

Abstract
Glycan biosynthesis on cell surface proteins and lipids is orchestrated by different classes of enzymes and proteins including the following: i. glycosyltransferases that add saccharides; ii. glycosidases that trim glycans; iii. conserved oligomeric golgi complex members that regulate intracellular transport; iv. enzymes aiding the biosynthesis of sugar–nucleotides; and v. sulfotransferases. This manuscript describes a pooled “glycoGene CRISPR” lentiviral library that targets 347 human genes involved in the above processes. Approximately 10 single-guide RNA (sgRNA) are included against each glycogene, with the putative editing site spanning the length of the target. A data analysis scheme is presented in order to determine glycosylation pathways regulating biological processes. As proof of principle, forward genetic screen results are presented to identify penetrating glycogenes that regulate the binding of P-/E-selectin, anti-sialyl Lewis-X monoclonal antibody HECA-452 and selected lectins (phaseolus vulgaris leucoagglutinin, vicia villosa lectin, peanut agglutinin) to HL-60 promyelocytic cells. Besides validating previously established biology, the study identifies three enzymes, PAPSS1, SLC35B2 and TPST2, as key molecules regulating sulfation of the major P-selectin glycoprotein ligand-1 in leukocytes. Approximately 80–90% of the sgRNA used in this study displayed high editing efficiency, and the CRISPR library picked up entire gene sets regulating specific biosynthetic pathways rather than only isolated genes. These data suggest that the glycoGene CRISPR library contains high-efficiency sgRNA. Further, this resource could be useful for the rapid screening of glycosylation-related genes and pathways that control lectin recognition in a variety of contexts.
Keywords: CRISPR-Cas9, forward genetic screen , gene editing, glycoscience, selectin
Introduction
Glycosylation is a common, complex and prominent posttranslational modification (PTM) of various extracellular proteins and lipids, and also intracellular macromolecules. Glycan structures formed as a result regulate almost all biological processes including, but not limited to, protein folding, molecular half-life in blood circulation, cell adhesion, cell signaling and transcription, host–pathogen interactions, inflammation, immunity, tumorigenesis and cancer metastasis (Moremen et al. 2012; Varki 2017). Underlying the importance of this PTM, defects in glycosylation result in a family of disorders that are collectively called the congenital disorders of glycosylation (Peanne et al. 2018).
Recent years have witnessed an emphasis on the development of molecular tools to study glycosylation from the systems level, as these complement classical biochemical methods that dissect individual steps (Neelamegham and Liu 2011; Neelamegham and Mahal 2016). In this regard, forward genetic screens have emerged as a powerful tool to dissect the major players involved in specific biological processes. While there are a number of readily available genome-wide CRISPR libraries (Shalem et al. 2014; Zhou et al. 2014), none of these specifically focused on dissecting the human glycosylation process. This is however important since genome-wide screens may miss important steps regulating glycosylation as they focus broadly on ~20,000 human genes. In addition, smaller libraries result in lower cost due to smaller cell culture scales, sorting expenses and reduced next-generation sequencing requirements. Narimatsu et al. (2018) described a single-guide RNA (sgRNA) plasmid set that targets 183 human glycosyltransferases. While valuable, only a limited number of sgRNA were screened against each of the genes, and the method used to generate this resource is labor intensive as sgRNA are individually screened. Other efforts to use CRISPR-Cas9 technology for human glycosylation-related studies focus on only a few key enzymes (Mondal et al. 2016; Stolfa et al. 2016), and this also does not capture the complexity of the full system.
To address the above gaps, this manuscript describes a new pooled “glycoGene CRISPR” library targeting 347 glycosylation-related genes or “glycogenes” (Supplementary Table SI). Editing is driven by Streptococcus pyogenes Cas9 nuclease. This system allows the timed regulation of gene expression, as it is based on our recently described doxycycline (Dox)-regulated self-inactivating CRISPR (SiC) vectors (Kelkar et al. 2020). It targets most of the human glycosyltransferases involved in the biosynthesis of different types of glycoconjugates including N-glycans, O-glycans, glycolipids, glycosaminoglycans, glycosylphosphatidylinositol-anchored carbohydrates, O-fucose, O-N-acetylglucosamine (GlcNAc), O-glucose, O-mannose and epidermal growth factor domain-specific O-linked GlcNAcTs. In addition, the library includes sgRNA targeting proteins/enzymes involved in intracellular protein transport, sulfotransferases, enzymes involved in nucleotide–sugar biosynthesis, nucleotide–sugar transporters and a range of glycosidases. A unique feature of the library is that it contains ~9–11 sgRNAs per glycogene that are computationally designed to have high on-target and low off-target editing efficiency. These sgRNA span the length of the glycogenes with an average distribution of 40-40-20 spanning the beginning, middle and end of the average molecular target. Thus, in principle, the library may be used to examine the effect of protein knockout vs. protein truncation in a site-specific manner. The merits and disadvantages of using validated sgRNA vs. forward genetic screens are presented in Table I.
Table I.
