

Abstract
Paenibacillus sp. 32352 is a soil-dwelling bacterium capable of producing an enzyme, Pn3Pase that degrades the capsular polysaccharide of Streptococcus pneumoniae serotype 3 (Pn3P). Recent reports on Pn3Pase have demonstrated its initial characterization and potential for protection against highly virulent S. pneumoniae serotype 3 infections. Initial experiments revealed this enzyme functions as an exo-β1,4-glucuronidase cleaving the β(1,4) linkage between glucuronic acid and glucose. However, the catalytic mechanism of this enzyme is still unknown. Here, we report the detailed biochemical analysis of Pn3Pase. Pn3Pase shows no significant sequence similarity to known glycoside hydrolase (GH) families, thus this novel enzyme establishes a new carbohydrate-active enzyme (CAZy) GH family. Site-directed mutagenesis studies revealed two catalytic residues along with truncation mutants defining essential domains for function. Pn3Pase and its mutants were screened for activity, substrate binding and kinetics. Additionally, nuclear magnetic resonance spectroscopy analysis revealed that Pn3Pase acts through a retaining mechanism. This study exhibits Pn3Pase activity at the structural and mechanistic level to establish the new CAZy GH family GH169 belonging to the large GH-A clan. This study will also serve toward generating Pn3Pase derivatives with optimal activity and pharmacokinetics aiding in the use of Pn3Pase as a novel therapeutic approach against type 3 S. pneumoniae infections.
Keywords: capsular polysaccharide, carbohydrate-active enzyme, glycoside hydrolase, Streptococcus pneumoniae
Introduction
A number of early studies demonstrated an enzyme found in Bacillus circulans was capable of degrading the capsular polysaccharide (CPS) of highly virulent type 3 Streptococcus pneumoniae (Pn3P) (Avery and Dubos 1930; Dubos and Avery 1931; Sickles and Shaw 1934). However, upon sequencing the genome of this bacterial strain “Bacillus circulans Jordan strain 32352” we discovered this bacterium rather belonged within the Paenibacillus genus and was reestablished as Paenibacillus sp. 32352 (Middleton et al. 2017). As previously reported, this soil-dwelling Paenibacillus bacterium is capable of producing a glycoside hydrolase (GH), Pn3Pase, which degrades the linear repeating disaccharide polymer -3)βGlcA(1-4)βGlc(1- that makes up Pn3P (Avery and Dubos 1930; Dubos and Avery 1931; Sickles and Shaw 1934). Recently, we reported further on Pn3Pase identifying and cloning the Pn3Pase gene from Paenibacillus sp. 32352, determining optimal activity parameters and characterizing oligosaccharide products from Pn3P degradation (Middleton et al. 2018b). Further, we have reported on Pn3Pase potential as a therapeutic agent against type 3 pneumococcal infections (Middleton et al. 2018a).
With the reclassification of the Paenibacillus bacteria and exploration into the Pn3Pase gene it appeared, this GH did not belong to any existing carbohydrate-active enzyme (CAZy) (Lombard et al. 2014) (http://www.cazy.org) family. Sequence alignments revealed no significant homology to current CAZy GH families over the entire length of a known catalytic domain; however, InterPro analysis suggests local similarities to family GH39 (Mitchell et al. 2019). Additionally, Phyre2 analysis suggests sequence similarities to glucuronidases of family GH79 (Kelley et al. 2015). Sequence alignments also suggest that Pn3Pase may belong to the GH-A clan and thus may function with a retaining mechanism, as other GH-A clan members. Further, this enzyme has a primary sequence without homologs in the database, and it is also functionally unique. To our knowledge, this is the only enzyme that demonstrates Pn3P hydrolysis activity. Taken together, this information suggested that Pn3Pase may establish and belong to a new GH family.
Given that Pn3Pase has potential as a therapeutic agent against the highly infectious serotype 3 S. pneumoniae, determining the structure function relationship of this enzyme is crucial to generating Pn3Pase derivatives with optimal activity and pharmacokinetics. Apart from the study exploring Pn3Pase as a therapeutic (Middleton et al. 2018a) and the work on elucidating the Pn3Pase gene for recombinant protein expression, (Middleton et al. 2018b) little work has been done on this enzyme. We do not yet know enzymatic activity sites/domains, mechanism of action, substrate binding specificity, kinetic parameters or structure.
In the continuation of our work characterizing this important enzyme, here we present the detailed biochemical analysis of Pn3Pase. This work led to the establishment of a new GH family designated GH169 of which Pn3Pase is its founding member. Site-directed mutagenesis revealed the nucleophile and acid/base catalytic residues, while C-terminal truncations indicated domains crucial for activity. Binding and kinetic assays revealed this enzyme’s activity toward Pn3P and its substrate specificity. Nuclear magnetic resonance (NMR) analysis demonstrated that Pn3Pase utilizes a retaining mechanism similar to other GH-A clan members. Taken together, this work provides the biochemical basis for the Pn3P hydrolysis catalyzed by Pn3Pase. Additionally, this work will aid in developing Pn3Pase as a therapeutic agent against the highly pathogenic type 3 pneumococcus infection.
