

ABSTRACT
Introduction: COVID-19 pandemic has caused huge loss of human lives and extensive socio-economic damages. The immuno-pathology of this disease is neither clearly understood nor there are effective drugs for severe cases of COVID-19. Repurposing of available drugs for the treatment of COVID-19 is imperative.
Areas Covered: This review has gathered the evidence from PubMed, Google Scholar, WHO, and other reliable websites on COVID-19 and summarized the existing knowledge of the immuno-pathology of COVID-19. We elucidated how vitamin D through its diverse actions on immune effector cells, epithelial cells, or renin-angiotensin-aldosterone system could have a modulatory role on the pathogenic mechanisms of COVID-19. The epidemiological evidence associating vitamin D deficiency with the severity and incidence of COVID-19 is also presented. However, the evidence of clinical benefit to patients of COVID-19 from randomized controlled trials with vitamin D has not come as yet.
Expert opinion: It is now established that fatality of COVID-19 is primarily determined by hyperactivation of the host’s innate immune system in response to SARS-CoV-2 invasion, and thus the research on the immuno-modulatory and other roles of vitamin D against viral infections should be pursued vigorously. This would be also useful for future pandemics caused by other novel viruses.
KEYWORDS: COVID-19, cytokine, immuno-modulation, innate immunity, interferon, SARS-CoV-2, vitamin D
1. Introduction
The novel coronavirus pandemic (COVID-19) has posed significant threats to all countries across the globe in terms of loss of human lives and depletion of economic resources. The viral outbreak originated in the Wuhan city of China in the middle of December 2019 and spread through most of the countries in the world affecting 61.8 million people and taking a toll of more than 1.4 million human lives as of 1 December 2020 [1]. The disease is caused by SARS-CoV-2, a single-stranded (+) RNA virus belonging to the β-coronavirus sub-type, and its high degree of genomic homology with several types of bat coronaviruses indicates that SARS-CoV-2 probably originated in bat, but human to human transmission of the disease is responsible for the global spread of COVID-19 [2–4].
SARS-CoV, a novel Coronavirus, was identified in 2002 as the pathogenic agent of the Severe Acute Respiratory Syndrome (SARS) outbreak that occurred in the Guangdong Province of China [5]. Again a novel human coronavirus, named Middle East Respiratory Syndrome-CoV (MERS-CoV), emerged in the Middle East in 2012 causing a series of highly pathogenic respiratory tract infections in Saudi Arabia and other countries in the Middle East [6,7]. Since December 2019, severe pneumonia-like cases of unknown origin were observed in Wuhan city (China). The pathogen was identified as a novel enveloped RNA β coronavirus that was named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) or initially as 2019-nCoV [8]. Several theories have highlighted the zoonotic origin of these viruses suddenly jumping from animals to humans with high virulence and complications. In the case of SARS-CoV-2, the communicability and modes of spread are far wide than its previous counterparts. However, the basic mechanisms of viral colonization in the respiratory tract and elsewhere and replication processes are very similar.
Several important aspects of COVID-19 include a variable spectrum of symptoms and signs, a high rate of transmission, and disparate outcomes in terms of severity and mortality in different nations [9–11]. Additionally, the disease can be asymptomatic in a fairly large number of people, and thus, the control of the disease spread through the identification of patients followed by isolation and quarantine becomes extremely difficult. Further, recent epidemiological studies worldwide have shown that the presence of co-morbidities like diabetes mellitus (DM), hypertension and cardiac dysfunction, asthma, chronic obstructive pulmonary disease (COPD), chronic kidney disease (CKD), cancer, and old age lead to most of the COVID-19-related deaths [10,12,13]. Although the vast majority of the initial reports of COVID-19 deaths indicated atypical pneumonia with severe respiratory distress as the major cause of mortality in this disease, subsequent studies attributed many deaths of COVID-19 to vascular dysfunction, coagulopathy with thrombosis or myocardial injury, and cardiac dysfunctions [14–19]. Thus, the molecular mechanisms underlying COVID-19 pathogenesis, complications, and mortality have become quite controversial. In the absence of specific anti-viral drugs for COVID-19 and with mass vaccination programs yet to cover sizable portions of the populations at risk, the identification of new drug targets and re-purposing of existing drugs will be required to combat this pandemic. A deeper understanding of the immuno-pathology of COVID-19 is thus imperative. In general, viral invasion and replication in diverse tissues may be directly responsible for the pathophysiology of COVID-19 or the latter may be triggered by an abnormal response of the host in the form of immune-dysfunction and cytokine storm or the dysfunction of the renin-angiotensin–aldosterone system or RAAS [20–22]. In the current review, we will analyze the pathogenic mechanisms of COVID-19 from existing literature, and then attempt to assess if vitamin D through its diverse genomic and non-genomic actions could provide some therapeutic benefits in this disease condition. The potential beneficial effects of vitamin D in COVID-19 have been argued upon by several researchers, but a more critical view is necessary [23,24].
While writing this narrative review, we have searched PubMed, Google Scholar, WHO website, and other reliable websites using keywords like COVID-19, SARS-CoV-2, COVID-19 and innate immunity, COVID-19 and adaptive immune response, SARS-CoV-2 and RAAS, vitamin D, vitamin D and immuno-modulation, vitamin D and RAAS, vitamin D and COVID-19, and also examined many cross-references from the primary sources. Between July 2020 and February 2021, we screened 219 full-length articles published in English language and additional one article published in Chinese language for which we relied on the abstract in English, and finally, we selected 175 publications to complete our review.
2. Pathogenesis and pathology of COVID-19
2.1. SARS-CoV-2: invasion and multiplication in different organs
SARS-CoV-2 invades the epithelial cells of the respiratory tract, lungs, and gastrointestinal tract, vascular endothelial cells and also cells of different organs like kidney, heart, liver, and possibly brain [9,25,26]. The virus with the help of the viral spike protein (S) binds with angiotensin II converting enzyme 2 (ACE2) receptors present on the membranes of the host cells, and this process requires the priming of S protein by two proteolytic cleavages with the help of a host-cell serine protease called TMPRSS2 [27,28]. Several other proteases (host or virus-derived) are involved in the binding and internalization of the virus and its subsequent maturation within the cells [29]. Within the cells, the positive single-strand viral genomic RNA is first translated to produce two polyproteins which are cleaved to generate several non-structural proteins of SARS-CoV-2 that assemble to form the replication/transcription machinery that helps to produce finally positive-strand genomic RNA and many subgenomic RNAs (+) coding for viral structural proteins [25]. A primary target of SARS-CoV-2 invasion is the respiratory tract and lungs which leads to initial symptoms of sore throat, and dry cough, and the virus is detectable in sputum and nasopharyngeal swabs [26,30]. In around 80% of the infected patients, the disease will be mild and mostly restricted to the upper and conducting airways. However, in many cases, the patients would progress to a more severe form of the disease-causing pneumonia, hypoxemia, and respiratory distress. Apart from the respiratory tract involvement, many COVID-19 patients present with nausea, vomiting, diarrhea, abdominal pain, and so on, presumably because of the invasion of gastro-intestinal epithelial cells by the virus which is detectable in fecal samples [26,30]. There is evidence that many other organs and vascular endothelial cells are invaded by SARS-CoV-2 which is understandable given the expression profiles of ACE2 in a wide range of tissues [9,16,26]. Clinicians and researchers have attempted to describe the different phases of progression of COVID-19 based on clinical presentations or radiological findings or immuno-pathogenic mechanisms or a combination of these, and despite differences in terminologies and overlapping features and pathogenic mechanisms among the different stages of COVID-19, a common pattern is gradually being recognized [31–35]. Thus, the different stages of the disease have been described as the asymptomatic (RT-qPCR positivity for SARS-CoV-2 without any clinical symptoms), mild (features limited to upper respiratory tract infection), moderate (pneumonia with pulmonary lesions detectable by CT lungs but without any obvious hypoxemia), severe (pneumonia with hypoxemia) and critical (acute respiratory distress syndrome or ARDS, myocardial damage, acute kidney injury, shock, etc.) [32]. This staging mostly corresponds with that suggested by WHO [36]. An impressive staging based on immuno-pathogenic progression with the corresponding clinical features is suggested as Stage I (early infection; predominantly viral response phase with nonspecific symptoms of malaise, fever, dry cough, etc.), Stage II (pulmonary phase IIa without hypoxia and IIb with hypoxia; an overlap of viral response phase and host inflammatory response phase) and Stage III (hyperinflammation phase with ARDS, cardiac failure, shock, etc.) [34]. Other stagings of disease progression suggest four stages: Stage I (viral entry and replication, asymptomatic), Stage II (viral replication and spread; clinically mild to moderate disease), Stage III (multi-organ inflammation; clinically severe disease) and Stage IV (endothelial damage, thrombosis, multi-organ damage; critical cases) [35]. However, it is to be emphasized that many aspects of COVID-19 pathogenesis are still unknown, and it is not certain whether the chronology of progression of the disease pathogenesis can be really phased in such a discrete manner.
2.2. Immune-response of the host against SARS-CoV-2
The pathophysiology of COVID-19 involves a complex interaction between the SARS-CoV-2 and the body immune system and thus the disease spectrum varies from asymptomatic cases to severe respiratory failure. Physical barrier integrity, appropriate innate and adaptive immune responses are integral in defense against viral diseases. The entry of any virus triggers the innate immune system to eliminate the virus by different mechanisms. The activation of membrane-bound Toll-like receptors (TLRs) results in the production of both lipid signaling molecules such as prostaglandins and protein (or peptide) signaling molecules such as cytokines and chemokines, etc., and together these produce inflammatory reactions, recruit macrophages and dendritic cells and stimulate both humoral and cell-mediated immune response. Some of the TLRs which are present in the endoplasmic reticulum membrane like TLR3, TLR 7, TLR8, TLR9, and possibly TLR2 and TLR4 which can appear in the endosomes may recognize pathogen-associated molecular patterns present in RNA or DNA viruses [37,38]. Apart from TLRs, there are other pattern-recognition receptors in the cytosol which are important components of innate immunity against viral infections such as the retinoic-acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 or MDA5, etc. [38–40]. The innate immune response plays a crucial role in controlling the viral infection by activating several transcription factors that enhance the expressions of multiple genes such as type I interferons (IFNs), IFN-stimulated genes, and genes for cytokines and chemokines [39]. However, the viruses have developed ingenious mechanisms to subvert the innate immune response of the host by interfering with TLR-signaling pathways, suppressing IFN production by inhibiting the activation of interferon regulatory factor 3 (IRF-3), inhibiting expressions of IFN-stimulated genes, blocking the recognition of viral genome by the intra-cellular sensor proteins like RIG-I or MDA5 or producing chemokine-mimicking molecules that bind to chemokine receptors and trigger incongruous signals [38–43]. Although the innate immune response is important for controlling viral infection, a prolonged and abnormal immune response can lead to extensive tissue damage in multiple organs which could be extremely detrimental to the host.
