

Abstract
Nature uses multinuclear metal clusters to catalyse a number of important multielectron redox reactions. Examples that employ complex Fe–S clusters in catalysis include the Fe–Mo cofactor (FeMoco) of nitrogenase and its V and all-Fe variants, and the [FeFe] and [NiFe] hydrogenases. This Perspective begins with a focus on the catalytic H-cluster of [FeFe] hydrogenase, which is highly active in producing molecular H2. There has been much recent progress in characterizing the enzyme-catalysed assembly of the H-cluster, including information gleaned from spectroscopy combined with in vitro isotopic labelling of this cluster using chemical synthesis. We then compare the lessons learned from H-cluster biosynthesis to what is known about the bioassembly of the binuclear active site of [NiFe] hydrogenase and the nitrogenase active site cluster FeMoco.
Several complex enzymes based on Fe–S clusters play crucial roles in biological redox processes and serve as aspirational paradigms for renewable energy catalysis1. Nitrogenases are expressed in a specific group of microorganisms called diazotrophs (of which Azotobacter vinelandii is the best studied organism) and carry out the reduction of atmospheric N2 to NH3. In this way, the enzymes supply the vast majority of natural bioavailable nitrogen in the biosphere, which is supplemented by anthropogenic processes such as the Haber–Bosch process2. Hydrogenases catalyse the interconversion of H+ and H2, a process crucial for metabolism of their host organisms. These highly active enzymes also serve as aspirational examples for H2 production and utilization in renewable energy applications3,4. Hydrogenases are found in many microorganisms from archaea and bacteria to some eukaryotes and can be categorized into three subgroups based on the metal content of their catalytic sites: [FeFe] hydrogenases, [NiFe] hydrogenases and [Fe] hydrogenases. This Perspective covers the bioassembly of the active sites in [FeFe] and [NiFe] hydrogenases, which catalyse the redox interconversion 2H+ + 2e− ⇌ H2. Both active sites feature Fe atoms ligated by CO and CN− ligands and the question of how these toxic inorganic diatomics are safely formed and vectored into the assembly of complex Fe–S clusters is of great interest. [NiFe] hydrogenases in all organisms feature, aside from their active sites, three additional Fe–S clusters or connect to additional Fe–S-cluster-containing subunits that serve as an electron-transport chain3. By contrast, [FeFe] hydrogenase enzymes all feature a catalytic 6Fe ‘H-cluster’ but have varying arrangements of accessory Fe–S clusters. For example, [FeFe] hydrogenase from Clostridium pasteurianum (CpI) contains three [4Fe–4S] clusters and one [2Fe–2S] cluster in its electron-transport chain5. By contrast, [FeFe] hydrogenase from Desulfovibrio desulfuricans (DdH) uses two additional [4Fe–4S] clusters6, while the enzyme from green algae Chlamydomonas reinhardtii (CrHydA1) features the H-cluster as its only Fe–S centre7. Thus, the H-cluster in the CrHydA1 enzyme is the most easily studied because it can be probed without interferences from other Fe–S clusters. For other enzymes, electron paramagnetic resonance (EPR) spectra feature overlapping signals for the H-cluster and accessory clusters.
The [FeFe] hydrogenase H-cluster
We begin this Perspective with a focus on the bioassembly of the catalytic 6Fe H-cluster of [FeFe] hydrogenase, which produces H2 at a very high rate (~104 s−1)8. The H-cluster consists of a canonical [4Fe–4S]H cluster linked by a Cys thiolate to a unique binuclear [2Fe]H cluster (FIG. 1; the subscript H denotes part of the H-cluster). Each Fe in the organometallic [2Fe]H cluster has terminal CN− and CO ligands, with an additional CO bridging the two Fe centres5,6,9. Lastly, there is an additional bridging azadithiolate (adt2−, HN(CH2S−)2) ligand, of which the secondary amine is thought to relay H+ to/from a coordination site on the Fe that is distal to the [4Fe–4S]H cluster10–13. In active states of the enzyme, this site can either be vacant or be occupied by H− or H2 ligands. For example, the site is vacant in the oxidized Hox state and occupied by H− in the Hhyd state. As we see, these two prominent EPR-active forms have spectroscopic signatures that allow us to rapidly compare native and semi-synthetic enzymes to deduce the enzyme’s structure and biosynthesis.
Fig. 1 |. The [FeFe] hydrogenase H-cluster and its biosynthetic origins.
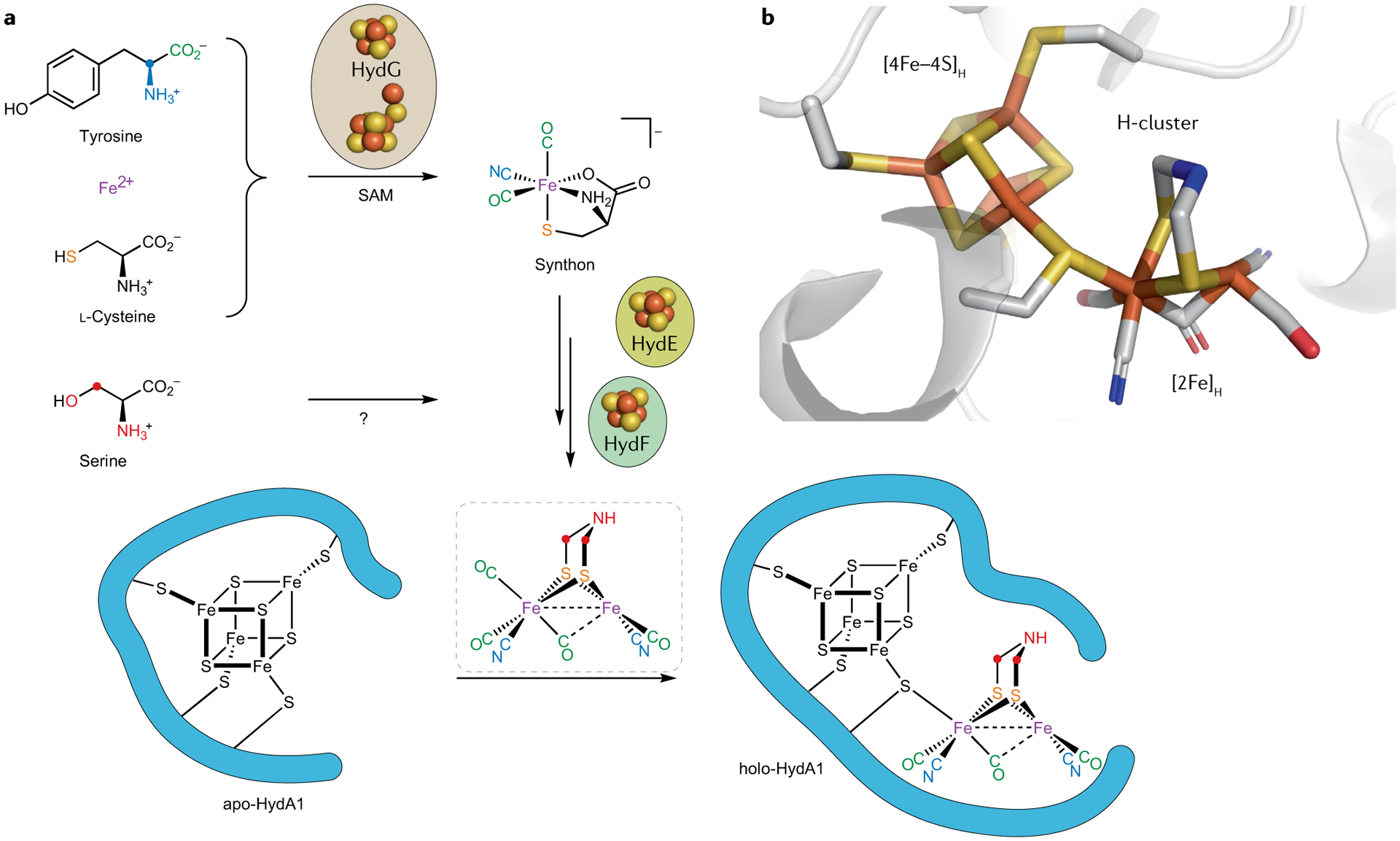
a | The present model of the [FeFe] hydrogenase H-cluster bioassembly accounts for the molecular origins of all atoms in the [2Fe]H subcluster. b | The X-ray structure of the H-cluster in Clostridium pasteurianum [FeFe] hydrogenase (CpI) (Protein Data Bank identifier: 4XDC)9. SAM, S-adenosylmethionine.
