

Abstract
Studies on the shell color and banding polymorphism of the grove snail Cepaea nemoralis and the sister taxon Cepaea hortensis have provided compelling evidence for the fundamental role of natural selection in promoting and maintaining intraspecific variation. More recently, Cepaea has been the focus of citizen science projects on shell color evolution in relation to climate change and urbanization. C. nemoralis is particularly useful for studies on the genetics of shell polymorphism and the evolution of “supergenes,” as well as evo-devo studies of shell biomineralization, because it is relatively easily maintained in captivity. However, an absence of genomic resources for C. nemoralis has generally hindered detailed genetic and molecular investigations. We therefore generated ∼23× coverage long-read data for the ∼3.5 Gb genome, and produced a draft assembly composed of 28,537 contigs with the N50 length of 333 kb. Genome completeness, estimated by BUSCO using the metazoa dataset, was 91%. Repetitive regions cover over 77% of the genome. A total of 43,519 protein-coding genes were predicted in the assembled genome, and 97.3% of these were functionally annotated from either sequence homology or protein signature searches. This first assembled and annotated genome sequence for a helicoid snail, a large group that includes edible species, agricultural pests, and parasite hosts, will be a core resource for identifying the loci that determine the shell polymorphism, as well as in a wide range of analyses in evolutionary and developmental biology, and snail biology in general.
Keywords: mollusks, shell pigmentation, supergene, de novo assembly and annotation, PacBio sequencing
Introduction
Studies on the shell color and banding polymorphism of the grove snail Cepaea nemoralis (Figure 1), and its sister taxon Cepaea hortensis, played a prominent role in demonstrating how selective forces and random processes drive or maintain morphological variation, and contributed to the establishment of the field of ecological genetics (Jones et al. 1977; Cook 1998; Ozgo 2008). Alongside the peppered moth, the shell polymorphism of Cepaea snails is still the classic text book example used to illustrate natural selection and micro‐evolution. Recently, C. nemoralis has been the focus of citizen science projects which studied shell color evolution in association to climate change and urbanization (Silvertown et al. 2011; Kerstes et al. 2019). Being relatively easily maintained and bred in captivity, this snail is also particularly appropriate for evo‐devo studies of shell biomineralization (Mann and Jackson 2014; Jackson and Degnan 2016) and pigmentation (Kerkvliet et al. 2017; Affenzeller et al. 2020).
Figure 1.

Genetically determined color polymorphism in C. nemoralis. Morphs from left to right: (1) yellow unbanded, white lip; (2) brown mid-banded, white lip; (3) pink unbanded, normal lip; (4) yellow five-banded, white lip; (5) brown unbanded, normal lip; (6) pink five-banded, normal lip; (7) yellow three-banded (10305), normal lip. Image credit: Angus Davison.
Previous work has shown that the shell polymorphism is controlled by a series of nine or more loci, of which five or more are tightly linked in a single “supergene” (Cook 1998; Gonzalez et al. 2019). This, combined with the advantages mentioned above, means that Cepaea has great potential to provide insights into supergene evolution and the role of genome structure in adaptation. However, progress in understanding the genetic basis of its color pattern formation has been slow, in contrast to other classical systems such as mimicry in Heliconius butterflies (Nadeau et al. 2016) and industrial melanism in the peppered moth (Van ‘t Hof et al. 2016). Although some advancement toward identifying the supergene has been made recently (Richards et al. 2013; Kerkvliet et al. 2017), a lack of genomic resources has largely prevented further analyses.
Here, we present a draft assembly and annotation of the C. nemoralis genome, the first available genome for helicoid snails (Wade et al. 2007) and the second for a terrestrial mollusk, after the giant African snail Achatina fulica (Guo et al. 2019). Helicoidea is a large group of stylommatophoran land snails that includes not only important models for studies of shell formation and chirality (e.g. the genus Euhadra, see Davison 2020), but also several edible species (e.g. including Cepaea, but especially the genera Helix and Cornu) and agricultural pests. In addition, many Helicoidea are intermediate hosts of various parasites (e.g.Gérard et al. 2020), and therefore are important subjects in studies of human and animal disease prevention.
Despite great ecological, economical, and medical importance, stylommatophoran land snails have been underrepresented in whole genome sequencing projects (Yang et al. 2020), mainly because of their large repetitive genomes (C-values between 1.68 and 4.00, see http://www.genomesize.com/). Usually, sequencing coverage above 30× is recommended to overcome this problem (Dominguez Del Angel et al. 2018), but this is often financially challenging for individual research groups. Here, we took advantage of recent technological and computational breakthroughs to produce the draft assembly of C. nemoralis genome based on lower coverage PacBio sequencing. Even though the assembly presented here is rather fragmented, it should be a key resource for researchers working on diverse aspects of land snail biology, including the identification of genes involved in developmental processes, e.g. shell formation and color patterning. Furthermore, it will open up new research avenues for understanding such important biological processes as adaptation to urban environments and climate change, interactions with parasites, and reproduction.
