

Summary
DNA methyl transferase-1 or DNMT1 maintains DNA methylation in the genome and is important for regulating gene expression in cells. Aberrant changes in DNMT1 activity and DNA methylation are commonly observed in cancers and many other diseases. Recently, a number of long intergenic non-protein-coding RNAs or lincRNAs have been shown to play a role in regulating DNMT1 activity. CCDC26 is a nuclear lincRNA that is frequently mutated in cancers and is a hotbed for disease-associated single nucleotide changes. However, the functional mechanism of CCDC26 is not understood. Here, we show that this lincRNA is concentrated on the nuclear periphery. Strikingly, in the absence of CCDC26 lincRNA, DNMT1 is mis-located in the cytoplasm, and the genomic DNA is significantly hypomethylated. This is accompanied by double-stranded DNA breaks and increased cell death. These results point to a previously unrecognized mechanism of lincRNA-mediated subcellular localization of DNMT1 and regulation of DNA methylation.
Subject areas: Molecular Biology, Cell Biology
Graphical abstract
Highlights
-
•
LincRNA CCDC26 influences cellular localization of the enzyme DNMT1
-
•
In the absence of CCDC26, the genomic DNA is significantly hypomethylated
-
•
Removal of CCDC26 leads to double-stranded DNA breaks and increased cell death
Molecular Biology; Cell Biology
Introduction
In the mammalian genome, DNA is often methylated at cytosines in CpG dinucleotides. DNA methylation is one of the important epigenetic modifications needed for transcriptional regulation of genes (Jaenisch and Bird, 2003). This modification plays a crucial role in many vital cellular processes such as heterochromatin formation, X-chromosomal inactivation, and genomic stability (Csankovszki et al., 2001; Nan et al., 1998; Watt and Molloy, 1988). Unsurprisingly, aberrant DNA methylation is implicated in many diseases and developmental defects (Baets et al., 2015; Daskalos et al., 2009; Eden et al., 2003; Gaudet et al., 2003; Heller et al., 2016; Klein et al., 2013; Morgan et al., 2018; Pakneshan et al., 2004; Schmelz et al., 2005; Stirzaker et al., 1997; Wen et al., 2018; Yu et al., 2013). Regulation of DNA methylation is therefore crucial throughout mammalian existence.
In mammals, DNMT3a, DNMT3b, and DNMT1 are DNA methyltransferases that are responsible for establishing and maintaining genomic methylation in cells (Lyko, 2018). DNMT3a and DNMT3b are primarily involved in establishing de novo DNA methylation patterns in the genome. In the very early stages of development, DNA methylation is completely eradicated in primordial germ cells, resulting in an epigenetically “blank canvas.” The DNMT3s then restore DNA methylation in a non-CpG-specific and ubiquitous manner (Doherty et al., 2002; Otani et al., 2009; Rasmussen and Helin, 2016). In contrast, DNMT1 plays a more predominant role in maintaining post-replicative methylation patterns, by preferentially binding hemi-methylated DNA, and methylating the newly synthesized daughter strand (Lyko, 2018). At late S-phase of the cell cycle, DNMT1 is targeted to replication foci, a process dependent on an additional ubiquitin-like protein, with PHD and RING finger domains 1 (UHRF1) (Berkyurek et al., 2014; Bostick et al., 2007; Bronner et al., 2019). Evidence suggests that during the replication process, DNMT1 also interacts with histone-modifying enzymes such as histone deacetylase HDAC2 as well as histone methyltransferases, EZH2 and G9a (Esteve et al., 2006; Rountree et al., 2000; Wang et al., 2017).
Aberrant DNA methylation is suspected to play a role in many cancers, e.g., hepatocellular carcinoma, glioblastoma, breast cancer, squamous cell lung cancer, thyroid cancer, and leukemia, (Daskalos et al., 2009; Eden et al., 2003; Gaudet et al., 2003; Heller et al., 2016; Morgan et al., 2018; Pakneshan et al., 2004; Schmelz et al., 2005; Stirzaker et al., 1997; Wen et al., 2018; Yu et al., 2013), and DNMT mutations are also the cause of developmental diseases such as hereditary sensory and autonomic neuropathy type 1E (HSAN1E) (Baets et al., 2015; Klein et al., 2013).
In somatic cells, DNMT1 is the most abundant and most active methyl transferase. It is a predominantly nuclear protein with an N-terminal nuclear localization signal (NLS) stretching between 177 and 205 amino acid residues (Alvarez-Ponce et al., 2018). The N-terminus of DNMT1 also contains domains required for its interaction with partner proteins, including DMAP1, HP1, G9a, and PCNA (Esteve et al., 2006; Fuks et al., 2003; Iida et al., 2002; Rountree et al., 2000). The central region of DNMT1 is needed for its targeting to replication foci (Leonhardt et al., 1992), whereas the C-terminus comprises the catalytic domain required for methyl-transferase activity (Song et al., 2012).
A number of studies have shown that DNA methylation is regulated by non-protein-coding RNAs or ncRNAs (Berghoff et al., 2013; Imamura et al., 2004; Jeffery and Nakielny, 2004; Yu et al., 2008). Also, DNMT1 function is often influenced by its interactions with ncRNAs. Long ncRNAs or lncRNAs such as KCNQ1OT1 (Mohammad et al., 2010), Dali (Chalei et al., 2014), lincRNA-p21 (Bao et al., 2015), PARTICLE (O'Leary et al., 2017), ecCEBP (Di Ruscio et al., 2013), DACOR1 (Somasundaram et al., 2018), and HOXA11-AS1 (Guo et al., 2019) are shown to interact with DNMT1 and modulate its activity. Here we report an interaction between DNMT1 and a long intergenic non-coding RNA (lincRNA), CCDC26. LincRNA CCDC26 is transcribed from a 328-kilobase gene on chromosome 8, from 8q24.21 locus neighboring the proto-oncogene c-MYC. The 8q24 locus is a hotbed for disease-associated mutations including cancer-associated SNPs and copy-number alterations (Wilson and Kanhere, 2021). It is of specific interest in acute myeloid leukemia (AML) because of the high-frequency occurrence of AML-associated mutations and variants in CCDC26 gene (Duployez et al., 2018; Izadifard et al., 2018; Kuhn et al., 2012; Radtke et al., 2009). These observations suggest that CCDC26 might play an important role in driving cancer progression. Previous studies show that CCDC26 might be involved in regulating apoptosis and differentiation in myeloid cells (Yin et al., 2006; Hirano et al., 2015). However, the functional mechanism of CCDC26 remains elusive.
In this study, we aimed to understand the function of CCDC26 and its role in cancer. Here, we show that CCDC26 interacts with DNMT1 and is predominantly localized on the nuclear periphery. In the absence of CCDC26, DNMT1 is mis-localized in the cytoplasm, leading to DNA hypomethylation and apoptosis similar to that observed on inhibition of DNMT1 in myeloid leukemia cells. As a result, we observe genome-wide changes in gene expression. LincRNA-mediated DNMT1 mis-localization has not been previously reported and has significant implications to DNA methylation regulation, as well as cancer and RNA biology.
Results
LincRNA CCDC26 is a myeloid-specific RNA expressed from second TSS
We first sought to understand the gene structure and expression pattern of CCDC26. Previous investigations (Hirano et al., 2015; Yin et al., 2006) on various cell types showed that CCDC26 is highly expressed in myeloid leukemias. We further examined this by measuring CCDC26 levels in a number of additional leukemia and non-leukemia lines (Figure S1A). In agreement with previous results, we observed that CCDC26 is highly expressed in myeloid cells and is present at much lower levels in other cell types. We also observed that among myeloid cells the highest level of expression is in chronic myeloid cells, K562 (Figure S1A).
According to current gene annotations, altogether there are four isoforms of CCDC26 that are transcribed from human chromosome 8 from two distinct transcription start sites, TSS1 and TSS2 (Figure 1A). These isoforms show alternative splicing patterns and contain combinations of six exons. Isoforms-1 (1666bp) and -2 (1649bp) are transcribed from an independent transcription start site (TSS2) and differ only by an additional 17 nucleotide sequence at the 3′ end of exon 4 in Isoform-1. Isoform-3 (1495bp), which is also transcribed from TSS2, lacks exon 4 completely. Isoform 4 (1718bp) is transcribed from TSS1, a start site that lies upstream of TSS2.
Figure 1.

LincRNA CCDC26 knockout results in slower growth and increased apoptosis and DNA damage in K562 cells
(A) A schematic diagram illustrating the four currently known isoforms of CCDC26, adapted from the UCSC Genome Browser view of the CCDC26 locus (NCBI RefSeq tracks, GRCh37/hg19 assembly. Ideogram for chromosome 8 is shown at the top. The red baron ideogram indicates CCDC26 position. CCDC26 isoforms are transcribed from one of two transcription start sites (TSS-1 and TSS-2) located on Chr8q24 locus.
