

ABSTRACT
The therapeutic potential of targeting CD19 in B cell malignancies has garnered attention in the past decade, resulting in the introduction of novel immunotherapy agents. Encouraging clinical data have been reported for T cell-based targeting agents, such as anti-CD19/CD3 bispecific T-cell engager blinatumomab and chimeric antigen receptor (CAR)-T therapies, for acute lymphoblastic leukemia and B cell non-Hodgkin lymphoma (B-NHL). However, clinical use of both blinatumomab and CAR-T therapies has been limited due to unfavorable pharmacokinetics (PK), significant toxicity associated with cytokine release syndrome and neurotoxicity, and manufacturing challenges. We present here a fully human CD19xCD3 bispecific antibody (TNB-486) for the treatment of B-NHL that could address the limitations of the current approved treatments. In the presence of CD19+ target cells and T cells, TNB-486 induces tumor cell lysis with minimal cytokine release, when compared to a positive control. In vivo, TNB-486 clears CD19+ tumor cells in immunocompromised mice in the presence of human peripheral blood mononuclear cells in multiple models. Additionally, the PK of TNB-486 in mice or cynomolgus monkeys is similar to conventional antibodies. This new T cell engaging bispecific antibody targeting CD19 represents a novel therapeutic that induces potent T cell-mediated tumor-cell cytotoxicity uncoupled from high levels of cytokine release, making it an attractive candidate for B-NHL therapy.
KEYWORDS: Bispecific antibody, T-cell engager, CD19XCD3, cytokine release syndrome, TNB-486, low cytokine release
Introduction
B cell non-Hodgkin lymphoma (B-NHL) affects approximately 1.5 million patients worldwide and presents with diverse clinical manifestations dependent on the subtype. Among the major subtypes of B-NHL, diffuse large B cell lymphoma (DLBCL) and follicular lymphoma represent the most common aggressive and indolent subtypes of B-NHL, respectively, and together account for over 50% of all cases.1 Chemotherapy coupled with Rituxan®, a monoclonal antibody (mAb) targeting CD20, has remained the first-line treatment for the majority of the subtypes. Despite a high overall response rate (>90%), most patients relapse and treatment of relapsed/refractory B-NHL remains a challenge owing to the complexity of the disease subtypes, patient characteristics and variable responses between subtypes to initial therapy.2,3
Based on the success of Rituxan®, lineage markers gained popularity as targets for the treatment of hematologic malignancies. CD19 is a B cell restricted surface receptor present on all B cells, including neoplastic B cells. The expression of CD19 is broader than CD20, from the pro-B cell to plasmablast stage, and is lost upon differentiation into plasma cells, making it a popular target for B cell malignancies.4,5 Efforts to target CD19 have included multiple immunotherapy modalities such as mAbs and their derivatives, e.g., antibody-drug conjugates (ADCs), T cell engaging bispecific antibodies (T-BsAbs), and chimeric antigen receptor (CAR)-T cells.5–7 Of these, mAbs have shown limited success, likely owing to lower antigen density of CD19 on malignant B cells compared to CD20,8 the inherently limited antibody-dependent cell-mediated cytotoxicity or complement-dependent cytotoxicity activities of mAbs, or rapid internalization of CD19 upon antibody crosslinking.5 CD19-targeted ADCs have also faced hurdles due to toxicity associated with the cytotoxic payload or inhibition of internalization due to high CD21 expression.9,10
Immunotherapy modalities that harness the cytolytic potential of T cells to target and kill tumor cells, such as T-BsAbs or CAR-T cells have shown promise in the treatment of chemotherapy-resistant or relapsed hematologic malignancies. Blinatumomab (Blincyto®), an antibody-based Bispecific T cell Engager (BiTE) that targets CD3 and CD19, is approved by the U.S. Food and Drug Administration (FDA) for B cell acute lymphoblastic leukemia (B-ALL) and shows durable responses in relapsed/refractory DLBCL.11,12 Axicabtagene ciloleucel (Yescarta®) and tisagenlecleucel (Kymriah®) are CD19 targeted CAR-T cell products that are FDA-approved as third-line therapies for B-NHL. However, both T-BsAbs and CAR-T cells have significant toxicity hurdles due to cytokine release syndrome (CRS) and neurotoxicity,13 which result in limited or modified dose regimens combined with clinical management of the toxicities.14,15
T-BsAbs that demonstrate efficient tumor cell cytotoxicity coupled with reduced cytokine release in vitro have been described and could prove beneficial in the clinic by reducing CRS-related toxicity.16–18 Previous work from our group has highlighted a novel CD3-engaging arm which, when paired with a tumor-targeting arm, shows reduced cytokine secretion with comparable tumor cell lysis relative to a T-BsAb that contains a higher affinity anti-CD3 arm.16
This study describes TNB-486, a novel fully human CD19xCD3 bispecific antibody designed for the treatment of B-NHL. TNB-486 engages CD19 and CD3, leading to activation of resting polyclonal CD4+ and CD8+ T cells that result in efficient lysis of CD19+ tumor cells, but with markedly lower cytokine release. The preclinical characterization of TNB-486 presented here outlines the anti-tumor efficacy of TNB-486 in in vitro, in vivo and ex vivo models of B cell malignancies.
Results
CD19 expression on normal and malignant B cells
Cell surface expression of CD19 on tumor cell lines derived from various B cell malignancies was confirmed by flow cytometry. The number of CD19 molecules on the cell surface (antigen density) of multiple CD19-positive cell lines representing Burkitt’s lymphoma (BL), DLBCL or ALL were quantified by flow cytometry analysis using a commercially available monoclonal CD19 antibody (Figure 1a). Similarly, CD19 expression was also determined on B cells in peripheral blood mononuclear cells (PBMCs) from healthy individuals, and PBMCs or dissociated tumor cells (DTCs) from chronic lymphocytic leukemia (CLL) or DLBCL patients (Figure 1b). The CD19 antigen density on tumor cell lines and patient-derived samples ranged from 6 × 103 to 161 × 103 and 8 × 103 to 98 x 103, respectively. In contrast, normal PBMCs had a more uniform expression ranging between 21–32 × 103 CD19 molecules per cell (Figure 1b).
Figure 1.
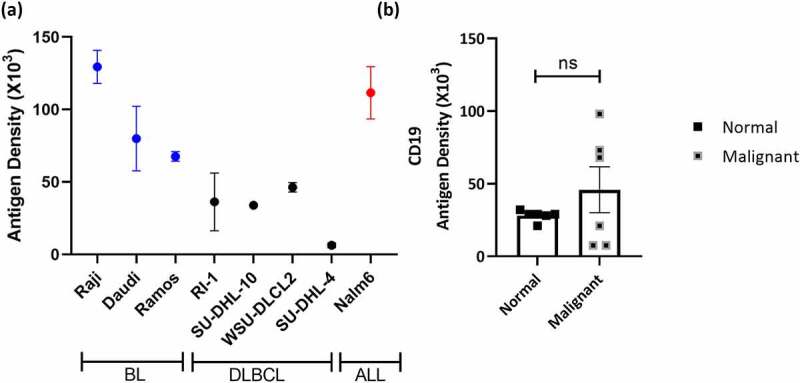
CD19 Expression on normal and malignant B cells (a) Antigen density of CD19 was measured on the indicated human B cell lines by quantitative flow cytometry. Error bars indicate standard error mean (SEM) of 2–4 independent experiments. (b) Antigen density of CD19 was enumerated on normal human PBMCs and malignant B cells from 4 CLL and 2 DLBCL DTCs or PBMCs. Error bars represent SEM of independent samples. Unpaired Student’s t-test was used to determine significance
Generation and characterization of a CD19xCD3 bispecific antibody
Due to the complex extracellular structure of CD19,19,20 protein-based immunization strategies have shown limited success, leading to low numbers of unique antibody clones against CD19. The handful of available clones are primarily derived from mouse hybridomas and require humanization.21–23 Some humanized murine antibodies have a higher risk of immunogenicity in the clinic, and thus fully human antibodies are a preferred alternative. Fully human heavy-chain only antibodies targeting CD19 were generated by immunization of UniRats® followed by next-generation sequencing (NGS)-based antibody repertoire discovery approach.24 A lead anti-CD19 UniAb® was identified by screening multiple candidates for cell surface binding to CD19+ tumor cells, as well as high-affinity binding to CD19 protein by Octet. Antibodies against CD3 were generated in OmniFlic® animals and have been previously described.16,25
T-BsAbs were generated by pairing the lead anti-CD19 VH (variable heavy region of the antibody) with a high-affinity CD3-binding arm (as a Positive Control, PC) or a low-affinity CD3-binding arm (Lead antibody, TNB-486) using knobs-in-holes technology as previously described.16 TNB-486 is a fully human asymmetric bispecific monoclonal IgG4 antibody. The structure is presented in Figure 2a. The PC T-BsAb that contains a high-affinity CD3-binding arm was used in all relevant experiments. A negative control antibody (NC) that contains the same anti-CD3 VH as TNB-486 but an irrelevant tumor binding arm was used where applicable. TNB-486 shows robust expression, desirable biophysical properties, and low aggregation propensity after thermal stress (Table 1, Figure S1).
Figure 2.
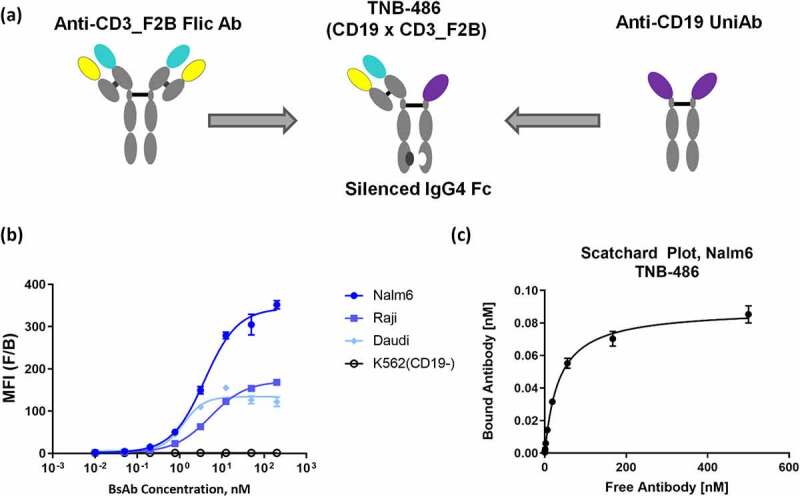
TNB-486 is a fully human bispecific antibody engaging CD19 and CD3 (a) TNB-486 was constructed using knobs-into-holes technology. The format of the antibody is depicted. (b) Cell binding dose curves of TNB-486 on three CD19+ B cell lines Daudi, Raji and Nalm-6, and one CD19- cell line, K562 is shown. (c) Cell surface affinity of TNB-486 to CD19 expressed on Nalm-6 cells was determined by Scatchard analysis
Table 1.