Results
Pn3Pase domain analysis and production
We previously reported on the Paenibacillus sp. 32352 gene, Pbac_3551, which encodes for the Pn3Pase protein (Middleton et al. 2018b). The translated protein sequence of Pn3Pase contains 1545 amino acids and yields a mature protein of 164.1 kDa. InterPro sequence analysis (Mitchell et al. 2019) of the full protein revealed putative domains (Figure 1A, B). There was homology between residues 180 and 353 to GH superfamily, with suggested homology to GH family 39 at the N-terminal region. Other predicted regions of interests were homology to galactose-binding-like superfamily, which for the purpose of this study we took to be the potential carbohydrate-binding module (CBM) (Figure 1B), a domain of unknown function 1080 (DUF1080), which has structural similarity to a β1,3–1,4 glucanase in GH family 16, and a concanavalin A-like lectin/glucanase superfamily (Figure 1B). Alignments with existing GHs in the CAZy database revealed short segments of homology with other carbohydrate-active enzymes; however, no significant homology across the full length of Pn3Pase exists. Pn3Pase was run against CAZy HMMs built for GH families (HMMer2,3), which yielded scores insufficient for inclusion in any existing CAZy GH family. The only two borderline hits, GH39 and GH79, showed local similarity that did not extend along the length of the catalytic domains. However, the alignments with GH39 and GH79 showed the conservation of the subsequence surrounding the known acid/base residue of clan GH-A (amino acid E in GNEPN) and the known nucleophile (amino acid E in VSEYGW), with the later only visible in the alignment with GH39 (Supplementary data 1) (Henrissat et al. 1996). Altogether these low score and incomplete alignments suggested that Pn3Pase would establish a new GH family related to clan GH-A, have potential catalytic residues at amino acid sites 196 and 306, and suggested that Pn3Pase works through a retaining mechanism. Enzymes belonging to clan GH-A function through retaining mechanisms with their catalytic domain having a (β/α)8 barrel fold (Henrissat and Bairoch 1996) suggesting Pn3Pase may also have these features.
Fig. 1.
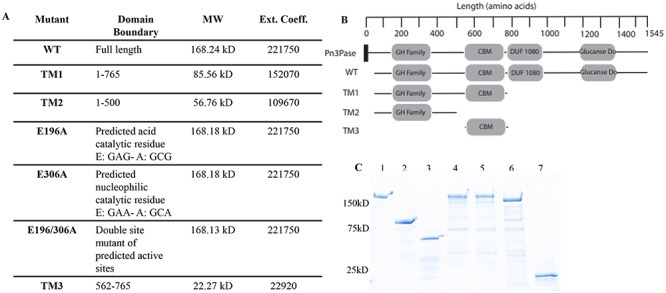
Characterization of Pn3Pase mutants. (A) The description of Pn3Pase and derivatives used in this study. Extinction coefficients were determined using ExPASy ProtParam tool, reference Gasteiger et al. (2003). (B) Schematic of predicted domains (InterPro). Pn3Pase is full-length native protein, WT is recombinant Pn3Pase, TM1,2,3 are truncation mutants (TM). CBM, carbohydrate binding module homologous to galactose binding domains. Signal peptide is represented by black rectangle. (C) Stain free SDS-PAGE gel of Pn3Pase mutants: 1) WT, 2) TM1, 3) TM2, 4) E196A, 5) E306A, 6) E196/306A and 7) TM3.
Moving forward with these preliminary evidences, we performed site-directed mutagenesis and deletion studies to confirm the identity of catalytic residues and of domains important for function (Figure 1). Figure 1A and B describe the Pn3Pase derivatives, and their predicted domains used in this study. Wild Type (WT) Pn3Pase and mutant coding sequences were amplified and cloned into pET-DEST42 vector with a C-terminal His6tag, and expressed in E. coli. Recombinant proteins were purified using affinity chromatography followed by size exclusion chromatography, and purity was assessed using SDS-PAGE gel (Figure 1C). Bands at appropriate molecular weights (Figure 1A) for each protein were observed (Figure 1C). Oligonucleotides used to generate the mutants that are displayed in Table I, and site-directed mutants were confirmed through sequencing.
Table I.
Oligonucleotides for each protein used in this study
| Oligonucleotide | Sequence 5′-3′ |
|---|---|
| WT_F | ggggacaagtttgtacaaaaaagcaggcttcgaaggagatagaaccatggcacccgtgaatctggaagc |
| WT_R | ggggaccactttgtacaagaaagctgggtgctccacgatcaccttattcgataacg |
| TM1_R | ggggaccactttgtacaagaaagctgggtgttcggcaaaaacctttacatcg |
| TM2_R | ggggaccactttgtacaagaaagctgggtgggtgtagaaagtgcggcc |
| E196A_F | gggcaacgcgccgaacc |
| E196A_R | ggttcggcgcgttgccc |
| E306A_F | tgttagcgcatacggctggaag |
| E306A_R | cttccagccgtatgcgctaaca |
| TM3_F | ggggacaagtttgtacaaaaaagcaggcttcgaaggagatagaaccatgaatatcgtgtctgccggc |
| TM3_R | ggggaccactttgtacaagaaagctgggtgttcggcaaaaacctttacatcg |
Biochemical characterization of Pn3Pase mutants
To assess domains important for function and predicted catalytic residues, we performed activity assays utilizing the p-hydroxybenzoic acid hydrazide (PAHBAH) method (Blakeney and Mutton 1980). The method labels the reducing ends of sugars and would reveal if Pn3P hydrolysis was occurring through each Pn3Pase derivative. Unmodified Pn3Pase and its derivatives were first assayed under previously established conditions utilizing 10 μg/mL Pn3P substrate in phosphate-buffered saline (PBS) with 30 nM enzyme for 30–120 min (Figure 2A). As expected WT reached Pn3P hydrolysis saturation within 30 min with labeled reducing ends not significantly increasing at 60 or 120 min (Figure 2A). Interestingly, TM1 and E196A mutant began displaying significant hydrolysis activity at 30 min, which continued to increase over the time course. However, no other Pn3Pase mutant showed hydrolase activity even after 4 h (Figure 2A). To determine if these derivatives truly exhibited no activity against Pn3P, we increased the enzyme concentration to 2x and 4x that of the original concentration (Figure 2B, C). Again, no derivatives except TM1 and E196A displayed Pn3P hydrolase activity even at increased enzyme concentrations. TM1 displayed increased levels of activity compared with E196A, which was especially evident at 120 nM where TM1 reached hydrolysis levels similar to WT after 120 min (Figure 3C).