(B) A plot showing the RNA levels of different CCDC26 isoforms, relative to GAPDH, in K562 cells, measured using qRT-PCR. Error bars represent the mean ± standard deviation.
(C) A plot showing expression level of CCDC26 RNA relative to GAPDH in WT K562, KO.1, and KO.2 cells measured using qRT-PCR. CCDC26 levels were significantly reduced in both knockouts. Error bars represent the mean ± standard deviation.
(D) Growth curve for WT K562, KO.1, and KO.2 cells showing slower growth in the latter cell lines. Values represent the mean ± standard deviation. ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001 (unpaired, two-tailed t test, n = 3).
(E) Growth curve for KO.1 expressing CCDC26 from exogeneous plasmid vector and KO.1 expressing empty vector. Values represent the mean ± standard deviation. ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001 (unpaired, two-tailed t test, n = 3).
(F) A plot showing the percentage of apoptotic WT, KO.1, and KO.2 cells in the sub-G1 phase of the cell cycle according to propidium iodide FACS analysis. Values represent the mean ± standard deviation. ∗p < 0.05, ∗∗p < 0.005; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001 (unpaired, two-tailed t test, n = 3).
(G) The ApoTox-Glo assay allows for detection of apoptosis in cells. The Caspase-Glo 3/7 reagent results in cell lysis, leading to caspase cleavage and the release of the luciferase substrate amino-luciferin, causing the luciferase reaction to emit light, indicating apoptosis. Luminescence was detected and measured for WT and CCDC26 KO cells to determine the level of apoptosis in cells. Values represent the mean ± standard deviation. ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001 (unpaired, two-tailed t test, n = 5).
(H) Confocal images demonstrating the results of anti-γ-H2AX immunofluorescence. WT, KO.1, and KO.2 cells were stained with DAPI nuclear stain (blue) and anti-γ-H2AX antibody (cyan). Increased numbers of γ-H2AX foci are present in the KO cells. Scale bar, 25 um.
To understand which CCDC26 isoforms are expressed in K562 cells, we carried out qRT-PCR measurements using isoform-specific primers (Figure 1B and Table S1). In these cells, the four isoforms of CCDC26 are not uniformly expressed. Isoforms starting from TSS2 i.e. isoforms-1, -2, and -3 account for more than 95% of the total CCDC26 transcripts in the cell. Among the three isoforms starting at TSS2, Isoform-1 and Isoform-2 alone make 80% of CCDC26, whereas Isoform-3 is detected at considerably lower levels, accounting for only ∼15% of CCDC26 expression. On the other hand, Isoform-4, which is expressed from another transcription start site, TSS1, is barely detectable (Figure 1B).
LincRNA CCDC26 depletion leads to DNA damage and apoptosis
To further understand the role of CCDC26 in myeloid cells, we carried out CRISPR-Cas9-mediated knockout (KO) of this lincRNA in K562 cells. Given that more than 99% of CCDC26 is transcribed from TSS2, we simultaneously used two small guide RNAs to mutate TSS2 (Figure S1B). Following single-cell clonal expansion, we established two cell lines, KO.1 and KO.2, which exhibit ∼99% and ∼88% reduction in CCDC26 levels, respectively (Figure 1C).
We first analyzed the effect of CCDC26 knockout on cell growth. Interestingly, CCDC26 depletion resulted in a significantly reduced rate of growth in both KO cell lines (Figure 1D). Cells were counted every 24 h across a 72 h period, and growth curves were subsequently plotted. Although wild-type (WT) cells double in number approximately every 24 h as previously reported (Murray et al., 1993), the number of KO cells only increase 2-fold approximately every 48 h (Figure 1D). A previous study (Hirano et al., 2015) also observed slower growth rate upon shRNA mediated knockdown of CCDC26. However, this effect was only observed under high serum conditions. The effect of CCDC26 removal under normal growth conditions has not been reported before. Our results confirm that, even under normal conditions, CCDC26 removal leads to reduced cell growth (Figure 1D). To further confirm this phenotype and to rule out the possibility that the slow growth in KO cells is a result of clonal selection after CRISPR knockouts, we compared the growth rates of KOs with clonal population transfected with CRISPR/Cas9 plasmid on its own without sgRNAs. However, this CRISPR control displayed similar growth rate as K562 cells (Figure S1C), supporting our observation that the slower growth rate observed in KOs is a likely consequence of CCDC26 knockout and not a result of clonal isolation. We also reintroduced CCDC26 in the knockouts using an exogenous plasmid vector. Overexpression of CCDC26 in the knockouts increased cell growth rate as compared with knockouts that expressed an empty vector (Figures 1E and S1D), further supporting our observation that the cell growth changes are due to removal of CCDC26.
The slow growth observed in KO cells could be either because of changes in cell cycle or because of an increased rate of cell death. Previously, CCDC26 has been implicated in apoptosis (Hirano et al., 2015; Yin et al., 2006); however, its effect on cell cycle has not been investigated. To understand the reason behind slow growth rate in KO cells, we subsequently analyzed cell-cycle progression in WT and KOs by incorporation of propidium iodide and subsequent FACS analysis. This method measures the number of DNA strands to determine cell cycle progression. We did not observe any significant changes in any of the key cell-cycle stages (Figures S1E–S1G). However, cell cycle analysis showed an increased population of CCDC26 KO cells in the sub-G1 state, which is indicative of apoptosis (Figure 1F). Cell cytotoxicity and apoptosis assays that measure caspase levels also confirmed that in comparison to WT, CCDC26 KO cells were less viable and more apoptotic (Figure 1G). The process of apoptosis is often linked to DNA damage. Hence, we explored the possibility of increased DNA damage in CCDC26 KO cells. We tested for increased presence of histone variant γ-H2AX (Figure 1H). Histone variant H2AX is key in the cellular response to DNA damage, as its C-terminal tail is rapidly phosphorylated at a Serine residue following the occurrence of a DNA double-strand break (DSB). Phosphorylated H2AX or γ-H2AX is used as a DSB marker and can be readily detected by anti- γ-H2AX antibody (Rogakou et al., 1998). A visible increase in γ-H2AX foci was observed in both CCDC26 KO lines as compared with WT cells (Figure 1H and Tables S2 and S3). Together, these results indicated that removal of CCDC26 results in DNA damage, apoptosis, and slow growth.
Absence of CCDC26 leads to DNMT1 mis-localization and DNA hypomethylation
We further sought to understand the mechanism behind CCDC26-mediated DNA damage. We first enquired if this RNA influences genomic DNA and chromatin in any other way. Many lncRNAs are involved in regulation of chromatin modifications (Di Ruscio et al., 2013; Mohammad et al., 2010; Pandey et al., 2008; Rinn et al., 2007). We speculated that CCDC26 might also function by regulating changes in epigenetic modifications. In order to verify this, we tested global genomic levels of multiple common histone modifications such as H3K27me3, H3K27ac, H3K9me3, H3K9ac, and H4K16ac using immunoblotting (Figure S2). However, we did not see any significant changes in genomic levels of tested histone modifications in CCDC26 KO cells. Using immunoblotting, we also tested levels of histone-modifying enzymes such as EZH2, G9a, and HDAC2 (Figure S2) that are involved in catalyzing these modifications. However, similar to histone modifications, levels of these catalytic proteins did not show any changes. Although this observation does not rule out possibility of site-specific variations in histone modifications, these results indicate that absence of CCDC26 did not lead to any significant changes in overall levels of histone modifications or the enzymes that catalyze these histone modifications.
In addition to histone modifications, DNA methylation is an important epigenetic modification. Importantly, DNA methylation is also shown to be regulated by various lncRNAs (Bao et al., 2015; Chalei et al., 2014; Di Ruscio et al., 2013; Gao et al., 2020; Guo et al., 2019; Merry et al., 2015; Mohammad et al., 2010; O'Leary et al., 2017; Sun et al., 2016; Wang et al., 2015). To test if DNA methylation levels have changed in the KOs, we carried out immunofluorescence measurements using anti-5-methyl cytosine antibody (Figures 2A, S3A, Tables S2 and S3). Surprisingly, in KO lines, the 5-methyl cytosine signal was considerably weaker as compared with WT cells, indicating genomic DNA was hypomethylated (Figures 2A and S3A).
Figure 2.

CCDC26 knockout results in mis-localization of DNMT1 in the cytosol and global DNA hypomethylation
(A) Confocal images demonstrating the results of anti-5mC immunofluorescence. WT, KO.1, and KO.2 cells were stained with DAPI nuclear stain (blue) and anti-5mC antibody (yellow). Reduced levels of 5mC fluorescence are observed in both KO cell lines. Scale bar, 50 um.
(B) Total protein levels of DNMT1, DNMT3a, and DNMT3b measured relative to GAPDH by immunoblotting are unchanged in WT and CCDC26 KO cells.
(C) Confocal images demonstrating the results of anti-DNMT1 immunofluorescence. WT, KO.1, and KO.2 cells were stained with DAPI nuclear stain (blue) and anti-DNMT1 antibody (red). The outline of the cell membrane can be seen with the addition of the brightfield lens in the right-hand panels. DNMT1 is nuclear in WT cells and is largely cytosolic in the KO cells. Scale bar, 5 um.
