

SUMMARY
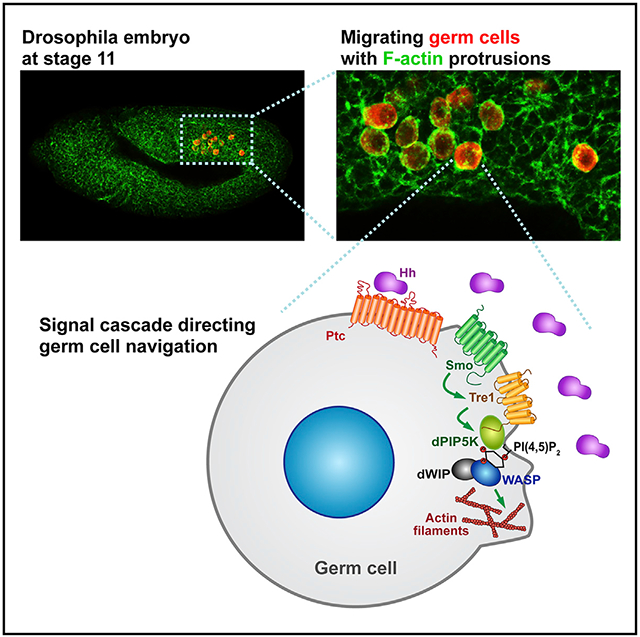
The Tre1 G-protein coupled receptor (GPCR) was discovered to be required for Drosophila germ cell (GC) coalescence almost two decades ago, yet the molecular events both upstream and downstream of Tre1 activation remain poorly understood. To gain insight into these events, we describe a bona fide null allele and both untagged and tagged versions of Tre1. We find that the primary defect with complete Tre1 loss is the failure of GCs to properly navigate, with GC mis-migration occurring from early stages. We find that Tre1 localizes with F-actin at the migration front, along with PI(4,5)P2; dPIP5K, an enzyme that generates PI(4,5)P2; and dWIP, a protein that binds activated Wiskott-Aldrich syndrome protein (WASP), which stimulates F-actin polymerization. We show that Tre1 is required for polarized accumulation of F-actin, PI(4,5)P2, and dPIP5K. Smoothened also localizes with F-actin at the migration front, and Hh, through Smo, increases levels of Tre1 at the plasma membrane and Tre1’s association with dPIP5K.
Graphical Abstract
In brief
Kim et al. uncover molecular and cellular events upstream and downstream of the Tre1 G-protein coupled receptor (GPCR), which is required for germ cell navigation in Drosophila. Hedgehog signaling through Smoothened localizes Tre1 to activate F-actin assembly through dPIP5K, PI(4,5)P2, and WASP.
INTRODUCTION
Directional cell migration plays pivotal roles in shaping and positioning developing organs. This highly complex process requires multi-dimensional controls on signal transduction, intracellular trafficking, cytoskeletal and membrane dynamics, and cell mechanics. Drosophila embryonic germ cells (GCs) are ideal for studying cell migration in vivo due to the wide range of genetic tools and an easily observable, stereotypical migration pattern (Kunwar et al., 2006). During the initial stages of development, the approximately 30 GCs form at the posterior pole of the embryo and are then passively internalized into the posterior midgut pocket. Subsequently, GCs actively traverse the midgut endoderm as individual cells (Figure 1A). Each cell then migrates through the mesoderm dorsally and away from the midgut (Figure 1B). The cells continue to migrate antero-laterally with the somatic gonad precursors (SGPs) (Figures 1C and 1D), which eventually coalesce to form the embryonic gonad (Figure 1E; Jaglarz and Howard, 1995; Kunwar et al., 2006; Santos and Lehmann, 2004). Unlike systems where persistent migration in a single direction is observed (e.g., Drosophila border cells [Peercy and Starz-Gaiano, 2020] and zebrafsh lateral line [Olson and Nechiporuk, 2018]), migration of individual GCs is often locally somewhat stochastic. Nonetheless, the net movements are directional, with most GCs arriving and coalescing with the somatic gonad by the beginning of embryonic stage 14 (~11 h post-fertilization).
Figure 1. Tre1 is required to direct GC navigation from very early stages.

(A–E) In the cartoons illustrating embryos at each developmental stage, GCs are marked in green and SGPs in red. The cartoons were adapted from Kunwar et al. (2006). For the corresponding fluorescent images, GCs were stained for the GC-specific cytoplasmic protein Vasa (green) and SGPs for the SGP-specific nuclear protein Eya (red). Note that SGPs are dispersed at multiple locations surrounding and ahead of GCs. In the fully coalesced gonad (E, lower panel), Eya expression is observed in only a subset of SGPs. Arrows indicate the overall migration directions of the GC populations. The anterior-posterior (A-P) and dorsal-ventral (D-V) axes are indicated.
(F) Vasa staining in late-stage WT embryo.
(G–M) Vasa staining in stage 16 (except stage 12 in L) embryos missing maternal and/or zygotic Tre1 function. Open arrowheads indicate GC destination of the somatic gonad.
(N) Vasa staining in a WT stage 10 embryo.
(O–Q) Vasa staining in stage 10 embryos missing both maternal and zygotic – (m-z-). Black arrows highlight early mis-migrating GCs.
(R–W) Vasa staining of GCs in rescue controls (R and S) and nos-Gal4 driven attempted rescue of Tre1 function with untagged UAS-Tre1 (T) and UAS-Tre1-C-terminally tagged forms (U–W).
(X and Y) Vasa staining of otherwise WT embryos expressing UAS-C-terminally tagged forms of Tre1 with the nos-Gal4 driver.
(Z) Quantification of late-stage embryos with different numbers of mis-migrated GCs (color-coded with yellow, blue, or red) in all genotypes tested.
The statistical analysis of the phenotypes is provided in Table S1.
Although GCs are actively attracted to the SGPs after departing the midgut, the source of earlier migratory signals is unknown, and the cue provided by the SGPs remains controversial. Hedgehog (Hh) has been proposed as the attractive SGP guidance molecule based on the following: (1) Hh expression in SGP cells; (2) GC migration defects with loss of the Hh signaling genes hh, smoothened (smo), and patched (ptc); and (3) mis-migration of GCs toward regions ectopically expressing Hh (Deshpande and Schedl, 2005; Deshpande et al., 2001). Renault et al. (2009) have challenged this role for Hh based on (1) the argument that Hh is also expressed in other non-SGP mesoderm; (2) the lack of detectable Ptc (Hh receptor) in GCs; and (3) their failure to observe GC migration defects when ectopically expressing Hh, expressing mutant versions of cubitus interruptus (CI), or using the same or comparable genetic backgrounds as those used by the original group. Whereas an independent party confirmed the originally reported migration defects associated with ectopic Hh (Deshpande et al., 2013), other reports continue to contest these findings (Kenwrick et al., 2019). Consequently, the molecular roles for other genes required for GC migration, such as HMG coenzyme A reductase (hmgcr) and Multi drug resistance 49 (Mdr49), have been heavily debated; one argument is that these genes are required for the production of the mature accessible Hh ligand (Deshpande et al., 2009, 2013, 2016; Deshpande and Schedl, 2005), and the other is that these genes are required to generate a signal completely unrelated to Hh (Barton et al., 2016; Renault et al., 2009; Ricardo and Lehmann, 2009; Van Doren et al., 1998).
GCs are also passively repelled from regions of the embryo due to depletion of putative GC attractants. The lipid phosphate phosphatases, Wunen and Wunen2, provide this repelling function as GCs are scattered throughout embryos mutant for the wunen genes and are excluded from regions ectopically expressing Wunen proteins (Mukherjee et al., 2013; Renault et al., 2010; Sano et al., 2005; Zhang et al., 1997). The requirement for molecules such as Wunen and Wunen2 suggests that the GC attractant(s) could be widely expressed and must therefore be spatially limited for normal GC migration.
The G-protein coupled receptor (GPCR) Trapped in endoderm 1 (Tre1) is critical for GCs to arrive at their correct final destinations in the somatic gonad (Kunwar et al., 2003, 2008). Loss-of-function phenotypes in other tissues have revealed roles for Tre1 in both cell migration and cell polarization. Hemocytes, Drosophila immune cells, require Tre1 for their extravasation and migration toward wounded tissues (Thuma et al., 2018), and neuroblasts require Tre1 to polarize for oriented cell divisions (Yoshiura et al., 2012). Indeed, a role for Tre1 in relocalizing E-cadherin and the Rho1 GTPase to the “rear” of the cell to allow GCs to polarize, disassociate from one another, and subsequently migrate through the midgut endoderm was proposed (Kunwar et al., 2008; LeBlanc and Lehmann, 2017): the conserved Tre1 asparagine, proline, isoleucine, isoleucine, tyrosine (NPIIY) motif polarizes Rho1 and ECadherin (ECad) for GC dissociation, and the conserved Tre1 asparagine, arginine, tyrosine (NRY) domain is needed for the G-protein-dependent migratory response to guidance cues (LeBlanc and Lehmann, 2017).
In a more recent detailed analysis of wild-type (WT) and Tre1 mutant phenotypes, the Lehmann lab made several observations that contradict their original findings and instead support a model wherein all of the GC phenotypes of Tre1 mutants can be explained simply by a failure to sense and respond to guidance cues (Lin et al., 2020). Included in their findings was the observation that, as opposed to their previous view that Tre1 generates Rho1 polarity, Tre1 regulates only the orientation of an intrinsic Rho1 polarity. Whereas polarized Rho1 activity (and the consequent downstream localization of activated Myosin II [MyoII]) is quite stable in WT, active Rho1 and activated MyoII rapidly change position in Tre1 mutant GCs. The group further showed that relocalization of ECad does not require Tre1 and that overexpression of ECad (or of another cell adhesion molecule) has no effect on GC dispersal. Finally, they propose a model wherein the random movement of Tre1 mutant cells within the endoderm impedes the dissociation of these cells. Thus, GCs are not “trapped in endoderm” because they cannot move; rather, they simply keep bumping into one another, making it challenging for the cells to separate and escape through the endoderm. Thus, the current consensus is that Tre1 functions as the receptor for one or more guidance cues required for GC navigation (Coffman et al., 2002; LeBlanc and Lehmann, 2017; Lin et al., 2020; this study). How Tre1 orients the mechanics of migration, as well as the identity of the guidance cue(s), remains unknown.
In this study, we take advantage of a complete null allele and tagged forms of Tre1 to characterize both the signaling cascade downstream of Tre1 during GC migration and the upstream events regulating Tre1 activation. We demonstrate that Tre1 localizes to the leading edge of migrating GCs, where we propose it drives F-actin assembly by modulating the levels of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) at the cell membrane. Our findings support a model wherein Drosophila PI4P 5-kinase (dPIP5K) and Wiskott-Aldrich syndrome protein (WASP) function downstream of Tre1 activation to generate PI(4,5)P2 and polymerize actin, respectively. We show that both Ptc and Smo are expressed in GCs and that Smo also localizes at the F-actin-enriched protrusions in migrating GCs. We find that Hh is not only expressed in SGPs, but also endocytosed in migrating GCs from very early stages. We show that in S2R+ cells expressing Smo, the addition of Hh decreases intracellular vesicular pools of Tre1 and increases both plasma membrane (PM) localization of Tre1 and its association with dPIP5K. Our findings thus suggest a molecular mechanism for how the Tre1 GPCR activates the cellular machinery to direct cell navigation in vivo and provide supporting evidence for a role for Hh signaling in guiding this process.
