

Abstract
Comprehensive cancer data sets recently generated by the Clinical Proteomic Tumor Analysis Consortium (CPTAC) offer great potential for advancing our understanding of how to combat cancer. These data sets include DNA, RNA, protein, and clinical characterization for tumor and normal samples from large cohorts of many different cancer types. The raw data are publicly available at various Cancer Research Data Commons. However, widespread reuse of these data sets is also facilitated by easy access to the processed quantitative data tables. We have created a data application programming interface (API) to distribute these processed tables, implemented as a Python package called cptac. We implement it such that users who prefer to work in R can easily use our package for data access and then transfer the data into R for analysis. Our package distributes the finalized processed CPTAC data sets in a consistent, up-to-date format. This consistency makes it easy to integrate the data with common graphing, statistical, and machine-learning packages for advanced analysis. Additionally, consistent formatting across all cancer types promotes the investigation of pan-cancer trends. The data API structure of directly streaming data within a programming environment enhances the reproducibility. Finally, with the accompanying tutorials, this package provides a novel resource for cancer research education. View the software documentation at https://paynelab.github.io/cptac/. View the GitHub repository at https://github.com/PayneLab/cptac.
Keywords: data dissemination, data access, cancer, proteomics, genomics, proteogenomics, mass spectrometry, CPTAC, Python, R, reproducibility
Introduction
Large consortia, like the Clinical Proteomic Tumor Analysis Consortium (CPTAC), drive science forward by creating coordinated and structured data sets on a scale that is typically not possible with individual investigators. They amass both a number of samples and a diversity of measurements that require a large collaborative effort. In addition to the primary analysis done by the consortium and published as flagship manuscripts,1−7 these data sets are designed to be a resource to the scientific community to explore new questions or apply novel methodologies.8−12
Proteogenomic cancer data are of interest to a wide interdisciplinary audience, and different scientists may want to interact with different data products, for example, raw instrument data versus summarized quantitative tables. Funding agencies have focused on building resources for the dissemination of voluminous raw data files, for example, NCI Genomic Data Commons.13,14 These data warehouses address the logistical and technological challenges of storing and disseminating terabytes of sequencing and mass spectrometry data; however, raw data repositories have a limited audience, as the reanalysis of raw instrument files requires domain-specific knowledge and significant computational resources.
A growing audience of scientists want to directly interact with quantitative proteogenomics data tables and not the raw data. Currently there is not a common method for projects, large or small, to share these data tables in an open and computable format. Although some quantitative data may be shared through the large data warehouses,15 this mechanism has several drawbacks. First, the final data tables used in a publication are usually highly curated and processed by harmonization, batch correction, normalization, filtering, and so on. The detailed attention in these computational adjustments should not be lost; the public should have access to the exact data tables used in a publication. Second, the Data Commons model, as currently designed, separates multiomics data sets into different Data Commons instances, requiring users to have prior knowledge of which data sets belong together and where they are stored. Finally, computational convenience should be a driving factor in the data storage mechanism, meaning that data should be easy to programmatically access and quickly use in computation. Thus an alternative dissemination mechanism that facilitates accessing and utilizing these data tables is needed to serve a broader scientific community. A convenient method for disseminating coordinated data sets is the data Application Programming Interface (API) model, where data are streamed directly into a programming environment.16
As an illustration of the need for higher level data distribution methods, consider the widespread use of Jupyter Notebooks17,18 and other similar programming environments19 for sharing research. These tools are ideal for both explaining the context of an analysis and showing the exact methodology. However, for this method of sharing research to be successful, it needs an accompanying flexible data distribution method. If a shared notebook uses files that are stored on the original researcher’s computer or are too large to be conveniently streamed, then it will be much less useful to others who wish to replicate and extend that analysis. The data API model solves this problem by ensuring that the exact version of a data set is globally and universally accessible.
We present here a data API for proteogenomic cancer data generated by CPTAC, representing six tumor types. We provide access to the finalized data tables that were associated with the flagship publication for each cancer type. All tumor samples are characterized with genomic, transcriptomic, proteomic, and clinical data. The API streams proteogenomic data directly into a pandas DataFrame within a Python programming environment, dramatically improving the simplicity of data access. By using native DataFrame variables, the proteogenomic data easily integrate with common Python libraries for machine learning, graphing, and statistics. By leveraging the interoperability of R and Python, the data and analysis methods of the API can also be easily accessed via R scripts. Along with the API, we released an extensive set of tutorials to demonstrate common data analysis methods in proteogenomic and pan-cancer studies.
Methods
Overview
When reading these methods, it is important to keep in mind the distinction between the data files and the software API used to access them. In our project, these two entities are entirely separate and can be independently changed/updated.