(D) Immunoblotting for DNMT1 on nuclear and cytosolic protein fractions shows a shift in the subcellular localization of DNMT1. DNMT1 is almost exclusively nuclear in the WT cells but appears both nuclear and cytosolic in CCDC26 KO cells. EZH2 and GAPDH are used as nuclear and cytosolic markers, respectively (nuc = nuclear protein fraction; cyt = cytosolic protein fraction).
A potential explanation for hypomethylation of genomic DNA in CCDC26 KO lines could be reduced levels of DNA methyltransferase proteins. However, immunoblotting in the CCDC26 knockouts, showed no significant changes in the levels of the three DNA methyltransferase proteins, DNMT3a, DNMT3b, and DNMT1 (Figures 2B and S3B). Therefore, we speculated that DNA-binding capacity or the enzymatic activity of a DNA methyltransferase might have changed in the absence of CCDC26. To detect changes in the association of DNMTs with DNA, we performed anti-DNMT immunofluorescence. We observed that, in KOs, the subcellular localization of DNMT1 is predominantly cytosolic in contrast to WT where DNMT1 is, as expected, localized in the nucleus (Figure 2C). This was supported by immunoblotting measurements using anti-DNMT1 antibody on nuclear and cytosolic fractions of KOs as compared with WT (Figure 2D). However, mis-localization was not observed in case of DNMT3a and DNMT3b or other nuclear proteins such as HDAC2 (Figures S3C and S3D).
Genes repressed by DNMT1 are upregulated in KO cells
We further hypothesized that if DNMT1 protein mis-localizes in the cytosol, then DNMT1 would be unable to carry out its primary function of methylating genomic DNA in the nucleus. Consequently, these cells should behave similarly to cells lacking DNMT1. To confirm this, we investigated expression levels of a selection of seven genes, previously shown to be significantly impacted by DNMT1 and methylation levels in myeloid cells (Figure 3A). These were protein tyrosine phosphatase non-receptor type 6 (PTPN6) (Li et al., 2017; Wang et al., 2017), cyclin-dependent kinase inhibitor 1A (CDKN1A) (Milutinovic et al., 2004; Schmelz et al., 2005), cyclin-dependent kinase inhibitor 2B (CDKN2B) (Herman et al., 1996; Yu et al., 2013), CD9 (Kim et al., 2012), VAV1 (Fernandez-Zapico et al., 2005; Ilan and Katzav, 2012), and JUNB (Fiskus et al., 2009; Yang et al., 2003), all of which have previously demonstrated upregulation in response to DNMT1 downregulation or DNA hypomethylation. We also measured levels of IGF1, as it has previously been reported that IGF1 is repressed as a result of DNMT1 inhibition (Pastural et al., 2007). qRT-PCRs demonstrated that out of seven genes we tested, five (PTPN6, CDKN1A, CDKN2B, CD9, and VAV1) were significantly upregulated in both KO cell lines as reported in past studies on DNMT1 KD or inhibition. JUNB, albeit not significantly, also demonstrated upregulation. As expected, IGF1 was significantly downregulated in both KO cell lines. This result demonstrates that gene expression changes in CCDC26 KO cells are very similar to cells in which DNMT1 has been knocked down or inhibited. Presumably this is due to the unavailability of DNMT1 in the CCDC26 KOs, given its predominantly cytoplasmic localization in these cells.
Figure 3.

DNMT1 and methylation-regulated genes show expression changes in CCDC26 KO cells
(A) A plot showing expression levels of various genes whose expression has previously been shown to be impacted by DNMT1 depletion or DNA hypomethylation in myeloid leukemia. Levels are measured relative to GAPDH in WT K562, KO.1, and KO.2 cells by qRT-PCR. Values represent the mean ± standard deviation. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS = Not significant (unpaired, two-tailed t test).
(B and C) (B) Heatmaps showing GC distribution around of Transcription Start Sites (TSS) of 2-fold downregulated and upregulated genes in KO as well as same number of randomly selected control set of genes. (C) Metagene plots of average enrichment of DNA methylation K562 cells at 2-fold downregulated genes (blue) as compared with upregulated genes (green) in JARID2 KO cells and random set of genes as control (black). The plots are centered on TSS of genes, and distance from TSS is indicated on the x axes.
(D) Confocal images demonstrating the results of anti-5mC immunofluorescence on cells treated with either 0uM, 5uM, or 10uM DNMT1 inhibitor DAC. Cells were stained with DAPI nuclear stain (blue) and anti-5mC antibody (yellow). Reduced levels of 5mC fluorescence are observed in cells treated with 5uM and 10uM DAC. Scale bar, 50 um.
(E) A plot showing expression levels of various genes (as in A) whose expression has previously been shown to be impacted by DNMT1 depletion or DNA hypomethylation in myeloid leukemia. Levels are measured relative to GAPDH in cells treated with 0-uM, 5-uM, and 10-uM DNMT1 inhibitor DAC. Values represent the mean ± standard deviation. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS = Not significant (unpaired, two-tailed t test).
To confirm if these genes were upregulated as a result of DNA methylation changes at their promoters, we measured DNA methylation levels at the promoters of PTPN6, VAV1, CD9, and CDKN1A genes that were significantly upregulated in CCDC26 KOs as compared with WT. DNA methylation levels were measured using bisulfite conversion and followed by a pyrosequencing assay. We observed that, with the exception of CDKN1A, promoter methylation was reduced at the tested genes in both KOs (Tables S4 and S5 and Figure S4A).
In order to verify this at the genomic level, using RNA-seq we measured genome-wide changes in gene expression. RNA-seq analysis showed that, in total, 287 genes showed more than 2-fold changes in RNA levels in both KO lines. Among these, 146 were upregulated and 141 were downregulated. It was previously shown that DNA methylation of CpG rich promoters leads to transcriptional repression (Kass et al., 1997; Li et al., 1993; Siegfried et al., 1999; Venolia and Gartler, 1983; Walsh et al., 1998). If gene expression changes in CCDC26 KO are due to mis-localization of DNMT1, promoter methylation-mediated repression of genes should be relieved in these cells. In other words, promoters of the genes upregulated in the KOs, should be GC-rich and should show a higher level of DNA methylation in WT cells. In order to verify this, we plotted GC density at differentially expressed genes against a random set of genes (Figure 3B). We observed that both up- and downregulated genes showed a much higher density of GC nucleotides compared with the control set of genes. In addition, we utilized previously published genome-wide DNA methylation levels in K562 cells to verify if the promoters of affected genes are methylated. As suspected, genes upregulated in KO cells show a much higher level of DNA methylation in K562 cells (Figure 3C), supporting the idea that gene expression changes we see in the KOs are due to changes in DNA methylation levels imposed by mis-localization of DNMT1 in the cytoplasm.
In order to further confirm that genes affected in the KO are regulated by DNMT1, we treated K562 cells with DNMT1 inhibitor, 5-aza-2′-deoxycytidine, also known as decitabine (DAC). DAC is a DNMT1 inhibitor that functions by covalently trapping DNMT1 to the DNA, thereby rendering it non-functional (Stresemann and Lyko, 2008). K562 WT cells were grown in the presence 0uM, 5uM, and 10uM DAC for 48 h. Treatment with both 5uM and 10uM DAC concentrations significantly reduced global 5mC levels (Figure 3D). DAC has also been shown to cause reductions in DNMT1 levels by inducing its proteasomal degradation (Ghoshal et al., 2005, 2018; Patel et al., 2010). Western blotting for DNMT1 also demonstrated decreased protein levels (Figure S4B).
Following confirmation of DNMT1 inhibition and global hypomethylation by DAC (Figure 3D), we next examined whether this elicited similar effects to that seen in CCDC26 KO. Similar to the KOs, γ-H2AX immunofluorescence increased upon DNMT1 inhibition, indicating an increase in DNA damage in cells treated with both 5uM and 10uM DAC for 48 h (Figure S4C). Significantly, qRT-PCRs for DNMT1-regulated genes (Figure 3A) showed similar patterns of expression after DAC treatment as upon CCDC26 KO (Figure 3E). Importantly, changes in gene expression were more pronounced in cells treated with 10uM DAC as compared with 5uM DAC (Figure 3E). These results support that reduction in DNMT1 can lead to DNA hypomethylation and subsequently DNA damage.
Apoptosis and DNA damage are a consequence of DNMT1 mis-localization
We next sought to establish the sequence of events that results in DNMT1 mis-localizing to the cytosol and DNA hypomethylation in CCDC26 KO cells. In order to fully understand the functional mechanism of CCDC26, it is important to examine whether cytosolic localization of DNMT1 is a consequence of DNA damage and apoptosis.