Nature uses a set of three Fe–S proteins — HydE, HydF and HydG — to construct the unusual [2Fe]H cluster and link it to the [4Fe–4S]H cluster, which is installed by the regular Fe–S cluster assembly machinery encoded by, for example, the isc operon7,14–16. In recent years, we have combined spectroscopic methods with in vitro maturation of the H-cluster, employing the purified apo-hydrogenase CrHydA1 (as expressed in Escherichia coli with only the [4Fe–4S]H subcluster), along with cell lysates enriched in the three maturase proteins as well as with a cocktail of low-molecular-weight cofactors and precursors, all mixed together in a test tube under anaerobic conditions17–19. With the in vitro-maturation approach, we can conveniently introduce isotopic labels into the maturation pathway by using labelled substrates and cofactors during enzymatic assembly of the H-cluster or, alternatively, in kinetically-resolved studies of individual maturation enzymes. This approach has proven powerful and has allowed us to identify the molecular sources of each atom in the [2Fe]H cluster and to isotopically label the H-cluster to enable further spectroscopic and mechanistic studies (FIG. 1a). Many of the intermediates in the maturation pathway are paramagnetic, such that magnetically-active nuclei near unpaired spin density are readily detected because they give rise to electron–nuclear hyperfine interactions. We have used both continuous-wave (CW) and pulse EPR techniques in these studies, with pulse methods such as hyperfine sublevel correlation (HYSCORE) and electron–nuclear double resonance (ENDOR) allowing us to probe weaker hyperfine interactions than are evident in conventional CW EPR (see REFS20–22 for the principles and applications of pulse EPR techniques). The information gleaned from the spectroscopy is then translated into chemical insights into the maturation pathway.
Initial Fe–CO and Fe–CN bonding
Of the three H-cluster maturases, HydG has been the most studied and best understood enzyme. It is a member of the radical S-adenosylmethionine (rSAM) superfamily of enzymes23–25 that homolytically cleave an S-adenosylmethionine (SAM) coenzyme bound to a [4Fe–4S] cluster. The resulting C-centred 5′-deoxyadenosyl radical26,27 typically initiates transformation of a substrate through stereoselective and regioselective H-atom abstraction. The substrate of HydG is free tyrosine28, the amino group of which undergoes H-atom abstraction by the 5′-deoxyadenosyl radical29 (FIG. 2a), leading to homolysis of the tyrosine Cα–Cβ bond and a transient EPR-active 4-hydroxybenzyl (4-OB·) radical, along with dehydroglycine (DHG)30. The DHG further fragments to form CN− and CO (REFS31–34), which are observable in transient Fourier transform infrared (FTIR) spectra35 (FIG. 2b). We note that, although toxic, at low levels, CO plays an important signalling role. In humans, this signalling provides protective stress and immune responses to injuries, preventing the development of high-fat-diet-induced obesity and regulating metabolic states in various cancer cells or tumour microenvironments36. However, the CO used in signalling is produced by the oxidative degradation of haem by haem oxygenase37, a very different process to the rSAM HydG generation of CO/CN− stoichiometrically through tyrosine radical fission.
Fig. 2 |. Reactions carried out by the radical SAM maturase HydG.
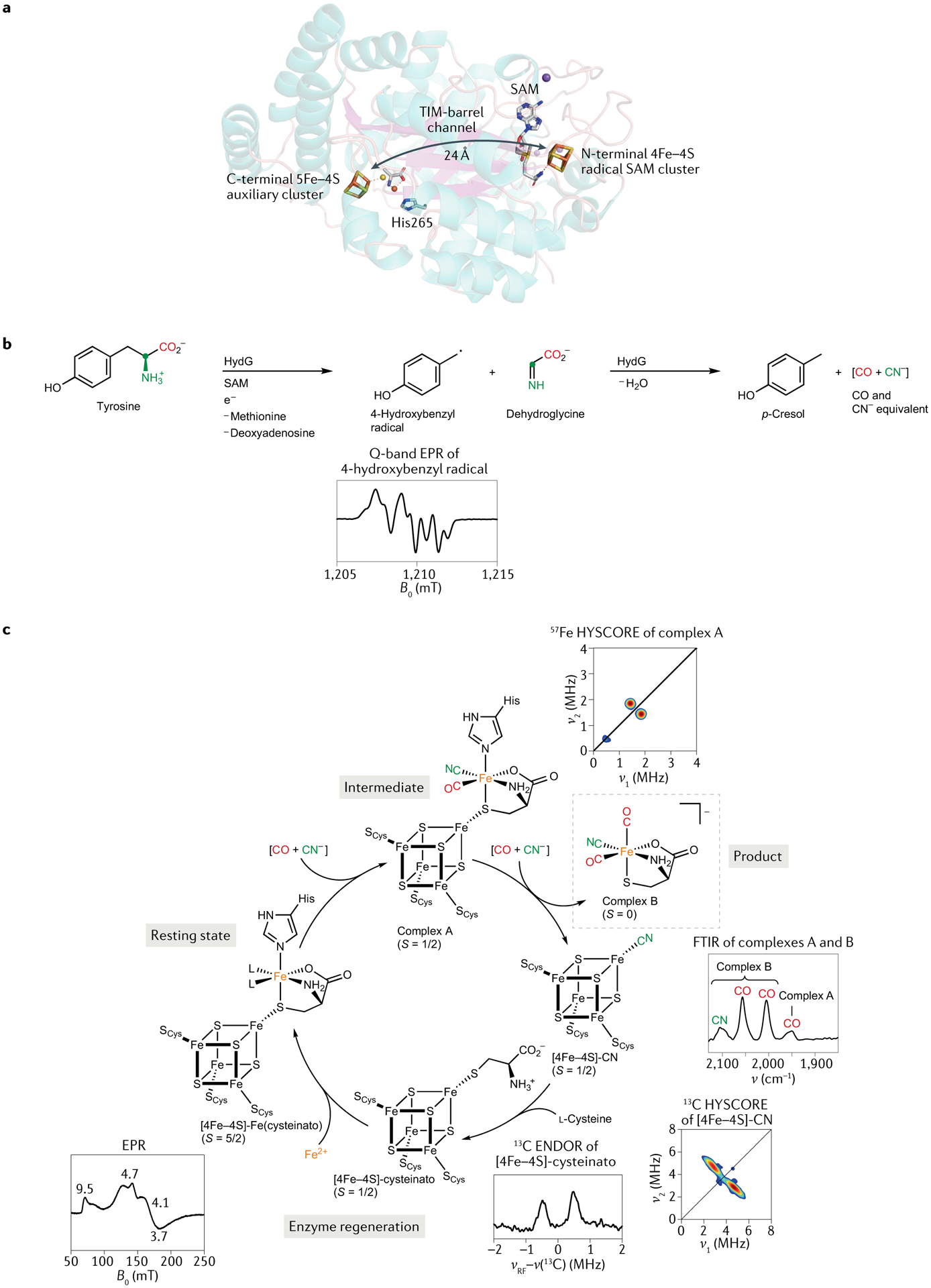
a | The X-ray structure of HydG, which features two Fe–S clusters (Protein Data Bank identifier: 4WCX)38. b | Tyrosine is subjected to radical reactions that see it fragment into the electron paramagnetic resonance (EPR)-active 4-hydroxybenzyl radical (4-OB·) and dehydroglycine (DHG). These further convert to p-cresol, CO and CN−. c | The organometallic synthon forms at the auxiliary 5Fe–4S cluster, as evidenced by FTIR spectroscopy. ENDOR, electron–nuclear double resonance; FTIR, Fourier transform infrared; HYSCORE, hyperfine sublevel correlation; SAM, S-adenosylmethionine. Part b adapted with permission from REF.30, AAAS. Part c adapted with permission from REF.35, AAAS.