Two approaches to study glycogene function
| Validated guides | Forward genetic screen | |
|---|---|---|
| Advantage | • Requires less bioinformatics knowledge | • sgRNA libraries can be rapidly designed using established bioinformatics pipelines |
| • Good approach when there is one or a few targets to study. Well suited for hypothesis validation | • Same library can be reused for multiple screens. Ideal for hypothesis generation | |
| Disadvantage | • Guide validation can be expensive and time consuming as individual genes have to be screened | • Quality of results depends on the quality of sgRNA |
| • Guide validated in one system may not work in others due to epigenetic differences | • Some genes may be missed when library size is large |
sgRNA, single-guide RNA.
In this manuscript, we used the “glycoGene CRISPR” library to identify enzymes regulating the binding of specific common lectins- and carbohydrate-binding monoclonal antibodies (mAbs). The results showed that >80–90% of the sgRNA targeting individual glycogenes resulted in loss of lectin/mAb-binding function in cell-based assays. All data, thus far, suggest that the sgRNA present have high gene editing efficiency. Thus, the glycoGene CRISPR resource could be valuable for the study of human glycosylation processes in vitro and in vivo.
Results and discussion
Generating glycoGene CRISPR library transduced HL60s
A high-efficiency CRISPR library was designed to target 347 glycogenes regulating various glycan biosynthetic steps, using the Azimuth algorithm to define sgRNA potency scores for each guide (Doench et al. 2016; Supplementary Table SI). These sgRNA were cloned into a Dox-regulated lentiviral vector either downstream of the U6 promoter in “U6-Dox-sgRNA” or following the H1 promoter in “H1-Dox-sgRNA” (Figure 1A). This library includes ~10 sgRNA against each target gene, with sgRNA editing sites spanning the length of the protein coding sequence (Supplementary Table SII). To test this system, an isogenic HL60 clone was established that stably over-expresses S. pyogenes Cas9 (“HL60-Cas9”; Figure 1B), using the lentiviral SiC-V1-scr (Figure 1A). This construct contains a fluorescent dTomato reporter, and thus the HL60-Cas9 cells exhibit red fluorescence. Lentivirus generated using the glycoGene CRISPR library (“U6-Dox-sgRNA” construct) was applied to these cells at various concentrations (Supplementary Figure S1), with the final selected cell line being infected at a multiplicity of infection (MOI) of ~0.4 (Figure 1B). Thus, most glycoGene CRISPR expressing cells contained ~1 sgRNA/cell. Editing was initiated by addition of 1 μg/mL Dox 2 days after glycoGene CRISPR library transduction. Cas9 was subsequently knocked out at day 4 by electroporating with a synthetic guide (“Cas9G7”) that self-inactivates the nuclease (Kelkar et al. 2020). By day 6, four distinct cell populations were evident with one-third of the cells lacking Cas9 activity based on loss of dTomato signal while simultaneously stably expressing sgRNA based on the blue fluorescence protein (BFP) reporter expressed by U6-Dox-sgRNA. These BFP+dTomato− HL60 cells were sorted on day 12 to obtain the “HL60-lib cells” used in this study. sgRNA representation in these cells followed a bell-shaped profile (Supplementary Figure S2).
Fig. 1.
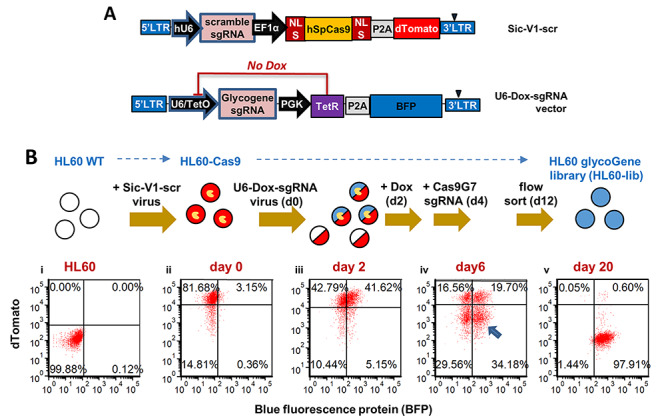
Creation of GlycoGene CRISPR library transduced cells. A. Schematic of lentiviral vectors used in the CRISPR screen. dTomato acts as a surrogate reporter of Cas9 nuclease activity in SiC-V1-scr. GlycoGene CRISPR library was cloned in a doxycycline-inducible vector with BFP fluorescence reporter. B. An HL60-Cas9 cell line was established by transduction with SiC-V1-scr vector, and single cell flow sorting for isogenic clone expressing high levels of dTomato reporter (Bi–ii). A virus pool synthesized from the glycoGene CRISPR sgRNA library (U6-Dox-sgRNA) was applied to these cells. Two days later, 42% of the cells were BFP positive (Biii). One microgram per milliliter Dox was added starting at day 2 to initiate gene editing. Dox was removed and Cas9G7 sgRNA electroporated on day 4 to inactivate Cas9 activity. dTomato−BFP+ cells (arrow) were FACS sorted at day 12 in order to establish the stable HL-60 glycoGene cell library (called “HL60-lib”). Flow cytometry plots represent: i. wild-type HL60; ii. HL60-Cas9; iii. HL60-Cas9 with glycoGene library; iv. cells after Cas9 nuclease editing on day 6; and v. HL60-lib cells on day 20. dTomato fluorescence of HL60-lib on day 20 (Bv) is similar to wild-type HL60s (Bi) confirming complete knockdown of Cas9 activity. (This figure is available in black and white in print and in color at Glycobiology online)
To test the efficacy of the sgRNA library, additional three color sorts were also performed on day 12 to obtain BFP+dTomato− HL60 cells that exhibited altered lectin/mAb-binding preference. The specific populations we enriched include cells displaying reduced E- and P-selectin binding (Figure 2A and B), reduced HECA-452 expression, low phaseolus vulgaris leucoagglutinin (PHA-L) binding (Figure 2C and D), enhanced vicia villosa lectin (VVA) binding (Figure 2E) and reduced PNA (Peanut Agglutinin) binding (Figure 2F). Since a distinct cell population was not obtained in a single sort due to the low number of genes contributing to the selected function, 1–2 additional sorts were performed within 2–3 weeks to enrich the various “lectin-selected” cell populations (Supplementary Figure S3).
