

Abstract
Select dimeric chromenones exhibit low micromolar cyctotoxicity toward lymphoma and leukemia cell lines, L5178Y and HL60, respectively. The bioactive dimeric chromenones were identified from a focused library of structurally-simplified derivatives of naturally-occurring dimeric chromenones and tetrahydroxanthones that was prepared as part of this study. The simple dimeric chromenone scaffolds contain no stereogenic centers, are easily synthesized, and may be utilized as lead compounds in cancer research and drug discovery.
Keywords: Natural Products, Cancer, Drug Discovery, Drug Design, Medicinal Chemistry
Graphical Abstract
A focused family of naturally-inspired chromenone dimers has been synthesized and studied for anticancer activity. Select dimeric chromenones prepared in this study have were found to have moderate cytotoxcity toward L5178Y and HL60 cells. These compounds may serve as promising leads for the identification of new ligands and biological targets.
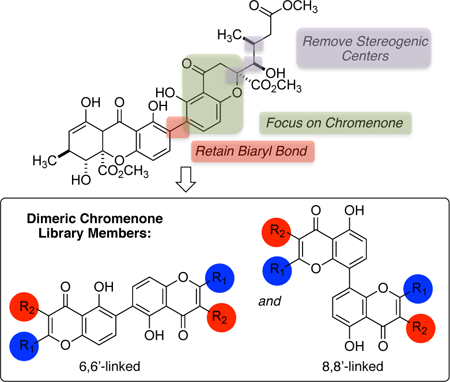
Introduction
Naturally occurring dimeric chromanones and tetrahydroxanthones often possess enticing biological activities.1For example, the dimeric chromanone gonytolide A2 (1) promotes an innate immune response while dimeric tetrahydroxanthones phomoxanthone A3 (2), dicerandrol C4 (3), and penexanthone A5 (4) are cytotoxic toward various cancer cell lines (Figure 1). Rugulotrosin A6 (5) and phomalevone C7 (6) retain antibacterial properties. Despite the promising bioactivity of these dimeric oxygen heterocycles, naturally occurring chromanone and/or xanthone dimers remain an understudied collection of compounds in terms of drug discovery and chemical probe development.
Figure 1.
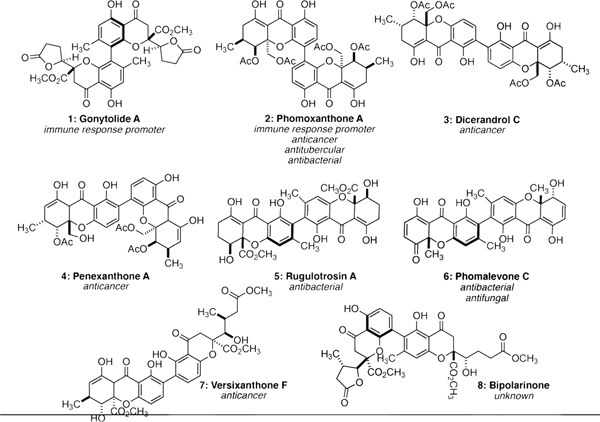
Naturally occurring dimeric chromanones and tetrahydroxanthones.
The limited investigations dedicated toward advancing their biological performance in the context of drug discovery can be, at least in part, attributed to the synthetic challenges that naturally occurring dimeric chromanones and tetrahydroxanthones present.8 The chromanone and/or xanthone units of these dimers can be bonded in several scenarios, including scaffolds that contain 8,8’-, 6,6’-, and 6,8’-linked chromenone/xanthone units (for the purposes of this paper a chromanone numbering scheme has been adopted, A-C, Figure 2). As a consequence of the sterically congested nature of the biaryl bonds connecting the monomers, the methodologies available for their construction are limited and often plagued by low yields and narrow substrate scopes.8d,9 In addition to the biaryl bond, the multiple stereogenic centers present in these families of natural products intensify the synthetic challenge. The 2-stereogenic center is a particularly challenging aspect in the synthesis of dimeric chromanone and xanthone derivatives. There are few methodologies to establish the absolute stereochemistry at the 2-position and they are limited in substrate scope.10
Figure 2.

Connectivity patterns of dimeric chromenones and xanthones.
Bioactive natural products serve as important lead compounds in drug development. It is estimated that between 1940–2014 49% of small molecules approved for cancer treatment were natural products or compounds derived from natural products.11 Given limitations associated with many natural occurring molecules (e.g., limited supply, challenging syntheses), including the dimeric chromanones and tetrahydroxanthones depicted in Figure 1, it is lucrative to pursue the construction and study of structurally simplified bioactive derivatives for the purposes of drug development. Indeed, this notion is at the heart of Wender’s function-oriented synthesis approach: identify more accessible scaffolds that retain the structural features necessary for bioactivity.12 The concept of biology-oriented synthesis includes the study of natural product families as inspirational starting points for drug candidates, chemical probes, and new targets.13 Moreover, in a recent and particularly inspiring study, Cravatt and coworkers demonstrated that fragment based drug discovery using simplified bioactive molecule libraries based upon privileged cores enables both ligand and target discovery in human cells.14 In the studies, simplified bioactive molecules serve as probes for the identification of new small molecule targets and lead compounds in the development of scaffolds for protein binding.
Chromanone and xanthone heterocycles have been designated as privileged structures in drug development as many of these compounds elicit a powerful range of biological responses (e.g., anticancer, antimalarial, antimicrobial, see Figure 1).1 Given the promise of simplified bioactive compound libraries in fragment based drug discovery,13–15 we hypothesized that structurally simplified dimeric chromenones would retain biological activity and ultimately inspire the discovery of new therapeutic agents and the identification of new protein targets. To probe the validity of our hypothesis, we synthesized a focused family of naturally-inspired dimeric chromenones for study in cancer therapy. Herein, we report generalizable methods to prepare structure simplified compounds that resemble various bioactive chromenone and xanthone natural products, including both 8,8’-dimers, and 6,6’-dimers, and the results of their in vitro phenotypic screen in six cancer cell lines.
Results and Discussion
Although we are not aware of a broad systematic structure activity relationship study of naturally occurring dimeric chromenones and tetrahydroxanthones, there is a significant amount of data available in the literature that can be used to deduce the beginnings of the relationship of structure to activity for these families of molecules.1–8 This data allowed for the production of a 1st generation library, built from the following observations. It has been documented that the dimeric chromenones and xanthones can carry more potent bioactive activity than their monomeric counterparts.2,16 It is proposed that g-lactone chromenones (e.g., 1) and the tetrahydroxanthones (e.g., 2-6) are biosynthetically related (Scheme 1).17 Specifically, the carbonyl carbon of the enolized C ring of the tetrahydroxanthone 9 may serve as an electrophilic site for an intramolecular cyclization of the alcohol, resulting in the release from the B ring to form g-lactone chromanone 10. The formation of ring opened variants (11), such as versixanthone F18 (7) and bipolarinone19 (8), plausibly result from methanolysis during isolation. Notably, the ring-opened lactone form (e.g., 7) can possess more potent cytotoxicity than the corresponding ring closed forms.18 Finally, the results of recently published studies conducted by Kikuchi and coworkers aided in our dimeric chromenone library development.20 They found simplified gonytolide derivatives built without stereogenic centers exhibit biological responses at only slightly reduced levels when compared to naturally occurring gonytolide A.
Scheme 1.

Plausible relationship of tetrahydroxanthone 9, γ-lactone chromanone 10, and ring-opened chromanone 11.
Following from the literature observations described above, the design of our structurally-simplified naturally-inspired chromanone library focused on the following features: (1) retention of the biaryl bond, (2) inclusion of both 6,6’-linked and 8,8’-linked dimeric chromenone cores, (3) removal of the C-ring, (4) removal of the stereogenic centers, and (5) introduction of a focused selection of aryl and aliphatic groups at carbons 2 and 3 (Scheme 2).
Scheme 2.

Design of naturally-inspired, structurally-simplified dimeric chromenone derivatives.
While a significant amount of information is available in the literature about chromenone synthesis, to our surprise in both 6,6’-linked, and 8,8’-linked dimers, most of these methods are very substrate specific and not generalizable to include a variety of functional groups. Therefore, our synthetic efforts toward both the 6,6’-and 8,8’-linked dimers focused on the development of a general approach to quickly prepare a library of the desired category of compounds that possess the structural features we identified (above) to be probed.
The synthesis of a family of 6,6’-linked dimeric chromenones began with the construction of monomeric chromenones 19a-e (Scheme 3). After several failed attempts to directly introduce the iodo substituent ortho to the methoxy group on the chromenone, the introduction of the iodo substituent prior to chromenone formation was pursued and the following series of steps proved to be reliable in accessing 19. The mono-tosylation of commercially available 2’6’-dihydroxyacetophenone (16) was achieved with careful stoichiometric control of the addition of tosyl chloride (TsCl) and diisopropylethyl amine. The regioselective iodination of the resultant tosylate was carried out with Niodosuccinimine (NIS) and trifluoroacetic acid (TFA) to give rise to 17 in good yield. Under the influence of iodomethane and K2CO3 methylation of the hydroxy group occurred. A subsequent detosylation was effected by addition of 20% aq. NaOH in t-butanol, resulting in the formation of common intermediate 18. The 2-substituted chromenones 19a-e were prepared by first treating 18 with the desired diesters with NaH and then following with an acid mediated cyclization.
Scheme 3.

Synthesis of monomer 19.
a) TosCl, diispropylethylamine, CH2Cl2, 23 °C, 48 h, 70%; b) NIS, TFA, CH2Cl2, 23 °C, 48 h, 72%; c) CH3I, K2CO3, Acetone, reflux, 24 h, 82%; d) 20% aq. NaOH, t-BuOH, reflux, 24 h, 98%; e) NaH, CH3CH2O2R1, 23 °C, 24 h; then CH3OH, aq. HCl 40–60%.
With the family of iodochromenones 19a-e prepared, efforts were directed toward dimer formation via transition metal catalyzed cross coupling. Initial studies focused on effecting the desired biaryl bonding forming event under the influence of Suzuki-Miyaura reaction conditions (Scheme 4). Unfortunately, all attempted Suzuki-type couplings were unsuccessful.9 Neither borylation to prepare 20 nor dimerization to prepare 21 led to the formation of desired product. Frequently, dehalogenation of 19 was observed under the Suzuki reaction conditions. It was fortunate that a Stille cross coupling approach, depicted in Scheme 5, proved general and enabled the synthesis of dimeric chromenones 21a-e.8c The deprotection of 21 was easily affected in high yield in the presence of BBr3.
Scheme 4.

Dimerization of 19 to yield 6,6’ linked chromenones.
Scheme 5.

Synthesis of 8,8’-linked dimers.
a) i) NaH, CH3CH2O2R1, 23 °C, 24 h, ii) CH3OH, aq. HCl 40–60%; b) I2, C6H5I(OCOF3)2, CH2Cl2, 23 °C, 24 h, 58–76%; c) Pd(OAc)2, S-Phos, B2Pin2, K3PO4, CH2Cl2, H2O (4:1) reflux, 18 h, 24–60%; d) BBr3, CH2Cl2, 0°C, 6 h, 46–82%
The members of the 8,8’-linked library of dimeric chromenones were accessible in four steps from commercially available acetophenone derivative 23 (Scheme 5). The sequence begins with the cyclization of 23 and appropriate diester reaction partners for the generalized syntheses of 2-substituted chromenones 24a-e. Chromenones 24 were often acid sensitive and susceptible to bisiodinations under many standard literature iodination conditions (e.g., NIS, TFA). As a result, a novel method was developed for the efficient and regioselective iodination of 24 using [bis(trifluoroacetoxy)iodo]benzene and I2. The resultant iodochromenones 25 were dimerized, typically giving rise to 26 in good yield, when subjected to Suzuki-Miyaura coupling conditions. The subsequent demethylation of 26 was achieved under the influence of BBr3, generating the desired dimers 15a-e.
To better mimic naturally occurring dimeric chromenones and tetrahydroxanthones, like phomoxanthone A, the synthesis of 8,8’-linked dimeric chromenone analogs with hydrogen bond acceptor groups in the 2- and 3-positions was pursued (Scheme 6 and 7). The chromenone dimer 30, containing an acetate in the 3-position, was prepared beginning with a Claisen condensation of acetophenone derivative 23 with ethyl acetate followed by the treatment of the resulting diketone with acetic anhydride.3 The monomeric chromenone 27 was then subjected to p-iodination using NIS in dichloromethane to give rise to iodochromenone 28. The coupling of 28 under Suzuki-Miyaura reaction conditions afforded 29 in 59% yield. The demethylation of 29 went smoothly under standard reaction conditions, BBr3.
