

SUMMARY
Using intravital imaging, we report that bone marrow (BM) plasma cells (PCs) are motile. BM PCs exhibit a unique migration pattern, characterized by intermittent periods of high motility and longer stretches of confined migration or arrest. BM PCs accumulate into clusters, which have reduced cell motility. APRIL promotes cluster formation and overall PC motility in the BM. Although CXCL12 and its receptor, CXCR4, promote PC motility in the BM, VLA4 activity promotes arrest. However, blocking either pathway promotes PC egress from the BM. Under steady-state conditions, BM PCs recirculate to other bones and spleen. In older mice, overall PC motility and recirculation increase, and this is correlated with increased CXCR4 expression, which depends on PC age or maturation rather than mouse age. Altogether, these results suggest that changes in PC motility and CXCR4 expression are linked with survival of long-lived PCs in the BM.
Graphical Abstract
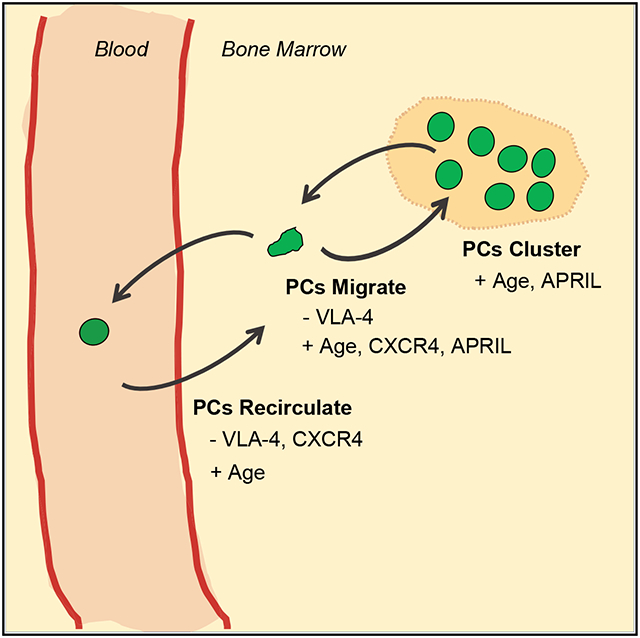
In Brief
Benet et al. report that bone marrow (BM) plasma cells (PCs) are motile and found in clusters organized by the survival factor APRIL. They identify CXCR4 and VLA4 as key factors regulating PC motility and retention in the BM. PCs recirculate to other bones. PC motility increases with age.
INTRODUCTION
Plasma cells (PCs) are central to humoral immunological memory, which provides prophylactic antibody protection against pathogens (Khodadadi et al., 2019; Lightman et al., 2019). PCs are terminally differentiated B cells that can have both T cell-dependent and T cell-independent generation. PCs are CD138high and B220−, with high expression of the transcription factor Blimp1; they secrete antibodies and are considered non-proliferative. PC maturation is a continuum, with less mature, proliferative short-lived PCs (CD138+ B220+ or plasma-blasts [PBs]) giving rise to more mature PCs or long-lived PCs (LLPCs). These LLPCs are operationally defined based on their longevity in the body, generally express the most mature PC phenotype, and can provide lifelong humoral protection. In humans, the bone marrow (BM) is a major site of LLPCs, and by some estimates, more than 50% of BM PCs are long lived (Manz et al., 1997; Slifka et al., 1998).
Survival of PCs depends on various extrinsic factors produced in the bone marrow (BM). These extrinsic factors include membrane-bound ligands like CD80/86 and secreted proteins such as APRIL, interleukin-6 (IL-6), BAFF, and CXCL12 (or SDF-1); CXCL12 also acts as the master chemokine for BM tropic mononuclear cells (Lightman et al., 2019). Many types of BM stromal cells, both hematopoietic and non-hematopoietic, have been implicated in the production of these factors and in supporting LLPC viability. Various mesenchymal BM cells can produce CXCL12 (Tikhonova et al., 2019), whereas APRIL and other soluble factors are produced by various hematopoietic subsets, including granulocytes, megakaryocytes, and myeloid cells (Khodadadi et al., 2019).
It is unclear how the BM niche of PCs is organized. Initial imaging has shown that PCs are sessile and non-migratory in the BM (Zehentmeier et al., 2014). This has led many to speculate about the nature of the PC BM niche and how these accessory niche cells are coordinated (Chu et al., 2011; Khodadadi et al., 2019; Lightman et al., 2019). However, histological dissection of the PC niche did not reveal a clear structure or specific composition of cell types in direct association with PCs (Zehentmeier et al., 2014). Here we report that PCs in the BM are motile and partially clustered, with unique migratory patterns that change with age. By better understanding the structure, regulation, and dynamics of the BM PC niche, we may better understand how to maintain lifelong humoral immune protection, particularly in old age.
RESULTS
Most BM PC are motile on timescales of hours
To better understand the dynamics of BM PC niche, we imaged PCs in the tibial BM of Blimp1-YFP reporter mice (16–36 weeks old) (Figure 1A). Time-lapse intravital tibial imaging and cell tracking revealed that BM PCs were motile compared with sessile autofluorescent cells (Figure 1B; Video S1), which we previously characterized as CD169+ macrophages (Spaulding et al., 2016). In this reporter (Fooksman et al., 2010), more than 95% BM cells expressing high levels of yellow fluorescent protein (YFPhigh) are phenotypically PCs (CD138high, B220−, CD19low, Ki67−-, and major histocompatibility complex [MHC] class II−) (Figure S1A) and secrete antibody by ELISPOT (McCarron et al., 2017). Although 5% of YFPhigh BM cells are effector CD8+ T cells, they expressed 10-fold lower levels of YFP than PCs (Figure S1B) and moved ~10 μm/min; thus, they were excluded from PC analysis based on cell size and YFP intensity. Using Blimp1-YFP+ CD4-cre+Rosa26LSL-tdTomato mice, YFP+ Tomato+ double-positive T cells were extremely dim, small, and too fast to track using our standard PC analysis (Figures S1C-S1F).
Figure 1. BM PCs are motile at hourlong timescales.

(A) Intravital image of tibial BM in XY showing compact bone (blue), autofluorescent macrophages (MPs, orange), and Blimp1-YFP+ PCs (green).
(B) Plotted 2-dimensional (x and y axes) cell tracks of PCs and MPs with a common origin.
(C) Displacement of PCs and MPs at different timescales, with the percentage of moving cells (displacement > 20 μm) in parentheses.
(D) Track velocity analysis.
(E) Displacement velocity.
(F) Frequency histogram of instantaneous velocities.
(G) MSD plots for MPs and PCs. Error reflect SEM.
(H) Comparison of track velocities versus ranges of mouse ages.
(I) Samples of BM PC tracks exhibiting periods of both high and low displacement.
(J) Analysis of all PC tracks with durations longer than 4 h.
(K) Comparison of high and low displacement periods.
Data in (B)–(K) except (H) were pooled from movies of three separate mice aged 16–36 weeks (n = ~1,200 cells, ~300 cells/mouse). Bars reflect means. Mann-Whitney t tests were conducted. ****p < 0.0001.
When imaging PCs in the BM at steady state for 30 min, 95% PCs had limited motility based on a displacement threshold of 20 μm, a distance of two cell diameters (Figure 1C). However, by establishing stable, long-term imaging for up to 12 h, we found 54% of PCs in the BM displaced at least 20 μm and were considered motile when tracked for periods longer than 30 min. When tracked for at least 4 h, ~78% of PCs were motile. On average, BM PCs were traveling at an average speed (or track velocity) of 1 μm/min (Figure 1D) and a displacement velocity of 0.2 μm/min (Figure 1E), but cells reached instantaneous speeds of up to 6 μm/min (Figure 1F). We did not observe directionality of migration (Figure S2A), and the mean squared displacement (MSD) was consistent with a random walk at hourlong timescales (Figure 1G). We did not observe a decrease in the number of PCs over time (Figure S2B), which could indicate signs of inflammation during imaging (Slocombe et al., 2013).
