

Abstract
Pleurotus mushrooms are among the most cultivated fungi in the world and are highly valuable for food, medicine, and biotechnology industries. Furthermore, Pleurotus species are carnivorous fungi; they can rapidly paralyze and kill nematodes when nutrient-deprived. The predator–prey interactions between Pleurotus and nematodes are still widely unexplored. Moreover, the molecular mechanisms and the genes involved in the carnivorous behavior of Pleurotus mushrooms remain a mystery. We are attempting to understand the interactions between Pleurotus mushrooms and their nematode prey through genetic and genomic analyses. Two single spores (ss2 and ss5) isolated from a fruiting body of Pleurotus pulmonarius exhibited significant differences in growth and toxicity against nematodes. Thus, using PacBio long reads, we assembled and annotated two high-quality genomes for these two isolates of P. pulmonarius. Each of these assemblies contains 23 scaffolds, including 6 (ss2) and 8 (ss5) telomere-to-telomere scaffolds, and they are among the most complete assembled genomes of the Pleurotus species. Comparative analyses identified the genomic differences between the two P. pulmonarius strains. In sum, this work provides a genomic resource that will be invaluable for better understanding the Italian oyster mushroom P. pulmonarius.
Keywords: Pleurotus pulmonarius, oyster mushroom, nematophagous fungi
Introduction
Pleurotus mushrooms are among the most cultivated and consumed edible fungi in the world (Gregori et al. 2007; Yin et al. 2014). While most fungi are difficult to cultivate, Pleurotus can be grown commercially to high yield (Banik and Nandi 2004). In addition to their nutritional value, these mushrooms are a natural source of prebiotics (Aida et al. 2009) and antioxidants (Khatun et al. 2015) and are thus of great interest to the food industry. Also, Pleurotus species contain remarkable medicinal properties (Golak-Siwulska et al. 2018). For example, Pleurotus pulmonarius exhibits anti-inflammatory (Smiderle et al. 2008; Nguyen et al. 2016), analgesic, and antitumor activities (Zhang et al. 2007). This fungus has also shown its potential in bioremediation since it is able to degrade contaminants such as aromatic pollutants (Rodrı’guez et al. 2004) and biocides (Law et al. 2003). More recent studies have focused on the potential outcomes for the biotechnology of P. pulmonarius. For instance, this fungus can produce useful enzymes even while degrading waste products (Inácio et al. 2015) as well as being consumed after being part of biofuel production (Chen et al. 2020), therefore underlying the versatility of this fungus.
One of the most unknown characteristics of Pleurotus mushrooms is their ability to kill and feed on living nematodes (Thorn and Barron 1984) and therefore their potential for the biocontrol of parasitic nematodes (Degenkolb and Vilcinskas 2016; Sivanandhan et al. 2017; Castañeda-Ramírez et al. 2020). Although a few compounds produced by Pleurotus have been reported to cause paralysis in nematode worms (Kwok et al. 1992; Satou et al. 2008), a more recent study has suggested that the true identity of the toxins targeting nematodes remains to be discovered (Lee et al. 2020).
The advance of sequencing techniques has led to the publication of the genome of several Pleurotus species; however, only a few high-quality assemblies have been produced for Pleurotus so far. Only the latest assemblies of Pleurotus ostreatus (Riley et al. 2014; Qu et al. 2016; Wang et al. 2018) show the overall quality and contiguity necessary for more comprehensive genetic analyses of these mushrooms. In contrast, most of the Pleurotus genome assemblies are very fragmented, including Pleurotus eryngii, Pleurotus tuoliensis (Zhang et al. 2018), Pleurotus platypus and Pleurotus citrinopileatus (Li et al. 2018) ranging from 106 to 10,689 scaffolds. In addition, other Pleurotus species, such as P. pulmonarius, have not yet been sequenced despite their importance for different industries.
We thus studied the genomes of two haploid monokaryotic strains derived from spores of a wild isolate of P. pulmonarius collected in Taiwan. These two haploid monokaryons (ss2 and ss5) exhibit significant phenotypic differences in their growth and toxicity against nematodes. Using PacBio long reads, we produced two high-quality annotated genomes for P. pulmonarius. Moreover, our assemblies represent the first available P. pulmonarius genomes and are among the most complete assembled Pleurotus. Through this report we also provide genomic comparisons between the two assemblies, identifying highly dissimilar regions that might contribute to their observed phenotypic differences. We believe that the two high-quality genomes will be useful genomic resources that will contribute to a better understanding of P. pulmonarius and pave the way for future comparative genomic analyses of Pleurotus mushrooms.