TNB-486 has favorable protein biophysical characteristics. Thermal stability of TNB-486 was assessed by determining the melting temperatures (Tonset,Tm1 and Tm2). Further, aggregation propensity was assessed by SEC-UPLC before and after exposure to heat stress. Percent High Molecular Weight (%HMW) species are shown at T = 0 and after 30 days
| Yield (g/L) | Thermal Stability (%HMW): 5⁰C | Thermal Stability (%HMW): 25⁰C | Tonset (⁰C) | Tm1 (⁰C) | Tm2 (⁰C) | ||
|---|---|---|---|---|---|---|---|
| Td0 |
Td30 |
Td0 |
Td30 |
||||
| 3.3 | 0.5 | 0.5 | 0.5 | 0.5 | 57.4 | 67.7 | 76.9 |
Cell binding dose curves of TNB-486 across a panel of CD19-positive and -negative cell lines are shown in Figure 2b. The EC50 of cell binding on the Daudi, Raji and Nalm-6 cell lines ranged from 1.2 to 5.2 nM. The cell surface affinity of TNB-486 on target expressing cells was determined by Scatchard analysis. Consistent with EC50 values obtained in Figure 2b, the KD of the anti-CD19 arm to Nalm-6 cells was 1.8 nM (± 0.54 nM). The binding of the anti-CD3 arm to human T cells has been previously published.16
In vitro functional assessment of TNB-486
The ability of TNB-486 to activate CD4+ and CD8+ T cells in the presence of CD19+ tumor cells was assessed by CD69 expression. Consistent with the mode of action of T-BsAbs, in the presence of CD19+ RI-1 tumor cells, TNB-486 activated both CD4+ and CD8+ T cells to a similar extent as the PC (Figure 3a). Approximately 95% of total CD8+ T cells and 55% of total CD4+ T cells were CD69+ following incubation of pan T cells with CD19+ RI-1 tumor cells at saturating concentrations of TNB-486. The EC50 of TNB-486-induced activation was 103.6 pM and 121.8 pM, for CD4+ and CD8+ T cells, respectively, compared to EC50 ~ 1–2 pM for the PC in both T cell subtypes. In a similar assay setup that included an incubation time of 5 days, TNB-486 induced proliferation of CD4+ and CD8+ T cells to the same extent as the PC, but with a higher EC50 value (Figure 3b). The EC50s for TNB-486-mediated proliferation was 62.9 pM among CD4+ carboxyfluorescein succinimidyl ester (CFSE)+ T cell subset and 46.9 pM for CD8+ CFSE+ T cell subset, compared to EC50s ≤ 1 pM for the PC. The NC did not mediate activation or proliferation of T cells, demonstrating target-dependent activation of T cells by TNB-486. In addition, TNB-486-mediated T cell activation was accompanied by the release of cytotoxic granules, perforin and granzyme B, in the supernatant of co-cultures of T cells and RI-1 tumor cells (Figure 3c). The maximum concentration of the cytotoxic granules release was similar to that of the PC. As expected, cytotoxic granule release coincided with flow-cytometry based measurements of tumor cell lysis in the co-culture assay (data not shown).
Figure 3.
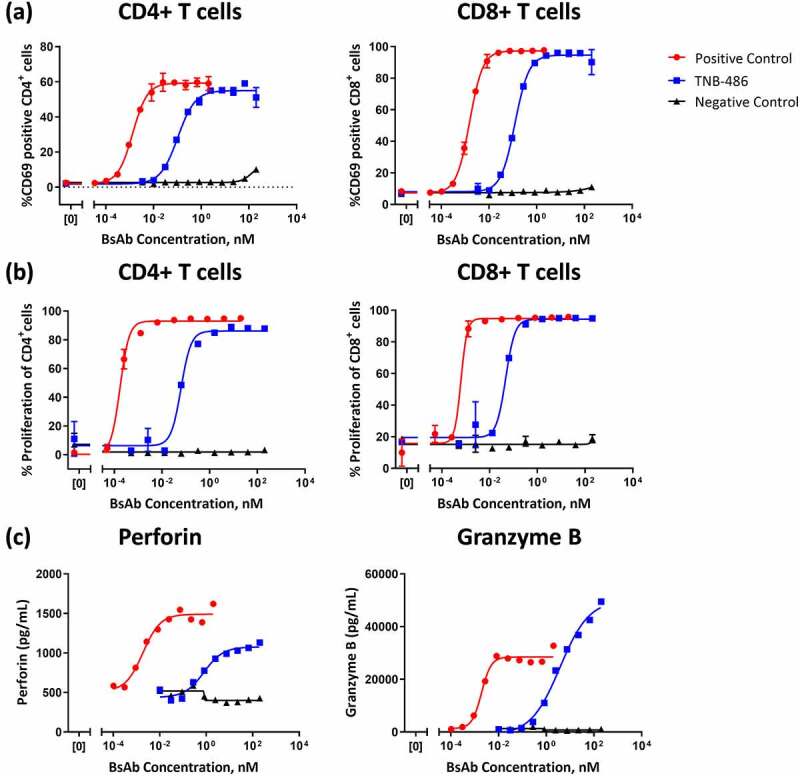
TNB-486 mediates T cell activation, proliferation, and cytotoxic granule release. CD19+ RI-1 tumor cells were incubated with T cells from a single healthy donor at an E:T ratio of 10:1 and serially diluted indicated test antibody (a) Activation of CD4+ or CD8+ T cells were determined by flow cytometric measurement of the activation marker CD69 after 24 hours of co-culture (b) T cell proliferation was measured by labeling T cells with CFSE and monitoring CD4 or CD8 proliferation after 5 days of co-culture (c) Perforin and granzyme B levels were measured in the cell culture supernatants of co-culture after 48 hours by ELISA
Since CD19 is expressed on both normal and neoplastic B cells, TNB-486 is expected to mediate lysis of normal B cells in peripheral blood. To test this, T cells and B cells sorted from PBMCs of healthy human donors were incubated with TNB-486 at a ratio of 10:1 and the depletion of B cells was evaluated over 4 days. By 24 hours of incubation, TNB-486 induced ~40% T cell-dependent cellular cytotoxicity (TDCC) of B cells in the culture. Percent lysis increased to 65% by 48 hours and reached over 90% by 96 hours. Of note, the EC50 of lysis did not vary substantially between 48 hours and 96 hours (EC50s ranging from 0.93 to 1.35 nM) and therefore, all cytotoxicity experiments henceforth were performed at 48 hours (Figure 4a). Next, the TDCC of PBMCs from three independent donors was evaluated. Figure 4b shows that in the presence of TNB-486, autologous T cells can mediate lysis of B cells in the culture by 48 hours. In addition, compared to the PC, TNB-486 induced lower levels of interleukin (IL)-2, interferon (IFN)γ, IL-6, IL-10 and tumor necrosis factor (TNF) in the culture supernatant (Figure S2). To confirm that this time point was the most relevant for evaluating TDCC with CD19+ tumor cells as well, TDCC experiments were conducted by incubating TNB-486 with Nalm-6 CD19+ tumor cell line with pan T cells from PBMCs. After 48 and 72 hours of co-culture, cytotoxicity and cytokine release were evaluated. While the maximum %cytotoxicity increases over time, the EC50 does not change between 48 and 72 hours (0.11 nM vs 0.18 nM for TNB-486 at 48 vs 72 hours, respectively) (Figure S3). TNB-486-mediated induction of IL-2 and IFNγ levels remained low at both timepoints (~100 pg/mL and 60 pg/mL of IL-2 at 48 and 72 hours, respectively; and ~600 pg/mL of IFNγ at both 48 and 72 hours). Based on this data, the 48-hour timepoint gives a conservative measure of cytokine release as well as cytotoxicity in this in vitro system and was used for further TDCC experiments.
Figure 4.
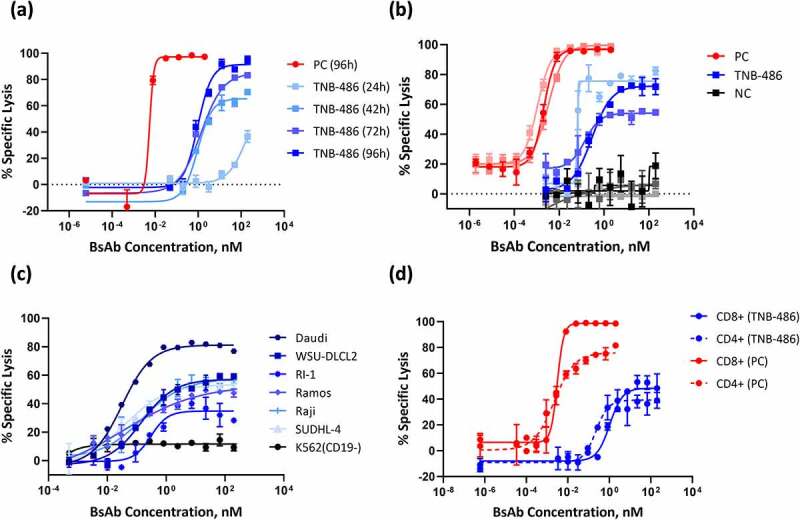
TNB-486 induces lysis of CD19+ B cells (a) T cells and B cells sorted from normal PBMCs were co-cultured at an E:T ratio of 10:1 and incubated with varying doses of TNB-486. Cytotoxicity of B cells was evaluated by flow cytometry at 24 h, 42 h, 72 h and 96 h. (b) PBMCs from three independent donors (the shades of blue represent three independent donors for TNB-486, shades of red and gray/black represent the three donors for PC and NC, respectively) were incubated with increasing doses of TNB-486 without adjusting the E:T ratio. Cytotoxicity of B cells was measured by flow cytometry at 48 h. (c) CD19+ tumor cells Daudi, Raji, Ramos, SU-DHL-4, WSU-DLCL2 and RI-1 or a CD19- K562 cell line were co-cultured with T cells from a healthy donor (at an E:T ratio of 5:1) and increasing doses of TNB-486. TDCC was measured at 48 hours by enumerating the live CD20+ B cells by flow cytometry (d) TNB-486 mediated cytotoxicity of CD19+ RI-1 tumor cells and CD4+ or CD8+ T cells as effectors was determined as in (C)
To evaluate the T cell redirecting properties of TNB-486, TDCC experiments were performed by incubating TNB-486 in the presence of resting CD3+ human T cells and a variety of CD19+ tumor cells for 48 hours. At an effector to target ratio (E:T) of 5:1, TNB-486 induced concentration-dependent TDCC of multiple CD19+ tumor cell lines derived from leukemia or lymphoma origin, namely, Ramos, Daudi, Raji, Nalm-6, WSU-DLCL2, SU-DHL-4, or RI-1 (or RIVA). Among these, RI-1 is a DLBCL cell line known to be resistant to rituximab,26 and WSU-DLCL2 is derived from a patient with aggressive lymphoma and is resistant and refractory to chemotherapy agents.27 The maximum average percent lysis induced by TNB-486 ranged from 35% to 82% between the different tumor cell lines (Figure 4c). In contrast, a concentration-dependent TDCC was not observed in the CD19- K562 tumor cell line, illustrating the specificity of TNB-486 to CD19 expressing tumor cells.
Upon treatment with T-BsAbs, both CD4+ and CD8+ T cells are known to contribute to cytotoxicity of tumor cells.28 To assess this, TNB-486 was incubated with either CD4+ or CD8+ T cells in the presence of RI-1 tumor cells and cytotoxicity was measured at 48 hours. As shown in Figure 4d, cytotoxicity was observed when either CD4+ or CD8+ T cells were used as the source of effector cells. The maximal percent cytotoxicity induced by TNB-486 was similar when CD4+ or CD8+ cells were used as effectors (39.2% and 48.4% lysis, respectively) but lower than that of the PC (75.6% and 98.6%, respectively).
