

Abstract
Neuronal nicotinic receptors containing α4 and β2 subunits assemble in two pentameric stoichiometries, (α4)3(β2)2 and (α4)2(β2)3, each with distinct pharmacological signatures; (α4)3(β2)2 receptors are strongly potentiated by the drug NS9283, whereas (α4)2(β2)3 receptors are unaffected. Despite this stoichiometry-selective pharmacology, the molecular identity of the target for NS9283 remains elusive. Here, studying (α4)3(β2)2 receptors, we show that mutations at either the principal face of the β2 subunit or the complementary face of the α4 subunit prevent NS9283 potentiation of ACh-elicited single-channel currents, suggesting the drug targets the β2–α4 pseudo-agonist sites, the α4–α4 agonist site, or both sites. To distinguish among these possibilities, we generated concatemeric receptors with mutations at specified subunit interfaces, and monitored the ability of NS9283 to potentiate ACh-elicited single-channel currents. We find that a mutation at the principal face of the β2 subunit at either β2–α4 pseudo-agonist site suppresses potentiation, whereas mutation at the complementary face of the α4 subunit at the α4–α4 agonist site allows a significant potentiation. Thus, monitoring potentiation of single concatemeric receptor channels reveals that the β2–α4 pseudo-agonist sites are required for stoichiometry-selective drug action. Together with the recently determined structure of the (α4)3(β2)2 receptor, the findings have implications for structure-guided drug design.
Keywords: Neurotransmitters, Nicotine addiction, Protein interfaces, PAM, Ligand-gated ion channels
Introduction
Nicotinic acetylcholine receptors (AChRs) are found in the central and peripheral nervous systems, and trigger moment-to-moment neuronal excitation and modulate release of neurotransmitters [1, 2]. They are implicated in a variety of neurological disorders [2, 3], participate in nicotine addiction [4–6], and are targets for a host of inhibitors and activators, both man-made and occurring in nature. AChRs belong to the large family of Cys-loop receptors and are pentamers formed from homologous subunits of which there are many different subtypes. At the neuromuscular junction, the AChR contains four types of subunits that assemble in a fixed stoichiometry and arrangement [7], whereas, in the brain, there are many different types of subunits that assemble in a variety of combinations and stoichiometry [1]. The majority of [3H] nicotine binding in the brain is associated with receptors containing α4 and β2 subunits [8–11]. These subunits assemble in two pentameric stoichiometries, one with three and the other with two α4 subunits [12–15]. These stoichiometric variants exhibit distinct functional and pharmacological properties. In particular, receptors with three α4 subunits are strongly potentiated by the drug NS9283, whereas receptors with two α4 subunits are unaffected [14, 16]. This subunit-selective pharmacology could arise through creation of a drug-binding site through exchange of an α4 for a β2 subunit, or through changes in inter-subunit interactions. To identify structures required for drug potentiation, studies of receptors with known stoichiometry and subunit arrangement are necessary.
Previously, we generated enriched populations of receptors with either three α4 and two β2 subunits or two α4 and three β2 subunits by transfecting cells with biased ratios of cDNAs encoding the subunits, and recorded ACh-elicited single-channel currents from receptors with each stoichiometry [14]. In addition, we established that pentameric concatemers recapitulated single-channel biophysical properties of receptors formed from the biased ratios of unlinked subunits. In particular, in the presence of ACh alone, concatemeric receptors with three nonconsecutive α4 subunits and two β2 subunits activate predominantly as single-channel openings flanked by long closings, whereas, in the presence of ACh and NS9283, they activate in a series of many channel openings in quick succession [14]. Here, studying receptors formed from biased ratios of the subunits, we find that a mutation at either the principal face of the β2 subunit or the complementary face of the α4 subunit prevents potentiation by NS9283. However, in both cases, the mutation was present in multiple copies per receptor. Using concatemeric receptors formed from five subunits linked head to tail, we introduce a single copy of each mutant subunit per receptor, record ACh-elicited single currents, and identify the type, number, and location of subunits required for drug potentiation.
Materials and methods
Mutagenesis and expression of human (α4)3(β2)2 AChRs formed from unlinked and linked subunits
cDNAs encoding unlinked α4 and β2 subunits, or linked β2–α4–β2–α4–α4 subunits, and the chaperone protein 14-3-3 were individually sub-cloned into a modified pCI mammalian expression vector (Promega), as previously described [17, 18]. The 14-3-3 chaperone increases expression of α4β2 AChRs formed by unlinked as well as linked subunits [17, 19, 20]. BOSC 23 cells, a cell line derived from HEK 293 cells [21], were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) containing 10% fetal bovine serum, and transfected by calcium phosphate precipitation, as previously described [22–24]. For experiments with receptors formed from unlinked subunits, the stoichiometry was biased towards (α4)3(β2)2 by transfecting cells with a 10:1:10 ratio of α4, β2, and 14-3-3 cDNAs. Varying the ratio of α4 to β2 subunit cDNAs biases the receptor population toward a single subunit stoichiometry in both mammalian cell lines [15, 25] and Xenopus laevis oocytes [13]. The amounts of transfected α4 and β2 cDNAs were, respectively, 3 and 0.3 μg for each 35 mm culture dish of cells. A cDNA encoding green fluorescent protein was included in all transfections. Transfections were carried out for 4–16 h, followed by medium exchange. Single-channel recordings were made 48–72 h post-transfection. For experiments with receptors formed from linked β2–α4–β2–α4–α4 subunits, the total amount of cDNA was 1–10 μg per 35 mm culture dish. Furthermore, following transfection at 37 °C, the cells were incubated at 30 °C until use, which increases the expression of receptors on the cell surface [26]. Single-channel recordings from cells expressing receptors formed from linked subunits were made 72–96 h post-transfection. Mutations were installed in cDNAs encoding unlinked and linked subunits and confirmed by sequencing as described previously [13, 27, 28].
Drugs
Acetylcholine (ACh) was purchased from Sigma-Aldrich (St Louis, MO, USA), and 3-[3-(3-Pyridinyl)-1,2,4-oxadiazol-5-yl]benzonitrile (NS9283) from Tocris (UK).
Patch-clamp recordings
Single-channel recordings were obtained in the cell-attached patch configuration at a membrane potential of − 70 mV and a temperature of 20 °C, as previously described [14–16, 29, 30]. For all experiments, the extracellular bathing solution contained (mM): 142 KCl, 5.4 NaCl, 1.8 CaCl2, 1.7 MgCl2, and 10 HEPES, adjusted to pH 7.4 with NaOH. The pipette solution contained (mM): 80 KF, 20 KCl, 40 K-aspartate, 2 MgCl2, 1 EGTA, and 10 HEPES, adjusted to pH 7.4 with KOH [31, 32]. Concentrated stock solutions of ACh were made in pipette solution and stored at − 80 °C until the day of each experiment. A concentrated stock solution of NS9283 was prepared in DMSO, stored at − 80 °C, and added to the pipette solution the day of each experiment. Pipette solution without NS9283 contained an equivalent volume of DMSO. Patch pipettes were pulled from glass capillary tubes (no. 7052, Garner Glass) and coated with Sylgard (Dow Corning).
Data analysis
Single-channel currents were recorded using an Axopatch 200B patch-clamp amplifier (molecular devices), with a gain of 100 mV/pA and the internal Bessel filter at 10 kHz. Currents were sampled at intervals of 20 μs using a PCI-6111E acquisition card (National Instruments), and recorded to hard disk using the program Acquire (Bruxton Corporation). Channel opening and closing transitions were determined using the program TAC 4.2.0 (Bruxton Corporation), which digitally filters the data (Gaussian response; final effective bandwidth 5 kHz), interpolates the digitized points using a cubic spline function, and detects channel openings using the half-amplitude threshold criterion, as previously described [33]. Following detection, each single-channel open dwell time was plotted against its time of occurrence during the recording to assess the stability of the channel opening frequency and gating kinetics.
To determine single-channel current amplitudes, the variable amplitude option in TAC was used, whereas, to determine open and closed dwell times, the fixed amplitude option was used. Dwell time histograms were plotted using a logarithmic abscissa and square root ordinate [34], with a uniformly imposed dead time of 40 μs, and the sum of exponentials was fitted to the data by maximum likelihood using the program TACFit 4.2.0 [33]. Clusters of channel openings were identified as a series of closely spaced openings preceded and followed by closed intervals longer than a specified critical duration (τcrit). This duration was taken as the point of intersection between consecutive components in the closed time histogram and ranged between 1 and 50 ms. A cluster duration, therefore, comprises the total open time of a series of openings plus that of the intervening closings briefer than τcrit.
The effect of NS9283 on the probability a channel will re-open was quantified by plotting the fraction of channel opening episodes with greater than N openings against the number of openings per episode. A channel opening episode was defined as a series of one or more openings separated by closings shorter than τcrit. The re-opening distributions were fitted with exponential decay functions, and an F test was used to determine whether a single or a biexponential decay best described the re-opening data; a single-exponential decay was preferred unless the sum-of-squares’ F test had a p value less than 0.01. Both the exponential fitting and F test were carried out using the Prism software package (GraphPad Software). In addition, the mean number of openings per episode was calculated as the reciprocal of the decay constant, and an F test was used to determine whether the fitted decay constants and fractional areas differed significantly between pairs of recordings under different experimental conditions. Parameters were considered significantly different if the p value was less than 0.01.
