

Abstract
Patients with systemic lupus erythematosus (SLE) frequently show symptoms of central nervous system (CNS) involvement, termed neuropsychiatric SLE (NPSLE). The CNS manifestations of SLE are diverse and have a broad spectrum of severity and prognostic implications. Patients with NPSLE typically present with nonspecific symptoms, such as headache and cognitive impairment, but might also experience devastating features, such as memory loss, seizures and stroke. Some features of NPSLE, in particular those related to coagulopathy, have been characterized and an evidence-based treatment algorithm is available. The cognitive and affective manifestations of NPSLE, however, remain poorly understood. Various immune effectors have been evaluated as contributors to its pathogenesis, including brain-reactive autoantibodies, cytokines and cell-mediated inflammation. Additional brain-intrinsic elements (such as resident microglia, the blood–brain barrier and other neurovascular interfaces) are important facilitators of NPSLE. As yet, however, no unifying model has been found to underlie the pathogenesis of NPSLE, suggesting that this disease has multiple contributors and perhaps several distinct aetiologies. This heterogeneity presents a challenge for clinicians who have traditionally relied on empirical judgement in choosing treatment modalities for patients with NPSLE. Improved understanding of this manifestation of SLE might yield further options for managing this disease.
The nervous system is one of the major organs affected in patients with systemic lupus erythematosus (SLE). Research interest in neuropsychiatric SLE (NPSLE; formerly termed lupus cerebritis) has seen major growth during the past 5 years, which is largely attributable to the understanding that NPSLE develops along unique pathogenetic pathways compared with other SLE manifestations. One challenge that clinicians often face in the diagnosis and management of patients with NPSLE is that its presentation can be highly variable, ranging from common and nonspecific features, such as headache, cognitive abnormalities and mood disorders, to rare presentations including Guillain–Barré syndrome and autonomic dysfunction1.
The true prevalence of NPSLE is unknown, but published estimates suggest that it affects between 12% and 95% of patients with SLE1–4 (Table 1). This wide range is probably due to differences in study design, inclusion and exclusion criteria and inconsistency in the attribution of neuropsychiatric presentations to SLE. For example, the protocols of some studies exclude headache because it is essentially ubiquitous in the general population. Nevertheless, even when minor neuropsychiatric symptoms are excluded, CNS disease can be conservatively estimated to occur in >20% of patients with SLE3. In fact, NPSLE is a major source of morbidity in the SLE population, and its mortality is second only to that of lupus nephritis.
Table 1 |.
Prevalence of neuropsychiatric features in patients with SLE
| Feature | Source | Prevalence (%) | Refs |
|---|---|---|---|
| Diffuse psychiatric or neuropsychological syndromes | |||
| Psychosis | CNS | 0.6–11.0 | 1,6,7,206 |
| Anxiety disorder | CNS | 6.4–40.0 | 1,6,206,207 |
| Acute confusional state | CNS | 0.9–7.0 | 1,6,7,206 |
| Mood disorder | CNS | 7.4–65.0 | 1,6,7,206,207 |
| Cognitive impairment | CNS | 6.6–80.0 | 1,6,7,206 |
| Neurological syndromes | |||
| Headache (including migraine and benign intracranial hypertension) | CNS | 12.2–28.3 | 1,6,7,206 |
| Seizure disorders | CNS | 7.0–20.0 | 1,6,7,206 |
| Cerebrovascular disease | CNS | 8.0–15.0 | 1,6,7,206 |
| Movement disorder (chorea) | CNS | 0.9 | 1,6,206,208 |
| Myelopathy | CNS | 0.9–3.9 | 1,6,7 |
| Demyelinating syndrome | CNS | 0.9–2.7 | 1,6,7,206,209,210 |
| Aseptic meningitis | CNS | 0.3–2.7 | 1,6,7,206 |
| Acute inflammatory demyelinating polyradiculopathy (Guillain–Barré syndrome) | PNS | 0.08–1.20 | 1,6,7 |
| Autonomic disorder | PNS | 0.08–1.30 | 1,6,7 |
| Mononeuropathy, single or multiplex | PNS | 0.9–6.9 | 1,6,7 |
| Plexopathy | PNS | NR | 6,7 |
| Polyneuropathy | PNS | 1.5–5.4 | 1,6,7 |
| Myasthenia gravis | PNS | 0.2 | 1,6 |
| Cranial neuropathy | PNS | 1.0 | 1,6,7 |
Some central nervous system (CNS) and most peripheral nervous system (PNS) manifestations are uncommonly studied, with uncertain and/or rare prevalence. NR, not reported; SLE, systemic lupus erythematosus. References cited here discuss the particular presentation and/or its prevalence in SLE.
Our understanding of NPSLE is confounded by several complexities: its presentation is diverse and often elusive; it probably encompasses a variety of pathogenetic mechanisms (including thrombosis, autoantibodies, cytokines and cell-mediated inflammation); and its management options are still inadequately optimized. Nevertheless, SLE research in general has greatly benefited from extraordinary progress in the understanding of its immunological mechanisms over the past decade, as well as from advances in investigational techniques that have accelerated our ability to study this complex and multifactorial disease. NPSLE in particular, owing to its heterogeneous and intricate pathogenetic mechanisms, has experienced a resurgence of interest and renewed investigational efforts.
In this Review, we present the current understanding of NPSLE and explore new ideas and directions in the study and management of this complex condition.
Clinical manifestations
NPSLE encompasses a variety of neurological and psychiatric signs and symptoms that are often hard to distinguish from SLE-unrelated events. The central nervous system (CNS) manifestations of SLE range from subtle cognitive dysfunction, which occurs in up to 50% of patients with SLE (even in those with mild disease and without overt NPSLE manifestations5), to acute confusional states, psychosis, seizure disorders and stroke1. In 1999, an ad hoc committee of the ACR released a consensus statement in which 19 neuropsychiatric syndromes that can occur in patients with SLE were defined6. Of these, 12 syndromes were related to the CNS and 7 were peripheral nervous system manifestations. These were further classified into diffuse psychiatric or neuropsychological syndromes and focal neurological syndromes (Table 1).
Focal NPSLE represents localized CNS involvement, which is generally attributable to episodes of either venous thrombosis or arterial ischaemia. These events are thought to account for ~20% of NPSLE cases7,8, but reported rates range from 3% to 43%, probably because of differences in the stringency of attribution models for NPSLE in the various studies9,10. These events are predominantly attributable to thromboembolic phenomena that occur in the context of SLE-related hypercoagulable states and are clearly correlated with the presence of anti-phospholipid (aPL) antibodies4,7–16. True vasculitis as an underlying cause of cerebrovascular disease, however, is a rare cause of focal or diffuse NPSLE manifestations17.
Although headache, mood disorders, anxiety and mild cognitive dysfunction are the most frequent neuropsychiatric complaints in patients with SLE, they typically do not reflect disease activity in the CNS7. Cerebrovascular disease, seizure disorders, acute confusional states and neuropathies are the most common presentations of NPSLE7,9,18. These myriad presentations and their varying intensities suggest that multiple pathogenetic mechanisms are involved in NPSLE, similar to our current understanding of the extracranial manifestations of SLE12.
Attribution
Linking neuropsychiatric manifestations directly to SLE can often present a challenge, as the symptoms themselves can be mild and nonspecific (such as headaches and ‘brain fog’ or mild cognitive dysfunction) and no specific objective histological, laboratory or imaging findings can confirm the diagnosis. Many of the neuropsychiatric presentations found in patients with SLE also occur frequently in the general population. Moreover, risk factors for these features are associated with conditions that are frequently comorbid with SLE, such as cerebrovascular disease in patients with hypertension, diabetes or hyperlipidaemia. Some NPSLE manifestations, primarily the affective ones, can be explained by either the emotional burden of having a chronic disease or the adverse effects of commonly used medications such as corticosteroids. Many SLE-related neuropsychiatric events occur at disease onset or within the first 1–2 years after diagnosis10,19. Similarly, although neuropsychiatric presentations occurring in close proximity to SLE diagnosis or flare are probably a primary manifestation of SLE (that is, directly caused by the disease rather than being attributable to secondary causes), they can occur independently of overt SLE activity, which again complicates the diagnostic process.
Neuroimaging has inconsistent utility in identifying primary CNS involvement in SLE. The current standard of care is MRI, which can identify intracranial abnormalities and assess their chronicity and evolution in addition to ruling out other causes of the neuropsychiatric symptoms such as abscesses, infectious meningitis and mycotic aneurysms. MRI is especially sensitive for identifying haemorrhagic and ischaemic infarction and transverse myelitis but does not currently have the spatial resolution needed to detect microvascular involvement (which is known to be present in up to 42% of patients with SLE who have neurological manifestations)20,21. In circumstances when conventional MRI is insufficient to identify a suspect lesion, we recommend the use of advanced imaging modalities if available, such as quantitative MRI, single photon emission computed tomography (SPECT) or PET19. The radiological modalities in current use or in development that have shown potential in NPSLE have been reviewed in depth elsewhere22. However, the specificity of neuroimaging for NPSLE (60–82% for MRI19) remains inadequate even for these advanced modalities, and this circumstance, along with the not-infrequent absence of overt CNS lesions in patients with suspected NPSLE, relegates the current utility of neuroimaging to adjunct use. Neuroimaging is helpful mostly to rule out other possible causes of the presentation but cannot be the sole arbiter of NPSLE diagnosis, which still greatly relies on the skilled clinician.
Establishing whether particular neuropsychiatric symptoms are primary manifestations of SLE (which would suggest a need for increased immunosuppression) or secondary to other factors (such as high-dose corticosteroid treatment or psychological disease burden) certainly has implications for the treatment of affected patients. Several models have been devised to improve the attribution of neuropsychiatric symptoms to SLE (reviewed elsewhere)23. These models take into account factors that have been identified as associated with NPSLE, such as a temporal relationship between the neuropsychiatric syndrome onset and diagnosis or change in activity of SLE, minor versus major syndrome (minor syndromes, such as headaches, are common in the general population, whereas major syndromes are rare but more specific for NPSLE), viable alternative explanations and risk factors specific to SLE, such as aPL antibodies or anti-ribosomal P protein antibodies7,12,19,24–26. However, despite providing a fairly reliable bedside diagnostic tool (positive and negative predictive values are both in the range 70–85%)12,13,23–26, these models are based on statistical predictions and correlation with expert opinion as the gold standard as opposed to objective evidence of NPSLE, which leaves room for persistent diagnostic uncertainty. Therefore, until more reliable and specific diagnostic tools are available, the attribution of neuropsychiatric symptoms to SLE remains a moving target reliant on the clinician’s assessment, index of suspicion and clinical reasoning. Importantly, a careful multidisciplinary approach, including neuropsychological, radiological and laboratory evaluations in addition to the rheumatology work-up, increases the specificity of NPSLE diagnosis and is an important avenue to consider22,27.
Pathogenesis
Animal models of NPSLE
As in many neurological conditions, animal models are the mainstay of basic research in NPSLE because human brain tissue is difficult to come by. Several mouse models are used to study NPSLE, each with its own strengths and weaknesses (Table 2). Most of these models recapitulate diffuse but not focal symptoms of NPSLE alongside a lupus phenotype of varying severity. Additionally, in adoptive transfer studies, neuropathic antibodies introduced into the CNS of otherwise healthy non-autoimmune mice have been used to demonstrate the direct pathogenic effects of specific autoantibodies associated with human NPSLE28–30.
Table 2 |.
Animal models of NPSLE
| Mouse model | Advantages | Disadvantages |
|---|---|---|
| MRL/MpJ-Faslpr/lpr |
|
Complex pathogenic contributors |
| NZB × NZW F1 offspring |
|
|
| 564Igi | Emphasizes the contribution of a particular anti-nuclear antibody (directed against RNA) and increased type I interferon activity to disease development |
Minimizes effects of the adaptive immune response due to an invariant IgG clone |
| C57Bl/6-Faslpr/lpr | The same Fas mutation as in MRL/lpr mice, but on the C57Bl/6 background, which allows for more effective genetic manipulation using existing mutant strains | Disease develops in an attenuated fashion and with incomplete penetrance compared with that in MRL/lpr mice |
IgG, immunoglobulin G; NPSLE, neuropsychiatric systemic lupus erythematosus; NZB, New Zealand black; NZW, New Zealand white; SLE, systemic lupus erythematosus. Other models of SLE with neuropsychiatric disease exist but are less often employed owing to various confounding variables, such as anatomical abnormalities or technical considerations.
The two leading animal models of NPSLE are the New Zealand black/New Zealand white F1 (NZB × NZW F1) and MRL/MpJ-Faslpr/lpr (MRL/lpr) mice. NZB × NZW F1 mice manifest systemic and serological features resembling SLE, such as high titres of anti-nuclear and anti-double-stranded DNA (dsDNA) antibodies31. These mice also develop lupus-like behavioural manifestations along with a CNS cellular infiltrate that peaks between 10 and 18 months of age32. The first major study of neuropsychiatric disease in NZB × NZW F1 mice evaluated postural response and placing reflex behaviours. These behaviours correlate with thalamic and limbic brain regions, consistent with the anatomical structures associated with affective symptoms in patients with diffuse NPSLE32. Subsequently, NZB × NZW F1 mice were shown to develop an aberrant neuroprogenitor cell population in the corpus callosum, along with accelerated immunopathology in the same region33.
