

Abstract
During the first half of the viral life cycle, HIV-1 reverse transcribes its RNA genome and integrates the double-stranded DNA copy into a host cell chromosome. Despite progress in characterizing and inhibiting these processes, in situ mechanistic and structural studies remain challenging. This is because these operations are executed by individual viral preintegration complexes deep within cells. We therefore reconstituted and imaged the early stages of HIV-1 replication in a cell-free system. HIV-1 cores released from permeabilized virions supported efficient, capsid-dependent endogenous reverse transcription to produce double-stranded DNA genomes, which looped out from ruptured capsid walls. Concerted integration of both viral DNA ends into a target plasmid then proceeded in a cell extract-dependent reaction. This reconstituted system uncovers the role of the capsid in templating replication.
Graphical Abstract
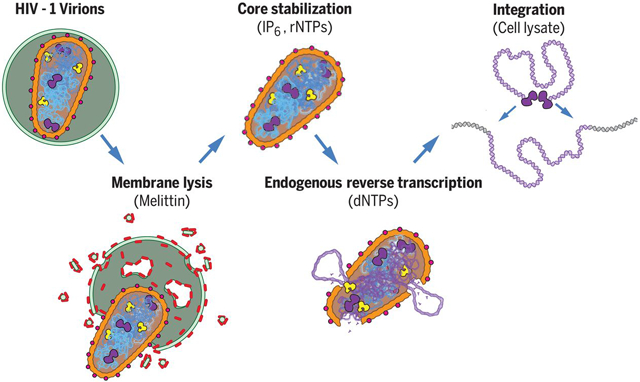
One Sentence Summary:
We report the reconstitution and electron cryotomographic imaging of HIV-1 cores performing concerted reverse transcription and integration in a cell-free system.
Reverse transcription and integration are the signature events of retrovirus replication. Reverse transcription creates a double-stranded DNA copy of the positive-sense viral RNA genome, and integration archives that copy within the genome of the infected cell, thereby confounding viral eradication. Both processes are targets of successful HIV-1 anti-retroviral therapies (1-3), and the associated enzymatic activities have been characterized by elegant structural, biochemical, and molecular virological analyses (4-6). Nevertheless, mechanistic studies of the native processes remain challenging because viral core particles perform their key transformations deep within the cell cytoplasm and nucleus. In principle, informative mechanistic and imaging analyses could be performed in vitro, but the coupled processes of reverse transcription and concerted integration have not yet been recapitulated outside of the cell. To address this limitation, we reconstituted these processes in a cell-free system, using purified HIV-1 virions as the source of viral genomes and enzymes.
Optimized Endogenous Reverse Transcription
HIV-1 infections begin when the viral Envelope (Env) protein mediates fusion of the viral and target cell membranes, thereby releasing the viral core particle into the cytoplasm. Core particles contain conical outer capsids comprising ~1500 CA protein subunits, arranged in a fullerene cone geometry composed of ~240 CA hexamers and 12 CA pentamers (7, 8). The capsid encloses the factors required for replication, including the viral RNA genome (2 copies/virion), NC protein (nucleocapsid, ~3000 copies), RT (reverse transcriptase, ~150 copies) and IN (integrase, ~150 copies) (9-11). Endogenous reverse transcription (ERT) of the viral RNA genome can initiate from a packaged host tRNALys,3 primer when the viral membrane is permeabilized and dNTPs are added (12). We used this system as a starting point for optimizing ERT efficiency, using HIV-1SG3 virions that lacked the viral Env protein but were otherwise native and fully infectious when pseudotyped with viral envelope proteins (13-15). The viral membrane can be permeabilized without detergent using the pore-forming melittin peptide from bee venom (12, 16), and concentrations of melittin, buffer, and salt were optimized empirically (see Materials and Methods). Polyphosphorylated small molecules, including nucleotide triphosphates (rNTPs and dNTPs) and inositol hexakisphosphate (IP6), can bind and stabilize the hexameric CA building blocks of the viral capsid and increase ERT efficiency (17-19). We thus also included IP6 at native cellular concentrations (40 μM) (20), and rNTPs and dNTPs at concentrations found in CD4+ T cell cytoplasms (21, 22). We also tested the effect of adding cell extracts, which have been reported to enhance ERT (23).
Successive stages of reverse transcription were monitored by quantitiative PCR (qPCR) using primers that detected: 1) an early initiation product that preceded minus strand strong stop DNA formation (MSSS, see Fig. 1A), 2) an intermediate minus strand product that required first strand transfer (FST), or 3) a late product that required second strand transfer (SST) and initiation of dsDNA synthesis. At early time points, each input core could support formation of two single-stranded (MSSS) and intermediate (FST) transcripts because there are two strands of viral RNA per core. At later time points, qPCR measurements of these two transcripts could rise as high as four per input core because later replication steps create complementary DNA strands and also introduce second copies of the LTR binding sites for the qPCR primers used to detect these transcripts (Fig. 1A). A double-stranded version of the qPCR primer binding site used to detect late (SST) products is only made once, however, and we presume that by this point in the replication cycle there will be fewer than two viral templates/core owing to strand switching (24). ERT efficiencies were therefore calculated using qPCR to quantify late (SST) ERT products, assuming a theoretical yield of 1 late product per core, and estimating input viral core numbers by quantifying virion CA levels and assuming an average of 3000 CA molecules per virion and 1.33 cores/virion (9, 11, 25).
Fig. 1. Endogenous Reverse Transcription.

(A) Different steps in HIV-1 reverse transcription, and primer positions used to assess them. (B) Time course showing accumulation of early, intermediate, and late ERT products, as quantified by qPCR. Note that synthesis of the viral plus strand cDNA introduces second copies of the binding sites for the primers used to detect early (MSSS) and intermediate (FST), which accounts for the apparent increase in signal for those two products seen between 4 and 6 hours. (C) ERT product accumulation under different reaction conditions (10 h, 37C incubations except where noted, and see Materials and Methods for other details). The RT inhibitor efavirenz (10 μM) and an inactive RT mutant enzyme (D185A) were used as negative controls. p values are from a one-way ANOVA test with Tukey’s multiple comparisons test: p<0.01: **, p<0.001: ***, p<0.0001: ****. Graphs and error bars show mean ± SD from three qPCR measurements from a representative experiment, selected from three independent experiments.
ERT products increased with time, and appeared in a temporal order that matched the well-characterized stages and timing of HIV replication in cells (26), with early reverse transcripts appearing rapidly (0.25-1 h), intermediate FST products appearing after a short delay, and late reverse transcripts beginning to accumulate after a 3 h lag and reaching a maximum after ~8-10 h (Fig. 1B). ERT was reduced to background levels when the incubation was performed in the presence of the RT inhibitor efavirenz, or when HIV-1 virions contained RT enzymes with the inactivating D185A point mutation (Fig. 1C). As expected, ERT products did not form in reactions that lacked either melittin or dNTPs. IP6 was also required, indicating a role for stable CA hexamers. Removing rNTPs reduced ERT efficiency modestly, and had a more detrimental effect when IP6 levels were lowered from 40 to 2 μM. Thus, rNTPs also contributed to ERT efficiency, particularly when IP6 levels were limiting. Finally, cell extract addition reproducibly increased ERT efficiency, from 0.39±0.09 to 0.48±0.08 late transcripts per core, implying that the extract contained factor(s) that could stimulate ERT. The ~50% efficiency of this optimized ERT reaction was remarkably high given that production of late dsDNA transcripts requires multiple steps. The viral RT/RNase H enzymes must copy sequences from both strands of the 9712 nt genome, utilize two different RNA primers (tRNALys,3 and the polypurine track), execute at least two different strand transfer steps, and degrade the genomic RNA template.