Results
Overview
The present work identifies sites required for NS9283 potentiation through a combination of single-channel recording, mutations of candidate drug sites, and concatemeric receptors. First, we establish experimental conditions to demonstrate, and analysis methods to quantify, potentiation of (α4)3(β2)2 receptors by NS9283 at the level of single-channel currents. Second, we define concentrations of ACh and NS9283 that maximize potentiation. Third, we determine the impact on potentiation of mutations at either the principal face of the β2 or the complementary face of the α4 subunit in receptors formed from unlinked subunits. Finally, we determine the impact on potentiation of a single copy of each mutation in concatemeric receptors composed of five subunits linked head to tail.
NS9283 potentiation at the single-channel level
To generate an enriched population of AChRs with three α4 and two β2 subunits, we transfected BOSC 23 cells, a variant of the 293 HEK cell line, with a 10:1 ratio of cDNAs encoding the α4 and β2 subunits, as described previously [14]. Using a patch pipette filled with extracellular solution containing a known concentration of ACh, we established a giga-ohm seal to a cell and recorded single-channel currents in the cell-attached patch configuration. The recordings reveal a single conductance class of channel openings with 4.1 ± 0.3 pA unitary current amplitude (Fig. 1a), an electrical signature indicating receptors with three α4 and two β2 subunits [14]. In the presence of ACh alone, the majority of channel openings appear as solitary current pulses flanked by long periods of baseline current (Fig. 1b), while a minority appears as several pulses in quick succession, which we call bursts (Fig. 1d). By contrast, in the presence of ACh and NS9283, the majority of channel openings appear as a series of current pulses in quick succession (Fig. 1c), which we call clusters (Fig. 1d), while a minority appears as solitary current pulses flanked by long periods of baseline current. Thus, qualitatively, NS9283 potentiates (α4)3(β2)2 receptors by enhancing the ability of a receptor channel that just closed to re-open [14].
Fig. 1.
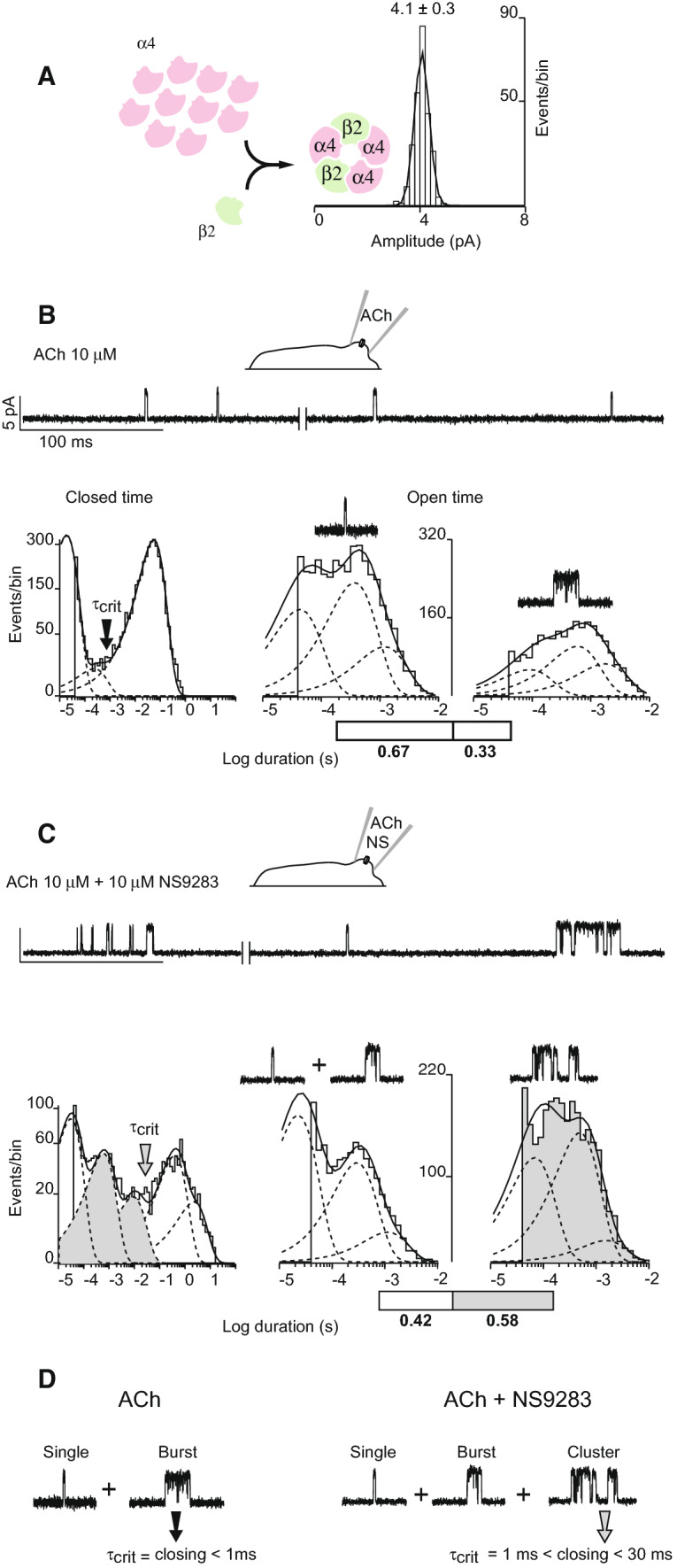
NS9283 potentiates single-channel currents through (α4)3(β2)2 AChRs. a BOSC 23 cells transfected with a tenfold excess of α4 over β2 subunit cDNAs predominantly express (α4)3(β2)2 AChRs. Amplitude histogram from a cell-attached patch recording in the presence of 10 μM ACh reveals a single conductance class of channel openings with 4.1 pA unitary current amplitude (holding potential − 70 mV). b Single-channel currents were recorded with 10 μM ACh in the recording pipette (holding potential − 70 mV; Gaussian filter 4 kHz). Left panel shows a closed duration histogram fitted by the sum of exponentials. The point of intersection of adjacent components (arrow) yields a critical closed time τcrit of 0.7 ms. Center panel shows a histogram of channel openings separated by closings longer than τcrit fitted by the sum of three exponential components (dashed curves); an exemplar channel opening is shown. Right panel shows a histogram of channel openings separated by closings briefer than τcrit; an exemplar channel opening episode is shown. c Single-channel currents were recorded in the presence of 10 μM ACh and 10 μM NS9283 as in b. Left panel shows a closed duration histogram fitted by the sum of exponentials. The point of intersection of adjacent components (arrow) yields a critical closed time τcrit of 30 ms that was used to distinguish episodes of un-potentiated from potentiated channel openings. Closed duration components that selectively flank potentiated channel openings are shaded. Middle panel shows a histogram of channel openings separated by closings longer than τcrit; exemplar channel openings are shown. Right panel shows a histogram of channel openings separated by closings briefer than τcrit; exemplar channel opening episodes are shown. Exponential components corresponding to potentiated openings are shaded. Percentage of channel openings that are potentiated is indicated by the shaded portion of the bar. d Panel summarizes the classes of channel opening episodes recorded in the presence of ACh without (left traces) or with NS9283 (right traces)
Dwell time analysis of un-potentiated channel openings
To quantify potentiation, we developed methods to distinguish un-potentiated from potentiated channel openings. The first step was to analyze histograms of closed dwell times from recordings obtained in the presence of ACh alone. A representative histogram, obtained from a recording in the presence of 10 μM ACh, is well fitted by the sum of exponentials: two major components with brief and long mean durations, and a third minor component with intermediate duration. The components with brief and intermediate mean durations represent closings within bursts of openings by the same receptor channel, whereas the component with longest mean duration represents closings between both solitary and bursts of openings primarily from different receptor channels (Fig. 1b). To distinguish solitary from bursts of channel openings, we established a discriminating closed time, τcrit, from the point of intersection between the components with longest and intermediate mean durations. For the recording illustrated in Fig. 1b, the discriminating closed time was 0.7 ms. However, owing to different numbers of receptors in each patch, the component with long mean duration varied, and in patches with low opening frequency, or in which the opening frequency declined as the recording progressed, the discriminating closed time extended to 1 ms. Thus, for uniformity, we applied a discriminating closed time of 1 ms to distinguish solitary from bursts of channel openings. After assigning each channel opening to one of the two classes, we find that some 67% of openings are solitary channel openings, while 33% are bursts of channel openings. In addition, for both classes of channel openings, the open time histogram contains three exponential components with mean durations and relative areas that are similar between the two classes (Fig. 1b). Thus, although the two classes of openings are readily distinguished by their flanking closed dwell times, their open time distributions are very similar. This analysis of channel openings in the presence of ACh alone provides a frame of reference to distinguish potentiated channel openings in the presence of ACh and NS9283.
