

Abstract
Pfeiffer syndrome is a rare inherited craniofacial disorder. Upper airway obstruction is common among patients with Pfeiffer syndrome due to craniosynostosis. They may also present with lower respiratory tract obstruction due to a rare congenital airway malformation called tracheal cartilaginous sleeve (TCS). We report the case of a patient with Pfeiffer syndrome who presented with recurrent bronchopneumonia, discovered incidentally to have TCS via direct visualisation during tracheostomy. Relevant literature for this rare clinical condition are reviewed and discussed. Clinicians should be aware of TCS when encountering patients with craniosynostosis who present with recurrent lower respiratory tract infections. Careful and meticulous investigations should be performed to look for TCS, especially in patients with craniosynostosis.
Keywords: ear, nose and throat/otolaryngology, paediatrics, congenital disorders
Background
Pfeiffer syndrome is a rare genetic disorder among Western and Asian population.1 2 It was originally described in 1964 by Rudolf Pfeiffer, characterised by premature fusion of certain cranial bones (craniosynostosis), midface hypoplasia with beaked nasal tip, ocular proptosis, abnormally broad and medially deviated thumbs and great toes, and conductive hearing loss.3 Patients with Pfeiffer syndrome are usually associated with upper airway obstruction due to craniosynostosis. However, lower airway anomalies should also be identified as they may also present with lower respiratory tract obstruction due to tracheal cartilaginous sleeve (TCS), a rare congenital airway malformation in which there is a continuous tracheal cartilaginous cylinder instead of discrete cartilaginous rings.4 It is essential to intervene early for TCS by performing tracheostomy and vigilant surveillance of the airway to prevent complications related to granulation tissue.4
Case presentation
A boy was born at term with features of Pfeiffer syndrome, including sagittal craniosynostosis with scaphocephaly, hypertelorism, midfacial hypoplasia, prominent thumb and big toes, and fixed extension of bilateral elbow. He had recurrent episodes of pneumonia associated with upper airway obstruction since age 3 months that required non-invasive ventilation in the paediatric intensive care unit. He underwent bronchoscopy procedure for airway assessment at the age of 8 months during his second admission due to symptoms of upper airway obstruction. Airway assessment was performed relatively late due to the late referral to the paediatrics respiratory and paediatrics otorhinolaryngology team who detected the features of choanal stenosis in the child. The finding of bronchoscopy was bilateral choanal stenosis in which the left side was much smaller compared with the right side. Other structures of the larynx were normal. Tracheoscopy was performed using a flexible endoscope; however, no abnormality of the trachea was detected up until the carina at that point in time.
Due to his condition of having persistent nasal blockage and inability to wean off oxygen, the bilateral choanal stenosis was repaired at the age of 11 months, which was 3 months after the first airway assessment. The repair was performed late due to recurrent bouts of respiratory distress with episodes of desaturation secondary to pneumonia, which delayed the procedure. Postoperatively, he was kept admitted for a few days for observation and reassessment before discharge. Unfortunately, even after the repair of the choanal stenosis, he still had signs and symptoms of upper airway obstruction associated with pneumonia, requiring hospital admissions. The parents were counselled regarding the need for tracheostomy, but they refused. He was later discharged home with nocturnal continuous positive airway pressure (CPAP). With nocturnal CPAP, his symptoms improved partially. The oxygen saturation was maintained, but intermittently he was noted to have nasal blockage and snoring, especially during sleep. The parents had to perform regular bilateral nasal suctioning using suction tube size 5–6 French about six times daily to clear the nasal discharge. He was also noted to have poor weight gain.
At the age of 2 years, he presented with a history of cough and runny nose associated with low-grade fever of 3 days’ duration. He was noted to be less active and showed increased breathing effort 2 days prior to presentation to the emergency department. He was in severe respiratory distress with pulse oximetry reading of 54% on room air, respiratory rate of 44 breaths/min with marked subcostal and suprasternal recession. Lung auscultation revealed bilaterally reduced breath sounds with crepitations. Due to impending respiratory failure with severe bronchopneumonia, he was intubated and ventilated with high ventilator setting. He received a few courses of antibiotics in view of persistent high temperature and was ventilated for 19 days. His parents were counselled again regarding the need for tracheostomy in view of the prolonged ventilation and recurrent lung infections and they finally agreed.
Investigations and treatment
Intraoperatively, the trachea was noted to be stiff and narrow, with no palpable tracheal rings (figure 1a). Rigid tracheoscopy was performed and it was noted that the tracheal rings were absent up to the carina (figure 1b). Bilateral main bronchus was also narrow.