In HydG, the potentially toxic CO and CN− fragments are immediately passivated by rapid binding to an Fe site in a unique 5Fe auxiliary Fe–S cluster, which, in the X-ray structure of HydG, is located at the other end of a 24 Å channel from the SAM cluster38,39. The CO and CN− molecules produced by the first tyrosine scission both bind a ‘dangler’ Fe (REF.40), which is additionally ligated by a conserved His residue and a cysteine cofactor. The thiolate in the latter non-peptidic ligand bridges this unique Fe to a [4Fe–4S] cluster41,42 (FIG. 2a). Scission of a second tyrosine molecule again affords CO and CN−, but whereas the former binds the dangler Fe, the latter binds the [4Fe–4S] cluster to give an EPR-detectable [4Fe–4S]-CN species41. This binding releases the HydG product, an organometallic {Fe(CO)2(CN)(cysteinato)} synthon and precursor in the assembly of the [2Fe]H cluster (FIG. 2c). By introducing 57Fe into HydG and using 13C-labelled and 15N-labelled tyrosine, one can use CW and pulse EPR to track the incorporation of these isotopes into the paramagnetic Hox and Hhyd forms of the H-cluster (FIG. 1a) assembled in vitro35,43,44. This magnetic-isotope editing allows us to measure the specific nuclear hyperfine interactions between the 13C, 15N and 57Fe centres and the unpaired electron spin density residing on the H-cluster in these two paramagnetic states. Moreover, use of 13C-labelled and 15N-labelled tyrosine as the substrate produces clear isotopic shifts in time-resolved FTIR bands assigned to the Fe-bound CO and CN− ligands35.
It is notable that cysteine is a component of the organometallic HydG product {Fe(CO)2(CN)(cysteinato)}. The [4Fe–4S]-CN co-product converts to [4Fe–4S]-cysteinato, of which the pendant amino acid and the aforementioned proximal His residue make up a binding site for a fifth Fe. The introduction of this Fe completes the catalytic cycle. Aside from the cysteinato’s role in binding Fe, could it also be possible that this ligand is the source of the S atoms in the adt2− cofactor? And, if so, does the rest of the adt2− bridge also originate from cysteine? This has recently been addressed in a collaboration between our group and the Rauchfuss group. In this work, we performed in vitro maturation without HydG and tyrosine, instead using a synthetic {Fe(CO)2(CN)(cysteinato)} source, termed syn-B, to test whether the proposed final product of HydG can be effectively introduced as a synthetic analogue45 (FIG. 3a). This approach is reminiscent of prior work in which the synthetic 2Fe complex [Fe2(CO)4(CN)2(adt)]2− was linked to [4Fe–4S]-containing apo-hydrogenase to form fully active hydrogenase in the absence of maturase enzymes (or with only HydF in the first publication)9,46–48. Our distinct semi-synthesis approach takes this a step further back in the biosynthesis to the point where the mononuclear Fe complex is assembled. This approach affords a [FeFe] hydrogenase, whose Hox EPR signal and high activity for H2 evolution are comparable to that of the enzyme maturated in vitro with HydE, HydF and HydG present. All these measurements are, reassuringly, also similar to those for the native enzyme. If one uses a source of {Fe(CO)2(13CN)(cysteinato)}, the final product gives rise to the same 13C pulse EPR spectrum as that obtained earlier using 13C-labelled tyrosine and HydG as part of the standard in vitro method. The precursor syn-B does not only offer possibilities for isotope substitution at the diatomic ligands but also at the cysteinato ligand. However, 13C and/or 15N labels in this ligand are not incorporated into the adt2− bridge, with the expected hyperfine coupling being absent from pulse EPR signals. Parallel mass spectrometry experiments revealed the presence of 13C3-pyruvate in the maturation mixture, indicating S–13C bond cleavage of the 13C3,15N-cysteinato ligand within labelled syn-B. One could, in principle, explore possible couplings to a magnetically active 33S nucleus introduced by using 33S-cysteine, but 33S is difficult to obtain (it is 0.7% abundant) and is an I = 3/2 nucleus with a relatively low magnetic moment, along with quadrupolar broadening. An alternative approach is to incorporate selenocysteine into a synthetic carrier and to use extended X-ray absorption fine structure spectroscopy to characterize the structure of the resultant H-cluster (FIG. 3b). Here, we observe two Se–Fe interactions, measured at both the Se and Fe X-ray absorption edges, and the consensus Se–Fe distance of 2.43 Å is consistent with two Se atoms being incorporated into adt2− at the locations usually occupied by S (FIG. 3). We, thus, concluded that each of the two chalcogen atoms in adt2− are from cysteine/selenocysteine in the {Fe(CO)2(CN)(cysteinate/selenocysteinato)} carrier, with the rest of cysteine being clipped off and converted into pyruvate (13C3-pyruvate when using 13C3,15N-cysteine). This demonstrates that the S–Fe bond, first formed when cysteine binds the dangler Fe to give the resting state of the HydG, remains intact throughout the assembly all the way to the mature H-cluster45. But these results leave open another question: what are the molecular sources of C and N components of the adt2− bridge?
Fig. 3 |. Transfer of S/Se from the HydG synthon to make the adt2− bridge.