Dwell time analysis of potentiated channel openings
In the presence of submaximal concentrations of ACh and NS9283, a mixture of potentiated and un-potentiated channel openings is expected. Un-potentiated channel openings comprise either solitary or bursts of openings, as just described, whereas potentiated openings comprise clusters of many successive channel openings. Thus, we devised a three-step procedure to distinguish potentiated from un-potentiated channel openings. In the first step, openings separated by closings briefer than 1 ms were joined to yield composite events composed of N openings and N − 1 closings; a portion of these composite events corresponds to un-potentiated openings, whereas the remaining portion corresponds to potentiated openings (Fig. 1c; Table 1). In the second step, to distinguish composite events that are potentiated from those that are un-potentiated, the closed duration histogram was again fitted by the sum of exponentials, and a discriminating closed time of 30 ms was determined from the point of intersection between successive exponential components, by analogy to the analysis described for recordings in the presence of ACh alone; composite events flanked by closings briefer than 30 ms were classified as potentiated, whereas composite events flanked by closings longer than 30 ms were classified as un-potentiated (Fig. 1d). In the third step, the two classes of composite events were separated, closings briefer than 1 ms that were masked initially were re-introduced, and a histogram of open durations was generated for each class of channel openings (Fig. 1c, shaded and un-shaded histograms). The number of channel openings in the potentiated and un-potentiated classes was then determined from the total number of openings in the histogram for each class. The analysis reveals that, in the presence of 10 μM ACh and 10 μM NS9283, some 58% of channel openings are potentiated (Fig. 1c, shaded portion of the bar; Table 1). In addition, openings in both the potentiated and un-potentiated classes contain three exponential components with mean durations that are similar between the two classes. Thus, NS9283 does not affect open-to-closed transitions, but, instead, increases both the probability and rate of closed-to-open transitions.
Table 1.
Potentiated versus un-potentiated channel openings for various types of AChRs in the presence of ACh and NS9283
| Receptor type (number of patches) | [ACh] μM | [NS9283] μM | Total openings (n) | Proportion of openings | SE | |
|---|---|---|---|---|---|---|
| Un-potentiated | Potentiated | |||||
| (α4)3(β2)2 (n = 3) | 10 | 10 | 4895 | 0.42 | 0.58 | ± 0.01 |
| (α4)3(β2)2 (n = 6) | 1 | 30 | 2214 | 0.39 | 0.61 | ± 0.04 |
| (α4)3(β2)2 (n = 4) | 10 | 30 | 3462 | 0.22 | 0.78 | ± 0.06 |
| (α4)3(β2)2 (n = 6) | 50 | 30 | 27,025 | 0.26 | 0.74 | ± 0.03 |
| (α4)3(β2)2 (n = 4) | 100 | 30 | 1885 | 0.45 | 0.55 | ± 0.02 |
| (α4)3(β2W176A)2 (n = 6) | 50 | 30 | 1752 | 0.90 | 0.10 | ± 0.01 |
| β2α4β2α4α4 (n = 6) | 50 | 30 | 2776 | 0.19 | 0.81 | ± 0.04 |
| β2W176Aα4β2α4α4 (n = 6) | 50 | 30 | 1403 | 0.84 | 0.16 | ± 0.01 |
| β2α4β2W176Aα4α4 (n = 8) | 50 | 30 | 810 | 0.71 | 0.29 | ± 0.04 |
| β2W176Aα4β2W176Aα4α4 (n = 10) | 50 | 30 | 473 | 0.86 | 0.14 | ± 0.05 |
| β2α4β2α4H142Vα4 (n = 8) | 50 | 30 | 2080 | 0.45 | 0.55 | ± 0.09 |
Averaged proportions of potentiated versus un-potentiated openings are given for the indicated number of patches (n) along with ± the standard error (SE). For each experimental condition, the total number of openings from n patches is indicated. The table summarizes results displayed in Figs. 1, 2, 3, and 4
Potentiation by NS9283 as a function of ACh concentration
To determine the functional consequences of mutations of candidate targets for NS9283, we first established experimental conditions that maximize potentiation. Thus, we recorded single-channel currents in the presence of a maximally effective concentration of NS9283, 30 μM, but with a range of ACh concentrations. We then quantified the percentage of potentiated and un-potentiated channel openings, as just described. In the presence of 1 μM ACh alone, a minimum concentration to elicit channel opening, the majority of openings are solitary events flanked by long closings (Fig. 2a), while a minority comprises bursts of several openings in quick succession. Because both classes of channel openings are un-potentiated, for clarity, they are combined into a single class here and in subsequent sections (Fig. 2a, un-shaded bar). In the presence of 1 μM ACh and NS9283, the majority of channel openings appear as clusters of many openings flanked by brief closings, and a minority appears as solitary or bursts of openings flanked by long closings (Fig. 2b). After applying discriminating closed times to distinguish potentiated from un-potentiated channel openings, we find that potentiated openings comprise some 61% of all openings (Fig. 2b, shaded portion of the bar; Table 1). Furthermore, open duration histograms for both potentiated and un-potentiated openings contain three exponential components, which, again, mirror those observed in the presence of ACh alone (Fig. 2a). Thus, in the presence of a minimal concentration of ACh and a maximal concentration of NS9283, a substantial percentage of the channel openings are potentiated.
Fig. 2.

Dependence of NS9283 potentiation on ACh concentration. a–i Single-channel currents from (α4)3(β2)2 AChRs were recorded in the presence of the indicated concentrations of ACh, without or with 30 μM NS9283 (holding potential − 70 mV, Gaussian filter 4 kHz). To the right of each trace are histograms of un-potentiated and potentiated channel openings, respectively, determined as described in the text and Fig. 1, fitted by the sum of three exponentials. Exponential components of potentiated channel openings are shaded. Percentage of channel openings that are potentiated is indicated by the shaded portion of the bar
Increasing the ACh concentration, while maintaining a maximal concentration of NS9283, yields channel openings and closings that qualitatively mirror those in the presence of lower concentrations of ACh (Fig. 2c–i). In the presence of 10 and 50 μM ACh, the percentage of potentiated channel openings increases to 78 and 74%, respectively (Fig. 2d, f; Table 1). However, in the presence of 100 μM ACh, the percentage of potentiated channel openings declines to 55% (Fig. 2i; Table 1); this decline may arise from enhanced desensitization of potentiated relative to un-potentiated channel openings owing to the increased ACh concentration. These results establish the concentrations of ACh and NS9283 that maximize potentiation.
Mutations in α4 and β2 subunits block potentiation
The recent cryo-EM structure of the (α4)3(β2)2 receptor is shown in Fig. 3a, b. There are two α4–β2 subunit interfaces that form orthosteric agonist-binding sites, one α4–α4 subunit interface that forms a third orthosteric site, and two β2–α4 subunit interfaces that form pseudo-agonist sites. The agonist nicotine, included during preparation of the receptor protein for cryo-EM, is present at the α4–β2 and α4–α4 interfaces, but not at the β2–α4 interfaces. When each subunit interface is viewed from the side, the subunit on the left forms the principal face, while the subunit on the right forms the complementary face. The principal face contains structural motifs known as loops A, B, and C, from which conserved aromatic residues extend into the binding pocket. The complementary face contains loops D, E, F, and G, from which aromatic, hydrophobic, polar, and anionic residues extend into the pocket. Furthermore, each pair of α4–β2 and β2–α4 subunits is asymmetric owing to differences in the flanking subunits; one α4–β2 pair is flanked by β2 and α4 subunits and the other pair is flanked α4 and α4 subunits; one β2–α4 pair is flanked by α4 and α4 subunits and the other pair is flanked by α4 and β2 subunits (Fig. 3a, right panel).
Fig. 3.

For AChRs formed from unlinked subunits, mutations in both β2 and α4 subunits block NS9283 potentiation. a, b Recent cryo-EM structure of the (α4)3(β2)2 AChR (PDB code 6CNK). a Side (left) and top (right) views of the (α4)3(β2)2 AChR complex. b Left panel shows the structure of an β2–α4 subunit interface within the (α4)3(β2)2 AChR. Right panel shows a close-up view of the β2–α4 subunit interface with residues subjected to mutation, β2W176 and α4H142, highlighted as spheres. c–e Single-channel currents from wild-type or mutant (α4)3(β2)2 AChRs were recorded in the presence of 50 μM ACh and 30 μM NS9283 (holding potential − 70 mV, Gaussian filter 4 kHz). Red asterisks indicate the locations of mutations. To the right of each trace is a histogram of cluster durations, corresponding to successive channel openings and intervening closings, fitted by the sum of exponentials (dashed curves); the component with longest mean duration, present for the wild-type AChR, is reduced or absent for the mutant AChRs. To the right of the cluster duration histograms are histograms of channel openings classified as either un-potentiated (un-shaded) or potentiated (shaded) fitted by the sum of three exponentials. Above each histogram, the shaded portion of the bar indicates the percentage of channel openings that are potentiated
To identify subunit interfaces required for NS9283 potentiation, we generated mutations at either the complementary face of the α4 subunit or the principal face of the β2 subunit. Previous work showed that the mutation α4H142V, either alone or combined with mutations of two nearby residues, prevented NS9283 potentiation of ACh-elicited macroscopic currents [35–37]. Our choice of the mutation β2W176A was based on the structures of the (α4)3(β2)2 and (α4)2(β2)3 receptors in which β2W176 establishes close contact with α4H142 at each β2–α4 subunit interface (Fig. 3b, right panel). Each mutant subunit was co-transfected with the complementary wild-type subunit using biased ratios of subunit cDNAs to promote the expression of (α4)3(β2)2 receptors. In the presence of 50 μM ACh and 30 μM NS9283, receptors comprised of wild-type subunits open in clusters of many channel openings flanked by brief closings, with some 74% of channel openings classified as potentiated (Fig. 3c, shaded bar and histogram; Table 1), as in Fig. 2f. In addition, to provide a second measure of potentiation, a histogram of cluster durations was generated; clusters were defined as a series of channel openings separated by closings shorter than 30 ms. The histogram of cluster durations is well described as the sum of four major exponential components, with the component with longest mean duration representing maximally potentiated channel openings (Fig. 3c).
