

Key Points
ASFV MGF-505-7R inhibits cGAS–STING signaling.
MGF-505-7R–deficient ASFV was fully attenuated in pigs at a dose of 10 HAD50.
Visual Abstract
Abstract
African swine fever virus (ASFV) is a devastating infectious disease in pigs, severely threatening the global pig industry. To efficiently infect animals, ASFV must evade or inhibit fundamental elements of the innate immune system, namely the type I IFN response. In this study, we identified that ASFV MGF-505-7R protein exerts a negative regulatory effect on STING-dependent antiviral responses. MGF-505-7R interacted with STING and inhibited the cGAS–STING signaling pathway at STING level. MGF-505-7R overexpression either degraded STING or STING expression was reduced in ASFV-infected cells via autophagy, whereas STING expression was elevated in MGF-505-7R–deficient ASFV-infected cells. We further found that MGF-505-7R promoted the expression of the autophagy-related protein ULK1 to degrade STING, whereas ULK1 was elevated in MGF-505-7R–deficient ASFV-infected cells. Moreover, MGF-505-7R–deficient ASFV induced more IFN-β production than wild-type ASFV and was attenuated in replication compared with wild-type ASFV. The replicative ability of MGF-505-7R–deficient ASFV was also attenuated compared with wild-type. Importantly, MGF-505-7R–deficient ASFV was fully attenuated in pigs. Our results showed for the first time, to our knowledge, a relationship involving the cGAS–STING pathway and ASFV MGF-505-7R, contributing to uncover the molecular mechanisms of ASFV virulence and to the rational development of ASFV vaccines.
Introduction
African swine fever virus (ASFV) is a domestic swine pathogenic DNA arbovirus that is the sole member of family Asfarviridae (1). ASFV is a large, icosahedral virus that contains a linear dsDNA genome (170–190 kbp) encoding ∼165 genes (2). ASFV infection causes virulent disease in domestic pigs and European wild boars with mortality rates approaching 100%. However, there is no vaccine to prevent ASFV infection, and there are no drugs currently approved to treat ASFV infectious diseases (3). A previous study has demonstrated that ASFV proteins could efficiently suppress cellular and organismal defenses, which are pivotal for establishing immune evasion (4).
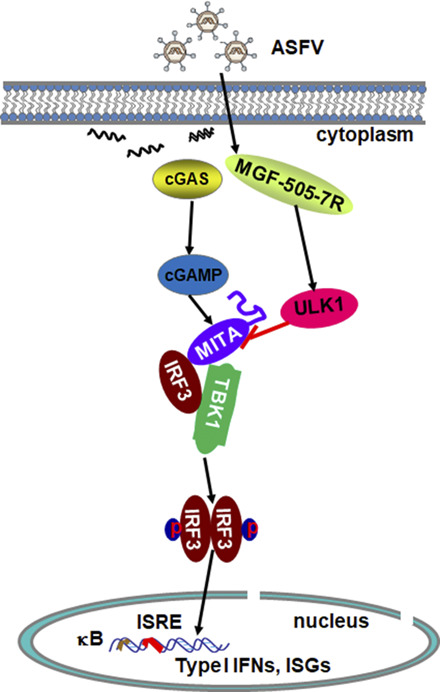
Innate immune response provides the first line of host defenses against invading microbial pathogens (5). Upon DNA viruses infecting a permissive cell, viral DNA is detected by cytosolic sensors. Although various cytosolic DNA sensors have been identified, it is widely accepted that a nucleotidyltransferase family member, called cyclic GMP–AMP (cGAMP) synthase (cGAS), has been used to detect cytosolic DNA in various cell types (6). After sensing viral DNA, cGAS catalyzes synthesis of the second messenger molecule cGAMP, which binds to the adaptor STING (also called MITA, MPYS, and ERIS), a critical adaptor in virus-triggered IFN induction pathways and innate antiviral responses (7–12). STING is a 379-aa protein, which consists of four N-terminal transmembrane domains (aa 1–137) and a C-terminal domain (aa 138–397). STING is localized to the endoplasmic reticulum, mitochondria, and mitochondrial-associated membrane via its N-terminal transmembrane domains, whereas its C-terminal domain remains in the cytosol to bind the second messenger cGAMP. Viral infection induces dimerization/aggregation and subsequent signaling to downstream effectors through its C-terminal tail (aa 340–379), which serves as a scaffold to assemble IRF3 in close proximity to TBK1, leading to TBK1-dependent phosphorylation of IRF3 and induction of downstream genes (12, 13). It has been reported that STING plays an important role in DNA virus–mediated type I IFN induction (8).
Recently, it has been demonstrated that DP96R of ASFV China 2018/1 strain subverts type I IFN production in the cGAS sensing pathway (14). Moreover, ASFV Armenia/07 inhibits the cGAS–STING pathway by impairing STING activation during ASFV infection (15). However, the molecular mechanisms by which ASFV proteins target the cGAS–STING axis for immune evasion are not completely understood. In this study, we aimed to identify ASFV proteins that inhibit the cGAS–STING pathway. We found that ASFV MGF-505-7R acted as an important inhibitor of the cGAS–STING DNA-sensing pathway. Mechanistically, ASFV MGF-505-7R interacted with STING and impaired the expression of STING by upregulating ULK1 expression. Our findings reveal a novel strategy through which ASFV evades host innate immunity and provides a new candidate gene to develop ASFV vaccines.
Materials and Methods
Cells and viruses
Porcine alveolar macrophages (PAM) cells were prepared by bronchoalveolar lavage as previously described (16) and grown in DMEM supplemented with 2 mM l-glutamine, 100 U/ml gentamicin, nonessential amino acids, and 10% porcine serum. 293T cells, Ma-104 cells, and HeLa cells were obtained from the American Type Culture Collection and grown in DMEM supplemented with 2 mM l-glutamine, 100 U/ml gentamicin, nonessential amino acids, and 10% FBS. The ASFV isolates CN/GS/2018 were propagated on PAM as previously described (15). STING knockout PK-15 cells were provided by X. Qin (Lanzhou Veterinary Research Institute).
Constructs
IFN-β promoter, ISRE luciferase reporter plasmids, mammalian expression plasmids for hemagglutinin (HA)–tagged cGAS, STING, TBK1, and IRF3 were as previously described (17). Mammalian expression plasmid for Flag-tagged ULK1 was constructed by standard molecular biology techniques. For construction of Flag-MGF-505-7R, a DNA fragment containing the full-length MGF-505-7R of ASFV was amplified by PCR from ASFV cDNAs and subcloned into pRK-Flag vector.
Abs and reagents
Rabbit anti–MGF-505-7R and anti-P30 sera were raised against recombinant ASFV MGF-505-7R and P30 proteins. Polyclonal rabbit anti-TBK1, phospho-TBK1, ULK1, IRF3, phospho-IRF3, LC3, and STING were purchased from Cell Signaling Technology. Monoclonal mouse anti-HA, β-actin, and Flag were purchased from Sigma-Aldrich. Alexa Fluor 488–conjugated goat anti-mouse IgG (H+L) and Alexa Fluor 594–conjugated goat anti-rabbit IgG (H+L) Abs were purchased from Cell Signaling Technology. 3-MA and MG132 were purchased from Sigma-Aldrich. H151 and 2′3′-cGAMP were purchased from MedChemExpress.
cGAMP and H151 treatment
PAM cells were treated with defined concentrations of 2′3′-cGAMP (10 μg/ml) or STING inhibitor H151 (1 μM) in digitonin permeabilization buffer (50 mM HEPES [pH 7.0], 100 mM KCl, 85 mM sucrose, 3 mM MgCl2, 0.2% BSA, 1 mM ATP, 0.1 mM DTT, and 10 μg/ml digitonin) (18). Cells were incubated for 30 min at 37°C and then washed with PBS and fresh media, or virus inoculation was then added as indicated. Concerning H151-treated samples, cells were incubated for 2 h with fresh DMEM before being infected.