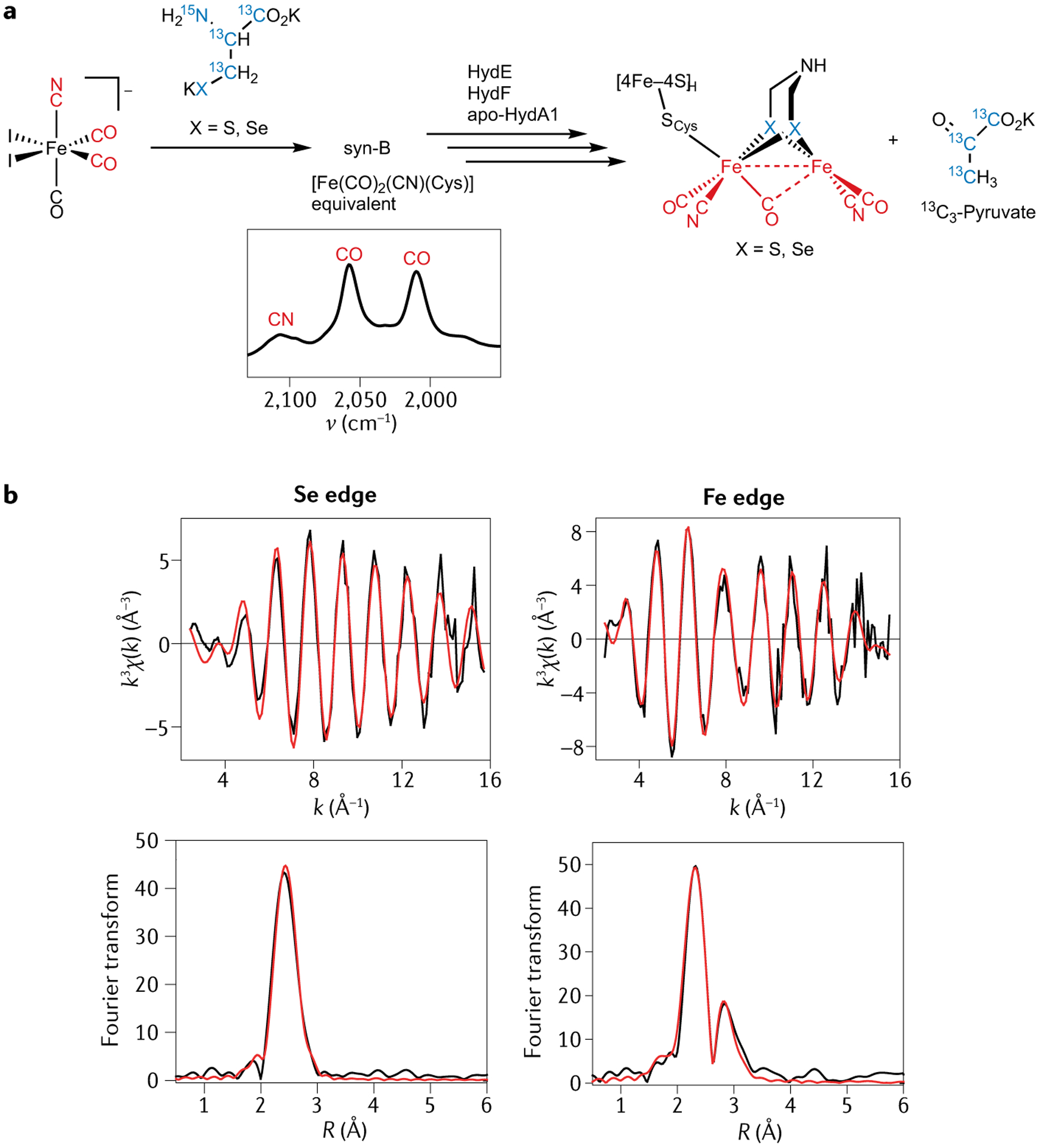
a | The semi-synthetic maturation of the H-cluster with the bridging S/Se sourced from cysteine/selenocysteine introduced by a synthetic HydG synthon carrier. b | Se and Fe edge extended X-ray absorption fine structure data show the incorporation of Se into the [2Fe]H subcluster. Adapted with permission from REF.45, PNAS.
The CNC moiety of the azadithiolate
Given the conclusion that only the S atom of cysteine ends up incorporated into the adt2− bridge, we set out to find the source of the CNC atoms in the centre of the bridge. A distinct possibility is one or more amino acids, so we first added a fully 13C,15N-labelled algal amino acid mixture to the maturation media and observed a set of 13C and 15N peaks in the HYSCORE data of the Hox state49. This motivated a systematic survey of subsets of labelled amino acids with their possible incorporation assayed by HYSCORE, which told us that labelled serine affords an enzyme with a fully 15N-labelled and 13C-labelled adt2− bridge. Using 13C3,15N-serine affords an enzyme whose Hox state gives rise to an ENDOR spectrum with a well-defined pair of 15N transitions and two distinct 13C couplings, consistent with a central 15N and two magnetically inequivalent 13C sites originating from the labelled amino acid (FIG. 4a). Moreover, these 15N and 13C couplings closely match those observed from semisynthesis of the enzyme using synthetic [Fe2(CO)4(CN)2(adt)]2− featuring a 13C2,15N-adt2− bridge50,51. Furthermore, by separately using the three singly 13C-labelled serine isotopologues and comparing 13C ENDOR data of the resulting product to that of the 13C3,15N-serine sample, we showed that the C3 atom of serine is the source of the two C atoms of the adt2− bridge (FIG. 4b). We also observed a 2H ENDOR signal in another Hox sample prepared with C3-2H2 serine in the maturation media. Given hat only C3 of serine is utilized, it is clear that two such serine molecules are needed for the synthesis of each adt2− bridge49.
Fig. 4 |. The CNC component of the azadithiolate is sourced from serine.
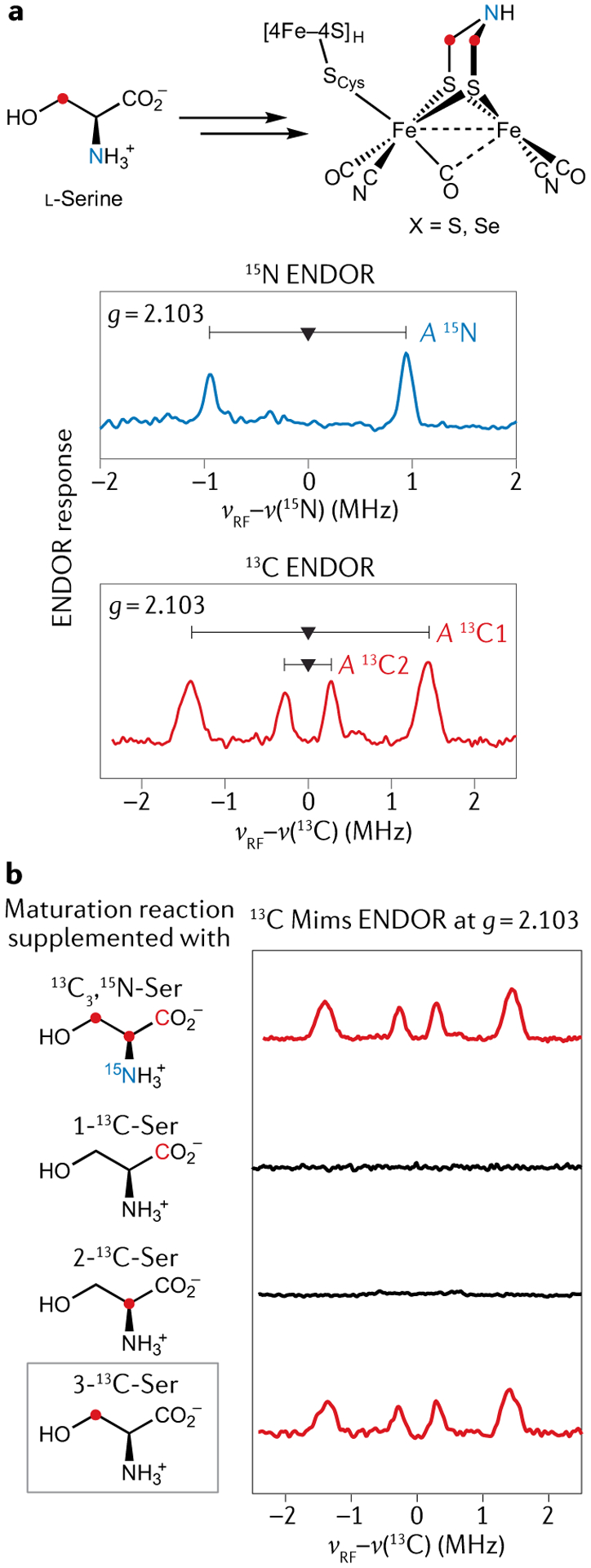
a | 15N and 13C electron–nuclear double resonance (ENDOR) spectra arising from the Hox H-cluster electron paramagnetic resonance signal of a HydA1 [FeFe] hydrogenase sample produced by in vitro maturation with added 13C3,15N-labelled serine. b | 13C ENDOR spectra of the Hox state of HydA1 [FeFe] hydrogenase samples produced via in vitro maturation with added 13C3 15N-labelled, 1-13C-labelled, 2-13C-labelled and 3-13C-labelled serine, respectively, showing that the C atoms of the azadithiolate (adt2−) bridge originate from the 3-C of serine. Part b adapted with permission from REF.49, Royal Society of Chemistry.