Effects of Viral Capsid Stability and Capsid Inhibitors
The HIV-1 capsid performs multiple functions, one of which may be to enhance the efficiency of reverse transcription (27, 28). Consistent with such a role, CA mutations and small molecule inhibitors that alter viral capsid stability can inhibit viral reverse transcription in infected cells (29, 30). Similarly, the IP6 and rNTP dependence of our ERT reactions suggested that the viral capsid might also be playing an important role in our system. To test this idea, we examined the effects of CA point mutations reported to either stabilize (E45A), moderately destabilize (Q63,67A) or severely destabilize the capsid (E28,29A) (29, 31, 32). The expected effects on capsid stability were confirmed by using negative stain transmission electron microscopy (TEM) to quantify the numbers of wild type and mutant cores with apparently intact capsids following 10 h incubations under standard ERT conditions, but with three different IP6 concentrations (Fig. 2A, Fig. S1).
Fig. 2. Effects of Capsid Stability on ERT Efficiency.

(A) Stability of HIV-1 capsids composed of wild type (WT) or the indicated CA mutant subunits. Capsid stability was assessed by treating virions with melittin to release cores into lysate-free ERT buffers that contained different IP6 levels, and then quantifying the numbers of apparently intact capsids (insets; scale bars, 100 nm) by imaging with negative stain transmission electron microscopy (TEM). Graphs report the mean and standard deviation (n=3) of intact cores per 900 μm2 under the different conditions (see Materials and Methods and Fig. S1 for details). (B) ERT levels from cores with WT or mutant CA proteins at different IP6 concentrations. Grey bars highlight IP6 concentrations that optimized transcript numbers. Standard “non-lysate” conditions were used for these experiments, except for variations in IP6 levels.
ERT efficiencies were also measured for each CA construct across a wide range of IP6 concentrations (Fig. 2B). These experiments were performed in the absence of cell extract so that IP6 concentrations could be controlled precisely. Wild type capsids exhibited maximal ERT over an IP6 concentration range of 40-80 μM, which approximates the estimated cellular concentration (~40 μM) (20). ERT efficiencies were reduced at both higher and lower IP6 concentrations, ultimately reaching background levels in the absence of exogenous IP6. The different CA mutants exhibited similar behaviors, but with differing optimal IP6 concentrations that correlated inversely with intrinsic capsid stability. IP6 stabilizes capsids by binding within the central pores of the hexameric (and likely also pentameric) CA building blocks (18). Thus, IP6-bound CA rings promote ERT, possibly by stabilizing higher-order capsid lattice assemblies. Conversely, the inhibition of ERT observed at high IP6 levels, which was particularly evident for the hyperstable CA E45A mutant, indicates that capsid overstabilization is also detrimental, as has similarly been reported for HIV-1 infections in cultured primary CD4+ T cells (29). Hence, a degree of capsid flexibility also appears to be required for efficient reverse transcription. The CA E28,29A mutant, which is severely impaired in forming intact capsids (31), initiated DNA synthesis efficiently when IP6 levels were high, even under conditions where no intact capsids were observed. Nevertheless, late transcripts were not produced under these conditions (Fig. 2B). Thus, local patches of capsid lattice, perhaps even just CA hexamers, appear to stimulate the production of early ERT products, and stable capsid lattices may be particularly important for efficient strand transfer or other later-stage steps in viral replication.
As another test of capsid involvement in ERT, we examined the effects of a potent small molecule inhibitor that binds the viral capsid (30, 33). GS-CA1 and its close analog GS-6702 bind in a pocket located between the N- and C-terminal domains of adjacent CA subunits within the hexameric ring and can thereby stabilize the CA hexamer. The inhibitors impair different capsid-dependent functions throughout different stages of the viral life cycle with distinct potencies, including virion production and maturation (GS-CA1 IC50 0.1-1 nM), reverse transcription (IC50 5-25 nM), nuclear entry (IC50 0.5-5 nM), and integration (IC50 0.05-0.5 nM) (30, 33). In our system, GS-CA1 strongly inhibited ERT in either the presence or absence of lysate (Fig. 3A), with an IC50 (10-100 nM) that matched GS-CA1 inhibition of viral reverse transcription in cells (30). A CA mutation (M66I) in the GS-CA1 binding site that confers drug resistance in cultured virus (30) also conferred GS-CA1 resistance in the ERT assay (Fig. 3B). Thus, GS-CA1 inhibition of viral reverse transcription and drug resistance were both recapitulated in vitro. The GS-CA1 inhibition patterns mimicked those seen for the severely capsid-destabilizing E28,E29A mutant at high IP6 concentrations in that ERT initiated with reasonable efficiency even at high drug concentrations, whereas production of late transcripts was potently blocked. In contrast, the control inhibitor efavirenz, which acts most potently during RT initiation (34), blocked formation of early transcripts (Fig. 3A,B).
Fig. 3. Effects of the Capsid Inhibitor GS-CA1.

(A, B) ERT levels in the presence of different concentrations of GS-CA1, for cores containing WT (A) or a drug-resistant CA mutant M66I (B). Standard ERT conditions were used for these experiments. (C) Cryo-EM images of cores observed after 4 h under standard ERT conditions in the absence or presence of 100 nM GS-CA1. Scale bars, 50 nm. The capsid lattices are rendered as hexamer units, each colored by cross-correlation value as determined by sub-tomogram averaging, with red denoting low correlations. Full galleries are shown in Fig. S2. (D) Number of CA hexamers mapped per observable core in the absence or presence of 100 nM GS-CA1. Data are composite from two independent experiments that agreed well. Center lines show the medians, mean values are denoted by ×, box limits indicate the 25th and 75th percentiles, whiskers extend to minimum and maximum values, and circles indicate outliers, as determined in GraphPad Prism. Note that fully intact capsids contain an average of 240 hexamers.
Unlike the E28,29A capsid-destabilizing mutation, however, cores with recognizable capsids were readily observed by negative stain TEM imaging of GS-CA1-treated wild type samples. To determine how GS-CA1 treatment affected the integrity of the capsid structure at the subunit level, we used electron cryotomography (ECT) and sub-tomogram lattice mapping to visualize individual cores and map their constituent hexameric CA rings (Fig. S2A,B). All GS-CA1-treated capsids (18/18) had undergone substantial disassembly after 4 h incubations (Fig. 3C,D). In contrast, a majority of cores examined (8/14) in untreated control samples contained intact or nearly intact capsids. Thus, GS-CA1 appeared to accelerate loss of capsid integrity. Furthermore, comparison of surface curvature in the remaining capsid fragments indicated that GS-CA1 flattened the capsid lattice (Fig. S2C). The GS-CA1 binding pocket (also called the ‘NTD-CTD interface’) adopts multiple different configurations that help to generate capsid lattice curvature (8, 35-37). GS-CA1 binding to the NTD-CTD interface may restrict the flexibility of the CA subunits, which can explain the observed effect on lattice curvature. We propose that this effect, in turn, introduces lattice strain and promotes capsid fracturing. A similar capsid fracturing model has been proposed for PF74 (38), a less potent capsid inhibitor that also binds within the NTD-CTD interface.