Materials and methods
Fungal strains and DNA extraction
Spores of P. pulmonarius were collected from a fruiting body. Two of the monokaryon strains (ss2 and ss5) each derived from a single basidiospore were established in the laboratory for analysis. Fungi were cultured on YMG medium (yeast extract, malt extract, and glucose) at 25°C. To extract their DNA, fungal mycelium was inoculated in 100 ml of YMG liquid medium at 25°C for 3 days. DNA was extracted using the cetyltrimethylammonium bromide and purified with chloroform, isopropanol, and phenol-chloroform. The genome of ss2 and ss5 was sequenced from the Pacbio RSII platform performed at Ramaciotti Centre for Genomics (Sydeny, Australia) using a PacBio SMRTBell Template Prep Kit 1.0 SPv3.
Fresh cultures of the P. pulmonarius strains were grown from mother cultures on YMG at 25°C for 7 days and then transferred to potato dextrose agar (PDA) and low nutrient medium (LNM) plates (5-cm diameter) for phenotypic characterization (in biological triplicates). After 13 days of growth, 30 adult C. elegans nematodes (strain N2) were added to the LNM plates and the number of paralyzed worms on each place was computed after 10 min.
Genome sequencing and assembly
Long PacBio reads were sequenced from each P. pulmonarius strain with the Pacbio RSII platform. A total of ∼0.5 M reads with a mean length of 9419 bp were obtained from ss2, while ∼0.7 M reads with a mean length of 8422 bp were obtained from ss5; these corresponded to 122× genome coverage for ss2 and 142× genome coverage for ss5. The Pacbio reads were used to build two preliminary assemblies: one using Canu (v1.7) (Koren et al. 2017) and the other using Falcon (pbalign v0.0.2) (Chin et al. 2016). Canu was executed with parameters genomeSize=40m useGrid=false maxThreads=8, where the estimated genome size of 40 Mbp was calculated as the average of the genome sizes of P. eryngii (44.61 Mbp) and P. ostreatus (34.3–35.6 Mbp). Falcon was run using an adapted version of the configuration file for fungal genomes assembling provided at: https://pb-falcon.readthedocs.io/en/latest/parameters.html, where Genome_size was set to 40 Mb and memory-related options were changed to fit the requirements of PSC Bridges (Towns et al. 2014; Nystrom et al. 2015). Next, we used Sourmash (v2.0.0) (Titus Brown and Irber 2016) to identify and remove contaminants from each preliminary assembly. The decontaminated assemblies were subsequently polished using Quiver (genomicconsensus v2.3.2) (Chin et al. 2013) with the original raw PacBio reads, mapped to the decontaminated assembly using pbalign (v0.0.2), as input. Further information about the preliminary assemblies together with the results obtained from each of the abovementioned steps is found in Supplementary Table S1.
The polished assemblies obtained with Canu were more accurate at the telomeric regions, while the Falcon assemblies were more contiguous. Hence, we merged the Canu and Falcon assemblies of each P. pulmonarius strain using Quickmerge (v0.3) (Chakraborty et al. 2016). Quickmerge uses the information of a donor assembly to fill in the gaps of a reference assembly; accordingly, we tested both Canu as reference and Falcon as donor (Canu-Falcon) and vice versa (Falcon–Canu). Since the Canu–Falcon assemblies showed better contiguity and overall statistics (Supplementary Table S2), they were selected for further assembling steps. Nucmer (Mummer4) (Marçais et al. 2018) was used to detect redundant contigs in the Canu–Falcon assemblies (function maxmatch where the Canu–Falcon assembly was used both as reference and query). As a result, two small contigs (28,260 and 15,057 bp in size) were removed from the ss2 assembly, while only one contig (22,621 bp in size) was removed from the assembly of ss5. Finally, the contigs from the two assemblies were sorted by size and renamed.
Genome annotation
The assemblies of P. pulmonarius were annotated with funannotate (v1.5.2) (Palmer and Stajich 2016) using the following steps and options. First, the genomes were softmasked using funannotate mask, calling RepeatMasker (Smit et al. 2013). The masked genomes were used as input for funannotate train (options: –max_intronlen 2000 –stranded no), together with RNA-seq data from P. ostreatus [strain PC9 (Alfaro et al. 2016), downloaded from the Joint Genome Institute (JGI) at https://mycocosm.jgi.doe.gov/PleosPC9_1/PleosPC9_1.info.html (Grigoriev et al. 2012)]. During this step, Trinity (Grabherr et al. 2011) and PASA (Haas et al. 2003) were run to generate preliminary gene models. Then, funannotate predict was used to generate consensus gene models from the preliminary gene models and known transcripts and proteins of P. ostreatus (use as evidence for gene prediction). Funannotate predict calls the following programs: AUGUSTUS (Stanke and Waack 2003), GeneMark (Borodovsky and McIninch 1993), and EVidenceModeler (Haas et al. 2008). In the final step of the annotation, funannotate annotate was run to add functional annotation to the final gene models including: InterPro and PFAM domains, GO ontology terms, fungal transcription factors, COGs, secondary metabolites [AntiSMASH (Medema et al. 2011) was run using funnanotate remote], CAZYmes, secreted proteins, proteases (MEROPS), and BUSCO groups.