Materials and methods
Estimation of genome size by flow cytometry
The haploid chromosome number in C. nemoralis is 22 (Page 1978). We performed flow cytometry analysis to estimate the haploid genome size using zebrafish Danio rerio as a reference and the “CyStain PI Absolute P” reagent kit (Sysmex Europe, Germany). Briefly, zebrafish tail and snail foot tissues were chopped with a sharp razor blade in 500 µL ice-cold nuclei extraction buffer in a petri dish and incubated for 1 min. Then, the tissues were incubated for 30 minutes in 2.0 mL of staining buffer containing the fluorescent dye propidium iodide (50 µg/mL), RNAse (10 µg/mL), 0.1% dithiothreitol, and 1% polyvinylpyrolidone. The processed sample was passed through a nylon 50 µm filter. The DNA content of stained nuclei was determined using CyFlow-Cube-6 flow cytometer (Sysmex Europe, Germany) as an average of three replicates.
Sample preparation
A single mid-banded hyalozonate snail with yellow ground color was used for the construction of the reference genome. This individual (C981) is the offspring of cross #13 described in Gonzalez et al. (2019), partially inbred, with additional information on and DNA from five generations of the relatives available for future work. High-molecular-weight genomic DNA (HMW-gDNA) was extracted from frozen snail foot tissue using the CTAB (cetyl trimethylammonium bromide) protocol as described in Richards et al. (2013) and Gonzalez et al. (2019). In brief, slices of snail tissue were incubated at 65°C in extraction solution (3% CTAB, 100 mM Tris‐HCl, pH 7.5, 25 mM EDTA, pH 8, 2 M NaCl) with 0.2 mg/mL proteinase K and 80 μg/mL RNase. Upon lysis, a chloroform extraction was performed, then three volumes of CTAB dilution solution were added (1% CTAB, 50 mm Tris‐HCl, pH 7.5, 10 mM EDTA, pH 8). Samples were mixed until a precipitate appeared, then the supernatant was removed. The pellet was washed twice in 0.4 M NaCl in TE (0.4 M NaCl, 10 mM Tris‐HCl, pH 7.5, 1 mm EDTA, pH 8), redissolved in 1.42 M NaCl in TE (1.42 M NaCl, 10 mM Tris‐HCl, pH 7.5, 1 mM EDTA, pH 8), then precipitated in ethanol, spooled out, washed in 70% ethanol, and air dried. The integrity of extracted HMW-gDNA was evaluated by performing pulsed-field agarose gel electrophoresis, whereas the purity and concentration were measured by spectrophotometry (with Nanodrop 2000, Thermo Fisher Scientific Inc.) and fluorometry (with Qubit 3.0, Thermo Fisher Scientific Inc.), respectively.
Whole genome sequencing and quality control
We sequenced the genome of C. nemoralis using PacBio single-molecule real-time (SMRT) and Illumina platforms. PacBio library preparation and sequencing were performed at Leiden Genome Technology Center (Leiden, the Netherlands). Without additional shearing, 4 µg of HMW-gDNA was converted into a SMRTbell library using “Procedure & Checklist—Preparing >30 kb Libraries Using SMRTbell Express Template Preparation Kit” (Pacific Biosciences). The insert size of the final library was then determined on Fragment Analyzer (Agilent Technologies). To increase the sequencing read length, an additional damage repair was performed on the library. The library was annealed with sequencing primer V4 and binding was done using binding kit version 3. The library was sequenced with 20 h movie-time using Sequel Sequencing kit v3.0 chemistry on 12 PacBio Sequel SMRT cells (PacBio Sequel System, RRID: SCR_017989), generating 7,202,997 subreads, or 80 Gb of sequence data (i.e. 23× genome coverage). The polymerase read length N50 (18,196 bp) was only slightly higher than the subread length N50 (16,882 bp), indicating that the majority of data consists of continuous long reads (CLRs). In addition, 17,390 circular consensus sequencing (CCS) reads of >99% accuracy were generated as well.
For Illumina sequencing, HMW-gDNA was sheared with the Covaris M220 (Covaris Inc., Woburn, MA, USA), set to 500-bp fragment size. A paired-end library was prepared using NEBNext Ultra II DNA Library Prep Kit (New England Biolabs) and sequenced on the Illumina NovaSeq 6000 Sequencing System (RRID: SCR_016387). Illumina sequencing was performed at BaseClear B.V. (Leiden, the Netherlands). Initial quality assessment was based on data passing the Illumina Chastity filtering. Subsequently, reads containing PhiX control signal were removed using an in-house filtering protocol. In addition, reads containing (partial) adapters were clipped (up to a minimum read length of 50 bp). The second quality assessment of the remaining reads was done with FASTQC v0.11.5 (Andrews 2014). We obtained ∼400 million of filtered 150 bp paired-end reads, or 120 Gb of sequence data, representing ∼34× coverage of a 3.5 Gb genome.
Heterozygosity estimation
Illumina paired-end reads were used to estimate heterozygosity of the sequenced individual by k-mer analysis. We used Jellyfish v2.3.0 (Jellyfish, RRID: SCR 005491) (Marcais and Kingsford 2011) to count canonical 31-mers from the sequencing data and to produce the k-mer count histogram with max coverage threshold set to 1,000,000. The latter was analyzed by GenomeScope (Vurture et al. 2017) to estimate the heterozygosity.
