

Abstract
Elevated cardiovascular (CV) risk including stroke, heart failure, and heart attack is present even after normalization of blood pressure (BP) in hypertensive patients. Underlying immune cell activation is a likely culprit. Although immune cells are important for protection against invading pathogens, their chronic overactivation may lead to tissue damage and high BP. Triggers that may initiate immune activation include viral infections, autoimmunity, and lifestyle factors such as excess dietary salt. These conditions activate the immune system either directly or through their impact on the gut microbiome, which ultimately produces chronic inflammation and hypertension. T cells are central to the immune responses contributing to hypertension. They are activated in part by binding specific antigens that are presented in major histocompatibility complex molecules on professional antigen-presenting cells, and they generate repertoires of rearranged T cell receptors. Activated T cells infiltrate tissues and produce cytokines including interleukin 17A, which promote renal and vascular dysfunction and end-organ damage leading to hypertension. In this comprehensive review, we highlight environmental, genetic, and microbial associated mechanisms contributing to both innate and adaptive immune cell activation leading to hypertension. Targeting the underlying chronic immune cell activation in hypertension has the potential to mitigate the excess cardiovascular risk associated with this common and deadly disease.
Keywords: Inflammation, hypertension, immunity, dendritic cells, T cells, immune system
Subject Terms: Inflammation, Hypertension
Introduction
Hypertension is the worldwide leading cause of mortality and disability, accounting for nearly half of all strokes, heart failure, myocardial infarction, kidney damage, increased maternal mortality, and cognitive dysfunction.1–6 By calendar year 2000, the worldwide prevalence of hypertension was estimated as 31.1%, affecting 1.39 billion people. By 2016, an elevated BP was ranked as the leading risk factor for global burden of disease in both developed and underdeveloped countries.7 The annual increase in the worldwide prevalence of hypertension has accelerated over the last decade, becoming responsible for 10.8 million or 19.2% of all attributable deaths in 2019.8 This increase is in part due to the aging population, particularly in Western, high-salt consuming societies, since about 70% of adults develop hypertension by age 70. Recent recognition of the prognostic significance of lower levels of BP elevation led the American Heart Association and American College of Cardiology to reclassify hypertension as starting at 130/80 mmHg.9, 10 According to this reclassification, nearly half of the adult United States population currently suffers from hypertension.
Major advances in the pharmacological treatment of an elevated BP occurred over the last five decades. However, despite the effort of major national and international societies and public health organizations, rates of control of BP have been dismal. In the United States, where hypertension accounts for $46 billion in annual health care costs, data from the National Health and Nutrition Examination Survey show that control rates increased from 31.8% in 1999–2000 to a maximum which barely exceeded half all hypertensives (53.8%) in 2013–2014, and unfortunately declined again most recently to 43.7% in 2017–2018 (or to 38.9% if applying the cutoffs in the new AHA-ACC guideline).11 These values in the community at large are very disappointing because in certain health care systems, it has been shown that control can be achieved in >80% of the patients.12 The reasons for poor rates of control of hypertension include those pertaining to the health care system: a) overestimation of office BP by improper recording techniques, which may occur in up to one-third of apparent resistant hypertensive patients in primary care;13 b) lack of recognition of the white-coat phenomenon (i.e., uncontrolled hypertension during the office visit but controlled the rest of the day) in about a third of apparently resistant patients14, 15 which although suspected due to lack of target organ damage or from discordance between home and office BP can only be diagnosed with a 24-hour ambulatory monitor, infrequently available in community health care settings; c) lack of recognition of the pressor effect of illicit drugs or medications to treat concomitant disorders, including but not limited to nonspecific and COX-2-selective nonsteroidal anti-inflammatory agents, sympathomimetics (decongestants, diet pills, and cocaine), stimulants (methylphenidate, dextroamphetamine, amphetamine, methamphetamine, and modafinil), excessive alcohol consumption, oral contraceptives, cyclosporine, erythropoietin, VEGF inhibitors, and licorice-containing products;16 d) undertreatment, as shown in a study of 150,000 uncontrolled hypertensive subjects among whom only 30% were on at least three antihypertensive agents and only 15% on a regimen considered optimal;17 and e) underdiagnosis of secondary forms of hypertension.
There are also social determinants of lack of control and patients’ medication adherence such as stable income, housing, availability of healthy food, transportation, education, access to health care, health insurance, and barriers owing to racial bias. Medication adherence is also affected by the fact that hypertension is mostly an asymptomatic disease, whereas its treatment may produce untoward symptoms. Recent methods to investigate patients’ adherence include administration of medications in the office followed by a 24-hour ambulatory recording,18 and measurement of drug concentrations by HPLC-MS in urine or serum. A pooled analysis of nine HPLC-MS trials in apparently resistant hypertension estimated that poor adherence ranged from 13 to 46% of the subjects and full non-adherence from 2 to 35%.19
Taken together, the data above may suggest that solving the issue of uncontrolled hypertension falls within the realms of public policy, public health, healthcare organizations, health education, and the social sciences only. However, this would ignore three facts that belong exclusively in the biological realm and may contribute to either lack of control of hypertension or to suboptimal protection from hypertensive target organ damage by current therapies. These include; a) the existence of a group of subjects with truly resistant (or even refractory) hypertension, despite optimal treatment,20 b) the fact that half the hypertensive population belongs to the salt-sensitive phenotype or phenotypes, in whom salt may be a CV risk factor independent of BP, particularly in terms of target organ damage,21 and c) the puzzling observation that the residual risk of well treated and controlled hypertensive subjects, assessed by longitudinal study of outcomes, is higher than that of untreated normotensive subjects at the same BP level.22 The goal of this review is to highlight the role of inflammation as an important underlying mechanism that confers CV risk, even in controlled hypertension (Figure 1).
Figure 1:

Proposed Role of Immune Activation in Hypertension.
Resistant and Refractory Hypertension
Truly resistant hypertension is defined as uncontrolled BP while on three antihypertensive agents of different classes including a diuretic, or as BP controlled with four or more agents. A more severe subgroup of patients is defined as having refractory hypertension when they remain uncontrolled after 6 months of treatment by a hypertension specialist, usually despite six or more antihypertensive agents. Both resistant and refractory hypertension are more common among African American individuals living in the southern United States “stroke belt”. After excluding all causes of pseudo-resistance described above, the prevalence of truly resistant hypertension is about 6% of all hypertensive patients and that of refractory hypertension about 5–10% of all individuals with resistant hypertension.23 The intuitive explanation for lack of BP control in these patients is that the prevailing pressor system (or deficient depressor system) underlying their BP elevation has not been addressed by any of the mechanisms of action of the employed medications. A historical precedent for this assumption exists because spironolactone, an agent previously used mostly as a potassium sparing diuretic, became the most useful add-on drug for resistant hypertension, once it was understood that many of these subjects had aldosterone levels inappropriately high for their salt balance or level of activation of the renin-angiotensin system.24
Analogously, recent recognition that some patients with severe hypertension do not respond to spironolactone but exhibit depressor responses to amiloride suggests that another as of yet uncommonly treated mechanism underlying their hypertension is a mineralocorticoid-independent excessive activation of the epithelial sodium channel (ENaC). These patients have a variant single nucleotide polymorphism (SNP) in the CYP4A11 gene, which codes for a monooxygenase.25 Deletion of the monooxygenase Cyp4a10 in mice leads to diminished expression of renal epoxygenases, with ensuing decreases in vasodilator and natriuretic epoxyeicosatrienoic acids (EETs) and development of salt-sensitive hypertension.26 A mineralocorticoid-independent activation of ENaC is also supported by the facts that BP reduction produced by spironolactone in refractory subjects was half that observed in resistant ones, despite their equal renin and aldosterone plasma levels, and that in severe, low renin hypertension in Blacks, responses to amiloride were twice as potent as those to spironolactone.20, 27 It is therefore likely that lack of BP control in a large number of hypertensive subjects may be due to still unrecognized, therefore untreated, additional mechanisms. A possible role for these drugs in dampening the inflammation associated with hypertension is discussed below.
Salt Sensitivity of Blood Pressure
Salt sensitivity of blood pressure (SSBP) is a phenotype of humans and other mammals whereby a certain proportion of the population exhibits changes in BP that parallel changes in salt intake or salt balance. In normotensive humans, about one quarter of subjects exhibit significant increases in BP with salt loading and decreases in BP with salt depletion. In hypertensive humans, the prevalence is about 50% and even higher in certain subpopulations (e.g., African Americans 75–80%). The SSBP phenotype has genetic and environmental components. The former was suspected from studies in siblings and twins but finally demonstrated by the ability to inbreed purely salt sensitive (SS) and salt resistant (SR) strains of rodents, such as the Dahl rats.28 The latter was supported by a major effect of aging in increasing SSBP over time, as well as additional effects of race, sex and measures of adiposity.29 In the dietary approaches to stop hypertension (DASH) low sodium trial, women sustained approximately 2 mmHg greater BP reduction than men and the prevalence of SSBP in women increases significantly with menopause, which has been attributed to loss of beneficial effects of estrogens on the nitric oxide vasodilator system.30
There is an ongoing controversy on whether the primary abnormality in SSBP is a defect in the sodium excretory functions of the kidney or in the ability of vascular smooth muscle cells to normally respond to a salt load with vasodilation. The renal excretory hypothesis prevailed for many years, based on the concept of the infinite gain of the renal function curve proposed by Guyton and coworkers. Within this construct, derived from mathematical modeling, it was impossible to sustain the pressor effect of any hypertensive stimulus if a normal kidney managed to excrete enough salt to restore normotension.31 Consistent with this hypothesis, abnormalities in about 100 genes, the products of which participate somehow in regulation of sodium excretion have been described.21 However, differences in cardiac output or plasma volume in response to salt loading of SS and SR humans or animals have not been observed, and they are a pre-requisite to accept that ensuing total body autoregulation is responsible for the increased total peripheral resistance of essential hypertension. The vascular hypothesis is based on the observation that when humans are exposed to a salt load, the SR subjects sustain rapid vasodilation for maintenance of normal BP whereas the SS ones fail to vasodilate and develop salt-dependent hypertension. These changes have been observed as rapidly as 24 hours, and without differences in cardiac output between the SS and SR groups, making it highly unlikely that autoregulation underlies the vasoconstriction.32
Regardless of its mechanism, SSBP is a CV risk factor with a similar pattern of target organ damage in rodents and humans, characterized by predominant renal damage, stroke, and left ventricular hypertrophy, somewhat different from the predominant atherosclerotic disease observed in SR forms of hypertension. The role of SSBP as a risk factor is independent of BP, and results in increased CV morbidity and mortality,33, 34 which cannot be tackled therapeutically owing to lack of understanding of its causation. The genetic abnormalities mentioned above involve multiple vasoactive and natriuretic regulatory systems, including renal transporters and metabolic-, angiogenic- and inflammation-related substances. Because many of these systems interact, it has not been possible to date to define a primary abnormality. It is also possible that SSBP is not one but several phenotypes instead, with different polygenic causation. The question of how salt ultimately raises BP and produces target organ damage in SS subjects is obviously not settled. However, recent developments about the effects of sodium on immune function may be important in this regard as discussed below.
Residual Cardiovascular Risk
Long term follow-up of thousands of well treated and well controlled hypertensive patients has clearly shown that their risk for CV complications is not reduced by antihypertensive treatment to the level expected from epidemiological observations in normotensive subjects with the same level of BP. This non-reversible excess residual risk was found to be 50% for any CV event, 46% for coronary disease, 75% for stroke and 62% for CV death.35 In this study, residual risk for coronary events was greater for calcium channel blockers, whereas that for stroke was greater for angiotensin converting enzyme inhibitors, suggesting that at least in part, residual risk may depend on specific effects of pharmacological agents with different mechanisms of action on protection from organ damage or its lack thereof.
Also, high-risk individuals with most severe BP elevation benefit the most from therapy in terms of absolute number of events because antihypertensive agents produce the same relative risk reduction across any level of severity of hypertension. Paradoxically, these high-risk subjects with the most benefit from therapy are also left with the worst residual risk. It has therefore been proposed that pre-existing or non-reversible target organ damage may be a major determinant of residual risk,36 which is supported by the observation that up to one third of optimally treated hypertensive patients have silent asymptomatic cardiac abnormalities.37 However, when investigators of the Framingham Offspring Cohort analyzed the multivariate calculated hazard ratios for residual risk excluding and including a covariate score representing the preexisting burden of CV disease (left ventricular hypertrophy, systolic dysfunction, carotid ultrasound abnormality, peripheral artery disease, and microalbuminuria), they showed that preexisting target organ damage contributed at most 20% to the variability of the residual risk.38 In other words, about 80% of the residual excess risk of optimally treated hypertensive patients remains unexplained. This strongly suggests that some fundamental pathophysiological mechanism that underlies the organ damage of hypertension is not reversed, or even remains ongoing despite normalization of BP, because currently available antihypertensive therapies do not address it. In this regard, we discuss below the role of inflammation in hypertensive target organ damage, which we believe is both a cause and consequence of tissue injury in hypertension.
Inflammation in Hypertension
Inflammation is a critical component of immune activation normally required to eliminate invading pathogens. However, chronic low-grade inflammation contributes to the pathogenesis of an array of human diseases. Immune cells and their cytokines have been associated with BP elevation and end-organ damage for several decades, yet no therapies targeting inflammation in hypertension have been developed, and there is a paucity of clinical trials to understand the impact of anti-inflammatory agents on hypertension. In the recent CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcome Study) trial, inhibition of interleukin (IL)-1β using the human monoclonal antibody canakinumab reduced inflammatory markers including high sensitivity C-reactive protein (hsCRP) and IL-6 and CV events, but did not reduce development of incident hypertension or modify the relationship between hypertension and CV events. This suggests that the CV benefit of canakinumab was directly due to reduction in inflammation, not hypertension.39–42 While these studies seem inconsistent with prior evidence suggesting a role of inflammation in the causation of hypertension, a secondary analysis of the CANTOS trial revealed a trend for greater reductions in major adverse cardiac events in people belonging to the highest BP quartile, suggesting that perhaps those with the most severe hypertension had the highest levels of inflammation.43 Further studies are necessary to determine if finer stratification of hypertension status or the use of other inflammatory markers might identify those likely to benefit the most from IL-1β inhibition. In keeping with this, we recently performed an RNA sequence analysis of circulating monocytes from normotensive and hypertensive humans. We identified 60 genes that were differentially expressed in monocytes from hypertensive humans, many of which are related to IL-1β and IL-18, products of the inflammasome.44 Together, these studies suggest a potentially important role for the inflammasome in the pathogenesis of hypertension and cardiovascular disease.
The Inflammasome in Hypertension
During hypertension, the potent vasoactive molecules such as endothelin-1, aldosterone, and angiotensin II (Ang II) act as priming stimuli able to activate the inflammasome (Figure 2).45 Triggering of NLRP3 (NOD-like receptor family pyrin domain containing 3) activation and inflammasome assembly occur through either adenosine triphosphate (ATP) induced K+ efflux or generation of reactive oxygen species (ROS), which are considered “classical” activators of the NLRP3 inflammasome during hypertension.46 The NLRP3 inflammasome plays a pivotal role in the chronic, uncontrolled inflammation that is present in hypertension and other CV diseases, as supported by the fact that NF-κB activity is increased in experimental and human hypertension, with consequent increases in tissue and circulating levels of the proinflammatory cytokines IL-1β and IL-18.47, 48 These cytokines exert effects on immune cells, including monocytes, macrophages, and dendritic cells (DCs) and also nonimmune cells, such as vascular endothelial and smooth muscle cells.46 Moreover, NLRP3 gene polymorphisms have been associated with hypertension, and elevated IL-1β expression has been observed in patients with essential hypertension.49–51
Figure 2: The NLRP3 Inflammasome in Hypertension.