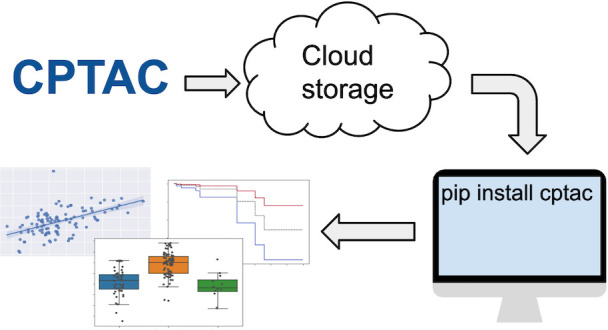
The CPTAC Python package, called cptac, is a data API that facilitates access to and utilization of cancer proteogenomic data within Python scripts (Figure 1). Similar to other Data-as-a-Service applications, the cptac package gives users on-demand access to structured data. The API parses, loads, integrates, and manipulates the cancer proteogenomic data from the CPTAC consortium. When data are accessed via the API, they are presented to the user as pandas DataFrames. This data structure conveniently and seamlessly integrates with most major machine learning, graphing, and data analytics libraries in Python.
Figure 1.

CPTAC data API. (top) Cohorts in CPTAC have the same fundamental multiomics and clinical data. (bottom) The cptac Python module is simple to install and use within a Python programming environment. Image courtesy of Nathan Johnson and Darcy Zacchilli. Copyright 2021.
The software code for the cptac package is open source and available at https://github.com/PayneLab/cptac. Formal versions are tagged on GitHub and released as software updates through the Python Package Index (PyPI; https://pypi.org/project/cptac/). Thus developers can work with the code of the API by forking the GitHub repository, but users of the data should install the package on their local computer with “pip install cptac”. See below for more detailed installation instructions. Users should follow the tutorials at https://paynelab.github.io/cptac/#documentation for explicit code demonstrating the use of the API. Developers should follow the instructions in the /devdocs folder on the GitHub repository for software design and implementation requirements.
Installation and Software Dependencies
We strive to make installation of the package as simple and easy as possible. Users must have Python 3.6 or greater installed to run the package. They can then install our package in a single step, using the pip program: pip install cptac. This downloads the package from where it is posted on PyPI and installs it on the user’s computer. The package is then ready to use.
The package depends on several other Python libraries including numpy, pandas, requests, and others. pip will automatically install these dependencies when it installs cptac because they are all also available on PyPI. Thus the user does not have to worry about managing the package’s software dependencies.
For additional installation details, consult the Installation section in the package documentation (https://paynelab.github.io/cptac/#installation).
Data Tables
The data accessible with the cptac package are the published final data tables for each CPTAC tumor type, namely, the published data tables associated with flagship publications of the CPTAC consortium for seven cancer types: colon,5 serous ovarian,4 breast,6 endometrial,2 renal clear cell,1 head and neck squamous cell carcinoma,7 and lung adenocarcinoma.3 The data set for every tumor type contains genomic, transcriptomic, proteomic, and phosphoproteomic measurements on the tumor samples. Each patient in the data set is described by a variety of clinical data including demographic, treatment, and outcome.
Package Construction and Organization
The methods of tumor collection and data generation within CPTAC follow a consistent, organized structure (Figure 1A). Our package has taken advantage of this consistency by creating an abstract class “Dataset”, which defines common variables and get functions to access all data tables that a data set will have, for example, get_clinical(), get_CNV(), get_proteomics(), get_somatic_mutation(), and so on. In addition to simple data access, the cptac package assists users in merging data across DataFrames using native join functions as part of the abstract Dataset class. These work on any combination of omics data and metadata, for example, join_metadata_to_mutations(). The join functions facilitate all merging to ensure that the returned DataFrames are properly keyed and indexed. Each tumor type is coded as a class object that inherits from the abstract Dataset class, that is, a derived class. Thus each derived class only needs to parse its specific data files into pandas DataFrames matching the format required by the abstract Dataset container; then, the rest of the functionality (i.e. get and join functions) is automatically integrated. This construction greatly simplifies the process of adding new tumor types.
To minimize the number of DataFrames and keep data descriptors present in the same variables as data values, we have implemented a multilevel index for some omics DataFrames. For example, proteomics and transcriptomics DataFrames reference protein/RNA isoforms both by their common name and by their unique database identifier. Although common names are used as the primary indexing key, the unique database identifiers are used as the secondary key to differentiate between isoforms. A second data type that uses a multi-index is post-translational modification (PTM) proteomics data such as phosphorylation. Here the multi-index comprises the gene name, database identifier, modified amino acid residue(s), and the MS-identified peptide sequence. This is necessary because multiple peptides may be observed in a data set with the same phosphorylated residue, often arising because of incomplete tryptic digestion.