The induction of the innate immune response in COVID-19 has been studied in some detail, which shows similarities with earlier observations made during SARS and MERS outbreaks [44–46]. Experimental studies, in vitro with lung tissue and in vivo with animal models, and analysis of postmortem lungs of COVID-19 patients suggest that invasion of cells by SARS-CoV-2 causes an impaired IFN response, but the cells (epithelial cells of lung alveoli and other organs) secrete a variety of pro-inflammatory cytokines and chemokines [47]. The impaired IFN response in SARS-CoV-2 infection occurs because the virus uses multiple escape mechanisms to deceive the sensor proteins of the innate immune system, and further several SARS-CoV-2 proteins (both non-structural and structural proteins such as nsp1, nsp3, nsp4, nsp7, nsp16, ORF3b, ORF6a, M, E proteins, etc.) have been shown to interfere with IFN expression and/or signaling functions at multiple levels [48,49]. These mechanisms are probably very similar between SARS-CoV and SARS-CoV-2 [48,50]. This impaired IFN response with consequent failure of the anti-viral defense of the cells causes an exuberant replication of the virus leading to invasion of new alveolar cells by the virus, secretion of pro-inflammatory cytokines and chemokines, infiltration of lungs by monocytes, monocyte-derived macrophages, and neutrophils with further secretion of pro-inflammatory mediators, and all these processes combine to bring in the hyperinflammation phase with cytolysis of alveolar cells by multiple mechanisms [49,51,52]. However, it is to be emphasized that impaired IFN response in SARS-CoV-2 infection is not so severe as with SARS-CoV and MERS-CoV, and studies have shown that IFN-α2 can be detected in plasma of COVID-19 patients with non-detectable levels associated with worse clinical course [48,53]. Likewise, another study performed meta-transcriptome sequencing in broncho-alveolar lavage fluid of COVID-19 patients, analyzed the expression profiles of viral and host genes, and observed several folds increase in the expressions of IFN-stimulated genes of the host cells than that in negative controls especially for those genes involved in inflammation [54,55]. Although a coherent picture of the innate immune response in COVID-19 is still not available, a moderately impaired and delayed IFN response with an associated robust secretion of proinflammatory chemokines and cytokines is the common scenario following SARS-CoV-2 invasion of lungs. The infected alveolar epithelial cells and various infiltrating cells from the circulation, especially the monocytes and macrophages, take part in the release of inflammatory chemokines and cytokines following SARS-CoV-2 invasion of lungs. The hyperinflammatory response has been speculated to involve complex signaling pathways that include different types of pattern recognition receptors TLRs, RIG-I, MDA-5, etc., their downstream adaptors and effectors leading to activation of inflammosomes, nuclear factor – kB (NF-kB), signal transducer and activator of transcription (STAT), etc. [48,49,52,56]. In agreement with this pathogenic process, the analysis of sera showed increased levels of cytokines like TNF-α, IL-2, IL-7, IL-1β, IL-6, IL-8, IL-10, and granulocyte-colony stimulating factor (G-CSF), as well as chemokines such as monocyte chemoattractant protein 1 (MCP1), IFN-gamma-inducible protein 10 (IP10), and macrophage inflammatory protein 1α (MIP1α), etc., in patients of COVID-19 with higher levels of some of these parameters correlating with clinical severity of the disease [12,30,46,56–59]. Very high levels of cytokines and chemokines released from the infected lungs in severely ill COVID-19 subjects have been termed ‘the cytokine storm’ which is presumably responsible for ARDS and multiple organ failure (acute kidney failure, acute heart failure, rhabdomyolysis, etc.) resulting in death in many cases.
The adaptive immune response against SARS-CoV-2 is yet to be fully elucidated, though in general for all viral infections cytotoxic CD8+ T cells eliminate the infected cells and helper CD4+ T cells promote the B cells to secrete antibodies or recruit macrophages at the site of viral invasion or assist the cytotoxic T cells. In COVID-19, lymphopenia is observed in peripheral circulation with decreases in CD4+ and CD8+ T cells, as well as B cells, and the decrease is more conspicuous in severe cases [12,60,61]. A few studies have suggested that peripheral lymphopenia in COVID-19 is primarily because of a decrease in CD4+ T cells [62]. Several studies performed a detailed immuno-profiling of peripheral blood mononuclear cells of moderate and severe cases of COVID-19 and demonstrated in peripheral circulation lymphopenia with decreased number of T cells and natural killer or NK cells but with an increased proportion of mature NK cells which had antigenic phenotypes as CD57+ CD56dim or NKG2C+CD57+ CD56dim [59,63]. NK cells not only can kill the virally infected cells but may take part in adaptive immune response mediated by T cells. The CD56bright (cytokine-producing cells) and CD56dim (cytotoxic) NK cells as well as CD4+ and CD8+ T cells were described to be in activated or exhausted state in peripheral circulation of COVID-19 patients [59,63]. The decrease in circulating lymphocyte count in COVID-19 could be partly attributed to the migration of lymphocytes in different tissues invaded by the virus, and an adaptive immune response to SARS-CoV-2 through specific CD4+ and CD8+ T cells recognizing multiple epitopes in structural and non-structural proteins of the virus have been clearly demonstrated in convalescent COVID-19 patients [64–66]. Another study likewise demonstrated strong and SARS-CoV-2 specific CD4+ and CD8+ T cell response in recovered patients of COVID-19 with a stronger and broader T cell response in patients with the severe form of the disease [67]. However, T cell response in subjects who had a milder form of COVID-19 showed a proportionately higher CD8+ T cell response compared to CD4+ response when challenged with SARS-CoV-2 peptides in the ex-vivo assay [67]. More interestingly, these studies have shown the existence of memory T cells in peripheral circulation responding to SARS-CoV-2 proteins in subjects who were never exposed to this virus, and these memory T-cells also cross-react with several other coronaviruses endemic in the human population causing mild respiratory tract illnesses [65,66,68]. However, it is to be emphasized that adaptive immunity in COVID-19 is a complex phenomenon, and an elaborate study demonstrated three different immunotypes in COVID-19 patients with different CD4+ and CD8+ T cell responses [69]. Another arm of the adaptive immune response is the production of neutralizing antibodies by B cells against SARS-CoV-2 which needs to be explored further. In a Chinese cohort study with mild cases of COVID-19, neutralizing antibodies have been detected with aged patients having a higher titer of antibodies than the younger patients [70]. Likewise, neutralizing antibodies specific to several epitopes located in the receptor-binding domain and other regions of viral S protein have been identified in the plasma of subjects who recovered from mild to moderate COVID-19 [71]. Another study also reported the isolation of neutralizing antibodies directed toward multiple epitopes of S protein from memory B lymphocytes from the peripheral circulation of patients of COVID-19 [72]. The presence of neutralizing antibodies specific to a receptor-binding domain (RBD) and other regions of S protein were detected in the sera of a large number of convalescent COVID-19 patients in another study, and this study further demonstrated the presence of specific B lymphocytes responding to RBD in the peripheral circulation of some of these patients [73]. In the peripheral circulation of COVID-19 patients, other studies have also corroborated the generation of neutralizing antibodies of various types and primed B cells to SARS-CoV-2 protein epitopes, present notably in RBD of S protein, and some of these B cells probably persist as memory B cells even after the level of antibodies in the blood goes down considerably [72,74,75]. The existence of these memory T and B cells after SARS-CoV-2 infection may prevent a further re-infection by SARS-CoV-2 later on, but this is yet to be established. Overall, it appears that both innate and adaptive immunity may play an important role in viral elimination during the course of the disease and prevention of a re-infection, while an exuberant immune response may lead to fatal consequences through extensive tissue damage.
2.3. RAAS dysfunction in COVID-19: involvement of ACE2
The renin–angiotensin–aldosterone system or RAAS regulates the physiology of fluid and electrolyte retention, blood flow, and blood pressure. ACE2 degrades angiotensin II to angiotensin (1–7) and thereby prevents the excessive effects of angiotensin II such as vasoconstriction, sodium and water retention, cardiomyocyte hypertrophy and cardiac fibrosis, lung inflammation, pulmonary edema, and pulmonary hypertension [22,28]. Besides, angiotensin (1-7) derived from angiotensin II has vasodilatory, anti-inflammatory, and various cardio-protective functions mediated through Mas receptors and other mechanisms [22,28,76,77]. Functional dysregulation of RAS accompanied by enhanced ACE/Angiotensin II expression (mRNA and protein) levels in lung tissue and reduced levels of ACE2 and angiotensin (1–7) in serum and lungs has been reported to contribute to ischemia-reperfusion-induced Acute Lung Injury (ALI) in mice [78]. In LPS induced acute lung injury (ALI) the role of angiotensin II acting through its receptor angiotensin II receptor type I (AT1) has been confirmed in multiple studies [79,80].