Scheme 6.

Synthesis of 8,8’ linked dimers 29 and 30.
a) i) NaH, EtOAc, 23 °C, 24 h, 90%; b) NaOAc, Ac2O, reflux, 24 h 78%; c) NIS, TFA, CH2Cl2, 45 °C, 2 h, 51%; c) Pd(OAc)2, S-Phos, B2Pin2, K3PO4, CH2Cl2, H2O (4:1) reflux, 18 h, 59%; d) BBr3, CH2Cl2, 0°C, 6 h, 62%
Scheme 7.

Synthesis of 8,8’-linked dimers 35.
a) i) NaH, EtOAc, 23 °C, 24 h, 90%; b) CH2Cl2, 23 °C, 5 d, 66–81%; c) I2, C6H5I(OCOF3)2, CH2Cl2, 23 °C, 18 h, 51–62%; d) Pd(OAc)2, S-Phos, B2Pin2, K3PO4, CH2Cl2, H2O (4:1) reflux, 18 h 40–53%
The synthesis of 35a-c, analogs containing substitution in the 2-position of the chromenone core that has a matching stereochemistry and oxidation state of many naturally occurring tetrahydroxanthones, such as phomoxanthone A, was carried out (Scheme 7). Attempts to directly oxidize the allylic methyl group in 27 were unsuccessful. Taken inspiration from the synthesis of 30, the diketone 31 was then reacted with appropriately-substituted acid halides 32 to generate the desired chiral substituted chromenones 33a-c. This is a novel method developed to introduce chirality to these chromenones at this center. Chromenones 33 were then regioselectively iodinated under the influence of [bis(trifluoroacetoxy)-iodo]benzene and I2 giving rise to 34a-c. Upon treatment with palladium acetate, bis(pinacolato)diboron, and an appropriate phosphine ligand, the dimerization of 34a-c was achieved to afford desired chromenones 35a-c. The dimethoxy dimers 35, unfortunately, could not be demethylated under common literature conditions due to the decomposition of the materials.
With a small series of 6,6’-linked and 8,8’-linked dimeric chromenones in hand, cytotoxicity screening was conducted in Jurkat cell lines. The goal of this screening was to establish: (1) whether such simple dimeric chromenones would possess any cytotoxicity, (2) if there was a difference in biological activity of the 6,6’-linked compared to the 8,8’-linked, (3) if the substituent in the 2-position influenced biological activity, and (4) if the phenol group affected cytotoxicity. The results of the screening are depicted in Figure 3 and include the following observations. Select simple dimeric chromenones exhibit moderate cytotoxicity in Jurkat cell lines (15e = 20 μM). When all the data is considered together, the 8,8’-linked dimers tend to be more cytotoxic than the 6,6’-linked dimers. The substituent in the 2-position had a rather dramatic effect on cytotoxicity. Specifically, compounds containing the methyl isobutyrate (22c, 26c, 15c) and tetrahydrofuran (22e and 15e) substituents gave rise to the highest levels of cytotoxicity in this screen. Finally, the presence of a phenol group did affect cytotoxicity. With the most cytotoxic compounds in this series, the presence of the phenol group typically resulted in increased cytotoxicity (e.g, 22c v. 14c, 26c v. 15c, 26e v. 15e).
Figure 3.

IC50 Values of Select Dimeric Chromenones
The most cytotoxic of the simple dimeric chromenones (14c, 15c, 15e, 22e, 26c) and the more functionalized chromenones (29, 30, 35a-c) were subject to further anticancer screening in six cell lines (Jurkat, L5178Y, HL60, KB, MCF7, and SKOV3 cells, Table 1). Select simplified chromenone dimers possessed moderate cyctotoxicities. The most active compounds in our series were methyl isobutyrate-derived dimeric chromenones 15c and 26c. Both 15c and 26c showed activity toward L5178Y and HL60 cells in the low micromolar range. 26c had an IC50 = 14 μM against L5178Y cell lines and IC50 = 24 μM against HL60 cells. Compound 15c was active against HL60 and L5178Y cells with IC50 = 15 μM and 23 μM, respectively. The dimeric chromenones described in this work were not significantly active (IC50 >50 μM) toward MCF7 and SKOV3 cell lines.
Table 1.
Chromenone Cytotoxicity Studies (IC50: μM)
| Compound | Jurkat | L5178Y | HL60 | KB |
|---|---|---|---|---|
| 2[a] | 0.5 | 0.3 | -- | 0.99 |
| PXB[a][b] | na | na | na | 4.1 |
| 3[a] | na | 10 | na | na |
| 7[a] | na | na | 1.00 | na |
| 22c | 39 ± 1.1 | 40 ± 1.0 | 37 ± 1.0 | 41 ± 1.1 |
| 15c | 28 ± 1.4 | 23 ± 1.1 | 15 ± 1.1 | >50 |
| 15e | 20 ± 1.0 | >50 | >50 | >50 |
| 26c | 32 ± 1.6 | 14 ± 1.1 | 24 ± 1.1 | ND |
| 22e | 28 ± 1.3 | 48 ± 1.0 | >50 | >50 |
| 29 | >50 | >50 | 35 ± 1.0 | >50 |
| 30 | 37 ± 1.0 | >50 | >50 | >50 |
| 35c | >50 | 45 ± 1.2 | >50 | >50 |
= Literature values
= Phomoxanthone B
ND= Not Determined
na = Not Available
Conclusions
A focused library of structure simplified dimeric chromenones, inspired by naturally occurring dimeric chromanones and tetrahydroxanthones, has been synthesized and studied in several cancer cell lines. The sequences used to synthesize the library are robust and generalizable to accommodate a wide range of substrates so as to permit the generation of a library of dimeric chromenone derivatives for study. The synthetic effort in the library development required invention of two new methodologies: (i) the regioselective p-iodination of sensitive substrates under neutral conditions and (ii) a novel approach for the introduction of a chiral center to generate naturally inspired dimeric chromenones.
The results of the anticancer testing demonstrate that select members of the library give rise to promising levels of biological activity. Specifically, the 8,8’-linked dimeric chromenones 15c and 26c containing methyl butyrate substituents are cytotoxic toward both L5178Y and HL60 cells, with IC50 values in the low micromolar range. The cytotoxic dimeric chromenones identified in this study are promising starting points for anticancer drug discovery and our investigations are currently focused on creating more potent analogs, and applying the more cytotoxic analogs as chemical probes to identify new small molecule targets.
Experimental Section
Ethyl acetate, dichloromethane, and hexanes were used as received. Toluene was dried over 4Å molecular sieves prior to use. All other reagents were used directly as received from the manufacturer unless otherwise noted. Preparative silica gel chromatography was performed using SiliaFlash F60 silica gel (40 – 63 μm). Analytical thin layer chromatography was performed using Analtech 250 μm silica gel HLF plates and visualized under UV 254nm. All 1H NMR spectra were acquired using a Bruker BioSpin 500MHz Avance III Digital NMR spectrometer and calibrated using the solvent signal (CDCl3 7.26 ppm). Multiplicities were determined using MNova software. All 13C NMR spectra were acquired using a Bruker BioSpin 126MHz Avance III Digital NMR spectrometer and calibrated using the solvent signal (CDCl3 77.16 ppm). Infrared spectra were acquired using a Bruker Vertex 70 with an ATR accessory. High resolution mass spectra were acquired using an Agilent 6520 Q-TOF mass spectrometer.
General procedure for the Chromenone Synthesis (General Procedure A): A solution of the 2’-hydroxy-6’-methoxyacetophenone (1 mmol ) and the diester (1.1 mmol, 1.1 equiv) in dry THF (5 mL) under nitrogen atmosphere was cooled to 0 °C. Into this mixture was added NaH (60 % in oil, 3.5 equiv). The temperature was allowed to rise to room temperature and stirred overnight. The crude mixture was quenched with MeOH (2.5 mL) and acidified (pH 0–1) with concentrated HCl. The resultant heterogenous mixture was stirred at room temperature an additional 24 h. MeOH was removed by rotary evaporation and the residue was diluted with EtOAc. The two layers were separated and the aqueous layer was washed with EtOAc (25 mL × 3). The combined organic solution was dried using anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the crude product. The crude material was then purified on silica gel.
General procedure for aryl stannanes syntheses (General Procedure B). To a 20 mL reaction vial was added the aryl halide (0.1 mmol), (nBuSn)2 (2.0 equiv), Pddba2 (0.1 equiv), PtBu3HBF4 (0.2 equiv), LiCl (5.0 equiv), and dry dioxane (1 mL). Nitrogen gas was then bubbled through the resulting mixture and the reaction was allowed to stirred at 50˚C for 4 h. The crude mixture was then filtered through celite and rinsed with EtOAc. The crude material was then concentrated under reduced pressure and purified by silica flash chromatography. Products were obtained as oils.
General procedure for Stille Coupling (General Procedure C): To a 20mL reaction vial was added the aryl tin monomer (0.1 mmol) and a magnetic stir bar. The vial was then placed under nitrogen atmosphere using a glovebox and CuCl (5.0 equiv) and CuCl2 (1.0 equiv) was added. The vial was sealed followed by addition of dry DMA (1 mL) and N2 was bubbled through. The reaction was allowed to stir at room temperature overnight. The mixture was quenched with NH4Cl (20 mL) and extracted with EtOAc (5 × 20 mL). The combined organic solution was dried using anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the crude product.
BBr3-mediated demethylations (General Procedure D): A solution of the methoxy dimer (0.02 mmol) in dry DCM (1.0 mL) under nitrogen atmosphere was cooled to –78 °C. The BBr3 solution (1 M in heptane, 0.40 mmol, 20 equiv.) was slowly syringed into the reaction flask. The temperature was stirred at –78 °C for 2 h. The crude mixture is quenched with sat. aq. NH4Cl (4 mL) and diluted with DCM (5 mL). The two layers were separated and the aq. layer was washed with DCM (10 mL × 3). The combined organic solution was dried using anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the crude product. The crude material was then purified on silica gel.
General procedure for the PIFA iodination (General Procedure E): A solution of the chromenone/xanthone (1.0 mmol) in dry DCM at room temperature was treated with PIFA (1.2 equiv) and I2 (0.6 equiv). The mixture was then stirred under nitrogen at room temperature for 24 h. The solution was then quenched using saturated aqueous sodium bisulfite (10 mL) and extracted using DCM (25 mL × 3). The combined organic solution was dried using anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the crude product. The crude material was then purified on silica gel.
General procedure for Suzuki Coupling (General Procedure F): A flame-dried round bottom flask containing magnetic stir bar was charged with the degassed DCM/H2O solvent (4:1 (v/v)). The flask was then charged with the aryl halide (0.1 mmol), Pd(OAc)2 (0.01 mmol, 10 mmol %), S-Phos (0.06 mmol, 0.6 equiv), B2Pin2 (0.06 mmol, 0.6 equiv), K3PO4 (0.3 mmol, 3 equiv). The mixture is heated under reflux (70 oC) for 18 hours and cooled to room temperature. The solids are then filtered off. The filtrate was diluted with DCM and water. The layers were separated and the aqueous layer was washed with DCM (20 mL × 3). The combined organic solution was dried using anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the crude product. The crude material was then purified on silica gel.
General procedure for the syntheses of chiral compounds (General Procedure G): Into a round bottom flask containing 1-(2-hydroxy-6-methoxyphenyl)butane-1,3-dione (2 mmol) in anhydrous DCM (25 mL) was added anhydrous Et3N (0.45 mL, 6 mmol). The mixture was then cooled to 0 °C and the appropriate acid halide (2.5 mmol) was added. The mixture was then allowed to warmed to room temperature and stirred overnight. The mixture was then quenched with water (25 mL) and the two layers were separated. The aqueous layer was extracted with DCM (2 × 25 mL) and the combined organic solution was dried using anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the crude product as a dark brown oil. The crude product was then taken up with 25 mL anhydrous DCM. Into this mixture was added anhydrous Et3N (0.45 mL, 6 mmol). The mixture was then stirred at room temperature for 36 h for complete reaction. The mixture was quenched with water and two layers were separated. The aqueous layer was extracted with DCM (2 × 25 mL) and the combined organic solution was dried using anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the crude product as a dark black oil. The crude product was then purified on silica gel.