Virus titration
Wild-type ASFV CN/GS/2018 and MGF-505-7R–deficient viruses were quantified using the hemadsorption (HAD) assay as described previously (19) with minor modifications. PAM cells were seeded in 96-well plates. The samples were then added to the plates and titrated in triplicate using 10-fold serial dilutions. HAD was determined on day 7 postinoculation, and 50% HAD doses (HAD50) were calculated using the method of Reed and Muench (20).
Samples from the gene-deleted ASFV-infected cell supernatants were quantified by testing their 50% tissue culture infectious dose. PAMs were seeded into 96-well plates, and 3 d later, 10-fold serially diluted samples were added into each well in triplicate. After 7 d of culture, the fluorescent protein expression was assessed using fluorescence microscopy. The 50% tissue culture infectious dose was calculated using the method of Reed and Muench (20).
Transfection and reporter gene assays
293T cells (1 × 105) were seeded into 24-well plates and transfected on the following day by the standard calcium phosphate precipitation method. In the same experiment, empty control plasmid was added to ensure that each transfection received the same amount of total DNA. To normalize for transfection efficiency, 0.01 μg of pRL-TK Renilla luciferase reporter gene plasmid was added to each transfection. Luciferase assays were performed using a dual-specific luciferase assay kit (Promega), and firefly luciferase activity was normalized based on Renilla luciferase activity.
RNA interference experiments
Small interfering RNAs corresponding to the porcine ULK1 target sequence were purchased from Sangon Biotech (Shanghai, China). PAM cells (1 × 105) were transfected with control or ULK–RNA interference (RNAi) (2 μg). Twenty-four hours after transfection, cells were left uninfected or infected with ASFV at a multiplicity of infection (MOI) of 0.01 for the indicated times. Expression of proteins was detected by immunoblot using the indicated Abs.
Confocal microscopy
HeLa cells were transfected with the indicated HA-STING (1 μg) and Flag-MGF-505-7R (1 μg) plasmids using Lipofectamine 2000 (Invitrogen). At 24 h after transfection, the cells were retransfected with B-DNA (1 μg/ml) for 12 h, or Ma-104 cells were infected with ASFV (MOI = 0.1) for 8 h and then fixed with 4% paraformaldehyde for 10 min at room temperature and permeabilized with 0.1% Triton X-100 for 15 min. The cells were incubated with anti-Flag tag or anti-STING rabbit polyclonal Ab for 2 h and then incubated with anti-HA tag or anti–MGF-505-7R mouse mAb. The cells were then incubated with goat anti-mouse IgG (whole molecule)–FITC Ab (F2012; Sigma) and goat anti-rabbit IgG (whole molecule)–tetramethyl rhodamine isocyanate Ab (T6778; Sigma). Cells were stained with DAPI for 15 min and examined with a Leica SP2 confocal system (Leica Microsystems).
RT-PCR assay
PMA cells were seeded in p60 plates (6 × 106 cells/plate) for 24 h. Then, cells were transfected with B-DNA for 12 h before ASFV infection at the indicated times. 293T cells were transfected with empty vector or MGF-707-7R for 24 h and then retransfected with B-DNA for 12 h. Total RNA was extracted from PMA or 293T cells using TRIzol reagent, according to the manufacturer’s instructions (Sigma). Reverse transcription reactions for mRNAs were performed using PrimeScript RT reagent kit (Takara). To determine relative mRNA abundance, quantitative PCR was performed with Powerup SYBR Green Master Mix (Applied Biosystems) on an ABI StepOnePlus system, according to the manufacturer’s protocol. Data were analyzed with StepOnePlus software. PCR primer sequence information is provided in Table I. Human and porcine GAPDH were used for normalization.
Table I. Primers and oligonucleotide list.
| Primers | Sequences (5′-3′) |
|---|---|
| Human IFNB-F | 5′-CAGCAATTTTCAGTGTCAGAAGCT-3′ |
| Human IFNB-R | 5′-CAGTGACTGTACTCCTTGGCCTT-3′ |
| Human TNF-α-F | 5′-GCCGCATCGCCGTCTCCTAC-3′ |
| Human TNF-α-R | 5′-CCTCAGCCCCCTCTGGGGTC-3′ |
| Human IL-6-F | 5′-TTCTCCACAAGCGCCTTCGGTC-3′ |
| Human IL-6-R | 5′-TCTGTGTGGGGCGGCTACATCT-3′ |
| Human cGAS-F | 5′-CCGCCAGTAGTGCTTGGTTT-3′ |
| Human cGAS-R | 5′-GTTTCAGGAAAAGGCCGCAA-3′ |
| Human STING-F | 5′-ACTCTTCTGCCGGACACTTG-3′ |
| Human STING-R | 5′-GCAGGTTCCTGGTAGGCAAT-3′ |
| Human CXCL-10-F | 5′-GGTGAGAAGAGATGTCTGAATCC-3′ |
| Human CXCL-10-R | 5′-GTCCATCCTTGGAAGCACTGCA-3′ |
| Human GAPDH-F | 5′-AAAATCAAGTGGGGCGATGCT-3′ |
| Human GAPDH-R | 5′-GGGCAGAGATGATGACCCTTT-3′ |
| Porcine IFNB-F | 5′-CACTGGCTGGAATGAAACCG-3′ |
| Porcine IFNB-R | 5′-AATGGTCATGTCTCCCCTGG-3′ |
| Porcine TNF-α-F | 5′-GCCCAAGGACTCAGATCATC-3′ |
| Porcine TNF-α-R | 5′-GGCATTGGCATACCCACTCT-3′ |
| Porcine IL-6-F | 5′-CTGCTTCTGGTGATGGCTACTG-3′ |
| Porcine IL-6-R | 5′-GGCATCACCTTTGGCATCTT-3′ |
| Porcine CXCL-10-F | 5′-CCCACATGTTGAGATCATTGC-3′ |
| Porcine CXCL-10-R | 5′-CATCCTTATCAGTAGTGCCG-3′ |
| Porcine GAPDH-F | 5′-ACATGGCCTCCAAGGAGTAAGA-3′ |
| Porcine GAPDH-R | 5′-GATCGAGTTGGGGCTGTGACT-3′ |
| Porcine cGAS-F | 5′-GGAGCCCTGCAGTAACACTT-3′ |
| Porcine cGAS-R | 5′-GCTCCAAGCCACTGACTGAT-3′ |
| Porcine STING-F | 5′-CTTGGTCCTTCTAGGTGCCTG-3′ |
| Porcine STING-R | 5′-CACCGGAGCGTGTATTCTGG-3′ |
F, forward; R, reverse.
Afterward, ASFV DNA copy number was evaluated through relative quantification as previously described (21). Briefly, ASFV DNA (105 cells, 3 μl blood, or 0.7 mg tissues samples) was extracted using the QIAamp All Nucleic Acid Kit MDx Kit (QIAGEN). After the DNA extraction procedure, the cartridge was processed for the RT-PCR. The target for amplification of the ASFV genome was the conserved p72 gene segment, using the following primers: 5′-ctgctcatggtatcaatcttatcga-3′ and 5′-gataccacaagatc(ag)gccgt-3′. A TaqMan probe (5′-[6-carboxy-fluorescein]-ccacgggaggaataccaacccagtg-3′-[6-carboxy-tetramethyl-rhodamine]; Applied Biosystems) was designed from an alignment of 54 available ASFV sequences for the 3′-end of p72. Analysis was performed using MxPro software, and the quantitative PCR procedure included the following steps: denaturation (95°C), annealing (58°C), and elongation (72°C). The quantity of ASFV genome was calculated using the standard curve and expressed as genome copies per milliliter.
