

Summary
Background
High-grade serous ovarian cancers show increased replication stress, rendering cells vulnerable to ATR inhibition because of near universal loss of the G1/S checkpoint (through deleterious TP53 mutations), premature S phase entry (due to CCNE1 amplification, RB1 loss, or CDKN2A mRNA downregulation), alterations of homologous recombination repair genes, and expression of oncogenic drivers (through MYC amplification and other mechanisms). We hypothesised that the combination of the selective ATR inhibitor, berzosertib, and gemcitabine could show acceptable toxicity and superior efficacy to gemcitabine alone in high-grade serous ovarian cancer.
Methods
In this multicentre, open-label, randomised, phase 2 study, 11 different centres in the US Experimental Therapeutics Clinical Trials Network enrolled women (aged ≥18 years) with recurrent, platinum-resistant high-grade serous ovarian cancer (determined histologically) and Eastern Cooperative Oncology Group performance status of 0 or 1, who had unlimited previous lines of cytotoxic therapy in the platinum-sensitive setting but no more than one line of cytotoxic therapy in the platinum-resistant setting. Eligible patients were randomly assigned (1:1) to receive intravenous gemcitabine (1000 mg/m2) on day 1 and day 8, or gemcitabine plus intravenous berzosertib (210 mg/m2) on day 2 and day 9 of a 21-day cycle until disease progression or intolerable toxicity. Randomisation was done centrally using the Theradex Interactive Web Response System, stratified by platinum-free interval, and with a permuted block size of six. Following central randomisation, patients and investigators were not masked to treatment assignment. The primary endpoint was investigator-assessed progression-free survival, and analyses included all patients who received at least one dose of the study drugs. The study is registered with ClinicalTrials.gov, NCT02595892, and is active but closed to enrolment.
Findings
Between Feb 14, 2017, and Sept 7, 2018, 88 patients were assessed for eligibility, of whom 70 were randomly assigned to treatment with gemcitabine alone (36 patients) or gemcitabine plus berzosertib (34 patients). At the data cutoff date (Feb 21, 2020), the median follow-up was 53·2 weeks (25·6–81·8) in the gemcitabine plus berzosertib group and 43·0 weeks (IQR 23·2–69·1) in the gemcitabine alone group. Median progression-free survival was 22·9 weeks (17·9–72·0) for gemcitabine plus berzosertib and 14·7 weeks (90% CI 9·7–36·7) for gemcitabine alone (hazard ratio 0.57, 90% CI 0.33–0.98; one-sided log-rank test p=0·044). The most common treatment-related grade 3 or 4 adverse events were decreased neutrophil count (14 [39%] of 36 patients in the gemcitabine alone group vs 16 [47%] of 34 patients in the gemcitabine plus berzosertib group) and decreased platelet count (two [6%] vs eight [24%]). Serious adverse events were observed in ten (28%) patients in the gemcitabine alone group and nine (26%) patients in the gemcitabine plus berzosertib group. There was one treatment-related death in the gemcitabine alone group due to sepsis and one treatment-related death in the gemcitabine plus berzosertib group due to pneumonitis.
Interpretation
To our knowledge, this is the first randomised study of an ATR inhibitor in any tumour type. This study shows a benefit of adding berzosertib to gemcitabine in platinum-resistant high-grade serous ovarian cancer. This combination warrants further investigation in this setting.
Funding
US National Cancer Institute.
Introduction
Replication stress is defined as the slowing or stalling of replication fork progression during DNA synthesis.1 Sources of replication stress include loss of G1/S checkpoint (eg, due to deleterious TP53 mutations), premature entry into the S phase (eg, due to RB1 loss or CCNE1 amplification), oncogenic drive (eg, due to KRAS activating mutations or MYC amplification), and DNA repair deficiencies (eg, deficiencies in homologous recombination repair and nucleotide excision repair).2–4 All these mechanisms of increased replication stress are prevalent in high-grade serous ovarian cancer, the most common histological subtype of epithelial ovarian cancer.5–7 Specifically, large-scale genomic studies have shown that high-grade serous ovarian cancers have near universal loss of the G1/S checkpoint (via deleterious TP53 mutations); premature S phase entry due to CCNE1 amplification in about 20% of tumours, RB1 loss in about 11% of tumours, or CDKN2A mRNA downregulation in about 32% of tumours; oncogenic driver activation via amplification of the MYC oncogene in up to 40% of tumours or NF1 loss in about 10% of tumours; and DNA repair deficiencies, mainly due to homologous recombination repair alterations in about 50% of tumours and nucleotide excision repair alterations in 4–8% of tumours.5–7
The cellular response to replication stress involves activation of the ATR kinase that mediates the activation of DNA repair pathways and stabilisation of replication forks.1,8 ATR is a member of the PI3K-related kinase family of proteins and is the master kinase that regulates cellular responses to stalled and collapsed replication forks, mainly by activating DNA repair proteins and suppressing dormant origin firing. Activation of ATR triggers the intra-S phase and the G2 checkpoints through phosphorylation of CHK1, which in turn phosphorylates WEE1 and CDC25 phosphatases to inhibit cell cycle progression through the coordinated suppression of cyclin-dependent kinase activity.1–3,9 ATR also facilitates DNA repair through phosphorylation of several proteins involved in homologous recombination repair and interstrand crosslink repair, including BRCA2, RAD51, FANCD2, and FANCE.1,8
Given the prevalence of DNA replication stress in high-grade serous ovarian cancer, inhibition of ATR might be an effective strategy against these tumours. Furthermore, treatment with gemcitabine, a drug used routinely in platinum-resistant ovarian cancer, induces additional replication stress and dependence on ATR through two mechanisms: inhibition of DNA repair by incorporating gemcitabine nucleotides into the DNA, and inhibition of ribonucleotide reductase leading to depletion of the deoxyribonucleotide pool required for replication and repair.10–13 Preclinical models have shown a synergistic antitumour effect with gemcitabine and the ATR inhibitor berzosertib, with maximal effect when berzosertib is administered 24 h after starting gemcitabine.14 This schedule dependence is attributed to the fact that 24 h after gemcitabine administration coincides with the highest accumulation of cells in S phase, with a concomitant increase in ATR activity (as measured by phosphorylated CHK1) and thus maximal susceptibility to ATR inhibition.14 Therefore, in this study, we aimed to evaluate whether berzosertib would enhance the activity of gemcitabine and show acceptable toxicity and superior efficacy compared with gemcitabine alone in patients with recurrent, platinum-resistant high-grade serous ovarian, primary peritoneal, or fallopian tube cancers.
Methods
Study design and participants
In this multicentre, open-label, investigator-initiated, randomised, phase 2 study, we enrolled patients at 11 different cancer centres in the USA through the Experimental Therapeutics Clinical Trials Network (appendix p 10).