Finally, the cptac package contains extra functionality within the utils subpackage, accessed as “from cptac import utils” or “import cptac.utils”. The utils subpackage implements commonly used functions and is continually expanding. To help users identify interacting proteins, a set of utility functions access pathway information from BioPlex, STRING, UniProt, and WikiPathways. The utils submodule also provides several wrappers for common statistical tests, like t tests and linear regression, with automatic correction of the p-value cutoff for multiple hypothesis testing. Finally, utils automates the identification of frequently mutated genes.
Streaming Data through the API
The cptac module gives users access to a large amount of data; the disk space for a single cancer type is 50–100 MB. Because PyPI restricts the package size and because a user may not want all of the data for all cancer types, the data files are not stored directly within the package or its GitHub repository. Initially, the package contains only the URLs needed to access the data. The user must request to download the data set for a particular cancer type, whereon the package uses one of these URLs to download an index file for that data set. This index file contains a list of all versions of the data set and a list of files, URLs, and MD5 hashes contained in each version. After each file is downloaded, the package hashes it and checks against the hash in the index to make sure none of the data was corrupted in the download process. After a user has downloaded a data set, they can load the data into variables in their Python program, for example, “dataset = cptac.Colon()”. The current implementation of the cptac package utilizes Box as a remote storage server. However, the software architecture has isolated the code involving remote data streaming, which makes it trivial to change the remote location.
New Data or API Releases
Before each data set’s flagship publication is finalized, the finalized data tables change periodically as various pipelines are compared and optimized. This is tracked by the consortium as versioned data releases, for example, 1.0 or 2.0 and so on. The package keeps track of these internal release versions. When a manuscript is published, these previous data versions are not recommended but are served by the package as a historical record. The API automatically monitors whether a user is working with the most recent release as follows: Each time the user loads a data set within their Python code, the package checks to make sure that the user has the current index for that data set. If necessary, it downloads a new version of the index from the server. Then, it checks whether the user is using the latest version recorded in the index. If the user did not request the latest version, then the package warns them that they are using an out-of-date version. It then loads whichever data version the user requested.
Note that when new data versions are released and downloaded, the package maintains any prior versions that were downloaded to a user’s computer. When a user loads a data set, they can tell the package to load one of these older versions. Thus if changes in a new version affect the results of a user’s analyses, then they can go back and look at the old version to explicitly compare the two outputs.
The package also makes sure that the software API itself is up to date. Periodically we release new versions of the package to add or improve functionality. Each time the user imports the package, it downloads a small file from the server that contains the latest release version number. It then compares this with the version number of the installed copy of the package. If they do not match, then it informs the user that their copy of the package is out of date and tells them how to update it using pip.
Because Internet connectivity is not universal, the package first checks to see whether there is a connection prior to comparing index files for API or data versions. If it is not possible to download the files needed to check whether the indices and package are up to date, then the package automatically skips this version check and works with whatever it has installed locally.
Error Handling
The table joining and other data manipulation functions in our package provide novel opportunities for users to make mistakes when working with data tables. If they request an operation that causes problems severe enough that the operation needs to be canceled, then the package raises an exception that informs them why it cannot do what they asked. If the operation can be completed but may cause issues for the user, then the package issues a warning to alert them of the potential problem.
The package uses the standard Python methods for raising exceptions and issuing warnings. However, the standard way that Python prints warnings and exceptions, with the full stack trace and associated information, can be intimidating for users without a computer programming background. To make these messages easier to decipher, our package defines custom sys.excepthook and warnings.showwarning functions so that whenever the package raises an exception or issues a warning, it will be printed in a concise, approachable format for users. Warnings and exceptions from outside of the package are printed in the normal fashion. However, in an IPython notebook environment, we cannot control how warnings and exceptions are displayed, so this feature is a specific enhancement for users accessing the package through the command line or scripts.
Results
To promote reproducibility, transparency, and collaboration in the analysis of CPTAC proteogenomic data, we created a data API that explicitly links data access and data analysis. The API gives access to the CPTAC data from seven cancer types: colon,5 serous ovarian,4 breast,6 endometrial,2 renal clear cell,1 head and neck squamous cell carcinoma, and lung adenocarcinoma.3 As new data sets are published, they will become publicly available through the API. Data for each cancer type contain information on DNA, RNA and proteins and clinical information (Figure 1). DNA data are derived from the whole genome and whole exome sequencing of tumor and blood normal and are processed to yield somatic mutation calls and somatic copy number variation calls. RNA data are from RNA-seq and are processed to yield transcriptomics and frequently circular RNA and micro-RNA. Protein data are from mass spectrometry-based proteomics and contain global proteomics and phosphoproteomics. Clinical data contain patient demographic information, descriptive data for the tumor, and patient follow-up information.