2-Acetyl-3-hydroxy 4-methylbenzenesulfonate.
To a flame-dried round bottom flask was added dihydroxyacetophenone (15g, 98.6 mmol) and dry DCM (500 mL). The solution was put under N2 cooled to 0˚C in an ice bath followed by the addition of N,N,-diisopropylethylamine (20.6mL, 1.2 equiv) and portion-wise addition of 4-toluenesulfonyl chloride (20.7g, 1.1 equiv). The reaction was brought to room temperature and left to stir for three days. The reaction was washed with deionized water (2 × 100 mL), NaHCO3 (1 × 100 mL), and brine (1 × 100mL). The combined organic solution was dried using anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the crude product. The crude material was then purified by flash chromatography (20% EtOAc/hexanes). Product obtained as white solid (62%). Rf = 0.47 (EtOAc/hex 1:4); M.p. 74–75˚C; 1H NMR (500 MHz, CDCl3) δ 12.44 (s, 1H), 7.73 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 7.9 Hz, 2H), 7.29 (d, J = 8.3 Hz, 2H), 6.89 (dd, J = 8.5, 1.1 Hz, 1H), 6.46 (dd, J = 8.1, 1.1 Hz, 1H), 2.69 (s, 3H), 2.47 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 204.11, 163.76, 149.91, 146.52, 135.20, 132.45, 130.39, 128.89, 117.80, 115.63, 113.27, 32.90, 22.11; vmax = 2925, 1635, 1493, 1371, 1189, 821, 769 cm−1; HRMS (ESI): calcd for C15H15O5S [M+H+]: 307.0635; found: 307.0633.
2-Acetyl-3-hydroxy-4-iodophenyl-methylbenzenesulfonate (17).
To a flame-dried round bottom flask was added 2-acetyl-3-hydroxy 4methylbenzenesulfonate (17.8g, 58.1 mmol) and dry DCM (550 mL). The solution was put under N2 cooled to 0˚C in an ice bath followed by the addition of trifluoroacetic acid (53 mL) and portion-wise addition of N-iodosuccinimide (14.3g, 1.1 equiv). The reaction was brought to room temperature and reacted for four days. The crude mixture was quenched with sodium sulfite (100 mL) and washed with NaHCO3 (1 × 100 mL) and brine (1 × 100mL). The combined organic solution was dried using anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the crude product. The crude material was then purified by flash chromatography (80% DCM/hexanes). Product obtained as yellow solid (86%). Rf = 0.49 (EtOAc/hex 1:4); M.p. 118–120˚C; 1H NMR (500 MHz, CDCl3) δ 13.39 (s, 1H), 7.79 (d, J = 8.6 Hz, 1H), 7.73 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 7.9 Hz, 2H), 6.30 (d, J = 8.6 Hz, 1H), 2.71 (s, 3H), 2.48 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 203.91, 162.22, 150.28, 146.82, 144.31, 132.16, 130.53, 128.93, 115.65, 115.02, 85.07, 32.66, 22.16; IR (neat): vmax = 1627, 1410, 1356, 1170, 1090, 810, 754 cm−1; HRMS (ESI): calcd for C15H14IO5S [M+H+]: 432.9601; found: 432.9604.
1-(6-Hydroxy-3-iodo-2-methoxy)-acetophenone (18).
To a solution of 2-acetyl-3-hydroxy-4-iodophenyl-methylbenzenesulfonate (17.5 g, 40.5 mmol) in acetone (250 mL) was added K2CO3 (11.2 g, 2 equiv). The reaction was stirred at room temperature for 30 min before the dropwiseaddition of MeI (5.0 mL, 2 equiv) and was then brought to reflux for 24 h. The crude mixture was brought to room temperature and filtered followed by extraction with DCM (2 × 100 mL) and concentrated under reduced pressure. The crude product was used immediately in the next step. To a round bottom flask was added 2-acetyl-4-iodophenyl-3-methoxymethylbenzenesulfonate (18 g), 20% NaOH (67 mL), and t-BuOH (112 mL). The mixture was brought to reflux for 24 h. The reaction was then brought to room temperature and neutralized with 2N HCl (70 mL). The crude mixture was extracted with DCM (2 × 100 mL) and washed with deionized water (1 × 100 mL) and brine (1 × 100 mL). The combined organic layers were dried using anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the crude product. The crude material was then purified by flash chromatography (60% DCM/hexanes). Product obtained as yellow solid (80%). Rf = 0.56 (EtOAc/hex 1:9); M.p. 54–55˚C; 1H NMR (500 MHz, CDCl3) δ 12.58 (s, 1H), 7.79 (d, J = 8.9 Hz, 1H), 6.61 (d, J = 8.9 Hz, 1H), 3.84 (s, 3H), 2.76 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 204.95, 164.37, 161.64, 145.60, 117.21, 116.56, 78.89, 62.78, 31.89; IR (neat): vmax = 2940, 1621, 1447, 1323, 1210, 829 cm−1 HRMS (ESI): calcd for C9H10IO3 [M+H+]: 292.9669; found: 292.9668.
Methyl 4-(6-iodo-5-methoxy-4-oxo-4H-chromen-2-yl)butanoate (19a).
Prepared from common starting material 1-(6-hydroxy-3-iodo-2-methoxy)acetophenone and the corresponding diester as outlined in Procedure A. Product obtained as an off-white solid (62%). Rf = 0.24 (EtOAc/hex 1:2); M.p. 87–89˚C; 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 8.9 Hz, 1H), 7.01 (d, J = 8.9 Hz, 1H), 6.12 (s, 1H), 3.91 (s, 3H), 3.69 (s, 3H), 2.67 – 2.60 (t, J = 7.2 Hz, 2H), 2.43 (t, J = 7.2 Hz, 2H), 2.06 (p, J = 7.3 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 176.42, 173.34, 166.87, 159.09, 158.63, 142.65, 119.31, 116.23, 111.72, 88.83, 62.27, 52.06, 33.23, 33.13, 22.08; IR (neat): vmax = 1734, 1639, 1403, 1335, 1138, 1035, 844, 744 cm−1; HRMS (ESI): calcd for C15H16IO5 [M+H+]: 403.0037; found: 403.0027.
Methyl 3-(6-iodo-5-methoxy-4-oxo-4H-chromen-2-yl)propanoate (19b).
Prepared from common starting material 1-(6-hydroxy-3-iodo-2methoxy)-acetophenone and the corresponding diester as outlined in Procedure A. Product obtained as a yellow solid (30%). Rf = 0.25 (EtOAc/hex 1:2); M.p. 103–104˚C; 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 8.9 Hz, 1H), 7.00 (d, J = 8.9 Hz, 1H), 3.91 (s, 3H), 3.72 (s, 3H), 2.92 (t, J = 7.5 Hz, 2H), 2.74 (t, J = 7.4 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 176.35, 172.21, 165.88, 159.14, 158.56, 142.73, 119.32, 116.16, 111.57, 88.93, 62.29, 52.40, 30.92, 29.18; IR (neat): vmax = 1731, 1649, 1411, 1352, 1168, 1048, 845, 793 cm−1; HRMS (ESI): calcd for C14H14IO5 [M+H+]: 388.9880; found: 388.9874.
Methyl 2-(6-iodo-5-methoxy-4-oxo-4H-chromen-2-yl)-2methylpropanoate (19c).
Prepared from common starting material 1-(6hydroxy-3-iodo-2-methoxy)-acetophenone and the corresponding diester as outlined in Procedure A. Product obtained as an off-white solid (50%). Rf = 0.43 (EtOAc/hex 1:2); M.p. 114–115˚C; 1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 8.9 Hz, 1H), 7.01 (d, J = 8.9 Hz, 1H), 6.27 (s, 1H), 3.92 (s, 3H), 3.72 (s, 3H), 1.58 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 176.73, 173.83, 168.57, 159.08, 158.48, 142.90, 119.16, 116.24, 110.00, 88.90, 62.29, 53.20, 47.69, 23.82; IR (neat): vmax = 1729, 1649, 1461, 1346, 1149, 1035, 827 cm−1; HRMS (ESI): calcd for C15H16IO5 [M+H+]: 403.0037; found: 403.0027.
Methyl 6-iodo-5-methoxy-4-oxo-4H-chromene-2-carboxylate (19d).
Prepared from common starting material 1-(6-hydroxy-3-iodo-2-methoxy)acetophenone and the corresponding diester as outlined in Procedure A. Product obtained as a white solid (86%). Rf = 0.27 (EtOAc/hex 1:4); M.p. 153–155˚C; 1H NMR (500 MHz, CDCl3) δ 8.07 (d, J = 9.0 Hz, 1H), 7.19 (d, J = 8.9 Hz, 1H), 7.04 (s, 1H), 4.00 (s, 3H), 3.92 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 176.45, 161.04, 159.17, 158.00, 150.87, 143.91, 120.11, 116.88, 116.46, 89.87, 62.37, 53.90; IR (neat): vmax = 1731, 1639, 1409, 1368, 1258, 1125, 1041, 840, 770 cm−1; HRMS (ESI): calcd for C12H10IO5 [M+H+]: 360.9567; found: 360.9551.
6-Iodo-5-methoxy-2-(tetrahydrofuran-2-yl)-4H-chromen-4-one (19e).
Prepared from common starting material 1-(6-hydroxy-3-iodo-2-methoxy)-acetophenone and the corresponding diester as outlined in Procedure A. Product obtained as yellow solid (74%). Rf=0.36 (EtOAc/Hex 2:3); M.p. 9495˚C; 1H NMR (500 MHz CDCl3) δ 7.98 (d, J = 8.9 Hz, 1H), 7.02 (d, J = 8.9 Hz, 1H), 6.36 (d, J = 0.9 Hz, 1H), 4.80 – 4.73 (m, 1H), 4.10 – 4.04 (m, 1H), 3.99 – 3.93 (m, 1H), 3.91 (s, 3H), 2.42 – 2.29 (m, 1H), 2.14 – 2.05 (m, 1H), 2.05 – 1.97 (m, 2H); 13C NMR (126 MHz, Chloroform-d) δ 176.37, 167.73, 159.02, 158.30, 142.54, 119.47, 116.09, 109.17, 88.73, 76.59, 69.57, 62.12, 31.12, 25.54; IR (neat): vmax = 2930, 1657, 1431, 1329, 1044, 856, 787 cm−1; HRMS (ESI): calcd for C14H13IO4 [M+H+]: 372.9859; found: 372.9915.
Methyl 4-(5-methoxy-4-oxo-6-(tributylstannyl)-4H-chromen-2-yl)butanoate (21a).
Prepared from corresponding aryl halide as outlined in Procedure B. Product obtained as a colorless oil (96%). Rf = 0.22 (EtOAc/hex 1:4); 1H NMR (500 MHz, CDCl3) δ 7.59 (d, J = 8.2 Hz, 1H), 7.17 (d, J = 8.1 Hz, 1H), 6.10 (s, 1H), 3.85 (s, 3H), 3.69 (s, 3H), 2.63 (t, J = 7.5 Hz, 2H), 2.43 (t, J = 7.3 Hz, 2H), 2.10 – 2.03 (m, 2H), 1.57 – 1.47 (m, 6H), 1.38 – 1.27 (m, 6H), 1.14 – 1.07 (m, 6H), 0.88 (t, J = 7.3 Hz, 9H). 13C NMR (126 MHz, CDCl3) δ 177.82, 173.47, 166.34, 164.32, 159.55, 141.38, 132.13, 117.26, 114.25, 112.00, 63.06, 52.05, 33.27, 33.22, 29.44, 27.71, 22.21, 14.02, 10.50; HRMS (ESI): calcd for C27H43O5Sn [M+H+]: 567.2127; found: 567.2125.
Methyl 3-(5-methoxy-4-oxo-6-(tributylstannyl)-4H-chromen-2-yl)propanoate (21b).