RESULTS
Tre1 is required in GCs for their navigation
To fully characterize the role of Tre1 in GCs, we first generated a complete null allele of Tre1 by homologous recombination (Tre1KO), since the existing allele previously described as null (Kunwar et al., 2003) produces mRNA with an intact open reading frame (ORF) (Figures S1A-S1E and S1J-S1M) and turns out to have notably less penetrant GC migration defects than Tre1KO when homozygous (data not shown). The Tre1KO allele removes almost the entire Tre1 ORF, leaving only the two upstream non-coding exons and a very small 3′ portion of the final exon (Figure S1I; (Larkin et al., 2020)). Tre1KO maternal and zygotic mutants exhibited severe migration defects, with ~90% of embryos having at least six (and often more) GCs failing to reach the somatic gonad (Figures 1F, 1G, 1M, and 1Z). The Tre1KO allele failed to complement previously characterized Tre1 alleles Tre1ΔEP5 and Tre1sctt (Figures 1J, 1K, 1P, 1Q, and 1Z). Our analysis of the Tre1KO null allele also reveals that Tre1 is required only maternally for GC migration (Figures 1G-1I and 1Z), unlike findings reported with previously existing Tre1 alleles (Kamps et al., 2010; Kunwar et al., 2003).
GCs in Tre1KO embryos were found throughout the posterior half of the embryo at late stages (Figure 1G), with some GCs even on the outside of the embryo (Figure 1M). This broad scattering of GCs indicates that without Tre1 activity, GCs can actively migrate across the midgut endoderm but cannot properly navigate to the gonad. Supporting this idea, Tre1KO GCs showed aberrant migration even at very early stages. Instead of migrating only posteriorly through the midgut endoderm, GCs were frequently found in a more anterior-dorsal position within the midgut pocket, often within or exiting the nascent hindgut (Figures 1L and 1N-1Q). Thus, Tre1 is required for GC navigation from very early stages, consistent with the recent observation that the failure to cross the midgut endoderm is due to a failure of oriented navigation (Lin et al., 2020) and in contrast to earlier studies suggesting a failure to polarize to allow for migration (Kunwar et al., 2008; LeBlanc and Lehmann, 2017).
To verify that the navigation defects observed in Tre1KO were attributable to loss of Tre1 specifically in GCs, we asked if GC-specific expression of untagged or tagged forms of the Tre1 ORF could rescue the GC navigation defects of Tre1KO. This experiment also allowed us to learn where the tagged functional forms of Tre1 localize. Expression of full-length Tre1 (UAS-Tre1; provided through the fathers) driven by GC-specific Nanos-Gal4 (nos-Gal4; provided through the mothers) rescued the GC navigation defects of Tre1KO embryos (Figures 1R-1T and 1Z). Thus, although endogenous zygotic Tre1 expression does not rescue maternal loss, GC-specific expression driven at early stages can. We found that GC-specific expression of Tre1 tagged with FLAG or HA at the C terminus (UAS-Tre1-FLAG or UAS-Tre1-HA) also rescued GC navigation in Tre1KO mutants (Figures 1U, 1V, and 1Z), although not quite as well as untagged Tre1. In contrast, Tre1 tagged with GFP (UAS-Tre1-GFP) completely failed to rescue (Figures 1W and 1Z). Moreover, nos-Gal4-driven expression of Tre1-GFP in otherwise WT embryos also resulted in strong GC navigation defects, a phenotype not observed with GC expression of other tagged or untagged versions of Tre1 (Figures 1X-1Z), suggesting that Tre1-GFP functions as a dominant negative. Altogether, these data show that Tre1 is required cell autonomously for GC navigation and that it is the maternal mRNA/protein that normally supplies this function.
Tre1 colocalizes with F-actin during GC migration
Migrating GCs generate F-actin-enriched protrusions at their migration fronts (Jaglarz and Howard, 1995; LeBlanc and Lehmann, 2017); the localization of F-actin to the migration front is observed even early before GCs have begun to cross the midgut endoderm (Lin et al., 2020). To learn where Tre1 localizes with respect to F-actin, we co-stained stage 11 embryos (when GCs are most spread out) expressing tagged forms of Tre1 in GCs with antibodies recognizing the tags (αFLAG or αGFP) and with fluorescently labeled Phalloidin. Both forms of Tre1 showed significant overlap with F-actin. Tre1-FLAG, the tagged version that best rescued GC navigation, colocalized with F-actin in a PM domain at the presumed migration front (Figures 2A-2C′). Tre1-GFP, the tagged version that caused mis-migration of GCs even in a WT background, colocalized with F-actin around the entire PM and in large intracellular puncta near the cell surface (Figures 2D-2F). As a control for this colocalization, we expressed the PM marker UAS-Transmembrane-GFP (TMEM-GFP) in GCs. Whereas GFP from this construct was detected more broadly along the GC PM, high-level F-actin accumulation was observed in only a limited PM domain, as in WT GCs (Figures S2B-S2B‴). To quantify the Tre1-FLAG and F-actin colocalization versus the control TMEM-GFP and F-actin colocalization, we measured the fluorescence intensity around the cell periphery in the different channels (Figure 2C) and determined the Pearson’s correlation coefficient for relative staining intensity. For Tre1-FLAG and F-actin, the average value was 0.73 ± 0.16, whereas the value was only 0.21 ± 0.25 for Tre1-FLAG and Vasa (which stains GC cytoplasm but also overlaps the membrane staining because of the resolution of our confocal imaging) (Figure 2C′). With TMEM-GFP and F-actin, the average Pearson’s correlation coefficient was 0.47 ± 0.20, whereas it was 0.70 ± 0.20 for the TMEM-GFP and Vasa (Figure S2C), indicating that F-actin intensity is not simply higher where the PM (and cell) is thicker or vice versa. Altogether, these findings indicate that Tre1 normally localizes where F-actin assembles in migrating GCs.
Figure 2. Tre1, PI(4,5)P2, and dPIP5K localize at F-actin-enriched protrusions in migrating GCs.

(A–B″) GCs (blue, Vasa staining) from stage 11 embryos expressing Tre1-FLAG show the localization of Tre1-FLAG (green) at protrusive regions (arrowhead) enriched with F-actin (red). The embryo is in a dorsal view, and the A-P axis is indicated. Scale bars, 10 μm. See also Video S1.
(C and C′) The line scan of fluorescent signals on the entire GC circumference in (B″) and the Pearson’s correlation coefficients between fluorescent signals from GCs in (A″).
(D–E″) GCs expressing Tre1-GFP show high-level F-actin colocalization with Tre1-GFP at the PM and in intracellular punctae (arrowhead). The embryo is in a lateral view, and the A-P and D-V axes are indicated. Scale bars, 10 μm.
(F) The line scan of fluorescent signals on the dotted line in (E″). GCs boxed in (A″) and (D″) are shown in magnified views in (B)–(B″) and (E)–(E″), respectively.
(G) Western blots of extracts from S2R+ cells expressing different tagged forms of Tre1 at different times post-treatment of the cells with cycloheximide (CHX) and probed with antibodies to the tags and to α-tubulin for loading control.
(H) Graph showing relative levels of each tagged Tre1 protein over several hours from three independent experiments for each tagged Tre1.
(I–K″) S2R+ cells expressing different C-terminally tagged forms of Tre1 with staining for the Tre1 tags (green), F-actin (red), and nucleus (blue, DAPI). Although some limited colocalization at the cell periphery is observed with all three tagged versions, note the numerous large puncta of intracellular colocalization of Tre1-GFP and F-actin (arrowheads) not observed with Tre1-FLAG or Tre1-HA. Scale bars, 10 μm. See also Video S1 Figure S2.
(L–M″) In stage 11 GCs (blue, Vasa) expressing a GFP-tagged PI(4,5)P2 sensor (PLCγ-PH, green), the PI(4,5)P2 sensor was enriched at F-actin (red) protrusions (arrowhead). The embryo in dorsal view and the A-P axis are indicated.
(N and N′) The line scan of fluorescent signals on the entire GC circumference in (M″) and the Pearson’s correlation coefficients between fluorescent signals from GCs in (L″).
(O–P″) GCs with co-expression of Tre1-GFP (green) and an mCherry-tagged PI(4,5)P2 sensor (PLCδ-PH, red) reveal overlap throughout the PM and in intracellular punctae (arrowhead). The embryo in dorsal view and the A-P axis are indicated. GCs boxed in (L″) and (O″) are shown in magnified views in (M)–(M″) and (P)–(P″), respectively. Scale bars, 10 μm.
(Q) The line scan of fluorescent signals on the dotted line in (P″).
(R–S′) Staining of WT stage 11 GCs (Vasa, red) with dPIP5K antiserum (green). The embryo is in a lateral view and the A-P and D-V axes are indicated. dPIP5K is enriched at GC boundary (arrowhead).
(T–U″) Venus-tagged dPIP5K (green) expression in GCs (Vasa, blue) reveals high accumulation of dPIP5K at membrane protrusions (arrowhead) with F-actin (red). The embryo is in a lateral view and the A-P and D-V axes are indicated. GCs boxed in (R′) and (T″) are shown in magnified views in (S) and (S′) and (U)–(U″), respectively.
(V and V′) The line scan of fluorescent signals on the entire GC circumference in (U″) and the Pearson’s correlation coefficients between fluorescent signals from GCs in (T″). Scale bars, 10 μm. See also Figures S3-S5 and Video S2.
(W–AB) Vasa staining of stage 16/17 (W) WT embryo, (X–Z) embryos missing zygotic function of dpip5k or both sktl and dpip5k, and (AA and AB) embryos in which the maternal expression of dpip5k or sktl were knocked down by RNAi.
(AC–AE) Expression of a kinase-dead (KD) form of dPIP5K disrupts GC migration in embryos from Tre1 heterozygous mothers (AE) but does not affect GC migration in embryos from WT mothers (AD). Open arrowheads indicate GC destination of the somatic gonad.
(AF) Quantification of embryos with different numbers of mis-migrated GCs in all genotypes from (W) to (AE).
The statistical analysis of the phenotypes is provided in Table S2.
Tre1-GFP consistently presented much stronger GC signal intensity compared to Tre1-FLAG or Tre1-HA. Thus, we speculated that Tre1-GFP might not turn over as readily as other tagged forms due to its relatively larger C-terminal tag (Figure S2A) and, we propose, could continuously activate F-actin assembly. Indeed, when the synthesis of new proteins was blocked in Drosophila S2R+ cells with cycloheximide, the amount of Tre1-GFP remained stable, whereas levels of Tre1-FLAG or Tre1-HA decreased over several hours, with Tre1-HA—the form most challenging to detect in embryos—showing the shortest half-life (Figures 2G and 2H). We also examined Tre1 and F-actin accumulation in the S2R+ cells expressing the different tagged forms of Tre1. In all cases, Tre1 was found in numerous small vesicular structures within the cells, with limited and variable accumulation at the cell periphery (Figures 2I-2K). With all three constructs, some limited amount of F-actin was also detected at the cell periphery (Figures 2I′-2K′). Tre1-GFP also induced extensive F-actin accumulation in large intracellular aggregates that overlapped with the GFP staining, a feature not observed with either Tre1-FLAG or Tre1-HA (Figures 2I″-2K″). These data suggest that the site of Tre1 accumulation determines where F-actin assembles and that Tre1-GFP continuously activates F-actin assembly by failing to turn over and thus functions as a dominant negative.
F-actin assembly in GCs through PI(4,5)P2 synthesis
We next asked how Tre1 regulates actin assembly. Phosphoinositides (PIs) are versatile signaling molecules that coordinate diverse cellular processes including actin cytoskeleton dynamics (Di Paolo and De Camilli, 2006). To determine if GCs utilize PIs for directional migration, we first examined localization of GFP-labeled sensors that specifically bind to each PI. Among the six PIs tested, only the PI(4,5)P2 sensors localized to the PM near where F-actin assembly occurs during GC migration (Figures S3A-S3F′). Consistent with a potential involvement for PI(4,5)P2 in GC navigation, tagged PI(4,5)P2 sensors co-localized with F-actin at GC protrusions (Figures 2L-2N;GFP-PLCγ-PH) and were enriched with Tre1-GFP in GCs (Figures 2O-2Q; PLCδ-PH-mCherry). Again, measuring levels of the GFP-PLCγ-PH PI(4,5)P2 sensor and F-actin in the periphery of GCs revealed a high Pearson’s correlation coefficient (0.82 ± 0.11), whereas a similar comparison between the PI(4,5)P2 sensor and Vasa had a lower correlation coefficient (0.40 ± 0.31; Figure 2N′). Furthermore, live imaging of migrating GCs reveals that both F-actin and the GFP-PLCγ-PH PI(4,5)P2 sensor are transiently enriched at the leading front of GCs that are migrating entirely within the plane captured in Videos S1 and S2. Although we observed GC colocalization of both Tre1 and F-actin and the PI(4,5)P2 sensors and F-actin, the position of high-level accumulation was never polarized in the same direction in all GCs captured within a given field. GCs migrate in response to cues provided by the SGPs (Deshpande et al., 2001; Kenwrick et al., 2019). Co-staining of GCs and SGPs (Figure 1B) indicates that at this stage, the two cell types are in very close proximity, but the SGPs are not positioned such that all migrating GCs will be receiving the cue from the same direction at any given time. This observation can explain why GC migration is somewhat stochastic at the local level.