Fig. 2.
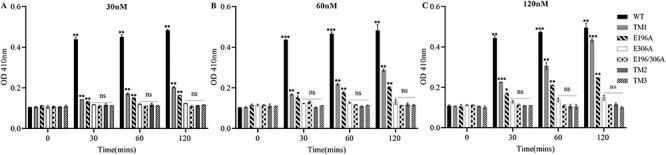
Activity characteristics of Pn3Pase mutants. p-hydroxybenzoic acid hydrazide assay was used to determine substrate degradation. All samples had 0.1 mg/mL Pn3P substrate and (A) 30 nM, (B) 60 nM and (C) 120 nM enzyme incubated for 0, 30, 60 and 120 min. Legend for all graphs is the same and to the right of (C). Significance for each data is determined using Student’s t test comparing data to corresponding time zero data.
Fig. 3.
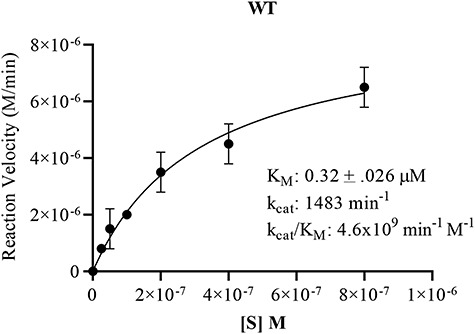
Substrate saturation curve for WT Pn3Pase. Michaelis–Menten kinetics were determined plotting substrate concentration against initial velocities for WT Pn3Pase. For kinetics experiments, WT received 1 μg/mL (5.97 nM) enzyme, respectively. Substrate concentrations were 800, 400, 200, 100, 50, 25 and 0 nM. Initial velocities were determined as the slope of the line in amount of product formed between 0 and 8 min. Amount product formed was determined using tetrasaccharide as standards. Kinetic parameters were determined using nonlinear regression (GraphPad Prism). Standard deviation of data was determined through independent duplicates.
To continue the biochemical assessment of Pn3Pase activity, we measured Michaelis–Menten kinetic parameters utilizing PAHBAH assay. Since WT was the only enzyme to display significant activity levels at early time points with low enzyme and substrate levels, we only carried on with this protein. Michaelis–Menten parameters of WT Pn3Pase were determined using different concentrations of Pn3P substrate ranging from 0–800 μM with 1 μg/mL (5.9 nM) enzyme. WT Pn3Pase displayed activity kinetics with a kcat of 1483 min−1 and KM of 0.32 ± 0.026 μM (Figure 3). The catalytic efficiency defined by the specificity constant kcat/KM was calculated as 4.6 × 109min−1 M−1.
Taken together, these results suggest that the glutamic acid residues at 196 and 306 positions function as catalytic residues. Further, both E196 and E306 are conserved glutamic acids in Pn3Pase and its close homologs as is the case for clan GH-A GHs (Davies and Henrissat 1995) making them the acid/base and the nucleophilic residues, respectively. Additionally, amino acids and domains after residue 765 do not appear to be crucial for function, while the putative CBM is required for function. Although TM1 and E196A are the only Pn3Pase mutants to display function, they hydrolyze at reduced efficiency compared with WT Pn3Pase.
Pn3Pase binding affinity to Pn3P
To determine Pn3Pase binding affinity for its substrate, we utilized biolayer interferometry (BLI). Pn3P was biotinylated, bound to streptavidin biosensors, and Pn3Pase proteins were used as ligand (Figure 4). We used only the noncatalytic Pn3Pase (low catalytic activity for E196A) mutants for this assay as WT and TM1 significantly hydrolyzed the substrate during the course of the experiment and therefore showed little to no binding (data not shown). The site mutants E196A, E306A and E196/306A had high affinities with KD (equilibrium dissociation constant) of 418, 451 and 357 nM, respectively. TM2 binding curves at 100 and 250 nM are both present but overlapped and appeared as a single curve. TM2 had a KD roughly 10-fold higher than site mutants at 3.7 μM, which appears to be due to a slower kon (second-order rate constant of the binding reaction) rate. This result is to be expected as TM2 mutant lacks the putative CBM, which has been suggested to play a large role in substrate binding (Klontz et al. 2019). Importantly, this previous study also suggests that both the GH and CBMs are involved in binding elucidating why we observed binding even in the absence of a CBM albeit at a lower affinity than noncatalytic full-length mutants (Klontz et al. 2019). In addition, we observed no apparent binding of TM3 to Pn3P substrate (Figure 4). As TM3 is the putative CBM mutant we expected to see binding; however, it has been suggested some CBMs display weak binding interactions as could be the case for our protein (Volkov et al. 2004).
Fig. 4.
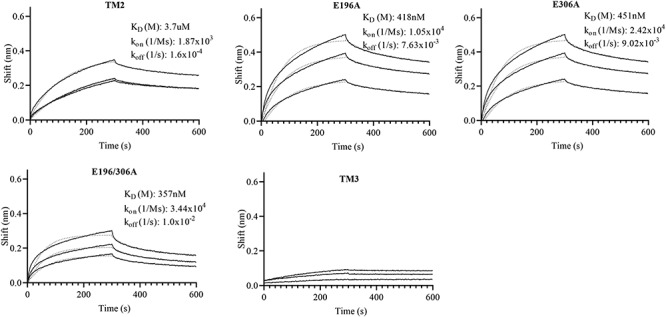
Binding characterization of Pn3Pase and mutants to Pn3P. Binding rates were determined using biolayer interferometry. Biotinylated Pn3 polysaccharide (20 μg/mL) was attached to streptavidin-coated sensor for 150 sec then returned to baseline buffer. Sensor was then exposed to Pn3Pase or mutants (100, 250 and 500 nm) for 300 sec. Dissociation was measured over 300 sec to determine KD. KD, kon and koff rates were determined as the average of the three binding curves. All three binding curves are present for TM2 mutant, 100 nm and 250 nm curves are overlapping. Fits are shown as dotted lines.