For receptors containing the mutant α4H142V, the two measures of NS9283 potentiation are markedly reduced (Fig. 3d). The percentage of channel openings classified as potentiated falls below our limits of detection, and the exponential component of clusters with longest mean duration is eliminated. Because the mutation is present at the α4–α4 subunit interface, as well as the two β2–α4 interfaces, one or both types of interfaces are required for NS9283 potentiation.
For receptors containing the mutant β2W176A, the two measures of potentiation are also markedly reduced (Fig. 3e; Table 1). The percentage of channel openings classified as potentiated falls from 74 to 10%, and the exponential component of clusters with longest mean duration appears as a small tail rather than a distinct component. Because the mutant β2 subunit is present at only β2–α4 subunit interfaces, one or both these interfaces are required for NS9283 potentiation.
Mutations of individual α4 and β2 subunits in pentameric concatemers
Previously, we showed that NS9283 strongly potentiated ACh-elicited single-channel currents from concatemeric receptors composed of three nonconsecutive α4 and two β2 subunits [14]. Here, in the presence of optimal concentrations of ACh and NS9283, the same concatemeric receptors activate as clusters of many channel openings separated by brief closings (Fig. 4a, b), which mirror potentiated channel openings by (α4)3(β2)2 receptors formed from unlinked subunits. The analysis to quantify the percentage of potentiated and un-potentiated channel openings reveals that, for concatemeric receptors in the presence of optimal concentrations of ACh and NS9283, some 81% of channel openings are potentiated (Fig. 4b; Table 1). In addition, the histogram of cluster durations contains a fourth, prolonged component representing maximally potentiated channel openings. Thus, receptors formed from concatemeric subunits recapitulate key measures of potentiation observed for receptors formed from unlinked subunits.
Fig. 4.

Potentiation of (α4)3(β2)2 AChRs formed from linked subunits is blocked by mutation in the β2 but not the α4 subunit. a Schematic diagram of the plasmid encoding the five linked subunits that form the (α4)3(β2)2 AChR; subunits are labeled 1–5 indicating the order of subunit linkage. b–f Single-channel currents from the indicated wild-type or mutant (α4)3(β2)2 AChRs were recorded in the presence of 50 μM ACh and 30 μM NS9283 (holding potential − 70 mV, Gaussian filter 4 kHz). Red asterisks indicate the locations of mutations. To the right of each trace is a histogram of cluster durations, corresponding to successive channel openings and intervening closings, fitted by the sum of exponentials; the component with longest mean duration, present for the wild-type (b) and α4H142V mutant AChRs (f), is absent for the β2W176A mutant AChR (c–e). To the right of the cluster duration histograms are histograms of channel openings classified as either un-potentiated (un-shaded) or potentiated (shaded) fitted by the sum of three exponentials. The shaded portion of the bar above each histogram indicates the percentage of channel openings that are potentiated
We then installed the β2W176A mutation at either of the two β2–α4 subunit interfaces of the concatemer. Recordings in the presence of optimal concentrations of ACh and NS9283 show predominantly solitary channel openings flanked by long closings, and the percentage of potentiated channel openings markedly decreases; when the β2W176A mutation is present at one of the β2–α4 interfaces, the percentage of potentiated channel openings decreases to 16%, whereas when the mutation is present at the other β2–α4 interface, the percentage of potentiated channel openings decreases to 29% (Fig. 4c, d; Table 1). In addition, for each of the receptors containing a single β2W176A mutation, the fourth, prolonged component of clusters is eliminated. In a third construct containing the β2W176A mutation at both β2–α4 subunit interfaces, the percentage of potentiated channel openings decreases to 14%, and the fourth, prolonged component of clusters is eliminated (Fig. 4e; Table 1).
When a mutation is generated in only one subunit of a concatemer, the particular subunit interface to which the mutation localizes depends on whether the subunits assemble in a clockwise or anticlockwise direction. However, in constructs containing a mutant β2 subunit, an α4 subunit is present on both sides, so that regardless of the direction in which the subunits assemble, the mutation localizes to a β2–α4 interface. Thus, mutating the principal face of the β2 subunit markedly suppresses potentiation by NS9283, showing that both β2–α4 subunit interfaces are required for potentiation.
We next installed the α4H142V mutation into the concatemer at the presumed α4–α4 subunit interface, and in the presence of optimal concentrations of ACh and NS9283, observed plentiful clusters of channel openings (Fig. 4f). The percentage of channel openings classified as potentiated decreases compared to that for the concatemer comprised of wild subunits, but some 55% of channel openings remain drug potentiated (Fig. 4f, shaded bar and histogram; Table 1). In addition, a prolonged component of cluster durations is present with a mean duration similar to that for the concatemer composed of wild-type subunits. If the subunits assembled in a clockwise direction, the α4H142V mutation would be located at the α4–α4 interface, whereas, if they assembled in an anticlockwise direction, the mutation would be located at the β2–α4 interface. Nevertheless, receptors containing the α4H142V mutation still potentiate, suggesting one of two scenarios. If the mutation is at the α4–α4 interface, this interface is not required for potentiation; or if the mutation is at the β2–α4 interface, the mutated residue is not required.
Channel re-opening as a measure of drug potentiation: receptors formed from unlinked subunits
To illustrate channel re-opening as a measure of drug potentiation, Fig. 5a shows recording segments from the wild-type (α4)3(β2)2 receptor formed from unlinked subunits in the presence of either ACh alone or ACh plus NS9283. In the presence of ACh alone, one-channel opening episode comprises three successive openings and another just one opening, whereas, in the presence of ACh and NS9283, one-channel opening episode comprises 7 openings and another 17 openings. Thus, to further quantify potentiation, we plotted the fraction of channel opening episodes with greater than N openings against the number of openings per episode, in either the presence of ACh alone or in the presence of ACh and NS9283. For a recording in the presence of ACh alone, the re-opening plot decays biexponentially; the mean of the major component is 0.7 re-openings per episode, while the mean of the minor component is 2.8 re-openings per episode (Fig. 5b; Table 2). However, in the presence of ACh and NS9283, the mean of the major component is ten re-openings per episode, while the mean of the minor component is two re-openings per episode. In addition, NS9283 alters the fractional weights of the two components; the weight of the component with fewest openings per episode declines from 71 to 59%, while that of the component with greatest openings per episode increases from 29 to 41% (Table 2). Application of the F test reveals that the means and fractional weights of the two components are significantly different without versus with NS9283 (“Materials and methods”; Table 2). Because the component with a mean of ten re-openings per episode is observed only in the presence of ACh and NS9283, we conclude that it corresponds to potentiated channel openings. These determinations of channel re-opening in Fig. 5b mirror those reported previously for wild-type (α4)3(β2)2 receptors formed from unlinked subunits [14].
Fig. 5.