Capsid Uncoating
Many aspects of HIV-1 capsid uncoating are not well understood, including the timing, morphological transformations, and molecular trigger(s) (reviewed in (28, 39-42)). These uncertainties result, at least in part, from the challenges associated with TEM imaging of replicating viral cores within infected cells. To address this limitation, we used ECT and lattice mapping to image the viral capsids at the subunit level. Cores with mappable hexamers released from mellitin-treated virions in the absence of dNTPs and other reaction components (negative control) had predominantly intact or nearly intact capsids (45/55, 82%) (Fig. S3A). In contrast, cores incubated under standard ERT conditions (37C, 8-10 h) exhibited a greater distribution of capsid morphologies (Fig. S3B,C). In these samples, cores surrounded by intact or nearly intact capsids still predominated amongst the cores with mappable capsid hexamers (53/94, 56%) (Fig. S3B). We also observed a large number of partially disassembled capsid shells (34/94, 36%), with a continuum of lattice completeness that ranged from capsids with small ruptures, to capsid remnants that were missing more than half of their hexamers (Fig. S3B). The morphologies of these capsid disassembly intermediates were particularly striking and unexpected in two ways. First, many capsids had lost local “patches” of their lattices, rather than disassembling in an all or none fashion (Fig. 4). Second, we could often discern strands of nucleic acids looping out of the resulting capsid openings (Fig. 4, and movies S1-S3). It was possible to trace these continuous polynucleotide strands as they emanated from the core interior, exited through the gaps, and then looped back into the core. Loops were observed in the presence or absence of exogenous DNA, indicating that they were viral nucleic acids. We surmise that the looping strands corresponded to double-stranded cDNA transcripts, perhaps associated with NC protein, because the loops were only observed at late time points (8-10 h), when most input cores had already completed reverse transcription. The longest loop shown in Fig. 4 (top) would correspond to ~0.5-0.6 kb dsDNA, and the longest we observed in all of our images was ~1.5 kb, implying that the majority of the 9.7 kb genome remained tightly packed within the capsid. Viral capsid dissociation is promoted by reverse transcription (26, 43), and our results support the proposal that increased pressure imposed by forming a relatively rigid dsDNA genome can rupture the viral capsid locally (44). Nevertheless, the capsid shell can remain largely intact during reverse transcription. This “local lattice rupture” model can reconcile reports that capsids become permeable to protein markers soon after entering the cytoplasm (43), yet remain largely intact all the way into the nucleus (26).
Fig. 4. Imaging Cores During Viral Replication.

(A) Tomographic slices of ruptured cores observed after 8-10 h under standard ERT conditions. Polynucleotide loops (magenta) were segmented using the tools in imod (58). Red spheres indicate apparent protein densities associated with the loops within the core. Scale bars, 50 nm. (B) Composite representations illustrating the three-dimensional arrangements of loops and capsid lattice maps. The capsid lattice maps are rendered as hexamer units, each colored by cross-correlation value as determined by sub-tomogram averaging, with red denoting low correlations.
Integration
We also investigated whether our cell-free reactions could support integration of the reverse-transcribed viral dsDNA into a target DNA plasmid. Integration of the 3’ end of the viral genome was detected using a PCR-based assay in which one primer annealed to the viral DNA and paired with one of two other primers that annealed to plasmid DNA sequences (Figs. S4-S5). This approach enriched for target plasmid integration events, and detected integration in either possible orientation (45). In these experiments, the different stages of ERT again proceeded sequentially, with late dsDNA products accumulating maximally after 8-10 h at 37C (Fig. 5A, upper panel). In the integration component of the assay, only low background qPCR signals were detected for the first 6 h, before abruptly increasing >5000-fold at the 8 and 10 h time points, indicating that fully reverse transcribed viral dsDNA integrated into the target plasmid (Fig. 5A, lower panel). In control experiments, integration signals were reduced to background levels in samples that contained efavirenz or an inactivating point mutation within IN (D116A) (Fig. 5B). As expected, efavirenz prevented the production of dsDNA integration substrates, whereas the IN D116A mutation did not alter ERT efficiency, indicating this mutation specifically inhibited only the integration step. Integration also decreased in a dose dependent fashion in the presence of increasing concentrations of the IN inhibitor raltegravir. ERT efficiencies were not altered by raltegravir treatment, indicating that the inhibitor also acted specifically at the integration step, and the IC50 (1-10 nM) value matched that seen in infected cells and in vitro assays (2-7 nM) (46, 47). Most importantly, integration also required cell extract (Fig. 5B), indicating that a host factor(s) was required to promote the reaction.
Fig. 5. Reconstituted Integration.

(A) Time courses showing accumulation of ERT (upper panel) and integration products (lower panel). The time courses show the sequential formation of early, intermediate and late ERT products, followed by integration. Note that integration products first appeared at t=8 h, in parallel with peak production of late ERT products. Error bars here and in (B) show mean ± SD from three qPCR measurements from a representative experiment, selected from three independent experiments. (B) Quantification of ERT (upper panel) and integration products under different reaction conditions (see Materials and Methods for details). Negative controls show the effects of no incubation (t=0), raltegravir (IN inhibitor, 1 nM-100 μM), efavirenz (RT inhibitor, 10 μM), or an inactivating IN mutation (D116A). (C) Circular pK184 plasmid map showing the position and frequency of integration events determined by deep sequencing of read junctions between the 3’ end of the viral genome and the target plasmid (orange bars, ln scale). The graphic shows composite data from three independent integration and deep sequencing reactions, with integration in both possible orientations picked up by the two designated primers. (D) Favored DNA sequences for HIV integration, analyzed using WebLogo as described in Materials and Methods. HIV integration in the target DNA sequence occurs between positions 0 and −1 on the sequenced strand and between positions 4 and 5 for the complementary strand. A representative logo from one of three experiments (sample 47, Supplemental Table S1) is shown. Bases and their heights show the level of conservation at each target base position (perfect conservation = 2 bits).
Integration efficiencies again exhibited strong biphasic dependence on IP6 levels and capsid stability (Fig. S6). Integration from wild type capsids was strongly reduced at low levels of exogenous IP6 where these capsids were unstable and late ERT product levels were very low, yet proceeded efficiently at low IP6 levels when the capsids contained the stabilizing E45A CA mutant. Integration levels also diminished at very high IP6 levels, where the wild type capsids were hyperstabilized and late ERT product levels were again reduced. Integration was even strongly impaired at moderately elevated IP6 levels where significant levels of late ERT products were present (e.g., 320 μM IP6), indicating that a final capsid “uncoating” reaction needs to occur prior to integration, as also occurs in the nucleus of HIV-1 infected cells (26).