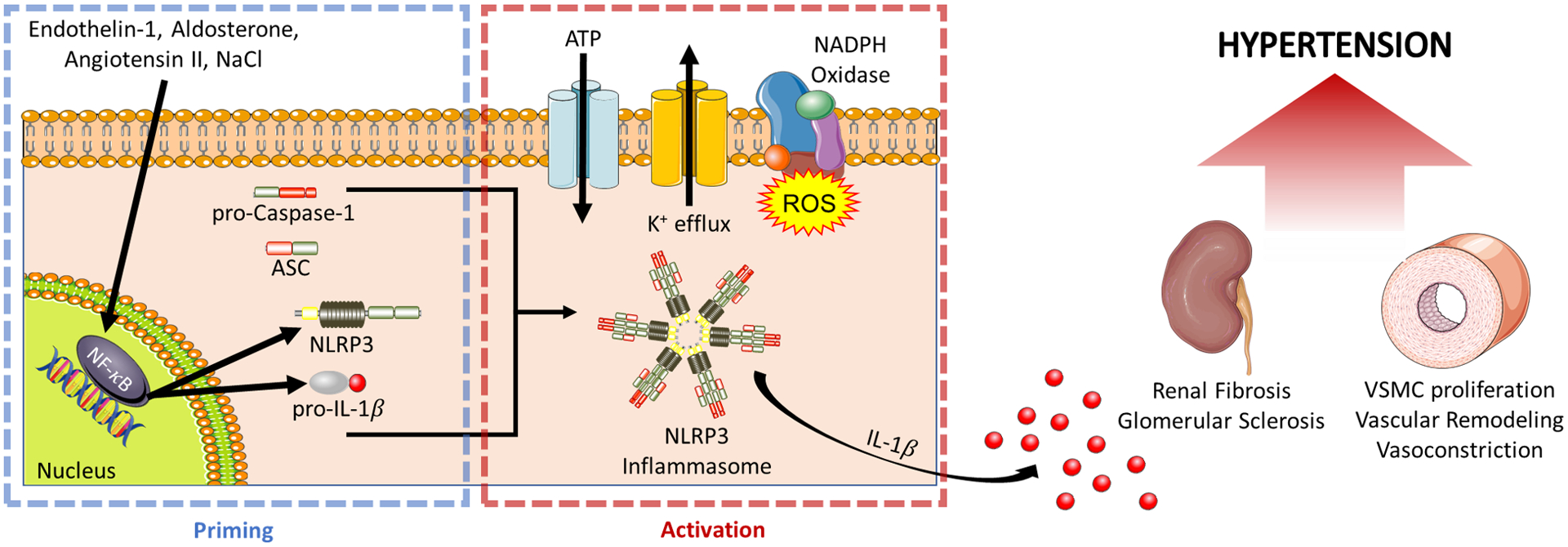
The NLRP3 inflammasome begins its activation through a priming step mediated by the transcription factor NF-κB, which upregulates inflammasome components NLRP3 and pro-IL-1β. Multiple endogenous vasoactive molecules including Endothelin-1, Aldosterone, Angiotensin II, and NaCl can serve as priming stimuli. A second activation step is needed to trigger the formation of the components into the NLRP3 inflammasome signaling complex. Once assembled, caspase-1 proteolytically cleaves pro-IL-1β into the released IL-1β. NLRP3 inflammasome activation has been shown to play a role in the induction of renal injury and vascular remodeling, ultimately leading to hypertension. (Illustration Credit: Ben Smith)
Actions on the vasculature and kidney contribute to the role of the inflammasome in CV disease. A phenotypic switch in vascular smooth muscle cells (VSMCs) plays a major pathophysiological role in vascular disease, including hypertension, as an important initiating factor in vascular remodeling. Deletion of the NLRP3 gene attenuated Ang II-induced vascular inflammation and most importantly, it inhibited vascular remodeling and hypertension in mice.52 Also, NLRP3 deficiency in spontaneously hypertensive rats (SHR) attenuated VSMC phenotypic transformation and proliferation, which in turn reduced BP.53 Moreover, inhibition of serum and glucocorticoid- regulated kinase (SGK1), with consequent negative regulation of NLRP3, reduced the CV inflammatory response associated with Ang II-hypertension.54
Renal inflammation is another typical feature of hypertension and strongly correlates to the severity of increased BP.55, 56 Inhibition of NF-κB with resultant negative regulation of NLRP3 expression abolished the hypertension and renal cortex inflammation of SHR.57 Mice deficient in NLRP3 were protected against Ang II-induced podocyte injury and mitochondrial dysfunction,58 and genetic deletion of NLRP3 attenuated aldosterone-induced podocyte injury and glomerular sclerosis.59, 60 On the other hand, excessive aldosterone-induced NLRP3 inflammasome activation in podocytes was abolished by treatment with antioxidants and eplerenone.59 The renal NLRP3 inflammasome also has a role during salt-sensitive hypertension,61, 62 perhaps because NaCl is a priming stimulus for its activation.63 Consistent with this, inhibition of NLRP3 reduced BP, renal fibrosis, and inflammation in the murine deoxycorticosterone acetate (DOCA)-salt model.62 Finally, ongoing studies of our group aim to investigate a possible role for immune-specific inflammasome expression in hypertension, particularly in antigen presenting myeloid cells.
GWAS studies of hypertension suggest a pathogenic role for inflammation
Familial studies have provided estimates of substantial heritability of BP at approximately 30–60%.64 While a limited number of monogenic disorders involving mutations in approximately 30 genes have been identified to promote hypertension development, these disorders are exceedingly rare.65 Most genetic contribution to hypertension is therefore believed to be polygenic. Recent approaches have centered on large-scale evaluation of common genetic variations through the use of genome-wide association studies (GWAS). Beginning in 2007, GWAS of hypertension first became large enough to identify genome-wide significant associations of about 8 genomic loci.66, 67 Further studies have confirmed and extended these findings with increasingly larger populations. To date, these studies have revealed nearly 1,000 genetic variants associated with hypertension and BP.68 Though individual single nucleotide polymorphisms (SNPs) make small overall contributions to alterations in BP, together these SNPs explain about 27% of the estimated heritability of BP.65 Clearly, additional heritability remains to be discovered, however the large number of identified polymorphisms and genetic loci associating with hypertension from GWAS provide tremendous potential to enhance our understanding of the genetic contributions and novel pathogenic mediators of the disease.
While GWAS have identified SNPs in genetic loci with roles in classical mechanisms of hypertension such as vasoconstriction, sodium reabsorption, and sympathetic nervous system activity, identified genetic loci are also associated with inflammation and immunity. A recent meta-analysis by Rodriguez-Iturbe et al. demonstrated that of 97 distinct genes containing SNPs associated with hypertension from multiple GWAS, 81 of these have direct or indirect roles in inflammation and/or immunity. This includes SNPs associated with major histocompatibility complex alleles which are classically linked with autoimmunity, and SNPs associated with genes involved in myeloid and/or T cell activation such as GATA-binding protein 4 (GATA4), RELA proto-oncogene NFκB subunit, SGK1, and nuclear factor of activated T cells 5 (NFAT5).69 These findings suggest an important role for inflammation and immunity in hypertension. However, one of the challenges in determining the importance of hypertension-associated polymorphisms is in determining causality. Most lead SNPs in GWAS studies are in intergenic and noncoding regions, making determination of their potential functional role challenging. As a result, only a very limited number of SNPs and associated genetic loci have undergone direct testing for causal or mechanistic roles in hypertension.65
Meta-analyses of GWAS results combined with integration of clinical, transcriptomic, epigenomic, and/or protein interaction networks have helped prioritize candidate causal SNPs.68 One such approach by Huan et al identified several genes including SH2B3, ATXN2, NMT1, and NSF as key drivers of hypertension. Of these, SH2B3 is of particular interest as an intracellular adaptor protein that negatively regulates inflammatory cytokine and growth factor signaling. A polymorphism in SH2B3, rs3184504, is strongly associated with hypertension in multiple GWAS studies, and also with a variety of autoimmune diseases.66, 70–72 This polymorphism is a missense SNP that converts an arginine encoded by the major allele to a tryptophan encoded by the minor allele within the plekstrin homology domain of the protein. Although the effect of this polymorphism on SH2B3 protein function and hypertension development is unknown, genetic deletion of Sh2b3 in mice results in increased Ang II-induced blood pressure, enhanced renal and endothelial injury, and enhanced T cell IFNγ production. Increases in BP were most prominent with selective deficiency of SH2B3 in bone marrow-derived cells.73 These findings suggest that SH2B3 plays an important role to limit BP elevation and end-organ damage through reduced inflammatory signaling and T cell activation. Interestingly however, deletion of a 6 base pair region in the SH2 domain of SH2B3 in rats led to a decrease in BP,74 suggesting that genetic alterations in discrete portions of the protein may have differing effects. These findings indicate the importance of determining the direct effect of the rs3184504 missense SNP in hypertension, as well as the multitude of other identified inflammation-related SNPs and genetic loci that to date have remained untested.
In summary, GWAS studies have provided powerful insights into the genetic basis of hypertension and suggested an important role for inflammation and immunity. The large number of genetic loci identified to date has the potential to provide key insights into pathophysiological mechanisms. Because hypertension is a complex, multi-system disease, it necessitates testing for causal roles of individual SNPs and genetic loci in in vivo models. Use of technology such as CRISPR-Cas9 to directly introduce polymorphisms into mice and other animal models holds the potential to test causal roles and determine mechanisms for SNPs identified in humans as a reverse translational approach. Combining these approaches with bioinformatic methodologies to prioritize SNPs most likely to play causal roles has the potential to greatly inform our fundamental understanding of the pathogenesis of hypertension and the search for new therapies.
Myeloid Immune Cell Populations in Hypertension
Monocytes and macrophages:
The importance of innate immunity in the development of hypertension and its associated end-organ damage has been demonstrated over the last several decades.75, 76 Seminal studies investigated the role of myeloid cells, specifically monocytes, macrophages, and DCs, in antigen presentation and production of pro-inflammatory cytokines that promote hypertension (Figure 3).77–81 Ablation of myelomonocytic cells expressing lysozyme M (LysM iDTR) by low-dose administration of diphtheria toxin completely abrogated the rise of BP in response to a pressor dose of Ang II. Moreover, deletion of LysM+ monocytes prevented both vascular endothelial and smooth muscle dysfunction through a reduction in vascular superoxide formation.81 In a translational study, Loperena et al showed that human aortic endothelial cells undergoing uniaxial hypertensive stretch (10% stretch; 1 Hz) for 48 hours promoted conversion of human classical monocytes (CD14++/CD16−) to an intermediate monocyte phenotype (CD14+/CD16++) which produces pro-inflammatory cytokines (IL-6, IL-1β, IL-23, and tumor necrosis factor alpha, TNF-α) and drive T cell proliferation from the same human volunteers. This demonstrated a critical role for immune and endothelial cell crosstalk in the promotion of inflammation during hypertensive stimuli (Figure 4).79 This phenotypic conversion to intermediate monocytes was hydrogen peroxide and IL-6 dependent, as shown by its prevention by scavenging of hydrogen peroxide with catalase or neutralization of IL-6. Interestingly, 10% stretch of human aortic endothelial cells promotes the upregulation of cluster of differentiation 209 (CD209; DC-SGN), demonstrating a critical role of the endothelium in monocyte and monocyte-derived DC activation in an ex vivo model of hypertension.79 In keeping with this, we demonstrated that in response to exposure to high salt (190 mM NaCl) in vitro, human monocytes are activated by accumulation of highly reactive isolevuglandins (IsoLGs), with upregulation of co-stimulatory molecules (CD83 and CD86), and secretion of the pro-inflammatory cytokines IL-1β, TNF-α, and IL-6.82 This sodium dependent activation of monocytes was mediated by the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. Immunodeficient (NOD-scid IL2Rγ null; NSG) mice fed a high-salt (4%) chow diet prior to receiving adoptive joint transfer of human monocytes and T cells demonstrated a profound T cell proliferation and activation of T cells by IFN-γ and IL-17A production compared to mice fed a normal salt diet. Taken together, these studies demonstrate a critical role of monocytes and macrophages in the development of hypertension and end-organ damage.
Figure 3: Innate and adaptive immune cells that have been implicated in hypertension pathophysiology.

Cells of the innate and adaptive immune system secrete factors including reactive oxygen species (ROS), hydrogen peroxide (H2O2), and cytokines that promote or inhibit hypertension and hypertensive end-organ damage. The role of eosinophils and B cells, which secrete immunoglobulins such as IgG, is still unclear. (Illustration Credit: Ben Smith)
Figure 4: Aldosterone-independent activation of ENaC in immune cells.