MRL/lpr mice are similar to NZB × NZW F1 mice in that both models develop neuropsychiatric features as well as systemic (non-neurological) manifestations of SLE. One disadvantage of the NZB × NZW F1 mouse model, however, is the late onset and slow progression of overt neuropsychiatric disease. By contrast, MRL/lpr mice, which develop SLE as a consequence of a loss-of-function mutation in the Fas gene superimposed on a complex MRL background, show an accelerated course of disease, which peaks at a median of 16 weeks of age34. The MRL/lpr mouse consistently exhibits a range of cognitive and affective manifestations, including memory deficits and depression-like behaviour, both of which resemble traits observed in patients with NPSLE. Moreover, MRL/lpr mice develop many lupus-associated anti-nuclear31 and brain-reactive35 autoantibodies. Although both MRL/lpr and NZB × NZW F1 mice have prominent serological phenotypes, their circulating autoantibody profiles have not been directly compared to assess differences in brain-reactive antibody specificities or neuropathic potential.
Several other, less frequently employed, models of NPSLE are useful for exploring the contribution of individual pathways to disease development. One such mouse is the 564Igi strain, which has an invariant immunoglobulin G (IgG) knock-in with RNA auto-reactivity leading to the development of a lupus-like phenotype, albeit one with slow disease progression36–38. Behavioural deficits and increased neuronal death also occur in B6 mice carrying the same Fas mutation found in MRL/lpr mice (B6-lpr/lpr mice), although this phenotype is probably attenuated compared with that of the MRL/lpr strain39. Of note, none of the mutations and genetic risk factors seen in the above models of NPSLE commonly occur in patients with SLE. Rather, these models recapitulate key clinical manifestations and potential immunological effectors of this disease, which enable the study of specific underlying pathways and therapeutic targets.
Neuroimmune interfaces
The CNS has long been thought to be an immune-privileged site owing to the presence of a highly restrictive and tightly regulated blood–brain barrier (BBB) that prevents passive transfer of most immune mediators from the circulation to the CNS. Therefore, a question (and one that remains to be fully answered) of importance to the discussion of CNS pathology in SLE is whether breach of the BBB is truly central to the pathogenesis of NPSLE, as it has long been considered. Although focal NPSLE is most frequently a consequence of cerebrovascular disease40,41, in which BBB damage results from the mechanical disruption of brain vascular integrity, reperfusion and/or local inflammation42–47, the mechanisms underlying increased BBB permeability in diffuse NPSLE are not yet clear.
Much of what is believed about the loss of BBB integrity in SLE is based on empirical inference and surrogate markers. The presence of serum albumin within the CNS (the cerebrospinal fluid (CSF) serum albumin quotient, or Qalb) has long been employed as the gold-standard surrogate marker of serum leakage into the CNS48,49. Serum and CSF positivity for autoantibodies that might mediate direct neuronal damage, such as anti-N-methyl-d-aspartate receptor (NMDAR)50–52, anti-Smith53 and anti-ribosomal P protein antibodies54–56, correlate with the development of diffuse NPSLE, as well as with an elevated Qalb in some patients52,53, suggesting that these antibodies do penetrate the BBB (which is typically antibody impermeable). As seropositivity for these autoantibodies does not reliably predict NPSLE development57, an additional ‘hit’, such as excessive stress or an underlying infection, might induce temporary BBB disruption and thereby facilitate brain injury by serum-derived auto antibodies. Similarly, in animal models, the pathogenicity of neuropathic autoantibodies has been demonstrated, although always in the context of BBB disruption or bypassing; for example, injections of either adrenaline or lipopolysaccharide (LPS) are used to model stress or infection, respectively29,58–60. Thus, increased permeability of the BBB remains a strong candidate for the mechanism and route by which pathogenic antibodies reach the brain in human NPSLE.
Despite the logical inference that BBB disruption occurs in NPSLE, and the long history of the finding of an elevated Qalb in patients with NPSLE as described above, few studies have specifically probed the matter. Albumin exits from the brain vasculature into the parenchyma by diffusion as well as active and vesicular transport61, which potentially increases its intrathecal concentration without necessarily indicating vascular leakage per se (accordingly, this mechanism remains speculative at this time). Additionally, although brain-reactive autoantibodies within the CNS of patients with SLE might have originated in serum, their presence could also indicate direct intrathecal antibody production by plasma cells. Furthermore, the cerebral vasculature is not the only perfused site within the skull; the meningeal barrier and the choroid plexus (the site of the blood–CSF barrier) also warrant attention as potential sites of neuroimmune interactions. Finally, two fluid-transport mechanisms have been identified in the CNS in the past 5 years: the glymphatic pathway, a perivascular conduit running along the cerebral vasculature that enables coordinated movement of interstitial fluid and CSF62, and the intradural lymphatic network63. Both these systems represent previously under-recognized potential avenues for leukocyte transport into and out of the CNS. The roles of these fluid channels in neuro-immunity in general (and NPSLE in particular) have yet to be fully explored but are promising avenues of investigation (Fig. 1).
Fig. 1 |. Neuroimmune interfaces and postulated mechanisms by which they can be breached.
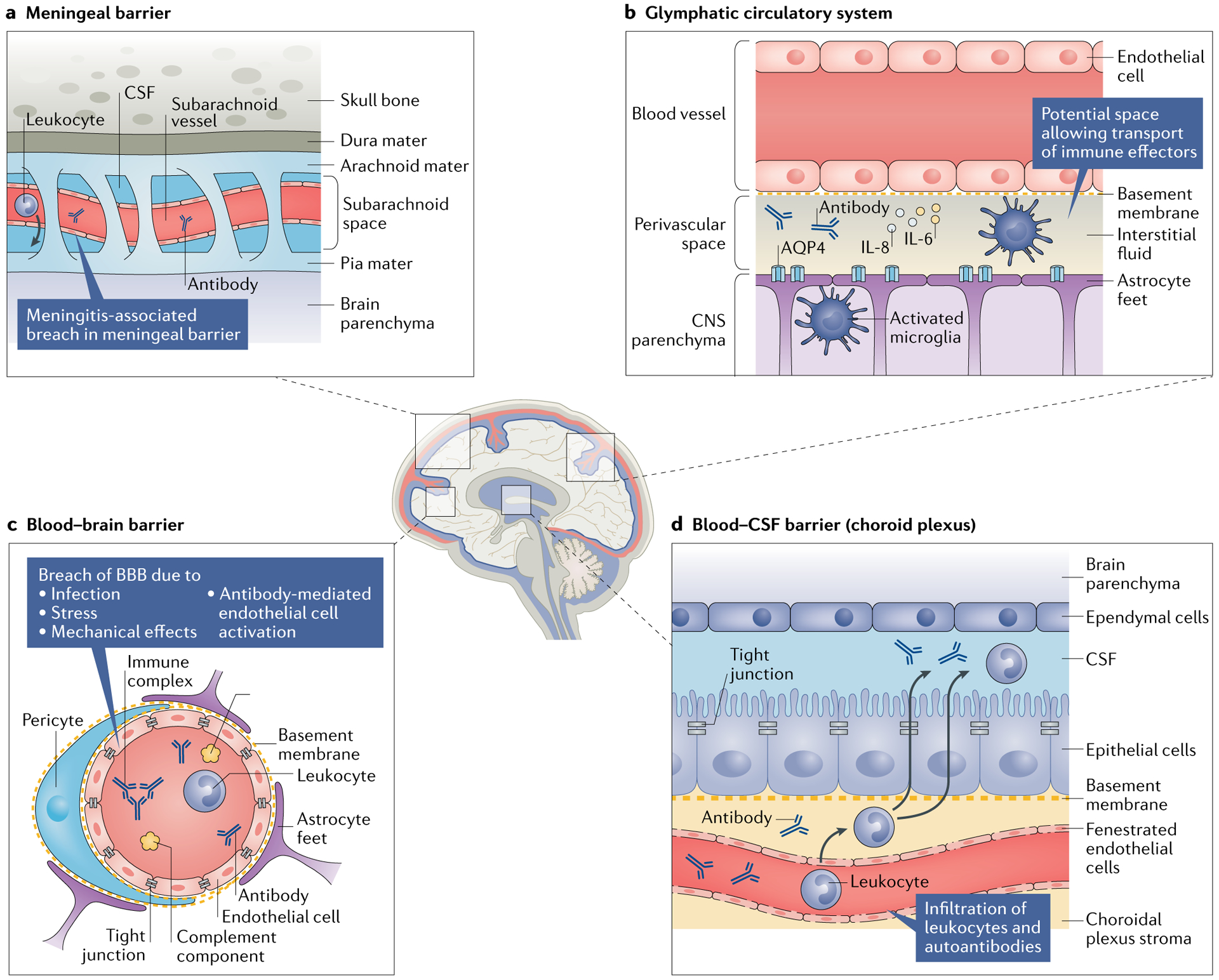
Damage to the blood–brain barrier (BBB) might enable neuropathic antibodies in the serum of patients with systemic lupus erythematosus to enter the central nervous system (CNS); however, several other interfaces could also serve as sites of leukocyte and pathogenic antibody transfer into the CNS. a | The arachnoid epithelium serves as the meningeal barrier between the cerebrospinal fluid (CSF) in the subarachnoid space and the blood in cerebral veins. Meningitis (both aseptic and infectious) can cause inflammation in the subarachnoid space, potentially leading to a breach of this barrier that enables circulating pathogenic antibodies, leukocytes and pro-inflammatory cytokines to enter into the CSF. b | The glymphatic pathway is a perivascular pseudolymphatic system that provides a conduit for interstitial fluid in the brain parenchyma. Brain antigens (such as microtubule-associated protein 2) might be recognized by indwelling antigen-presenting cells in the perivascular space, which can migrate to the cervical lymph nodes and initiate an adaptive response. c | Breach of the BBB has been observed under several circumstances, including infection (simulated by lipopolysaccharide administration), stress (simulated by adrenaline infusion), the mechanical and inflammatory sequelae of vascular occlusion and antibody-mediated endothelial cell activation. d | The fenestrated capillary plexus within the choroid plexus enables ready access of antibodies and leukocytes to the choroidal plexus stroma (a previously characterized site of immunosurveillance) and potentially into the CSF. AQP4, aquaporin 4.
In addition to disruption of the BBB, which can enable serum-borne effectors to penetrate the CNS, other brain structures might serve as locations of serum–CSF interactions. For example, the meningeal barrier can be disrupted in patients with aseptic meningitis either as a primary manifestation of SLE64,65 or by NSAID use, which is common in patients with SLE66. Patients with SLE are also at an increased risk of infectious meningitis owing to the frequent need for corticosteroids and other immunosuppressive treatments in the management of SLE67.
The choroid plexus is another potential site of immune penetrance into the CNS. This structure is regionally situated deep within the CNS, floating within the ventricles and bathed in CSF. The choroid plexus is a secretory epithelial structure surrounding a highly vascularized capillary plexus; uniquely, its endothelium is fenestrated and includes interepithelial tight junctions that serve a barrier function. Additionally, the choroid plexus is the site of CSF production and is the default source of any solutes therein61. The choroid plexus is thought to be the site through which inflammation is initiated in multiple sclerosis68 and is implicated in age-related inflammatory changes associated with cognitive decline69. Studies of the choroid plexus in patients with SLE have revealed evidence of disease involvement, particularly immune complex deposition70–73, although this finding might be nonspecific. Anecdotal evidence in the form of case reports have associated MRI enhancement of the choroid plexus with the onset of cognitive dysfunction74. Of note, in MRL/lpr mice, the choroid plexus epithelium has been identified as a route of entry into the CSF for pathogenic autoantibodies and leukocytes and is a primary site of neuropathology75–79. The contributions of the various brain barriers to the pathogenesis of NPSLE has been extensively reviewed elsewhere80.
Autoantibodies
A hallmark of SLE is the formation of autoantibodies, several of which are implicated in NPSLE manifestations. Indeed, a substantial number of the autoantibodies identified in patients with NPSLE are believed to contribute to the pathogenesis of the disease. Of these, aPL antibodies are thought to be directly (although not exclusively) related to focal NPSLE via autoantibody-mediated thrombosis; others, such as anti-NMDAR and anti-ribosomal P protein antibodies, are considered to target specific brain parenchymal structures and might explain diffuse NPSLE presentations (Table 3).
Table 3 |.
Autoantibodies in NPSLE
| Antibody | Effects in humans | Data from preclinical models | Refs |
|---|---|---|---|
Anti-phospholipid antibodies:
|
|
Intracerebroventricular injection causes neurobehavioural deficits in mice | 16,81–92 |
| Anti-N-methyl-d-aspartate receptor antibodies | CSF positivity is correlated with NPSLE in some patients | Induce cognitive and affective symptoms in mice when introduced intrathecally | 50,58–60,96–103 |
| Anti-ribosomal P protein antibodies | Seropositivity has long been correlated with NPSLE |
|
30,54–56,105–115 |
| Anti-aquaporin 4 antibodies | Associated with demyelination in neuromyelitis optica, found not only in NPSLE but also in SLE without neuropsychiatric features | ND | 116–124 |
| Anti-endothelial cell antibodies | Present in a majority of patients with NPSLE and in many with SLE | Might induce endothelial cell activation and expression of adhesion molecules or cytokines | 125–128 |
| Anti-MAP2 antibodies |
|
MAP2 is exclusively found in neurons | 129,130 |
| Anti-suprabasin antibodies |
|
|
131–133 |
BBB, blood–brain barrier; CNS, central nervous system; CSF, cerebrospinal fluid; MAP2, microtubule-associated protein 2; ND, not discussed; NPSLE, neuropsychiatric systemic lupus erythematosus; SLE, systemic lupus erythematosus.