We emphasize that our software mirrors the data tables associated with the flagship publication for each CPTAC data set. Although the overall experimental design and data acquisition for each cancer type were consistent, the data processing pipeline used to produce quantitative data matrices was subtly distinct, for example, different protein quantification algorithms. Users interested in specific details of sample acquisition and data processing should consult the original publications. When performing pan-cancer analysis, users should design their analysis such that it is not affected by these differences between data sets, such as by looking for a particular trend in multiple cancer types but not comparing quantitative values. Access to the raw mass spectrometry and genomic data are provided by NCI’s Cancer Research Data Commons (i.e., Proteome Data Commons and Genome Data Commons) and the CPTAC’s Data Coordinating Center.
The data API is designed to provide frictionless access to quantitative data tables; its goal is to provide the data in the most convenient format with the least effort. The focus on quantitative data tables and not raw instrument data has several important benefits. First, these relatively small-sized files do not necessitate a voluminous server or special considerations for downloading, which enables the API to work on standard computers with standard Internet connectivity. Second, these processed tables are also completely publicly available and do not contain any private germline information, further facilitating data access. Third, quantitative tables can be provided directly within the programming environment as a simple matrix variable, meaning that a user’s first interaction with data is as a properly parsed and loaded Python/pandas DataFrame (Figure 2A). In addition to single table access, the API also has native join functions which will merge multiomics data types (Figure 2B). This simple access dramatically improves a user’s ability for interaction, exploration, and visualization (Figure 2C). Fourth, quantitative data files are the starting point of hypothesis testing and data analysis. Providing direct access to these data improves the transparency and reproducibility by ensuring that all analysts are accessing the exact same data. This type of universal synchronization is not typically seen when analysts keep their own local versions of data files, which are frequently altered and resaved as renormalized or filtered versions of the original. Moreover, independent file versions often lack a detailed provenance of data manipulation. However, with a data streaming API, all data access is within a programming environment, and any data manipulation is directly exposed in code following the data access. Finally, the data API represents a dramatically simpler method for users to access and analyze data. All data for the CPTAC cohorts are available in the same format from the same simple software interface. There is no need to visit multiple repositories, for example, sequencing and mass spectrometry data at multiple NCI Data Commons.
Figure 2.

Getting data from the cptac API. (A) The data API makes accessing CPTAC data simple and returns data in a native pandas DataFrame. (B) Merging different data types is facilitated by a suite of join functions in the API. (C) Joined mutation and proteomics data from panel B are shown with a boxplot from the seaborn Python graphing module. The example is drawn from use case 2 in the cptac documentation (https://paynelab.github.io/cptac/usecase02_clinical_attributes.html)
Versions
The data API is designed to keep track of formal versions of data. This is accomplished through a strict separation of the software code to load the data, and the actual data files. As with many large-scale projects, the data tables used in CPTAC analyses are periodically updated. Such adjustments are common in data analysis, and using a formal data version helps to properly track changes. These updates are often prompted by new patient survival or treatment information from follow-up visits. Distributing these updates throughout the consortium in a coordinated manner is best done through an official data release. Other reasons that prompt a data release include when the consortium has refined a software pipeline involved in generating the quantitative data tables (e.g., a different algorithm for processing raw sequencing or mass spectrometry data). The data set versions used in the CPTAC manuscripts are considered the final versions, and previous versions, although accessible, are not recommended for use. The end user can access these different versions when loading data into their Python environment. (See the Methods.) When working with the data API, the default version is the current published version; however, a user can specify a different version.
Tutorials and User Support
One of the most important goals of the cptac package is to promote reanalysis of the consortium’s data sets. To facilitate this, we have created a comprehensive set of tutorials and use cases (Table 1). The tutorials cover the basic mechanics of accessing data through the API and manipulating and merging data tables. The use cases are short examples that use the package to investigate real, biologically meaningful questions; they demonstrate how to use the API for hypothesis-driven biological discovery. Because the CPTAC data sets contain a variety of diverse data types, use cases often explore scientific questions that utilize integrated multiomics data.
Table 1. User Documentationa.
| Tutorial 1: Data intro | Goes over the basics of how to install the package and access data |
| Tutorial 2: pandas | More in-depth description of how to work with the tables using pandas |
| Tutorial 3: Joining DataFrames | Shows how to use built-in functions from cptac to join tables of different data types |
| Tutorial 4: MultiIndex | Some tables provided by the package use multilevel column indexes in cases where multiple keys are required to uniquely identify each column. This tutorial describes unique aspects of working with these tables |
| Tutorial 5: Updates | An explanation of how to access and work with data version updates and package version updates |
| Tutorial 6: Python and R | How access the Python API within R |
| Use case 1: Multiomic integration | Data access and integration for multiple omics data types |
| Use case 2: Clinical covariates | Explores metadata for correlation between clinical attributes |
| Use case 3: Clinical and acetylation | Compares acetylation levels between tumor subtypes |
| Use case 4: Mutations and omics | Studies the effects of DNA mutations on protein abundance |
| Use case 5: Enrichment analysis | Uses the GSEApyb module to find enriched pathways |
| Use case 6: Derived molecular | Identifies correlation between proteomics and attributes derived from molecular data, for example, MSI status |
| Use case 7: Trans genetic effect | Studies the effect of DNA mutations on the expression of a different protein |
| Use case 8: Outliers | Uses the Blacksheepc module to study outliers in expression values |
| Use case 9: Clinical outcomes | Uses patient follow-up data to look for correlations between clinical and molecular data and patient survival |
| Use case 10: Pathway overlay | Integrates quantitative molecular data with Reactome pathway maps |
A list of tutorials and use cases to help users explore the data API is available at https://paynelab.github.io/cptac/#documentation.
