

Abstract
The connecting peptide (C-peptide) is a hormone with promising health benefits in ameliorating diabetes-related complications, yet mechanisms remain elusive. Emerging studies point to a possible dependence of peptide activity on bioavailable metals, particularly Cu(II) and Zn(II). However, little is known about the chemical nature of the interactions, hindering advances in its therapeutic applications. This work uncovers the Cu(II)-binding site in C-peptide that may be key to understanding its metal-dependent function. A combination of spectroscopic studies reveal that Cu(II) and Zn(II) bind to C-peptide at specific residues in the N-terminal region of the peptide and that Cu(II) is able to displace Zn(II) for C-peptide binding. The data point to a Cu(II)-binding site consisting of 1N3O square-planar coordination that is entropically driven. Furthermore, the entire random coil peptide sequence is needed for specific metal binding as mutations and truncations reshuffle the coordinating residues. These results expand our understanding of how metals influence hormone activity and facilitate the discovery and validation of both new and established paradigms in peptide biology.
Graphical Abstract
INTRODUCTION
Endogenous peptides play a wide array of roles in biology from catalysts1,2 to host defense3,4 to extracellular signaling.5–7 The proper function of these biomolecules is integral to healthy physiology as indicated by the number of diseases associated with their misregulation. Several reports have implicated metal ions in inducing aberrant functions in these peptides, such as promoting the pathological aggregation of the amyloid-β or α-synuclein peptides in association with Alzheimer’s and Parkinson’s diseases, respectively.8–11 However, instances have also reported the necessity of these metals in other peptides; for instance, Zn(II) stabilizes the hexameric form of insulin for efficient storage in pancreatic vesicles.12,13 This intricate balance in the bioinorganic chemistry of these peptides warrants further investigation to elucidate their native function and promote their therapeutic applications.
While a number of bioactive peptides hold promise for clinical use such as in hormone therapies, progress has been hampered by a lack of understanding of the secondary factors, including metal ions, that may influence their activity. One such example is the connecting peptide (C-peptide) for which metals have a putative role in its activation. C-peptide is co-released from β-cells in equimolar amounts with respect to insulin in response to elevated glucose in the blood.14–18 While initially considered inert, C-peptide has emerged as a potential candidate for peptide-based therapy in diabetes for its diverse beneficial functions, most notably ameliorating vasculopathies and kidney dysfunction.19–22 However, progress has been slowed by the lack of mechanistic clarity of its activity. Early work showed that C-peptide increases blood flow in several tissue types,23–26 and later work by Spence and co-workers attributed these effects to the ability of the peptide to induce ATP release from red blood cells.27–29 Interestingly, ATP release is enhanced by the presence of divalent metal ions including Zn(II).29 Further work from their laboratory showed that albumin is required for the transport of both Zn(II) and C-peptide from pancreatic β-cells to red blood cells.30 Another study reported a protective effect of C-peptide in preventing islet amyloid polypeptide (IAPP) aggregation in the pancreas but only in the presence of Zn(II).31 Recently, we uncovered a possible role for metals in mediating the internalization of the peptide; specifically, copper could inhibit the internalization of the peptide into HEK293 cells. Additionally, we determined that, in solution, Cu(II) directly interacts with C-peptide.32
Cu(II) binding was an intriguing discovery given that the peptide sequence does not contain classical copper binding residues, potentially pointing to the anchoring of Cu(II) through the terminal amine as seen with other peptides. Precisely defining this binding site poses challenges that are inherent to the structural aspects of the peptide. First, the short 31-mer peptide is primarily a random coil,33,34 complicating the use of standard biophysical approaches for structural elucidation. In addition, our earlier work did not show any metal-specific alterations in the overall structure that could otherwise be used to probe the interaction.32 Second, the peptide sequence (EAEDLQVGQVELGGGPGAGSLQPLALEGSLQ) is low in complexity, bearing only nine different residues, with glycine appearing seven times. Elucidating the metal binding site thus requires the question to be approached via multiple techniques to dissect the various pieces that contribute to the interaction.
In this study, we sought to determine how and where Cu(II) binds to C-peptide and, given the reports on Zn(II)-dependent effects, whether the binding site is shared with Zn(II). We found that Cu(II) binds to carboxylate groups at the N-terminus of C-peptide along with the terminal amine comprising a 1N3O square-planar geometry that is entropically driven. Moreover, this specific interaction requires the presence of the intact peptide, as both truncated and mutated analogues exhibit a reshuffling of the binding site. Consistent with our previous findings, Cu(II) exhibits higher-affinity binding to C-peptide than Zn(II) and consequently out-competes as well as precludes Zn(II) binding. The data suggest a model whereby Cu(II) binds preferentially at the N-terminal amine of the peptide and the E3 and D4 residues yet requires the remaining regions for site specificity. The elucidated Cu(II) binding site within C-peptide represents a unique ligand set in peptide biology and further expands the possibilities for metal/peptide hormone interactions.
RESULTS AND DISCUSSION
Assessing Copper and Zinc Competition for Direct C-peptide Binding.
Spence and colleagues demonstrated a metal-dependent physiological role of C-peptide: the incubation of erythrocytes with C-peptide induces ATP release but only if supplemented with Zn(II).27,29 Analysis via ESI-MS showed that Zn(II) directly interacts with C-peptide and that this interaction is most likely at one of the acidic residues.27 Given our previous observations that both Zn(II) and Cu(II) could directly interact with C-peptide in solution and alter the cellular behavior of the peptide,32 we sought to characterize the Cu(II)/C-peptide interactions, compare these observations to the previously studied Zn(II)/C-peptide adduct, and determine if the two metals could compete for C-peptide binding.
The mass spectra of apo C-peptide exhibit doubly and triply charged molecular ions corresponding to m/z values of 1510.27 ([M + 2H]2+) and 1007.18 ([M + 3H]3+), respectively (Figure 1A). Additional peaks corresponding to C-peptide with sodium bound such as in [M + H + Na]2+ were also observed (m/z = 1521.26) (Figure 1A). The addition of 1 equiv of Zn(II) results in shifts in the m/z values and indicates the presence of a Zn(II)/C-peptide adduct at a 1:1 metal-to-peptide ratio (m/z = 1541.22 [M + Zn]2+ and 1027.81 [M + H + Zn]3+) (Figure 1B). Spectra obtained via high-resolution ESI-MS confirmed the presence of the adduct by the expected isotopic distribution pattern for zinc as described by Tsednee et al.35 These m/z values agree well with the previously reported masses obtained by the Spence group.29 Similarly to Zn(II), the addition of 1 equiv of Cu(II) to C-peptide results in the appearances of the doubly and triply charged 1:1 metal-to-peptide adducts at m/z values of 1540.72 ([M + Cu]2+) and 1027.49 ([M + H + Cu]3+), respectively (Figure 1C). The experimental isotopic pattern is in good agreement with the theoretical isotopic distribution for Cu(II).35 These observations suggest that both Zn(II) and Cu(II) bind C-peptide in a 1:1 metal-to-peptide stoichiometry in the gas phase.
Figure 1.
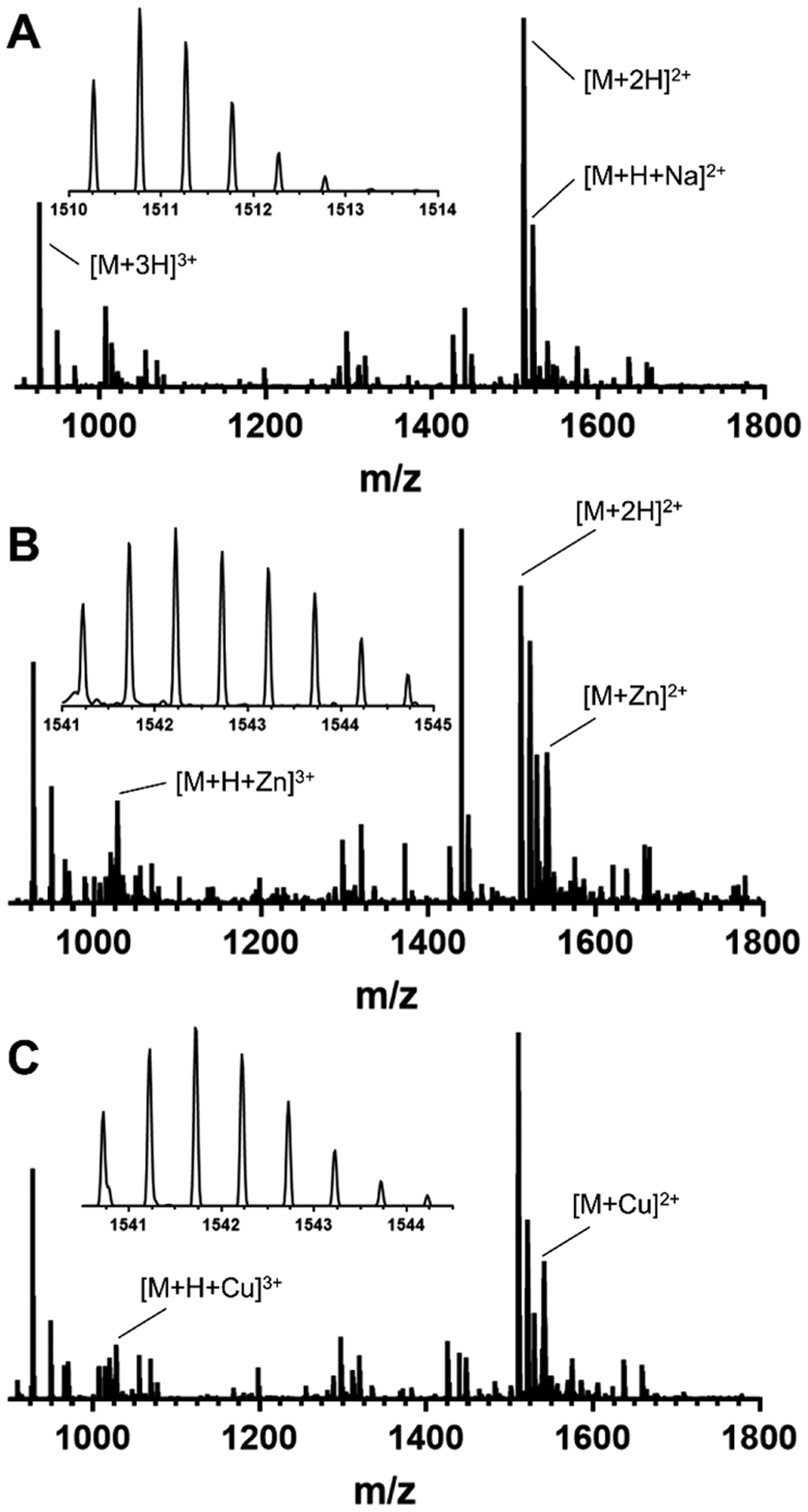
ESI-MS of (A) apo C-peptide, (B) Zn(II)/C-peptide, and (C) Cu(II)/C-peptide. The spectra demonstrate direct interactions between the metal ions and the peptide with a 1:1 metal-to-peptide ratio. All solutions were prepared in nanopure water at 50 μM C-peptide in the absence or presence of 1 equiv of the metal salt (CuSO4 or ZnClO4).
To determine whether Cu(II) could outcompete Zn(II) for C-peptide binding, an equimolar amount of Cu(II) was added to a solution of the Zn(II)/C-peptide adduct, and the mass spectra of the resulting species were analyzed. The Cu(II)/C-peptide adduct clearly emerged, as indicated by the distinct Cu(II) isotopic distribution and the monoisotopic mass of 1540.72 m/z ([M + Cu]2+), whereas no remaining peaks from the Zn(II):C-peptide adduct were observed (Figure 2). These data indicate that Cu(II) binds to C-peptide with a greater affinity than Zn(II), and either the two metals compete for the same site or Cu(II) binding induces conformational changes that preclude Zn(II) binding.
Figure 2.