Over the course of imaging PCs in the BM of numerous mice, we observed heterogeneity in the motility of BM PCs, which correlated with the age of the mice. The overall track speed significantly increased with age (Figure 1H; Video S1). Therefore, we used age-matched mice and, when applicable, analyzed changes in PC behavior versus age.
BM PC motility is intermittent and heterogeneous over time
Under steady-state conditions, most lymphocytes typically migrate at a consistent cadence or speed, which varies based on their activation state and anatomical location (Germain et al., 2012). In contrast, BM PCs (in 16- to 36-week-old mice) were mostly sessile with intermittent intervals of high displacement over time (Figure 1I; Video S2) and speeds up to 6 μm/min (Figure 1F). We classified these high and low displacement intervals using a rolling average of track displacement at 30-minute intervals (Figures S2C and S2D). When applied to cell tracks, most PC tracks had mixed and highly variable migration behaviors, exhibiting durations of both high and low displacement rates per cell (Figure 1J). On average, the duration was 53 ± 34 min for high displacement and 263 ± 105 min for low displacement, indicating that overall, PCs spend more time arrested than moving in the BM (Figure 1K).
BM PC motility is reduced in PC clusters
Within the BM, PCs were not uniformly distributed within the parenchyma but appeared to be clustered. Using a custom algorithm to classify regions of high PC density (Figure S2E), we identified regions of the BM with a high density of PCs as being clustered (Figure 2A; Video S3, part A). Using this classifier, in the BM of 16- to 36-week-old mice, 40% of PCs were considered clustered, with around 150 clusters per cubic millimeter of BM and each cluster consisting of ~12 cells per cluster on average (Figure 2B). In contrast, 6- to 8-week-old mice had less PC clustering (Figure 2C). Within clusters, PCs had lower motility compared with PCs outside of these regions based on track velocity (Figure 2D), displacement velocity (Figure 2E), track straightness (Figure 2F), and MSD (Figure 2G). Some PCs could be observed entering or exiting clusters during imaging. While in these clusters, PCs had reduced motility compared with when they were outside the cluster (Figure 2H), suggesting local extrinsic factors regulate their motility. The overall density of clusters (percentage of cells in clusters) was unchanged over 6–7 h of imaging (Figure 2I), and on a per-cluster basis, most clusters were stable in terms of the number of PCs localized per cluster (Figure 2J). However, we observed that some clusters dissipate over time (Figure 2K; Video S3, part B) and new clusters form in areas that had few PCs at the start of imaging (Figure 2L; Video S3, part C). This increase in PC density also correlated with reduced motility by MSD and displacement velocity of those PCs in the cluster (Figures 2M and 2N).
Figure 2. BM PC motility is reduced in PC clusters.

(A) Representative intravital 3D flattened image of raw (left) and processed (right) PCs to classify PCs and clusters.
(B) Percentage of all PCs in clusters (top), number of clusters normalized to imaging volume (middle), and number of PCs per cluster (bottom).
(C) Comparison of parameters in (B) at different mouse ages (n = 1 mouse/dot, 3 independent experiments).
(D–G) Comparison of PC tracks entirely inside or outside clusters.
(D) Track velocity.
(E) Displacement velocity.
(F) Track straightness.
(G) MSD.
(H) Per-cell analysis of track speed of mixed PC tracks that entered and exited clusters.
(I) Analysis of the percentage of PCs in clusters (as in B) over time.
(J) Number of PCs per individual cluster.
(K) Example spots of defined PCs before and after cluster dissipation.
(L–N) Spots of defined PCs before and after cluster formation (L), and (M) their respective MSD and (N) paired track velocity. Scale bar is 20 μm.
For (B)–(J) except (C), PCs from three 15- to 36-week-old mice were pooled and analyzed. All bars represent mean, and error is SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 by Mann-Whitney tests, except (H), (N), which used paired t test analysis.
APRIL contributes to PC organization and motility
We suspected that this PC clustering may be organized by survival niches and soluble factors, such as APRIL. In April−/− mice (knockout [KO] mice), BM PC numbers were reduced by 90%, with a 50% reduction in April+/− (hetereozygous [HET] mice). Based on BM chimeras, we found that hematopoietically produced APRIL is required to maintain the BM PC population (Figures 3A and 3B). We bred Blimp1-YFP reporter into the APRIL KO background to observe the role of APRIL on PC dynamics and organization and saw a marked decrease in PC clustering in APRIL HET and KO mice compared with wild-type (WT) mice (Figures 3C-3E). Moreover, PC motility, based on track velocity, displacement velocity, and MSD, was reduced in HET and KO mice compared with WT mice (Figures 3F-3H). Altogether, these data indicate that APRIL promotes PC organization and motility in the BM.
Figure 3. APRIL contributes to PC organization and motility.

(A) Comparisons of PC frequencies and total numbers in BM and spleen with April+/+ (WT), April+/− (Het), and April−/− (KO) backgrounds.
(B) Analysis of PC frequencies and total cell numbers in the BM of recipient mice 8 weeks following irradiation and reconstitution after donor BM, annotated as donor → recipient notation.
(C) Sample tiled BM images of PCs (green) and autofluorescence (red) from Blimp-1 YFP bred into WT, Het, and KO backgrounds, analyzed in (D) and (E).
(D) Analysis of the percentage of PCs in clusters per mouse in each background.
(E) Comparison of the number of PC clusters, normalized to 1 mm3 of BM parenchyma.
(F–H) Comparison of PC dynamics based on (F) track velocity, (G) displacement velocity, and (H) MSD analysis of displacement. Analysis is pooled from 3 or more mice in each April genetic background.
For cell tracking, n = ~1,000 tracks, with ~300 cells/mouse, for (F)–(H). For plots in (A)–(E), n = 1 mouse/dot. All bars represent mean, and error is SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by Mann-Whitney t tests.
CXCR4 is required for PC motility
Because CXCR4 signaling is critical for PC BM engraftment (Hargreaves et al., 2001), we tested whether signaling controlled PC dynamics in the BM parenchyma. Mice were imaged before and following treatment with AMD3100, a CXCR4 antagonist, which triggered PC arrest within 15 min after injection (Figure 4A; Video S4). PCs after AMD3100 treatment had reduced motility based on track velocity, displacement velocity, and MSD slopes (Figures 4B-4D). Typically, even sessile PCs showed some dynamic pseudopod activity, consistent with extracellular sensing or interplay between stop and go signals. However, AMD3100-treated PCs showed no pseudopod activity and were more spherical (Figure 4E), suggesting CXCR4 signaling is required for PC pseudopodal activity and PC polarity. The effects of AMD3100 reversed after 4–6 h, consistent with the half-life of AMD3100 in vivo (Beck et al., 2014; Hendrix et al., 2000), and cells began to regain some motility. Mice that were treated with pertussis toxin, which irreversibly ablates Gαi-mediated signaling, including via CXCR4, had reduced PC motility (Figures S3A and S3B). Overall, these results indicate that CXCR4 signaling promotes PC motility within the BM parenchyma.
Figure 4. CXCR4 is required for BM PC motility.

(A–E) Blimp1-YFP mice were injected intravenously (i.v.) with CXCR4 inhibitor AMD3100. Cell movement was tracked by intravital imaging of the tibia for 2–3 h before treatment (blue) and for 2–3 h immediately after treatment (red). (A) Examples of PC track movement of 3 individual PCs, color coded for before (blue) and after (red) AMD3100 treatment (left), with corresponding measurements of instantaneous velocity for each cell over time (right). (B–E) Pooled measurement of PC motility before and after AMD3100 treatment from 2 independent experiments, with ~200 cells/mouse.
(B) Track velocity.
(C) Displacement velocity.
(D) MSD.
(E) Example images of PC morphology and analysis of sphericity of PCs. Scale bar is 5 μm.