GSEApy module is available at https://gseapy.readthedocs.io/en/latest/introduction.html.
Blacksheep module is available at https://blacksheep.readthedocs.io/en/master/.
The tutorials and use cases are written in interactive Python notebooks (Jupyter Notebooks). This format allows explanatory text, code, and output to be seamlessly integrated into a single document. The notebooks can be viewed as static web pages on our GitHub site (https://paynelab.github.io/cptac/#documentation). That site also has directions for users who wish to download the original notebooks and run them interactively or access interactive web versions hosted by Binder. In addition to the Python-based notebooks, tutorial 6 demonstrates how to access the API within the R programming environment.
Along with our tutorials and use cases, we also provide user support through our GitHub issues page. This page allows users to submit bug reports, feature requests, and other feedback about our package. Past questions and answers are publicly available for others to view for reference.
Integration with R
The API is natively written in Python; however, there are options that facilitate access in other software languages. Users who prefer to work in R can easily use cptac to access the data they want and then transfer that data into R for analysis. A familiar way to transfer the tables from Python into R would be to load the data in Python using our package, save the tables to tab-delimited plain text files, and then read those files into R. A more seamless method is to use the R package reticulate to interface with cptac. reticulate allows a user to load and interface with Python packages and objects from an R environment and then convert Python objects into native R objects for subsequent use. Users interested in this course of action should consult tutorial 6.
Integrating with External Bioinformatics tools
To enhance usability and functionality, the data API connects to several bioinformatics tools. The first category of tools are those that have a Python implementation. Working with these third-party packages is facilitated by our use of DataFrames to encapsulate omics data, as many Python packages are written for pandas. Gene set analysis is a frequent step in omics data analysis to find common biological functions among a specified set of genes. One of the first of these tools was GSEA,20 which has been reimplemented in Python in the gseapy module (https://pypi.org/project/gseapy/). A tutorial demonstrating the use of gseapy is available in the cptac documentation. Another common task for CPTAC data is survival analysis. Several Python packages implement analyses like the Kaplan–Meier curves or the Cox proportional hazard test. These tests identify whether time to an event is affected by a specified variable, for example, protein abundance, tumor grade, and so on (Figure 3). The cptac tutorials demonstrate the lifelines package (https://pypi.org/project/lifelines/) using data from the API.
Figure 3.

Survival analysis with CPTAC data. Using the lifelines Python module, we identify variables that impact patient survival in ovarian cancer. A Kaplan–Meier curve is shown to separate time to death based on: (A) the FIGO stage, (B) the protein expression of PODXL, and (C) RAC2. A Cox proportional hazard assessment is shown in panel D. The images were generated using the code in use case 9 of the cptac documentation (https://paynelab.github.io/cptac/usecase09_clinical_outcomes.html).
A second type of external tool that can be integrated with the data API is bioinformatics databases. Many large databases like UniProt have web services that allow for programmatic access to specific information. To integrate our CPTAC data with these, the API wraps REST calls to the database. For example, the API can give users a list of interacting proteins from both UniProt and STRING by wrapping a call to their respective web service. Some other databases either do not have a convenient REST-API or have a small enough database that it can be loaded in full. Proteins belonging to biological pathways are curated by Wikipathways. We have downloaded this open-source information and integrated it into the data API, which users can access through simple function calls.
Discussion
Frictionless data access has become an important goal for science, as it promotes collaboration, transparency, reproducibility, and data reuse. In the past decade, attitudes and expectations in the scientific community have changed with respect to data sharing. Because data reuse extends the value of the financial investment beyond the original grant holder, various funding agencies have begun to set benchmarks for program success based on these more inclusive definitions of impact, as opposed to simple citation metrics. Although the sharing of raw data has a robust infrastructure for many primary data types, processed data tables are frequently still not shared in a simple and convenient manner. Here we present a data API for cancer proteogenomic data associated with the CPTAC consortium. By adopting the goals of the Data-as-a-Service model, our API radically improves access to these data.