Pn3Pase reaction mechanism
Next, we determined whether Pn3Pase acts through a mechanism that yields either a retention or inversion of the stereochemistry at the GlcA anomeric reducing end (Figure 5). We used 1H NMR spectroscopy to monitor the hydrolysis of the β-GlcA(1-4)β-Glc- glycosidic linkage in Pn3P by Pn3Pase over time. Chemical shifts of Pn3P were based on previously published data (Middleton et al. 2018b). Since the β-H1 signal of free reducing GlcA (4.60 ppm) is obscured by the overlapping water signal at 37°C and further reduced by the water suppression NMR pulse program, the GlcA H5 signals were used to monitor the reaction (Figure 5). The Pn3P starting material shows the glycosidically linked β-GlcA H5 signal at 3.9 ppm (bottom trace, Figure 5). Immediately after the addition of enzyme, a signal corresponding to the H5 of the free reducing β-anomer of GlcA (3.85 ppm) is produced and continues to grow over time (Figure 5). Only after around 10 min, does a small signal appear corresponding to the H5 of the free reducing α-GlcA (4.04 ppm) (Figure 5, top trace), which is due to mutarotation. This experiment confirms that Pn3Pase functions through a retaining mechanism and supports an activity mechanism that functions through a nucleophile and an acid/base catalytic residue (Davies and Henrissat 1995) as other clan GH-A members further suggesting Pn3Pase may belong to this GH clan. Taken together these results demonstrate that Pn3Pase acts via a retaining mechanism to hydrolyze Pn3P.
Fig. 5.
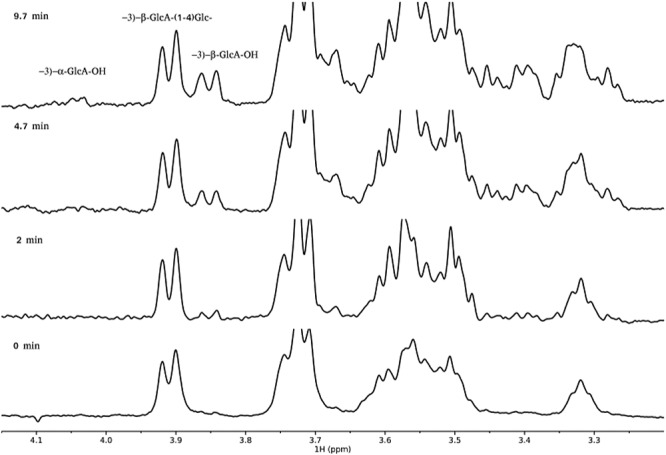
Reaction mechanism of Pn3Pase. Region of the 600 MHz proton spectrum of Pn3P, [−3)-β-D-GlcA(1-4)-β-D-Glc(1-]n in PBS buffer with 10% D2O at 37°C identifying doublet signals from GlcA-H5. Bottom trace at time zero, additional spectra at times indicated.
Discussion
The CPS of many pathogenic bacteria are prominent features serving many functions such as assisting in adhesion and colonization and inhibition of opsonophagocytosis rendering them major virulence factors (Magee and Yother 2001, Moxon and Kroll 1990). Unencapsulated mutants often fail to colonize and rarely cause infections due to efficient opsonophagocytotic clearance by host cells (Magee and Yother 2001, Nelson et al. 2007). Among S. pneumoniae species, serotype 3 is one of the most virulent (Briles et al. 1992; Martens et al. 2004; Weinberger et al. 2010) and continues to be of concern despite its inclusion in the current conjugate vaccine against pneumococcal infections (Gruber et al. 2012; Richter et al. 2013; Wantuch and Avci 2018; Wantuch and Avci 2019). Armed with this knowledge, the benefits of utilizing an alternative therapy against type 3 pneumococcal infection become apparent. Indeed, we have begun investigations into utilizing Pn3Pase, described in this study, as a therapeutic agent against highly virulent type 3 infections (Middleton et al. 2018a). Pn3Pase’s unique ability to hydrolyze the CPS of type 3 S. pneumoniae makes it a potential therapeutic agent to reduce bacterial colonization and infection. Information from this current study will aid in further developing Pn3Pase as an alternative to conventional treatments such as antibiotics, establishing the biochemical characteristics necessary for optimum therapeutic potential. Additionally, recent work on a mucin-selective protease StcE (Malaker et al. 2019) suggests a possible role for the noncatalytic Pn3Pase mutants in purifying capsule. Type 3 is among the pneumococcal serotypes that have exhibited capsule shedding (Kietzman et al. 2016). Nonactive mutants of Pn3Pase may be useful in isolating shed capsule in vivo to assess capsule shedding in bacterial virulence as well as for serotyping in clinics.