Figure 1.
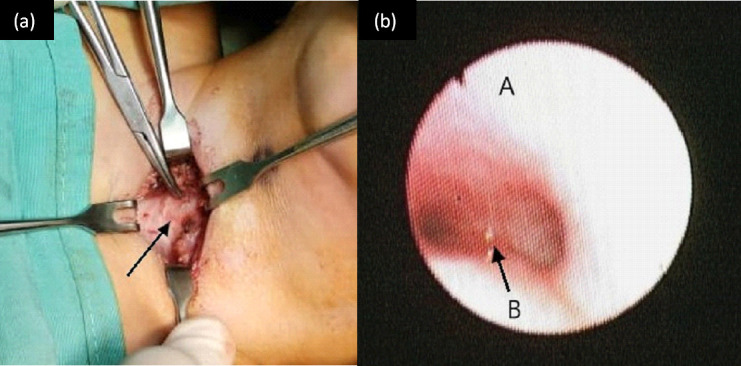
(a) Intraoperative finding of absent tracheal ring during tracheostomy procedure. The trachea is annotated by a black arrow. Figure 1 (b) Endoscopic findings of the trachea showing smooth tracheal wall with no mucosal indentation, (A is anterioar tracheal wall and B is carina area).
Contrast CT scan of thorax was then performed postoperatively and it was noted that there was a long segment of narrowing with circumferential hyperdense soft tissue surrounding the tracheal lumen, measuring 0.9 cm in length, giving rise to an hourglass appearance on sagittal view (figure 2). The left main bronchus shows a small calibre with similar circumferential soft tissue thickening, 2.2 cm in length and 0.3 cm in narrowest diameter. The left upper and left lower bronchi and the right main bronchus are normal. The presence of the circumferential uninterrupted cartilage of the distal trachea narrowing is suggestive of congenital TCS.
Figure 2.
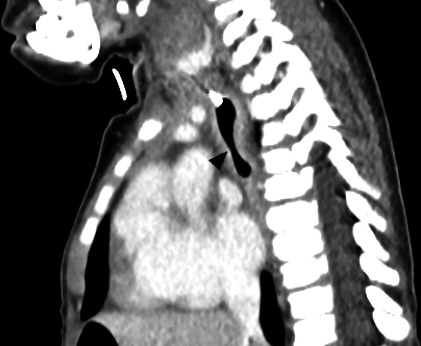
CT thorax showing hourglass appearance of the trachea (arrowhead) on sagittal view.
Outcome and follow-up
About 2 days post tracheostomy, it was noted there was ease in bronchial toileting with improvement of his general condition where he was able to sleep well with no breathing difficulties. He recovered from the bronchopneumonia, CPAP and oxygen was able to be weaned off and was discharged home 26 days post tracheostomy.
During clinic review 2 weeks after he was discharged, he was well and gaining weight accordingly.
Discussion
Pfeiffer syndrome is a rare genetic disorder in the Western and Asian population.1 The incidence of Pfeiffer syndrome is approximately 1 in 100 000 worldwide.2 Pfeiffer syndrome is a rare autosomal dominant genetic disorder, which is also known to be a part of a condition caused by mutation in fibroblast growth factor receptor (FGFR) genes, including Apert syndrome, Crouzon syndrome, Beare-Stevenson syndrome, FGFR2-related isolated coronal synostosis and Jackson-Weiss syndrome to name a few.2 Based on a classification developed by Dr Cohen in 1993, the three types of Pfeiffer syndrome are defined as: type 1, patients with mild expression of the classic findings; type 2 and type 3 are more severely affected, characterised by severe ocular proptosis and midfacial hypoplasia, with type 2 including the cloverleaf skull deformity, which is not present in type 3. In type 2 and type 3 infants, it is often associated with hydrocephalus, abnormal fixation and ankylosis of elbow joints as well as visceral anomalies.2 Type 2 and type 3 Pfeiffer syndrome are usually associated with neurodevelopmental abnormalities and have shortened life expectancy.3 Our patient was initially characterised as type 1 Pfeiffer syndrome in which he has craniosynostosis and midfacial hypoplasia, which includes bilateral choanal stenosis, prominent thumbs and great toes, as well as fixed extension of bilateral elbows. There was non-communicating hydrocephalus and ventriculoperitoneal shunt was inserted at the age of 9 months. After the discovery of the TCS, this patient was later characterised as having type 3 Pfeiffer syndrome. As expected in Pfeiffer syndrome, he had global developmental delay, which corresponded to the developmental age of 12 months at the age of 2 years. FGFR gene study was not performed due to financial constraint.