Deep sequencing reactions confirmed authentic integration of the 3’ end of the viral dsDNA genome into the target plasmid (Fig. 5C, Figs. S5, S7, Table S1). Three independent reactions generated a total of 8.2×106 deep sequencing reads in which the 3’ end of the viral genome abutted plasmid DNA sequences. Collectively, these reads showed integration at nearly every base step of the entire 2423 nt target plasmid, in both possible orientations and with large read numbers at most base steps (mean 3397, median 307). Thus, many thousands of independent integration events occurred during each reaction. Integration frequencies varied dramatically at different sites, reflecting HIV-1 intasome sequence preferences (48-50), and the sequence preferences observed in vitro resembled those reported in HIV-1 infected cells (Fig. 5D) (48-50).
We also observed reads that corresponded to autointegration of the 3’ end of the genome back into the middle of the viral genome (Fig. S8). These reads were much less abundant (1.1×104 reads), presumably because autointegration events occurred less frequently and were not exponentially amplified by our PCR protocol. Autointegration sites clustered about the center of the viral DNA and exhibited a strong orientation bias, which can be explained by the optimal DNA genome topology for concerted integration of both viral genome termini. Analogous off pathway autointegration reactions also occur during viral infections, and their frequencies can be modulated by cellular host factors (51-53).
Creation of a viable provirus requires concerted integration of both viral termini across a 5 bp staggered double-stranded break in the target DNA (Fig. S4). Our cell-free system supported such concerted integration, as evaluated by amplifying, cloning, and sequencing individual plasmids that carried both integrated viral dsDNA termini (Fig. S9). A selection of 99 independent clones from 12 different repetitions of this experiment were sequenced, and concerted integration of both viral termini was confirmed in every case. We also observed the expected nucleotide sequences at both ends of the viral DNA, with 5 bp target site duplications in the target DNA, and with target site sequence preferences that again resembled those reported for HIV-infected cells (48-50). Moreover, a polymorphic sequence marker was transferred from the original 3’ viral RNA LTR element to the 5’ end of the dsDNA genome, again consistent with the established mechanisms for viral replication (Fig. 1A).
Discussion
Here, we have reconstituted the major early steps of the HIV-1 life cycle in a cell-free system. ERT was highly efficient, with approximately half of the input cores yielding late RT products, which required copying ten thousand nucleotides, degrading the viral RNA template, and executing two distinct sets of priming and strand transfer reactions. The virus has thus evolved mechanisms for performing each of the different replication steps very efficiently, apparently in response to the strong selective pressure imposed by the fact that productive infections are typically initiated by a single founder virus during sexual transmission (54). Importantly, the viral CA protein is not an inert bystander during these processes, but rather contributes directly to ERT efficiency. This activity requires intact CA rings and/or capsid lattice sections of the appropriate stability and geometry.
Our imaging studies revealed several interesting features of capsid-dependent ERT. Most fundamentally, we found that many viral capsids remained largely intact during the ERT reaction, and those that did uncoat did not do so in an all-or-none fashion, as might have been anticipated for a highly cooperative structure. Instead, we observed a continuum of disassembly intermediates. The most striking of these were largely intact capsids in which viral nucleic acid strands extruded through lattice openings. Thus, the initial stages of capsid uncoating may simply involve the loss of relatively small surface patches to accommodate the growing viral double-stranded cDNA, whose stiffness and persistence length will be greater than that of the templating single-stranded RNA genome. Such largely intact capsids could, in principle, explain the recent observation that reverse transcribing viral cores maintain the majority of their CA subunits as they move into the nucleus, complete reverse transcription, and traffic to sites of integration before uncoating fully (26). Local lattice ruptures might also help accommodate radial constriction of the capsid as the viral core passes through the narrow inner channel of the nuclear pore, which is currently a puzzling paradox in the field. Our experiments in soluble cell extracts demonstrate, however, that viral cores need not pass through nuclear pores to complete reverse transcription, uncoat, or integrate.
Although we have not yet systematically synchronized and imaged the different stages of reverse transcription, the extruded polynucleotide loops that we observed at late time points are consistent with a model in which much of the viral cDNA is not held in a strict orientation, but where the viral genome termini reside in close proximity. This configuration could facilitate strand transfer reactions during replication and then position the termini for intasome formation and concerted integration. Association of the genome ends could occur when the RNA is first packaged, although at present packaging is only known to require sequences located near the 5’ end of the viral RNA genome (55, 56). Alternatively, the termini could associate during first strand transfer, when the minus strand strong stop cDNA copy of the 5’ end of the viral RNA genome reanneals to the 3’ end of the viral RNA genome. We envision that an extended capsid interior surface could “cage” RT to prevent dissociation (and promote reassociation), organize the genome termini and/or facilitate key steps in viral replication such as priming, strand transfer, and intasome formation. Our experiments indicate that in order to function properly, this capsid surface must adopt the proper geometry yet retain some dynamic flexibility, but also that the entire capsid need not remain fully closed. Finally, the viral NC protein likely also facilitates replication and integration (57). Thus, layered contacts between the different core components appear to be sufficiently stable to ameliorate highly cooperative capsid uncoating and ensure that supramolecular viral complexes remain intact as they traffic through the cytoplasm and nucleus to sites of integration.
Our ERT reactions produced complete dsDNA reverse transcripts that were properly configured to access and utilize the packaged IN enzymes. Although we did not quantify integration efficiencies in absolute terms, thousands of integration events occurred in each reaction and concerted integration was observed, indicating that native-like intasomes and synapses were formed (4). Integration was also dependent on an exogenous soluble cellular factor(s), whose identity will be of interest. Finally, integration did not proceed efficiently when the capsid was hyperstablized, indicating that the CA lattice must rearrange or uncoat completely for integration to occur, as has been observed in the context of a viral infection (26). We anticipate that our cell-free system will enable systematic analyses of these, and other key steps in viral replication and integration, and thereby elucidate the transformations that occur as HIV-1 proceeds through the first half of the viral life cycle.
Materials and Methods
Cell Culture, Virion Production, and Cell Lysates
Human 293T and HeLa cells were obtained from the American Type Culture Collection and grown in Dulbecco modified Eagle medium (ThermoFisher) supplemented with 10% fetal calf serum (Atlanta Biologicals) at 37C in 5% CO2. Cell cultures were confirmed to be negative for mycoplasma in routine tests (Lonza) every six months. Viral particles were produced by Calcium Phosphate transfection (Takara) of 293T cells with the pSG3Δenv plasmid, obtained from the NIH AIDS Reagent Program (Catalog # 11051). Virions produced from this construct lack the viral Env protein, but are fully infectious when pseudotyped with envelope glycoproteins from HIV-1 or VSV ((13-15) and confirmed for our construct). For virion production, 2.5×106 293T cells were seeded on 10 cm plates and transfected 24 h later. 18 h post transfection, media was removed and replaced with fresh media. 48 h post transfection, virion-containing media was removed and filtered through a 0.45 μM membrane, layered on a 20% sucrose cushion in HS buffer (10 mM Hepes pH 7.4 and 140 mM NaCl), and centrifuged for 2 h at 28,000 rpm in a SW32 Ti rotor (Beckman). After centrifugation, media was decanted and virion-containing pellets were resuspended in 100 μl HS buffer per 10 cm plate of transfected cells, aliquoted, flash frozen in liquid nitrogen, and stored at −80C. Virion CA concentrations were determined by HIV-1 p24 ELISA assay (XpressBio). Typical yields were 40-100 pmol CA per 10 cm plate.