Genome analysis and comparison
General assembly statistics such as N50, length, and number of scaffolds were computed directly from the fasta files using the Perl script count_fasta_residues.pl (https://github.com/SchwarzEM/ems_perl/blob/master/fasta/count_fasta_residues.pl). BUSCO completeness was assessed using BUSCO 3.0.1 (Simão et al. 2015) against the basidiomycota dataset basidiomycota_odb9. We used the protocol described in Coghlan et al. (2018) to make repeat libraries and subsequently look for repetitive elements in each genome. We further identified and classified transposable elements (TEs) using the one_code_to_find_them_all script (Bailly-Bechet et al. 2014). In addition, we searched for telomeric regions characterized by the telomeric repeating unit TTAGGG (Pérez et al. 2009). Raw PacBio reads were mapped to each genome using pbalign and SAMtools (Li et al. 2009) to compute local coverage.
To find structural differences between the two genomes, we used megablast (Morgulis et al. 2008) to precisely define the boundaries for regions of similarity and dissimilarity that were grossly visible in plots by Circos (Krzywinski et al. 2009) and Dgenies (relying on minimap2) (Cabanettes and Klopp 2018). Other comparisons between genomes were derived from the results of funannotate compare (Palmer and Stajich 2016).
Data availability
The final assembled and annotated genomes of P. pulmonarius spores ss2 and ss5 were uploaded to NCBI and are available with accession codes: GCA_012980525.1 and GCA_012980535.1, respectively. In addition, all the strains used in this study will be deposited in FGSC and are available upon request.
Supplementary materials are available at figshare DOI: https://doi.org/10.25387/g3.13078253.
Results and discussion
Phenotypic differences between the two meiotic progenies of a P. pulmonarius mushroom
A wild P. pulmonarius mushroom (heterokaryon) was collected near Taipei, Taiwan, and its basidiospores were collected and individually dissected using a dissecting microscope. Two monokaryotic strains derived from two basidiospores (referred as ss2 and ss5 strains from here on) were selected for genome sequencing because they exhibited prominently different phenotypes. For example, the growth of the two strains on LNM and on the rich PDA was visibly dissimilar (Figure 1A). On nutrient-rich medium (PDA), ss2 developed a denser, but smaller mycelium colony compared to that of ss5. On LNM, ss2 developed a smaller colony that exhibited similar hyphal density compared to that of ss5. In both conditions, the growth of ss5 was much faster than ss2. We further discovered that ss2 and ss5 exhibited differences in toxicity toward the nematode Caenorhabditis elegans. All nematodes in contact with the mycelium of ss5 were paralyzed after 10 min exposed to the fungal hyphae, which was comparable to previous studies of Pleurotus species (Thorn and Barron 1984; Lee et al. 2020). In contrast, the mycelium of ss2 showed a much weaker toxicity toward C. elegans, being able to paralyze only ∼40% of the nematode population tested (Figure 1B). In view of these results, we decided to further analyze the genetic differences of the two P. pulmonarius spores by sequencing their genomes.
Figure 1.

Phenotypic characterization of P. pulmonarius mycelia from ss2 and ss5. (A) Mycelia developed after 13 days of growth for each of the studied spores on PDA and LNM. (B) Percentage of C. elegans (over a total of 30 nematodes) paralyzed after 10 min of contact with each P. pulmonarius strain.
High-quality genome assemblies of P. pulmonarius
Long PacBio reads were sequenced from both P. pulmonarius strains, ss2 and ss5, to build two high-quality genome assemblies. The genome size of the assembled P. pulmonarius is ∼39.2 Mb for ss2 and ∼39.9 Mb for ss5 (Table 1). These sizes are consistent with genome sizes observed in other related Pleurotus species: for example P. ostreatus with about 35 Mb (Riley et al. 2014; Qu et al. 2016; Wang et al. 2018) and P. eryngii with 49.9 Mb (Zhang et al. 2018). Both of our genomes are distributed in 23 scaffolds ranging in size from 5.55 Mb to 11 kb (in ss2) and 5.06 Mb to 21 kb (in ss5), and with N50 values of 3.2 and 3.4 Mb (respectively), showing high contiguity. These N50 values are higher than those of four other available Pleurotus assemblies: P. platypus (N50 = 62 kb); P. citrinopileatus (N50 = 9.7 kb) (Li et al. 2018); P. tuoliensis (N50 = 1.17 Mb); and P. eryngii (N50 = 563 kb) (Zhang et al. 2018). Moreover, these N50 values are comparable to two of the best-quality Pleurotus genome assemblies: P. ostreatus strain PC15 (N50 = 3.27 Mb) (Riley et al. 2014) and P. ostreatus strain CCMSSC00389 (N50 = 3.07 Mb) (Wang et al. 2018). We also computed the BUSCO completeness of both assemblies against the basidiomycota dataset; both assemblies showed high completeness (97.3% for ss2 and 96% for ss5). Overall, the P. pulmonarius assemblies presented here are among the most complete and contiguous of the Pleurotus species, only matched by the latest assemblies of P. ostreatus [strains PC15 (Riley et al. 2014) and CCMSSC00389 (Wang et al. 2018)].