Anti-phospholipid antibodies.
Anti-phospholipid syndrome (APS) is characterized by thrombosis of the venous or arterial circulation and/or adverse pregnancy outcomes in the presence of persistently elevated titres of one or more aPL antibodies, including lupus anticoagulant (which represents multiple antibodies that inhibit phospholipid-dependent coagulation), anti-cardiolipin antibodies and anti-β2-glycoprotein 1 antibodies. Although this syndrome can occur in isolation, it is much more prevalent in patients with SLE than in the general population (10–44% versus 0.1–5.0%)81. The specific role of aPL antibodies in thrombus formation is still not fully characterized, although they are known to activate endothelial cells, platelets and monocytes, which are then thought to shed prothrombic microparticles81,82.
The CNS is more susceptible than most tissues to thrombus formation, which accounts for the increased risk of stroke and transient ischaemic attack seen in aPL-antibody-positive patients83. In addition, aPL antibodies accelerate atherosclerosis84, which is an independent risk factor for cerebrovascular ischaemia. The risk of stroke in individuals aged <50 years is about eightfold higher in aPL-antibody-positive than in aPL-antibody-negative individuals85. Among patients with SLE, those with aPL antibodies are about twice as likely as aPL-antibody-negative people to develop NPSLE9. Indeed, the presence of these antibodies has long been considered a strong risk factor for NPSLE11,15,19. Interestingly, aPL antibody positivity has also been linked with NPSLE syndromes that are not necessarily directly related to thrombosis, such as seizures, chorea, cognitive dysfunction and myelopathy16,86–91, which suggests that these autoantibodies have a pathogenic role beyond their prothrombotic effects. In vitro, aPL antibodies bind to neurons and other CNS cells, and intrathecal transfer of immunoglobulins from aPL-antibody-positive patients induces cognitive deficits in recipient mice92, thereby supporting a direct effect of these antibodies on the brain. Nevertheless, most of the NPSLE manifestations attributed to aPL antibody seropositivity are thought to be the sequelae of ischaemic events in brain regions such as the amygdala, hippocampus and frontal cortex4. In addition, local vascular injury caused by a thrombotic event can damage the integrity of the BBB, enabling peripheral effectors (such as circulating neuropathic autoantibodies and leukocytes) to enter the CNS44,93.
Anti-NMDAR antibodies.
Autoantibodies mediate several pathological manifestations of SLE. The most well-known association relates to anti-dsDNA antibodies, which are important mediators of lupus nephritis94,95. A subset of anti-dsDNA antibodies cross-reacts with the NR2A and NR2B subunits of NMDARs; binding of these autoantibodies to NMDARs leads to neuronal death, primarily via increased neuronal calcium influx, which mimics glutamate excitotoxicity96,97. These cross-reactive anti-NMDAR antibodies occur in patients with SLE and are frequently associated with NPSLE50,51,60,98. Although serum levels of anti-NMDAR antibodies are not consistently correlated with NPSLE activity25,99–101, CSF titres of these antibodies are higher in patients with active diffuse NPSLE than in individuals with focal NPSLE or non-inflammatory CNS disease99,102. However, a substantial population of patients with non-neuropsychiatric SLE is also anti-NMDAR antibody positive103.
Passive transfer of sera containing anti-NMDAR antibodies from patients with SLE to healthy BALB/c mice induced features of NPSLE, although only after additional disruption of the BBB29. Similarly, immunization of healthy BALB/c mice with the NMDAR-derived DWEYS pentapeptide induced the development of anti-NMDAR antibodies, although these autoantibodies were likewise unable to induce CNS symptoms unless the mice also underwent pharmacological BBB disruption59. Interestingly, LPS treatment (which induces inflammation mimicking a response to infection) led to localization of anti-NMDAR antibodies within the hippocampus, where they caused cognitive impairment, whereas administration of adrenaline (which mimics a stress response) resulted in localization of the antibodies to the amygdala, where they caused affective disturbances29,58,59. These studies collectively indicate that some method by which anti-NMDAR antibodies can enter the CNS is required to induce pathology.
Beyond direct neuronal excitotoxic effects, an intriguing report has described a novel pathogenic effect of anti-NMDAR antibodies in a passive transfer mouse model. Anti-NMDAR antibodies impaired dendritic arborization, which induced deficiencies in spatial recognition; this effect was dependent on locally activated microglia and the presence of complement component C1q. Interestingly, this particular pathogenic mechanism could be attenuated by angiotensin-converting enzyme inhibitors, which deactivate microglia104.
Anti-ribosomal P protein antibodies.
Anti-ribosomal P protein antibodies are highly specific for SLE and are present in up to 46% of patients with SLE105. High titres of these autoantibodies are associated with psychosis54–56,106 and a wide range of NPSLE syndromes, including depression, seizure, coma, transverse myelopathy and aseptic meningitis107,108. Adoptive transfer studies of anti-ribosomal P protein antibodies from patients with SLE in non-autoimmune mice models support a role for these antibodies in depression109 and memory impairment30. Immunohistochemical evaluation of brains from mice injected with anti-ribosomal P protein antibodies found that these antibodies bind to the hippocampus, cingulate cortex and piriform cortex, which are all part of the limbic system and are implicated in mood107,109. Interestingly, binding of anti-ribosomal P protein antibodies to olfactory structures caused a reduced sense of smell92,110, which might represent another mechanism by which anti-ribosomal P protein antibodies cause depression111,112. This observation suggests that interconnectivity exists between the environment (smell), immunity and mental health.
Anti-ribosomal P protein antibodies bind to the carboxy-terminal regions of three ribosomal P proteins, P1, P2 and P0, at a binding site termed the P epitope113. These autoantibodies cross-react with a P epitope found on neuronal surface P antigen (NSPA), a large integral plasma membrane protein found exclusively in neurons that acts as an ubiquitin ligase that regulates the function of NMDAR at the synaptic region. NSPA is also involved in synaptic transmission and plasticity related to memory in the hippocampus and mediates the deleterious effects of anti-ribosomal P protein antibodies on these processes114.
In vitro, anti-ribosomal P protein antibodies bind directly to human peripheral blood monocytes to induce TNF production115. The release of TNF could promote local CNS inflammation and result in BBB impairment, which enables infiltration of the antibodies into the CNS where they cause direct neuronal damage.
Anti-aquaporin 4 antibodies.
Aquaporin 4 (AQP4) is a water channel protein that is expressed on astrocytic foot processes surrounding blood vessels, thereby controlling the flow of water into and out of the brain116. Anti-AQP4 antibodies cause astrocyte toxicity, particularly in the optic nerve and white matter structures of the spinal cord, and are thought to underlie the pathogenesis of neuromyelitis optica (NMO)116–118. The diagnoses of NMO and SLE overlap to some extent, as some patients with NMO eventually develop SLE, and a minority of patients with SLE develop the demyelinating lesions typical of NMO. Anti-AQP4 antibodies occur in 70–90% of patients with NMO119,120. In one study, anti-AQP4 antibodies were detected in 3% of all patients with NPSLE121. However, they were present in 27% of patients with NPSLE who had demyelinating lesions121. Similar to other circulating neuronal antibodies, anti-AQP4 antibodies must penetrate the BBB to induce injury (in Lewis rats)122. Interestingly, and specifically in patients with NMO, an additional autoantibody directed against glucose-regulated protein 78 (which is a component of the brain cerebral vasculature) can disrupt the BBB, allowing the penetration of anti-AQP4 antibodies into the CNS123. Importantly, some patients with SLE remain positive for anti-AQP4 antibodies for many years without developing NMO or any other neurological symptoms124.
Anti-endothelial cell antibodies.
Anti-endothelial cell antibodies (AECAs) are found in >60% of patients with NPSLE (compared with ~30% of patients with SLE who do not have neuropsychiatric involvement) and are associated with psychosis and depression125,126. AECAs induce the expression of adhesion molecules in endothelial cells, which enhances leukocyte rolling and adhesion to vessel walls, in addition to inducing endothelial cell secretion of various cytokines, such as IL-6 and IL-8 (reF.127). This activation of endothelial cells might contribute to cerebral vasculopathy, which, in turn, mediates the neuropsychiatric symptoms of SLE. Finally, molecular cloning studies have shown that some AECAs cross-react with the ribosomal P epitope128, although other studies have not found any such association between AECAs and anti-ribosomal P protein antibodies125.
Anti-microtubule-associated protein 2 antibodies.
Microtubule-associated protein 2 (MAP2) is a cytoskeletal protein found exclusively in neurons. Anti-MAP2 antibodies have been found in the sera of patients with NPSLE, and, although NPSLE can occur in their absence, their presence in the CSF of patients with SLE correlates closely with the presence of neurological symptoms129,130. However, the pathogenic roles and biomarker capabilities of these antibodies need to be further explored.
Anti-suprabasin antibodies.
Suprabasin is a protein secreted specifically in stratified epithelium and is thought to be an epidermal differentiation marker131,132. The CSF of patients with NPSLE was compared with that of patients with non-neuropsychiatric SLE, multiple sclerosis and normal-pressure hydrocephalus in a pro-teomic study that aimed to identify the target antigens of circulating immune complexes. Titres of anti-suprabasin antibodies were higher in the patients with NPSLE than in any of the other groups. In vitro, exposure of astrocytes to anti-suprabasin antibodies activated senescence and autophagy pathways, which might provide some insight into the pathogenesis of the neuropsychiatric symptoms mediated by these antibodies133.
Cell-mediated inflammation
Evidence obtained in both mouse models and humans, albeit preliminary, indicates that cell-mediated inflammation is involved in NPSLE. MRL/lpr mice show increased rolling and adhesion of leukocytes in the cerebral vasculature, which is attenuated by α-integrin and vascular cell adhesion molecule 1 (VCAM1) blockade134. The leukocyte population within the cerebral vasculature (although not that in the brain parenchyma) contained substantial numbers of double-negative T cells76,134. This T cell subset is important in SLE, particularly in renal disease activity135,136, but its role in NPSLE has not been defined. In addition, MRL/lpr mice show prominent cellular infiltration through the choroid plexus, including of CD4+ T helper cells and plasma cells that can mediate CNS inflammation79,80,137. These observations could explain the presence of intrathecal pathogenic autoantibodies, even without major damage to the BBB79,80,137.
Macrophages are instrumental in SLE138, and activated macrophages are found in affected organs (such as the skin and kidneys) of MRL/lpr mice139,140. Within the brain, resident microglia are the predominant immune cells of the CNS and are potent cytokine producers. Levels of type I interferons are elevated in the serum and hippocampus of patients with SLE and induce microglial activation and aberrant synaptic pruning in mouse models of NPSLE38. Indeed, activated microglia are a feature of several mouse models of lupus49,141,142, and we and others have shown that inhibition of microglial activation attenuates the phenotype of NPSLE in these mice38,104,143.
Information on the nature of the inflammatory process in human NPSLE is scarce, mainly owing to the difficulty of obtaining sufficient brain tissue samples. In the few studies that did evaluate brain pathology in patients with NPSLE, the most striking feature was vasculopathy rather than robust cellular infiltration17,20,144. However, inflammation-like changes were seen in ~7% of patients with NPSLE who underwent brain MRI20, whereas in a different MRI study, selective contrast enhancement of the choroid plexus, seen in two patients with NPSLE145, was reminiscent of the local inflammation found in the choroid plexus of the MRL/lpr mice described above. These MRI findings point to the presence of local CNS inflammatory processes, which are presumably driven by immune cells, but the extent of this cellular inflammation in human NPSLE needs to be further explored.
Cytokine-mediated inflammation
In the MRL/lpr mouse model, early neuropsychiatric changes are accompanied by increased serum levels of multiple cytokines, including IFNγ, IL-10, CXC-chemokine ligand 10 (CXCL10; also known as IP10) and CC-chemokine ligand 2 (CCL2; also known as MCP1), to name several146. Similarly, patients’ serum and intrathecal cytokine environments have been extensively characterized in the search for potential biomarkers. As in SLE itself, IFNα has been implicated as a leading effector of NPSLE pathogenesis. In early work, increased CSF levels of IFNα were found in patients with NPSLE, as well as evidence of IFNα production within the CNS147,148. Additionally, antibodies obtained from the CSF of patients with SLE induce IFNα expression in vitro149. Moreover, therapeutic administration of type I interferons induces psychiatric symptoms, including sickness behaviour associated with depression150,151.
Although studies that looked for associations between IFNα expression and NPSLE have reported equivocal results5,152,153, IFNα is directly involved in aberrant synaptic pruning in a lupus-prone mouse model. Neuropsychiatric manifestations observed in this model were reversible with IFNα inhibition, indicating that IFNα is important in the pathogenesis of NPSLE38. TNF-like weak inducer of apoptosis (TWEAK) has shown promise in mouse models of NPSLE49,154,155, although this cytokine seems to be elevated in the CSF of patients with SLE regardless of whether they have neuropsychiatric symptoms156. Elevated intrathecal levels of IL-6 have been demonstrated in patients with conditions linked to diffuse NPSLE, such as acute confusional state and psychosis; this cytokine could be an interesting diagnostic tool once infection and stroke have been ruled out157–159. Increased levels of various other cytokines are also correlated with the development of NPSLE, including IL-8 (reF.152) and IFNγ160,161, which warrant additional study (Table 4).
Table 4 |.