(F–L) Each dot represents a mouse, and each color represents mice from 3 independent experiments.
(F) Analysis of surface CXCR4 levels on PCs (CD138high B220−) or B cells (B220high) of BM cells in 6-, 15-, 32-, and 50-week-old mice.
(G) Enumeration of BM PCs in mice in (F).
(H) Analysis of CXCR4 as a function of BM PC numbers pooled from mice of different ages, as labeled in the legend.
(I) Total (intracellular and surface) CXCR4 levels in BM PCs in 6- and 15-week-old mice.
(J) Cxcr4 RNA, normalized to β-actin in fluorescence-activated cell-sorted PCs and B cells in 6- and 15-week-old mice.
(K) Experimental schema for comparing age-matched, de novo-generated, NP-specific (YFP+) PCs and age-matched recipients following immunization with NP-KLH.
(L) Analysis of CXCR4 surface levels on NP-specific (YFP+ CD138high) BM PCs, with PC and mouse ages marked.
(M) Pooled single-cell analysis of CXCR4 versus BLIMP values for PBs and PCs from experiments in (K) and (L). Linear fit (red) was found to be a significant (non-zero) positive slope. All bars reflect mean, errors are SEM. Comparisons were conducted by Mann-Whitney t test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To see whether CXCR4 surface expression was involved in age-associated changes in PC motility, we analyzed CXCR4 levels in mice aged 6, 15, 30 or 50 weeks. We found that as mice aged, CXCR4 levels on BM PCs increased (Figure 4F). Although these CXCR4 values varied from mouse to mouse within an age group, so did the number of BM PCs in each group and with mouse age (Figure 4G). However, there was a positive correlation (single exponential fit, r2 = 0.52) of CXCR4 expression with the number of PCs (Figure 4H). Surface CXCR4 levels could reflect differences in recycling because of signaling; however, total CXCR4 protein levels in PCs (both surface and intracellular) were higher in 15-week-old mice compared with 6-week-old mice, suggesting increases result from enhanced production rather than trafficking (Figure 4I). CXCR4 protein and mRNA levels in BM PCs increased with age, whereas expression on BM B cells was unaffected by age (Figures 4F and 4J), indicating changes in expression were specific to PC lineage and possibly related to intrinsic aging of the PC (i.e., LLPC).
To see whether increasing CXCR4 levels were related to the intrinsic age of the PC or the age of the mouse, we transferred nitrophenyl (NP)-specific transgenic (B1–8high) B cells (co-expressing Blimp1-YFP reporter) and immunized mice using NP conjugated to Keyhole limpet hemocyanin (KLH) with alum to generate de novo NP-specific YFP+ PCs that could be tracked over time. Using recipients of different ages and different durations after immunization, we sought to assess the contribution of both intrinsic and extrinsic age-related factors to CXCR4 expression on PCs (Figure 4K). We found that CXCR4 levels increased with the age of the YFP+ PCs; 8-week-old PCs had the highest CXCR4 expression compared with 4- or 2-week-old PCs, whereas 4-week-old PCs in 8- or 16-week-old mice had similar CXCR4 expression (Figure 4L). Moreover, CXCR4 expression increased with Blimp-1 YFP expression, which correlates with PC maturation (Fooksman et al., 2010; Kallies et al., 2007), further indicating that CXCR4 upregulation is intrinsic to PC age (Figure 4M).
CXCL12 promotes PC motility in the BM
To corroborate the effects of blocking CXCR4 on BM PC motility, we tested the role of its ligand, CXCL12, on PC motility. Adult mice were treated acutely with a CXCL12-blocking antibody or isotype (Beck et al., 2014) and imaged after treatment. After treatment with anti-CXCL12, PCs had reduced motility compared with isotype-treated mice, based on track velocity, displacement velocity, and MSD slope (Figures 5A-5C; Video S5). Transferred fluorescently labeled naive B cells showed reduced motility following treatment, consistent with previous studies of immature B cells (Beck et al., 2014; data not shown).
Figure 5. CXCL12 promotes PC motility in the BM.

(A–C) Blimp 1-YFP mice were injected i.v. with either CXCL12 neutralizing antibody or isotype control antibody. Cell movement was tracked by intravital imaging of the tibia for at least 4 h after treatment.
(A) Track velocity of PCs.
(B) Displacement velocity of PCs.
(C) MSD of PCs over time. Numbers depict the average mean motility coefficient of 3 and 2 individual experiments for CXCL12 neutralizing antibody and isotype control antibody treatments, respectively. Data are pooled from 3 and 2 individual experiments for CXCL12 neutralizing antibody and isotype control antibody treatments, respectively, with ~300 cells/mouse.
(D) Transwell migration analysis of BM B cells and PCs, analyzed and normalized to input bulk BM from 6- and 15-week-old mice, in the presence or absence of CXCL12 in the lower chamber and with or without AMD3100 pretreatment.
(E) Analysis of CXCL12 protein in BM supernatant by ELISA.
(F) Analysis of Cxcl12 total RNA in 6-, 15-, 32-, and 50-week-old mice normalized to a β-actin transcript.
(G) Analysis of Cxcl12 α, β, and γ isoforms, shown as the percentage of the total Cxcl12 transcript.
Data in (D)–(F) were pooled from 2 independent experiments, each dot represents 1 mouse. In (G), technical replicates (3 per sample) and biological replicates (n = 4 mice total) from two experiments were averaged. All quantification plots show mean. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by unpaired Mann-Whitney U test.
To visualize CXCL12 gradients in the BM, we bred Blimp1-YFP Cxcl12DsRed mice that fluorescently labeled Cxcl12-producing cells, including perivascular cells, endothelial cells, and osteoblasts (Ding and Morrison, 2013; Pitt et al., 2015). Within the BM, the network of DsRed+ cells is dense (Figure S3C) and DsRed fluorescence intensity was highly uniform despite large variations in the production of Cxcl12 mRNA among different mesenchymal subsets (Tikhonova et al., 2019), indicating the reporter was not sensitive to these differences in expression. Thus, as could be expected, we found that PCs were in constant contact with a dense DsRed+ network (Figure S3D; Video S6) and could not discern differences in PC motility in relation to cell distance to the DsRed+ network (Figure S3E).
To see whether differences in PC motility in the BM were related to CXCL12 sensitivity, we conducted transwell chemotaxis assays. We titrated CXCL12 levels and found a bell-shaped curve of transmigration rates (Figure S3F). Using total BM from 6- or 15-week-old mice as input, PCs from 15-week-old mice were more motile than those in younger mice (Figure 5D), whereas BM B cells were similar, suggesting cell-type-specific differences in motility. Although both CXCR4 and CXCR7 (or Ackr3) receptors can respond to the CXCL12 ligand, Cxcr4 mRNA levels were 20-fold higher than Ackr3 on PCs (Cornelis et al., 2020), and pretreatment with AMD3100 (which blocks CXCR4, but not CXCR7) ablated PC transmigration and motility in vivo (Figures 4A-4D), indicating the response to CXCL12 was driven by CXCR4. We analyzed CXCL12 levels in BM supernatant by ELISA, which decreased as mice aged from 6 to 50 weeks (Figure 5E). However, Cxcl12 total mRNA levels were similar (Figure 5F), as were the relative distributions of the three Cxcl12 isotypes α, β, and γ (Figure 5G), which have known differences in heparin binding that could affect ELISA measurements of free CXCL12 in the BM. These results suggest that CXCL12 protein production or secretion were altered in older mice, leading to reduced levelsin the BM.