Prepared from corresponding aryl halide as outlined in Procedure B. Product obtained as a colorless oil (95%). Rf = 0.22 (EtOAc/hex 1:4); 1H NMR (500 MHz, CDCl3) δ 7.59 (d, J = 8.2 Hz, 1H), 7.16 (d, J = 8.2 Hz, 1H), 6.12 (s, 1H), 3.84 (s, 3H), 3.72 (s, 3H), 2.92 (t, J = 7.5 Hz, 2H), 2.75 (t, J = 7.4 Hz, 2H), 1.57 – 1.47 (m, 6H), 1.38 – 1.27 (m, 6H), 1.14 – 1.06 (m, 6H), 0.88 (t, J = 7.3 Hz, 9H). 13C NMR (126 MHz, CDCl3) δ 177.74, 172.39, 165.33, 164.33, 159.47, 141.45, 132.25, 117.25, 114.17, 111.79, 63.06, 52.37, 31.08, 29.44, 29.23, 27.70, 14.02, 10.50; HRMS (ESI): calcd for C26H41O5Sn [M+H+]: 553.1970; found: 553.1970.
Methyl 2-(5-methoxy-4-oxo-6-(tributylstannyl)-4H-chromen-2-yl)-2methylpropanoate (21c).
Prepared from corresponding aryl halide as outlined in Procedure B. Product obtained as a colorless oil (95%). Rf = 0.47 (EtOAc/hex 1:4); 1H NMR (500 MHz, CDCl3) δ 7.59 (d, J = 8.2 Hz, 1H), 7.17 (d, J = 8.2 Hz, 1H), 6.26 (s, 1H), 3.86 (s, 3H), 3.72 (s, 3H), 1.58 (s, 6H), 1.54 – 1.48 (m, 7H), 1.37 – 1.29 (m, 6H), 1.14 – 1.07 (m, 6H), 0.88 (t, J = 7.3 Hz, 9H). 13C NMR (126 MHz, CDCl3) δ 178.09, 174.13, 168.09, 164.28, 159.39, 141.63, 132.25, 117.12, 114.26, 110.17, 63.05, 53.15, 47.66, 29.44, 27.71, 23.90, 14.02, 10.50; HRMS (ESI): calcd for C27H43O5Sn [M+H+]: 567.2127; found: 567.2125.
Methyl 5-methoxy-4-oxo-6-(tributylstannyl)-4H-chromene-2-carboxylate (21d).
Prepared from corresponding aryl halide as outlined in Procedure B. Product obtained as a yellow oil (99%). Rf = 0.56 (EtOAc/hex 1:4); 1H NMR (500 MHz, CDCl3) δ 7.68 (d, J = 8.2 Hz, 1H), 7.35 (d, J = 8.2 Hz, 1H), 7.03 (s, 1H), 4.00 (s, 3H), 3.85 (s, 3H), 1.58 – 1.47 (m, 6H), 1.37 – 1.29 (m, 6H), 1.15 – 1.08 (m, 6H), 0.88 (t, J = 7.3 Hz, 9H). 13C NMR (126 MHz, CDCl3) δ 177.80, 164.29, 161.43, 158.91, 150.54, 142.66, 133.49, 118.09, 116.82, 114.87, 63.14, 53.77, 29.42, 27.69, 14.01, 10.56; IR (neat): vmax = 2924, 1745, 1656, 1404, 1359, 1253, 1108, 1045, 873, 774 cm−1; HRMS (ESI): calcd for C24H37O5Sn [M+H+]: 525.1657; found: 525.1656.
5-Methoxy-2-(tetrahydrofuran-2-yl)-6-(tributylstannyl)-4H-chromen-4-one (21e).
Prepared from corresponding aryl halide as outlined in Procedure B. Product obtained as yellow oil (87%). Rf=0.67 (EtOAc/Hex 2:3); 1H NMR (500 MHz, Chloroform-d) δ 7.58 (d, J = 8.1 Hz, 1H), 7.17 (d, J = 8.2, 1.4 Hz, 1H), 6.33 (s, 1H), 4.77 (dd, J = 8.0, 5.1 Hz, 1H), 4.10 – 3.91 (m, 2H), 3.84 (s, 3H), 2.39 – 2.06 (m, 2H), 2.03 – 1.98 (m, 2H), 1.54 – 1.49 (m, 5H), 1.37 – 1.28 (m, 7H), 1.13 – 1.07 (m, 5H), 0.90 – 0.85 (m,9H); 13C NMR (126 MHz, Chloroform-d) δ 178.00, 167.44, 164.45, 159.43, 141.49, 132.26, 114.34, 109.64, 76.96, 69.76, 63.11, 31.35, 29.57, 29.49, 29.41, 27.75, 25.77, 10.55; IR (neat): vmax = 2923, 1656, 1402, 1321, 1050, 862 cm−1; HRMS (ESI): calcd for C26H40O4Sn[M+H+]: 537.1949; found: 537.2004.
Dimethyl 4,4’-(5,5’-dimethoxy-4,4’-dioxo-4H,4’H-[6,6’-bichromene]2,2’-diyl)dibutyrate (22a).
Prepared from corresponding aryl stannane as outlined in the Procedure C. Product obtained as a white solid (28%). Rf = 0.59 (EtOAc/hex 2:1); M.p. 57–58˚C; 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J = 9.0 Hz, 1H), 7.16 (d, J = 9.0 Hz, 1H), 6.10 (s, 1H), 3.96 (s, 3H), 3.69 (s, 3H), 2.64 (t, J = 7.6 Hz, 2H), 2.43 (t, J = 7.3 Hz, 2H), 2.06 (p, J = 7.3 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 176.87, 173.36, 166.94, 156.82, 155.45, 134.28, 125.30, 119.81, 115.02, 111.74, 62.20, 52.08, 33.24, 33.15, 22.12; IR (neat): vmax = 1741, 1642, 1408, 1341, 1296, 1143, 1041, 882, 751 cm−1; calcd for [M+H+]: 560.1839.
Dimethyl 3,3’-(5,5’-dimethoxy-4,4’-dioxo-4H,4’H-[6,6’-bichromene]2,2’-diyl)dipropionate (22b).
Prepared from corresponding aryl stannane as outlined in the Procedure C. Product obtained as a white solid (26%). Rf = 0.33 (EtOAc/hex 1:1); M.p. 112–113˚C; 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J = 9.0 Hz, 1H), 7.15 (d, J = 9.0 Hz, 1H), 6.11 (s, 1H), 3.96 (s, 3H), 3.72 (s, 3H), 2.93 (t, J = 7.3 Hz, 2H), 2.74 (t, J = 7.3 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 176.77, 172.21, 165.93, 156.73, 155.46, 134.33, 125.38, 119.80, 114.95, 111.56, 62.19, 52.40, 30.93, 29.16; IR (neat): vmax = 1736, 1647, 1409, 1367, 1295, 1176, 1056, 830, 790 cm−1; calcd for [M+H+]: 523.1526.
Dimethyl 2,2’-(5,5’-dimethoxy-4,4’-dioxo-4H,4’H-[6,6’-bichromene]2,2’-diyl)bis(2-methylpropanoate) (22c).
Prepared from corresponding aryl stannane as outlined in the Procedure C Product obtained as a white solid (24%). Rf = 0.50 (EtOAc/hex 1:2); M.p. 109–110˚C; 1H NMR (500 MHz, CDCl3) δ 7.62 (d, J = 9.0 Hz, 1H), 7.16 (d, J = 9.0 Hz, 1H), 6.25 (s, 1H), 3.97 (s, 3H), 3.72 (s, 3H), 1.58 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 177.15, 173.86, 168.63, 156.65, 155.40, 134.51, 125.40, 119.66, 115.03, 110.00, 62.20, 53.20, 47.68, 23.84; IR (neat): vmax = 1733, 1651, 1407, 1350, 1255, 1152, 1045, 822 cm−1; calcd for [M+H+]: 551.1839.
Dimethyl 5,5’-dimethoxy-4,4’-dioxo-4H,4’H-[6,6’-bichromene]-2,2’dicarboxylate (22d).
Prepared from corresponding aryl stannane as outlined in the Procedure C. Product obtained as a white solid (35%). Rf = 0.21 (EtOAc/hex 1:4); M.p. 151–153˚C;1H NMR (500 MHz, CDCl3) δ 7.71 (d, J = 9.1 Hz, 1H), 7.35 (d, J = 9.1 Hz, 1H), 7.02 (s, 1H), 4.00 (s, 3H), 3.97 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 176.93, 161.06, 156.19, 155.47, 150.94, 135.53, 126.25, 120.65, 116.37, 115.76, 62.28, 53.90; IR (neat): vmax = 1744, 1651, 1455, 1314, 1257, 1121, 1044, 832, 776 cm−1; calcd for [M+H+]: 467.0900.
5-Methoxychromone-2-tetrahydrofuran dimer (22e).
Prepared from corresponding aryl stannane as outlined in the Procedure C. Product obtained as white solid (16%). Rf=0.33 (EtOAc/Hex 2:3); M.p. 72–73˚C; 1H NMR (500 MHz CDCl3) δ 7.61 (d, J= 9.0, 1.4 Hz, 1H), 7.16 (d, J= 9.0, 1.0 Hz, 1H), 6.33 (s, 1H), 4.87 – 4.64 (m, 1H), 4.12 – 4.01 (m, 1H), 4.01 – 3.88 (m, 4H), 2.40 – 2.04 (m, 2H), 2.04 – 1.97 (m, 2H);13C NMR (126 MHz, Chloroform-d) δ 176.79, 167.81, 156.46, 155.32, 134.13, 125.18, 119.93, 114.86, 109.13, 76.57, 69.55, 62.01, 31.11, 25.53; IR (neat): vmax = 1655, 1405, 1337, 1231, 1105, 1055, 855 cm−1; HRMS (ESI): calcd for C28H26O8[M+H+]: 491.1628;
Dimethyl 4,4’-(5,5’-dihydroxy-4,4’-dioxo-4H,4’H-[6,6’-bichromene]2,2’-diyl)dibutyrate (14a).
Prepared from the corresponding methoxy dimer as described in the Procedure D. Product obtained as an off-white solid (88%). Rf = 0.42 (EtOAc/hex 1:2); M.p. 63–64˚C; 1H NMR (500 MHz, CDCl3) δ 13.17 (s, 1H), 7.58 (d, J = 9.0 Hz, 1H), 6.87 (d, J = 9.0 Hz, 1H), 6.16 (s, 1H), 3.69 (s, 2H), 2.70 (t, J = 7.6 Hz, 2H), 2.44 (t, J = 7.2 Hz, 2H), 2.08 (p, J = 7.3 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 183.31, 173.24, 170.74, 156.41, 155.40, 135.60, 115.61, 111.44, 109.04, 107.90, 77.61, 77.36, 77.11, 52.14, 33.83, 33.10, 22.22; IR (neat): vmax = 1737, 1650, 1410, 1307, 1231, 1168, 1033, 963, 844 cm−1; calcd for [M+H+]: 523.1526.
Dimethyl 3,3’-(5,5’-dihydroxy-4,4’-dioxo-4H,4’H-[6,6’-bichromene]2,2’-diyl)dipropionate (14b).
Prepared from the corresponding methoxy dimer as described in the Procedure D. Product obtained as an off-white solid (89%). Rf = 0.35 (EtOAc/hex 1:2); M.p. 102–103˚ 1H NMR (500 MHz, CDCl3) δ 13.12 (s, 1H), 7.58 (d, J = 9.3 Hz, 1H), 6.86 (d, J = 9.0 Hz, 1H), 6.18 (s, 1H), 3.73 (s, 3H), 2.99 (t, J = 7.4 Hz, 2H), 2.77 (t, J = 7.4 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 183.27, 172.03, 169.62, 156.43, 155.33, 135.66, 115.73, 111.46, 109.02, 107.85, 52.50, 30.90, 29.75; IR (neat): vmax = 1740, 1647, 1447, 1381, 1272, 1194, 1036, 970, 854 cm−1; calcd for [M+H+]: 495.1213.
Dimethyl 2,2’-(5,5’-dihydroxy-4,4’-dioxo-4H,4’H-[6,6’-bichromene]2,2’-diyl)bis(2-methylpropanoate) (14c).
Prepared from the corresponding methoxy dimer as described in the Procedure D. Product obtained as an off-white solid (91%). Rf = 0.40 (EtOAc/hex 1:4); M.p. 129131˚C; 1H NMR (500 MHz, CDCl3) δ 13.04 (s, 1H), 7.60 (d, J = 9.0 Hz, 1H), 6.87 (d, J = 9.0 Hz, 1H), 6.31 (s, 1H), 3.73 (s, 3H), 1.60 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 183.62, 173.49, 172.14, 156.33, 155.24, 135.85, 115.73, 111.40, 107.96, 107.44, 53.35, 48.23, 23.95; IR (neat): vmax = 1743, 1656, 1443, 1387, 1256, 1153, 1091, 953, 852 cm−1; calcd for [M+H+]: 523.1526.