TNB-486 consistently mediated TDCC in PBMCs derived from several donors. In TDCC experiments with two different CD19+ tumor cells (RI-1 or Raji) and T cells from three independent donors, TNB-486 resulted in T-cell-mediated tumor cell lysis irrespective of the donor. The EC50s of TNB-486 mediated cell lysis were 23.5 ± 11.5 pM or 0.9 ± 0.2 nM for Raji cells or RI-1, respectively, compared to that of the PC with an EC50 of 2.2 ± 2.5 pM in both cell lines. Importantly, at concentrations sufficient to achieve maximum tumor lysis, the levels of IL-2 induced by TNB-486 was markedly lower than that induced by the PC (Figure 5). IFNγ and TNF also displayed a similar trend where TNB-486 induced lower cytokine levels compared to the PC. IL-6 and IL-10 levels were similar between TNB-486 and PC, but with a markedly higher EC50 for TNB-486 (Table 2). In similar TDCC experiments with Raji tumor cells and T cells, TNB-486 was compared with blinatumomab and the PC. TNB-486 induced similar maximum percentage of lysis as that of blinatumomab, which was similar to the PC used in this study, but with a higher EC50. The equivalent cytotoxicity was accompanied by reduced cytokine secretion in TNB-486-treated samples compared to blinatumomab (Figure S4).
Figure 5.
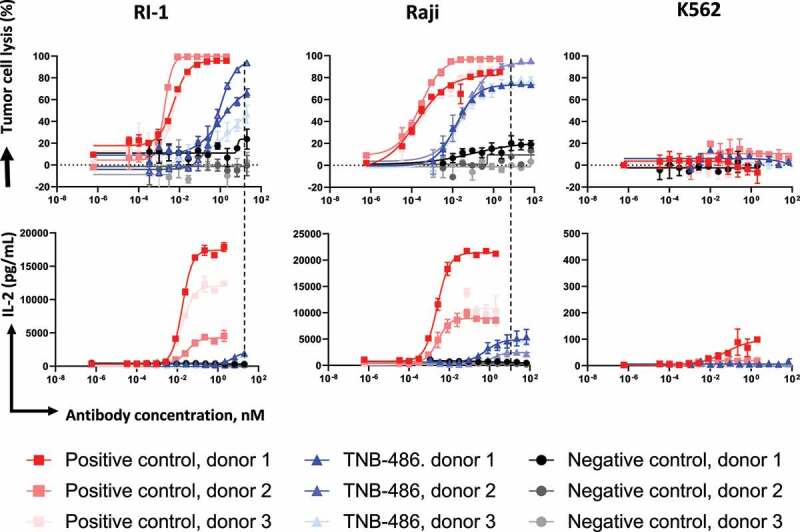
TNB-486 induced lysis is accompanied by low cytokine release. TDCC of TNB-486 was evaluated as in (Figure 4) but using T cells from three independent donors. Raji or RI-1 tumor cells were used as target cells. IL-2 levels were measured from culture supernatants of the co-culture assay by MSD
Table 2.
TNB-486 induces lower cytokine release compared to the PC. The TDCC of TNB-486 was assessed as described in Figure 5. Supernatants from the co-culture were used to measure cytokine release. The EC50s of cytokine release are shown in Table 2(A) and the maximum cytokine released is shown in Table 2(B)
| (A) EC50 values of dose curves for TNB-486 or PC mediated cytokines | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (pM), TNB-486 |
EC50 (pM), PC |
|||||||||||
| Target Cell |
RI-1 |
Raji |
RI-1 |
Raji |
||||||||
| Donor ID | D2546 | D2921 | D0628 | D2546 | D2921 | D0628 | D2546 | D2921 | D0628 | D2546 | D2921 | D0628 |
| IFNγ | 1534 | N/A | 1411 | N/A | 2164 | 358 | 7 | N/A | 5 | 8 | 3 | 5 |
| IL-2 | 5364 | N/A | 6192 | N/A | 1410 | 570 | 18 | 31 | 17 | 3 | 3 | 4 |
| IL-6 | 3487 | N/A | 2961 | N/A | N/A | N/A | 15 | N/A | 10 | N/A | N/A | N/A |
| IL-10 | 1423 | 735 | 1369 | N/A | N/A | 322 | 3 | N/A | N/A | 16 | N/A | N/A |
| TNF | 2620 | 708 | 3051 | N/A | 1225 | 490 | 18 | 14 | 13 | 1 | 2 | N/A |
| (B) Maximum cytokine levels induced by TNB-486 or PC (related to Figure 5) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Maximum Induction (pg/mL), TNB-486 |
Maximum Induction (pg/mL), PC |
|||||||||||
| Target Cell |
RI-1 |
Raji |
RI-1 |
Raji |
||||||||
| Donor ID | D2546 | D2921 | D0628 | D2546 | D2921 | D0628 | D2546 | D2921 | D0628 | D2546 | D2921 | D0628 |
| IFNγ | 42492 | 13647 | 24080 | 18188 | 18373 | 6416 | 65646 | 22860 | 28701 | 80441 | 43053 | 17758 |
| IL-2 | 2061 | 443 | 2094 | 4929 | 2456 | 4494 | 17874 | 4537 | 12430 | 21217 | 8567 | 10378 |
| IL-6 | 246 | 3 | 193 | 39 | 47 | 19 | 1124 | 131 | 554 | 189 | 79 | 27 |
| IL-10 | 1294 | 74 | 1804 | 326 | 56 | 213 | 1448 | 101 | 1795 | 910 | 99 | 360 |
| TNF | 1473 | 632 | 1372 | 742 | 500 | 500 | 8174 | 2533 | 4367 | 5747 | 1828 | 1159 |
Evaluation of cytotoxicity in patient-derived samples was performed by obtaining either PBMCs or DTCs from B-CLL or DLBCL patients. Depletion of CD20 positive B cells was used as a readout for lysis. Depending on the number of viable cell numbers recovered from each of the frozen samples, the experiment was conducted with an adjusted E:T ratio of 10:1 or unadjusted E:T ratio. Data in Table 3 shows that TNB-486 induced the autologous T cell killing of >10% B cells in 6 of 8 samples tested.
Table 3.
TNB-486 mediates lysis of tumor cells derived from leukemia or lymphoma patients. Lysis of CD19+ cells from ex vivo patient derived PBMCs or dissociated tumor cells (DTCs) was evaluated by flow cytometry. Average % lysis of tumor cells is shown
|
Ex Vivo Sample Type |
Effector:Target Ratio |
CD19 Antigen Density (x103)d |
Average % lysis of tumor cellse |
| Lymphoma, NOS | 1.707b | 7.6 | 2.3a |
| NHL, Extranodal Marginal Zone B cell (MALT: Mucosa-Associated Lymphoid Tissue) | 0.01b | NT | -1.4 |
| NHL, Extranodal Marginal Zone B cell (MALT: Mucosa-Associated Lymphoid Tissue) PBMC | 12.54b | 20.7 | 55.7 |
| NHL, Diffuse Large B cell Lymphoma | 25.95b | 7.6 | 25.4a |
| CLL PBMC | 10c | 70.3 | 18.2 |
| CLL PBMC | 10c | 20.4 | 14.9 |
| NHL | 2 b | - | 28.2 |
| CLL PBMCf | 10c | 97.9 | 44.4 |
| 0.064b | 17.5 |
aAssay incubation time was 24 hours
bEffector to target cell ratio was left unaltered
cEffector to target cell ratio was adjusted to indicated value
dAntibody used for antigen density estimate; Biolegend Cat#302208 Clone#HIB19
eNormalized to control samples without antibody
fSame sample was tested with and without modifying E:T ratio as indicated in column 2.
In vivo anti-tumor efficacy of TNB-486 in murine xenograft models
The in vivo anti-tumor efficacy of TNB-486 was assessed against human tumor cell lines xenografted into immunocompromised NOG (NOD.Cg-Prkdcscid IL2rgtm1Sug/JicTac) mice in three different models. In the first study using a disseminated model of Burkitt lymphoma, NOG mice were engrafted with luciferase-expressing Raji tumor cells and human PBMCs, followed by treatment with TNB-486. The doses of TNB-486 were 10 ng, 100 ng, 1 µg, or 10 µg per mouse (or 10 ng PC or 10 µg NC, respectively) administered intravenously (IV) every 5 days. Bioluminescent imaging was used to monitor tumor burden. After two treatments at the respective dose levels, TNB-486-treated animals showed a dose-dependent tumor reduction compared to the NC, which did not show any reduction in tumor growth (Figure 6a). At doses 100 ng, 1 μg, or 100 μg/mouse, TNB-486 cleared tumors similar to the PC at 10 ng/mouse. The 10 ng/mouse dose of TNB-486 showed tumor growth inhibition (TGI) at 8 days post-implantation (dpi), but the TGI was not sustained as seen at 14 dpi, indicating that higher doses of TNB-486 compared to the PC are required to achieve a similar efficacy, which is consistent with the in vitro TDCC experiments.
Figure 6.
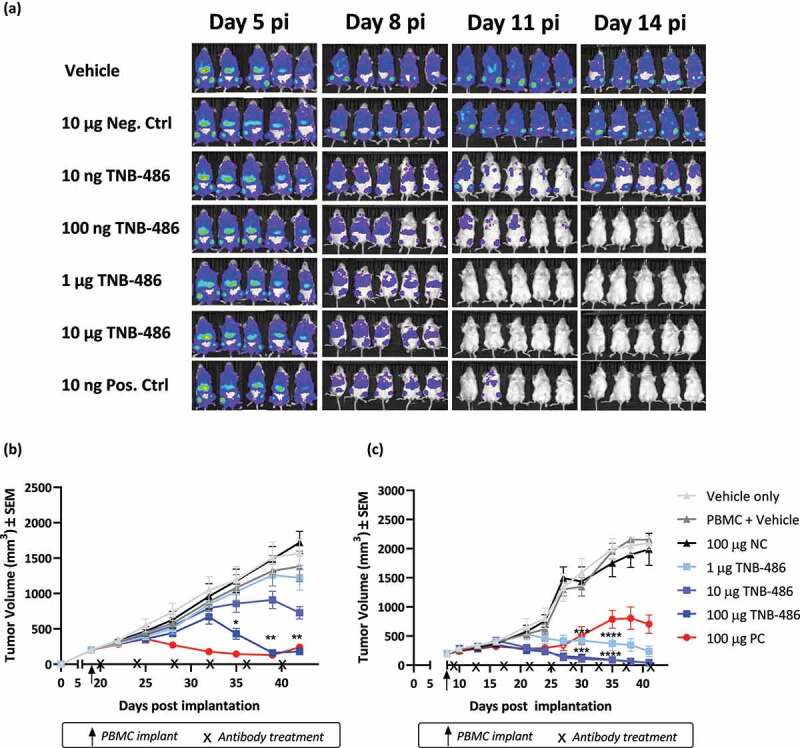
TNB-486 induces tumor regression in multiple xenograft models of established tumors. (a) 1 × 106 Raji-luc cells were injected i.v via tail vein into CIEA-NOG mice (N = 5/group). On day 6 post-implantation (pi), 10 × 106 human PBMCs were injected i.v. TNB-486, NC or PC, at the respective doses shown, was injected on days 7, 14 and 21. GvHD/GvT onset was observed by day 15 pi and hence data points after day 14 are not shown. (b and c) CIEA-NOG mice (N = 10/group) received 10 × 106 SUDHL-10 cells (b) or Nalm-6 cells (c) with 50% Matrigel subcutaneously in the lower right flank. When tumor volume reached 200 mm3, mice received 10 × 106 human PBMCs i. v followed by antibody treatment Q4D as shown in the figure. Statistical analyses were performed by Two-way ANOVA on GraphPad Prism. TNB-486 treated animals were compared with PBMC + vehicle treated animals. A value of p < .05 was considered significant
Efficacy was also measured in a xenograft model using the SU-DHL-10 (DLBCL) tumor cell line, where immunocompromised NOG mice received tumor cells subcutaneously, followed by PBMC engraftment and treatment with TNB-486. By day 40 post-implantation (pi), TNB-486 displayed a dose-dependent TGI (10.89%, 35.68% and 88.52%) with doses 1, 10, or 100 μg/animal, respectively, compared to the 100 μg/animal dose of PC, which resulted in a TGI of 90.74% (Figure 6b). In a similar subcutaneously injected tumor model using the Nalm-6 (ALL) cell line and resting PBMCs as the source of effector cells, TNB-486 treatment resulted in significant TGI at all doses by 35 dpi (79.36%, 94.99%, and 94.86% TGI, at doses 1, 10, or 100 μg/animal, respectively, compared to 55.86% TGI in the 100 μg/animal dose of PC) (Figure 6c). Interestingly, the PC showed reduced efficacy compared to TNB-486, suggesting that strongly activating anti-CD3 containing antibodies could allow relapse of tumor over time.