To further confirm that DNMT1 mis-localization is a result of CCDC26 KO and not a consequence of DNA damage, it was critical to establish that this type of movement of DNMT1 is not a general response to DNA damage and apoptosis. To investigate this, DNA damage was induced in WT cells using cisplatin. Cisplatin is a platinum-based drug that forms bonds with, and ultimately crosslinks, bases within and between DNA strands. This can distort the double helix, interfere with both DNA replication and transcription, and consequently induce DNA damage and apoptosis (Goodsell, 2006). To begin, the amount of cisplatin and treatment time required to induce DNA damage but prior to complete cell death was optimized (Figure S5A). Microscopic observations demonstrated that after 24 h of cisplatin treatment, cells treated with 5uM or 10uM of the drug still appeared viable. DNA damage was also confirmed in the cells treated with 5uM and 10uM cisplatin by monitoring γ-H2AX foci using immunofluorescence (Figure S5B).
To assess DNMT1 localization in DNA-damage-induced cells, DNMT1 immunofluorescence was performed on cisplatin-treated cells. This experiment exhibited no significant differences between 0uM control cells and the drug-treated cells. Similar to WT K562 cells, in cisplatin-treated cells, DNMT1 appeared primarily nuclear, demonstrating a diffused pattern of distribution throughout (Figure 4A). Accordingly, there was also no substantial change in the levels of 5mC immunofluorescence between control and cisplatin-treated cells (Figure 4B). This suggests that DNMT1 mis-localization in cytoplasm is not a general consequence of DNA damage. As further confirmation, the expression levels of genes that are up- or downregulated in response to CCDC26 KO and DNMT1 inhibition (as shown in Figure 3A) were also tested in cisplatin-treated cells. Some genes (IGF1 and CDKN1A) demonstrated similar changes in expression patterns in response to cisplatin as they did in response to DNMT inhibition and CCDC26 KO. However, for majority of genes, expression was either not significantly altered or they demonstrated an opposite change in expression (Figure S5C). This suggests that the expression changes seen in the KOs are response to DNMT1 mis-localization and DNA hypomethylation but not a consequence of DNA damage. Interestingly, CDKN1A gene whose promoter methylation was not affected by CCDC26 KO (Figure S4A) was the only gene that was significantly upregulated in cisplatin-treated cells, indicating that changes in CDKN1A expression were in response to DNA damage downstream of DNA hypomethylation.
Figure 4.

Cisplatin-induced DNA damage does not result in subcellular mis-localization of DNMT1
(A) Confocal images demonstrating the results of anti-DNMT1 immunofluorescence. Cells treated with 0 uM, 5 uM, and 10 uM cisplatin were stained with DAPI nuclear stain (blue) and anti-DNMT1 antibody (red). The outline of the cell membrane can be seen with the addition of the brightfield lens in the right-hand panels. DNMT1 appears nuclear in cells treated with different cisplatin concentrations. Scale bar, 5 um.
(B) Confocal images demonstrating the results of anti-5mC immunofluorescence on cells treated with either 0 uM, 5 uM, or 10 uM cisplatin. Cells were stained with DAPI nuclear stain (blue) and anti-5mC antibody (yellow). Similar levels of 5mC fluorescence are observed in cells treated with different cisplatin concentrations. Scale bar, 50 um.
CCDC26 is a nuclear lincRNA and interacts with DNMT1
To further investigate the relationship between CCDC26 and DNMT1, we investigated the possibility that CCDC26 interacts with DNMT1, thereby influencing its cellular localization. We first endeavored to find out the cellular localization of CCDC26 RNA. A large proportion of lncRNAs that have shown a predominantly nuclear localization are either associated with chromatin or enriched in nuclear sub-compartments and organelles (Clemson et al., 2009; Hutchinson et al., 2007; Werner and Ruthenburg, 2015). The subcellular localization of a lncRNA can give a clue to its functional mechanism. Previous results showed that CCDC26 might be marginally enriched in nucleus (Hirano et al., 2015). In this previous publication, all CCDC26 isoforms were not tested for their localization. It is possible that only selected isoforms are nuclear, thus influencing CCDC26 function. To understand the localization, we measured the levels of all four CCDC26 isoforms using qRT-PCRs in nuclear and cytosolic RNA fractions (Figure 5A). snoRNA U105 and GAPDH were used as gene markers to assess the quality of nuclear and cytosolic fractions, respectively (Figure S6A). Actin was used as a housekeeping control gene against which CCDC26 expression could be measured, given its similar levels in both the nucleus and cytosol (Figure S6B). Consistent with previous publication (Hirano et al., 2015), we found that all CCDC26 isoforms were much more enriched in the nucleus as compared with the cytosol (Figure 5A). To determine the location of CCDC26 in the nucleus, we also performed fluorescence in situ hybridization (RNA-FISH) on WT and KO cells. A fluorescently labeled probe specific to exon 6 of CCDC26 was generated and used for RNA FISH, followed by analysis using confocal microscopy. Interestingly, microscopic images (Figure 5B) showed that CCDC26 is predominantly located within the nucleus, demonstrating an enrichment at the periphery of the nucleus. The absence of fluorescent CCDC26 signal in the KO cells indicated that the signal was specific (Figure 5B).
Figure 5.

CCDC26 is a nuclear lincRNA and interacts with DNMT1
(A) A plot showing the levels of different CCDC26 isoforms, relative to Actin, in nuclear and cytosolic fractions of WT K562 cells, measured using qRT-PCR. Greater presence of all CCDC26 isoforms was found in the nuclear fraction. Error bars represent the mean ± standard deviation.
(B) Confocal images demonstrating the results of RNA FISH using a CCDC26-specific fluorescent probe. WT, KO.1, and KO.2 cells were stained with DAPI nuclear stain (blue) and a CCDC26 probe (green). The outline of the cell membrane can be seen with the addition of the brightfield lens in the right-hand panels. CCDC26 is primarily localized in the nucleus of the cell, specifically at the nuclear periphery. Scale bar, 5 um.
(C) Protein-RNA complexes pulled down with either anti-IgG, anti-DNMT1, or no antibody were immunoblotted with anti-DNMT1, to ensure that the DNMT1 protein was correctly pulled down for RNA immunoprecipitation. RNA pulled-down in each IP was purified, converted to cDNA, and subjected to qRT-PCR with CCDC26 and KCNQ1OT1 primers, to determine how much RNA was pulled down relative to the input in each instance. Values represent the mean ± standard deviation. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS = Not significant (unpaired, two-tailed t test).
It is important to establish whether DNMT1 mis-localization could be due to a direct interaction between CCDC26 and DNMT1 or an indirect effect of CCDC26 knockout. DNMT1 has previously been shown to bind and undergo regulation by multiple lncRNAs (Bao et al., 2015; Chalei et al., 2014; Di Ruscio et al., 2013; Gao et al., 2020; Guo et al., 2019; Merry et al., 2015; Mohammad et al., 2010; O'Leary et al., 2017; Sun et al., 2016; Wang et al., 2015). It has also been suggested that DNMT1 has higher affinity for RNA than DNA (Di Ruscio et al., 2013; Merry et al., 2015). Nuclear localization of CCDC26 points to a possibility that this lincRNA might also interact with DNMT1 in the nucleus. To further confirm this, we performed DNMT1 RNA immunoprecipitation (RIP) using anti-DNMT1 antibody (Figure 5C). An anti-IgG antibody was used to produce a control sample. The protein-bound-RNA pulled down with anti-DNMT1 and anti-IgG antibodies was used to perform qRT-PCRs with primers specific to CCDC26, and the RNA levels were measured relative to the input. Anti-DNMT1 pulled-down RNA showed approximately three times more enrichment of CCDC26 compared with the IgG control (Figure 5C). Moreover, enrichment of CCDC26 in the RIP assay is comparable to another long non-coding RNA, KCNQ1OT1 (Figure 5C), which was previously shown to interact with DNMT1(Mohammad et al., 2010). This raises a possibility that CCDC26 directly or indirectly interacts with DNMT1. However, the exact nature of CCDC26 and DNMT1 interaction needs to be confirmed with a more detailed investigation.
However, we can get additional support for this interaction by analyzing previously studied protein-RNA interactions that have been studied using RIP or variations of this method (Bai et al., 2019; Di Ruscio et al., 2013; Hendrickson et al., 2016; Hui et al., 2019; Xu et al., 2019). At least two datasets exploring DNMT1-RNA interactions in myeloid cells have been published (Di Ruscio et al., 2013; Hendrickson et al., 2016). We first adopted a bioinformatics approach where we re-mapped previously published RIP-seq datasets to the CCDC26 locus. The first dataset that we analyzed was generated using another myeloid line, HL60 (Di Ruscio et al., 2013). These data were generated by first pulling down cellular RNAs that bind to DNMT1 using anti-DNMT1 antibody and then sequencing and mapping these RNAs to the human genome. These data showed that CCDC26 is highly enriched in DNMT1-RIP assays when compared with an IgG RIP control (Figure S6C). Furthermore, analysis of another dataset, produced by a variation of the RIP method, formaldehyde-RIP-seq (fRIP-seq), also showed high enrichment of CCDC26 in DNMT1-bound RNAs in K562 cells (Hendrickson et al., 2016), further confirming an interaction between DNMT1-CCDC26 (Figure S6D).