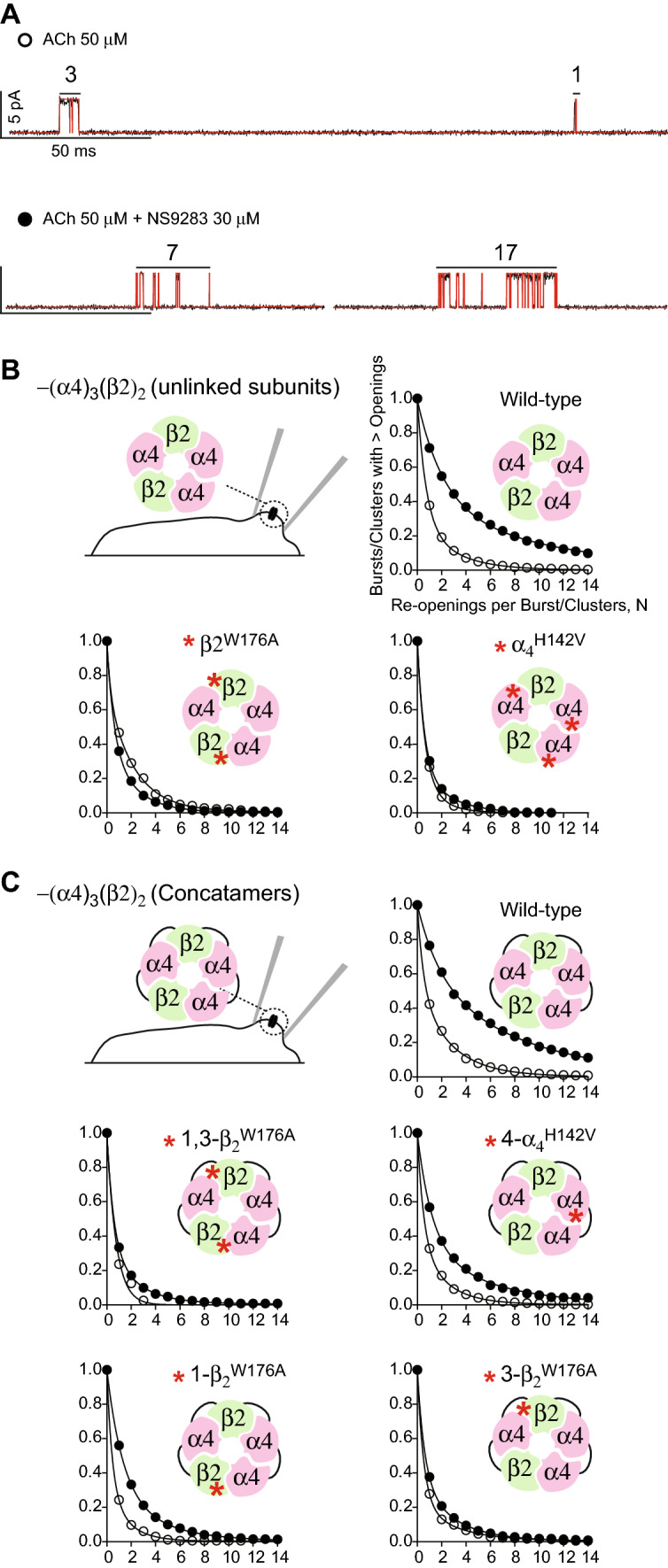
Enhanced channel re-opening by NS9283 is blocked by mutation in the β2 but not the α4 subunit. a Traces of single-channel currents and detected channel openings in the presence of 50 μM ACh (upper) or 50 μM ACh and 30 μM NS9283 (lower). Red vertical lines indicate detected channel openings. b Plots of the fraction of channel opening episodes with greater than N openings against the number of openings per episode. Data shown are for the indicated wild-type or mutant AChRs formed from unlinked subunits, in the presence of ACh (open symbols) or ACh and NS9283 (filled symbols), fitted by the sum of two exponentials. Red asterisks indicate the locations of mutations. Note that, for the wild-type AChR, NS9283 markedly enhances channel re-opening over that in the presence of ACh alone, whereas, for the mutant AChRs, channel re-opening is similar in the presence of ACh alone and ACh plus NS9283. c Plots of channel re-opening as in b for the indicated wild-type or mutant AChRs formed from linked subunits in the presence of ACh (open symbols) or ACh plus NS9283 (filled symbols) fitted by the sum of two exponentials. For the wild-type AChR formed from linked subunits, NS9283 markedly enhances channel re-opening over that in the presence of ACh alone, whereas, for AChRs containing the β2W176A mutation, at either position 3 or positions 1 and 3, channel re-opening is similar in the presence of ACh alone and ACh plus NS9283. However, for AChRs containing one α4H142V mutant subunit, channel re-opening, though reduced, is still significant
Table 2.
NS9283 promotes ACh-elicited channel re-opening
| Receptor type (number of patches) | [ACh] μM | [NS9283] μM | Best model (p < 0.01) | Mean re-opening (95% IC) [% phase (95% IC)] | Significance (p < 0.01) | |||
|---|---|---|---|---|---|---|---|---|
| First phase | Second phase | First phase | Second phase | % First phase | ||||
| (α4)3(β2)2 (n = 6) | 50 | 30 | Biphasic |
2 (1.9–2.2) [59 (56–62)] |
10 (9.6–10.6) [41 (38–44)] |
Yes | Yes | Yes |
| (α4)3(β2)2 (n = 4) | 50 | Biphasic |
0.7 (0.69–0.73) [71 (69–72)] |
2.8 (2.7–2.9) [29 (28–31)] |
||||
| (α4)3(β2W176A)2 (n = 6) | 50 | 30 | Biphasic |
0.57 (0.5–0.6) [60 (56–64)] |
2.2 (2.0–2.3) [40 (36–44)] |
Yes | n.d. | n.d. |
| (α4)3(β2W176A)2 (n = 4) | 50 | One phase |
0.63 (0.57–0.71) [1] |
|||||
| (α4H142V)3(β2)2 (n = 17) | 50 | 30 | Biphasic |
0.58 (0.5–0.6) [75 (66–81)] |
2.6 (2.0–3.6) [25 (19–34)] |
No | No | No |
| (α4H142V)3(β2)2 (n = 4) | 50 | Biphasic |
0.58 (0.56–0.6) [75 (72–78)] |
1.6 (1.5–1.7) [25 (22–28)] |
||||
| β2α4β2α4α4 (n = 6) | 50 | 30 | Biphasic |
1.7 (1.5–1.9) [37 (34–40)] |
7.7 (7.4–8.0) [63 (60–66)] |
Yes | Yes | Yes |
| β2α4β2α4α4 (n = 5) | 50 | Biphasic |
0.5 (0.4–0.6) [47 (43–52)] |
2.7 (2.5–2.8) [53 (48–57)] |
||||
| β2W176Aα4β2α4α4 (n = 6) | 50 | 30 | Biphasic |
1.3 (1.2–1.4) [70 (64–75)] |
3.9 (3.5–4.4) [30 (25–36)] |
Yes | Yes | No |
| β2W176Aα4β2α4α4 (n = 4) | 50 | Biphasic |
0.4 (0.2–0.5) [67 (46–82)] |
1.6 (1.2–2.4) [33 (18–54)] |
||||
| β2α4β2W176Aα4α4 (n = 8) | 50 | 30 | Biphasic |
0.58 (0.52–0.64) [63 (58–68)] |
2.9 (2.5–3.2) [37 (32–42)] |
No | No | Yes |
| β2α4β2W176Aα4α4 (n = 6) | 50 | Biphasic |
0.5 (0.45–0.57) [77 (72–82)] |
3 (2.5–3.7) [23 (18–28)] |
||||
| β2W176Aα4β2W176Aα4α4 (n = 10) | 50 | 30 | Biphasic |
0.55 (0.5–0.6) [65 (60–70)] |
2.3 (2.1–2.5) [35 (30–40)] |
No | n.d. | n.d. |
| β2W176Aα4β2W176Aα4α4 (n = 7) | 50 | One phase |
0.76 (0.4–1.2) [1] |
|||||
| β2α4β2α4H142Vα4 (n = 8) | 50 | 30 | Biphasic |
1.2 (1.0–1.3) [63 (57–69)] |
5.4 (4.7–6.2) [37 (43–31)] |
Yes | Yes | No |
| β2α4β2α4H142Vα4 (n = 9) | 50 | Biphasic |
0.5 (0.4–0.6) [62 (56–68)] |
2.2 (1.9–2.4) [38 (32–44)] |
||||
Mean number of re-openings and the relative percentage of the corresponding phase are given along with the 95% confidence interval (95% IC). An F test was used to determine whether a monophasic or biphasic model best described the data, and to determine whether the fitted decay rates and fractional areas differed significantly without versus with NS9283. The table summarizes results displayed in Fig. 5
For receptors containing either the α4H142V or β2W176A mutations in the presence of ACh alone, channel re-opening is similar to that observed for the wild-type receptor (Fig. 5b). However, for the α4H142V mutant receptor in the presence of ACh and NS9283, channel re-opening is similar to that in the presence of ACh alone, and statistical analyses show that the fitted parameters do not differ without or with NS9283 (Table 2). Similarly, for the β2W176A mutant receptor in the presence of ACh and NS9283, channel re-opening does not increase relative to that in the presence ACh alone; in fact, channel re-opening is modestly reduced in the presence of NS9283 (Table 2). These results, obtained for receptors formed from unlinked subunits, confirm that a mutation in either the α4 or the β2 subunit suppresses potentiation. However, because the receptors are formed from unlinked subunits, each mutation is present at multiple subunit interfaces per receptor.
Channel re-opening as a measure of drug potentiation: receptors formed from linked subunits
In the presence of ACh alone, the pentameric (α4)3(β2)2 concatemer composed of wild-type subunits exhibits two exponential components of channel re-opening with means of 0.5 and 2.7 re-openings per episode (Fig. 5c; Table 2), similar to that observed for wild-type receptors formed from unlinked subunits (Fig. 5b). In the presence of ACh and NS9283, the two components show means of 1.7 and 7.7 re-openings per opening episode, which again are similar to those observed for wild-type receptors formed from unlinked subunits (Table 2). Statistical comparison of the re-opening plots, without versus with NS9283, reveals that the decay rates and relative weights of the two components differ significantly with p < 0.01 (Table 2), as observed for receptors formed from unlinked subunits. For the concatemer with the β2W176A mutation at either of the β2–α4 subunit interfaces, channel re-opening in the presence of NS9283 approaches that in the presence of ACh alone, but this depends on the subunit interface that contains the mutation. When the β2W176A mutation is present at one of the β2–α4 interfaces, the major and minor components show means of 0.6 and 2.9, which do not differ significantly from those in the presence of ACh alone (Table 2), indicating essentially complete suppression of potentiation. On the other hand, when the mutation is at the other β2–α4 interface, the means are 1.3 and 3.9 re-openings per episode (Table 2), which differ significantly from that in the presence of ACh alone, so that although potentiation is reduced it remains significant. Furthermore, when the β2W176A is present at both β2–α4 subunit interfaces, re-opening is reduced further, with means of 0.6 and 2.3 re-openings per episode for the major and minor components; however, statistical comparison of the fitted parameters is precluded, because in the presence of ACh alone, re-opening is described by a single-exponential decay, whereas, in the presence of ACh and NS9283, re-opening is described as a double exponential decay (Table 2). By contrast, for the concatemer containing the α4H142V mutation at the presumed α4–α4 subunit interface, NS9283 still increases channel re-opening, although the extent of the increase is reduced compared to that of the wild-type concatemer; the major and minor components show means of 1.2 and 5.4 re-openings per episode, which differ significantly from that in the presence of ACh alone (Table 2). Thus, as measured by channel re-opening, the β2W176A mutation suppresses potentiation to a greater extent than the α4H142V mutation.