In this current work, we observed unique binding characteristics in our truncation mutants compared with noncatalytic full-length proteins. TM3 mutant did not exhibit any significant binding to Pn3P substrate as well as having abolished activity. TM3 being noncatalytic was expected (Boraston et al. 2004). It was previously noted that substrate binding for CBMs varies in a wide range with some having relatively weak interactions (Volkov et al. 2004) as could be the case in Pn3Pase’s putative CBM. TM2 recorded binding to Pn3P with approximately 10-fold lower affinity than full-length proteins. An important role of the CBM is to target the substrate and bring it into closer proximity with the enzyme (Hervé et al. 2010) that increases the hydrolysis of the substrate (Shoseyov et al. 2006). TM2 lacks the putative CBM, which paired with the published work above may explain the lower binding affinity and no enzymatic activity. With no CBM, TM2 does not appear to bring Pn3P into close proximity with the hydrolase domain nor bind the substrate long enough for hydrolysis to occur. Several studies have suggested that removal of the CBM dramatically decreases enzymatic activity (Coutinho et al. 1993; Carrard and Linder 1999; Shoseyov et al. 2006).
TM1 and E196A were the only mutants to display catalytic activity. We were unable to observe substrate binding with WT or TM1 due to degradation activity. Compared with WT, TM1 and E196A have significantly lower catalytic activities. In TM1, it is possible that this could be due to structure and removal of the C terminal domains. InterPro analysis suggests full-length Pn3Pase has 2 C-terminal domains that may possess carbohydrate-binding-like properties, the DUF1080 and a ConA-like/glucanase superfamily domain. Reviews have suggested proteins that possess hydrolytic activity, i.e. GHs, can have one or more CBMs (Shoseyov et al. 2006). If this is the case for Pn3Pase, TM1, being half the size of WT, could have lower catalytic activity due to the removal of these other potential CBM-like domains. Additionally, the C terminal domains could be important for overall tertiary structure that could aid in activity. TM1 mutant does, however, demonstrate the minimum structure requirements for Pn3Pase activity, the GH domain and putative CBM. All other C terminal domains do not appear to be essential for hydrolase activity. Our results also suggest the correct assignment of E196 as the acid/base catalytic residue and E306 as the nucleophile. This conclusion is apparent by E196A drastically decreased activity compared with WT and E306A complete loss of function. Prior work on GHs has shown that mutating these catalytic residues does not always lead to a complete ablation of activity (Armstrong and Davies 2020). With catalysis taking place at the glucuronic acid residue bond, it is possible that the carboxylate of GlcA may be able to act as the acid/base residue in the E196A mutant thereby leading to partial activity rescue (Figure 6). Further, members of clan GH-A have conserved glutamic acid residues functioning as the acid/base and nucleophile catalytic residues as appears to be the case with Pn3Pase (Henrissat et al. 1996)
Fig. 6.
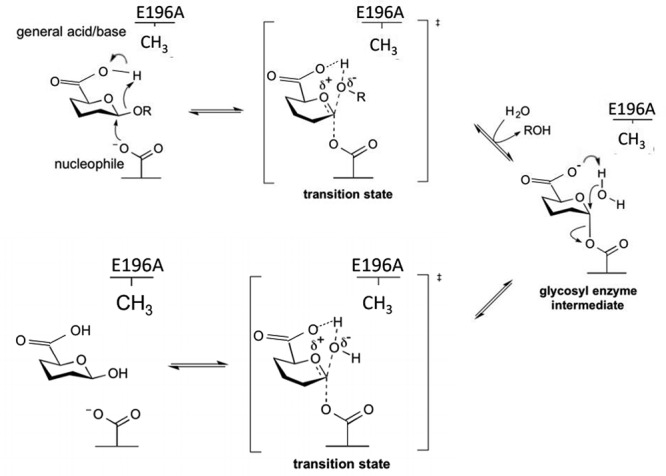
Proposed reaction mechanism for Pn3Pase and its E196A mutant (adapted from CAZypedia’s reaction mechanism for a retaining β-glycoside hydrolase). Pn3Pase operates with a double displacement mechanism which involves a glucuronyl-enzyme intermediate. The second step is the nucleophile attack of the glucuronyl-enzyme intermediate by a water residue, which is normally activated by E196. In the E196A mutant the water is likely activated, albeit less efficiently, by the carboxylate of the GlcA accounting for the small retention of activity when the acid//base catalytic residue is mutated.
A blast search using Pn3Pase sequence against the nonredundant protein sequence database of the NCBI shows that a small number of close homologs exist, all of which are essentially found in Paenibacillus sequences. Proteins with more distant similarity were also found (Supplementary data 2), but their distance to the only characterized GH169 member (Pn3Pase) is such that it is appropriate to place them into the nonclassified CAZy section of the GHs until a sufficient taxonomical diversity is captured in family GH169. The reason behind the small size of the family is unclear; perhaps, it simply means that the degradation of Pn3P is not widespread and that the more distant relatives of Pn3Pase (those outside the red box in the Supplementary data 2) may act on a different substrate.
Taken together, the work in this current study establishes the initial biochemical analysis of the GH Pn3Pase, which led to the establishment of GH family GH169. Despite our best efforts we have yet to obtain a crystal structure of this enzyme. Due to the large sequence divergence between Pn3Pase and other clan GH-A members, we were also unable to build a homology model that had meaningful features. However, similarity with clan GH-A predicts, Pn3Pase would have a β/α8 barrel fold (TIM barrel), but the exact structural details that govern substrate recognition cannot be reliably predicted. Continuing work with Pn3Pase will involve a crystal structure to complement the work in this study and shed more light into the structure–function relationship of Pn3Pase with Pn3P. Additionally, further structural analysis will reveal if the correlations between activity and domains observed in this study are accurate and confirm the assignment of residues E196 and E306 as the acid/base and nucleophile. Further structural investigations will also aid in developing Pn3Pase as a therapeutic against type 3 pneumococcal infections.