Discussion
In the last three decades, following the development of whole-genome technologies, lncRNAs have gained importance (Derrien et al., 2012; Hangauer et al., 2013; Iyer et al., 2015; Okazaki et al., 2002), with many examples demonstrating their role in transcription regulation.
Here we have performed functional analysis of lincRNA, CCDC26 and demonstrated its importance in regulating global DNA methylation. Our data show that, in the absence of CCDC26, the genome is hypomethylated, which leads to increase in apoptosis and DNA damage and cell growth inhibition. In the past, non-coding RNAs have been shown to impact DNA methylation levels through transcriptional and post-transcriptional regulation of DNMT genes (Chen et al., 2015; Cheng et al., 2018; Di Ruscio et al., 2013; Merry et al., 2015; Mohammad et al., 2010; Wang et al., 2018). However, DNMT expression levels were unchanged in CCDC26 KOs, indicating that the mechanism behind the observed DNA hypomethylation is different. Strikingly, in the absence of lincRNA CCDC26, a large proportion of DNMT1 protein is mis-localized in the cytosol. In KO cells, the mis-localization of DNMT1 in cytoplasm is most likely responsible for the observed hypomethylated state of the genome and cell death. This connection between CCDC26 and DNMT1 can provide the missing link between frequent mutations in CCDC26 locus and leukemia. Deletion of DNMT1 is shown to prevent MLL-AF9 leukemia. In addition, it has been reported that the absence of DNMT1 induces apoptosis in hematopoietic stem cells. This is similar to what we observe in this study when CCDC26 is removed. In corollary, direct or indirect role of CCDC26 in retaining DNMT1 in nucleus and also maintaining cell proliferation can explain why CCDC26 is often upregulated in AML (Duployez et al., 2018; Izadifard et al., 2018; Kuhn et al., 2012; Radtke et al., 2009).
LncRNA-mediated regulation of cellular localization, although not reported in case of DNMT1, has been previously reported in case of other proteins. In some instances, this has been shown to occur via a direct interaction; for example, lncRNA TP53TG1 binds the transcription factor, YBX1, thereby preventing its nuclear trafficking (Diaz-Lagares et al., 2016). In other instances, this can also occur via an indirect effect; an interaction between lncRNA CRYBG3 and actin for example is sufficient to prevent translocation of myelin and lymphocyte protein (MAL) into the nucleus (Pei et al., 2018). Similarly, NFkB-interacting LncRNA (NKILA) binds and prevents phosphorylation of the inhibitory IB subunit. This blocks its degradation, which subsequently prevents the active p65 subunit of NFkB from re-localizing from the cytosol to the nucleus (Liu et al., 2015). However, we did not observe any changes in DNMT1 stability upon CCDC26 KO (Figure S7). Moreover, we demonstrate an interaction between DNMT1 and CCDC26, suggesting that this lincRNA might play a direct or indirect role in localizing DNMT1 in the nucleus.
Numerous DNMT1-interacting lncRNAs have been identified previously (Bao et al., 2015; Chalei et al., 2014; Di Ruscio et al., 2013; Gao et al., 2020; Guo et al., 2019; Merry et al., 2015; Mohammad et al., 2010; O'Leary et al., 2017; Sun et al., 2016; Wang et al., 2015); however, these largely demonstrate effects on DNA methylation at localized genes or regions, as opposed to the global effect observed here. In addition, lincRNA-mediated DNMT1 localization has not been reported before. Based on these results, we provide a model (Figure 6) suggesting that lincRNAs, in this case CCDC26, can regulate sub-cellular localization of DNMT1 via direct or indirect interaction in the nucleus. This provides a means of regulating global genomic DNA methylation, and disruption of the lincRNA can result in hypomethylation and apoptosis.
Figure 6.

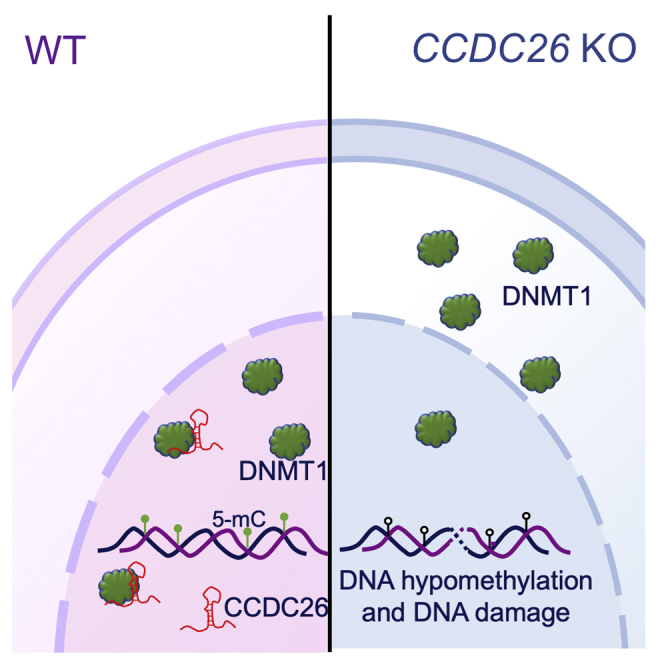
Model for CCDC26-mediated DNMT1 regulation
In WT K562 cells, DNMT1 directly or indirectly interacts with CCDC26. DNMT1 is almost exclusively localized in the nucleus where it maintains DNA methylation patterns as cells replicate. In the absence of CCDC26, DNMT1 is re-localized to the cytoplasm and cells become hypomethylated.
A major question that remains in this instance is how CCDC26 orchestrates nuclear localization of DNMT1 and the mechanism that causes its cytosolic mis-localization in the absence of CCDC26. Independent of lincRNAs, in rare circumstances, DNMT1 localization in the cytoplasm has been demonstrated and can provide clues regarding the mechanism behind lincRNA-mediated DNMT1 localization. Arguably the best studied of these instances is during preimplantation in early development. An oocyte-specific form of DNMT1, DNMT1o, demonstrates a preferential localization within the cytosol during preimplantation development. DNMT1o lacks 118 amino acids at the N-terminus compared with somatic DNMT1. It has been postulated that an alternative, extended region of the N-terminus is critical, and complex folding of this area likely plays a large part in overriding the NLS. This is required for demethylation of the embryonic genome, upon which lineage specific methylation patterns are established (Cardoso and Leonhardt, 1999). In instances other than during embryonic development, cytosolic localization of DNMT1 tends to be aberrant; for example, it has been associated with several neurological disorders including hereditary sensory and autonomic neuropathy type 1E (HSAN1E) (Baets et al., 2015), Alzheimer disease (Mastroeni et al., 2013) and Parkinson disease (Desplats et al., 2011), as well as cancer tumorigenesis (Arzenani et al., 2011; Hodge et al., 2007). The reasons behind aberrant cytosolic localization of DNMT1 is not entirely clear. Various mechanisms including changes to post-translational modifications of DNMT1 (Hodge et al., 2007), HDAC inhibition (Arzenani et al., 2011), mutations within the RFTS domain (Baets et al., 2015), and disruption to nucleo-cytoplasmic transport systems across the nuclear membrane (Mastroeni et al., 2013) have been reported. Given that previous reports of cytosolic DNMT1 have often involved the N-terminal domain of DNMT1 (Baets et al., 2015; Cardoso and Leonhardt, 1999; Hodge et al., 2007), where the NLS resides, it can be speculated that this region of the protein may be affected by CCDC26, possibly through lincRNA-mediated post-translational modifications or protein folding.
Limitations of the study
Although our study shows that CCDC26 influences DNMT1 localization, to gain further insights into how CCDC26 functions, we will have to address a number of questions and will need to carry out a more detailed investigation. Our RIP assay indicates that CCDC26 is similarly enriched in DNMT1 pulldown as other ncRNAs that are previously reported to bind to DNMT1. However, we will need to confirm if this is a direct or indirect interaction. We can speculate that the interaction of CCDC26 with DNMT1 leads to changes in the configuration of DNMT1's nuclear localization domain that are needed for retaining DNMT1 in the nucleus. RNA-binding domain of DNMT1 is previously mapped, and it overlaps its C-terminal enzymatic domain (Di Ruscio et al., 2013). We will need to verify that CCDC26 binds to the same domain, and also, we will need to investigate the relationship between CCDC26 binding domain and nuclear localization domain of DNMT1. If CCDC26 and DNMT1 interact in vivo we expect them to colocalize in the cell. Our microscopy observations show that both DNMT1 and CCDC26 are nuclear. But, in addition, we have also observed that CCDC26 also accumulates in the nuclear periphery. We have not studied the significance of accumulation of CCDC26 in the nuclear periphery. However, our observation that CCDC26 knockout does not affect localization of other nuclear proteins such as EZH2 and HDAC2 shows that the role of CCDC26 is specific and it does not affect all nuclear proteins. Lastly, it is important to see if DNMT1 mis-localization is reversible and if re-introducing CCDC26 in the cells will re-localize DNMT1 in the nucleus. Currently, many of these questions are out of scope of this study but will need to be addressed in the future.