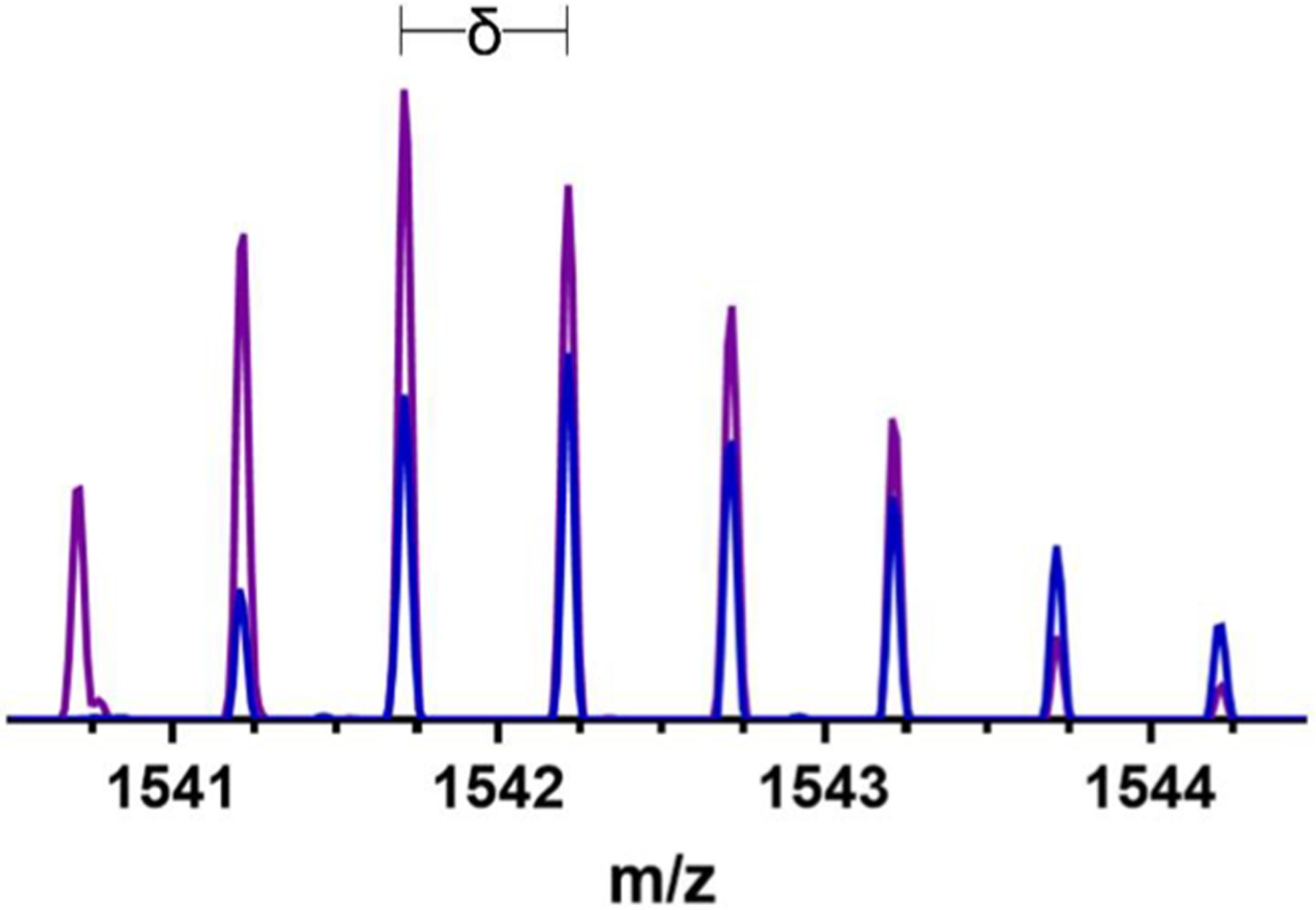
ESI-MS of C-peptide incubated with 1 equiv of Zn(II) (blue) and the resulting spectrum when 1 equiv of Cu(II) (purple) is added to the same solution. δ signifies the isotopic mass shift of the most intense peak present for C-peptide. Comparison of the spectra demonstrate the ability of Cu(II) to outcompete Zn(II) for C-peptide binding. Solutions were prepared in nanopure water at 50 μM C-peptide.
Determining which of these binding hypotheses are correct requires a more precise understanding of the binding sites. We sought to further probe the competition and binding location(s) of the metals via 1H NMR spectroscopy, and we assessed for any characteristic shifts in resonances induced by the metals. In comparison to the spectra of apo C-peptide at pH 7.4 (Figure 3A and Figure S1A), the addition of Zn(II) results in subtle shifts of proton resonances throughout the spectrum, most visibly at 2.7, 3.7, 8.45, and 8.7 ppm (Figure 3B and Figure S1B), which could be attributed to CHβ protons on D4, glycine protons, and the backbone amide protons from D4 and E3, respectively. On the other hand, the addition of Cu(II) to C-peptide obliterates or weakens some signals (Figure 3C and Figure S1C). Because Cu(II) is paramagnetic, this loss of signal is most likely due to the paramagnetic broadening of protons in close proximity to the metal ion. Signal loss is specifically observed where peaks are expected for a set of aliphatic protons (1.4 ppm), the CHβ protons on D4 (2.7 ppm), glycine protons (3.7 ppm), and backbone amide protons from D4 (8.45 ppm) and E3 (8.7 ppm), and this obliteration of signal requires the saturation of the peptide with Cu(II) (Figure S2).
Figure 3.
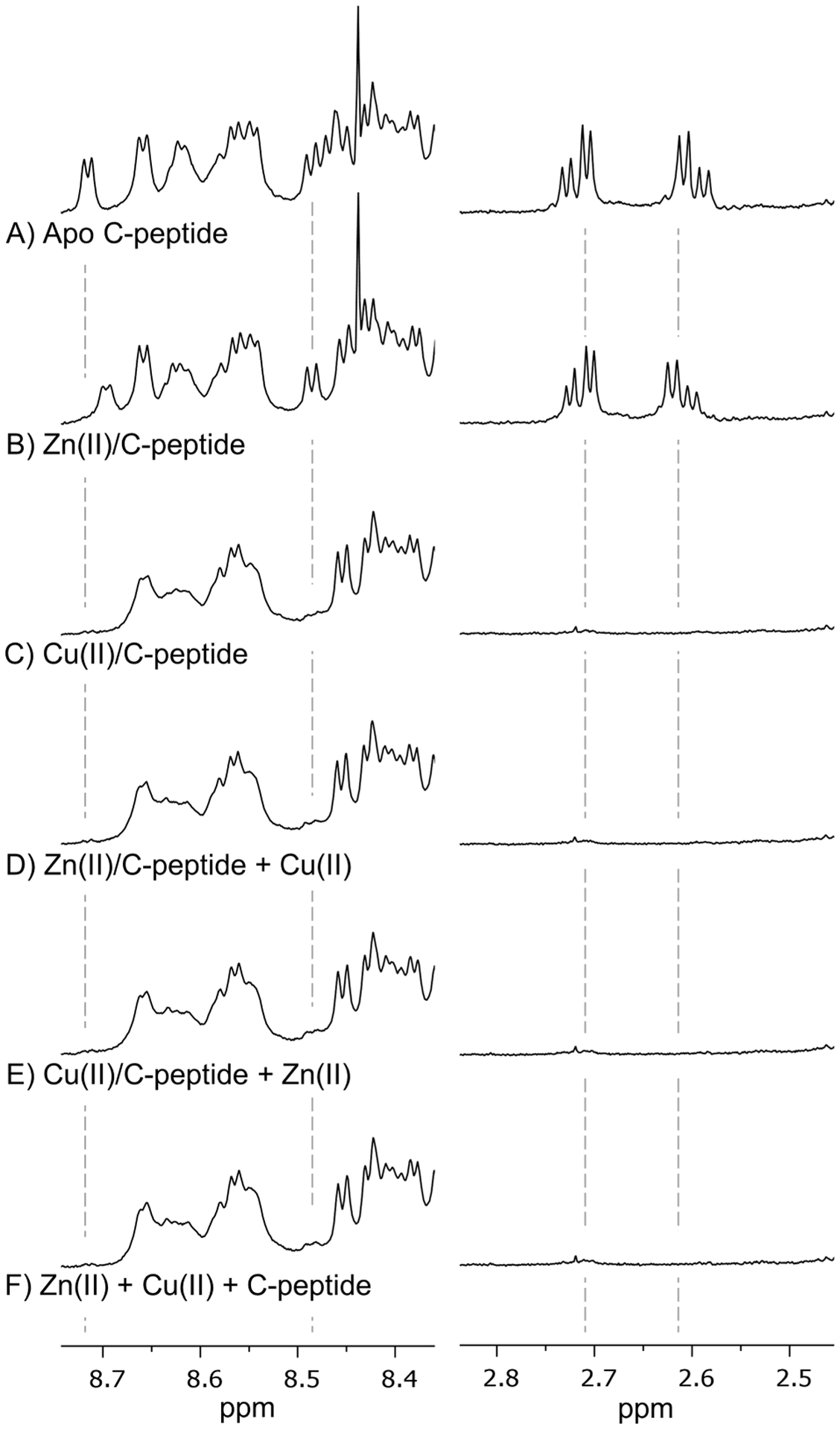
Highlighted regions of the 1H NMR spectra of (A) apo C-peptide, (B) Zn(II)/C-peptide, (C) Cu(II)/C-peptide, (D) equimolar Cu(II) added to Zn(II)/C-peptide, (E) equimolar Zn(II) added to Cu(II)/C-peptide, and (F) Cu(II) and Zn(II) added to apo C-peptide simultaneously. Full spectra are shown in Figure S1. The addition of Zn(II) induces shifts in proton resonances for CHβ protons on D4 (2.6 ppm) and backbone amide protons from D4 (8.45 ppm) and E3 (8.7 ppm), while Cu(II) obliterates proton resonances within these regions. The spectra of Cu(II) competition with Zn(II) (D–F) result in spectra that resemble Cu(II)/C-peptide and not Zn(II)/C-peptide, indicating that Cu(II) displaces Zn(II) for binding C-peptide. Solutions were prepared at 1.5 mM peptide in 95:5 (v/v) H2O/D2O with 10 mM Tris-d11 at pH 7.4, and spectra were collected at 800 MHz and 10 °C.
Using these resonances as markers, we probed the competition between Cu(II) and Zn(II) by 1H NMR. A stoichiometric amount of Cu(II) was added to the preformed Zn(II)/C-peptide complex or the reverse (Zn(II) added to the preformed Cu(II)/C-peptide complex) to determine if either metal could displace the other. In both scenarios, the resulting spectra are identical to that of Cu(II)/C-peptide with negligible resonances that correspond to Zn(II)/C-peptide and indicate that only the Cu(II)-bound form persists (Figure 3D,E and Figure S1D,E). Similarly, the simultaneous addition of stoichiometric equivalents of Cu(II) and Zn(II) to C-peptide results in a spectrum that is identical to the spectrum of Cu(II)/C-peptide (Figure 3F and Figure S1F). These NMR competition experiments show that Cu(II) can preferentially bind to and displace Zn(II) from C-peptide. Moreover, the lack of any observed bimetallic C-peptide complexes in the solutions containing both Cu(II) and Zn(II) suggest that both Cu(II) and Zn(II) bind to the same region of C-peptide, offering the possibility of the metal-mediated modulation of peptide activity.
Determining the Copper- and Zinc-Binding Regions.
To locate the metal binding region, the proton resonances of metal-bound and apo C-peptide were assigned through a combination of 2D NMR experiments (Tables S1–S5). 1H–1H TOCSY NMR allows for the classification of resonances into spin systems, which can then be correlated to specific amino acids (Figure S3). However, given that the 31-mer C-peptide sequence consists of only nine different amino acids (7 G, 6 L, 4 E, 4 Q, 3 A, 2 P, 2 S, 2 V, and 1 D), the sequence-specific assignment required the implementation of 1H–1H NOESY spectroscopy via backbone NHi–CHαi and CHαi–NHi+1 correlations (Figure S3). Having assigned the 1H chemical shifts, regional variations in the resonances were assessed by plotting the difference between the chemical shifts of metal-bound C-peptide and apo C-peptide, Δδ = δ(metal-bound C-peptide) − δ(apo C-peptide) (Figures S4 and S5, results summarized in Figure 4).