Fig. 2.
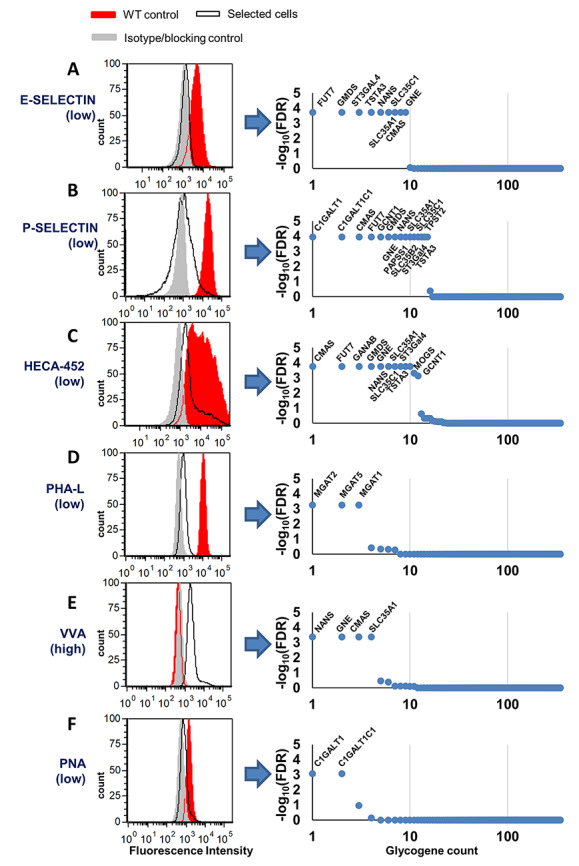
GlycoGene screening for altered lectin binding using MAGeCK. A. HL60 cells containing the full glycoGene library (i.e. HL60-lib, BFP+dTomato−) were sorted on day 12. On the same day, three color sorts were also performed to select for BFP+dTomato− cells exhibiting low E-selectin IgG binding (A), low P-selectin IgG binding (B), low HECA-452/sialyl Lewis-X expression (C), low PHA-L binding (D), high VVA binding (E) and low PNA binding (F). Either one or two additional sorts were performed to obtain “lectin-selected” cell populations (left panels). Red histogram marks the binding of HL60-Cas9 control, gray histogram marks the negative control (isotype/blocking antibody) and black line marks the final lectin-selected population. sgRNA enriched in the lectin-selected population were identified using MAGeCK following deep sequencing (right panels). Genes identified with FDR < 10−3 are marked in the individual panels, next to the relevant data point. (This figure is available in black and white in print and in color at Glycobiology online)
Workflow to identify regulators of lectin binding
Genomic DNA was isolated from HL60-lib cells that exhibited nominal lectin-binding properties, and also various enriched populations with altered carbohydrate expression profiles. Regions surrounding the sgRNA sequence in the isolated genomic DNA were polymerase chain reaction (PCR) amplified, barcoded, mixed and then subjected to deep sequencing. Supplementary Table SIII presents raw count data.