Coimmunoprecipitation and immunoblotting analyses
For the transient transfection coimmunoprecipitation experiments, the 293T cells were transfected with the appropriate plasmid. Twenty-four hours after transfection, the cells were harvested and lysed in 1 ml of lysis buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 1% Triton, 1 mM EDTA, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mM PMSF). For each immunoprecipitation reaction, 0.4 ml of cell lysate was incubated with 0.5 μg of the indicated Ab or control IgG and 40 μl of protein G agarose beads (Santa Cruz Biotechnology) at 4°C. After a 4-h incubation, the beads were washed three times with 1 ml of lysis buffer containing 0.5M NaCl. Samples were resolved by NaDodSO4 polyacrylamide gels (SDS-PAGE) and transferred to Immobilon-P membranes (MilliporeSigma). The membranes were incubated with the indicated specific primary Abs diluted in TBS supplemented with 1% milk. Membranes were washed three times with TBS and exposed for 1 h to specific peroxidase-conjugated secondary Abs. Chemiluminescence detection was performed by ECL prime (MilliporeSigma). For the endogenous coimmunoprecipitation experiments, PMA cells were uninfected or infected with ASFV (MOI: 0.01) for the indicated times. The subsequent procedures were performed as described above.
CRISPR–Cas9 knockout
Double-stranded oligonucleotides corresponding to the target sequences were cloned into the lenti-CRISPR-V2 vector and cotransfected packaging plasmids into 293T cells. Lentiviral particles were collected and used to transduce 293T cells. The infected 293T cells were selected with puromycin (1 μg/ml) for 2 wk before additional experiments were performed. The following sequences were targeted for human cGAS cDNA: 5′-GCCCCCATTCTCGTACGGAG-3′ and human STING cDNA: 5′-GCTCACTGCACCCCGTAGCA-3′.
ASFV MGF-505-7R–transduced stable PAM cells
The 293T cells were transfected with two packaging plasmids (pGAG-Pol and pVSV-G) together with a control or ASFV MGF-505-7R retroviral plasmid. Twenty-four hours later, cells were incubated with new medium without antibiotics for another 24 h. The recombinant virus–containing medium was filtered and then added to PAM cells in the presence of polybrene (8 μg/ml). The infected cells were selected with puromycin (0.5 μg/ml) for 1 d before additional experiments.
Plasmid design for traditional recombination
Plasmid pUC19 lacking its multiple cloning was used as a backbone; the recombination cassette was inserted at the SalI and NdeI restriction sites after the T7 promoter. The recombination cassette contains a left recombination arm that is 1000 bp upstream of open reading frame 7R identical to ASFV CN/GS/2018 nucleotide positions 39695–40694, followed by the p72 promoter identical to ASFV CN/GS/2018 nucleotide positions on the negative strand 105677–105465, followed by eGFP, and a SV40 termination sequence, followed by a right recombination arm that is 1000 bp downstream of 7R identical to ASFV CN/GS/2018 nucleotide positions 42270–43269.
CRISPR/Cas9 transfection
CRISPR/Cas9 experiments were conducted after 1 h of virus adsorption at 37°C under 5% CO2, the inoculum was then discarded, and the indicated plasmids were transfected with Fugene HD, following the manufacturer’s protocol (available: http://www.promega.com/techserv/tools/FugeneHdTool/). A 3:1 Fugene:DNA ratio was used with 3.3 μg of DNA and 9.9 μl of Fugene HD. The complex was mixed carefully by pipetting and incubated for 10 min; 150 μl of the complex was added to the cells dropwise. Cells were then incubated at 37°C under 5% CO2 and observed for the presence of eGFP and frozen 24 h posttransfection, thawed, and titrated.
Complete sequencing of ASFV genomes using next-generation sequencing
PAM cells were seeded as described and infected with ASFV or ASFV-Δ7R; once the cytopathic effect was evident throughout the monolayer, DNA was isolated as described above from cells infected with ASFV. The extracted DNA was then used to completely sequence the viral DNA as previously described (22). In brief, viral DNA was sheared using enzymatic reactions assessed for the distribution of size fragmentation, and then identifying barcodes were ligated to the DNA fragments using an adapter sequence. Using a Pippin Prep (Sage Science, Beverly, MA), the required size range of the library was collected and normalized. We then used this DNA library for next-generation sequencing using the NextSeq (Illumina, San Diego, CA), following the manufacturer’s protocol. Sequence analysis was performed using CLC Genomics Workbench software (CLC bio, Waltham, MA).
Biosafety statement and facility
All experiments with live ASFVs were conducted within the enhanced biosafety level 3 facilities in the Lanzhou Veterinary Research Institute of the Chinese Academy of Agricultural Sciences approved by the Ministry of Agriculture and Rural Affairs and China National Accreditation Service for Conformity Assessment.
Ethics statement
All animals were handled in strict accordance with good animal practice according to the Animal Ethics Procedures and Guidelines of the People’s Republic of China, and the study was approved by the Animal Ethics Committee of Lanzhou Veterinary Research Institute of the Chinese Academy of Agricultural Sciences.
Animal experiments
ASFV-Δ7R was assessed for its virulence relative to the parental ASFV CN/GS/2018 virus using 80–90-pound commercially bred swine. Five pigs were inoculated i.m. with either 10 HAD50 of ASFV-Δ7R or ASFV CN/GS/2018. Blood and tissues samples collected from pigs (n = 3) that were inoculated i.m. with either 10 HAD50 of ASFV-Δ7R or ASFV CN/GS/2018 for 6 d were used for viral DNA and cytokines expression detection.
ELISA
Porcine sera were collected and assayed for porcine IFN-β using porcine IFN-β ELISA kit (Solarbio), as described by the manufacturer.
Statistical analysis
All experiments were performed independently at least three times. Statistical analyses were performed using unpaired, two-tailed Student t test. A p value <0.05 was considered to be statistically significant.
Results
ASFV inhibits dsDNA-triggered induction of downstream antiviral genes
To verify whether ASFV inhibits dsDNA-triggered induction of downstream antiviral genes, PAM cells were pretreated with B-DNA and then infected with ASFV. Afterward, we analyzed the B-DNA–triggered transcriptional levels of the Ifnb1, Tnfa, Il6, and Cxcl10 genes. The results suggested that ASFV impaired B-DNA–triggered transcription levels of the Ifnb1, Tnfa, Il6, and Cxcl10 genes in ASFV-infected PAM cells (Fig. 1A–D). To confirm the roles of ASFV in dsDNA-triggered signaling, we examined the activation of downstream signaling components involved in virus-triggered type I IFN induction. As shown in Fig. 1E, B-DNA–triggered phosphorylation of TBK1 and IRF3, which are hallmarks of activation of virus-triggered IFN induction pathways, was lower in ASFV-infected PAM cells in comparison with control cells. Taken together, these results suggested that ASFV inhibits dsDNA-triggered induction of downstream antiviral genes.
FIGURE 1.

ASFV inhibits B-DNA–induced signaling pathway. (A–D) Effects of ASFV on B-DNA–triggered transcription of IFN-β (A), TNF-α (B), IL-6 (C), and Cxcl-10 (D) genes in PAM cells. PAM cells were seeded in 12-well plates for 24 h. Then, cells were transfected with B-DNA (1 μg/ml) for 12 h. Cells were uninfected or infected with ASFV (MOI: 0.01) for 12 h before RT-PCR experiments. (E) ASFV inhibits the B-DNA–induced phosphorylation of TBK1 and IRF3. PAM cells were seeded in 12-well plates for 24 h. Then, cells were transfected with B-DNA (1 μg/ml) for 12 h. Cells were uninfected or infected with ASFV (MOI: 0.01) for the indicated times. Cell lysates were analyzed by immunoblotting with the indicated Abs. All the experiments were repeated three times with similar results. Data are shown as mean ± SD; n = 3. *p < 0.05, **p < 0.01. RT-PCR, real-time PCR.