Eligible patients were women aged 18 years or older with life expectancy of more than 6 months, who had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 (with Karnofsky Performance Status score ≥70), and histologically determined high-grade serous carcinoma of the ovary, fallopian tube, or primary peritoneum (collectively referred to as ovarian cancer) that was platinum resistant, defined as progression within 6 months after the last platinum regimen. Patients were required to have measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, and adequate haematological function (white blood cell count ≥3000 cells per μL, absolute neutrophil count ≥1500 cells per μL, and platelets ≥100 000 per μL), renal function (serum creatinine concentration supper limit of normal [ULN] or creatinine clearance ≥60 mL/min), and hepatic function (total bilirubin concentration ≤ULN and alanine and aspartate aminotransferase concentration ≥2·5 × ULN). Patients could have received unlimited lines of cytotoxic therapy when their cancer was platinum sensitive, but no more than one line of cytotoxic therapy was allowed in the platinum-resistant setting. Previous hormonal therapies, immunotherapy, and antiangiogenic therapies were allowed and did not count as lines if given as monotherapy. Previous receipt of PARP inhibitors was allowed, and in an approved amendment to the protocol (June 14, 2018), it was clarified that previous PARP inhibitor use did not count as a separate line of treatment if it was given as maintenance after platinum therapy. Previous carboplatin plus gemcitabine therapy was allowed if there was no disease progression within 12 months after completion of that regimen. In addition, availability of archival formalin-fixed, paraffin embedded tumour specimens was required for participation, for doing exploratory correlative studies.
Patients were ineligible if they had primary platinum-refractory disease, known brain metastases, ongoing grade 2 or worse adverse events related to previous treatment, if they were taking any inhibitors or inducers of cytochrome P450 3A4 enzyme, or if they had any major comorbidities (including, but not limited to ongoing or active infection, symptomatic congestive heart failure, unstable angina pectoris, cardiac arrhythmia, or psychiatric illness or difficulty in social situations that would limit compliance with the study requirements). Previous treatments targeting the ATR/CHK1 pathway and previous administration of gemcitabine as monotherapy were not permitted. A full list of inclusion and exclusion criteria can be found in the protocol (appendix).
Written informed consent was obtained from all patients or their guardians before enrolment in the study. The clinical trial was approved by the National Cancer Institute (NCI) Central Institutional Review Board and the US Food and Drug Administration. All procedures were done in accordance with the Declaration of Helsinki. The full protocol is available in the appendix.
Randomisation and masking
Eligible patients were randomly assigned (1:1) to receive either gemcitabine alone or gemcitabine plus berzosertib. Randomisation was stratified by platinum-free interval (≤3 months vs >3 months but <6 months [as in the AURELIA study15]). Patient randomisation was done centrally and independently through the Theradex Interactive Web Response System. The study statistician (S-CC) generated the allocation sequence using stratified permuted blocks with a block size of six. Patients and investigators were not masked to treatment assignment.
Procedures
Eligible patients received intravenous gemcitabine (1000 mg/m2 during 30 min) on day 1 and day 8 of each 21-day cycle, either alone or in combination with intravenous berzosertib (210 mg/m2 during 1 h) on day 2 and day 9 of each 21-day cycle. The gemcitabine and berzosertib dose and the timing of administration was the recommended phase 2 dose that was established in a previous dose-escalation study.16 Berzosertib was administered with corticosteroid and antihistamine premedcations to protect against acute hypersensitivity. Haematopoietic growth factor support was allowed.
Treatment continued until disease progression, unacceptable toxicity, patient refusal to participate in the study, intercurrent illness that prevented further administration of treatment, or general or specific changes in the participant’s condition that rendered the treatment unacceptable in the opinion of the treating investigator. Toxicity-related dose delays of up to 4 weeks were allowed. Depending on the grade of the adverse event, gemcitabine could be reduced to 750 mg/m2 or 500 mg/m2, and berzosertib could be reduced to 165 mg/m2 or 120 mg/m2. Generally, for grade 3 gastrointestinal-related adverse events and other grade 4 non-haematological adverse events, treatment with both drugs was delayed until adverse events resolved to grade 2 or less, and the dose of both gemcitabine and berzosertib was reduced by one dose level each. For other grade 3 non-haematological adverse events, treatment was delayed until they resolved to grade 2 or less; gemcitabine treatment was resumed at a reduced dose level but berzosertib was resumed at the previous dose. If, after the dose reduction of gemcitabine, grade 3 non-haematological adverse events recurred, or if there was persistent grade 2 or lower non-haematological adverse event that resulted in a need for a dose delay of more than 2 weeks, then berzosertib was also reduced by one dose level. For grade 3 haematological adverse events, treatment was delayed until complete blood count recovery. For grade 4 neutropenia or grade 3 thrombocytopenia, treatment was delayed until count recovery and gemcitabine was reduced by one dose level. For grade 4 neutropenia persisting for more than 7 days, grade 4 thrombocytopenia, or febrile neutropenia, treatment was delayed until they were resolved to grade 2 or less, and gemcitabine treatment was resumed at a reduced dose level. If these adverse events recurred after the reduction in gemcitabine, then berzosertib was also reduced by one dose level. Crossover from the gemcitabine group to the gemcitabine plus berzosertib group was allowed for patients who developed disease progression according to RECIST 1.1 criteria.
Tumour reassessment was through CT or MRI of the chest, abdomen, and pelvis every 9 weeks for the first year and every 12 weeks after the first year. Assessments could be done up to 7 days before or after the designated timepoint, and were done by the investigator using RECIST version 1.1. Safety and adverse events were assessed on day 1 and day 8 of each cycle and were graded according to the NCI Common Terminology Criteria for Adverse Events version 4.0. Laboratory assessments (complete blood count, blood urea nitrogen, creatinine, sodium, potassium, calcium, carbon dioxide, alkaline phosphatase, aspartate aminotransferase, alanine aminotransferase, total bilirubin, total protein, and albumin) were done on day 1 and day 8 of each cycle, and ovarian cancer-related tumour marker (CA-125) was tested on day 1 of each cycle. After stopping study treatment, patients were followed up every 3 months for 1 year and every 6 months thereafter, for 3 years after removal from study treatment or until death, whichever occurred first.
Outcomes
The primary endpoint was progression-free survival, defined as the time from randomisation until the date of documented, investigator-assessed RECIST-based progressive disease or death (regardless of cause). Secondary endpoints were overall survival (defined as the time from randomisation to death, regardless of cause); objective response rate (defined as the percentage of participants with a complete or partial response as per RECIST version 1.1) in the whole population and in crossover patients; clinical benefit rate (defined as the percentage of patients showing complete response, partial response, or stable disease for 4 months or more); duration of response (measured from the time that criteria were met for complete or partial response [whichever came first] until the first date that progressive disease was recorded); proportion of patients with a reduction in CA-125 by more than 50%; progression-free survival at 6 months; and the safety profile of each group.
Statistical analysis
The study was designed to have 80% power to detect an improvement of median progression-free survival from 15 weeks with gemcitabine alone to 27·3 weeks with gemcitabine plus berzosertib using the stratified log-rank test (with the platinum-free interval as the stratification factor) at a one-sided alpha level of 0·1. Randomly assigning 64 patients (32 patients in each group) was assumed to achieve the required 50 progression-free survival events, allowing for a 5% dropout. Anticipating that 10% of participants would not be evaluable, the maximum total number of patients that needed to be enrolled was 70.
Progression-free survival, overall survival, and safety analyses were done in all patients who received at least one dose of the study drugs. Analysis of all other secondary efficacy endpoints were also done in all patients who received at least one dose of the study drug, including those with measurable disease who were not assessable for objective response because of early discontinuation of protocol therapy.