Table 1.
Genome features of P. pulmonarius strains ss2 and ss5
| General features | P. pulmonarius ss2 | P. pulmonarius ss5 |
|---|---|---|
| Genome size (bp) | 39,235,597 | 39,866,212 |
| Number of scaffolds | 23 | 23 |
| GC content (%) | 51 | 51.04 |
| Scaffold N50 (bp) | 3,175,356 | 3,374,720 |
| Scaffold N90 (bp) | 1,418,296 | 1,759,240 |
| Scaffold max size (bp) | 5,554,685 | 5,063,380 |
| Scaffol min size (bp) | 10,862 | 21,250 |
| Number of genes | 10,848 | 11,139 |
| Number of tRNA genes | 208 | 186 |
| BUSCO completeness (%) | 97.3 | 96 |
The distribution and size of the scaffolds comprising the assembled genomes of P. pulmonarius is shown in Figure 2. First, we looked at the depth coverage at each genome position for both assemblies. While ss2 shows an average depth of 97.7 reads (Figure 2A), the average depth at each genome position of ss5 is only 86.36 reads (Figure 2B). High depth regions correspond mostly with the ends of the scaffolds for both genomes, whereas low depth is observed in some of the smaller scaffolds of both assemblies (scaffolds ss2.20 and ss2.22 for ss2 and ss5.18, ss5.19 and ss5.20 for ss5). Out of the 23 scaffolds of ss2, 6 scaffolds represent telomere-to-telomere complete chromosomes, while another 14 scaffolds show only one telomere at one end (Figure 2A). The genome of ss5 is distributed in eight whole chromosomes and eight half-chromosomes (Figure 2B). In addition, we observed telomeric repeats in several of the small scaffolds; this suggests that they may correspond to the other ends of some half-chromosomes but failed to be assembled into these larger contigs due to their repetitive nature. These results are in line with the total number of chromosomes estimated in other Pleurotus species such as P. ostreatus (11 chromosomes) (Larraya et al. 2000; Pérez et al. 2009) and P. tuoliensis (12 chromosomes) (Gao et al. 2018). Next, we generated a custom-built repeat library for each genome to study the distribution of TEs. About 7.3% (6.4%) of the genome of P. pulmonarius ss2 (ss5) consists of TEs (Table 2). The repetitive sequences of our P. pulmonarius genome assemblies mainly contain class I elements, retrotransposons, accounting for ∼90% and ∼80% of the repetitive genome fraction and about 78% and 85% of the total repeats in ss2 and ss5, respectively. TE density and distribution are also observed in Figure 2; these are highly variable, as has been previously reported for other Pleurotus species (Castanera et al. 2016). We observed clusters of TEs in both genomes mostly aligning with regions of relative low gene density (Figure 2). However, we did not observe clear evidence of centromeres, which are often characterized by clusters of TEs in fungi (Stajich et al. 2010). Detailed information about each of the classified TE identified in ss2 and ss5 is found in Supplementary Tables S3 and S4, respectively. Gene density was computed for each assembly (Figure 2) in sliding windows of 100 kb. A total of 10,848 genes were predicted in ss2 using funannotate (Palmer and Stajich 2016) and 11,139 were found in ss5 genome. These values are within the range of close Pleurotus species, such as P. ostreatus [11,603 genes in PC15 and 12,206 in PC9 (Castanera et al. 2016)] and P. eryngii [15,960 genes in strain ATCC 90797 (Gao et al. 2018)] as described in jgi (Grigoriev et al. 2012). On average about 28 genes per 100-kb sliding window were annotated in both ss2 and ss5; however, genes are not evenly distributed across the genomes presenting areas of low and high gene density for most scaffolds (Figure 2). Moreover, few genes were predicted in the smaller scaffolds of both genomes, once again suggesting that these scaffolds may correspond to misplaced telomeric or centromeric regions of chromosomes. The predicted function of genes in both genomes, cataloged using the cluster of orthologous groups (COGs) database, is found in Figure 2C. As hinted by the difference in total number of annotated genes between the genomes, ss5 contains more genes than ss2 for most functional categories. However, significant differences in the overall distribution of genes by function between the two genomes were not observed (Figure 2C).
Figure 2.