De novo genome assembly
The reference genome of C. nemoralis was constructed from PacBio CLRs of >5 kb (a total of 4.8 million reads, or 73.7 Gb of sequence data) using three different assembly packages. First, we used Flye v2.4.2 (Flye, RRID: SCR_017016) (Kolmogorov et al. 2019) with default parameters for raw PacBio reads to construct a 4.2 Gb genome assembly with 70,762 contigs and a contig N50 length of 105 kb. Then, we used Canu v1.8 (Canu, RRID: SCR_015880) (Koren et al. 2017) with parameters adjusted for low coverage and fast overlap (corMhapFilterThreshold = 0.0000000002 corMhapOptions = “–threshold 0.80 –num-hashes 512 –num-min-matches 3 –ordered-sketch-size 1000 –ordered-kmer-size 14 –min-olap-length 2500 –repeat-idf-scale 50” mhapBlockSize = 500 ovlMerDistinct = 0.975 correctedErrorRate = 0.105 corMinCoverage = 0 corMhapSensitivity=high minReadLength = 5000 minOverlapLength = 2500 corOutCoverage = 200) to produce a 4.9 Gb assembly with 66,503 contigs and N50 of 111 kb. Finally, we used the output of Canu trimming stage (4.1 million reads, or 61.5 Gb) and the CCS reads as an input for wtdbg2 v2.4 (WTDBG2, RRID: SCR_017225) (Ruan and Li 2020), which was run with preset parameters for CCS reads and options “–edge-min 2 –rescue-low-cov-edges.” The resultant Wtdbg2 assembly is 3.5 Gb and contained 64,453 contigs with N50 of 132 kb. Assembly statistics assessed using Quast v5.02 (QUAST, RRID: SCR_001228) (Gurevich et al. 2013) are shown in Table 1.
Table 1.
Statistics for different stages of genome assembly
| Assembly | Total size, bp | GC (%) | Contig number | No. of contigs > 10 kb | Max. contig length, bp | Contig N50, bp | Contig L50 |
|---|---|---|---|---|---|---|---|
| Flye | 4,193,822,794 | 41.38 | 70,762 | 54,326 | 1,622,356 | 105,488 | 10,998 |
| Canu | 4,892,999,477 | 41.09 | 66,503 | 64,936 | 1,255,215 | 110,511 | 12,124 |
| Wtdbg2 | 3,512,271,831 | 41.16 | 64,453 | 52,879 | 1,739,336 | 131,562 | 7,053 |
| Flye_red | 3,362,274,305 | 41.36 | 42,275 | 37,759 | 1,622,356 | 121,492 | 7,997 |
| Canu_red | 3,565,230,412 | 41.16 | 38,076 | 37,950 | 1,255,215 | 132,263 | 8,271 |
| Wtdbg2_red | 3,163,782,079 | 41.16 | 44,228 | 39,951 | 1,739,336 | 148,882 | 5,957 |
| Final | 3,490,924,950 | 41.25 | 28,537 | 26,580 | 3,510,238 | 333,110 | 3,035 |
Duplicated contigs were removed from all three assemblies using purge_dups v1.0.0 (Guan et al. 2020) with default parameters. These reduced assemblies are characterized by smaller size and contig numbers, and improved contig N50 lengths (Table 1). Next, to produce a more contiguous assembly, we merged the three reduced assemblies in two consecutive steps. First, we aligned Canu_red and Flye_red assemblies using MUMmer v4.0.0 (MUMmer, RRID: SCR_018171) (Kurtz et al. 2004) with nucmer parameters “–maxmatch -c 300 -l 100” and delta-filter parameters “-r -q -l 10000,” and merged them using Quickmerge (Chakraborty et al. 2016) (parameters “-hco 10 -c 3 -l 130000 -ml 10000”), with Flye_red as a query and Canu_red as reference input. The resultant assembly was used as reference input to merge with the Wtdbg2_red assembly in the second step, with parameters “-hco 10 -c 3 -l 200000 -ml 10000.” Finally, the assembly was polished twice: (1) using the arrow algorithm from PacBo GenomicConsensus package (https://github.com/pacificbiosciences/genomicconsensus/) and PacBio subreads and (2) using Pilon v1.23 (Pilon, RRID: SCR_014731) (Walker et al. 2014) with highly accurate Illumina short reads and parameters “–changes –diploid –fix bases –nostrays.”
Genome assembly quality evaluation
The final assembly was evaluated in three different ways. First, to assess potential contamination in the sequences, we used BlobTools v1.0.1 (Blobtools, RRID: SCR_017618) (Laetsch and Blaxter 2017) with default parameters. The NCBI nonredundant nucleotide database and the UniProt reference proteome database (both downloaded on October 29, 2019) were used for the taxonomy classification step. Hit files were generated by sequence similarity searches against these databases using BLASTn v2.9.0+ (BLASTN, RRID: SCR 001598) (Camacho et al. 2009) and Diamond (DIAMOND, RRID: SCR_016071) (Buchfink et al. 2015), respectively. Second, we aligned raw Illumina paired-end reads to the assembly with BWA v0.7.16a (BWA, RRID: SCR_010910) (Li and Durbin 2009). Finally, we evaluated gene content completeness using BUSCO v4.0.2 (BUSCO, RRID: SCR_015008) (Simão et al. 2015) with the metazoa_odb10 dataset consisting of 954 BUSCOs (Benchmarking Universal Single-Copy Orthologs) from 65 species.