The radical SAM enzyme HydE
We have, so far, built up a detailed picture of the reaction mechanism and final product of HydG, yet, the other rSAM enzyme, HydE, has remained relatively uncharacterized. It has been suggested that HydE is involved in building the adt2− ligand, and SAM turnover studies have been interpreted to support a thiol-containing compound as its substrate52. More recently, other groups have shown that HydE reacts with 1,3-thiazolidine to form a new C5′–S bond53. Although the relevance of this reaction to H-cluster synthesis seemed unclear, our recent work using the synthetic HydG product surrogate syn-B reveals a parallel HydE reaction that is on-pathway, where HydE generates the 5′-deoxyadenosyl radical that specifically attacks the cysteine S of the organometallic synthon formed by HydG, which we described putatively as {Feii(CO)2(CN)(cysteinato)}. The C5′–S bond forms concomitant with reduction of the low-spin Feii to Fei (REF.54), which, in turn, leads to cleavage of the internal cysteine C3–S bond, showing that HydE is responsible for the specific cysteine cleavage revealed earlier by maturation with the HydG-free system augmented with syn-B45. HydE’s production of a mononuclear {Fei(CO)2(CN)S} species is proposed to enable the subsequent formation of a dinuclear complex {Fei2(CO)4(CN)2(μ-S)2}, which then only needs the addition of the serine-derived CNC moiety to complete the [2Fe]H subcluster. A remaining question is: does the final maturase HydF have a role in this last step of [2Fe]H subcluster synthesis and/or its final linkage to [4Fe–4S]H subcluster to complete the active H-cluster? We conclude our discussion of [FeFe] hydrogenase with this open question and now describe what is known about the assembly of [NiFe] hydrogenase enzymes.
The [NiFe] hydrogenase cluster
The [NiFe] class of hydrogenase harbours a heterobimetallic {(Cys)2Ni(μ-Cys)2Fe(CO)(CN)2} active site that, relative to [FeFe] hydrogenase, is more tightly anchored to its polypeptide backbone. Instead of featuring just one Cys ligand, [NiFe] hydrogenases have four: two Cys thiolates bind Ni terminally (one of these is replaced by selenocysteinate in the related enzyme [NiFeSe] hydrogenase), while another two bridge it to Fe. Thus, the elaborate machinery responsible for the installation of the non-peptidic dithiolate adt2− in [FeFe] hydrogenase is not required here. Nevertheless, both enzymes feature CO and CN− ligands bound to a low-spin Feii centre4. It is, therefore, of interest to compare the bioassembly of these two clusters, not least the way in which the diatomic ligands are synthesized. The present model of [NiFe] binuclear cluster bioassembly involves a few sequential steps, including the biogenesis of the CO and CN− ligands, formation of the {Fe(CO)(CN)2} synthon and the insertion of Ni (REFS4,55) (FIG. 5a). In E. coli, the necessary machinery is encoded by the hyp operon (HypABCDE and HypF). Although the {Fe(CO)(CN)2} unit in [NiFe] hydrogenases is similar to the {Fe(CO)2(CN)} unit in the [FeFe] congener, little similarity can be found in their bioassembly pathways. The CN− ligand in [NiFe] hydrogenase is derived from carbamoyl phosphate (an intermediate metabolite in the urea cycle) through HypEF-complex-catalysed S-carbamoylation and subsequent ATP-dependent dehydration56 (FIG. 5b). The biogenesis of the CO ligand has long been elusive but has recently been unravelled. However, the new mechanism is only for the aerobic bioassembly of [NiFe] hydrogenase; the source of CO in anaerobic organisms still remains enigmatic. The central C1 pool is the aerobic source of the CO ligand57 and the purified protein HypX, an additional maturase required in the aerobic assembly, acts on N10-formyltetrahydrofolate to release CO, which is proposed to occur through a formyl-coenzyme A intermediate58 (FIG. 5c). Thus, the mechanism of CO/CN− synthesis is dramatically different from that used in the assembly of the [FeFe] hydrogenase H-cluster. Notably, the chemistry leading to both CO and CN− ligands is mediated by enzymes that do not contain any metallocofactors, in sharp contrast to the case of HydG-catalysed tyrosine cleavage, in which the nascent CO and CN− ligands are trapped by a labile Fe site inside this Fe–S protein to directly generate the organometallic synthon. This requires a different shuffle mechanism of these toxic ligands in the assembly of the [NiFe] centre. Indeed, HypE serves as the CN− carrier by converting a cysteine thiol into a thiocyanate group. This process occurs twice and, in each case, the CN− ligand is transferred from the HypE-SCN adduct to an Fe centre coordinated by the HypCD protein complex59,60. The CO ligand is proposed to be attached to the same Fe, although the delivery of CO to finish the {Fe(CO)(CN)2} synthon is not fully understood. In any case, the {Fe(CO) (CN)2} synthon is then transferred into the apo-form of the [NiFe] hydrogenase large subunit, which triggers the selective insertion of Ni into the active site, as facilitated by metallochaperone proteins HypAB and SlyD61,62. On completion, the holo-protein undergoes proteolysis, conformational changes and translocation to form the fully functional [NiFe] hydrogenase.
Fig. 5 |. Aerobic bioassembly of the [NiFe] hydrogenase (Cys)2Ni(μ-Cys)2Fe(CO)(CN)2 active site.
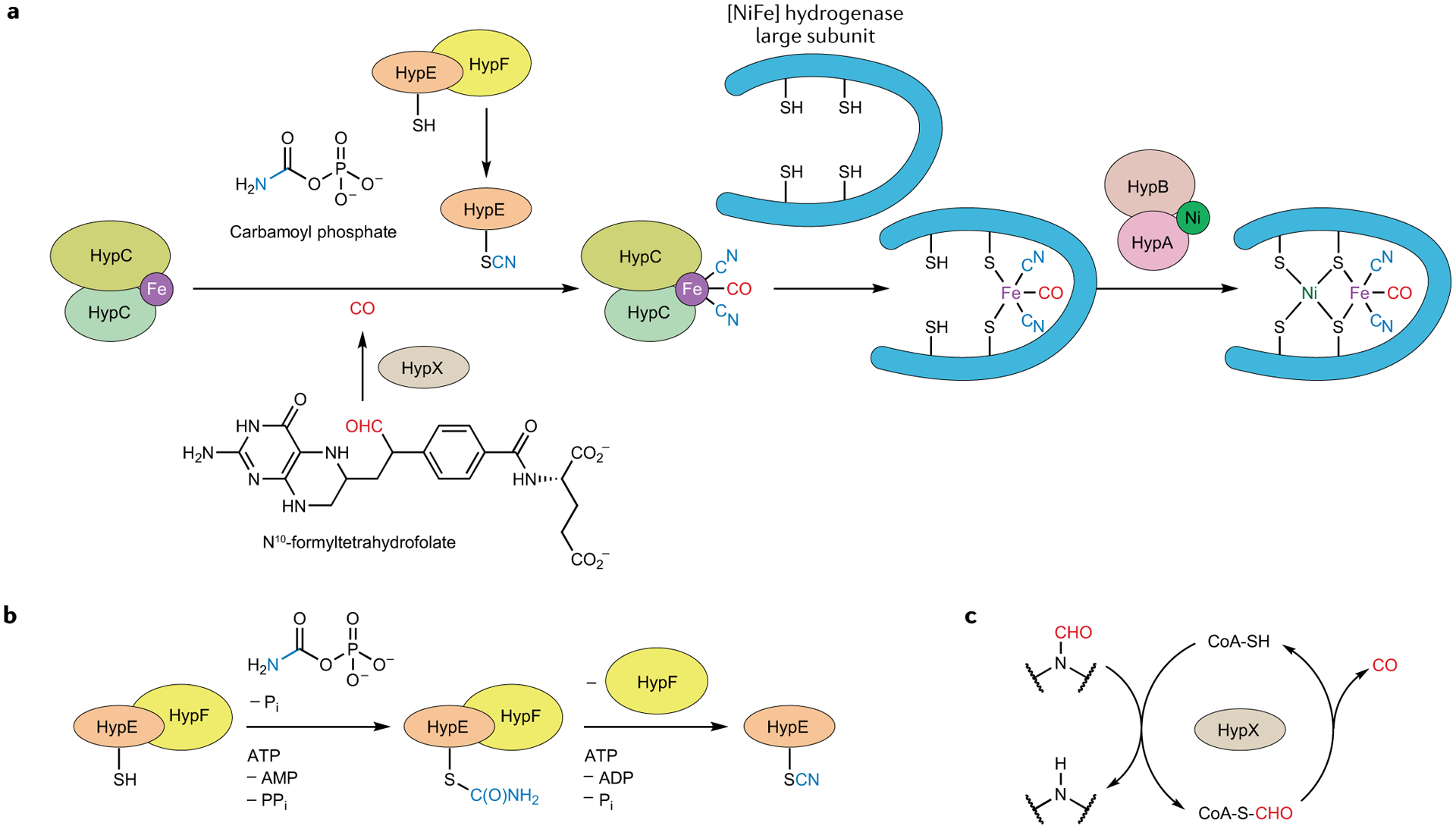
a | Fe acquires its CO and CN− ligands from organic sources, before the {Fe(CO)(CN)2} fragment is delivered to the hydrogenase large subunit. The four Cys ligands then also bind Ni (REF.55). b | Biosynthesis of the CN− ligand by the HypEF protein complex56. c | The HypX protein helps convert a formamide into the CO ligand via a thioformate intermediate58.