Pharmacokinetics (PK) of TNB-486 in non-human species
The PK of TNB-486 was evaluated in BALB/c mice following a single tail vein injection at 1 or 10 mg/kg. Non-compartmental analysis and two compartmental analyses showed that TNB-486 PK was linear across the dose ranges tested. Group mean clearance (CL) ranged from 13.0 to 15.5 mL/day/kg and group mean half-life (t1/2) ranged from 2.6 to 4.1 days (Table 4). Since TNB-486 does not cross-react with CD19 or CD3 in rodent species, the observed linear PK was expected.
Table 4.
The PK parameters of TNB-486 is similar to conventional antibodies. The PK parameters of TNB-486 were evaluated in mice or monkeys and the values are shown below
| Parameter | Units |
Balb/c mice |
Cynomolgus monkey |
|||
| |
|
1 mg/kg |
10 mg/kg |
0.1 mg/kg |
1 mg/kg |
10 mg/kg |
| Cmax | µg/mL | 16.1 | 183 | 1.8 | 16.1 | 183 |
| Cmax/D | (µg/mL)/(µg/kg) | 0.0161 | 0.018 | 0.018 | 0.0161 | 0.018 |
| t1/2 | days | 11.4 | 12.9 | 12 | 11.4 | 12.9 |
| AUCinf | µg/mL*days | 168 | 1599 | 13.8 | 168 | 1599 |
| AUCinf/D | (µg/mL*days)/ (µg/kg) | 0.17 | 0.16 | 0.14 | 0.17 | 0.16 |
| % Extrap | % | 41.9 | 41.4 | 32.6 | 41.9 | 41.4 |
| CL | mL/day/kg | 5.99 | 6.43 | 7.41 | 5.99 | 6.43 |
| Vss | mL/kg | 95.6 | 121 | 116 | 95.6 | 121 |
PK parameters in cynomolgus monkeys were similarly measured after a single IV bolus dose of 0.1, 1 or 10 mg/kg. CL ranged from 5.99 to 7.41 mL/day/kg and group mean terminal half-life (t1/2) ranged from 11.4 to 12.9 days (Table 4). Although the anti-CD19 arm binds to cynomolgus monkey CD19, it did not affect TNB-486 PK in monkeys at the tested dose levels. Taken together, the observed linear PK in mice or monkeys is consistent with nonspecific clearance mechanisms dominating PK. Figures 7a and 7b show the serum concentrations of TNB-486 in mice and cynomolgus monkeys, respectively, at the specified doses. Cross-reactivity of the CD19 VH to cynomolgus monkey B cells did not affect serum chemistry, PK or immune cell subset frequencies in the blood of cynomolgus monkeys (data not shown).
Figure 7.
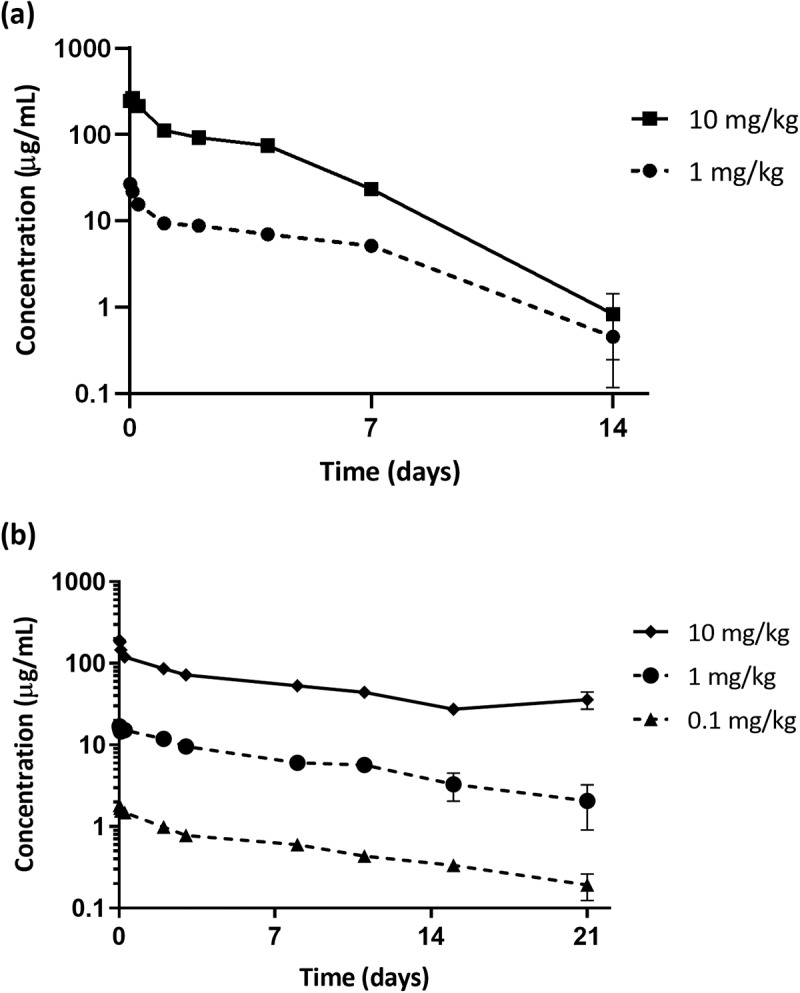
TNB-486 pharmacokinetics in mice and cynomolgus monkeys (a) PK parameters of TNB-486 was evaluated in normal Balb/c mice at 1 or 10 mg/kg (b) PK parameters of TNB-486 was evaluated in cynomolgus monkeys at 0.1, 1 or 10 mg/kg. Serum concentration of TNB-486 in mice were determined by an IgG4 specific AlphaLISA kit (mice) or antigen specific ELISA (cynomolgus monkeys)
Discussion
MAbs and derivatives such as ADCs targeting CD19 have been explored with limited clinical success. Rapid internalization of CD19 upon antibody binding limits the success of mAbs, and, in particular, ADCs have encountered challenges due to compensatory tumor evasion mechanisms.6,9 T cell redirecting bispecific antibodies such as TNB-486 rely on cross-linking of T cells and target-expressing cells, thereby clustering T cell receptors (TCRs) to initiate a signaling cascade that results in activation, proliferation and cytokine/cytotoxic granule release by the T cells to lyse target cells.29 This polyclonal and Major Histocompatibility Complex (MHC)-independent activation of T cells overcomes common evasion mechanisms used by cancer cells, such as down modulation of peptide-MHC antigen presentation or loss of antigenic epitopes.
Current CD19 targeting T cell therapies such as T-BsAbs/BiTEs and CAR-Ts are efficacious in the treatment of B cell malignancies, but have drawbacks such as CRS and neurotoxicity,7,30,31 in addition to other challenges related to dosing of BiTEs or accessibility of CAR-Ts. Another tumor evasion mechanism with CD19-targeted CAR-Ts is antigen loss associated with exon skipping.32,33 This appears to be likely due to immune pressure from long-term persistence of immunostimulatory domain-containing CAR-Ts compared to BiTEs.34 In contrast, there was no CD19 loss observed in patients refractory to blinatumomab, a BiTE targeting CD19 and CD3.35 The approval of blinatumomab has validated the T-BsAb approach for targeting CD19 in certain subsets of B cell lymphomas, but safety concerns have limited its use beyond ALL.36 Of note, a CD19xCD3 T-BsAb was halted in development due to severe adverse events, including a Grade 5 adverse event related to neurotoxicity and/or CRS likely resulting from over-stimulation of T cells arising from the high-affinity engagement of CD3.37,38 Therefore, safer off-the shelf T cell redirecting therapies targeting CD19 will change the course of therapies available for B-NHL.
Approaches to mitigate or manage CRS in current T-BsAb-based therapies generally belong to two categories: 1) Development of a newer-generation of T-BsAbs that intrinsically lower cytokine release compared to existing molecules; or 2) Mitigation of CRS in the clinic by step-dosing,39 or management of CRS with anti-cytokine mAbs and, corticosteroids.14,40 While the latter method is the most used currently, next-generation T-BsAbs using CD3 binding domains optimized for low cytokine release and reduced toxicity without compromising efficacy show substantial promise in the clinic.41,42
Novel anti-CD3 antibodies or affinity engineering of existing anti-CD3 antibodies have been generated by our group and others, respectively.16,17,42 While affinity modification of the anti-CD3 arm can result in reduced cytokine release,17 it is also possible that such engineering can result in unexpected immunogenicity or loss of binding specificity due to additional mutations introduced in the CDR.43 TNB-486 is a fully human bispecific antibody derived from novel humanized rats without a need for humanization via CDR grafting. This could reduce the immunogenicity risk in humans compared to other rodent-derived antibodies that require humanization. The CD3 binding moiety in TNB-486 has shown similar activity when combined with a BCMA (B cell maturation antigen) binding arm where efficient tumor cell lysis is accompanied by minimal cytokine release.16 Similar to our BCMAxCD3 molecule, TNB-486 displayed efficient tumor cell lysis in multiple experimental formats, but did so while inducing lower levels of the cytokines IL-2, IFNγ, IL-6, IL-10 and TNF compared to a PC with a higher affinity anti-CD3 arm (Figure 4, 5 and Table 2 and Trinklein et al.).16 The ability of TNB-486 to induce in vitro TDCC of multiple CD19+ cell lines derived from different subtypes of lymphoma cells, namely Burkitt’s lymphoma, DLBCL, leukemia, including two cell lines that are refractory to chemotherapy (WSU-DLCL2) or rituximab (RI-1), suggests that TNB-486 has the potential to treat R/R B-NHL.26,27
Cytotoxicity induced via the perforin-granzyme pathway accounts for the majority of T cell-mediated target cell killing induced by T-BsAbs.44 Consistent with this mechanism, TNB-486 induced the secretion of perforin and granzyme B in vitro, confirming the predominant mode of action of TNB-486-induced cytotoxicity. Interestingly, release of perforins and granzymes is similar between the PC and TNB-486, but cytokine production by TNB-486 is greatly reduced (Figure 5). The decoupling of cytokine release from cytotoxicity has been described in the context of pMHC-TCR complex formation at the T cell and target cell interface.45 Cytotoxic T lymphocytes (CTLs) exposed to very low pMHC densities result in equivalent target cell cytotoxicity compared to those exposed to high pMHC densities, but are unable to induce cytokine secretion. This dual activation threshold determines the duration and the successful formation of a mature immunological synapse, which is critical for efficient T cell-mediated cytokine release, but not for T cell-mediated cytotoxicity.46,47 Likewise, we believe that low-affinity engagement of CD3 by TNB-486 mirrors an immature immunological synapse with polarized lytic granule release and cytotoxicity, but is delayed in forming a complete synapse or unable to do so, thereby resulting in reduced cytokine release. In a previous study, the F2B anti-CD3 arm, the F1F anti-CD3 arm (used in PC) and clone OKT3, a widely used anti-CD3 antibody clone, were evaluated for their ability to bind to CD3ε, CD3δε or CD3γε heterodimers. It was shown that the F1F- and F2B-containing CD3 binding molecules bind to CD3δε but not CD3γε, whereas OKT3 binds to both CD3δε and CD3γε heterodimers. Additionally, F1F-containing antibodies can bind cynomolgus T cells, whereas F2B-containing antibodies cannot.16 Therefore, the efficient cytotoxicity combined with low cytokine release appears to be a feature unique to the F2B-containing T-BsAbs and is likely due to a combination of the low affinity and a potential novel epitope. Studies to fully dissect the mechanism of decoupling cytokine release from cytotoxicity at a cellular level are underway.