SARS-CoV-2 may interfere with the functions of RAAS by binding with ACE2 through the mediation of viral S protein, but the overall effect of RAAS dysfunction in COVID-19 is still not clearly defined. For example, apparently, the over-expression of ACE-2 may tend to allow enhanced entry of SARS-CoV-2 into lung epithelial or other cells making such subjects vulnerable to COVID-19, and this possibility initially was suggested for patients under treatment with angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin II receptor blockers (ARBs). However, no consistent results of ACE2 over-expression or its enhanced activity following ACEIs or ARBs treatment could be obtained in animal or human studies, and likewise increased vulnerability of subjects using ACEIs or ARBs to COVID-19 could not be established [21,81,82]. On the other hand, the entry of SARS-CoV-2 appears also to subsequently downregulate ACE2 expression by multiple mechanisms [21,77]. This may, in turn, lead to excess angiotensin II activity with cardiovascular dysfunction or lung inflammation and edema including acute respiratory distress syndrome or ARDS [21,22,79]. The diminished production of angiotensin (1-7) under such a condition of downregulation of ACE2 may further aggravate the cardiac dysfunction, but all these possibilities so far have not been proven with any degree of certainty. The fact that the excess angiotensin II activity may aggravate lung inflammation and edema probably explains why many studies have reported better clinical outcomes in hypertensive COVID-19 patients on ACEIs or ARBs therapy [83,84]. Further, there is an interesting possibility that ACEIs and ARBs may produce differential beneficial effects in COVID-19 clinical outcome [85]. In the presence of ARBs, a larger proportion of angiotensin II would be available for degradation by ACE2 producing the vasodilatory and cardioprotective angiotensin (1-7), while ACEIs will inhibit the production of angiotensin II leading in consequence of a decreased availability of angiotensin-(1-7) [85]. The significance of this hypothesis in COVID-19 outcome in patients on ARBs or ACEIs therapy needs further exploration. However, it may be said at this point that there is sufficient ground to believe that RAAS dysfunction plays a critical role in modulating the severity of COVID-19 in relation to lung inflammation and cardiovascular damage. This also partly explains why hypertensive patients with an existing backdrop of RAAS dysfunction often present as severe or critical cases of COVID-19. An interesting link between hypertension and severe cases of COVID-19 has been suggested through the mediation of heat-shock protein 60 (Hsp60) [86]. Hsp60, a chaperonin located pre-dominantly in mitochondria, can be secreted outside by different tissues under pathological conditions, and the circulating Hsp60 can elicit a pro-inflammatory response by activating TLR-signaling pathways [87,88]. In chronic hypertension with pressure overload the cardiomyocytes can overexpress and secrete Hsp60 which can trigger a pro-inflammatory response in the heart as well other tissues, and thus this process can further aggravate the hyperinflammation phase of COVID-19 [86].
2.4. Pathology of COVID-19
The study of macroscopic and microscopic pathology of COVID-19 which is essential in understanding and verifying the pathogenic mechanisms has been hampered by different restrictions imposed by regulatory bodies concerning postmortem examinations. However, postmortem histopathologic features of respiratory tract and lungs in COVID-19 started emerging, initially from single-case or single-institutional studies but later on from multi-centric studies and multiple such reports are now available. In general, trachea-bronchitis, diffuse alveolar damage (DAD), inflammation and microthrombi formation in pulmonary vasculature are typical features [19,89,90]. However, the pathological features are a bit variegated presumably because the autopsy samples were obtained in different phases of lung damage and also different pathogenic mechanisms were involved in this process. Thus, in postmortem lungs of COVID-19 patients desquamation and necrosis of alveolar cells, pulmonary edema, proteinaceous exudates formation in the alveoli, fibrinoid alveolar buds, hyaline membrane formation, alveolar hemorrhage, infiltration of lung parenchyma by leucocytes predominantly lymphocytes, atypical hyperplasia of pneumocytes, multi-nucleated giant-cell formation, fibroblastic proliferation in the lung interstitium, multiple microthrombi (platelet and thrombin-rich) within small vessels and capillaries of pulmonary vasculature with features of endothelial inflammation, etc., are present in variable combinations [19,60,89–92]. Increased levels of D-dimers in the blood of COVID-19 patients indicate the presence of coagulopathy, but the clinical and laboratory findings of typical disseminated intravascular coagulopathy (DIC) are not often seen in COVID-19, and thus a special type of coagulopathy affecting the pulmonary vasculature has been suggested in COVID-19 which is named as pulmonary intravascular coagulopathy or PIC [17,93].
Cardiovascular disease (CVD) is a common co-morbidity associated with many fatal cases of COVID-19, and cardiac damage is considered as a major mechanism of death in this disease condition [94,95]. However, postmortem reports of cardiac pathology in COVID-19 are limited and variable. Most autopsy studies have reported dilatation of the heart macroscopically, but in histopathological examinations, focal inflammatory myocarditis with lymphocytic infiltrations and scattered areas of necrotic cardiomyocytes in some cases, or macrophage infiltration of myocardium without evidence of damage to cardiomyocytes, or focal areas of necrotic cardiomyocytes without significant lymphocytic infiltration in the surroundings have all been reported in COVID-19 [96,97]. Lymphocytic myocarditis of varying degrees or simple infiltration of the myocardium with macrophages has been reported by other groups from autopsy studies of COVID-19 patients [98]. In many patients who have survived COVID-19, myocarditis has been diagnosed based on resonance imaging studies supported by markers of cardiac damage in serum [98]. The presence of SARS-CoV-2 within the myocardium or other layers of the heart has not been established with certainty so far. The cardiac pathology results in clinical features like arrhythmia, heart failure, acute coronary syndrome, etc., in many patients of COVID-19 especially in those with preexisting cardiovascular co-morbidity.
SARS-CoV-2 can easily invade vascular endothelium because of the presence of ACE2 receptors on these cells and many of the damaging effects of SARS-CoV-2 such as hypercoagulable state with thrombotic manifestations and inflammatory response in various organs may actually result from endothelial dysfunction [99]. Limited postmortem studies specifically examined the damage to vascular endothelium in several organs and observed endothelitis with the accumulation of neutrophils and peripheral mononuclear cells, apoptosis of endothelial cells, and the presence of virus-derived inclusion bodies within endothelium [16]. Other postmortem studies indicated pulmonary embolism and microthrombi in small vessels and capillaries in lungs, kidneys, and other organs in COVID-19, but not the evidence of vasculitis in most cases [92,100]. A meta-analysis of available autopsy reports of COVID-19 patients shows that most extensive histopathological observations have been made with respect to lungs and these are by and large consistent, but for other organs less extensive and variable findings have been reported [101].
Overall, it appears that the pathophysiology and pathogenesis of COVID-19 are varied and involves a complex interplay of viral invasion, the body immune response, and RAAS alterations, but it would not be easy to identify the relative contribution of different pathways to the overall severity and mortality of COVID-19. Secondary infections and tissue damage resulting from hypoxemia and shock can further complicate the picture. Nevertheless, it would be interesting to explore those compounds which may modulate these pathways through multiple mechanisms producing clinically beneficial effects. Vitamin D because of its diverse protective roles could be an ideal candidate in inhibiting the development of COVID-19 complications.
3. Vitamin D: many faces
3.1. Chemistry and cellular functions of vitamin D
Vitamin D, now generally considered as a hormone, can be synthesized in our body with the help of sunlight, and it also may be obtained from dietary sources. Vitamin D2 (ergocalciferol) is derived from ergosterol and is often used commercially for the enrichment of food, while vitamin D3 (cholecalciferol) is synthesized in the body from 7-dehydroscholesterol under the influence of the B portion of ultra-violet rays (UVB) of the sun. Vitamin D3 or vitamin D2 is first hydroxylated in the liver to form 25-hydroxy derivative which subsequently undergoes another oxidation in the kidney by 1 alpha-hydroxylase to be converted to 1,25-dihydroxy derivative (the active form of vitamin D). We will use the term vitamin D in this review to indicate generally the active form or 1,25-dihydroxyvitamin D3 unless stated otherwise in a particular context. 1,25-dihydroxyvitamin D3 is the ligand for the vitamin D receptor (VDR) which is a nuclear receptor acting as a transcription factor binding to sites in the DNA called vitamin D response elements (VDREs) [102,103]. Thus, vitamin D increases the expressions of a large number of genes involved in maintaining bone integrity and maturation and differentiation of many cells including immune cells. The effects of vitamin D are exerted not only through its nuclear hormone receptors but also through its membrane receptors, membrane-associated, rapid-response, steroid-binding protein (1,25 MARRS) [104]. The membrane-bound forms of VDR, identical to nuclear receptors, trigger rapid signal transduction pathways, and these are responsible for non-genomic actions of vitamin D [104]. In its genomic actions, vitamin D regulates the transcription of various genes by forming a heterodimer with retinoid X receptor (RXR) and subsequently translocating to vitamin D response element in the DNA [105,106]. Apart from playing an important role in bone mineralization and calcium homeostasis, genomic and non-genomic actions of vitamin D encompass a wider variety of effects out of which anti-inflammatory, anti-oxidant, immunomodulatory, anticoagulant effects in the context of immune response and COVID-19 are reviewed here.
3.2. Vitamin D: effects on epithelial defense and immuno-modulation
Human β-defensins and the cathelicidin (containing an anti-microbial domain LL-37) act as a primary defense against bacterial and viral infection. These peptides, which are produced by neutrophils and mucosal cells including that of the respiratory tract, have anti-microbial actions against bacteria, fungi, and viruses, and they also promote innate and adaptive immunity [107]. Vitamin D has been shown to increase the productions of β-defensins and the cathelicidin in the respiratory mucosa [107–109]. The epithelial and endothelial barrier functions are maintained by tight junctions (TJs) and adherens junctions (AJs). A recent study has demonstrated that in the lungs of VDR-deficient mice (VDR -/-), the expression levels of various proteins of TJs and AJs, especially claudins, β-catenin, and VE-cadherin, are significantly impaired, and this phenomenon is likely to cause increased alveolar permeability [110]. This is supported by another earlier study demonstrating more severe lung injury and increased alveolar permeability and decreased expressions of proteins of TJs in VDR knock-out mice compared to that in wild-type animals following lipopolysaccharide (LPS) administration [111].
Thus, vitamin D/VDR signaling has an important role in epithelial permeability properties and defense mechanisms. In another study with an LPS-induced model of lung injury, vitamin D was shown to promote the proliferation of alveolar epithelial cell type 2 (AT-II) and inhibit the apoptosis of AT-II cells and their conversion to mesenchymal cells [112]. On other aspects of innate immunity, vitamin D has important effects as shown in cell-based studies and animal models. Thus, the active form of vitamin D through VDR signaling decreases the expression levels of TLR2 and TLR4 and attenuates the LPS-induced production of TNF α and CD142 (pro-coagulatory tissue factor) in human monocytes [113]. The increased levels of IL-2, IL-6, and TNF-β in the serum of pigs challenged with porcine rotavirus could be suppressed by vitamin D supplementation, which occurs through activation of RIG-I signaling pathways [114]. Another study demonstrated that both 1,25-dihydroxyvitamin D3 and 25-hydroxyvitamin D3 could inhibit the production of IL-6 and TNF-α and upregulate the expression of mitogen-activated protein kinase phosphatase-1 (MKP-1) in human monocytes upon challenge with LPS [115]. Likewise, in human primary bronchial cells infected with rhinovirus, an active form of vitamin D inhibits replication and release of the virus, enhanced virus-induced interferon-stimulated gene expressions (mRNA) and cathelicidin mRNA expression, but decreased the production of IL-6 and IL-8 [116]. Further, calcitriol or the active form of vitamin D3 shows antiviral activity against hepatitis C virus in hepatoma cells in culture by enhancing the production of IFN-β and increasing the expression of interferon-stimulated myxovirus A protein (MxA) [117]. Likewise, when mononuclear cells from patients of multiple sclerosis were treated in vitro with IFN-β in the presence of active form of vitamin D, the interferon response was enhanced as shown by the increased induction of phosphorylated STAT-1 and MxA protein [118]. In Huh-7.5 cells, vitamin D enhanced the action of IFN-α in suppressing the replication of hepatitis C virus through induction of ISG expression, but vitamin D alone did not produce any effect on viral replication [119]. Interestingly, this study demonstrated that VDR formed a complex with STAT1 constitutively, but upon treatment with vitamin D and IFN-α this interaction was abolished which allowed STAT1 in the phosphorylated form to mediate the IFN-α signaling and ISG expression [119]. On the other hand, other reports suggested that vitamin D impaired the expressions of interferon-stimulated antiviral proteins, and also decreased the production of IFN-β and CXCL induced by respiratory syncytial virus in human airway epithelial cells but without increasing viral pathogenicity [120]. These varied reports indicate that the role of vitamin D on innate immunity is modulatory, complex, and contextual.
