

ABSTRACT
South Asian (SA) Canadian immigrants have a higher risk of developing certain immune-mediated inflammatory diseases compared to non-migrant SAs. We sought to investigate the effect of migration on the gut metagenome and to identify microbiological associations between migration and conditions that may influence the development of immune-mediated inflammatory diseases. Metagenomic analysis of 58 first-generation (GEN1) SA immigrants and 38 unrelated Canadian born children-of-immigrants (GEN2) determined that the time lived in Canada was associated with continued changes in gut microbial communities. Migration of GEN1 to Canada early in life results in a gut community with similarities to GEN2 SA Canadians and non-SA North Americans. Conversely, GEN1 immigrants who arrived recently to Canada exhibited pronounced differences from GEN2, while displaying microbial similarities to a non-migrating SA cohort. Multivariate analysis identified that community composition was primarily influenced by high abundance taxa. Prevotella copri dominated in GEN1 and non-migrant SAs. Clostridia and functionally related Bacteroidia spp. replaced P. copri dominance over generations in Canada. Mutually exclusive Dialister species occurred at differing relative abundances over time and generations in Canada. This shift in species composition is accompanied by a change in genes associated with carbohydrate utilization and short-chain fatty acid production. Total energy derived from carbohydrates compared to protein consumption was significantly higher for GEN1 recent immigrants, which may influence the functional requirements of the gut community. This study demonstrates the associations between migration and the gut microbiome, which may be further associated with the altered risk of immune-mediated inflammatory diseases observed for SA Canadians.
KEYWORDS: Gut metagenome, immune-mediated inflammatory disease, immigration, scfa, prevotella, dialister
Introduction
Diversity of the gut microbiome has been examined in a variety of divergent populations1, revealing associations with culture, geography, diet, lifestyle, and migration.2–8 Immune-mediated inflammatory diseases, which are a significant factor reducing the quality of life and increasing mortality, are particularly prevalent in westernized regions such as North America.9–11 These diseases include Type 1 diabetes, Type 2 diabetes mellitus, asthma, allergies, and inflammatory bowel disease, including ulcerative colitis and Crohn’s disease. Risk factors promoting immune-mediated inflammatory diseases include a diet rich in saturated fats, trans-fats, and refined sugars, particularly for obese and diabetic individuals.11 Along with North America, India has recently become another epicenter of type 2 diabetes mellitus incidence, with onset now occurring for people with a lower BMI and at a younger age.12,13 The incidence rate of inflammatory bowel disease in India is also rising, approaching the levels observed in European and North American countries, nations currently with the highest prevalence rates.14–16
Of the 1.2 million immigrants that arrived in Canada between 2011 and 2016, India and Pakistan were among the top five most prevalent countries of birth.17 Previous studies identified a higher incidence of type 2 diabetes mellitus in South Asian (SA) immigrant Canadians, compared to multi-generation Canadians, and non-SA immigrants.18 Type 2 diabetes mellitus in SA immigrants has been associated with time since migration to Canada, suggesting that the likelihood of disease increases as dietary acculturation occurs.19 The incidence of inflammatory bowel disease is currently low for first-generation SA immigrants, regardless of time spent living in Canada.18 After a generation was born in Canada, the incidence rate increased, becoming similar between Canadian-born children of SA immigrants and multi-generational Canadians.18,20
Over the last half century, there has been a shift from the consumption of coarse grains such as sorghum, barley, rye, maize, and millet, to the consumption of rice and wheat in SA countries.21 Consumption of simple sugars and dairy fat, such as ghee, has also risen.21 Carbohydrate consumption is typically high for SAs, particularly in the case of vegetarians, while the intake of fiber is low among both vegetarians and non-vegetarians.20,22,23 In the United States, low and high carbohydrate consumption were both associated with increased mortality rates, while moderate carbohydrate consumption resulted in the lowest risk of metabolic disease development, especially when plant-derived proteins were most often consumed.24 For SA immigrants, an increase in the consumption of fats and dietary cholesterol and a decrease in carbohydrates, fiber, and folate were directly associated with length of residence in North America.25,26
Diet influences the gut microbiota, affecting both the Firmicutes/Bacteroidetes (F/B) and Prevotella/Bacteroides (P/B) ratios.5,7,27 A low-fat, high-fiber diet has previously been associated with a reduced risk of obesity and inflammatory diseases, including type 2 diabetes mellitus and inflammatory bowel disease, and a high ratio of P/B.28–31 However, this is not always consistent, and other researchers have observed differing trends between obesity and the F/B, or P/B ratios.32–36 A higher relative abundance of Prevotella has been observed in Asian populations, populations consuming non-westernized diets, and vegetarians compared to omnivores.4,6,7,27,37,38 Fiber and non-digestible carbohydrates are fermented by bacteria in the lower gut, resulting in the production of many compounds, including short-chain fatty acids (SCFA).39 SCFAs are consumed by enterocytes in the intestine, suppressing gut inflammation and reducing the incidence of related diseases.40,41 In the absence of fiber, certain polysaccharide degrading gut bacteria can utilize mucus glycoproteins (mucin glycans) as nutrients, which results in an eroded colonic mucus barrier and inflammation.42 Western diets may alter the P/B ratio, affecting the amount and type of SCFA produced, potentially contributing to a proinflammatory state in the gut.43
We sought to investigate the effect of migration on the gut metagenome of Canadians of SA ancestry. Specifically, we were interested in determining taxonomic and functional differences in the gut microbiome of first-generation SA immigrants (GEN1) compared to Canadian-born children of unrelated SA immigrants (GEN2), and how these differences may influence the incidence rate of various immune-mediated inflammatory diseases for each generation. Furthermore, we hope to identify changes in the gut microbiome of SA Canadians as a function of time in Canada, compared to the gut microbiome of an Indian and a North American population, constituting of persons who have not migrated from their respective countries of birth. This will allow us to better identify characteristics of microbiota that are susceptible to change over time, and those that are a stable reflection of early life exposure. It has been previously observed that migration to North America can significantly impact gut microbiota for other first- and second-generation immigrants.7 We hope to further this research by showing that these changes occur across many immigrant communities, while exploring how the SA Canadian community is specifically affected, leading to the high immune-mediated inflammatory disease prevalence rates previously observed for this population.
Results
Participant characteristics and dietary intake measurements are consistent between generations, estimated socioeconomic status influenced by time in Canada
The metagenomes of 58 first-generation and 38 second-generation Canadians, age 18–35, who identify ethnically as South Asian (SA) were sequenced (Table 1, Figure S1). First-generation (GEN1) Canadians were defined as those individuals who migrated to Canada, while second-generation (GEN2) Canadians are the offspring of unrelated GEN1 migrants. Ethnicity was self-declared by participants.
Table 1.
Participant Characteristics
| Characteristics | GEN1 (n = 58) | GEN2 (n = 38) |
|---|---|---|
| Age, mean (SD) | 24.3 (4.5) | 23.3 (3.8) |
| Male Sex, count (%) | 30 (52) | 20 (53) |
| BMI, mean (SD) | 24.8 (5.8) | 24.7 (6.2) |
| Immigration Age, mean (SD) | 16 (9.4) | NA |
| Immigration as Adult (>18), count (%) | 24 (43) | NA |
| Self-Reported Ethnicity, count (%) | ||
| Indian | 20 (34) | 6 (15) |
| Pakistani | 7 (12) | 5 (13) |
| Tamil | 5 (8) | 4 (10) |
| Punjabi | 2 (3) | 6 (16) |
| Sri Lankan | 1 (2) | 6 (16) |
| Bangladeshi | 6 (10) | 1 (3) |
| Bengali | 3 (5) | 2 (5) |
| Gujarati | 3 (5) | 1 (3) |
Generation (GEN), years after immigration (YAI), and immigration as an adult were examined. Immigration as an adult was defined as participants who immigrated over the age of 18. YAI was examined as both a continuous and categorical variable, divided into tertiles; recent immigrants (3 months to 4 years), moderately recent immigrants (4 to 14 years), and early immigrants (greater than 14 years since immigration) (Figure S1). There was no significant correlation between generation or YAI and self-declared ethnicity. A greater frequency of GEN2 Canadians participated in the study at a younger age. YAI and immigration age were significantly correlated (ANOVA, p < .01, adjusted R2 = 0.75). There was no significant difference between Body Mass Index (BMI) or Waist Circumference (WC) between generations, YAI, immigration as an adult, or age (Table S1A and S1B).
Socioeconomic status for each participant was estimated by identifying the value of socioeconomic status factors for the participants’ respective dissemination areas using the 2016 Canadian Census. There was no significant difference in these estimated socioeconomic status values between generations, combined with or as independent factors (Table S1C). Estimated median total household income was observably lower for both GEN1 and GEN2, compared to the median values of all the dissemination areas belonging to the census districts in which participants resided. The difference between our cohort and the median census district values was particularly apparent for recent and moderately recent YAI groups (Figure S1). Population density was also higher for both GEN1 and GEN2 compared to the median values of the census district regions. Due in part to the age restrictions of the study, we observed that the majority of GEN1 recent immigrants were adults when they immigrated, while none of the GEN1 early immigrants were adults when they immigrated. No significant effect of immigration as an adult on socioeconomic status was identified.
No significant differences were observed between GEN1 and GEN2 nutrition, based on food frequency questionnaire data. On average, participants consumed significantly higher amounts of omega-3 fat, protein, total sugar, and sodium (Figure S1), and significantly lower vitamin D, fruit servings, and vegetable servings than the recommended daily allowances. GEN1, but not GEN2, participants also consumed significantly lower calcium and fiber compared to the recommended daily allowance values.
High interpersonal variability in human stool microbiota at species level
We performed Illumina metagenomic sequencing on stool samples from each study participant and obtained a mean of 18,196,704 ± 1,045,084 150 bp paired-end reads per participant. After removing low-quality sequences, PCR duplicates, and human DNA, a mean of 81.0 ± 5.63% of the sequence data remained for metagenomic analysis (median = 82.4%, minimum = 17.5%, maximum = 86.2%).44,45 Nonpareil analysis estimated that the metagenomes were sequenced at a depth resulting in a mean coverage of 86.5 ± 4.75% of the complete metagenome.
Across the 96 participant samples, we observed eight unique phyla of bacteria, archaea, and viruses in the metagenomes after removing rare species. The most abundant phyla were Firmicutes, Bacteroidetes, and Actinobacteria (Table S1D). Bacteroidetes and Firmicutes were inversely correlated (Pearson r = −0.89), while the most abundant species were Prevotella copri, Eubacterium rectale, and Faecalibacterium prausnitzii, and the most prevalent species was Subdoligranulum sp., which was identified in all participants.
Abundant taxa strongly influence community composition
The species composition of GEN1 and GEN2 SA Canadians was compared using Bray–Curtis PCoA plots (Figure S2, Table S2A). While the estimated alpha diversity did not differ significantly between generations when tested independently (Table S1E), beta-diversity (MetaPhlAn2 normalized) distances were significantly associated with the most abundant taxa (Table S1F) within a sample and the Chao1 alpha-diversity (ADONIS R2 = 0.37, p < .05 and R2 = 0.019, p < .05, respectively). When the data were then re-scaled using cumulative sum scaling normalization (Figure 1) 46,47 we continued to identify a significant effect of the maximum abundant taxa, but with a reduced impact (ADONIS R2 = 0.22, p < .05). Sequencing coverage and the percentage of human DNA did not significantly affect the community, while total mapped genes and identified gene families did (Table S2B).
Figure 1.
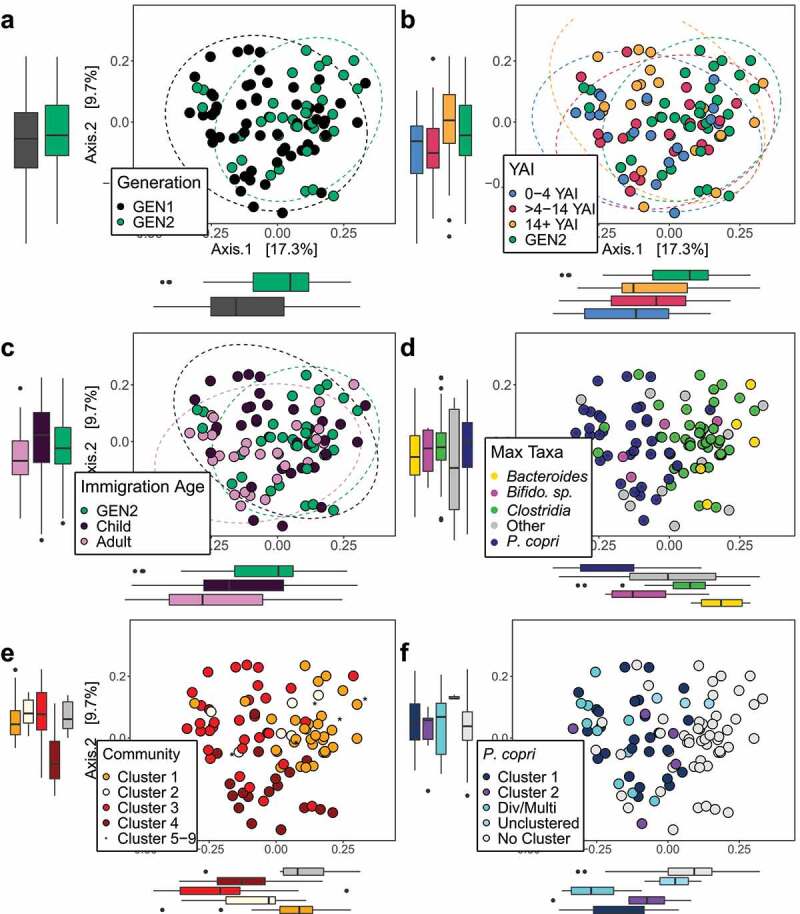
Bray–Curtis PCoA plots of community dissimilarities shows separation of participants by (a) generation, (b) years after immigration (YAI) groups, (c) immigration as an adult (IA), and (d) maximum abundant taxa. Boxplots show the dissimilarity distances for each group, on each primary axis. (b) The recent immigrant group (0–4 YAI) and (c) those who immigrated as adults show the greatest dissimilarity from GEN2 (Pairwise ADONIS, p < .01). The samples are primarily separating by (d) maximum abundant taxa (ADONIS, R2 = 0.22, p < .05). Participants also show Bray–Curtis PCoA separation by the (e) four main species-composition community clusters identified using UPGMA-GMD hierarchical clustering and by the (f) two main P. copri pangenome clusters. Participants with divergent or multi-strain (Div/Multi) P. copri contained highly polymorphic P. copri that mapped outside of the pangenome species complex, suggesting the presence of unique strains or a greater proportion of multiple P. copri strains within these communities. ‘Unclustered’ samples contained P. copri pangenomes that formed individual UPGMA-GMD clusters. ‘No Cluster’ samples contained a P. copri relative abundance median <1% and were filtered out when following the default PanPhlAn analysis parameters
We determined that participants were grouped into nine UPGMA-GMD-based clusters based on metagenomic community composition, with four major clusters having two or more participants and encompassed 96% of the samples (Figure 1, Table S3A). We measured the association between clusters and categorical variables and found that the clusters are most strongly associated with the maximum taxa (Goodman–Kruskal Tau test of association, Table S3B). PanPhlAn analysis and UPGMA-GMD clustering identified two major P. copri pangenome clusters, labeled as Cluster 1 and 2. ‘No Cluster’ samples contained a P. copri relative abundance average and median of 2.12% and 0.13%, respectively, and were automatically filtered out when following the default PanPhlAn analysis parameters (Table S3C). ‘Unclustered’ samples contained P. copri pangenomes that formed individual UPGMA-GMD clusters. We identified a third group of dissimilar P. copri identified in P. copri maximum taxa participants, but with gene content so divergent that they were also filtered out when following the default PanPhlAn analysis parameters. StrainPhlAn was used to determine whether this was due to strain specific divergence or the presence of a multi-strain species complex. StrainPhlAn determined a P. copri average polymorphism rate of 2.4 ± 1.7% for all participants and 4.1 ± 1.2% for the divergent cluster. The P. copri average polymorphism rate was 2.2 ± 1.7% for GEN2 and 2.6 ± 1.7% for GEN1, with the highest average polymorphism rate reported at 2.8 ± 2.0% for GEN1 recent immigrants compared to other YAI groups. Average polymorphism values for other species of interest are shown in Table 2.