VLA4 promotes PC arrest in the BM
Chemokine signaling can trigger integrin activation in a process known as inside-out signaling (Springer and Dustin, 2012). Because VLA4 (α4β1) is the major integrin regulating entry and retention in the BM for hematopoietic cells (Beck et al., 2014), we tested whether VLA4 was critical for PC motility. Mice were acutely treated with the dual α9β1/α4β1 integrin inhibitor N-benzenesulfonyl proline-based compound BOP (Cao et al., 2016) and imaged pre- and post-treatment. In contrast to CXCR4 blockade, VLA4 inhibition increased PC motility minutes after treatment (Figure 6A; Video S7) based on track velocity (Figure 6B), displacement velocity (Figure 6C), and MSD slope (Figure 6D). Similarly, treatment with an anti-α4 blocking antibody increased PC motility over time (Figures S3G-S3K).
Figure 6. VLA4 promotes PC arrest in the BM.

(A–D) Blimp1-YFP mice were injected i.v. with VLA4 inhibitor BOP. Cell movement was tracked by intravital imaging of the tibia for 2–3 h before treatment (blue) and for 5–6 h immediately after treatment (red).
(A) Examples of movement of 3 individual PCs shown by colored trajectories before (blue) and after (red) BOP treatment.
(B) Track velocity of PCs.
(C) Displacement velocity of PCs.
(D) MSD of PCs over time. Slopes indicate the average mean motility coefficient of 2 individual experiments. Data are pooled from 2 individual mouse experiments, with ~300 cells/mouse. All quantification plots show mean. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by unpaired Mann-Whitney U test.
(E–I) In vitro live-cell imaging and analysis of PB/PC motility and adhesion on various substrates with or without BOP treatment.
(E) Interference reflection microscopy (IRM) footprint in gray, Blimp1-YFP+ (green) PB/PC cell, and tracks (red lines) on ICAM-1, VCAM-1, fibronectin, or ICAM-1+VCAM-1 (20:80 mix) substrates.
(F) Analysis of frequency of PBs with adhesion (based on an IRM pattern) on various substrates with or without BOP pretreatment (dots indicate the average for each independent experiment).
(G) PB track velocity from imaging ± BOP treatment.
(H) PB track displacement (20-minute fixed movies) ± BOP treatment.
(I) Track velocity from BM PCs, ex vivo imaging ± BOP treatment.
Each dot in (G)–(I) is a single-cell track, pooled from 2–3 independent experiments per mouse per condition. All quantification plots show mean. Errors are SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by Mann-Whitney test.
To understand how inhibition of VLA4 could increase PC motility in vivo, we used in vitro live-cell confocal imaging on substrate-coated glass. We employed interference reflection microscopy (Figure 6E), which illuminates cell adhesion to the substrate to exclude rolling or floating movements in the media from genuine crawling (Fooksman et al., 2010). We analyzed lipopolysaccaride-induced PB adhesion and motility based on track speed and displacement on various substrates and found that although PBs were motile on ICAM-1 and fibronectin, they were arrested on VCAM-1 with tight adhesion (Figures 6E-6H; Video S8). As expected, treatment with BOP blocked VLA4-mediated adhesion to fibronectin and VCAM-1, but not to ICAM-1. We hypothesized that in the BM, PCs would be engaging multiple types of adhesion ligands simultaneously and BOP treatment may increase motility by biasing engagement from VCAM-1 to ICAM-1 molecules. To test this idea, we generated mixed ICAM-1/VCAM-1 substrates and found that PBs adhered and exhibited low motility. However, acute treatment with BOP increased PB motility on these substrates, as well as reducing adhesion (Video S9). These effects were confirmed using BM PCs from Blimp1-YFP mice, which showed similar changes in motility with and without BOP treatment (Figure 6I), consistent with the increased motility seen in PBs generated in vitro. Altogether, these data suggest that BOP treatment may promote increased motility in vivo by altering the adhesion substrates that BM PCs use, such as ICAM-1, leading to faster motility.
Chemokine and integrin inhibition promote BM PC mobilization
Blocking CXCR4 and VLA4 together is known to mobilize hematopoietic stem cells, among other cell types, from the BM (Cao et al., 2016). Because inhibition of these pathways altered PC motility, we tested whether any of these treatments could mobilize PCs (CD138high B220−) in contrast to newly minted pre-PCs or PBs (CD138+ B220+), which are found in the blood at steady-state conditions. WT mice treated with AMD3100 or BOP had no statistically significant increase in the blood at 4 h after treatment; however, combined CXCR4 and VLA4 inhibition increased blood PCs by 3-fold (Figure 7A). To see whether some increased blood PCs were emigrating from the BM, we assessed PCs numbers in the BM by imaging and saw similar decreases in BM PCs over time after treatment (Figure 7B). Similar results were obtained when mice were treated with a VLA4 blocking antibody that targets the α4 chain in combination with AMD3100 (Figure 7C), which also led to increased BM egress, measured by time-lapse intravital imaging (Figures S4A and S4B). By 24 h after treatment, blockade of VLA4 alone, with either BOP or antibody, mobilized BM PCs 3- to 4-fold, while blockade in combination with AMD3100 resulted in 16-fold mobilization (Figures S4C and S4D). Acute treatment with anti-CXCL12 also increased PCs in the blood at 24 h after treatment (Figure 7D). Although single-agent AMD3100 was a poor mobilizer of BM PCs, it may result from the short half-life and rapid deactivation that occur in vivo. In contrast, acute, inducible, genetic deletion of the Cxcr4 allele in a mixed BM chimera reduced PCs from the BM in a cell-intrinsic manner (Figures S4E and S4F), suggesting that more potent inhibitors of CXCR4 could mobilize PCs as a single agent and confirming that CXCR4 is required for PC retention in the BM.
Figure 7. BM PC mobilization and steady-state recirculation patterns.

(A) Number of PCs in the blood of C57BL/6 mice i.v. treated with PBS, 80 μg of AMD3100, 250 μg of BOP, or both AMD3100 and BOP for 4 h. Bars represent mean, and error is SEM (each dot is an independent mouse, n = 4–7 mice/group, 3 experiments).
(B) Percentage of PCs detected in intravital two-photon imaging compared with the initial imaging frame of the tibial BM (2–4 mice per group).
(C) Number of PCs in the blood of C57BL/6 mice i.v. treated with PBS, 80 of μg AMD3100, 300 of μg αVLA4 antibody, or both AMD3100 and αVLA4 antibody for 4 h. Bars represent mean, and error is SEM (n = 2–4 mice/group, 4 experiments).
(D) Fold change of PCs in the blood of C57BL/6 mice treated with 80 μg of anti-CXCL12 antibody or isotype. Bars represent mean, and error is SEM (n = 4–5 mice/group, 4 experiments, normalized to isotype).
(E) Number of PCs in the blood of C57BL/6 mice i.v. treated with PBS, FTY720 (FTY), AMD+BOP as before, and FTY720+AMD+BOP.
(F–I) Direct tibial labeling of PCs. (F) Specificity of labeling at 1 day post treatent. with 5 μg of 4HT on gated PCs. Kinetics of recirculation of (G) labeled PCs and (H) B cells (left), and their ratios of labeled cells in the CL to the injected tibia (right). (I) Comparison of ratios of the percentage of tomato+-labeled PCs in the CL versus the injected paired tibia of 14- to 18-week-old mice (closed circle, data shown in G) and 6- to 8-week-old mice (open circle).
In (F)–(H), n = 1–3 mice/group, 3 experiments. All bars represent mean, and error is SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Finally, because S1P gradients between blood and BM sinusoids could potentially contribute to PC egress (Pereira et al., 2010), we tested whether blocking S1P signaling with FTY720 (which targets S1PR1, S1PR3, S1PR4, and S1PR5 receptors) could inhibit PC egress triggered by AMD3100+BOP treatment. We found no change in PC egress with single-agent FTY720 or co-treatment with AMD3100+BOP (Figures 7B, 7E, and S4G), suggesting S1P was not controlling PC egress from the BM.