Antigen-presenting cells like macrophages, dendritic cells, etc., and activated T cells all have VDR, and vitamin D acts as a suppressor of adaptive immunity via multiple mechanisms as observed in a large number of experimental systems [121–125]. Thus, vitamin D has been shown to have important effects on CD4+ T-helper cells (Th1 and Th2) and regulatory T cells (Treg). Vitamin D in its active form suppresses the immune response mediated by T helper cells (Th1) by inhibiting the production of inflammatory cytokines IL-2 and interferon-gamma (INF-γ) [124]. Furthermore, 1,25-dihydroxyvitamin D enhances the cytokine production by Th2 helper T cells which in turn can suppress the functions of Th1 cells [124,126,127]. In addition, vitamin D induces the regulatory T cells which prevent inflammatory response and suppress the Th17 cells which secrete IL-17 and are involved in some auto-immune diseases [123,124]. Further experiments showed that induction of Treg cells by 1,25-dihydroxyvitamin D especially in the presence of IL-2 was associated with a robust increase in the expressions of CTLA-4 and FoxP3, and likewise, vitamin D and IL-2 in combination caused suppression of T cells secreting IL-17 and IFN-γ [125]. Thus, overall these studies carried out in vitro and in vivo in mouse models indicate that vitamin D has a complex role in the differentiation and proliferation of different subsets of T cells involved in adaptive immunity.
3.3. Vitamin D and functions of RAAS
LPS induced ALI in rats is associated with increased expression levels (mRNA and protein) of renin, ACE, and AT1 receptors, enhanced production of angiotensin II and impaired expression of ACE2 (mRNA as well as protein) in pulmonary epithelial and pulmonary microvascular endothelial cells, and all these effects are reversed by the active form of vitamin D3 [128]. This study claimed that vitamin D per se did not affect the expression changes of ACE, ACE2, renin, angiotensin II and AT1 receptors in the absence of LPS treatment [128]. However, others demonstrated that in the brain of both wild-type rats and spontaneously hypertensive rats (SHR), 1,25-dihydroxyvitamin D increased the protein expressions of ACE2, but ACE and AT1 receptor expressions were inhibited significantly only in SHR [129]. Although in this study treatment with active form of vitamin D was able to increase the ACE2 protein expression in the brain of normal rats, angiotensin II or angiotensin (1-7) levels in the brain remained similar to that of untreated rats, and thus the biological significance of vitamin D effects on ACE2 expression in normal rat brain was not obvious [129]. On the other hand, in SHR, vitamin D effects on ACE and ACE2 expressions in the brain resulted in the decrease in angiotensin II and a rise in the angiotensin (1-7) levels in the organ [129]. Likewise, in another study of experimental ALI induced in rats, calcitriol significantly prevented the disease manifestations and the impaired expressions of both ACE2 and VDR caused by LPS [130]. In a model of chronic vitamin D deficiency in mice, pulmonary fibrosis with deposition of extra-cellular matrix developed with increased expression level of renin, and these changes could be prevented by AT1 receptor blocker or an antagonist of renin [131]. Further, the role of vitamin D and VDR signaling in RAAS dysfunction and cardiovascular diseases have been examined in experimental animals. VDR null mice showed increased expression of renin in the kidney and enhanced plasma level of angiotensin II and also developed hypertension, cardiac dysfunction, and abnormal drinking behavior [132]. This study further showed that inhibition of synthesis of active form of vitamin D3 (1,25-dihydroxyvitamin D3) in wild mice caused overexpression of renin, while the administration of 1,25-dihydroxyvitamin D3 inhibited the expression of renin in kidney tissue [132]. The increased expressions (mRNA and protein) of renin in kidney and heart associated with cardiac hypertrophy in VDR (-/-) rats were also reported in another study [133]. Thus, experimental evidence in animal models suggests that vitamin D can modulate RAAS at different levels to prevent both acute lung injury and cardiac dysfunction and hypertension. In particular, the effects of vitamin D3 on ACE2expression could be significant in ARDS because ACE2 knock-out mice show more severe forms of pulmonary edema, inflammation and hypoxemia than control mice in different models of severe acute lung injury induced by acid-aspiration, endotoxin or sepsis [134].
3.4. Vitamin D effects on ARDS and cardiovascular diseases in human
The experimental evidence suggests that vitamin D could be beneficial in preventing acute lung injury and ARDS as well as cardiovascular diseases by modulating the epithelial barrier functions and proliferation and apoptosis of epithelial cells in the alveoli and regulating the innate immunity and/or RAAS. However, some of the experimental studies, especially with cultured cells, have used active form of vitamin D in much higher concentrations than the physiological circulating level. Thus, the results in human subjects of the beneficial effects of vitamin D in ARDS or cardiovascular dysfunction are not unequivocal. For ARDS from various causes, several clinical studies have shown an association of hypovitaminosis D with this condition [135,136]. Hypovitaminosis D is also associated with adverse clinical outcome in patients of community-acquired pneumonia [137]. A large case–control study, however, failed to demonstrate any correlation between vitamin D deficiency and the development of ARDS [138]. Epidemiological evidence shows some association of hypovitaminosis D with hypertension and cardiovascular diseases, but the meta-analysis of randomized controlled trials (RCTs) do not show any clear benefit of vitamin D supplementation in those conditions [139,140]. As mentioned already, endothelial dysfunction is an important element of COVID-19 pathophysiology, and there is evidence that reactive hyperemia index (RHI) which is a good indicator of endothelial function is decreased in vitamin D deficient subjects [141]. Moreover, supplementation with vitamin D improves endothelial functions in chronic kidney disease as measured by increased brachial artery flow-mediated dilation and decreased levels of endothelial biomarkers [142]. However, it is an open question if this action of vitamin D will have a protective effect against SARS-CoV-2 mediated endothelitis.
4. Vitamin D and COVID-19: epidemiological evidence
A large number of apparently healthy adults have been reported to be with low levels of vitamin D, measured as 25-hydroxyvitamin D, and this makes them vulnerable to a variety of diseases [143,144]. Institutionalized persons, those on regular duties at night and many elderly people have limited exposure to sunlight and may develop insufficiency or deficiency of vitamin D. Besides, serum vitamin D measured as 25-hydroxyvitamin D concentration tends to decrease with age and in winter season, and women are more vulnerable to develop vitamin D deficiency [145]. The association of lower serum vitamin D levels with COVID-19 has been reported from multiple studies. For example, a Swiss cohort study showed a significantly decreased level of serum vitamin D in SARS-CoV-2 positive cases compared to that in negative cases [146]. Similarly, over the age of 65 years, there is a clear association of hypovitaminosis D with COVID-19 cases which also leads to worse clinical morbidity [147]. Hypovitaminosis D was shown in a large percentage of COVID-19 patients with acute respiratory failure, and severe deficiency of vitamin D was associated with significantly higher mortality [148]. In another study with 149 patients reported that lower serum vitamin D levels were associated with more than 93% cases of severe COVID-19, and vitamin D level is an independent predictor of mortality [149]. Likewise, in a study with 154 asymptomatic and severe cases of COVID-19, markedly low levels of vitamin D have been reported in severe cases with increased serum levels of pro-inflammatory cytokines, and higher mortality from the disease was observed in vitamin D-deficient subjects [150]. In a retrospective cohort study, likely- deficient vitamin D status was associated with increased risk of COVID-19; the patients were examined for vitamin D status in the year before COVID-19 testing [151]. A study presented an inverse association between mean serum vitamin D with COVID-19 positive cases per million as well as mortality from the disease per million in many European countries [152]. Some indirect evidence also implicates vitamin D deficiency with increased incidence or severity of COVID-19. When mortality from COVID-19 was plotted to degrees latitude, it was observed that countries below latitude 35° North have lower mortality from COVID-19, and these populations are likely to get sufficient sunlight to synthesize vitamin D in contrast to those residing in countries above the latitude 35° [153]. Further, co-morbidities like diabetes, hypertension, obesity, chronic kidney disease, and so on, which tend to aggravate COVID-19 are also associated with hypovitaminosis D [154–157]. The Black people in the UK and Wales are stated to be more vulnerable to COVID-19 death presumably because of their darker skin preventing the synthesis of vitamin D with the help of sunlight [158].
This growing body of direct and circumstantial evidence has led to the initiation of multiple interventional studies with vitamin D supplementation as a therapeutic measure against COVID-19. For example, 76 consecutive COVID-19 patients were randomized and divided in two groups of 26 receiving standard COVID-19 therapy prevalent in that hospital and 50 receiving standard therapy with vitamin D supplementation (oral 25-hydroxyvitamin D3); vitamin D supplementation significantly decreased the clinical severity and need for ICU admission of COVID-19 patients [159]. In other interventional studies with different designs and in subjects of different age groups, supplementation with vitamin D alone or in combination with vitamin B12 and magnesium decreased the severity of COVID-19 or mortality from the disease [160–162]. In a large-scale study in UK, 444 COVID-19 patients’ records (primary cohort) were analyzed retrospectively which showed that mortality of COVID-19 was not affected by baseline serum vitamin D (25-hydroxyvitamin D) level, but supplementation with high – dose vitamin D reduced the mortality from the disease [163]. The same study further recruited a ‘validation cohort’ of 541 COVID-19 patients to conduct an observational study of booster vitamin D therapy which showed that vitamin D supplementation significantly decreased the mortality from the disease irrespective of baseline level of serum vitamin D [163]. In a small study of four COVID-19 patients with vitamin D deficiency, it was observed that high-dose vitamin D therapy (ergocalciferol 50,000 IU daily for 5 days) instead of standard-dose (cholecalciferol 1000 IU daily for 5 days) supplementation decreased the clinical severity of the disease with improvement in serum vitamin D status [164]. In a retrospective analysis of 91 patients of COVID-19 of which 50 had two or more co-morbidities, the beneficial effect of high vitamin D supplementation therapy on clinical outcome was significantly amplified by the presence of the co-morbidities [165]. In a randomized, placebo-controlled study with asymptomatic or mild COVID-19 cases with vitamin D deficiency, a larger proportion of participants became qRT-PCR negative for SARS-CoV-2 within 3 weeks after high-dose vitamin D supplementation [166]. Based on experimental data and epidemiological leads, a recent review has suggested using vitamin D in loading doses (10,000 IU daily) for a few weeks followed by a daily dose of 5000 IU to reduce the risk of COVID-19 [167]. The potential beneficial effects of vitamin D at the mechanistic level are summarized in (Figure 1).