Table 2.
Species Average Polymorphism Rates
| Species | Percent average polymorphism ± s.d. |
|---|---|
| P. copri all participants | 2.48 ± 1.67 |
| P. copri divergent cluster | 4.06 ± 1.16 |
| P. copri GEN1 | 2.61 ± 1.67 |
| P. copri GEN1 recent immigrants | 2.83 ± 1.97 |
| P. copri GEN2 | 2.22 ± 1.69 |
| Bacteroidia spp. | 0.50 ± 0.14 |
| B. fragilis | 0.56 ± 0.35 |
| B. plebeius | 0.48 ± 0.36 |
| B. dorei | 0.63 ± 0.36 |
| B. ovatus | 0.47 ± 0.27 |
| B. stercoris | 0.46 ± 0.35 |
| B. uniformis | 0.53 ± 0.41 |
| B. vulgatus | 0.63 ± 0.41 |
| B. intestinalis | 0.17 ± 0.14 |
| B. longum | 0.63 ± 0.35 |
| E. rectale | 0.91 ± 0.35 |
| F. prausnitzii | 4.57 ± 1.15 |
| D. invisus | 0.90 ± 0.48 |
| D. succinatiphilus | 1.29 ± 0.76 |
We examined P. copri and Prevotella sp. metagenome assembled genomes (MAGs) using Anvi’o in order to compare the Prevotella genomes identified in this study with the genome clades (A, B, C, D) identified by Tett et al., 2019.48 Samples without identifiable P. copri, labeled as ‘No Cluster’ in the previously described analysis, were not analyzed. Within the remaining data, we identified two P. copri genome bins, referred to as ‘Bin 1ʹ and ‘Bin 2ʹ. Genome average nucleotide identity (ANI) clustered P. copri Bin 1 with reference genomes from Clades C and D, while P. copri Bin 2 clustered with reference genomes from Clade A (Figure S3). The Prevotella sp. genome bins primarily clustered outside of the reference clades, except for ‘Bin 3ʹ, which clustered with a reference genome from Clade B. The average relative abundance and variability (rate of polymorphism) were identified for ‘Bin 1ʹ and ‘Bin 2ʹ in each sample (Table S3A). These two P. copri bins were identified in all examined participants at differing abundances. We observed that the proportion of Bin 1 was 1.9x higher than that of Bin 2 in the Div/Multi cluster, compared to an average of 0.6x higher for all other samples in Clusters 1, 2, and Unclustered samples. There was no observable association between abundance of the different bins between GEN1 and GEN2.
We also investigated the association of each metadata factor of interest with the microbial composition, both individually and combined, using Redundancy Analysis (Figure 2). As with the ADONIS results, maximum abundant taxa, as well as generation, BMI, YAI, immigration as an adult or child, and Chao1 were all significantly associated with the species composition when tested either individually or as a mixed model (p < .05) (Table S3E). The Principal Component Analysis biplot shows that BMI, YAI, and immigration as an adult significantly influence species abundances in a similar direction.
Figure 2.
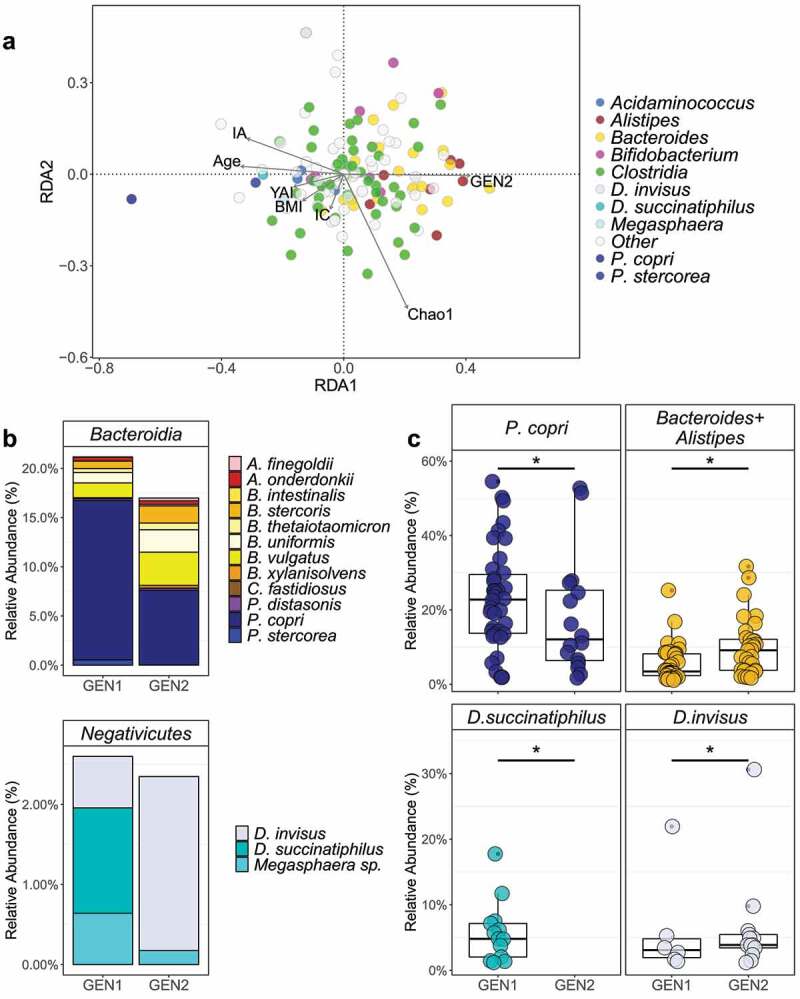
(a) The RDA biplot shows the significant effect of many factors (labeled solid arrows), including generation, age, YAI, BMI, Chao1, immigration as an adult (IA), and immigration as a child (IC) on the relative abundance of each microbial species. Unlike the PCoA plots, each point represents a species, not a participant. (b) The relative abundance averages and (c) dispersion of Bacteroidia and Negativicutes species identified as significantly associated with GEN1 or GEN2 (Table 3 and Table S3D). Prevotella, Bacteroides, and Alistipes species were often found to co-occur, while D. succinatiphilus and D. invisus were never identified in the same participant
Shifting from P. copri and D. succinatiphilus to Bacteroides spp. and D. invisus for GEN1 to GEN2 Canadians
We examined whether specific taxa were differentially abundant between generations via multiple methods (Figure 2, Table 3, Figure S4, Table S3D). Within the Negativicutes class, D. succinatiphilus was significantly more abundant in GEN1, while D. invisus was significantly more abundant in GEN2. Within the Bacteroidia class, multiple species from the genus Bacteroides were significantly more abundant in the GEN2, while P. copri and P. stercorea were significantly more abundant in GEN1. Different species from the class Clostridia, Akkermansia muciniphila, and Bifidobacterium catenulatum were also found to be significantly enriched in either GEN1 or GEN2.
Table 3.
Differentially Abundant Species Between Generations
| Species | Class | Enriched | GEN1 (mean RA%) |
GEN2 (mean RA%) |
LEfSE LDA Score |
FitDO Odds Ratio | FitZigLogFC |
|---|---|---|---|---|---|---|---|
| Bifidobacterium catenulatum | Actinobacteria | GEN2 | 8.01E-02 | 1.09E-01 | ns | 0.15*** | 2.70*** |
| Alistipes finegoldii | Bacteroidia | GEN2 | 3.95E-02 | 2.81E-01 | ns | 0.00*** | 2.65*** |
| Alistipes onderdonkii | Bacteroidia | GEN2 | 3.58E-01 | 3.62E-01 | ns | 0.50** | 1.47*** |
| Bacteroides intestinalis | Bacteroidia | GEN2 | 3.25E-02 | 1.58E-01 | ns | 0.14*** | 2.33*** |
| Bacteroides stercoris | Bacteroidia | GEN2 | 7.57E-01 | 1.74E+00 | 3.35** | 1.80** | ns |
| Bacteroides thetaiotaomicron | Bacteroidia | GEN2 | 3.95E-01 | 6.79E-01 | ns | 0.55** | 1.41*** |
| Bacteroides uniformis | Bacteroidia | GEN2 | 1.06E+00 | 2.28E+00 | 3.54* | 0.59*** | ns |
| Bacteroides vulgatus | Bacteroidia | GEN2 | 1.50E+00 | 3.38E+00 | 3.90** | 0.62*** | ns |
| Bacteroides xylanisolvens | Bacteroidia | GEN2 | 1.03E-01 | 2.65E-01 | 2.89* | 0.28*** | ns |
| Coprobacter fastidiosus | Bacteroidia | GEN2 | 2.09E-04 | 3.45E-02 | 2.614* | ns | 2.81*** |
| Parabacteroides distasonis | Bacteroidia | GEN2 | 1.77E-01 | 2.01E-01 | ns | 0.24*** | 2.04*** |
| Prevotella copri | Bacteroidia | GEN1 | 1.62E+01 | 7.56E+00 | 4.48* | 1.78*** | ns |
| Prevotella stercorea | Bacteroidia | GEN1 | 5.46E-01 | 4.72E-02 | ns | 8.52*** | −2.22*** |
| Clostridium hathewayi | Clostridia | GEN2 | 8.71E-03 | 3.93E-02 | 2.23* | ns | 2.59*** |
| Coprococcus sp. ART55 1 | Clostridia | GEN1 | 1.78E+00 | 9.12E-01 | ns | 1.77** | −1.35** |
| Roseburia unclassified | Clostridia | GEN2 | 2.42E-02 | 2.95E-01 | 3.20** | 0.00*** | 2.89*** |
| Ruminococcus sp. 5 1 39BFAA | Clostridia | GEN2 | 1.31E+00 | 2.28E+00 | 3.75** | 0.69** | ns |
| Dialister invisus | Negativicutes | GEN2 | 6.44E-01 | 2.17E+00 | 3.99** | 0.31*** | 2.04*** |
| Dialister succinatiphilus | Negativicutes | GEN1 | 1.32E+00 | 0.00E+00 | 3.83*** | Inf*** | −2.79*** |
| Megasphaera unclassified | Negativicutes | GEN1 | 6.41E-01 | 1.76E-01 | 3.23* | 2.66*** | ns |
| Akkermansia muciniphila | Verrucomicrobia | GEN2 | 3.84E-01 | 6.72E-01 | ns | 0.40*** | 1.40*** |
0.05 ≥ * > 0.01 ≥ ** > 0.001 ≥ ***; adjusted for multiple comparisons; ns = not significant
When BMI and Chao1 index values, which were significantly associated with beta diversity, were added to the differential analysis model comparing GEN1 and GEN2, P. copri was no longer associated with generation, suggesting the involvement of BMI and Chao1 values on the relative abundance of P. copri (Table S4A). Additionally, we found that Megamonas funiformis was significantly enriched in overweight and obese participants, compared to those with a normal BMI, when controlling for age, sex, and YAI (Table S4B).
All participants' samples contained either a species of Bacteroides, Alistipes, or P. copri, and 73% of participants had all three taxa. The relative abundance of P. copri was inversely proportional to these other Bacteroidia species. In contrast, no samples contained both D. invisus and D. succinatiphilus, while 50% of participants had one of the two species. Again, the relative abundances of these species are inversely proportional by generation, with no GEN2 participants reporting the presence of D. succinatiphilus.
Effect of time spent in Canada on the abundance of Dialister and Bacteroides spp
The effect of years after immigration (YAI, i.e., time since immigration) and immigration as an adult were compared between GEN1 and GEN2 (Figure 1). The Bray–Curtis beta diversity PCoA plot shows observable clustering by YAI group and by the immigration age. As with the generation analysis, the maximum taxa, as well as the Chao1 index, and immigration age were significantly associated with beta diversity (p < .05) (Table S2C). The Bray–Curtis distances differ significantly between the recent immigrant group (0–4 YAI) and GEN2 (ADONIS adjusted p-value = 0.002), and between immigration as an adult and GEN2, and immigration as an adult and immigration as a child (ADONIS adjust p-values 0.0006 and 0.03, respectively) (Table S2D). This difference was not observed between GEN2 and moderately recent (mid) or early immigrant groups, or between GEN2 and immigration as a child.
The change in the relative abundance of species between the YAI recent immigrant groups and GEN2 was specifically assessed. From classes Negativicutes and Bacteroidia, our results identified D. succinatiphilus and Acidaminococcus fermentans to be significantly enriched in GEN1 recent immigrants, and B. thetaiotaomicron and B. cellulosilyticus to be significantly reduced (Table S4C). D. succinatiphilus was significantly enriched in both participants who immigrated as children or as adults, compared to GEN2.
Negatively co-occurring species segregate into distinct microbial community types
We looked for species co-occurrence and relative abundance correlation via network analysis (Figure S5, and Table S5A). Within the largest network, consisting of both positively and negatively correlated species, we identified 10 network clusters. Clostridium spp. were identified within the top 10 most interconnected species, acting as hub species. To examine these relationships more closely, we created separate positive and negative correlation networks. Secondary positive correlation networks were identified to both contain correlated Negativicutes species including the positive correlation between D. succinatiphilus and M. funiformis.