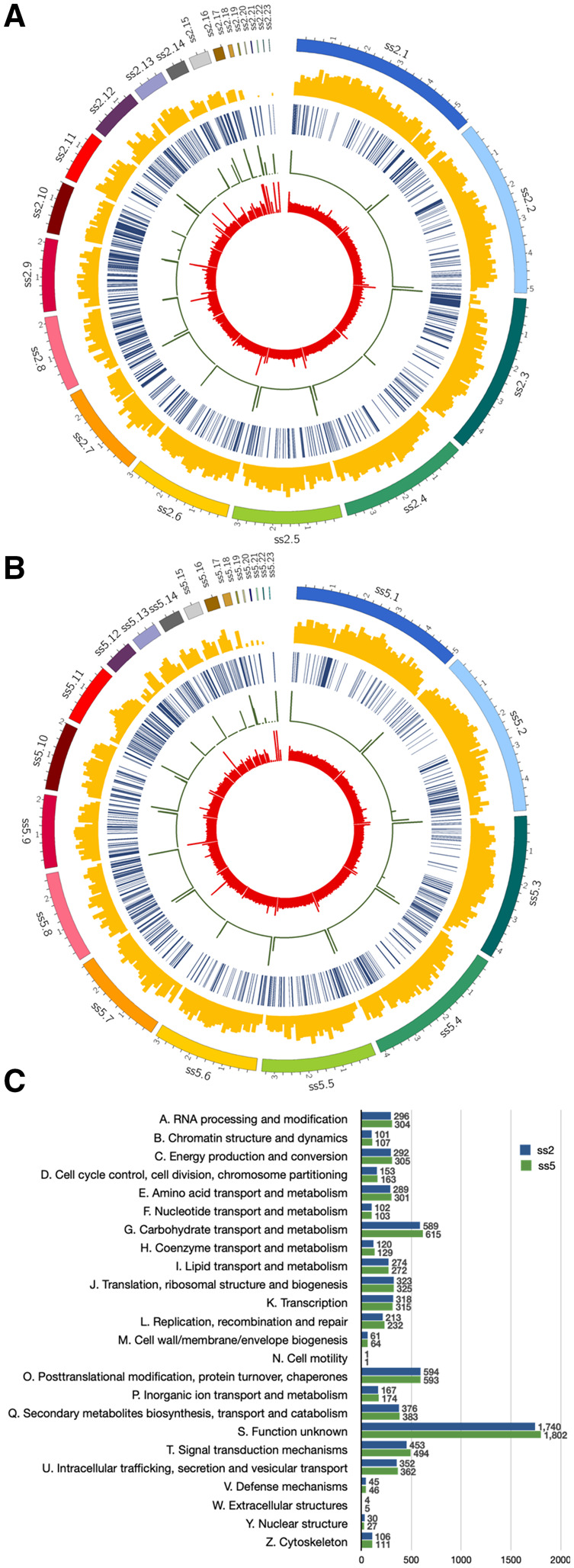
Genome architecture of P. pulmonarius. Tracks (outer to inner) represent the distribution of genomic features: (1) positions (in Mb) of the assembled scaffolds, with numbers indicating the order of scaffolds by size; (2) gene density (along 100-kb sliding windows, ranging between 0 and 50 genes); (3) distribution of TEs along the genome; (4) telomere repeat frequency (along 10 kb sliding window, ranging between 10 and 30 repeats); and (5) mean base pair depth coverage (along a 10-kb sliding window, ranging between 0 and 300 depth) for strains ss2 (A) and ss5 (B). (C) Predicted function of genes in each P. pulmonarius assembly catalogued using the COGs database.
Table 2.
Classification of TEs identified in P. pulmonarius monokaryon strains ss2 and ss5
| Family |
P. pulmonarius ss2
|
P. pulmonarius ss5
|
||||
|---|---|---|---|---|---|---|
| Fragments | Copies | Total bp | Fragments | Copies | Total bp | |
| Class I | ||||||
| LINE/I | 1 | 1 | 414 | 1 | 0 | 0 |
| LINE/L1 | 107 | 5 | 11,665 | 19 | 11 | 23,464 |
| LINE/L1-Tx1 | 3 | 1 | 2451 | 21 | 8 | 23,115 |
| LINE/L2 | 157 | 102 | 651,829 | 101 | 93 | 184,053 |
| LINE/R1 | 64 | 41 | 21,126 | 0 | 0 | 0 |
| LINE/Tad1 | 48 | 37 | 56,788 | 20 | 14 | 28,805 |
| Other LINE | 5 | 1 | 834 | 0 | 0 | 0 |
| LTR/Caulimovirus | 0 | 0 | 0 | 55 | 21 | 23,920 |
| LTR/Copia | 227 | 134 | 299,643 | 425 | 167 | 292,397 |
| LTR/Gypsy | 947 | 525 | 1,413,722 | 1117 | 641 | 1,380,124 |
| LTR/Ngaro | 128 | 33 | 87,204 | 60 | 36 | 61,347 |
| LTR/Pao | 0 | 0 | 0 | 12 | 4 | 4632 |
| Other LTR | 16 | 16 | 5979 | 18 | 18 | 6256 |
| Class II | ||||||
| DNA/CMC-EnSpm | 142 | 82 | 88,249 | 126 | 47 | 93,887 |
| DNA/Crypton | 5 | 4 | 25,088 | 0 | 0 | 0 |
| DNA/Crypton-V | 0 | 0 | 0 | 54 | 51 | 316,697 |
| DNA/Dada | 0 | 0 | 0 | 12 | 10 | 9340 |
| DNA/hAT-Ac | 10 | 7 | 6624 | 4 | 1 | 786 |
| DNA/Maverick | 152 | 52 | 41,933 | 39 | 20 | 51,514 |
| DNA/MULE-MuDR | 77 | 17 | 41,283 | 26 | 3 | 1870 |
| DNA/PIF-Harbinger | 19 | 9 | 8138 | 13 | 3 | 1103 |
| DNA/RC | 95 | 59 | 78,957 | 33 | 22 | 44,417 |
| DNA/TcMar-Ant1 | 11 | 7 | 5873 | 3 | 1 | 981 |
| DNA/TcMar-Fot1 | 10 | 7 | 9998 | 8 | 6 | 9088 |
| DNA/TcMar-Pogo | 2 | 2 | 1284 | 3 | 1 | 540 |
| DNA/TcMar-Tc1 | 3 | 3 | 2578 | 0 | 0 | 0 |
| Other DNA | 5 | 3 | 5289 | 37 | 9 | 7742 |
| Total repeats | 2218 | 1132 | 286,0970 | 2189 | 1169 | 2,559,822 |
Genomic differences between P. pulmonarius strains
Using our high-quality assemblies of P. pulmonarius, we conducted a preliminary analysis of the main differences between the genomes of ss2 and ss5. As can be observed in the dot plot of Figure 3A, our assemblies aligned almost perfectly to each other, only showing lower identity at the ends of the scaffolds and a few regions of low similarity. Moreover, we observed that the end of scaffold10 of ss2 and the beginning of scaffold9 of ss5 do not correspond to any of the major scaffolds of the opposite assembly (Figure 3A). A closer look to the similarities between both assemblies is found in Figure 3B, showing high similarity regions (>95%) between the most relevant scaffolds of each genome (>500 kb in size). In this comparison, the missing beginning of scaffold 9 in ss5 and end of scaffold 10 in ss2 become even more apparent. We observed some possible chromosomal translocation in scaffold 2 of ss5 consisting of the majority of scaffold 1 and a piece of scaffold 10 from ss2; however, due to the presence of many TEs in the genome, we cannot rule out the possibility that this is a result of misassembly. Further experimental analysis would be necessary to confirm this phenomenon. Finally, we looked for genes that are present only in one of the genomes. A prominent cluster of unique genes at the beginning of scaffold 9 in ss5 contained 159 genes that are missing in ss2 (Supplementary Table S6). Similarly, we also observed that ss2 harbored 66 genes on scaffold 10 that were not present in ss5 (Supplementary Table S5).
Figure 3.

Comparison between the assembled genomes of P. pulmonarius, strains ss2 and ss5. (A) Dot-plot alignment of ss5 (query) against ss2 (reference). (B) Correspondence between regions of high similarity (>95%, length >10 kb) among the most relevant scaffolds (>500 kb in size) of the two assembled genomes. Tracks (outer to inner) represent the distribution of genomic features in each assembly: (1) positions (in Mb) of the assembled scaffolds; (2) number of genes unique in each assembly (along 100-kb sliding windows, ranging between 0 and 35 genes); and (3) telomere repeat frequency (along 10-kb sliding window, ranging between 10 and 30 repeats).
Conclusions
In this study, we conducted PacBio genome sequencing for two spores harvested from a dikaryotic strain of P. pulmonarius and assembled two high-quality genomes. We observed significant phenotypic differences in terms of growth and toxicity against nematodes between the two monokaryotic strains. Motivated by these results, we conducted genomic analyses of these two P. pulmonarius strains. Their two assemblies are the first available genome resources for P. pulmonarius, and their quality places them among the best Pleurotus genome assemblies. Comparison of these two assemblies showed dissimilar regions that may contribute to the phenotypic differences of these two strains. The de novo assemblies presented here will be crucial genomic resources for studying the biology of P. pulmonarius; they will also enable comparative genomic studies in Pleurotus mushrooms, which contains several species of most cultivated mushrooms around the globe.
Acknowledgments
The authors thank Ting-Fang Wang for comments on this article.
Funding
Work here was supported in part by startup and research allocations from NSF XSEDE (TG-MCB180039 and TG-MCB190010) to E.M.S. and Academia Sinica Career Development Award AS-CDA-106-L03 to Y.-P.H.
Conflicts of interest: None declared.