Materials and methods
Production of recombinant Pn3Pase and mutants
WT recombinant Pn3Pase (DDBJ/ENA/GenBank accession number: MZNT01000000) was produced as previously described (Middleton et al. 2018b). Briefly, the coding region of Pbac_3331 (Pn3Pase) was amplified from Paenibacillus sp. 32352 genomic deoxyribonucleic acid (DNA) into pDONR221 using BP clonase reaction (Thermo Fisher Scientific). Primers for cloning WT and all mutants are listed in Table I. After transformation into DH5α cells and DNA sequence confirmation, LR clonase reaction (Thermo Fisher Scientific) was performed to insert into pET-DEST42 destination vector for the expression of a carboxy-terminal His6-tagged fusion protein in E. coli BL21(DE3) cells. C-terminal truncation mutants, TM1, TM2 and TM3 were produced by first amplifying DNA in polymerase chain reaction (PCR) reaction containing 25 μL 2x master mix, 2.5 μL each of forward and reverse primers, 2.5 μL template Pbac. genomic DNA, and brought up to 50 μL with water. PCR reaction conditions were 98°C 120 s, followed by 20 cycles of 98°C for 15 s, 55°C for 15 s and 72°C for 6 min. PCR products were visualized on 1% agarose gel and excised. DNA was extracted from gel using EZNA gel extraction kit (Omega Bio-tek). Using extracted DNA, mutants were then produced following the same BP and LR clonase reactions as WT above. Single and double site mutant proteins were produced using the QuikChange XL Site Directed Mutagenesis Kit (Agilent). PCR reactions contained 5 μL 10x buffer, 5 μL Pbac_3551 DNA in pDONR221 vector, 2.5 μL each of forward and reverse primers, 1 μL dNTPs, 3 μL QuikSolution, 1 μL PfuUltra HF and brought up to 50 μL with water. PCR reaction conditions were 95°C for 60 s followed by 18 cycles of 95°C for 50 s, 60°C for 50 s and 68°C for 9 min. Following PCR Dpn1, reaction from kit was performed followed by transformation into XL10 competent cells and DNA sequence confirmation. LR clonase reaction was done same as above.
Purification of enzymes
BL21 cells transformed with pET-DEST42-Pn3Pase or mutant plasmid were grown in LB medium supplemented with 100 μg/mL ampicillin at 37°C while cell density was monitored at absorbance 600 nm. Once OD 600 nm reached 1, cells were transferred to 20°C, protein expression was induced by adding Isopropyl β-D-1-thiogalactopyranoside to a final concentration of 1 mM, and the cell culture was allowed to incubated for 24 h. Cells were harvested by centrifugation. Cells were resuspended in PBS (pH 7.2) with 10 μg/mL deoxyribonuclease and lysed using EmulsiFlex-C5 homogenizer (Avestin). Lysate was cleared by centrifugation at 18,000 xg for 45 min at 4°C and passed through a 0.45-um filter. Proteins were purified by Ni2+-NTA resin at 4°C and eluted with 300 mM imidazole. Elution was run through FPLC Superdex 200 sizing column (GE LifeSciences) with PBS as running buffer. Protein concentration was determined using NanoDrop (Thermo Fisher Scientific) using extinction coefficients for each protein determined using ExPASy ProtParam tool (Gasteiger et al. 2003). Purity was assessed by visualizing proteins on stain-free tris-glycine gel (BioRad) using gel doc EZ imager (BioRad).
Enzyme activity assays
Recombinant Pn3Pase and mutant hydrolysis activity was determined by measuring the increase in reducing ends using p-hydroxybenzoic acid hydrazide (PAHBAH) method (Blakeney and Mutton 1980). A reaction mixture (100 μL) containing 10 μg Pn3P (American Type Culture Collection 172-X) and 30 nM, 60 nM or 120 nM enzyme in PBS was incubated at 37°C for 30, 60 or 120 min then heated at 100°C for 5 min to stop reaction. Time 0, used as a negative control, was the same reaction mixtures containing 30 nM, 60 nM or 120 nM of heat-killed enzyme. A volume of 40 μL of the reaction was mixed with 120 μL of 1% (w/v) PAHBAH-hydrocholoric acid (HCl) solution in duplicate, heated at 100°C for 5 min. Absorbance at 410 nm was measured on Biotek synergy H1 microplate reader in a clear flat bottom 96-well microplate. Statistical analysis was determined using Student’s t test and compared all data points with their corresponding time zero data.
Enzyme kinetics
The Michaelis–Menten constant (KM) and the maximum velocity (Vmax) of WT Pn3Pase were measured using Pn3P as substrate (average molecular weight: 400,000 Da). Pn3P was found to be 400 kDa as determined by size exclusion using the Sephacryl S-300 column (GE Healthcare) using the refractive index of defined mass dextran standards for comparison (Li et al. 2015). The substrate was used at seven concentrations for WT (800, 400, 200, 100, 50, 25 and 0 nM) in PBS. WT Pn3Pase was added at 1 μg/mL (5.9 nM). Reactions were heated at 37°C, and aliquots were taken/stopped at 0, 4, 8, 12, 16 and 20 min (corresponding to approximately 10% of total hydrolysis yielding mostly tetrasaccharides) by heating reactions at 100°C for 5 min. The amount of product formed was measured using PAHBAH-HCl assay as described above with tetrasaccharides, obtained from enzymatic degradation of Pn3P and used to generate a standard curve for data fitting. Initial velocity was determined using the amount of product formed in the linear region of absorbance. Initial velocities of each substrate concentration were curve fitted using nonlinear regression with Michaelis–Menten model on GraphPad Prism to determine KM and Vmax. kcat was likewise determined using GraphPad Prism as Vmax/ET with ET set as total enzyme concentration 5.9 nM. The catalytic efficiency kcat/Km was determined using these values in min−1 M−1. Tetrasaccharides used for standard curve were generated as previously described (Middleton et al. 2018b). Standard deviation of data was determined through independent experimental duplicates.