All study participants carried at least one of the highly abundant and prevalent species, P. copri, Bacteroides uniformis, or Bifidobacterium longum. In fact, there was a strong negative association between P. copri and B. uniformis and B. longum, as well as the hub species C. bolteae (Figure 3). A Dirichlet-Multinomial Mixture (DMM) model identified two community types, with the top drivers including B. longum and B. uniformis for one community type, and P. copri as the top community driver for the other. The Dirichlet Component Value for P. copri was twice as high as any other contributing species value.
Figure 3.
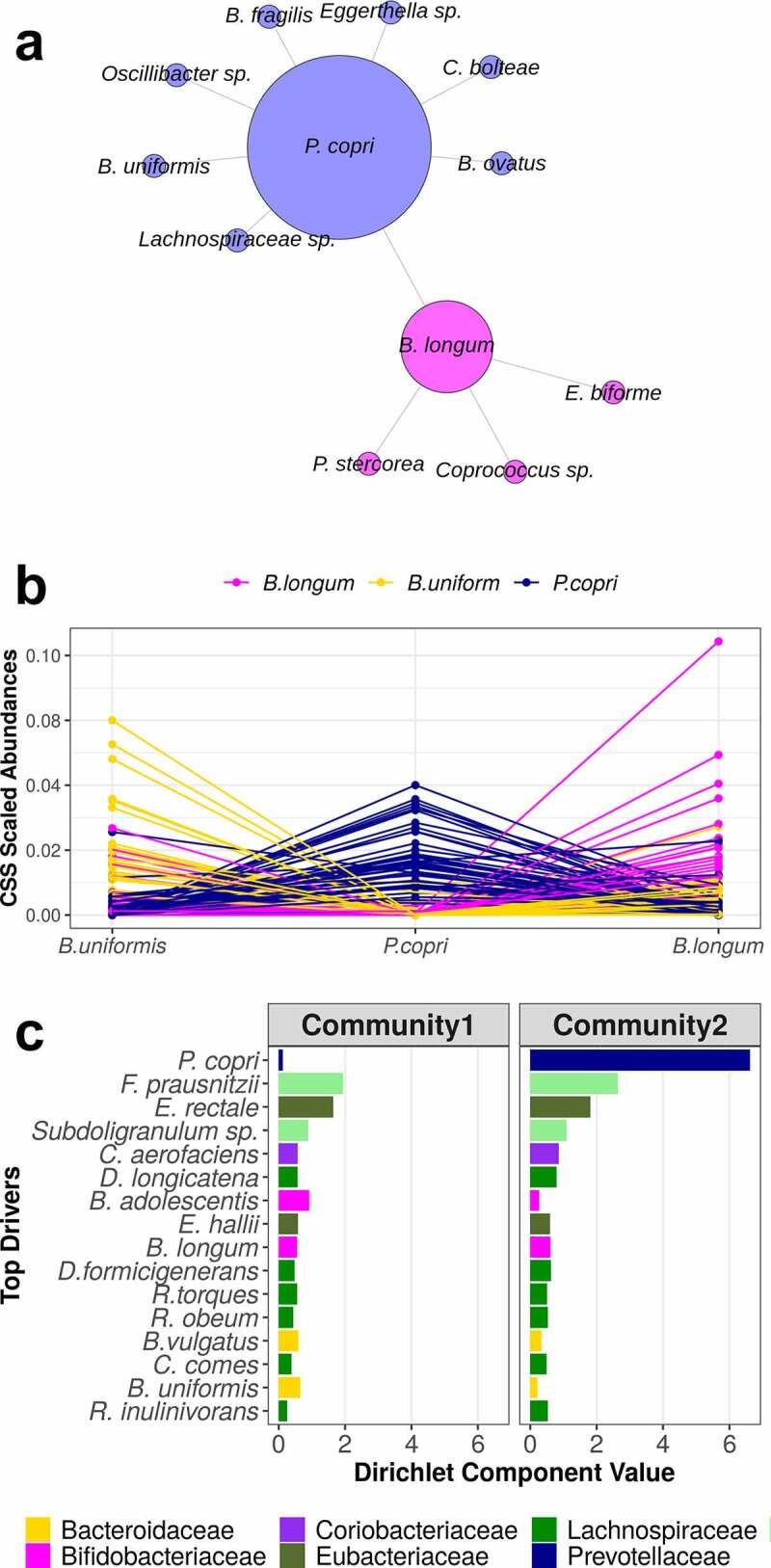
(a) A co-occurrence network of negatively correlated species. Each point represents a species, and the size of the point reflects the number of connections. The colors represent the walk-trap clusters identified in the network. The abundance of P. copri is negatively correlated with the abundance of B. longum and B. uniformis, among others. (b) Either P. copri, B. longum, or B. uniformis is present in every participant. Each point shows the abundance of the species, by participant (lines connecting participants), and the color of the point is the most abundant of these three species in each participant. Rarely do we observe equal abundance among these species, resulting in the negative correlations observed. (c) Dirichlet Multinomial Mixture (DMM) modeling identified two community clusters. The top community drivers (95 percentile) include these negatively correlated species. The highest driving component was determined as P. copri, suggesting this species has the greatest effect on community structure
Examining the transition from an Indian metagenome to ‘westernized’ metagenome
An Indian cohort (NCBI BioProject PRJNA397112) and an American Caucasian cohort (https://ibdmdb.org) were used as comparators to our Canadian SA immigrant data set. The sequence reads were retrieved and processed using the same method as the Canadian data. Of the two regions represented in the Indian cohort, Bhopal is an urban city in North-Central India whose participants consumed primarily a plant-based diet, and Kerala is a rural state in South-Western India whose participants consumed an omnivorous diet.37 The American participants all consumed an omnivorous diet. As with the Canadian data, the subset of participants used for this analysis self-declared to not have any known diseases and were considered healthy.
The Bray–Curtis beta diversity PCoA plot shows a separation by maximum abundant taxa on the primary axis, and by cohort on the secondary axis (Figure 4, Figure S2, Table S2E). As with the previous analysis, the maximum abundant taxa had the greatest effect on clustering on the cumulative sum scaling normalized data (ADONIS, R2 = 0.26, p = .001), followed by the effect of population-YAI and population-generation group comparisons (ADONIS, R2 = 0.13, p < .05, and R2 = 0.11, p < .05, respectively). A pairwise ADONIS test revealed that all population cohorts and YAI groups differed significantly from each other (p < .01) (Table S2F).
Figure 4.
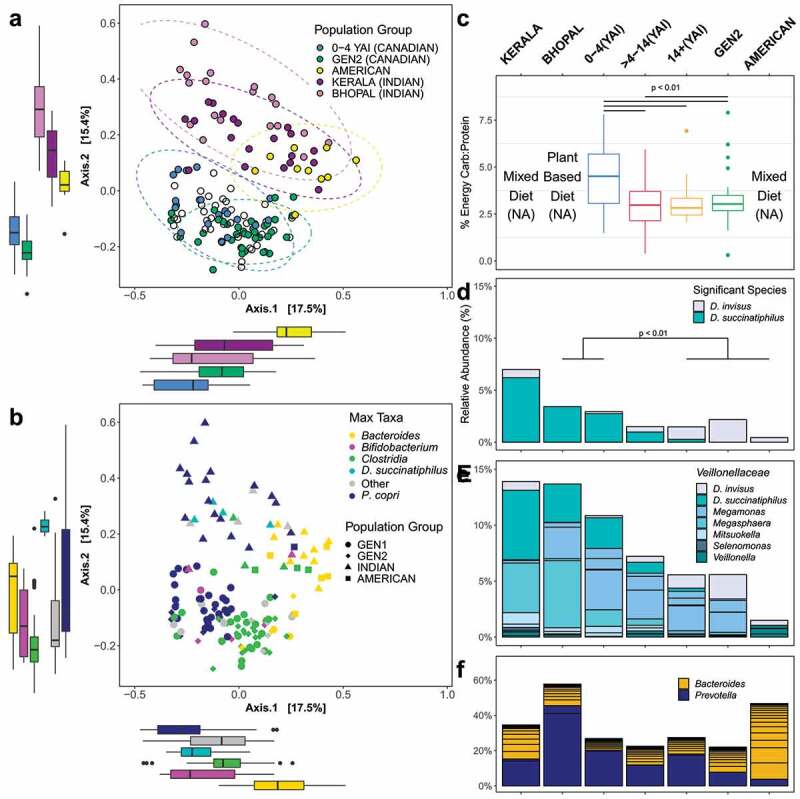
In the Bray–Curtis PCoA plots we observed (a) separation of the Canadian SA cohort from both the Indian and American cohorts. The open circles represent samples from GEN1 moderately recent (mid) and early immigrant groups. Boxplots show the dissimilarity distances for each group, on each axis. (b) This separation was primarily driven by the maximum taxa, with P. copri, Bacteroides spp., or D. succinatiphilus dominating the majority of the communities in the Indian cohort, and Bacteroides spp. or Clostridia spp. dominating the majority of communities in the American cohort. (c) A significantly higher percent of energy was derived from carbohydrates compared to proteins in the GEN1 recent immigrant group, compared to other YAI groups and GEN2, as determined by the food frequency questionnaire (FFQ). Participant macronutrient data was not available (NA) for external data sets. (d) D. succinatiphilus was significantly enriched (log-fold change (LFC) > 3, p < .001) in the Indian cohort and recent SA Canadian immigrants, compared to the early immigrants and GEN2. D. invisus is significantly depleted, particularly for the residents of Bhopal, who consumed primarily a plant-based diet (LFC < −1, p < .001). We extracted all the species of (e) Veillonellaceae (class Negativicutes), (f) Prevotella, and Bacteroides from the data to observe trends in the abundances of these taxa across all cohorts and YAI groups
Using the food frequency questionnaire data, we determined that the percent energy derived from carbohydrates compared to proteins was significantly higher in GEN1 recent immigrants compared to other YAI groups and GEN2 (Figure 4, Table S1G). The difference in the relative abundance of species between both geographic regions of the Indian cohort, each YAI group of GEN1, GEN2, and the American cohort was assessed (Table S4D). D. succinatiphilus was significantly enriched for Bhopal, Kerala, and GEN1 recent immigrants when compared to GEN2 and the American cohort. D. invisus was enriched in GEN2 compared to residents of Bhopal and GEN1 recent immigrants, but not when compared to residents of Kerala. We observed an overall decreasing relative abundance of most Veillonellaceae and Prevotella species in the westernized populations (American cohort and GEN2 Canadians) relative to the Indian cohort. Due to the large effect of population on beta diversity, we did not examine the potential metagenomic functional differences between these population cohorts.
Functional redundancy identified in gut microbiota
The presence and relative abundance of gene families were identified using HUMAnN2. These gene families were normalized and re-grouped into metabolic enzyme (MetaCyc) gene groups. We did not identify any gene families that were differentially abundant between generations in the SA Canadian cohort despite differences in the species composition, suggesting functional redundancy. To gain a greater insight into the functional similarities among microbial taxa, we created a positive Spearman correlation undirected network based on gene family presence for each species. This analysis links species based on their shared metabolic profiles (Table S5B).
The largest network contained six clusters made up of 60 species from the Firmicutes phylum (Figure 5). The two largest clusters were made up of many species from the class Clostridia, while D. succinatiphilus and D. invisus formed their own functional cluster. The second largest network contained two clusters made up of 37 species from the Bacteroidetes phylum. One cluster contained species of Alistipes, Bacteroides, P. copri, among others, while P. copri is positioned on the outside of the network, suggesting a greater proportion of unique features.
Figure 5.
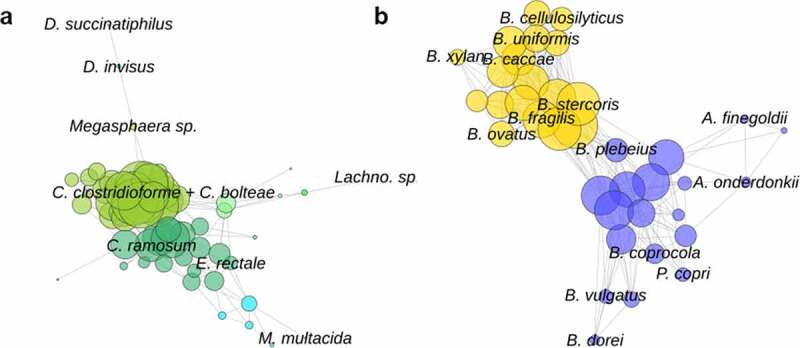
(a) The largest Spearman correlation network of gene family presence, consisting of Firmicutes species, and (b) the second largest network consisting of Bacteroidetes species. Each point represents a species, the size of the point reflects the number of connections. Colors separate the network clusters. Certain species of interest are labeled on the figure. Species with functional differences, such as D. succinatiphilus and D. invisus, are located on the periphery of the networks
Carbohydrate Metabolism of Bacteroidia, Negativicutes, and Clostridia spp
To understand the metabolic relationships between species from the phyla Firmicutes and Bacteroidetes, we sub-sampled the Canadian data set, extracted species of interest, and examined the MetaCyc enzyme gene families involved in the fermentation of simple carbohydrates and complex non-digestible fibers (i.e., polysaccharides) to the synthesis of short-chain fatty acids (SCFA) (Table S6A and S6B).
We used the presence of the enzyme families of interest to cluster the species into hierarchical groups by UPGMA (Figure 6, Figure S6). Many Bacteroides species harbored multiple gene families involved in these fermentation processes within a single species, while Negativicutes species and P. copri often contained only a single-gene family per fermentation process; missing many of the genes required to complete the fermentation pathway. We examined the average normalized Copies Per Million of these carbohydrate degradation gene families by generation (Table S6C), specifically focusing on the potential for cellulose, pectin, and xylan degradation. Although the differences were not significant, we observed a greater potential for xylan degradation in GEN1 and a greater potential for pectin and mucin glycan degradation in GEN2. P. copri was the most abundant endo-1,4-B-xylanase containing species, followed by B. uniformis. Potential mucin degrading enzymes were identified across the metagenome, present in B. longum and many Bacteroides, Alistipes, Clostridia, and Negativicutes species. Pectinesterase was present in P. copri but not in B. uniformis or B. longum.
Figure 6.