HeLa cell lysates were prepared by seeding 2.8×106 cells on a 15 cm plate. After 48 h, media was removed and cells were washed with PBS, removed from the plate with a cell lifter (Sigma Aldrich), placed in a micro-centrifuge tube, and centrifuged for 1 min at 1500 rpm (4C). After centrifugation, excess PBS was removed and cells were resuspended in 200 μl of lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 50 μg/ml melittin) per 15 cm plate and incubated on ice for 15 min. Lysates were then spun in a micro-centrifuge for 10 min at 17,000 rpm. The soluble fraction was collected, aliquoted, flash frozen in liquid nitrogen, and stored at −80C.
Lysates prepared in this fashion stimulated ERT (Fig. 1C) and were required for integration (Fig. 5B). Preliminary characterization of the extract activities indicates that flow throughs from lysates passed through Centricon filters with cutoffs of 3K, 50K, and 100K did not support the integration reaction, implying that the lysate is not simply supplying a small molecule like Mg2+ or IP6. Integration activity was also lost when the lysates were heated to either 65C (20 min) or 95C (5 min), implying that at least one key factor is likely to be a structured biomolecule.
Endogenous Reverse Transcription (ERT) Assay
Prior to assaying for ERT, virion preparations were treated with cyanase (RiboSolutions) to digest any plasmid DNA carried-over from transfection. Briefly, virions were diluted to 20 nM CA in cyanase digestion buffer (50 mM Tris pH 8.0, 20 mM NaCl, and 12 mM MgCl2) in a total volume of 800 μl. 1 μl cyanase was added and the reaction was incubated at 37C for 30 min. 60 μl of cyanase inactivation resin (RiboSolutions) was then added and incubated with constant inversion at 37C for 20 min. Virions were then centrifuged for 1 min at 5000×g and virion containing supernatant was removed and stored on ice. Background level measurements in the ERT and integration assays were greatly reduced by cyanase treatment, demonstrating the importance of removing carry-over DNA from transfection.
ERT was initiated by combining 20 μl of virions with 20 μl of 2×ERT Buffer (50 mM Tris pH 7.4, 150 mM NaCl, 12 μM dCTP, 9.2 μM dGTP, 16 μM dTTP, 10.4 μM dATP, 50 μg/ml melittin, 80 μM IP6, 360 μM CTP, 3.5 mM GTP, 1.4 mM UTP, 13.4 mM ATP, 6.67 mM MgCl2, and 0.6 ng/μl of the pK184 target plasmid) and 5 μl of HeLa cell lysate or lysis buffer control, then incubated at 37C for 10 h (denoted “Standard Reaction Conditions” or “Standard Reaction Conditions -Lysate”, respectively), or otherwise for time courses as indicated in figures. rNTPs and dNTPs were purchased from Promega and their concentrations were chosen to correspond to those estimated for T lymphocyte cytoplasms (21, 22). IP6 was purchased from Sigma, and the optimal concentration was 40-80 μM, based upon yields of early (MSSS) DNA transcripts (e.g., see Fig. 2A). 40 μM IP6 was chosen for our standard condition because this is the estimated intracellular concentration (20). Melittin was purchased from Sigma, titrated, and used at a concentration that balanced the optimal yields of early (MSSS) transcripts, late (SST) transcripts, 3’ integration products and membrane-free cores. We found that Triton X-100 could work nearly as well as melittin in the ERT and integration assays, but the optimal concentration range was narrower and had greater variability in product yields. Mg2+ concentrations were also optimized, using early (MSSS) transcripts as the readout. Product formation was maximal in the range of 8-10 mM Mg2+, and we chose a concentration (9.33 mM) that equalled the total rNTP concentration in the final ERT buffer conditions (i.e., rNTPs were added at 18.66 mM in 2× ERT buffer then diluted 1:1 with virions to 9.33 mM; MgCl2 was added at 12 mM during Cyanase digestion and 6.67 mM in 2× ERT buffer, which when combined 1:1 resulted in a final concentration of 9.33 mM). 20 μl of 0.5 M EDTA was added to stop ERT. Final ERT DNA products were purified using a Qiaquick PCR purification kit (Qiagen) and eluted from the column in 120 μl elution buffer (Qiagen). Drugs at final concentrations were as follows; GS-CA1 (Gilead) in DMSO at 1 nM to 10 μM, raltegravir (NIH AIDS Reagent Program) in water at 1 nM to 100 μM, and efavirenz (NIH AIDS Reagent Program) in DMSO at 10 μM.
Two-Step PCR 3’ Integration Assay
The plasmid pK184 (added to the 2×ERT Buffer, see above) was used as an integration target for the full-length viral cDNA produced in the ERT reaction. Purified reaction products (see above) were amplified by PCR using Phusion Polymerase (NEB), diluted 100-fold and measured by quantitative PCR (qPCR). Briefly, 5 μl of purified ERT product was combined with 10 μl Phusion HF Buffer, 200 μM each dNTP, 0.24 μM each of three primers (pK184-IN-FWD, pk184-IN-REV, and DC3014), 0.5 μl Phusion Polymerase, and water to a final volume of 50 μl. PCR cycles were as follows: Denaturation at 95C for 5 min; 23 cycles of 95C for 30 sec, 58C for 30 sec, and 72C for 90 sec; and a final extension at 72C for 5 min followed by a hold at 4C. Final PCR products were purified with a Qiaquick PCR purification kit (Qiagen) according to the manufacturer’s instructions and eluted in 120 μl elution buffer and diluted 100-fold for qPCR analysis of 3’ integration. Note that subsequent to these studies, re-optimization of the DC3014 primer further reduced the background PCR signal in the integration quantification assay by about 10-fold; the sequence of the improved primer (designated DC3014c) is provided in Supplemental Table 2.
To obtain 100 ng DNA for deep sequencing, 8 (for experimental repetitions 046 and 048) or 24 (for 047) identical PCR reactions were performed, combined, purified with a Qiaquick PCR purification kit (Qiagen), eluted in 50 μl elution buffer and submitted for deep sequencing analysis. Experimental repetitions 046 and 047 used 23 PCR amplification cycles, whereas 048 was amplified for 35 cycles. These samples were analyzed by deep sequencing to identify 3’ integration target site sequences.