BM PCs recirculate in steady state
Because these treatments could trigger egress of PCs (CD138high B220−) from the BM, we asked whether recirculation was a normal feature of BM PC behavior under steady-state conditions. To do so, we established two methods for labeling cells in one bone and tracking migration patterns throughout the body. First, using mice expressing Rosa26ERT2-Cre/LSL-tdTomato alleles, we intratibially (IT) injected low-dose, low-volume 4-hydroxy tamoxifen (4HT), the active form of tamoxifen, which triggered recombination and Tomato expression in the injected bone (~10% of cells) 10-fold less than in the proximal femur bone, spleen, proximal popliteal lymph node, and contralateral tibia (CL), indicating that labeling was highly targeted (Figure 7F). This approach labels all cell types in the BM, including polyclonal B cells that could eventually differentiate into Tomato+ PCs. However, in a 2-week kinetic, we expected this to be a rare event. We IT-injected 4HT into mice and tracked kinetics of migration of Tomato-labeled B cells and PCs over time to the CL and spleen. By day 3, labeled PCs had a sharp increase in the other bones and spleen, and by day 14, frequencies of Tomato+ PCs were equilibrated throughout the body, consistent with recirculation (Figure 7G). Similarly, we observed recirculation for labeled (B220+) B cells, although B cells accumulated in the spleen by day 14, consistent with the expansion and egress of maturing labeled B cells from the BM (Figure 7H). We compared recirculation rates in 14- to 18-week-old mice versus 6- to 8-week-old mice and found that PC recirculation rates were higher in older mice (Figure 7I).
To verify these data, we established a second, less invasive, labeling approach using mice expressing photoactivatable green fluorescent protein (PA-GFP) (Victora et al., 2010). Using these mice, the tibia was surgically exposed as before and photoactivated using two-photon excitation at 840 nm through the tibia compact bone, which converted PA-GFP into GFP in all cell types (Upadhaya et al., 2020). Because converted-GFP protein has a 36-h half-life in resting B cells (Victora et al., 2010), GFP fluorescence is rapidly reduced with division. Thus, this approach has a limited window and is not amenable to highly proliferating cells such as PBs. Using this technique, we were able to label about 10% of tibia BM cells on day 0, including PCs, whereas few GFP+ cells were detectable in other bones, blood, or spleen immediately after photoactivation was completed (Figure S5). Mice were sutured and recovered for 48 h, at which point they were sacrificed to assess the distribution GFP+ PCs throughout the body. Although GFP fluorescence was decreased by 48 h (Figure S5A), we found increased numbers of GFP+ PCs (CD138high B220−) in the marrows of the contralateral leg (femurs and tibia pooled) and spleen 48 h later, whereas they decreased in the photoactivated tibia (Figure S5B). Altogether, these data indicate that under steady-state conditions in mice, BM PCs are recirculating to other bone niches and to the spleen, suggesting a shared pool of LLPCs throughout the body.
DISCUSSION
This study reveals that homeostatic BM PCs are motile, both within the parenchyma and recirculating throughout the body. By tracking individual PCs over hours, we could observe cells undergoing intermittent periods of high and low motility, which were inversely correlated with PC density or PC clustering. Our observations of PC motility at first appear to conflict with previous reports that have shown the PCs are arrested in the BM (Reismann et al., 2017; Zehentmeier et al., 2014). However, the difference between our conclusions arises directly from the longer observation period in our study. First, in both datasets, imaging PCs for 30–45 min reveals limited motility or displacement compared with other cells. However, by following PCs for 4–12 h continuously, we were able to confirm that PCs exhibit significant displacement over time. It is not clear whether differences in the manner in which PC were introduced in earlier studies, either adoptive transfer (Zehentmeier et al., 2014) or visualized by surgical implant (Reismann et al., 2017), also contributed to different conclusions. Moreover, our studies solely relied on Blimp1-YFP to visualize PCs in the BM. It remains to be seen whether other independent groups using the same or complementary reporter tools will reach similar imaging findings at long timescales.
Because we found that most PCs (~78%) migrated in our long time-lapse intravital imaging, and most PCs in the BM (>50%) are reported to be long lived (Manz et al., 1997; Slifka et al., 1998), we can conclude that we are visualizing LLPC migration in the BM. These BM PC speeds are slower than PBs in the lymph node after immunization (Fooksman et al., 2010) partly due to the BM microenvironment, which is hypoxic. T cells were also less motile in the BM than we found in the lymph node. Although previous studies have shown that mature PCs are less responsive to chemokines than PBs in vitro (Hauser et al., 2002; Kallies et al., 2004), we found that resting BM PCs before transwell assay increases their chemotaxis. In addition, because they express higher CXCR4 levels than B cells, we found more transmigration of BM PCs than BM B cells to CXCL12.
Motility of PCs in the BM is controlled partly by CXCR4 and VLA4 machinery, which seems to control migration and arrest, respectively. Nevertheless, blocking either or both seemingly enhanced PC egress from the BM. We found that PC binding to VCAM-1 promotes cell arrest and tight adhesion, whereas PCs are more dynamic on fibronectin and ICAM-1 ligands. The distribution of these ligands (i.e., in clusters), or their utilization via intrinsic mechanisms may regulate the ability of PCs to switch among arrest, migration, and egress from the BM. There are limited studies of migration of cells in the BM parenchyma. One notable exception is a comprehensive study from Beck and colleagues characterizing immature B cells in the BM (Beck et al., 2014). That study found that immature B cells are highly motile in the BM in a CXCR4- and VLA4-dependent manner. Inhibition of these pathways caused arrest of immature B cells, leading to their egress from the BM. Although we see some differences in motility of PCs, it is clear that both cell types require CXCR4 and VLA4 for motility and retention.
We found that within the BM of adult mice, PCs are partially organized into clusters, confirming a previous histological study (Mokhtari et al., 2015). Although most clusters were stable, we observed occasional formation and dissolution of PC clusters in situ, suggesting the PC niche does not consist of fixed anatomical structures within the BM. Moreover, we discovered that APRIL promoted cluster formation. Because APRIL is hematopoietically derived, possibly by a mix of myeloid and granulocytic cells (Matthes et al., 2011), these APRIL-secreting cells may dynamically coalesce in regions of the BM, recruiting migrating PCs into clusters. In this context, one might speculate that APRIL acts as a stop signal in these clusters; however, we saw decreased PC motility in APRIL-deficient hosts, suggesting the opposite. APRIL may promote PC motility through direct signaling (Cornelis et al., 2020) or indirectly via enhanced fitness or survival. In either case, we see that PC motility is linked to PC survival, and vice versa.
In addition to their migration and organization in the BM, PC migration patterns involve recirculation to other bones and to the spleen. Regular migrations between organs may allow PCs to access new niches and new stores of survival factors. Recirculation may also allow PCs to be recruited to sites of inflammation via CXCR3 or escape bones that have experienced localized infection or damage that could impair long-term survival of these cells (Slocombe et al., 2013). Future studies would be able to determine whether BM PCs are retained in the red pulp of the spleen or merely passing through during recirculation to other bones.
Finally, we reported that with age, PCs become more motile, as measured by speeds in the BM parenchyma and egress into the blood. These changes in behavior were linked with cell-intrinsic increases in CXCR4 expression, which promotes motility. We hypothesize that enhanced PC motility, via CXCR4 upregulation, and enhanced survival through higher CD138 (McCarron et al., 2017) work in concert to promote LLPC survival over decades.