Cytokines in NPSLE
| Cytokine | Effects | Refs |
|---|---|---|
| TNF |
|
115,211–214 |
| BAFF |
|
215,216,189 |
| IFNα |
|
149–151,217–219 |
| TWEAK |
|
49,156,220 |
| IFNγ |
|
160 |
| IL-6 |
|
152,160,211,221 |
| IL-8 |
|
152,160,211 |
BAFF, B cell activating factor; BBB, blood–brain barrier; CSF, cerebrospinal fluid; NPSLE, neuropsychiatric systemic lupus erythematosus; SLE, systemic lupus erythematosus; TH17 cell, T helper 17 cell; TWEAK, TNF-like weak inducer of apoptosis.
Each of the aforementioned cytokines might prove to be promising targets in the treatment of neuropsychiatric and systemic manifestations of SLE. However, the need to measure cytokine levels in CSF is a major obstacle to the identification of appropriate targets. CSF solute levels are subject to exquisitely complicated regulation61 that results in considerable fluctuation, which makes it challenging to derive accurate CNS levels of any given solute from CSF analyses. Additionally, the additional routes of fluid movement described above (namely, the glymphatic162 and CNS lymphatic systems) further confound CSF analysis as the sole method of evaluating intrathecal molecular composition. The development of new and creative means of evaluating intrathecal solute levels is required.
Complement activity
The role of complement activation in NPSLE is another area of exciting developments in both mouse models and human SLE163. In MRL/lpr mice, complement blockade decreased the expression of inflammatory cytokine and adhesion molecule genes and diminished caspase-mediated apoptosis164. A histopathological evaluation of brain tissue from patients with NPSLE found, in addition to increased vascular pathology and ischaemic changes, strong evidence of local activation of the complement cascade in at least some patients20. The levels of complement components in the CSF probably represent both intrathecal production and leakage from the systemic circulation. After correction for Qalb (to assess intrathecal complement production), the available studies report conflicting results as to whether complement activity is increased in patients with NPSLE165,166. A subsequent study that evaluated CSF levels of C3 indicated an increase specifically in C3 of systemic origin in patients with an acute confusional state, compared with patients who had other diffuse and focal presentations of NPSLE, or SLE without neuropsychiatric features159. Similarly, CSF levels of C5a and C5 were closely correlated with Qalb in patients with NPSLE167. Thus, BBB integrity might be a major driver of CSF complement levels, but whether the observed increases in levels of complement components derived from the systemic circulation are involved in the pathogenesis of NPSLE is yet to be elucidated. C5a itself has been postulated to be involved in disruption of the BBB, which could explain its apparent serological consumption when the BBB is breached in NPSLE167. However, this hypothesis needs to be further studied.
As mentioned above, the importance of C1q in synaptic pruning by microglia has been demonstrated in a mouse model of NPSLE mediated by passive transfer of anti-NMDAR antibodies, but this mechanism has yet to be explored in human disease104. Of note, no clinical trials of complement-related therapies for SLE are currently included in the ClinicalTrials.gov database; nevertheless, anecdotal evidence suggests the potential efficacy of a complement-targeted approach, particularly using eculizumab (a recombinant monoclonal antibody that targets complement component C5), in patients with catastrophic APS168–170 and recurrent NMO171, perhaps hinting at a potential therapeutic role of complement inhibition in specific manifestations of NPSLE.
Brain structural changes
An intriguing area of investigation is the structural changes that occur in the brains of patients with SLE. These changes might explain why neurological manifestations are often discrepant with disease activity, as chronic structural changes would be expected to persist during disease quiescence. Furthermore, some patients with SLE experience cognitive impairment even in the absence of a diagnosis of NPSLE or evidence of active inflammation, suggesting that this symptom is associated with an underlying SLE-related chronic neurological pathology5. Local scarring or damage from a thrombotic event can explain manifestations such as seizures, chorea, myelopathy and cognitive decline. However, these and other manifestations might also occur in patients without a history of microvascular or macrovascular thrombosis and at times when the systemic, non-neurological disease is well controlled.
Although this observation of temporal discrepancy is still unexplained, a number of published findings are of interest and perhaps relevant. Synaptic loss in the brains of patients with SLE might be due to increased IFNα-induced microglial activation38. A similar neuronal deficit in patients with NPSLE might be related to increased CSF levels of the neurite outgrowth inhibitor reticulon 4 (also known as nogo protein); CSF levels of nogo are also elevated in MRL/lpr mice, and nogo inhibits neuronal recovery following damage in the adult CNS172. Inhibition of this pathway promoted myelin repair, in addition to improving cognition and memory, in the MRL/lpr mouse model172. A fascinating study published in 2018 showed (in mouse models) that microglia might retain the ‘memory’ of prior systemic inflammatory stimuli, which alters their subsequent responses to local pathology such as ischaemia or amyloid plaque accumulation173. This memory is generated by epigenetic changes in the microglia that persist for at least 6 months following the systemic trigger. In this study, the aberrant microglial response was shaped by acute systemic inflammatory events, but we consider that similar processes might be relevant to the chronic inflammation associated with SLE.
In patients with rheumatoid arthritis, functional studies of neuronal networks have shown changes in network association patterns that correlated with the degree of inflammation and with pain and fatigue174. Although this evaluation was not performed in patients with SLE, the results suggest that chronic inflammation leads to functional changes in brain connectivity and function, which could explain some manifestations of NPSLE or contribute to their severity. In addition, an elegant MRI study showed disturbed white matter microstructure in the brains of patients with SLE that correlated with disease duration and fatigue but not with SLE disease activity or damage, white matter intensities or depression175.
SLE often manifests at an early age, while the brain is still maturing. During this time, brain structural changes are likely to be particularly devastating, even life-trajectory-altering. Therefore, it is imperative that the pathogenic mechanisms behind these processes are further elucidated (and, if possible, halted) as soon as they are diagnosed. In addition, increased understanding of these changes would improve our ability to recognize and differentiate between acute inflammation-related neuropsychiatric manifestations of SLE, which can be treated with immunosuppressive agents, and chronic processes that might occur independently of systemic disease activity.
Genetic contribution to disease
Although SLE is thought to possess a strong genetic component, SLE is definitely not subject to Mendelian inheritance in the large majority of patients seen in clinical practice. In addition, the considerable variability in its presentation, together with the variety of environmental factors that are probably important in triggering the disease, its manifestations and its severity, make the genetic drivers of SLE hard to trace. NPSLE, with its sometimes vague presentations and difficulty in diagnosis, is similarly challenging to pin down. Nevertheless, several studies of SLE risk alleles identified in genome-wide association studies have found evidence of several single-nucleotide polymorphisms (SNPs) that occur more frequently in patients with NPSLE than in the general SLE population.
Several of these polymorphisms are associated with specific NPSLE syndromes. TREX1 (which encodes 3ʹ repair exonuclease, also known as DNase III) is linked to Aicardi–Goutiéres syndrome and increased expression of type I interferons, and SNPs in this gene are associated with NPSLE, particularly with seizures176–178. Similarly, the HLA-DRB1*04/*13 genotype and the SNP rs10181656 in STAT4 (encoding signal transducer and activator of transcription 4) have both been associated with strokes in SLE, independently of aPL antibody status and traditional cardiovascular risk factors179,180. Finally, the cumulative effect of SNPs in several genes associated with SLE (including HLADRB1, IRF5, STAT4, BLK, TNFAIP3, TNIP1, FCGR2B and TNFSF13) has been evaluated in Japanese patients with SLE. Having ten or more SNPs increased these patients’ risk of developing neurological manifestations twofold181.
Management
Management of NPSLE can be challenging on many levels, including diagnosis based on obscure symptoms, challenges in attributing them to SLE and the fairly limited and nonspecific treatment armamentarium currently available. Initially, a thorough investigation for alternative causes, such as infection, malignancy, metabolic disorders or adverse effects of medication, should be undertaken. Once these confounders and mimics have been ruled out and the symptoms are considered primary to SLE, the goals of management are twofold. First, symptomatic treatment should be provided: correction of hypertension and metabolic derangements; anti-epileptics for seizures; and anxiolytics, antidepressants, mood-stabilizers or antipsychotics for psychiatric manifestations, as appropriate. At the same time, treatment of the underlying SLE process should be administered according to whether the neuropsychiatric symptoms are attributable to a diffuse, inflammation-driven syndrome or a focal (localized) thromboembolic process19.
Inflammation-induced manifestations
Other than belimumab, no specific targeted biologic treatments are available for SLE. End-organ involvement in patients with SLE is usually treated with high doses of corticosteroids, cyclophosphamide, mycophenolate mofetil and azathioprine. A similar treatment strategy is also used in patients with NPSLE, in which the specific choice of the steroid-sparing agent is based on the clinician’s assessment of disease severity and their clinical experience. Unfortunately, high-level clinical evidence regarding the optimization of NPSLE treatment is strikingly absent. Only a single randomized controlled trial has compared intravenous cyclophosphamide infusions with bimonthly intravenous methylprednisolone in treating patients with severe NPSLE, and this study showed a much better response rate in the cyclophosphamide arm182. In addition, oral cyclophosphamide for 6 months followed by azathioprine maintenance therapy was effective in the treatment of lupus psychosis183. Although the evidence in favour of this approach is still anecdotal, the addition of rituximab (or another anti-CD20 monoclonal antibody) to the NPSLE treatment algorithm warrants consideration. The efficacy of rituximab was evaluated in ten patients with refractory NPSLE, who experienced substantial and rapid improvement in their clinical signs and symptoms, as well as in radiological findings184. Rituximab was also effective and fairly safe in a retrospective study of paediatric patients with NPSLE185. Perhaps of relevance is the observation that rituximab can be beneficial in other inflammatory neurological conditions, such as NMO, anti-NMDAR encephalitis and opsoclonus–myoclonus syndrome186–188. Of note, belimumab has not been evaluated in the treatment of major CNS manifestations189.
Thromboembolic manifestations
As with all aPL-antibody-related thrombosis, lifelong anticoagulation with warfarin is the mainstay of therapy190. The recommended international normalized ratio (INR) target in patients with APS is 2.5–3.0, and in patients with recurrence of thrombosis despite optimal warfarin therapy INR should be kept at 3.0–4.0 (reF.191). Statins can prevent endothelial cell activation secondary to aPL antibodies192, and antimalarial agents are protective against thrombosis in patients with SLE193. As the safety profile of these two medications is favourable, they should be considered in addition to warfarin treatment, especially in patients with recurrent thrombosis.
Low-dose aspirin is recommended in patients with cardiovascular risk factors. Of note, although randomized clinical trials are ongoing, currently the data are insufficient to recommend the use of direct oral anticoagulants (also known as novel oral anticoagulants) to prevent aPL-antibody-mediated thromboembolic events168. In patients with NPSLE and catastrophic APS, pulse corticosteroids, intravenous immunoglobulin infusions and/or plasmapheresis are recommended194. The addition of eculizumab to these treatments was effective in several case reports and small series168–170.
Future directions
The human brain tissue used for research into NPSLE is almost exclusively obtained post-mortem, usually after long-term treatment directed towards both systemic and neuropsychiatric manifestations of SLE, and shows in addition typical age-related neuropathology. Only limited systematic efforts have been made to include brain tissue from patients with NPSLE in brain tissue repositories, which typically focus on primary and CNS-limited disease processes. However, given our evolving understanding of NPSLE and the advances in model organisms, increased efforts to promote the specific recruitment of SLE brain tissue for translational research purposes, beyond local efforts that are already in place, would be useful.
A pressing need remains for additional targeted therapies for SLE, but the varied and complex underlying pathogenic pathways complicate the development of such therapies. Studies of drug treatments for NPSLE in particular, with its challenges in symptom attribution and often severe manifestations, are difficult to execute and therefore have been few and far between. However, a number of agents are currently being studied that might prove promising (Fig. 2).
Fig. 2 |. Pathogenetic mechanisms and potential treatment targets in diffuse NPSLE.