Although CD8+ T cells are known to be the most effective at mediating T cell-dependent cytotoxicity owing to their ability to store preformed cytolytic granules such as perforin and/or granzyme, CD4+ T cells also induce cytotoxicity, albeit in a delayed manner.26,36,37 Accordingly, TNB-486-mediated lysis occurred by recruitment of both CD4+ and CD8+ T cells to a similar extent (Figure 4c), whereas the positive control induced a higher percentage of lysis when CD8+ T cells were used. Along with the potent anti-tumor efficacy and low cytokine release, the linear PK and longer in vivo half-life of TNB-486 (Figure 7a, 7b and Table 1) could translate to a lower frequency and ease of dosing in the clinic compared to blinatumomab, which requires continuous IV infusion with a pump over multiple days depending on dosing regimen.48
The low magnitude of cytokine release observed in the TDCC experiments with TNB-486 differentiates it from other T-BsAbs and shows promise as a safer alternative to molecules co-targeting CD19 and CD3 that have high rates of CRS in the clinic. In addition to the safety issues arising from CRS, there is also a higher incidence of neurotoxicity associated with T cell-based therapies that target CD19.15,49 Currently, it is unclear whether neurotoxicity is attributed to on-target toxicity or to damage caused by excessive CRS. Since neurotoxicity is observed in patients treated with CAR-Ts and T-BsAbs specific for targets such as CD20, CD22, and BCMA,50–53 it appears to be more likely that CRS plays a critical role in neurotoxicity. There is also considerable evidence that CD19-targeted neurotoxicity is driven by hyperinflammation arising from CRS and the resulting disruption of the blood-brain barrier.54,55 Therefore, efforts to mitigate CRS would also indirectly reduce incidence of associated neurotoxicity, making TNB-486 an attractive candidate for treatment of B cell malignancies. Historically, combination therapies have been shown to be most effective in cancers. Therefore, safer, and efficacious T cell engagers will allow combinations with other existing treatments to further improve the B-NHL treatment landscape.
In conclusion, the results shown in this work demonstrate that TNB-486 binds CD19 and CD3, resulting in depletion of CD19-expressing cells in preclinical models with minimal cytokine release. The low cytokine release profile suggests that TNB-486 could have a wider therapeutic window compared to other CD19xCD3 T-BsAbs in the clinic or in development. In addition, TNB-486 may have a more favorable dosing schedule owing to the longer half-life compared to single-chain variable fragment-based T-BsAbs. The anti-tumor efficacy in multiple models, low cytokine release, and favorable pharmacokinetics together strongly support the testing of TNB-486 in clinical trials for the treatment of B cell malignancies.
Materials and methods
Immunizations, next-generation sequencing, clonotype analysis and cloning
Methods used for immunization, NGS analysis and cloning were previously described in Harris et al.25 Briefly, UniRat™ animals were immunized using Titermax/Ribi or Complete Freund’s Adjuvant in combination with recombinant protein antigens (Antibody Solutions, CA, USA) in a 48-day protocol or genetic immunizations (Aldevron and MfD Diagnostics GmbH, Freiburg, Germany). For protein immunizations, animals received boosts of 10 µg of recombinant protein injected into each leg with the appropriate adjuvant. In the case of genetic immunizations, gold particles were coated with vectors containing cDNA of the target antigen, which were subsequently administered with a gene gun subcutaneously every 7 days. Plasma samples were collected to assess serum titers against the antigen by Enzyme-Linked Immunosorbent Assay (ELISA).
After approximately 7 weeks (protein antigen) or 10 weeks (DNA antigen) of immunization, total RNA was isolated from the draining lymph nodes of each animal. Ig heavy chain sequences were amplified using first-strand cDNA synthesis and 5′ RACE by Polymerase Chain Reaction, following methods previously described in Harris et al.,25 and then purified by gel extraction.
Next-generation sequencing was completed using the MiSeq platform (Illumina) with 2 × 300 paired-end reads. Indexing labels were added by primer extension to enable multiplexing of samples. Approximately 100,000 paired reads covered each sample, and those that showed alignment of less than 20 nucleotides to a human Ig locus were discarded. The merged forward and reverse reads of VH regions were translated into open-reading frames. Subsequently, the framework and CDR regions were identified using IGBLAST (https://www.ncbi.nlm.nih.gov/igblast/). Agglomerative clustering was used to define clonotypes (defined by CDR3 protein sequences with at least 80% sequence similarity) within the samples. CDR3 clonotypes were ranked by the percent of total reads in a sample defined by that clonotype. The clonotypes with the greatest abundance were prioritized for high-throughput cloning into an expression vector containing a CH1-deleted human IgG1 Fc region. The resulting plasmids were validated by Sanger sequencing. Plasmids were transformed into E. coli grown in Luria-Bertani (LB) culture media and then purified to enable transient transfection of HEK 293 cells in 96-well format. After of expression, supernatants containing antibody were harvested and clarified by centrifugation.
Tumor cell lines
Nalm6 (catalog# CRL-3273), Raji (catalog# CCL-86), Daudi (catalog# CCL-213), Ramos (catalog# CRL-1596), K562 (catalog# CCL-243), SU-DHL-4 (catalog# CRL-2957) and SU-DHL-10 (catalog# CRL-2963) were purchased from American Type Culture Collection (ATCC) and maintained as per vendor’s instructions. RI-1 (catalog # ACC 585) and WSU-DLCL2 (catalog# ACC 575) were purchased from DSMZ (German Collection of Microorganisms and Cell Cultures GmbH) and maintained as per vendor’s instructions. All cell lines except K562 were grown in complete media containing Roswell Park Memorial Institute (RPMI)-1640 + 10% fetal bovine serum (FBS) + 1% PenStrep. K562 cell line was grown in Iscove’s Modified Dulbecco’s Medium+ 10% FBS + 1% PenStrep. All cells were grown at 37°C and 8% CO2.
Cell binding by flow cytometry
Clarified supernatants from 96-well transfections containing antibodies or purified antibodies were evaluated for cell binding by flow cytometry. All washes and dilutions of cells, antibodies, and reagents were performed using flow buffer (1X phosphate-buffered saline (PBS), 1% bovine serum albumin (BSA), 0.1% NaN3, pH 7.4). Staining was performed in a round-bottom 96-well plate (Corning) seeded at 100,000 cells/well and all incubations were performed at 4°C or on ice. CD19+ tumor cells were incubated for 30 minutes with pre-diluted test antibodies (-curves) or 1:5 diluted HEK 293 supernatants containing antibodies (for primary screens) in a total volume of 50 μL. The cells were washed twice with 200 µL flow buffer. The cells were then incubated for 30 minutes with detection antibody (Goat F(ab’)2 Anti-Human IgG-PE, Southern Biotech catalog # 2042–09) at a final concentration of 0.625 µg/mL in flow buffer. Following two washes, the cells were resuspended in a final volume of 150 µL of flow buffer. The cells were analyzed on a BD FACSCelesta or a Guava easyCyte 8-HT flow cytometer. At least 3000 events were collected, and phycoerythrin (PE) geometric mean fluorescence intensity was plotted as a fold over background represented by cells incubated with secondary detection antibody only.
Expression and purification of CD19xCD3 bispecific antibody
TNB-486, PC and NC were expressed in ExpiCHO cells, according to manufacturer’s instructions (Thermo Fisher Scientific, high titer protocol). Clarified supernatants were harvested and affinity purified using CaptureSelect™ CH1-XL resin (Thermo Fisher Scientific). Antibodies were further purified by anion exchange (Q Sepharose Fast Flow, Cytiva ID) to remove any product-related impurities. All proteins were analyzed by size exclusion chromatography-ultra-high performance liquid chromatography (SEC-UPLC) and sodium dodecyl sulfate Polyacrylamide Gel Electrophoresis to confirm their size and purity.
Biophysical characterization (Tm, Tonset)
The thermal denaturation experiment was performed using Capillary- Differential scanning calorimetry (DSC). Briefly, a temperature ramp of 1°C was performed from 20°C to 120°C and a 16 sec filtering period. DSC scans were analyzed using MicroCal Origin® software. After subtraction of the respective buffer scan, protein scans were normalized against the protein concentration. The Tonset (the temperature that immediately precedes the protein unfolding event), was manually determined. Non two-state peak fitting was used to determine the Tm values and to calculate the enthalpies for each peak present in the normalized scans.
Thermal stress and stability characterization
TNB-486 was concentrated to 10 mg/mL in 20 mM citrate and 0.1 M NaCl pH 6.2. Presence of high molecular weight species (%HMW) was determined by SEC-UPLC before and after temperature stress (1 month at 37°C).
Quantification of CD19 expression
To estimate antibodies bound per cell (ABC), cell lines were stained at a saturating concentration of commercially available PE-labeled anti-CD19 antibody and tested via flow cytometry using BD Quantibrite beads as per manufacturer’s instructions (BD catalog # 340495). Briefly, a PE-labeled CD19 antibody (BioLegend, clone HIB19) was used to establish saturating dose curves on the indicated cells. To generate the calibration curve, the BD Quantibrite calibration beads were also run on the flow cytometer, using the same settings used to acquire the cells. A standard curve was obtained by plotting log (Molecules of Equivalent Soluble Fluorochrome) vs log (Mean Fluorescent Intensity) of the beads. Using the dose-dependent mean fluorescent intensity values for the antibody and the linear regression equation obtained from the Quantibrite PE beads, the number of PE-conjugated antibodies bound to each cell, ABC was determined by extrapolation of the standard curve. Estimated antigen density (or CD19 molecules per cell) was obtained by multiplying ABC values by 2.56
Calculation of cell surface affinity by Scatchard plot
Cell surface affinity of TNB-486 to human CD19 was assessed using an ALL cell line expressing human CD19 (Nalm6). Antibodies bound per cell was determined as described earlier using Alexa Fluor 488-labeled TNB-486 (Alexa Fluor labeling kit by Thermo Fisher Scientific), and Bangs Lab Quantum™ Alexa Fluor® 488 was used for generating the standard curve. To calculate affinity of the molecule (KD), antibodies bound per cell was determined and used to further calculate free TNB-486 concentration using the formula:
Isolation of PBMCs
PBMCs were isolated from peripheral blood leukapheresis pack (StemCell Technologies) using standard Ficoll-Paque PLUS reagent (GE healthcare) as per manufacturer’s instructions. PBMCs were cryopreserved in CryoStor® freezing media (StemCell Technologies) in liquid nitrogen tanks until required for further analysis.