Repeat element annotation
A species-specific de novo library of transposable elements (TEs) and repeats was generated for C. nemoralis using RepeatModeler v2.0.1 (RepeatModeler, RRID: SCR_015027) (Smit and Hubley 2008–2015) and its integrated tools RECON (Bao and Eddy 2002), Tandem Repeats Finder v4.09 (Benson 1999), and RepeatScout v1.0.5 (RepeatScout, RRID: SCR_014653) (Price et al. 2005). This custom database (Supplemental File S1) was combined with the library of known repeats from other species v24.01 obtained from RepBase (Bao et al. 2015). The combined library was used to identify and soft-mask repetitive elements in the C. nemoralis genome with RepeatMasker v4.1.0 (RepeatMasker, RRID: SCR 012954) (Smit et al. 2013–2015), run with rmblastn v2.9.0+ as search engine.
Genome annotation
The annotation was performed on the soft-masked assembly to avoid missing (parts of) coding sequences due to overlap with masked areas of the genome. We used the MAKER v2.31.10 pipeline (MAKER, RRID: SCR_005309) (Cantarel et al. 2007; Campbell et al. 2014) in three consecutive rounds, combining ab initio gene predictions with sequence-based evidence. In the first round, the available transcriptome generated from foot and mantle tissues of four C. nemoralis snails (147,397 contigs, see Kerkvliet et al. 2017), as well as the protein dataset of A. fulica snail (23,726 predicted proteins, see Guo et al. 2019), were aligned to the genome with BLASTn (BLASTN, RRID: SCR_001598) and BLASTx (BLASTX, RRID: SCR_001653) algorithms from BLAST v2.9.0+ (NCBI BLAST, RRID: SCR_004870), respectively (est2genome and protein2genome options in MAKER configuration file). After further refinement of these alignments with respect to splice sites using Exonerate v2.4.0 (Exonerate, RRID: SCR_016088) (Slater and Birney 2005), MAKER generated gene models and calculated their annotation edit distance (AED) scores in order to assess the quality of gene prediction (i.e. the lower AED value the smaller the difference between the predicted protein and the transcript/protein evidence). Out of 308,927 genes models generated in the first round, 89% had an AED <0.5, indicating that the annotation is well-supported by transcript and/or protein evidence.
The second and third rounds of MAKER were performed on the gene models with AED < 0.4 obtained from the first and second runs, respectively. MAKER scripts maker2zff, fathom, forge, and hmm-assembler.pl were used to create snaphmm files (snaphmm option in maker configuration file) to train ab initio gene predictor SNAP (SNAP, RRID: SCR_002127) (Korf 2004). Another ab initio gene predictor, Augustus v3.3.3 (Augustus, RRID: SCR_008417) (Stanke et al. 2006), was self-trained running BUSCO v4.0.2 with the specific parameter (–long); the generated “retraining parameters” file for C. nemoralis was included in the second and third rounds of MAKER annotation. The third and final round of MAKER generated 173,620 gene models with AED <0.5. As the annotation was performed on the soft-masked assembly, many of these putative genes could be derived from repetitive sequences, explaining such a high number. Hence, we removed gene models with >50% overlap within a single repeat region as annotated by Repeat Masker (see above). This resulted in the final set of 43,519 predicted protein-coding genes (Supplementary Files S2 and S3) with average AED of 0.27.
We performed functional annotation of predicted proteins using three automated methods. First, we applied Diamond (Buchfink et al. 2015) BLASTp searches (–sensitive –max-target-seqs 1 –outfmt 6 qseqid sallseqid pident evalue bitscore -evalue 1e-5) against UniProt reference proteome database (v2019_09, composed of 561,176 Swiss-Prot and 180,179,667 TrEMBL entries) and the NCBI nonredundant protein database (downloaded on 26 May 2020 and composed of 287,467,303 entries). Second, we used KEGG Automatic Annotation Server (KAAS) (Moriya et al. 2007) with eukaryotic species set and the bi-directional best-hit method to assign KEGG orthology (Kanehisa et al. 2012) to gene models. Finally, we used InterProScan (Jones et al. 2014) and Blast2GO (Götz et al. 2008) functions in the OmicsBox to examine motifs, domains, and signatures in the protein sequences and to assign gene ontology (GO) terms to the gene models.
Data availability
This C. nemoralis whole genome sequencing project has been submitted to NCBI with BioProject accession number PRJNA646049. Sequencing reads from Illumina and PacBio platforms have been deposited at NCBI Sequence Read Archive (SRA) under the accession numbers SRX8724912 and SRX8724913, respectively. The assembled genome sequence has been deposited at DDBJ/ENA/GenBank under the accession JACEFZ000000000. The version described in this study is version JACEFZ010000000. Supplementary material is available on figshare (https://doi.org/10.25387/g3.13353083). Supplementary File S1 contains de novo library of repeats and TEs generated by RepeatModeler. Supplementary File S2 contains sequences of protein-coding genes predicted in the C. nemoralis genome with MAKER. Supplementary File S3 contains MAKER annotation results. Supplementary Table S1 contains GenomeScope results. Supplementary Table S2 contains full output of the BlobTools analysis. Supplementary Table S3 contains BUSCO results. Supplementary Table S4 contains RepeatMasker results. Supplementary Table S5 contains functional annotation of the predicted protein-coding genes. Supplementary Figure S1 describes main characteristics of the predicted protein-coding genes.