Quantitative PCR Analyses
Light Cycler 480 SYBR Green I Master (Roche) with a Light Cycler 480 (Roche) were used for all qPCR analyses. Briefly, 5 μl of either ERT product or diluted 3’ Integration PCR product, 500 nM each of two primers (MSSS-FWD + MSSS-REV, FST-FWD + FST-REV, Late RT-FWD + Late RT-REV, or AE3013 + IN990-REV for integration) and 10 μl of SYBR Green I Master Mix were combined in a total volume of 20 μl and analyzed by qPCR according to the manufacturer’s instructions. All qPCR samples were analyzed in triplicate. Standard curves for ERT products were generated by using a 10-fold dilution series of the pSG3Δenv plasmid for quantitation of ERT products. The number of DNA copies per core was determined from the ratio of viral cDNA copies (determined by qPCR) to the number of input core particles in each reaction (typically ~4×107). The number of HIV core particles in each reaction was determined from the input CA protein level (typically ~0.38 pmol, measured by ELISA, see above) and by assuming that there were 3000 Gag/CA molecules per virion and 1.33 cores per virion (9, 25). Yields were calculated by assuming a theoretical yield of one late RT product/core. In reality, RT may initiate on both of the packaged RNA strands, and it is not clear when the conversion to a single template occurs. Note also that the primers used to quantify early/minus strand strong stop (MSSS) and intermediate/first strand transfer (FST) products ultimately have two binding sites on the viral DNA template by the time the full dsDNA preintegration complex is formed. This effect is evident in the ERT time courses shown in Fig. 1A and 5A, where the qPCR products from these two primers rise late in the time course (e.g., compare the signals at 6 h and 8 h). These effects apparently explain why the qPCR measurements for the MSSS and FST primers can approach 2 copies/core in some experiments. Integration product levels were normalized relative to the qPCR signal observed for the t=0 time point. Three independent repetitions and analyses were performed, and representative examples are shown for the experiments in Figs. 1A, 1B, 2B, 3A, 5A, 5B and S7.
Negative Stain Transmission Electron Microscopy
ERT reaction mixtures (10 h, 37C, standard ERT conditions except that IP6 was varied as described, 3.5 μl) were applied onto the carbon side of Formvar/Carbon-coated, 300-mesh copper grids (Electron Microscopy Sciences) for 90 s, rinsed for 90 s by flotation on a drop of 100 mM KCl, blotted dry, stained for 90 s by floating on a drop of 2% (w/v) solution of uranyl acetate, blotted dry, and allowed to air dry. Samples were viewed on an FEI Tecnai F20 transmission electron microscope operating at 120 kV. Core quantification was performed at a nominal magnification of ×11,500, where viral capsids could be distinguished from similar size debris. For each grid, three well-stained squares were selected at random and capsids were counted manually while sweeping the perimeter of each square, covering an area of ~900 μm2. Quantification was done in three independent experiments, and in two of these experiments the grids were randomized and the experimenter was blinded to their identities.
Cryo-Electron Tomography and Lattice Mapping
ERT reaction mixtures were mixed with an equal volume of 10-nm BSA Gold Tracer (Electron Microscopy Sciences), 3.5 μl aliquots were applied onto glow-discharged 2/2 carbon C-flat or Quantifoil grids (Electron Microscopy Science), blotted to near dryness manually, and then plunge-frozen into liquid ethane. Cryotomograms were acquired using an FEI Titan Krios electron microscope operating at 300 kV and equipped with either a Falcon III camera or a K3/GIF with a slit width of 20 eV. Tilt series were collected using the data collection software Tomography 4.0 (FEI) with an angular range of −60° to +60°, an angular increment of either 1 or 2°, defocus values of 5 to 10 μm, and a nominal magnification of ×29,000 (Falcon III) or ×33,000 (K3), which correspond to a pixel (px) size of 2.92 (Falcon III) or 2.69 (K3) Å. Tilt series were aligned by using imod (58). Weighted back-projection or SIRT was used to reconstruct tomograms in imod.
Lattice mapping was performed based on previously described protocols (59, 60) using as search template a cryoEM map of the HIV-1 CA hexamer (EMD-3465) (8) down sampled to 25 Å resolution. Each capsid surface was modeled and oversampled as a mesh by using surface modeling tools in the Dynamo software package (58). Two sets of sub-volumes centered at the same positions but of different box size (48×48×48 px3 and 32×32×32 px3) were extracted from 4× binned tomograms and assigned polar Euler angles in reference to the modeled capsid surface; azimuthal angles were randomized. For each box size set, four rounds of azimuthal angle search were performed, applying 6-fold symmetry, a low-pass filter of 30 Å and using a soft-edged cylindrical alignment mask in Dynamo. Overlapping and misaligned particles, as well as those with very low cross-correlation values were discarded. The two sets were then compared, and only particles common to both sets were retained. These were re-extracted (box size of 64×64×64 px3) from 2× binned tomograms. To fill in gaps, the refined particle set was used to define a new surface model, which was oversampled and extracted (64×64×64 px3) from 2× binned tomograms, retaining only those that are >10 px away from particles in the refined set. Four rounds of template alignment were then performed on the combined set of particles, using progressively narrower angular and positional search ranges and a low-pass filter set to 25 Å. After each round, overlapping and misaligned particles, as well as those with very low cross-correlation values were discarded. Final lattice maps were generated and visualized in Chimera (61) as described (59, 60). Software used in this study were curated by SBGrid (62).
Deep Sequencing and Analysis of 3’ Integration Events
100 ng of the first PCR product from the 3’ integration assay (above) was submitted for Ion Torrent sequencing, performed at the DNA Sequencing Core Facility, University of Utah. Reads were screened for sequences with at least 20/23 matches to the extreme end of the HIV-1 3’ LTR using Seal (BBTools suite, Joint Genome Institute). To identify reads with junctions aligning to the pK184 target plasmid, these Seal reads were then remapped onto linearized pK184 reference genomes using BWA-MEM (63), again requiring at least 20/23 matches to the reference. To identify reads that could represent autointegration events, Seal reads with sequences matching the 3’ LTR were remapped onto a modified HIV-1 reference genome using BWA-MEM. Because the alignment software cannot distinguish identical sequences, the reference genome was modified by substituting the 5’ LTR with 633 Ns. Finally, subsets of reads from both types of alignments were checked manually to ensure that the alignment procedures identified integration sites and their orientations correctly. Alignments of integration events into pK184 and autointegration events into HIV-1 were visualized using the Integrative Genomics Viewer (64).
To score integration events at specific bases in the target plasmid, reads aligning to the HIV-1 3’ LTR and pK184 were parsed using their CIGAR string to identify the pK184 integration positions and orientations (Python code archived at GitHub repository DOI: 10.5281/zenodo.3966034). Because the alignment and integration analyses were performed on linearized pK184 reference genomes, the analyses were performed twice on different linearized pK184 reference genomes in which the plasmid “ends” were shifted to very different positions. After correcting for this shift, natural log-transformed merged integration counts were plotted using the circlize package in R (65). Autointegration events in HIV-1 were similarly scored by counting junction sites in reads that aligned to the 3’ LTR and to an additional site located between residues 1000 and 9000 of the HIV-1 genome. Autointegration counts were plotted using GraphPad Prism (66).