The nitrogenase FeMoco cluster
The nitrogenases are another important class of Fe–S enzyme, catalysing the crucial N2 reduction reaction: N2 + 8e− + 16ATP + 8H+ →2NH3 + H2 +16ADP +16Pi (REF.63). As with the hydrogenases, nitrogenases evolve H2, albeit as part of a larger overall reduction reaction that consumes ATP to afford NH3. The Mo nitrogenase enzyme NifDK has two types of Fe–S cluster, the 8Fe–7S P-cluster and the 8Fe–C–9S–Mo M-cluster that serves as the catalytic centre; alternative forms have V (VnfDGK) or Fe (AnfDGK) atoms instead of Mo (REFS64,65). There is also a specific protein bearing a 4Fe–4S cluster that serves as the obligatory electron source for the N2 reduction reaction. There has been much recent progress in characterizing the biosynthesis of the M-cluster, which relies on the chemistry of a key radical SAM enzyme NifB (REFS66–68) (FIG. 6). In addition to the rSAM [4Fe–4S] cluster, NifB houses two auxiliary 4Fe–4S clusters, K1 and K2, that are transformed into an 8Fe precursor to the M-cluster. The SAM cofactor plays two distinct roles here. First, there is a SAM-sourced transfer of a methyl group to one of the 4Fe–4S clusters. Second, rSAM chemistry initiated at the NifB SAM cluster gives a 5′-deoxyadenosyl radical from a second SAM, which then abstracts a H atom from a methyl group, ultimately forming the 8Fe–8S–C ‘L*-cluster’, wherein the μ6-coordinated interstitial carbide is from the methyl group69–71. Insertion of an additional (ninth) S atom completes the central belt72, forming the 8Fe–9S–C ‘L-cluster’, which is then transferred from NifB to the scaffold protein NifEN (or VnfEN), where Mo (or V) and homocitrate are inserted by the NifH protein to form the final M-cluster67,73. All that remains is to transfer the M-cluster to the specific catalytic nitrogenase protein NifDK, VnfDGK or AnfDGK.
Fig. 6 |. Bioassembly of the nitrogenase M-cluster.
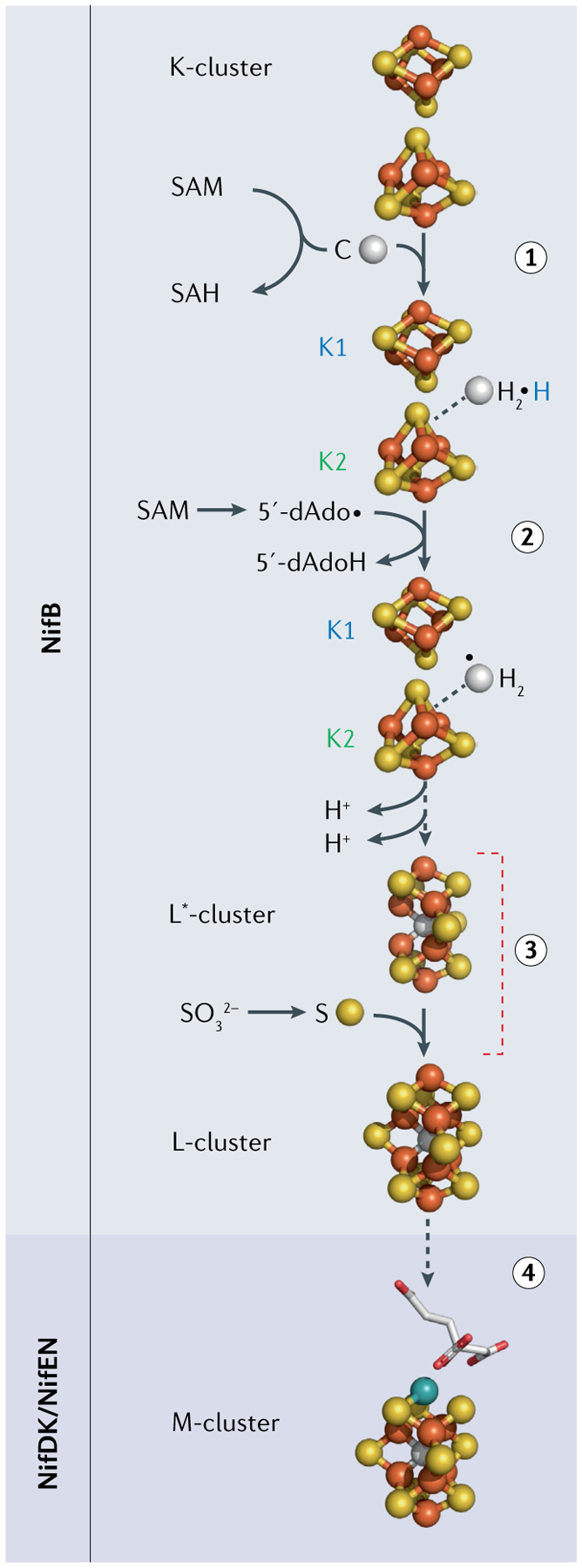
The key initial step occurs at the radical S-adenosylmethionine (SAM) enzyme NifB, where two 4Fe–4S clusters are stitched together via an SAM methyl insertion, followed by a radical SAM H-atom abstraction to make the 8Fe–8S L*-cluster (steps 1 and 2). Addition of a ninth S atom makes the final L-cluster (step 3), the target of the final specific metal substitution (V or Mo) capped by homocitrate (step 4). SAH, S-adenosylhomocysteine. Adapted from REF.75, CC BY 4.0.