Figure 4.
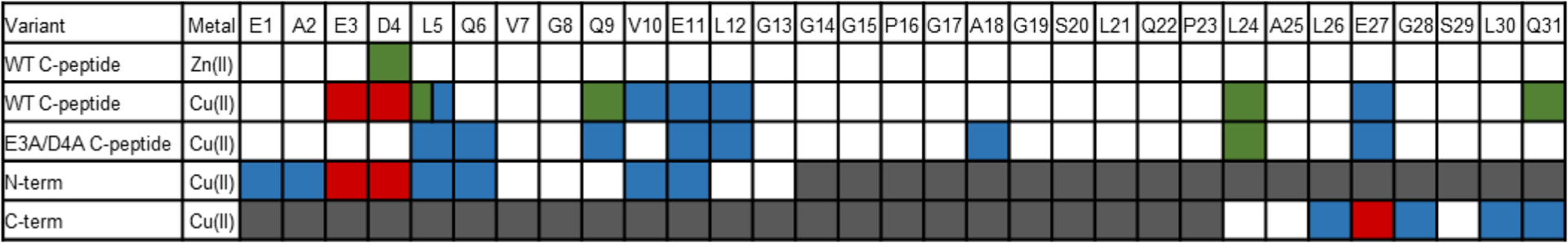
Visual depiction of the changes in proton resonances of C-peptide and its variants as a result of metal binding. Colored blocks indicate changes as follows: Δδ ≥ 0.03 (green), broadened signal (blue), obliterated signal (red), and truncated residues (gray). The corresponding spectra (Figures S3, S15, and S16) and Δδ tables (Tables S1–S5 and S7–S9) are provided in the Supporting Information. The changes in proton resonances indicate that the metals bind to C-peptide at E3 and D4; mutations (double mutant E3A/D4A) or truncations (N-term and C-term) lead to promiscuous metal binding.
In the Δδ plots for Zn(II)/C-peptide, the most notable changes are observed with the NH protons, specifically at residues E3, D4, L5, V10, and E11 (Figure S4A and Table S4), potentially indicating a structural change occurring in the N-terminus of C-peptide upon Zn(II) binding. No significant changes are observed in the chemical shifts of the side-chain protons except for the CHβ protons on D4, indicating this residue to be a possible binding locale (Figure S5A and Table S4). In contrast, the Δδ plots of Cu(II)/C-peptide show minimal changes in the NH and CHα resonances (Figure S4B and Table S5). However, consistent with the 1D 1H NMR spectrum, the 2D 1H–1H homonuclear NMR experiments confirm the loss of a portion of the side-chain resonances. As indicated in the Δδ plots and chemical shift assignments, all side-chain proton resonances of E3 and D4 were obliterated with several signal reductions and shifts in proton resonances (Figure 4, Figure S5B, and Table S5). The lack of signals suggest a binding locale of Cu(II) to the N-terminal region, specifically at E3 and D4, that imposes only modest, if any, structural changes to C-peptide. Furthermore, the CHδ protons of L5 and the side-chain proton resonances of V10, E11, L12, and E27 were reduced in signal intensity coupled with shifts in proton resonances of L5, Q9, L24, and Q31 (Figure 4 and Figure S5B) and indicate possible transient Cu(II) binding interactions or sufficient proximity to the Cu(II) center to experience altered proton relaxation rates. From the NMR spectra, the main Cu(II) and Zn(II) binding locales are at E3 and D4.
Characterizing the Coordination Environment of the Copper Center.
C-peptide does not contain classical Cu(II)-binding residue histidine. While the carboxylate side chains of the available aspartate and glutamate residues could serve as ligands, Cu(II) binding modes containing only oxygen donors are uncommon in native peptides and proteins. On the basis of electronic absorption spectroscopy studies, we previously hypothesized that Cu(II) could be binding to a combination of the backbone amide nitrogens and the carboxylate side chains for a mixed NxOy ligand set.32 To assess this hypothesis, we turned to electron paramagnetic resonance (EPR) spectroscopy, which probes the ligand environment around the paramagnetic Cu(II) (3d9, S = 1/2) center. For instance, if a nitrogen is directly bound to Cu(II), then the 14N-hyperfine coupling is strong enough (~45 MHz) to yield characteristic hyperfine splittings in the X-band EPR spectrum, although the absence of hyperfine splitting does not preclude direct nitrogen coordination as evidenced by previous studies performed with Cu(II)/amyloid-β complexes.36 The continuous wave (CW) X-band EPR spectrum of Cu(II)/C-peptide was acquired at 20 K in the presence of excess C-peptide and shows one axial Cu(II) component with g‖ = 2.229 and A‖ = 536 MHz, suggesting that all Cu(II) was bound by the peptide in a square-planar geometry. Using these parameters with the Peisach and Blumberg truth tables,37 the data indicates either 4O with −2 charge, 1N3O with nearly neutral charge, or 2N2O with nearly neutral charge for the coordination environment.
The possible binding modes may be further differentiated by determining whether any nitrogen hyperfine splittings are observed. Interestingly, no obvious 14N-hyperfine splitting at g⊥ (Figure 5A) suggests that the Cu(II) center is most likely in a 4O environment, although sometimes the 14N-hyperfine splitting pattern is difficult to resolve due to line-shape broadening.38 Furthermore, the high-resolution two-dimensional method consisting of hyperfine sublevel correlation spectroscopy (HYSCORE) detects only weak hyperfine coupling from remote backbone nitrogens (Figure 5B).39 If Cu(II) is coordinated to the side chains of E3 and D4, then the backbone amide nitrogens for each residue would be six and five bond lengths away, respectively, matching the experimental observations. However, it must be noted that nitrogen coordination is still possible because the lack of data does not preclude nitrogen coordination.36 Combined with the NMR data, the EPR data support a model for Cu(II) binding to C-peptide through the side-chain carboxylate oxygens of E3 and D4 in a square-planar geometry. To provide further insight into the coordinating ligands, we turned to vibrational spectroscopy.
Figure 5.
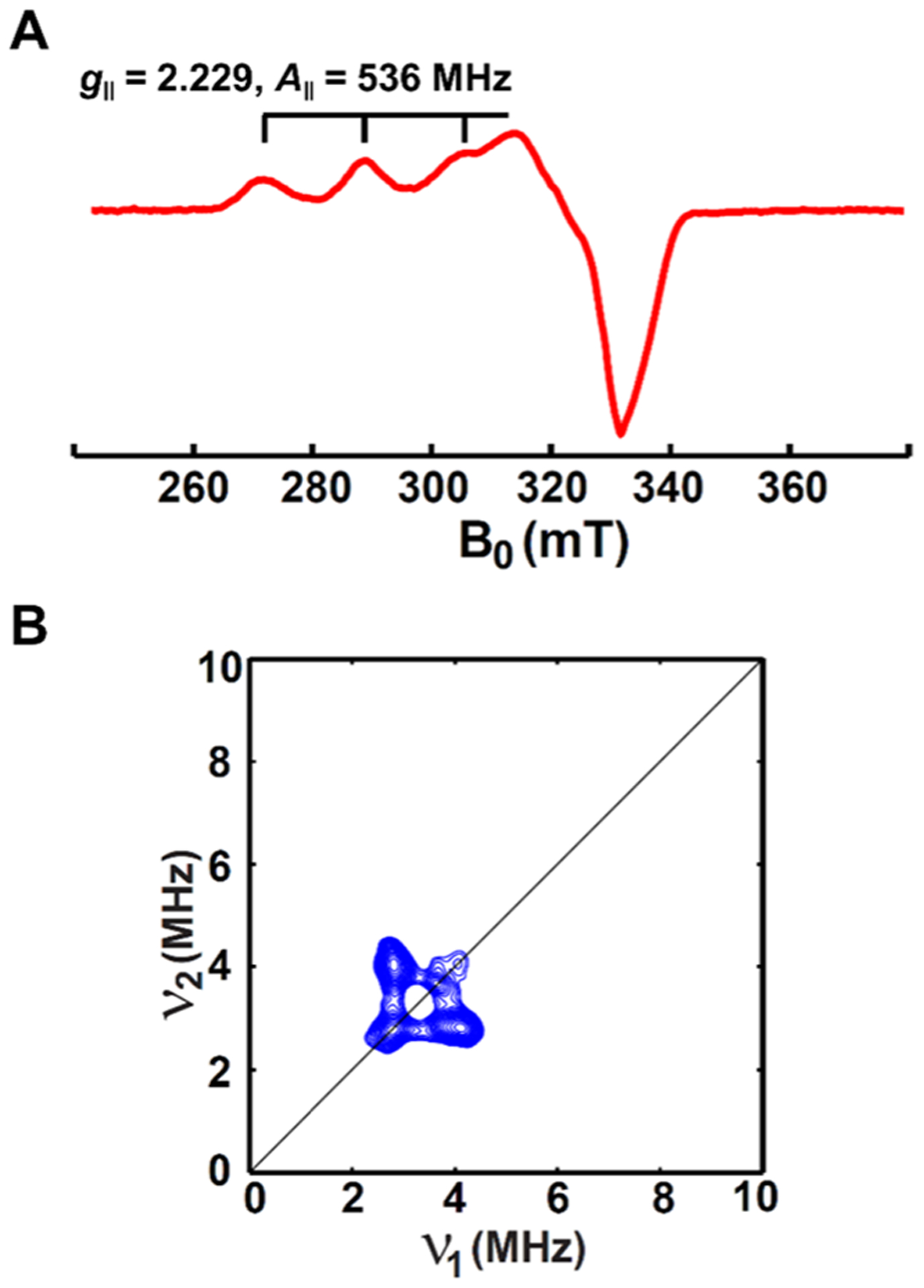
(A) CW X-band and (B) HYSCORE EPR spectra of Cu(II) bound to C-peptide obtained at 20 K. Samples were prepared with 0.5 mM Cu(II) and 1.0 mM C-peptide in 15 mM MOPS, pH 7.4, with 20% ethylene glycol as a glassing agent. The CW X-band and HYSCORE spectra indicate that Cu(II) is coordinated to two to four oxygens of C-peptide in a square-planar geometry but does not preclude coordination to nitrogen.
To identify the chemical nature of the oxygen-containing ligands, we implemented Fourier-transform infrared (FTIR) spectroscopy. In peptides and proteins, the position of the amide bands provides structural information. When bound to metals, the asymmetric (as) and symmetric (s) stretching frequencies of oxygen-containing ligands provide insight into how the metal ion is coordinated: larger Δ(νas − νs) (~200–300 cm−1) typically corresponds to unidentate metal coordination whereas smaller Δ(νas − νs) of less than 150 cm−1 corresponds to bidentate coordination.40–43 The FTIR spectrum of apo C-peptide shows amide I′ and amide II′ bands centered at 1645 and 1539 cm−1, respectively (Figure 6, Table 1). The position of the amide I′ band is indicative of a random coil peptide and in good agreement with previously published CD spectra.44 Also seen in apo C-peptide is the presence of a νs COO− band at 1401 cm−1. However, since the νas COO− band is buried under the amide I′ and amide II′ bands and cannot be observed, a difference spectrum of Cu(II)/C-peptide minus apo C-peptide was calculated to locate the asymmetric stretching band. Both spectra were normalized to δCH at 1451 cm−1.42 From the difference spectrum, the νas COO− bands are readily seen at 1597 and 1542 cm−1 along with the νs COO− bands at 1428 and 1379 cm−1, causing Δ(νas − νs) to range from 218 to 114 cm−1. From these Δ(νas − νs) values, the Cu(II) coordination may be a combination of unidentate and bidentate to the carboxylate side chains.