We also expressed tagged PI(4,5)P2 sensors in S2R+ cells along with the tagged forms of Tre1; colocalization was observed with Tre1-FLAG and Tre1-HA in limited regions, mostly along the PM (Figures S4A-S4C″). With Tre1-GFP, however, the PI(4,5)P2 sensor localized to the same large intracellular aggregates where both Tre1-GFP and F-actin colocalized (Figures 2K-2K″ and S4D-S4D″). Since PI(4,5)P2 can facilitate actin polymerization by recruiting and activating actin-regulating proteins (Senju and Lappalainen, 2019; Yin and Janmey, 2003), these observations suggest that PI(4,5)P2 mediates F-actin assembly downstream of localized Tre1 activation.
The major biochemical pathway to generate PI(4,5)P2 is phosphorylation on PI(4)P by Phosphatidylinositol-4-phosphate 5-kinases(PIP5Ks) (van den Bout and Divecha, 2009). We found that a Drosophila PIP5K, dPIP5K (encoded by the gene pip5k59b; Chakrabarti et al., 2015), localizes at the PM of migrating GCs and other surrounding cells (Figures 2R-2S′). To more carefully localize this enzyme in GCs, we drove expression of Venus-tagged dPIP5K in GCs with nos-Gal4 and visualized the Venus signal with antibody staining. We found that the tagged form of dPIP5K was concentrated at the F-actin-enriched protrusions of GCs (Figures 2T-2U″) with a high Pearson’s correlation coefficient (0.67 ± 0.18; Figures 2V and 2V′), suggesting that dPIP5K could be the enzyme that generates local pools of PI(4,5)P2 to activate actin polymerization. In further support of this idea, tagged forms of dPIP5K also colocalized with tagged forms of Tre1 in S2R+ cells in the same pattern as observed with the PI(4,5)P2 sensor and F-actin (Figures S4E-S4H″): some co-localization mostly at the PM with Tre1-FLAG and Tre1-HA, but extensive colocalization in intracellular aggregates with Tre1-GFP.
To test the functional requirement of dPIP5K in GC migration, we examined embryos mutant for dpip5k. dpip5k and skittles (sktl), dpip5k double-zygotic mutants (sktl encodes another Drosophila PIP5K), did not exhibit GC navigation defects (Figures 2W-2Z and 2AF), possibly due to maternal supplies. Supporting this idea, maternal knockdown of dpip5k expression using RNAi resulted in statistically significant (p = 1.16E–06), albeit low-level, GC migration defects (Figures 2AA, 2AB, and 2AF). GC expression of a kinase-dead version of dPIP5K, which suppresses endogenous kinase activity, synergistically increased GC navigation defects in embryos from Tre1KO heterozygous mothers (Figures 2AC-2AF). Together, these results suggest that activated Tre1 can recruit dPIP5K for localized PI(4,5)P2 synthesis to generate F-actin-enriched migratory protrusions.
Tre1KO GCs lose polarized localization of PI(4,5)P2, dPIP5K, and F-actin
If Tre1 recruits dPIP5K for localized PI(4,5)P2 synthesis and actin polymerization, we would expect that loss of Tre1 would affect the localization of these molecules. To test this hypothesis, we expressed Venus- or GFP-tagged dPIP5K, a PI(4,5)P2 sensor (GFP-PLCγ-PH), and an F-actin probe (MoeABD) and measured the fluorescent intensities along the PMs in at least 50 WT and 50 Tre1KO GCs. dPIP5K, PI(4,5)P2, and F-actin exhibited intense local concentrations in GCs, manifested as a single peak of fluorescent intensity, as shown in the examples (Figures 3A, 3D, and 3G), in about 60%–70% of WT GCs (Figures 3C, 3F, 3I, S5A, S5D, and S5G). This local concentration disappeared in Tre1KO mutant GCs, as shown by the lack of a prominent high-intensity peak in the examples (Figures 3B, 3E, and 3H) and only ~20%–30% of all mutant GCs showing a single intensity peak (Figures 3C, 3F, 3I, S5B, S5E, and S5H). Our observation that Tre1KO mutant GCs demonstrated significantly more uniform distributions of fluorescent signals through the entire cellular circumferences is also captured by the smaller variances of fluorescent intensities around the cell periphery in the mutants compared with WT GCs (Figures 3J, S5C, S5F, and S5I). This finding is consistent with the model that localized Tre1 activation of actin polymerization through recruitment of dPIP5K and synthesis of PI(4,5)P2 guides the directional migration of GCs. Indeed, activated GPCRs have been shown to recruit PIP5K following release of G proteins through adaptor proteins such as β-arrestin (Nelson et al., 2008).
Figure 3. Polarized accumulation of dPIP5K, PI(4,5)P2, and F-actin is lost in Tre1 mutant GCs.

(A–C) Localization of Venus-tagged dPIP5K in WT GCs and in GCs missing maternal Tre1. Scale bars in (A) and (B), 10 μm.
(D–F) Localization of GFP-tagged PI(4,5)P2 sensor (the PH domain of PLCγ) in WT GCs and in GCs missing maternal Tre1. Scale bars in (D) and (E), 10 μm.
(G–I) Localization of GFP-tagged F-actin sensor (the actin binding domain of Moesin) in WT GCs and in GCs missing maternal Tre1. All GCs are from stage 11 embryos. Trace to the right of each confocal image is from a single cell from each sample (asterisk). Counting of the peak numbers in the fluorescent intensity is shown in (C), (F), and (I). Scale bars in (G) and (H), 10 μm.
(J) The variance of fluorescent intensity to quantify polarized distribution of each marker on PM in each genetic background—at least four to six embryos per genotype were scored with a minimum of 50 total GCs. Mean and SEM are shown.
See also Figure S5.
WASP and WIP are expressed in GCs and are required for their navigation
Although the aforementioned pathway suggests a mechanism for how Tre1 can regulate PI(4,5)P2, it does not directly link Tre1 to actin assembly. A well-known actin regulator downstream of PI(4,5)P2 is WASP, an actin-nucleation-promoting factor and activator of the Arp2/3 complex. PI(4,5)P2 releases WASP from an auto-inhibitory conformation by binding to the Pleckstrin homology (PH) domain and exposing the VCA domain (Figures 4A and 4B). The exposed verprolin homology sequence (V) that binds monomeric actin and central (C) and acidic sequences (A) (VCA) domain activates Arp2/3-complex-mediated actin polymerization (Figure 4B) (Padrick and Rosen, 2010). In migrating GCs, WASP is expressed but not enriched at the F-actin protrusions; GC-specific expression of GFP-tagged WASP shows a similar localization (Figures 4C-4H′). Importantly, dWIP, which binds WASP in its open PI(4,5)P2-bound configuration and stabilizes active WASP protein, is enriched at the F-actin protrusions (Figures 4I-4K″; average Pearson’s correlation coefficient = 0.80 ± 0.18; Anton et al., 2007; Kim et al., 2007; Massarwa et al., 2007), consistent with local pools of PI(4,5)P2 activating WASP to drive actin assembly.
Figure 4. The requirement of WASP and dWIP in GC navigation.

(A) A signaling cascade from GPCR to WASP via PIP5K and PI(4,5)P2.
(B) WASP is activated by interaction with PI(4,5)P2 and WIP to induce Arp2/3-mediated actin polymerization. Cartoons here and in (A) were adapted from Min and Defea (2011).
(C–D″) Staining of endogenous WASP tagged with GFP (green) and F-Actin (red) in GCs (Vasa, blue) of a wasp GFP-trap line. The stage 11 embryo is in a dorsal view with the A-P axis indicated. Scale bars, 10 μm.
(E and E′) The line scan of fluorescent signals on the entire GC circumference in (D″) and the Pearson’s correlation coefficients between fluorescent signals from GCs in (C″). Scale bars, 10 μm.
(F–G″) F-Actin (red) and GFP-tagged WASP (green) expression driven by nos-Gal4 in GCs (blue, Vasa). The stage 11 embryo is in a dorsal view with the A-P axis indicated. WASP is not enriched at the F-actin (red) protrusions (arrowheads in D″ and G″). Scale bars, 10 μm.
(H and H′) The line scan of fluorescent signals on the entire GC circumference in (G″) and the Pearson’s correlation coefficients between fluorescent signals from GCs in (F″). Scale bars, 10 μm.
(I–J″) dWIP (green) antibody staining shows its colocalization with F-actin (red) at the protrusions (arrowhead) in GCs (blue, Vasa). The stage 11 embryo is in a lateral view, and the A-P and D-V axes are indicated. Scale bars, 10 μm.
(K and K′) The line scan of fluorescent signals on the entire GC circumference in (J″) and the Pearson’s correlation coefficients between fluorescent signals from GCs in (I″). GCs boxed in (C″), (F″), and (I″) are shown in magnified views in (D)–(D″), (G)–(G″), and (J)–(J″), respectively. Scale bars, 10 μm.
(L–S) Vasa staining of (L) WT embryo, (M and N) embryos homozygous for the wasp1 or wasp3 allele, (O) wasp zygotic mutant embryo carrying two null alleles in trans, (P and Q) embryo missing zygotic function of dpip5k or both dpip5k and wasp, and (R and S) embryos mutant for dwip or carrying a null allele of dwip over a deficiency that deletes dwip. Note that loss of zygotic dpip5k exacerbates the GC migration defect in wasp1/wasp3. Open arrowheads indicate GC destination of the somatic gonad.
(T) Quantification of embryos with different numbers of mis-migrated GCs in all genotypes from (L) to (S). The statistical analysis of the phenotypes is provided in Table S3.
To test if WASP and/or Drosophila WASP-interacting protein (dWIP) are involved in GC navigation, we examined GC localization in embryos missing zygotic function of the corresponding genes. Loss of wasp resulted in significant GC navigation defects (Figures 4L-4O and 4T), whereas loss of dwip resulted in lower-level GC migration defects (Figures 4R-4T). The defects observed with zygotic loss of wasp alone were intensified in dpip5k; wasp double mutants, supporting a functional requirement for both proteins in GC navigation and consistent with the proteins functioning in the same pathway (Figures 4P, 4Q, and 4T). Altogether, the localization data and the GC migration defects observed in mutants are consistent with roles for WASP and dWIP in activating actin polymerization downstream of Tre1 and PI(4,5)P2 in GC navigation.
Hh signaling and GC navigation
Whereas the requirement for Tre1 in GC navigation has been clearly demonstrated (this study; Kamps et al., 2010; LeBlanc and Lehmann, 2017), how Tre1 is activated in GCs remains unknown. Indeed, Tre1 is one of many orphan receptors with no known ligand (Hanlon and Andrew, 2015). As described in the Introduction, Hh has been proposed as an attractive guidance cue for GC migration (Deshpande et al., 2001, 2017; Deshpande and Schedl, 2005); this suggestion is controversial, however, due in part to questions regarding the expression of necessary downstream signaling components within the GCs (Renault et al., 2009). To address these issues, we re-examined the localization of the early Hh signaling components.