In summary, even though detailed molecular mechanism behind CCDC26-mediated DNMT1 localization still remains to be investigated, our study provides an insight into the role of CCDC26 in cancer as well as a novel lincRNA mechanism of DNMT1 regulation.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Aditi Kanhere (a.kanhere@liverpool.ac.uk).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The accession number for the RNA-sequencing data generated and reported in this paper is GEO: GSE105029.
Methods
All methods can be found in the accompanying transparent methods supplemental file.
Acknowledgments
We thank Dr J Woolley, Dr M Winch, and Dr E Petermann for useful discussions. We are also grateful to Dr Alessandro Di Maio for help with microscopy data analysis. During the course of study, AK was funded by SSfH fellowship from University of Birmingham. RJ was supported by BBSRC MIBTP fellowship. SW was supported by MRC Doctoral Training Program and University of Birmingham.
Author contributions
AK conceived the study. RH and AK wrote the manuscript. RH, SW, JH, CW, TL, and AK designed and carried out the experiments.
Declaration of interests
Authors declare no competing financial interest.
Published: April 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102273.
Supplemental information
References
- Alvarez-Ponce D., Torres-Sanchez M., Feyertag F., Kulkarni A., Nappi T. Molecular evolution of DNMT1 in vertebrates: duplications in marsupials followed by positive selection. PLoS One. 2018;13:e0195162. doi: 10.1371/journal.pone.0195162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arzenani M.K., Zade A.E., Ming Y., Vijverberg S.J., Zhang Z., Khan Z., Sadique S., Kallenbach L., Hu L., Vukojevic V. Genomic DNA hypomethylation by histone deacetylase inhibition implicates DNMT1 nuclear dynamics. Mol. Cell Biol. 2011;31:4119–4128. doi: 10.1128/MCB.01304-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baets J., Duan X., Wu Y., Smith G., Seeley W.W., Mademan I., McGrath N.M., Beadell N.C., Khoury J., Botuyan M.V. Defects of mutant DNMT1 are linked to a spectrum of neurological disorders. Brain. 2015;138:845–861. doi: 10.1093/brain/awv010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y., Wang W., Zhang Y., Zhang F., Zhang H. lncRNA MIAT suppression alleviates corneal angiogenesis through regulating miR-1246/ACE. Cell Cycle. 2019;18:661–669. doi: 10.1080/15384101.2019.1578143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X., Wu H., Zhu X., Guo X., Hutchins A.P., Luo Z., Song H., Chen Y., Lai K., Yin M. The p53-induced lincRNA-p21 derails somatic cell reprogramming by sustaining H3K9me3 and CpG methylation at pluripotency gene promoters. Cell Res. 2015;25:80–92. doi: 10.1038/cr.2014.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghoff E.G., Clark M.F., Chen S., Cajigas I., Leib D.E., Kohtz J.D. Evf2 (Dlx6as) lncRNA regulates ultraconserved enhancer methylation and the differential transcriptional control of adjacent genes. Development. 2013;140:4407–4416. doi: 10.1242/dev.099390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkyurek A.C., Suetake I., Arita K., Takeshita K., Nakagawa A., Shirakawa M., Tajima S. The DNA methyltransferase Dnmt1 directly interacts with the SET and RING finger-associated (SRA) domain of the multifunctional protein Uhrf1 to facilitate accession of the catalytic center to hemi-methylated DNA. J. Biol. Chem. 2014;289:379–386. doi: 10.1074/jbc.M113.523209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick M., Kim J.K., Esteve P.O., Clark A., Pradhan S., Jacobsen S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–1764. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- Bronner C., Alhosin M., Hamiche A., Mousli M. Coordinated dialogue between UHRF1 and DNMT1 to ensure faithful inheritance of methylated DNA patterns. Genes (Basel) 2019;10:65. doi: 10.3390/genes10010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso M.C., Leonhardt H. DNA methyltransferase is actively retained in the cytoplasm during early development. J. Cell Biol. 1999;147:25–32. doi: 10.1083/jcb.147.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalei V., Sansom S.N., Kong L., Lee S., Montiel J.F., Vance K.W., Ponting C.P. The long non-coding RNA Dali is an epigenetic regulator of neural differentiation. Elife. 2014;3:e04530. doi: 10.7554/eLife.04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Liu S., Tian L., Wu M., Ai F., Tang W., Zhao L., Ding J., Zhang L., Tang A. miR-124 and miR-506 inhibit colorectal cancer progression by targeting DNMT3B and DNMT1. Oncotarget. 2015;6:38139–38150. doi: 10.18632/oncotarget.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D., Deng J., Zhang B., He X., Meng Z., Li G., Ye H., Zheng S., Wei L., Deng X. LncRNA HOTAIR epigenetically suppresses miR-122 expression in hepatocellular carcinoma via DNA methylation. EBioMedicine. 2018;36:159–170. doi: 10.1016/j.ebiom.2018.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson C.M., Hutchinson J.N., Sara S.A., Ensminger A.W., Fox A.H., Chess A., Lawrence J.B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell. 2009;33:717–726. doi: 10.1016/j.molcel.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csankovszki G., Nagy A., Jaenisch R. Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J. Cell Biol. 2001;153:773–784. doi: 10.1083/jcb.153.4.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daskalos A., Nikolaidis G., Xinarianos G., Savvari P., Cassidy A., Zakopoulou R., Kotsinas A., Gorgoulis V., Field J.K., Liloglou T. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int. J. Cancer. 2009;124:81–87. doi: 10.1002/ijc.23849. [DOI] [PubMed] [Google Scholar]
- Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P., Spencer B., Coffee E., Patel P., Michael S., Patrick C., Adame A., Rockenstein E., Masliah E. Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011;286:9031–9037. doi: 10.1074/jbc.C110.212589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Ruscio A., Ebralidze A.K., Benoukraf T., Amabile G., Goff L.A., Terragni J., Figueroa M.E., De Figueiredo Pontes L.L., Alberich-Jorda M., Zhang P. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature. 2013;503:371–376. doi: 10.1038/nature12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Lagares A., Crujeiras A.B., Lopez-Serra P., Soler M., Setien F., Goyal A., Sandoval J., Hashimoto Y., Martinez-Cardus A., Gomez A. Epigenetic inactivation of the p53-induced long noncoding RNA TP53 target 1 in human cancer. Proc. Natl. Acad. Sci. U S A. 2016;113:E7535–E7544. doi: 10.1073/pnas.1608585113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty A.S., Bartolomei M.S., Schultz R.M. Regulation of stage-specific nuclear translocation of Dnmt1o during preimplantation mouse development. Dev. Biol. 2002;242:255–266. doi: 10.1006/dbio.2001.0534. [DOI] [PubMed] [Google Scholar]
- Duployez N., Boudry-Labis E., Roumier C., Boissel N., Petit A., Geffroy S., Helevaut N., Celli-Lebras K., Terre C., Fenneteau O. SNP-array lesions in core binding factor acute myeloid leukemia. Oncotarget. 2018;9:6478–6489. doi: 10.18632/oncotarget.24031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden A., Gaudet F., Waghmare A., Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300:455. doi: 10.1126/science.1083557. [DOI] [PubMed] [Google Scholar]
- Esteve P.O., Chin H.G., Smallwood A., Feehery G.R., Gangisetty O., Karpf A.R., Carey M.F., Pradhan S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006;20:3089–3103. doi: 10.1101/gad.1463706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Zapico M.E., Gonzalez-Paz N.C., Weiss E., Savoy D.N., Molina J.R., Fonseca R., Smyrk T.C., Chari S.T., Urrutia R., Billadeau D.D. Ectopic expression of VAV1 reveals an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell. 2005;7:39–49. doi: 10.1016/j.ccr.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Fiskus W., Buckley K., Rao R., Mandawat A., Yang Y., Joshi R., Wang Y., Balusu R., Chen J., Koul S. Panobinostat treatment depletes EZH2 and DNMT1 levels and enhances decitabine mediated de-repression of JunB and loss of survival of human acute leukemia cells. Cancer Biol. Ther. 2009;8:939–950. doi: 10.4161/cbt.8.10.8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F., Hurd P.J., Deplus R., Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31:2305–2312. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., Dai C., Yu X., Yin X.B., Liao W.J., Huang Y., Zhou F. Silencing of long non-coding RNA FOXD2-AS1 inhibits the progression of gallbladder cancer by mediating methylation-dependent induction of MLH1. Gene Therapy. 2020 doi: 10.1038/s41434-020-00187-w. [DOI] [PubMed] [Google Scholar]