FGFR-related craniosynostosis syndromes, such as Apert syndrome, Crouzon syndrome and Pfeiffer syndrome cause significant upper airway compromise due to craniofacial malformations. Several recent studies have explored the nasal and pharyngeal airway volumes of these syndromes.5–7 In Apert syndrome infants, the airway compromise is more apparent in the nasal cavity. As they grow up, the disproportionate growth of the mandible will result in reduced pharyngeal airway volume.6 As for Crouzon syndrome and Pfeiffer syndrome, both the nasal and pharyngeal airway volumes are significantly reduced.5 7 In Pfeiffer syndrome, the hypoplastic sphenoid contributed to the limited nasal pathway height and width, in addition to the pre-existing choanal stenosis. The upper airway compromise faced by patients with Pfeiffer syndromic is also contributed to by restricted mediolateral and anteroposterior dimensions seen in the entire course of the pharyngeal airways. Therefore, patients with Pfeiffer syndrome may benefit from reconstructive surgery, namely, mediolateral maxillary expansion as well as maxillomandibular advancement.5 Although options of craniofacial reconstructions can be offered to this patient, it was somehow unfortunate that the parents opted for a more conservative approach.
TCS is a rare congenital airway malformation in which there is a continuous, vertically fused tracheal cartilaginous cylinder instead of discrete cartilaginous rings. There are two types of TCS described in the literature: circumferential form and the one with posterior membranous septum.3 4 8 9 This condition is often associated with craniosynostosis syndrome, such as Apert syndrome, Crouzon syndrome and Pfeiffer syndrome.4 The extension of the abnormality may affect from the subglottis down to carina or bronchi. The occurrence of the TCS was contributed to by the FGFR mutations that promote the proliferation of tracheal cartilage progenitor cells within the mesenchyme. This condition led to a more rigid airway with poor airway clearance.10 11 This condition may affect the mechanical properties of the airway and disrupt the effectiveness of natural airway protective mechanisms. The distensibility of the normal trachea provided by the interring fibrous tissue as well as a pars membranacea is lacking in TCS.4 This condition may cause the tracheal lumen to be occluded, especially when sleeping with the neck flexed. In patients with TCS and craniosynostosis, the mortality rate is 90% at 2 years of age. Almost all patients with TCS are associated with premature death, with mean age of death of 3 years. It was postulated that if the TCS causes the airway to remain the same size and the patient outgrows their airway, it will lead to premature death.3
TCS is almost always diagnosed postmortem. It is usually discovered incidentally via endoscopic examination while trying to establish causes of persistent respiratory difficulties, direct visualisation of the trachea during tracheostomy or at autopsy.4 TCS is often identified late in patients with craniosynostosis with upper airway obstruction. The upper airway obstruction is initially thought to be the only cause of airway obstruction as in this patient who presented with multiple episodes of bronchopneumonia with upper airway obstruction, where he was initially discovered to have bilateral choanal stenosis. Even after the choanal stenosis repair, he presented with recurrent bronchopneumonia. This may be explained by the impaired ability to clear secretions due to the loss of natural, dynamic movement associated with the normal respiratory cycle caused by the rigidity of the tracheal column.8 The first airway assessment was performed on this patient at the age of 8 months; however, the diagnosis of TCS was probably missed because the endoscopic findings may be subtle and can be misinterpreted as fibrosis secondary to chronic tracheitis. Hence, it is of substantial importance to always look for the features of TCS during airway evaluation in order to avoid missing the diagnosis. On performing endoscopic examination, one should suspect the diagnosis of TCS when there is a smooth inner tracheal surface with absence of normal mucosal indentations of the tracheal rings with either normal, sparse or absent pars membranacea posteriorly.8 In this case, radiological imaging may help in visualising the possible type of TCS the patient may be having. As in our patient, the contrast CT scan showed the presence of the circumferential uninterrupted cartilage of the distal trachea narrowing. Therefore, this patient may be having circumferential form of TCS. Ultrasound imaging also has a role in screening patients at high risk for TCS and diagnosing TCS. The modality is considered to have significant advantages as it is non-invasive, has reduced risk of ionising radiation, as well as lower cost and greater portability.12
Tracheostomy can be beneficial to patients with TCS, especially to patients with craniosynostosis syndrome with upper airway obstruction. Tracheostomy also allows better bronchial toileting of the abnormal tracheal column. Some authors advocate tracheostomy as an intervention to give survival advantage and extend lifespan.3 Post-tracheostomy care is paramount in patients with TCS as they appear to be very sensitive to trauma as well as instrumentation. The formation of tracheal granulomas can cause obstruction of the airway; therefore, it is also important to perform frequent interval bronchoscopy with removal of granulation tissue.4
Learning points.