As a generalized adaptation of the Data-as-a-Service model, our API’s guiding philosophy is that data sets should be accessible within a programming environment. Previous work with a similar on-demand goal is often achieved with a REST-API, which frequently returns data in a structured JSON format. Unfortunately, a JSON object requires parsing and reshaping to obtain the practical data matrix object. Because a data matrix is the most common data type for many scientific domains, our API directly gives users a native DataFrame object. A second advantage of our data API implementation over a REST-API is that it obviates the need to host a Web server, making it simpler for creation and long-term maintenance. In our implementation, data are hosted on a robust commercial server, and the code used to stream the data is minimal and compartmentalized. Therefore, moving files to a different location, for example, switching from Box to Amazon, would require minimal code changes. Numerous scientific projects, large and small, could improve the dissemination of their data through this mechanism.
Acknowledgments
We acknowledge important testing and feedback on the API provided by members of the Payne lab and the Fenyö and Ruggles laboratories (New York University). We also thank Jason McDermott (Pacific Northwest National Laboratory) for initial conversations about the concept. This work was supported by the National Cancer Institute (NCI) CPTAC award (U24 CA210972) and by the Simmons Center for Cancer Research.
The authors declare no competing financial interest.
Notes
View the software documentation at https://paynelab.github.io/cptac/. View the GitHub repository at https://github.com/PayneLab/cptac.
References
- Clark D. J.; Dhanasekaran S. M.; Petralia F.; Pan J.; Song X.; Hu Y.; da Veiga Leprevost F.; Reva B.; Lih T.-S. M.; Chang H.-Y.; Ma W.; Huang C.; Ricketts C. J.; Chen L.; Krek A.; Li Y.; Rykunov D.; Li Q. K.; Chen L. S.; Ozbek U.; Vasaikar S.; Wu Y.; Yoo S.; Chowdhury S.; Wyczalkowski M. A.; Ji J.; Schnaubelt M.; Kong A.; Sethuraman S.; Avtonomov D. M.; Ao M.; Colaprico A.; Cao S.; Cho K.-C.; Kalayci S.; Ma S.; Liu W.; Ruggles K.; Calinawan A.; Gümüş Z. H.; Geiszler D.; Kawaler E.; Teo G. C.; Wen B.; Zhang Y.; Keegan S.; Li K.; Chen F.; Edwards N.; Pierorazio P. M.; Chen X. S.; Pavlovich C. P.; Hakimi A. A.; Brominski G.; Hsieh J. J.; Antczak A.; Omelchenko T.; Lubinski J.; Wiznerowicz M.; Linehan W. M.; Kinsinger C. R.; Thiagarajan M.; Boja E. S.; Mesri M.; Hiltke T.; Robles A. I.; Rodriguez H.; Qian J.; Fenyö D.; Zhang B.; Ding L.; Schadt E.; Chinnaiyan A. M.; Zhang Z.; Omenn G. S.; Cieslik M.; Chan D. W.; Nesvizhskii A. I.; Wang P.; Zhang H.; et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 2019, 179 (4), 964–983.e31. 10.1016/j.cell.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y.; Kawaler E. A.; Cui Zhou D.; Gritsenko M. A.; Huang C.; Blumenberg L.; Karpova A.; Petyuk V. A.; Savage S. R.; Satpathy S.; Liu W.; Wu Y.; Tsai C.-F.; Wen B.; Li Z.; Cao S.; Moon J.; Shi Z.; Cornwell M.; Wyczalkowski M. A.; Chu R. K.; Vasaikar S.; Zhou H.; Gao Q.; Moore R. J.; Li K.; Sethuraman S.; Monroe M. E.; Zhao R.; Heiman D.; Krug K.; Clauser K.; Kothadia R.; Maruvka Y.; Pico A. R.; Oliphant A. E.; Hoskins E. L.; Pugh S. L.; Beecroft S. J. I.; Adams D. W.; Jarman J. C.; Kong A.; Chang H.-Y.; Reva B.; Liao Y.; Rykunov D.; Colaprico A.; Chen X. S.; Czekański A.; Jędryka M.; Matkowski R.; Wiznerowicz M.; Hiltke T.; Boja E.; Kinsinger C. R.; Mesri M.; Robles A. I.; Rodriguez H.; Mutch D.; Fuh K.; Ellis M. J.; DeLair D.; Thiagarajan M.; Mani D. R.; Getz G.; Noble M.; Nesvizhskii A. I.; Wang P.; Anderson M. L.; Levine D. A.; Smith R. D.; Payne S. H.; Ruggles K. V.; Rodland K. D.; Ding L.; Zhang B.; Liu T.; Fenyö D.; et al. Proteogenomic Characterization of Endometrial Carcinoma. Cell 2020, 180 (4), 729–748.e26. 