Figure 1.
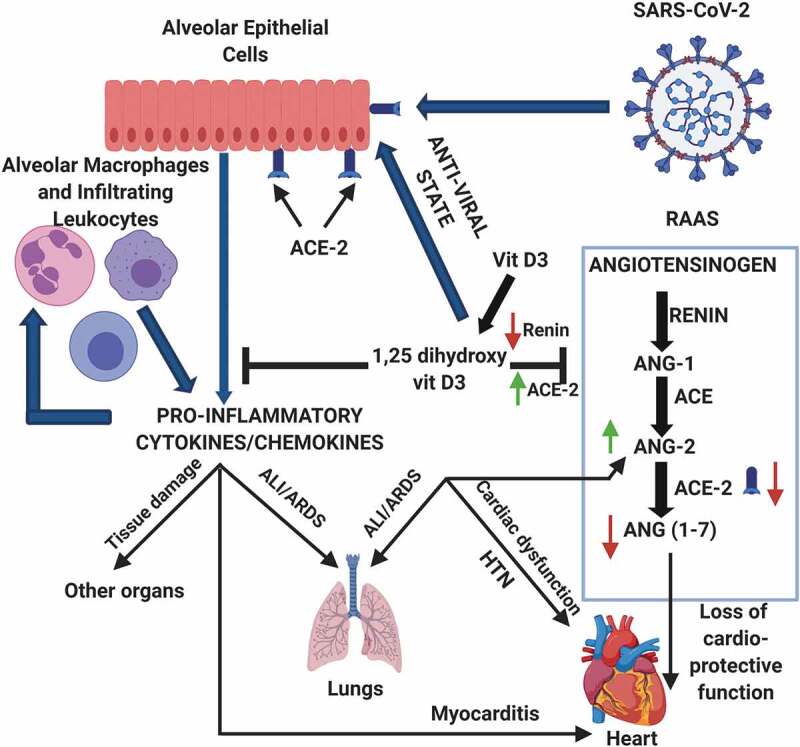
Potential protective mechanisms of vitamin D in COVID-19
The active form of vitamin D, 1,25-dihydroxyvitamin D, can prevent the ‘cytokine storm’ triggered by virally infected epithelial cells of the respiratory tract and lungs as well as infiltrating leucocytes; the vitamin also induces anti-viral state in the infected epithelial cells. Vitamin D can also prevent RAAS dysfunction caused by SARS-CoV-2. ANG-1, angiotensin I; ANG-2, angiotensin II; ANG (1–7), angiotensin (1–7); ALI, acute lung injury; ARDS, acute respiratory distress syndrome; HTN, hypertension; RAAS, renin-angiotensin-aldosterone system. Figure was created using BioRender software.
5. Conclusion
It appears from both epidemiological data and biochemical and immunological evidence discussed that vitamin D could be an important disease modifying-agent in COVID-19. The effects of vitamin D on innate immunity and RAAS are likely to be beneficial, but the overall suppression of adaptive immunity may interfere with body’s defense against the invading virus. Repurposing of available drugs is imperative in combating COVID-19 in the absence of specific anti-virals against SARS-CoV-2. Recently, several vaccines for COVID-19 have been approved in different countries, but the long-term toxicity of the vaccines and its efficacy against SARS-CoV-2 and its variants are not fully established. Additionally, the mass vaccination programs to halt the spread of the disease effectively across different populations will probably take many more months. Thus, vitamin D supplementation could be an effective strategy in combating COVID-19 in the present context. However, at the same time, proper randomized controlled trials should be conducted to determine the efficacy of this method and also the dosage schedule and the period of supplementation. Also, more mechanistic information should be gathered from experimental research to understand how vitamin D could modify the immuno-pathology of COVID-19.
6. Expert opinion
The innate immune response plays an important role in defense against viral invasion, and when overactive it causes severe damage to vital organs leading to death in many cases. This is true for infection with SARS-CoV-2 also where infected pulmonary epithelial cells, infiltrating monocytes and neutrophils and resident macrophages mount a hyperinflammatory reaction. Therefore, modulation of the innate immune response by drugs should be a viable option for combating viral diseases like COVID-19. Our current emphasis, however, is on vaccine development which relies on adaptive immune response. With great developments in structural protein chemistry, recombinant DNA technology, nucleic acid sequencing, viral genetics and bioinformatics, the development of a vaccine has become remarkably fast, but clinical testing for long-term efficacy and toxicity remains a slow and winding process through different guidelines of the regulatory bodies. Thus, mass vaccination is not really an effective procedure to contain a rapidly spreading viral pandemic, and COVID-19 pandemic, more than anything else, has proved this beyond doubt when two million across the globe died with devastating loss of economic resources before a vaccine could even be introduced. There is a growing concern that pandemics like COVID-19 may recur in near future, and thus other therapeutic options beyond vaccination must be considered for future pandemics. As far as anti-virals are concerned, the existing anti-virals may not be suitable for a novel virus or may take a long time for clinical validation for use against the new virus. This has happened with SARS-CoV-2, and the usefulness of remdesivir in COVID-19 could be established several months after the advent of the pandemic [168]. Thus, molecules that upregulate and modulate the innate immune response of the body should be actively identified and explored for clinically beneficial effects in combating multiple and novel viral diseases, but at present not too many such molecules are available. For the treatment of moderate to severe cases of COVID-19, the use of corticosteroids has already been recommended which have modulatory effects on both innate and adaptive immune response. Other drugs or procedures which can interfere with the host’s immune response have been used in COVID-19 as part of clinical trials or on experimental basis in different countries following approval from local regulatory bodies. Thus, monoclonal antibodies against IL-6 receptor and IL-1 receptor antagonists have been used in COVID-19 cases [169–171]. Other procedures that involve immuno-modulatory actions such as transplantation of mesenchymal stem cells (MSC) or infusion of immunoglobulins could be potential therapeutic approaches in COVID-19 [172,173]. However, anti-interleukin drugs, MSC or immunoglobulins would be usable in moderate to severe cases of COVID-19 or similar viral infections, and certainly cannot be used as prophylactic measures. Vitamin D because of its known immuno-modulatory actions and safety profile may be an ideal molecule to manipulate the innate immune system to derive clinical benefit both as a prophylactic drug or as an additional drug for the treatment of active viral diseases. Another future endeavor would be to synthesize chemical analogues of 1,25-dihydroxyvitamin D with better immuno-modulatory actions and less calcemic actions. Such analogues of 1,25-dihydroxyvitamin D having more potent anti-neoplastic or anti-inflammatory actions are being used in experimental models of cancer and auto-immune diseases [174,175]. This approach could be extremely relevant in the context of immuno-modulatory actions of vitamin D in the treatment of viral diseases.
Acknowledgements
The authors thank the management at Maharishi Markandeshwar University (Deemed to be) for their support.
Funding Statement
This paper was not funded.
Notes on contributors
AB1, UG2, SC2 conceptualized the article. UG2 and AB1 wrote the paper, and SC2 edited it thoroughly. SS3, SC4 and RVS5 did extensive literature survey and provided important inputs. RKR6 and LS7 provided critical comments on the content and organization of the manuscript.
REVIEW
Article highlights
SARS-CoV-2 invades respiratory and other epithelial cells.
Host’s hyperactive immune response triggers COVID-19 pathology.
Hyperactive immune response is manifested as a ‘Cytokine Storm’.
Renin-angiotensin-aldosterone system (RAAS) dysfunction also contributes to Covid-19 pathophysiology.
Vitamin D modulates host’s immune response to SARS-CoV-2
Vitamin D may correct RAAS dysfunction also in COVID-19.
Hypovitaminosis D is associated with COVID-19 incidence and severity.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.WHO Weekly Epidemiological and Operational updates . 2020. [cited 2020 Dec 1]. Available from:. https://www.who.int/publications/m/item/weekly-epidemiological-update—1-december-2020
- 2.Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sardar R, Satish D, Birla S, et al. Integrative analyses of SARS-CoV-2 genomes from different geographical locations reveal unique features potentially consequential to host-virus interaction, pathogenesis and clues for novel therapies. Heliyon. 2020;6(9):e04658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peiris JSM, Yuen KY, Osterhaus ADME, et al. The severe acute respiratory syndrome. N Engl J Med. 2003;349(25):2431–2441. [DOI] [PubMed] [Google Scholar]
- 6.Fehr AR, Channappanavar R, Perlman S.. Middle east respiratory syndrome: emergence of a pathogenic human coronavirus. Annu Rev Med. 2017;68(1):387–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharif-Yakan A, Kanj SS. Emergence of MERS-CoV in the Middle East: origins, transmission, treatment, and perspectives. PLoS Pathog. 2014;10(12):e1004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raoult D, Zumla A, Locatelli F, et al. Coronavirus infections: epidemiological, clinical and immunological features and hypotheses. Cell Stress. 2020;4(4):66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rothan HA, Byrareddy SN. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J Autoimmun. 2020;109:102433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chakrabarti SS, Kaur U, Banerjee A, et al. COVID-19 in India: are biological and environmental factors helping to stem the incidence and severity? Aging Dis. 2020;11(3):480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This early paper first suggested that cross-reactive anti-bodies and T-cells from previous exposure to endemic coronaviruses may modify the severity of COVID-19 in some populations.