Two data analysis methods were applied in order to determine sgRNA over-expressed in each of the “lectin-selected” cell populations with respect to the control HL60-lib. First, the Model-based Analysis of Genome-wide CRISPR-Cas9 Knockouts package (MAGeCK) identified sgRNAs having significantly higher representation in the lectin-selected pool compared to HL60-lib (Zhou et al. 2014; Supplementary Table SIV). Here, “total sample read count normalization” was performed prior to statistical testing. The false discovery rate (FDR < 10−3) parameter from this analysis identified putative glycogenes altering lectin/mAb binding (Figure 2 right panels). Second, we computed log-fold change (LFC) values in the selected cell populations, in order to determine sgRNA read counts for target genes with respect to nontarget sgRNA control (Figure 3). Here, LFC > 0 indicates sgRNA enrichment upon lectin/mAb-based selection. Corresponding target genes play a functional role in lectin/mAb binding. LFC ≤ 0 indicates specific guides that are not enriched as their absolute read counts would be similar to nontarget control sgRNA. This analysis showed that typically >80–90% of the sgRNA targeting specific glycogenes were increased upon sorting for specific lectin-binding phenotype. Thus, a majority of the sgRNA in the glycoGene CRISPR library displayed high on-target editing efficiency. Further, all the functional genes identified using MAGeCK were also observed to be important in the LFC calculations, thus validating the overall analysis approach (Supplementary Table SV).
Fig. 3.
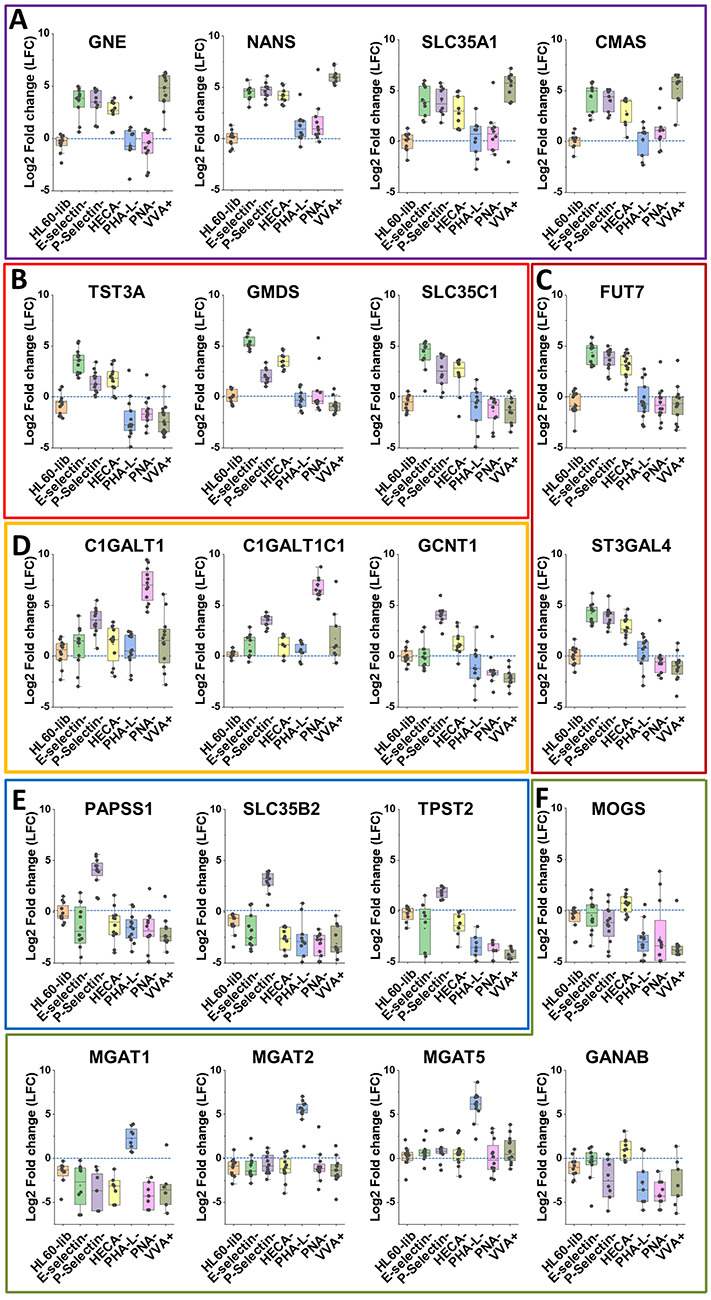
LFC analysis. Log2-fold change sgRNA data are presented for target gene vs. nontarget control for the unselected HL60-lib cells, and also the six lectin-selected cell types, i.e. LFC = log2[sgRNA count for target gene/sgRNA count for nontarget gene]. These data are presented for 20 different glycogenes that are involved in the following: A. CMP-sialic acid biosynthesis; B. GDP-fucose biosynthesis; C. α(1,3)fucosyltransferase FUT7 and α(2,3)sialyltransferase ST3Gal-4 activity; D. O-glycan construction; E. protein tyrosine sulfation; and F. N-glycan elaboration. Each dot represents one sgRNA. Enriched sgRNA display LFC > 0. Unaffected sgRNA have LFC ≤ 0, similar to the HL60-lib control. (This figure is available in black and white in print and in color at Glycobiology online)
Glycogenes regulating selectin recognition
Among the identified targets, genes involved in guanosine diphosphate (GDP)-fucose (GDP-fucose) and cytidine-5'-monophosphate-Neu5Ac (CMP-sialic acid) biosynthesis and corresponding nucleotide–sugar transport were critical for E-selectin, P-selectin and HECA-452 binding. This observation is consistent with the well-known roles of fucose and sialic acid during selectin recognition (Cummings and McEver 2015). Among the glycosyltransferases, the human α(1,3)fucosyltransferase FUT7 and α(2,3)sialyltransferase ST3Gal4 were found to be most penetrating (Buffone et al. 2013; Mondal et al. 2015). Enzymes involved in core-1 and core-2 glycan biosynthesis were necessary for P-selectin binding, and this is consistent with the dominant role of such glycans for P-selectin-dependent inflammatory leukocyte adhesion (Ellies et al. 1998; Lo et al. 2013). None of the enzymes involved in N-acetyllactosamine (LacNAc) and glycolipid biosynthesis were identified to be critical for E-selectin and HECA-452 recognition. However, we noted a role for N-glycan-forming glucosidases (GANAB, MOGS) and core-2 GlcNAcT (GCNT1) in regulating HECA-452 epitope expression. It is possible that deep sequencing of cells after the first sort can identify additional partial regulators of selectin binding, as studies with sequential sorts are only designed to identify critical or “penetrating” glycogenes.