High index of suspicion for tracheal cartilaginous sleeve (TCS) is of substantial importance, especially in patients with craniosynostosis condition. Hence, meticulous investigation for eliciting symptoms and signs of TCS should be performed as the diagnosis can be easily missed if not specifically looking for it.
In Pfeiffer syndrome, the rarity of airway anomaly (TCS) made it missed. Hence, it is vital to assess carefully the integrity of the upper and the lower airways for congenital airway anomalies to avoid late diagnosis leading to the respiratory complications.
In patients with TCS, early diagnosis and intervention by performing tracheostomy can improve the clinical condition as well as improving the survival of the patients.
It is also important to perform early and regular airway assessment in patient with craniosynostosis so that early intervention can be delivered to the patient.
Footnotes
Contributors: NM: Has written the case report and collected the relevant literature reviews for the purpose of writing the case report. HAL: The consultant paediatric pulmonologist who treated the patient described in the case report. Helped in writing this case report. FMZ: Consultant paediatric radiologist who was involved in the management of the patient described in the case report. BSG: The corresponding author and the consultant paediatric otorhinolaryngologist who is the surgeon in-charge and involved in managing the patient described in the case report.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer-reviewed.
References
- 1.Park MS, Yoo JE, Chung J, et al. A case of Pfeiffer syndrome. J Korean Med Sci 2006;21:374. 10.3346/jkms.2006.21.2.374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogels A, Fryns J-P. Pfeiffer syndrome. Orphanet J Rare Dis 2006;1:19. 10.1186/1750-1172-1-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hockstein NG, McDonald-McGinn D, Zackai E, et al. Tracheal anomalies in Pfeiffer syndrome. Arch Otolaryngol Head Neck Surg 2004;130:1298–302. 10.1001/archotol.130.11.1298 [DOI] [PubMed] [Google Scholar]
- 4.Lertsburapa K, Schroeder JW, Sullivan C. Tracheal cartilaginous sleeve in patients with craniosynostosis syndromes: a meta-analysis. J Pediatr Surg 2010;45:1438–44. 10.1016/j.jpedsurg.2009.09.005 [DOI] [PubMed] [Google Scholar]
- 5.Lu X, Forte AJ, Allam O. Nasopharyngeal Airway and Subcranial Space Analysis in Pfeiffer Syndrome. British Journal of Oral and Maxillofacial Surgery (Published Online First: 15 December 2020). [DOI] [PubMed]
- 6.Lu X, Forte AJ, Park KE. Morphological basis for airway surgical intervention in Apert syndrome. Annals of Plastic Surgery 2021. [DOI] [PubMed] [Google Scholar]
- 7.Forte AJ, Lu X, Hashim PW, et al. Analysis of airway and midface in Crouzon syndromes. Ann Plast Surg 2019;82:686–91. 10.1097/SAP.0000000000001740 [DOI] [PubMed] [Google Scholar]
- 8.Lin SY, Chen JC, Hotaling AJ, et al. Congenital tracheal cartilaginous sleeve. Laryngoscope 1995;105:1213–9. 10.1288/00005537-199511000-00014 [DOI] [PubMed] [Google Scholar]
- 9.Inglis AF, Kokesh J, Siebert J, et al. Vertically fused tracheal cartilage. An underrecognized anomaly. Arch Otolaryngol Head Neck Surg 1992;118:436–8. 10.1001/archotol.1992.01880040102017 [DOI] [PubMed] [Google Scholar]
- 10.Holmes G, O'Rourke C, Motch Perrine SM, et al. Midface and upper airway dysgenesis in FGFR2-related craniosynostosis involves multiple tissue-specific and cell cycle effects. Development 2018;145:dev166488. 10.1242/dev.166488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tiozzo C, De Langhe S, Carraro G, et al. Fibroblast growth factor 10 plays a causative role in the tracheal cartilage defects in a mouse model of Apert syndrome. Pediatr Res 2009;66:386–90. 10.1203/PDR.0b013e3181b45580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loy KA, Lam AS, Otjen JP, et al. Tracheal cartilaginous sleeve diagnosed on ultrasound in a child with Pfeiffer syndrome. Int J Pediatr Otorhinolaryngol 2020;138:110321. 10.1016/j.ijporl.2020.110321 [DOI] [PMC free article] [PubMed] [Google Scholar]