Dimethyl 5,5’-dihydroxy-4,4’-dioxo-4H,4’H-[6,6’-bichromene]-2,2’dicarboxylate (14d).
Prepared from the corresponding methoxy dimer as described in the Procedure D. Product obtained as an off-white solid (94%). Rf = 0.34 (EtOAc/hex 1:4); M.p. 149–151˚C; 1H NMR (500 MHz, CDCl3) δ 12.73 (s, 1H), 7.69 (d, J = 9.0 Hz, 1H), 7.09 (s, 1H), 7.06 (d, J = 9.0 Hz, 1H), 4.03 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 183.59, 160.42, 156.29, 154.75, 153.46, 136.99, 116.65, 113.85, 112.58, 108.76, 54.16; IR (neat): vmax = 1742, 1647, 1438, 1327, 1240, 1125, 1039, 974, 808 cm−1; calcd for [M+H+]: 439.0587.
5-Hydroxychromone-2-tetrahydrofuran dimer (14e).
Prepared from the corresponding methoxy dimer as described in the Procedure D. Product obtained as a yellow solid (22%). 1H NMR (500 MHz, CDCl3) δ 13.18 (s, 1H), 7.57 (d, J= 8.9 Hz, 1H), 6.86 (d, J= 9.0 Hz, 1H), 6.41 (s, 1H), 4.92 – 4.65 (m, 1H), 4.12 – 4.01 (m, 1H), 4.01 – 3.94 (m, 1H), 2.45 – 2.05 (m, 2H), 2.05 – 1.98 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 183.08, 171.58, 156.05, 154.76, 135.19, 115.23, 111.28, 107.45, 106.09, 76.49, 69.43, 31.17, 25.27; HRMS (ESI): calcd for C26H22O8[M+H+]: 463.1315
Methyl 4-(5-methoxy-4-oxo-4H-chromen-2-yl)butanoate (24a).
Prepared from 2’-hydroxy-6’-methoxyacetophenone and corresponding diester, as described in the general procedure A. Ethyl ester was cleanly converted to the methyl ester under the reaction conditions. The pure product was obtained as a yellow solid (60 %). Rf = 0.32 (EtOAc); m.p. 7678 °C; 1H NMR (500 MHz, CDCl3): δ 7.45 (t, J = 8.8 Hz, 1H), 6.91 (d, J = 9.4 Hz, 1H), 6.71 (d, J = 8.3 Hz, 1H), 6.01 (s, 1H), 3.90 (s, 3H), 3.61 (s, 3H), 2.54 (t, J = 7.6 Hz, 2H), 2.36 (t, J = 7.3 Hz, 2H), 1.99 ppm (q, J = 7.4 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 178.47, 173.49, 166.13, 160.10, 158.87, 133.86, 114.69, 112.04, 110.32, 106.62, 56.80, 52.06, 33.22, 22.13 ppm; IR (neat): vmax = 1721, 1645, 1600, 1473, 1442, 1429, 1336, 1176, 856, 768 cm−1; HRMS (ESI): calcd for C15H17O5 [M+H+]: 277.1071; found: 277.1063.
Methyl 3-(5-methoxy-4-oxo-4H-chromen-2-yl)propanoate (24b).
Prepared from 2’-hydroxy-6’-methoxyacetophenone and corresponding diester, as described in the general procedure A. Ethyl ester was cleanly converted to the methyl ester under the reaction conditions. The pure product was obtained as a yellow solid (47 %). Rf = 0.36 (EtOAc); m.p. 8789 °C; 1H NMR (500 MHz, CDCl3): δ 7.44 (t, J = 10 Hz, 1H), 6.89 (d, J = 5 Hz, 1H), 6.72 (d, J = 5 Hz, 1H), 6.00 (s, 1H), 3.89 (s, 3H), 3.64 (s, 3H), 2.83 (t, J = 8.3 Hz, 2H), 2.66 ppm (t, J = 8.4 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 178.05, 172.16, 164.93, 159.86, 158.55, 133.74, 114.47, 111.60, 110.03, 106.53, 56.60, 52.12, 30.83, 28.92 ppm; IR (neat): vmax = 1721, 1634, 1581, 1470, 1420, 1342, 1076, 855, 706 cm−1; HRMS (ESI): calcd for C14H15O5 [M+H+]: 263.0914; found: 263.0908.
Methyl 2,2-dimethyl-2-(5-methoxy-4-oxo-4H-chromen-2-yl)ethanoate (24c).
Prepared from 2’-hydroxy-6’-methoxyacetophenone and corresponding diester, as described in the general procedure A. The pure product was obtained as a yellow solid (40 %). Rf = 0.68 (EtOAc); m.p. 125–127 °C; 1H NMR (500 MHz, CDCl3): δ 7.51 (t, J = 8.4 Hz, 1H), 6.96 (d, J = 8.6 Hz, 1H), 6.78 (d, J = 8.6 Hz, 1H), 6.22 (s, 1H), 3.96 (s, 3H), 3.69 (s, 3H), 1.55 ppm (s, 6H); 13C NMR (125 MHz, CDCl3): δ 178.58, 174.09, 160.03, 158.68, 134.04, 114.53, 110.26, 110.23, 106.66, 56.81, 53.08, 47.55, 23.83 ppm; IR (neat): vmax = 1716, 1632, 1523, 1445, 1100, 965, 926, 827 cm−1; HRMS (ESI): calcd for C15H17O5 [M+H+]: 277.1071; found: 277.1076.
Methyl (5-methoxy-4-oxo-4H-chromen-2-yl)carboxyate (24d).
Prepared from 2’-hydroxy-6’-methoxyacetophenone and corresponding diester, as described in the general procedure A. The pure product was obtained as a yellow solid (60 %). Rf = 0.21 (EtOAc/hex 1:1); m.p. 125127 °C; 1H NMR (500 MHz, CDCl3): δ 7.65 (t, J = 8.4 Hz, 1H), 7.19 (d, J = 8.5 Hz, 1H), 7.05 (s, 1H), 6.88 (d, J = 8.2 Hz, 1H), 4.02 ppm (s, 6H); 13C NMR (125 MHz, CDCl3): δ 178.41, 161.38, 160.16, 158.33, 150.48, 135.19, 116.93, 115.56, 110.89, 107.26, 56.85, 53.78 ppm; IR (neat): vmax = 1746, 1611, 1630, 1522, 1470, 1422, 1275, 1103, 815 cm−1; HRMS (ESI): calcd for C12H11O5 [M+H+]: 235.0528; found: 235.0593.
5-Methoxy-2-(tetrahydrofuran-2-yl)-4H-chromen-4-one (24e).
Prepared from 2’-hydroxy-6’-methoxyacetophenone and corresponding furan ester, as described in the general procedure A. The pure product was obtained as an yellow waxy solid (54 %). Rf = 0.4 (EtOAc); m.p. 5659 °C; 1H NMR (500 MHz, CDCl3): δ 7.52(t, J = 8.6 Hz, 1H), 7.45 (d, J = 8.8 Hz, 1H), 6.99 (d, J = 8.2 Hz, 1H), 6.30 (s, 1H), 4.74 (m, 1H), 4.11 (m, 1H), 3.96 (s, 3H), 2.35 (m, 1H), 2.10 (m, 1H), 2.00(m, 2H) ppm; 13C NMR (125 MHz, CDCl3): δ 177.02, 162.19, 160.61, 156.51, 143.15, 115.88, 114.11, 110.13, 108.89, 73.60, 69.85, 56.97, 31.20, 25.95 ppm; IR (neat): vmax = 1720, 1544, 1480, 1422, 821, 747, 721 cm−1; HRMS (ESI): calcd for C14H15O4 [M+H+]: 247.0965; found: 247.0971.
Methyl 4-(8-iodo-5-methoxy-4-oxo-4H-chromen-2-yl)butanoate (25a).
Prepared from corresponding 2’-substituted chromenone as described in the general procedure E. The pure product was obtained as a brown solid (62 %). Rf = 0.30 (EtOAc); m.p. 89–91 °C; 1H NMR (500 MHz, CDCl3): δ 7.90 (d, J = 8.8 Hz, 1H), 6.60 (d, J = 8.6 Hz, 1H), 6.09 (s, 1H), 3.92 (s, 3H), 3.66 (s, 3H), 2.66 (t, J = 7.45 Hz, 2H), 2.45 (d, J = 7.2 Hz, 2 H), 2.15–2.05 ppm (m, 2H); 13C NMR (125 MHz, CDCl3): δ 203.87, 177.81, 173.22, 166.27, 160.50, 156.80, 143.00, 112.07, 108.78, 73.61, 56.89, 51.98, 41.15, 33.14, 32.98, 21.99 ppm; IR (neat): vmax = 1741, 1640, 1620, 1470, 1430, 1421, 1218, 1076, 820, 760 cm−1; HRMS (ESI): calcd for C15H16IO5 [M+H+]: 403.0037; found: 403.0018.
Methyl 4-(8-iodo-5-methoxy-4-oxo-4H-chromen-2-yl)propanoate (25b).
Prepared from corresponding 2’-substituted chromenone as described in the general procedure E. The pure product was obtained as a brown solid (68 %). Rf = 0.34 (EtOAc); m.p. 98–100 °C; 1H NMR (500 MHz, CDCl3): δ 7.93 (d, J = 8.8 Hz, 1H), 6.64 (d, J = 8.9 Hz, 1H), 6.15 (s, 1H), 3.96 (s, 3H), 3.72 (s, 3H), 2.98 (t, J = 7.6 Hz, 2H), 2.84 ppm (t, J = 7.05 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 177.89, 172.35, 165.27, 160.64, 156.82, 143.14, 115.63, 112.09, 108.92, 73.65, 57.0, 52.42, 30.92, 29.07 ppm; IR (neat): vmax = 1733, 1645, 1584, 1472, 1428, 1255, 1064, 846, 823 cm−1; HRMS (ESI): calcd for C14H14IO5 [M+H+]: 388.9880; found: 388.9870.
Methyl 2,2-dimethyl-2-(8-iodo-5-methoxy-4-oxo-4H-chromen-2yl)ethanoate (25c).
Prepared from corresponding 2’-substituted chromenone as described in the general procedure E. The pure product was obtained as a off-white solid (58 %). Rf = 0.34 (EtOAc); m.p. 134–136 °C; 1H NMR (500 MHz, CDCl3): δ 7.94 (d, J = 5 Hz, 1H), 6.64 (d, J = 10 Hz, 1H), 6.25 (s, 1H), 3.96 (s, 3H), 3.74 (s, 3H), 1.61 ppm (s, 6H); 13C NMR (125 MHz, CDCl3): δ 178.22, 173.89, 168.47, 160.55, 156,72, 143.24, 110.29, 108.89, 73.67, 57.03, 53.27, 47.84, 24.067, 20.39 ppm; IR (neat): vmax = 1732, 1612, 1555, 1560, 1226, 1107, 975, 920, 828 cm−1; HRMS (ESI): calcd for C15H16IO5 [M+H+]: 403.0037; found: 403.0028.
Methyl (8-iodo-5-methoxy-4-oxo-4H-chromen-2-yl)carboxylate (25d).
Prepared from corresponding 2’-substituted chromenone as described in the general procedure E. The pure product was obtained as an off-white solid (76 %). Rf = 0.57 (EtOAc); m.p. 122–125 °C; 1H NMR (500 MHz, CDCl3): δ 8.04 (d, J = 8.8 Hz, 1H), 7.04 (s, 1H), 6.69 (d, J = 8.8 Hz, 1H), 4.01, (s, 3H), 3.99 ppm (s, 3H); 13C NMR (125 MHz, CDCl3): δ 177.89, 160.97, 160.63, 156.63, 150.75, 144.41, 116.92, 116.49, 109.40, 73.75, 57.05, 53.89 ppm; IR (neat): vmax = 1723, 1655, 1634, 1586, 1475, 1422, 1388, 1275, 1113, 815 cm−1; HRMS (ESI): calcd for C12H10IO5 [M+H+]: 360.9567; found: 360.9565.
8-Iodo-5-methoxy-2-(tetrahydrofuran-2-yl)-4H-chromen-4-one (25e).