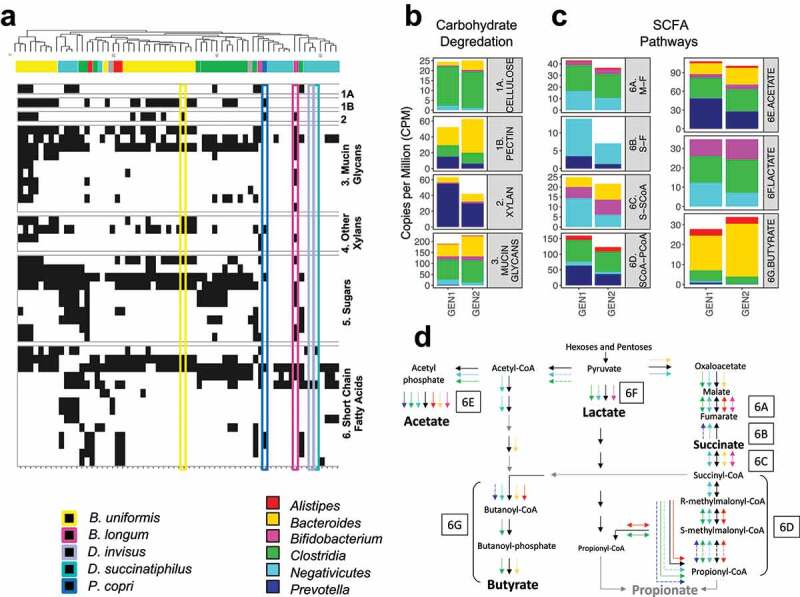
(a) Hierarchical clustering of carbohydrate degradation and SCFA fermentation enzyme gene families. The presence of a gene family for a given species is indicated by a black tile. The associated taxa are indicated by the filled color bar at the top, and the rectangular boxes highlight the species of interest (for a detailed version, refer to Figure S6A). The average normalized Copies Per Million per generation group for the enzyme gene families in (b) non-digestible carbohydrate degradation and (c) SCFA fermentation. Enzyme gene families are labeled 1A, 1B, 2 and 3 for carbohydrate degradation and 6A – 6G for SCFA fermentation. (d) The presence of gene families in each SCFA fermentation pathway and their associated taxa (complete annotated pathways Figure S6B). Solid arrows indicate gene families identified in more than one species within the taxa group, while dashed arrows indicate only one associated species. Color-coding of the arrows corresponds to that shown in the legend of Figure 6A. Gene families shown as gray arrows were not observed in the data set. Gene families shown as black arrows, but with no taxa association, were identified in species outside of our interest group. The boxed letters for each component of interest correspond to Figure 6C
The primary SCFAs produced from fermentation are acetate, butanoate (butyrate), lactate, and propionate-succinate.49 We calculated copies per million of these SCFA-associated genes averaged by generation, and visualized the SCFA production pathways (Figure 6, Figure S6, Table S6B). We did not identify any of the genes required for a propionate end-product or many genes required to complete other SCFA pathways. We observed that all of the succinate-propionate pathway genes were identified throughout various Negativicutes species, and different portions of the pathway were identified in selected species of Clostridia, Alistipes, Bacteroides, and P. copri. Lactate production gene families were identified primarily in Firmicutes, including Megamonas and Eubacterium. Butanoate production gene families were identified in Bacteroides species, as well as the Clostridia species that clustered in the Bacteroides dominant clade.
Discussion
The gut metagenome of multi-generational South Asian (SA) Canadian immigrants differs between generations and changes over time spent in Canada. The metagenome structure primarily differed by the maximum taxa within the gut. First-generation (GEN1) experienced a higher prevalence of P. copri maximum, a higher average abundance of P. copri, and the presence of D. succinatiphilus. Second generation (GEN2) experienced a higher prevalence of Bacteroides spp. or Clostridia spp. maximum, a higher average abundance of multiple Bacteroides spp., and a higher abundance of D. invisus. These differences were in turn reflected in differences in carbohydrate degradation and SCFA associated gene families.
We provide evidence that migration has an impact on the gut metagenome of multi-generation SA Canadians, resulting in changes in microbial taxonomic and genomic composition. We find that the gut community of GEN1 SA Canadians shifts from the structure that would have existed pre-migration, to a composition made up of a mix of stable taxa that were established during early life along with other taxa that are more characteristic of the general Canadian population (i.e., multi-generation North Americans). The extent of this shift appears to be dependent primarily on the time since immigration. This shift in microbial community structure likely contributes to the high prevalence of certain immune-mediated inflammatory diseases such as type 2 diabetes mellitus, which is observed in GEN1 SA Canadians with an incidence rate proportional to time lived in Canada.18,19 On the other hand, GEN2 SA Canadians’ gut communities have features which resemble multi-generation North Americans. This community structure likely contributes to the high prevalence of other immune-mediated inflammatory diseases, such as inflammatory bowel disease and asthma, which are prevalent in GEN2 SA Canadians, non-SA Canadians, and Americans alike.10,18
We determined that the participants’ metagenomes stratified primarily based on the most abundant taxa, which varied dramatically between individuals. P. copri was the most abundant taxa in early, mid, and recent GEN1 immigrants, while the relative abundance of Veillonellaceae decreased significantly over time since migration. This suggests that P. copri dominance may be determined by early life exposures and is more often maintained, while Veillonellaceae abundance may be more susceptible to change based on current exposures and environments. The P. copri dominant community has been described previously for other Asian and Mediterranean populations, associated with higher carbohydrate and lower fat diets, while the Bacteroides and Eubacterium dominant communities have been previously described for North American populations, westernized diets outside of North America, and omnivores compared to vegetarians.4–7,38 An analysis of the P. copri pangenome species complex revealed distinct subspecies clusters, including a divergent high diversity group of P. copri. A strain-based analysis suggests that this diversity is due to a higher proportion of multiple P. copri strains present within a given participant, as suggested by the higher percentage of polymorphic sites, which is consistent with previous studies that found multiple strains of P. copri within gut communities.48,50,51 Tett et al. 2019 observed that P. copri was nearly ubiquitous in ‘non-westernized’ populations (95.4% in non-westernized vs. 29.6% in westernized population) with over 60% of non-westernized individuals carrying all four previously examined P. copri clades. We identified two P. copri genomic bins in our data set that were found in differing proportions in all individuals containing P. copri. One of these bins clustered closely with Tett et al. 2019 Clade C and D reference genomes and was found to be in a higher proportion in the high diversity group, suggesting that a higher rate of polymorphism may be identified in P. copri associated with these clades. The other P. copri genomic bin is clustered closely with Clade A reference genomes. As with our data, co-presence of multiple P. copri clades has been previously identified in many individuals, with Clade A being the most prevalent in all individuals from both westernized and non-westernized locations.48
We did not observe a significant relationship between generation and P. copri diversity, but we did observe the greatest average percentage of P. copri polymorphic sites in GEN1 recent immigrants compared to GEN2 and GEN1 mid and early immigrants. This suggests that even though P. copri is observed as the maximum abundant species in certain GEN1 and GEN2 participants, multi-strain complexes may be more often present upon arrival and lost over time in Canada. The low average percentage of polymorphic sites identified in Bacteroides spp., consistent with previous research, suggests the presence of only a few strains per species co-occurring in the gut.50 We hypothesize that the role of the P. copri species complex may be replaced by multiple Bacteroides species acting in concert for GEN2. Since each participant was only sampled once, we were unable to determine whether strain replacement within Bacteroides species occurs over time based on functional necessity, as has been previously shown specifically for B. fragilis.41
We hypothesize that the shift from P. copri to Bacteroides spp. is influenced by a change in diet. Different plant materials contain different types of non-digestible carbohydrates, which pass through the small intestine into the large intestine where they are fermented by gut microbes.52–54 Fermentation of these carbohydrates, and the subsequent production of SCFA, is important for reducing metabolic and inflammatory disease symptoms.55 Prevotella has previously been identified as a core constituent in the Indian microbiome, with Bacteroides and Dialister associated with certain Indian sub-populations.3 The enrichment of D. succinatiphilus and P. copri observed in the Indian cohort and GEN1 Canadians could be due to the quantity and type of grains consumed, particularly for GEN1 recent immigrants who derived a greater proportion of energy from carbohydrates compared to protein, and for residents of Bhopal who consumed primarily a plant-based diet.56–58 The co-occurrence of P. copri and Dialister has been previously identified in first-generation Americans who immigrated from Korea and in rural Himalayan populations.8,59 In contrast, Canadian and American born individuals show highly similar community profiles, typically with low abundances of Prevotellaceae and high abundances of Bacteroidaceae, consistent with our observations.60
A limitation of comparing these Canadian data with preexisting data sets, is the possibility of inferring differences that exist simply due to differences in sample processing. Typically, the ‘kitome’ varies between DNA extraction kits and methods, but would generally result in false negatives due to poor cell lysis and would not produce high abundance false positives such as high abundance D. succinatiphilus identified in the Indian samples.61 Previous examination showed that estimated differential abundances were consistent across DNA extraction and sequencing methods, while differences in bioinformatic methodology produced inconsistent results.62 We have utilized the same analysis methods across all data sets and believe that despite other possible inconsistencies, multi-study examinations are relevant and important ways to connect the metagenomic data available worldwide.
The mutual exclusivity of D. succinatiphilus and D. invisus observed in this study has not been previously reported and requires further investigation. One clear functional difference between D. succinatiphilus and D. invisus is the ability of D. succinatiphilus to consume succinate. Succinate producers, including P. copri and multiple Bacteroides spp. appear ubiquitously in many gut communities.63–65 Other potential succinate consumers were identified in our study, including a known consumer, B. thetaiotaomicron. 66 The inverse relationship observed between time in Canada and abundance of D. succinatiphilus and B. thetaiotaomicron suggests a shift in succinate consumers.
While the role of P. copri in human health, gut inflammation, and the development of immune-mediated inflammatory diseases is still poorly understood,30,64 recent research suggests that probiotic modulation and the reduction of circulating succinate may be a therapeutic target to potentially treat obesity and certain immune-mediated inflammatory diseases.67,68 Our results show a possible link between P. copri abundance and BMI. Recently, P. copri abundance and the presence of multiple P. copri subtypes were shown to be positively correlated with beneficial cardiometabolic markers.69 We hypothesize that as P. copri strain diversity is lost over time in Canada, the modulation of succinate production and consumption in relation to D. succinatiphilus abundance, may be altered. This could affect BMI and potentially lead to the increased incidence of type 2 diabetes mellitus observed in GEN1 SA Canadians.18 Whether due to changes in diet or other unknown factors, it remains unclear why this change in species association of succinate production and consumption has occurred over time and generations in Canada, and what other roles D. succinatiphilus may play in the gut community unique to GEN1.
Bacteroides and Bifidobacterium species are known to possess multiple gene families capable of polysaccharide and monosaccharide degradation and can often switch between energy sources.70 The flexibility of what nutrients Bacteroides and Bifidobacterium can utilize may indicate that these species are less reliant on interspecies cross-feeding.71 We hypothesize that the negative correlation between the abundance of P. copri and both B. longum and B. uniformis, may be due to redundancy in certain functions, such as xylan degradation. Without dietary fibers, certain species, particularly from the genus Bacteroides, will ferment host mucin glycans, creating a potentially proinflammatory environment.42,72,73 Other degraders, such as P. copri, though capable of breaking down the xylan backbone of mucin, do not contain the enzymes required to further debranch the attached sugars (Figure 6a). Previous research determined that the P. copri 1,4-beta-xylanase was present in 94% in vegans and only 58% in omnivores,74 and that individuals who consume a diet rich in cellulose and xylan have gut communities with high abundances of Prevotella.75,76 This suggests that Prevotella-based xylan degradation is favored when exposed to consistent, high levels of complex carbohydrates, while B. longum and B. uniformis xylan degradation may be favored when the function is only required sporadically, and a range of other nutrient sources are often introduced to the gut.
We hypothesize that the observed susceptibility to inflammatory bowel disease in GEN2 SA Canadians, matching the high susceptibility of multi-generation Canadians and Americans,18 may be due to the higher relative abundance of Bacteroides in these cohorts, and their potential contribution to the degradation of mucin glycans in the gut in the absence of fiber-rich foods.77,78 The continued displacement of Prevotella with Bacteroides over generations, and the plausible effect on fiber degradation and SCFA metabolism has been previously described for immigrant communities in the United States 7,8. Our study recapitulates this observation, while further exploring the interaction between these potentially key taxa and the complete metagenome, and the functional redundancy between GEN1 and GEN2 associated species. In the future, it would be important to test these hypotheses in vitro to determine the xylan degrading capabilities of the Prevotella and Bacteroides species identified in this study.
The socioeconomic status of GEN1 and GEN2 SA Canadians likely affects the gut metagenome, which may contribute to the development of immune-mediated inflammatory diseases for certain individuals. Low-income rates are generally higher among immigrant Canadians compared to Canadian-born persons, particularly in urban populations.79 This study was limited to Canadians living in Toronto and the Greater Toronto Area, which is primarily composed of urban and suburban neighborhoods. Generally, a lower socioeconomic status is associated with type 2 diabetes mellitus prevalence for all Canadians.17,80,81 Globally, the incidence of inflammatory bowel disease may be associated with a higher socioeconomic status, but poor disease outcomes and mortality are generally associated with a lower socioeconomic status.82 Within Canada, children from low-income neighborhoods were more likely to utilize health services related to inflammatory bowel disease, and adults with inflammatory bowel disease were more likely to be unemployed compared to the general public.83,84 Additionally, an individual’s socioeconomic status has been previously shown to affect the composition of the gut microbiome.85
We observed that the estimated median total household income by participant dissemination area was lower, and the population density higher, for both GEN1 and GEN2 compared to the median values for the census districts in which participants resided. Since socioeconomic status was estimated by dissemination area and not reflective of personal socioeconomic status, these values were not incorporated into the primary analysis to avoid over generalizations and incorrect inferences. However, we did observe that socioeconomic status was lowest for GEN1 recent immigrants, who also showed the greatest differences in the overall structure and contents of the gut metagenome (Figure 1). In Canada, inadequate nutrient intake was more prevalent in low-income individuals.86 Dietary acculturation is largely influenced by the availability of traditional ingredients, availability of specific tools, income, and food preparation time.87,88 All GEN1 early immigrants arrived as children, presumably with families or guardians. We hypothesize that large differences may exist between the nutrition and dietary acculturation of those who immigrate as children and those who immigrated as adults, with socioeconomic status contributing to these differences. Furthermore, GEN2 contained a greater proportion of younger participants, which may influence the characterized community given the known changes that occur in the gut from childhood to adulthood.89 Combined, these differences may result in the differences we observe in the gut metagenome. To determine the contribution of immigration age, dietary acculturation, and socioeconomic status on the gut metagenome, we would require more representative sampling including age- and income-matched non-SA Canadians.