Figure 6.
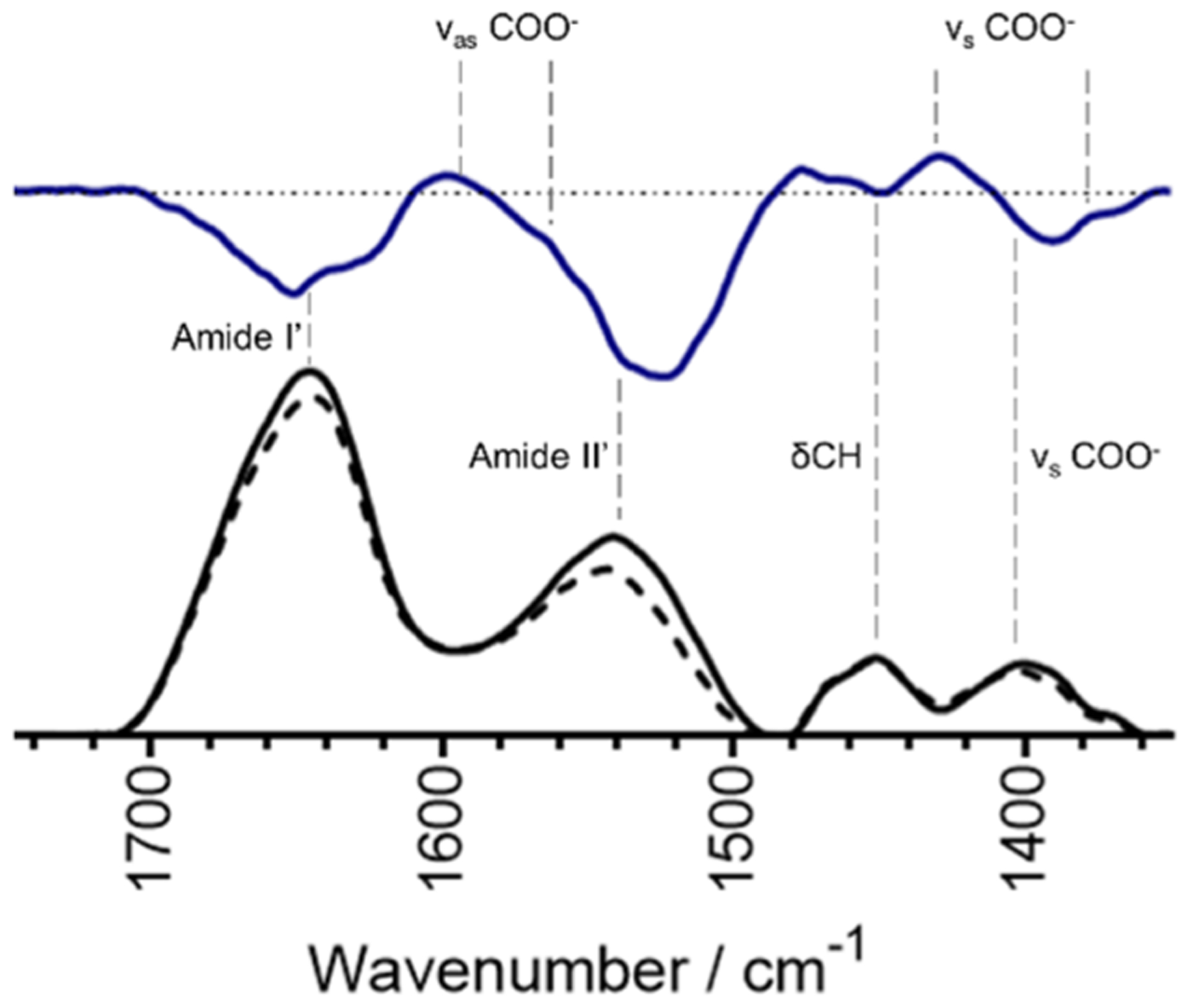
FTIR absorbance spectra of 1.2 mM apo C-peptide (solid black line) and 1.2 mM Cu(II)/peptide (dashed black line) at pH 7.4, with the difference spectrum (solid blue line). The dashed horizontal line for the difference spectra is set to zero. Vertical dashed lines indicate the bands associated with the given stretch or vibration. Spectra are buffer subtracted and normalized to the internal standard of the δCH centered at 1451 cm−1. Frequencies for antisymmetric and symmetric bending and stretching of carboxylate groups are visible and indicate a combination of bidentate and unidentate coordination to Cu(II).
Table 1.
Frequencies of Key Vibrations (cm−1)
| amide I′ | amide II′ | δCH | νas COO− | νs COO− | |
|---|---|---|---|---|---|
| apo | 1645 | 1539 | 1451 | n.d. | 1401 |
| +Cu(II) | 1645 | 1539 | 1451 | 1597/1542 | 1428/1374 |
The second derivative of FTIR spectra was employed to further observe subtle changes in the bands and features of the peptide (Figure S6). Of note in this transformation are the stretching frequencies below 1300 cm−1, indicative of C–H and Ser(OH) stretching and bending.45 In this region, the second derivatives of apo and Cu(II)-bound C-peptide are nearly identical, indicating that Cu(II) is not influencing the stretching and bending of CH and Ser(OH) bonds. The FTIR data thus further confirms that Cu(II) is binding to carboxylate ligands, likely the side chains of E3 and D4, in a combined unidentate and bidentate mode which would provide three of the four oxygens needed for axial coordination to Cu(II). Confirmation of the fourth ligand as the terminal amine proved challenging by FTIR due to its bending and stretching frequencies overshadowed by solvent and amide bands.45
To further validate that the identity of the fourth ligand is nitrogen, we turned to electronic absorption spectroscopy which directly probes the Cu(II) center. The addition of 1 equiv of Cu(II) to C-peptide in 15 mM MOPS at pH 7.4 was similar to the previously published spectrum where a large band centered at 220 nm is attributed to the peptide backbone and a small absorption band centered at 638 nm is attributed to the d–d transition on the metal center of Cu(II)/C-peptide (Figure 7).32 The high concentration of C-peptide (300 μM) was required to observe the weak d–d band, but at this concentration, no precipitation or aggregation was observed. The λmax of the d–d band at 638 nm is consistent with an amine coordinated to Cu(II).46 The only amine in C-peptide is the N-terminal amine and is a common ligand for anchoring Cu(II) to peptides and proteins. The data from NMR, FTIR, EPR, and electron absorption spectroscopies can now be interpreted together. NMR shows that Cu(II) binds C-peptide through E3 and D4, with FTIR indicating bidentate and unidentate coordination and electronic absorption indicating the presence of a coordinated amine. With this ligand set, the charge at the metal center would be zero and the EPR data is now interpreted to have 1N3O square-planar coordination. This ligand set would have a low affinity, and Cu(II) would be relatively labile, thus providing leverage for other ligands to chelate Cu(II) away from C-peptide and making the Cu(II)/C-peptide complex kinetically favored. Considering our earlier finding that Cu(II) inhibits C-peptide internalization, the chelation of Cu(II) from C-peptide could serve as a means to restore the C-peptide internalization activity.
Figure 7.
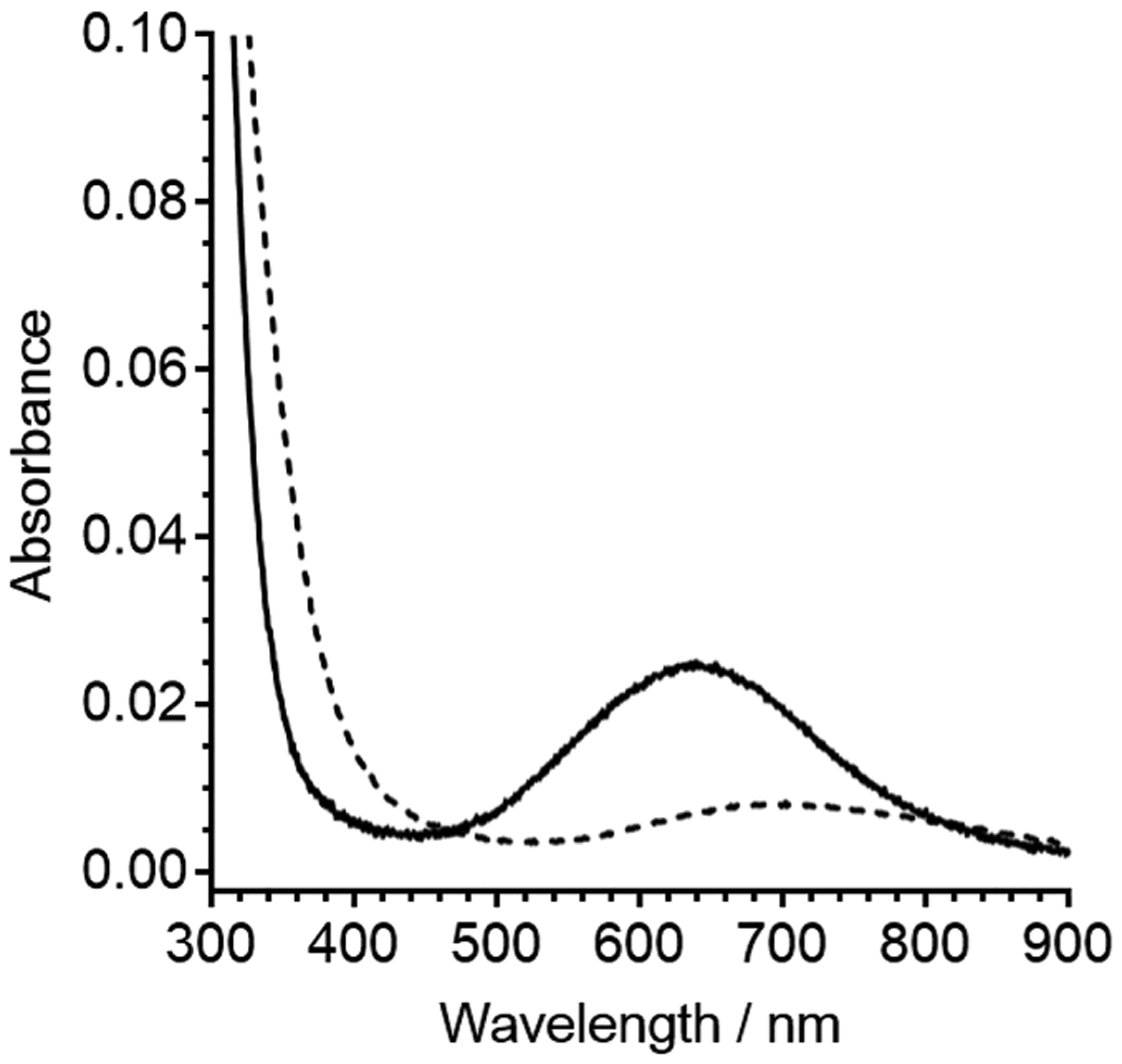
Spectra depicting the d–d band from 300 μM Cu(II)/C-peptide (solid line) and 300 μM Cu(II) in 15 mM MOPS at pH 7.4 shown as a reference (dashed line). The wavelengths of maximum absorption for the d–d band as determined by the first derivative of Cu(II)/C-peptide and Cu(II) in buffer are 638 and 696 nm, respectively (Figure S10). The energy of the d–d band for Cu(II)/C-peptide suggests that Cu(II) is bound to at least one nitrogen ligand.
Binding Thermodynamics of the Copper/C-peptide Interaction.