Binding assay
Binding affinity of recombinant proteins to Pn3P substrate was determined using BLI (FortèBio OctetRED-384). All proteins and Pn3P were suspended in buffer (PBS, 0.5% bovine serum albumin, 0.05% tween). Biotinylated Pn3P substrate (20 μg/mL) was immobilized on streptavidin biosensor tips (FortèBio) for 150 s after an initial baseline in running buffer for 60 s. Baseline signal was measured again for 60 s before biosensor tips were immersed in wells containing protein (0, 100, 250 or 500 nM) for 300 s. Dissociation was measured by returning biosensor tips to baseline for 300 s. Octet data analysis software was used to analyze data. Values of reference wells containing no protein (0 nm) were subtracted from data, and affinity values were calculated using local and partial fit curve function with a 1:1 binding model. kon, koff and KD values were determined as the average of the three substrate concentration binding curves. Binding curves were graphed using GraphPad Prism.
NMR for stereochemical analysis
1H NMR analysis was performed on reaction products released from Pn3Pase hydrolysis of Pn3P to determine reaction mechanism. Pn3P was resuspended as 1 mg/mL (2.6 mM) in PBS and 10% D2O and transferred to a 3 mm NMR tube. After collecting an initial spectrum at 37°C, the enzyme was added to the tube with a final concentration of 15 μg/mL, mixed, and data collection then resumed after 2 min. Data were collected continually, and four transients were summed every 8 sec. NMR spectra were acquired on an Agilent 600 MHz DD2 spectrometer equipped with a 3 mm cryoprobe and used the standard presaturation pulse sequence to reduce the water signal. Data were processed with Mnova software (Mestrelab, Inc.)
Generation of biotinylated Pn3P
Biotinylated Pn3P was prepared using hydrazide biotin. In total, 2 mg of 25 kDa Pn3P was dissolved in 500 μL of 0.1 M sodium borate buffer (pH 5.4). EZ-Link hydrazide-biotin (Thermo Fisher Scientific) (12 mg) was dissolved in 100 μL dimethyl sulfoxide and added to Pn3P solution. EDC (1-ethyl-3-(3-dimethylaminopropyl) carboiimide HCl) (Thermo Scientific) (1.5 mg) was added to the solution, vortexed and incubated before at 25°C for 3 h with agitation. Product was purified using Superdex 200 sizing column (GE LifeSciences).
Funding
National Institutes of Health (R01AI123383 to F.A.).
Supplementary Material
Abbreviations
- BLI
biolayer interferometry
- CAZy
carbohydrate-active enzyme
- CBM
carbohydrate-binding module
- CPS
capsular polysaccharide
- DNA
deoxyribonucleic acid
- GH
glycoside hydrolase
Contributor Information
Paeton L Wantuch, Department of Biochemistry & Molecular Biology, University of Georgia, 325 Riverbend Rd, Athens GA 30602, USA; Center for Molecular Medicine, University of Georgia, 325 Riverbend Rd, Athens GA 30602, USA.
Satya Jella, Center for Molecular Medicine, University of Georgia, 325 Riverbend Rd, Athens GA 30602, USA.
Jeremy A Duke, Department of Biochemistry & Molecular Biology, University of Georgia, 325 Riverbend Rd, Athens GA 30602, USA; Center for Molecular Medicine, University of Georgia, 325 Riverbend Rd, Athens GA 30602, USA.
Jarrod J Mousa, Center for Vaccines and Immunology, College of Veterinary Medicine, University of Georgia, 501 D.W. Brooks Dr Athens, Athens GA 30602, USA; Department of Infectious Diseases, College of Veterinary Medicine, University of Georgia, 501 D.W. Brooks Dr Athens, Athens GA 30602, USA.
Bernard Henrissat, Architecture et Fonction des Macromolécules Biologiques, CNRS, Aix-Marseille University, 163 Avenue de Luminy, Parc Scientifique et Technologique de Luminy, 13288 Marseille, France; USC1408 Architecture et Fonction des Macromolécules Biologiques, Institut National de la Recherche Agronomique, 163 Avenue de Luminy, Parc Scientifique et Technologique de Luminy, 13288 Marseille, France; Department of Biological Sciences, King Abdulaziz University, Al Jami`ah, Jeddah, 23218, Saudi Arabia.
John Glushka, Complex Carbohydrate Research Center, University of Georgia, 315 Riverbend Rd, Athens GA 30602, USA.
Fikri Y Avci, Department of Biochemistry & Molecular Biology, University of Georgia, 325 Riverbend Rd, Athens GA 30602, USA; Center for Molecular Medicine, University of Georgia, 325 Riverbend Rd, Athens GA 30602, USA.