These findings have important implications for public health, because they suggest that as PCs age, they are more fit and more readily able to migrate to and settle in limited niches, which could explain how LLPCs can survive and maintain serological memory for a lifetime. In contrast, as these LLPCs accumulate, the chances for survival diminish for a newly minted PC with reduced CXCR4 expression. Competition between LLPCs and new PCs may contribute an additional layer that leads to the weakened and short-lived de novo humoral responses seen in older adults. However, this adaptation may be beneficial, because it biases protection against long-standing pathogens and epitopes seen early in life rather than eliciting strong responses to new epitopes. These new epitopes may be self-derived and the result of aging, oxidation, and cellular damage (Zolla et al., 2015), and they could contribute to autoimmune diseases. Similar concepts have been proposed for age-associated B cells, which accumulate with age but have defective B cell receptor signaling, which may limit self-reactive antibody responses (Cancro, 2020). Future studies may shed light on how PC competition in the BM is regulated on cellular and molecular levels.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Inquiries for further information or requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David R. Fooksman (david.fooksman@einsteinmed.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The code supporting the current study have not been deposited in a public repository because the script requires Imaris and MATLAB software to run but are available from the corresponding author on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experiments were conducted as sex unbiased, with randomly assigned 2-5 mice per experimental group. All mice were bred and maintained in a specific-pathogen-free barrier facility with free access to food and water. Both male and female mice were used between 16-36 weeks of age for most experiments, unless specified otherwise. All mouse studies and protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at Albert Einstein College of Medicine. Wild-type C57BL6/J, Rosa26STOP-tdTomato (AI14) (Madisen et al., 2010), Rosa26-CreERT2 (Ventura et al., 2007), CD4-Cre (Lee et al., 2001), Cxcl12DsRed (Ding and Morrison, 2013), PA-GFP (Victora et al., 2010), APRIL−/− (Xiao et al., 2008), Blimp1-YFP (Fooksman et al., 2010) and B1-8hi (Shih et al., 2002) mice were used for most experiments and obtained from The Jackson Laboratory. Wild-type C57BL6 mice were used for PC-intrinsic-age-related CXCR4 level studies and obtained from the Charles River Laboratories.
METHOD DETAILS
Treatments and injections
For intratibial labeling experiments, mice were anesthetized with ketamine-xylazine, leg was shaved, and injected through the knee joint using a 30G insulin syringe with 5 μg/ml 4-hydroxy tamoxifen (Sigma #H7904) in an 8.3 μL volume.
For CXCR4 blockade treatment, mice were injected i.v. with 160 μg of AMD3100 (Tocris Bioscience). For VLA-4 blockade treatment, mice were administered i.v. with 250 μg of dual α9β1/α4β1 integrin inhibitor BOP (Tocris Bioscience) or i.v. with 300 μg of VLA-4 neutralizing antibody (PS/2, BioXCell). For CXCL12 neutralizing antibody treatment, mice were treated i.v. with 80 μg anti-CXCL12 (79014; R&D Systems) or isotype control antibody (11711; R&D Systems). All i.v. injections were verified by co-injecting i.v. with 5-10 μL Qdot 655 or Qdot 705 vascular labels (Invitrogen).
Cell transfer and immunization
To generate NP-specific PCs of different ages, B1-8hi B cells were isolated from spleen and lymph nodes by magnetic cell separation (MACS) using anti-CD43 beads (Miltenyi Biotec), and transferred intravenously to C57BL/6 recipient mice with 3-7x106 B1-8hi B cells or 0.5-1x106 Igλ+ B1-8hi B cells per mouse. One day after the transfer, recipient mice were immunized intraperitoneally with 50 μg of NP(25)-KLH (Biosearch Technologies) emulsified in alum (Imject Alum; Thermo Fisher Scientific) at 2:1 v:v ratio in 150 μL volume for 2-8 weeks.
In-vitro assays
For chemotaxis transwell assay, mononuclear cells were flushed from the BM, washed into RPMI media and rested for 30 minutes in RPMI supplemented with 0.5% fatty acid free media. Cells were then layered carefully on 5 μm transwell inserts (Corning). 4 hours later, the transmigrated fraction was taken for flow cytometric analysis. For CXCR4 surface recovery cells were freshly isolated from BM and allowed to rest for 2.5 hours in RPMI supplemented with 10% FCS at 37°C and then resuspended in 0.5% fatty acid free BSA in RPMI for transwell assay. Cells were treated with 100 ng/mL of CXCL12 to induce CXCR4 downregulation. For preparation of bone marrow plasma, both ends of femurs and tibias were cut before being put into 0.2 mL PCR tubes punctured at the bottom and nested in a 0.5 mL microcentrifuge tube followed by centrifuging at 16,000 g at 4°C for 1min. Then BM supernatant was collected and diluted 1:1000 in 1% BSA in PBS for CXCL12 ELISA.
Protocols for IRM in vitro imaging experiments are described previously (Fooksman et al., 2010). Time-lapse images were collected on a LeicaSP5 confocal microscope in the Einstein Analytical Imaging Facility.
For CXCL12 ELISA of bone marrow plasma, 96-well plates (Costar; Corning) were coated with anti-CXCL12 mAb (79018; R&D Systems) at 2 μg/mL in 100 mM bicarbonate/carbonate buffer at 4°C overnight. The plates were washed 4 times with 0.1% Tween 20 in PBS, and blocked with 200 μl/well 1% BSA in PBS at RT for 2 hr. Then plates were loaded with 100 μl/well diluted bone marrow plasma samples followed by their 2-fold serial dilutions, and incubated at RT for 2 hr. The plates were washed again, and added and incubated with 50 μl/well biotinylated anti-CXCL12 mAb (BAF310; R&D Systems) as detection antibody at RT for 2 hr. The plates were washed again, and incubated with 50 μl/well avidin-horseradish peroxidase (Bio-Rad) at RT for 2 hr. After the last wash, 50 μl/well TMB substrate was added for 5-10min at RT in dark, and reactions were stopped by adding 25 ml/well sulfuric acid before the measurement of absorbance at 450nm by a plate reader (Wallac 1420). CXCL12 standards (R&D Systems) were used to determine the absolute concentrations of CXCL12.
Intravital imaging and analysis
Surgical preparation for BM intra-tibial imaging has been described previously (Pitt et al., 2015). Mice were kept under anesthesia using isoflurane gas and for long term imaging, 5hrs or more, mice were hydrated with lactated ringers s.c. All imaging was conducted on an Olympus FVE-1200 upright microscope, using 25X 1.04 NA objective, and Deepsee MaiTai Ti-Sapphire pulsed laser (Spectra-Physics) tuned to 920nm. To maintain temperature and limit room light, the microscope was fitted with custom-built incubator chamber and heated 37°C platform. Time lapses were conducted every 3 minutes, or in some cases, every 90 s, where listed.
For photo-activation survival experiment, tibia was exposed as described above, imaged for 1-2 hours with 840nm light, to convert to GFP. To achieve large 3D areas of photo-conversion, multiple (5-10) small regions (512mm*512mm) were scanned sequentially at 0.4 μm/slice for around 50-100 μm depth below the bone surface. Mice were sutured and allowed to recover for 2 days before analysis.