We took advantage of a line carrying a Bac construct encoding a functional GFP-tagged version of Hh, which has been shown to recapitulate endogenous expression and protein localization (Chen et al., 2017). Interestingly, we observed not only the previously reported Hh expression in the co-migrating SGPs, but also high levels of Hh-GFP in large intracellular puncta within GCs (Figures 5A-5B″). Indeed, we observed Hh-positive puncta in GCs from stage 9, when they are in the endoderm pocket, through stage 14, when they coalesce with SGPs to form the gonad (Figure S6 shows single confocal slices revealing Hh puncta within GCs). smo mRNA is observed at the posterior end of Drosophila embryos where GCs (also called pole cells) form at very early stages [Berkeley Drosophila Genome Project], and both Smo and Ptc proteins are detectable in GCs (Figures 5C-5E″). We also observe variable low-level expression of βgal (UAS-lacZNZ) driven by a ptc-Gal4 driver that reflects reported ptc activation downstream of Hh, although levels are not as high as in surrounding somatic tissues (Figures 5F-5F″). Importantly, Smo is enriched near F-actin protrusions (Figures 5D-5D″). To more easily localize Ptc and Smo in only migrating GCs, we expressed either GFP-tagged Smo or GFP-tagged Ptc in GCs using the nos-Gal4 driver. GFP-Smo was highly concentrated with F-actin often at protrusions (average Pearson’s correlation value 0.64 ± 0.14; Figures 5G-5I′), suggesting that Smo could function to organize F-actin in the GCs just as Tre1 does. In contrast, GFP-Ptc staining did not coincide with the F-actin protrusions (average Pearson’s correlation value 0.25 ± 0.17; Figures 5J-5L′). Indeed, high levels of GFP-Ptc were often observed in large intracellular puncta, much like those seen with Hh in GCs, with relatively lower levels in the PM (Figures 5J and 5K). This finding suggests the possibility that Ptc-bound Hh is internalized in GCs, fully consistent with a role for Hh signaling in GC navigation.
Figure 5. Hh uptake and Smo localization in migrating GCs.

(A–B″) GFP-tagged Hh protein, expressed under the control of its endogenous promoter in a Bac transgene, is stained with anti-GFP antibody (green) (A and A″). Nuclear Eya (red) (A′ and A″) and cytoplasmic Vasa (blue) (A″) staining indicate SGPs and GCs, respectively. A GC boxed in (A″) is magnified in (B)–(B″). Arrowheads in (B″) indicate Hh proteins internalized and accumulated in GCs. Scale bars, 10 μm.
(C–E) Smo and Ptc are expressed in migrating GCs. Immunostaining with anti-Smo (C) or anti-Ptc (E) antibody reveals the presence of Smo and Ptc proteins in GCs. A GC boxed in (C″) is magnified in (D)–(D″) and shows colocalization of Smo and F-actin at the protrusion (arrowhead in D″). Note the segmented staining patterns with both antibodies through the embryos showing the developmental expression patterns of these Hh pathway components. Arrows indicate representative GCs showing Smo (C) or Ptc (E) expression. Scale bars, 10 μm.
(F–F″) Expression of the ptc-Gal4 shown by UAS-lacZ expression, which reports on canonical Hh signaling. A subset of migrating GCs exhibits lacZ expression shown by anti-β-Galactosidase antibody staining (arrowheads in F–F″). Scale bars, 10 μm.
(G–H″) GFP-Smo (green) and F-actin (red) colocalize to the leading edge of migrating GCs. Scale bars, 10 μm.
(I and I′) The line scan of fluorescent signals on the entire GC circumference in (H″) and the Pearson’s correlation coefficients between fluorescent signals from GCs in (G″).
(J–K″) GFP-Ptc (green) and F-actin (red) do not colocalize. Scale bars, 10 μm.
(L and L′) The line scan of fluorescent signals on the entire GC circumference (the cell at bottom) in (K″) and the Pearson’s correlation coefficients between fluorescent signals from GCs in (J″).
GCs boxed in (G″) and (J″) are shown in magnified views in (H)–(H″) and (K)–(K″), respectively. The A-P (dorsal view) and A-P/D-V axes (lateral view) are indicated. All GC images are from stage 11 embryos.
Complete loss of zygotic hh results in severe GC migration defects (Deshpande et al., 2001; our own findings that 100% of hh mutants have severe migration defects). However, these migration defects could be an indirect consequence of the segmental patterning defects of hh nulls. Supporting this idea, we find that loss of wg, which is also required for segmental patterning, also results in severe GC mis-migration. However, only the complete loss of hh and not of wg resulted in the early GC mis-migration we observe with loss of Tre1 (Figure S7). We also examined GC migration in embryos heterozygous for the Mrt allele of hh (hhMrt) (Deshpande and Schedl, 2005), which deletes 24 nt of the hh enhancer and results in ectopic hh expression in wing discs (Pérez et al., 2011). We observed significant GC mis-migration in hhMrt heterozygotes (Figures 6A, 6B, and 6I) with no accompanying segmental patterning defects, consistent with the migration defects observed by others with ectopic Hh (Deshpande et al., 2001, 2013). When Smo function was reduced by maternally driven RNAi, we also observed significant GC navigation defects in the absence of any of the segmentation defects associated with zygotic loss of Smo (Figures 6C-6F and 6I). Moreover, smo RNAi also resulted in the same early GC navigation defects and occurrence of GCs on the outside of the embryo as observed in Tre1 mutant embryos and in hh nulls (Figures 6J-6L), further supporting a role for Smo in GC migration.
Figure 6. Smo activity is required for GC navigation and the polarized localization of PI(4,5)P2.

(A–H) Stage 15/16 embryos of the genotype indicated in lower left of each panel stained with Vasa. Open triangles indicate position of GC coalescence in gonad.
(I) Quantification of embryos with different numbers of mis-migrated GCs in all genotypes from (A)–(H). The statistical analysis of the phenotypes is provided in Table S4.
(J–L) Early embryos stained with Vasa reveal GC mis-migration at very early stages and (L) at a later stage (arrows).
(M–R) PM localization of PI(4,5)P2 sensor in WT GCs (M and N), GFP-Smo-expressing GCs (O and P), and GFP-Ptc-expressing GCs (Q and R). All GCs are from stage 11 embryos. Scale bars, 10 μm.
(S) The variance of fluorescent intensity for the PI(4,5)P2 sensor in each genetic background—four to six embryos and at least 60 GCs were analyzed for each genotype. Mean and SEM are shown.
Based on the Smo requirement in GC navigation and its colocalization with F-actin, we next asked if it also localizes with molecules that function downstream of activated Tre1 to facilitate actin polymerization. Indeed, GFP-Smo colocalizes with the PI(4,5)P2 sensor PLCδPH-mCherry in migrating GCs (Figures 6M-6O′). The polarized localization of PI(4,5)P2 and F-actin in GFP-Smo embryos suggests that the GCs in these embryos should properly navigate to the somatic gonad, which we found to be the case (Figures 6G and 6I). Since GCs mis-migrate in hh, ptc, and smo mutant embryos (Deshpande and Schedl, 2005; Deshpande et al., 2001), we predicted that interfering with Smo signaling would also result in loss of PI(4,5)P2 localization. Smo signaling can be blocked by overexpression of its negative regulator, Ptc. As expected, overexpression of GFP-Ptc in GCs not only perturbed the localization of the PI(4,5)P2 sensor (Figures 6Q-6S), but also caused significant, low-level GC navigation defects (Figures 6H and 6I). Overall, these findings are consistent with a role for Hh signaling through Smo in guiding GC migration through the same actin polymerization mechanism as Tre1.
Hh-activated Smo recruits and/or stabilizes Tre1 in the PM
The observation that Smo localizes with PI(4,5)P2 and F-actin in migrating GCs prompted us to investigate how Tre1 integrates into this model. We hypothesize that activation of Smo recruits and/or stabilizes Tre1 in the PM and that Tre1 then recruits dPIP5k to synthesize PI(4,5)P2 to facilitate actin assembly. Classically, Smo accumulates on the PM in response to Hh, a response that can be recapitulated in S2R+ cells, which do not express endogenous Hh (Figures 7A-7D″; Flybase; Thurmond et al., 2019). We then asked if Hh-mediated PM localization of Smo also affected the localization of Tre1 and its downstream effectors. To address this question, we expressed Tre1-HA, GFP-Smo, and FLAG-dPIP5K and induced signaling by treating the cells with Hh-conditioned media. In these transiently transfected S2R+ cells, not all cells expressed all three proteins, allowing for internal controls within the same field of view. In cells lacking Smo (indicated by lack of blue GFP signal and “−”), Tre1 localized primarily to intracellular puncta with only low-level PM staining (Figures 7E-7G‴). Strikingly, in nearby cells where GFP-Smo had been activated by Hh (indicated by the presence of blue GFP on the PM and “+”), Tre1 localization was reduced in intracellular puncta and relatively higher at the PM (Figures 7E-7G″). dPIP5K exhibited constant localization on the PM in S2R+ cells regardless of the activation of Smo (Figures 7E″, 7F″, and 7G″; see also merged images). This membrane localization is also visible in Figures S4E-S4H″—except in the case of expression of Tre1-GFP, where it accumulates with Tre1-GFP in large intracellular puncta—and suggests that dPIP5K localization in S2R+ cells differs from its localization in GCs (Figures 2L-2P″). In any case, these results suggest that activation of Smo by Hh stabilizes Tre1 at the PM and/or causes Tre1 to translocate to the PM.
Figure 7. Smo activation increases Tre1 PM localization and association with dPIP5K in S2R+ cells.

(A–D″) Smo is derepressed and translocated to the PM in the presence of Hh in S2R+ cells. GFP-Smo localizes to intracellular vesicles in control medium (A and B′), whereas much of Smo localizes to the PM in cells treated with Hh-conditioned medium (C and D′). Scale bars, 10 μm.
(E–G%) S2R+ cells transfected with constructs expressing GFP-Smo (blue), Tre1-HA (red), and FLAG-dPIP5K (green) in the presence of Hh-conditioned media. Note that whereas Tre1-HA is largely in intracellular puncta in the cell without membrane-associated GFP-Smo ([−] cells marked in E‴, F‴, and G‴), Tre1-HA is also observed in the PM in the cell with membrane-associated GFP-Smo ([+] cells in E‴, F‴, and G‴). Scale bars, 10 μm.
(H and H′) Hh treatment increases Tre1 localization on PM. A sample blot showing the increased level of biotinylated (PM-localized) Tre1-FLAG in cells treated with Hh-conditioned media (H). Levels of biotinylated Tre1-FLAG were quantified from six independent experiments (H′).
(I and I′) Tre1 association with dPIP5K is increased by Hh treatment. (I) Co-IP of dPIP5K with Tre1-HA exposed to control or Hh-conditioned media. Comparisons of levels of dPIP5K associated with Tre1-HA with control or Hh-conditioned media were quantified from four independent experiments in (I′).
(J) Model for how Hh signaling directs GC navigation through Tre1. Localized Hh signaling relieves Ptc repression of Smo, which allows Smo to translocate from intracellular vesicles to the PM. At the PM, activated Smo increases/prolongs Tre1 localization. Activated Tre1 then recruits dPIP5K to synthesize local pools of PI(4,5)P2. In turn, PI(4,5)P2 activates WASP with dWIP to facilitate localized assembly of F-actin at the leading edge of migrating GCs. Polarized actin assembly and consequent protrusion formation leads directional migration of GCs.
To confirm that Hh signaling increases PM levels of Tre1, we used a biochemical approach. In this approach, we exclusively labeled proteins on the cell surface with cell-membrane-impermeable biotin and compared the amounts of biotinylated Tre1 in the absence and presence of Hh. Hh increased Tre1 on the PM by about 2-fold, on average (Figures 7H and 7H′). Correspondingly, we found that the physical association of Tre1 with dPIP5K increased with Hh treatment by about 3-fold under the same conditions (Figures 7I and 7I′). These observations support a model wherein Hh signaling through Smo localizes/stabilizes Tre1 on the PM to facilitate PI(4,5)P2 synthesis through dPIP5K (Figure 7J).