10.1016/j.cell.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillette M. A.; Satpathy S.; Cao S.; Dhanasekaran S. M.; Vasaikar S. V.; Krug K.; Petralia F.; Li Y.; Liang W.-W.; Reva B.; Krek A.; Ji J.; Song X.; Liu W.; Hong R.; Yao L.; Blumenberg L.; Savage S. R.; Wendl M. C.; Wen B.; Li K.; Tang L. C.; MacMullan M. A.; Avanessian S. C.; Kane M. H.; Newton C. J.; Cornwell M.; Kothadia R. B.; Ma W.; Yoo S.; Mannan R.; Vats P.; Kumar-Sinha C.; Kawaler E. A.; Omelchenko T.; Colaprico A.; Geffen Y.; Maruvka Y. E.; da Veiga Leprevost F.; Wiznerowicz M.; Gümüş Z. H.; Veluswamy R. R.; Hostetter G.; Heiman D. I.; Wyczalkowski M. A.; Hiltke T.; Mesri M.; Kinsinger C. R.; Boja E. S.; Omenn G. S.; Chinnaiyan A. M.; Rodriguez H.; Li Q. K.; Jewell S. D.; Thiagarajan M.; Getz G.; Zhang B.; Fenyö D.; Ruggles K. V.; Cieslik M. P.; Robles A. I.; Clauser K. R.; Govindan R.; Wang P.; Nesvizhskii A. I.; Ding L.; Mani D. R.; Carr S. A.; et al. Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 2020, 182 (1), 200–225.e35. 10.1016/j.cell.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proteogenomic Characterization of Ovarian HGSC Implicates Mitotic Kinases, Replication Stress in Observed Chromosomal Instability. Cell Rep. Med. 2020, 1 (1), 100004. 10.1016/j.xcrm.2020.100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasaikar S.; Huang C.; Wang X.; Petyuk V. A.; Savage S. R.; Wen B.; Dou Y.; Zhang Y.; Shi Z.; Arshad O. A.; Gritsenko M. A.; Zimmerman L. J.; McDermott J. E.; Clauss T. R.; Moore R. J.; Zhao R.; Monroe M. E.; Wang Y.-T.; Chambers M. C.; Slebos R. J. C.; Lau K. S.; Mo Q.; Ding L.; Ellis M.; Thiagarajan M.; Kinsinger C. R.; Rodriguez H.; Smith R. D.; Rodland K. D.; Liebler D. C.; Liu T.; Zhang B.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177 (4), 1035–1049.e19. 10.1016/j.cell.2019.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug K.; Jaehnig E. J.; Satpathy S.; Blumenberg L.; Karpova A.; Anurag M.; Miles G.; Mertins P.; Geffen Y.; Tang L. C.; Heiman D. I.; Cao S.; Maruvka Y. E.; Lei J. T.; Huang C.; Kothadia R. B.; Colaprico A.; Birger C.; Wang J.; Dou Y.; Wen B.; Shi Z.; Liao Y.; Wiznerowicz M.; Wyczalkowski M. A.; Chen X. S.; Kennedy J. J.; Paulovich A. G.; Thiagarajan M.; Kinsinger C. R.; Hiltke T.; Boja E. S.; Mesri M.; Robles A. I.; Rodriguez H.; Westbrook T. F.; Ding L.; Getz G.; Clauser K. R.; Fenyö D.; Ruggles K. V.; Zhang B.; Mani D. R.; Carr S. A.; Ellis M. J.; Gillette M. A.; et al. Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 2020, 183 (5), 1436–1456.e31. 10.1016/j.cell.2020.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.; Chen L.; Savage S. R.; Eguez R. V.; Dou Y.; Li Y.; da Veiga Leprevost F.; Jaehnig E. J.; Lei J. T.; Wen B.; Schnaubelt M.; Krug K.; Song X.; Cieślik M.; Chang H.-Y.; Wyczalkowski M. A.; Li K.; Colaprico A.; Li Q. K.; Clark D. J.; Hu Y.; Cao L.; Pan J.; Wang Y.; Cho K.-C.; Shi Z.; Liao Y.; Jiang W.; Anurag M.; Ji J.; Yoo S.; Zhou D. C.; Liang W.-W.; Wendl M.; Vats P.; Carr S. A.; Mani D. R.; Zhang Z.; Qian J.; Chen X. S.; Pico A. R.; Wang P.; Chinnaiyan A. M.; Ketchum K. A.; Kinsinger C. R.; Robles A. I.; An E.; Hiltke T.; Mesri M.; Thiagarajan M.; Weaver A. M.; Sikora A. G.; Lubiński J.; Wierzbicka M.; Wiznerowicz M.; Satpathy S.; Gillette M. A.; Miles G.; Ellis M. J.; Omenn G. S.; Rodriguez H.; Boja E. S.; Dhanasekaran S. M.; Ding L.; Nesvizhskii A. I.; El-Naggar A. K.; Chan D. W.; Zhang H.; Zhang B. Proteogenomic Insights into the Biology and Treatment of HPV-Negative Head and Neck Squamous Cell Carcinoma. Cancer Cell 2021, 10.1016/j.ccell.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X.; Xu X.; Wang Y.; Hawke D. H.; Yu S.; Han L.; Zhou Z.; Mojumdar K.; Jeong K. J.; Labrie M.; Tsang Y. H.; Zhang M.; Lu Y.; Hwu P.; Scott K. L.; Liang H.; Mills G. B. A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell 2018, 33 (5), 817–828.e7. 