Through the structural studies, we show that C-peptide binds Cu(II) with a 1N3O ligand set. To determine the driving force of complexation, we turned to isothermal titration calorimetry (ITC) to measure the binding thermodynamics, namely, the enthalpy, binding affinity, and stoichiometry. From these parameters, the free energy and entropy of Cu(II) binding to C-peptide can be determined. Dissecting the binding free energy into enthalpic and entropic contributions provides valuable information regarding the driving force of the reaction. In our previous work, we calculated an apparent binding constant of Cu(II) to C-peptide in the 15–40 nM range via competition with the chromophoric ligand, phenanthroline.32 Using this range as a starting point, Cu(II) was titrated into C-peptide in MOPS and MOPSO buffers, which were chosen for their small KCu(II)-buffer (Figure 8 and Figure S7),47 in triplicate, and the experimental fit parameters are listed in Table S6. (There is a dearth of buffers with small KCu(II)-buffer that will allow for the study of low-affinity peptides and proteins.) Additionally, since the two buffers have different protonation enthalpies,48 the number of protons that dissociate from C-peptide upon Cu(II) binding can be estimated from the slope of the protonation plot of ΔHITC + ΔHCu(II)-buffer vs ΔHbuffer-H (Figure S8; see Scheme S1 for the determination of ΔHCu(II)-MOPSO).49 The resulting thermograms are fit to the one-site binding model to provide the experimental binding thermodynamics summarized in Table S6. Moreover, these experiments revealed that approximately zero (0 ± 1) protons dissociate from C-peptide upon Cu(II) binding (Figure S8). This value, although it has a large error, is consistent with carboxylate ligands from the side chains of E3 and D4 and the partial protonation (~0.8 H+) of the terminal amine at pH 7.4. Taking into account the displacement of a negligible number of protons from C-peptide upon Cu(II) binding, the buffer-independent thermodynamics at pH 7.4 were calculated to be KCu(II)/C-peptide = 1 (±1) × 108 (Kd,Cu(II)/C-peptide = 10 nM), ΔG°Cu(II)/C-peptide = −11 ± 1 kcal mol−1, ΔHCu(II)/C-peptide = −2 ± 1 kcal mol−1, and ΔSCu(II)/C-peptide = 29 ± 3 cal mol−1 K−1 and are summarized in Table 2.
Figure 8.

Representative thermogram of 1.0 mM Cu(II) titrated into 100 μM C-peptide in 15 mM MOPS at pH 7.4 with a red fit line: n = 0.769 ± 0.008, KITC = 1.1 (±0.2) × 106, and ΔHITC = −1.27 ± 0.02 kcal mol−1. Buffer-independent thermodynamics are summarized in Table 2 and indicate a predominantly entropic driving force.
Table 2.
Buffer-Independent Thermodynamics of Cu(II) Binding to C-peptide at pH 7.4
| buffer | KCu(II)/C-peptide | ΔG°Cu(II)/C-peptide (kcal mol−1) | ΔHCu(II)/C-peptide (kcal mol−1) | −TΔSCu(II)/C-peptide (kcal mol−1) | ΔSCu(II)/C-peptide (cal mol−1 K−1) |
|---|---|---|---|---|---|
| MOPS | 2 (± 1) × 108 | −11.2 ± 0.4 | −2 ± 1 | −9 ± 1 | 30 ± 3 |
| MOPSO | 6 (± 3) × 107 | −10.5 ± 0.3 | −2 ± 1 | −9 ± 1 | 28 ± 3 |
The formation constant determined here for C-peptide is consistent with what we previously reported.32 The use of ITC allows for the decomposition of the free energy into its enthalpic and entropic contributions, and from this inspection, we see that Cu(II) binding is predominantly entropically driven. This favorable entropy may be the result of the desolvation of both C-peptide at the metal-binding site and the Cu(II) ion (the latter of which can have up to 10 water molecules surrounding it),50 as well as the chelate effect. Given the NMR data and our previously reported CD spectra,32 there is negligible structural rearrangement when Cu(II) binds, indicating a minimal entropic penalty from the peptide structure. The small enthalpic contribution may be from Cu(II) binding to carboxylate ligands (ΔHCu(II)−COO ≈ 1.2 kcal mol−1) coupled with binding to the amine and small, favorable intrapeptide interactions.48,51
The parameters elucidated in our studies may point to the Cu(II)/C-peptide complex being a relevant species in the body. First, the local environment surrounding tissues is believed to have fluctuating metal content. After transport through blood to target tissues, C-peptide may bind to Cu(II), thus inhibiting its internalization until either Cu(II) is chelated away by another higher-affinity ligand or displaced by another metal ion that does not inhibit internalization. This would be an example of a metal-mediate negative feedback mechanism. Second, there are numerous proteins within the blood, some of which have high Cu(II) affinities such as albumin. Work by Spence and colleagues have shown that albumin is needed for C-peptide transport to cells, but the characteristics of such a ternary complex remain elusive. Understanding the Cu(II)/C-peptide complex is the first step in further probing the ternary complex, which we are actively studying.
Previously, we showed via competition with the chromophoric ligand, Zincon, that Zn(II) binds to C-peptide with an apparent binding constant of 10–100 μM.32 However, titrations of Zn(II) into C-peptide by ITC showed no detectable binding in five buffered solutions: Tris, bisTris, PIPES, MOPS, and MOPSO (Figure S9). Of these buffers, the Zn(II) binding affinities to Tris and bisTris are reported to be log K = 2.27 and 2.38, respectively.48 The lack of an observed binding isotherm indicates a low Zn(II)/C-peptide affinity that cannot compete with the Zn(II)-buffer complexes under the ITC experimental conditions studied here and does not preclude Zn(II) from binding to C-peptide. The estimation of an upper limit of the Zn(II)/C-peptide affinity is log K ≈ 5 and is consistent with the low affinity of the Zn(II)/C-peptide complex that we previously reported.32
Metal Binding to C-peptide Variants and Truncations.
The combination of spectroscopic and calorimetric data points to a 1N3O Cu(II) binding site within the native, or wild-type (WT), C-peptide. In order to confirm the identity of the binding ligands as the side chains of E3 and D4, we synthesized three variants of C-peptide with these residues replaced with alanine: (1) a single-residue replacement at E3 (E3A), (2) a single-residue replacement at D4 (D4A), and (3) a double mutant with both E3 and D4 replaced (E3A/D4A). These C-peptide variants were then analyzed by electronic absorption spectroscopy and compared to WT C-peptide to screen for Cu(II) binding. As described above, Cu(II) bound to WT C-peptide featured a large band centered at 220 nm attributed to the peptide backbone and a small absorption band centered at 638 nm attributed to the d–d transition on the metal center of Cu(II)/C-peptide (Figure 9).32 Surprisingly, the addition of 1 equiv of Cu(II) to each C-peptide variant was similar to that of WT C-peptide (Figure 9) with λmax values for WT, E3A, D4A, and E3A/D4A C-peptide at 638, 637, 620, and 658 nm, respectively. The similar λmax values for each variant indicate similar environments and energy splitting at the Cu(II) site and can be inferred to have similar 1N3O coordinating atoms and metal geometries. While E3 and D4 are replaced with alanine in the mutants, the carboxylate groups of E1, E11, E27, and the C-terminus remain available, potentially serving as alternate ligands. This may illustrate that Cu(II) can promiscuously bind to several other locales on C-peptide. Of the variants tested, the largest Δλmax from WT C-peptide is from E3A/D4A C-peptide and was thus further investigated and compared to that of the native peptide.
Figure 9.
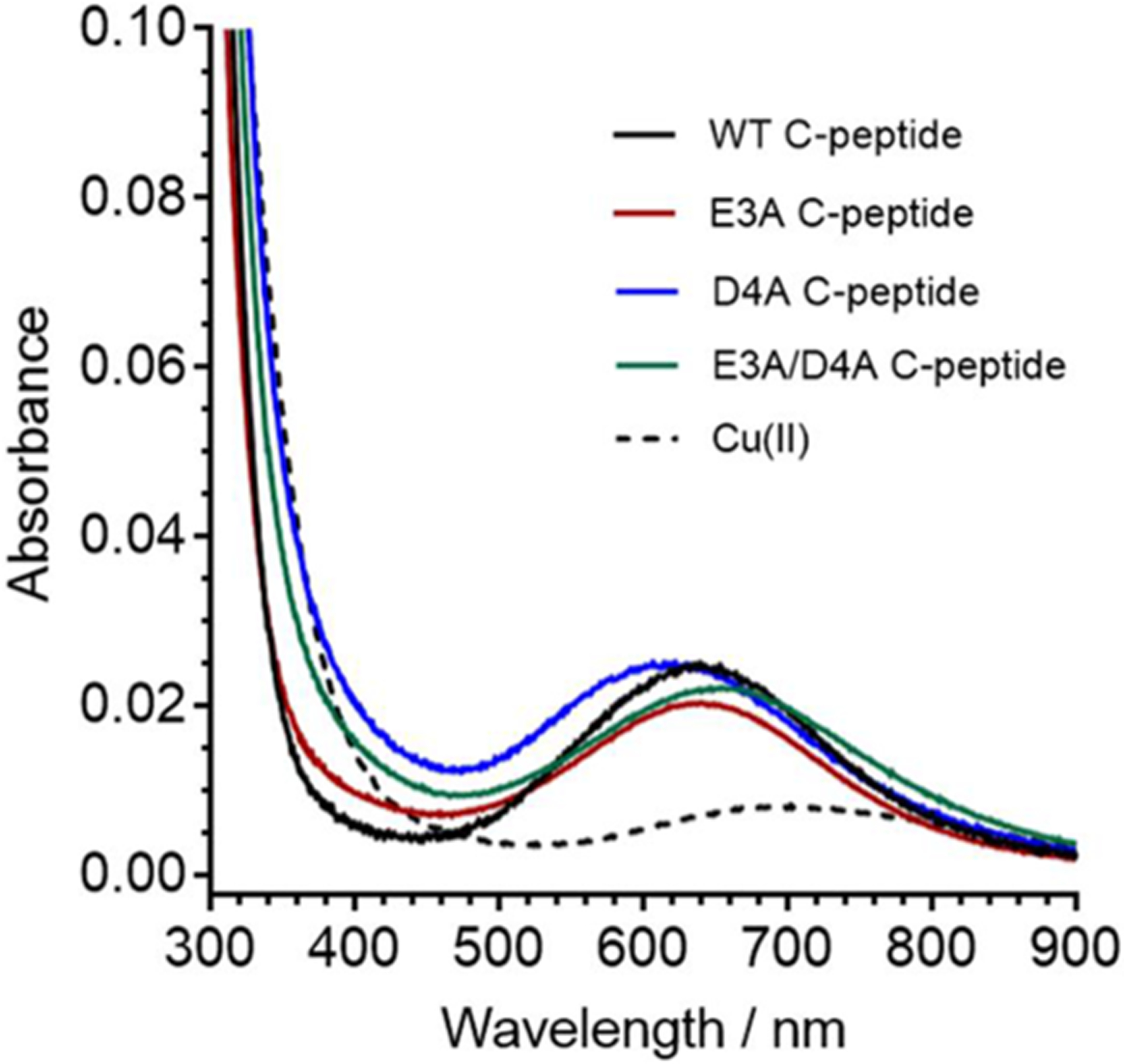
Spectra of C-peptide mutants depicting the d–d band from Cu(II)/peptide of 300 μM WT C-peptide (black), E3A C-peptide (red), D4A C-peptide (blue), E3A/D4A C-peptide (green), and 300 μM Cu(II) in 15 mM MOPS at pH 7.4 shown as reference (black dashed line). The wavelengths of maximum absorption for the d–d band as determined by the first derivative of WT, E3A, D4A, and E3A/D4A C-peptide and Cu in buffer are 638, 637, 620, 658, and 696 nm, respectively (Figure S10). The similar energy of the d–d band from Cu(II) suggests that the copper is bound to similar ligands in all peptides but that E3A/D4A C-peptide is red-shifted, most similar to buffer alone, and has the largest change in maximum absorption.