DISCUSSION
In this study, we provide evidence from both Tre1 loss-of-function analysis and analysis of an abnormally stable form of the protein (Tre1-GFP) that Tre1 activation results in the polarized accumulation of dPIP5K to generate localized PI(4,5)P2. Our findings further suggest a model wherein localized PI(4,5)P2 activates WASP to induce locally boosted Arp2/3-mediated actin polymerization at the migration front (Figure 7J). The failure of GCs to polarize dPIP5K, PI(4,5)P2, and F-actin explains the navigation defects of the Tre1KO mutants. Likewise, the altered distributions of PI(4,5)P2 and F-actin associated with Tre1-GFP can explain the GC navigation defects associated with expression of this highly stable form of Tre1. We also provide evidence that Hh-Smo signaling guides GC navigation. Smo, the signal transducer in the Hh signaling pathway, is enriched at the migrating front in GCs, colocalizing with PI(4,5)P2 and F-actin and, by extension, Tre1, dPIP5K, activated WASP, and WIP. When activated by Hh, Smo localizes to the PM. Activated Smo induces and/or stabilizes Tre1 localization at the PM and increases Tre1 binding to dPIP5K to presumably facilitate local PI(4,5)P2 synthesis for robust actin assembly. Taken together, these findings suggest a mechanism for directional cell migration whereby Hh signaling provides the directional guidance cue to migrating GCs through Smo to regulate actin cytoskeleton dynamics via Tre1.
Our model is consistent with the report showing that GCs harboring mutations in Tre1’s conserved motifs are found outside the midgut (LeBlanc and Lehmann, 2017) and that Tre1 null GCs are randomly migrating from very early stages (Lin et al., 2020), indicating that Tre1 mutant GCs are capable of migration but not navigation. Furthermore, GCs of Tre1sctt, which lacks key residues required for interactions with G-proteins, are unable to maintain directionally relevant F-actin protrusions and instead form abnormal protrusions in random directions (LeBlanc and Lehmann, 2017) (Figure 7J). Our work suggests a role for Tre1 in migrating GCs to locally elevate F-actin assembly through PI metabolism to direct persistence in migration (Figure 7J). GCs mutant for Tre1 in all the genotypes we tested were scattered throughout the embryos. More importantly, Tre1 mutant GCs move out of the midgut (often heading in the wrong direction) from very early stages, indicating that Tre1 function is not required to initiate or maintain migration but is instead required for directionality (Figure 1).
We also provide supporting evidence that Hh guides GCs via the Ptc and Smo signaling cascade (Deshpande and Schedl, 2005; Deshpande et al., 2001). We show that Hh uptake is observed in GCs from very early stages, even before these cells have begun to exit the endoderm (Figure S6). The early mis-migration of GCs observed with loss of Hh, Smo, and Tre1 suggests that Hh could be the guidance cue for all stages of GC navigation, even well before the association of GCs with the SGPs. The broad expression pattern of Hh in developing embryos, however, raises a reasonable question about its effectiveness as an attractant cue (Renault et al., 2009). This potential issue can be explained by the localized expression of positive regulators of Hh activity. The cofactors necessary for Hh secretion and trafficking are expressed in more limited spatial domains than Hh, including 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR), the enzyme that catalyzes the rate limiting step of cholesterol biosynthesis. Cholesterol modification of Hh is essential for the range and shape of Hh gradients (Jeong and McMahon, 2002). HMGCR, which is not only required for GC navigation but can also result in GC mis-migration when overexpressed (Kenwrick et al., 2019), is normally expressed throughout the mesoderm surrounding the midgut endoderm early (Van Doren et al., 1998; stage 9). Later, HMGCR is expressed more specifically in SGPs (Deshpande et al., 2009, 2013; Deshpande and Schedl, 2005; Van Doren et al., 1998), consistent with a role for this protein in limiting the spatial distributions of active Hh at all stages of GC migration (Deshpande et al., 2009, 2013; Deshpande and Schedl, 2005; Van Doren et al., 1998). Indeed, the finding that ectopic HMGCR expression can result in GC mis-migration even in the absence of Hh (challenging results to interpret given that GCs are already mis-migrating without Hh) can be explained by the observation that the binding of cholesterol and oxysterols to the extracellular N-terminal domain of Smo is sufficient to activate Smo signaling (Luchetti et al., 2016; Nachtergaele et al., 2013). Importantly, there is no evidence that cholesterol or the oxysterols (the oxygenated derivatives of cholesterol) are endogenous Smo ligands (Nachtergaele et al., 2013), suggesting that HMGCR is normally acting through its effects on Hh (and/or some yet-to-be-discovered cholesterol-dependent ligand).
Our findings further suggest that Hh signaling in GCs might not follow the canonical Hh signaling pathway. In the canonical model, ptc expression is transcriptionally upregulated in response to Hh signaling (Robbins et al., 2012), a regulatory event we did not explore in these studies other than to show that we can detect low-level Hh-dependent Ptc expression in WT GCs. The finding by others that expression of neither dominant-negative nor constitutively active forms of the Ci transcription factor affect GC migration (Renault et al., 2009) is consistent with the notion that the Hh signaling cascade in GCs is non-canonical. Thus, we propose that Tre1 is directly activated by Smo downstream of Hh signaling. This hypothesis is supported by evidence that Tre1 localization to the PM is induced and/or stabilized by Hh activation of Smo in S2R+ cells (Figure 7), which do not express ci transcripts (Brown et al., 2014). Additionally, both loss of Tre1 and Ptc overexpression in GCs result in similar PI(4,5)P2 polarization defects (Figures 3 and 6).
To our knowledge, the proposed non-canonical pathway of Smo signaling through Tre1 to regulate PI metabolism and cytoskeleton dynamics is novel. However, Smo has been shown to regulate the localization of other GPCRs during canonical Hh signaling. In vertebrate primary cilia, Shh-activated Smo relocates Gpr161, a negative regulator of Gli activation, from the cilia to the cell body in a Grk2- and β-arrestin-dependent manner (Mukhopadhyay et al., 2013; Pal et al., 2016). Conversely, an orphan GPCR, Gpr175, relocalizes to the primary cilia upon Hh stimulation in a Smo-dependent manner (Singh et al., 2015). Whereas it is clear that Smo can regulate the localization of other GPCRs, how this process occurs is not well understood, although there are several possible mechanisms. First, Smo may enhance exocytosis or suppress endocytosis. In Drosophila olfactory sensory neurons, odorant receptors are trafficked to primary cilia by the kinesin-like protein Costal-2 (Cos2) that is regulated by Hh- and Smo-dependent signaling (Sanchez et al., 2016). Second, Smo may physically bind and activate/stabilize GPCRs on the PM. Heterodimerization of certain GPCRs is required for proper localization and functional activation (Prinster et al., 2005). In this case, within GCs, Smo would be the functional ligand or stimulant for Tre1. In light of this scenario, it is relevant that Smo has been shown to not only bind PI(4)P upon activation, but also to require PI(4)P binding for its signaling function (Jiang et al., 2016; Yavari et al., 2010). These discoveries suggest that Smo may not only stabilize Tre1 at the membrane but may also provide a proximal source of the substrate for the dPIP5K that associates with Tre1 upon activation. The requirement for PI(4)P binding for Smo function also provides a feedback mechanism for the subsequent downregulation of signaling as the PI(4)P is converted to PI(4,5) P2 by dPIP5K. Alternatively, Smo may affect GPCR localization by regulating post-translational modification of Tre1 through Smo-engaged kinases like Fu, PKA, and Gprk2. In support of this possibility, Deshpande et al. (2001) reported that loss of fu or of pka causes GC migration defects. Finally, Smo and Tre1 may localize to the same secretory vesicles that get activated and fuse with the PM upon Hh activation. Indeed, our preliminary findings suggest that this could be the case, at least in S2R+ cells. Further study of how Smo regulates Tre1 localization in GCs will allow us to distinguish among these possibilities.
Tre1 is believed to exert its downstream effects through classical G-protein signaling. GCs lacking Gγ1 or Gβ13f are unable to migrate properly (Kunwar et al., 2008), and conserved residues in Tre1 that are necessary for Gα signaling are required for GC navigation (Kamps et al., 2010; LeBlanc and Lehmann, 2017). Although no single Gα protein has been identified as a binding partner of Tre1, overexpression of Gαo-GDP in GCs results in Tre1-like GC migration defects (LeBlanc and Lehmann, 2017), and Tre1 and Gαo interact to direct the orientation of neuroblast cell division (Yoshiura et al., 2012). Our observed interaction between Tre1 and dPIP5K (Figures 7I and 7I′) could be facilitated indirectly via the heterotrimeric G proteins or other intermediate molecules. One intriguing candidate for this is β-arrestin. Classically, β-arrestins are known for their role in receptor recycling; however, studies indicate that β-arrestins can also function as signaling scaffolds through local recruitment and activation of cellular enzymes. β-arrestin has been shown to bind and activate PIP5K enzymes to generate PI(4,5)P2 upon GPCR activation (Nelson et al., 2008). Although the sole β-arrestin (outside the visual system) in Drosophila, kurtz (krz), has been shown to be required for GC development, its role in GC migration remains to be revealed (Molnar et al., 2011). Understanding how Tre1 coordinates its downstream effectors like dPIP5K, Rho1, and the heterotrimeric G proteins during GC navigation will be important to better understand the complexity of GPCR signaling.
In conclusion, we propose that Tre1 functions as a master helmsman in GC navigation by integrating extracellular signals to induce intracellular changes. Future studies on Tre1—including its trafficking, post-translational modifications, and interacting proteins—will provide a more general understanding of GPCR function and regulation during in vivo cell migration. Our discoveries provide a better understanding of GC guidance mechanisms by potentially reconciling the controversial findings on the role of Hh signaling in GC migration. More broadly, aberrant Hh signaling has been implicated in the metastasis of many cancer types (Skoda et al., 2018), and PIP5Ks have been increasingly shown to have pathological relevance in cancer metastasis (Semenas et al., 2014; Yamaguchi et al., 2010). These studies suggest that PIP5K enzymes could be important downstream effectors in the migration of metastatic cancer cells in response to Hh signaling. Altogether, our work has broad implications in the fields of both developmental biology and cell migration.
STAR ★METHODS
RESOURCE AVAILABILITY
Lead contact
Any additional information and/or requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Deborah J. Andrew (dandrew@jhmi.edu)
Materials availability
All antibodies and fly lines generated in this study are available from the Lead Contact without restriction.
Data and code availability
This study did not generate/analyze datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila strains and cell culture
Drosophila crosses and embryo collections were performed at 25°C, unless otherwise indicated. The Drosophila lines used include: Oregon-R (wild-type control; Bloomington Drosophila Stock Center), Tre1KO (this study), Tre1ΔEP5, Tre1ΔEP19 (Ueno et al., 2001), Tre1sctt (Coffman et al., 2002), nos-Gal4 (Van Doren et al., 1998), UAS-Tre1, UAS-Tre1-Flag, UAS-Tre1-HA, UAS-Tre1-GFP (this study), UAS-GFP-PLCγPH (Pinal et al., 2006), UAS-PLCδPH-mCherry, tGPH (Bloomington Drosophila Stock Center), UAS-PX-Phox40-GFP, UAS-GFP-PH-FAPP1, UAS-GFP-PH-TAPP1, UAS-GFP-Atg18 (gifts from Amy Kiger), UAS-TMEM-GFP (Finley et al., 1998; Fox and Andrew, 2015), UAS-GFP-Smo, UAS-GFP-Ptc, UAS-lacZNZ (Bloomington Drosophila Stock Center), UAS-Venus-dPIP5K (this study), UAS-HA-dPIP5KKD, dpip5k18, dpip5k30 (Chakrabarti et al., 2015), sktlΔ15 (Hassan et al., 1998), TRiP.HMC05328 (UAS-dPIP5K RNAi stock), TRiP.GL00072 (UAS-sktl RNAi stock), wasp1, wasp3, waspMI05533-GFSTF.0, Df(2R) Exel7170 (Bloomington Drosophila Stock Center), UAS-GFP-WASP (Jin et al., 2011), dwipS1946 (Kim et al., 2007), y1w*;P{70FLP} 11 P{70I-SceI}2B nocSco /CyO,S2, TRiP.HMC03577 and TRiP.GL01472, (UAS-Smo RNAi stocks), wg1-8; pftz-lacZ, hhAC/TM3, Sb1, hh21/TM3, Sb1, and hhMrt/TM3, Sb1 (Bloomington Drosophila Stock Center). For Hh localization in SGPs and GCs, we used the Hh:GFP Bac provided by the Kornberg Lab (Chen et al., 2017). To express a UAS-transgene in embryonic GCs in rescue and overexpression experiments, virgins carrying nos-Gal4 were crossed with the UAS-transgene male flies. To knock down maternal contributions of different genes, we used maternal-Tub-Gal4 (Staller et al., 2013).