Discussion
The diversity of nicotinic AChR subunits, together with their ability to assemble in different combinations, enables a wide variety of functional and pharmacological signatures. A major challenge in the field is to understand how the combination and stoichiometry of subunits endow a receptor with its signature function and pharmacology. This understanding is required not only to understand receptor-mediated neuronal signaling, but also to design therapeutic drugs to target a particular signaling pathway. It is the overall context that frames the present study.
Previous studies showed that the drug NS9283 selectively potentiates heteromeric nicotinic AChRs containing either α2 or α4 subunits, but not α3 or α7 subunits [51]. Studies in laboratory animals showed that NS9283 enhances cognitive function, and the enhancement was blocked by the cholinergic antagonist mecamylamine [51]. NS9283 also attenuated nicotine self-administration and reinstatement, and it had no effect when sucrose was substituted for nicotine [38]. Cholinergic agonists such as nicotine and epibatidine are potent analgesics, but they suffer from side effects. However, administration of the synthetic nicotinic agonist ABT 595 together with NS9283 enhanced the potency of this agonist against nociception [39], providing a rationale for combination therapy. Owing to their selective ability to target AChRs with particular subunits, potentiators such as NS9283 are promising drugs to treat cognitive impairment, nicotine dependence, and nociception. Thus, defining molecular targets of AChR potentiators are essential steps toward developing more potent and target-selective drugs.
Seminal studies showed that when α4 and β2 subunits were co-expressed, the agonist dose–response relationship, based on recordings of voltage-clamped macroscopic current, exhibited high- and low-sensitivity components [12, 13, 15, 40]. The high-sensitivity component was shown to arise from receptors with two α4 and three β2 subunits, whereas the low-sensitivity component was shown to arise from receptors with three α4 and two β2 subunits [13, 17]. In addition, receptors with low sensitivity exhibited robust potentiation by the drug NS9283, whereas receptors with high agonist sensitivity were unaffected by the drug [14, 16, 35]. Thus, for a receptor with composition (α4)3(β2)2, a third α4 subunit in place of a β2 subunit endowed drug sensitivity. Insight into this stoichiometry-specific drug sensitivity emerged from mutagenesis studies, showing that mutations at the complementary face of the α4 subunit abolished potentiation [35–37]. An appealing interpretation was that NS9283 bound to the α4–α4 subunit interface, and the mutation prevented drug-binding and thus blocked potentiation. Thus, a co-agonist mechanism emerged in which the agonist binds to the two α4–β2 interfaces, and potentiation resulted from binding of NS9283 to the α4–α4 interface [36]. However, an equally plausible interpretation is that the agonist binds to the α4–β2 and α4–α4 interfaces, while NS9283 binds to the β2–α4 interfaces. The present work distinguishes between these two interpretations, showing that, although a third α4 subunit is necessary for NS9283 potentiation, a mutation at the presumed α4–α4 interface still permits potentiation. By contrast, a mutation at either of the two β2–α4 interfaces markedly curtails potentiation. Because the agonist does not bind to the β2–α4 interface, as shown by the recent crystal and cryo-EM structures of the α4β2 receptor with bound nicotine [41, 42], NS9283 could bind to the β2–α4 interfaces and potentiate through a mechanism analogous to benzodiazepine potentiation of GABAA receptors [43–46].
The β2–α4 subunit interface harbors four of the five conserved aromatic residues present at the α4–β2 subunit interface that forms the orthosteric ligand-binding site. However, the β2–α4 interface also harbors an Arg residue, analogous to a bound agonist, and thus may be considered a pseudo-agonist site [41, 42]. For receptors formed from unlinked subunits, when the mutant α4H142V subunit is co-expressed with the β2 subunit, the mutation is present at the α4–α4 as well as the two β2–α4 interfaces. By contrast, when the mutant β2W176A subunit is co-expressed with the α4 subunit, the mutation is present only at the β2–α4 interfaces. Because both the β2W176A and α4H142V mutations markedly curtail potentiation in receptors formed from unlinked subunits, a possible interpretation is that the subunit interface common to both mutations, β2–α4, is required for NS9283 potentiation. To test this possibility, we studied receptors formed from concatemeric subunits.
For a receptor comprised of five covalently linked subunits, there is the issue of whether the subunits assemble in a clockwise or counter-clockwise direction. A recent study based on the ability of NS9283 to potentiate concatemeric receptors with mutations in individual subunits suggested the subunits assembled in a counter-clockwise direction [47]. On the other hand, studies of the effects of single-residue mutations [18, 48–50], and the ability of agonists to protect against covalent reaction of a methanethiosulfonate reagent with a substituted cysteine [28], suggested the subunits assembled in a clockwise direction. Thus, to interpret our findings, we consider both scenarios of subunit assembly. In a β2–α4–β2–α4–α4 concatemer, if the subunits assemble clockwise, a mutation at the complementary face of the penultimate α4 subunit will be located at the α4–α4 interface, whereas if they assemble counter-clockwise, the mutation will be located at a β2–α4 interface. The two possible locations of the mutation arise because the mutant α4 subunit is flanked by a β2 subunit on one side and an α4 subunit on the other side. Conversely, a mutation at the principal face of a β2 subunit will always be located at a β2–α4 interface because each β2 subunit is flanked by two α4 subunits. Thus, our conclusion that the β2–α4 interface is required for potentiation is independent of the direction of subunit assembly.
Our studies of concatemeric receptors show that each of the β2–α4 subunit interfaces is required for potentiation, and that the contribution of each interface to potentiation is not equivalent. This functional asymmetry may originate from differences in the subunits that flank the β2–α4 interfaces, as suggested in previous work in which mutations at nominally equivalent α4–β2 subunit interfaces had different functional consequences [25, 49]. For the β2–α4 interface flanked by α4 and β2 subunits, the β2W176A mutation reduces the percentage of channel openings that are potentiated from 81 to 29. By contrast, for the β2–α4 interface flanked by two α4 subunits, the β2W176A mutation reduces the percentage of openings that are potentiated from 81 to 16. When the β2W176A mutation is present at both β2–α4 interfaces, the percentage of potentiated channel openings is reduced to 14, close to the reduction to 10% observed when the β2W176A mutation is incorporated into receptors formed from unlinked subunits. In these three mutant receptors, the residual potentiation may arise from an incomplete effect of mutating only one residue at the β2–α4 interface.
We also find that the mutation α4H142V prevents potentiation in receptors formed from unlinked subunits; in these receptors, the mutation is present at each of the β2–α4 interfaces, as well as the α4–α4 interface. However, when the α4H142V mutation is incorporated into the presumed α4–α4 interface of a concatemeric receptor, the percentage of channel openings that is potentiated is still 55%. In addition, two other measures of NS9283 potentiation are preserved; the exponential component of clusters with prolonged mean duration remains, and channel re-opening is increased relative to that in the presence of ACh alone. Further studies are required to determine whether the α4H142V mutation is located at the α4–α4 versus the β2–α4 interface. By contrast, our results show that potentiation is essentially eliminated when the β2W176A mutation is present at the β2–α4 interface.
Although our results demonstrate that NS9283 potentiation requires the β2–α4 interface, each receptor stoichiometry, (α4)3(β2)2 and (α4)2(β2)3, contains two copies of this interface. The major difference between the two stoichiometries is an α4 subunit in place of a β2 subunit. This exchange of a single subunit creates two subunit interfaces that differ between each stoichiometry: α4–α4 and α4–β2 interfaces in the (α4)3(β2)2 stoichiometry, versus α4–β2 and β2–β2 interfaces in the (α4)2(β2)3 stoichiometry. Thus, the contributions of the β2–α4 interfaces to NS9283 potentiation may depend on subunit interfaces novel to the (α4)3(β2)2 stoichiometry.
Studies of potentiation at the single-channel level provide novel insights into the underlying mechanism. We find that the open-channel lifetime contains three exponential components, indicating three stable open states, in both the presence and absence of NS9283. Thus, potentiation does not affect transitions from open to closed states. On the other hand, potentiation increases the probability a channel that just closed will re-open, as well as the speed with which it re-opens. Moreover, by distinguishing potentiated from un-potentiated channel openings, our single-channel measurements provide a quantitative measure of potentiation. In addition, potentiation is assessed from an increase in the mean duration of clusters of channel openings, as well as an increase in the number of channel re-openings per opening episode. Thus, by contrast to measurements of macroscopic currents, measurements of single-channel currents not only distinguish between un-potentiated and potentiated receptor channel openings, but they also provide several quantitative measures of potentiation.