Importantly, our studies identified the three genes controlling protein tyrosine sulfation that is necessary for P-selectin-dependent adhesion in HL60s: 3'-Phosphoadenosine 5'-Phosphosulfate Synthase 1 (PAPSS1), Adenosine 3'-phospho 5'-phosphosulfate transporter 1 (SLC35B2) and Tyrosylprotein Sulfotransferase 2 (TPST2). In this regard, PAPSS1 is one of two PAP synthase isoforms in humans, along with PAPSS2 (van den Boom et al. 2012). It is a bifunctional, cytosolic enzyme that contains both adenosine triphosphate (ATP) sulfurylase and adenosine-5'-phosphosulfate (APS) kinase activity (Girard et al. 1998). Thus, it both transfers sulfate to ATP to yield APS and then transfers the phosphate group from ATP to APS to yield 3'-phosphoadenylylsulfate (PAPS). SLC35B2 is one of two PAPS multi-pass, membrane spanning transporters, which, along with SLC35B3, regulates PAPS anti-port into the Golgi (Song 2013). Finally, TPST2 is also one of two human tyrosylprotein sulfotransferases. Previous work has established an important role for the TPSTs in regulating P-selectin binding to P-selectin glycoprotein ligand-1 (Wilkins et al. 1995; Westmuckett et al. 2011). This study identifies these putative functional genes in human leukocytes.
Glycogenes regulating lectin binding
Pilot studies were performed to determine if the glycoGene CRISPR library can be useful for arbitrary lectin-binding screens. Here, we confirmed that PHA-L binding is highly dependent on the Galβ1-4GlcNAcβ1-2(Galβ1-4GlcNAcβ1-6)Manα1 arm of N-glycans as knocking out MGAT1, MGAT2 and MGAT5 abolished lectin recognition. MGAT4 and MGAT3 function was not necessary for PHA-L binding. Interestingly, none of the enzymes involved in lipid-linked oligosaccharide biosynthesis were important for PHA-L binding. This warrants investigation as it suggests that the de novo synthesis of the dolichol-linked Glc3-Man9-GlcNAc2 oligosaccharide via a linear biosynthetic pathway may not be absolutely critical for N-glycan branching in knockouts.
Among the O-glycan binding lectins, the study examined the glycogenes upregulating VVA and reducing PNA binding. Here, PNA binding to HL60s was low to start with and this could be further reduced by abolishing O-glycan elaboration by knocking out core-1 glycan biosynthesis via either the core-1 galactosyltransferase (GalT) enzyme C1GALT1 or its chaperone C1GALT1C1. This is consistent with the observation that PNA predominantly binds type-III lactosamine chains (Galβ1,3GalNAcα), and that sgRNA targeting O-glycan core-1 biosynthesis work efficiently. As anticipated, VVA binding was dependent on sialic acid biosynthetic pathways, as this lectin recognizes GalNAcα-Ser/Thr, without sialylation. However, surprisingly, cells displaying high VVA binding were not enriched for sgRNA targeting either C1GALT1 or C1GALT1C1 (Steentoft et al. 2011; Stolfa et al. 2016; Chugh et al. 2018). We speculate that this may be due to the nature of the cell sorting selection method used to obtain VVA-high cells. Thus, the glyco-wide screening approach while efficient in many cases could sometimes miss key regulators previously identified using more targeted methods.