The effect of the gut microbiome on overall human health remains disputed, with previous studies often describing completely opposite trends in species abundances related to inflammation and obesity.2,33,35,36 The contribution of the metagenome to the development of immune-mediated inflammatory diseases is likely due to a complex interplay between nutrient degradation, utilization, and SCFA production associated with the entire functionality of the gut community.
Methods
Recruitment and characteristics of participants
Study subjects included female and male individuals who ethnically self-identified as South Asian (SA) and either were born in a SA country and immigrated to Canada (GEN1) or were the children-of-parents who immigrated to Canada (GEN2), self-reported as healthy, and were between 18 and 35 years old. Participant recruitment took place for the most part on the campus at the University of Toronto in collaboration with SA student groups and at SA community events and centers outside the university (Festival of South Asia, Little India Festival).
Our exclusion criteria consisted of (1) use of antibiotics, (2) travel to SA in the 3-month period prior to the start of the study, or (3) existing chronic inflammatory conditions. Sample collection occurred between 2016 and 2018. The study staff completed the Subject Screening and Demographics Questionnaire with the subject and recorded weight, height, and waist circumference. Personal identifiers, including name, sex, date of birth, contact information, and health history were collected. Subjects also completed a Food Frequency Questionnaire (FFQ) and General Health, Environment, and Lifestyle Assessment Questionnaire (GHELQ) on their own. After exclusions, a total of 96 adult subjects completed the study.
Body Mass Index (BMI) and Waist Circumference (WC) were analyzed to determine if there was a significant difference between generations, years after immigration (YAI) groups, sex, or participation age. BMI was grouped into health categories: underweight (BMI < 18.5 kg/m2), normal weight (BMI 18.5–24.9 kg/m2), overweight (BMI 25–29.9 kg/m2), and obese (BMI > 30 kg/m2). WC was grouped into health categories: healthy female (WC < 80 cm), unhealthy female (WC ≥ 80 cm), healthy male (WC < 94 cm), unhealthy male (WC ≥ 94 cm) (Figure S1E and S1F). Statistical analyses were performed using R Stats Software version 3.6.3.90 A one-way ANOVA was performed between generations for BMI and WC, for each sex separately. Ethnicity, age, and YAI were also assessed and added as covariables to the BMI and WC ANOVAs. For GEN2 participants, YAI was set as zero. Statistical significance was defined as p < .05 (Table S1B and S1C).
Stool collection
Participants used an at-home stool collection kit with instructions provided to collect stool samples. The instructions specified that the participants were to collect stool following the International Human Microbiome Standards protocol, utilizing a “FecesCatcher” and a 30 ml stool collection container. Once the stool was collected and contained, participants were instructed to place the collection device in a Ziploc bag and freeze at home. The stool was then submitted within 48 hours to the study in a frozen state and stored at −80 oC. Each participant collected and submitted a single stool sample.
Dietary analysis
We used the Canadianized Dietary History Questionnaire (CDHQ II), a validated FFQ. The CDHQ II has 134 food items and captures information on usual dietary intake over the past month, including cooking methods, serving sizes and dietary supplements.91 Participants were sent unique links to complete the food frequency questionnaire online. Once participants completed the questionnaire online, data were electronically coded and constructed in spreadsheet format through Diet*Calc software. Participant reports of usual intakes were calculated into mean daily intake estimates of each nutrient and food group captured by the CDHQ II.92
The objective of this analysis was to determine the association between birthplace (SA or Canada) and daily intakes of various nutrients. The primary outcomes were daily intakes of total energy (kcal), total fat, saturated fat, polyunsaturated fat, and omega-3 fat (grams), protein (grams), vitamin A (retinoic acid equivalents or RAE), vitamin C (micrograms or mcg), vitamin D (international units or IU), folate (daily folate equivalents or DFE), calcium (milligrams or mg), fiber (grams), servings of fruits, servings of vegetables, total sugar (grams), sodium (mg), and caffeine (mg). Hypothesized clinically relevant covariates included living at home (yes/no), participant age in years, sex (male/female), age when immigrated to Canada (years), and total daily energy intake (kcal).
For each nutrient determined from the Canadianized Dietary History Questionnaire (CDHQ II), a valid FFQ, a multiple linear regression model was used to determine the association between birthplace (SA or Canada) and daily intake of each nutrient. Each multiple linear regression model was adjusted for the following covariates: living at home (yes/no), participant age in years, sex (male/female), age when immigrated to Canada (years), and total daily energy intake (kcal). All models except for daily energy intake (kcal) were adjusted for total daily kcal intake. Statistical significance was defined as p < .05.
The percentage of daily energy from carbohydrates, proteins, and fats was determined by the food frequency questionnaire.92 The ratio between daily energy from carbohydrates (%) to proteins (%) was determined for each birthplace and YAI group (Figure 4). A one-way ANOVA was used to compare Years After Immigration (YAI) groups. Pairwise comparisons between each YAI group were performed using the TukeyHSD method, available through R (Table S1G). Statistical significance was defined as p < .05.
Socioeconomic status estimations
Socioeconomic status is typically measured by assessing a combination of education, income, and occupation.93 To estimate the socioeconomic status of the participants, the postal code of the participant’s residences was used to determine the value of various socioeconomic status factors in for their Dissemination Area (DA). The University of Toronto Libraries “Map and Data Library” was used to access the Postal Code Conversion File (PCCF) from the 2016 Canadian Census data.94 This conversion file was used to map the postal codes given by the participants to the dissemination areas from the Computing in the Humanities and Social Sciences (CHASS) Canadian Census Analyzer, also available through the “Map and Data Library”. Within the Canadian Census Analyzer, the 2016 Census data were accessed, and the following data were collected: Population Density (v6), Employment Rate (v5472), Education levels – No certificate (v4921), Secondary (v4922), Postsecondary (v4923), Median total income of households in 2015 (v1876), Average household size (v121). These values were then compared between participants, as described below. Median values for each of these factors were also determined as a comparator for the entire Census Division (CD) in which the participants resided. This included four census districts in Ontario, Canada: Toronto, Durham, Peel, and York.
Estimated Socioeconomic Status was compared between generations, YAI values, and YAI groups. Covariance and correlation between factors were determined using the cov and rcorr functions in R. socioeconomic status groups were compared as a mixed model ANOVA including all factors, including only factors that were not significantly correlated, and individually as separate one-way ANOVA analyses. These factors were Population Density (v6), Employment Rate (v5472), Education levels – No certificate (v4921), Secondary (v4922), Postsecondary (v4923), Median total income of households in 2015 (v1876), Average household size (v121). The relative proportion of postsecondary education to no certificate and secondary level was calculated per participant, to be used as a single value in the analysis. Statistical significance was defined as p < .05 with a Tukey-HSD correction for the YAI group multiple comparisons.
DNA extraction and metagenomic sequencing
DNA was extracted from stool samples following the International Human Microbiome Standards (IHMS) SOP 07 V1: Protocol H.95 DNA was quantified using Qubit BR DNA quantification kit (ThermoFisher Scientific). 500 ng of DNA was diluted to a final volume of 130 uL in 10 mM Tris-HCl pH 8 and sheared to 400 bp fragments using the Covaris S2 (Covaris) with the following specifications: Duty Cycle – 10%, Intensity – 4, Cycles per burst – 200, Time – 55 seconds. The sheared DNA was cleaned with Ampure XP magnetic beads (Beckman Coulter). 100 ng of Covaris sheared, cleaned DNA was input into the NEB Ultra II Prep kit and the standard protocol was followed (New England Biolabs), following the size selection guide for a 400 bp sheared fragment size, and five PCR cycles for the barcoding PCR reaction. The barcoded samples were quantified using Quant-iT PicoGreen dsDNA kit (ThermoFisher Scientific) and pooled to even concentrations. The pooled sequencing library was quantified using Qubit HS DNA quantification kit and diluted to 4 nM. The library was denatured and loaded on to the Illumina NextSeq 500 and sequenced using a 2 × 150 bp cycle kit (Illumina). The samples were sequenced in two batches, one to achieve high coverage and one to achieve moderate sequence depth. We did not identify significant differences in the predicted coverage of the metagenome between batches, so the data were normalized and combined for the analysis. No significant differences in the percentage of species identified or mapped reads were identified between batches after HUMAnN2 analysis.
Metagenomic sequence processing and bioinformatic analysis
The sequences were trimmed to remove adapters and low-quality sequences using Trimmomatic, following default parameters, an average quality minimum of 20, and a minimum sequence length of 125 bp.96 Cutadapt was used to remove any stretches of sequences that contained homopolymers of G base, an error that can occur in the Illumina NextSeq due to the two-color sequencing system (Martin, DOI:10.14806/ej.17.1.200). PCR duplicates were identified and removed using PrinSeq.97 Human sequences were identified using Bowtie2 and the hg19 human sequence database available through NCBI (BioProject: PRJNA31257).98 The unmapped, non-human reads were processed as the microbial metagenome. Read1 and Read2 were merged into one file and the metagenome coverage was estimated for 15 representative samples using NonPareil sequence redundancy analysis.99
HUMAnN2 software was used to identify the taxonomic and functional profiles of each community, using the MetaPhlAn2 program option for taxonomy and the UniRef90 database for function, following all default parameters.44,45,100 Resulting functional annotations were mapped to the MetaCyc gene family ontology.101 The pathways of interested were identified using the online MetaCyc database, the BioCyc Omics Viewers, and Smart Tables tool.102 The HUMAnN2 software was used to stratify the MetaCyc results to determine the taxonomic contribution for each gene family. The unmapped MetaCyc reaction gene families (undetermined species associations) were removed from the data set. Due to the lack of completeness of the metagenomes (proposed hypothetical average coverage ~85%, shown in results), the relative abundances of gene families were summed across all subjects and transformed into a binary table, representing the presence or absence of each gene families per organism, if detected in any participant’s metagenome (Table S6).
Data analysis was conducted in R.90 Processing of the species relative abundance table created by HUMANn2 was conducted using the PhyloSeq package 103,104 and the Tidyverse.105 The data were filtered to remove species with an abundance less than the median total abundance (0.81), and with a prevalence less than 2. This resulted in 30% of the unique species removed due to low prevalence (singletons) and 20% due to low abundance, but less than an average of 1% by relative abundance (minimum = 0%, maximum = 7.2%).
Years After Immigration (YAI) was examined as both a continuous and categorical variable. As a categorical variable, the GEN1 participants were divided into approximately even tertiles using the numeric YAI values. These tertiles were defined as recent immigrants (3 months to 4 YAI), moderately recent immigrants (4 to 14 YAI), and early immigrants (greater than 14 YAI). These tertiles were then treated as categorical variables and compared relative to GEN2, the Indian cohort, and the American cohort, when appropriate. As a continuous variable, the numeric YAI value was examined and a value of zero was assigned to those who had not immigrated themselves.
MetaCyc analysis of glycoside hydrolases
We extracted all glycoside hydrolases (EC 3.2.1) from the MetaCyc results, as well as all documented mono and disaccharide sugar degradation pathway gene families. We focused the results on polysaccharides commonly found in foods, such as cellulose, pectin, xylan, xyloglucan, and starch, and the mucin glycans found in the mucus layer of the gastrointestinal tract. Mucin glycans included in the analysis were O-linked N-acetylgalactosamine (GalNAc), N-acetylglucosamine (GlcNAc), mannose, xylose, and others.106 Enzymes involved in the degradation of mucin glycans were determined based on previous studies and overlap with the enzymes required to debranch the side chains on food-based xylans.78,107 We also included enzyme gene families involved in the major SCFA production pathways (acetate, lactate, butanoate, and propanoate-succinate) (Table S6). The CAZy database was used to validate the carbohydrate metabolism genes of interest, by manually cross-referencing the MetaCyc pathways with enzyme groups.108
The dendrogram clustering species by the MetaCyc Glycoside Hydrolase gene presence-absence profile (Figure 6a, Figure S6A) was created using the ‘dist’ and ‘hclust’ function with the method algorithm set as ‘euclidean’ and ‘average’, respectively. The ggplot function ‘ggdendrogram’ was then used to visualize the distance tree.90
Community composition and diversity analysis
The following methods are also described in complete detail as a knitr file (AnalysisMethods_SouthAsianCanadian_Metagenome.html). The analysis can be performed using files provided (DataFiles_SouthAsianCanadian_Metagenome.zip). The Bray–Curtis dissimilarity values between all samples were determined using the species relative abundance compositions from HUMAnN2 results, through Phyloseq, available in R. As specified in the results, the HUMAnN2 normalized or cumulative sum scaled values were used. Cumulative sum scaling was determined using the R packaged metagMisc:: phyloseq_transform_css, options for normalization and log transforming were selected. The Principal Coordinate values for each sample were then identified and plotted using Principal Coordinate Analysis (PcoA) plotting. Axis 1 and Axis 2 were visualized for each PcoA plot. If present, ellipses were determined using the ‘stat_ellipse’ function available in R, using the default multivariate t-distribution to draw the ellipse.
To approximate alpha diversity, the relative abundance data produced by HUMANn2 were transformed to mock coverage data, by inflating the normalized data to values out of 100,000 sequences per sample. Due to this approximation, alpha diversity, specifically Chao1 index values, was not investigated independently. Chao1 index values were only used as a co-variable in other analysis, as described.
The most abundant taxa (Max Taxa) were identified in each participant based on MetaPhlAn normalized relative abundances after filtering to remove low abundance and low prevalence species, as described above. For species identified as the most abundant in >3 samples, species name was specified (P. copri, E. rectale, and F. prausnitzii). If a species was identified in ≤3 samples but from a class otherwise not represented, species name was specified (D. invisus). If the species name was unknown or if ≤3 samples were associated with a certain species maximum, genus or family was specified (Bacteroides, Bifidobacterium, Erysipelotrichaceae, Megamonas, Ruminococcus, and Subdoligranulum). Singleton associations were labeled as ‘Other’. Max Taxa associations were used for ADONIS and RDA analysis, Max Taxa Group was used to label figures (Table S1F).
Beta diversity Bray–Curtis distances were determined through PhyloSeq. Normalized data produced by HUMANn2 were utilized, as well as cumulative sum scaled (css) data, which was determined using the R package MetagMisc and the function ‘phyloseq_transform_css’. Significant sources of variation were identified using the ‘adonis’ function (permutational ANOVA), run with 1000 permutations and a Benjamini-Hochberg false discovery rate correction, available through the Vegan package in R.109 Participant metadata including sex, age, BMI, WC, YAI, self-reported ethnicity, most abundant taxa in each participant sample, and an estimated within-sample alpha diversity index value (Chao1 value) were input into the model when available. The results were corrected for multiple comparison using the Benjamini-Hochberg-Yekutieli method. A pairwise ADONIS was determined using the package R package PairwiseAdonis.110 The effect of sequencing coverage, percentage of mapped human DNA, percentage of unmapped data, and number of identified gene families was also examined to determine the effect on community composition and Bray–Curtis distances. The same method was followed as described above, taking into account these factors of interest, while investigating and controlling for an interaction between data processing and participant metadata. Significance was defined as p < .01 (Table S2A – S2C).