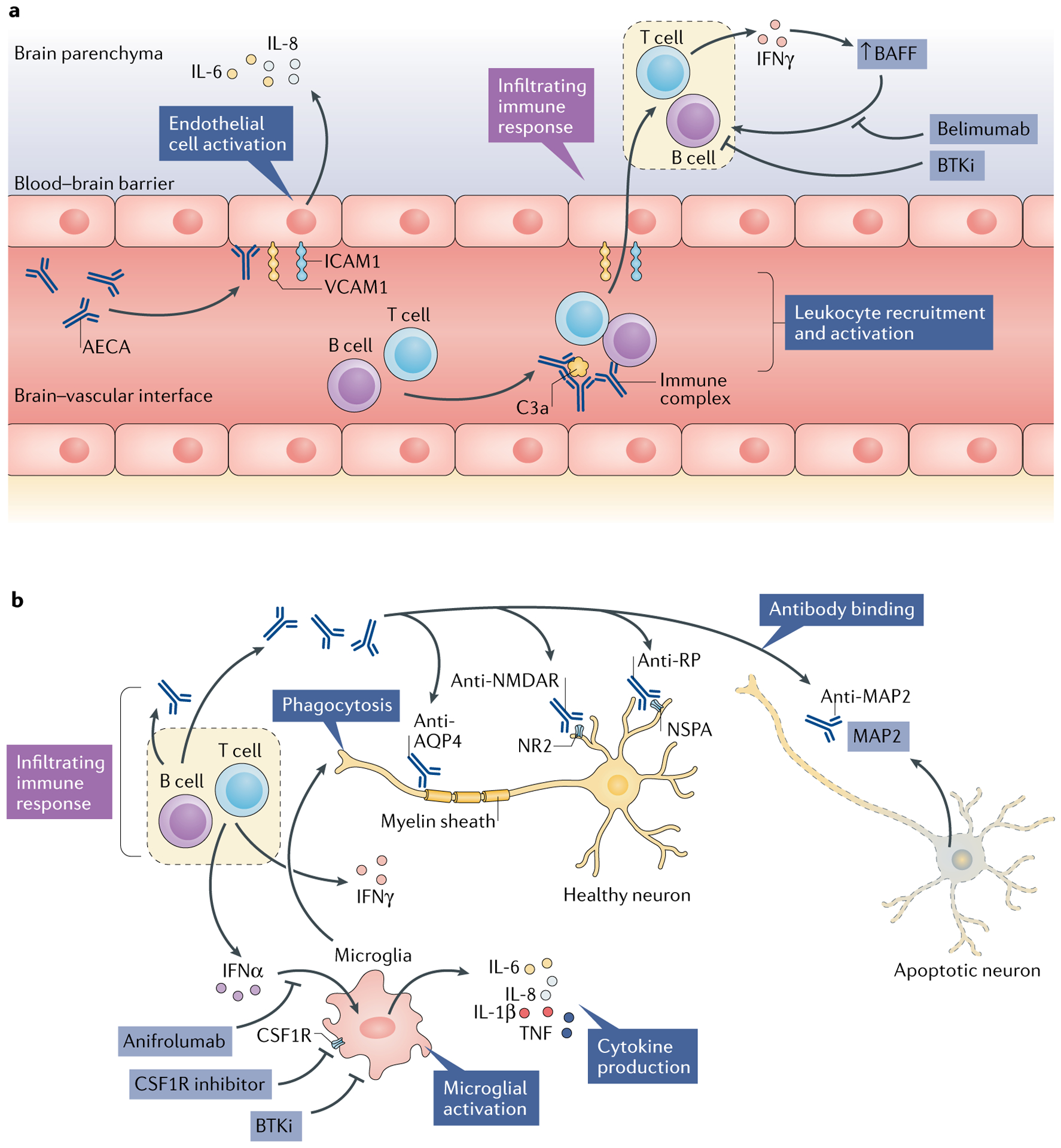
Several different but potentially complementary pathways and effectors in the central nervous system (CNS) microenvironment might be involved in the pathogenesis of neuropsychiatric systemic lupus erythematosus (NPSLE). a | Endothelial cells connected by tight junctions comprise the blood–brain barrier and are subject to activation by autoantibodies such as anti-endothelial cell antibodies (AECAs). Activated endothelial cells show upregulated expression of adhesion molecules (such as intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion molecule 1 (VCAM1)), which facilitate leukocyte infiltration into the CNS parenchyma. In addition, these activated endothelial cells secrete pro-inflammatory cytokines, including IL-6 and IL-8. Concurrently, immune complexes in the cerebral vasculature activate the complement system, which further promotes chemotaxis. The locally infiltrating leukocytes secrete pro-inflammatory cytokines, such as IFNγ, which in turn promotes B cell survival and activation via increasing local levels of B cell activating factor (BAFF). b | Microglia are consequently activated, most notably by interferons, the elevated intrathecal levels of which are commonly seen in patients with NPSLE. Activated microglia further propagate local cytokine and chemokine signalling cascades, in addition to direct phagocytic activity focused on neuronal surface signalling domains and synaptic termini. Finally, several neuropathic autoantibodies have been implicated in NPSLE, including antibodies to the NR2 subunit of the anti-N-methyl-d-aspartate receptor (NMDAR), anti-ribosomal P protein (RP; targeting neuronal surface P antigen (NSPA)) and anti-microtubule-associated protein 2 (MAP2) antibodies, which might have direct neurotoxic effects and provide a source of intrathecal immune complexes. Anti-aquaporin 4 (AQP4) antibodies, directed against the myelin sheath, are found in some patients with SLE who have demyelinating disease. The above pathogenetic mechanisms, along with studies in animal models, suggest that several drugs might prove to be effective in treating NPSLE, including belimumab (a BAFF inhibitor), anifrolumab (a type I interferon receptor antagonist), macrophage colony-stimulating factor-1 receptor (CSF1R) inhibitors (which block activation of microglia and infiltrating macrophages) and tyrosine-protein kinase BTK (also known as Bruton tyrosine kinase) inhibitors (BTKis), which interrupt inflammatory activation of B cells and macrophages or microglia.
Anifrolumab, a type I interferon receptor antagonist, has shown some success in a phase IIb trial in patients with SLE: this treatment led to a substantial reduction in moderate-to-severe disease activity, especially in patients who had a strong type I interferon signature at baseline195. Patients with severe NPSLE were excluded from this study; therefore, these data do not support the efficacy of this drug in NPSLE. However, as previously mentioned, type I interferon receptor inhibition decreased microglia-related synaptic loss and attenuated anxiety-like behaviour and cognitive deficits in 564Igi lupus-prone mice38. Therefore, type I interferon inhibition might have a future role in the treatment of NPSLE, most likely in patients with a strong type I interferon signature.
Macrophage colony-stimulating factor 1 receptor (CSF1R) is an important regulator of both macrophage and microglia function. Signalling through this receptor is crucial for macrophage and microglia development, survival and activation196. In MRL/lpr mice, inhibition of CSF1R signalling reduced the brain expression of pro-inflammatory cytokines and attenuated depression-like behaviour, although no improvement was seen in abnormal spatial recognition197.
Tyrosine-protein kinase BTK (also known as Bruton tyrosine kinase) is important in B cell development, survival and function and in crystallizable fragment (Fc) receptor and Toll-like receptor (TLR) signalling in macrophages and macrophage polarization198–200. Inhibition of BTK improves nephritis in several mouse models of lupus201–203. In addition, treatment with a highly selective BTK inhibitor (BI-BTK-1) decreased microglial activation, reduced choroid plexus cellular infiltration and improved cognitive functioning in MRL/lpr mice143. Of note, the BTK inhibitor ibrutinib is already approved for clinical use in haematological indications such as chronic lymphocytic leukaemia204. Results from ongoing early phase clinical trials of BTK inhibitors in patients with SLE are eagerly awaited.
In addition to the above investigational therapies, most of which are still in preliminary stages of development, other tools are being developed to aid the management and personalized treatment of NPSLE. For example, a multi-modal imaging-based approach for diagnosis of NPSLE has been developed that takes into account both structural and functional information205. Furthermore, biomarkers, mostly in the form of cytokine levels in serum or CSF, are still being pursued for diagnosis and surveillance purposes. Genetic investigations, including gene expression analyses, are also ongoing to accurately characterize the risk of NPSLE and its specific manifestations.
Conclusions
Neuropsychiatric disease is a major cause of morbidity and mortality in patients with SLE, yet many knowledge gaps remain in our basic understanding of NPSLE and its clinical management. As the signs and symptoms of NPSLE vary greatly and can often be nonspecific, it is often challenging to confidently attribute them to SLE; indeed, no ‘gold-standard’ diagnostic method exists. In addition, the pathogenesis of neuropsychiatric disease in patients with SLE is probably multifaceted, complex and possibly unique to specific individuals or subsets of patients. Creation of a unified treatment algorithm for specific syndromes therefore seems unlikely to be possible, further highlighting the importance of a skilled clinician in management of these presentations. Many important questions are still unanswered, such as the mechanisms through which particular autoantibodies exert pathogenic effects, the route or routes through which neuropathic antibodies penetrate the brain, the role of infiltrating leukocytes, the contributions of specific cytokines and chemokines to the neuropathological process and, importantly, the relationship between particular pathogenic factors and specific presentations of NPSLE. Promising research efforts into novel targeted therapies and improved diagnostic tools are continuing, but much work remains to be done to optimize our ability to diagnose, prognosticate and treat NPSLE.
Key points.
Management of neuropsychiatric symptoms in patients with systemic lupus erythematosus (SLE) remains challenging as evidence-based regimens are not generally available.
A pressing need in the management of neuropsychiatric SLE (NPSLE) is the appropriate attribution of symptoms to either primary inflammatory pathology or secondary consequences of the general SLE disease burden.
Research efforts are aggressively pursuing the identification of pathways involved in NPSLE development, along with new therapeutic targets.
Mechanisms at the neuroimmune interface are being studied and might extend beyond the cerebral circulation and the blood–brain barrier to include the blood– cerebrospinal fluid barrier and/or the meningeal barrier.
Novel therapies, including small-molecule inhibitors and biologic agents that target inflammatory pathways, are currently being explored to target NPSLE specifically.
Acknowledgements
N.S. was supported by the Hospital for Special Surgery Research Institute Rheumatology Training Program grant (T32 AR071302). A.D.S. was supported by the Albert Einstein College of Medicine Medical Scientist Training grant (T32-GM007822). C.P. was supported by an R01 grant from the US National Institute of Arthritis and Musculoskeletal Diseases (AR065594).
Competing interests
C.P. declares that he has received research funding from Biogen Idec for studies of the TNF-like weak inducer of apoptosis (TWEAK) pathway and from Boehringer Ingelheim for studies of tyrosine-protein kinase BTK inhibition in animal models of lupus. N.S. and A.D.S. declare no competing interests.
Footnotes
Reviewer information
Nature Reviews Rheumatology thanks S. Hirohata and the other anonymous reviewers, for their contribution to the peer review of this work.
References
- 1.Unterman A et al. Neuropsychiatric syndromes in systemic lupus erythematosus: a meta-analysis. Semin. Arthritis Rheum 41, 1–11 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Ainiala H, Loukkola J, Peltola J, Korpela M & Hietaharju A The prevalence of neuropsychiatric syndromes in systemic lupus erythematosus. Neurology 57, 496–500 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Bertsias GK & Boumpas DT Pathogenesis, diagnosis and management of neuropsychiatric SLE manifestations. Nat. Rev. Rheumatol 6, 358–367 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Borowoy AM et al. Neuropsychiatric lupus: the prevalence and autoantibody associations depend on the definition: results from the 1000 Faces of Lupus cohort. Semin. Arthritis Rheum 42, 179–185 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Kozora E et al. Immune function and brain abnormalities in patients with systemic lupus erythematosus without overt neuropsychiatric manifestations. Lupus 21, 402–411 (2012). [DOI] [PubMed] [Google Scholar]
- 6.The American College of Rheumatology. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 42, 599–608 (1999). [DOI] [PubMed] [Google Scholar]
- 7.Hanly JG et al. Prospective analysis of neuropsychiatric events in an international disease inception cohort of patients with systemic lupus erythematosus. Ann. Rheum. Dis 69, 529–535 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mok CC, Lau CS & Wong RW Neuropsychiatric manifestations and their clinical associations in southern Chinese patients with systemic lupus erythematosus. J. Rheumatol 28, 766–771 (2001). [PubMed] [Google Scholar]
- 9.Ho RC et al. A meta-analysis of serum and cerebrospinal fluid autoantibodies in neuropsychiatric systemic lupus erythematosus. Autoimmun. Rev 15, 124–138 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Steup-Beekman GM et al. Neuropsychiatric manifestations in patients with systemic lupus erythematosus: epidemiology and radiology pointing to an immune-mediated cause. Ann. Rheum. Dis 72 (Suppl. 2), ii76–ii79 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Bujan S et al. Contribution of the initial features of systemic lupus erythematosus to the clinical evolution and survival of a cohort of Mediterranean patients. Ann. Rheum. Dis 62, 859–865 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Govoni M et al. Factors and comorbidities associated with first neuropsychiatric event in systemic lupus erythematosus: does a risk profile exist? A large multicentre retrospective cross-sectional study on 959 Italian patients. Rheumatology (Oxford) 51, 157–168 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Govoni M et al. The diagnosis and clinical management of the neuropsychiatric manifestations of lupus. J. Autoimmun 74, 41–72 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Hanly JG et al. Cerebrovascular events in systemic lupus erythematosus: results from an international inception cohort study. Arthritis Care Res. 70, 1478–1487 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mikdashi J & Handwerger B Predictors of neuropsychiatric damage in systemic lupus erythematosus: data from the Maryland lupus cohort. Rheumatology (Oxford) 43, 1555–1560 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Brey RL, Gharavi AE & Lockshin MD Neurologic complications of antiphospholipid antibodies. Rheum. Dis. Clin. North Am 19, 833–850 (1993). [PubMed] [Google Scholar]
- 17.Ellis SG & Verity MA Central nervous system involvement in systemic lupus erythematosus: a review of neuropathologic findings in 57 cases, 1955–1977. Semin. Arthritis Rheum 8, 212–221 (1979). [DOI] [PubMed] [Google Scholar]