Literature cited
- Aida FMNA, Shuhaimi M, Yazid M, Maaruf AG.. 2009. Mushroom as a potential source of prebiotics: a review. Trends Food Sci Technol. 20:567–575. [Google Scholar]
- Alfaro M, Castanera R, Lavín JL, Grigoriev IV, Oguiza JA, et al. 2016. Comparative and transcriptional analysis of the predicted secretome in the lignocellulose-degrading basidiomycete fungus Pleurotus ostreatus. Environ Microbiol. 18:4710–4726. [DOI] [PubMed] [Google Scholar]
- Bailly-Bechet M, Haudry A, Lerat E.. 2014. “One code to find them all”: a perl tool to conveniently parse RepeatMasker output files. Mob DNA. 5:1–15.24382139 [Google Scholar]
- Banik S, Nandi R.. 2004. Effect of supplementation of rice straw with biogas residual slurry manure on the yield, protein and mineral contents of oyster mushroom. Ind Crops Prod. 20:311–319. [Google Scholar]
- Borodovsky M, McIninch J.. 1993. GENEMARK: parallel gene recognition for both DNA strands. Comput Chem. 17:123–133. [Google Scholar]
- Cabanettes F, Klopp C.. 2018. 2018 D-GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ. 6:e4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castañeda-Ramírez GSTorres-Acosta JFDJSánchez JEMendoza-De-Gives PGonzález-Cortázar M, . et al. 2020. The Possible Biotechnological Use of Edible Mushroom Bioproducts for Controlling Plant and Animal Parasitic Nematodes. BioMed Research International. 2020:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanera R, López-Varas L, Borgognone A, LaButti K, Lapidus A, et al. 2016. Transposable elements versus the fungal genome: impact on whole-genome architecture and transcriptional profiles. PLoS Genet. 12:e1006108–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty M, Baldwin-Brown JG, Long AD, Emerson JJ.. 2016. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 44:gkw654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Xiong S, Sundelin J, Martín C, Hultberg M.. 2020. Potential for combined production of food and biofuel: cultivation of Pleurotus pulmonarius on soft- and hardwood sawdusts. J Clean Prod. 266:122011. [Google Scholar]
- Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, et al. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 10:563–569. [DOI] [PubMed] [Google Scholar]
- Chin C, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, et al. 2016. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods. 13:1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan A, Tsai IJ, Berriman M.. 2018. Creation of a comprehensive repeat library for a newly sequenced parasitic worm genome. Protoc Exch. (Version1):1–6. [Google Scholar]
- Degenkolb T, Vilcinskas A.. 2016. Metabolites from nematophagous fungi and nematicidal natural products from fungi as alternatives for biological control. Part II: metabolites from nematophagous basidiomycetes and non-nematophagous fungi. Appl Microbiol Biotechnol. 100:3813–3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Qu J, Zhang J, Sonnenberg A, Chen Q, et al. 2018. A genetic linkage map of Pleurotus tuoliensis integrated with physical mapping of the de novo sequenced genome and the mating type loci. BMC Genomics. 19:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golak-Siwulska I, Kałużewicz A, Spiżewski T, Siwulski M, Sobieralski K.. 2018. Bioactive compounds and medicinal properties of Oyster mushrooms (Pleurotus sp). Folia Hortic. 30:191–201. [Google Scholar]
- Grabherr MG, Brian N, Haas Moran Yassour Joshua J, Levin Z, Thompson DA, Amit I, et al. 2011. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol. 29:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregori A, Švagelf M, Pohleven J.. 2007. Cultivation techniques and medicinal properties of Pleurotus spp. Food Technol Biotechnol. 45:238–249. [Google Scholar]
- Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M.. 2012. The Genome Portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 40:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith RK, et al. 2003. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31:5654–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, et al. 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9:R7–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inácio FD, Ferreira RO, de Araujo CAV, Peralta RM, de Souza CGM.. 2015. Production of enzymes and biotransformation of orange waste by oyster mushroom. Adv Microbiol. 05:1–8. [Google Scholar]
- Khatun S, Islam A, Cakilcioglu U, Guler P, Chatterjee NC.. 2015. Nutritional qualities and antioxidant activity of three edible oyster mushrooms (Pleurotus spp). NJAS Wageningen J Life Sci. 72–73:1–5. [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, et al. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, et al. 2009. Circos: an information aesthetic for comparative genomics. Genome Res. 19:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok OCH, Plattner R, Weisleder D, Wicklow DT.. 1992. A nematicidal toxin from Pleurotus ostreatus NRRL 3526. J Chem Ecol. 18:127–136. [DOI] [PubMed] [Google Scholar]
- Larraya LM, Pérez G, Ritter E, Pisabarro AG, Ramı’rez L.. 2000. Genetic linkage map of the edible basidiomycete Pleurotus ostreatus. Appl Environ Microbiol. 66:5290–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law WM, Lau WN, Lo KL, Wai LM, Chiu SW.. 2003. Removal of biocide pentachlorophenol in water system by the spent mushroom compost of Pleurotus pulmonarius. Chemosphere. 52:1531–1537. [DOI] [PubMed] [Google Scholar]
- Lee C-H, Chang H-W, Yang C-T, Wali N, Shie J-J, et al. 2020. Sensory cilia as the Achilles heel of nematodes when attacked by carnivorous mushrooms. Proc Natl Acad Sci USA. 117:6014–6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J. et al. ; 1000 Genome Project Data Processing Subgroup, 2009. The Sequence alignment/map format and SAMtools. Bioinformatics. 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wu S, Ma X, Chen W, Zhang J, et al. 2018. The Genome Sequences of 90 mushrooms. Sci Rep. 8:2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, et al. 2018. MUMmer4: a fast and versatile genome alignment system (A. E. Darling, Ed). PLoS Comput Biol. 14:e1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema MH, Blin K, Cimermancic P, De Jager V, Zakrzewski P, et al. 2011. AntiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39:339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgulis A, Coulouris G, Raytselis Y, Madden TL, Agarwala R, et al. 2008. Database indexing for production MegaBLAST searches. Bioinformatics. 24:1757–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TK, Im KH, Choi J, Shin PG, Lee TS.. 2016. Mycobiology anti-inflammatory effects of culinary mushroom Pleurotus pulmonarius. Mycobiology. 44:291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystrom NA, Levine MJ, Roskies RZ, Scott JR.. 2015. Bridges: a uniquely flexible HPC resource for new communities and data analytics. ACM Int. Conf. Proceeding Ser, July 2015.