For isolation of T cell subsets of B cells, PBMCs isolated by the Ficoll-Paque method described earlier were removed from liquid nitrogen storage and thawed in a bead bath set at 37°C. The cells were transferred dropwise into a 50 mL conical centrifuge tube containing culture media (RPMI-1640 + 10% FBS+ 1% PenStrep) and recovered by centrifugation at 500 x g for 10 minutes at room temperature. The PBMCs were then resuspended in culture medium (RPMI-1640 + 10% FBS + 1% PenStrep) and proceeded for T cell isolation either immediately or after resting overnight at 37°C in a CO2 buffered incubator. CD3+ T cells were isolated by negative selection using the Pan T cell isolation kit (Miltenyi) as per manufacturer’s protocol. CD4+ or CD8+ subsets were isolated from pan T cells by negative selection microbeads for CD8+ or CD4+, respectively. B cells were isolated form thawed and/or rested PBMCs using the Pan B cell isolation kit (Miltenyi). For some ex vivo sample cytotoxicity assays, the B-CLL cell isolation kit was used (Miltenyi) as per manufacturer’s protocol.
In vitro cytotoxicity assays
TDCC was determined by flow cytometric analysis of live CD20-positive tumor cells after 48 hours of co-culture with Pan T cells (Effector:Target is 5:1). After incubation, the supernatant was aliquoted into 96-well plates and stored at −80°C until needed for cytokine analysis by MesoScale Discovery (MSD). The cells were stained with Near Infrared(IR) Live/Dead marker (Thermo Fisher Scientific, 1:1000) and/or with human TrueStain Fc block (BioLegend, 1:20) followed by staining for different lineage markers with fluorescently labeled antibodies (BioLegend, anti-CD20 (clone 2H7), anti‑CD8a (clone RPA-T8) and anti-CD4 (clone OKT4), 5 ul/test). After incubation, cells were washed with flow buffer (1X PBS + 1% BSA + 0.1% NaN3) and resuspended at 1.00E+06 cells/mL in flow buffer. Equal volume of CountBright absolute counting beads (Invitrogen) was added to each well for uniform sample acquisition on a BD FACSCelesta flow cytometer. FlowJoTM version 10.6 was used for data analysis. To determine the percent of specific lysis of CD19+ B cells in normal cells, previously frozen PBMCs were thawed and incubated with serially diluted test antibodies without adjusting the E:T ratio. The averaged count of live CD20-positive tumor cells from the wells without antibody was used to set the background value. The percent of specific lysis was calculated as follows: percent specific lysis = (live target cell number without BsAb − live target cell number with BsAb)/(live target cell number without BsAb) × 100. To measure CD19+ B cell lysis over a time course of 24, 48, 72 and 96 hours, isolated pan T cells were used as effector cells and isolated B cells from the same donor were used as target cells at a E:T ratio of 10:1 and incubated with serially diluted test antibodies. The averaged count of live CD20-positive tumor cells from the wells without antibody was used to set the background value. The percent of specific lysis was calculated as described above.
Ex vivo cytotoxicity assay
The ability of TNB-486 to kill CD19+ tumor cells from CLL or DLBCL patient-derived samples (Discovery Life Sciences and IQ Biosciences) was determined by flow cytometric analysis of live CD20-positive cells after incubation for 24 or 48 hours in culture media. Cytotoxicity of TNB-486 is measured at 200 nM and the effector:target cell ratio was either set to 10:1 or left unchanged. The cells were stained with Near IR Live/Dead marker (Thermo Fisher Scientific, 1:1000) and with human TruStain Fc block (BioLegend, 1:20) followed by staining for different lineage markers with fluorescently labeled antibodies (BioLegend, anti-CD20 (clone 2H7), anti‑CD8a (clone RPA-T8) and anti-CD4 (clone OKT4)). After incubation, cells were washed with flow buffer (1X PBS + 1% BSA + 0.1% NaN3) and resuspended in 100 µL flow buffer. Samples were acquired on BD FACSCelesta flow cytometer and analyzed using FlowJoTM version 10.6.
Measurement of cytokines
Measurement of cytokines from the supernatant of in vitro TDCC assays or from serum of cynomolgus monkeys in the PK study was performed with the MSD U-plex platform. The preparation of standards and samples were performed as per manufacturer’s instructions (MSD U-plex platform, cat# K15067L-4 and K15068L-2) and analyzed on a Meso Quickplex SQ 120 instrument.
Measurement of cytotoxic granules
Measurement of granzyme B and perforin from the supernatant of samples and preparation of standards were performed as per manufacturers’ instructions (Invitrogen, Cat#BMS2027 and Cell Sciences Cat# CDK102A, respectively). The day prior to measuring perforin, the capture antibody was prepared in 1X PBS and 100 µL was seeded into each well of a Nunc flat-bottom 96-well plate (Invitrogen), covered with adhesive and stored at 4°C. All other reagents are provided in the kits inclusive of substrate solution and stop solution. The absorbance was read at 450 nm as the primary wavelength and 620 nm as the reference wavelength on a SpectraMax i3X plate reader (Molecular Devices).
Activation and proliferation of T cells
To measure activation of CD4+ and CD8+ T cells, isolated Pan T cells were incubated with diffuse large B cell lymphoma cells, RI-1 (E:T is 10:1), in the presence of TNB-486, positive control or negative control antibodies for 20 hours at 37°C and 8% CO2. After incubation, cells were stained with fluorescently labeled antibodies for CD8 (clone RPA-T8), CD4 (clone OKT4) and CD69 (clone FN50). To determine activation of T cells, the percent increase in CD69-positive population was measured by flow cytometry. Proliferation assay setup is similar except the isolated T cells were first labeled with 2 µM CFSE dye (Invitrogen) before co-culturing with RI-1 cells and test articles for 5 days. Proliferation was determined by calculating the %CFSE positive proliferating CD8+ and CD4+ T cells by BD FACSCelesta flow cytometer.
In vivo efficacy studies
Disseminated mouse xenograft efficacy study
Each immune-compromised female Central Institute for Experimental Animals- NOG (NOD.Cg-Prkdcscid IL2rgtm1Sug/JicTac) (CIEA-NOG) mouse (4–5 weeks, Taconic Biosciences) was injected IV via tail vein with 1 × 106 Raji-luc cells (Aragen Biosciences Inc). On day six post-implantation (pi), each mouse was adoptively transferred IV with 1 × 107 PBMCs from a single donor (StemCell Technologies). On day seven, the mice were randomized into six treatment groups or one control group that received equivalent volume of the vehicle. All six treatment groups contained 5 animals each and each animal received the following dose per injection: 1) 10 µg NC, 2) 10 ng PC, 3) 10 ng TNB-486. 4) 100 ng TNB-486, 5) 1 µg TNB-486, or 6) 10 µg TNB-486.Test article (TNB-486, PC or NC) was injected every 5 days (Q5D) on days 6, 11 and 16 in the respective groups and at respective doses. Bioluminescent imaging and body weight measurements were determined every 3 days for 3 weeks.
Subcutaneous mouse xenograft efficacy studies
CIEA-NOG mice (5–7 weeks, Taconic Biosciences) were subcutaneously injected with 10 × 106 SU-DHL-10 or Nalm-6 with 50% Matrigel in lower right flank. When tumor volume reached 200 mm3, the mice were randomized into seven groups. Group one did not receive any PBMCs or drug treatment (group 1 – vehicle only). Groups 2–7 were adoptively transferred IV with 1 × 107 PBMCs (StemCell Technologies) from a single donor. One day after PBMC injection, test article or vehicle was injected into each animal in groups 2–7 as follows: 2) vehicle, 3) 100 µg NC, 4) 1 µg TNB-486, 5) 10 µg TNB-486, 6) 100 µg TNB-486, or 7) 100 µg PC. Test article or vehicle was injected every 4 days in the respective groups and at respective doses for a total of 6 treatments. Twice per week, the length and width of each tumor were measured with digital calipers in two perpendicular dimensions, and the tumor volume was calculated using this formula: (width2 × length)/2. Clinical signs and body weight were also assessed twice per week. The murine studies were performed in accordance with protocols approved by the Aragen Bioscience’s Institutional Animal Care and Use Committee (IACUC).
Mouse PK evaluation
The PK of TNB-486 was evaluated in BALB/c mice. 18 male BALB/c mice were administered a single tail vein injection of 1 or 10 mg/kg of TNB-486 (n = 3/group * 6 groups). Serum samples were collected for 14 days post-dose at set times ranging from 30 minutes to 14 days. Serum TNB-486 concentrations were measured by Perkin Elmer’s Human Immunoglobulin G subclass 4 Pharmacokinetic Kit (Cat# AL304 C) as per manufacturer’s instructions. PK parameters were estimated using Phoenix WinNonlin version 7.0 software (Pharsight Corp). 2-compartmental analysis was used to determine the PK of TNB-486 at each dose level.
Cynomolgus PK evaluation
The PK of TNB-486 was evaluated in nine female cynomolgus monkeys following a single IV bolus dose of 0.1, 1 or 10 mg/kg dose (Altasciences Preclinical Seattle LLC). Serum samples were collected for 21 days post-dose at set times ranging from 30 minutes to 21 days. Serum TNB-486 concentrations were measured by an antigen-based ELISA. 3 µg/mL of recombinant huCD19-His (Acro Biosystems Cat# CD9-H52H2) was coated onto a 96-well, Nunc MaxiSorp™ flat-bottom plate (Thermo Fisher Scientific, Cat# 44–2404-21). Plates were sealed and incubated overnight at 4°C, protected from light. After overnight incubation, plates were washed 3 times with 180 µL of wash buffer (Tris-Buffered Saline + 0.05% tween20). Wash supernatant was removed completely after the final wash step, and 200 µL of assay buffer (wash buffer + 1% milk w/v) was added as a blocking reagent. Plates were sealed and then incubated at room temperature for 1 hour and subsequently washed 6 times with 180 µL of wash buffer. After washing, 100 µL of reference standard or sample analytes diluted in assay buffer were added to each well and incubated at room temperature for 2 hours. After another wash cycle with 9 washes, 100 µL of 1 µg/mL of an anti-idiotypic antibody against anti-CD3 of TNB-486 (CD3_F2B_antiID, generated by Teneobio) was added and incubated for 30 minutes at room temperature. Following 9 washes, 100 µL of 1:5000 diluted mouse anti-Rat IgG2a-HRP was added as a detection reagent (Southern Biotech Cat# 3065–05) and was incubated for 15–20 minutes at room temperature. Following this incubation and a wash cycle with 9 washes, 100 µL of Ultra 3,3′,5,5′-Tetramethylbenzidine (TMB) ELISA Substrate (Thermo Fisher Scientific cat# 34028) was added until a blue color developed (1–2 minutes). 100 µL of 2 N sulfuric acid was added to stop the reaction prior to absorbance measurements on a plate reader (SpectraMax i3X, Molecular Devices). The concentration of TNB-486 in the samples are directly proportional to absorbance values at 450 nm and were interpolated from a four-parameter logistic curve fit of the standard curve. PK parameters were estimated using Phoenix WinNonlin version 7.0 software (Pharsight Corp). 2-compartmental analysis was used to determine the PK of TNB-486 at each dose level.