- 18.Hanly JG et al. Prospective study of neuropsychiatric events in systemic lupus erythematosus. J. Rheumatol 36, 1449–1459 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Bertsias GK et al. EULAR recommendations for the management of systemic lupus erythematosus with neuropsychiatric manifestations: report of a task force of the EULAR standing committee for clinical affairs. Ann. Rheum. Dis 69, 2074–2082 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Cohen D et al. Brain histopathology in patients with systemic lupus erythematosus: identification of lesions associated with clinical neuropsychiatric lupus syndromes and the role of complement. Rheumatology (Oxford) 56, 77–86 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Luyendijk J et al. Neuropsychiatric systemic lupus erythematosus: lessons learned from magnetic resonance imaging. Arthritis Rheum. 63, 722–732 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Hanly JG, Kozora E, Beyea S & Birnbaum J Nervous system disease in systemic lupus erythematosus: current status and future directions. Arthritis Rheumatol. 71, 33–42 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Bortoluzzi A, Scire CA & Govoni M Attribution of neuropsychiatric manifestations to systemic lupus erythematosus. Front. Med 5, 68 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanly JG et al. Neuropsychiatric events at the time of diagnosis of systemic lupus erythematosus: an international inception cohort study. Arthritis Rheum. 56, 265–273 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Hanly JG et al. Short-term outcome of neuropsychiatric events in systemic lupus erythematosus upon enrollment into an international inception cohort study. Arthritis Rheum. 59, 721–729 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bortoluzzi A et al. Development and validation of a new algorithm for attribution of neuropsychiatric events in systemic lupus erythematosus. Rheumatology (Oxford) 54, 891–898 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Magro-Checa C et al. Value of multidisciplinary reassessment in attribution of neuropsychiatric events to systemic lupus erythematosus: prospective data from the Leiden NPSLE cohort. Rheumatology (Oxford) 56, 1676–1683 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Castillo-Gomez E et al. All naturally occurring autoantibodies against the NMDA receptor subunit NR1 have pathogenic potential irrespective of epitope and immunoglobulin class. Mol. Psychiatry 22, 1776–1784 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Kowal C et al. Human lupus autoantibodies against NMDA receptors mediate cognitive impairment. Proc. Natl Acad. Sci. USA 103, 19854–19859 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bravo-Zehnder M et al. Anti-ribosomal P protein autoantibodies from patients with neuropsychiatric lupus impair memory in mice. Arthritis Rheumatol. 67, 204–214 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Du Y, Sanam S, Kate K & Mohan C Animal models of lupus and lupus nephritis. Curr. Pharm. Des 21, 2320–2349 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Kier AB Clinical neurology and brain histopathology in NZB/NZW F1 lupus mice. J. Comp. Pathol 102, 165–177 (1990). [DOI] [PubMed] [Google Scholar]
- 33.Leung JW, Lau BW, Chan VS, Lau CS & So KF Abnormal increase of neuronal precursor cells and exacerbated neuroinflammation in the corpus callosum in murine model of systemic lupus erythematosus. Restor. Neurol. Neurosci 34, 443–453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ballok DA Neuroimmunopathology in a murine model of neuropsychiatric lupus. Brain Res. Rev 54, 67–79 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams S, Stafford P & Hoffman SA Diagnosis and early detection of CNS-SLE in MRL/lpr mice using peptide microarrays. BMC Immunol. 15, 23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han JH et al. Expression of an anti-RNA autoantibody in a mouse model of SLE increases neutrophil and monocyte numbers as well as IFN-I expression. Eur. J. Immunol 44, 215–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDonald G et al. Accelerated systemic autoimmunity in the absence of somatic hypermutation in 564Igi: a mouse model of systemic lupus with knocked-in heavy and light chain genes. Front. Immunol 8, 1094 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bialas AR et al. Microglia-dependent synapse loss in type I interferon-mediated lupus. Nature 546, 539–543 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Shi D et al. FTY720 attenuates behavioral deficits in a murine model of systemic lupus erythematosus. Brain Behav. Immun 70, 293–304 (2018). [DOI] [PubMed] [Google Scholar]
- 40.de Amorim LC, Maia FM & Rodrigues CE Stroke in systemic lupus erythematosus and antiphospholipid syndrome: risk factors, clinical manifestations, neuroimaging, and treatment. Lupus 26, 529–536 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Sarbu N et al. Brain abnormalities in newly diagnosed neuropsychiatric lupus: systematic MRI approach and correlation with clinical and laboratory data in a large multicenter cohort. Autoimmun. Rev 14, 153–159 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Merali Z, Huang K, Mikulis D, Silver F & Kassner A Evolution of blood–brain-barrier permeability after acute ischemic stroke. PLOS ONE 12, e0171558 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuntz M et al. Stroke-induced brain parenchymal injury drives blood–brain barrier early leakage kinetics: a combined in vivo/in vitro study. J. Cereb. Blood Flow Metab 34, 95–107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knowland D et al. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 82, 603–617 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rochfort KD & Cummins PM The blood-brain barrier endothelium: a target for pro-inflammatory cytokines. Biochem. Soc. Trans 43, 702–706 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Dimitrijevic OB, Stamatovic SM, Keep RF & Andjelkovic AV Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 38, 1345–1353 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Yepes M et al. A soluble Fn14-Fc decoy receptor reduces infarct volume in a murine model of cerebral ischemia. Am. J. Pathol 166, 511–520 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salahuddin TS, Kalimo H, Johansson BB & Olsson Y Observations on exudation of fibronectin, fibrinogen and albumin in the brain after carotid infusion of hyperosmolar solutions. An immunohistochemical study in the rat indicating long-lasting changes in the brain microenvironment and multifocal nerve cell injuries. Acta Neuropathol. 76, 1–10 (1988). [DOI] [PubMed] [Google Scholar]
- 49.Wen J et al. TNF-like weak inducer of apoptosis promotes blood brain barrier disruption and increases neuronal cell death in MRL/lpr mice. J. Autoimmun 60, 40–50 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fragoso-Loyo H et al. Serum and cerebrospinal fluid autoantibodies in patients with neuropsychiatric lupus erythematosus. Implications for diagnosis and pathogenesis. PLOS ONE 3, e3347 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Omdal R et al. Neuropsychiatric disturbances in SLE are associated with antibodies against NMDA receptors. Eur. J. Neurol 12, 392–398 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Hirohata S, Arinuma Y, Yanagida T & Yoshio T Blood-brain barrier damages and intrathecal synthesis of anti-N-methyl-D-aspartate receptor NR2 antibodies in diffuse psychiatric/neuropsychological syndromes in systemic lupus erythematosus. Arthritis Res. Ther 16, R77 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirohata S, Sakuma Y, Yanagida T & Yoshio T Association of cerebrospinal fluid anti-Sm antibodies with acute confusional state in systemic lupus erythematosus. Arthritis Res. Ther 16, 450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bonfa E et al. Association between lupus psychosis and anti-ribosomal P protein antibodies. N. Engl. J. Med 317, 265–271 (1987). [DOI] [PubMed] [Google Scholar]
- 55.Schneebaum AB et al. Association of psychiatric manifestations with antibodies to ribosomal P proteins in systemic lupus erythematosus. Am. J. Med 90, 54–62 (1991). [DOI] [PubMed] [Google Scholar]
- 56.Nojima Y et al. Correlation of antibodies to ribosomal P protein with psychosis in patients with systemic lupus erythematosus. Ann. Rheum. Dis 51, 1053–1055 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hammer C et al. Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol. Psychiatry 19, 1143–1149 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Huerta PT, Kowal C, DeGiorgio LA, Volpe BT & Diamond B Immunity and behavior: antibodies alter emotion. Proc. Natl Acad. Sci. USA 103, 678–683 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kowal C et al. Cognition and immunity; antibody impairs memory. Immunity 21, 179–188 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Lapteva L et al. Anti-N-methyl-D-aspartate receptor antibodies, cognitive dysfunction, and depression in systemic lupus erythematosus. Arthritis Rheum. 54, 2505–2514 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Tumani H, Huss A & Bachhuber F The cerebrospinal fluid and barriers - anatomic and physiologic considerations. Handb. Clin. Neurol 146, 21–32 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Yang L et al. Evaluating glymphatic pathway function utilizing clinically relevant intrathecal infusion of CSF tracer. J. Transl. Med 11, 107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Louveau A et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baizabal-Carvallo JF, Delgadillo-Marquez G, Estanol B & Garcia-Ramos G Clinical characteristics and outcomes of the meningitides in systemic lupus erythematosus. Eur. Neurol 61, 143–148 (2009). [DOI] [PubMed] [Google Scholar]
- 65.Kakati S, Barman B, Ahmed SU & Hussain M Neurological manifestations in systemic lupus erythematosus: a single centre study from North East India. J. Clin. Diagn. Res 11, OC05–OC09 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yelehe-Okouma M, Czmil-Garon J, Pape E, Petitpain N & Gillet P Drug-induced aseptic meningitis: a mini-review. Fund. Clin. Pharmacol 32, 252–260 (2018). [DOI] [PubMed] [Google Scholar]
- 67.van Veen KEB, Brouwer MC, van der Ende A & van de Beek D Bacterial meningitis in patients using immunosuppressive medication: a population-based prospective nationwide study. J. Neuroimmune Pharmacol 12, 213–218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reboldi A et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol 10, 514–523 (2009). [DOI] [PubMed] [Google Scholar]
- 69.Baruch K et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346, 89–93 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Atkins CJ, Kondon JJ, Quismorio FP & Friou GJ The choroid plexus in systemic lupus erythematosus. Ann. Intern. Med 76, 65–72 (1972). [DOI] [PubMed] [Google Scholar]
- 71.Sher JH & Pertschuk LP Immunoglobulin G deposits in the choroid plexus of a child with systemic lupus erythematosus. J. Pediatr 85, 385–387 (1974). [DOI] [PubMed] [Google Scholar]
- 72.Gershwin ME, Hyman LR & Steinberg AD The choroid plexus in CNS involvement of systemic lupus erythematosus. J. Pediatr 87, 588–590 (1975). [DOI] [PubMed] [Google Scholar]
- 73.Boyer RS, Sun NC, Verity A, Nies KM & Louie JS Immunoperoxidase staining of the choroid plexus in systemic lupus erythematosus. J. Rheumatol 7, 645–650 (1980). [PubMed] [Google Scholar]
- 74.Amaro E Jr & Scheinberg, M. Onset of cognitive dysfunction in systemic lupus erythematosus and selective involvement of the choroid plexus. J. Rheumatol 36, 2554–2555 (2009). [DOI] [PubMed] [Google Scholar]
- 75.Stock A, Wen J, Doerner J & Putterman C Neuropsychiatric lupus occurs independently of systemic autoimmunity. J. Immunol 194, 2 (2015). [Google Scholar]
- 76.James WG, Hutchinson P, Bullard DC & Hickey MJ Cerebral leucocyte infiltration in lupus-prone MRL/MpJ-fas lpr mice — roles of intercellular adhesion molecule-1 and P-selectin. Clin. Exp. Immunol 144, 299–308 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ma X, Foster J & Sakic B Distribution and prevalence of leukocyte phenotypes in brains of lupus-prone mice. J. Neuroimmunol 179, 26–36 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Ballok DA, Ma X, Denburg JA, Arsenault L & Sakic B Ibuprofen fails to prevent brain pathology in a model of neuropsychiatric lupus. J. Rheumatol 33, 2199–2213 (2006). [PMC free article] [PubMed] [Google Scholar]
- 79.Gelb S, Stock AD, Anzi S, Putterman C & Ben-Zvi A Mechanisms of neuropsychiatric lupus: the relative roles of the blood-cerebrospinal fluid barrier versus blood-brain barrier. J. Autoimmun 91, 34–44 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stock AD, Gelb S, Pasternak O, Ben-Zvi A & Putterman C The blood brain barrier and neuropsychiatric lupus: new perspectives in light of advances in understanding the neuroimmune interface. Autoimmun. Rev 16, 612–619 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schreiber K et al. Antiphospholipid syndrome. Nat. Rev. Dis. Primers 4, 18005 (2018). [DOI] [PubMed] [Google Scholar]
- 82.Gao C et al. Thrombotic role of blood and endothelial cells in uremia through phosphatidylserine exposure and microparticle release. PLOS ONE 10, e0142835b (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Giannakopoulos B & Krilis SA The pathogenesis of the antiphospholipid syndrome. N. Engl. J. Med 368, 1033–1044 (2013). [DOI] [PubMed] [Google Scholar]
- 84.Narshi CB, Giles IP & Rahman A The endothelium: an interface between autoimmunity and atherosclerosis in systemic lupus erythematosus? Lupus 20, 5–13 (2011). [DOI] [PubMed] [Google Scholar]
- 85.Kittner SJ & Gorelick PB Antiphospholipid antibodies and stroke: an epidemiological perspective. Stroke 23, I19–22 (1992). [PubMed] [Google Scholar]
- 86.Andrade RM et al. Seizures in patients with systemic lupus erythematosus: data from LUMINA, a multiethnic cohort (LUMINA LIV). Ann. Rheum. Dis 67, 829–834 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Appenzeller S, Cendes F & Costallat LT Epileptic seizures in systemic lupus erythematosus. Neurology 63, 1808–1812 (2004). [DOI] [PubMed] [Google Scholar]
- 88.McLaurin EY, Holliday SL, Williams P & Brey RL Predictors of cognitive dysfunction in patients with systemic lupus erythematosus. Neurology 64, 297–303 (2005). [DOI] [PubMed] [Google Scholar]
- 89.Mok MY et al. Antiphospholipid antibody profiles and their clinical associations in Chinese patients with systemic lupus erythematosus. J. Rheumatol 32, 622–628 (2005). [PubMed] [Google Scholar]