We next sought to determine the Cu(II) binding location on E3A/D4A C-peptide via 2D 1H–1H NOESY and TOCSY NMR spectroscopy, and we assessed the paramagnetic broadening of the proton resonances from Cu(II) interactions. Unlike WT C-peptide, no resonances were completely obliterated upon Cu(II) addition, but several residue side chains had reduced intensities (Figure 4, Figures S11 and S12, and Tables S7–S9). Since the resulting NMR spectra reflect the summation of all species in solution, this observation may suggest that Cu(II) is binding to different regions of C-peptide, resulting in different populations. Furthermore, the differences in the signal reduction pattern between E3A/D4A and WT C-peptide substantiate the claim that Cu(II) binds to E3 and D4 in WT C-peptide, and this binding gets reshuffled when those residues are replaced. Interestingly, in the E3A/D4A variant the proton resonances of the NH and CHα protons do not change substantially (Table S9) as compared to those of WT C-peptide (Table S5) and indicate little structural rearrangement of the peptide upon Cu(II) binding. Furthermore, ITC was performed to understand the thermodynamic differences between WT and E3A/D4A C-peptide (Figure S13). Interestingly, statistical analysis show that the thermodynamic data are similar (Table S10); however, the stoichiometry for E3A/D4A C-peptide is 0.5, indicating that one Cu(II) binds per two peptides. This substantiates a reshuffling of the Cu(II) binding site on E3A/D4A C-peptide different than that of WT C-peptide.
We next investigated the role of each segment of C-peptide to learn if a portion of the peptide would be suitable for Cu(II) binding or if the full-length peptide is needed.33 C-peptide is believed to be tripartite in function, where the N-terminal, flexible middle, and C-terminal portions differ in structure and bioactivity.33,52 Peptides were synthesized to contain either the N-terminal segment (N-term) comprising residues E1 to G13 or the C-terminal (C-term) segment comprising residues L24 to Q31. These two truncations leave out the glycine- and proline-rich middle segment, which does not contain carboxylate-bearing residues. We anticipated the N-term peptide to preserve the E3/D4 binding interaction as in WT C-peptide, whereas the C-term peptide would resemble the rearranged binding observed for the E3A/D4A C-peptide.
Upon Cu(II) addition to these truncated peptides, the electronic absorption spectra of N-term and C-term show distinct d–d bands at 623 and 658 nm, respectively, similar to the full-length peptides (Figure 10). The similarity in energies and intensities to WT C-peptide suggests that Cu(II) is bound to similar ligands with a similar geometry in both truncations. This is confirmed by the EPR experiments wherein the CW and HYSCORE spectra of the truncations closely resemble those of the WT C-peptide and have similar fitting parameters (Figures S14 and S15). This indicates that all three peptides coordinate Cu(II) in a 1N3O square-planar geometry. It is worth noting in the CW EPR spectrum of C-term that there is an extra set of features centered at ~290 mT that may point to another, albeit minor, population of Cu(II). Signal reduction analysis of the NMR resonances of N-term shows the obliteration of the proton resonance signals for E3 and D4 with broadened signal intensities from E1, A2, L5, Q6, V10, and E11 (Figure 4 and Figure S16). In the case of C-term, E27 proton resonances were obliterated and L26, G28, L30, and Q31 were reduced with Cu(II) present (Figure 4 and Figure S17). While the Cu(II) coordination sphere may thus be similar between the WT peptide and the truncated peptides, the NMR data may indicate that Cu(II) is binding to or interacting with more residues or that there are multiple populations of Cu(II)-containing adducts present in the truncated peptides as compared to in WT C-peptide. The resonance broadening in the C-term truncation also suggests that while Cu(II) may bind to the C-terminal portion of the peptide in a truncated form, it is not the predominant species in WT C-peptide. Interestingly, Cu(II) binding to the truncations seen by ITC is nearly identical to that of WT C-peptide and indicates similar binding thermodynamics (Figure S18 and Table S10).
Figure 10.
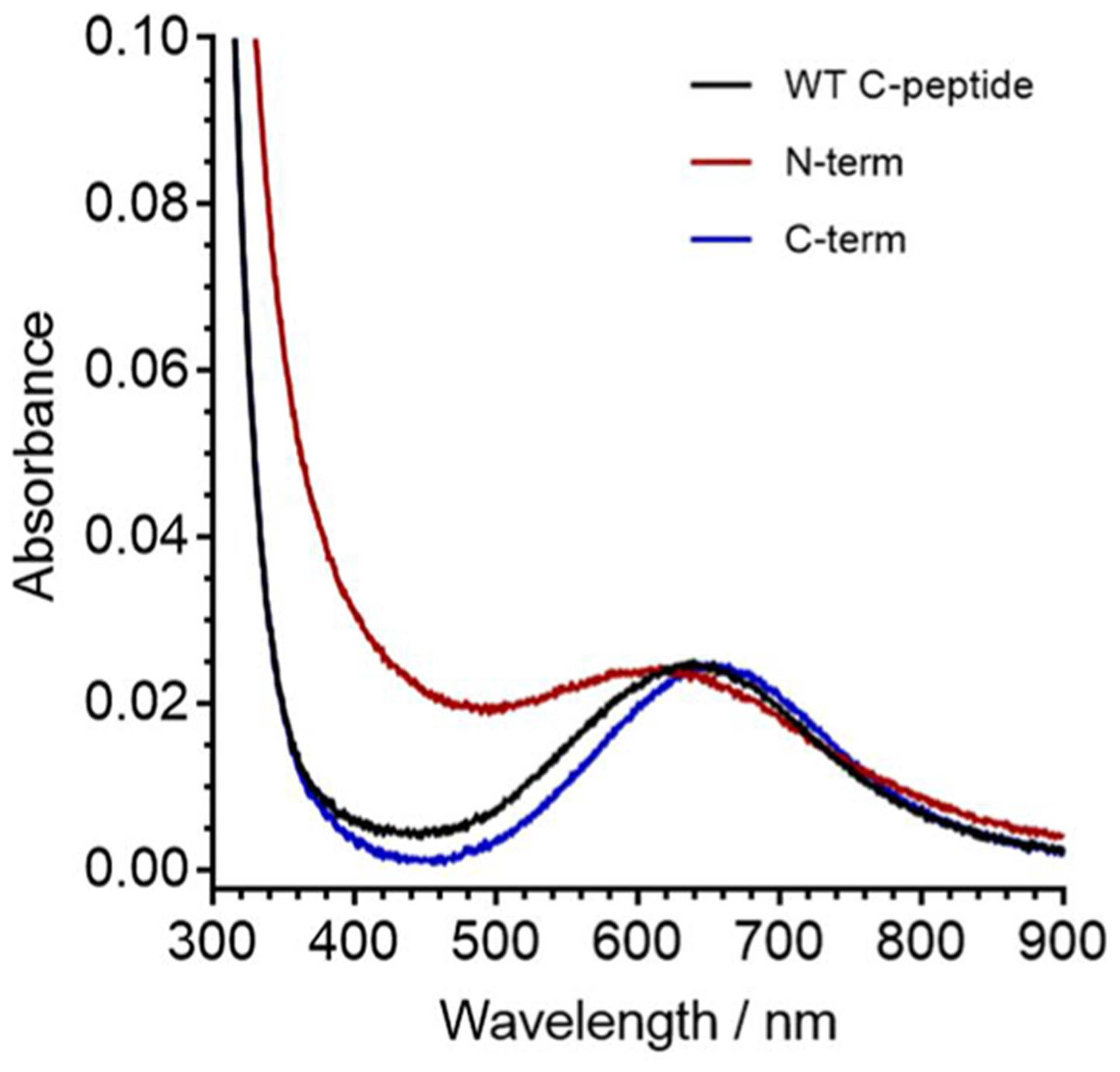
Spectra of C-peptide mutants depicting the d–d band from Cu(II)/peptide of 300 μM WT C-peptide (black), N-term (red), and C-term (blue) in 15 mM MOPS at pH 7.4. The wavelengths of maximum absorption for the d–d band of WT, N-term, and C-term C-peptide are 640, 623, and 658 nm, respectively. The similar energy of the d–d band from Cu(II) suggests that the copper is bound to similar ligands.
The differences in binding modes between the truncations and WT C-peptide may indicate that despite the full-length peptide being a random coil, all three segments are required for precise Cu(II) binding to the N-terminal region of the peptide. Cu(II) coordination to the terminal amine may serve as an anchor, as seen in other peptides, directing Cu(II) binding to the N-terminus in the WT C-peptide. Then, given the promiscuous binding to the N-term peptide, the middle glycine- and proline-rich segments of the full-length peptide may perhaps orient the two termini in such a way that occludes Cu(II) binding promiscuously to the other acidic residues, thus facilitating precise Cu(II) binding to E3 and D4 residues.
CONCLUSIONS
Previous work has shown that WT C-peptide interacts with a variety of transition metals, but it was Cu(II) that showed a decrease in internalization into HEK293 cells. In this study, we sought to determine the binding locale of Cu(II) to WT C-peptide along with the binding thermodynamics. Cu(II) binds at the terminal amine and carboxylates of E3 and D4 of WT C-peptide in a 1N3O square-planar geometry and is able to outcompete Zn(II) for binding. Cu(II) has a higher affinity for C-peptide and is entropically driven, most likely from desolvation of the peptide and metal ion coupled with the chelate effect. However, given the relatively low affinity for Cu(II), Cu(II)/C-peptide may be a kinetic product existing transiently in the body. Interestingly, the precision of Cu(II) binding to WT C-peptide is seen through the truncation experiments; both truncations bind Cu(II) differently, even given that one, N-term, contains the Cu(II) binding ligands in WT C-peptide. Even though WT C-peptide is a random coil and Cu(II) has no noticeable effect on secondary structure, the discrepancy in Cu(II) binding between the WT and truncated peptides may point to the importance of the middle segment of C-peptide in conformational behavior. The glycine- and proline-rich middle segment may orient the two termini, providing steric hindrance to inhibit Cu(II) binding to the C-terminus and facilitating only one species on the N-terminus, without which there is promiscuity in the Cu(II) binding. This study provides insight into the role that Cu(II) plays in C-peptide regulation and shows that the fold of C-peptide is important along with its metal cofactor. The metal-mediated negative feedback mechanism of random coil peptide hormones presents a different paradigm for hormone regulation.
Because the exact function and mechanism of action of C-peptide remain open questions, the relevance of the presence of the metal that we and others have observed may be a key missing piece that would help unravel such biological pathways. Elucidating the binding site and the consequences of mutating it offers powerful chemical information with immediate utility for future biological investigations. While this work has expanded our understanding of this particular peptide, we posit that several other bioactive peptides may indeed require metal cofactors for the function beyond the classical modes. Expanding our understanding of how metals may influence such activity will facilitate the discovery and validation of both new and established paradigms in peptide biology.
MATERIALS AND METHODS
Reagents.
All chemicals, reagents, and kits were purchased from Fisher Scientific or Millipore Sigma, unless otherwise noted. Specifically, dimethylformamide (DMF), 4-methylpiperidine, N,N-diisopropylethylamine (DIEA), N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU), dichloromethane (DCM), 2,2,2-trifluoroacetic acid (TFA), diethyl ether, formic acid (FA), methanol, dimethylsulfoxide (DMSO), CuCl2, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 2-amino-2-(hydroxymethyl)-1,3-propanediol (Tris), 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol (bisTris), HCl, NaOH, and ethylenediaminetetraacetic acid (EDTA) were purchased from ThermoFisher. ZnCl2, Tris(hydroxymethyl-d3)amino-d2-methane (deuterated Tris, Tris-d11), 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt (DSS), 3-(N-morpholino)propanesulfonic acid (MOPS), 3-morpholino-2-hydroxypropanesulfonic acid (MOPS), Chelex 100 sodium form, and 1,4-piperazinediethanesulfonic acid (PIPES) were purchased from Millipore Sigma. Wang resin preloaded with Fmoc-Gln(Trt)-OH and Fmoc-Gly-OH and Fmoc-protected amino acids were manufactured by ChemImpex but were purchased from ThermoFisher. Buffered solutions and metal salt solutions were made using Direct-Q3 deionized water (>18 MΩ, Millipore).
Peptide Synthesis.