- 11.Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang W, Berube J, McNamara M, et al. Lymphocyte subset counts in COVID-19 patients: a meta-analysis. Cytometry. 2020;97(8):772–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanyaolu A, Okorie C, Marinkovic A, et al. Comorbidity and its impact on patients with COVID-19. SN Compr Clin Med. 2020;1–8. DOI: 10.1007/s42399-020-00363-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nadkarni GN, Lala A, Bagiella E, et al. Anticoagulation, mortality, bleeding and pathology among patients hospitalized with COVID-19: a Single health system study. J Am Coll Cardiol. 2020;76(16):1815-1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madjid M, Safavi-Naeini P, Solomon SD, et al. Potential effects of coronaviruses on the cardiovascular system: a review. JAMA Cardiol. 2020;5(7):831–840. [DOI] [PubMed] [Google Scholar]
- 16.Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fogarty H, Townsend L, Cheallaigh CN, et al. COVID19 coagulopathy in Caucasian patients. Br J Haematol. 2020;189(6):1044–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calabrese F, Pezzuto F, Fortarezza F, et al. Pulmonary pathology and COVID-19: lessons from autopsy. The experience of European Pulmonary Pathologists. Virchows Arch. 2020;477(3):359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borczuk AC, Salvatore SP, Seshan SV, et al. COVID-19 pulmonary pathology: a multi-institutional autopsy cohort from Italy and New York City. Mod Pathol. 2020;33(11):2156–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Very thorough multi-centric post-mortem analysis of lung pathology of COVID-19; absolutely important to understand the pathology of COVID-19.
- 20.Tay MZ, Poh CK, Rénia L, et al. The Trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaduganathan M, Vardeny O, Michel T, et al. Renin-angiotensin-aldosterone system inhibitors in patients with Covid-19. N Engl J Med. 2020;382(17):1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaur U, Acharya K, Mondal R, et al. Should ACE2 be given a chance in COVID-19 therapeutics: a semi-systematic review of strategies enhancing ACE2. Eur J Pharmacol. 2020;887:173545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jakovac H. COVID-19 and vitamin D—Is there a link and an opportunity for intervention? Endocrinol Metab. 2020;318(5):589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slominski AT, Slominski RM, Goepfert PA, et al. Can vitamin D prevent or manage COVID-19 illness? Am J Physiol Endocrinol Metab. 2020;319(2):455–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Machhi J, Herskovitz J, Senan AM, et al. The Natural History, pathobiology, and clinical manifestations of SARS-CoV-2 infections. J Neuroimmune Pharmacol. 2020;15(3):359–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson KD, Harris C, Cain JK, et al. Pulmonary and extra-pulmonary clinical manifestations of COVID-19. Front Med. 2020;7:526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARSCoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• A very elegant experimental work to prove the mechanisms of SARS-CoV-2 entry in to cells; very important to identify anti-viral drug targets.
- 28.Wang JJ, Edin ML, Zeldin DC, et al. Good or bad: application of RAAS inhibitors in COVID-19 patients with cardiovascular comorbidities. Pharmacol Ther. 2020;215:107628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaur U, Chakrabarti SS, Ojha B, et al. Targeting host cell proteases to prevent SARS-CoV-2 invasion. Curr Drug Targets. 2021;22(2):2. [DOI] [PubMed] [Google Scholar]
- 30.Wang W, Xu Y, Gao R, et al. Detection of SARS-CoV-2 in different types of clinical specimens. JAMA. 2020;323(18):1843–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mason RJ. Pathogenesis of COVID-19 from a cell biology perspective. Eur Respir J. 2020;55(4):2000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuki K, Fujiogi M, Koutsogiannaki S. COVID-19 pathophysiology: a review. J Clim. 2020;215:108427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu CC, Ruan XZ, Chen XQ, et al. CT-based staging and prognosis of novel coronavirus (COVID-19) pneumonia: correlation with blood glucose levels. Eur Rev Med Pharmacol Sci. 2020;24(24):13056–13061. [DOI] [PubMed] [Google Scholar]
- 34.Siddiqi HK, Mehra MR. COVID-19 illness in native and immunosuppressed states: a clinical-therapeutic staging proposal. J Heart Lung Transplant. 2020;39(5):405–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cordon-Cardo C, Pujadas E, Wajnberg A, et al. COVID-19: staging of a new disease. Cancer Cell. 2020;38(5):594–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.World Health Organization . 2020. Clinical management of COVID-19: interim guidance, 2020. World Health Organization. [cited 2021 Feb 10]. Available from: https://apps.who.int/iris/handle/10665/332196. [Google Scholar]
- 37.Greiller CL, Martineau AR. Modulation of the immune response to respiratory viruses by Vitamin D. Nutr. 2015;7(6):4240–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katze MG, Fornek JL, Palermo RE, et al. Innate immune modulation by RNA viruses: emerging insights from functional genomics. Nat Rev Immunol. 2008;8(8):644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katze MG, He Y, Gale M. Viruses and interferon: a fight for supremacy. Nat Rev Immunol. 2002;2(9):675–687. [DOI] [PubMed] [Google Scholar]
- 40.Unterholzner L, Almine JF. Camouflage and interception: how pathogens evade detection by intracellular nucleic acid sensors. Immunol. 2018;156(3):217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alcami A. Viral mimicry of cytokines, chemokines and their receptors. Nat Rev Immunol. 2003;3(1):36–50. [DOI] [PubMed] [Google Scholar]
- 42.Hartman AL, Towner JS, Nichol STA. C-terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology. 2004;328(2):177–184. [DOI] [PubMed] [Google Scholar]
- 43.Kash JC, Muhlberger E, Carter V, et al. Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J Virol. 2006;80(6):3009–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li G, Fan Y, Lai Y, et al. Coronavirus infections and immune responses. J Med Virol. 2020;92(4):424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang YT, Landeras-Bueno S, Hsieh LE, et al. Spiking pandemic potential: structural and immunological aspects of SARS-CoV-2. Trends Microbiol. 2020;28(8):605–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang Y, Liu J, Zhang D, et al. Cytokine storm in COVID-19: the current evidence and treatment strategies. Front Immunol. 2020;11:1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia LF. Immune response, inflammation, and the clinical spectrum of COVID-19. Front Immunol. 2020;11:1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Birra D, Benucci M, Landolfi L, et al. COVID 19: a clue from innate immunity. Immunol Res. 2020;68(3):161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amor S, Fernandez Blanco L, Baker D. Innate immunity during SARS-CoV-2: evasion strategies and activation trigger hypoxia and vascular damage. Clin Exp Immunol. 2020;202(2):193–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fung SY, Yuen KS, Ye ZW, et al. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: lessons from other pathogenic viruses. Emerg Microbes Infect. 2020;9(1):558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pellegrini S, Uze G. An old cytokine against a new virus? J Interferon Cytokine Res. 2020;40(8):425–428. [DOI] [PubMed] [Google Scholar]
- 52.Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trouillet-Assant S, Viel S, Gaymard A, et al. Type I IFN immunoprofiling in COVID-19 patients. J Allergy Clin Immunol. 2020;146(1):206–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou Z, Ren L, Zhang L, et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe. 2020;28(6):883–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Catanzaro M, Fagiani F, Racchi M, et al. Immune response in COVID-19: addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct Target Ther. 2020;5(1):84. DOI: 10.1038/s41392-020-0191-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pedersen SV, Ho Y-C. SARS-CoV-2: a storm is raging. J Clin Invest. 2020;130(5):2202–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaur S, Bansal R, Kollimuttathuillam S, et al. The looming storm: blood and cytokines in COVID-19. Blood Rev. 2020;100743. DOI: 10.1016/j.blre.2020.100743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qin C, Zhou L, Hu Z, et al. Dysregulation of immune response in patients with COVID-19 in Wuhan. China Clin Infect Dis. 2020;71(15):762–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Varchetta S, Mele D, Oliviero B, et al. Unique immunological profile in patients with COVID-19. Cell Mol Immunol. 2020;1–9. DOI: 10.1038/s41423-020-00557-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu B, Fan CY, Wang AL, et al. Suppressed T cell-mediated immunity in patients with COVID-19: a clinical retrospective study in Wuhan, China. J Infect. 2020;81(1):51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Diao B, Wang C, Tan Y, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol. 2020;11:827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun HB, Zhang YM, Huang LG, et al. The changes of the peripheral CD4+ lymphocytes and inflammatory cytokines in patients with COVID-19. PLOS One. 2020;15(9):e0239532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maucourant C, Filipovic I, Ponzetta A, et al. Natural killer cell immunotypes related to COVID-19 disease severity. Sci Immunol. 2020;5(50):eabd6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ni L, Ye F, Cheng ML, et al. Detection of SARS-CoV-2-specific humoral and cellular immunity in COVID-19 convalescent individuals. Immunity. 2020;52(6):971–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bert NL, Tan AT, Kunasegaran K, et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 2020;584(7821):457–462. [DOI] [PubMed] [Google Scholar]
- 66.Grifoni A, Weiskopf D, Ramirez SI, et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. 2020;181(7):1489–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Very elegant work identifying the targets of T-cell response to SARS-CoV-2 and clearly demonstrating the presence of cross-reactive T cells in unexposed subjects; very important for understanding the immune-response in COVID-19 and epidemiology of the disease with respect to cross-immunity and herd immunity.
- 67.Peng Y, Mentzer AJ, Liu G, et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat Immunol. 2020;21(11):1336–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mateus J, Grifoni A, Tarke A, et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science. 2020;370(6512):89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• A very significant paper to understand the immune-response against SARS-CoV-2; the study demonstrated the possible presence of cross-immunity in the population.
- 69.Mathew D, Giles JR, Baxter AE, et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369(6508):eabc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu F, Liu M, Wang A, et al. Evaluating the association of clinical characteristics with neutralizing antibody levels in patients who have recovered from mild COVID-19 in Shanghai, China. JAMA Intern Med. 2020;180(10):1356–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rogers TF, Zhao F, Huang D, et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science. 2020;369(6506):956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu L, Wang P, Nair MS, et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature. 2020;584(7821):450–456. [DOI] [PubMed] [Google Scholar]; •• A meticulous study isolating memory B cells from COVID-19 patients, identifying neutralizing monoclonal antibodies secreted by such cells against SARS-CoV-2 followed by epitope mapping; information necessary for vaccine development.