Conclusions
The manuscript describes a freely available, comprehensive glycoGene CRISPR library that could be useful for human cell systems, both in vitro and in vivo. Upon generating knockouts, it may be used to identify glycogenes regulating lectin binding and carbohydrate recognition function in a variety of physiological and disease contexts, including tumorigenesis, metastasis, inflammation, thrombosis, neurological disorders, etc. In addition, this CRISPR library has been specifically designed to measure the effect of mutations along the length of the glycogene. Thus, it may be used to study the nature of multi-glycosyltransferase complexes, and the hierarchy of molecular organization in the Golgi. The availability of multiple guides is also useful, if epigenetic changes render portions of the chromatin inaccessible. Only penetrating glycogenes were studied in this manuscript since we focused on proteins that completely abolish specific lectin/mAb binding. Relaxing the selection criterion may enable the identification of partial regulators. Finally, the library system is designed to study the impact of glycogenes in vivo in a temporal manner. In this regard, it should be feasible to transplant human cells expressing spCas9 and the glycogene CRISPR library into appropriate animal systems, and then feeding dox-chow to initiate timed gene editing (Kelkar et al. 2020). Similar timed editing trials may also be possible ex vivo in different experimental setups, e.g. 3D-organoid models.
All data collected so far in our laboratory suggest that the sgRNA that compose the glycogene CRISPR library have high editing efficiency. Thus, while we did not quantify the efficiency of individual sgRNA, the data support the possibility that sgRNA listed here could also be individually applied in functional assays. In addition, ~10 guides are provided for each target gene, and an FDR-based bioinformatics pipeline is used to account for variability in gene editing efficiency. Using robust rank aggregation (Figure 2), it is clear that only a small subset of candidates have FDR < 0.001 and this gives confidence in the overall approach. Due to the high efficiency, in addition to picking up single glycogenes, the library could pick up entire pathways regulating glycosylation.
While library-based screening is ideal for hypothesis generation, secondary validation assays are critical for corroboration. Here, it is necessary both to quantify gene editing efficiency using sequencing methods and to check function using enzymatic/structure-based assays. To enable the selection of sgRNA against specific gene targets for such secondary screens, we provide an online tool at https://virtualglycome.org/gCRISPR/ with data on sgRNA sequence, target location and sgNA efficiency data when available. Using this tool, if one wishes to edit multiple, redundant or overlapping genes in a pathway, it should be possible to select sgRNA against the target genes of interest, complex with spCas9 to form ribonucleoprotein particles and electroporate into cells. Functional studies can then be performed with these modified cells, with targeted amplicon sequencing of genomic DNA being used to quantify gene editing efficiency. Alternatively, it is also possible to study the impact of multiple genes in a single cell by applying sgRNA containing virus at MOI > 1. Overall, the above examples illustrate the many potential applications of the glycogene CRISPR library. Thus, this could be a valuable resource for systems-level investigations of glycosylation.
Materials and methods
Supplementary material provides detailed sources of materials, stepwise methods for creation of the glycoGene CRISPR library/virus and other common molecular biology techniques.
Generation of HL60-lib and lectin/mAb-binding selected cells
One microliter of 200× concentrated glycoGene CRISPR lentivirus was added to 3 × 106 HL60-Cas9 cells in a six-well plate containing 3 mL media in the presence of 8 μg/mL polybrene (Buffone et al. 2013). Two days post transduction, 1 μg/mL Dox was added to the culture medium to activate gene editing. On day 4, Dox was removed and the Neon transfection kit was used to electroporate 1.5–2.5 × 106 cells suspended in 100 μL Resuspension Buffer R with 10 μg Cas9G7 sgRNA (Kelkar et al. 2020; Supplementary Material). The electroporation conditions were as follows: 1600 V, 10 ms, 3 pulses. Following electroporation, the cells were transferred into six-well plates containing 3 mL of complete IMDM culture medium (Iscove's Modified Dulbecco's Medium (IMDM) 1640 Medium, 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid with 10% fetal bovine serum, 1X Antibiotic–Antimycotic and 1% GlutaMAX™). Cells were scaled up till day 12, when fluorescence-activated cell sorting was undertaken to select HL60-lib cells containing sgRNA (BFP-positive) but lacking Cas9 (dTomato-negative).
Independent sorts were also undertaken on day 12 for selection of BFP+dTomato− cells that exhibit specific lectin-binding preference. To this end, 10 μg/mL fluorescent lectin, 2 μg/mL anti-human cutaneous lymphocyte antigen mAb (HECA-452-eFluor® 660) or 3–10 μg/mL E-/P-selectin-immunoglobulin G (IgG) preincubated with 1.5 μg/mL Alexa-647 conjugated Goat anti-Human IgG F(ab')₂ were added to 2 × 106/mL HL60s containing the sgRNA library on ice for 20 min. Three color flow sorting was then performed to enrich for BFP+dTomato− cells exhibiting specific lectin/mAb-binding preference. One additional cell sorts was performed during the following week to enrich for the cell population with minimum interaction with E-selectin, P-selectin, HECA, PHA-L and PNA. Two sorts were required in order to obtain a distinct high VVA-binding population. Gates used for each sort are shown in Supplementary Figure S3.