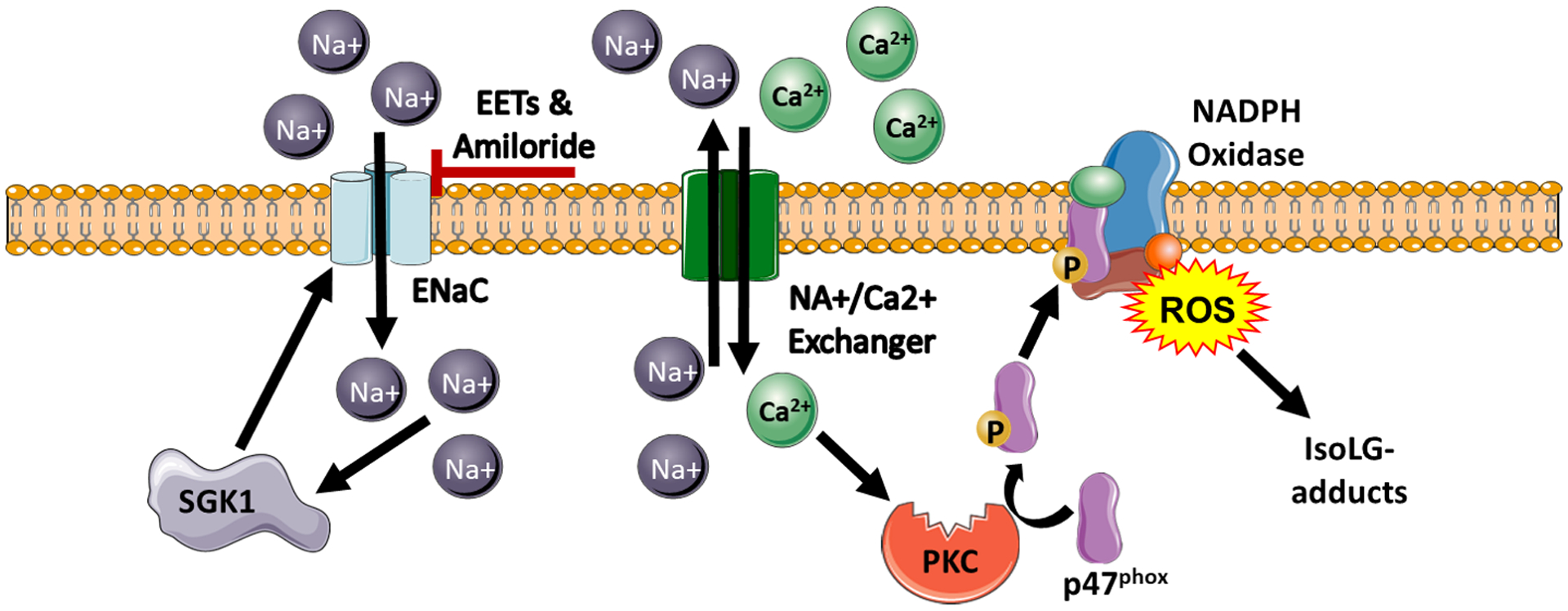
Extracellular Na+ activates the amiloride-inhibitable channel ENaC, which leads to influx in intracellular calcium via the sodium-calcium exchanger. This influx of Ca2+ activates protein kinase-C (PKC). PKC then phosphorylates p47phox leading to the assembly and activation of NADPH oxidase and subsequent production of superoxide and isoLG-adducted proteins. Increased intracellular sodium influx stimulates SGK1 mediates expression and assembly of ENaC in immune cells. Additionally, EETs inhibit ENaC channel activity in these cells. (Illustration Credit: Ben Smith)
Prohypertensive dendritic cells and the role of ENaC:
DCs have been demonstrated to play a critical role in the promotion of hypertension and its associated inflammation.77, 78, 80 We found that immunogenic IsoLG-modified peptides were presented by activated DCs which secreted IL-6 and IL-1β, and drove proliferation of T cells, hypertension, and of its associated end-organ damage. Scavenging of IsoLG-adducts with 2-hydroxybenzilamine (2-HOBA), abrogated the increase in systolic BP in response to both Ang II-induced and DOCA-salt hypertension.78 More recently, we found that genetic deletion of SGK1 in antigen presenting cells (APCs) and DCs prevented vascular dysfunction and the development of salt-sensitive hypertension through an NADPH-oxidase mechanism. Also, in vitro SGK1 inhibition with GSK650934, prevented the salt-dependent increase in IsoLG-adducts in CD11c+ cells.80 Moreover, Hevia et al found that cell-specific expression of the diphtheria toxin receptor under the control of the CD11c promoter prevented mice from developing Ang II plus a high-salt diet-induced hypertension. This was in part due to modulation of the intrarenal renin–angiotensin system components and natriuresis and tubular sodium transporters.83
We have shown that sodium enters DCs through ENaC, because such entry is blocked by amiloride, but not by diuretics that act on different transporters. Sodium is then exchanged for calcium via the sodium hydrogen exchanger. Also, sodium entry leads to increased expression of SGK1. Both calcium and SGK1-induced subunit assembly lead to activation of the NADPH-oxidase and accumulation of IsoLGs.77 SGK1 also enhances assembly of ENaC subunits into active channels, which closes a feed forward loop for activation of DCs by salt. These DCs, after exposure to high (190 mM) salt in vitro, prime an increase in systolic BP by a sub-pressor dose of Ang II, when adoptively transferred into naïve recipient mice77.
Our observations about a role for DC ENaC in salt-induced hypertension are consistent with previous knowledge about the role for this ubiquitous channel in human hypertension. For example, although the reason for increased prevalence of SSBP in African American subjects is not fully understood,84–86 these patients exhibit greater antihypertensive responses to amiloride compared to whites, suggesting overactivity of ENaC. In addition to the ENaC gain-of-function and loss-of-function mutations described in Mendelian disorders of hypertension or hypotension,87–97 numerous variants have been identified by gene sequencing among the three genes encoding ENaC subunits (dbSNP and TOPMed databases). Individuals who harbor gain-of-function variants may be at increased risk for salt-sensitive hypertension.98
Many human ENaC nonsynonymous single nucleotide polymorphisms (nsSNPs) affect channel function by altering the sodium self-inhibition response, whereby extracellular Na+ binds to and inhibits ENaC. Numerous sites have been identified in different domains of the extracellular regions of mouse and human ENaC where amino acid substitutions alter channel activity by changing the sodium self-inhibition response.99–107 Although many missense mutations are mostly silent or inhibit protein function,108–110 a number of variants have been identified that have a gain-of-function phenotype reflecting a loss of sodium self-inhibition. Also, siblings with a mild Liddle syndrome phenotype and ENaC gain-of-function mutation (αC479R) have been reported.98 The CYP-epoxygenase metabolite 11,12-epoxyeicosatrienoic acid (EET) inhibits ENaC by reducing channel open probability,111, 112 and SNPs in epoxygenases associate with SSBP. Also, the BP of African Americans who are homozygous for the variant CC allele of a SNP (rs3890011) in the CYP4A11 monooxygenase responds to amiloride but not spironolactone, suggesting an aldosterone-independent activation of their ENaC.25 All observations above, taken together, suggest that ENaC overactivity in immune cells and non-immune tissues may exert a major role in severe and in salt-sensitive hypertension.
The characterization and definition of myeloid cells has evolved through advances in technology in recent years. Using single cell RNA sequencing Villani et al. recently reclassified DC and monocyte populations in the circulation of normal human participants.111 They identified a new subset of DCs with surface expression of the receptor tyrosine kinase Axl, and sialic acid-binding Ig-like lectin-6 (SIGLEC-6) and termed them “AS DCs”. These AS DCs produce copious amounts of IL-1β, TNF-α and IL-6 in response to Toll-like receptor stimulation, potently driving both CD4+ and CD8+ T cell proliferation.111 They also reclassified human classical (CD14+) and non-classical (CD16+) monocytes into four different populations. The role of these novel monocyte and DC subsets in the development of inflammation and human hypertension is unknown and requires future investigation.
Anti-inflammatory myeloid cells:
Although the studies above demonstrate a crucial role of DCs in the promotion of inflammation and the development of hypertension, there are DC subsets that are anti-inflammatory and attenuate hypertension. Expression of A20 (a ubiquitin-editing protein involved in maintenance of immunological homeostasis) in DCs prevents Ang II-induced hypertension,113 with concomitant prevention of the activation and accumulation of T cells in the kidney.113 Another subset of myeloid cells, myeloid-derived suppressor cells (MDSCs), also prevent the increase in BP and its associated inflammatory response in multiple models of murine experimental hypertension.114 Adoptive transfer of these MSDCs improved vascular relaxation and prevented both renal and vascular immune cell infiltration in a murine model of hypertension.115 These studies demonstrate that myeloid cell subsets can provide a crucial immunological break to prevent inflammatory cascades in the development of hypertension.
Innate Lymphoid Cells and NK T cells:
Natural killer cells or innate lymphoid cells (ILCs) are activated and exert cytotoxic effects in hypertension. ILCs have been classified into three groups by their surface expression markers, cytokine production, and transcription factors.116 ILC1s, produce IFN-γ and the transcription factors T-Bet, NFIL3, and Runx3. ILC2s produce type 2 cytokines (IL-4, IL-5, IL-13 and IL-9) and the transcription factors RORα, BCL11B, GATA3, and GFI1. ILC3s express RORγT, AHR, and ID2 and produce type 3 cytokines, (IL-22, IL-17, and GM-CSF).116 A role for ILCs in hypertension remains to be determined.
Natural killer T cells (NK T cells) are innate lymphocytes that respond to the MHCI-related glycoprotein CD1d that specializes in presentation of lipid antigens. Although IsoLG-adducted proteins could represent lipid-modified antigens, we found that mice lacking Jα18 (Ja18−/− mice), an invariant α chain of the T cell receptor on NK T cells, developed similar degrees of hypertension in response to Ang II as those observed in wild type mice. Similarly, we found that the hypertensive response to Ang II in Cd1d−/− mice was identical to that observed in wild type mice.78 These studies indicate that NK T cells or lipid antigens do not play a role in Ang II–induced hypertension and that IsoLGs do not act via the CD1d/NK T cell axis.
Eosinophils:
Eosinophils, which play a role in a number of immune mediated diseases117 are another innate immune cell-type that has recently been associated with hypertension. We found that people with hypertension had increased eosinophil counts compared to normotensive participants,118 and that they were associated with increased body mass index. In contrast, virally suppressed HIV+ persons with hypertension had increases in the eosinophil maturation and differentiation factor IL-5 but their eosinophil counts, also elevated, were independent of body mass. These studies suggest that HIV infection is accompanied by adipose tissue dysfunction that mimics that of obesity. The ultimate mechanisms by which eosinophils contribute to hypertension has not been elucidated.
Complement and Hypertension:
The complement system is a lytic cascade of plasma and membrane bound proteins that plays key roles in homeostasis and host defense as an immune surveillance system. However, it is becoming increasingly recognized that complement also contributes to chronic inflammation. Complement activation leads to cleavage of complement proteins including C3 and C5 by C3 and C5 convertase, respectively.119 Complement proteins produced by antigen presenting cells and T cells regulate both innate and adaptive immune functions.120–123 Complement activation has been implicated in experimental and human hypertension with effects on various cell types important for vascular and renal function.124
Circulating levels of C3 have been linked to incident hypertension and blood pressure in humans.125–127 Elevations in renal complement levels precede the onset of albuminuria in Ang II-induced hypertension.128 C3 is increased in glomeruli of spontaneously hypertensive rats, and C3 inhibition blunts mesangial cell proliferation, indicating a role for C3 in renal remodeling.129 Additionally, mice lacking C3 show decreased renal fibrosis and tubular atrophy in a unilateral ureteral obstruction model.130 In a DOCA-salt model, perivascular adipose tissue was found to produce C3, which was required for pro-inflammatory macrophage phenotypes and adventitial remodeling.131, 132 These studies point to C3 as playing a critical role in renal and vascular damage. Circulating levels of C5a are also increased in human hypertensive subjects compared to controls.133 There are conflicting reports on the role of C5a receptor 1 (C5aR1) in experimental hypertension. Weiss et.al reported that mice deficient in C5aR1 display decreased albuminuria in a uni-nephrectomy and Ang II model despite no change in blood pressure. Paradoxically, cardiac fibrosis and heart weight are elevated in C5aR1-deficient mice.134 In contrast, Zhang et al. reported decreased cardiac fibrosis in C5aR1 deficient mice after Ang II infusion. Bone marrow transfer showed that hematopoietic C5aR1 expression is critical for protection from cardiac fibrosis.133 Intriguingly, mice deficient in both C3aR1 and C5aR1 have a blunted response to Ang II infusion which is associated with increased renal and circulating T regulatory cells.125 Meanwhile, inhibition of C3 and C5 convertases in the Dahl salt-sensitive rat model did not affect blood pressure or albuminuria.135 While the cell type specific effects of each element of the complement system remain to be fully elucidated, growing evidence suggests an important role for complement in hypertensive end-organ damage.
Inflammatory CD4+/CD8+ T cells and Cytokines in Hypertension
The critical role of T cells in hypertension was demonstrated in 2007 when Guzik et al. showed that recombinase activating gene 1 deficient (Rag1−/−) mice, which lack T and B lymphocytes, are protected from the full development of hypertension and end-organ damage. Importantly, the hypertensive phenotype was restored by adoptive transfer of T but not B lymphocytes.136 Although some recent investigations using the Rag1−/− mice from Jackson Laboratory (USA) fail to show this protective phenotype,137, 138 a large body of literature published since then (see below) using different mouse models as well as human samples has firmly established an important role for T cells in hypertension. Possible explanations for this change in the Rag1−/− mice are discussed in Madhur et al. 2020 and underscores the complexity and plasticity of the immune system.139 Briefly, these include an expansion of a population of natural killer cells that compensate for the lack of adaptive immune cells by releasing cytokines normally released by T cells as well as potential changes in the microbiome.
T cells are classified by their surface markers, cytokine profile, and lineage determining transcription factors, with each subset exhibiting precise functions in health and disease. Major T cell classes are CD4+ T helper (Th) cells, CD8+ T cytotoxic (Tc) cells, and CD4−CD8− T cells, which includes the innate-like gamma delta (γδ) T cells (Figure 3). In response to signals from antigen presenting cells or other environmental factors including the local cytokine milieu, CD4+ Th cells further differentiate into pro- or anti-inflammatory subtypes including: Th1 cells that produce interferon gamma (IFNγ) and respond to intracellular pathogens; Th2 cells that produce IL-4 and IL-5 and are involved in allergic disorders and parasitic infections; Th17 cells that produce IL-17A and IL-21 and are involved in autoimmunity and the response to extracellular pathogens; T follicular helper (Tfh) cells that produce IL-21 and signal to B cells to promote antigen-specific immunoglobulin production; and T regulatory cells (Tregs) that produce IL-10 and suppress immune responses. CD8+ T cells are similarly classified as Tc1, Tc2, Tc17, and CD8 Treg cells. With the exception of Th2 cells, which have not been well characterized in hypertension, and Treg cells which likely play a protective role (described below), the remaining Th subtypes (Th1, Tfh, and Th17) are primarily pathogenic in hypertension. Hypertension is associated with a skewing of Th cells into these pro-inflammatory subsets and away from the protective Treg phenotype. The signature cytokines of these pro-inflammatory T cells, namely IL-17A, IFNγ, and IL-21, have been shown to be elevated in hypertensive mouse models and in humans with hypertension.140–143
We first demonstrated a critical role for IL-17A in hypertension in 2010 by showing that IL-17A−/− mice develop blunted hypertension in response to Ang II infusion.144 It is important to note that while the reduction in blood pressure of ~20 mmHg seen in the IL-17A−/− mice compared to wild type (WT) mice is a clinically significant reduction, the IL-17A−/− mice are still hypertensive. Despite this residual hypertension, the end-organ damage normally caused by Ang II is virtually abolished in these mice. They are protected from increases in vascular superoxide production and inflammation, endothelial dysfunction, aortic stiffness, and glomerular injury.144–146
Mechanistically, IL-17A acts on multiple cell types to promote increases in blood pressure and target organ damage. In endothelial cells, IL-17A inhibits nitric oxide production via phosphorylation of the inhibitory site threonine 495 on endothelial nitric oxide synthase.147 In vascular smooth muscle cells, IL-17A in conjunction with tumor necrosis factor alpha induces inflammatory cytokine and chemokine expression, thus promoting the recruitment of other inflammatory cells to the vessel wall.144 IL-17A promotes aortic stiffening via increased collagen synthesis from aortic fibroblasts.145 While the effects of IL-17A on aortic stiffness appear to be BP-dependent, IL-17A has BP-independent effects on small vessel remodeling and stiffness. Orejudo et al. showed that IL-17A infusion increases BP and promotes inward hypertrophic remodeling and stiffness of small mesenteric arteries even when hydralazine and hydrochlorothiazide are used to prevent the BP increase.148 In the kidney, IL-17A acts on proximal and distal tubular cells to increase renal sodium transporter abundance or activity.146, 149 In fact, this is a potential mechanism by which a number of inflammatory cytokines regulate salt and water balance and blood pressure (reviewed in detail in Norlander and Madhur150). IL-17A also upregulates chemokines in the kidney and induces renal damage.151
As discussed above, salt-sensitivity of BP is an independent cardiovascular risk factor. In addition to its effects on DCs, salt can directly increase IL-17A production from Th17 cells and inhibit Treg cells in an SGK1 dependent manner.152–154 We showed that deletion of SGK1 specifically in T cells attenuates Ang II and DOCA-salt induced hypertension, prevents the upregulation of Th17 cells in the spleen in response to Ang II, and abrogates hypertension-induced renal and vascular inflammation and end-organ damage.155
In an elegant study by Amador et al. using a DOCA-salt-induced hypertensive rat model, the authors demonstrated an increase in Th17 cells and downregulation of Treg cells in the heart and kidneys.156 Interestingly, spironolactone treatment prevented the Th17 cell activation and increased numbers of Tregs. Antihypertensive therapy with reserpine, hydralazine, and hydrochlorothiazide, however, did not abrogate the Th17 response in this model, indicating that mineralocorticoid receptor activation, and not the BP per se, underlies the Th17/Treg imbalance.156
Mineralocorticoid receptor signaling also plays an important role in increasing T cell IFNγ production. While Th1 cells make IFNγ, the major T cell source of IFNγ in hypertension is CD8+ Tc1 cells.143, 157 Sun et al. showed that mineralocorticoid receptor deficiency specifically in T cells decreases Ang II-induced hypertension, attenuates renal and vascular damage, and mitigates hypertension-induced accumulation of IFNγ producing T cells, particularly CD8+ T cells, in the aorta and kidney.158 Treatment of WT mice with eplerenone, a mineralocorticoid receptor antagonist, attenuated Ang II-induced hypertension and the accumulation of IFNγ producing T cells.158 In cultured CD8+ T cells, these authors showed that the mineralocorticoid receptor interacted with the IFNγ regulating transcription factors NFAT1 and AP-1, suggesting a potential mechanism by which mineralocorticoid receptor activation increases IFNγ production in these cells.