References
- Armstrong Z, Davies GJ. 2020. Structure and function of Bs164 β-mannosidase from Bacteroides salyersiae the founding member of glycoside hydrolase family GH164. J Biol Chem. 295:4316–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery OT, Dubos R. 1930. The specific action of a bacterial enzyme on pneumococci of type III. Science. 72:151–152. [DOI] [PubMed] [Google Scholar]
- Blakeney AB, Mutton LL. 1980. A simple colorimetric method for the determination of sugars in fruit and vegetables. J Sci Food Agric. 31:889–897. [Google Scholar]
- Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. 2004. Carbohydrate-binding modules: Fine-tuning polysaccharide recognition. Biochem J. 382:769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briles DE, Crain MJ, Gray BM, Forman C, Yother J. 1992. Strong association between capsular type and virulence for mice among human isolates of Streptococcus pneumoniae. Infect Immun. 60:111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrard G, Linder M. 1999. Widely different off rates of two closely related cellulose-binding domains from Trichoderma reesei. Eur J Biochem. 262:637–643. [DOI] [PubMed] [Google Scholar]
- Coutinho JB, Gilkes NR, Kilburn DG, Warren RAJ, Miller RC Jr. 1993. The nature of the cellulose-binding domain effects the activities of a bacterial endoglucanase on different forms of cellulose. 113(2):211–217. [Google Scholar]
- Davies G, Henrissat B. 1995. Structures and mechanisms of glycosyl hydrolases. Structure. 3:853–859. [DOI] [PubMed] [Google Scholar]
- Dubos R, Avery OT. 1931. Decomposition of the capsular polysaccharide of pneumococcus type III by a bacterial enzyme. J Exp Med. 54:51–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. 2003. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31:3784–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber WC, Scott DA, Emini EA. 2012. Development and clinical evaluation of Prevnar 13, a 13-valent pneumocococcal CRM197 conjugate vaccine. Ann N Y Acad Sci. 1263:15–26. [DOI] [PubMed] [Google Scholar]
- Henrissat B, Bairoch A. 1996. Updating the sequence-based classification of glycosyl hydrolases. Biochem J. 316(Pt 2):695–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrissat B, Callebaut I, Fabrega S, Lehn P, Mornon JP, Davies G. 1996. Conserved catalytic machinery and the prediction of a common fold for several families of glycosyl hydrolases. Proc Natl Acad Sci U S A. 93:5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervé C, Rogowski A, Blake AW, Marcus SE, Gilbert HJ, Knox JP. 2010. Carbohydrate-binding modules promote the enzymatic deconstruction of intact plant cell walls by targeting and proximity effects. Proc Natl Acad Sci U S A. 107:15293–15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kietzman CC, Gao G, Mann B, Myers L, Tuomanen EI. 2016. Dynamic capsule restructuring by the main pneumococcal autolysin LytA in response to the epithelium. Nat Commun. 7:10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klontz EH, Trastoy B, Deredge D, Fields JK, Li C, Orwenyo J, Marina A, Beadenkopf R, Günther S, Flores J, et al. 2019. Molecular basis of broad spectrum N-glycan specificity and processing of therapeutic IgG monoclonal antibodies by endoglycosidase S2. ACS Cent Sci. 5:524–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Li L, Xue C, Middleton D, Linhardt RJ, Avci FY. 2015. Profiling pneumococcal type 3-derived oligosaccharides by high resolution liquid chromatography-tandem mass spectrometry. J Chromatogr A. 1397:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42:D490–D495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee AD, Yother J. 2001. Requirement for capsule in colonization by Streptococcus pneumoniae. Infect Immun. 69:3755–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaker SA, Pedram K, Ferracane MJ, Bensing BA, Krishnan V, Pett C, Yu J, Woods EC, Kramer JR, Westerlind U, et al. 2019. The mucin-selective protease StcE enables molecular and functional analysis of human cancer-associated mucins. Proc Natl Acad Sci U S A. 116:7278–7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens P, Worm SW, Lundgren B, Konradsen HB, Benfield T. 2004. Serotype-specific mortality from invasive Streptococcus pneumoniae disease revisited. BMC Infect Dis. 4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton DR, Lorenz W, Avci FY. 2017. Complete genome sequence of the bacterium. Genome Announc. 5: e00289-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton DR, Paschall AV, Duke JA, Avci FY. 2018a. Enzymatic hydrolysis of pneumococcal capsular polysaccharide renders the bacterium vulnerable to host defense. Infect Immun. 86 :e00316-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton DR, Zhang X, Wantuch PL, Ozdilek A, Liu X, LoPilato R, Gangasani N, Bridger R, Wells L, Linhardt RJ, et al. 2018b. Identification and characterization of the Streptococcus pneumoniae type 3 capsule-specific glycoside hydrolase of Paenibacillus species 32352. Glycobiology. 28:90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AL, Attwood TK, Babbitt PC, Blum M, Bork P, Bridge A, Brown SD, Chang HY, El-Gebali S, Fraser MI, et al. 2019. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47:D351–D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moxon ER, Kroll JS. 1990. The role of bacterial polysaccharide capsules as virulence factors. Curr Top Microbiol Immunol. 150:65–85. [DOI] [PubMed] [Google Scholar]
- Nelson AL, Roche AM, Gould JM, Chim K, Ratner AJ, Weiser JN. 2007. Capsule enhances pneumococcal colonization by limiting mucus-mediated clearance. Infect Immun. 75:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter SS, Heilmann KP, Dohrn CL, Riahi F, Diekema DJ, Doern GV. 2013. Pneumococcal serotypes before and after introduction of conjugate vaccines, United States, 1999-2011(1.). Emerg Infect Dis. 19:1074–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoseyov O, Shani Z, Levy I. 2006. Carbohydrate binding modules: biochemical properties and novel applications. Microbiol Mol Biol Rev. 70:283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickles GM, Shaw M. 1934. A systematic study of microorganisms which decompose the specific carbohydrates of the pneumococcus. J Bacteriol. 28:415–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov II, Lunina NA, Velikodvorskaia GA. 2004. Prospects for practical application of substrate-binding modules of glycosyl hydrolases. Prikl Biokhim Mikrobiol. 40:499–504. [PubMed] [Google Scholar]
- Wantuch PL, Avci FY. 2018. Current status and future directions of invasive pneumococcal diseases and prophylactic approaches to control them. Hum Vaccin Immunother. 14:2303–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wantuch PL, Avci FY. 2019. Invasive pneumococcal disease in relation to vaccine type serotypes. Hum Vaccin Immunother. 15: 874–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger DM, Harboe ZB, Sanders EA, Ndiritu M, Klugman KP, Rückinger S, Dagan R, Adegbola R, Cutts F, Johnson HL, et al. 2010. Association of serotype with risk of death due to pneumococcal pneumonia: A meta-analysis. Clin Infect Dis. 51:692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.