Patient characteristics, objective response rate, clinical benefit rate, proportion of patients with a reduction in CA-125, and adverse events were summarised by counts (percentages) and confidence intervals when appropriate. Treatment differences in clinical benefit rate were compared by Fisher’s exact test (two-sided). Progression-free survival and overall survival for each treatment group were summarised using the Kaplan-Meier method. The log-rank test was used to compare progression-free survival and overall survival between the two groups with a one-sided significance of 0·1. The Cox proportional hazards models were fit to estimate the treatment hazard ratio (HR) and the corresponding two-sided 90% CI, as well as the upper one-sided 90% CI. We used the test developed by Grambsch and Therneau17 based on Schoenfeld residuals, to examine the proportional hazards assumption; the proportional hazards assumption was not rejected. We also examined the treatment effects on progression-free survival in the two platinum-free interval strata using Kaplan-Meier curves and the Cox model (with a term for interaction between treatment and platinum-free interval) to assess whether it was suitable to fit the data with a Cox model stratified by platinum-free interval. Patients who withdrew treatment for reasons unrelated to progression were included and censored at their off-protocol treatment date in the progression-free survival analysis.
Exploratory post-hoc subgroup analysis of progression-free survival was graphically displayed with a forest plot to examine heterogeneity in treatment effects across subgroups.
For patients who crossed over, objective response rate was included as a secondary endpoint. Progression-free survival for crossover patients was a post-hoc analysis, and was measured from the time of progressive disease when receiving gemcitabine alone until they progressed on gemcitabine plus berzosertib.
Patients who started a non-protocol treatment before documented progressive disease were censored at the time that they started non-protocol therapy. By definition, crossover patients had events in the progression-free survival analysis and were included per their randomly assigned groups in the overall survival analysis. An interim futility analysis was planned at 25 progression-free survival events to stop enrolment in the case of futility, but it was not done because all patients had been enrolled before that point. Descriptive statistics were used to summarise objective response information after crossover.
All statistical analyses were done using R (version 3.6.2). This study is registered with ClinicalTrials.gov, NCT02595892.
Role of the funding source
The drug was provided by Merck KGaA/EMD Serono through the NCI. The NCI had a role in the study design, data collection, data analysis, data interpretation, and writing of the report. Two authors (LAD and ECK) are from the NCI. Merck KGaA/EMD Serono had no role in the study design, data collection, data analysis, data interpretation, or writing of the report. All authors had full access to all the data in the study, were involved in writing the report, and approved the final version for submission. PAK and UAM had final responsibility for the decision to submit for publication.
Results
Between Feb 14, 2017, and Sept 7, 2018, 88 patients were assessed for eligibility, of whom 70 were randomly assigned to treatment with either gemcitabine alone (36 patients) or gemcitabine plus berzosertib (34 patients; figure 1). Three patients, at some timepoints, had imaging scans without oral or intravenous and oral contrast. All 70 patients received treatment and were included in the efficacy and safety analyses. Baseline patient characteristics were balanced between the two groups (table 1). At the data cutoff date of Feb 21, 2020, all patients had discontinued treatment (figure 1). The median follow-up was 43·0 weeks (IQR 23·2–69·1) in the gemcitabine alone group and 53·2 weeks (25·6–81·8) in the gemcitabine plus berzosertib group.
Figure 1: Trial profile.
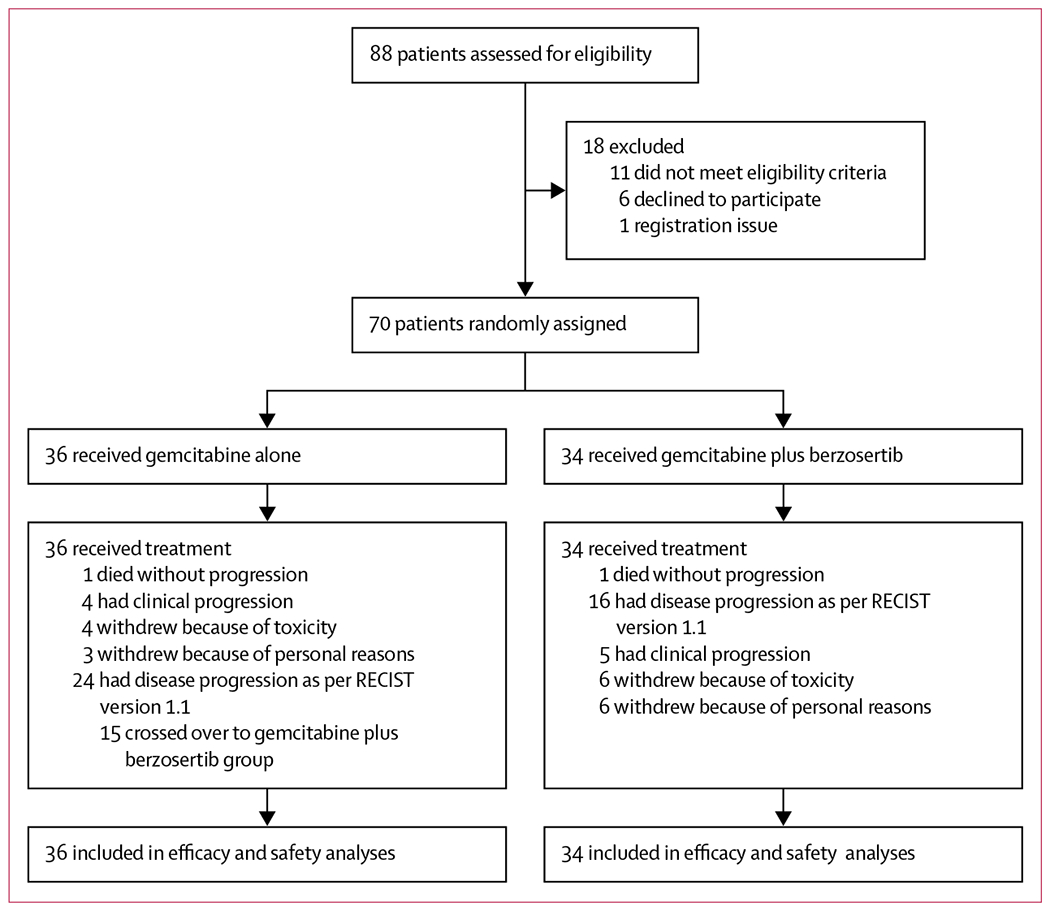
RECIST=Response Evaluation Criteria in Solid Tumors.
Table 1:
Baseline characteristics
| Gemcitabine alone (n=36) | Gemcitabine plus berzosertib (n=34) | |
|---|---|---|
| Race | ||
| White | 34 (94%) | 30 (88%) |
| Asian | 0 | 2 (6%) |
| Unknown | 2 (6%) | 2 (6%) |
| Ethnicity | ||
| Not Hispanic or Latino | 34 (94%) | 30 (88%) |
| Hispanic or Latino | 1 (3%) | 2 (6%) |
| Unknown | 1 (3%) | 2 (6%) |
| ECOG performance status | ||
| 0 | 22 (61%) | 20 (59%) |
| 1 | 14 (39%) | 14 (41%) |
| Stage at diagnosis | ||
| II | 0 (0%) | 2 (6%) |
| III | 16 (44%) | 18 (53%) |
| IV | 14 (39%) | 9 (26%) |
| Unknown | 6 (17%) | 5 (15%) |
| Platinum-free interval* | ||
| ≤3 months | 13 (36%) | 13 (38%) |
| >3 to <6 months | 23 (64%) | 21 (62%) |
| Previous therapy lines | ||
| 1 | 10 (28%) | 3 (9%) |
| 2 | 18 (50%) | 20 (59%) |
| 3 | 5 (14%) | 6 (18%) |
| 4 | 2 (6%) | 4 (12%) |
| 5 | 0 | 1 (3%) |
| 8 | 1 (3%) | 0 |
| Previous therapy lines in platinum-resistant setting | ||
| 1 | 11 (31%) | 14 (41%) |
| 0 | 25 (69%) | 20 (59%) |
| Previous PARP inhibitor | ||
| Yes | 7 (19%) | 11 (32%) |
| No | 29 (81%) | 23 (68%) |
| Previous antiangiogenic therapy | ||
| Yes | 9 (25%) | 10 (29%) |
| No | 27 (75%) | 24 (71%) |
| BRCA mutated | ||
| Yes | 5 (14%) | 6 (18%) |
| No | 25 (69%) | 21 (62%) |
| Unknown | 6 (17%) | 7 (21%) |
Stratification factor. ECOG=Eastern Cooperative Oncology Group.