- 73.Robbiani DF, Gaebler C, Muecksch F, et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature. 2020;584(7821):437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nguyen-Contant P, Embong K, Kanagaiah P, et al. S protein-reactive IgG and memory B cell production after human SARS-CoV-2 infection includes broad reactivity to the S2 subunit. mBio. 2020;11(5):e01991–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brouwer PJM, Caniels TG, van der Straten K, et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science. 2020;369(6504):643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang F, Yang J, Zhang Y, et al. Angiotensin-converting enzyme 2 and angiotensin 1-7: novel therapeutic targets. Nat Rev Cardiol. 2014;11(7):413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Groβ S, Jahn C, Cushman S, et al. SARS-CoV-2 receptor ACE2-dependent implications on the cardiovascular system: from basic science to clinical implications. J Mol Cell Cardiol. 2020;144:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen LN, Yang XH, Nissen DH, et al. Dysregulated renin-angiotensin system contributes to acute lung injury caused by hind-limb ischemia-reperfusion in mice. Shock. 2013;40(5):420–429. [DOI] [PubMed] [Google Scholar]
- 79.Wang F, Xia ZF, Chen XL, et al. Angiotensin II type-1 receptor antagonist attenuates LPS-induced acute lung injury. Cytokine. 2009;48(3):246–253. [DOI] [PubMed] [Google Scholar]
- 80.Liu L, Qiu HB, Yang Y, et al. Losartan, an antagonist of AT1 receptor for angiotensin II, attenuates lipopolysaccharide-induced acute lung injury in rat. Arch Biochem Biophys. 2009;481(1):131–136. [DOI] [PubMed] [Google Scholar]
- 81.Zhang J, Wang M, Ding W, et al. The interaction of RAAS inhibitors with COVID-19: current progress, perspective and future. Life Sci. 2020;257:118142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saha S, Chakrabarti S, Singh PK, et al. Physiological relevance of angiotensin converting enzyme 2 as a metabolic linker and therapeutic implication of mesenchymal stem cells in COVID-19 and Hypertension. Stem Cell Rev Rep. 2020;16(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Meng J, Xiao G, Zhang J, et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg Microbes Infect. 2020;9(1):757–760. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This is a good clinical study that shows how repurposing existing drugs could help us to combat COVID-19.
- 84.Pirola CJ, Sookoian S. Estimation of Renin-Angiotensin-Aldosterone-System (RAAS)-Inhibitor effect on COVID-19 outcome: a meta-analysis. J Infection. 2020;81(2):276–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jakovac H. COVID-19: is the ACE2 just a foe? Am J Physiol Lung Cell Mol Physiol. 2020;318(5):1025–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jakovac H. COVID-19 and hypertension: is the HSP60 culprit for the severe course and worse outcome? Am J Physiol Heart Circ Physiol. 2020;319(4):793–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Juwono J, Martinus RD. Does Hsp60 provide a link between mitochondrial stress and inflammation in diabetes mellitus? J Diabetes Rese. 2016;8017571. DOI: 10.1155/2016/8017571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blagonravov ML, Sklifasovskaya AP, Korshunova AY, et al. Heat Shock Protein HSP60 in left ventricular cardiomyocytes of hypertensive rats with and without insulin-dependent diabetes mellitus. Bull Exp Biol Med. 2020;170(1):10–14. [DOI] [PubMed] [Google Scholar]
- 89.Nicholson AG, Osborn M, Devaraj A, et al. COVID‐19 related lung pathology: old patterns in new clothing? Histopathology. 2020;77(2):169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carsana L, Sonzogni A, Nasr A, et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet. 2020;20(10):1135–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tian S, Hu W, Niu L, et al. Pulmonary pathology of early-phase 2019 novel coronavirus (COVID-19) pneumonia in two patients with lung cancer. J Thorac Oncol. 2020;15(5):700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Menter T, Haslbauer JD, Nienhold R, et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology. 2020;77(2):198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McGonagle D, O’Donnell JS, Sharif K, et al. Why the immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia are distinct from macrophage activation syndrome with disseminated intravascular coagulation. Lancet Rheum. 2020;2(7):437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nishiga M, Wang DW, Han Y, et al. COVID-19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat Rev Cardiol. 2020;17(9):543–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Guo T, Fan Y, Chen M, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5(7):811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fox SE, Li G, Akmatbekov A, et al. Unexpected features of cardiac pathology in COVID-19 infection. circulation. 2020;142(11):1123–1125. [DOI] [PubMed] [Google Scholar]
- 97.Basso C, Leone O, Rizzo S, et al. Pathological features of COVID-19-associated myocardial injury: a multicentre cardiovascular pathology study. Eur heart J. 2020;41(39):3827–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Knowlton KU. Pathogenesis of SARS-CoV-2 induced cardiac injury from the perspective of the virus. J Mol Cell Cardiol. 2020;147:12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gavriilaki E, Anyfanti P, Gavriilaki M, et al. Endothelial dysfunction in COVID-19: lessons learned from coronaviruses. Hypertens Heart. 2020;22. DOI: 10.1007/s11906-020-01078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hanley B, Naresh KN, Roufosse C, et al. Histopathological findings and viral tropism in UK patients with severe fatal COVID-19: a post-mortem study. Lancet. 2020;1:245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vasquez-Bonilla WO, Orozco R, Argueta V, et al. A review of the main histopathological findings in coronavirus disease 2019. Hum Pathol. 2020;105:74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nair R, Maseeh A. Vitamin D: the “sunshine” vitamin. J Pharmacol Pharmacother. 2012;3(2):118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Holick MF, MacLaughlin JA, Clark MB, et al. Photosynthesis of previtamin D3 in human skin and the physiologic consequences. Science. 1980;210(4466):203–205. [DOI] [PubMed] [Google Scholar]
- 104.Fleet JC. Rapid, membrane-initiated actions of 1,25dihydroxyvitamin D: what are they and what do they mean? J Nutr. 2004;134(12):3215–3218. [DOI] [PubMed] [Google Scholar]
- 105.Carlberg C. The vitamin D3 receptor in the context of the nuclear receptor superfamily: the central role of the retinoid X receptor. Endocrine. 1996;4(2):91–105. [DOI] [PubMed] [Google Scholar]
- 106.Yu VC, Delsert C, Andersen B, et al. RXR beta: a coregulator that enhances binding of retinoic acid, thyroid hormone, and vitamin D receptors to their cognate response elements. Cell. 1991;67(6):1251–1266. [DOI] [PubMed] [Google Scholar]
- 107.Tecle T, Tripathi S, Hartshorn KL. Defensins and cathelicidins in lung immunity. Innate Immun. 2010;16(3):151–159. [DOI] [PubMed] [Google Scholar]
- 108.Leaf DE, Croy HE, Abrahams SJ, et al. Cathelicidin antimicrobial protein, vitamin D, and risk of death in critically ill patients. Crit Care. 2015;19(1):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fabbri A, Infante M, Ricordi C. Vitamin D status: a key modulator of innate immunity and natural defense from acute viral respiratory infections. Eur Rev Med Pharmacol Sci. 2020;24(7):4048–4052. [DOI] [PubMed] [Google Scholar]
- 110.Chen H, Lu R, Zhang YG, et al. Vitamin D receptor deletion leads to the destruction of tight and adherens junctions in lungs. Tissue Barriers. 2018;6(4):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shi YY, Liu TJ, Fu JH, et al. Vitamin D/VDR signaling attenuates lipopolysaccharide-induced acute lung injury by maintaining the integrity of the pulmonary epithelial barrier. Mol Med Rep. 2016;13(2):1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zheng SX, Yang JX, Hu X, et al. Vitamin D attenuates lung injury via stimulating epithelial repair, reducing epithelial cell apoptosis and inhibits TGF-β induced epithelial to mesenchymal transition. Biochem Pharmacol. 2020;177:113955. [DOI] [PubMed] [Google Scholar]
- 113.Sadeghi K, Wessner B, Laggner U, et al. Vitamin D3 down-regulates monocyte TLR expression and triggers hyporesponsiveness to pathogen-associated molecular patterns. Eur J Immunol. 2020;36(2):361–370. [DOI] [PubMed] [Google Scholar]
- 114.Zhao Y, Yu B, Mao X, et al. Dietary vitamin D supplementation attenuates immune responses of pigs challenged with rotavirus potentially through the retinoic acid-inducible gene I signalling pathway. Br J Nutr. 2014;112(3):381–389. [DOI] [PubMed] [Google Scholar]
- 115.Zhang Y, Leung DYM, Richers BN, et al. Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J Immunol. 2012;188(5):2127–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Telcian AG, Zdrenghea MT, Edwards MR, et al. Vitamin D increases the antiviral activity of bronchial epithelial cells in vitro. Antiviral Res. 2017;137:93–101. [DOI] [PubMed] [Google Scholar]
- 117.Gal-Tanamy M, Bachmetov L, Ravid A, et al. Vitamin D: an innate antiviral agent suppressing hepatitis C virus in human hepatocytes. Hepatology. 2011;54(5):1570–1579. [DOI] [PubMed] [Google Scholar]
- 118.Feng X, Wang Z, Howlett-Prieto Q, et al. Vitamin D enhances responses to interferon-β in MS. Neurol Neuroimmunol Neuroinflamm. 2019;6(6):e622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lange CM, Gouttenoire J, Duong FHT, et al. Vitamin D receptor and Jak–STAT signaling crosstalk results in calcitriol-mediated increase of hepatocellular response to IFN-α. J Immunol. 6037-44;192. DOI: 10.1080/15384047.2016.1235669. [DOI] [PubMed] [Google Scholar]
- 120.Hansdottir S, Monick MM, Lovan N, et al. Vitamin D decreases respiratory syncytial virus induction of NF-κB–linked chemokines and cytokines in airway epithelium while maintaining the antiviral state. J Immunol. 2010;184(2):965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lemire JM, Adams JS, Kermani-Arab V, et al. 1,25-Dihydroxyvitamin D3 suppresses human T helper/inducer lymphocyte activity in vitro. J Immunol. 1985;134(5):3032–3035. [PubMed] [Google Scholar]
- 122.Lemire JM, Archer DC. 1,25-dihydroxyvitamin D3 prevents the in vivo induction of murine experimental autoimmune encephalomyelitis. J Clin Invest. 1991;87(3):1103–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cantorna MT. Mechanisms underlying the effect of vitamin D on the immune system. Proc Nutr Soc. 2010;69(3):286–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wei R, Christakos S. Mechanisms underlying the regulation of innate and adaptive immunity by vitamin D. Nutrients. 2015;7(10):8251–8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jeffrey LE, Burke F, Mura M, et al. 1,25-dihydroxyvitamin D3 and IL-2 combine to inhibit T Cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J Immunol. 2009;183(9):5458–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Caprio M, Infante M, Calanchini M, et al. Vitamin D: not just the bone. Evidence for beneficial pleiotropic extraskeletal effects. Eat Weight Disord. 2017;22(1):27–41. [DOI] [PubMed] [Google Scholar]