Wild-type human C-peptide, E3A, D4A, E3A/D4A C-peptide, and N-terminus (N-term, E1-G13) and C-terminus (C-term, L24-Q31) truncations were synthesized by means of a heated Fmoc-based solid-phase synthesis (SPPS). Peptides were synthesized on the 0.2 mmol scale. For the N-term truncation, Wang resin preloaded with Fmoc-Gly-OH was swelled overnight in five times the resin volume of DMF. For wild-type, E3A, D4A, E3A/D4A, and the C-terminus truncation, Wang resin preloaded with Fmoc-Gln-(Trt)-OH was swelled overnight in 5 times the resin volume of DMF. The following procedures were repeated in the synthesis of the target peptides. After swelling overnight, the resin was washed 10 times with DMF. Each residue was then deprotected with 3 times the resin volume of 25% 4-methylpiperidine in DMF. The first rinse was shaken by hand for 1 min, and the second rinse was placed on an orbital shaker for an additional 10 min. The resin was then washed 10 times with 2 times the resin volume of DMF. Amino acids (4 equiv) and HBTU (3.9 equiv) were both dissolved in minimal amounts of DMF, and 10 equiv of DIEA was added. The amino acid, HBTU, and DIEA solution was then added to the resin and incubated at 95 °C for 5 min, shaken, and incubated again at 95 °C for an additional 5 min. The solution was cooled for 2 min on an orbital shaker and then washed 10 times with 2 times the resin volume of DMF. The procedure was repeated starting from the Fmoc deprotection step for each additional amino acid. Following the final amino acid coupling, the resin was washed 10 times with DMF followed by an additional 10 times with DCM, after which the resin was dried out overnight. Cleavage of both the peptide and protecting groups from the resin consisted of dissolving the dried resin in a minimal amount of 95:5 TFA/water solution and shaking for 1–4 h. This solution was poured drop by drop into 40 mL of chilled diethyl ether to form a precipitate, followed by centrifugation at 3900 rpm for 10 min. The supernatant was decanted, and 40 mL of chilled diethyl was poured over the crude peptide, followed by centrifugation as described above. This process was repeated two additional times, and the resulting crude peptide was dried with a slow stream of air overnight.
Mass Spectrometry.
C-peptide solutions were prepared at 50 μM for each sample, with CuSO4 and ZnClO4 added at 1 equiv in nanopure water. For Zn(II) and Cu(II) competition experiments, an equimolar amount of ZnClO4 was added to a 50 μM C-peptide solution and divided into aliquots. Immediately, to 1 aliquot an equimolar amount of CuSO4 was added, which was then added to the Zn(II)/C-peptide solution. A Thermofisher LTQ XL Orbitrap was used for all experiments and operated in positive ion mode at a resolution set at 60 000 for MS scans. Source conditions were as follows: flow rate of 5 μL/min, spray voltage of 3.5 kV, capillary temperature of 275 °C, capillary voltage of 44 V, and tube lens offset voltage of 185 V. Acquired spectra were averaged over 50 scans before peak assignments.
NMR.
NMR experiments were performed on a Bruker Avance III 800 MHz spectrometer equipped with a CPTCI cryoprobe. Peptide samples were prepared at a final concentration of 1.5 mM and dissolved in 95:5 (v/v) H2O/D2O solution buffered at pH 7.4 with 10 mM deuterated Tris-d11. For metal-peptide spectra, the samples were prepared differently in order to minimize metal coordination by Tris buffer. Peptide samples were first dissolved in Milli-Q water and the pH was adjusted to 7.4, followed by the addition of a 1 mol equiv of metal chloride salt and then 10 mM Tris-d11 at pH 7.4. All NMR spectra were collected at 10 °C in 5 mm NMR tubes. The chemical shifts were measured downfield compared to the internal standard (sodium trimethylsilylpropanesulfonate (DSS)). Solvent suppression for 1D, TOCSY, and NOESY spectra was achieved by using excitation sculpting using 180 water-selective pulses with gradients. One-dimensional spectra were collected using 16K data points with 64 scans. Gradient-based phase-selective 1H–1H 2D spectra were collected with mixing times of 20 or 80 ms for TOCSY experiments and 100 or 200 ms for NOESY experiments. Two-dimensional spectra were collected using 4096 data points in t2 for 512 t1 values and averaged over 16 scans. NMR spectra were processed with MestreNova.
Combinations of 1D, TOCSY, and NOESY spectra were used to assign the proton resonances of peptides with and without the treatment of metals. 1H–1H TOCSY spectra were first used to assign the spin systems of individual amino acids. Subsequently, the sequence-specific assignments were determined by backbone NHi−CHai and CHai−NH + 1 correlations from the 1H–1H NOESY spectra.
EPR.
The X-band spectra presented in this work are pseudomodulated spectra using a modulation amplitude of 3.0 mT. X-band two-pulse electron spin–echo (ESE)-detected field-swept EPR spectra (π/2-τ-π-τ-echo) were collected at 20 K on the Bruker Biospin EleXsys 580 spectrometer by employing a split-ring (MS5) resonator. Experiment parameters were as follows: π/2 = 12 ns and τ = 300 ns. EPR samples were prepared by dissolving peptides of known mass in Milli-Q water and adjusting the pH to 7.4 so that the final concentration was 1.0 mM peptide. CuCl2 was subsequently added to the peptide solution for a final concentration of 0.5 mM, and MOPS at pH 7.4 was added for a final concentration of 15 mM. To this solution, 20% ethylene glycol was added to act as a glassing agent.
Hyperfine sublevel correlation spectroscopy (HYSCORE) is a two-dimensional pulse EPR technique which correlates nuclear spin-flip transition frequencies in a one-electron-spin manifold to those in another electron-spin manifold. HYSCORE spectra were recorded at 20 K on the Bruker Biospin EleXsys 580 spectrometer by employing a split-ring (MS5) resonator for the X-band. Pulse sequence π/2-τ-π/2-t1-π-t2-π/2-τ-echo was programmed with the PulseSPEL programmer via the XEPR interface. The pulse length for the inversion pulse (tπ) and the π/2 pulse (tπ/2) was 24 ns. Eight-step phase cycling was used. Time-domain spectra were baseline-corrected (third-order polynomial), apodized with a hamming window, zero-filled to 8-fold points, and fast Fourier transformed to yield the frequency-domain spectra. Particular spectrometer settings are given in the corresponding figure captions.
ITC.
Samples were prepared immediately prior to use. Peptides of known masses were dissolved in buffer that was treated with Chelex 100 and subsequently filtered. Metal chloride salts were dissolved in water and diluted to working concentrations in buffer. The buffer and pH of both samples were carefully matched prior to each experiment.
All ITC measurements were carried out in triplicate on a MicroCal VP-ITC. Titrations were conducted at 25.0 ± 0.1 °C with 25–30 injections of 10 μL of titrant and stirring at 307 rpm. For each figure containing ITC data, the raw, baseline-corrected ITC data is presented in the top panel as heat flow versus time. The integrated and concentration-normalized heat for each injection is presented in the bottom panel after subtraction of the heat of dilution from the final injections. The data in the bottom panel are fit to one site using a nonlinear least-squares analysis. The thermodynamics reported are the average of at least three experiments, and the error is the standard deviation. A post hoc analysis to determine the buffer-independent thermodynamics follows Grossoehme et al.49
FTIR.
Peptides of known masses were dissolved in 15 mM MOPS in D2O with corrected pH 7.4. CuCl2 was dissolved in Milli-Q water. Apo samples were prepared by diluting metal-free peptide to 1.2 mM, while Cu(II)/peptide samples were prepared by diluting peptide to 1.2 mM and adding CuCl2 to a final concentration of 1.2 mM. Two microliters of sample was deposited onto the diamond crystal and dried under a stream of argon. Spectra were collected on a Bruker Alpha FTIR spectrophotometer from 4000 to 600 cm−1 at a 2 cm−1 resolution and averaged for 128 scans. Spectra were subsequently processed with OPUS software to remove atmospheric humidity and CO2, baseline-adjusted, and smoothed at 21 points. The spectra from MOPS buffer were then subtracted from all spectra and intensities at the normalized CH vibration (1451 cm−1).
UV–Vis.
All measurements were recorded on a UV-1900 (Shimadzu) at room temperature using quartz cuvettes (Starna Cells) with a path length of 1 cm. For metal titrations into apo C-peptide and both its respective N- and C-terminus truncations, each peptide was dissolved in 15 mM MOPS at pH 7.4 to a final concentration of 100 μM. The CuCl2 solution was dissolved in Milli-Q water, and 50–500 μM CuCl2 was added to each peptide. After equilibration for 3 min at room temperature, each measurement was recorded, with water being the spectral reference. After data collection, the spectrum of MOPS buffer alone was subtracted, and the data were normalized in order to account for dilution.
Supplementary Material
ACKNOWLEDGMENTS
We thank Prof. Andrew J. Fisher for use of the isothermal titration calorimeter and Prof. Alan Balch for use of the FTIR. We also thank Danielle J. Asiain and Adam R. Hillaire for their assistance with sample preparation and all members of the Heffern laboratory for valuable discussions. We thank Richard Saylor of the Britt laboratory for help with EPR collection and the UC Davis Campus Mass Spectrometry Facility.
Funding
This work was supported by the NIH MIRA 1R35GM133684-01, the Hartwell Foundation’s Individual Biomedical Research Award, the U.C. Davis Campus Research Core Facilities (CRCF) Award (ID no. 181032), and NSF DBI-0722538 for use of the Bruker Avance III 800. M.C.H. is also thankful for the support of the ADVANCE CAMPOS Faculty Scholar Award.
ABBREVIATIONS
- MS
mass spectrometry
- NMR
nuclear magnetic resonance
- EPR
electron paramagnetic resonance
- ITC
isothermal titration calorimetry
- FTIR
Fourier-transform infrared spectroscopy
- N-term
N-terminal truncation of C-peptide
- C-term
C-terminal truncation of C-peptide
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.0c01212.
Additional figures, tables, and a scheme (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.inorgchem.0c01212
The authors declare no competing financial interest.