17 (50%) of 34 patients receiving gemcitabine plus berzosertib and 25 (69%) of 36 patients receiving gemcitabine alone had progression-free survival events. Median progression-free survival was 22·9 weeks (90% CI 17·9–72·0) in the gemcitabine plus berzosertib group versus 14·7 weeks (9·7–36·7) in the gemcitabine alone group (HR 0·57, 0·33–0·98; one-sided log-rank test p=0·044; upper one-sided 90% CI 0·87; figure 2A). In patients with a platinum-free interval of 3 months or less, median progression-free survival was 27·7 weeks (90% CI 15·7–not reached [NR]) in those receiving gemcitabine plus berzosertib (seven progression-free survival events) versus 9·0 weeks (8·7–NR) in those receiving gemcitabine alone (11 progression-free survival events; HR 0·29, 0·12–0·71; one-sided log-rank test p=0·0087; figure 2B). In patients with a platinum-free interval greater than 3 months, median progression-free survival was 18·6 weeks (9·4–NR) for those receiving gemcitabine plus berzosertib (ten progression-free survival events) versus 15·3 weeks (14·7–NR) for those receiving gemcitabine alone (14 progression-free survival events; HR 1·04, 0·51–2·12; one-sided log-rank test p=0·46; figure 2C). Exploratory post-hoc analyses of progression-free survival are shown in figure 3.
Figure 2: Progression-free survival in all patients who initiated protocol therapy (A), patients with a platinum-free interval of 3 months or less (B), and patients with a platinum-free interval of more than 3 months to less than 6 months (C).
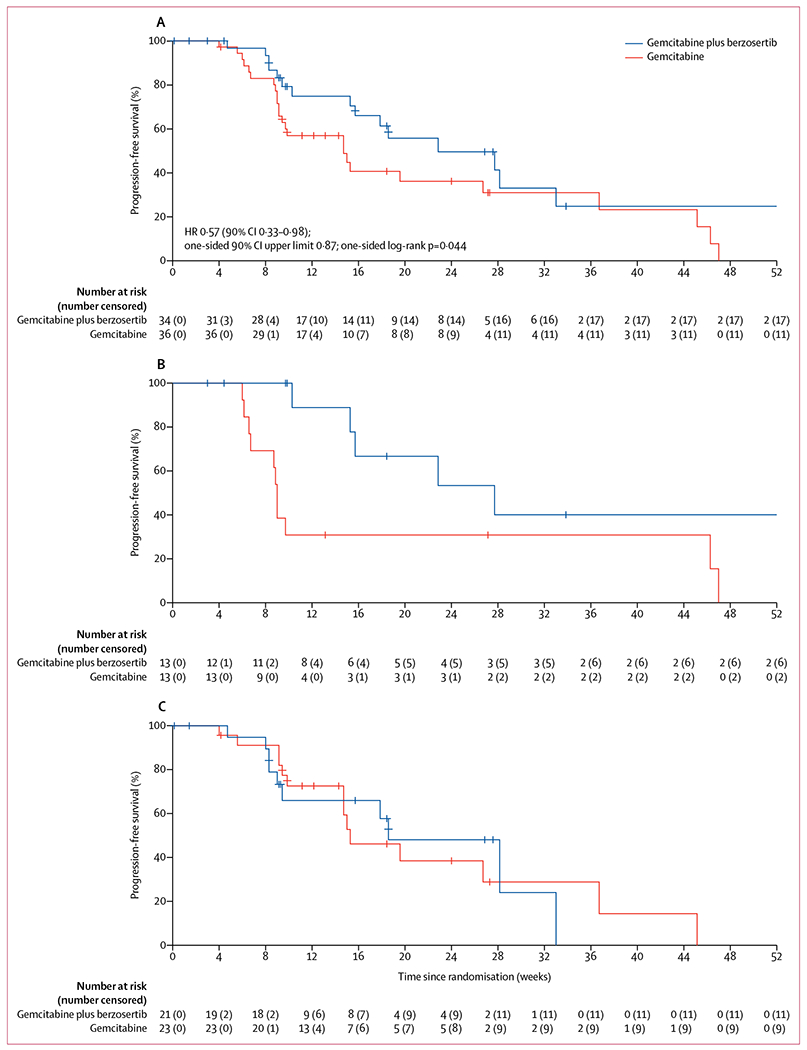
HR=hazard ratio.
Figure 3: Post-hoc subgroup analysis of progression-free survival.
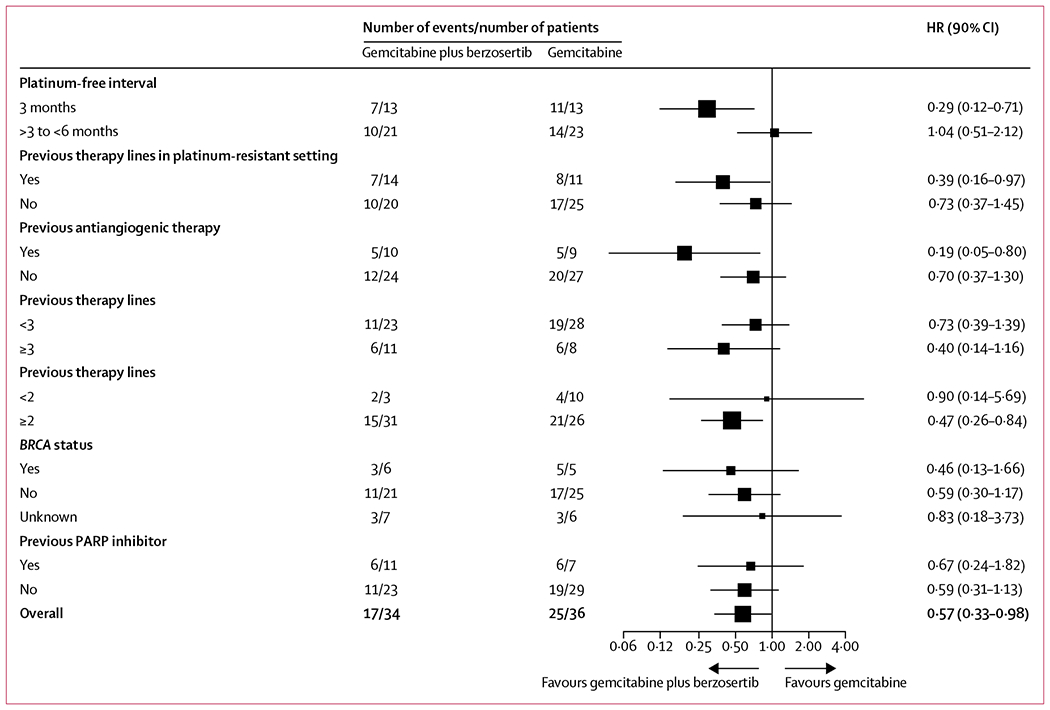
HR=hazard ratio.