MGF-505-7R inhibits cGAS-STING–induced activation of IFN-β promoter
To identify ASFV proteins that may inhibit cGAS-STING–triggered IFN-β promotor and ISRE activation, we first constructed 168 expression clones encoding individual ASFV proteins. This screen identified MGF-505-7R clones that could markedly inhibit cGAS-STING–mediated ISRE activation. In reporter assays, ectopic expression of MGF-505-7R inhibited cGAS-STING–induced activation of IFN-β promoter and ISRE in human embryonic kidney 293T cells (Fig. 2A, 2B). Further experiments indicated that MGF-505-7R overexpression inhibited cGAS-STING–triggered activation of the IFN-β promoter and ISRE in a dose-dependent manner in 293T cells (Fig. 2C, 2D). Consistently, ectopic expression of MGF-505-7R inhibited phosphorylation of TBK1 and IRF3 induced by B-DNA transfection in PAM cells (Fig. 2E). These results suggested that MGF-505-7R protein efficiently inhibited the cGAS-STING–mediated signaling pathway.
FIGURE 2.

ASFV MGF-505-7R inhibits cGAS-STING–triggered signaling pathway. (A and B) Effects of ASFV MGF-505-7R overexpression on cGAS-STING–triggered IFN-β promoter (A) and ISRE (B) activation. 293T cells (1 × 105) were transfected with IFN-β or ISRE reporter (0.1 μg) and MGF-505-7R expression (0.1 μg) plasmids for 24 h before luciferase assays were performed. Expression of cGAS, STING, and MGF-505-7R was analyzed by Western blotting. (C and D) Dose-dependent effects of MGF-505-7R on cGAS-STING–triggered IFN-β promoter (C) and ISRE (D) activation. The experiments were similarly performed as in (A). Expression of cGAS, STING, and MGF-505-7R was analyzed by Western blotting. (E) MGF-505-7R inhibits B-DNA–induced phosphorylation of TBK1 and IRF3. MGF-505-7R–overexpressing PAM cells (2 × 105) were transfected with B-DNA for the indicated times. Cell lysates were analyzed by immunoblotting with the indicated Abs. All the experiments were repeated three times with similar results. Graphs data are shown as means ± SD; n = 3. *p < 0.05, **p < 0.01. Luc, luciferase.
MGF-505-7R inhibits dsDNA-triggered induction of downstream antiviral genes
We next investigated the function of MGF-505-7R in dsDNA-triggered type I IFN production. In RT-PCR experiments, we observed that ectopic expression of MGF-505-7R inhibited B-DNA–induced transcription of Ifnb1, Tnfa, Il6, and Cxcl10 genes in 293T cells (Fig. 3A). Similarly, MGF-505-7R also inhibited transcription of Ifnb1, Tnfa, Il6, and Cxcl10 genes induced by B-DNA in PAM cells (Fig. 3B). We also found that B-DNA could induce low mRNA levels of STING and cGAS in 293T cells (Fig. 3C). In addition, we also found that B-DNA could not induce IFN-β expression in STING or cGAS knockout 293T cells (Fig. 3D–F). These results suggested that MGF-505-7R inhibits dsDNA-triggered induction of downstream antiviral genes.
FIGURE 3.

ASFV MGF-505-7R inhibits B-DNA–triggered downstream antiviral genes. (A) Effects of ASFV MGF-505-7R on B-DNA–triggered transcription of IFN-β, TNF-α, IL-6, and Cxcl-10 genes. MGF-505-7R–overexpressing 293T cells (2 × 105) were transfected with B-DNA (1 μg/ml) for 12 h before RT-PCR experiments were performed with the indicated primers. (B) Effects of ASFV MGF-505-7R on B-DNA–triggered transcription of IFN-β, TNF-α, IL-6, and Cxcl-10 genes in PAM. The experiments were similarly performed as in (A). (C) B-DNA–triggered transcription of STING and cGAS genes in 293T cells. 293T cells were transfected with B-DNA (1 μg/ml) for the indicated times before RT-PCR experiments were performed with the indicated primers. (D) Expression of cGAS gene in cGAS knockout 293T cells was analyzed by RT-PCR. (E) Expression of STING gene in STING knockout 293T cells was analyzed by RT-PCR. (F) Effects of cGAS or STING on B-DNA–triggered transcription of IFN-β gene. STING or cGAS knockout 293T cells (2 × 105) were transfected with B-DNA (1 μg/ml) for 12 h before RT-PCR experiments were performed with the indicated primers. All the experiments were repeated three times with similar results. Graph data are shown as mean ± SD; n = 3. *p < 0.05, **p < 0.01. RT-PCR, real-time PCR.
MGF-505-7R inhibits cGAS-STING–triggered signaling at the STING level
Various components are involved in cGAS-STING–triggered signaling pathways. As shown in Fig. 4A, MGF-505-7R inhibited IFN-β promoter activation mediated by STING and its upstream component cGAS but not its downstream component TBK1. Consistently, MGF-505-7R also inhibited ISRE activation mediated by overexpression of these components (Fig. 4B). Further, we found that ASFV MGF-505-7R was localized in the cytoplasm in ASFV MGF-505-7R–overexpressed HeLa cells, and the location did not change in HeLa cells treated with B-DNA (Fig. 4C). To explore the effects of STING on MGF-505-7R in PK-15 cells, PK-15 or STING knockout PK-15 cells were transfected with MGF-505-7R plasmid. The result showed that MGF-505-7R inhibited B-DNA–induced IFN-β and TNF-α in PK-15 cells but not in STING knockout PK-15 cells (Fig. 4D). These results suggested that ASFV MGF-505-7R inhibits cGAS–STING signaling pathway at STING level.
FIGURE 4.

ASFV MGF-505-7R targets at STING level. (A and B) Effects of MGF-505-7R on cGAS-STING–triggered IFN-β promoter (A) and ISRE (B) activation. 293T cells (1 × 105) were transfected with IFN-β or ISRE reporter (0.1 μg) and expression plasmids for MGF-505-7R and the indicated plasmids (0.1 μg each). Luciferase assays were performed 24 h after transfection. (C) MGF-505-7R locates at cytoplasm in HeLa cells. HeLa cells were transfected with MGF-505-7R for 24 h. Then, cells were treated with B-DNA for 12 h, fixed, and then stained with DAPI and anti–MGF-Flag Ab. Finally, the location of cells was observed by confocal microscopy. (D) The effects of STING on MGF-505-7R in PK-15 cells. PK-15 or STING knockout PK-15 cells were transfected with MGF-505-7R (2 μg) for 24 h. Then, the cells were retransfected with B-DNA (1 μg/ml) for 12 h before RT-PCR experiments were performed with the indicated primers. Expression of STING and MGF-505-7R was analyzed by Western blot. All the experiments were repeated three times with similar results. Data are shown as means ± SD; n = 3. **p < 0.01. KO, knockout; Luc, luciferase; 7R, Flag-MGF-505-7R; WT, wild-type.