Deep sequencing
Genomic DNA was isolated from ~5 × 106 cells (HL60-lib and lectin-selected) using PureLink™ Genomic DNA Mini Kit (Invitrogen™, Carlsbad, CA). A two-step PCR was applied to generate amplicons for deep sequencing. First, regions flanking the sgRNA were PCR amplified by mixing 1 μg of isolated genomic DNA with 1 μM gFwd primer, 1 μM gRev primer and 25 μL NEBNext® Ultra™ II Q5® Master Mix (New England Biolabs, Ipswich, MA) and water to make up a 50 μL reaction (primer sequences in Supplementary Table SVI). Product size was checked using 2% agarose gel electrophoresis, followed by PCR column purification using NucleoSpin® Gel and PCR Clean-up kit (Macherey-Nagel, Bethlehem, PA). Second, the Illumina P5 adapter was added to the 5'-end of the above PCR product and P7 adapters along with a sample specific barcode to the 3'-end, in a similar PCR as above using ~15 μL amplicon from first step as template, 1 μM iFwd and 1 μM iRev primer (Supplementary Table SVI). All PCR products were gel purified, pooled and sequenced using an illumina MiSeq platform (San Diego, CA): 150 bp paired end, 30 × 106 reads.
Bioinformatics analysis
Deep sequencing data were analyzed using two methods. First, the MAGeCK package was used to identify sgRNA responsible for altered lectin/mAb binding (Zhou et al. 2014). Briefly, MAGeCK determines the number of sgRNA in the HL60-lib and lectin-selected samples by using the sgRNA amplicon read.fastq data and a reference sgRNA sequence file containing all possible guide sequences. These data are normalized based on the total sample read count: sgRNAi,norm = 106 × (sgRNAi,/ΣsgRNAi), where sgRNAi is the raw read count of the ith sgRNA in a given cell sample. Statistical analysis was then performed to determine if sgRNA read count differences between samples (lectin-selected vs. HL60-lib) is significantly different. These enrichment likelihoods (P-values) along with the corresponding sgRNA log2-fold change values are used to rank the sgRNAs detected first from high- to low-fold change, then based on low to high P-value. This ranked list is used in an α-robust rank aggregation analysis to identify the gene knockouts that produce the relevant phenotype. The P-values computed from this analysis are adjusted for false discovery using the Benjamini–Hochberg procedure. These adjusted P-values (FDR values) are used to sort genes based on their relevance to the phenotype in ascending order.
Second, in the fold change calculations, we measured the ratio of target sgRNA counts in each cell type vs. nontarget sgRNA counts in the same cell. Here, we used sgRNA corresponding to carbohydrate sulfation (all carbohydrate sulfotransferases) and heparan sulfation (all HS6STs, HS3STs and HS2STs) enzymes as nontarget control. Representation of sgRNA corresponding to these genes is not expected to be altered for the lectins used in this study. These data are plotted as log2-fold change. Here, sgRNA enrichment results in log2-fold change > 0 and depletion in log2-fold change ≤ 0.
Supplementary Material
Acknowledgment
The authors gratefully thank Edward J. Sobczak and Srirangaraj Setlur for systems administration of the virtualGlycome server.
Contributor Information
Yuqi Zhu, Chemical and Biological Engineering, Biomedical Engineering and Medicine, University at Buffalo, State University of New York, Buffalo, NY 14260, USA.
Theodore Groth, Chemical and Biological Engineering, Biomedical Engineering and Medicine, University at Buffalo, State University of New York, Buffalo, NY 14260, USA.
Anju Kelkar, Chemical and Biological Engineering, Biomedical Engineering and Medicine, University at Buffalo, State University of New York, Buffalo, NY 14260, USA.
Yusen Zhou, Chemical and Biological Engineering, Biomedical Engineering and Medicine, University at Buffalo, State University of New York, Buffalo, NY 14260, USA.
Sriram Neelamegham, Chemical and Biological Engineering, Biomedical Engineering and Medicine, University at Buffalo, State University of New York, Buffalo, NY 14260, USA.
Funding
This work was supported by National Institutes of Health grants (HL103411, GM133195), and a New York Stem Cell training program award (DOH01-C30290GG-3450000). The glycoGene CRISPR libraries are distributed via Addgene. For the sgRNA selection tool and stepwise usage methods/protocols, please visit https://virtualGlycome.org/gCRISPR/.
Conflict of interest statement
None declared
Abbreviations
APS, adenosine-5'-phosphosulfate; ATP, adenosine triphosphate; Dox, doxycycline; FDR, false discovery rate; GlcNAc, N-acetylglucosamine; LacNAc, N-acetyllactosamine; mAbs, monoclonal antibodies; MAGeCK, Model-based Analysis of Genome-wide CRISPR-Cas9 Knockouts package; MOI, multiplicity of infection; PAPS, 3'-phosphoadenylylsulfate; PCR, polymerase chain reaction; PTM, posttranslational modification; sgRNA, single-guide RNA; SiC, self-inactivating CRISPR.