The redundancy analysis (RDA) was performed on cumulative sum scaling of normalized species relative abundances using the ‘rda’ function in Vegan, following the suggested procedure (Oksanen et al., 2019). The adjusted R square value was identified in the results and a one-way ANOVA was performed to identify factors that contributed significantly to the redundancy analysis (Table S3E). The p-values were adjusted for multiple comparisons using the Benjamini-Hochberg-Yekutieli method. A biplot was created from the RDA species summary was plotted using the first two principal components. The arrows indicate the direction and standard deviation of each significant variable. Significance was defined as p < .01 (figure 1f).
The relative abundances of species in the GEN1 and GEN2 communities were compared using MetagenomeSeq, which utilizes RNA-seq Limma software adapted for metagenomics. PhyloSeq was used to convert the data to MetagenomeSeq format. Zero-inflated Gaussian (FitZig) function in MetagenomeSeq was primarily used to identify differences between comparison groups, retaining results with a p-value <0.01 and a log-fold change (LFC) > 1. Participant age, sex, and YAI were controlled for and results were filtered to remove species that were not identified in the MetagenomeSeq: Effective Sample Size. Discovery odds ratio testing (MetagenomeSeq: FitDO), selecting for hits with a false discovery rate <0.01, and linear discriminant analysis (LDA) Effective Size (LefSe),111 following default parameters, were used to validate the results from FitZig. We retained species identified using two or more of these methods. Results were adjusted for multiple comparisons using the Benjamini-Hochberg-Yekutieli method. Specific comparisons with more than two groups were achieved using the ‘makeContrasts’ function in MetagenomeSeq (Table S4A).
The relative abundances of species in the GEN1, GEN2, Indian, and American communities were compared using MetagenomeSeq.47,112 PhyloSeq was used to convert the data to MetagenomeSeq format. Zero-inflated Gaussian (FitZig) was used to identify differences between comparison groups (Paulson, 2016), retaining results with a p-value <0.01 and a log-fold change (LFC) > 1. Participant age and sex were controlled for and results were filtered to remove species that were not identified in the MetagenomeSeq: Effective Sample Size. Results were adjusted for multiple comparisons using the Benjamini-Hochberg-Yekutieli method. The data were first examined by comparing the Indian and American cohorts with the Canadian cohort divided by YAI groups. Then, the Indian data were further divided into their two sampling locations, Bhopal and Kerala. Specific comparisons with more than two groups were achieved using the ‘makeContrasts’ function in MetagenomeSeq (Table S4D).
Comparator datasets
A metagenomic dataset from an Indian cohort (NCBI BioProject PRJNA397112) was downloaded from the Sequence Read Archive (SRA). Our inclusion criteria were participants a minimum of 18 years of age. A representative set of 40 age matched healthy participant samples was retrieved from the Indian BioProject, consisting of 20 women and 20 men sampled from two geographical regions. The raw FASTQ data were downloaded and analyzed with the same pipeline used to analyze the Canadian immigrant data set.
A metagenomic dataset from the United States, representing a westernized North American cohort (NCBI BioProject PRJNA398089), was downloaded from the Inflammatory Bowel Disease Multi’omics Database (IBDMDB) at (https://ibdmdb.org/tunnel/public/HMP2/WGS/1818/rawfiles). This data was used as a proxy for Canadians, since a diverse and representative Canadian metagenomic data set is not currently available. Canadians and Americans exhibit similar patterns in health lifestyle, particularly for Canadian who live close to the American border such as people living in Toronto and the Greater Toronto Area, such as these participants.113 The data set has been previously filtered to remove low-quality sequences and sequences that map to the human genome. The metagenomic data were selected to include participants who were a minimum of 18 years old, that self-identified as ‘non-IBD’ for their diagnosis, and ‘white’ for their ethnicity, according to the associated metadata file provided by the IBDMDB. A single data time point was selected for each participant randomly, resulting in a total of 10 samples, three samples from women and seven samples from men, added to the analysis to represent a ‘westernized’ metagenome. The FASTQ data were downloaded and analyzed with the same pipeline used to analyze the Canadian immigrant data set.
Dirichlet multinomial and cluster analysis
Dirichlet Multinomal Mixtures (DMM) determined community clusters through PhyloSeq from the species relative abundances, using an infinite mixture model. The DMM model was tested for a maximum of 10 community types, and the best fit was selected as 2 communities.114 The top community drivers were defined as those in the 95% percentile of determined Dirichlet Component values (Figure 3).
UPGMA clusters for each metagenomic community were identified from cumulative sum scaled species relative abundances Bray–Curtis distances, determined through PhyloSeq, and clustered using hclust with the method algorithm set as ‘average’.90 The cluster number was determined using the GMD package and the elbow batch method.115 The GK tau values, comparing clusters to maximum abundant taxa and other metadata factors of interest, were performed using the GoodmanKruskal package.116
Anvi’o analysis of P. copri metagenome assembled genomes
Anvi’o was used to create metagenome assembled genomes (MAGs) following their ‘Tutorial for Metagenomics Workflow’, described as follows.117 Quality filtered reads used in previous analysis were selected from samples containing P. copri, as identified in the MetaPhlAn2 and PanPhlAn analysis, shown as Groups 1, 2, Div/Multi, and Unclustered in Table S3A. Anvi’o was used to assemble these quality filtered reads into contigs for each sample using megahit, following default settings.118 The anvi’o script anvi-script-reformat-fasta were used to select contigs with a minimum length of 2500 bp, as suggested in the workflow. Bowtie2 was used to map contigs to each sample.98 A contigs database was then created and hmm profiles were identified using the anvi’o scripts anvi-gen-contigs-database and anvi-run-hmms, respectively. An anvi-profile was determined for each sample using the anvi’o script anvi-profile and Centrifuge was used to identify taxonomies.119 All sample profiles were merged using anvi’o anvi-merge and genomic bins were identified using anvi’o anvi-cluster-contigs with the driver set as Concoct, and default parameters were followed.120 The taxonomy for each contig was then identified using the anvi’o script anvi-estimate-scg-taxonomy following default parameters and ‘compute-scg-coverages’ specified. Genomic bins identified as ‘Prevotella copri’ were selected from all the MAGs identified in the samples, as well as genomic bins identified at the genus level as ‘Prevotella’ with at least 10 single copy genes (scg) associated with this taxonomic call. All other binning programs available through anvi’o were tested; however, these were unable to identify P. copri at the species level, and so analysis was not carried out with these other results. The bins identified in Concoct were selected using the anvi’o script anvi-split and set as ‘internal genomes’. External genomes were sourced from the Tett et al., 2019 P. copri reference data set, including P. copri genomes from Clades A, B, C, and D. Contigs were generated for these reference genomes using the anvi’o script anvi-gen-contigs-database using Prodigal and were set as ‘external genomes’.121 Anvi’o was used for pangenome analysis of these internal and external genomes using the anvi’o script anvi-pan-genome and Diamond 122 and genome similarity was identified using the anvi’o script anvi-compute-genome-similarity with pyANI set as the program.123 The average nucleotide identity (ANI) results showing ‘full percentage identity’ newick tree of Euclidean distances were used to visualize the similarity between the internal P. copri and Prevotella genomes and the reference genomes. The anvi’o script anvi-summarize was used to identify the relative abundance and variability of each P. copri bin across each sample.
Relative abundance and functional network analysis
We used the R package ‘igraph’ to construct the networks of relative abundance correlation values and of gene family presence-absence. We followed an established protocol.124,125 For the relative abundance correlation network, we determined that the data followed a non-normal distribution, and so we used Spearman correlation values and the ‘graph adjacency’ function to create undirected networks. The correlation coefficient critical value was determined to be 0.4, selected based on the approximate midpoint of the sigmoidal curve when the correlation coefficient was plotted against number of clusters (Figure S5A). This value determines the degree of co-occurrence association between pairs of taxa required for retention of the edge in the resulting network. Clusters were identified using ‘clusters’ function in ‘igraph’. Sub-networks were identified using the ‘induced subgraph’ function, from all network clusters with more than three species. The walk-trap algorithm was then used to identify clusters within each sub-network.
For the gene family network, we selected all Negativicutes (phylum Firmicutes) and all Bacteroides (phylum Bacteroidetes) species from the Canadian cohort. We also extracted potentially relevant species from previous analyses, including all maximum abundance taxa (Figure 1c), species determined to be at significantly different relative abundances between GEN1 and GEN2 (Table 3, Figure 1), and hub species in the relative abundance networks (complete list of species: Table S6A). A correlation coefficient critical value was determined to be 0.75, selected again based on the approximate midpoint of the sigmoidal curve (Figure S5). By selecting a single correlation cutoff for all species in the metagenome, we accept that poorly characterized species and highly dissimilar singleton species to be filtered out from this analysis. Clusters were identified using ‘clusters’ function in ‘igraph’. Sub-networks were identified using the ‘induced subgraph’ function, from all network clusters with more than three species. The walk-trap algorithm was then used to identify clusters within each sub-network. The three largest relative abundance correlation subnetworks were visualized (Figure S5).
PanPhlAn pangenome analysis
PanPhlAn was used to estimate the pangenome of Prevotella copri within the Canadian data set, following all default settings. The P. copri reference genomes REF_G000157935 and REF_G000435255 from the PanPhlAn database were used. The resulting pangenome profiles from each sample were clustered using ‘hclust’ available through R, with the method algorithm set as ‘average’ for the UPGMA method. The cluster number was determined using the GMD package. We defined samples as containing the ‘Div/Multi’ when P. copri was identified as a maximum species in the previous MetaPhlAn analysis but were not included in the pangenome output based on the standard thresholds. We verified that these samples all contained at least 5% P. copri sequence data as determined by the initial PanPhlAn read mapping, with an average of 15% across all samples in this ‘Div/Multi’ group. The ‘Unclustered’ cluster contained highly diverse pangenome profiles, resulting in clusters with two or less samples each. Samples that were listed as ‘No Cluster’ contained an average and median of 2.12% and 0.13% , respectively, and P. copri was not identified as the maximum species based on the previous MetaPhlAn analysis.
StrainPhlAn strain analysis
StrainPhlAn3 was used to estimate the presence of multiple Prevotella copri strains within the Canadian data set following all default settings. The MetaPhlAn mpa_v30_CHOCOPhlAn_201901 database was used to map reads to marker sequences, 17 P. copri reference genomes from Tett et al., 2019 (NCBI-BioProject PRJNA559898) strain analysis were used to generate the alignments and phylogenetic trees. The percentage of polymorphic sites was output in the polymorphic.txt file by the StrainPhlAn command. The average percentage of polymorphic sites was determined for each PanPhlAn P. copri cluster. The StrainPhlAn polymorphic average for Bacteroides spp. was determined for species identified in at least 70% or participants or with an average relative abundance >1% and were not filtered out by StrainPhlAn due to poorly inferred phylogeny. This included B. dorei, B. ovatus, B. plebeius, B. stercoris, B. uniformis, and B. vulgatus.
Amplicon validation set
The following methods are also described in complete detail as a knitr file (Comparison_V4_Metagenomics_SouthAsianCanadian.html). A validation set of samples was randomly selected (n = 14). The V4 region of the 16S rRNA gene was amplified and sequenced for these samples. These results shown in Comparison_V4_Metagenomics_SouthAsianCanadian.html were used to compare and validate the metagenomic data.
Supplementary Material
Acknowledgements
We thank all members of the GEMINI Research Team (Michelle Smith, Ikbel Naouar, Jayne Danska, Philippe Poussier, Andrew Paterson, Gary Bader, Geoffrey Nguyen, Sylva Donaldson, Allison Hall, Kathleen Wilson, Naomi Schwartz, Anne Griffiths, John Parkinson, Michelle Ouzounis, Thomas Waters, and Aleixo Muise) for fruitful discussions and support. We thank the team members at the Centre for the Analysis of Genome Evolution and Function (Lijie Yuan and Yunchen Gong) for their support in sample preparation, sequencing, and data management. We thank the laboratory technicians from the Croitoru lab involved in sample handling preparation (Mitra Noori). We thank Gary Chao for support in sample collection and participant interviews.
Funding Statement
This project has received funding from the Connaught Funds and CIHR EGCD Team Grant.
List of Abbreviations
BMI Body Mass Index
GEN1/GEN2 Generation 1 and Generation 2 respectively
IA Immigration as an Adult
PCoA Principal Coordinate Analysis
RDA Redundancy Analysis
SA South Asian
SCFA Short-Chain Fatty Acids
UPGMA Unweighted Paired Group Method with Arithmetic mean
WC Waist Circumference
YAI Years After Immigration
Ethics Approval and Consent to Participate
This study was approved for human subject by the University of Toronto Research Ethics Board (protocol 31593). Written informed consent to participate was obtained from all study participants prior to enrollment in the study.
Consent for Publication
Written informed consent that collected data would be used for publication was obtained from all study participants prior to enrollment in the study.
Disclosure of Interest
The authors report no conflict of interest.
Availability of Data and Material
The metagenomic sequencing data have been submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under submission number SUB7709855. The accession number for the sequencing data reported in this paper is BioProject PRJNA644666. All data generated or analyzed during this study are included in this published article (and its supplementary information files) or through the SRA. Additional files include a knitr document describing all methods used for data analysis.