- 90.Sanna G et al. Neuropsychiatric manifestations in systemic lupus erythematosus: prevalence and association with antiphospholipid antibodies. J. Rheumatol 30, 985–992 (2003). [PubMed] [Google Scholar]
- 91.Tomietto P et al. General and specific factors associated with severity of cognitive impairment in systemic lupus erythematosus. Arthritis Rheum. 57, 1461–1472 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Katzav A et al. Antibody-specific behavioral effects: intracerebroventricular injection of antiphospholipid antibodies induces hyperactive behavior while anti-ribosomal-P antibodies induces depression and smell deficits in mice. J. Neuroimmunol 272, 10–15 (2014). [DOI] [PubMed] [Google Scholar]
- 93.Chi OZ, Hunter C, Liu X & Weiss HR Effects of exogenous excitatory amino acid neurotransmitters on blood-brain barrier disruption in focal cerebral ischemia. Neurochem. Res 34, 1249–1254 (2009). [DOI] [PubMed] [Google Scholar]
- 94.Du H, Chen M, Zhang Y, Zhao MH & Wang HY Cross-reaction of anti-DNA autoantibodies with membrane proteins of human glomerular mesangial cells in sera from patients with lupus nephritis. Clin. Exp. Immunol 145, 21–27 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhao Z et al. Cross-reactivity of human lupus anti-DNA antibodies with alpha-actinin and nephritogenic potential. Arthritis Rheum. 52, 522–530 (2005). [DOI] [PubMed] [Google Scholar]
- 96.DeGiorgio LA et al. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat. Med 7, 1189–1193 (2001). [DOI] [PubMed] [Google Scholar]
- 97.Faust TW et al. Neurotoxic lupus autoantibodies alter brain function through two distinct mechanisms. Proc. Natl Acad. Sci. USA 107, 18569–18574 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gao HX et al. Depression is an early disease manifestation in lupus-prone MRL/lpr mice. J. Neuroimmunol 207, 45–56 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arinuma Y, Yanagida T & Hirohata S Association of cerebrospinal fluid anti-NR2 glutamate receptor antibodies with diffuse neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 58, 1130–1135 (2008). [DOI] [PubMed] [Google Scholar]
- 100.Kozora E et al. Antibodies against N-methyl-D-aspartate receptors in patients with systemic lupus erythematosus without major neuropsychiatric syndromes. J. Neurol. Sci 295, 87–91 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Petri M et al. Depression and cognitive impairment in newly diagnosed systemic lupus erythematosus. J. Rheumatol 37, 2032–2038 (2010). [DOI] [PubMed] [Google Scholar]
- 102.Brimberg L et al. Antibodies as mediators of brain pathology. Trends Immunol. 36, 709–724 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Husebye ES et al. Autoantibodies to a NR2A peptide of the glutamate/NMDA receptor in sera of patients with systemic lupus erythematosus. Ann. Rheum. Dis 64, 1210–1213 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nestor J et al. Lupus antibodies induce behavioral changes mediated by microglia and blocked by ACE inhibitors. J. Exp. Med 215, 2554–2566 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Eber T, Chapman J & Shoenfeld Y Anti-ribosomal P-protein and its role in psychiatric manifestations of systemic lupus erythematosus: myth or reality? Lupus 14, 571–575 (2005). [DOI] [PubMed] [Google Scholar]
- 106.Tzioufas AG et al. The clinical relevance of antibodies to ribosomal-P common epitope in two targeted systemic lupus erythematosus populations: a large cohort of consecutive patients and patients with active central nervous system disease. Ann. Rheum. Dis 59, 99–104 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Moscavitch SD, Szyper-Kravitz M & Shoenfeld Y Autoimmune pathology accounts for common manifestations in a wide range of neuropsychiatric disorders: the olfactory and immune system interrelationship. Clin. Immunol 130, 235–243 (2009). [DOI] [PubMed] [Google Scholar]
- 108.Yoshio T et al. Quantification of antiribosomal P0 protein antibodies by ELISA with recombinant P0 fusion protein and their association with central nervous system disease in systemic lupus erythematosus. J. Rheumatol 22, 1681–1687 (1995). [PubMed] [Google Scholar]
- 109.Katzav A et al. Induction of autoimmune depression in mice by anti-ribosomal P antibodies via the limbic system. Arthritis Rheum. 56, 938–948 (2007). [DOI] [PubMed] [Google Scholar]
- 110.Katzav A et al. Anti-P ribosomal antibodies induce defect in smell capability in a model of CNS-SLE (depression). J. Autoimmun 31, 393–398 (2008). [DOI] [PubMed] [Google Scholar]
- 111.Perricone C et al. Smell and autoimmunity: a comprehensive review. Clin. Rev. Allergy Immunol 45, 87–96 (2013). [DOI] [PubMed] [Google Scholar]
- 112.Song C & Leonard BE The olfactory bulbectomised rat as a model of depression. Neurosci. Biobehav. Rev 29, 627–647 (2005). [DOI] [PubMed] [Google Scholar]
- 113.Elkon KB, Parnassa AP & Foster CL Lupus autoantibodies target ribosomal P proteins. J. Exp. Med 162, 459–471 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Segovia-Miranda F et al. Pathogenicity of lupus anti-ribosomal P antibodies: role of cross-reacting neuronal surface P antigen in glutamatergic transmission and plasticity in a mouse model. Arthritis Rheumatol. 67, 1598–1610 (2015). [DOI] [PubMed] [Google Scholar]
- 115.Nagai T, Yanagida T & Hirohata S Anti-ribosomal P protein antibody induces Th1 responses by enhancing the production of IL-12 in activated monocytes. Mod. Rheumatol 21, 57–62 (2011). [DOI] [PubMed] [Google Scholar]
- 116.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS & Hinson SR IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med 202, 473–477 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dellavance A et al. Anti-aquaporin-4 antibodies in the context of assorted immune-mediated diseases. Eur. J. Neurol 19, 248–252 (2012). [DOI] [PubMed] [Google Scholar]
- 118.Verkman AS, Phuan PW, Asavapanumas N & Tradtrantip L Biology of AQP4 and anti-AQP4 antibody: therapeutic implications for NMO. Brain Pathol. 23, 684–695 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Waters P et al. Aquaporin-4 antibodies in neuromyelitis optica and longitudinally extensive transverse myelitis. Arch. Neurol 65, 913–919 (2008). [DOI] [PubMed] [Google Scholar]
- 120.Waters PJ et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 78, 665–671 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mader S et al. Understanding the antibody repertoire in neuropsychiatric systemic lupus erythematosus and neuromyelitis optica spectrum disorder: do they share common targets? Arthritis Rheumatol. 70, 277–286 (2018). [DOI] [PubMed] [Google Scholar]
- 122.Bradl M et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann. Neurol 66, 630–643 (2009). [DOI] [PubMed] [Google Scholar]
- 123.Shimizu F et al. Glucose-regulated protein 78 autoantibody associates with blood-brain barrier disruption in neuromyelitis optica. Sci. Transl. Med 9, eaai9111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Alexopoulos H et al. Anti-aquaporin-4 autoantibodies in systemic lupus erythematosus persist for years and induce astrocytic cytotoxicity but not CNS disease. J. Neuroimmunol 289, 8–11 (2015). [DOI] [PubMed] [Google Scholar]
- 125.Conti F et al. Autoantibody profile in systemic lupus erythematosus with psychiatric manifestations: a role for anti-endothelial-cell antibodies. Arthritis Res. Ther 6, R366–R372 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Song J, Park YB, Lee WK, Lee KH & Lee SK Clinical associations of anti-endothelial cell antibodies in patients with systemic lupus erythematosus. Rheumatol. Int 20, 1–7 (2000). [DOI] [PubMed] [Google Scholar]
- 127.Nara H, Okamoto H, Minota S & Yoshio T Mouse monoclonal anti-human thrombomodulin antibodies bind to and activate endothelial cells through NF-κB activation in vitro. Arthritis Rheum. 54, 1629–1637 (2006). [DOI] [PubMed] [Google Scholar]
- 128.Frampton G et al. Identification of candidate endothelial cell autoantigens in systemic lupus erythematosus using a molecular cloning strategy: a role for ribosomal P protein P0 as an endothelial cell autoantigen. Rheumatology (Oxford) 39, 1114–1120 (2000). [DOI] [PubMed] [Google Scholar]
- 129.Williams RC Jr., Sugiura K & Tan EM Antibodies to microtubule-associated protein 2 in patients with neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 50, 1239–1247 (2004). [DOI] [PubMed] [Google Scholar]
- 130.Yamada Y et al. Antibodies to microtubule-associated protein-2 in the cerebrospinal fluid are a useful diagnostic biomarker for neuropsychiatric systemic lupus erythematosus. Mod. Rheum 26, 562–568 (2016). [DOI] [PubMed] [Google Scholar]
- 131.Matsui T et al. Identification of novel keratinocyte-secreted peptides dermokine-alpha/-beta and a new stratified epithelium-secreted protein gene complex on human chromosome 19q13.1. Genomics 84, 384–397 (2004). [DOI] [PubMed] [Google Scholar]
- 132.Park GT, Lim SE, Jang SI & Morasso MI Suprabasin, a novel epidermal differentiation marker and potential cornified envelope precursor. J. Biol. Chem 277, 45195–45202 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ichinose K et al. Novel anti-suprabasin antibodies may contribute to the pathogenesis of neuropsychiatric systemic lupus erythematosus. Clin. Immunol 193, 123–130 (2018). [DOI] [PubMed] [Google Scholar]
- 134.James WG, Bullard DC & Hickey MJ Critical role of the alpha 4 integrin/VCAM-1 pathway in cerebral leukocyte trafficking in lupus-prone MRL/fas (lpr) mice. J. Immunol 170, 520–527 (2003). [DOI] [PubMed] [Google Scholar]
- 135.Crispin JC et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J. Immunol 181, 8761–8766 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shivakumar S, Tsokos GC & Datta SK T cell receptor alpha/beta expressing double-negative (CD4−/CD8−) and CD4+T helper cells in humans augment the production of pathogenic anti-DNA autoantibodies associated with lupus nephritis. J. Immunol 143, 103–112 (1989). [PubMed] [Google Scholar]
- 137.Jain S, Stock A, Macian F & Putterman C A distinct T follicular helper cell subset infiltrates the brain in murine neuropsychiatric lupus. Front. Immunol 9, 487 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Crispin JC et al. Pathogenesis of human systemic lupus erythematosus: recent advances. Trends Mol. Med 16, 47–57 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chalmers SA et al. Macrophage depletion ameliorates nephritis induced by pathogenic antibodies. J. Autoimmun 57, 42–52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Menke J et al. Sunlight triggers cutaneous lupus through a CSF-1-dependent mechanism in MRL-Fas(lpr) mice. J. Immunol 181, 7367–7379 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Crupi R et al. Reduced adult neurogenesis and altered emotional behaviors in autoimmune-prone B cell activating factor transgenic mice. Biol. Psychiatry 67, 558–566 (2010). [DOI] [PubMed] [Google Scholar]
- 142.Mondal TK, Saha SK, Miller VM, Seegal RF & Lawrence DA Autoantibody-mediated neuroinflammation: pathogenesis of neuropsychiatric systemic lupus erythematosus in the NZM88 murine model. Brain Behav. Immun 22, 949–959 (2008). [DOI] [PubMed] [Google Scholar]
- 143.Chalmers SA et al. Highly selective inhibition of Bruton’s tyrosine kinase attenuates skin and brain disease in murine lupus. Arthritis Res. Ther 20, 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hanly JG, Walsh NM & Sangalang V Brain pathology in systemic lupus erythematosus. J. Rheumatol 19, 732–741 (1992). [PubMed] [Google Scholar]
- 145.Duprez T, Nzeusseu A, Peeters A & Houssiau FA Selective involvement of the choroid plexus on cerebral magnetic resonance images: a new radiological sign in patients with systemic lupus erythematosus with neurological symptoms. J. Rheumatol 28, 387–391 (2001). [PubMed] [Google Scholar]
- 146.Li Y et al. Behavioral deficits are accompanied by immunological and neurochemical changes in a mouse model for neuropsychiatric lupus (NP-SLE). Int. J. Mol. Sci 16, 15150–15171 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shiozawa S, Kuroki Y, Kim M, Hirohata S & Ogino T Interferon-alpha in lupus psychosis. Arthritis Rheum. 35, 417–422 (1992). [DOI] [PubMed] [Google Scholar]
- 148.Fragoso-Loyo H, Atisha-Fregoso Y, Llorente L & Sanchez-Guerrero J Inflammatory profile in cerebrospinal fluid of patients with headache as a manifestation of neuropsychiatric systemic lupus erythematosus. Rheumatology (Oxford) 52, 2218–2222 (2013). [DOI] [PubMed] [Google Scholar]
- 149.Santer DM, Yoshio T, Minota S, Moller T & Elkon KB Potent induction of IFN-α and chemokines by autoantibodies in the cerebrospinal fluid of patients with neuropsychiatric lupus. J. Immunol 182, 1192–1201 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ronnblom L, Alm GV & Eloranta ML Type I interferon and lupus. Curr. Opin. Rheumatol 21, 471–477 (2009). [DOI] [PubMed] [Google Scholar]
- 151.Karageorgas TP, Tseronis DD & Mavragani CP Activation of type I interferon pathway in systemic lupus erythematosus: association with distinct clinical phenotypes. J. Biomed. Biotechnol 2011, 273907 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yoshio T et al. IL-6, IL-8, IP-10, MCP-1 and G-CSF are significantly increased in cerebrospinal fluid but not in sera of patients with central neuropsychiatric lupus erythematosus. Lupus 25, 997–1003 (2016). [DOI] [PubMed] [Google Scholar]