Results and discussion
Genome size and heterozigosity estimation
We used flow cytometry to determine that the haploid genome size of C. nemoralis is 2.06 times larger than that of the zebrafish (C-value ∼1.7, see Vinogradov 1998; Ciudad et al. 2002) and is therefore ∼3.5 picogram, or ∼3.42 Gb. When taking the total length of the most recent zebrafish genome assembly of 1.68 Gb (cf. Genome Reference Consortium, https://www.ncbi.nlm.nih.gov/grc/zebrafish/data, last accessed on 13-01-2021) as a reference, the genome size of C. nemoralis is calculated at ∼3.46 Gb. This fits within the range of estimated genome sizes for others members of the family Helicidae (C-values between 2.88 and 4.00, see http://www.genomesize.com/, last accessed on 13-01-2021). The 31-mer based estimate of genome size provided by GenomeScope (∼3.1 Gb, see Figure 2 and Supplementary Table S1) is smaller than the flow cytometry estimate. Such discrepancy is often found in repeat-rich genomes (e.g.Edwards et al. 2018), because high-frequency repeats are difficult to model accurately, leading to an underestimation of total repeat length and therefore genome size. The heterozygosity of the individual C981 (Gonzalez et al. 2019) genome estimated by GenomeScope (Vurture et al. 2017) is ∼1.42%, consistent with the high heterozygosity of other sequenced mollusks (e.g.Guo et al. 2019; Kenny et al. 2020).
Figure 2.
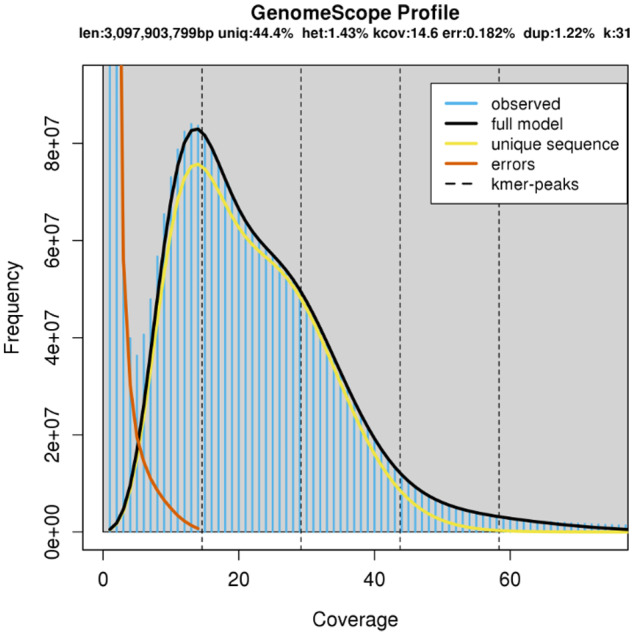
GenomeScope k-mer profile plot for the genome of C. nemoralis individual C981, based on 31-mers in Illumina reads. The observed k-mer frequency distribution is depicted in blue, whereas the GenomeScope fit model is shown as a black line. The unique and putative error k-mer distributions are plotted in yellow and red, respectively.
Genome assembly and quality evaluation
We used 4.8 million PacBio long reads, or 73.7 Gb of sequence data, to assemble the genome of C. nemoralis. The assembly was polished with PacBio subreads and with highly accurate Illumina short reads. The final genome assembly has total length of 3.5 Gb and is composed of 28,537 contigs with N50 length of 333 kb (Table 1). The mapping rate of Illumina reads agains the final assembly was rather high, with about 99.3% of the reads aligned, and 93.5% properly paired (i.e. both reads of the pair mapped to the same contig).
Blobtools analyses indicated no substantial contamination with bacterial DNA (Figure 3 and Supplementary Table S2). About 75% of the contigs were assigned to Mollusca, whereas ∼20% were assigned to Chordata and Arthropoda. Closer examination of such cases revealed that the assignment to these two orders is due to a chance blast match with relatively high similarity over a small region of the contig (i.e. top hit is to a vertebrate/arthropod species but multiple other hits with a slightly lower bit score are to a mollusk species).
Figure 3.

BlobPlot of the C. nemoralis genome assembly. Each contig is represented by a circle, colored according to the best match to taxonomic annotation (e.g. Mollusca, Chordata, and so on) and distributed according to the proportion GC (x-axis) and read coverage (y-axis). The upper- and right-hand panels show the distribution of the total span (kb) of contigs for a given coverage (right panel) or GC (upper panel) bin.
Finally, assembly completeness was assessed with BUSCO v4.0.2 (Simão et al. 2015), the tool that looks for Benchmarking Universal Single-Copy Orthologs (BUSCOs) that should be present in a metazoan genome. Out of the 954 metazoan BUSCOs, 832 (87.2%) were identified in the draft assembly of C. nemoralis genome as complete (709, or 74.3% as single copy, and 123, or 12.9% as duplicated), 36 (3.8%) as fragmented, and only 86 (9.0%) as missing (Supplementary Table S3). High levels of duplicated genes indicate that, despite haplotig removal, some genomic regions were assembled as separate contigs, most likely due to the high heterozygosity of the genome.
Genome annotation
We estimated the total repeat content of the C. nemoralis genome to be around 77% (Figure 4), comparable to the 71% found in A. fulica (Guo et al. 2019) and expected for such a large genome. Nearly 45% of the genome can be attributed to TEs: nonLTR retrotransposons such as LINEs (long interspersed nuclear elements) and SINEs (short interspersed nuclear elements), LTR (long terminal repeat) retrotransposons, and DNA transposons; ∼6.4% of the repeats were predicted to be small RNAs (i.e. transfer RNAs and small nuclear RNAs), satellites, simple and low-complexity repeats (Table 2 and Supplementary Table S4).
Figure 4.