- Gaudet F., Hodgson J.G., Eden A., Jackson-Grusby L., Dausman J., Gray J.W., Leonhardt H., Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- Ghoshal K., Datta J., Majumder S., Bai S., Kutay H., Motiwala T., Jacob S.T. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol. Cell Biol. 2005;25:4727–4741. doi: 10.1128/MCB.25.11.4727-4741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ghoshal K., Datta J., Majumder S., Bai S., Kutay H., Motiwala T., Jacob S.T. Correction for Ghoshal et al., "5-Aza-Deoxycytidine Induces Selective Degradation of DNA Methyltransferase 1 by a Proteasomal Pathway That Requires the KEN Box, Bromo-Adjacent Homology Domain, and Nuclear Localization Signal". Mol. Cell Biol. 2018;38:e00539-17. doi: 10.1128/MCB.00539-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodsell D.S. The molecular perspective: Cisplatin. Stem Cells. 2006;24:514–515. doi: 10.1634/stemcells.2006-CSC2. [DOI] [PubMed] [Google Scholar]
- Guo X., Chen Z., Zhao L., Cheng D., Song W., Zhang X. Long non-coding RNA-HAGLR suppressed tumor growth of lung adenocarcinoma through epigenetically silencing E2F1. Exp. Cell Res. 2019;382:111461. doi: 10.1016/j.yexcr.2019.06.006. [DOI] [PubMed] [Google Scholar]
- Hangauer M.J., Vaughn I.W., McManus M.T. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013;9:e1003569. doi: 10.1371/journal.pgen.1003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller G., Topakian T., Altenberger C., Cerny-Reiterer S., Herndlhofer S., Ziegler B., Datlinger P., Byrgazov K., Bock C., Mannhalter C. Next-generation sequencing identifies major DNA methylation changes during progression of Ph+ chronic myeloid leukemia. Leukemia. 2016;30:1861–1868. doi: 10.1038/leu.2016.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson G., Kelley D.R., Tenen D., Bernstein B., Rinn J.L. Widespread RNA binding by chromatin-associated proteins. Genome Biol. 2016;17:28. doi: 10.1186/s13059-016-0878-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman J.G., Jen J., Merlo A., Baylin S.B. Hypermethylation-associated inactivation indicates a tumor suppressor role for p15INK4B. Cancer Res. 1996;56:722–727. [PubMed] [Google Scholar]
- Hirano T., Yoshikawa R., Harada H., Harada Y., Ishida A., Yamazaki T. Long noncoding RNA, CCDC26, controls myeloid leukemia cell growth through regulation of KIT expression. Mol. Cancer. 2015;14:90. doi: 10.1186/s12943-015-0364-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge D.R., Cho E., Copeland T.D., Guszczynski T., Yang E., Seth A.K., Farrar W.L. IL-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (DNMT1) via phosphorylation of the nuclear localization sequence by the AKT kinase. Cancer Genomics Proteomics. 2007;4:387–398. [PubMed] [Google Scholar]
- Hui B., Ji H., Xu Y., Wang J., Ma Z., Zhang C., Wang K., Zhou Y. RREB1-induced upregulation of the lncRNA AGAP2-AS1 regulates the proliferation and migration of pancreatic cancer partly through suppressing ANKRD1 and ANGPTL4. Cell Death Dis. 2019;10:207. doi: 10.1038/s41419-019-1384-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson J.N., Ensminger A.W., Clemson C.M., Lynch C.R., Lawrence J.B., Chess A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics. 2007;8:39. doi: 10.1186/1471-2164-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida T., Suetake I., Tajima S., Morioka H., Ohta S., Obuse C., Tsurimoto T. PCNA clamp facilitates action of DNA cytosine methyltransferase 1 on hemimethylated DNA. Genes Cells. 2002;7:997–1007. doi: 10.1046/j.1365-2443.2002.00584.x. [DOI] [PubMed] [Google Scholar]
- Ilan L., Katzav S. Human Vav1 expression in hematopoietic and cancer cell lines is regulated by c-Myb and by CpG methylation. PLoS One. 2012;7:e29939. doi: 10.1371/journal.pone.0029939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura T., Yamamoto S., Ohgane J., Hattori N., Tanaka S., Shiota K. Non-coding RNA directed DNA demethylation of Sphk1 CpG island. Biochem. Biophys. Res. Commun. 2004;322:593–600. doi: 10.1016/j.bbrc.2004.07.159. [DOI] [PubMed] [Google Scholar]
- Iyer M.K., Niknafs Y.S., Malik R., Singhal U., Sahu A., Hosono Y., Barrette T.R., Prensner J.R., Evans J.R., Zhao S. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izadifard M., Pashaiefar H., Yaghmaie M., Montazeri M., Sadraie M., Momeny M., Jalili M., Ahmadvand M., Ghaffari S.H., Mohammadi S. Expression analysis of PVT1, CCDC26, and CCAT1 long noncoding RNAs in acute myeloid leukemia patients. Genet. Test Mol. Biomarkers. 2018;22:593–598. doi: 10.1089/gtmb.2018.0143. [DOI] [PubMed] [Google Scholar]
- Jaenisch R., Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Jeffery L., Nakielny S. Components of the DNA methylation system of chromatin control are RNA-binding proteins. J. Biol. Chem. 2004;279:49479–49487. doi: 10.1074/jbc.M409070200. [DOI] [PubMed] [Google Scholar]
- Kass S.U., Landsberger N., Wolffe A.P. DNA methylation directs a time-dependent repression of transcription initiation. Curr. Biol. 1997;7:157–165. doi: 10.1016/s0960-9822(97)70086-1. [DOI] [PubMed] [Google Scholar]
- Kim K.M., Small D., Scott R.J. Gene expression profiling of human myeloid leukemic MV4-11 cells treated with 5-Aza-2’-deoxycytidine. J. Cancer Ther. 2012;3:177–182. [Google Scholar]
- Klein C.J., Bird T., Ertekin-Taner N., Lincoln S., Hjorth R., Wu Y., Kwok J., Mer G., Dyck P.J., Nicholson G.A. DNMT1 mutation hot spot causes varied phenotypes of HSAN1 with dementia and hearing loss. Neurology. 2013;80:824–828. doi: 10.1212/WNL.0b013e318284076d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M.W., Radtke I., Bullinger L., Goorha S., Cheng J., Edelmann J., Gohlke J., Su X., Paschka P., Pounds S. High-resolution genomic profiling of adult and pediatric core-binding factor acute myeloid leukemia reveals new recurrent genomic alterations. Blood. 2012;119:67–75. doi: 10.1182/blood-2011-09-380444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt H., Page A.W., Weier H.U., Bestor T.H. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell. 1992;71:865–873. doi: 10.1016/0092-8674(92)90561-p. [DOI] [PubMed] [Google Scholar]
- Li E., Beard C., Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- Li Y., Liu X., Guo X., Liu X., Luo J. DNA methyltransferase 1 mediated aberrant methylation and silencing of SHP-1 gene in chronic myelogenous leukemia cells. Leuk. Res. 2017;58:9–13. doi: 10.1016/j.leukres.2017.03.014. [DOI] [PubMed] [Google Scholar]
- Liu B., Sun L., Liu Q., Gong C., Yao Y., Lv X., Lin L., Yao H., Su F., Li D. A cytoplasmic NF-kappaB interacting long noncoding RNA blocks IkappaB phosphorylation and suppresses breast cancer metastasis. Cancer Cell. 2015;27:370–381. doi: 10.1016/j.ccell.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018;19:81–92. doi: 10.1038/nrg.2017.80. [DOI] [PubMed] [Google Scholar]
- Mastroeni D., Chouliaras L., Grover A., Liang W.S., Hauns K., Rogers J., Coleman P.D. Reduced RAN expression and disrupted transport between cytoplasm and nucleus; a key event in Alzheimer's disease pathophysiology. PLoS One. 2013;8:e53349. doi: 10.1371/journal.pone.0053349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merry C.R., Forrest M.E., Sabers J.N., Beard L., Gao X.H., Hatzoglou M., Jackson M.W., Wang Z., Markowitz S.D., Khalil A.M. DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum. Mol. Genet. 2015;24:6240–6253. doi: 10.1093/hmg/ddv343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milutinovic S., Brown S.E., Zhuang Q., Szyf M. DNA methyltransferase 1 knock down induces gene expression by a mechanism independent of DNA methylation and histone deacetylation. J. Biol. Chem. 2004;279:27915–27927. doi: 10.1074/jbc.M312823200. [DOI] [PubMed] [Google Scholar]
- Mohammad F., Mondal T., Guseva N., Pandey G.K., Kanduri C. Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development. 2010;137:2493–2499. doi: 10.1242/dev.048181. [DOI] [PubMed] [Google Scholar]
- Morgan A.E., Davies T.J., Mc Auley M.T. The role of DNA methylation in ageing and cancer. Proc. Nutr. Soc. 2018;77:412–422. doi: 10.1017/S0029665118000150. [DOI] [PubMed] [Google Scholar]