At the data cutoff date, 24 (71%) of 34 patients in the gemcitabine plus berzosertib group and 29 (81%) of 36 patients in the gemcitabine alone group had died. Median overall survival was 59·4 weeks (90% CI 33·7–84·4) in the gemcitabine plus berzosertib group versus 43·0 weeks (34·4–67·9) in the gemcitabine alone group (HR 0·84, 0·53–1·32; one-sided log-rank test p=0·26; appendix p 1). In patients with a platinum-free interval of 3 months or less, median overall survival was 84·4 weeks (59·4–NR) in the gemcitabine plus berzosertib group (eight patients died) versus 40·4 weeks (27·6–92·4) in the gemcitabine alone group (11 patients died; HR 0·42, 0·19–0·94; one-sided log-rank test p=0·034; appendix p 1). In patients with a platinum-free interval of more than 3 months, median overall survival was 39·0 weeks (31·3–80·6) in the gemcitabine plus berzosertib group (16 patients died) versus 59·9 weeks (34·6–98·1) in the gemcitabine alone group (18 patients died; HR 1·29, 0·73–2·31; one-sided log-rank test p=0·23; appendix p 1).
Secondary endpoints of objective response, duration of response, 6-month progression-free survival, and clinical benefit rate were measured for all 70 patients who received at least one dose of the study drug (table 2). 18 (26%) of 70 patients had a decrease in CA-125 of more than 50%, including nine (25%) of 36 patients in the gemcitabine alone group and nine (27%) of 34 patients in the gemcitabine plus berzosertib group.
Table 2:
Secondary endpoints
| Objective response rate |
Clinical benefit rate |
6-month progression-free survival |
|||||
|---|---|---|---|---|---|---|---|
| Gemcitabine alone (n=36) | Gemcitabine plus berzosertib (n=34) | Gemcitabine alone (n=36) | Gemcitabine plus berzosertib (n=34) | p value* | Gemcitabine alone (n=36) | Gemcitabine plus berzosertib (n=34) | |
| All patients | 4 (11%) | 1 (3%) | 9/36 (25%, 14–40) | 12/34 (35%, 22–51) | 0·44 | 36% (24–55) | 50% (35–71) |
| Platinum-free interval ≤3 months | 1/13 (8%)† | 1/13 (8%)‡ | 3/13 (23%, 7–49) | 7/13 (54%, 29–78) | 0·23 | 31% (16–61) | 53% (31–91) |
| Platinum-free interval 3–6 months | 3/23 (13%)§ | 0/21 | 6/23 (26%, 12–45) | 5/21 (24%, 10–44) | 1·0 | 39% (23–65) | 48% (30–77) |
Data are n (%), n/N (%), n/N (%, 90% CI), or % (90% CI).
Two-sided Fisher’s exact test.
Duration of response 10 weeks.
Duration of response 7 weeks.
Duration of response 6 weeks, 18 weeks, and 27 weeks.
Clinical benefit rate was defined as the proportion of patients showing complete response, partial response, or stable disease for 4 months or more.
Ten (67%) of 15 patients who crossed over from the gemcitabine alone group to the gemcitabine plus berzosertib group had a disease evaluation scan after crossover. Among these ten patients, one (10%) had stable disease and then withdrew because of an adverse event, one (10%) had stable disease and then had clinical progression, and the other eight (80%) had progressive disease. In a post-hoc analysis, median progression-free survival for these patients was 19 weeks (90% CI 8·7–NR) after crossover (ie, after progressive disease when receiving gemcitabine alone; appendix p 2).
The most common treatment-related grade 3 or 4 adverse events were decreased neutrophil count (14 [39%] of 36 patients in the gemcitabine alone group vs 16 [47%] of 34 patients in the gemcitabine plus berzosertib group) and decreased platelet count (two [6%] vs eight [24%]; table 3; appendix pp 3–7). No clinically significant bleeding was observed in the combination group, but one case was observed in the gemcitabine alone group.
Table 3:
Treatment-related adverse events
| Gemcitabine (n=36) |
Gemcitabine plus berzosertib (n=34) |
|||||
|---|---|---|---|---|---|---|
| Grades 1–2 | Grade 3 | Grade 4 | Grades 1–2 | Grade 3 | Grade 4 | |
| Blood and lymphatic system disorders | ||||||
| Anaemia | 21 (58%) | 4 (11%) | 0 | 18 (53%) | 5 (15%) | 0 |
| Febrile neutropenia | 1 (3%) | 1 (3%) | 0 | 0 | 1 (3%) | 0 |
| Haemolytic uraemic syndrome | 0 | 1 (3%) | 0 | 0 | 0 | 0 |
| Thrombocytosis | 0 | 0 | 0 | 1 (3%) | 1 (3%) | 0 |
| Lymphocyte count decreased | 5 (14%) | 1 (3%) | 0 | 0 | 1 (3%) | 0 |
| Neutrophil count decreased | 7 (19%) | 10 (28%) | 4 (11%) | 8 (24%) | 12 (35%) | 4 (12%) |
| Platelet count decreased | 4 (11%) | 0 | 2 (6%) | 9 (26%) | 2 (6%) | 6 (18%) |
| White blood cell decreased | 4 (11%) | 4 (11%) | 0 | 5 (15%) | 3 (9%) | 0 |
| Cardiac disorders | ||||||
| Myocardial infarction | 0 | 1 (3%) | 0 | 0 | 0 | 0 |
| Endocrine disorders | ||||||
| Addisonian crisis | 0 | 0 | 0 | 0 | 1 (3%) | 0 |
| Gastrointestinal disorders | ||||||
| Abdominal pain | 1 (3%) | 0 | 0 | 0 | 1 (3%) | 0 |
| Constipation | 4 (11%) | 0 | 0 | 6 (18%) | 0 | 0 |
| Diarrhoea | 6 (17%) | 0 | 0 | 8 (24%) | 1 (3%) | 0 |
| Gastric haemorrhage | 0 | 1 (3%) | 0 | 0 | 0 | 0 |
| Mucositis oral | 1 (3%) | 0 | 0 | 6 (18%) | 0 | 0 |
| Nausea | 17 (47%) | 1 (3%) | 0 | 19 (56%) | 1 (3%) | 0 |
| Vomiting | 7 (19%) | 0 | 0 | 12 (35%) | 1 (3%) | 0 |
| General disorders and administration site conditions | ||||||
| Chills | 2 (6%) | 0 | 0 | 6 (18%) | 0 | 0 |
| Oedema limbs | 6 (17%) | 1 (3%) | 0 | 1 (3%) | 0 | 0 |
| Fatigue | 18 (50%) | 3 (8%) | 0 | 19 (56%) | 3 (9%) | 0 |
| Fever | 3 (8%) | 0 | 0 | 8 (24%) | 0 | 0 |
| Flu-like symptoms | 4 (11%) | 0 | 0 | 4 (12%) | 0 | 0 |
| Injury, poisoning, and procedural complications | ||||||
| Fall | 0 | 1 (3%) | 0 | 0 | 0 | 0 |
| Investigations | ||||||
| Alanine aminotransferase increased | 7 (19%) | 0 | 0 | 10 (29%) | 0 | 0 |
| Aspartate aminotransferase increased | 8 (22%) | 0 | 0 | 12 (35%) | 0 | 0 |
| Cardiac troponin T increased | 0 | 1 (3%) | 0 | 0 | 0 | 0 |
| Metabolism and nutrition disorders | ||||||
| Anorexia | 5 (14%) | 0 | 0 | 7 (21%) | 0 | 0 |
| Musculoskeletal and connective tissue disorders | ||||||
| Generalised muscle weakness | 1 (3%) | 0 | 0 | 2 (6%) | 1 (3%) | 0 |
| Myalgia | 3 (8%) | 0 | 0 | 5 (15%) | 0 | 0 |
| Nervous system disorders | ||||||
| Headache | 3 (8%) | 0 | 0 | 7 (21%) | 1 (3%) | 0 |
| Respiratory, thoracic, and mediastinal disorders | ||||||
| Dyspnoea | 2 (6%) | 1 (3%) | 0 | 5 (15%) | 0 | 0 |
| Skin and subcutaneous tissue disorders | ||||||
| Maculopapular rash | 5 (14%) | 0 | 0 | 4 (12%) | 0 | 0 |
| Vascular disorders | ||||||
| Capillary leak syndrome | 0 | 1 (3%) | 0 | 0 | 0 | 0 |
Grade 1 and 2 adverse events occurring in at least 10% of patients and all grade 3 and 4 adverse events are shown. There was one treatment-related death in the gemcitabine alone group due to sepsis and one treatment-related death in the gemcitabine plus berzosertib group due to pneumonitis. There were no other deaths due to adverse events.