Authors Contributions
Conceptualization, JLG, KC, DSG
Methodology Development, GC, PWW
Formal Analysis, JKC
Investigation, PWW
Resources, JLG, DSG
Data Curation, JKC, GC, SV, PWW, DSG
Writing –Original Draft, JKC, DSG
Writing –Review & Editing, JKC, EA, DSG, GC, JLG, KC, EB
Visualization, JLG, DSG
Supervision, DSG, JLG
Project Administration, GC, PWW
Funding Acquisition, JLG, KC, DSG
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, Beghini F, Manghi P, Tett A, Ghensi P, et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell. 2019;176(3):649–662.e20. doi: 10.1016/j.cell.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chávez-Carbajal A, Nirmalkar K, Pérez-Lizaur A, Hernández-Quiroz F, Ramírez-del-alto S, García-Mena J, Hernández-Guerrero H-GC.. Gut Microbiota and Predicted Metabolic Pathways in a Sample of Mexican Women Affected by Obesity and Obesity Plus Metabolic Syndrome. Int J Mol Sci. 2019;20(2):438. doi: 10.3390/ijms20020438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dehingia M, Thangjam Devi K, Talukdar NC, Talukdar R, Reddy N, Mande SS, Deka M, Khan MR. Gut bacterial diversity of the tribes of India and comparison with the worldwide data. Sci Rep. 2015;5(1):18563. doi: 10.1038/srep18563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kabeerdoss J, Shobana Devi R, Regina Mary R, Ramakrishna BS. Faecal microbiota composition in vegetarians: comparison with omnivores in a cohort of young women in southern India. Br J Nutr. 2012;108(6):953–29. doi: 10.1017/S0007114511006362. [DOI] [PubMed] [Google Scholar]
- 5.Nakayama J, Yamamoto A, Palermo-Conde LA, Higashi K, Sonomoto K, Tan J, Lee Y-K. Impact of Westernized Diet on Gut Microbiota in Children on Leyte Island. Front Microbiol Internet] 2017. [cited 2019 April5]; 8. doi: 10.3389/fmicb.2017.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schnorr SL, Candela M, Rampelli S, Centanni M, Consolandi C, Basaglia G, Turroni S, Biagi E, Peano C, Severgnini M, et al. Gut microbiome of the Hadza hunter-gatherers. Nat Commun. 2014;5(1):3654. doi: 10.1038/ncomms4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vangay P, Johnson AJ, Ward TL, Al-Ghalith GA, Shields-Cutler RR, Hillmann BM, Lucas SK, Beura LK, Thompson EA, Till LM, et al. US Immigration Westernizes the Human Gut Microbiome. Cell. 2018;175(4):962–972.e10. doi: 10.1016/j.cell.2018.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters BA, Yi SS, Beasley JM, Cobbs EN, Choi HS, Beggs DB, Hayes RB, Ahn J. US nativity and dietary acculturation impact the gut microbiome in a diverse US population. Isme J. 2020;14(7):1639–1650. doi: 10.1038/s41396-020-0630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Gabalawy H, Guenther LC, Bernstein CN. Epidemiology of Immune-Mediated Inflammatory Diseases: incidence, Prevalence, Natural History, and Comorbidities. J Rheumatol Suppl. 2010;85(0):2–10. doi: 10.3899/jrheum.091461. [DOI] [PubMed] [Google Scholar]
- 10.Lerner A, Jeremias P, Matthias T. The World Incidence and Prevalence of Autoimmune Diseases is Increasing. Int J Celiac Dis. 2016;3(4):151–155. doi: 10.12691/ijcd-3-4-8. [DOI] [Google Scholar]
- 11.Pahwa R, Goyal A, Bansal P, Ishwarlal J. Chronic Inflammation. NCBI Bookshelf. Treasure Island; FL. StatPearls Publishing; 2019. [Google Scholar]
- 12.Kong APS, Xu G, Brown N, So W-Y, Ma RCW, Chan JCN. Diabetes and its comorbidities—where East meets West. Nat Rev Endocrinol. 2013;9(9):537–547. doi: 10.1038/nrendo.2013.102. [DOI] [PubMed] [Google Scholar]
- 13.Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14(2):88–98. doi: 10.1038/nrendo.2017.151. [DOI] [PubMed] [Google Scholar]
- 14.Burisch J, Munkholm P. The epidemiology of inflammatory bowel disease. Scand J Gastroenterol. 2015;50(8):942–951. doi: 10.3109/00365521.2015.1014407. [DOI] [PubMed] [Google Scholar]
- 15.Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, Panaccione R, Ghosh S, Wu JCY, Chan FKL, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. The Lancet. 2017;390(10114):2769–2778. doi: 10.1016/S0140-6736(17)32448-0. [DOI] [PubMed] [Google Scholar]
- 16.Singh P, Ananthakrishnan A, Ahuja V. Pivot to Asia: inflammatory bowel disease burden. Intest Res. 2017;15(1):138. doi: 10.5217/ir.2017.15.1.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Statistics Canada . Immigration population in Canada, 2016. Census of Population [Internet]. 2016 [cited 2019 May1]; Available from: https://www150.statcan.gc.ca/n1/pub/11-627-m/11-627-m2017028-eng.htm
- 18.Benchimol EI, Manuel DG, To T, Mack DR, Nguyen GC, Gommerman JL, Croitoru K, Mojaverian N, Wang X, Quach P, et al. Asthma, Type 1 and Type 2 Diabetes Mellitus, and Inflammatory Bowel Disease amongst South Asian Immigrants to Canada and Their Children: a Population-Based Cohort Study. Plos One. 2015;10(4):e0123599. doi: 10.1371/journal.pone.0123599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Creatore MI, Moineddin R, Booth G, Manuel DH, DesMeules M, McDermott S, Glazier RH. Age- and sex-related prevalence of diabetes mellitus among immigrants to Ontario, Canada. Can Med Assoc J. 2010;182(8):781–789. doi: 10.1503/cmaj.091551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Misra R, Faiz O, Munkholm P, Burisch J, Arebi N. Epidemiology of inflammatory bowel disease in racial and ethnic migrant groups. World J Gastroenterol. 2018;24(3):424–437. doi: 10.3748/wjg.v24.i3.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Popkin BM, Horton S, Kim S, Mahal A, Shuigao J. Trends in Diet, Nutritional Status, and Diet-related Noncommunicable Diseases in China and India: the Economic Costs of the Nutrition Transition. Nutr Rev. 2009;59(12):379–390. doi: 10.1111/j.1753-4887.2001.tb06967.x. [DOI] [PubMed] [Google Scholar]
- 22.Martyn-Nemeth P, Quinn L, Menon U, Shrestha S, Patel C, Shah G. Dietary Profiles of First-Generation South Asian Indian Adolescents in the United States. J Immigr Minor Health. 2017;19(2):309–317. doi: 10.1007/s10903-016-0382-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shridhar K, Dhillon PK, Bowen L, Kinra S, Bharathi AV, Prabhakaran D, Reddy KS, Ebrahim S. Nutritional profile of Indian vegetarian diets – the Indian Migration Study (IMS). Nutr J. 2014;13(1):55. doi: 10.1186/1475-2891-13-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seidelmann SB, Claggett B, Cheng S, Henglin M, Shah A, Steffen LM, Folsom AR, Rimm EB, Willett WC, Solomon SD. Dietary carbohydrate intake and mortality: a prospective cohort study and meta-analysis. Lancet Public Health. 2018;3(9):e419–28. doi: 10.1016/S2468-2667(18)30135-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Talegawkar SA, Kandula NR, Gadgil MD, Desai D, Kanaya AM. Dietary intakes among South Asian adults differ by length of residence in the USA. Public Health Nutr. 2016;19(2):348–355. doi: 10.1017/S1368980015001512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelemen LE, Anand SS, Vuksan V, Yi Q, Teo KK, Devanesen S, Yusuf S. Development and evaluation of cultural food frequency questionnaires for South Asians, Chinese, and Europeans in North America. J Am Diet Assoc. 2003;103(9):1178–1184. doi: 10.1016/S0002-8223(03)00985-4. [DOI] [PubMed] [Google Scholar]
- 27.Nakayama J, Watanabe K, Jiang J, Matsuda K, Chao S-H, Haryono P, La-ongkham O, Sarwoko M-A, Sujaya IN, Zhao L, et al. Diversity in gut bacterial community of school-age children in Asia. Sci Rep. 2015;5(1):8397. doi: 10.1038/srep08397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol. 2015;12(4):205–217. doi: 10.1038/nrgastro.2015.34. [DOI] [PubMed] [Google Scholar]
- 29.Egshatyan L, Kashtanova D, Popenko A, Tkacheva O, Tyakht A, Alexeev D, Karamnova N, Kostryukova E, Babenko V, Vakhitova M, et al. Gut microbiota and diet in patients with different glucose tolerance. Endocr Connect. 2016;5(1):1–9. doi: 10.1530/EC-15-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kovatcheva-Datchary P, Nilsson A, Akrami R, Lee YS, De Vadder F, Arora T, Hallen A, Martens E, Björck I, Bäckhed BF. Dietary Fiber-Induced Improvement in Glucose Metabolism Is Associated with Increased Abundance of Prevotella. Cell Metab. 2015;22(6):971–982. doi: 10.1016/j.cmet.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 31.Zimmet P, Alberti KG, Magliano DJ, Bennett PH. Diabetes mellitus statistics on prevalence and mortality: facts and fallacies. Nat Rev Endocrinol. 2016;12(10):616–622. doi: 10.1038/nrendo.2016.105. [DOI] [PubMed] [Google Scholar]
- 32.Castaner O, Schröder H. Response to: comment on “The Gut Microbiome Profile in Obesity: a Systematic Review”. Int J Endocrinol. 2018;2018:1–9. doi: 10.1155/2018/9109451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cockburn DW, Suh C, Medina KP, Duvall RM, Wawrzak Z, Henrissat B, Koropatkin NM. Novel carbohydrate binding modules in the surface anchored α-amylase of Eubacterium rectale provide a molecular rationale for the range of starches used by this organism in the human gut. Mol Microbiol. 2018;107(2):16. doi: 10.1111/mmi.13881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marques FZ, Nelson E, Chu P-Y, Horlock D, Fiedler A, Ziemann M, Tan JK, Kuruppu S, Rajapakse NW, El-Osta A, et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation. 2017;135(10):964–977. doi: 10.1161/CIRCULATIONAHA.116.024545. [DOI] [PubMed] [Google Scholar]
- 35.Pittayanon R, Lau JT, Yuan Y, Leontiadis GI, Tse F, Surette M, Moayyedi P. Gut Microbiota in Patients With Irritable Bowel Syndrome—a Systematic Review. Gastroenterology. 2019;157(1):97-108. [DOI] [PubMed] [Google Scholar]
- 36.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 37.Dhakan DB, Maji A, Sharma AK, Saxena R, Pulikkan J, Grace T, Gomez A, Scaria J, Amato KR, Sharma VK. The unique composition of Indian gut microbiome, gene catalogue, and associated fecal metabolome deciphered using multi-omics approaches. GigaScience [Internet] 2019. [cited 2019 April10]; 8(3). 10.1093/gigascience/giz004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shankar V, Gouda M, Moncivaiz J, Gordon A, Reo NV, Hussein L, Paliy O, Manichanh C. Differences in Gut Metabolites and Microbial Composition and Functions between Egyptian and U.S. Children Are Consistent with Their Diets. mSystems [Internet] 2017. [cited 2019 April5]; 2(1). 10.1128/mSystems.00169-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaoutari AE, Armougom F, Gordon JI, Raoult D, Henrissat B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol. 2013;11(7):497–504. doi: 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- 40.Kim CH. Microbiota or short-chain fatty acids: which regulates diabetes? Cell Mol Immunol. 2018;15(2):88–91. doi: 10.1038/cmi.2017.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, Fu H, Xue X, Lu C, Ma J, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science. 2018;359(6380):1151–1156. doi: 10.1126/science.aao5774. [DOI] [PubMed] [Google Scholar]
- 42.Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell. 2016;167(5):1339–1353.e21. doi: 10.1016/j.cell.2016.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chambers ES, Preston T, Frost G, Morrison DJ. Role of Gut Microbiota-Generated Short-Chain Fatty Acids in Metabolic and Cardiovascular Health. Curr Nutr Rep. 2018;7(4):198–206. doi: 10.1007/s13668-018-0248-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, et al. Metabolic Reconstruction for Metagenomic Data and Its Application to the Human Microbiome. PLoS Comput Biol. 2012;8(6):e1002358. doi: 10.1371/journal.pcbi.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Franzosa EA, Morgan XC, Segata N, Waldron L, Reyes J, Earl AM, Giannoukos G, Boylan MR, Ciulla D, Gevers D, et al. Relating the metatranscriptome and metagenome of the human gut. Proc Natl Acad Sci. 2014;111(22):E2329–38. doi: 10.1073/pnas.1319284111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mikryukov V metagMisc: miscellaneous functions for metagenomic analysis. 2019.