S2 and S2R+ cells were cultured in Schneider’s medium (GIBCO) supplemented with 10% fetal bovine serum (FBS) (GIBCO) and penicillin/streptomycin (Sigma). S2 cells stably expressing Hh-N (S2-HhN; a gift from J. Jiang) were selected with 200 μg/ml hygromycin B (Invitrogen). Effectene (QIAGEN) was used to transfect cells to express proteins according to the manufacturer’s protocol.
Tre1KO and UAS expression lines
The Tre1KO flies were generated by homologous recombination following the protocol of Rong and Golic (2001), replacing the region spanning from ~540 nt upstream of the ATG start codon of Tre1 up to the residues encoding the last 23 residues of the Tre1 ORF. The UAS constructs to express Tre1, dPIP5K, PLCδPH, and Smo were generated by PCR and Gateway Cloning system (Invitrogen) into either the pTW, pTWG, pTWH or pTWF vectors. Template DNAs used for PCR were as follows: RE07751 (Tre1, Drosophila Genomics Resource Center [DGRC]), FI05352 (dPIP5K, DGRC), genomic DNA extracted from the UAS-PLCδPH-mCherry line (PLCδPH), and genomic DNA extracted from the UAS-Smo-GFP line (Smo).
METHOD DETAILS
Embryo in situ hybridization, immunostaining and imaging
Whole-mount in situ hybridization and fluorescence in situ hybridization were performed as described (Knirr et al., 1999; Lehmann and Tautz, 1994) using an anti-sense digoxygenin-labeled Tre1 RNA probe generated from cDNA RE07751. For immunostaining, embryos were fixed with 4% formaldehyde and devitellinized with methanol. For F-actin staining with phalloidin, the vitelline membrane was removed by hand after fixation. The dilution ratios of primary antibodies follow: rabbit α-Vasa (1:100, Santa Cruz), rabbit α-GFP (1:500, Molecular Probes), rabbit α-βGalactosidase (1:100, Invitrogen), rat α-Vasa (1:100, DSHB), rat α-dPIP5K (1:100, a gift from P. Raghu), rat α-dWIP (1:50, a gift from E. Chen), mouse α-Flag (1:200, Sigma-Aldrich), chicken α-GFP (1:500, Abcam), mouse α-Eya (1:10, DSHB), mouse α-Smo (1:10, DSHB) and mouse α-Ptc (1:10, DSHB). Fluorescence- or biotin-conjugated secondary antibodies (Life technologies) were used at 1:200. To visualize filamentous actin, Alexa Fluor 488- or Alexa Fluor 568-conjugated phalloidin (ThermoFisher) were used at 1:200. DIC images were obtained on an Axiophot microscope (Zeiss) equipped with ProgRes CapturePro (Jenoptik). Fluorescent images were obtained on an LSM 700 Meta confocal microscope (Zeiss) equipped with Zen software (Zeiss) and processed using Adobe Photoshop CS.
For live imaging of migrating GCs, embryos were collected and dechorionated in 50% bleach. Subsequently, embryos were washed in water and covered with a layer of Halocarbon oil 700/27 (2:1; Sigma) on a microslide. Time lapse image acquisition was carried out on an LSM 700 Meta confocal microscope (Zeiss).
Cultured cell immunostaining and imaging
For immunofluorescent staining of culture cells, cells were fixed with 4% formaldehyde in PBS, washed in PBST (PBS with 0.1% Triton X-100) and PBSTB (PBST with 0.2% BSA) consecutively. The primary antibodies were used in PBSTB with the dilution ratios that follow: mouse α-Flag (1:1000), rabbit α-HA (1:500), rabbit α-GFP (1:1000), chicken α-GFP (1:1000). To stain cell surface GFP-Smo proteins exclusively, chicken α-GFP was treated in PBS without detergent. Secondary Alexa Fluor 488-, Alexa Fluor 568-, or Alexa Fluor 647-conjugated antibodies were used at 1:400. To visualized F-actin, Alexa Fluor 488- or Alexa Fluor 568-conjugated phalloidin was used at 1:500 in PBST. DAPI (0.1 μg/ml) was used to stain nuclei. Fluorescent images were obtained on an LSM 700 Meta confocal microscope (Zeiss), acquired with Zen software (Zeiss), and processed using Adobe Photoshop.
The preparation and treatment of Hh-conditioned medium was performed as described previously (Chen et al., 2010). Hh-conditioned medium was prepared by inducing the secreted Hh, HhN, expression in the S2-HhN cells with 0.7 mM CuSO4 in the absence of hygromycin B for 24-48 hours. The collected Hh-conditioned medium was diluted with fresh medium at 6:4 ratio and applied to S2R+ cells for 24-48 hr.
Co-immunoprecipitation
S2R+ cells were transfected to express proteins of interest. After harvesting cells by centrifugation, cells were washed with cold PBS and lysed in Lysis buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% NP-40) containing a protease/phosphatase inhibitor cocktail (ThermoFisher). After centrifugation, supernatants were incubated with the indicated antibodies at 4°C for 2-3 hr. Protein A/G agarose resin (Pierce) was used to precipitate the antibodies. Immunoprecipitated proteins were analyzed by SDS-PAGE and western blot.
Cycloheximide chase of Tre1
S2R+ cells were transfected with constructs for expression of tagged Tre1 proteins. Cells were treated with 50 μg/ml cycloheximide at 48 hr after transfection and harvested at individual time points. The amounts of Tre1 proteins in whole cell lysates prepared in Lysis buffer containing the protease/phosphatase inhibitor cocktail were analyzed by SDS-PAGE and western blot.
Biotinylation of cell surface proteins
S2R+ cells were transfected to express GFP-Smo and Tre1-Flag. After treatment of control or Hh-conditioned medium, cells were washed with cold PBS (pH 8.0) and incubated with 1 mg/ml cell-impermeable Sulfo-NHS-biotin (ThermoFisher) in PBS for 30 min at room temperature. Biotinylation was terminated by washing cells with 100 mM glycine in PBS 3 times. Cells were harvested and lysed in PBS containing 1% NP-40 and the protease/phosphatase inhibitor cocktail. Streptavidin-agarose resin (Pierce) was used to precipitate biotinylated proteins from the cell lysates. Precipitated proteins were analyzed by SDS-PAGE and western blot.
QUANTIFICATION AND STATISTICAL ANALYSIS
To quantify GC migration phenotypes, embryos were stained with α-Vasa antibody. In embryos older than stage 14, GCs separated from the somatic gonads were counted as mis-migrating cells. The statistical significance between genotypes was calculated using a G-test of maximum likelihood of statistical significance (http://www.biostathandbook.com/gtestind.html). n in each graph (Figures 1Z, 2AF, 4T, and 6I) represents the number of embryos in which GCs were counted.
To quantify fluorescent signals of dPIP5K, the PI(4,5)P2 sensor, and the F-actin probe in GC, the signal intensities on the entire plasma membrane were measured by the ImageJ program (https://imagej.nih.gov/ij/) and normalized with background signals measured from control areas. Normalized data were used to calculate the variance of the fluorescent signals in each cell by Microsoft Excel. Variance is defined as the average of the squared differences from the mean and indicates how far each number in the set is from the mean. Since the majority of WT GCs present a single peak that stands out in the fluorescent signals, variance can be used as an indicator of fluorescence signal polarization. Microsoft Excel and GraphPad Prism softwares were used to generate graphs and calculate statistical values. n in each graph (Figures 3J and 6S) represents the number of GCs in which the fluorescent signals were measured. Dispersion and precision measures (mean, SD, SEM) are described in the figure legends.