We previously showed that IFNγ deficient mice are protected from Ang II-induced hypertension, and that like IL-17A, IFNγ has effects on renal sodium transporters.149, 157 Furthermore, we showed that IFNγ production, particularly from CD8+ T cells, likely underlies the increased hypertension and renal and vascular damage caused by deficiency of the lymphocyte adaptor protein, LNK/Sh2b3, which is described in more detail below.157 However, it is important to note that the effects of IFNγ signaling on BP and target organ damage are somewhat inconsistent between studies, suggesting that IFNγ may have both beneficial and detrimental effects depending on the context. For example, Marko et al. found that IFNγ receptor deficiency had no effect on BP in response to a much higher dose of Ang II than used in our studies. Importantly however, despite a lack of BP protection, IFNγ receptor deficiency resulted in reduced cardiac hypertrophy, inflammation, fibrosis, and arrhythmogenic electrical remodeling. In the kidney, IFNγ receptor deficiency reduced inflammation and tubulointerstitial damage.159 In a hypertensive chronic kidney disease model characterized by a high dose of Ang II infusion combined with uninephrectomy, Zhang et al. did not observe an effect of IFNγ deficiency on BP or albuminuria.160 In contrast, Garcia et al. showed that IFNγ may actually play a protective role in the heart. In an aldosterone-dependent hypertensive model, these authors showed that although IFNγ deficiency decreased BP, it was associated with increased cardiac hypertrophy and worse diastolic dysfunction.
Although IFNγ signaling seems to have mixed effects on target organ damage, CD8+ T cells do seem to play an important role in hypertension, which may be mediated in part by their other functions involving both secreted factors and direct cell contact. Youn et al. profiled T cells from humans with and without hypertension and found an increase in immunosenescent, pro-inflammatory CD8+ T cells in hypertensive subjects. These cells are characterized by loss of the surface marker CD28 and presence of CD57. These authors also noted an increase in CD8+ T cells expressing perforin, granzyme B, IFNγ, and/or TNFα. Interestingly, hypertensive subjects also had higher circulating levels of granzyme B and CD4+ T cells expressing perforin.161 How perforin and granzyme B might contribute to hypertension pathology is not entirely clear. Perforin creates pores in target cell membranes, and granzymes cleave both extracellular and intracellular proteins. These effects could result in matrix remodeling, cell detachment, and cell death. Granzyme B can cause rapid caspase-dependent apoptosis. In keeping with this, Shen et al. found that Granzyme B deficiency protected against Ang II-induced cardiac hypertrophy, fibrosis, and inflammation.162 In vitro, Granzyme B cleaved vascular endothelial (VE)-cadherin resulting in disruption of endothelial barrier function. A similar process may underlie the vascular remodeling and microvascular rarefaction that occurs in hypertension. Trott et al. demonstrated that CD8 deficient mice exhibit a blunted hypertensive response to Ang II and are protected against Ang II-induced endothelial dysfunction as well as vascular remodeling and microvascular rarefaction in the kidney.163 Furthermore, following Ang II infusion, WT mice demonstrate an impaired ability to excrete an intraperitoneal saline load, but this ability was preserved in CD8−/− mice, suggesting that CD8+ T cells contribute to renal sodium retention. The mechanism for this may be related to the effect of CD8+ T cells on vascular remodeling/rarefaction, the effect of IFNγ on renal sodium transporters,149 and/or direct effects of CD8+ T cells on renal tubular cells to increase sodium transporters. In support of the latter, CD8+ T cells were shown to stimulate the sodium chloride co-transporter, NCC, in distal convoluted tubule cells through a mechanism that involves direct cell-cell contact.164
IL-21, produced by Th17 and Tfh cells, appears to function upstream of T cell IL-17A and IFNγ production. We showed that mice deficient in IL-21 do not exhibit Ang II-induced increases in IL-17A from CD4+ T cells or IFNγ from CD8+ T cells and are protected from hypertensive end-organ damage.141 In humans, peripheral blood CD4+ T cell production of IL-21 correlated with CD4+ T cell production of IL-17A and with systolic BP at the time of blood draw.141 Importantly, we and others demonstrated that neutralizing antibodies to IL-17A, the IL-17A receptor, or IL-21 administered after the onset of hypertension in rodents, lowers BP and reduces target organ damage.141, 156, 165 These data suggest that anti-cytokine therapies targeting the IL-21/IL-17A pathway may be potential adjunct therapies for reducing the inflammatory damage associated with hypertension. While several therapeutics have recently been developed to target IL-17A signaling in the setting of autoimmune diseases,166, 167 no long-term studies evaluating the effect of these drugs on hypertension or other cardiovascular endpoints has been conducted. However, shorter studies do show promise. For example, von Stebut et al. demonstrated in a randomized, double-blind, placebo-controlled exploratory trial that anti-IL-17A treatment with secukinumab improves endothelial function in patients with moderate-to-severe plaque psoriasis after 52 weeks.168 Mehta and colleagues showed in a prospective observational study that biologic therapy including inhibitors of TNFα, IL-23, and IL-17A is associated with favorable modulation of coronary plaque characteristics in one year.169 Larger and longer clinical trials to investigate therapies aimed at modulating pro-inflammatory T cell phenotype and function may offer new strategies to mitigate the inflammatory end-organ damage and residual cardiovascular risk associated with hypertension.
Regulatory T cells in hypertension
Tregs are a Th cell subset defined by their immunosuppressive function and ability to promote immunological tolerance and homeostasis. Natural Tregs are the most well studied and are a subset of CD4+ T cells initially defined by the presence of cell surface marker CD25 and absence of CD127. Foxp3 was later identified as the canonical transcription factor and master regulator of natural Treg differentiation and function. The discovery of Foxp3 also permitted more definitive identification of these immunosuppressive cells.170 Tregs are functionally marked by the ability to suppress the activation of conventional effector T cells to limit excessive immune responses and promote tolerance to self-antigens. Genetic deletion of Foxp3 in mice or inactivating mutations in humans lead to Treg deficiency and spontaneous autoimmune pathology termed scurfy in mice and autoimmune immunodysregulation, polyendocrinopathy, and enteropathy X-linked (IPEX) syndrome in humans.171, 172 Evidence suggests that loss of Treg number and/or function contributes to a variety of autoimmune diseases through defects in immune tolerance.173 As a result, approaches of adoptive Treg administration and Treg activation are currently in clinical trials for the treatment of a variety of autoimmune diseases.174
Given the emerging role of inflammation and immunity in hypertension, and the immunosuppressive functions of Tregs, multiple studies have investigated the role of Tregs in the pathogenesis of hypertension. Animal models of hypertension have demonstrated decreased Tregs in the blood and kidneys coincident with elevations in BP.125, 175 In humans, some studies have demonstrated decreases in circulating Tregs in those with hypertension,176 while others have shown no difference in circulating Treg numbers.125, 177 To date, most studies testing roles for Tregs in hypertension have performed adoptive transfer of CD4+CD25+ splenic Tregs in animal models. While multiple studies have demonstrated reductions in BP after Treg adoptive transfer,163, 175, 178 these reductions have not been observed uniformly.179–182 Reasons for these differences may relate to different Treg isolation and preparation methodologies and/or the duration and timing of Treg transfer and BP monitoring. Although Treg adoptive transfer has not consistently decreased BP, more robust reductions in hypertension-induced end-organ damage such as vascular stiffness and cardiac hypertrophy have been observed,175, 178–182 suggesting particular benefit to limit end-organ damage. As an alternative approach to adoptive transfer, Treg expansion using low dose IL-2 infusion has been tested, which both reduced BP and renal injury in a mouse model of lupus.183 While approaches to transfer or activate Tregs suggest potential beneficial roles in hypertension, a more limited number of studies have tested the effect of Treg depletion. These studies have primarily used anti-CD25 antibodies to deplete Tregs and have shown either increased BP184 or no effect.185 Interestingly, a recent study by Belanger et al. showed that anti-CD25-mediated Treg depletion increased BP in female but not male mice, suggesting that sex differences alter Treg function in the regulation of BP.186 Mechanisms of the observed beneficial effects of Tregs in hypertension remain unclear, however at least some of the benefit may be through improved vascular endothelial function via Treg production of the anti-inflammatory cytokine IL-10.187–189 In addition, Treg injection in hypertensive mice has been shown to reduce renal neutrophil content through enhanced CD39-dependent neutrophil apoptosis.184
On the whole, studies performed to date suggest that Tregs likely play protective roles in hypertension with variable effects to limit BP elevations but more consistent beneficial roles to limit hypertension-related end-organ damage. These findings suggest a potential for therapeutic use of Treg activation and/or expansion in hypertension. However, improved understanding of the fundamental biology and role of Tregs in hypertension is needed. Given that CD25 can be expressed at least transiently in activated conventional T cells,173 adoptive transfer of CD4+CD25+ cells can include a limited number of conventional T cells in addition to Tregs, which may explain some of the negative results with Treg adoptive transfer described above. Future studies using more specific markers to identify Foxp3+ cells such as GFP from Foxp3-GFP expressing mice can help clarify the effect of Treg transfer on BP.190 Similarly, using diphtheria toxin to specifically deplete Tregs in mice with insertion of the diphtheria toxin receptor into the Foxp3 locus can permit more specific Treg depletion than anti-CD25 antibodies.191 In addition, recent evidence has demonstrated that some Treg subsets may play pathogenic roles in diseases such as type 1 diabetes,192 lung fibrosis,193, 194 and ischemic heart failure195 through reducing angiogenesis and/or promoting fibrosis. Combined with findings that Treg subsets may have distinct functions in different tissues such as skin, adipose tissue, and colon,196, 197 these studies suggest that particular subsets of Tregs may exhibit subtype- and/or context-specific roles in hypertension. Identifying hypertension-associated Treg subsets in humans and animal models will be critical to determine roles of these distinct cell populations in the pathogenesis of hypertension. These efforts to advance our fundamental understanding of Treg function in hypertension have the potential to advance new therapeutic options targeting activation and/or expansion of these cells as a novel therapy.
Gamma delta T cells in hypertension
Most T cells (96–99%) possess a conventional α/β T cell receptor (TCR). However, a small subset of unconventional, innate-like T cells expresses the γδ TCR (1–4%). These cells are generally negative for the CD4 and CD8 surface receptors. Like other T cells, there is considerable heterogeneity among γδ T cells in terms of surface marker expression, cytokine secretion, and function. Unlike conventional T cells, these cells don’t recognize specific protein antigens bound to classic MHCI and MHCII and are activated instead by factors such as non-protein phosphoantigens, isoprenoid pyrophosphates, and heat shock protein-derived peptides without antigen processing and MHC presentation. These cells localize in non-lymphoid tissues where there are poised to respond quickly. A subset of γδ T cells make IL-17A and in certain pathological conditions, are the most prominent source of IL-17A. We found by flow cytometry combined with intracellular staining that both γδ T cells and Th17 cells contribute approximately equally to IL-17A production in the kidney and aorta of Ang II-infused animals.165 Consistent with an important role for γδ T cells in hypertension, Caillon et al. demonstrated that mice with genetic deletion or pharmacologic depletion of γδ T cells exhibit blunted hypertension and preserved endothelial function in response to Ang II.198 Thus, γδ T cells may represent a novel therapeutic target for the treatment of hypertension.
B cells in hypertension
While we have made significant advances over the past two decades in exploring the role of innate immune cells and T cells in hypertension, very little is known about the role of B cells and immunoglobulins in hypertension. This is an important area since B cell involvement and specific immunoglobulin production would suggest an antigen-mediated humoral immune response and open up additional avenues for immune modulating therapies in the management of hypertension. For over 50 years, modest elevations in specific subsets of serum immunoglobulins, particularly IgG, have been observed in hypertensive animals and humans,141, 199–202 but the precise targets of these immunoglobulins and whether they play a causal role in hypertension is unknown. Interestingly, Khamis et al. showed that elevated total serum IgG levels are actually an independent predictor of freedom from adverse cardiovascular events in hypertensive patients, suggesting that IgG may play a protective role.203
As described above, Guzik et al. demonstrated in 2007 that Rag1−/− mice, that are deficient in T and B cells, develop blunted experimental hypertension and that adoptive transfer of T but not B cells restores the hypertensive response.136 This finding led the scientific community to focus almost exclusively on T cells, while B cells were largely ignored. In 2015, Chan et al. revisited the role of B cells in hypertension and demonstrated that pharmacologic depletion of B cells using anti-CD20 antibodies or genetic deletion of B-cell activating factor receptor (BAFF-R) in mice reduced Ang II-induced hypertension and vascular end-organ damage.202 Although not exclusively expressed on B cells, BAFF-R plays a critical role in normal B cell maturation and survival. Subsequently, Dingwell et al. demonstrated that mice unable to produce functional B cells due to deletion of the gene for the heavy chain joining region (JHT) have a modest reduction in baseline BP.204 However, these authors did not study the response to hypertensive stimuli in JHT mice. Using a similar model consisting of deletion of the IgM heavy chain (μMT−/−) as well as activation-induced cytidine deaminase (Aicda−/−) mice that are unable to produce high affinity class-switched immunoglobulins, we recently showed that neither B cells nor high affinity class-switched immunoglobulins are necessary for experimental hypertension.205
One potential explanation for these disparate findings is the type of B cells targeted in the various studies. B cells are divided into 2 major classes: B1 and B2. B1 cells are innate-like cells found predominantly in the peritoneal and pleural cavities that produce natural IgM antibodies. B2 cells are the conventional B cells. Peritoneal B1 cells are relatively resistant to anti-CD20 treatment.206 In addition, BAFF-R−/− mice have a preferential reduction in B2 B cells.207 Thus, both of the models used by Chan et al. only confers partial B cell depletion, primarily of the B2 cell subtype, with some retention of B1 cells. In contrast, our models are characterized by virtually absent or defective B1 and B2 cells. It is therefore possible that innate-like B1 cells play a protective role in hypertension that might be masked by global B cell deficiency. However, further studies are needed to test this hypothesis. Alternatively, B cell activation and immunoglobulin production may not be causal to the pathophysiology of hypertension and instead may simply be a biomarker reflective of immune activation. In keeping with this, it is important to note that IL-21 produced by Tfh cells is a potent inducer of germinal center B cell proliferation and differentiation into immunoglobulin secreting plasma cells. Thus, the increased immunoglobulins observed in hypertension may be reflective of increased IL-21 signaling. Nevertheless, it is important to determine whether specific B cell subsets have an immunomodulatory role in hypertension, and thus further studies are critically needed in this area. Given the limitations and risk of compensatory changes that can occur with genetically modified animals, methods that involve acute pharmacological depletion or inducible genetic deletion of individual B cell subsets will be very valuable and informative.