On the other hand, whether NS9283 affects slower processes such as desensitization or deactivation is best assessed through the measurements of macroscopic currents. In fact, measurements of the time courses of desensitization onset and recovery show that NS9283 does not affect onset, and promotes only a modest slowing of recovery [16]. However, measurements of deactivation following a brief pulse of agonist reveal that NS9283 slows deactivation [16]. In accord with a slowing of deactivation, our results show that in the presence of ACh alone, the receptor channels open primarily as single isolated openings, whereas, in the presence of NS9283, they open in clusters of many successive openings. Thus, the overall findings from macroscopic and single-channel current measurements suggest NS9283 stabilizes activatable receptor states.
Our results suggest NS9283 potentiates (α4)3(β2)2 receptors through binding to the β2–α4 subunit interfaces. In support, W176 from the principal face of the β2 subunit contacts H142 from the complementary face of the α4 subunit [42], and together the two residues could contribute to an interfacial drug-binding site. In addition, NS9283 did not compete against [3H]-cytisine binding to rat cortical tissue [51], a region rich in (α4)3(β2)2 receptors [40], suggesting it does not bind to the orthosteric sites. Nevertheless, the β2–α4 subunit interfaces might not be the binding sites for NS9283, but may instead serve as transduction elements for potentiation. Among the family of nicotinic receptors, and the larger family of pentameric ligand-gated channels, sites for drug modulation have been identified in a variety of locations: the extracellular domain [35, 43, 45, 52–54], transmembrane domain [55–57], and the junction between the two domains [35]. Atomic scale structures of drug–receptor complexes will be required to distinguish whether the focal determinants we have identified mediate drug-binding or transduction of drug-binding to potentiation.
Acknowledgements
This research was supported by NIH Grant NS31744 to SMS.
Abbreviations
- nAChR
Nicotinic acetylcholine receptor
- ACh
Acetylcholine
Author contributions
SMS, SM, and IB conceived and coordinated the research; SM performed the research; SM and SMS analyzed the results; SMS and SM wrote the paper. All authors edited and approved the final version.
Compliance with ethical standards
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Millar NS, Gotti C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology. 2009;56:237–246. doi: 10.1016/j.neuropharm.2008.07.041. [DOI] [PubMed] [Google Scholar]
- 2.Albuquerque EX, Pereira EFR, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hurst R, Rollema H, Bertrand D. Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol Ther. 2013;137:22–54. doi: 10.1016/j.pharmthera.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 4.Taly A, Corringer P-J, Guedin D, Lestage P, Changeux J-P. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov. 2009;8:733–750. doi: 10.1038/nrd2927. [DOI] [PubMed] [Google Scholar]
- 5.Laviolette SR, van der Kooy D. The neurobiology of nicotine addiction: bridging the gap from molecules to behaviour. Nat Rev Neurosci. 2004;5:55–65. doi: 10.1038/nrn1298. [DOI] [PubMed] [Google Scholar]
- 6.Picciotto MR, Kenny PJ. Molecular mechanisms underlying behaviors related to nicotine addiction. Cold Spring Harb Perspect Med. 2013;3:a012112. doi: 10.1101/cshperspect.a012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 8.Marubio LM, del Mar Arroyo-Jimenez M, Cordero-Erausquin M, Lena C, Le Novere N, de Kerchove d’Exaerde A, Huchet M, Damaj MI, Changeux JP. Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature. 1999;398:805–810. doi: 10.1038/19756. [DOI] [PubMed] [Google Scholar]
- 9.Picciotto MR, Zoli M, Léna C, Bessis A, Lallemand Y, Le Novère N, Vincent P, Pich EM, Brûlet P, Changeux JP. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature. 1995;374:65–67. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]
- 10.Ross SA, Wong JY, Clifford JJ, Kinsella A, Massalas JS, Horne MK, Scheffer IE, Kola I, Waddington JL, Berkovic SF, Drago J. Phenotypic characterization of an alpha 4 neuronal nicotinic acetylcholine receptor subunit knock-out mouse. J Neurosci. 2000;20:6431–6441. doi: 10.1523/JNEUROSCI.20-17-06431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zoli M, Léna C, Picciotto MR, Changeux JP. Identification of four classes of brain nicotinic receptors using beta2 mutant mice. J Neurosci. 1998;18:4461–4472. doi: 10.1523/JNEUROSCI.18-12-04461.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zwart R, Vijverberg HP. Four pharmacologically distinct subtypes of alpha4beta2 nicotinic acetylcholine receptor expressed in Xenopus laevis oocytes. Mol Pharmacol. 1998;54:1124–1131. doi: 10.1124/mol.54.6.1124. [DOI] [PubMed] [Google Scholar]
- 13.Moroni M, Zwart R, Sher E, Cassels BK, Bermudez I. alpha4beta2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol Pharmacol. 2006;70:755–768. doi: 10.1124/mol.106.023044. [DOI] [PubMed] [Google Scholar]
- 14.Mazzaferro S, Bermudez I, Sine SM. α4β2 nicotinic acetylcholine receptors: relationships between subunit stoichiometry and function at the single channel level. J Biol Chem. 2017;292:2729–2740. doi: 10.1074/jbc.M116.764183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson ME, Kuryatov A, Choi CH, Zhou Y, Lindstrom J. Alternate stoichiometries of alpha4beta2 nicotinic acetylcholine receptors. Mol Pharmacol. 2003;63:332–341. doi: 10.1124/mol.63.2.332. [DOI] [PubMed] [Google Scholar]
- 16.Grupe M, Jensen AA, Ahring PK, Christensen JK, Grunnet M. Unravelling the mechanism of action of NS9283, a positive allosteric modulator of (α4)3(β2)2 nicotinic ACh receptors. Br J Pharmacol. 2013;168:2000–2010. doi: 10.1111/bph.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carbone A-L, Moroni M, Groot-Kormelink P-J, Bermudez I. Pentameric concatenated (alpha4)(2)(beta2)(3) and (alpha4)(3)(beta2)(2) nicotinic acetylcholine receptors: subunit arrangement determines functional expression. Br J Pharmacol. 2009;156:970–981. doi: 10.1111/j.1476-5381.2008.00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mazzaferro S, Benallegue N, Carbone A, Gasparri F, Vijayan R, Biggin PC, Moroni M, Bermudez I. Additional acetylcholine (ACh) binding site at α4/α4 interface of (α4β2)2α4 nicotinic receptor influences agonist sensitivity. J Biol Chem. 2011;286:31043–31054. doi: 10.1074/jbc.M111.262014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeanclos EM, Lin L, Treuil MW, Rao J, DeCoster MA, Anand R. The chaperone protein 14-3-3η interacts with the nicotinic acetylcholine receptor α4 subunit. J Biol Chem. 2001;276:28281–28290. doi: 10.1074/jbc.M011549200. [DOI] [PubMed] [Google Scholar]
- 20.Exley R, Moroni M, Sasdelli F, Houlihan LM, Lukas RJ, Sher E, Zwart R, Bermudez I. Chaperone protein 14-3-3 and protein kinase a increase the relative abundance of low agonist sensitivity human α4β2 nicotinic acetylcholine receptors in Xenopus oocytes. J Neurochem. 2006;98:876–885. doi: 10.1111/j.1471-4159.2006.03915.x. [DOI] [PubMed] [Google Scholar]
- 21.Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sine SM. Molecular dissection of subunit interfaces in the acetylcholine receptor: identification of residues that determine curare selectivity. Proc Natl Acad Sci USA. 1993;90:9436–9440. doi: 10.1073/pnas.90.20.9436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sine SM, Quiram P, Papanikolaou F, Kreienkamp HJ, Taylor P. Conserved tyrosines in the alpha subunit of the nicotinic acetylcholine receptor stabilize quaternary ammonium groups of agonists and curariform antagonists. J Biol Chem. 1994;269:8808–8816. [PubMed] [Google Scholar]
- 24.Bouzat C, Bren N, Sine SM. Structural basis of the different gating kinetics of fetal and adult acetylcholine receptors. Neuron. 1994;13:1395–1402. doi: 10.1016/0896-6273(94)90424-3. [DOI] [PubMed] [Google Scholar]
- 25.New K, Del Villar SG, Mazzaferro S, Alcaino C, Bermudez I. The fifth subunit of the (α4β2)2 β2 nicotinic ACh receptor modulates maximal ACh responses. Br J Pharmacol. 2018;175:1822–1837. doi: 10.1111/bph.13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooper ST, Harkness PC, Baker ER, Millar NS. Up-regulation of cell-surface α4β2 neuronal nicotinic receptors by lower temperature and expression of chimeric subunits. J Biol Chem. 1999;274:27145–27152. doi: 10.1074/jbc.274.38.27145. [DOI] [PubMed] [Google Scholar]