MGF-505-7R interacts with STING
To investigate the molecular mechanisms of MGF-505-7R in an innate immune response, we first determined whether MGF-505-7R is associated with components involved in DNA virus–triggered signaling pathways. The results indicated that MGF-505-7R was associated with STING but not with cGAS, TBK1, or IRF3 (Fig. 5A). To further determine whether MGF-505-7R protein interacts with cellular STING in the context of ASFV infection, ASFV-infected PAM cell lysates were immunoprecipitated with an anti–MGF-505-7R polyclonal Ab and probed for the presence of STING with anti-STING polyclonal Ab. STING was readily detected in ASFV-infected PAM cells (Fig. 5B), indicating that STING indeed interacts with endogenous MGF-505-7R protein in ASFV-infected PAM cells. To examine the colocalization of MGF-505-7R protein with STING, 293T cells were cotransfected with plasmids expressing STING and MGF-505-7R proteins, and the results showed that MGF-505-7R could localize with STING by confocal microscopy (Fig. 5C). During ASFV infection, confocal microscopy revealed that MGF-505-7R and STING were well colocalized in the cytoplasm (Fig. 5D). To investigate the domains responsible for the MGF-505-7R–STING interaction, we performed coimmunoprecipitation experiments with a series of truncations of MGF-505-7R and STING. We found that the first transmembrane domain of MGF-505-7R (1–175 aa) and the N-terminal transmembrane domains of STING (1–153 aa) were required for their interaction (Fig. 5E, 5F). In addition, we also found that MGF-505-7R and MGF-505-7R (1–175 aa) but not MGF-505-7R (176–350 aa) and MGF-505-7R (351–527 aa) inhibited cGAS-STING–triggered activation of the IFN-β promoter in 293T cells (Fig. 5G). Collectively, these findings confirmed that STING is an interacting partner of ASFV MGF-505-7R protein.
FIGURE 5.

ASFV MGF-505-7R interacts with STING. (A) MGF-505-7R interacted with STING but not with cGAS, TBK1, and IRF3 in the overexpression system. 293T cells (2 × 106) were transfected with the indicated plasmids (5 μg each). Coimmunoprecipitation and immunoblot analyses were performed with the indicated Abs. The expression of transfected proteins was analyzed by immunoblotting with anti-HA or anti-Flag Abs. (B) Endogenous associations between MGF-505-7R and STING. PAM cells were left infected or infected with ASFV for the indicated times before coimmunoprecipitation and immunoblot analysis. (C) Colocalization of MGF-505-7R with STING. HeLa cells were transfected with HA-STING (1 μg) and Flag-MGF-505-7R (1 μg) plasmids. Twenty hours after transfection, cells were retransfected with B-DNA (1 μg/ml) for 12 h before confocal microscopy. (D) Confocal microscopy of Ma-104 cells infected with ASFV for 8 h. (E and F) Domain mapping of MGF-505-7R and STING interaction. 293T cells were transfected with the indicated truncation plasmids before coimmunoprecipitation and immunoblot analysis with the indicated Abs. HA-STINGs: HA-STING, HA-STING (1–160 aa), HA-STING (1–240 aa), HA-STING (111–379 aa), and HA-STING (153–370 aa). (G) Effects of ASFV MGF-505-7R or its mutants overexpression on cGAS-STING–triggered IFN-β promoter activation. 293T cells (1 × 105) were transfected with IFN-β reporter (0.1 μg) and MGF-505-7R or MGF-505-7R’s mutants expression (0.1 μg) plasmids for 24 h before luciferase assays were performed. All the experiments were repeated three times with similar results. Data are shown as means ± SD; n = 3. **p < 0.01. EV, empty vector; αH, anti-HA; HC, H chain; Luc, luciferase; WT, wild-type.
MGF-505-7R degrades STING by autophagy pathway
We next investigated how ASFV MGF-505-7R regulates STING in cGAS-STING–triggered signaling pathway. To investigate whether MGF-505-7R affects the expression of STING, 293T cells were cotransfected with expression plasmids encoding MGF-505-7R and STING. Immunoblotting revealed that MGF-505-7R overexpression resulted in the decreased expression of STING (Fig. 6A), indicating that MGF-505-7R degrades STING. To explore the effect of ASFV infection on STING expression, PAM cells were infected with ASFV at the indicated times. The results showed that ASFV infection inhibited the expression of STING protein (Fig. 6B). Protein degradation is one of the main strategies involved for turning off protein functions in biological processes. At least three systems exist for protein degradation, including the ubiquitin–proteasome, lysosomal, and autophagosome pathways. To investigate the mechanisms responsible for the role of MGF-505-7R on the stability of STING, 293T cells were transfected with the indicated plasmids and then treated with various inhibitors for protein degradation pathways. MGF-505-7R–mediated degradation of STING was completely inhibited by the autophagosome inhibitor 3-MA but not by the lysosomal inhibitor NH4Cl or proteasome inhibitor MG132 (Fig. 6C). Consistently, we also found that ASFV-mediated degradation of STING was completely inhibited by 3-MA (Fig. 6D). Further, we found that LC3II protein, which is commonly used as a marker of autophagic activity within cells, was increased by transfecting with MGF-505-7R in 293T cells (Fig. 6E). To explore the effects of MGF-505-7R on STING dimerization, 293T cells were transfected with the indicated plasmids before treating or untreating with cGAMP. The results showed that MGF-505-7R inhibited STING dimerization in the absence or presence of cGAMP (Fig. 6F). Collectively, these results suggested that ASFV MGF-505-7R degrades STING via the autophagy pathway.
FIGURE 6.

ASFV MGF-505-7R degrades STING by autophagy pathway. (A) Effects of ASFV MGF-505-7R on the expression of STING in 293T cells. 293T cells (2 × 105) were transfected with HA-STING (1 μg) and Flag-MGF-505-7R (0, 0.25, 0.5, and 1.0 μg) for 24 h. Cell lysates were analyzed by immunoblotting with the indicated Abs. (B) Effects of ASFV on endogenous STING in PAM cells. PAM cells were left uninfected or infected with ASFV for the indicated times and then analyzed by immunoblotting with the indicated Abs. (C) Effects of inhibitors on MGF-505-7R–mediated destabilization of STING. 293T cells (4 × 105) were transfected with the indicated plasmids. Eighteen hours after transfection, the cells were treated with the indicated inhibitors for 6 h before immunoblot analysis was performed. (D) Effects of inhibitors on ASFV-mediated destabilization of STING. PAM cells were treated with MG132 (100 μM), NH4CL (25 mM), or 3-MA (500 ng/ml) for 1 h. And then, the cells were left uninfected or infected with ASFV for 6 h and then analyzed by immunoblotting with the indicated Abs. (E) Effects of MGF-505-7R on LC3 protein in 293T cells. 293T cells transfected with MGF-505-7R (0, 1, 2, and 4 μg) for 24 h before immunoblotting analysis was performed using the indicated Abs. (F) Effects of MGF-505-7R on STING dimerization. 293T cells were transfected with the indicated plasmids (each 5 μg) for 18 h. The cells were treated or untreated with cGAMP (100 nM) for 6 h. Coimmunoprecipitation and immunoblotting analyses were performed with the indicated Abs. All the experiments were repeated three times with similar results.
MGF-505-7R degrades STING by upregulating ULK1 expression
A previous study reported that ULK1 was able to phosphorylate STING on S366 to facilitate STING degradation (23), and the finding prompted us to investigate whether ULK1 mediates MGF-505-7R degradation of STING. To determine whether ASFV MGF-505-7R is associated with ULK1, coimmunoprecipitation experiments were performed with 293T cells transiently coexpressing ASFV MGF-505-7R and Flag-tagged ULK1 plasmids. The results showed that ASFV MGF-505-7R interacted with ULK1 (Fig. 7A). MGF-505-7R interacted with endogenous ULK1 in ASFV-infected PAM cells (Fig. 7B). We also found that MGF-505-7R increased the expression of ULK1 in a dose-dependent manner in 293T cells (Fig. 7C). Afterward, ULK1 inhibited the expression of STING in a dose dependent in 293T cells (Fig. 7D). In addition, we also found that ULK1 increased MGF-505-7R degradation of STING (Fig. 7E). To explore the effects of ULK1 on STING and MGF-505-7R expression, we detected the STING and MGF-505-7R expression in ASFV-infected ULK1 knockdown PAM. The results showed that STING levels were increased and MGF-505-7R was reduced in ULK1 knockdown PAM cells compared with controls (Fig. 7F). These data indicated that MGF-505-7R degrades STING by upregulating ULK1 expression.