Autoimmune Collagen Vascular Disease and Hypertension
Numerous collagen vascular diseases are associated with a significant increase in risk of CV disease. Specifically, patients with rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) experience increased CV events. Initial observations revealed a significantly increased risk of death from CV and cerebrovascular diseases in patients with RA.208 They exhibit early atherosclerosis as measured by carotid artery ultrasound and also augmented coronary-artery calcium scores when compared to control subjects.209, 210 Young women with SLE aged 35–44 are 52 times more likely to experience a myocardial infarction when compared to matched controls from the Framingham Offspring Study.211 Inflammation plays an important role in the development of CVD in both RA and SLE subjects, since their CVD is associated with systemic inflammation (elevated inflammatory markers and cytokine secretion). Importantly, subclinical atherosclerosis in RA correlates with elevated erythrocyte sedimentation rate, TNF-α, and IL-6 levels.210, 212 and atherosclerosis development in SLE is associated with augmented type I interferon levels.213
Hypertension is one of a multitude of risk factors for CV disease in patients with systemic autoimmunity. There is mounting evidence that hypertension is a direct and specific effect of immune activation independent of additional organ involvement. In SLE, hypertension occurs independently of renal disease and of markers of systemic inflammation. In a study of 235 patients with SLE, hypertension could not be correlated with renal disease or serum complement levels.214 The importance of hypertension as a primary driver of CV disease in collagen vascular diseases has been highlighted by numerous studies. In a prospective study of 229 subjects with SLE, the requirement for antihypertensive treatment was associated with coronary artery disease.215 Moreover, a recent study by Tselios et al. showed that BP reduction below 130/80 in patients with SLE reduces atherosclerotic vascular events.216 Hypertension is also highly prevalent in RA.217 Panoulas et. al. described the presence of hypertension in over 70.5% of RA patients as defined by systolic BP≥140 mmHg or diastolic BP≥90 mmHg.218
Additional evidence for a role of inflammation in the hypertension observed in collagen vascular diseases arises from the study of animal models of SLE. The NZBWF1 mouse strain develops systemic autoimmunity characterized by immune complex formation and autoantibody production. Female mice develop systemic hypertension at 6–9 months of age and have been extensively studied as a model of SLE associated hypertension. Importantly, cells of both the adaptive and innate immune system have been implicated in the pathogenesis of hypertension in this model. B-cell depletion by different methods revealed the importance these cells in the development of SLE-associated hypertension. For example, treatment of NZBWF1 animals with anti-CD20 abrogates the development of hypertension219 and treatment with bortezomib, a proteasome inhibitor that exhibits plasma cell toxicity, also attenuates mean arterial pressure in these SLE prone mice.220
A role for oxidative stress in the immune activation observed in the hypertension of autoimmune disorders has been demonstrated by treatment of NZBWF1 animals with the superoxide scavenger tempol, or the NADPH oxidase inhibitor apocynin; both reduced BP and renal injury in this model.221 This occurred in the absence of a reduction in SLE disease activity as measured by the accumulation of anti-double-stranded DNA antibodies. We have shown that scavenging of isolevuglandins (isoLGs), a product of lipid peroxidation, with the specific scavenger 2-HOBA attenuates SLE associated hypertension in both the NZBWF1 and B6.SLE123 models of SLE. In contrast to the above, 2-HOBA reduced SLE severity as measured by anti-double stranded DNA antibodies and renal immune complex deposition.222 These data suggest that specific products of ROS-mediated lipid peroxidation contribute to the genesis of hypertension in SLE.
Treatment of collagen vascular diseases is based on global immunosuppressants despite the fact that their mechanisms are heterogeneous. Investigation of the factors driving disease heterogeneity and their associations with CVD will allow for future therapeutic approaches that target such specific factors.
Viral Infections and Hypertension
Numerous studies have found that people with hypertension have increased susceptibility to viral infections with worse outcomes. Hypertension is among the comorbidities that worsen the prognosis for the recent coronavirus pandemic (COVID-19).223 Other studies have linked hypertension with incidence and outcomes of influenza infection.224, 225 Perhaps the most studied viral infection and its relation to hypertension is that by the human immunodeficiency virus (HIV).
COVID-19:
The recent coronavirus disease 2019 (COVID-19) pandemic, caused by the novel severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2), has led to an unprecedented medical crisis worldwide. People with hypertension are at a heightened risk for severity and mortality due to COVID-19. Because SARS-CoV-2 uses the angiotensin-converting enzyme 2 (ACE2) receptor to infect cells, there has been a debate as to whether use of antihypertensive drugs including the Angiotensin receptor blockers (ARBs) and ACE inhibitors, which increase expression of ACE2, would increase the risk for infectivity and severity of COVID-19 in people with hypertension.226 However, a recent study by Trump et al found that augmented immune cell activation might explain the adverse COVID-19 outcomes in patients with hypertension and that ACE inhibitors could be the more beneficial anti-hypertensive treatment during COVID-19.227 They found that in people with hypertension, there is a delayed viral clearance and an exacerbated airway inflammation in patients with COVID-19. Treatment with ACE inhibitors was associated with a reduced COVID-19-related inflammation and with increased cell intrinsic antiviral responses, whereas ARB treatment related to enhanced epithelial–immune cell interactions.227 Macrophages and neutrophils of patients with hypertension, exhibited higher expression of the pro-inflammatory cytokines CCL3 and CCL4 and the chemokine receptor CCR1.227 These studies suggest that the worse outcomes in patients with COVID-19 who have hypertension may stem from an exaggerated immune response.
HIV:
Studies of blood pressure in people infected with HIV may provide a unique window into the role of the immune system in human hypertension. Over 36 million adults are now living with HIV. Currently, 25 million people living with HIV (PLWH) are on Antiretroviral Therapy (ART) and 2 million start ART each year. As a result of ART, PLWH are living longer and healthier lives with less morbidity and mortality from opportunistic infections. According to a 2018 meta-analysis, the risk of CVD in PLWH is ~2.5-fold higher than HIV-uninfected adults of similar age.228 According to one recent study of the largest cohort of PLWH in North America, ~42% of the variability of CVD is attributable to high office BP.229 More than one-third (35%) of PLWH on ART have hypertension, and they develop CVD at lower office BP levels than HIV-uninfected adults.230
Several findings support that immune activation and chronic inflammation contribute to hypertension and CVD in PLWH. Macrophage derived serum IL-6 and soluble CD14 are both elevated in PLWH and precede incident hypertension.231 Soluble CD163, a marker of monocyte activation, is also elevated and has been associated with vascular damage in this population.232 T-cell secreted cytokines such as IL-17A and IFNγ and, the eosinophil maturation cytokine, IL-5, are significantly elevated in hypertensive PLWH.118
HIV-specific proteins are known to directly trigger inflammatory cascades. The transcriptional regulator Tat induces expression of endothelial cellular adhesion molecules (ICAM, VCAM and E-selectin) and primes the vasculature for inflammatory cell migration.233 Additionally, Tat upregulates T-cell derived interleukin-17A expression, and elevated serum IL-17A is associated with hypertension in PLWH.118 Expression of the NLRP3 inflammasome is upregulated in the peripheral blood immune compartment of PLWH compared to HIV-uninfected controls,234 which may contribute to their hypertension. Moreover, CD4+ T-cells of PLWH increase during the first two years after ART initiation in parallel manner to a concomitant rapid increase in BP.235 Eosinophil counts are associated with hypertension in PLWH but not in HIV-uninfected controls after adjustment for traditional risk factors.118 Dysfunctional production of the T-cell derived, eosinophil maturation factor IL-5 has been suggested as a possible underlying mechanism; although the macrophage/monocyte derived macrophage inflammatory protein 1 alpha (MIP-1a), and others, may also contribute.
In summary, PLWH are at increased risk of hypertension and CVD. Neither traditional risk factors such as obesity and age nor HIV-specific factors such as ART type, lipodystrophy and HIV viral load account for this risk. Recent insight into the complex interplay of these factors and HIV-specific inflammatory pathways have shed light on the pathophysiology of hypertension in PLWH. However, further studies are needed to outline a unifying theory on mechanisms and so identify potentially actionable targets.
The Gut Microbiome, Inflammation and Hypertension
The gut microbiome, made of trillions of microbes and their genetic material, is now widely acknowledged to affect host physiology and disease.236 Of all microbes, bacteria and their metabolites are the best understood for their role in CV disorders like hypertension.237 Evidence from human studies indicates that hypertension is associated with different microbial composition in terms of taxa, gut-derived metabolites, and intestinal mucosa structure. Hypertensive individuals have reduced microbial richness, diversity, and display a microbial shift with greater abundance in pathogenic taxa compared to normotensive individuals.238,239 Beneficial metabolites such as short-chain fatty acids are decreased in hypertension, and the gut barrier integrity is compromised.240 Animal models have played a substantial role in elucidating mechanisms of the microbiome beyond associations. For instance, fecal microbial transplantation from hypertensive subjects into germ-free mice elicits hypertension suggesting a direct role of the gut microbiome in modulating host BP.241, 242
The gastrointestinal tract houses the largest immune organ of the body, the gut-associated lymphoid tissue. The gut microbiome regulates inflammatory pathways that contribute to the pathogenesis of hypertension. Metabolites produced by bacteria in the gut including trimethylamine-N-oxide (TMAO), lipopolysaccharide (LPS), and short-chain fatty acids (SCFAs) are known mediators of these inflammatory pathways.237 SCFAs attenuate hypertension, gut dysbiosis, and restore a balance between Th17 and Treg cells in animal models of hypertension.243,244 They promote Th1 but prevent Th17 cell differentiation through G-protein coupled receptor 43 (GPR43).245,246 The gut microbiome is highly dynamic and can be influenced by factors such as dietary composition. For instance, a diet rich in fiber confers antihypertensive benefits whereas a high-salt diet increases BP and causes a shift in microbial composition.239,247 In mice, a high salt diet decreases the Firmicutes to Bacteroidetes ratio, promotes gut barrier dysfunction, and disrupts inflammatory responses.239,248 Interestingly, high salt increases Th17 cells by mechanisms directly dependent on the gut microbiome,248 and exacerbates gut inflammation by increasing the expression of inflammatory markers such as IFN-γ and some Toll-like receptors in the colon.249 Mucosal DCs, which normally regulate gut homeostasis tightly,250 influence gut dysbiosis and hypertension in response to a high salt stimulus when activated.239
Sex Differences
There is an established sexual dimorphism in hypertension. Hypertension is more prevalent in men than in women younger than 60 years of age; whereas the opposite trend is present in older subjects.251 While both men and women exhibit SSBP, men have higher mortality from SSBP for unclear reasons.33, 252 Experimental animal studies indicate that SSBP is more pronounced in males than females.253 It is not known if sex/gender differences exist in monocyte activation and how this impacts SSBP. Gender differences have been identified in Na+ storage between muscle and skin: men exhibit higher Na+ content in skin than in muscle, whereas women accumulate Na+ to greater levels in muscle.254
The observed associations between HIV and hypertension appear to be similar in males and females. Few published data exist for HIV-infected women, though, since most studies of HIV and hypertension have mainly enrolled homosexual men. Studies report that HIV-infected women on long-term ART have significantly elevated sCD14 and sCD163 as compared to HIV-infected males,255 and higher sCD14 levels are strongly linked with higher blood pressure in people living with HIV.256 Further studies are needed in HIV-infected women and should investigate both the pre-menopausal and post-menopausal period.
There are also sex differences in factors that influence gut microbiota such as salt. Women exhibit greater BP changes in response to high-salt consumption.257 While direct evidence on sex differences in the gut microbiota in humans is lacking, studies in preclinical models have shown sex-dependent differences in the gut microbiome. In mice, there is differential abundance in taxa and metabolites between males and females and there is a role for sex hormones in modulating gut microbiota composition.258,259 Testosterone and estrogens play a substantial role in sex differences and both hormones are differentially affected by gut-related mechanisms.260,261,262
Germ-free mice also exhibit sex differences in vascular function, an important regulator of BP; for example young male germ-free mice exhibited increased vascular stiffness that was not observed in females.263 More studies on the impact of sex differences in the gut microbiome on hypertension are warranted because of the therapeutic implications for management of sex-specific diseases such as hypertensive disorders of pregnancy. Growing evidence suggests a role for gut microbiome disruption in the pathophysiology of adverse pregnancy outcomes such as preterm birth and preeclampsia.264 Preeclampsia is associated with gut dysbiosis with an increase in Proteobacteria and greater fecal and plasma TMAO and LPS compared to healthy pregnant controls.265 LPS activates an inflammatory response through Toll-like receptor 4 and is used to induce preeclampsia-like phenotypes in rodents.266 Better characterization of the involvement of the gut microbiome in hypertensive disorders of pregnancy may improve our understanding of their elusive etiologies. Furthermore, preeclampsia is an established risk factor for adverse maternal and offspring CV outcomes later in life,267 and there is growing evidence that maternal dysbiosis can be transferred to the fetus in utero, programming the offspring for susceptibility to illness.268,269 The specific role of the gut microbiome in determining maternal and offspring prognosis needs further investigation.
Conclusion and Perspectives
The inability to control hypertension in some subjects who are compliant with medications, the effect of salt to raise BP in certain individuals regardless of hypertension status, and the presence of residual CV risk in those with treated and controlled hypertension strongly suggest that current pharmacological therapies leave some underlying pathogenic mechanism of the disorder unaddressed. In this review, we summarize the now overwhelming evidence that immune-mediated inflammation may be such a mechanism and discuss the genetic and environmental risk factors, such as viral infections, high salt diet, and the microbiome, which can modulate inflammation and CV risk (Figure 5). In some individuals, the magnitude of vascular and renal inflammation may preclude total resolution of vasoconstriction or normalization of sodium handling by current therapies, hence leading to refractory uncontrolled hypertension. In the majority of non-refractory hypertensive patients, current therapies may control BP, but the unaddressed underlying inflammatory processes may perpetuate target organ damage and explain their excess residual CV risk.
Figure 5: Risk factors contributing to inflammation and hypertension.

Risk factors including viral infections, high salt diet, genetics, and the microbiome lead to an inflammatory milieu that contributes to hypertension.
Therefore, the ultimate aim of investigating immunity in hypertension is to develop therapies that can be tested for control of BP or for reduction of CV risk in clinical trials. Conventional immunosuppressants, such as mycophenolate mofetil, reduce BP in experimental models and humans and more recently, the anti-IL-1β monoclonal antibody canakinumab reduced CV events in patients with established atherosclerotic disease. However, ubiquitous suppression of immunity by these type of agents increases infectious complications, limiting their widespread use for a disease that affects almost half of the adult population of the world. Thus, a more granular understanding of the immune mechanisms of hypertension may allow for more targeted and personalized therapeutic approaches. In addition, approaches that restore the balance of pro-inflammatory and anti-inflammatory immune cells in hypertension without causing global immunosuppression will be important to develop and test in clinical trials. Rigorous and routine measurement of BP and CV events should be considered in current and future trials of anti-inflammatory medications being tested for other diseases to determine their potential in decreasing CV events in at-risk populations. Furthermore, more studies using existing drugs such as amiloride and spironolactone which seem to have anti-inflammatory effects will help determine if they should perhaps be used more widely or selectively in certain populations such as those with salt-sensitive hypertension or hypertension driven by excess aldosterone, respectively. Finally, novel technologies, such as targeted nanoparticles, may be able to inhibit or overexpress certain factors in specific subsets of immune cells, thus delivering precise and targeted therapies without the risks of global immunosuppression. Accomplishing these goals will require a coordinated effort among cardiovascular physiologists, immunologists, biomedical engineers, and clinical trialists. It is our hope that these efforts will continue and that one-day, immune modulation will be added to our arsenal to eradicate this deadly epidemic.