To determine the average number of peaks of signal intensity with the markers described above, the signal intensity data from ImageJ scans of the entire plasma membrane of all WT and Tre1 mutant GCs were first binned so that every cell had 36 average pixel intensity measurements. The highest value was then shifted to the middle position for every cell. The number of peaks was calculated by normalizing the data; for all cells, the data for each cell was normalized to its average and was also normalized to the average of all wild-type values, so that the data reflected that the knockout values in the mutants were lower than the wild-type values. This is more easily seen in the line plots for the wild-type and knockout cells (Figure S4). Peaks were defined as five or more consecutive values that were greater than 110 (e.g., greater than 110% of the average) (Figures 3C, 3F, and 3I). All calculations were performed in Microsoft Excel.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Vasa | Santa Cruz | Cat# sc-30210, RRID:AB_793874 |
| Rat monoclonal anti-Vasa | Developmental Studies Hybridoma Bank | Cat# anti-vasa, RRID:AB_760351 |
| Mouse monoclonal anti-Eya | Developmental Studies Hybridoma Bank | Cat#eya10H6, RRID:AB_528232 |
| Mouse monoclonal anti-Smo | Developmental Studies Hybridoma Bank | Cat# 20C6, RRID:AB_528472 |
| Mouse monoclonal anti-Ptc | Developmental Studies Hybridoma Bank | Cat# Apa1, RRID:AB_528441 |
| Rabbit polyclonal anti-βGalactosidase | Invitrogen | Cat# A-11132; RRID:AB_221539 |
| Mouse monoclonal anti-Flag | Sigma-Aldrich | Cat# F3165, RRID:AB_259529 |
| Rabbit polyclonal anti-HA | ThermoFisher | Cat# PA1-985, RRID:AB_559366 |
| Rabbit polyclonal anti-GFP | Molecular Probes | Cat# A-11122, RRID:AB_221569 |
| Chicken polyclonal anti-GFP | Abcam | Cat# ab13970, RRID:AB_300798 |
| Rat polyclonal anti-dPIP5K | Gift from Padinjat Raghu (NCBS, India) | Chakrabarti et al., 2015 |
| Rat polyclonal anti-dWIP | Gift from E. Chen (UT Southwestern) | Kim et al., 2007 |
| Alexa Fluor 488-conjugated goat anti-mouse IgG | Invitrogen | Cat# A32723; RRID: AB_2633275 |
| Alexa Fluor 488-conjugated goat anti-rabbit IgG | Invitrogen | Cat# A32731;RRID: AB_2633280 |
| Alexa Fluor 488-conjugated goat anti-rat IgG | Invitrogen | Cat# A11006; RRID: AB_2534074 |
| Alexa Fluor 488-conjugated goat antichicken IgY | Invitrogen | Cat# A11039; RRID: AB_2534096 |
| Alexa Fluor 568-conjugated goat anti-rabbit IgG | Invitrogen | Cat# A-11011; RRID: AB_143157 |
| Alexa Fluor 647-conjugated goat anti-rabbit IgG | Invitrogen | Cat# A32733; RRID: AB_2633282 |
| Alexa Fluor 647-conjugated goat antichicken IgY | Invitrogen | Cat# A-21449; RRID: AB_2535866 |
| Biotin-conjugated goat anti-mouse IgG (H+L) | Jackson ImmunoResearch | Cat# 115-065-003, RRID:AB_2338557 |
| Biotin-conjugated goat anti-rabbit IgG (H+L) | Jackson ImmunoResearch | Cat# 111-065-003, RRID:AB_2337959 |
| Chemicals, peptides, and recombinant proteins | ||
| Cycloheximide | Sigma-Aldrich | Cat# 01810 |
| Protein A/G-agarose resin | Pierce | Cat# 20421 |
| Halt Protease and phosphatase inhibitor cocktail | ThermoFisher | Cat# 78444 |
| Alexa Fluor 488-phalloidin | ThermoFisher | Cat# A12379 |
| Alexa Fluor 568-phalloidin | ThermoFisher | Cat#A12380 |
| DAPI | ThermoFisher | Cat# 62247 |
| EZ-Link Sulfo-NHS-Biotin | ThermoFisher | Cat# 21217 |
| Streptavidin-agarose resin | Pierce | Cat# 20357 |
| Experimental models: cell lines | ||
| D.melanogaster: S2-HhN | Gift from J. Jiang (UT Southwestern) | Chen et al., 2010 |
| D.melanogaster: S2R+ | Gift from E. Chen (UT Southwestern) | RRID:CVCL_Z831 |
| Experimental models: organisms/strains | ||
| D.melanogaster: Oregon-R | Bloomington Drosophila Stock Center | RRID: BDSC_2376 |
| D.melanogaster: Tre1ΔEP5 | Gift from R. Lehmann | Ueno et al., 2001 |
| D.melanogaster: Tre1ΔEP19 | Gift from R. Lehman | Ueno et al., 2001 |
| D.melanogaster: Tre1sctt | Gift from C. Coffman | Coffman et al., 2002 |
| D.melanogaster: Tre1KO | This study | N/A |
| D.melanogaster: dpip5k18 | Gift from Padinjat Raghu (NCBS, India) | Chakrabarti et al., 2015 |
| D.melanogaster: dpip5k30 | Gift from Padinjat Raghu (NCBS, India) | Chakrabarti et al., 2015 |
| D.melanogaster: sktlΔ15 | Gift from J. Brill (Univ. of Toronto) | Hassan et al., 1998 |
| D.melanogaster: wasp1 | Bloomington Drosophila Stock Center | RRID: BDSC_51657 |
| D.melanogaster: wasp3 | Bloomington Drosophila Stock Center | RRID: BDSC_39725 |
| D.melanogaster: dwipS1946 | Gift from E. Chen (UT Southwestern) | Kim et al., 2007 |
| D.melanogaster: wgl-8;pftz-lacZ | Bloomington Drosophila Stock Center | RRID: BDSC_5351 |
| D.melanogaster: Df(2R)Exel7170 | Bloomington Drosophila Stock Center | RRID: BDSC_7901 |
| D.melanogaster: UAS-Tre1 | This study | N/A |
| D.melanogaster: UAS-Tre1-HA | This study | N/A |
| D.melanogaster: UAS-Tre1-Flag | This study | N/A |
| D.melanogaster: UAS-Tre1-GFP | This study | N/A |
| D.melanogaster: UAS-Venus-dPIP5K | This study | N/A |
| D.melanogaster: UAS-HA-dPIP5KKD | Gift from Padinjat Raghu (NCBS, India) | Chakrabarti et al., 2015 |
| D.melanogaster: TRiP.HMC05328 | Bloominton Drosophila Stock Center | RRID: BDSC_62855 |
| D.melanogaster: TRiP.GL00072 | Bloominton Drosophila Stock Center | RRID: BDSC_35198 |
| D.melanogaster: TRiP.HMC03577 | Bloominton Drosophila Stock Center | RRID: BDSC_53348 |
| D.melanogaster: TRiP.GL01472 | Bloominton Drosophila Stock Center | RRID: BDSC_43134 |
| D.melanogaster: UAS-GEP-PLCγ-PH | Gift from M. Baylies (Sloan Kettering Institute) | Pinal et al., 2006 |
| D.melanogaster: UAS-PLCδ-PH-mCherry | Bloominton Drosophila Stock Center | RRID: BDSC_51658 |
| D.melanogaster: tGPH | Bloomington Drosophila Stock Center | RRID:BDSC_8164 |
| D.melanogaster: UAS-GFP-Atg18 | Gift from A. Kiger (UC San Diego) | N/A |
| D.melanogaster: UAS-GFP-PH-TAPP1 | Gift from A. Kiger (UC San Diego) | N/A |
| D.melanogaster: UAS-GFP-PH-FAPP1 | Gift from A. Kiger (UC San Diego) | N/A |
| D.melanogaster: UAS-PX-Phox40-GFP | Gift from A. Kiger (UC San Diego) | N/A |
| D.melanogaster: UAS-TMEM-GFP | S. Hayashi | Fox and Andrew, 2015 |
| D.melanogaster: UAS-lacZNZ | Bloominton Drosophila Stock Center | RRID: BDSC_3956 |
| D.melanogaster: waspMI05533-GFSTF.0 | Bloominton Drosophila Stock Center | RRID: BDSC_60289 |
| D.melanogaster: UAS-GEP-WASP | Gift from E. Chen (UT Southwestern) | Jin et al., 2011 |
| D.melanogaster: y1w*;P{70FLP}11 P{70I-SceI}2B nocSco /CyO,S2 | Bloominton Drosophila Stock Center | RRID: BDSC_6934 |
| D.melanogaster: ptc-Gal4 | Bloominton Drosophila Stock Center | RRID: BDSC_2017 |
| D.melanogaster: y1 w*; nos-Gal4 | Gift from R. Lehmann | Van Doren et al., 1998 |
| Recombinant DNA | ||
| UAS-Tre1 | This study | N/A |
| UAS-Tre1-Flag | This study | N/A |
| UAS-Tre1-HA | This study | N/A |
| UAS-Tre1-GFP | This study | N/A |
| UAS-Venus-dPIP5K | This study | N/A |
| UAS-HA-dPIP5K | This study | N/A |
| UAS-Flag-dPIP5K | This study | N/A |
| UAS-GFP-dPIP5K | This study | N/A |
| UAS-Flag-PLCδPH | This study | N/A |
| UAS-GFP-PLCδPH | This study | N/A |
| UAS-GFP-Smo | This study | N/A |
| Software and algorithms | ||
| Zen | Zeiss | https://www.zeiss.com/microscopy/us/microscope-cameras.html |
| ProgRes CapturePro | Jenoptik | https://www.jenoptik.com/products/cameras-and-imaging-modules/microscope-cameras/progres-camera |
| Photoshop | Adobe | https://www.adobe.com/products/photoshop.html |
| Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Excel | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| G-test of goodness-of-fit | Handbook of Biological Statistics | http://www.biostathandbook.com/gtestind.html |
| Zeiss LSM 700 confocal microscope | Zeiss | https://www.zeiss.com/microscopy/us/products/confocal-microscopes.html |
| Zeiss Axiophot fluorescent microscope | Zeiss | https://www.micro-shop.zeiss.com/de/de |
| Program to visualize Tre1 structures | Protter Interactive Protein Feature Visualization Program | http://wlab.ethz.ch/protter/start/ |
Highlights.
Tre1 localizes with dPIP5K, PI(4,5)P2, and F-actin and is required for their localization
Hh and its receptor Ptc accumulate in intracellular structures within migrating GCs
Smoothened also colocalizes with F-actin in GCs and is required for GC navigation
Hh, through Smo, increases Tre1 at the plasma membrane and its association with dPIP5K
ACKNOWLEDGMENTS
We thank M. Baylies, J. Brill, E. Chen, C. Coffman, S. Hayashi, A. Kiger, T. Kornberg, R. Lehmann, P. Raghu, and C. Samakovlis for fly lines; J. Jiang for the cell line expressing Hh; and T. Kornberg for the Bac line expressing GFP-tagged Hh. We thank S.Y. Chung, R. Loganathan, M. Wells, and T. Kornberg for helpful suggestions during this study. We thank M. de Cuevas for reading and commenting on the manuscript. We also thank the anonymous reviewers whose critiques vastly improved this paper. Funding for this study is from NIH RO1DE013899 awarded to D.J.A., R35GM118177 awarded to P.N.D., and F31DE022233 awarded to C.D.H.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2021.108799.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Antón IM, Jones GE, Wandosell F, Geha R, and Ramesh N (2007). WASP-interacting protein (WIP): working in polymerisation and much more. Trends Cell Biol. 17, 555–562. [DOI] [PubMed] [Google Scholar]
- Barton LJ, LeBlanc MG, and Lehmann R (2016). Finding their way: themes in germ cell migration. Curr. Opin. Cell Biol 42, 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JB, Boley N, Eisman R, May GE, Stoiber MH, Duff MO, Booth BW, Wen J, Park S, Suzuki AM, et al. (2014). Diversity and dynamics of the Drosophila transcriptome. Nature 512, 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti P, Kolay S, Yadav S, Kumari K, Nair A, Trivedi D, and Raghu P (2015). A dPIP5K dependent pool of phosphatidylinositol 4,5 bisphosphate (PIP2) is required for G-protein coupled signal transduction in Drosophila photoreceptors. PLoS Genet. 11, e1004948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Li S, Tong C, Zhao Y, Wang B, Liu Y, Jia J, and Jiang J (2010). G protein-coupled receptor kinase 2 promotes high-level Hedgehog signaling by regulating the active state of Smo through kinase-dependent and kinase-independent mechanisms in Drosophila. Genes Dev. 24, 2054–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Huang H, Hatori R, and Kornberg TB (2017). Essential basal cytonemes take up Hedgehog in the Drosophila wing imaginal disc. Development 144, 3134–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman CR, Strohm RC, Oakley FD, Yamada Y, Przychodzin D, and Boswell RE (2002). Identification of X-linked genes required for migration and programmed cell death of Drosophila melanogaster germ cells. Genetics 162, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande G, and Schedl P (2005). HMGCoA reductase potentiates hedgehog signaling in Drosophila melanogaster. Dev. Cell 9, 629–638. [DOI] [PubMed] [Google Scholar]
- Deshpande G, Swanhart L, Chiang P, and Schedl P (2001). Hedgehog signaling in germ cell migration. Cell 106, 759–769. [DOI] [PubMed] [Google Scholar]
- Deshpande G, Godishala A, and Schedl P (2009). Ggamma1, a downstream target for the hmgcr-isoprenoid biosynthetic pathway, is required for releasing the Hedgehog ligand and directing germ cell migration. PLoS Genet. 5, e1000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande G, Zhou K, Wan JY, Friedrich J, Jourjine N, Smith D, and Schedl P (2013). The hedgehog pathway gene shifted functions together with the hmgcr-dependent isoprenoid biosynthetic pathway to orchestrate germ cell migration. PLoS Genet. 9, e1003720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande G, Manry D, Jourjine N, Mogila V, Mozes H, Bialistoky T, Gerlitz O, and Schedl P (2016). Role of the ABC transporter Mdr49 in Hedgehog signaling and germ cell migration. Development 143, 2111–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande G, Barr J, Gerlitz O, Lebedeva L, Shidlovskii Y, and Schedl P (2017). Cells on the move: Modulation of guidance cues during germ cell migration. Fly (Austin) 11, 200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, and De Camilli P (2006). Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657. [DOI] [PubMed] [Google Scholar]