There was one treatment-related death in the gemcitabine alone group due to sepsis and one treatment-related death in the gemcitabine plus berzosertib group due to pneumonitis. No other deaths due to adverse events (while on protocol treatment) were observed. Pneumonitis has a known association with gemcitabine. One patient developed pneumonitis in the gemcitabine alone group (grade 2), one developed grade 2 pneumonitis after crossing over to the gemcitabine plus berzosertib group (this patient had five cycles of gemcitabine alone, crossed over to gemcitabine plus berzosertib, and was diagnosed with pneumonitis after she received the first dose of berzosertib), and three patients developed pneumonitis in the gemcitabine plus berzosertib group (two grade 2 who discontinued protocol therapy because of pneumonitis and one for whom it was the cause of death).
Four (11%) of 36 patients in the gemcitabine alone group and six (18%) of 34 patients in the gemcitabine plus berzosertib group discontinued treatment. In the gemcitabine alone group, one patient each discontinued because of neutropenia, capillary leak syndrome, oedema, and acute renal failure. In the gemcitabine plus berzosertib group, two patients discontinued because of fatigue, one because of abdominal pain, one because of small bowel obstruction, and as stated, two patients in the combination group discontinued because of pneumonitis.
Serious adverse events were observed in ten (28%) of 36 participants in the gemcitabine alone group (predominantly fever or febrile neutropenia; two [6%] patients), and in nine (26%) of 34 participants in the gemcitabine plus berzosertib group (predominantly thrombocytopenia; four [12%] patients). All serious adverse events were deemed to be drug-related. 13 (36%) patients in the gemcitabine alone group and 13 (38%) patients in the gemcitabine plus berzosertib group had a dose reduction (appendix pp 8–9).
Discussion
The results of this multicentre, open-label, randomised, phase 2 trial show that the addition of the ATR inhibitor, berzosertib, to gemcitabine in platinum-resistant, high-grade serous ovarian cancer increased progression-free survival compared with gemcitabine alone. To our knowledge, this is the first randomised study of ATR inhibitor therapy in any tumour type, and the first suggesting a benefit of adding an ATR inhibitor to standard chemotherapy in ovarian cancer or any tumour type. Building on the mechanistic rationale behind using an ATR inhibitor in high-grade serous ovarian cancer (a tumour with high replication stress and thus high dependency on ATR), the results of its combination with gemcitabine (which further enhances replication stress and dependency on ATR), might be applicable to additional tumour types with high replication stress (eg, uterine serous cancers or small cell lung cancers).18,19 Furthermore, ATR inhibitor combinations with other drugs that enhance replication stress, such as topoisomerase-I inhibitors, PARP inhibitors, and PI3K inhibitors, should be considered.20,21 Of note, in this study, berzosertib was administered 24 h after gemcitabine, based on preclinical studies.14 These data highlight the importance of preclinical studies in determining the optimal sequence and schedule of administration of ATR inhibitors in combination with chemotherapy.
The management of patients with platinum-resistant ovarian cancer is a substantial unmet medical need. Nonplatinum cytotoxic chemotherapy (with or without the antiangiogenic drug bevacizumab) remains the standard of care for these patients, who typically have a poor prognosis and no targeted treatment options except PARP inhibitors if their tumours are BRCA-mutation positive.22 Weekly gemcitabine, administered in cycles of either 2 weeks on and 1 week off, or 3 weeks on and 1 week off, has shown similar activity to pegylated liposomal doxorubicin in two randomised phase 3 studies and is a standard regimen for platinum-resistant ovarian cancer.23,24 The median progression-free survival of 14·7 weeks observed with gemcitabine alone in this study is consistent with that previously reported for gemcitabine (about 3 months) and other standard cytotoxic drugs, such as pegylated liposomal doxorubicin (also about 3 months), as monotherapies in platinum-resistant ovarian cancer.23–25 The median progression-free survival of 22·9 weeks and the HR of 0·57 for the gemcitabine plus berzosertib group are promising, considering that this study allowed unlimited lines of previous therapies and up to one line in the platinum-resistant setting. This approach is in contrast to the AURELIA study,15 in which no patient could have received more than two previous lines of therapy and 107 (60%) of 179 patients in the chemotherapy plus bevacizumab group had received only one previous line of therapy (ie, first-line chemotherapy only), compared with only three (9%) of the 34 patients in the gemcitabine plus berzosertib group in this study.
A higher objective response rate was observed in the gemcitabine alone group than in the gemcitabine plus berzosertib group, whereas progression-free survival was better in the gemcitabine plus berzosertib group. These results are not uncommon in platinum-resistant ovarian cancer. In the CLIO study reported in 2019,26 in which olaparib was compared with standard chemotherapy in platinum-resistant ovarian cancer, the objective response rate was higher in patients assigned to olaparib than in patients who received chemotherapy (18% vs 6%), but no significant difference was observed in progression-free survival (median 2·9 months for olaparib vs 3·4 months for chemotherapy; HR 1·18). Furthermore, in a randomised phase 3 study of topotecan versus pegylated liposomal doxorubicin in recurrent ovarian cancer,27 the objective response rate in the platinum-resistant cohort was 12·3% for pegylated liposomal doxorubicin and 6·5% for topotecan, whereas the median progression-free survival was 9·1 weeks for pegylated liposomal doxorubicin compared with 13·6 weeks for topotecan.
The progression-free survival benefit of adding berzosertib to gemcitabine was notable in patients with a platinum-free interval of 3 months or less but not for those with a platinum-free interval greater than 3 months. These findings might reflect the fact that tumours of patients with a platinum-free interval of three months or less are enriched for biomarkers of replicative stress (such as CCNE1 amplification), which are likely to be predisposed to respond to ATR inhibition; this hypothesis is being investigated in ongoing correlative work. Furthermore, post-hoc analysis of progression-free survival in other subgroups indicated that there was a benefit of adding berzosertib to gemcitabine in more heavily pretreated patients, including patients with at least two previous lines of therapy and patients with one previous line of therapy in the platinum-resistant setting, although the benefit was not seen in patients who had received at least three lines of therapy. Given that ATR-mediated stabilisation of the replication fork is one of the known mechanisms of resistance to platinum and other DNA-damaging chemotherapy drugs in high-grade serous ovarian cancer,28,29 more heavily pretreated tumours might be more likely to have developed resistance through that mechani sm and therefore be more susceptible to ATR inhibition. Tumours that have been exposed to antiangiogenic therapy might be even more dependent on resistance through ATR-mediated stabilisation of the replication fork because anti-angiogenic drugs downregulate homologous recombination repair,30,31 thereby negating the restoration of homologous recombination repair as a mechanism of resistance in these tumours. This factor might explain the effect of the addition of berzosertib in patients who had previously received antiangiogenic therapy. All these mechanistic hypotheses are being evaluated in ongoing correlative work, which includes targeted next-generation sequencing to identify genomic determinants of response and assessment of biomarkers of replication stress and ATR activation in these tumours.