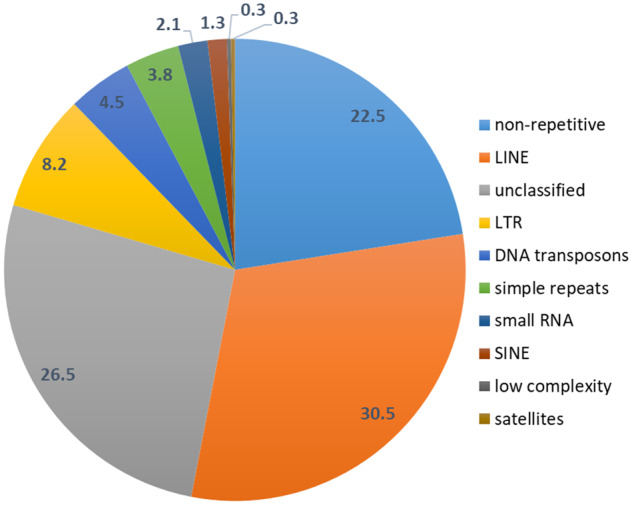
Repetitive content of the assembled C. nemoralis genome as identified by RepeatMasker. Numbers indicate percentages of the genome size. NonLTR retrotransposons of the LINE type and LTR retrotransposons, as well as unclassified sequences, dominate the repetitive content.
Table 2.
Major types of repeat elements identified in the C. nemoralis genome assembly
| Repeat class | Repeat element type | No. of elements | Total length, bp | % of genome |
|---|---|---|---|---|
| LINE | RTE/Bov-B | 1,922,793 | 657,283,360 | 18.83 |
| R1/LOA/Jockey | 378,298 | 203,267,666 | 5.82 | |
| L2/CR1/Rex | 91,512 | 44,303,678 | 1.27 | |
| R2/R4/NeSL | 15,845 | 6,537,107 | 0.19 | |
| SINE | Penelope | 88,259 | 27,930,778 | 0.80 |
| LTR | Gypsy/DIRS1 | 209,476 | 286,647,606 | 8.21 |
| DNA transposon | Tc1-IS630-Pogo | 162,488 | 58,611,608 | 1.68 |
| hobo-Activator | 147,068 | 50,294,093 | 1.44 |
We annotated the genome using MAKER v2.31.10 (Cantarel et al. 2007; Campbell et al. 2014), by supplementing the ab initio gene predictions with the C. nemoralis transcriptome (Kerkvliet et al. 2017) and the protein dataset of the snail A. fulica (Guo et al. 2019), and two additional rounds of further refinement of gene models with multiple tools integrated into the MAKER pipeline. The final assembly contains 43,519 predicted protein-coding genes (Supplementary Files S2 and S3). Length distribution for genes, exons, and introns is comparable to those of other mollusks (Guo et al. 2019) (Table 3 and Supplementary Figure S1). About 93.1% of the predicted genes have multiple exons (4.7 on average), which is slightly lower than in other mollusks (Kenny et al. 2020). This could be explained by some degree of fragmentation in the gene models, especially those in small contigs. In addition, 97.3% of the predicted protein-coding genes had a hit to at least one of the databases (Table 4) and were functionally annotated (Supplementary Table S5).
Table 3.
Characteristics of the annotated genes in the C. nemoralis assembly
| Feature | Value |
|---|---|
| Number of protein-coding genes | 43,519 |
| Mean gene locus size (bp) | 9,296 |
| Mean transcript size (bp) | 1,492 |
| Mean exon size (bp) | 315 |
| Mean intron size (bp) | 2,094 |
| Number of multi-exon genes | 40,534 |
| Number of single-exon genes | 2,985 |
| Number of distinct exons | 205,715 |
| Mean number of distinct exons per gene | 4.7 |
Table 4.
Summary of functional annotation
| Database | Number of hits | % |
|---|---|---|
| NCBI nonredundant protein (NR) | 37,991 | 87.3 |
| UniProt (Swiss-Prot and TrEMBL) | 37,510 | 86.2 |
| KEGG orthology | 9,342 | 21.5 |
| InterPro | 40,086 | 92.1 |
| GO terms (InterProScan) | 19,288 | 44.3 |
| GO terms (Blast2GO) | 14,866 | 34.2 |
| Nonredundant hits | 42,337 | 97.3 |
| Unannotated | 1,182 | 2.7 |
Conclusions and perspectives
We performed whole-genome assembly of C. nemoralis using a combination of PacBio long-read technology and Illumina short-read sequencing. This ∼3.5 Gb draft assembly is composed of 28,537 contigs with the N50 length of 333 kb; repetitive regions cover over 77% of the genome. BUSCO analysis showed that only 9.0% of metazoan orthologs were missing, indicating high genome completeness. More than 43,000 protein-coding genes were identified in the genome, and more than 97.0% of these were functionally annotated from either sequence homology or protein signature searches. To our best knowledge, this is the largest gastropod genome sequenced and assembled to date. Compared to other gastropods (e.g. Guo et al. 2019; Gomes-dos-Santos et al. 2020; Sun et al. 2020), the genome of C. nemoralis is characterized by a very high content of repetitive sequences.