Cloning Products of Concerted Integration
99 independent plasmids carrying concerted viral integration sites from 12 different integration reactions were cloned in two different ways: 53 clones were obtained following “repair” of the integration sites (described below), and 46 clones were obtained by simply amplifying the backbone without repair, implying that at least in some cases at least one of the integration sites must have been repaired in the extract. The repair protocol was as follows: 1) To remove the 5’ flap generated by integrase during integration, 10 μl of ERT product were incubated with 0.5 μl Thermostable FEN1 (NEB), 10 μl of Phusion HF Buffer, and 24.5 μl water at 65C for 5 min. 2) To fill in the 5 base integration target site gap, 200 μM each dNTP and 0.5 μl of Phusion Polymerase (NEB) was added, mixed, and incubated at 65C for 5 min. 3) To ligate the final phosphodiester bond and generate a complete double stranded DNA suitable for PCR amplification, T4 DNA ligase (NEB, 0.5 μl) was added and the sample was incubated at 37C for 30 min.
Whether or not samples were repaired, plasmid backbones that carried the two viral termini were amplified by adding 240 nM of each primer (SG3-U3-XhoI-REV and SG3-U5-XhoI-FWD) to the sample to PCR amplify the two ends of the HIV genome along with the entire pK184 plasmid backbone (see Fig. S9). PCR cycles were as follows: Denaturation at 95C for 5 min; 30 cycles of 95C for 30 sec, 58C for 30 sec, and 72C for 2 min; and a final extension at 72C for 5 min followed by a hold at 4C. Final PCR products were purified with a Qiaquick PCR purification kit (Qiagen), digested with XhoI, ligated with T4 DNA Ligase (NEB) and transformed in E. coli TOP10 cells. Plasmids were recovered from kanamycin-resistant colonies, and sequenced to verify concerted integration. In total, 151 plasmids recovered from 12 independent reactions were sequenced. Integration sites were judged to be necessarily independent if they did not match any of the other integration sites recovered from the same experiment (i.e., any duplicated integration sites from individual experiments were removed prior to analysis). Of the 103 remaining independent concerted integration events, all showed transfer of the 3’ U3T15 to the 5’ LTR, 99 showed the expected viral termini and 5’ target site duplications, and four showed aberrant target site duplications of 22 bp (one case) and 65 bp (three cases, all at the same site).
Generation of Integration Consensus Sites
To generate sequence logos showing target base preferences from our deep sequencing analysess of 3’ integration events, we first scaled integration counts to correct for PCR amplification bias using the gradients in read coverage around the pK184 target plasmid to guide the scaling. Read coverage depths, which decreased as a function of distance from the primer binding sites, were binned and plotted separately for forward- and reverse-oriented insertions into pK184. Forward and reverse coverage depth plots were used to generate LOWESS scaling curves and these curves were then applied to normalized integration count data from each independent experiment (using 50000 as a normalization multiplier for forward and reverse integrations from samples 46 and 47, and 15000 for forward and 25000 for reverse integrations from sample 48 – these normalization multipliers were chosen to keep the total number of sequence strings analyzed for each sample below 1 million). Integration frequencies at each base step between 200 and 2200 base pairs away from each primer were then analyzed (this 2000 bp segment was chosen to exclude regions of low coverage immediately adjacent or far away from the primers). The scaled sequence strings were submitted to WebLogo 3 (67, 68) to compute sequence logos. To generate the sequence logo for concerted integration, the sequences strings from the the 99 independent clones described above were submitted to WebLogo 3.
As shown in Figs. 5 and S9, the consensus integration sequence motifs arising from both of our different sets of experiments differ slightly and are not perfectly palindromic, but are nevertheless clearly related to one another and to those reported previously (48-50). We have not interpreted our data further because our DNA target plasmid was limited in size and was not chromatinized, and because we do not know whether our extracts contain any, or all, of the host factors that might favor site selection in cells. We note, however, that our cell free system should provide a well-controlled platform for analyzing the effects of candidate host factors on integration site selection.
Supplementary Material
Acknowledgments:
We thank Brian Lohman in the Genomics and Bioinformatics Analysis group at the Huntsman Cancer Institute for performing integration site sequencing alignments, Yueping Wan for assistance with large-scale virus production, Miguel Pereira for measuring the infectivity of pseudotyped ΔEnvSG3 virions, Kelly Dryden for expert EM assistance, Gilead Sciences for providing GS-CA1 inhibitor, and Janet Iwasa for creating the overview figure for the print summary. Oligonucleotides were synthesized by the University of Utah DNA/Peptide Core Facility, sequencing was performed at the University of Utah DNA sequencing Core Facility, and EM data were collected at the University of Virginia Molecular Electron Microscopy Core facility, which is supported in part by the School of Medicine and built with NIH grant G20-RR31199. The Titan Krios (SIG S10-RR025067), Falcon II/3EC direct detector (SIG S10-OD018149), and K3/GIF (U24-GM116790) were purchased in part or in full with the indicated NIH grants.
Funding: We acknowledge funding from NIH P50 AI150464 (JSJ, OP, WIS) and the University of Virginia School of Medicine Pinn Scholar Program (OP).
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: All plasmids used in this study are available at Addgene (see Supplemental Table S2). 3’ integration site deep sequencing data have been deposited in the NCBI Sequence Read Archive (SRA) database under Accession: PRJNA649355. Python scripts used for sequence analyses are available from the archived GitHub repository DOI: 10.5281/zenodo.3966034.