- Murray N.R., Baumgardner G.P., Burns D.J., Fields A.P. Protein kinase C isotypes in human erythroleukemia (K562) cell proliferation and differentiation. Evidence that beta II protein kinase C is required for proliferation. J. Biol. Chem. 1993;268:15847–15853. [PubMed] [Google Scholar]
- Nan X., Ng H.H., Johnson C.A., Laherty C.D., Turner B.M., Eisenman R.N., Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- O'Leary V.B., Hain S., Maugg D., Smida J., Azimzadeh O., Tapio S., Ovsepian S.V., Atkinson M.J. Long non-coding RNA PARTICLE bridges histone and DNA methylation. Sci. Rep. 2017;7:1790. doi: 10.1038/s41598-017-01875-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki Y., Furuno M., Kasukawa T., Adachi J., Bono H., Kondo S., Nikaido I., Osato N., Saito R., Suzuki H. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- Otani J., Nankumo T., Arita K., Inamoto S., Ariyoshi M., Shirakawa M. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 2009;10:1235–1241. doi: 10.1038/embor.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakneshan P., Tetu B., Rabbani S.A. Demethylation of urokinase promoter as a prognostic marker in patients with breast carcinoma. Clin. Cancer Res. 2004;10:3035–3041. doi: 10.1158/1078-0432.ccr-03-0545. [DOI] [PubMed] [Google Scholar]
- Pandey R.R., Mondal T., Mohammad F., Enroth S., Redrup L., Komorowski J., Nagano T., Mancini-Dinardo D., Kanduri C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell. 2008;32:232–246. doi: 10.1016/j.molcel.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Pastural E., Takahashi N., Dong W.F., Bainbridge M., Hull A., Pearson D., Huang S., Lowsky R., DeCoteau J.F., Geyer C.R. RIZ1 repression is associated with insulin-like growth factor-1 signaling activation in chronic myeloid leukemia cell lines. Oncogene. 2007;26:1586–1594. doi: 10.1038/sj.onc.1209959. [DOI] [PubMed] [Google Scholar]
- Patel K., Dickson J., Din S., Macleod K., Jodrell D., Ramsahoye B. Targeting of 5-aza-2'-deoxycytidine residues by chromatin-associated DNMT1 induces proteasomal degradation of the free enzyme. Nucleic Acids Res. 2010;38:4313–4324. doi: 10.1093/nar/gkq187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei H., Hu W., Guo Z., Chen H., Ma J., Mao W., Li B., Wang A., Wan J., Zhang J. Long noncoding RNA CRYBG3 blocks cytokinesis by directly binding G-actin. Cancer Res. 2018;78:4563–4572. doi: 10.1158/0008-5472.CAN-18-0988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke I., Mullighan C.G., Ishii M., Su X., Cheng J., Ma J., Ganti R., Cai Z., Goorha S., Pounds S.B. Genomic analysis reveals few genetic alterations in pediatric acute myeloid leukemia. Proc. Natl. Acad. Sci. U S A. 2009;106:12944–12949. doi: 10.1073/pnas.0903142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen K.D., Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016;30:733–750. doi: 10.1101/gad.276568.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn J.L., Kertesz M., Wang J.K., Squazzo S.L., Xu X., Brugmann S.A., Goodnough L.H., Helms J.A., Farnham P.J., Segal E. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Rountree M.R., Bachman K.E., Baylin S.B. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 2000;25:269–277. doi: 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- Schmelz K., Wagner M., Dorken B., Tamm I. 5-Aza-2'-deoxycytidine induces p21WAF expression by demethylation of p73 leading to p53-independent apoptosis in myeloid leukemia. Int. J. Cancer. 2005;114:683–695. doi: 10.1002/ijc.20797. [DOI] [PubMed] [Google Scholar]
- Siegfried Z., Eden S., Mendelsohn M., Feng X., Tsuberi B.Z., Cedar H. DNA methylation represses transcription in vivo. Nat. Genet. 1999;22:203–206. doi: 10.1038/9727. [DOI] [PubMed] [Google Scholar]
- Somasundaram S., Forrest M.E., Moinova H., Cohen A., Varadan V., LaFramboise T., Markowitz S., Khalil A.M. The DNMT1-associated lincRNA DACOR1 reprograms genome-wide DNA methylation in colon cancer. Clin. Epigenetics. 2018;10:127. doi: 10.1186/s13148-018-0555-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J., Teplova M., Ishibe-Murakami S., Patel D.J. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science. 2012;335:709–712. doi: 10.1126/science.1214453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirzaker C., Millar D.S., Paul C.L., Warnecke P.M., Harrison J., Vincent P.C., Frommer M., Clark S.J. Extensive DNA methylation spanning the Rb promoter in retinoblastoma tumors. Cancer Res. 1997;57:2229–2237. [PubMed] [Google Scholar]
- Stresemann C., Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer. 2008;123:8–13. doi: 10.1002/ijc.23607. [DOI] [PubMed] [Google Scholar]
- Sun M., Nie F., Wang Y., Zhang Z., Hou J., He D., Xie M., Xu L., De W., Wang Z. LncRNA HOXA11-AS promotes proliferation and invasion of gastric cancer by scaffolding the chromatin modification factors PRC2, LSD1, and DNMT1. Cancer Res. 2016;76:6299–6310. doi: 10.1158/0008-5472.CAN-16-0356. [DOI] [PubMed] [Google Scholar]
- Venolia L., Gartler S.M. Comparison of transformation efficiency of human active and inactive X-chromosomal DNA. Nature. 1983;302:82–83. doi: 10.1038/302082a0. [DOI] [PubMed] [Google Scholar]
- Walsh C.P., Chaillet J.R., Bestor T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998;20:116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- Wang J., Hua L., Guo M., Yang L., Liu X., Li Y., Shang X., Luo J. Notable roles of EZH2 and DNMT1 in epigenetic dormancy of the SHP1 gene during the progression of chronic myeloid leukaemia. Oncol. Lett. 2017;13:4979–4985. doi: 10.3892/ol.2017.6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Zhao Y., Bao X., Zhu X., Kwok Y.K., Sun K., Chen X., Huang Y., Jauch R., Esteban M.A. LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res. 2015;25:335–350. doi: 10.1038/cr.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.X., Guo G.C., Qian X.K., Dou D.W., Zhang Z., Xu X.D., Duan X., Pei X.H. miR-506 attenuates methylation of lncRNA MEG3 to inhibit migration and invasion of breast cancer cell lines via targeting SP1 and SP3. Cancer Cell Int. 2018;18:171. doi: 10.1186/s12935-018-0642-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt F., Molloy P.L. Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev. 1988;2:1136–1143. doi: 10.1101/gad.2.9.1136. [DOI] [PubMed] [Google Scholar]
- Wen L.Z., Ding K., Wang Z.R., Ding C.H., Lei S.J., Liu J.P., Yin C., Hu P.F., Ding J., Chen W.S. SHP-1 acts as a tumor suppressor in hepatocarcinogenesis and HCC progression. Cancer Res. 2018;78:4680–4691. doi: 10.1158/0008-5472.CAN-17-3896. [DOI] [PubMed] [Google Scholar]
- Werner M.S., Ruthenburg A.J. Nuclear fractionation reveals thousands of chromatin-tethered noncoding RNAs adjacent to active genes. Cell Rep. 2015;12:1089–1098. doi: 10.1016/j.celrep.2015.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C., Kanhere A. 8q24.21 locus: a paradigm to link non-coding RNAs, genome polymorphisms and cancer. Int. J. Mol. Sci. 2021;22(3):1094. doi: 10.3390/ijms22031094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M., Xu X., Pan B., Chen X., Lin K., Zeng K., Liu X., Xu T., Sun L., Qin J. LncRNA SATB2-AS1 inhibits tumor metastasis and affects the tumor immune cell microenvironment in colorectal cancer by regulating SATB2. Mol. Cancer. 2019;18:135. doi: 10.1186/s12943-019-1063-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.Y., Liu T.C., Chang J.G., Lin P.M., Lin S.F. JunB gene expression is inactivated by methylation in chronic myeloid leukemia. Blood. 2003;101:3205–3211. doi: 10.1182/blood-2002-05-1598. [DOI] [PubMed] [Google Scholar]
- Yin W., Rossin A., Clifford J.L., Gronemeyer H. Co-resistance to retinoic acid and TRAIL by insertion mutagenesis into RAM. Oncogene. 2006;25:3735–3744. doi: 10.1038/sj.onc.1209410. [DOI] [PubMed] [Google Scholar]
- Yu J., Peng Y., Wu L.C., Xie Z., Deng Y., Hughes T., He S., Mo X., Chiu M., Wang Q.E. Curcumin down-regulates DNA methyltransferase 1 and plays an anti-leukemic role in acute myeloid leukemia. PLoS One. 2013;8:e55934. doi: 10.1371/journal.pone.0055934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W., Gius D., Onyango P., Muldoon-Jacobs K., Karp J., Feinberg A.P., Cui H. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature. 2008;451:202–206. doi: 10.1038/nature06468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-sequencing data generated and reported in this paper is GEO: GSE105029.