Prepared from corresponding 2’-substituted chromenone as described in the general procedure E. The pure product was obtained as a yellow solid (70 %). Rf = 0.42 (EtOAc); m.p. 155–158 °C; 1H NMR (500 MHz, CDCl3): δ 7.94 (d, J = 8.8 Hz, 1H), 6.64 (d, J = 8.2 Hz, 1H), 6.34 (s, 1H), 4.83 (m, 1H), 4.11 (m, 1H), 3.98 (m, 1H), 3.95 (s, 3H), 2.37 (m, 1H), 2.20 (m, 1H), 2.10 (m, 1H), 2.01 ppm (m, 1H); 13C NMR (125 MHz, CDCl3): δ 178.02, 167.19, 160.65, 156.80, 143.15, 115.88, 110.13, 108.89, 76.90, 73.60, 69.85, 56.97, 31.20, 25.95 ppm; IR (neat): vmax = 1648, 1512, 1455, 1225, 825, 790, 785 cm−1; HRMS (ESI): calcd for C14H14IO4 [M+H+]: 372.9931; found: 372.9924.
Dimethyl 3,3’-(5,5’-dimethoxy-4,4’-dioxo-4H,4’H-[8,8’-bichromene]2,2’-diyl)dibutanoate (26a).
Prepared from the corresponding aryl iodide under Suzuki coupling conditions as described in the general procedure F. The pure product was obtained as a brown waxy solid (55 %). Rf = 0.24 (EtOAc/MeOH 9:1); m.p. 62–68 °C; 1H NMR (500 MHz, CDCl3): δ 7.52 (d, J = 10 Hz, 1H), 6.90 (d, J = 10 Hz, 1H), 6.09 (s, 1H), 4.04 (s, 3H), 3.62 (s, 3H), 2.39 (t, J = 8.8 Hz, 2H), 2.22 (d, J = 8.6Hz, 2 H), 1.75 ppm (m, J = 8.9 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 178.42, 173.14, 165.88, 160.03, 155.98, 135.55, 118.41, 114.62, 112.00, 106.46, 56.87, 52.02, 33.04, 32.94, 21.75 ppm; IR (neat): vmax = 1731, 1647, 1593, 1436, 1262, 1168, 1096, 1072, 847, 817 cm−1; HRMS (ESI): calcd for C30H31O10 [M+H+]: 551.1912; found: 551.1907.
Dimethyl 3,3’-(5,5’-dimethoxy-4,4’-dioxo-4H,4’H-[8,8’-bichromene]2,2’-diyl)dipropanoate (26b).
Prepared from the corresponding aryl iodide under Suzuki coupling conditions as described in the general procedure F. The pure product was obtained as a yellow waxy oil (60 %). Rf = 0.32 (EtOAc/MeOH 9:1); 1H NMR (500 MHz, CDCl3): δ 7.50 (d, J = 8.6 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 6.09 (s, 1H), 4.03 (s, 3H), 3.60 (s, 3H), 2.68 (t, J = 8.4 Hz, 2H), 2.41 ppm (t, J = 8.8 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 178.25, 171.26, 164.82, 160.00, 155.82, 135.57, 118.27, 114.45, 111.80, 106.52, 56.88, 52.26, 30.33, 28.74 ppm; IR (neat): vmax = 1734, 1644, 1610, 1593, 1578, 1492, 1389, 1198, 1098, 1076, 981, 859 cm−1; HRMS (ESI): calcd for C28H27O10 [M+H+]: 523.1599; found: 523.1592.
Dimethyl 2,2’-(5,5’-dimethoxy-4,4’-dioxo-4H,4’H-[8,8’-bichromene]2,2’-diyl)bis(2-methylpropanoate) (26c).
Prepared from the corresponding aryl iodide under Suzuki coupling conditions as described in the general procedure F. The pure product was obtained as a colorless waxy oil (24 %). Rf = 0.30 (EtOAc/MeOH 9.5:0.5); 1H NMR (500 MHz, CDCl3): δ 7.49 (d, J = 8.5 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 6.21 (s, 1H), 3.03 (s, 3H), 3.48 (s, 3H), 1.29 ppm (s, 6H); 13C NMR (125 MHz, CDCl3): δ 178.54, 173.47, 167.78, 159.95, 155.87, 135.56, 118.02, 110.44, 106.23, 56.91, 52.89, 47.61, 23.75, 19.99 ppm; IR (neat): vmax = 1728, 1614, 1625, 1578, 1450, 1198, 1098, 1076, 928, 859 cm−1; HRMS (ESI): calcd for C30H31O10[M+H+]: 551.1912; found: 551.1896
Dimethyl 5,5’-dimethoxy-4,4’-dioxo-4H,4’H-[8,8’-bichromene]-2,2’dicarboxylate (26d).
Prepared from the corresponding aryl iodide under Suzuki coupling conditions as described in the general procedure F. The pure product was obtained as a yellow oil (35%). Rf = 0.24 (EtOAc/MeOH 9.5:0.5); 1H NMR (500 MHz, CDCl3): δ 7.67 (d, J = 8.5 Hz, 1H), 7.02 (s, 1H), 6.96 (d, J = 8.6 Hz, 1H), 4.05, (s, 3H), 3.72 ppm (s, 3H); 13C NMR (125 MHz, CDCl3): δ 179.25, 160.12, 159.63, 157.63, 150.14, 144.41, 121.45, 117.92, 116.49, 110.40, 56.05, 53.80 ppm; IR (neat): vmax = 1728, 1652, 1596, 1582, 1248, 1130, 1074, 1057, 815, 786 cm−1; HRMS (ESI): calcd for C24H19O10 [M+H+]: 467.0973; found: 467.0981.
5,5’-Dimethoxy-2,2’-bis(tetrahydrofuran-2-yl)-4H,4’H-[8,8’-bichromene]-4,4’-dione (26e).
Prepared from the corresponding aryl iodide under Suzuki coupling conditions as described in the general procedure F. The pure product was obtained as a waxy yellow oil (55 %). Rf = 0.16 (EtOAc/MeOH 9.5:0.5); 1H NMR (500 MHz, CDCl3): δ 7.53 (d, J = 10 Hz, 1H), 6.91 (d, J = 10 Hz, 1H), 6.29 (s, 1H), 4.44 (m, 1H), 4.04 (s, 3H), 3.73 (m, 1H), 3.63 (m, 1H), 2.00 (m, 1H), 1.74 −1.62 ppm (m, 3H); 13C NMR (125 MHz, CDCl3): δ 178.57, 166.89, 160.10, 155.99, 135.33, 118.77, 110.15, 106.49, 69.79, 69.72, 56.94, 30.90, 25.92 ppm; IR (neat): vmax = 1655, 1601, 1512, 1411, 1220, 821, 785 cm−1; HRMS (ESI): calcd for C28H27O8 [M+H+]: 491.1700; found: 491.1690.
Dimethyl 3,3’-(5,5’-dihydroxy-4,4’-dioxo-4H,4’H-[8,8’-bichromene]2,2’-diyl)dibutanoate (15a).
Prepared from the corresponding methoxy dimer as described in the general procedure D. However, the reaction was run for 6 hours at 0°C. The pure product was obtained as a yellow oil (60 %). Rf = 0.21 (EtOAc); 1H NMR (500 MHz, CDCl3): δ 12.11 (s, 1H), 7.55 (d, J = 8.6 Hz, 1H), 6.92 (d, J = 8.8 Hz, 1H), 6.11 (s, 1H), 3.61 (s, 3H), 2.38 (t, J = 8.8 Hz, 2H), 2.22 (d, J = 8.6Hz, 2 H), 1.75 ppm (m, J = 8.9 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 177.42, 174.13, 166.88, 162.03, 156.98, 132.55, 119.41, 111.62, 112.00, 106.46, 52.02, 33.04, 32.94, 21.75 ppm; IR (neat): vmax = 3418, 1723, 1548, 1478, 1466, 826, 723, 703 cm−1; HRMS (ESI): calcd for C28H27O10 [M+H+]: 523.1599; found: 523.1567.
Dimethyl 3,3’-(5,5’-dihydroxy-4,4’-dioxo-4H,4’H-[8,8’-bichromene]2,2’-diyl)dipropanoate (15b).
Prepared from the corresponding methoxy dimer as described in the general procedure D. However, the reaction was run for 6 h at 0 °C. The pure product was obtained as a waxy yellow oil (82 %). Rf = 0.21 (EtOAc); 1H NMR (500 MHz, CDCl3): δ 12.72 (s, 1H), 7.50 (d, J = 5 Hz, 1H), 6.89 (d, J = 5 Hz, 1H), 6.18 (s, 1H), 3.63 (s, 3H), 2.83 (t, J = 8.6 Hz, 2H), 2.52 ppm (t, J = 8.9 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 183.89, 171.88, 168.95, 161.23, 153.98, 137.55, 114.90, 111.78, 110.94, 109.11, 52.42, 30.38, 29.44 ppm; IR (neat): vmax = 3220, 1730, 1649, 1612, 1579, 1415, 1238, 1169, 968, 783, 630 cm−1; HRMS (ESI): calcd for C26H23O10 [M+H+]: 495.1286; found: 495.1273.
Dimethyl 2,2’-(5,5’-dihydroxy-4,4’-dioxo-4H,4’H-[8,8’-bichromene]2,2’-diyl)bis(2-methylpropanoate) (15c).
Prepared from the corresponding methoxy dimer as described in the general procedure D. However, the reaction was run for 6 h at 0 °C. The pure product was obtained as a waxy yellow solid (46 %). Rf = 0.10 (EtOAc/MeOH 9.9:0.01); 1H NMR (500 MHz, CDCl3): δ 12.61 (s, 1H), 7.50 (d, J = 8.5 Hz, 1H), 6.87 (d, J = 8.5 Hz, 1H), 6.28 (s, 1H), 3.57 (s, 3H), 1.40 ppm (s, 6H); 13C NMR (125 MHz, CDCl3): δ 184.12, 173.22, 171.65, 160.99, 153.86, 137.67, 114.47, 111.43, 110.81, 107.70, 53.13, 48.29, 23.98 ppm; IR (neat): vmax = 3318, 1734, 1646, 1620, 1578, 1411, 1055, 1013, 915, 828 cm−1; HRMS (ESI): calcd for C28H27O10 [M+H+]: 523.1599; found: 523.1622
Dimethyl 5,5’-dihydroxy-4,4’-dioxo-4H,4’H-[8,8’-bichromene]-2,2’dicarboxylate (15d).
Prepared from the corresponding methoxy dimer as described in the general procedure D. However, the reaction was run for 6 h at 0 °C. The pure product was obtained as a yellow oil (47 %). Rf = 0.16 (EtOAc); 1H NMR (500 MHz, CDCl3): δ 7.68 (d, J = 8.5 Hz, 1H), 7.05 (s, 1H), 6.92 (d, J = 8.6 Hz, 1H), 3.78 ppm (s, 3H); 13C NMR (125 MHz, CDCl3): δ 178.25, 161.12, 160.43, 156.61, 151.14, 142.41, 120.45, 118.92, 116.50, 111.40, 50.80 ppm; IR (neat): vmax = 2946, 1755, 1610, 1556, 1520, 1100, 1086, 1047, 811, 768 cm−1; HRMS (ESI): calcd for C22H15O10 [M+H+]: 439.0660; found: 439.071.
5,5’-Dihydroxy-2,2’-bis(tetrahydrofuran-2-yl)-4H,4’H-[8,8’bichromene]-4,4’-dione (15e).
Prepared from the corresponding methoxy dimer as described in the general procedure D. However, the reaction was run for 6 h at 0 °C. The pure product was obtained as a yellow solid (82 %). Rf = 0.37 (EtOAc/MeOH 9.5:0.5); m.p.172–175 °C; 1H NMR (500 MHz, CDCl3): δ 12.56 (s, 1H), 7.55 (d, J = 8.6 Hz, 1H), 6.91 (d, J = 8.5 Hz, 1H), 6.40 (s, 1H), 4.37 (m, 1H), 3.32 (m, 1), 1.74 −1.62 ppm (m, 4H); 13C NMR (125 MHz, CDCl3): δ 184.11, 161.13, 153.61, 137.44, 111.97, 41.14, 33.54, 33.47, 33.36, 33.29, 29.94, 28.25 ppm; IR (neat): vmax = 3475, 1649, 1591, 1471, 1410, 1240, 820, 800 cm−1; HRMS (ESI): calcd for C26H23O8 [M+H+]: 463.1387; found: 463.1375.
3-Acetyl-5-methoxy-2-methyl-4H-1-benzopyran-4-one (27).