All image analysis was conducted using Imaris software 9.2 to track cells, drift correction and using custom scripts to calculate PC clusters, described in Figure S3. For drift correction, sessile macrophages or bone features were tracked using semi-automated or manual methods to correct XYZ registrations over time. Ratio and subtracted channels were used to isolate plasma cell fluorescence from background and bleed-through channel noise. All imaging and conditions were independently repeated by at least 2 authors to confirm reproducibility.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests were performed using GraphPad Prism (versions 7 and 8). Specific tests used are listed in the figure legends with p values. As imaging data can have high significance due to the large sample sizes, we routinely compared groups from multiple experiments independently and compared averages of averages from multiple experiments to see if differences were not artifacts of the large sample size.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-B220 APC-Cy7 (clone RA3-6B2) | BioLegend | Cat# 103224; RRID:AB_313007 |
| Rat monoclonal anti-CD3 eFluor 450 (clone 17A2) | eBiosciences | Cat# 48-0032-82; RRID:AB_1272193 |
| Rat monoclonal anti-CD16/CD32 (clone 2.4G2) | Bio X Cell | Cat# CUS-HB-197; RRID:AB_2687830 |
| Rat monoclonal anti-CD19 PE (clone 6D5) | BioLegend | Cat# 115507; RRID:AB_313642 |
| Rat monoclonal anti-CD38 PerCP-Cy5.5 (clone 90) | BD Biosciences | Cat# 562770; RRID:AB_2737782 |
| Rat monoclonal anti-CD138 APC (clone 281-2) | BD Biosciences | Cat# 558626; RRID:AB_1645216 |
| Rat monoclonal anti-CD138 PE (clone 281-2) | BD Biosciences | Cat# 553714; RRID:AB_395000 |
| Rat monoclonal anti-CD184 PE (clone 2B11) | BD Biosciences | Cat# 551966; RRID:AB_394305 |
| Rat monoclonal anti-CD184 BV421 (clone 2B11) | BD Biosciences | Cat# 562738; RRID:AB_2737757 |
| Mouse monoclonal anti-CXCL12/SDF-1 (clone 79014) | R&D Systems | Cat# MAB310500; RRID:AB_2276927 |
| Mouse monoclonal anti-CXCL12/SDF-1 (clone 79018) | R&D Systems | Cat# MAB350100; RRID:AB_2088149 |
| Mouse monoclonal IgG1 Isotype Control (clone 11711) | R&D Systems | Cat# MAB002; RRID:AB_357344 |
| Goat polyclonal anti-CXCL12/SDF-1 Biotinylated | R&D Systems | Cat# BAF310; RRID:AB_356384 |
| Rat monoclonal anti-Ig kappa light chain PE (clone 187.1) | BD Biosciences | Cat# 559940; RRID:AB_397384 |
| Rat monoclonal anti-Ki-67 Alexa Fluor 647 (clone 16A8) | BioLegend | Cat# 652408; RRID:AB_652408 |
| Mouse monoclonal anti-MHC Class II I-Ab PE (clone AF6-120.1) | eBiosciences | Cat# 12-5320-82; RRID:AB_2572619 |
| Goat polyclonal anti-Mouse IgG (H+L) HRP | Jackson ImmunoResearch | Cat# 115-035-146; RRID:AB_2307392 |
| Rat monoclonal anti-VLA-4 (clone PS/2) | Bio X Cell | Cat# BE0071; RRID:AB_1107657 |
| Chemicals, peptides, and recombinant proteins | ||
| AMD 3100 octahydrochloride | Tocris Bioscience | Cat# 3299 |
| Bovine Serum Albumin, Fraction V (Modified Cohn) | Sigma-Aldrich | Cat# 12-660-100GM |
| BOP | Tocris Bioscience | Cat# 6047 |
| EDTA (0.5M), pH 8.0, RNase-free | Thermo Fisher Scientific | Cat# AM9261 |
| Fibronectin, Bovine Plasma | Sigma-Aldrich | Cat# 341631-1MG |
| Fingolimod (FTY720) | Cayman Chemical | Cat# 10006292 |
| GIBCO Fetal Bovine Serum | Thermo Fisher Scientific | Cat# 10-437-028 |
| Imject Alum Adjuvant | Thermo Fisher Scientific | Cat# 77161 |
| Isothesia (Isoflurane) Solution | Covetrus | Cat# 029405 |
| Lactated Ringer’s Solution | Henry Schein | Cat# 14792 |
| Lonza ACK Lysing Buffer | Thermo Fisher Scientific | Cat# BW10548E |
| Lipopolysaccharides from Escherichia coli O111:B4 | Sigma | Cat# L-2630 |
| NP(24)-KLH | Biosearch Technologies | Cat# N-5060-25 |
| PBS, 10X Powder, pH 7.4 | Thermo Fisher Scientific | Cat# BP665-1 |
| Pertussis Toxin | Sigma-Aldrich | Cat# 516560 |
| Polyinosinic-polycytidylic Acid (Poly(I:C)) | Invivogen | Cat# tlrl-picw |
| Qtracker 655 Vascular Labels | Thermo Fisher Scientific | Cat# Q21021MP |
| Qtracker 705 Vascular Labels | Thermo Fisher Scientific | Cat# Q21061MP |
| Recombinant Murine IL-4 | PeproTech | Cat# 214-14 |
| Recombinant Mouse CXCL12/SDF-1 Alpha | R&D Systems | Cat# 460-SD-050 |
| Recombinant Mouse ICAM-1/CD54 Fc Chimera Protein | R&D Systems | Cat# 796-IC-050 |
| Recombinant Mouse VCAM-1/CD106 Fc Chimera Protein | R&D Systems | Cat# 643-VM-050 |
| RPMI 1640 Medium | Thermo Fisher Scientific | Cat# SH3009601 |
| TMB | MilliporeSigma | Cat# 613544 |
| 2-Mercaptoethanol | Thermo Fisher Scientific | Cat# 21985023 |
| 4-Hydroxytamoxifen | Sigma-Aldrich | Cat# H7904 |
| Critical commercial assays | ||
| BD Cytofix/Cytoperm Fixation/Permeabilization Kit | BD Biosciences | Cat# 554714 |
| Corning Transwell polycarbonate membrane cell culture inserts | Sigma-Aldrich | Cat# CLS3421 |
| High-Capacity RNA-to-cDNA Kit | Applied Biosystems | Cat# 4388950 |
| Live/Dead Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | Cat# L34957 |
| RNeasy Plus Mini Kit | QIAGEN | Cat# 74134 |
| TaqMan Universal Master Mix II, with UNG | Applied Biosystems | Cat# 4440046 |
| TaqMan Gene Expression Assay (FAM) [CXCL12; ActB] | Applied Biosystems | Cat# 4331182 |
| UltraComp eBeads Compensation Beads | Thermo Fisher Scientific | Cat# 01-2222-41 |
| Experimental models: organisms/strains | ||
| Mouse: APRIL-KO | The Jackson Laboratory | JAX stock# 022971 |
| Mouse: Blimp1-YFP | The Jackson Laboratory | JAX stock# 008828 |
| Mouse: B1-8hi | The Jackson Laboratory | JAX stock# 007775 |
| Mouse: C57BL/6 | Charles River Laboratories | NCI stock# 556 |
| Mouse: C57BL6/J | The Jackson Laboratory | JAX stock# 000664 |
| Mouse: CD4-Cre | The Jackson Laboratory | JAX stock# 022071 |
| Mouse: Cxcl12DsRed | The Jackson Laboratory | JAX stock# 022458 |
| Mouse: UBC PA-GFP | The Jackson Laboratory | JAX stock# 022486 |
| Mouse: Rosa26-CreERT2 | The Jackson Laboratory | JAX stock# 008463 |
| Mouse: Rosa26Stop-tdTomato (AI14) | The Jackson Laboratory | JAX stock# 007914 |
| Oligonucleotides | ||
| Common forward primer for Cxcl12: CCA ACC TGT GCC CTT CAG ATT G | Integrated DNA Technologies | N/A |
| Reverse primer for Cxcl12 isoform α: CAT ATG CTA TGG CGG AGT GTC | Integrated DNA Technologies | N/A |
| Reverse primer for Cxcl12 isoform β: CTT CAG CCC TGC TCA GGA GC | Integrated DNA Technologies | N/A |
| Reverse primer for Cxcl12 isoform γ: ACT GCG GTC CAT CGG CAG G | Integrated DNA Technologies | N/A |
| Software and algorithms | ||
| Adobe After Effects | Adobe | N/A |
| Adobe Illustrator | Adobe | RRID: SCR_010279 |
| Excel | Microsoft | RRID: SCR_016137 |
| FlowJo | Treestar | RRID: SCR_008520 |
| Imaris | Bitplane | RRID: SCR_007370 |
| MATLAB | MathWorks | RRID: SCR_001622 |
| Prism | Graphpad | RRID: SCR_002798 |
| Python | Python | RRID: SCR_008394 |
| Volocity | Quorum Technologies | RRID: SCR_002668 |
Highlights.