Funding Sources
This study was supported by the National Institutes of Health grants DP2HL137166 (MSM), K01HL130497 (AK), R01HL147818 (TRK and AK), K08 HL153786 (MRA), T32HL144446 (AP), F32HL142937 (JPV), P30DK079307 (TRK), K08 HL153789 (DP), K23 HL152926 (JRK) and the American Heart Association EIA34480023 (MSM).
Non-standard Abbreviations and Acronyms
- 2-HOBA
2-hydroxybenzilamine
- AHA-ACC
American Heart Association-American College of Cardiology
- AHR
aryl hydrocarbon receptor
- Aicda
activation-induced cytidine deaminase
- Ang II
angiotensin II
- AP-1
activator protein 1
- APCs
antigen presenting cells
- ART
antiretroviral Therapy
- ASC
apoptosis-associated speck-like adaptor protein
- ATP
adenosine triphosphate
- BAFF-R
B-cell activating factor receptor
- BCL11B
B cell leukemia 11b
- BP
blood pressure
- CANTOS
Canakinumab Anti-inflammatory Thrombosis Outcome Study
- CARD
C-terminal caspase recruitment domain
- COX
cyclooxygenase
- CV
cardiovascular
- CYP4A11
cytochrome P450 4A11
- DAMPs
damage associated molecular patterns
- DASH
dietary approaches to stop hypertension
- DC
dendritic cells
- DOCA
deoxycorticosterone acetate
- EETs
epoxyeicosatrienoic acids
- ENaC
epithelial sodium channel
- Foxp3
forkhead box P3
- GATA3
GATA-binding protein 3
- GATA4
GATA-binding protein 4
- GFI1
growth factor independent protein
- GFP
green fluorescent protein
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- GPR43
G-protein coupled receptor 43
- GWAS
genome-wide association studies
- HIV
human immunodeficiency virus
- HIV
human immunodeficiency virus
- HPLC-MS
high pressure liquid chromatography–mass spectrometry
- HsCRP
high sensitivity C-reactive protein
- ICAM
intercellular adhesion molecule
- ID2
DNA-binding protein inhibitor 2
- IFN-γ
interferon gamma
- IgG
immunoglobulin G
- IL
interleukin
- ILCs
innate lymphoid cells
- IPEX
enteropathy X-linked
- IsoLGs
isolevuglandins
- K+
potassium
- LPS
lipopolysaccharide
- LysM iDTR
myelomonocytic cells expressing lysozyme M
- MDSCs
myeloid-derived suppressor cells
- MHCI
major histocompatibility complex I
- MIP-1a
macrophage inflammatory protein 1 alpha
- NaCl
sodium chloride
- NADPH
nicotinamide adenine dinucleotide phosphate
- NCC
sodium chloride co-transporter
- NF-κB
nuclear factor-kappaB
- NFAT
nuclear factor of activated T cells
- NFIL3
nuclear factor, interleukin 3 regulated
- NK T
natural killer T cells
- NLR
nucleotide-binding oligomerization domain leucine-rich repeat
- NLRP3
NOD-like receptor family pyrin domain containing 3
- NOD
nucleotide oligomerization domain
- nsSNPs
nonsynonymous single nucleotide polymorphisms
- PAMPs
pathogen associated molecular patterns
- PLWH
people living with HIV
- pro-IL-1β
pro-interleukin-1beta
- PRR
pattern recognition receptors
- RA
rheumatoid arthritis
- RORα
retinoic acid related orphan receptor alpha
- RORγT
retinoic acid related orphan receptor gamma
- ROS
reactive oxygen species
- Runx3
runt-related transcription factor 3
- SCFAs
short-chain fatty acids
- SGK1
serum and glucocorticoid- regulated kinase
- SHR
spontaneously hypertensive rats
- SIGLEC-6
sialic acid-binding Ig-like lectin-6
- SLE
systemic lupus erythematosus
- SNP
single nucleotide polymorphism
- SR
salt resistant
- SS
salt sensitive
- SSBP
salt sensitivity of blood pressure
- Tc
T cytotoxic
- TCR
T cell receptor
- Tfh
T follicular helper
- Th
T helper
- TLRs
toll-like receptors
- TMAO
trimethylamine-N-oxide
- TNF-α
tumor necrosis factor alpha
- Tregs
T regulatory cells
- VCAM
vascular cell adhesion molecule
- VEGF
vascular endothelial growth factor
- VSMCs
vascular smooth muscle cells
- WT
wild type
Footnotes
Disclosures: None
REFERENCES
- 1.Nwankwo T, Yoon SS, Burt V, Gu Q. Hypertension among adults in the united states: National health and nutrition examination survey, 2011–2012. NCHS data brief. 2013:1–8 [PubMed] [Google Scholar]
- 2.Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: Analysis of worldwide data. Lancet. 2005;365:217–223 [DOI] [PubMed] [Google Scholar]
- 3.Murray CJ, Lopez AD. Measuring the global burden of disease. N Engl J Med. 2013;369:448–457 [DOI] [PubMed] [Google Scholar]
- 4.Rapsomaniki E, Timmis A, George J, et al. Blood pressure and incidence of twelve cardiovascular diseases: Lifetime risks, healthy life-years lost, and age-specific associations in 1.25 million people. Lancet. 2014;383:1899–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collaborators GBDRF. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990–2017: A systematic analysis for the global burden of disease study 2017. Lancet. 2018;392:1923–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawes CM, Vander Hoorn S, Rodgers A, International Society of H. Global burden of blood-pressure-related disease, 2001. Lancet. 2008;371:1513–1518 [DOI] [PubMed] [Google Scholar]
- 7.Collaborators GBDRF, Forouzanfar MH, Alexander L, et al. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks in 188 countries, 1990–2013: A systematic analysis for the global burden of disease study 2013. Lancet. 2015;386:2287–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collaborators GBDE. Global, regional, and national burden of epilepsy, 1990–2016: A systematic analysis for the global burden of disease study 2016. The Lancet. Neurology. 2019;18:357–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whelton PK, Carey RM, Aronow WS, et al. 2017 acc/aha/aapa/abc/acpm/ags/apha/ash/aspc/nma/pcna guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: Executive summary: A report of the american college of cardiology/american heart association task force on clinical practice guidelines. Hypertension. 2018;71:1269–1324 [DOI] [PubMed] [Google Scholar]
- 10.Lewington S, Clarke R, Qizilbash N, Peto R, Collins R, Prospective Studies C. Age-specific relevance of usual blood pressure to vascular mortality: A meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. 2002;360:1903–1913 [DOI] [PubMed] [Google Scholar]
- 11.Muntner P, Hardy ST, Fine LJ, Jaeger BC, Wozniak G, Levitan EB, Colantonio LD. Trends in blood pressure control among us adults with hypertension, 1999–2000 to 2017–2018. Jama. 2020;324:1190–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jaffe MG, Young JD. The kaiser permanente northern california story: Improving hypertension control from 44% to 90% in 13 years (2000 to 2013). Journal of clinical hypertension. 2016;18:260–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhatt H, Siddiqui M, Judd E, Oparil S, Calhoun D. Prevalence of pseudoresistant hypertension due to inaccurate blood pressure measurement. Journal of the American Society of Hypertension : JASH. 2016;10:493–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de la Sierra A, Segura J, Banegas JR, Gorostidi M, de la Cruz JJ, Armario P, Oliveras A, Ruilope LM. Clinical features of 8295 patients with resistant hypertension classified on the basis of ambulatory blood pressure monitoring. Hypertension. 2011;57:898–902 [DOI] [PubMed] [Google Scholar]
- 15.Muxfeldt ES, Bloch KV, Nogueira Ada R, Salles GF. True resistant hypertension: Is it possible to be recognized in the office? American journal of hypertension. 2005;18:1534–1540 [DOI] [PubMed] [Google Scholar]
- 16.Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: Diagnosis, evaluation, and treatment. A scientific statement from the american heart association professional education committee of the council for high blood pressure research. Hypertension. 2008;51:1403–1419 [DOI] [PubMed] [Google Scholar]
- 17.Egan BM, Zhao Y, Li J, Brzezinski WA, Todoran TM, Brook RD, Calhoun DA. Prevalence of optimal treatment regimens in patients with apparent treatment-resistant hypertension based on office blood pressure in a community-based practice network. Hypertension. 2013;62:691–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Persu A, Jin Y, Baelen M, et al. Eligibility for renal denervation: Experience at 11 european expert centers. Hypertension. 2014;63:1319–1325 [DOI] [PubMed] [Google Scholar]
- 19.Berra E, Azizi M, Capron A, Hoieggen A, Rabbia F, Kjeldsen SE, Staessen JA, Wallemacq P, Persu A. Evaluation of adherence should become an integral part of assessment of patients with apparently treatment-resistant hypertension. Hypertension. 2016;68:297–306 [DOI] [PubMed] [Google Scholar]
- 20.Acelajado MC, Pisoni R, Dudenbostel T, Dell’Italia LJ, Cartmill F, Zhang B, Cofield SS, Oparil S, Calhoun DA. Refractory hypertension: Definition, prevalence, and patient characteristics. Journal of clinical hypertension. 2012;14:7–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elijovich F, Weinberger MH, Anderson CA, et al. Salt sensitivity of blood pressure: A scientific statement from the american heart association. Hypertension. 2016;68:e7–e46 [DOI] [PubMed] [Google Scholar]
- 22.Blacher J, Evans A, Arveiler D, et al. Residual coronary risk in men aged 50–59 years treated for hypertension and hyperlipidaemia in the population: The prime study. Journal of hypertension. 2004;22:415–423 [DOI] [PubMed] [Google Scholar]
- 23.Grigoryan L, Pavlik VN, Hyman DJ. Characteristics, drug combinations and dosages of primary care patients with uncontrolled ambulatory blood pressure and high medication adherence. Journal of the American Society of Hypertension : JASH. 2013;7:471–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Acelajado MC, Hughes ZH, Oparil S, Calhoun DA. Treatment of resistant and refractory hypertension. Circulation research. 2019;124:1061–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laffer CL, Elijovich F, Eckert GJ, Tu W, Pratt JH, Brown NJ. Genetic variation in cyp4a11 and blood pressure response to mineralocorticoid receptor antagonism or enac inhibition: An exploratory pilot study in african americans. J Am Soc Hypertens. 2014;8:475–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakagawa K, Holla VR, Wei Y, et al. Salt-sensitive hypertension is associated with dysfunctional cyp4a10 gene and kidney epithelial sodium channel. The Journal of clinical investigation. 2006;116:1696–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saha C, Eckert GJ, Ambrosius WT, Chun TY, Wagner MA, Zhao Q, Pratt JH. Improvement in blood pressure with inhibition of the epithelial sodium channel in blacks with hypertension. Hypertension. 2005;46:481–487 [DOI] [PubMed] [Google Scholar]
- 28.Dahl LK, Heine M, Tassinari L. Role of genetic factors in susceptibility to experimental hypertension due to chronic excess salt ingestion. Nature. 1962;194:480–482 [DOI] [PubMed] [Google Scholar]
- 29.Vollmer WM, Sacks FM, Ard J, et al. Effects of diet and sodium intake on blood pressure: Subgroup analysis of the dash-sodium trial. Annals of internal medicine. 2001;135:1019–1028 [DOI] [PubMed] [Google Scholar]
- 30.Schulman IH, Aranda P, Raij L, Veronesi M, Aranda FJ, Martin R. Surgical menopause increases salt sensitivity of blood pressure. Hypertension. 2006;47:1168–1174 [DOI] [PubMed] [Google Scholar]
- 31.Guyton AC. The surprising kidney-fluid mechanism for pressure control--its infinite gain! Hypertension. 1990;16:725–730 [DOI] [PubMed] [Google Scholar]
- 32.Laffer CL, Scott RC 3rd, Titze JM, Luft FC, Elijovich F. Hemodynamics and salt-and-water balance link sodium storage and vascular dysfunction in salt-sensitive subjects. Hypertension. 2016;68:195–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weinberger MH, Fineberg NS, Fineberg SE, Weinberger M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension. 2001;37:429–432 [DOI] [PubMed] [Google Scholar]
- 34.Morimoto A, Uzu T, Fujii T, Nishimura M, Kuroda S, Nakamura S, Inenaga T, Kimura G. Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet. 1997;350:1734–1737 [DOI] [PubMed] [Google Scholar]
- 35.Blacher J, Evans A, Arveiler D, et al. Residual cardiovascular risk in treated hypertension and hyperlipidaemia: The prime study. Journal of human hypertension. 2010;24:19–26 [DOI] [PubMed] [Google Scholar]
- 36.Zanchetti A, Thomopoulos C, Parati G. Randomized controlled trials of blood pressure lowering in hypertension: A critical reappraisal. Circulation research. 2015;116:1058–1073 [DOI] [PubMed] [Google Scholar]
- 37.Nadir MA, Rekhraj S, Wei L, Lim TK, Davidson J, MacDonald TM, Lang CC, Dow E, Struthers AD. Improving the primary prevention of cardiovascular events by using biomarkers to identify individuals with silent heart disease. Journal of the American College of Cardiology. 2012;60:960–968 [DOI] [PubMed] [Google Scholar]
- 38.Lieb W, Enserro DM, Sullivan LM, Vasan RS. Residual cardiovascular risk in individuals on blood pressure-lowering treatment. Journal of the American Heart Association. 2015;4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131 [DOI] [PubMed] [Google Scholar]
- 40.Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, Libby P, Glynn RJ, Ridker PM. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. 2019;139:1289–1299 [DOI] [PubMed] [Google Scholar]
- 41.Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ, Group CT. Relationship of c-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the cantos randomised controlled trial. Lancet. 2018;391:319–328 [DOI] [PubMed] [Google Scholar]
- 42.Ridker PM, Libby P, MacFadyen JG, et al. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: Analyses from the canakinumab anti-inflammatory thrombosis outcomes study (cantos). European heart journal. 2018;39:3499–3507 [DOI] [PubMed] [Google Scholar]
- 43.Rothman AM, MacFadyen J, Thuren T, Webb A, Harrison DG, Guzik TJ, Libby P, Glynn RJ, Ridker PM. Effects of interleukin-1beta inhibition on blood pressure, incident hypertension, and residual inflammatory risk: A secondary analysis of cantos. Hypertension. 2020;75:477–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alexander MR, Norlander AE, Elijovich F, Atreya RV, Gaye A, Gnecco JS, Laffer CL, Galindo CL, Madhur MS. Human monocyte transcriptional profiling identifies il-18 receptor accessory protein and lactoferrin as novel immune targets in hypertension. Br J Pharmacol. 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Caillon A, Schiffrin EL. Role of inflammation and immunity in hypertension: Recent epidemiological, laboratory, and clinical evidence. Curr Hypertens Rep. 2016;18:21. [DOI] [PubMed] [Google Scholar]
- 46.Krishnan SM, Sobey CG, Latz E, Mansell A, Drummond GR. Il-1beta and il-18: Inflammatory markers or mediators of hypertension? Br J Pharmacol. 2014;171:5589–5602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dalekos GN, Elisaf M, Bairaktari E, Tsolas O, Siamopoulos KC. Increased serum levels of interleukin-1beta in the systemic circulation of patients with essential hypertension: Additional risk factor for atherogenesis in hypertensive patients? J Lab Clin Med. 1997;129:300–308 [DOI] [PubMed] [Google Scholar]
- 48.Rabkin SW. The role of interleukin 18 in the pathogenesis of hypertension-induced vascular disease. Nat Clin Pract Cardiovasc Med. 2009;6:192–199 [DOI] [PubMed] [Google Scholar]