- Palmer J, Stajich JE.. 2016. Funannotate: A Fungal Genome Annotation and Comparative Genomics Pipeline (v1.5.2). Zenodo doi: 105281/zenodo3354704.
- Pérez G, Pangilinan J, Pisabarro AG, Ramírez L, 2009. Telomere organization in the ligninolytic basidiomycete pleurotus ostreatus. Appl Environ Microbiol. 75:1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J, Zhao M, Hsiang T, Feng X, Zhang J, et al. 2016. Identification and characterization of small noncoding RNAs in genome sequences of the edible fungus Pleurotus ostreatus. Biomed Res Int. 2016:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley R, Salamov AA, Brown DW, Nagy LG, Floudas D, et al. 2014. Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc Natl Acad Sci USA. 111:9923–9928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrı’guez E, Nuero O, Guillén F, Martı’nez AT, Martı’nez MJ, 2004. Degradation of phenolic and non-phenolic aromatic pollutants by four Pleurotus species: the role of laccase and versatile peroxidase. Soil Biol Biochem. 36:909–916. [Google Scholar]
- Satou T, Kaneko K, Li W, Koike K.. 2008. The toxin produced by Pleurotus ostreatus reduces the head size of nematodes. Biol Pharm Bull. 31:574–576. [DOI] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31:3210–3212. [DOI] [PubMed] [Google Scholar]
- Sivanandhan S, Khusro A, Paulraj MG, Ignacimuthu S, Al-Dhabi NA.. 2017. Biocontrol properties of basidiomycetes: an overview. J Fungi. 3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiderle FR, Olsen LM, Carbonero ER, Baggio CH, Freitas CS, et al. 2008. Anti-inflammatory and analgesic properties in a rodent model of a (1→3),(1→6)-linked β-glucan isolated from Pleurotus pulmonarius. Eur J Pharmacol. 597:86–91. [DOI] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P.. 2013. RepeatMasker Open-4.0.
- Stajich JE, Wilke SK, Ahren D, Au CH, Birren BW, et al. 2010. Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc Natl Acad Sci USA. 107:11889–11894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Waack S.. 2003. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 19:ii215–225. [DOI] [PubMed] [Google Scholar]
- Thorn RG, Barron GL.. 1984. Carnivorous mushrooms. Science. 224:76–78. [DOI] [PubMed] [Google Scholar]
- Titus Brown C, Irber L.. 2016. sourmash: a library for MinHash sketching of DNA. JOSS. 1:27. [Google Scholar]
- Towns J, Cockerill T, Dahan M, Foster I, Gaither K, et al. 2014. XSEDE: accelerating scientific discovery. Comput Sci Eng. 16:62–74. [Google Scholar]
- Wang L, Gao W, Wu X, Zhao M, Qu J, et al. 2018. Genome-wide characterization and expression analyses of pleurotus ostreatus MYB transcription factors during developmental stages and under heat stress based on de novo sequenced genome. Int J Mol Sci. 19: 2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Liu Y, Li H, Zhao S, Wang S, et al. 2014. Genetic diversity of Pleurotus pulmonarius revealed by RAPD, ISSR, and SRAP fingerprinting. Curr Microbiol. 68:397–403. [DOI] [PubMed] [Google Scholar]
- Zhang M, Cui SW, Cheung PCK, Wang Q.. 2007. Antitumor polysaccharides from mushrooms: a review on their isolation process, structural characteristics and antitumor activity. Trends Food Sci Technol. 18:4–19. [Google Scholar]
- Zhang Z, Wen J, Li J, Ma X, Yu Y, et al. 2018. The evolution of genomic and epigenomic features in two Pleurotus fungi. Sci Rep. 8:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The final assembled and annotated genomes of P. pulmonarius spores ss2 and ss5 were uploaded to NCBI and are available with accession codes: GCA_012980525.1 and GCA_012980535.1, respectively. In addition, all the strains used in this study will be deposited in FGSC and are available upon request.
Supplementary materials are available at figshare DOI: https://doi.org/10.25387/g3.13078253.