Into a round bottom flask containing 1-(2-hydroxy-6-methoxyphenyl)butane-1,3dione (2 g, 9.6 mmol) in acetic anhydride (15 mL) was added NaOAc (0.85 g, 10 mmol). The insoluble mixture was heated at reflux for 24 h. The solution was cooled to room temperature and diluted with water (25 mL) and stirred at room temperature for 15 min. The mixture was then extracted with DCM (3 × 50 mL) and the combined organic solution was dried using anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the crude product as a yellow flaky solid. The crude product was then purified on silica column to give the desired product as a flaky white solid (1.73 g, 78 %). Rf = 0.16 (EtOAc/hex 1:1); m.p. 144–146 °C; 1H NMR (500 MHz, CDCl3): δ 7.55 (t, J = 8 Hz, 1H), 6.98 (d, J = 5 Hz, 1H), 6.82 (d, J = 5 Hz, 1H), 3.98 (s, 3H), 2.60 (s, 3H), 2.43 ppm (s, 3H); 13C NMR (125 MHz, CDCl3): δ 201.33, 176.30, 166.50, 160.30, 157.73, 134.48, 125.40, 114.51, 110.09, 107.14,, 32.52, 33.03, 19.56 ppm; IR (neat): vmax = 1734, 1624, 1532, 1410, 1400, 1021, 1015, 840, 786 cm−1; HRMS (ESI): calcd for C13H13O4 [M+H+]: 233.0808; found: 233.0798.
3-Acetyl-8-iodo-5-methoxy-2-methyl-4H-1-benzopyran-4-one (28).
Prepared from corresponding 2’, 3’-substituted chromenone as described in the general procedure E. The pure product was obtained as a pale white solid (67 %). Rf = 0.31 (EtOAc); m.p. 189–191 °C; 1H NMR (500 MHz, CDCl3): δ 7.97 (d, J = 8.8 Hz, 1H), 6.67 (d, J = 8.9 Hz, 1H), 3.99 (s, 3H), 2.60 (s, 3H), 2.52ppm (s, 3H); 13C NMR (125 MHz, CDCl3): δ 200.70, 175.94, 166.79, 160.79, 156.07, 143.69, 125.34, 115.51, 109.31, 73.28, 57.02, 32.43, 19.48 ppm; IR (neat): vmax = 1762, 1684, 1552, 1430, 1384, 1228, 1022, 1005, 845, 722 cm−1; HRMS (ESI): calcd for C13H12IO4 [M+H+]: 358.9775; found: 358.9770.
3,3’-Diacetyl-5,5’-dimethoxy-2,2’-dimethyl-4H,4’H-[8,8’-bichromene]-4,4’-dione (29).
The corresponding aryl iodide was homodimerized by subjecting it to Suzuki coupling as described in general procedure B. The pure product was obtained as a white solid (59 %). Rf = 0.50 (EtOAc/hex 9:1); m.p.176–178 °C; 1H NMR (500 MHz, CDCl3): δ 7.55 (d, J = 10 Hz, 1H), 6.94 (d, J = 10 Hz, 1H), 4.06 (s, 3H), 2.61 (s, 3H), 2.22 ppm (s, 3H); 13C NMR (125 MHz, CDCl3): δ 200.83, 176.30, 166.22, 160.34, 154.80, 136.24, 125.31, 117.72, 114.61, 107.07, 56.95, 32.53, 30.04, 19.45 ppm; IR (neat): vmax = 1732, 1642, 1582, 1444, 1400, 1122, 1075, 914, 755 cm1; HRMS (ESI): calcd for C26H23O8 [M+H+]: 463.1387; found: 463.1371.
3-Acetyl -5-hydroxy-2-methyl-4H-1-benzopyran-4-one dimer (30).
Prepared from the corresponding methoxy dimer as described in the general procedure D. However, the reagents were mixed at 0 °C and the reaction was run overnight at room temperature. The pure product was obtained as a yellow solid (62 %). Rf = 0.17 (EtOAc); m.p. 124–126 °C; 1H NMR (500 MHz, CDCl3): δ 12.67 (s, 1 H) 7.54 (d, J = 5 Hz, 1H), 6.94 (d, J = 5 Hz, 1H), 2.64 (s, 3H), 2.36 ppm (s, 3H); 13C NMR (125 MHz, CDCl3): δ 199.37, 182.08, 170.58, 161.83, 153.08, 138.36, 114.78, 112.84, 110.99, 32.95, 30.31, 20.60 ppm; IR (neat): vmax = 3288, 1686, 1646, 1588, 1478, 1416, 1400, 1132, 1065, 814, 781cm−1; HRMS (ESI): calcd for C24H19O8 [M+H+]: 435.1074; found: 435.1068.
1-(2-Hydroxy-6-methoxyphenyl)butane-1,3-dione (31).
A solution of the 2’-hydroxy-6’-methoxyacetophenone (10 g, 60 mmol ) and the EtOAc (5.2 g, 10.1 mmol) in dry THF (200 mL) under nitrogen atmosphere was cooled to 0 °C. Into this mixture was added NaH (60 % in oil, 8.4 g, 3.5 equiv) in 5 portions. The temperature was allowed to rise to room temperature and stirred overnight. The crude mixture was quenched with 0.1 N HCl and the pH was adjusted to pH 1. Most of the THF was then removed by rotary evaporation and the resulting mixture was diluted with EtOAc (150 mL). The two layers were separated and the aq. layer was washed with EtOAc (3 × 150 mL). The combined organic solution was dried using anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the crude product. The crude product was then dried thoroughly in vacuo to remove all EtOAc. The resulting residue was then triturated with hexanes to obtain the corresponding 1-(2-hydroxy-6-methoxyphenyl)butane-1,3dione (11.2 g, 90 %). This product was not purified any further.
3-Acetyl-5-methoxy-2-(methylacetyl)-4H-1-benzopyran-4-one (33a).
This compound was prepared as described in the general procedure G. 1(2-hydroxy-6-methoxyphenyl)butane-1,3-dione was reacted with commercially available acetoxyacetyl chloride. The crude product was purified on silica column to give the desired compound (72 %) as a brown solid. Rf = 0.17 (EtOAc/hex 1:1); m.p. 122–125 °C; 1H NMR (500 MHz, CDCl3): δ 7.58 (t, J = 8.4 Hz, 1H), 7.00 (d, J = 9.3 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 5.20 (s, 3 2H), 3.99 (s, 3H), 2.52 (s, 3H), 2.17 ppm (s, 3H); 13C NMR (125 MHz, CDCl3): δ 159.80, 176.49, 171.15, 170.36, 160.32, 157.56, 134.89, 122.37, 114.21, 110.14, 107.51, 70.67, 56.87, 20.88, 20.16 ppm; IR (neat): vmax = 1736, 1697, 1609, 1474, 1442, 1429, 1411, 1221, 1059, 937, 809, 791 cm−1; HRMS (ESI): calcd for C15H15O6 [M+H+]: 291.0863; found: 291.0860.
(S)-1-(3-acetyl-5-methoxy-4-oxo-4H-chromen-2-yl)ethyl acetate (33b).
This compound is prepared as described in the general procedure G. 1-(2hydroxy-6-methoxyphenyl)butane-1,3-dione was reacted with (S)-2acetoxylactyl chloride. The crude product was purified on silica column to give the desired compound (81 %) as a brown solid. Rf = 0.16 (EtOAc/hex 1:1); m.p. 75–78 °C; [α]20 = 0.59 (c = 1.0 CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.56 (t, J = 8.4 Hz, 1H), 7.00 (d, J = 9.2 Hz, 1H), 6.83 (d, J = 8.3 Hz, 1H), 5.73 (q, J = 6.8, 1H), 3.99 (s, 3H), 2.45 (s, 3H), 2.07 (s, 3H), 1.59 ppm (d, J = 6.9, 3H); 13C NMR (125 MHz, CDCl3): δ 200.67, 176.07, 170.92, 169.04, 160.35, 157.70, 134.65, 123.39, 114.31, 110.11, 107.33, 75.87, 56.89, 21.05, 19.88, 16.73 ppm; IR (neat): vmax = 1739, 1686, 1650, 1610, 1475, 1390, 1234, 1033, 819 cm−1; HRMS (ESI): calcd for C16H17O6 [M+H+]: 305.1020; found: 305.1016.
(S)-1-(3-Acetyl-5-methoxy-4-oxo-4H-chromen-2-yl)-2-methylpropyl acetate (33c).
This compound is prepared as described in the general procedure G. 1-(2-hydroxy-6-methoxyphenyl)butane-1,3-dione was reacted with (S)-2-(acetyloxy)-3-methyl-butanoyl chloride. The crude product was purified on silica column to give the desired compound (66 %) as a waxy brown solid. Rf = 0.21 (EtOAc/hex 1:1); m.p. 47–49 °C; [α]20 = 44 (c = 1.0 CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.59 (t, J = 8.4 Hz, 1H), 7.00 (d, J = 9.3 Hz, 1H), 6.86 (d, J = 8.4 Hz, 1H), 6.01 (d, J = 3.2 Hz ), 4.01 (s, 3H), 2.51 (s, 3H), 2.17 (s, 3H), 1.11 (d, J = 6.9 Hz, 3H), 0.94 ppm (d, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 198.49, 175.55, 170.78, 169.05, 160.32, 157.47, 134.51, 123.09, 114.32, 109.90, 107.32, 83.29, 56.82, 29.21, 20.90, 20.33, 19.99, 16.82 ppm; IR (neat): vmax = 1739, 1650, 1686, 1650, 1610, 1476, 1370, 1234, 1073, 1033, 799, 771, 752 cm−1; HRMS (ESI): calcd for C18H21O6 [M+H+]: 333.1333; found: 333.1320.
3-Acetyl-5-methoxy-2-(methylacetyl)-4H-1-benzopyran-4-one (34a).
Prepared from the corresponding starting material as described in the general procedure E. The pure product was obtained as a yellow solid (62%). Rf = 0.27 (EtOAc); m.p. 182–184 °C; 1H NMR (500 MHz, CDCl3): δ 7.99 (d, J = 8.8 Hz, 1H), 6.69 (d, J = 8.9 Hz, 1H), 5.17 (s, 2H), 3.99 (s, 3H), 2.56 (s, 3H), 2.17 ppm (s, 3H); 13C NMR (125 MHz, CDCl3): δ 195.40, 176.11, 170.48, 160.76, 156.00, 144.06, 122.38, 115.22, 109.63, 73.25, 70.51, 57.07, 20.86, 20.05 ppm; IR (neat): vmax = 1701, 1655, 1600, 1422, 1410, 1410, 1244, 927, 800, 742 cm−1; HRMS (ESI): calcd for C15H14IO6 [M+H+]: 416.9830; found: 418.9819.
(S)-1-(3-Acetyl-8-iodo-5-methoxy-4-oxo-4H-chromen-2-yl)ethyl acetate (34b).
Prepared from the corresponding starting material as described in the general procedure E. The pure product was obtained as a yellow solid (54 %). Rf = 0.24 (EtOAc); m.p. 147–150 °C; [α]20 = −48 (c = 1.0 CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.99 (d, J = 8.8 Hz, 1H), 6.67 (d, J = 8.9 Hz, 1H), 5.69 (q, J = 6.90 Hz, 1H), 3.98 (s, 3H), 2.52 (s, 3H), 2.07 (s, 3H), 1.57 ppm (d, J = 7.0 Hz 3H); 13C NMR (125 MHz, CDCl3): δ 195.40, 176.11, 170.48, 160.76, 156.00, 144.06, 122.38, 115.22, 109.63, 73.25, 70.51, 57.07, 20.86, 20.05 ppm; IR (neat): vmax = 1721, 1698, 1635, 1569, 1392, 1365, 1275, 1076, 817, 609 cm−1; HRMS (ESI): calcd for C16H16IO6 [M+H+]: 430.9986; found: 430.9974.
(S)-1-(3-Acetyl-8-iodo-5-methoxy-4-oxo-4H-chromen-2-yl)-2-methylpropyl acetate (34c).