- 47.Paulson JN, Stine OC, Bravo HC, Pop M. Differential abundance analyis for microbial marker-gene surveys. Nature Methods. 201. 3;10:1200-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tett A, Huang KD, Asnicar F, Fehlner-Peach H, Pasolli E, Karcher N, Armanini F, Manghi P, Bonham K, Zolfo M, et al. The Prevotella copri Complex Comprises Four Distinct Clades Underrepresented in Westernized Populations. Cell Host Microbe. 2019;26(5):666–679.e7. doi: 10.1016/j.chom.2019.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed BF. From Dietary Fiber to Host Physiology: short-Chain Fatty Acids as Key Bacterial Metabolites. Cell. 2016;165(6):1332–1345. doi: 10.1016/j.cell.2016.05.041. [DOI] [PubMed] [Google Scholar]
- 50.Truong DT, Tett A, Pasolli E, Huttenhower C, Segata N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017;27(4):626–638. doi: 10.1101/gr.216242.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fehlner-Peach H, Magnabosco C, Raghavan V, Scher JU, Tett A, Cox LM, Gottsegen C, Watters A, Wiltshire-Gordon JD, Segata N, et al. Distinct Polysaccharide Utilization Profiles of Human Intestinal Prevotella copri Isolates. Cell Host Microbe. 2019;26(5):680–690.e5. doi: 10.1016/j.chom.2019.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Venkataraman A, Sieber JR, Schmidt AW, Waldron C, Theis KR, Schmidt TM. Variable responses of human microbiomes to dietary supplementation with resistant starch. Microbiome. 2016;4(1):33. doi: 10.1186/s40168-016-0178-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bernstein A, Titgemeier B, Kirkpatrick K, Golubic M, Roizen M. Major Cereal Grain Fibers and Psyllium in Relation to Cardiovascular Health. Nutrients. 2013;5(5):1471–1487. doi: 10.3390/nu5051471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alexander C, Swanson KS, Fahey GC, Garleb KA. Perspective: physiologic Importance of Short-Chain Fatty Acids from Nondigestible Carbohydrate Fermentation. Adv Nutr. 2019;10(4):576–589. doi: 10.1093/advances/nmz004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mariño E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, McKenzie C, Kranich J, Oliveira AC, Rossello FJ, et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol. 2017;18(5):552–562. doi: 10.1038/ni.3713. [DOI] [PubMed] [Google Scholar]
- 56.Martínez I, Lattimer JM, Hubach KL, Case JA, Yang J, Weber CG, Louk JA, Rose DJ, Kyureghian G, Peterson DA, et al. Gut microbiome composition is linked to whole grain-induced immunological improvements. Isme J. 2013;7(2):269–280. doi: 10.1038/ismej.2012.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen T, Long W, Zhang C, Liu S, Zhao L, Hamaker BR. Fiber-utilizing capacity varies in Prevotella- versus Bacteroides-dominated gut microbiota. Sci Rep 2017; 7(1). doi: 10.1038/s41598-017-02995-4. Available from http://www.nature.com/articles/s41598-017-02995-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De Paepe K, Verspreet J, Verbeke K, Raes J, Courtin CM. Van de Wiele T. Introducing insoluble wheat bran as a gut microbiota niche in an in vitro dynamic gut model stimulates propionate and butyrate production and induces colon region specific shifts in the luminal and mucosal microbial community: long-term wheat bran intervention in the SHIME. Environ Microbiol. 2018;20:3406–3426. doi: 10.1111/1462-2920.14381. [DOI] [PubMed] [Google Scholar]
- 59.Jha AR, Davenport ER, Gautam Y, Bhandari D, Tandukar S, Ng KM, Fragiadakis GK, Holmes S, Gautam GP, Leach J, et al. Gut microbiome transition across a lifestyle gradient in Himalaya. PLOS Biol. 2018;16(11):e2005396. doi: 10.1371/journal.pbio.2005396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smits SA, Leach J, Sonnenburg ED, Gonzalez CG, Lichtman JS, Reid G, Knight R, Manjurano A, Changalucha J, Elias JE, et al. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science. 2017;357(6353):802–806. doi: 10.1126/science.aan4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim D, Hofstaedter CE, Zhao C, Mattei L, Tanes C, Clarke E, Lauder A, Sherrill-Mix S, Chehoud C, Kelsen J, et al. Optimizing methods and dodging pitfalls in microbiome research. Microbiome. 2017;5(1):52. doi: 10.1186/s40168-017-0267-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Szamosi JC, Forbes JD, Copeland JK, Knox NC, Shekarriz S, Rossi L, Graham M, Bonner C, Guttman DS, Van Domselaar G, et al. Assessment of Inter-Laboratory Variation in the Characterization and Analysis of the Mucosal Microbiota in Crohn’s Disease and Ulcerative Colitis. Front Microbiol. 2020;11:2028. doi: 10.3389/fmicb.2020.02028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reichardt N, Duncan SH, Young P, Belenguer A, McWilliam Leitch C, Scott KP, Flint HJ, Louis P. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. Isme J. 2014;8(6):1323–1335. doi: 10.1038/ismej.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Vadder F, Kovatcheva-Datchary P, Zitoun C, Duchampt A, Bäckhed F, Mithieux MG. Microbiota-Produced Succinate Improves Glucose Homeostasis via Intestinal Gluconeogenesis. Cell Metab. 2016;24(1):151–157. doi: 10.1016/j.cmet.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 65.Fernández-Veledo S, Vendrell J. Gut microbiota-derived succinate: friend or foe in human metabolic diseases? Rev Endocr Metab Disord. 2019;20(4):439–447. doi: 10.1007/s11154-019-09513-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ikeyama N, Murakami T, Toyoda A, Mori H, Iino T, Ohkuma M, Sakamoto M. Microbial interaction between the succinate-utilizing bacterium Phascolarctobacterium faecium and the gut commensal Bacteroides thetaiotaomicron. MicrobiologyOpen [Internet] 2020. [cited 2020 November4]; 9(10). 10.1002/mbo3.1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Connors J, Dawe N, Van Limbergen J. The Role of Succinate in the Regulation of Intestinal Inflammation. Nutrients. 2018;11(1):25. doi: 10.3390/nu11010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Serena C, Ceperuelo-Mallafré V, Keiran N, Queipo-Ortuño MI, Bernal R, Gomez-Huelgas R, Urpi-Sarda M, Sabater M, Pérez-Brocal V, Andrés-Lacueva C, et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. Isme J. 2018;12(7):1642–1657. doi: 10.1038/s41396-018-0068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Asnicar F, Berry SE, Valdes AM, Nguyen LH, Piccinno G, Drew DA, Leeming E, Gibson R, Le Roy C, Khatib HA, et al. Microbiome connections with host metabolism and habitual diet from 1,098 deeply phenotyped individuals. Nat Med Internet] 2021. [cited 2021 January19]; 27(2):321–332 doi: 10.1038/s41591-020-01183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Flint HJ, Scott KP, Duncan SH, Louis P, Forano E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes. 2012;3(4):289–306. doi: 10.4161/gmic.19897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Larsen OFA, Claassen E. The mechanistic link between health and gut microbiota diversity. Sci Rep. 2018;8(1):2183. doi: 10.1038/s41598-018-20141-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Makki K, Deehan EC, Walter J, The BF. Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe. 2018;23(6):705–715. doi: 10.1016/j.chom.2018.05.012. [DOI] [PubMed] [Google Scholar]
- 73.Schroeder BO, Birchenough GMH, Ståhlman M, Arike L, Johansson MEV, Hansson GC, Bäckhed F. Bifidobacteria or Fiber Protects against Diet-Induced Microbiota-Mediated Colonic Mucus Deterioration. Cell Host Microbe. 2018;23(1):27–40.e7. doi: 10.1016/j.chom.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.De Filippis F, Pasolli E, Tett A, Tarallo S, Naccarati A, De Angelis M, Neviani E, Cocolin L, Gobbetti M, Segata N, et al. Distinct Genetic and Functional Traits of Human Intestinal Prevotella copri Strains Are Associated with Different Habitual Diets. Cell Host Microbe. 2019;25(3):444–453.e3. doi: 10.1016/j.chom.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 75.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci. 2010;107(33):14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.De Filippis F, Pellegrini N, Laghi L, Gobbetti M, Ercolini D. Unusual sub-genus associations of faecal Prevotella and Bacteroides with specific dietary patterns. Microbiome. 2016;4(1):57. doi: 10.1186/s40168-016-0202-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hansson GC. Mucins and the Microbiome. Annu Rev Biochem. 2020. annurev-biochem-011520-105053;89(1):769–793. doi: 10.1146/annurev-biochem-011520-105053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tailford LE, Crost EH, Kavanaugh D, Juge N. Mucin glycan foraging in the human gut microbiome. Front Genet [cited 2020 June17]; 6. Available from. 2015;. . http://www.frontiersin.org/Nutrigenomics/10.3389/fgene.2015.00081/abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Picot WG, Hou F, Statistics Canada, Social Analysis and Modelling Division. Immigration, low income and income inequality in Canada: what’s new in the 2000s? [Internet]. 2015. [cited 2020 June17]. Available from: http://www.deslibris.ca/ID/246290
- 80.Ke C, Sohal P, Qian H, Quan H, Khan NA. Diabetes in the young: a population-based study of South Asian, Chinese and White people. Diabet Med. 2015;32(4):487–496. doi: 10.1111/dme.12657. [DOI] [PubMed] [Google Scholar]
- 81.Shah BR. Utilization of physician services for diabetic patients from ethnic minorities. J Public Health. 2008;30(3):327–331. doi: 10.1093/pubmed/fdn042. [DOI] [PubMed] [Google Scholar]
- 82.Wardle RA, Wardle AJ, Charadva C, Ghosh S, Moran GW. Literature review: impacts of socioeconomic status on the risk of inflammatory bowel disease and its outcomes. Eur J Gastroenterol Hepatol. 2017;29(8):879–884. doi: 10.1097/MEG.0000000000000899. [DOI] [PubMed] [Google Scholar]
- 83.Benchimol EI, To T, Griffiths AM, Rabeneck L, Guttmann A. Outcomes of Pediatric Inflammatory Bowel Disease: socioeconomic Status Disparity in a Universal-Access Healthcare System. J Pediatr. 2011;158(6):960–967.e4. doi: 10.1016/j.jpeds.2010.11.039. [DOI] [PubMed] [Google Scholar]
- 84.Bernstein CN, Kraut A, Blanchard JF, Rawsthorne P, Yu N, Walld R. The Relationship Between Inflammatory Bowel Disease and Socioeconomic Variables. The American Journal of Gastroenterology. 2001;96:7:2117-2125. [DOI] [PubMed] [Google Scholar]
- 85.Bowyer R, Jackson M, Le Roy C, Ni Lochlainn M, Spector T, Dowd J, Steves SC. Socioeconomic Status and the Gut Microbiome: a TwinsUK Cohort Study. Microorganisms. 2019;7(1):17. doi: 10.3390/microorganisms7010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tarasuk V, Fitzpatrick S, Ward H. Nutrition inequities in Canada. Appl Physiol Nutr Metab. 2010;35(2):172–179. doi: 10.1139/H10-002. [DOI] [PubMed] [Google Scholar]
- 87.Lesser IA, Gasevic D, Lear SA. The Association between Acculturation and Dietary Patterns of South Asian Immigrants. PLoS ONE. 2014;9(2):e88495. doi: 10.1371/journal.pone.0088495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sanou D, O’Reilly E, Ngnie-Teta I, Batal M, Mondain N, Andrew C, Newbold BK, Bourgeault IL. Acculturation and Nutritional Health of Immigrants in Canada: a Scoping Review. J Immigr Minor Health. 2014;16(1):24–34. doi: 10.1007/s10903-013-9823-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.R Core Team. R : A Language of Environment for Statistical Computing. R Found Stat Comput 2019
- 91.Epidemiology and Genomics Research Program . Health NIo. Diet History Questionnaire: canadian Version [Internet]. [cited 2020 Jan 24]. 2018; Available from: https://epi.grants.cancer.gov/DHQ/forms/canadian/
- 92.Epidemiology and Genomics Research Program . Health NIo. Diet History Questionnaire: diet*Calc Software [Internet]. [cited 2020 Jan 24]. 2018; Available from: https://epi.grants.cancer.gov/DHQ/dietcalc/
- 93.Baker EH. Socioeconomic Status, Definition. Wiley Blackwell Encycl Health Illn Behav Soc 2014
- 94.Postal CodeOM Conversion File (PCCF) , Reference Guide.:23.
- 95.Dore J, Ehrlich S, Levenez F, Pelletier E, Bertrand A, Bork P, Costea P, Sunagawa S, Guarner F, Manichanh C, et al. HMS_SOP 07 V1: standard operating procedure for fecal samples DNA extraction, Protocol H [Internet]. [cited 2019 July 5]. 2015; Available from: http://www.microbiome‐standards.org
- 96.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27(6):863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rodriguez-R LM, Konstantinidis KT. Nonpareil: a redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics. 2014;30(5):629–635. doi: 10.1093/bioinformatics/btt584. [DOI] [PubMed] [Google Scholar]
- 100.Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH. the UniProt Consortium. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015;31:926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Caspi R, Foerster H, Fulcher CA, Kaipa P, Krummenacker M, Latendresse M, Paley S, Rhee SY, Shearer AG, Tissier C, et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2007;36(Database):D623–31. doi: 10.1093/nar/gkm900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Caspi R, Billington R, Ferrer L, Foerster H, Fulcher CA, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016;44(D1):D471–80. doi: 10.1093/nar/gkv1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McMurdie PJ, Paulson JN. Biomformat: an interface package for the BIOM file format. [Internet]. [cited 2019 July 5]. 2019. Available from: https://github.com/joey711/biomformat
- 104.McMurdie PJ, Holmes S, Watson M. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8(4):e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wickham H tidyverse: easily Install and Load the “Tidyverse” [Internet]. [cited 2019 July 5]. 2017. Available from: https://CRAN.R-project.org/package=tidyverse
- 106.Bergstrom KSB, Xia L. Mucin-type O-glycans and their roles in intestinal homeostasis. Glycobiology. 2013;23(9):1026–1037. doi: 10.1093/glycob/cwt045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Martens EC, Chiang HC, Gordon JI. Mucosal Glycan Foraging Enhances Fitness and Transmission of a Saccharolytic Human Gut Bacterial Symbiont. Cell Host Microbe. 2008;4(5):447–457. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(D1):D490–5. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, et al. vegan: community Ecology Package [Internet]. [cited 2019 July 5]. 2018. Available from: https://CRAN.R-project.org/package=vegan
- 110.Martinez Arbizu P PairwiseAdonis: pairwise Multilevel Comparison using Adonis. 2017.
- 111.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47–e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Krueger PM, Bhaloo T, Rosenau PV. Health Lifestyles in the United States and Canada: are We Really So Different? Soc Sci Q. 2009;90(5):1380–1402. doi: 10.1111/j.1540-6237.2009.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Holmes I, Harris K, Quince C, Gilbert JA. Dirichlet Multinomial Mixtures: generative Models for Microbial Metagenomics. PLoS ONE. 2012;7(2):e30126. doi: 10.1371/journal.pone.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhao X, Valen E, Parker BJ, Sandelin A, Bähler J. Systematic Clustering of Transcription Start Site Landscapes. PLoS ONE. 2011;6(8):e23409. doi: 10.1371/journal.pone.0023409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pearson R. GoodmanKruskal: association Analysis for Categorical Variables. [Internet]. [cited 2019 July 5] 2016. Available from: https://CRAN.R-project.org/package=GoodmanKruskal
- 117.Eren AM, Kiefl E, Shaiber A, Veseli I, Miller SE, Schechter MS, Fink I, Pan JN, Yousef M, Fogarty EC, et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat Microbiol. 2021;6(1):3–6. doi: 10.1038/s41564-020-00834-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31(10):1674–1676. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 119.Kim D, Song L, Breitwieser FP, Salzberg SL. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016;26(12):1721–1729. doi: 10.1101/gr.210641.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Alneberg J, Bjarnason BS, De Bruijn I, Schirmer M, Quick J, Ijaz UZ, Lahti L, Loman NJ, Andersson AF, Quince C. Binning metagenomic contigs by coverage and composition. Nat Methods. 2014;11(11):1144–1146. doi: 10.1038/nmeth.3103. [DOI] [PubMed] [Google Scholar]
- 121.Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11(1):119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12(1):59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 123.Pritchard L, Glover RH, Humphris S, Elphinstone JG, Toth IK. Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal Methods. 2016;8(1):12–24. doi: 10.1039/C5AY02550H. [DOI] [Google Scholar]
- 124.Layeghifard M, Hwang DM, Guttman DS. Constructing and Analyzing Microbiome Networks in R. In: Beiko R, Hsiao W, Parkinson J (eds) Microbiome Analysis. Methods in Molecular Biology, vol 1849. : Humana Press, New York; 2018. page 243–66. [DOI] [PubMed] [Google Scholar]
- 125.Scholz M, Ward DV, Pasolli E, Tolio T, Zolfo M, Asnicar F, Truong DT, Tett A, Morrow AL, Segata N. Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat Methods. 2016;13(5):435–438. doi: 10.1038/nmeth.3802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The metagenomic sequencing data have been submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under submission number SUB7709855. The accession number for the sequencing data reported in this paper is BioProject PRJNA644666. All data generated or analyzed during this study are included in this published article (and its supplementary information files) or through the SRA. Additional files include a knitr document describing all methods used for data analysis.