The addition of berzosertib to gemcitabine was well tolerated with a similar percentage of patients having treatment-related grade 3 or worse adverse events and dose reductions in the two groups. In terms of myelo-suppression, thrombocytopenia was more common in the gemcitabine plus berzosertib group than in the gemcitabine alone group, but no clinically significant bleeding was observed; anaemia and neutropenia were similar in the two groups. Berzosertib was administered with corticosteroid and antihistamine premedications to protect against acute hypersensitivity, resulting in only two patients having infusion-related reactions in the experimental group, which were easily manageable. Pneumonitis, a known adverse effect of gemcitabine, was observed in one patient in the gemcitabine alone group, in one patient after crossing over to the gemcitabine plus berzosertib group, and in three patients in the combination group, including one death. It is impossible to conclude whether berzosertib increases the incidence or severity of gemcitabine-induced pneumonitis on the basis of these data; of note, pneumonitis has not been reported in other studies of berzosertib alone or in combination. Nonetheless, the development of pneumonitis should be carefully monitored in future studies of the gemcitabine plus berzosertib combination. No other safety signals or unexpected adverse events were observed in this study.
One of the main limitations of this study is the small sample size; this trial was a hypothesis-generating study powered to detect an improvement in median progression-free survival at a one-sided significance level of 0·1. The study was open label, although it is reassuring that the standard gemcitabine alone group showed a median progression-free survival that was consistent with that previously reported for platinum-sresistant ovarian cancer. These limitations notwithstanding, our findings warrant further investigation of gemcitabine plus berzosertib in a phase 3 trial in platinum-resistant ovarian cancer and support stratification based on platinum-free interval, number of previous lines of therapies, and previous antiangiogenic therapy. Finally, our results support future assessment of ATR inhibitor therapy in combination with gemcitabine in additional tumour types, especially in tumours that are predicted to have high replication stress.
Supplementary Material
Research in context.
Evidence before this study
We searched PubMed from database inception to Dec 28, 2019, to identify publications that included the search terms “ATR”, “randomised”, “randomized”, “cancer”, and “ovarian cancer”. We also searched PubMed for publications describing clinical evaluation of berzosertib using the terms “berzosertib”, “M6620”, or “VX-970” and assessment of other ATR inhibitors using the terms “ATR inhibitor”, “M4344”, “VX-803”, “AZD6738”, “ceralasertib”, and “BAY1895344”. We did not use any language restrictions. These searches did not identify any previous randomised trials that have investigated the use of ATR inhibitors, alone or in combination, either in ovarian cancer or in any other tumour type. Given the prevalence of DNA replication stress in high-grade serous ovarian cancer, inhibition of ATR might be an effective strategy against these tumours. Furthermore, treatment with gemcitabine, a drug used routinely in platinum-resistant ovarian cancer, induces additional replication stress (through inhibition of DNA repair and inhibition of ribonucleotide reductase, leading to depletion of the deoxyribonucleotide pool), thereby leading to higher dependency on ATR inhibition. Preclinical models have shown a synergistic antitumour effect with gemcitabine and the ATR inhibitor berzosertib, prompting our evaluation of whether berzosertib enhances the activity of gemcitabine.
Added value of the study
The results of this phase 2 trial showed that the addition of the ATR inhibitor, berzosertib, to gemcitabine in platinum-resistant high-grade serous ovarian cancer increased progression-free survival compared with gemcitabine alone. To our knowledge, this is the first randomised study of ATR inhibitor therapy in any tumour type and the first to show the benefit of adding an ATR inhibitor to standard chemotherapy in ovarian cancer or in any other tumour type.
Implications of all the available evidence
The results of this study support further investigation of gemcitabine plus berzosertib in a phase 3 trial in platinum-resistant ovarian cancer. Furthermore, when considered together with the mechanistic rationale behind using an ATR inhibitor in high-grade serous ovarian cancer (a tumour with high replication stress and thus high dependency on ATR) in combination with gemcitabine (a drug that further enhances replication stress and dependency on ATR), these results might be applicable to other tumour types with high replication stress (eg, uterine serous cancers or small cell lung cancers), as well as ATR inhibitor combinations with other drugs that enhance replication stress, such as topoisomerase-I inhibitors, PARP inhibitors, and PI3K inhibitors.
Acknowledgments
We thank all patients and their families for their participation in the trial. This study was funded by the National Cancer Institute Cancer Therapy Evaluation Program and conducted with the support of the Experimental Therapeutics Clinical Trials Network. Dana-Farber/Harvard Cancer Center served as the lead site for the study, supported by the National Institutes of Health grant UM1 CA186709 (with GIS as principal investigator). LAD and ECK are employees of the National Institutes of Health and members of the National Cancer Institute Cancer Therapy Evaluation Program in the Investigational Drug Branch (LAD) and the Clinical Investigations Branch (ECK).
Declaration of interests
PAK reports personal fees from GlaxoSmithKline, Tesaro, Merck, AstraZeneca, and Bayer, outside the submitted work. RTP reports personal fees and grants from Merck during the conduct of this study. LRD reports grants, personal fees, and other support (served as local Principal Investigator or Co-Principal Investigator for studies in her institution) from Genentech, Roche, and Merck; grants from Cerulean, NextGen, AbbVie, Tesaro, Pfizer, Aduro BioTech, Syndax, Ludwig, Morab, LEAP Therapeutics, Eisai, Advaxis, and Lycera; grants and other support (served as local Principal Investigator or Co-Principal Investigator for studies in her institution) from GlaxoSmithKline and Novartis; grants and personal fees from MorphoTek and Inovio; and personal fees from AstraZeneca, Advance Medical, UpToDate, Cue Biopharma, the British Journal of Obstetrics and Gynaecology, Parexel, the State of California, Elsevier, Amercian Society for Clinical Oncology, Expert review, ClearView Health Care, the US National Cancer Institute, and JB Learning, all outside the submitted work. MAC reports other support (served as clinical trial local Principal Investigator) from Tesaro, Janssen, Leap Therapeutics, Advaxis, Aprea Therapeutics, Hoffman-LaRoche, grants from AstraZeneca; non-financial support from Syndax; and personal fees from AbbVie and AstraZeneca, all outside the submitted work. SF reports grants from the US National Institutes of Health during the conduct of the study; and other support (clinical trial research support paid to the institution) from AstraZeneca, Abbisko, Anaeropharma Science, Arrien Pharmaceuticals, BeiGene, BioAtla, Boehringer Ingelheim, Eli Lilli, Hookipa Biotech, Huya Bioscience International, Macrogenetics, Medivir, Millennium Pharmaceuticals, Nerviano Medical Sciences, NeuPharma, the US National Institutes of Health, the US National Cancer Institute, Novartis, OncoMed Pharmaceuticals, Parexel International, Sellas Life Sciences Group, Soricimed Biopharma, and Tolero Pharmaceuticals, outside the submitted work. MTM reports personal fees from Tesaro and Eisai outside the submitted work, and grant funding from philanthropy for a phase 1B/2 clinical trial with the drug supplied by Verastem. RJS reports grants from the US National Cancer Institute during the conduct of the study; and personal fees from Incyte, Flatiron, Immunogen, Clovis, and Celsion, outside the submitted work. ADD’A reports personal fees and non-financial support from Vertex; non-financial support from AstraZeneca; and grants, personal fees, and non-financial support from Merck (EMD Serono), outside the submitted work. GIS reports grants from the US National Institutes of Health and National Cancer Institute during the conduct of the study; grants and personal fees from Lilly, Merck (EMD Serono), Sierra Oncology, and Pfizer; grants from Array Pharmaceuticals; and personal fees from G1 Therapeutics, Bicycle Therapeutics, Fusion Pharmaceutical, Bayer, Cybrexa Therapeutics, Astex, Almac, Ipsen, Roche, Angiex, Daiichi Sankyo, Seattle Genetics, Artios, Boehringer Ingelheim, Concarlo, and Atrin, outside the submitted work; has a patent on a dosage regimen for sapacitabine and seliciclib issued to Geoffrey Shapiro and Cyclacel Pharmaceuticals; has a patent on compositions and methods for predicting response and resistance to CDK4/6 inhibition pending to Geoffrey Shapiro and Liam Cornell; and conducts research on M6620 (VX-970) under a research agreement sponsored by Merck (EMD Serono) and participates in other trials on M6620 within the National Cancer Institute Cancer Therapy Evaluation Program. UAM reports personal fees from Astrazeneca, Myriad Genetics, Clovis, Merck, Eli Lilly, Mersana, Geneos, Fuji Film, 2X Oncology, Cerulean, Immunogen, and Novartis, outside the submitted work. All other authors declare no competing interests.
