrate and amino acid gut metabolites.
The effect of diet-based microbiota metabolites on the plasma metabolome is modest.
Acknowledgements:
Crohn’s & Colitis Foundation (G.D.W, J.D.L, and F.D.B), P30 DK 050306 Human-Microbial Analytic and Repository Core of the Center for Molecular Studies in Digestive and Liver Disease (GDW), and the PennCHOP Microbiome Program (G.D.W and F.D.B) and the Penn Center for Nutritional Science and Medicine (G.D.W., J.D.L, and H.L.). The project described was supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1TR000003. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: J.B. is on scientific advisory boards of Janssen Research and Prolacta Bioscience. M.A.F is a co-founder of Federation Bio. J.D.L. has received honorarium from Nestle Health Science for consulting and for participation in medical education events. F.D.R, J.D.L, and G.D.W are co-inventors on Patent No. US 10,058,576 B2 “Compositions and Methods Comprising a Defined Microbiome and Methods of Use Thereof”.
References
- Baxter NT, Schmidt AW, Venkataraman A, Kim KS, Waldron C, and Schmidt TM (2019). Dynamics of Human Gut Microbiota and Short-Chain Fatty Acids in Response to Dietary Interventions with Three Fermentable Fibers. MBio 10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15), 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, and Hazen SL (2017). Targeting of microbe-derived metabolites to improve human health: The next frontier for drug discovery. J Biol Chem 292(21), 8560–8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, and Huson DH (2015). Fast and sensitive protein alignment using DIAMOND. Nat Methods 12(1), 59–60. [DOI] [PubMed] [Google Scholar]
- Cantarel BL, Lombard V, and Henrissat B (2012). Complex carbohydrate utilization by the healthy human microbiome. PLoS One 7(6), e28742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Nwe PK, Yang Y, Rosen CE, Bielecka AA, Kuchroo M, Cline GW, Kruse AC, Ring AM, Crawford JM, et al. (2019). A Forward Chemical Genetic Screen Reveals Gut Microbiota Metabolites That Modulate Host Physiology. Cell 177(5), 1217–1231 e1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LJ, Esterhazy D, Kim SH, Lemetre C, Aguilar RR, Gordon EA, Pickard AJ, Cross JR, Emiliano AB, Han SM, et al. (2017). Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature 549(7670), 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosimo DA, Kohn JA, Luo PM, Piscotta FJ, Han SM, Pickard AJ, Rao A, Cross JR, Cohen LJ, and Brady SF (2019). Mapping Interactions of Microbial Metabolites with Human G-Protein-Coupled Receptors. Cell Host Microbe 26(2), 273–282 e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crost EH, Tailford LE, Le Gall G, Fons M, Henrissat B, and Juge N (2013). Utilisation of mucin glycans by the human gut symbiont Ruminococcus gnavus is strain-dependent. PLoS One 8(10), e76341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Materna AC, Friedman J, Campos-Baptista MI, Blackburn MC, Perrotta A, Erdman SE, and Alm EJ (2014). Host lifestyle affects human microbiota on daily timescales. Genome Biol 15(7), R89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. (2013). Diet rapidly and reproducibly alters the human gut microbiome. Nature [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, et al. (2016). A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 167(5), 1339–1353 e1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd D, Spitzer MH, Van Treuren W, Merrill BD, Hryckowian AJ, Higginbottom SK, Le A, Cowan TM, Nolan GP, Fischbach MA, et al. (2017). A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 551(7682), 648–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia MS, and Fischbach MA (2015). HUMAN MICROBIOTA. Small molecules from the human microbiota. Science 349(6246), 1254766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kaoutari A, Armougom F, Gordon JI, Raoult D, and Henrissat B (2013). The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol 11(7), 497–504. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, and Distrutti E (2015). Bile Acid-Activated Receptors, Intestinal Microbiota, and the Treatment of Metabolic Disorders. Trends Mol Med 21(11), 702–714. [DOI] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature [DOI] [PubMed] [Google Scholar]
- Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, and Gordon JI (2011). Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A 108(15), 6252–6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke MT, Kenny DJ, Cassilly CD, Vlamakis H, Xavier RJ, and Clardy J (2019). Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc Natl Acad Sci U S A 116(26), 12672–12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, Kirn TJ, Bushman FD, and Artis D (2010). Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal immunology 3(2), 148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, et al. (2013). Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155(7), 1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AJ, Vangay P, Al-Ghalith GA, Hillmann BM, Ward TL, Shields-Cutler RR, Kim AD, Shmagel AK, Syed AN, Personalized Microbiome Class S, et al. (2019). Daily Sampling Reveals Personalized Diet-Microbiome Associations in Humans. Cell Host Microbe 25(6), 789–802 e785. [DOI] [PubMed] [Google Scholar]
- Koh A, Molinaro A, Stahlman M, Khan MT, Schmidt C, Manneras-Holm L, Wu H, Carreras A, Jeong H, Olofsson LE, et al. (2018). Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell 175(4), 947–961 e917. [DOI] [PubMed] [Google Scholar]
- Kovatcheva-Datchary P, Nilsson A, Akrami R, Lee YS, De Vadder F, Arora T, Hallen A, Martens E, Bjorck I, and Backhed F (2015). Dietary Fiber-Induced Improvement in Glucose Metabolism Is Associated with Increased Abundance of Prevotella. Cell Metab 22(6), 971–982. [DOI] [PubMed] [Google Scholar]
- Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, Bittinger K, Bailey A, Friedman ES, Hoffmann C, et al. (2015). Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe 18(4), 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14), 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis P, and Flint HJ (2017). Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol 19(1), 29–41. [DOI] [PubMed] [Google Scholar]
- McDonald D, Ackermann G, Khailova L, Baird C, Heyland D, Kozar R, Lemieux M, Derenski K, King J, Vis-Kampen C, et al. (2016). Extreme Dysbiosis of the Microbiome in Critical Illness. mSphere 1(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Fleming L, Olin-Sandoval V, Campbell K, and Ralser M (2015). Remaining Mysteries of Molecular Biology: The Role of Polyamines in the Cell. J Mol Biol 427(21), 3389–3406. [DOI] [PubMed] [Google Scholar]
- Nagalingam NA, and Lynch SV (2012). Role of the microbiota in inflammatory bowel diseases. Inflamm Bowel Dis 18(5), 968–984. [DOI] [PubMed] [Google Scholar]
- Ng KM, Aranda-Diaz A, Tropini C, Frankel MR, Van Treuren W, O’Laughlin CT, Merrill BD, Yu FB, Pruss KM, Oliveira RA, et al. (2019). Recovery of the Gut Microbiota after Antibiotics Depends on Host Diet, Community Context, and Environmental Reservoirs. Cell Host Microbe 26(5), 650–665 e654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SC, Bernstein CN, Vatn MH, Lakatos PL, Loftus EV Jr., Tysk C, O’Morain C, Moum B, Colombel JF, Epidemiology, et al. (2013). Geographical variability and environmental risk factors in inflammatory bowel disease. Gut 62(4), 630–649. [DOI] [PubMed] [Google Scholar]
- NHANES, −. What We Eat in America: Nutrient intakes from food by gender and age. National Health and Nutrition Examination Survey (NHANES) 2009–10., Centers for Disease Control and Prevention (CDC). National Center for Health Statistics (NCHS). National Health and Nutrition Examination Survey Data Hyattsville, MD: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention. http://www.ars.usda.gov/Sp2userfiles/Place/12355000/Pdf/0910/Table_1_Nin_Gen_09.Pdf. . [Google Scholar]
- Ni J, Shen TD, Chen EZ, Bittinger K, Bailey A, Roggiani M, Sirota-Madi A, Friedman ES, Chau L, Lin A, et al. (2017). A role for bacterial urease in gut dysbiosis and Crohn’s disease. Sci Transl Med 9(416). [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe SJ, Li JV, Lahti L, Ou J, Carbonero F, Mohammed K, Posma JM, Kinross J, Wahl E, Ruder E, et al. (2015). Fat, fibre and cancer risk in African Americans and rural Africans. Nat Commun 6, 6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, and Kanehisa M (1999). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27(1), 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Png CW, Linden SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, McGuckin MA, and Florin TH (2010). Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol 105(11), 2420–2428. [DOI] [PubMed] [Google Scholar]