- 153.Fragoso-Loyo H, Atisha-Fregoso Y, Nunez-Alvarez CA, Llorente L & Sanchez-Guerrero J Utility of interferon-alpha as a biomarker in central neuropsychiatric involvement in systemic lupus erythematosus. J. Rheumatol 39, 504–509 (2012). [DOI] [PubMed] [Google Scholar]
- 154.Wen J et al. Inhibiting TWEAK (TNF-like weak inducer of apoptosis) signaling ameliorates blood brain barrier integrity and neuronal damage in neuropsychiatric lupus prone MRL/lpr mice. J. Immunol 192, 1 (2014). [Google Scholar]
- 155.Wen J et al. Neuropsychiatric disease in murine lupus is dependent on the TWEAK/Fn14 pathway. J. Autoimmun 43, 44–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fragoso-Loyo H, Atisha-Fregoso Y, Nunez-Alvarez CA & Llorente L Utility of TWEAK to assess neuropsychiatric disease activity in systemic lupus erythematosus. Lupus 25, 364–369 (2016). [DOI] [PubMed] [Google Scholar]
- 157.Katsumata Y et al. Diagnostic reliability of cerebral spinal fluid tests for acute confusional state (delirium) in patients with systemic lupus erythematosus: interleukin 6 (IL-6), IL-8, interferon-alpha, IgG index, and Q-albumin. J. Rheumatol 34, 2010–2017 (2007). [PubMed] [Google Scholar]
- 158.Hirohata S et al. Accuracy of cerebrospinal fluid IL-6 testing for diagnosis of lupus psychosis. A multicenter retrospective study. Clin. Rheumatol 28, 1319–1323 (2009). [DOI] [PubMed] [Google Scholar]
- 159.Asano T et al. Evaluation of blood-brain barrier function by quotient alpha2 macroglobulin and its relationship with interleukin-6 and complement component 3 levels in neuropsychiatric systemic lupus erythematosus. PLOS ONE 12, e0186414 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ichinose K et al. Distinguishing the cerebrospinal fluid cytokine profile in neuropsychiatric systemic lupus erythematosus from other autoimmune neurological diseases. Clin. Immunol 157, 114–120 (2015). [DOI] [PubMed] [Google Scholar]
- 161.Wang JB et al. Role of IL-1β, IL-6, IL-8 and IFN-γ in pathogenesis of central nervous system neuropsychiatric systemic lupus erythematous. Int. J. Clin. Exp. Med 8, 16658–16663 (2015). [PMC free article] [PubMed] [Google Scholar]
- 162.Plog BA & Nedergaard M The glymphatic system in central nervous system health and disease: past, present, and future. Annu. Rev. Pathol 13, 379–394 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gilkeson GS Complement-targeted therapies in lupus. Curr. Treatm Opt. Rheumatol 1, 10–18 (2015). [Google Scholar]
- 164.Alexander JJ, Jacob A, Bao L, Macdonald RL & Quigg RJ Complement-dependent apoptosis and inflammatory gene changes in murine lupus cerebritis. J. Immunol 175, 8312–8319 (2005). [DOI] [PubMed] [Google Scholar]
- 165.Jongen PJ, Boerbooms AM, Lamers KJ, Raes BC & Vierwinden G Diffuse CNS involvement in systemic lupus erythematosus: intrathecal synthesis of the 4th component of complement. Neurology 40, 1593–1596 (1990). [DOI] [PubMed] [Google Scholar]
- 166.Jongen PJ et al. Cerebrospinal fluid C3 and C4 indexes in immunological disorders of the central nervous system. Acta Neurol. Scand 101, 116–121 (2000). [DOI] [PubMed] [Google Scholar]
- 167.Sakuma Y, Nagai T, Yoshio T & Hirohata S Differential activation mechanisms of serum C5a in lupus nephritis and neuropsychiatric systemic lupus erythematosus. Mod. Rheumatol 27, 292–297 (2017). [DOI] [PubMed] [Google Scholar]
- 168.Dobrowolski C & Erkan D Treatment of antiphospholipid syndrome beyond anticoagulation. Clin. Immunol 10.1016/j.clim.2018.03.001 (2018). [DOI] [PubMed] [Google Scholar]
- 169.Kronbichler A et al. Efficacy of eculizumab in a patient with immunoadsorption-dependent catastrophic antiphospholipid syndrome: a case report. Medicine 93, e143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Shapira I, Andrade D, Allen SL & Salmon JE Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 64, 2719–2723 (2012). [DOI] [PubMed] [Google Scholar]
- 171.Paul F, Murphy O, Pardo S & Levy M Investigational drugs in development to prevent neuromyelitis optica relapses. Expert Opin. Investig. Drugs 27, 265–271 (2018). [DOI] [PubMed] [Google Scholar]
- 172.Lei HW et al. Neuropsychiatric involvement in lupus is associated with the Nogo-a/NgR1 pathway. J. Neuroimmunol 311, 22–28 (2017). [DOI] [PubMed] [Google Scholar]
- 173.Wendeln AC et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Schrepf A et al. A multi-modal MRI study of the central response to inflammation in rheumatoid arthritis. Nat. Commun 9, 2243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Nystedt J et al. Altered white matter microstructure in lupus patients: a diffusion tensor imaging study. Arthritis Res. Ther 20, 21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ceccarelli F et al. Genetic factors in systemic lupus erythematosus: contribution to disease phenotype. J. Immunol. Res 2015, 745647 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.de Vries B et al. TREX1 gene variant in neuropsychiatric systemic lupus erythematosus. Ann. Rheum. Dis 69, 1886–1887 (2010). [DOI] [PubMed] [Google Scholar]
- 178.Namjou B et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. 12, 270–279 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lundstrom E et al. HLA-DRB1*04/*13 alleles are associated with vascular disease and antiphospholipid antibodies in systemic lupus erythematosus. Ann. Rheum. Dis 72, 1018–1025 (2013). [DOI] [PubMed] [Google Scholar]
- 180.Rullo OJ & Tsao BP Recent insights into the genetic basis of systemic lupus erythematosus. Ann. Rheum. Dis 72 (Suppl. 2), ii56–ii61 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Koga M et al. Cumulative association of eight susceptibility genes with systemic lupus erythematosus in a Japanese female population. J. Hum. Gen 56, 503–507 (2011). [DOI] [PubMed] [Google Scholar]
- 182.Barile-Fabris L et al. Controlled clinical trial of IV cyclophosphamide versus IV methylprednisolone in severe neurological manifestations in systemic lupus erythematosus. Ann. Rheum. Dis 64, 620–625 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mok CC, Lau CS & Wong RW Treatment of lupus psychosis with oral cyclophosphamide followed by azathioprine maintenance: an open-label study. Am. J. Med 115, 59–62 (2003). [DOI] [PubMed] [Google Scholar]
- 184.Tokunaga M et al. Efficacy of rituximab (anti-CD20) for refractory systemic lupus erythematosus involving the central nervous system. Ann. Rheum. Dis 66, 470–475 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Dale RC et al. Utility and safety of rituximab in pediatric autoimmune and inflammatory CNS disease. Neurology 83, 142–150 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jacob A et al. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch. Neurol 65, 1443–1448 (2008). [DOI] [PubMed] [Google Scholar]
- 187.Pranzatelli MR et al. Rituximab (anti-CD20) adjunctive therapy for opsoclonus-myoclonus syndrome. J. Pediatr. Hematol. Oncol 28, 585–593 (2006). [DOI] [PubMed] [Google Scholar]
- 188.Titulaer MJ et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 12, 157–165 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Manzi S et al. Effects of belimumab, a B lymphocyte stimulator-specific inhibitor, on disease activity across multiple organ domains in patients with systemic lupus erythematosus: combined results from two phase III trials. Ann. Rheum. Dis 71, 1833–1838 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hanly JG in Systemic Lupus Erythematosus 5th edn (ed. Lahita RG) 727–746 (Elsevier, 2005). [Google Scholar]
- 191.Erkan D, Salmon J & Lockshin M in Kelley and Firestein’s Textbook of Rheumatology (ed. Firestein GS) 1389–1399 (Elsevier, 2017). [Google Scholar]
- 192.Meroni PL et al. Statins prevent endothelial cell activation induced by antiphospholipid (anti-β2-glycoprotein I) antibodies: effect on the proadhesive and proinflammatory phenotype. Arthritis Rheum. 44, 2870–2878 (2001). [DOI] [PubMed] [Google Scholar]
- 193.Jung H et al. The protective effect of antimalarial drugs on thrombovascular events in systemic lupus erythematosus. Arthritis Rheum. 62, 863–868 (2010). [DOI] [PubMed] [Google Scholar]
- 194.Cervera R CAPS Registry. Lupus 21, 755–757 (2012). [DOI] [PubMed] [Google Scholar]
- 195.Furie R et al. Anifrolumab, an anti-interferon-alpha receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. 69, 376–386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Chitu V & Stanley ER Colony-stimulating factor-1 in immunity and inflammation. Curr. Opin. Immunol 18, 39–48 (2006). [DOI] [PubMed] [Google Scholar]
- 197.Chalmers SA et al. CSF-1R inhibition attenuates renal and neuropsychiatric disease in murine lupus. Clin. Immunol 185, 100–108 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hendriks RW Drug discovery: new Btk inhibitor holds promise. Nat. Chem. Biol 7, 4–5 (2011). [DOI] [PubMed] [Google Scholar]
- 199.Jongstra-Bilen J et al. Dual functions of Bruton’s tyrosine kinase and Tec kinase during Fcγ receptor-induced signaling and phagocytosis. J. Immunol 181, 288–298 (2008). [DOI] [PubMed] [Google Scholar]
- 200.Ni Gabhann J et al. Btk regulates macrophage polarization in response to lipopolysaccharide. PLOS ONE 9, e85834 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Chalmers SA et al. Therapeutic blockade of immune complex-mediated glomerulonephritis by highly selective inhibition of Bruton’s tyrosine kinase. Sci. Rep 6, 26164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Mina-Osorio P et al. Suppression of glomerulonephritis in lupus-prone NZB × NZW mice by RN486, a selective inhibitor of Bruton’s tyrosine kinase. Arthritis Rheum. 65, 2380–2391 (2013). [DOI] [PubMed] [Google Scholar]
- 203.Rankin AL et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J. Immunol 191, 4540–4550 (2013). [DOI] [PubMed] [Google Scholar]
- 204.Byrd JC et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med 369, 32–42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zardi EM, Taccone A, Marigliano B, Margiotta DP & Afeltra A Neuropsychiatric systemic lupus erythematosus: tools for the diagnosis. Autoimmun. Rev 13, 831–839 (2014). [DOI] [PubMed] [Google Scholar]
- 206.Kivity S, Agmon-Levin N, Zandman-Goddard G, Chapman J & Shoenfeld Y Neuropsychiatric lupus: a mosaic of clinical presentations. BMC Med. 13, 43 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhang L, Fu T, Yin R, Zhang Q & Shen B Prevalence of depression and anxiety in systemic lupus erythematosus: a systematic review and meta-analysis. BMC Psychiatry 17, 70 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Torreggiani S et al. Chorea, a little-known manifestation in systemic lupus erythematosus: short literature review and four case reports. Pediatr. Rheumatol 11, 36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Li XY, Xiao HB & Pai P Myelitis in systemic lupus erythematosus. J. Clin. Neurosci 44, 18–22 (2017). [DOI] [PubMed] [Google Scholar]
- 210.Piga M et al. Demyelinating syndrome in SLE encompasses different subtypes: do we need new classification criteria? Pooled results from systematic literature review and monocentric cohort analysis. Autoimmun. Rev 16, 244–252 (2017). [DOI] [PubMed] [Google Scholar]
- 211.Fragoso-Loyo H et al. Interleukin-6 and chemokines in the neuropsychiatric manifestations of systemic lupus erythematosus. Arthritis Rheum. 56, 1242–1250 (2007). [DOI] [PubMed] [Google Scholar]
- 212.Dellalibera-Joviliano R, Dos Reis ML, Cunha Fde Q & Donadi EA Kinins and cytokines in plasma and cerebrospinal fluid of patients with neuropsychiatric lupus. J. Rheumatol 30, 485–492 (2003). [PubMed] [Google Scholar]
- 213.Baraczka K, Nekam K, Pozsonyi T, Szuts I & Ormos G Investigation of cytokine (tumor necrosis factor-alpha, interleukin-6, interleukin-10) concentrations in the cerebrospinal fluid of female patients with multiple sclerosis and systemic lupus erythematosus. Eur. J. Neurol 11, 37–42 (2004). [DOI] [PubMed] [Google Scholar]
- 214.Quaresma MV et al. Anti-TNF-α and hydralazine drug-induced lupus. An. Bras. Dermatol 90, 125–129 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Groom JR et al. BAFF and MyD88 signals promote a lupus like disease independent of T cells. J. Exp. Med 204, 1959–1971 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Zhang J et al. Cutting edge: a role for B lymphocyte stimulator in systemic lupus erythematosus. J. Immunol 166, 6–10 (2001). [DOI] [PubMed] [Google Scholar]
- 217.Crow MK Type I interferon in the pathogenesis of lupus. J. Immunol 192, 5459–5468 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Baechler EC, Gregersen PK & Behrens TW The emerging role of interferon in human systemic lupus erythematosus. Curr. Opin. Immunol 16, 801–807 (2004). [DOI] [PubMed] [Google Scholar]
- 219.Wichers M & Maes M The psychoneuroimmuno-pathophysiology of cytokine-induced depression in humans. Int. J. Neuropsychopharmacol 5, 375–388 (2002). [DOI] [PubMed] [Google Scholar]
- 220.Stock AD, Wen J & Putterman C Neuropsychiatric lupus, the blood brain barrier, and the TWEAK/Fn14 pathway. Front. Immunol 4, 484 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Ronnblom L & Elkon KB Cytokines as therapeutic targets in SLE. Nat. Rev. Rheumatol 6, 339–347 (2010). [DOI] [PubMed] [Google Scholar]