Footnotes
See Online for appendix
Data sharing
Upon email request, we will provide fully de-identified analysis datasets for adverse events listings (multiple records per subject), and for baseline characteristics, including treatment and stratification assignments, and the primary (progression-free survival) and secondary (overall survival, objective response rate, clinical benefit rate, duration of response) outcome endpoints (one record per subject).
References
- 1.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer 2012; 12: 801–17 [DOI] [PubMed] [Google Scholar]
- 2.Flynn RL, Zou L. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem Sci 2011; 36: 133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434: 907–13. [DOI] [PubMed] [Google Scholar]
- 4.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov 2015; 5: 1137–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474: 609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ceccaldi R, O’Connor KW, Mouw KW, et al. A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res 2015; 75: 628–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mouw KW, D’Andrea AD, Konstantinopoulos PA. Nucleotide excision repair (NER) alterations as evolving biomarkers and therapeutic targets in epithelial cancers. Oncoscience 2015; 2: 942–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee KY, Chung KY, Koo HS. The involvement of FANCM, FANCI, and checkpoint proteins in the interstrand DNA crosslink repair pathway is conserved in C elegans. DNA Repair 2010; 9: 374–82. [DOI] [PubMed] [Google Scholar]
- 9.Reaper PM, Griffiths MR, Long JM, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 2011; 7: 428–30. [DOI] [PubMed] [Google Scholar]
- 10.Fordham SE, Blair HJ, Elstob CJ, et al. Inhibition of ATR acutely sensitizes acute myeloid leukemia cells to nucleoside analogs that target ribonucleotide reductase. Blood Adv 2018; 2: 1157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karnitz LM, Flatten KS, Wagner JM, et al. Gemcitabine-induced activation of checkpoint signaling pathways that affect tumor cell survival. Mol Pharmacol 2005; 68: 1636–44. [DOI] [PubMed] [Google Scholar]
- 12.Liu S, Ge Y, Wang T, et al. Inhibition of ATR potentiates the cytotoxic effect of gemcitabine on pancreatic cancer cells through enhancement of DNA damage and abrogation of ribonucleotide reductase induction by gemcitabine. Oncol Rep 2017; 37: 3377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallez Y, Dunlop CR, Johnson TI, et al. The ATR Inhibitor AZD6738 Synergizes with gemcitabine in vitro and in vivo to induce pancreatic ductal adenocarcinoma regression. Mol Cancer Ther 2018; 17: 1670–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.EMD Serono. M6620 investigator’s brochure, 2018.
- 15.Pujade-Lauraine E, Hilpert F, Weber B, et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA open-label randomized phase III trial. J Clin Oncol 2014; 32: 1302–08. [DOI] [PubMed] [Google Scholar]
- 16.Plummer ER, Dean EJ, Evans TRJ, et al. Phase I trial of first-in-class ATR inhibitor VX-970 in combination with gemcitabine (Gem) in advanced solid tumors (NCT02157792). Proc Am Soc Clin Oncol 2016; 34 (suppl 15): 2513. [Google Scholar]
- 17.Grambsch PM, Therneau TM. Proportional hazards tests and diagnostics based on weighted residuals. Biometrika 1994; 81: 515–26. [Google Scholar]
- 18.Kandoth C, Schultz N, Cherniack AD, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013; 497: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015; 524: 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huntoon CJ, Flatten KS, Wahner Hendrickson AE, et al. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res 2013; 73: 3683–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konstantinopoulos PA, Barry WT, Birrer M, et al. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: a dose-escalation and dose-expansion phase 1b trial. Lancet Oncol 2019; 20: 570–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, Karlan BY. Ovarian cancer. Nat Rev Dis Primers 2016; 2: 16061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferrandina G, Ludovisi M, Lorusso D, et al. Phase III trial of gemcitabine compared with pegylated liposomal doxorubicin in progressive or recurrent ovarian cancer. J Clin Oncol 2008; 26: 890–96. [DOI] [PubMed] [Google Scholar]
- 24.Mutch DG, Orlando M, Goss T, et al. Randomized phase III trial of gemcitabine compared with pegylated liposomal doxorubicin in patients with platinum-resistant ovarian cancer. J Clin Oncol 2007; 25: 2811–18. [DOI] [PubMed] [Google Scholar]
- 25.Kurzeder C, Bover I, Marmé F, et al. Double-blind, placebo-controlled, randomized phase III trial evaluating pertuzumab combined with chemotherapy for low tumor human epidermal growth factor receptor 3 mRNA-expressing platinum-resistant ovarian cancer (PENELOPE). J Clin Oncol 2016; 34: 2516–25. [DOI] [PubMed] [Google Scholar]
- 26.Vanderstichele A, Nieuwenhuysen EV, Han S, et al. Randomized phase II CLIO study on olaparib monotherapy versus chemotherapy in platinum-resistant ovarian cancer. Proc Am Soc Clin Oncol 2019; 37 (suppl 15): 5507 [Google Scholar]
- 27.Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ. Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol 2001; 19: 3312–22. [DOI] [PubMed] [Google Scholar]
- 28.Hill SJ, Decker B, Roberts EA, et al. Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discov 2018; 8: 1404–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao H, Ji F, Helleday T, Ying S. Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep 2018; 19: e46263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bindra RS, Schaffer PJ, Meng A, et al. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol Cell Biol 2004; 24: 8504–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lim JJ, Yang K, Taylor-Harding B, Wiedemeyer WR, Buckanovich RJ. VEGFR3 inhibition chemosensitizes ovarian cancer stemlike cells through down-regulation of BRCA1 and BRCA2. Neoplasia 2014; 16: 343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.