- Sartor RB, and Wu GD (2017). Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology 152(2), 327–339 e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, and Huttenhower C (2012). Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 9(8), 811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits SA, Leach J, Sonnenburg ED, Gonzalez CG, Lichtman JS, Reid G, Knight R, Manjurano A, Changalucha J, Elias JE, et al. (2017). Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science 357(6353), 802–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, and Sonnenburg JL (2016). Diet-induced extinctions in the gut microbiota compound over generations. Nature 529(7585), 212–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, Buhler JD, and Gordon JI (2005). Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 307(5717), 1955–1959. [DOI] [PubMed] [Google Scholar]
- Venkatesh M, Mukherjee S, Wang H, Li H, Sun K, Benechet AP, Qiu Z, Maher L, Redinbo MR, Phillips RS, et al. (2014). Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 41(2), 296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vital M, Howe AC, and Tiedje JM (2014). Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. MBio 5(2), e00889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Armet AM, Finlay BB, and Shanahan F (2020). Establishing or Exaggerating Causality for the Gut Microbiome: Lessons from Human Microbiota-Associated Rodents. Cell 180(2), 221–232. [DOI] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. (2011). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472(7341), 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vazquez-Fresno R, Sajed T, Johnson D, Li C, Karu N, et al. (2018). HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res 46(D1), D608–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarska M, Luo C, Kolde R, d’Hennezel E, Annand JW, Heim CE, Krastel P, Schmitt EK, Omar AS, Creasey EA, et al. (2017). Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe 22(1), 25–37 e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science 334(6052), 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GD, Compher C, Chen EZ, Smith SA, Shah RD, Bittinger K, Chehoud C, Albenberg LG, Nessel L, Gilroy E, et al. (2016). Comparative metabolomics in vegans and omnivores reveal constraints on diet-dependent gut microbiota metabolite production. Gut 65(1), 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GD, Lewis JD, Hoffmann C, Chen YY, Knight R, Bittinger K, Hwang J, Chen J, Berkowsky R, Nessel L, et al. (2010). Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol 10, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, Ben-Yacov O, Lador D, Avnit-Sagi T, Lotan-Pompan M, et al. (2015). Personalized Nutrition by Prediction of Glycemic Responses. Cell 163(5), 1079–1094. [DOI] [PubMed] [Google Scholar]
- Zhang LS, and Davies SS (2016). Microbial metabolism of dietary components to bioactive metabolites: opportunities for new therapeutic interventions. Genome Med 8(1), 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Tang H, and Ye Y (2012). RAPSearch2: a fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics 28(1), 125–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Winter MG, Byndloss MX, Spiga L, Duerkop BA, Hughes ER, Buttner L, de Lima Romao E, Behrendt CL, Lopez CA, et al. (2018). Precision editing of the gut microbiota ameliorates colitis. Nature 553(7687), 208–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Chassaing B, Singh V, Pellizzon M, Ricci M, Fythe MD, Kumar MV, and Gewirtz AT (2018). Fiber-Mediated Nourishment of Gut Microbiota Protects against Diet-Induced Obesity by Restoring IL-22-Mediated Colonic Health. Cell Host Microbe 23(1), 41–53 e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2: Patterns in stool metabolites across the four study phases (Diet, Antibiotics+PEG, Early Recovery, Late Recovery), Related to Figure 6. “1” indicates an increase in metabolite relative abundance, “2” indicates no change, and “3” indicates a decrease.
Table S5: The number of total reads per sample after quality control step (Trimmomatic) and removing the reads that align to the human genome, the number of reads that align to the KEGG orthology database, the number of reads that map to the enzyme category EC:3.2.1.- within the KEGG orthology alignments, as well as the number of reads that align to genes responsible for IPA and butyrate production. Related to STAR Methods (Bioinformatics processing).
Data Availability Statement
Shotgun metagenomic sequence files are available at the Sequence Read Archive (SRA) under accession number PRJNA675301. Metabolomics data are available at the National Metabolomics Data Repository (www.metabolomicsworkbench.org) under accession number PR001024. The code generated during this study are available at Github at https://github.com/PennChopMicrobiomeProgram/FARMM.