FIGURE 7.

ASFV MGF-505-7R degrades STING by upregulating ULK1 expression. (A) MGF-505-7R interacted with ULK1 in the overexpression system. 293T cells (2 × 106) were transfected with the indicated plasmids (5 μg each). Coimmunoprecipitation and immunoblotting analyses were performed with the indicated Abs. The expression of transfected proteins was analyzed by immunoblotting with anti–MGF-505-7R or anti-Flag Abs. (B) Endogenous associations between MGF-505-7R and ULK1. PAM cells were uninfected or infected with ASFV (MOI: 0.01) for the indicated times before coimmunoprecipitation and immunoblot analysis. (C) MGF-505-7R increased the expression of ULK1 protein. 293T cells (2 × 105) were transfected with HA-ULK1 (1 μg) and Flag-MGF-505-7R (0, 0.25, 0.5, and 1.0 μg) for 24 h. Cell lysates were analyzed by immunoblotting with the indicated Abs. (D) ULK1 inhibits the expression of STING protein. The experiments were similarly performed as in (C). (E) ULK1 increases MGF-505-7R degradation of STING. 293T cells (2 × 105) were transfected with the indicated plasmids for 24 h. Cell lysates were analyzed by immunoblotting with the indicated Abs. (F) Effects of ULK1 knockdown on STING and MGF-505-7R. PAM cells were transfected with control–RNAi or ULK1–RNAi (2 μg). Twenty-four hours after transfection, cells were left uninfected or infected with ASFV (MOI: 0.01) for the indicated times before immunoblotting was performed with the indicated Abs. All the experiments were repeated three times with similar results.
STING and MGF-505-7R knockout inhibit ASFV replication
To determine the role of STING in ASFV replication, PAM cells were pretreated with STING inhibitor (H151) or STING activator (cGAMP) and then infected with ASFV. The results showed that MGF-505-7R expression was increased in STING-inhibited PAM cells, whereas p-TBK1 levels were inhibited in STING-inhibited PAM cells (Fig. 8A). Similarly, we also found that inhibition of STING by H151 increased ASFV replication (Fig. 8B). By contrast, activating STING by cGAMP presented the opposite effect (Fig. 8C, 8D). These results suggested that STING inhibits ASFV replication.
FIGURE 8.

STING inhibits ASFV replication. (A) Effects of inhibition of STING on ASFV MGF-505-7R. PAM cells were treated or untreated with STING inhibitor (H151). Cells were left infected or infected with ASFV for the indicated times. Cell lysates were analyzed by immunoblotting with the indicated Abs. (B) Effects of STING inhibition on ASFV genomic copy number. PAM cells were treated or untreated with STING inhibitor (H151). Cells were left infected or infected with ASFV for 8 h before RT-PCR experiments were performed with the indicated primers. (C) Effects of activation of STING on ASFV MGF-505-7R. The experiments were similarly performed as in (A). (D) Effects of activation of STING on ASFV genomic copies. The experiments were similarly performed as in (B). (E) Effects of ASFV MGF-505-7R knockout (ASFV-Δ7R) on expression of endogenous ASFV MGF-505-7R. (F) In vitro replication characteristics of ASFV-Δ7R and parental ASFV. PAM cell cultures were infected with each of the viruses (MOI: 0.01), and viral replication was detected at the indicated times postinfection. (G) Effects of ASFV-Δ7R on transcription of IFN-β genes. PAM cells were infected with parental ASFV or ASFV-Δ7R for 24 h before RT-PCR analysis. (H) Effects of ASFV-Δ7R on STING, ULK1, and P30 expression. The cells were infected with parental ASFV or ASFV-Δ7R for the indicated times before immunoblot analysis. (I) Effects of ASFV-Δ7R on phosphorylation of TBK1 and IRF3. The experiments were similarly performed as in (H). All the experiments were repeated three times with similar results. Data are shown as mean ± SD; n = 3. *p < 0.05, **p < 0.01.
To examine the role of endogenous MGF-505-7R in regulation of cGAS-STING–triggered signaling, we generated ASFV MGF-505-7R–deficient virus (ASFV-Δ7R) by CRISPR-Cas9 and DNA homologous recombination technique. Immunoblotting analysis confirmed that MGF-505-7R was undetectable in ASFV-Δ7R–infected PAM cells (Fig. 8E). In vitro replication characteristics of ASFV-Δ7R were evaluated in PAM cell cultures, the cell targeted by ASFV and compared relative to parental ASFV. The results showed that deletion of the MGF-505-7R gene decreased the ability of ASFV-Δ7R, relative to the parental ASFV CN/GS/2018 isolate, to replicate in PAM cells culture (Fig. 8F). We next investigated the roles of endogenous MGF-505-7R in antiviral response to ASFV. The results indicated that ASFV, but not MGF-505-7R–deficient ASFV, inhibited IFN-β transcription compared with mock (Fig. 8G). In addition, we also found that STING expression was inhibited in ASFV-infected PAM cells but not in ASFV-Δ7R–infected PAM cells, whereas ULK1 and P30 expression was inhibited in ASFV ASFV-Δ7R–infected PAM cells compared with that in cells infected with parental ASFV (Fig. 8H). Similarly, the phosphorylation of TBK1 and IRF3 levels were increased in ASFV-Δ7R–infected PAM cells compared with ASFV-infected PAM cells (Fig. 8I). Taken together, these results suggested that ASFV MGF-505-7R deficient increased IFN-β signaling.
Virulence evaluation of ASFV with MGF-505-7R gene deletion
To investigate whether MGF-505-7R–deleted viruses were attenuated in pigs, we i.m. injected groups of six 7-wk-old pigs with 10 HAD50 of the gene-deleted virus and observed the pigs for 3 wk. As shown in Fig. 9A, all of the pigs inoculated with ASFV CN/GS/2018 died within 15 d, whereas all of the pigs inoculated with ASFV-Δ7R remained healthy and survived the duration of the 3-wk observation period. We found that serum chemokines IFN-β was reduced in ASFV-infected pigs but not in ASFV-Δ7R–infected pigs (Fig. 9B). Consistent with the above results, the IFN-β transcription level in ASFV-Δ7R–infected tissues was increased in comparison with parental ASFV (Fig. 9C). In addition, we also found that ASFV-Δ7R replication in the blood and tissues of pigs was decreased in comparison with parental ASFV (Fig. 9D). Collectively, these data suggest that ASFV-Δ7R is dramatically attenuated in pigs.
FIGURE 9.

Assessment of ASFV-Δ7R virulence in swine. (A) Survival rates of pigs (n = 5 per group) after i.m. inoculation with parental ASFV or ASFV-Δ7R (10 HAD50 per animal). (B) Effects of ASFV-Δ7R on serum levels of IFN-β induced by i.m. inoculation with parental ASFV or ASFV-Δ7R (10 HAD50 per animal) for 6 d (n = 3 per group). (C) Effects of ASFV-Δ7R on IFN-β transcription levels in different tissues (spleen, lung, and lymph) (n = 3 per group). The experiments were similarly performed as in (B). (D) Effects of ASFV-Δ7R or parental ASFV on virus replication in blood and tissues (n = 3 per group). The experiments were similarly performed as in (B). All the experiments were repeated three times with similar results. *p < 0.05, **p < 0.01.