- 27.Moroni M, Vijayan R, Carbone A, Zwart R, Biggin PC, Bermudez I. Non-agonist-binding subunit interfaces confer distinct functional signatures to the alternate stoichiometries of the alpha4beta2 nicotinic receptor: an alpha4–alpha4 interface is required for Zn2+ potentiation. J Neurosci. 2008;28:6884–6894. doi: 10.1523/JNEUROSCI.1228-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mazzaferro S, Gasparri F, New K, Alcaino C, Faundez M, Vasquez PI, Vijayan R, Biggin PC, Bermudez I. Non-equivalent ligand selectivity of agonist sites in (α4β2)2α4 nicotinic acetylcholine receptors: a key determinant of agonist efficacy. J Biol Chem. 2014;289:21795–21806. doi: 10.1074/jbc.M114.555136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rayes D, Spitzmaul G, Sine SM, Bouzat C. Single-channel kinetic analysis of chimeric alpha7-5HT3A receptors. Mol Pharmacol. 2005;68:1475–1483. doi: 10.1124/mol.105.015438. [DOI] [PubMed] [Google Scholar]
- 30.Bouzat C, Bartos M, Corradi J, Sine SM. The interface between extracellular and transmembrane domains of homomeric Cys-loop receptors governs open-channel lifetime and rate of desensitization. J Neurosci. 2008;28:7808–7819. doi: 10.1523/JNEUROSCI.0448-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sine SM, Claudio T, Sigworth FJ. Activation of Torpedo acetylcholine receptors expressed in mouse fibroblasts. Single channel current kinetics reveal distinct agonist binding affinities. J Gen Physiol. 1990;96:395–437. doi: 10.1085/jgp.96.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mukhtasimova N, DaCosta CJB, Sine SM. Improved resolution of single channel dwell times reveals mechanisms of binding, priming, and gating in muscle AChR. J Gen Physiol. 2016;148:43–63. doi: 10.1085/jgp.201611584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colquhoun D, Sigworth FL. Single channel recording. US: Springer; 1983. Fitting and statistical analysis of single channel records; pp. 483–587. [Google Scholar]
- 34.Sigworth FJ, Sine SM. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J. 1987;52:1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olsen JA, Kastrup JS, Peters D, Gajhede M, Balle T, Ahring PK. Two distinct allosteric binding sites at α4β2 nicotinic acetylcholine receptors revealed by NS206 and NS9283 give unique insights to binding activity-associated linkage at Cys-loop receptors. J Biol Chem. 2013;288:35997–36006. doi: 10.1074/jbc.M113.498618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olsen JA, Ahring PK, Kastrup JS, Gajhede M, Balle T. Structural and functional studies of the modulator NS9283 reveal agonist-like mechanism of action at α4β2 nicotinic acetylcholine receptors. J Biol Chem. 2014;289:24911–24921. doi: 10.1074/jbc.M114.568097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z-J, Deba F, Mohamed TS, Chiara DC, Ramos K, Hamouda AK. Unraveling amino acid residues critical for allosteric potentiation of (α4)3(β2)2-type nicotinic acetylcholine receptor responses. J Biol Chem. 2017;292:9988–10001. doi: 10.1074/jbc.M116.771246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maurer JJ, Sandager-Nielsen K, Schmidt HD. Attenuation of nicotine taking and seeking in rats by the stoichiometry-selective alpha4beta2 nicotinic acetylcholine receptor positive allosteric modulator NS9283. Psychopharmacology. 2017;234:475–484. doi: 10.1007/s00213-016-4475-7. [DOI] [PubMed] [Google Scholar]
- 39.Zhu CZ, Chin CL, Rustay NR, Zhong C, Mikusa J, Chandran P, Salyers A, Gomez E, Simler G, Lewis LG, Gauvin D, Baker S, Pai M, Tovcimak A, Brown J, Komater V, Fox GB, Decker MW, Jacobson PB, Gopalakrishnan M, Lee CH, Honore P (2011) Potentiation of analgesic efficacy but not side effects: co-administration of an α4β2 neuronal nicotinic acetylcholine receptor agonist and its positive allosteric modulator in experimental models of pain in rats. In: Biochemical pharmacology, pp 967–976 [DOI] [PubMed]
- 40.DeDominicis KE, Sahibzada N, Olson TT, Xiao Y, Wolfe BB, Kellar KJ, Yasuda RP. The (α4)3 (β2)2 stoichiometry of the nicotinic acetylcholine receptor predominates in the rat motor cortex. Mol Pharmacol. 2017;92:327–337. doi: 10.1124/mol.116.106880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morales-Perez CL, Noviello CM, Hibbs RE. X-ray structure of the human α4β2 nicotinic receptor. Nature. 2016;538:411–415. doi: 10.1038/nature19785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walsh RM, Roh SH, Gharpure A, Morales-Perez CL, Teng J, Hibbs RE. Structural principles of distinct assemblies of the human α4β2 nicotinic receptor. Nature. 2018;557:261–265. doi: 10.1038/s41586-018-0081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sigel E, Buhr A. The benzodiazepine binding site of GABA(A) receptors. Trends Pharmacol Sci. 1997;18:425–429. doi: 10.1016/S0165-6147(97)01118-8. [DOI] [PubMed] [Google Scholar]
- 44.Sigel E, Steinmann ME. Structure, function, and modulation of GABAA receptors. J Biol Chem. 2012;287:40224–40231. doi: 10.1074/jbc.R112.386664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sigel EP, Luscher B. A closer look at the high affinity benzodiazepine binding site on GABAA receptors. Curr Top Med Chem. 2011;11:241–246. doi: 10.2174/156802611794863562. [DOI] [PubMed] [Google Scholar]
- 46.Zhu S, Noviello CM, Teng J, Walsh RM, Kim JJ, Hibbs RE. Structure of a human synaptic GABAA receptor. Nature. 2018;512:270. doi: 10.1038/s41586-018-0255-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahring PK, Liao VWY, Balle T. Concatenated nicotinic acetylcholine receptors: a gift or a curse? J Gen Physiol. 2018;150:453–473. doi: 10.1085/jgp.201711846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Benallegue N, Mazzaferro S, Alcaino C, Bermudez I. The additional ACh binding site at the α4(+)/α4(−) interface of the (α4β2)2α4 nicotinic ACh receptor contributes to desensitization. Br J Pharmacol. 2013;170:304–316. doi: 10.1111/bph.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lucero LM, Weltzin MM, Eaton JB, Cooper JF, Lindstrom JM, Lukas RJ, Whiteaker P. Differential α4(+)/(−)β2 agonist-binding site contributions to α4β2 nicotinic acetylcholine receptor function within and between isoforms. J Biol Chem. 2016;291:2444–2459. doi: 10.1074/jbc.M115.684373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eaton JB, Lucero LM, Stratton H, Chang Y, Cooper JF, Lindstrom JM, Lukas RJ, Whiteaker P. The unique α4(+)/(−) α4 agonist binding site in (α4)3(β2)2 subtype nicotinic acetylcholine receptors permits differential agonist desensitization pharmacology versus the (4)2(2)3 subtype. J Pharmacol Exp Ther. 2013;348:46–58. doi: 10.1124/jpet.113.208389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Timmermann DB, Sandager-Nielsen K, Dyhring T, Smith M, Jacobsen AM, Nielsen E, Grunnet M, Christensen JK, Peters D, Kohlhaas K, Olsen GM, Ahring PK. Augmentation of cognitive function by NS9283, a stoichiometry-dependent positive allosteric modulator of α2- and α4-containing nicotinic acetylcholine receptors. Br J Pharmacol. 2012;167:164–182. doi: 10.1111/j.1476-5381.2012.01989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seo S, Henry JT, Lewis AH, Wang N, Levandoski MM. The positive allosteric modulator morantel binds at noncanonical subunit interfaces of neuronal nicotinic acetylcholine receptors. J Neurosci. 2009;29:8734–8742. doi: 10.1523/JNEUROSCI.1859-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cesa LC, Higgins CA, Sando SR, Kuo DW, Levandoski MM. Specificity determinants of allosteric modulation in the neuronal nicotinic acetylcholine receptor: a fine line between inhibition and potentiation. Mol Pharmacol. 2012;81:239–249. doi: 10.1124/mol.111.076059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weltzin MM, Schulte MK. Desformylflustrabromine modulates α4β2 neuronal nicotinic acetylcholine receptor high- and low-sensitivity isoforms at allosteric clefts containing the β2 subunit. J Pharmacol Exp Ther. 2015;354:184–194. doi: 10.1124/jpet.115.223933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dacosta CJB, Sine SM. Stoichiometry for drug potentiation of a pentameric ion channel. Proc Natl Acad Sci USA. 2013;110:6595–6600. doi: 10.1073/pnas.1301909110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Young GT, Zwart R, Walker AS, Sher E, Millar NS. Potentiation of alpha7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc Natl Acad Sci USA. 2008;105:14686–14691. doi: 10.1073/pnas.0804372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alcaino C, Musgaard M, Minguez T, Mazzaferro S, Faundez M, Iturriaga-Vasquez P, Biggin PC, Bermudez I. Role of the cys loop and transmembrane domain in the allosteric modulation of α4β2 nicotinic acetylcholine receptors. J Biol Chem. 2017;292:551–562. doi: 10.1074/jbc.M116.751206. [DOI] [PMC free article] [PubMed] [Google Scholar]