- 127.Infante M, Ricordi C, Sanchez J, et al. Influence of Vitamin D on islet autoimmunity and beta-Cell function in Type 1 Diabetes. Nutrients. 2019;11(9):2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xu J, Yang J, Chen J, et al. Vitamin D alleviates lipopolysaccharide-induced acute lung injury via regulation of the renin-angiotensin system. Mol Med Repo. 2017;16(5):7432–7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Li YC, Kong J, Wei M, et al. 1,25-Dihydroxyvitamin D3 is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110(2):229–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yang J, Xu J, Zhang H. Effect of vitamin D on ACE2 and vitamin D receptor expression in rats with LPS-induced acute lung injury. Chin J Emergency Med. 2016. DOI: 10.3760/CMA.J.1671-0282.2016.12.016 [DOI] [Google Scholar]
- 131.Shi Y, Liu T, Yao L, et al. Chronic vitamin D deficiency induces lung fibrosis through activation of the renin-angiotensin system. Sci Rep. 2017;7(1):3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li YC, Kong J, Wei M, et al. 1,25-Dihydroxyvitamin D (3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110(2):229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xiang W, Kong J, Chen S, et al. Cardiac hypertrophy in vitamin D receptor knockout mice: role of the systemic and cardiac renin-angiotensin systems. Am J Physiol Endocrinol Metab. 2005;288(1):125–132. [DOI] [PubMed] [Google Scholar]
- 134.Imai Y, Kuba K, Rao S, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dancer RCA, Parekh D, Lax S, et al. Vitamin D deficiency contributes directly to the acute respiratory distress syndrome (ARDS). Thorax. 2015;70(7):617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Park S, Lee MG, Hong SB, et al. Effect of vitamin D deficiency in Korean patients with acute respiratory distress syndrome. Korean J Intern Med. 2019;34(3):685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Remmelts HHF, Van De Garde EMW, Meijvis SCA. Peelen ELGCA et al. Addition of vitamin D status to prognostic scores improves the prediction of outcome in community-acquired pneumonia. Clin Infect Dis. 2012;55(11):1488–1494. [DOI] [PubMed] [Google Scholar]
- 138.Barnett N, Zhao Z, Koyama T, et al. Vitamin D deficiency and risk of acute lung injury in severe sepsis and severe trauma: a case-control study. Ann Intensive Care. 2014;4(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mihos CG, De La Cruz J, Hernandez A, et al. Vitamin D deficiency and supplementation in cardiovascular disorders. Cardiol Rev. 2017;25(4):189–196. [DOI] [PubMed] [Google Scholar]
- 140.McGreevy C, Williams D. New insights about vitamin D and cardiovascular disease: a narrative review. Ann Intern Med. 2011;155(12):820–826. [DOI] [PubMed] [Google Scholar]
- 141.Caprio M, Mammi C, Rosano GMC, et al. a novel player in endothelial function and dysfunction. Arch Med Sci. 2012;8:4–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang Q, Zhang M, Wang H, et al. Vitamin D supplementation improves endothelial dysfunction in patients with non-dialysis chronic kidney disease. Int Urol Nephrol. 2018;50(5):923–927. [DOI] [PubMed] [Google Scholar]
- 143.Tangpricha V, Pearce EN, Chen TC, et al. Vitamin D insufficiency among free-living healthy young adults. Am J Med. 2002;112(8):659–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Holick MF, Chen TC. Vitamin D deficiency: a worldwide problem with health consequences. Am J Clin Nutr. 2008;87(87):1080S–60S. [DOI] [PubMed] [Google Scholar]
- 145.Vasarhelyi B, Satori A, Olajos F, et al. Low vitamin D levels among patients at Semmelweis University: retrospective analysis during a one-year period. Orv Hetil. 2011;152(32):1272–1277. [DOI] [PubMed] [Google Scholar]
- 146.D’Avolio A, Avataneo V, Manca A, et al. 25-Hydroxyvitamin D concentrations are lower in patients with positive PCR for SARS-CoV-2. Nutrients. 2020;12(5):1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Baktash V, Hosack T, Patel N, et al. Vitamin D status and outcomes for hospitalised older patients with COVID-19. Post Graduate Med J. 2020;postgradmedj-2020-138712. DOI: 10.1136/postgradmedj-2020-138712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Carpagnano GE, Lecce VD, Quaranta VN, et al. Vitamin D deficiency as a predictor of poor prognosis in patients with acute respiratory failure due to COVID-19. J Endocrinol Invest. 2020;1-7. DOI: 10.1007/s40618-020-01370-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Karahan S, Katkat F. Impact of Serum 25(OH) Vitamin D level on mortality in patients with COVID-19 in Turkey. J Nutr Health Aging. 2020. DOI: 10.1007/s12603-020-1479-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jain A, Chaurasia R, Sengar NS, et al. Analysis of vitamin D level among asymptomatic and critically ill COVID-19 patients and its correlation with inflammatory markers. Sci Rep. 2020;10(1):20191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Meltzer DO, Best TJ, Zhang H, et al. Association of Vitamin D status and other clinical characteristics with COVID-19 test results. JAMA Netw Open. 2020;3(9):e2019722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ilie PC, Stefanescu S, Smith L. The role of vitamin D in the prevention of coronavirus disease 2019 infection and mortality. Aging Clin Exp Res. 2020;32(7):1195–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rhodes JM, Subhramanian S, Laird E, et al. Editorial: low population mortality from COVID‐19 in countries south of latitude 35 degrees North supports vitamin D as a factor determining severity. Aliment PharmacolTher. 2020;51(12):1434–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Holick MF. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am J Clin Nutr. 2004;80(6):1678S–1688S. [DOI] [PubMed] [Google Scholar]
- 155.Berridge MJ. Vitamin D deficiency and diabetes. Biochem J. 2017;474(8):1321–1332. [DOI] [PubMed] [Google Scholar]
- 156.Nakashima A, Yokoyama K, Yokoo T, et al. Role of vitamin D in diabetes mellitus and chronic kidney disease. World J Diabetes. 2016;7(5):89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Migliaccio S, Nisio AD, Mele C, et al. Obesity and hypovitaminosis D: causality or casualty? Int J Obes Suppl. 2019;9(1):20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mitchell F. Vitamin-D and COVID-19: do deficient risk a poorer outcome? Lancet Diabetes Endocrinol. 2020;8(7):570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Castillo ME, Costa LME, Barrios JMV, et al. Effect of calcifediol treatment and best available therapy versus best available therapy on intensive care unit admission and mortality among patients hospitalized for COVID-19: a pilot randomized clinical study. J Steroid Biochem Mol Biol. 2020;203:105751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Annweiler C, Hanotte B, de I’eprevier CG, et al. Vitamin D and survival in COVID-19 patients: a quasi-experimental study. J Steroid Biochem Mol Biol. 2020;204:105771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tan CW, Ho LP, Kalimuddin S, et al. Cohort study to evaluate the effect of vitamin D, magnesium, and vitamin B12 in combination on progression to severe outcomes in older patients with coronavirus (COVID-19). Nutrition. 2020;79-80:111017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Annweiler G, Corvaisier M, Gautier J, et al. Vitamin D supplementation associated to better survival in hospitalized Frail Elderly COVID-19 Patients: the GERIA-COVID quasi-experimental study. Nutrients. 2020;12(11):3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ling SF, Broad E, Murphy R, et al. High-Dose cholecalciferol booster therapy is associated with a reduced risk of mortality in patients with COVID-19: a cross-sectional multi-centre observational study. Nutrients. 2020;12(12):3799. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• A large-scale well-designed study which indicates the therapeutic potential of vitamin D in COVID-19.
- 164.Ohaegbulam KC, Swalih M, Patel P, et al. Vitamin D supplementation in COVID-19 patients: a clinical case series. Am J Ther. 2020;27(5):485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Giannini S, Passeri G, Tripepi G, et al. Effectiveness of in-hospital cholecalciferol use on clinical outcomes in comorbid COVID-19 patients: a hypothesis-generating study. Nutrients. 2021;13(1):219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rastogi A, Bhansali A, Khare N, et al. Short term, high-dose vitamin D supplementation for COVID-19 disease: a randomised, placebo-controlled, study (SHADE study). Postgrad Med J. 2020;postgradmedj-2020-139065. DOI: 10.1136/postgradmedj-2020-139065. [DOI] [PubMed] [Google Scholar]
- 167.Grant WB, Lahore H, McDonnell SL, et al. Evidence that Vitamin D supplementation could reduce risk of influenza and COVID-19 infections and deaths. Nutrients. 2020;12(4):988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid-19 – final report. N Engl J Med. 2020;383(19):1813–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cauchois R, Koubi M, Delarbre D, et al. Early IL-1 receptor blockade in severe inflammatory respiratory failure complicating COVID-19. Proc Natl Acad Sci USA. 2020;117(32):18951–18953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cavalli G, Larcher A, Tomelleri A, et al. Interleukin-1 and interleukin-6 inhibition compared with standard management in patients with COVID-19 and hyperinflammation: a cohort study. Lancet Rheumatol. 2021. DOI: 10.1016/s2665-9913(21)00012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Alijotas-Reig J, Esteve-Valverde E, Belizna C, et al. Immunomodulatory therapy for the management of severe COVID-19. Beyond the anti-viral therapy: a comprehensive review. Autoimmun Rev. 2020;19(7):102569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Leng Z, Zhu R, Hou W, et al. Transplantation of ACE2 - Mesenchymal stem cells improves the outcome of patients with COVID-19 pneumonia. Aging Dis. 2020;11(2):216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mascolo S, Carleo MA, Contieri M, et al. SARS-CoV-2 and inflammatory responses: from mechanisms to the potential therapeutic use of intravenous immunoglobulin. J Med Virol. 2020. DOI: 10.1002/jmv.26651. [DOI] [PubMed] [Google Scholar]
- 174.Brown AJ, Slatoplosky E. Vitamin D analogs: therapeutic applications and mechanisms for selectivity. Mol Aspects Med. 2008;29(6):433–452. [DOI] [PubMed] [Google Scholar]
- 175.Chen J, Tang Z, Slominski AT, et al. Vitamin D and its analogs as anticancer and anti-inflammatory agents. Eur J Med Chem. 2020;207:112738. [DOI] [PubMed] [Google Scholar]