Prepared from the corresponding starting material as described in the general procedure E. The pure product was obtained as a yellow solid (51 %). Rf = 0.18 (EtOAc); m.p. 132–134 °C; [α]20 = −52 (c = 1.0 CHCl3); 1HNMR (500Hz, CDCl3): δ 7.97(d, J = 8.8 Hz, 1H), 6.67(d, J = 8.9 Hz, 1H), 5.93 (d, J = 3.2 Hz ), 3.97 (s, 3H), 2.55 (s, 3H), 2.48–2.42 (m, 1H), 2.14 (s, 3H), 1.07 (d, J = 6.7 Hz, 3H), 0.90 ppm (d, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 198.11, 175.26, 170.87, 169.21, 160.85, 155.92, 143.80, 123.18, 115.37, 109.56, 83.26, 73.09, 57.09, 29.31, 20.97, 20.39, 19.94, 16.90 ppm; IR (neat): vmax = 1730, 1662, 1631, 1610, 1410, 1224, 1010, 762, 765, 722 cm−1; HRMS (ESI): calcd for C18H20IO6 [M+H+]: 459.0299; found: 459.0286.
(3,3’-Diacetyl-5-methoxy-4,4’-dioxo-4H,4’H-[8,8’-bichromene]-2,2’diyl)bis(methylene) diacetate (35a).
Prepared from the corresponding aryl iodide as described in the general procedure F. The pure product was obtained as a white solid (48 %). Rf = 0.56 (EtOAc/MeOH 9:1); m.p. 190192 °C; 1H NMR (500 MHz, CDCl3): δ 7.58 (d, J = 8.8 Hz, 1H), 6.96 (d, J = 8.7 Hz, 1H), 5.20 (s, 2H), 4.07 (s, 3H), 2.32 (s, 3H), 2.16 ppm (s, 3H); 13C NMR (125 MHz, CDCl3): δ 195.45, 176.45, 171.14, 169.98, 160.40, 154.64, 136.68, 122.38, 117.50, 114.35, 107.45, 70.61, 57.02, 20.85, 20.05 ppm; IR (neat): vmax = 1721, 1621, 1551, 1420, 1403, 1021, 920, 852, 767 cm−1; HRMS (ESI): calcd for C30H27O12 [M+H+]: 579.1487; found: 579.1472.
(1S,1’S)-(3,3’-diacetyl-5-methoxy-4,4’-dioxo-4H,4’H-[8,8’bichromene]-2,2’-diyl)bis(ethane-1,1-diyl) diacetate (35b).
Prepared from the corresponding aryl iodide as described in the general procedure F. The pure product was obtained as a white brown solid (52 %). Rf = 0.60 (EtOAc/MeOH 9:1); m.p. 153–155 °C; [α]20 = −71 (c = 1.0 CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.57 (d, J = 8.6 Hz, 1H), 6.95 (d, J = 8.6Hz, 1H), 5.71 (q, J = 5.30 Hz, 1H), 4.06 (s, 3H), 2.23 (s, 3H), 2.06 (s, 3H), 1.59 ppm (d, J = 6.8 Hz 3H); 13C NMR (125 MHz, CDCl3): δ 200.37, 176.11, 170.95, 168.66, 160.40, 154.75, 136.36, 123.48, 117.70, 114.40, 107.28, 75.74, 57.02, 21.01, 19.67, 16.69 ppm; IR (neat): vmax = 1718, 1643, 1622, 1607, 1551, 1418, 1078, 903, 760, 745 cm−1; HRMS (ESI): calcd for C32H31O12 [M+H+]: 607.1810; found: 607.1804.
(1S,1’S)-(3,3’-diacetyl-5-methoxy-4,4’-dioxo-4H,4’H-[8,8’bichromene]-2,2’-diyl)bis(2-methylpropane-1,1-diyl) diacetate (35c).
Prepared from the corresponding aryl iodide as described in the general procedure F. The pure product was obtained as a waxy brown solid (40 %). Rf = 0.35 (EtOAc/MeOH 9.5:0.5); m.p. 86–88 °C; [α]20 = −26 (c = 1.0 CHCl3); 1H NMR (500 MHz, CDCl3):δ 7.56 (d, J = 8.6 Hz, 1H), 6.94(d, J = 8.7 Hz, 1H), 5.95 (d, J = 3.3Hz ), 4.05 (s, 3H), 2.47 (m, 1H), 2.25 (s, 3H), 2.13 (s, 3H), 1.08 (d, J = 6.9 Hz, 3H), 0.89 ppm (d, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 198.19, 175.62, 170.87, 168.634, 160.42, 154.57, 136.29, 123.23, 117.53, 107.34, 83.21, 57.01, 29.28, 25.19, 20.94, 19.83, 16.84 ppm; IR (neat): vmax = 1744, 1632, 1611, 1607, 1455, 1483, 1070, 754, 761, 741 cm−1; HRMS (ESI): calcd for C36H39O12 [M+H+]: 663.2436; found: 663.2430
Cell culture:
Jurkat and L5178Y cells were cultured in RPMI 1640 (ATCC), HL60 cells were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM, ATCC), KB and MCF7 cells were cultured in Eagles Minimum Essential Medium (EMEM, ATCC), and SKOV3 cells in McCoy’s 5a Medium (ATCC) supplemented with 10 % Heat Inactivated Fetal Bovine Serum (FBS), and Penicillin and Streptomycin (P/S) at 37 oC in a 5 % CO2 incubator.
Cytotoxicity assays:
Proliferating cells were seeded at approximately 20 × 103 cells per 50 μL per well into 96 well plates using a MultiFlo FX multimode dispenser (BioTek). On reaching ~40% cell density, the cells were then incubated with varying concentrations (195 nM to 200 μM final) of test samples using a Biomek FXP Automated Workstation (Beckman Coulter). 48 hours post drug incubation, cell viability was determined using Promega’s CellTiter-Glo Luminescent Cell Viability Assay according to the manufacturer’s protocol on a plate reader (EnVision from PerkinElmer) in Luminescence mode. In each experiment, each unique condition (i.e., different sample type and concentration) was tested in triplicate. The percentage of test sample induced toxicity was calculated with respect to DMSO treated cells, and graphed to give dose response curves. IC50 values for each treatment were calculated using GraphPad Prism 8.0 Software.
Supplementary Material
Acknowledgements
The National Institutes of Health are gratefully acknowledged for providing funds for our studies (5 R35 GM124804). We are thankful that E.V. was able to participate in these studies as part of a National Science Foundation Research Experience for Undergraduates at WPI (1659529).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1]. For recent reviews on dimeric chromanones and tetrahydroxanthones, see:; a) Wezeman T, Brase S, Masters K. Nat. Prod. Rep 2015, 32, 628; [DOI] [PubMed] [Google Scholar]; b) Masters K, Brase S, Chem. Rev 2012, 112, 3717–3776. [DOI] [PubMed] [Google Scholar]
- [2].Kikuchi H, Isobe M, Sekiya M, Abe Y, Hoshikawa T, Ueda K, Kurata S, Katou Y, Oshima Y Org. Lett 2011, 13, 4624–4627. [DOI] [PubMed] [Google Scholar]
- [3].a) Isaka M, Jaturapat A, Rukserre K, Damwisetkanjana K, Tanticharoen M, Thebtaranonth Y. J. Nat. Prod 2001, 64, 1015–1018; [DOI] [PubMed] [Google Scholar]; b) Ronsberg D, Debbab A, Mandi A, Vasylyeva V, Bohler P, Stork B, Engelke L, Hamcher A, Sawadogo R, Diederich M, Wray V, Lin M W.. Kassack C. Janiak S. Scheu, Wesselborg S, Kurtan T, Aly A, Proksch P. J. Org. Chem 2013, 78, 12409–12425. [DOI] [PubMed] [Google Scholar]
- [4].Wagenaar M, Clardy J. J. Nat. Prod 2001, 64, 1006–1009 [DOI] [PubMed] [Google Scholar]
- [5].Cao S, McMillin D, Tamyo G, Delmore J, Mitsiades C, Clardy J. J. Nat. Prod 2012, 75, 793–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Steward M, Capon R, White J, Lacey E, Tennant S, Gill J, Shaddock M. J. Nat. Prod 2004, 67, 728–730. [DOI] [PubMed] [Google Scholar]
- [7].Shim S, Baltrusaitis J, Gloer J, Wicklow D. J. Nat. Prod 2011, 74, 395401. [DOI] [PubMed] [Google Scholar]
- [8].For recent syntheses of naturally occurring dimeric chromenones and tetrahydroxanthones, see: [Google Scholar]; a) Wu X, Iwata T, Scharf A, Reichl K, Porco J. J. Am. Chem. Soc 2018, 140, 5969–5975; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ganapathy D, Reiner J, Valdomir G, Senthilkumar S, Tietze L. Chem. - Eur. J 2017, 23, 2299–2302; [DOI] [PubMed] [Google Scholar]; c) Qin T, Iwata T, Ransom T, Beutler J, Porco J. J. Am. Chem. Soc 2015, 137, 15225–15233; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Qin T, Skraba-Joiner S, Khalil Z, Johnson R, Capon R, Porco J. Nature Chem 2015, 7, 234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Geiger L, Nieger M, Brase S. Adv. Synth. Catal 2017, 359, 3421–3427. [Google Scholar]
- [10].For a review see: Nibbs A, Scheidt K. Eur. J Org. Chem 2012, 449–462. For select examples of enantioselective 2-alkylchromanone synthesis see: [DOI] [PMC free article] [PubMed] [Google Scholar]; a) Rao A, Gaitonde A, Prakash S, Rao Tetrahedron Lett S.. 1994, 35, 6347–6350, [Google Scholar]; b) Kawasaki N, Kakuda H, Goto M, Kawabata S, Kometani T. Tetrahedron: Asymmetry 2003, 14, 1529–1534, Biddle M, Lin M, Scheidt K. J. Am. Chem. Soc 2007, 129, 3830–3831, [Google Scholar]; c) Termath A, Sebode H, Schlundt W, Stemmler R, Netscher T, Bonrath W, Schmalz H. Chem. – Eur. J 2014, 20, 12051–12055; [DOI] [PubMed] [Google Scholar]; d) Brown M, Degrado S, Hoveyda A. Angew. Chem. Int. Ed 2005, 44, 5306–5320, [DOI] [PubMed] [Google Scholar]; e) Vila C, Hornillos V, Fananas-Mastral M, Feringa B. Chem. Commun 2013, 49, 5933–5935, [DOI] [PubMed] [Google Scholar]; f) Hardman-Baldwin A, Visco M, Wieting J, Stern C, Kondo S, Mattson A. Org. Lett 2016, 18, 3766–3769, [DOI] [PubMed] [Google Scholar]; g) Fischer T, Bamberger J, Gomez-Martinez M, Piekarski D, Mancheno O. Angew. Chem. Int. Ed 2019, 58, 3217–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Newman D, Cragg G. J. Nat. Prod 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]
- [12].Wender P, Verma V, Paxton T, Pillow T. Acc. Chem. Res 2007, 41, 40–49 [DOI] [PubMed] [Google Scholar]
- [13].Wetzel S, Bon R, Kumar K, Waldmann H. Angew. Chem. Int. Ed 2011, 50, 10800–10826. [DOI] [PubMed] [Google Scholar]
- [14].Parker C, Galmozzi A, Wang Y, Correia B, Sasaki K, Joslyn C, Kim A, Cavallaro C, Lawrence R, Johnson S, Narvaiza I, Saez E, Cravatt B. Cell 2017. 168, 527–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pahl A, Waldmann H, Kumar K. Chimia 2017, 71, 653–660. [DOI] [PubMed] [Google Scholar]
- [16].Zhang W, Krohn K, Zia-Ullah U. Florke G. Pescitelli, Bari DL, Antus S, Kurtan T, Rheinheimer J, Draeger S, Schulz B. Chem. Eur. J 2008, 14, 4913–4923. [DOI] [PubMed] [Google Scholar]
- [17].Tabata N, Tomoda H, Matsuzaki K, Omura S. J. Am. Chem. Soc 1993, 115, 8558–8564. [Google Scholar]
- [18].Wu G, Yu G, Kurtan T, Mandi A, Peng J, Mo X, Liu M, Li H, Sun X, Li J, Zhu T, Gu Q, Li D. J. Nat. Prod 2015, 78, 2691–2698. [DOI] [PubMed] [Google Scholar]
- [19].Arunpanichlert J, Rukachaisirikul V, Tadpetch K, Phongpaichit S, Hutadilok-Towatana N, Sapaphon O, Sakayaro J. Phytochemistry Lett 2012, 5, 604–608. [Google Scholar]
- [20].Kikuch H, Hoshikawa T, Kurata S, Katou Y, Oshima Y. J. Nat. Prod 2016, 79, 1259–1266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.