- 49.Omi T, Kumada M, Kamesaki T, et al. An intronic variable number of tandem repeat polymorphisms of the cold-induced autoinflammatory syndrome 1 (cias1) gene modifies gene expression and is associated with essential hypertension. Eur J Hum Genet. 2006;14:1295–1305 [DOI] [PubMed] [Google Scholar]
- 50.Xu L, Li S, Liu Z, Jiang S, Wang J, Guo M, Zhao X, Song W, Liu S. The nlrp3 rs10754558 polymorphism is a risk factor for preeclampsia in a chinese han population. J Matern Fetal Neonatal Med. 2019;32:1792–1799 [DOI] [PubMed] [Google Scholar]
- 51.Dorffel Y, Latsch C, Stuhlmuller B, Schreiber S, Scholze S, Burmester GR, Scholze J. Preactivated peripheral blood monocytes in patients with essential hypertension. Hypertension. 1999;34:113–117 [DOI] [PubMed] [Google Scholar]
- 52.Ren XS, Tong Y, Ling L, et al. Nlrp3 gene deletion attenuates angiotensin ii-induced phenotypic transformation of vascular smooth muscle cells and vascular remodeling. Cell Physiol Biochem. 2017;44:2269–2280 [DOI] [PubMed] [Google Scholar]
- 53.Sun HJ, Ren XS, Xiong XQ, et al. Nlrp3 inflammasome activation contributes to vsmc phenotypic transformation and proliferation in hypertension. Cell Death Dis. 2017;8:e3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gan W, Ren J, Li T, Lv S, Li C, Liu Z, Yang M. The sgk1 inhibitor emd638683, prevents angiotensin ii-induced cardiac inflammation and fibrosis by blocking nlrp3 inflammasome activation. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1–10 [DOI] [PubMed] [Google Scholar]
- 55.Rodriguez-Iturbe B, Pons H, Johnson RJ. Role of the immune system in hypertension. Physiol Rev. 2017;97:1127–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rodriguez-Iturbe B, Zhan CD, Quiroz Y, Sindhu RK, Vaziri ND. Antioxidant-rich diet relieves hypertension and reduces renal immune infiltration in spontaneously hypertensive rats. Hypertension. 2003;41:341–346 [DOI] [PubMed] [Google Scholar]
- 57.Chen D, Xiong XQ, Zang YH, et al. Bcl6 attenuates renal inflammation via negative regulation of nlrp3 transcription. Cell Death Dis. 2017;8:e3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao M, Bai M, Ding G, Zhang Y, Huang S, Jia Z, Zhang A. Angiotensin ii stimulates the nlrp3 inflammasome to induce podocyte injury and mitochondrial dysfunction. Kidney Dis (Basel). 2018;4:83–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kadoya H, Satoh M, Sasaki T, Taniguchi S, Takahashi M, Kashihara N. Excess aldosterone is a critical danger signal for inflammasome activation in the development of renal fibrosis in mice. FASEB J. 2015;29:3899–3910 [DOI] [PubMed] [Google Scholar]
- 60.Bai M, Chen Y, Zhao M, Zhang Y, He JC, Huang S, Jia Z, Zhang A. Nlrp3 inflammasome activation contributes to aldosterone-induced podocyte injury. Am J Physiol Renal Physiol. 2017;312:F556–F564 [DOI] [PubMed] [Google Scholar]
- 61.Krishnan SM, Dowling JK, Ling YH, et al. Inflammasome activity is essential for one kidney/deoxycorticosterone acetate/salt-induced hypertension in mice. Br J Pharmacol. 2016;173:752–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krishnan SM, Ling YH, Huuskes BM, et al. Pharmacological inhibition of the nlrp3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc Res. 2019;115:776–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prager P, Hollborn M, Steffen A, Wiedemann P, Kohen L, Bringmann A. P2y1 receptor signaling contributes to high salt-induced priming of the nlrp3 inflammasome in retinal pigment epithelial cells. PLoS One. 2016;11:e0165653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shih P-aB O Connor DT. Hereditary determinants of human hypertension: Strategies in the setting of genetic complexity. Hypertension (Dallas, Tex. : 1979). 2008;51:1456–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lip S, Padmanabhan S. Genomics of blood pressure and hypertension: Extending the mosaic theory toward stratification. The Canadian journal of cardiology. 2020;36:694–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Levy D, Ehret GB, Rice K, et al. Genome-wide association study of blood pressure and hypertension. Nature genetics. 2009;41:677–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Newton-Cheh C, Johnson T, Gateva V, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nature genetics. 2009;41:666–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ng FL, Warren HR, Caulfield MJ. Hypertension genomics and cardiovascular prevention. Ann Transl Med. 2018;6:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rodriguez-Iturbe B, Johnson RJ. Genetic polymorphisms in hypertension: Are we missing the immune connection? American journal of hypertension. 2019;32:113–122 [DOI] [PubMed] [Google Scholar]
- 70.Huan T, Meng Q, Saleh MA, et al. Integrative network analysis reveals molecular mechanisms of blood pressure regulation. Molecular systems biology. 2015;11:799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ehret GB, Munroe PB, Rice KM, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gudbjartsson DF, Bjornsdottir US, Halapi E, et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nature genetics. 2009;41:342–347 [DOI] [PubMed] [Google Scholar]
- 73.Saleh MA, McMaster WG, Wu J, et al. Lymphocyte adaptor protein lnk deficiency exacerbates hypertension and end-organ inflammation. J Clin Invest. 2015;125:1189–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rudemiller NP, Lund H, Priestley JR, Endres BT, Prokop JW, Jacob HJ, Geurts AM, Cohen EP, Mattson DL. Mutation of sh2b3 (lnk), a genome-wide association study candidate for hypertension, attenuates dahl salt-sensitive hypertension via inflammatory modulation. Hypertension. 2015;65:1111–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circulation research. 2015;116:1022–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Patrick DM, Van Beusecum JP, Kirabo A. The role of inflammation in hypertension: Novel concepts. Current opinion in physiology. 2021;19:92–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barbaro NR, Foss JD, Kryshtal DO, et al. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 2017;21:1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kirabo A, Fontana V, de Faria AP, et al. Dc isoketal-modified proteins activate t cells and promote hypertension. The Journal of clinical investigation. 2014;124:4642–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Loperena R, Van Beusecum JP, Itani HA, et al. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: Roles of stat3, interleukin 6 and hydrogen peroxide. Cardiovasc Res. 2018;114:1547–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Van Beusecum JP, Barbaro NR, McDowell Z, et al. High salt activates cd11c(+) antigen-presenting cells via sgk (serum glucocorticoid kinase) 1 to promote renal inflammation and salt-sensitive hypertension. Hypertension. 2019;74:555–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wenzel P, Knorr M, Kossmann S, et al. Lysozyme m-positive monocytes mediate angiotensin ii-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124:1370–1381 [DOI] [PubMed] [Google Scholar]
- 82.Ruggeri Barbaro N, Van Beusecum J, Xiao L, et al. Sodium activates human monocytes via the nadph oxidase and isolevuglandin formation. Cardiovasc Res. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hevia D, Araos P, Prado C, et al. Myeloid cd11c(+) antigen-presenting cells ablation prevents hypertension in response to angiotensin ii plus high-salt diet. Hypertension. 2018;71:709–718 [DOI] [PubMed] [Google Scholar]
- 84.Chen J. Sodium sensitivity of blood pressure in chinese populations. Curr Hypertens Rep. 2010;12:127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shukri MZ, Tan JW, Manosroi W, et al. Biological sex modulates the adrenal and blood pressure responses to angiotensin ii. Hypertension. 2018;71:1083–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Elliott P, Dyer A, Stamler R. The intersalt study: Results for 24 hour sodium and potassium, by age and sex. Intersalt co-operative research group. J Hum Hypertens. 1989;3:323–330 [PubMed] [Google Scholar]
- 87.Shimkets RA, Warnock DG, Bositis CM, et al. Liddle’s syndrome: Heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79:407–414 [DOI] [PubMed] [Google Scholar]
- 88.Snyder PM, Price MP, McDonald FJ, Adams CM, Volk KA, Zeiher BG, Stokes JB, Welsh MJ. Mechanism by which liddle’s syndrome mutations increase activity of a human epithelial na+ channel. Cell. 1995;83:969–978 [DOI] [PubMed] [Google Scholar]
- 89.Hansson JH, Nelson-Williams C, Suzuki H, et al. Hypertension caused by a truncated epithelial sodium channel gamma subunit: Genetic heterogeneity of liddle syndrome. Nature genetics. 1995;11:76–82 [DOI] [PubMed] [Google Scholar]
- 90.Hansson JH, Schild L, Lu Y, Wilson TA, Gautschi I, Shimkets R, Nelson-Williams C, Rossier BC, Lifton RP. A de novo missense mutation of the beta subunit of the epithelial sodium channel causes hypertension and liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:11495–11499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schild L, Canessa CM, Shimkets RA, Gautschi I, Lifton RP, Rossier BC. A mutation in the epithelial sodium channel causing liddle disease increases channel activity in the xenopus laevis oocyte expression system. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:5699–5703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tamura H, Schild L, Enomoto N, Matsui N, Marumo F, Rossier BC. Liddle disease caused by a missense mutation of beta subunit of the epithelial sodium channel gene. The Journal of clinical investigation. 1996;97:1780–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chang SS, Grunder S, Hanukoglu A, et al. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet. 1996;12:248–253 [DOI] [PubMed] [Google Scholar]
- 94.Strautnieks SS, Thompson RJ, Gardiner RM, Chung E. A novel splice-site mutation in the gamma subunit of the epithelial sodium channel gene in three pseudohypoaldosteronism type 1 families. Nature genetics. 1996;13:248–250 [DOI] [PubMed] [Google Scholar]
- 95.Soundararajan R, Pearce D, Hughey RP, Kleyman TR. Role of epithelial sodium channels and their regulators in hypertension. The Journal of biological chemistry. 2010;285:30363–30369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sheng S, Hallows KR, Kleyman TR. Epithelial na+ channels. In: Alpern RJ, Caplan MJ, Moe OW, eds. Seldin and giebisch’s the kidney: Physiology & pathophysiology New York, NY: Academic Press; 2012:983–1017. [Google Scholar]
- 97.Rossier BC. Epithelial sodium channel (enac) and the control of blood pressure. Current opinion in pharmacology. 2014;15:33–46 [DOI] [PubMed] [Google Scholar]
- 98.Salih M, Gautschi I, van Bemmelen MX, Di Benedetto M, Brooks AS, Lugtenberg D, Schild L, Hoorn EJ. A missense mutation in the extracellular domain of alphaenac causes liddle syndrome. Journal of the American Society of Nephrology : JASN. 2017;28:3291–3299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sheng S, Bruns JB, Kleyman TR. Extracellular histidine residues crucial for na+ self-inhibition of epithelial na+ channels. J Biol Chem. 2004;279:9743–9749 [DOI] [PubMed] [Google Scholar]
- 100.Sheng S, Carattino MD, Bruns JB, Hughey RP, Kleyman TR. Furin cleavage activates the epithelial na+ channel by relieving na+ self-inhibition. Am J Physiol Renal Physiol. 2006;290:F1488–1496 [DOI] [PubMed] [Google Scholar]
- 101.Sheng S, Maarouf AB, Bruns JB, Hughey RP, Kleyman TR. Functional role of extracellular loop cysteine residues of the epithelial na+ channel in na+ self-inhibition. J Biol Chem. 2007;282:20180–20190 [DOI] [PubMed] [Google Scholar]
- 102.Shi S, Blobner BM, Kashlan OB, Kleyman TR. Extracellular finger domain modulates the response of the epithelial sodium channel to shear stress. The Journal of biological chemistry. 2012;287:15439–15444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shi S, Ghosh DD, Okumura S, Carattino MD, Kashlan OB, Sheng S, Kleyman TR. Base of the thumb domain modulates epithelial sodium channel gating. The Journal of biological chemistry. 2011;286:14753–14761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shi S, Kleyman TR. Gamma subunit second transmembrane domain contributes to epithelial sodium channel gating and amiloride block. American journal of physiology. Renal physiology. 2013;305:F1585–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Maarouf AB, Sheng N, Chen J, Winarski KL, Okumura S, Carattino MD, Boyd CR, Kleyman TR, Sheng S. Novel determinants of epithelial sodium channel gating within extracellular thumb domains. The Journal of biological chemistry. 2009;284:7756–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Winarski KL, Sheng N, Chen J, Kleyman TR, Sheng S. Extracellular allosteric regulatory subdomain within the gamma subunit of the epithelial na+ channel. The Journal of biological chemistry. 2010;285:26088–26096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kashlan OB, Adelman JL, Okumura S, Blobner BM, Zuzek Z, Hughey RP, Kleyman TR, Grabe M. Constraint-based, homology model of the extracellular domain of the epithelial na+ channel alpha subunit reveals a mechanism of channel activation by proteases. The Journal of biological chemistry. 2011;286:649–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Allali-Hassani A, Wasney GA, Chau I, et al. A survey of proteins encoded by nonsynonymous single nucleotide polymorphisms reveals a significant fraction with altered stability and activity. Biochem J. 2009;424:15–26 [DOI] [PubMed] [Google Scholar]
- 109.Pakula AA, Sauer RT. Genetic analysis of protein stability and function. Annu Rev Genet. 1989;23:289–310 [DOI] [PubMed] [Google Scholar]
- 110.Wang Z, Moult J. Snps, protein structure, and disease. Hum Mutat. 2001;17:263–270 [DOI] [PubMed] [Google Scholar]
- 111.Wei Y, Lin DH, Kemp R, Yaddanapudi GS, Nasjletti A, Falck JR, Wang WH. Arachidonic acid inhibits epithelial na channel via cytochrome p450 (cyp) epoxygenase-dependent metabolic pathways. J Gen Physiol. 2004;124:719–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sun P, Antoun J, Lin DH, Yue P, Gotlinger KH, Capdevila J, Wang WH. Cyp2c44 epoxygenase is essential for preventing the renal sodium absorption during increasing dietary potassium intake. Hypertension. 2012;59:339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lu X, Rudemiller NP, Wen Y, et al. A20 in myeloid cells protects against hypertension by inhibiting dendritic cell-mediated t-cell activation. Circ Res. 2019;125:1055–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shah KH, Shi P, Giani JF, et al. Myeloid suppressor cells accumulate and regulate blood pressure in hypertension. Circ Res. 2015;117:858–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chiasson VL, Bounds KR, Chatterjee P, Manandhar L, Pakanati AR, Hernandez M, Aziz B, Mitchell BM. Myeloid-derived suppressor cells ameliorate cyclosporine a-induced hypertension in mice. Hypertension. 2018;71:199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vivier E, Artis D, Colonna M, et al. Innate lymphoid cells: 10 years on. Cell. 2018;174:1054–1066 [DOI] [PubMed] [Google Scholar]