REFERENCES
- (1).Hamley IW Small Bioactive Peptides for Biomaterials Design and Therapeutics. Chem. Rev 2017, 117, 14015–14041. [DOI] [PubMed] [Google Scholar]
- (2).Zozulia O; Dolan MA; Korendovych IV Catalytic Peptide Assemblies. Chem. Soc. Rev 2018, 47 (10), 3621–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zasloff M Antimicrobial Peptides of Multicellular Organisms. Nature 2002, 415 (6870), 389–395. [DOI] [PubMed] [Google Scholar]
- (4).Jeżowska-Bojczuk M; Stokowa-Sołtys K Peptides Having Antimicrobial Activity and Their Complexes with Transition Metal Ions. Eur. J. Med. Chem 2018, 143, 997–1009. [DOI] [PubMed] [Google Scholar]
- (5).Stevenson MJ; Uyeda KS; Harder NHO; Heffern MC Metal-Dependent Hormone Function: The Emerging Interdisciplinary Field of Metalloendocrinology. Metallomics 2019, 11 (1), 85–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nyberg F; Hallberg M Growth Hormone and Cognitive Function. Nat. Rev. Endocrinol 2013, 9 (6), 357–365. [DOI] [PubMed] [Google Scholar]
- (7).Sangkhae V; Nemeth E Regulation of the Iron Homeostatic Hormone Hepcidin. Adv. Nutr 2017, 8 (1), 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Travaglia A; Pietropaolo A; La Mendola D; Nicoletti VG; Rizzarelli E The Inorganic Perspectives of Neurotrophins and Alzheimer’s Disease. J. Inorg. Biochem 2012, 111, 130–137. [DOI] [PubMed] [Google Scholar]
- (9).Tõugu V; Karafin A; Palumaa P Binding of Zinc(II) and Copper(II) to the Full-Length Alzheimer’s Amyloid-β Peptide. J. Neurochem 2008, 104 (5), 1249–1259. [DOI] [PubMed] [Google Scholar]
- (10).Sharp P Iron in Eukarya. Metallobiology; 2014; pp 282–302. [Google Scholar]
- (11).Viles JH Metal Ions and Amyloid Fiber Formation in Neurodegenerative Diseases. Copper, Zinc and Iron in Alzheimer’s, Parkinson’s and Prion Diseases. Coord. Chem. Rev 2012, 256 (19–20), 2271–2284. [Google Scholar]
- (12).Norouzi S; Adulcikas J; Sohal SS; Myers S Zinc Stimulates Glucose Oxidation and Glycemic Control by Modulating the Insulin Signaling Pathway in Human and Mouse Skeletal Muscle Cell Lines. PLoS One 2018, 13, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Meyer JA; Spence DM A Perspective on the Role of Metals in Diabetes: Past Findings and Possible Future Directions. Metallomics 2009, 1 (1), 32–41. [Google Scholar]
- (14).Davidson HW; Rhodes CJ; Hutton JC Intraorganellar Calcium and PH Control Proinsulin Cleavage in the Pancreatic Beta Cell via Two Distinct Site-Specific Endopeptidases. Nature 1988, 333 (6168), 93–96. [DOI] [PubMed] [Google Scholar]
- (15).Steiner DF On the Role of the Proinsulin C-Peptide. Diabetes 1978, 27 (Supplement_1), 145–148. [DOI] [PubMed] [Google Scholar]
- (16).Wilcox G Insulin and Insulin Resistance. Clin. Biochem. Rev 2005, 26 (2), 19–39. [PMC free article] [PubMed] [Google Scholar]
- (17).Izumi T; Kasai K; Gomi H Secretory Vesicle Docking to the Plasma Membrane: Molecular Mechanism and Functional Significance. Diabetes, Obes. Metab 2007, 9 (SUPPL. 2), 109–117. [DOI] [PubMed] [Google Scholar]
- (18).Castle JD; Cameron RS; Arvan P; von Zastrow M; Rudnick G Similarities and Differences among Neuroendocrine, Exocrine, and Endocytic Vesicles. Ann. N. Y. Acad. Sci 1987, 493, 448–460. [DOI] [PubMed] [Google Scholar]
- (19).Johansson BL; Sjöberg S; Wahren J The Influence of Human C-Peptide on Renal Function and Glucose Utilization in Type 1 (Insulin-Dependent) Diabetic Patients. Diabetologia 1992, 35 (2), 121–128. [DOI] [PubMed] [Google Scholar]
- (20).Johansson J; Ekberg K; Shafqat J; Henriksson M; Chibalin A; Wahren J; Jörnvall H Molecular Effects of Proinsulin C-Peptide. Biochem. Biophys. Res. Commun 2002, 295 (5), 1035–1040. [DOI] [PubMed] [Google Scholar]
- (21).Ekberg K; Brismar T; Johansson BL; Jonsson B; Lindström P; Wahren J Amelioration of Sensory Nerve Dysfunction by C-Peptide in Patients with Type 1 Diabetes. Diabetes 2003, 52 (2), 536–541. [DOI] [PubMed] [Google Scholar]
- (22).Bhatt MP; Lim YC; Ha KS C-Peptide Replacement Therapy as an Emerging Strategy for Preventing Diabetic Vasculopathy. Cardiovasc. Res 2014, 104 (2), 234–244. [DOI] [PubMed] [Google Scholar]
- (23).Forst T; Kunt T Effects of C-Peptide on Microvascular Blood Flow and Blood Hemorheology. Exp. Diabesity Res 2004, 5 (1), 51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zierath J; Handberg A; Tally M; Wallberg-Henriksson H C-Peptide Stimulates Glucose Transport in Isolated Human Skeletal Muscle Independent of Insulin Receptor and Tyrosine Kinase Activation. Diabetologia 1996, 39 (3), 306–313. [DOI] [PubMed] [Google Scholar]
- (25).Stevens MJ; Zhang W; Li F; Sima AAF C-Peptide Corrects Endoneurial Blood Flow but Not Oxidative Stress in Type 1 BB/W or Rats. Am. J. Physiol. - Endocrinol. Metab 2004, 287, E497–E505. [DOI] [PubMed] [Google Scholar]
- (26).Jensen ME; Messina EJ C-Peptide Induces a Concentration-Dependent Dilation of Skeletal Muscle Arterioles Only in Presence of Insulin. Am. J. Physiol. - Hear. Circ. Physiol 1999, 276, H1223–H1228. [DOI] [PubMed] [Google Scholar]
- (27).Keltner Z; Meyer JA; Johnson EM; Palumbo AM; Spence DM; Reid GE Mass Spectrometric Characterization and Activity of Zinc-Activated Proinsulin C-Peptide and C-Peptide Mutants. Analyst 2010, 135 (2), 278–288. [DOI] [PubMed] [Google Scholar]
- (28).Meyer JA; Froelich JM; Reid GE; Karunarathne WKA; Spence DM Metal-Activated C-Peptide Facilitates Glucose Clearance and the Release of a Nitric Oxide Stimulus via the GLUT1 Transporter. Diabetologia 2007, 51 (1), 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Meyer JA; Subasinghe W; Sima AAF; Keltner Z; Reid GE; Daleke D; Spence DM Zinc-Activated C-Peptide Resistance to the Type 2 Diabetic Erythrocyte Is Associated with Hyperglycemia-Induced Phosphatidylserine Externalization and Reversed by Metformin. Mol. BioSyst 2009, 5 (10), 1157–1162. [DOI] [PubMed] [Google Scholar]
- (30).Liu Y; Chen C; Summers S; Medawala W; Spence DM C-Peptide and Zinc Delivery to Erythrocytes Requires the Presence of Albumin: Implications in Diabetes Explored with a 3D-Printed Fluidic Device. Integr. Biol. (United Kingdom) 2015, 7 (5), 534–543. [DOI] [PubMed] [Google Scholar]
- (31).Ge X; Kakinen A; Gurzov EN; Yang W; Pang L; Pilkington EH; Govindan-Nedumpully P; Chen P; Separovic F; Davis TP; Ke PC; Ding F Zinc-Coordination and C-Peptide Complexation: A Potential Mechanism for the Endogenous Inhibition of IAPP Aggregation. Chem. Commun 2017, 53 (68), 9394–9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Stevenson MJ; Farran IC; Uyeda KS; San Juan JA; Heffern MC Analysis of Metal Effects on C-Peptide Structure and Internalization. ChemBioChem 2019, 20 (19), 2447–2453. [DOI] [PubMed] [Google Scholar]
- (33).Landreh M; Johansson J; Wahren J; Jörnvall H The Structure, Molecular Interactions and Bioactivities of Proinsulin C-Peptide Correlate with a Tripartite Molecule. Biomol. Concepts 2014, 5 (2), 109–118. [DOI] [PubMed] [Google Scholar]
- (34).Mares-Guia TR; Maigret B; Martins NF; Maia ALT; Vilela L; Ramos CHI; Neto LJ; Juliano MA; dos Mares-Guia ML; Santoro MM Molecular Dynamics and Circular Dichroism Studies of Human and Rat C-Peptides. J. Mol. Graphics Modell 2006, 25 (4), 532–542. [DOI] [PubMed] [Google Scholar]
- (35).Tsednee M; Huang Y-C; Chen Y-R; Yeh K-C Identification of Metal Species by ESI-MS/MS through Release of Free Metals from the Corresponding Metal-Ligand Complexes. Sci. Rep 2016, 6 (1), 26785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Dorlet P; Gambarelli S; Faller P; Hureau C Pulse EPR Spectroscopy Reveals the Coordination Sphere of Copper(II) Ions in the 1–16 Amyloid-β Peptide: A Key Role of the First Two N-Terminus Residues. Angew. Chem., Int. Ed 2009, 48 (49), 9273–9276. [DOI] [PubMed] [Google Scholar]
- (37).Peisach J; Blumberg WE Structural Implications Derived from the Analysis of Electron Paramagnetic Resonance Spectra of Natural and Artificial Copper Proteins. Arch. Biochem. Biophys 1974, 165 (2), 691–708. [DOI] [PubMed] [Google Scholar]
- (38).Rao G; Bansal S; Law WX; O’Dowd B; Dikanov SA; Oldfield E Pulsed Electron Paramagnetic Resonance Insights into the Ligand Environment of Copper in Drosophila Lysyl Oxidase. Biochemistry 2017, 56 (29), 3770–3779. [DOI] [PubMed] [Google Scholar]
- (39).Murphy DM; Caretti I; Carter E; Fallis IA; Göbel MC; Landon J; Doorslaer SV; Willock DJ Visualizing Diastereomeric Interactions of Chiral Amine–Chiral Copper Salen Adducts by EPR Spectroscopy and DFT. Inorg. Chem 2011, 50 (15), 6944–6955. [DOI] [PubMed] [Google Scholar]
- (40).Nara M; Tanokura M Infrared Spectroscopic Study of the Metal-Coordination Structures of Calcium-Binding Proteins. Biochem. Biophys. Res. Commun 2008, 369 (1), 225–239. [DOI] [PubMed] [Google Scholar]
- (41).Deacon GB Relationships between the Carbon-Oxygen Stretching Frequencies of Carboxylato Complexes and the Type of Carboxylate Coordination. Coord. Chem. Rev 1980, 33 (3), 227–250. [Google Scholar]
- (42).Gerbino E; Mobili P; Tymczyszyn E; Fausto R; Gómez-Zavaglia A FTIR Spectroscopy Structural Analysis of the Interaction between Lactobacillus Kefir S-Layers and Metal Ions. J. Mol. Struct 2011, 987 (1–3), 186–192. [Google Scholar]
- (43).Tackett JE FT-IR Characterization of Metal Acetates in Aqueous Solution. Appl. Spectrosc 1989, 43 (3), 483–489. [Google Scholar]
- (44).Walker JM; Kleinschmidt JH Methods in Molecular Biology: Lipid–Protein Interactions; 2013. [DOI] [PubMed]
- (45).Barth A Infrared Spectroscopy of Proteins. Biochim. Biophys. Acta, Bioenerg 2007, 1767 (9), 1073–1101. [DOI] [PubMed] [Google Scholar]
- (46).Sigel H; Martin RB Coordinating Properties of the Amide Bond. Stability and Structure of Metal Ion Complexes of Peptides and Related Ligands. Chem. Rev 1982, 82 (4), 385–426. [Google Scholar]
- (47).Magyar JS; Godwin HA Spectropotentiometric Analysis of Metal Binding to Structural Zinc-Binding Sites: Accounting Quantitatively for PH and Metal Ion Buffering Effects. Anal. Biochem 2003, 320, 39–54. [DOI] [PubMed] [Google Scholar]
- (48).NIST Critically Selected Stability Constants of Metal Complexes, version 8.0.
- (49).Grossoehme NE; Spuches AM; Wilcox DE Application of Isothermal Titration Calorimetry in Bioinorganic Chemistry. J. Biol. Inorg. Chem 2010, 15 (8), 1183–1191. [DOI] [PubMed] [Google Scholar]
- (50).Marcus Y A Simple Empirical Model Describing the Thermodynamics of Hydration of Ions of Widely Varying Charges, Sizes, and Shapes. Biophys. Chem 1994, 51 (2–3), 111–127. [Google Scholar]
- (51).Daniele PG; Zerbinati O; Aruga R; Ostacoli G Thermodynamic and Spectrophotometric Study of Copper(II) and Cadmium(II) Homo- and Hetero-Nuclear Complexes with L-Histidylglycine in an Aqueous Medium. J. Chem. Soc., Dalton Trans 1988, 3 (5), 1115–1120. [Google Scholar]
- (52).Henriksson M; Nordling E; Melles E; Shafqat J; Ståhlberg M; Ekberg K; Persson B; Bergman T; Wahren J; Johansson J; Jörnvall H Separate Functional Features of Proinsulin C-Peptide. Cell. Mol. Life Sci 2005, 62 (15), 1772–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.