Discussion
In recent years, how DNA viruses trigger innate immune responses has been extensively investigated. In this study, we investigated whether the cGAS–STING axis is targeted by ASFV proteins for immune evasion. Using expression screens, we identified MGF-505-7R as a negative regulator of STING-mediated induction of downstream antiviral genes and innate antiviral responses.
Several lines of evidence suggest that MGF-505-7R directly targets STING for inhibition of innate antiviral responses. First, overexpression of MGF-505-7R inhibited cGAS- and STING- but not TBK1-triggered IFN-β promoter and ISRE activation. Second, MGF-505-7R interacted with STING. Third, expression of Ifnb1, Tnfa, Il6, and Cxcl10 genes was markedly reduced in MGF-505-7R–overexpressing cells in comparison with control cells.
Pathogenic ASFV genomes contain either 9 or 10 multigene family 505 (MGF530 and MGF505) genes. Homology searches revealed that MGF505 has no homology to other known genes. MGF505 genes have a conserved motif of 100 aa (28% amino acid identity) at the N terminus in common (24, 25). Among MGF members, amino acid similarity ranges from 46 to 74% for MGF505 proteins (26). The absence of IFN-α in Pr4-infected macrophages suggests that MGF360/530 genes either directly or indirectly suppress a type I IFN response (2). ASFV MGF-505-7R gene is a member of MGF505 (2), so it may be a potential candidate as an IFN evasion protein. Further research showed that MGF505-7R (A528R) inhibits the type I IFN induction through inhibiting both IRF3 and NF-κB transcription factors (4), but the mechanism by which MGF-505-7R suppress the induction of IFNs is not well elucidated. In this study, we found that MGF-505-7R interacted with STING and degraded STING.
STING is a prototype downstream effector, which can be modulated by several viral products to counteract the cGAS–STING pathway and type I IFN production (15). Although several viruses’ proteins have been reported to inhibit the cGAS–STING pathways, the mechanisms involving these proteins are distinct. For instance, the Kaposi sarcoma-associated herpesvirus open reading frame 52 and LANA directly inhibit cGAS enzymatic activity (27, 28), whereas HSV-1 ICP27 inhibits TBK1 activity (29). Human CMV UL82 inhibited the translocation of STING from the endoplasmic reticulum to perinuclear microsomes by disrupting the STING–iRhom2–TRAPβ translocation complex and also impairs the recruitment of TBK1 and IRF3 to the STING complex (30). Recent studies have shown that ASFV DP96R protein inhibits antiviral immune response by decreasing the phosphorylation of TBK1, and ASFV Armenia/07 virulent strain controls IFN-β production through the cGAS–STING pathway (14, 15). Our results showed that ASFV MGF-505-7R inhibited cGAS–STING pathway by degradation of STING through upregulating ULK1 expression. A previous study showed that STING degradation depends on ATG5 and WIP2 (31), but whether MGF-505-7R degrades STING through ATG5 and WIP2 needs further study. The findings show for the first time, to our knowledge, the involvement of ASFV protein, the cGAS–STING, and autophagy pathway.
Currently, there is no commercially available vaccine against ASFV. The use of attenuated strains is currently the most plausible approach to develop an effective ASFV vaccine. Rational development of attenuated strains by genetic manipulation is a valid alternative and perhaps safer methodology compared with the use of naturally attenuated isolates. Deletions of single genes or a group of genes by genetic manipulation have been shown to be virulence-attenuated strains that induce protection against the virulent parental virus (32–38). In this study, we found that deletion of ASFV MGF-505-7R from the highly virulent ASFV CN/GS/2018 produces complete viral attenuation in swine. Deletion of the 9GL gene (potentiated by the additional deletion of the UK gene), deletion of a group of six genes from the MGF360 and 530, and deletion of I177L have been shown to completely abolish virulence in the highly virulent ASFV Georgia isolate (34, 37–39). As the ASFV Georgia isolate has not been efficiently attenuated by deletion of other single genes except for I177L, which has been associated with attenuation in other ASFV isolates (34, 37, 40), the attenuation observed by deleting the MGF-505-7R gene is an important discovery. Previous several studies have shown that development of attenuated ASFV recombinant viruses by genetic manipulations of target genes is an effective approach for vaccine development.
Based on our findings and previously published results, we propose a working model of ASFV MGF-505-7R–mediated regulation of STING activity in innate antiviral responses to ASFV. During ASFV infection, ASFV MGF-505-7R interacted with ULK1 and increased ULK1 expression. And then ULK1 degraded STING to inhibit the cGAS–STING pathway. This study may provide new understanding regarding immune evasion mechanisms involving ASFV and guide future development of countermeasures against ASFV spreading global.
Acknowledgments
We thank Prof. Hong-Bing Shu (Wuhan University) for providing plasmids.
This article is featured in Top Reads, p.1679
This work was supported by grants from the National Science and Technology Infrastructure Program (2018YFC0840402), National Natural Science Foundation of China Grants (31941002), and Special Fund for Basic Scientific Research of Chinese Academy of Agricultural Sciences (Y2019YJ07-01).
- ASFV
- African swine fever virus
- cGAMP
- cyclic GMP–AMP
- cGAS
- cGAMP synthase
- HA
- hemagglutinin
- HAD
- hemadsorption
- HAD50
- 50% HAD dose
- MOI
- multiplicity of infection
- PAM
- porcine alveolar macrophage
- RNAi
- RNA interference.
Disclosures
The authors have no financial conflicts of interest.