Supplementary Material
Acknowledgments
We thank iQ Biosciences for the in vitro cytotoxicity study comparison with blinatumomab, Aragen Biosciences for performing the murine in vivo studies, and Altasciences Preclinical Seattle LLC for the cynomolgus monkey PK study. We also thank Maya Leabman and Rong Deng for mouse and cynomolgus monkey PK data analyses.
Disclosure statement
All authors are current or former employees of Teneobio, Inc. with equity interests.
Author contributions
U.S.R., H.K.M.C., N.D.T., K.P., and H.S.U. contributed to the study design, analysis of results and manuscript preparation; NGS-based repertoire analysis was completed by N.D.T., K.H., and A.A.B.; molecular biology was conducted by K.H., L.M.D., K.D., and H.O.; U.S., H.K.M.C., B.J., and H.S.U. contributed to expression, purification and biophysical characterization of antibodies; lead discovery experiments, namely ELISAs and cell binding flow cytometry assays were designed and run by K.H., H.O., K.D., D.P., and U.S.R; U.S.R., and K.P., designed and executed in vitro cell-based assays; H.K.M.C., N.D.T., S.I., B.B., and W.v.S. contributed to the design and analysis of in vivo experiments; B.B., D.P., H.K.M.C., U.S.R., and K.P. contributed to the planning and execution of ex vivo experiments; U.S.R., H.K.M.C., and K.P., prepared figures; B.B., U.S., S.I., W.v.S, and R.B. contributed to data analysis and critical review of the data; the manuscript was written by U.S.R., H.K.M.C., and K.P.; all authors read and approved the submitted version.
List of abbreviations.
| µg | Microgram |
| µL | Microliter |
| µM | Micromolar |
| ABC | Antibody Bound per Cell |
| ADC | Antibody-Drug Conjugate |
| ALL | Acute Lymphoblastic Leukemia |
| AUCinf | Area Under the Curve from Zero to Infinity |
| B-ALL | B cell Acute Lymphoblastic Leukemia |
| BCMA | B cell Maturation Antigen |
| BiTE | Bispecific T cell Engager |
| B-NHL | B cell Non-Hodgkin’s Lymphoma |
| BSA | Bovine Serum Albumin |
| BsAb | Bispecific Antibody |
| CAR-T | Chimeric Antigen Receptor T cells |
| CD | Cluster of Differentiation |
| CDR | Complementarity-Determining Regions |
| CFSE | Carboxyfluorescein Succinimidyl Ester |
| CH1 | Constant Heavy Chain 1 |
| CIEA | Central Institute for Experimental Animals |
| CL | Clearance |
| CLL | Chronic Lymphoblastic Leukemia |
| Cmax | Maximum Serum Concentration |
| CO2 | Carbon dioxide |
| CRS | Cytokine Release Syndrome |
| CTL | Cytotoxic T Lymphocyte |
| DLBCL | Diffuse Large B Cell Lymphoma |
| dpi | Days Post Implantation |
| DTC | Dissociated Tumor Cells |
| E:T | Effector:Target |
| EC50 | Half Maximal Effective Concentration |
| ELISA | Enzyme Linked Immunosorbent Assay |
| FBS | Fetal Bovine Serum |
| Fc | Fragment Crystallizable |
| FDA | Food and Drug Administration |
| g | Grams |
| IACUC | Institutional Animal Care and Use Committee |
| IFN | Interferon |
| IL | Interleukin |
| IR | Infrared |
| IV | Intravenous |
| KD | Equilibrium Dissociation Constant |
| kg | Kilogram |
| L | Liter |
| M | Molarity |
| mAb | Monoclonal Antibody |
| mg | Milligram |
| MHC | Major Histocompatibility Complex |
| mL | Milliliter |
| mM | Millimolar |
| MSD | Meso Scale Discovery |
| N | Normality |
| NaCl | Sodium chloride |
| NaN3 | Sodium Azide |
| NC | Negative Control |
| ng | Nanogram |
| NGS | Next-Generation Sequencing |
| nM | Nanomolar |
| nm | Nanometer |
| NOD | Non-Obese Diabetic |
| NOG | NOD.Cg-Prkdcscid IL2rgtm1Sug/JicTac |
| ns | Not Significant |
| PBMC | Peripheral Blood Mononuclear Cell |
| PBS | Phosphate-Buffered Saline |
| PC | Positive Control |
| PE | Phycoerythrin |
| pg | Picogram |
| pi | Post Implantation |
| PK | Pharmacokinetics |
| pM | Picomolar |
| pMHC | peptide-Major Histocompatibility Complex |
| Q5D | Every 5 Days |
| RACE | Rapid Amplification of cDNA Ends |
| RPMI | Roswell Park Memorial Institute |
| SD | Standard Deviation |
| SEC | Size Exclusion Chromatography |
| SEM | Standard Error Mean |
| t1/2 | Half-Life |
| T-BsAb | T cell-engaging Bispecific Antibody |
| TCR | T cell Receptor |
| TDCC | T cell Dependent Cellular Cytotoxicity |
| TGI | Tumor Growth Inhibition |
| Tm | Melting Temperature |
| TNF | Tumor Necrosis Factor |
| Tonset | Onset Temperature |
| UPLC | Ultra Performance Liquid Chromatography |
| VH | Variable Domain |
| VSS | Volume of distribution at Steady State |
Ethics statement
PBMCs from healthy, deidentified donors were isolated from leukapheresis pack (StemCell Technologies). Human PBMCs were collected in accordance with scientific, ethical, and regulatory guidelines. Animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Rat maintenance and immunizations were carried out by certified animal facilities in the U.S. (Antibody Solutions, Sunnyvale, CA) and Germany (Aldevron and MfD Diagnostics GmbH, Freiburg, Germany) in accordance with national and international guidelines, with protocols reviewed by IACUC boards in the U.S. and comparable government boards in Germany. Mouse and cynomolgus monkey PK studies as well as mouse xenograft studies were performed by AAALAC accredited facilities following internal approval by their IACUC boards (Aragen Biosciences; Altasciences Preclinical Seattle LLC).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–15. doi: 10.1182/blood-2016-01-643569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaudhari K, Rizvi S, Syed BA.. Non-Hodgkin lymphoma therapy landscape. Nat Rev Drug Discov. 2019;18(9):663–64. doi: 10.1038/d41573-019-00051-6. [DOI] [PubMed] [Google Scholar]
- 3.Hagemeister F. Rituximab for the Treatment of Non-Hodgkin’s Lymphoma and Chronic Lymphocytic Leukaemia. Drugs. 2010;70(3):261–72. doi: 10.2165/11532180-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Blanc V, Bousseau A, Caron A, Carrez C, Lutz RJ, Lambert JM. SAR3419: an Anti-CD19-maytansinoid immunoconjugate for the treatment of B-cell malignancies. Clin Cancer Res. 2011;17(20):6448–58. doi: 10.1158/1078-0432.CCR-11-0485. [DOI] [PubMed] [Google Scholar]
- 5.Naddafi F, Anti-CD DF. 19 monoclonal antibodies: a new approach to lymphoma therapy. Int J Mol Cell Med. 2015;4:143–51. [PMC free article] [PubMed] [Google Scholar]
- 6.Hammer O. CD19 as an attractive target for antibody-based therapy. mAbs. 2012;4(5):571–77. doi: 10.4161/mabs.21338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watkins MP, Bartlett NL. CD19-targeted immunotherapies for treatment of patients with non-Hodgkin B-cell lymphomas. Expert Opin Investig Drugs. 2018;27(7):601–11. doi: 10.1080/13543784.2018.1492549. [DOI] [PubMed] [Google Scholar]
- 8.Horna P, Nowakowski G, Endell J, Boxhammer R. Comparative assessment of surface CD19 and CD20 expression on B-cell lymphomas from clinical biopsies: implications for targeted therapies. Blood. 2019;134(Supplement_1):5345–5345. doi: 10.1182/blood-2019-129600. [DOI] [Google Scholar]
- 9.Ingle GS, Chan P, Elliott JM, Chang WS, Koeppen H, Stephan J-P, Scales SJ. High CD21 expression inhibits internalization of anti-CD19 antibodies and cytotoxicity of an anti-CD19-drug conjugate. Br J Haematol. 2008;140(1):46–58. doi: 10.1111/j.1365-2141.2007.06883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolska-Washer A, Robak T. Safety and tolerability of antibody-drug conjugates in cancer. Drug Saf. 2019;42(2):295–314. doi: 10.1007/s40264-018-0775-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Topp MS, Kufer P, Gökbuget N, Goebeler M, Klinger M, Neumann S, Horst H-A, Raff T, Viardot A, Schmid M, et al. Targeted therapy with the t-cell–engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in b-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;29(18):2493–98. doi: 10.1200/JCO.2010.32.7270. [DOI] [PubMed] [Google Scholar]
- 12.Goebeler M-E, Bargou R. Blinatumomab: a CD19/CD3 bispecific T cell engager (BiTE) with unique anti-tumor efficacy. Leuk Lymphoma. 2016;57(5):1021–32. doi: 10.3109/10428194.2016.1161185. [DOI] [PubMed] [Google Scholar]
- 13.Maude SL, Barrett D, Teachey DT, Grupp SA. Managing Cytokine Release Syndrome Associated With Novel T Cell-Engaging Therapies. Cancer J Sudbury Mass. 2014;20(2):119–22. doi: 10.1097/PPO.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, Grupp SA, Mackall CL. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–95. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dholaria BR, Bachmeier C, Locke F. Mechanisms and Management of Chimeric Antigen Receptor T-Cell Therapy Related Toxicities. BioDrugs Clin Immunother Biopharm Gene Ther. 2019;33:45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trinklein ND, Pham D, Schellenberger U, Buelow B, Boudreau A, Choudhry P, Clarke SC, Dang K, Harris KE, Iyer S, et al. Efficient tumor killing and minimal cytokine release with novel T-cell agonist bispecific antibodies. mAbs. 2019;11(4):639–52. doi: 10.1080/19420862.2019.1574521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zafra CLZD, Fajardo F, Zhong W, Bernett MJ, Muchhal US, Moore GL, Stevens J, Case R, Pearson JT, Liu S, et al. Targeting Multiple Myeloma with AMG 424, a Novel Anti-CD38/CD3 Bispecific T-cell–recruiting Antibody Optimized for Cytotoxicity and Cytokine Release. Clin Cancer Res. 2019;25(13):3921–33. doi: 10.1158/1078-0432.CCR-18-2752. [DOI] [PubMed] [Google Scholar]
- 18.Baliga R, Li K, Manlusoc M, Hinton PR, Ng DC, Tran MH, Shan B, Lu H, Saini A, Rahman S, et al. Abstract 5664: a bispecific IgM antibody format for enhanced T cell dependent killing with minimal cytokine release. Cancer Res. 2020;80(16 Supplement):5664–5664. [Google Scholar]
- 19.Lobner E, Wachernig A, Gudipati V, Mayrhofer P, Salzer B, Lehner M, Huppa JB, Kunert R. Getting CD19 Into Shape: expression of Natively Folded “Difficult-to- Express” CD19 for Staining and Stimulation of CAR-T Cells. Front Bioeng Biotechnol. 2020;8:49. doi: 10.3389/fbioe.2020.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teplyakov A, Obmolova G, Luo J, Gilliland GL. Crystal structure of B-cell co-receptor CD19 in complex with antibody B43 reveals an unexpected fold. Proteins. 2018;86(5):495–500. doi: 10.1002/prot.25485. [DOI] [PubMed] [Google Scholar]