10.1016/j.ccell.2018.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan C. J.; Kennedy S.; Bajrami I.; Matallanas D.; Lord C. J. A Compendium of Co-Regulated Protein Complexes in Breast Cancer Reveals Collateral Loss Events. Cell Syst 2017, 5 (4), 399–409.e5. 10.1016/j.cels.2017.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves E.; Fragoulis A.; Garcia-Alonso L.; Cramer T.; Saez-Rodriguez J.; Beltrao P. Widespread Post-Transcriptional Attenuation of Genomic Copy-Number Variation in Cancer. Cell Syst 2017, 5 (4), 386–398.e4. 10.1016/j.cels.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P.; Heins Z. J.; Muller J. T.; Katsnelson L.; de Bruijn I.; Abeshouse A. A.; Schultz N.; Fenyö D.; Gao J. Integration and Analysis of CPTAC Proteomics Data in the Context of Cancer Genomics in the CBioPortal. Mol. Cell Proteomics 2019, 18 (9), 1893–1898. 10.1074/mcp.TIR119.001673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong M.; Yu C.; Zhan D.; Zhang M.; Zhen B.; Zhu W.; Wang Y.; Wu C.; He F.; Qin J.; Li T. Molecular Subtyping of Cancer and Nomination of Kinase Candidates for Inhibition with Phosphoproteomics: Reanalysis of CPTAC Ovarian Cancer. EBioMedicine 2019, 40, 305–317. 10.1016/j.ebiom.2018.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Deutsch E. W.; Wang R.; Csordas A.; Reisinger F.; Ríos D.; Dianes J. A.; Sun Z.; Farrah T.; Bandeira N.; Binz P.-A.; Xenarios I.; Eisenacher M.; Mayer G.; Gatto L.; Campos A.; Chalkley R. J.; Kraus H.-J.; Albar J. P.; Martinez-Bartolomé S.; Apweiler R.; Omenn G. S.; Martens L.; Jones A. R.; Hermjakob H. ProteomeXchange Provides Globally Coordinated Proteomics Data Submission and Dissemination. Nat. Biotechnol. 2014, 32 (3), 223–226. 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman R. L.; Heath A. P.; Ferretti V.; Varmus H. E.; Lowy D. R.; Kibbe W. A.; Staudt L. M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375 (12), 1109–1112. 10.1056/NEJMp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Csordas A.; Bai J.; Bernal-Llinares M.; Hewapathirana S.; Kundu D. J.; Inuganti A.; Griss J.; Mayer G.; Eisenacher M.; Pérez E.; Uszkoreit J.; Pfeuffer J.; Sachsenberg T.; Yilmaz S.; Tiwary S.; Cox J.; Audain E.; Walzer M.; Jarnuczak A. F.; Ternent T.; Brazma A.; Vizcaíno J. A. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47 (D1), D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L.; Jin Z.; Yang S.; Xu Y.; Zhu Y.; Ji Y. TCGA-Assembler 2: Software Pipeline for Retrieval and Processing of TCGA/CPTAC Data. Bioinformatics 2018, 34 (9), 1615–1617. 10.1093/bioinformatics/btx812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez K. M.; Pritchard L.; Reinke S. N.; Broadhurst D. I. Toward Collaborative Open Data Science in Metabolomics Using Jupyter Notebooks and Cloud Computing. Metabolomics 2019, 15 (10), 125. 10.1007/s11306-019-1588-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rule A.; Birmingham A.; Zuniga C.; Altintas I.; Huang S.-C.; Knight R.; Moshiri N.; Nguyen M. H.; Rosenthal S. B.; Pérez F.; Rose P. W. Ten Simple Rules for Writing and Sharing Computational Analyses in Jupyter Notebooks. PLoS Comput. Biol. 2019, 15 (7), e1007007. 10.1371/journal.pcbi.1007007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rule A.; Tabard A.; Hollan J. D. Exploration and Explanation in Computational Notebooks. Proceedings of the 2018 CHI Conference on Human Factors in Computing Systems - CHI ’18 2018, 1–12. 10.1145/3173574.3173606. [DOI] [Google Scholar]
- Subramanian A.; Tamayo P.; Mootha V. K.; Mukherjee S.; Ebert B. L.; Gillette M. A.; Paulovich A.; Pomeroy S. L.; Golub T. R.; Lander E. S.; Mesirov J. P. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (43), 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]