References
- 1. Zhao, D. , Liu R., Zhang X., Li F., Wang J., Zhang J., Liu X., Wang L., Zhang J., Wu X., et al. 2019. Replication and virulence in pigs of the first African swine fever virus isolated in China. Emerg. Microbes Infect. 8: 438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Afonso, C. L. , Piccone M. E., Zaffuto K. M., Neilan J., Kutish G. F., Lu Z., Balinsky C. A., Gibb T. R., Bean T. J., Zsak L., Rock D. L.. 2004. African swine fever virus multigene family 360 and 530 genes affect host interferon response. J. Virol. 78: 1858–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thomson, G. R. 1985. The epidemiology of African swine fever: the role of free-living hosts in Africa. Onderstepoort J. Vet. Res. 52: 201–209. [PubMed] [Google Scholar]
- 4. Correia, S. , Ventura S., Parkhouse R. M.. 2013. Identification and utility of innate immune system evasion mechanisms of ASFV. Virus Res. 173: 87–100. [DOI] [PubMed] [Google Scholar]
- 5. Takeuchi, O. , Akira S.. 2010. Pattern recognition receptors and inflammation. Cell 140: 805–820. [DOI] [PubMed] [Google Scholar]
- 6. Sun, L. , Wu J., Du F., Chen X., Chen Z. J.. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Abe, T. , Harashima A., Xia T., Konno H., Konno K., Morales A., Ahn J., Gutman D., Barber G. N.. 2013. STING recognition of cytoplasmic DNA instigates cellular defense. Mol. Cell 50: 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ishikawa, H. , Barber G. N.. 2008. Sting is an endoplasmic reticulum adaptor that facilitates innate immune signalling. [Published erratum appears in 2008 Nature 456: 274.] Nature 455: 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ouyang, S. , Song X., Wang Y., Ru H., Shaw N., Jiang Y., Niu F., Zhu Y., Qiu W., Parvatiyar K., et al. 2012. Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity 36: 1073–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shu, C. , Yi G., Watts T., Kao C. C., Li P.. 2012. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat. Struct. Mol. Biol. 19: 722–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang, X. , Shi H., Wu J., Zhang X., Sun L., Chen C., Chen Z. J.. 2013. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 51: 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhong, B. , Yang Y., Li S., Wang Y. Y., Li Y., Diao F., Lei C., He X., Zhang L., Tien P., Shu H. B.. 2008. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29: 538–550. [DOI] [PubMed] [Google Scholar]
- 13. Tanaka, Y. , Chen Z. J.. 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 5: ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang, X. , Wu J., Wu Y., Chen H., Zhang S., Li J., Xin T., Jia H., Hou S., Jiang Y., et al. 2018. Inhibition of cGAS-STING-TBK1 signaling pathway by DP96R of ASFV China 2018/1. Biochem. Biophys. Res. Commun. 506: 437–443. [DOI] [PubMed] [Google Scholar]
- 15. García-Belmonte, R. , Pérez-Núñez D., Pittau M., Richt J. A., Revilla Y.. 2019. African swine fever virus Armenia/07 virulent strain controls interferon beta production through the cGAS-STING pathway. J. Virol. 93: e02298-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carrascosa, A. L. , Santarén J. F., Viñuela E.. 1982. Production and titration of African swine fever virus in porcine alveolar macrophages. J. Virol. Methods 3: 303–310. [DOI] [PubMed] [Google Scholar]
- 17. Luo, W. W. , Li S., Li C., Lian H., Yang Q., Zhong B., Shu H. B.. 2016. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat. Immunol. 17: 1057–1066. [DOI] [PubMed] [Google Scholar]
- 18. Li, X. D. , Wu J., Gao D., Wang H., Sun L., Chen Z. J.. 2013. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341: 1390–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Malmquist, W. A. , Hay D.. 1960. Hemadsorption and cytopathic effect produced by African Swine Fever virus in swine bone marrow and buffy coat cultures. Am. J. Vet. Res. 21: 104–108. [PubMed] [Google Scholar]
- 20. Muench, H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27: 493–497. [Google Scholar]
- 21. King, D. P. , Reid S. M., Hutchings G. H., Grierson S. S., Wilkinson P. J., Dixon L. K., Bastos A. D., Drew T. W.. 2003. Development of a TaqMan PCR assay with internal amplification control for the detection of African swine fever virus. J. Virol. Methods 107: 53–61. [DOI] [PubMed] [Google Scholar]
- 22. Krug, P. W. , Holinka L. G., O’Donnell V., Reese B., Sanford B., Fernandez-Sainz I., Gladue D. P., Arzt J., Rodriguez L., Risatti G. R., Borca M. V.. 2015. The progressive adaptation of a georgian isolate of African swine fever virus to vero cells leads to a gradual attenuation of virulence in swine corresponding to major modifications of the viral genome. J. Virol. 89: 2324–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Konno, H. , Konno K., Barber G. N.. 2013. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155: 688–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. González, A. , Calvo V., Almazán F., Almendral J. M., Ramírez J. C., de la Vega I., Blasco R., Viñuela E.. 1990. Multigene families in African swine fever virus: family 360. J. Virol. 64: 2073–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yozawa, T. , Kutish G. F., Afonso C. L., Lu Z., Rock D. L.. 1994. Two novel multigene families, 530 and 300, in the terminal variable regions of African swine fever virus genome. Virology 202: 997–1002. [DOI] [PubMed] [Google Scholar]
- 26. Zsak, L. , Lu Z., Burrage T. G., Neilan J. G., Kutish G. F., Moore D. M., Rock D. L.. 2001. African swine fever virus multigene family 360 and 530 genes are novel macrophage host range determinants. J. Virol. 75: 3066–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu, J. J. , Li W., Shao Y., Avey D., Fu B., Gillen J., Hand T., Ma S., Liu X., Miley W., et al. 2015. Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 18: 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang, G. , Chan B., Samarina N., Abere B., Weidner-Glunde M., Buch A., Pich A., Brinkmann M. M., Schulz T. F.. 2016. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc. Natl. Acad. Sci. USA 113: E1034–E1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Christensen, M. H. , Jensen S. B., Miettinen J. J., Luecke S., Prabakaran T., Reinert L. S., Mettenleiter T., Chen Z. J., Knipe D. M., Sandri-Goldin R. M., et al. 2016. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 35: 1385–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fu, Y. Z. , Su S., Gao Y. Q., Wang P. P., Huang Z. F., Hu M. M., Luo W. W., Li S., Luo M. H., Wang Y. Y., Shu H. B.. 2017. Human cytomegalovirus tegument protein UL82 inhibits STING-mediated signaling to evade antiviral immunity. Cell Host Microbe 21: 231–243. [DOI] [PubMed] [Google Scholar]
- 31. Gui, X. , Yang H., Li T., Tan X., Shi P., Li M., Du F., Chen Z. J.. 2019. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567: 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lewis, T. , Zsak L., Burrage T. G., Lu Z., Kutish G. F., Neilan J. G., Rock D. L.. 2000. An African swine fever virus ERV1-ALR homologue, 9GL, affects virion maturation and viral growth in macrophages and viral virulence in swine. J. Virol. 74: 1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moore, D. M. , Zsak L., Neilan J. G., Lu Z., Rock D. L.. 1998. The African swine fever virus thymidine kinase gene is required for efficient replication in swine macrophages and for virulence in swine. J. Virol. 72: 10310–10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. O’Donnell, V. , Holinka L. G., Krug P. W., Gladue D. P., Carlson J., Sanford B., Alfano M., Kramer E., Lu Z., Arzt J., et al. 2015. African swine fever virus Georgia 2007 with a deletion of virulence-associated gene 9GL (B119L), when administered at low doses, leads to virus attenuation in swine and induces an effective protection against homologous challenge. J. Virol. 89: 8556–8566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zsak, L. , Lu Z., Kutish G. F., Neilan J. G., Rock D. L.. 1996. An African swine fever virus virulence-associated gene NL-S with similarity to the herpes simplex virus ICP34.5 gene. J. Virol. 70: 8865–8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zsak, L. , Caler E., Lu Z., Kutish G. F., Neilan J. G., Rock D. L.. 1998. A nonessential African swine fever virus gene UK is a significant virulence determinant in domestic swine. J. Virol. 72: 1028–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Borca, M. V. , Ramirez-Medina E., Silva E., Vuono E., Rai A., Pruitt S., Holinka L. G., Velazquez-Salinas L., Zhu J., Gladue D. P.. 2020. Development of a highly effective African swine fever virus vaccine by deletion of the I177L gene results in sterile immunity against the current epidemic Eurasia strain. J. Virol. 94: e02017-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O’Donnell, V. , Holinka L. G., Gladue D. P., Sanford B., Krug P. W., Lu X., Arzt J., Reese B., Carrillo C., Risatti G. R., Borca M. V.. 2015. African swine fever virus Georgia isolate harboring deletions of MGF360 and MGF505 genes is attenuated in swine and confers protection against challenge with virulent parental virus. J. Virol. 89: 6048–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. O’Donnell, V. , Risatti G. R., Holinka L. G., Krug P. W., Carlson J., Velazquez-Salinas L., Azzinaro P. A., Gladue D. P., Borca M. V.. 2016. Simultaneous deletion of the 9GL and UK genes from the African swine fever virus Georgia 2007 isolate offers increased safety and protection against homologous challenge. J. Virol. 91: e01760-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ramirez-Medina, E. , Vuono E., O’Donnell V., Holinka L. G., Silva E., Rai A., Pruitt S., Carrillo C., Gladue D. P., Borca M. V.. 2019. Differential effect of the deletion of African swine fever virus virulence-associated genes in the induction of attenuation of the highly virulent Georgia strain. Viruses 11: 599. [DOI] [PMC free article] [PubMed] [Google Scholar]