Closing summary
Although the [FeFe] and [NiFe] hydrogenases catalyse the same reaction and share some organometallic structural elements, their assembly processes are quite different. Specifically, the formation and handling of the toxic CO and CN− species are very different in the two biosyntheses. The construction of [FeFe] hydrogenase sees CO and CN− formed from amino acids through an rSAM reaction in HydG, with these diatomics then being stabilized by binding to a specific Fe site at a second Fe–S cluster of the same enzyme to give a {Fe(CO)2(CN)(cysteinato)} synthon. In [NiFe] hydrogenase, a comparable {Fe(CO)(CN)2} synthon is formed from the interplay of multiple protein complexes that are essentially metal-free. It is, therefore, interesting to see how nature has evolved two different solutions to one problem. Circumstances often lead to nature forming the same product in different ways, as we highlighted earlier in the case of biosynthesis of signalling CO through haem oxidation.
In comparing the complexity of the biosynthesis of the two redox-active hydrogenases, the reader will note the many steps required to prepare the adt2− cofactor, a ligand unique to the [FeFe] class. The biosynthesis of this cofactor, which is unstable in metal-free form, is challenging and may require additional enzymes in a process that is not yet fully understood. In the [NiFe] enzymes, the H+ relay role is, instead, played by a proteinaceous Arg residue74 (although the terminally bound Cys thiolates have also been suggested). The nitrogenase assembly shares the crucial need for rSAM enzymes with the [FeFe] hydrogenase H-cluster assembly, but, in this case, the rSAM enzyme NifB carries out distinctive chemistry, using SAM both as a methyl donor and as a radical coenzyme to cement two [4Fe–4S] clusters together with an SAM-methyl-derived bridging carbide. Thus, the organometallic intermediate formed by NifB is very different to the Fe(CO)2(CN)-containing intermediate formed by HydG, although both enzymes harness the special power of rSAM chemistry.
Acknowledgements
This work is funded by the National Institutes of Health (1R35GM126961 to R.D.B.).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Peters JW & Broderick JB Emerging paradigms for complex iron–sulfur cofactor assembly and insertion. Annu. Rev. Biochem 81, 429–450 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Ribbe MW, Hu Y, Hodgson KO & Hedman B Biosynthesis of nitrogenase metalloclusters. Chem. Rev 114, 4063–4080 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vignais PM & Billoud B Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev 107, 4206–4272 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Lubitz W, Ogata H, Rudiger O & Reijerse E Hydrogenases. Chem. Rev 114, 4081–4148 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Peters JW, Lanzilotta WN, Lemon BJ & Seefeldt LC X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 angstrom resolution. Science 282, 1853–1858 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Nicolet Y, Piras C, Legrand P, Hatchikian CE & Fontecilla-Camps JC Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site Fe binuclear center. Structure 7, 13–23 (1999). [DOI] [PubMed] [Google Scholar]
- 7.Mulder DW et al. Stepwise [FeFe]-hydrogenase H-cluster assembly revealed in the structure of HydAΔEFG. Nature 465, 248–251 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Pandey K, Islam ST, Happe T & Armstrong FA Frequency and potential dependence of reversible electrocatalytic hydrogen interconversion by [FeFe]-hydrogenases. Proc. Natl Acad. Sci. USA 114, 3843–3848 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esselborn J et al. A structural view of synthetic cofactor integration into [FeFe]-hydrogenases. Chem. Sci 7, 959–968 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mulder DW, Guo Y, Ratzloff MW & King PW Identification of a catalytic iron–hydride at the H-cluster of [FeFe]-hydrogenase. J. Am. Chem. Soc 139, 83–86 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Reijerse EJ et al. Direct observation of an iron-bound terminal hydride in [FeFe]-hydrogenase by nuclear resonance vibrational spectroscopy. J. Am. Chem. Soc 139, 4306–4309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sommer C et al. Proton coupled electronic rearrangement within the H-cluster as an essential step in the catalytic cycle of [FeFe] hydrogenases. J. Am. Chem. Soc 139, 1440–1443 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Pelmenschikov V et al. Reaction coordinate leading to H2 production in [FeFe]-hydrogenase identified by nuclear resonance vibrational spectroscopy and density functional theory. J. Am. Chem. Soc 139, 16894–16902 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mulder DW et al. Insights into [FeFe]-hydrogenase structure, mechanism, and maturation. Structure 19, 1038–1052 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Shepard EM et al. [FeFe]-hydrogenase maturation. Biochemistry 53, 4090–4104 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Peters JW et al. [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim. Biophys. Acta 1853, 1350–1369, (2015). [DOI] [PubMed] [Google Scholar]
- 17.Boyer ME, Stapleton JA, Kuchenreuther JM, Wang CW & Swartz JR Cell-free synthesis and maturation of [FeFe] hydrogenases. Biotechnol. Bioeng 99, 59–67 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Kuchenreuther JM, Britt RD & Swartz JR New insights into [FeFe] hydrogenase activation and maturase function. PLoS ONE 7, e45850 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuchenreuther JM, Shiigi SA & Swartz JR Cell-free synthesis of the H-cluster: a model for the in vitro assembly of metalloprotein metal centers. Methods Mol. Biol 1122, 49–72 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Schweiger A & Jeschke G Principles of Pulse Electron Paramagnetic Resonance (Oxford Univ. Press, 2001). [Google Scholar]
- 21.Hanson G & Berliner L (eds) High resolution EPR: Applications to Metalloenzymes and Metals in Medicine (Springer, 2009). [Google Scholar]
- 22.Hagen WR Biomolecular EPR Spectroscopy (CRC, 2009). [Google Scholar]
- 23.Broderick JB, Duffus BR, Duschene KS & Shepard EM Radical S-adenosylmethionine enzymes. Chem. Rev 114, 4229–4317 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landgraf BJ, McCarthy EL & Booker SJ Radical S-adenosylmethionine enzymes in human health and disease. Annu. Rev. Biochem 85, 485–514 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Booker SJ & Grove TL Mechanistic and functional versatility of radical SAM enzymes. F1000 Biol. Rep 2, 52 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sayler RI et al. Trapping and electron paramagnetic resonance characterization of the 5′dAdo· radical in a radical S-adenosyl methionine enzyme reaction with a non-native substrate. ACS Cent. Sci 5, 1777–1785 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang H et al. The elusive 5′-deoxyadenosyl radical: captured and characterized by electron paramagnetic resonance and electron nuclear double resonance spectroscopies. J. Am. Chem. Soc 141, 12139–12146 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pilet E et al. The role of the maturase HydG in [FeFe]-hydrogenase active site synthesis and assembly. FEBS Lett. 583, 506–511 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Nicolet Y, Zeppieri L, Amara P & Fontecilla-Camps JC Crystal structure of tryptophan lyase (NosL): evidence for radical formation at the amino group of tryptophan. Angew. Chem. Int. Ed 53, 11840–11844 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Kuchenreuther JM et al. A radical intermediate in tyrosine scission to the CO and CN− ligands of FeFe hydrogenase. Science 342, 472–475 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Shepard EM et al. [FeFe]-hydrogenase maturation: HydG-catalyzed synthesis of carbon monoxide. J. Am. Chem. Soc 132, 9247–9249 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Driesener RC et al. [FeFe]-hydrogenase cyanide ligands derived from S-adenosylmethionine-dependent cleavage of tyrosine. Angew. Chem. Int. Ed 122, 1731–1734 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Driesener RC et al. Biochemical and kinetic characterization of radical S-adenosyl-l-methionine enzyme HydG. Biochemistry 52, 8696–8707 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Pagnier A, Martin L, Zeppieri L, Nicolet Y & Fontecilla-Camps JC CO and CN− syntheses by [FeFe]-hydrogenase maturase HydG are catalytically differentiated events. Proc. Natl Acad. Sci. USA 113, 104–109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuchenreuther JM et al. The HydG enzyme generates an Fe(CO)2(CN) synthon in assembly of the FeFe hydrogenase H-cluster. Science 343, 424–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wegiel B et al. Carbon monoxide expedites metabolic exhaustion to inhibit tumor growth. Cancer Res. 73, 7009–7021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsui T, Unno M & Ikeda-Saito M Heme oxygenase reveals its strategy for catalyzing three successive oxygenation reactions. Acc. Chem. Res 43, 240–247 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Dinis P et al. X-ray crystallographic and EPR spectroscopic analysis of HydG, a maturase in [FeFe]-hydrogenase H-cluster assembly. Proc. Natl Acad. Sci. USA 112, 1362–1367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nicolet Y et al. Crystal structure of HydG from Carboxydothermus hydrogenoformans: a trifunctional [FeFe]-hydrogenase maturase. ChemBioChem 16, 397–402 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Rao G, Tao L, Suess DLM & Britt RDA [4Fe–4S]-Fe(CO)(CN)-l-cysteine intermediate is the first organometallic precursor in [FeFe] hydrogenase H-cluster bioassembly. Nat. Chem 10, 555–560 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suess DL et al. Cysteine as a ligand platform in the biosynthesis of the FeFe hydrogenase H cluster. Proc. Natl Acad. Sci. USA 112, 11455–11460 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suess DL et al. The radical SAM enzyme HydG requires cysteine and a dangler iron for generating an organometallic precursor to the [FeFe]-hydrogenase H-cluster. J. Am. Chem. Soc 138, 1146–1149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Myers WK et al. The cyanide ligands of [FeFe] hydrogenase: pulse EPR studies of 13C and 15N-labeled H-cluster. J. Am. Chem. Soc 136, 12237–12240 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rao G & Britt RD Electronic structure of two catalytic states of the [FeFe] hydrogenase H-cluster as probed by pulse electron paramagnetic resonance spectroscopy. Inorg. Chem 57, 10935–10944 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rao G et al. The binuclear cluster of [FeFe] hydrogenase is formed with sulfur donated by cysteine of an [Fe(Cys)(CO)2(CN)] organometallic precursor. Proc. Natl Acad. Sci. USA 116, 20850–20855 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berggren G et al. Biomimetic assembly and activation of [FeFe]-hydrogenases. Nature 499, 66–69 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gilbert-Wilson R et al. Spectroscopic investigations of [FeFe] hydrogenase maturated with [57Fe2(adt)(CN)2(CO)4]2−. J. Am. Chem. Soc 137, 8998–9005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Esselborn J et al. Spontaneous activation of [FeFe]-hydrogenases by an inorganic [2Fe] active site mimic. Nat. Chem. Biol 9, 607–609 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rao G, Tao L & Britt RD Serine is the molecular source of the NH(CH2)2 bridgehead moiety of the in vitro assembled [FeFe] hydrogenase H-cluster. Chem. Sci 11, 1241–1247 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adamska-Venkatesh A et al. Spectroscopic characterization of the bridging amine in the active site of [FeFe] hydrogenase using isotopologues of the H-cluster. J. Am. Chem. Soc 137, 12744–12747 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Reijerse EJ et al. Asymmetry in the ligand coordination sphere of the [FeFe] hydrogenase active site is reflected in the magnetic spin interactions of the aza-propanedithiolate ligand. J. Phys. Chem. Lett 10, 6794–6799 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Betz JN et al. [FeFe]-hydrogenase maturation: insights into the role HydE plays in dithiomethylamine biosynthesis. Biochemistry 54, 1807–1818 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rohac R et al. Carbon–sulfur bond-forming reaction catalysed by the radical SAM enzyme HydE. Nat. Chem 8, 491–500 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Tao L et al. Radical SAM enzyme HydE generates adenosylated Fe(i) intermediates en route to the [FeFe]-hydrogenase catalytic H-cluster. J. Am. Chem. Soc 142, 10841–10848 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lacasse MJ & Zamble DB [NiFe]-hydrogenase maturation. Biochemistry 55, 1689–1701 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Reissmann S et al. Taming of a poison: biosynthesis of the NiFe-hydrogenase cyanide ligands. Science 299, 1067–1070 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Bürstel I et al. CO synthesized from the central one-carbon pool as source for the iron carbonyl in O2-tolerant [NiFe]-hydrogenase. Proc. Natl Acad. Sci. USA 113, 14722–14726 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schulz A-C et al. Formyltetrahydrofolate decarbonylase synthesizes the active site CO ligand of O2-tolerant [NiFe] hydrogenase. J. Am. Chem. Soc 142, 1457–1464 (2020). [DOI] [PubMed] [Google Scholar]
- 59.Bürstel I et al. A universal scaffold for synthesis of the Fe(CN)2(CO) moiety of [NiFe] hydrogenase. J. Biol. Chem 287, 38845–38853 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stripp ST et al. HypD is the scaffold protein for Fe-(CN)2CO cofactor assembly in [NiFe]-hydrogenase maturation. Biochemistry 52, 3289–3296 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Khorasani-Motlagh M, Noroozifar M, Kerman K & Zamble DB Complex formation between the Escherichia coli [NiFe]-hydrogenase nickel maturation factors. BioMetals 32, 521–532 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Lacasse MJ, Douglas CD & Zamble DB Mechanism of selective nickel transfer from HypB to HypA, Escherichia coli [NiFe]-hydrogenase accessory proteins. Biochemistry 55, 6821–6831 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Hoffman BM, Lukoyanov D, Yang ZY, Dean DR & Seefeldt LC Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem. Rev 114, 4041–4062 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mus F, Alleman AB, Pence N, Seefeldt LC & Peters JW Exploring the alternatives of biological nitrogen fixation. Metallomics 10, 523–538 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Jasniewski AJ, Lee CC, Ribbe MW & Hu Y Reactivity, mechanism, and assembly of the alternative nitrogenases. Chem. Rev 10.1021/acs.chemrev.9b00704 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wiig JA, Hu Y & Ribbe MW NifEN-B complex of Azotobacter vinelandii is fully functional in nitrogenase FeMo cofactor assembly. Proc. Natl Acad. Sci. USA 108, 8623–8627 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fay AW et al. Spectroscopic characterization of the isolated iron–molybdenum cofactor (FeMoco) precursor from the protein NifEN. Angew. Chem. Int. Ed 50, 7787–7790 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilcoxen J et al. Electron paramagnetic resonance characterization of three iron–sulfur clusters present in the nitrogenase cofactor maturase NifB from Methanocaldococcus infernus. J. Am. Chem. Soc 138, 7468–7471 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wiig JA, Hu Y, Chung Lee C & Ribbe MW Radical SAM-dependent carbon insertion into the nitrogenase M-cluster. Science 337, 1672–1675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jasniewski AJ et al. Spectroscopic characterization of an eight-iron nitrogenase cofactor precursor that lacks the “9th sulfur”. Angew. Chem. Int. Ed 58, 14703–14707 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wiig JA, Hu Y & Ribbe MW Refining the pathway of carbide insertion into the nitrogenase M-cluster. Nat. Commun 6, 8034 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tanifuji K et al. Tracing the ‘ninth sulfur’ of the nitrogenase cofactor via a semi-synthetic approach. Nat. Chem 10, 568–572 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaiser JT, Hu Y, Wiig JA, Rees DC & Ribbe MW Structure of precursor-bound NifEN: a nitrogenase FeMo cofactor maturase/insertase. Science 331, 91–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Evans RM et al. Mechanism of hydrogen activation by [NiFe] hydrogenases. Nat. Chem. Biol 12, 46–50 (2015). [DOI] [PubMed] [Google Scholar]
- 75.Rettberg LA et al. Identity and function of an essential nitrogen ligand of the nitrogenase cofactor biosynthesis protein NifB. Nat. Commun 11, 1757 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]