References and Notes
- 1.Saag MS et al. , Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2018 Recommendations of the International Antiviral Society-USA Panel. JAMA 320, 379–396 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Clercq E, The nucleoside reverse transcriptase inhibitors, nonnucleoside reverse transcriptase inhibitors, and protease inhibitors in the treatment of HIV infections (AIDS). Adv Pharmacol 67, 317–358 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Arts EJ, Hazuda DJ, HIV-1 antiretroviral drug therapy. Cold Spring Harb Perspect Med 2, a007161 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lesbats P, Engelman AN, Cherepanov P, Retroviral DNA Integration. Chem Rev 116, 12730–12757 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coffin JM, Fan H, The Discovery of Reverse Transcriptase. Annu Rev Virol 3, 29–51 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Sarafianos SG et al. , Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J Mol Biol 385, 693–713 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pornillos O, Ganser-Pornillos BK, Maturation of retroviruses. Curr Opin Virol 36, 47–55 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mattei S, Glass B, Hagen WJ, Krausslich HG, Briggs JA, The structure and flexibility of conical HIV-1 capsids determined within intact virions. Science 354, 1434–1437 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Briggs JA et al. , The stoichiometry of Gag protein in HIV-1. Nat Struct Mol Biol 11, 672–675 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Swanson CM, Malim MH, SnapShot: HIV-1 proteins. Cell 133, 742, 742 e741 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Carlson LA et al. , Three-dimensional analysis of budding sites and released virus suggests a revised model for HIV-1 morphogenesis. Cell Host Microbe 4, 592–599 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yong WH, Wyman S, Levy JA, Optimal conditions for synthesizing complementary DNA in the HIV-1 endogenous reverse transcriptase reaction. AIDS 4, 199–206 (1990). [DOI] [PubMed] [Google Scholar]
- 13.Ghosh SK et al. , A molecular clone of HIV-1 tropic and cytopathic for human and chimpanzee lymphocytes. Virology 194, 858–864 (1993). [DOI] [PubMed] [Google Scholar]
- 14.Wei X et al. , Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 46, 1896–1905 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei X et al. , Antibody neutralization and escape by HIV-1. Nature 422, 307–312 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Boone LR, Skalka A, Two species of full-length cDNA are synthesized in high yield by melittin-treated avian retrovirus particles. Proc Natl Acad Sci USA 77, 847–851 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mallery DL et al. , IP6 is an HIV pocket factor that prevents capsid collapse and promotes DNA synthesis. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dick RA et al. , Inositol phosphates are assembly co-factors for HIV-1. Nature 560, 509–512 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacques DA et al. , HIV-1 uses dynamic capsid pores to import nucleotides and fuel encapsidated DNA synthesis. Nature 536, 349–353 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Letcher AJ, Schell MJ, Irvine RF, Do mammals make all their own inositol hexakisphosphate? Biochem J 416, 263–270 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diamond TL et al. , Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem 279, 51545–51553 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kennedy EM et al. , Ribonucleoside triphosphates as substrate of human immunodeficiency virus type 1 reverse transcriptase in human macrophages. J Biol Chem 285, 39380–39391 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warrilow D et al. , Cell factors stimulate human immunodeficiency virus type 1 reverse transcription in vitro. J Virol 82, 1425–1437 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rawson JMO, Nikolaitchik OA, Keele BF, Pathak VK, Hu WS, Recombination is required for efficient HIV-1 replication and the maintenance of viral genome integrity. Nucleic Acids Res 46, 10535–10545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Briggs JA, Wilk T, Welker R, Krausslich HG, Fuller SD, Structural organization of authentic, mature HIV-1 virions and cores. EMBO J 22, 1707–1715 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burdick RC et al. , HIV-1 uncoats in the nucleus near sites of integration. Proc Natl Acad Sci USA 117, 5486–5493 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.James LC, The HIV-1 Capsid: More than Just a Delivery Package. Adv Exp Med Biol 1215, 69–83 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Campbell EM, Hope TJ, HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat Rev Microbiol 13, 471–483 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forshey BM, von Schwedler U, Sundquist WI, Aiken C, Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol 76, 5667–5677 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yant SR et al. , A highly potent long-acting small-molecule HIV-1 capsid inhibitor with efficacy in a humanized mouse model. Nature medicine 25, 1377–1384 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.von Schwedler UK, Stray KM, Garrus JE, Sundquist WI, Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J Virol 77, 5439–5450 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI, Assembly properties of the human immunodeficiency virus type 1 CA protein. J Virol 78, 2545–2552 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Link JO et al. , Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature (2020) epublication ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hooker CW, Lott WB, Harrich D, Inhibitors of human immunodeficiency virus type 1 reverse transcriptase target distinct phases of early reverse transcription. J Virol 75, 3095–3104 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pornillos O et al. , X-ray structures of the hexameric building block of the HIV capsid. Cell 137, 1282–1292 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pornillos O, Ganser-Pornillos BK, Yeager M, Atomic-level modelling of the HIV capsid. Nature 469, 424–427 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao G et al. , Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497, 643–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marquez CL et al. , Kinetics of HIV-1 capsid uncoating revealed by single-molecule analysis. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Novikova M, Zhang Y, Freed EO, Peng K, Multiple Roles of HIV-1 Capsid during the Virus Replication Cycle. Virol Sin 34, 119–134 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dick RA, Mallery DL, Vogt VM, James LC, IP6 Regulation of HIV Capsid Assembly, Stability, and Uncoating. Viruses 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ambrose Z, Aiken C, HIV-1 uncoating: connection to nuclear entry and regulation by host proteins. Virology 454-455, 371–379 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hilditch L, Towers GJ, A model for cofactor use during HIV-1 reverse transcription and nuclear entry. Curr Opin Virol 4, 32–36 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mamede JI, Cianci GC, Anderson MR, Hope TJ, Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc Natl Acad Sci USA 114, E7169–E7178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rankovic S, Varadarajan J, Ramalho R, Aiken C, Rousso I, Reverse Transcription Mechanically Initiates HIV-1 Capsid Disassembly. J Virol 91, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Engelman A, Oztop I, Vandegraaff N, Raghavendra NK, Quantitative analysis of HIV-1 preintegration complexes. Methods 47, 283–290 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Canducci F et al. , In vitro phenotypes to elvitegravir and dolutegravir in primary macrophages and lymphocytes of clonal recombinant viral variants selected in patients failing raltegravir. J Antimicrob Chemother 68, 2525–2532 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Temesgen Z, Siraj DS, Raltegravir: first in class HIV integrase inhibitor. Ther Clin Risk Manag 4, 493–500 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holman AG, Coffin JM, Symmetrical base preferences surrounding HIV-1, avian sarcoma/leukosis virus, and murine leukemia virus integration sites. Proc Natl Acad Sci USA 102, 6103–6107 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD, HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res 17, 1186–1194 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Serrao E, Ballandras-Colas A, Cherepanov P, Maertens GN, Engelman AN, Key determinants of target DNA recognition by retroviral intasomes. Retrovirology 12, 39 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Farnet CM, Haseltine WA, Circularization of human immunodeficiency virus type 1 DNA in vitro. J Virol 65, 6942–6952 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen H, Engelman A, The barrier-to-autointegration protein is a host factor for HIV type 1 integration. Proc Natl Acad Sci USA 95, 15270–15274 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yan N, Cherepanov P, Daigle JE, Engelman A, Lieberman J, The SET complex acts as a barrier to autointegration of HIV-1. PLoS Pathog 5, e1000327 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keele BF et al. , Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci USA 105, 7552–7557 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rein A, RNA Packaging in HIV. Trends Microbiol 27, 715–723 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bieniasz P, Telesnitsky A, Multiple, Switchable Protein:RNA Interactions Regulate Human Immunodeficiency Virus Type 1 Assembly. Annu Rev Virol 5, 165–183 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Rene B, Mauffret O, Fosse P, Retroviral nucleocapsid proteins and DNA strand transfers. Biochim Open 7, 10–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Castano-Diez D, Kudryashev M, Arheit M, Stahlberg H, Dynamo: a flexible, user-friendly development tool for subtomogram averaging of cryo-EM data in high-performance computing environments. J Struct Biol 178, 139–151 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Skorupka KA et al. , Hierarchical assembly governs TRIM5alpha recognition of HIV-1 and retroviral capsids. Sci Adv 5, eaaw3631 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qu K et al. , Structure and architecture of immature and mature murine leukemia virus capsids. Proc Natl Acad Sci USA 115, E11751–E11760 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pettersen EF et al. , UCSF Chimera - A Visualization System for Exploratory Research and Analysis. Journal of computational chemistry 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Morin A et al. , Collaboration gets the most out of software. Elife 2, e01456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H, Durbin R, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robinson JT et al. , Integrative genomics viewer. Nature biotechnology 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gu Z, Gu L, Eils R, Schlesner M, Brors B, circlize Implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Wickham H, ggplot2: Elegant Graphics for Data Analysis. (Springer, 2016). [Google Scholar]
- 67.Crooks GE, Hon G, Chandonia JM, Brenner SE, WebLogo: a sequence logo generator. Genome Res 14, 1188–1190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schneider TD, Stephens RM, Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 18, 6097–6100 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.