Despite its large size and the abundance of repeats, the assembly presented here is of high quality, and will be a valuable resource for the land snail research community. In particular, it will facilitate the identification of genes that drive the extraordinary diversity of shell colors and patterns in C. nemoralis, and the sister species C. hortensis, as well as comparative work in other stylommatophoran snails. In addition, the genome assembly described here will directly enable a wide range of studies on various aspects of terrestrial snail biology, from early development and biomineralization to physiology, behavior, and population genomics.
Acknowledgments
We thank Susan Kloet and Rolf Vossen from Leiden Genome Technology Centre for advice on PacBio sequencing and technical support.
Funding
Funding was provided by the Netherlands Organization for Scientific Research NWO (ALWOP.386 grant).
Conflicts of interest: None declared.
Literature cited
- Affenzeller S, Wolkenstein K, Frauendorf H, Jackson DJ.. 2020. Challenging the concept that eumelanin is the polymorphic brown banded pigment in Cepaea nemoralis. Sci Rep. 10:2442.doi:10.1038/s41598-020-59185-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. 2014. FastQC: a quality-control tool for highthroughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. (Accessed: 2020 December 2).
- Bao Z, Eddy SR.. 2002. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12:1269–1276. doi:10.1101/gr.88502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao W, Kojima KK, Kohany O.. 2015. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6:11.doi:10.1186/s13100-015-0041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27:573–580. doi:10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, Huson DH.. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 12:59–60. doi:10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, et al. 2009. BLAST+: architecture and applications. BMC Bioinformatics. 10:421. doi:10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MS, Holt C, Moore B, Yandell M.. 2014. Genome annotation and curation using MAKER and MAKER-P. Curr Protoc Bioinformatics. 48:4.11.1–4.11.39. doi:10.1002/0471250953.bi0411s48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, et al. 2007. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18:188–196. doi:10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty M, Baldwin-Brown JG, Long AD, Emerson JJ.. 2016. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 44:e147.doi:10.1093/nar/gkw654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciudad J, Cid E, Velasco A, Lara JM, Aijón J, et al. 2002. Flow cytometry measurement of the DNA contents of G0/G1 diploid cells from three different teleost fish species. Cytometry. 48:20–25. doi:10.1002/cyto.10100. [DOI] [PubMed] [Google Scholar]
- Cook LM. 1998. A two-stage model for Cepaea polymorphism. Philos Trans R Soc Lond B Biol Sci. 353:1577–1593. doi:10.1098/rstb.1998.0311. [Google Scholar]
- Davison A. 2020. Flipping shells! Unwinding LR asymmetry in mirror-image molluscs. Trends Genet. 36:189–202. doi:10.1016/j.tig.2019.12.003. [DOI] [PubMed] [Google Scholar]
- Dominguez Del Angel V, Hjerde E, Sterck L, Capella-Gutierrez S, Notredame C, et al. 2018. Ten steps to get started in genome assembly and annotation. F1000Res. 7:148. doi:10.12688/f1000research.13598.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RJ, Tuipulotu DE, Amos TG, O'Meally D, Richardson MF, et al. 2018. Draft genome assembly of the invasive cane toad, Rhinella marina. Gigascience. 7:giy095. doi:10.1093/gigascience/giy095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gérard C, Ansart A, Decanter N, Martin MC, Dahirel M.. 2020. Brachylaima spp. (Trematoda) parasitizing Cornu aspersum (Gastropoda) in France with potential risk of human consumption. Parasite. 27:15.doi:10.1051/parasite/2020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes-dos-Santos A, Lopes-Lima M, Castro LFC, Froufe E.. 2020. Molluscan genomics: the road so far and the way forward. Hydrobiologia. 847:1705–1726. doi:10.1007/s10750-019-04111-1. [Google Scholar]
- Gonzalez DR, Aramendia AC, Davison A.. 2019. Recombination within the Cepaea nemoralis supergene is confounded by incomplete penetrance and epistasis. Heredity. 123:153–161. doi:10.1038/s41437-019-0190-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, et al. 2008. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36:3420–3435., doi:10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan D, McCarthy SA, Wood J, Howe K, Wang Y, et al. 2020. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 36:2896–2898. doi:10.1093/bioinformatics/btaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Zhang Y, Liu Q, Huang Y, Mao G, et al. 2019. A chromosomal-level genome assembly for the giant African snail Achatina fulica. Gigascience. 8:giz124. doi:10.1093/gigascience/giz124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G.. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 29:1072–1075. doi:10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DJ, Degnan BM.. 2016. The importance of evo-devo to an integrated understanding of molluscan biomineralisation. J Struct Biol. 196:67–74. doi:10.1016/j.jsb.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Jones JS, Leith BH, Rawlings P.. 1977. Polymorphism in Cepaea: a problem with too many solutions? Annu Rev Ecol Syst. 8:109–143. doi:10.1146/annurev.es.08.110177.000545. [Google Scholar]