- 21.Meeker TC, Miller RA, Link MP, Bindl J, Warnke R, Levy R. A unique human B lymphocyte antigen defined by a monoclonal antibody. Hybridoma. 1984;3(4):305–20. doi: 10.1089/hyb.1984.3.305. [DOI] [PubMed] [Google Scholar]
- 22.Ghetie MA, Picker LJ, Richardson JA, Tucker K, Uhr JW, Vitetta ES. Anti-CD19 inhibits the growth of human B-cell tumor lines in vitro and of Daudi cells in SCID mice by inducing cell cycle arrest. Blood. 1994;83(5):1329–36. doi: 10.1182/blood.V83.5.1329.1329. [DOI] [PubMed] [Google Scholar]
- 23.Zola H, MacArdle PJ, Bradford T, Weedon H, Yasui H, Kurosawa Y, et al. Preparation and characterization of a chimeric CD19 monoclonal antibody. Immunol Cell Biol. 1991;69(Pt 6):411–22. [DOI] [PubMed] [Google Scholar]
- 24.Clarke SC, Ma B, Trinklein ND, Schellenberger U, Osborn MJ, Ouisse L-H, Boudreau A, Davison LM, Harris KE, Ugamraj HS, et al. Multispecific antibody development platform based on human heavy chain antibodies. Front Immunol. 2018;9:3037. doi: 10.3389/fimmu.2018.03037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harris KE, Aldred SF, Davison LM, Ogana HAN, Boudreau A, Brüggemann M, Osborn M, Ma B, Buelow B, Clarke SC, et al. Sequence-based discovery demonstrates that fixed light chain human transgenic rats produce a diverse repertoire of antigen-specific antibodies. Front Immunol. 2018;9:889. doi: 10.3389/fimmu.2018.00889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Due H, Brøndum RF, Young KH, Bøgsted M, Dybkær K. MicroRNAs associated to single drug components of R-CHOP identifies diffuse large B-cell lymphoma patients with poor outcome and adds prognostic value to the international prognostic index. BMC Cancer. 2020;20(1):237. doi: 10.1186/s12885-020-6643-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Katib AM, Smith MR, Kamanda WS, Pettit GR, Hamdan M, Mohamed AN, Chelladurai B, Mohammad RM, et al. Bryostatin 1 down-regulates mdr1 and potentiates vincristine cytotoxicity in diffuse large cell lymphoma xenografts. Clin Cancer Res Off J Am Assoc Cancer Res. 1998;4(5):1305–14. [PubMed] [Google Scholar]
- 28.Porakishvili N, Kardava L, Jewell AP, Yong K, Glennie MJ, Akbar A, Lydyard PM. Cytotoxic CD4+ T cells in patients with B cell chronic lymphocytic leukemia kill via a perforin-mediated pathway. Haematologica. 2004;89:435–43. [PubMed] [Google Scholar]
- 29.Wu Z, Cheung N-KV. T cell engaging bispecific antibody (T-BsAb): from technology to therapeutics. Pharmacol Ther. 2018;182:161–75. doi: 10.1016/j.pharmthera.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, Nichols KE, Suppa EK, Kalos M, Berg RA, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121(26):5154–57. doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maloney DG. Anti-CD19 CAR T cell therapy for lymphoma — off to the races! Nat Rev Clin Oncol. 2019;16(5):279–80. doi: 10.1038/s41571-019-0183-7. [DOI] [PubMed] [Google Scholar]
- 32.Asnani M, Hayer KE, Naqvi AS, Zheng S, Yang SY, Oldridge D, Ibrahim F, Maragkakis M, Gazzara MR, Black KL, et al. Retention of CD19 intron 2 contributes to CART-19 resistance in leukemias with subclonal frameshift mutations in CD19. Leukemia. 2020;34(4):1202–07. doi: 10.1038/s41375-019-0580-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, Levine JE, Qayed M, Grupp SA, Boyer M, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. 2018;24(10):1504–06. doi: 10.1038/s41591-018-0146-z. [DOI] [PubMed] [Google Scholar]
- 34.Jacoby E. Relapse and Resistance to CAR-T Cells and Blinatumomab in Hematologic Malignancies. Clin Hematol Int. 2019;1(2):79–84. doi: 10.2991/chi.d.190219.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jabbour E, Düll J, Yilmaz M, Khoury JD, Ravandi F, Jain N, Einsele H, Garcia‐Manero G, Konopleva M, Short NJ, et al. Outcome of patients with relapsed/refractory acute lymphoblastic leukemia after blinatumomab failure: no change in the level of CD19 expression. Am J Hematol. 2018;93(3):371–74. doi: 10.1002/ajh.24987. [DOI] [PubMed] [Google Scholar]
- 36.Bukhari A, Lee ST. Blinatumomab: a novel therapy for the treatment of non-Hodgkin’s lymphoma. Expert Rev Hematol. 2019;12(11):909–18. doi: 10.1080/17474086.2019.1676717. [DOI] [PubMed] [Google Scholar]
- 37.Affimed Places Cancer Immunotherapy Candidate AFM11 on Clinical Hold after Patient Death [Internet]. GEN - Genet. Eng. Biotechnol. News2018 [cited 2020 Dec 11]; Available from: https://www.genengnews.com/news/affimed-places-cancer-immunotherapy-candidate-afm11-on-clinical-hold-after-patient-death/ [Google Scholar]
- 38.Reusch U, Duell J, Ellwanger K, Herbrecht C, Knackmuss SH, Fucek I, Eser M, McAleese F, Molkenthin V, Gall FL, et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19(+) tumor cells. mAbs. 2015;7(3):584–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bartlett NL, Sehn LH, Assouline SE, Bosch F, Magid Diefenbach CS, Flinn I, Hong J, Kim WS, Matasar MJ, Nastoupil LJ, et al. Managing cytokine release syndrome (CRS) and neurotoxicity with step-fractionated dosing of mosunetuzumab in relapsed/refractory (R/R) B-cell non-Hodgkin lymphoma (NHL). J Clin Oncol. 2019;37(15_suppl):7518–7518. doi: 10.1200/JCO.2019.37.15_suppl.7518. [DOI] [Google Scholar]
- 40.Le RQ, Li L, Yuan W, Shord SS, Nie L, Habtemariam BA, Przepiorka D, Farrell AT, Pazdur R. FDA approval summary: tocilizumab for treatment of chimeric antigen receptor t cell-induced severe or life-threatening cytokine release syndrome. Oncologist. 2018;23(8):943–47. doi: 10.1634/theoncologist.2018-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teneobio, Inc . A Multicenter, Phase 1, Open-label, Dose-escalation and Expansion Study of TNB-383b, a Bispecific Antibody Targeting BCMA in Subjects With Relapsed or Refractory Multiple Myeloma. clinicaltrials.gov. 2020. Available from: https://clinicaltrials.gov/ct2/show/NCT03933735. [Google Scholar]
- 42.TNB383B.0001: A Multicenter, Phase 1, Open-Label, Dose-Escalation And Expansion Study of TNB-383B, a Bispecific Antibody Targeting BCMA in Subjects with Relapsed or Refractory Multiple Myeloma. Blood American Society of Hematology. [Google Scholar]
- 43.Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies. mAbs. 2010;2(3):256–65. doi: 10.4161/mabs.2.3.11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Renner C, Held G, Ohnesorge S, Bauer S, Gerlach K, Pfitzenmeier J-P, Pfreundschuh M. Role of perforin, granzymes and the proliferative state of the target cells in apoptosis and necrosis mediated by bispecific-antibody-activated cytotoxic T cells. Cancer Immunol Immunother CII. 1997;44(2):70–76. doi: 10.1007/s002620050357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Irvine DJ. Function-specific variations in the immunological synapses formed by cytotoxic T cells. Proc Natl Acad Sci. 2003;100(24):13739–40. doi: 10.1073/pnas.2536626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. T cell killing does not require the formation of a stable mature immunological synapse. Nat Immunol. 2004;5(5):524–30. doi: 10.1038/ni1058. [DOI] [PubMed] [Google Scholar]
- 47.Riquelme E, Carreño LJ, González PA, Kalergis AM. The duration of TCR/pMHC interactions regulates CTL effector function and tumor-killing capacity. Eur J Immunol. 2009;39(8):2259–69. doi: 10.1002/eji.200939341. [DOI] [PubMed] [Google Scholar]
- 48.Amgen Inc . BLINCYTO®: FDA Package Insert. US Food Drug Adm. Website. 2021. Feb 25 Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125557s013lbl.pdf. [Google Scholar]
- 49.Stein AS, Schiller G, Benjamin R, Jia C, Zhang A, Zhu M, Zimmerman Z, Topp MS. Neurologic adverse events in patients with relapsed/refractory acute lymphoblastic leukemia treated with blinatumomab: management and mitigating factors. Ann Hematol. 2019;98(1):159–67. doi: 10.1007/s00277-018-3497-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shalabi H, Wolters PL, Martin S, Tamula MA, Roderick MC, Struemph K, Kane E, Yates B, Delbrook C, Mackall CL, et al. Systematic evaluation of neurotoxicity in children and young adults undergoing CD22 chimeric antigen receptor-T cell therapy. J Immunother (1991). 2018;41(7):350–58. Hagerstown Md 1997. doi: 10.1097/CJI.0000000000000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu Y, Shen R, Hao R, Wang S, Ho M. Highlights of antibody engineering and therapeutics 2019 in San Diego, USA: bispecific antibody design and clinical applications. Antib Ther. 2020;3(2):146–54. doi: 10.1093/abt/tbaa012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garfall AL, Lancaster E, Stadtmauer EA, Lacey SF, Dengel K, Ambrose DE, Chen F, Gupta M, Kulikovskaya I, Vogl DT, et al. Posterior reversible encephalopathy syndrome (pres) after infusion of anti-bcma car t cells (CART-BCMA) for multiple myeloma: successful treatment with cyclophosphamide. Blood. 2016;128(22):5702–5702. doi: 10.1182/blood.V128.22.5702.5702. [DOI] [Google Scholar]
- 53.Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, Vogl DT, Weiss BM, Dengel K, Nelson A, et al. B cell maturation antigen–specific CAR T cells are clinically active in multiple myeloma. J Clin Invest. 2019;129(6):2210–21. doi: 10.1172/JCI126397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gust J, Hay KA, Hanafi L-A, Li D, Myerson D, Gonzalez-Cuyar LF, Yeung C, Liles WC, Wurfel M, Lopez JA, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017;7(12):1404–19. doi: 10.1158/2159-8290.CD-17-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siegler EL, Kenderian SS. Neurotoxicity and cytokine release syndrome after chimeric antigen receptor t cell therapy: insights into mechanisms and novel therapies. Front Immunol. 2020;11. doi: 10.3389/fimmu.2020.01973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pannu KK, Joe ET, Iyer SB. Performance evaluation of quantiBRITE phycoerythrin beads. Cytometry. 2001;45:250–58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.