Plasma cells are motile in the bone marrow, regulated by CXCR4 and VLA4
Plasma cells are partly organized into clusters in the bone marrow, which depends on APRIL
Plasma cells recirculate to other bones in steady state
Plasma cell motility, clustering, and recirculation increase with age
ACKNOWLEDGMENTS
We thank Michael Dustin, Jeffrey Segall, and Gregoire Lauvau for careful review of our manuscript and Susan Schwab for sharing a key mouse strain. This work was supported by R01HL141491 (to D.R.F.) and F32HL149155 (to Z.B.), with support from Albert Einstein NCI Cancer Center grant P30CA013330.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2021.108733.
REFERENCES
- Beck TC, Gomes AC, Cyster JG, and Pereira JP (2014). CXCR4 and a cell-extrinsic mechanism control immature B lymphocyte egress from bone marrow. J. Exp. Med 211, 2567–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancro MP (2020). Age-Associated B Cells. Annu. Rev. Immunol 38, 315–340. [DOI] [PubMed] [Google Scholar]
- Cao B, Zhang Z, Grassinger J, Williams B, Heazlewood CK, Churches QI, James SA, Li S, Papayannopoulou T, and Nilsson SK (2016). Therapeutic targeting and rapid mobilization of endosteal HSC using a small molecule integrin antagonist. Nat. Commun 7, 11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu VT, Fröhlich A, Steinhauser G, Scheel T, Roch T, Fillatreau S, Lee JJ, Löhning M, and Berek C (2011). Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat. Immunol 12, 151–159. [DOI] [PubMed] [Google Scholar]
- Cornelis R, Hahne S, Taddeo A, Petkau G, Malko D, Durek P, Thiem M, Heiberger L, Peter L, Mohr E, et al. (2020). Stromal cell-contact dependent PI3K and APRIL induced NF-κB signaling prevent mitochondrial- and ER stress induced death of memory plasma cells. Cell Rep. 32, 107982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, and Morrison SJ (2013). Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fooksman DR, Schwickert TA, Victora GD, Dustin ML, Nussenzweig MC, and Skokos D (2010). Development and migration of plasma cells in the mouse lymph node. Immunity 33, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain RN, Robey EA, and Cahalan MD (2012). A decade of imaging cellular motility and interaction dynamics in the immune system. Science 336, 1676–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves DC, Hyman PL, Lu TT, Ngo VN, Bidgol A, Suzuki G, Zou YR, Littman DR, and Cyster JG (2001). A coordinated change in chemokine responsiveness guides plasma cell movements. J. Exp. Med 194, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser AE, Debes GF, Arce S, Cassese G, Hamann A, Radbruch A, and Manz RA (2002). Chemotactic responsiveness toward ligands for CXCR3 and CXCR4 is regulated on plasma blasts during the time course of a memory immune response. J. Immunol 169, 1277–1282. [DOI] [PubMed] [Google Scholar]
- Hendrix CW, Flexner C, MacFarland RT, Giandomenico C, Fuchs EJ, Redpath E, Bridger G, and Henson GW (2000). Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrob. Agents Chemother 44, 1667–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, and Nutt SL (2004). Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J. Exp. Med. 200, 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallies A, Hasbold J, Fairfax K, Pridans C, Emslie D, McKenzie BS, Lew AM, Corcoran LM, Hodgkin PD, Tarlinton DM, and Nutt SL (2007). Initiation of plasma-cell differentiation is independent of the transcription factor Blimp-1. Immunity 26, 555–566. [DOI] [PubMed] [Google Scholar]
- Khodadadi L, Cheng Q, Radbruch A, and Hiepe F (2019). The Maintenance of Memory Plasma Cells. Front. Immunol 10, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Pérez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, et al. (2001). A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15, 763–774. [DOI] [PubMed] [Google Scholar]
- Lightman SM, Utley A, and Lee KP (2019). Survival of Long-Lived Plasma Cells (LLPC): Piecing Together the Puzzle. Front. Immunol 10, 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manz RA, Thiel A, and Radbruch A (1997). Lifetime of plasma cells in the bone marrow. Nature 388, 133–134. [DOI] [PubMed] [Google Scholar]
- Matthes T, Dunand-Sauthier I, Santiago-Raber ML, Krause KH, Donze O, Passweg J, McKee T, and Huard B (2011). Production of the plasma-cell survival factor a proliferation-inducing ligand (APRIL) peaks in myeloid precursor cells from human bone marrow. Blood 118, 1838–1844. [DOI] [PubMed] [Google Scholar]
- McCarron MJ, Park PW, and Fooksman DR (2017). CD138 mediates selection of mature plasma cells by regulating their survival. Blood 129, 2749–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokhtari Z, Mech F, Zehentmeier S, Hauser AE, and Figge MT (2015). Quantitative image analysis of cell colocalization in murine bone marrow. Cytometry A 87, 503–512. [DOI] [PubMed] [Google Scholar]
- Pereira JP, Xu Y, and Cyster JG (2010). A role for S1P and S1P1 in immature-B cell egress from mouse bone marrow. PLoS ONE 5, e9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, Sanchez-Martin M, Tsirigos A, Littman DR, Ferrando AA, et al. (2015). CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell 27, 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reismann D, Stefanowski J, Günther R, Rakhymzhan A, Matthys R, Nützi R, Zehentmeier S, Schmidt-Bleek K, Petkau G, Chang HD, et al. (2017). Longitudinal intravital imaging of the femoral bone marrow reveals plasticity within marrow vasculature. Nat. Commun 8, 2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih TA, Roederer M, and Nussenzweig MC (2002). Role of antigen receptor affinity in T cell-independent antibody responses in vivo. Nat. Immunol 3, 399–406. [DOI] [PubMed] [Google Scholar]
- Slifka MK, Antia R, Whitmire JK, and Ahmed R (1998). Humoral immunity due to long-lived plasma cells. Immunity 8, 363–372. [DOI] [PubMed] [Google Scholar]
- Slocombe T, Brown S, Miles K, Gray M, Barr TA, and Gray D (2013). Plasma cell homeostasis: the effects of chronic antigen stimulation and inflammation. J. Immunol 191, 3128–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding E, Fooksman D, Moore JM, Saidi A, Feintuch CM, Reizis B, Chorro L, Daily J, and Lauvau G (2016). STING-Licensed Macrophages Prime Type I IFN Production by Plasmacytoid Dendritic Cells in the Bone Marrow during Severe Plasmodium yoelii Malaria. PLoS Pathog. 12, e1005975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer TA, and Dustin ML (2012). Integrin inside-out signaling and the immunological synapse. Curr. Opin. Cell Biol 24, 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Domínguez Á, Pinho S, Akhmetzyanova I, Gao J, Witkowski M, et al. (2019). The bone marrow microenvironment at single-cell resolution. Nature 569, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhaya S, Krichevsky O, Akhmetzyanova I, Sawai CM, Fooksman DR, and Reizis B (2020). Intravital Imaging Reveals Motility of Adult Hematopoietic Stem Cells in the Bone Marrow Niche. Cell Stem Cell 27, 336–345.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, and Jacks T (2007). Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665. [DOI] [PubMed] [Google Scholar]
- Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, and Nussenzweig MC (2010). Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 143, 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Motomura S, and Podack ER (2008). APRIL (TNFSF13) regulates collagen-induced arthritis, IL-17 production and Th2 response. Eur. J. Immunol 38, 3450–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehentmeier S, Roth K, Cseresnyes Z, Sercan Ö, Horn K, Niesner RA, Chang HD, Radbruch A, and Hauser AE (2014). Static and dynamic components synergize to form a stable survival niche for bone marrow plasma cells. Eur. J. Immunol 44, 2306–2317. [DOI] [PubMed] [Google Scholar]
- Zolla V, Nizamutdinova IT, Scharf B, Clement CC, Maejima D, Akl T, Nagai T, Luciani P, Leroux JC, Halin C, et al. (2015). Aging-related anatomical and biochemical changes in lymphatic collectors impair lymph transport, fluid homeostasis, and pathogen clearance. Aging Cell 14, 582–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code supporting the current study have not been deposited in a public repository because the script requires Imaris and MATLAB software to run but are available from the corresponding author on request.