- Jones P, Binns D, Chang H-Y, Fraser M, Li W, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics. 30:1236–1240. doi:10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M.. 2012. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40:D109–D114. doi:10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny NJ, McCarthy SA, Dudchenko O, James K, Betteridge E, et al. 2020. The gene-rich genome of the scallop Pecten maximus. Gigascience. 9:giaa037. doi:10.1093/gigascience/giaa037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkvliet J, de Boer T, Schilthuizen M, Kraaijeveld K.. 2017. Candidate genes for shell colour polymorphism in Cepaea nemoralis. PeerJ. 5:e3715.doi:10.7717/peerj.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerstes NAG, Breeschoten T, Kalkman VJ, Schilthuizen M.. 2019. Snail shell colour evolution in urban heat islands detected via citizen science. Commun Biol. 2:264.doi:10.1038/s42003-019-0511-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov M, Yuan J, Lin Y, Pevzner PA.. 2019. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 37:540–546. doi:10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, et al. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27:722–736. doi:10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. 2004. Gene finding in novel genomes. BMC Bioinformatics 5:59.doi:10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, et al. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi:10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laetsch DR, Blaxter ML.. 2017. BlobTools: interrogation of genome assemblies. F1000Res. 6:1287.doi:10.12688/f1000research.12232.1. [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25:1754–1760. doi:10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann K, Jackson DJ.. 2014. Characterization of the pigmented shell-forming proteome of the common grove snail Cepaea nemoralis. BMC Genomics. 15:249.doi:10.1186/1471-2164-15-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcais G, Kingsford C.. 2011. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27:764–770. doi:10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M.. 2007. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35:W182–W185. doi:10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau NJ, Pardo-Diaz C, Whibley A, Supple MA, Saenko SV, et al. 2016. The gene cortex controls mimicry and crypsis in butterflies and moths. Nature. 534:106–110. doi:10.1038/nature17961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgo M. 2008. Current problems in the research of Cepaea polymorphism. Folia Malacol. 16:55–60. [Google Scholar]
- Page C. 1978. The karyotype of the land snail Cepaea nemoralis (L). Heredity. 41:321–325. [Google Scholar]
- Price AL, Jones NC, Pevzner PA.. 2005. De novo identification of repeat families in large genomes. Bioinformatics. 21:i351–i358. doi:10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- Richards PM, Liu MM, Lowe N, Davey JW, Blaxter ML, et al. 2013. RAD-Seq derived markers flank the shell colour and banding loci of the Cepaea nemoralis supergene. Mol Ecol. 22:3077–3089. doi:10.1111/mec.12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan J, Li H.. 2020. Fast and accurate long-read assembly with wtdbg2. Nat Methods 17:155–158. doi:10.1038/s41592-019-0669-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvertown J, Cook L, Cameron R, Dodd M, McConway K, et al. 2011. Citizen science reveals unexpected continental-scale evolutionary change in a model organism. PLoS One. 6:e18927. doi:10.1371/journal.pone.0018927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31:3210–3212. doi:10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Slater GSC, Birney E.. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 6:31.doi:10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R.. 2008. –2015. RepeatModeler Open-1.0. (Accessed: January 2020). http://www.repeatmasker.org.
- Smit AFA, Hubley R, Green P.. 2013. –2015. RepeatMasker Open-4.0. (Accessed: January 2020). http://www.repeatmasker.org.
- Stanke M, Schöffmann O, Morgenstern B, Waack S.. 2006. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics. 7:62.doi:10.1186/1471-2105-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Chen C, Miyamoto N, Li R, Sigwart JD, et al. 2020. The Scaly-foot Snail genome and implications for the origins of biomineralised armour. Nat Commun. 11:1657. doi:10.1038/s41467-020-15522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van‘t Hof AE, Campagne P, Rigden DJ, Yung CJ, Lingley J, et al. 2016. The industrial melanism mutation in British peppered moths is a transposable element. Nature. 534:102–105. doi:10.1038/nature17951. [DOI] [PubMed] [Google Scholar]
- Vinogradov AE. 1998. Genome size and GC-percent in vertebrates as determined by flow cytometry: the triangular relationship. Cytometry. 31:100–109. doi:10.1002/(sici)1097-0320(19980201)31:2<100::aid-cyto5>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Vurture G, Sedlazeck M, Nattestad M, Underwood CJ, Fang H, et al. 2017. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 33:2202–2204. doi:10.1093/bioinformatics/btx153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade CM, Hudelot C, Davison A, Naggs F, Mordan PB.. 2007. Molecular phylogeny of the helicoid land snails (Pulmonata: Stylommatophora: Helicoidea), with special emphasis on the Camaenidae. J Molluscan Stud. 73:411–415. doi: 10.1093/mollus/eym030. [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9:e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Zhang L, Hu J, Wang J, Bao Z, et al. 2020. The evo-devo of molluscs: insights from a genomic perspective. Evol Dev. 14:e12336. doi:10.1111/ede.12336. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This C. nemoralis whole genome sequencing project has been submitted to NCBI with BioProject accession number PRJNA646049. Sequencing reads from Illumina and PacBio platforms have been deposited at NCBI Sequence Read Archive (SRA) under the accession numbers SRX8724912 and SRX8724913, respectively. The assembled genome sequence has been deposited at DDBJ/ENA/GenBank under the accession JACEFZ000000000. The version described in this study is version JACEFZ010000000. Supplementary material is available on figshare (https://doi.org/10.25387/g3.13353083). Supplementary File S1 contains de novo library of repeats and TEs generated by RepeatModeler. Supplementary File S2 contains sequences of protein-coding genes predicted in the C. nemoralis genome with MAKER. Supplementary File S3 contains MAKER annotation results. Supplementary Table S1 contains GenomeScope results. Supplementary Table S2 contains full output of the BlobTools analysis. Supplementary Table S3 contains BUSCO results. Supplementary Table S4 contains RepeatMasker results. Supplementary Table S5 contains functional annotation of the predicted protein-coding genes. Supplementary Figure S1 describes main characteristics of the predicted protein-coding genes.