- Finley KD, Edeen PT, Foss M, Gross E, Ghbeish N, Palmer RH, Taylor BJ, and McKeown M (1998). Dissatisfaction encodes a tailless-like nuclear receptor expressed in a subset of CNS neurons controlling Drosophila sexual behavior. Neuron 21, 1363–1374. [DOI] [PubMed] [Google Scholar]
- Fox RM, and Andrew DJ (2015). Changes in organelle position and epithelial architecture associated with loss of CrebA. Biol. Open 4, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon CD, and Andrew DJ (2015). Outside-in signaling–a brief review of GPCR signaling with a focus on the Drosophila GPCR family. J. Cell Sci 128, 3533–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan BA, Prokopenko SN, Breuer S, Zhang B, Paululat A, and Bellen HJ (1998). skittles, a Drosophila phosphatidylinositol 4-phosphate 5-kinase, is required for cell viability, germline development and bristle morphology, but not for neurotransmitter release. Genetics 150, 1527–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaglarz MK, and Howard KR (1995). The active migration of Drosophila primordial germ cells. Development 121, 3495–3503. [DOI] [PubMed] [Google Scholar]
- Jeong J, and McMahon AP (2002). Cholesterol modification of Hedgehog family proteins. J. Clin. Invest 110, 591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K, Liu Y, Fan J, Zhang J, Li XA, Evers BM, Zhu H, and Jia J (2016). PI(4)P Promotes Phosphorylation and Conformational Change of Smoothened through Interaction with Its C-terminal Tail. PLoS Biol. 14, e1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Duan R, Luo F, Zhang G, Hong SN, and Chen EH (2011). Competition between Blown fuse and WASP for WIP binding regulates the dynamics of WASP-dependent actin polymerization in vivo. Dev. Cell 20, 623–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamps AR, Pruitt MM, Herriges JC, and Coffman CR (2010). An evolutionarily conserved arginine is essential for Tre1 G protein-coupled receptor function during germ cell migration in Drosophila melanogaster. PLoS ONE 5, e11839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenwrick K, Mukherjee A, and Renault AD (2019). Hmgcr promotes a long-range signal to attract Drosophila germ cells independently of Hedgehog. J. Cell Sci 132, jcs232637. [DOI] [PubMed] [Google Scholar]
- Kim S, Shilagardi K, Zhang S, Hong SN, Sens KL, Bo J, Gonzalez GA, and Chen EH (2007). A critical function for the actin cytoskeleton in targeted exocytosis of prefusion vesicles during myoblast fusion. Dev. Cell 12, 571–586. [DOI] [PubMed] [Google Scholar]
- Knirr S, Azpiazu N, and Frasch M (1999). The role of the NK-homeobox gene slouch (S59) in somatic muscle patterning. Development 126, 4525–4535. [DOI] [PubMed] [Google Scholar]
- Kunwar PS, Starz-Gaiano M, Bainton RJ, Heberlein U, and Lehmann R (2003). Tre1, a G protein-coupled receptor, directs transepithelial migration of Drosophila germ cells. PLoS Biol. 1, E80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunwar PS, Siekhaus DE, and Lehmann R (2006). In vivo migration: a germ cell perspective. Annu. Rev. Cell Dev. Biol 22, 237–265. [DOI] [PubMed] [Google Scholar]
- Kunwar PS, Sano H, Renault AD, Barbosa V, Fuse N, and Lehmann R (2008). Tre1 GPCR initiates germ cell transepithelial migration by regulating Drosophila melanogaster E-cadherin. J. Cell Biol 183, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin A, Marygold S, Antonazzo G, Attrill H, dos Santos G, Garapati P, Goodman J, Sian Gramates L, Millburn G, Strelets V, et al. ; FlyBase Consortium (2020). FlyBase: updates to the Drosophila melanogaster knowledge base. Nucleic Acids Research 49, D899–D907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc MG, and Lehmann R (2017). Domain-specific control ofgerm cell polarity and migration by multifunction Tre1 GPCR. J. Cell Biol 216, 2945–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann R, and Tautz D (1994). In situ hybridization to RNA. Methods Cell Biol. 44, 575–598. [DOI] [PubMed] [Google Scholar]
- Lin B, Luo J, and Lehmann R (2020). Collectively stabilizing and orienting posterior migratory forces disperses cell clusters in vivo. Nat. Commun 11, 4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchetti G, Sircar R, Kong JH, Nachtergaele S, Sagner A, Byrne EF, Covey DF, Siebold C, and Rohatgi R (2016). Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. eLife 5, e20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massarwa R, Carmon S, Shilo BZ, and Schejter ED (2007). WIP/WASp-based actin-polymerization machinery is essential for myoblast fusion in Drosophila. Dev. Cell 12, 557–569. [DOI] [PubMed] [Google Scholar]
- Min J, and Defea K (2011). β-arrestin-dependent actin reorganization: bringing the right players together at the leading edge. Mol. Pharmacol 80, 760–768. [DOI] [PubMed] [Google Scholar]
- Molnar C, Ruiz-Gómez A, Martin M, Rojo-Berciano S, Mayor F, and de Celis JF (2011). Role of the Drosophila non-visual β-arrestin kurtz in hedgehog signalling. PLoS Genet. 7, e1001335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, Neher RA, and Renault AD (2013). Quantifying the range of a lipid phosphate signal in vivo. J. Cell Sci 126, 5453–5464. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ, and Jackson PK (2013). The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 152, 210–223. [DOI] [PubMed] [Google Scholar]
- Nachtergaele S, Whalen DM, Mydock LK, Zhao Z, Malinauskas T, Krishnan K, Ingham PW, Covey DF, Siebold C, and Rohatgi R (2013). Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. eLife 2, e01340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CD, Kovacs JJ, Nobles KN, Whalen EJ, and Lefkowitz RJ (2008). Beta-arrestin scaffolding of phosphatidylinositol 4-phosphate 5-kinase Ialpha promotes agonist-stimulated sequestration of the beta2-adrenergic receptor. J. Biol. Chem 283, 21093–21101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson HM, and Nechiporuk AV (2018). Using Zebrafish to Study Collective Cell Migration in Development and Disease. Front. Cell Dev. Biol 6, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padrick SB, and Rosen MK (2010). Physical mechanisms of signal integration by WASP family proteins. Annu. Rev. Biochem 79, 707–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal K, Hwang SH, Somatilaka B, Badgandi H, Jackson PK, DeFea K, and Mukhopadhyay S (2016). Smoothened determines β-arrestin-mediated removal of the G protein-coupled receptor Gpr161 from the primary cilium. J. Cell Biol 212, 861–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peercy BE, and Starz-Gaiano M (2020). Clustered cell migration: Modeling the model system of Drosophila border cells. Semin. Cell Dev. Biol 100, 167–176. [DOI] [PubMed] [Google Scholar]
- Pérez L, Barrio L, Cano D, Fiuza UM, Muzzopappa M, and Milán M (2011). Enhancer-PRE communication contributes to the expansion of gene expression domains in proliferating primordia. Development 138, 3125–3134. [DOI] [PubMed] [Google Scholar]
- Pinal N, Goberdhan DC, Collinson L, Fujita Y, Cox IM, Wilson C, and Pichaud F (2006). Regulated and polarized PtdIns(3,4,5)P3 accumulation is essential for apical membrane morphogenesis in photoreceptor epithelial cells. Curr. Biol 16, 140–149. [DOI] [PubMed] [Google Scholar]
- Prinster SC, Hague C, and Hall RA (2005). Heterodimerization of g protein-coupled receptors: specificity and functional significance. Pharmacol. Rev 57, 289–298. [DOI] [PubMed] [Google Scholar]
- Renault AD, Ricardo S, Kunwar PS, Santos A, Starz-Gaiano M, Stein JA, and Lehmann R (2009). Hedgehog does not guide migrating Drosophila germ cells. Dev. Biol 328, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault AD, Kunwar PS, and Lehmann R (2010). Lipid phosphate phosphatase activity regulates dispersal and bilateral sorting of embryonic germ cells in Drosophila. Development 137, 1815–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricardo S, and Lehmann R (2009). An ABC transporter controls export of a Drosophila germ cell attractant. Science 323, 943–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins DJ, Fei DL, and Riobo NA (2012). The Hedgehog signal transduction network. Sci. Signal 5, re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong YS, and Golic KG (2001). A targeted gene knockout in Drosophila. Genetics 157, 1307–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez GM, Alkhori L, Hatano E, Schultz SW, Kuzhandaivel A, Jafari S, Granseth B, and Alenius M (2016). Hedgehog Signaling Regulates the Ciliary Transport of Odorant Receptors in Drosophila. Cell Rep. 14, 464–470. [DOI] [PubMed] [Google Scholar]
- Sano H, Renault AD, and Lehmann R (2005). Control of lateral migration and germ cell elimination by the Drosophila melanogaster lipid phosphate phosphatases Wunen and Wunen 2. J. Cell Biol 171, 675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos AC, and Lehmann R (2004). Germ cell specification and migration in Drosophila and beyond. Curr. Biol 14, R578–R589. [DOI] [PubMed] [Google Scholar]
- Semenas J, Hedblom A, Miftakhova RR, Sarwar M, Larsson R, Shcherbina L, Johansson ME, Härkönen P, Sterner O, and Persson JL (2014). The role of PI3K/AKT-related PIP5K1α and the discovery of its selective inhibitor for treatment of advanced prostate cancer. Proc. Natl. Acad. Sci. USA 111, E3689–E3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senju Y, and Lappalainen P (2019). Regulation of actin dynamics by PI(4,5)P2 in cell migration and endocytosis. Curr. Opin. Cell Biol 56, 7–13. [DOI] [PubMed] [Google Scholar]
- Singh J, Wen X, and Scales SJ (2015). The Orphan G Protein-coupled Receptor Gpr175 (Tpra40) Enhances Hedgehog Signaling by Modulating cAMP Levels. J. Biol. Chem 290, 29663–29675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoda AM, Simovic D, Karin V, Kardum V, Vranic S, and Serman L (2018). The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci 18, 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staller MV, Yan D, Randklev S, Bragdon MD, Wunderlich ZB, Tao R, Perkins LA, Depace AH, and Perrimon N (2013). Depleting gene activities in early Drosophila embryos with the “maternal-Gal4-shRNA” system. Genetics 193, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuma L, Carter D, Weavers H, and Martin P (2018). Drosophila immune cells extravasate from vessels to wounds using Tre1 GPCR and Rho signaling. J. Cell Biol 217, 3045–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurmond J, Goodman JL, Strelets VB, Attrill H, Gramates LS, Marygold SJ, Matthews BB, Millburn G, Antonazzo G, Trovisco V, et al. ; FlyBase Consortium (2019). FlyBase 2.0: the next generation. Nucleic Acids Res. 47, D759–D765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno K, Ohta M, Morita H, Mikuni Y, Nakajima S, Yamamoto K, and Isono K (2001). Trehalose sensitivity in Drosophila correlates with mutations in and expression of the gustatory receptor gene Gr5a. Curr. Biol 11, 1451–1455. [DOI] [PubMed] [Google Scholar]
- van den Bout I, and Divecha N (2009). PIP5K-driven PtdIns(4,5)P2 synthesis: regulation and cellular functions. J. Cell Sci 122, 3837–3850. [DOI] [PubMed] [Google Scholar]
- Van Doren M, Broihier HT, Moore LA, and Lehmann R (1998). HMG-CoA reductase guides migrating primordial germ cells. Nature 396, 466–469. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Yoshida S, Muroi E, Kawamura M, Kouchi Z, Nakamura Y, Sakai R, and Fukami K (2010). Phosphatidylinositol 4,5-bisphosphate and PIP5-kinase lalpha are required for invadopodia formation in human breast cancer cells. Cancer Sci. 101, 1632–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavari A, Nagaraj R, Owusu-Ansah E, Folick A, Ngo K, Hillman T, Call G, Rohatgi R, Scott MP, and Banerjee U (2010). Role of lipid metabolism in smoothened derepression in hedgehog signaling. Dev. Cell 19, 54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HL, and Janmey PA (2003). Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Physiol 65, 761–789. [DOI] [PubMed] [Google Scholar]
- Yoshiura S, Ohta N, and Matsuzaki F (2012). Tre1 GPCR signaling orients stem cell divisions in the Drosophila central nervous system. Dev. Cell 22, 79–91. [DOI] [PubMed] [Google Scholar]
- Zhang N, Zhang J, Purcell KJ, Cheng Y, and Howard K (1997). The Drosophila protein Wunen repels migrating germ cells. Nature 385, 64–67. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets or code.