References
- Buffone A Jr, Mondal N, Gupta R, McHugh KP, Lau JT, Neelamegham S. 2013. Silencing alpha1,3-fucosyltransferases in human leukocytes reveals a role for FUT9 enzyme during E-selectin-mediated cell adhesion. J Biol Chem. 288:1620–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugh S, Barkeer S, Rachagani S, Nimmakayala RK, Perumal N, Pothuraju R, Atri P, Mahapatra S, Thapa I, Talmon GA et al. 2018. Disruption of C1galt1 gene promotes development and metastasis of pancreatic adenocarcinomas in mice. Gastroenterology. 155:1608–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings RD, McEver RP. 2015. C-type lectins. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH et al., editors. Essentials of glycobiology. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. p. 435–452. [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R et al. 2016. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 34:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellies LG, Tsuboi S, Petryniak B, Lowe JB, Fukuda M, Marth JD. 1998. Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity. 9:881–890. [DOI] [PubMed] [Google Scholar]
- Girard JP, Baekkevold ES, Amalric F. 1998. Sulfation in high endothelial venules: Cloning and expression of the human PAPS synthetase. FASEB J. 12:603–612. [DOI] [PubMed] [Google Scholar]
- Kelkar A, Zhu Y, Groth T, Stolfa G, Stablewski AB, Singhi N, Nemeth M, Neelamegham S. 2020. Doxycycline-dependent self-inactivation of CRISPR-Cas9 to temporally regulate on- and off-target editing. Mol Ther. 28:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CY, Antonopoulos A, Gupta R, Qu J, Dell A, Haslam SM, Neelamegham S. 2013. Competition between core-2 GlcNAc-transferase and ST6GalNAc-transferase regulates the synthesis of the leukocyte selectin ligand on human P-selectin glycoprotein ligand-1. J Biol Chem. 288:13974–13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal N, Buffone A, Stolfa G, Antonopoulos A, Lau JTY, Haslam SM, Dell A, Neelamegham S. 2015. ST3Gal-4 is the primary sialyltransferase regulating the synthesis of E-, P-, and L-selectin ligands on human myeloid leukocytes. Blood. 125:687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal N, Stolfa G, Antonopoulos A, Zhu Y, Wang SS, Buffone A Jr, Atilla-Gokcumen GE, Haslam SM, Dell A, Neelamegham S. 2016. Glycosphingolipids on human myeloid cells stabilize E-selectin-dependent rolling in the multistep leukocyte adhesion cascade. Arterioscler Thromb Vasc Biol. 36:718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moremen KW, Tiemeyer M, Nairn AV. 2012. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat Rev Mol Cell Biol. 13:448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narimatsu Y, Joshi HJ, Yang Z, Gomes C, Chen YH, Lorenzetti FC, Furukawa S, Schjoldager KT, Hansen L, Clausen H et al. 2018. A validated gRNA library for CRISPR/Cas9 targeting of the human glycosyltransferase genome. Glycobiology. 28:295–305. [DOI] [PubMed] [Google Scholar]
- Neelamegham S, Liu G. 2011. Systems glycobiology: Biochemical reaction networks regulating glycan structure and function. Glycobiology. 21:1541–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelamegham S, Mahal LK. 2016. Multi-level regulation of cellular glycosylation: From genes to transcript to enzyme to structure. Curr Opin Struct Biol. 40:145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peanne R, de Lonlay P, Foulquier F, Kornak U, Lefeber DJ, Morava E, Perez B, Seta N, Thiel C, Van Schaftingen E et al. 2018. Congenital disorders of glycosylation (CDG): Quo vadis? Eur J Med Genet. 61:643–663. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG et al. 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 343:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z. 2013. Roles of the nucleotide sugar transporters (SLC35 family) in health and disease. Mol Aspects Med. 34:590–600. [DOI] [PubMed] [Google Scholar]
- Steentoft C, Vakhrushev SY, Vester-Christensen MB, Schjoldager KT, Kong Y, Bennett EP, Mandel U, Wandall H, Levery SB, Clausen H. 2011. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat Methods. 8:977–982. [DOI] [PubMed] [Google Scholar]
- Stolfa G, Mondal N, Zhu YQ, Yu XH, Buffone A, Neelamegham S. 2016. Using CRISPR-Cas9 to quantify the contributions of O-glycans, N-glycans and glycosphingolipids to human leukocyte-endothelium adhesion. Sci Rep. 6:30392, doi:10.1038/srep30392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom J, Heider D, Martin SR, Pastore A, Mueller JW. 2012. 3'-Phosphoadenosine 5'-phosphosulfate (PAPS) synthases, naturally fragile enzymes specifically stabilized by nucleotide binding. J Biol Chem. 287:17645–17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. 2017. Biological roles of glycans. Glycobiology. 27:3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmuckett AD, Thacker KM, Moore KL. 2011. Tyrosine sulfation of native mouse Psgl-1 is required for optimal leukocyte rolling on P-selectin in vivo. PLoS One. 6:e20406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins PP, Moore KL, McEver RP, Cummings RD. 1995. Tyrosine sulfation of P-selectin glycoprotein ligand-1 is required for high affinity binding to P-selectin. J Biol Chem. 270:22677–22680. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, Wei W. 2014. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 509:487–491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.