- 117.Jacobsen EA, Helmers RA, Lee JJ, Lee NA. The expanding role(s) of eosinophils in health and disease. Blood. 2012;120:3882–3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Masenga SK, Elijovich F, Hamooya BM, et al. Elevated eosinophils as a feature of inflammation associated with hypertension in virally suppressed people living with hiv. J Am Heart Assoc. 2020;9:e011450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kolev M, Friec GL, Kemper C. Complement-tapping into new sites and effector systems. 2014 [DOI] [PubMed]
- 120.Carroll MC, Isenman DE. Regulation of humoral immunity by complement. 2012 [DOI] [PMC free article] [PubMed]
- 121.West EE, Kolev M, Kemper C. Complement and the regulation of t cell responses. 2018 [DOI] [PMC free article] [PubMed]
- 122.Kwan WH, van der Touw W, Heeger PS. Complement regulation of t cell immunity. Immunol Res. 2012;54:247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20:34–50 [DOI] [PubMed] [Google Scholar]
- 124.Wenzel UO, Bode M, Köhl J, Ehmke H. Pathogenic role of complement in arterial hypertension and hypertensive end organ damage. 2017 [DOI] [PubMed]
- 125.Chen XH, Ruan CC, Ge Q, et al. Deficiency of complement c3a and c5a receptors prevents angiotensin ii-induced hypertension via regulatory t cells. Circ Res. 2018;122:970–983 [DOI] [PubMed] [Google Scholar]
- 126.Nilsson B, Hamad OA, Ahlstrom H, Kullberg J, Johansson L, Lindhagen L, Haenni A, Ekdahl KN, Lind L. C3 and c4 are strongly related to adipose tissue variables and cardiovascular risk factors. Eur J Clin Invest. 2014;44:587–596 [DOI] [PubMed] [Google Scholar]
- 127.Engstrom G, Hedblad B, Berglund G, Janzon L, Lindgarde F. Plasma levels of complement c3 is associated with development of hypertension: A longitudinal cohort study. J Hum Hypertens. 2007;21:276–282 [DOI] [PubMed] [Google Scholar]
- 128.Shagdarsuren E, Wellner M, Braesen JH, et al. Complement activation in angiotensin ii-induced organ damage. Circ Res. 2005;97:716–724 [DOI] [PubMed] [Google Scholar]
- 129.Ikeda K, Fukuda N, Ueno T, Endo M, Kobayashi N, Soma M, Matsumoto K. Role of complement 3a in the growth of mesangial cells from stroke-prone spontaneously hypertensive rats. Clin Exp Hypertens. 2014;36:58–63 [DOI] [PubMed] [Google Scholar]
- 130.Zhou X, Fukuda N, Matsuda H, et al. Complement 3 activates the renal renin-angiotensin system by induction of epithelial-to-mesenchymal transition of the nephrotubulus in mice. Am J Physiol Renal Physiol. 2013;305:F957–967 [DOI] [PubMed] [Google Scholar]
- 131.Ruan CC, Zhu DL, Chen QZ, Chen J, Guo SJ, Li XD, Gao PJ. Perivascular adipose tissue-derived complement 3 is required for adventitial fibroblast functions and adventitial remodeling in deoxycorticosterone acetate-salt hypertensive rats. Arterioscler Thromb Vasc Biol. 2010;30:2568–2574 [DOI] [PubMed] [Google Scholar]
- 132.Ruan CC, Ge Q, Li Y, et al. Complement-mediated macrophage polarization in perivascular adipose tissue contributes to vascular injury in deoxycorticosterone acetate-salt mice. Arterioscler Thromb Vasc Biol. 2015;35:598–606 [DOI] [PubMed] [Google Scholar]
- 133.Zhang C, Li Y, Wang C, et al. Complement 5a receptor mediates angiotensin ii-induced cardiac inflammation and remodeling. Arterioscler Thromb Vasc Biol. 2014;34:1240–1248 [DOI] [PubMed] [Google Scholar]
- 134.Weiss S, Rosendahl A, Czesla D, et al. The complement receptor c5ar1 contributes to renal damage but protects the heart in angiotensin ii-induced hypertension. Am J Physiol Renal Physiol. 2016;310:F1356–1365 [DOI] [PubMed] [Google Scholar]
- 135.Regal JF, Laule CF, McCutcheon L, Root KM, Lund H, Hashmat S, Mattson DL. The complement system in hypertension and renal damage in the dahl ss rat. Physiol Rep. 2018;6:e13655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the t cell in the genesis of angiotensin ii induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ji H, Pai AV, West CA, Wu X, Speth RC, Sandberg K. Loss of resistance to angiotensin ii-induced hypertension in the jackson laboratory recombination-activating gene null mouse on the c57bl/6j background. Hypertension. 2017;69:1121–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Seniuk A, Thiele JL, Stubbe A, Oser P, Rosendahl A, Bode M, Meyer-Schwesinger C, Wenzel UO, Ehmke H. B6.Rag1 knockout mice generated at the jackson laboratory in 2009 show a robust wild-type hypertensive phenotype in response to ang ii (angiotensin ii). Hypertension. 2020;75:1110–1116 [DOI] [PubMed] [Google Scholar]
- 139.Madhur MS, Kirabo A, Guzik TJ, Harrison DG. From rags to riches: Moving beyond rag1 in studies of hypertension. Hypertension. 2020;75:930–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Higaki A, Mahmoud AUM, Paradis P, Schiffrin EL. Role of interleukin-23/interleukin-17 axis in t-cell mediated actions in hypertension. Cardiovasc Res. 2020 [DOI] [PubMed] [Google Scholar]
- 141.Dale BL, Pandey AK, Chen Y, et al. Critical role of interleukin 21 and t follicular helper cells in hypertension and vascular dysfunction. JCI insight. 2019;5:e129278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ding R, Gao W, He Z, Liao M, Wu F, Zou S, Ma L, Liang C, Wu Z. Effect of serum interleukin 21 on the development of coronary artery disease. APMIS. 2014;122:842–847 [DOI] [PubMed] [Google Scholar]
- 143.Itani HA, McMaster WG Jr., Saleh MA, et al. Activation of human t cells in hypertension: Studies of humanized mice and hypertensive humans. Hypertension. 2016;68:123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin ii-induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, Madhur MS, Chen W, Harrison DG. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res. 2014;114:616–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Norlander AE, Saleh MA, Kamat NV, et al. Interleukin-17a regulates renal sodium transporters and renal injury in angiotensin ii-induced hypertension. Hypertension. 2016;68:167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97:696–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Orejudo M, Garcia-Redondo AB, Rodrigues-Diez RR, et al. Interleukin-17a induces vascular remodeling of small arteries and blood pressure elevation. Clin Sci (Lond). 2020;134:513–527 [DOI] [PubMed] [Google Scholar]
- 149.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, Delpire E, Harrison DG, McDonough AA. Renal transporter activation during angiotensin-ii hypertension is blunted in interferon-gamma−/− and interleukin-17a−/− mice. Hypertension. 2015;65:569–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Norlander AE, Madhur MS. Inflammatory cytokines regulate renal sodium transporters: How, where, and why? Am J Physiol Renal Physiol. 2017:ajprenal 00465 02016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Orejudo M, Rodrigues-Diez RR, Rodrigues-Diez R, et al. Interleukin 17a participates in renal inflammation associated to experimental and human hypertension. Front Pharmacol. 2019;10:1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, Kuchroo VK. Induction of pathogenic th17 cells by inducible salt-sensing kinase sgk1. Nature. 2013;496:513–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA. Sodium chloride drives autoimmune disease by the induction of pathogenic th17 cells. Nature. 2013;496:518–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hernandez AL, Kitz A, Wu C, et al. Sodium chloride inhibits the suppressive function of foxp3+ regulatory t cells. J Clin Invest. 2015;125:4212–4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Norlander AE, Saleh MA, Pandey AK, et al. A salt-sensing kinase in t lymphocytes, sgk1, drives hypertension and hypertensive end-organ damage. JCI insight. 2017;2:e92801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Amador CA, Barrientos V, Pena J, et al. Spironolactone decreases doca-salt-induced organ damage by blocking the activation of t helper 17 and the downregulation of regulatory t lymphocytes. Hypertension. 2014;63:797–803 [DOI] [PubMed] [Google Scholar]
- 157.Saleh MA, McMaster WG, Wu J, et al. Lymphocyte adaptor protein lnk deficiency exacerbates hypertension and end-organ inflammation. J Clin Invest. 2015;125:1189–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sun XN, Li C, Liu Y, et al. T-cell mineralocorticoid receptor controls blood pressure by regulating interferon-gamma. Circ Res. 2017;120:1584–1597 [DOI] [PubMed] [Google Scholar]
- 159.Marko L, Kvakan H, Park JK, et al. Interferon-gamma signaling inhibition ameliorates angiotensin ii-induced cardiac damage. Hypertension. 2012;60:1430–1436 [DOI] [PubMed] [Google Scholar]
- 160.Zhang J, Patel MB, Griffiths R, et al. Tumor necrosis factor-alpha produced in the kidney contributes to angiotensin ii-dependent hypertension. Hypertension. 2014;64:1275–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Youn JC, Yu HT, Lim BJ, et al. Immunosenescent cd8+ t cells and c-x-c chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension. 2013;62:126–133 [DOI] [PubMed] [Google Scholar]
- 162.Shen Y, Cheng F, Sharma M, et al. Granzyme b deficiency protects against angiotensin ii-induced cardiac fibrosis. Am J Pathol. 2016;186:87–100 [DOI] [PubMed] [Google Scholar]
- 163.Trott DW, Thabet SR, Kirabo A, et al. Oligoclonal cd8+ t cells play a critical role in the development of hypertension. Hypertension. 2014;64:1108–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Liu Y, Rafferty TM, Rhee SW, Webber JS, Song L, Ko B, Hoover RS, He B, Mu S. Cd8(+) t cells stimulate na-cl co-transporter ncc in distal convoluted tubules leading to salt-sensitive hypertension. Nature communications. 2017;8:14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Saleh MA, Norlander AE, Madhur MS. Inhibition of interleukin-17a, but not interleukin-17f, signaling lowers blood pressure, and reduces end-organ inflammation in angiotensin ii–induced hypertension. JACC: Basic to Translational Science. 2016;1:606–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hueber W, Patel DD, Dryja T, et al. Effects of ain457, a fully human antibody to interleukin-17a, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. 2010;2:52ra72. [DOI] [PubMed] [Google Scholar]
- 167.Papp KA, Langley RG, Sigurgeirsson B, et al. Efficacy and safety of secukinumab in the treatment of moderate-to-severe plaque psoriasis: A randomized, double-blind, placebo-controlled phase ii dose-ranging study. Br J Dermatol. 2013;168:412–421 [DOI] [PubMed] [Google Scholar]
- 168.von Stebut E, Reich K, Thaci D, et al. Impact of secukinumab on endothelial dysfunction and other cardiovascular disease parameters in psoriasis patients over 52 weeks. J Invest Dermatol. 2019;139:1054–1062 [DOI] [PubMed] [Google Scholar]
- 169.Elnabawi YA, Dey AK, Goyal A, et al. Coronary artery plaque characteristics and treatment with biologic therapy in severe psoriasis: Results from a prospective observational study. Cardiovasc Res. 2019;115:721–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory t cells and immune tolerance. Cell. 2008;133:775–787 [DOI] [PubMed] [Google Scholar]
- 171.Ochs HD, Gambineri E, Torgerson TR. Ipex, foxp3 and regulatory t-cells: A model for autoimmunity. Immunologic research. 2007;38:112–121 [DOI] [PubMed] [Google Scholar]
- 172.Godfrey VL, Wilkinson JE, Russell LB. X-linked lymphoreticular disease in the scurfy (sf) mutant mouse. The American journal of pathology. 1991;138:1379–1387 [PMC free article] [PubMed] [Google Scholar]
- 173.Vignali DAA, Collison LW, Workman CJ. How regulatory t cells work. Nat Rev Immunol. 2008;8:523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Raffin C, Vo LT, Bluestone JA. T(reg) cell-based therapies: Challenges and perspectives. Nat Rev Immunol. 2020;20:158–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent angiotensin ii-induced hypertension and vascular injury. Hypertension. 2011;57:469–476 [DOI] [PubMed] [Google Scholar]
- 176.Gackowska L, Michalkiewicz J, Helmin-Basa A, Klosowski M, Niemirska A, Obrycki L, Kubiszewska I, Wierzbicka A, Litwin M. Regulatory t-cell subset distribution in children with primary hypertension is associated with hypertension severity and hypertensive target organ damage. Journal of hypertension. 2019 [DOI] [PubMed] [Google Scholar]
- 177.Chen ZY, Chen F, Wang YG, Wang DH, Jang LL, Cheng LX. Down-regulation of helios expression in tregs from patients with hypertension. Curr Med Sci. 2018;38:58–63 [DOI] [PubMed] [Google Scholar]
- 178.Mian MO, Barhoumi T, Briet M, Paradis P, Schiffrin EL. Deficiency of t-regulatory cells exaggerates angiotensin ii-induced microvascular injury by enhancing immune responses. Journal of hypertension. 2016;34:97–108 [DOI] [PubMed] [Google Scholar]
- 179.Kvakan H, Kleinewietfeld M, Qadri F, et al. Regulatory t cells ameliorate angiotensin ii-induced cardiac damage. Circulation. 2009;119:2904–2912 [DOI] [PubMed] [Google Scholar]
- 180.Emmerson A, Trevelin SC, Mongue-Din H, et al. Nox2 in regulatory t cells promotes angiotensin ii-induced cardiovascular remodeling. J Clin Invest. 2018;128:3088–3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kasal DA, Barhoumi T, Li MW, et al. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension. 2012;59:324–330 [DOI] [PubMed] [Google Scholar]
- 182.Matrougui K, Abd Elmageed Z, Kassan M, et al. Natural regulatory t cells control coronary arteriolar endothelial dysfunction in hypertensive mice. Am J Pathol. 2011;178:434–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Taylor EB, Sasser JM, Maeda KJ, Ryan MJ. Expansion of regulatory t cells using low-dose interleukin-2 attenuates hypertension in an experimental model of systemic lupus erythematosus. American journal of physiology. Renal physiology. 2019;317:F1274–f1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Fabbiano S, Menacho-Márquez M, Robles-Valero J, et al. Immunosuppression-independent role of regulatory t cells against hypertension-driven renal dysfunctions. Mol Cell Biol. 2015;35:3528–3546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ait-Oufella H, Wang Y, Herbin O, et al. Natural regulatory t cells limit angiotensin ii-induced aneurysm formation and rupture in mice. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:2374–2379 [DOI] [PubMed] [Google Scholar]
- 186.Belanger Kasey M, Crislip GR, Gillis Ellen E, et al. Greater t regulatory cells in females attenuate doca-salt–induced increases in blood pressure versus males. Hypertension. 2020;75:1615–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Iulita MF, Duchemin S, Vallerand D, et al. Cd4(+) regulatory t lymphocytes prevent impaired cerebral blood flow in angiotensin ii-induced hypertension. Journal of the American Heart Association. 2019;8:e009372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Didion SP, Kinzenbaw DA, Schrader LI, Chu Y, Faraci FM. Endogenous interleukin-10 inhibits angiotensin ii-induced vascular dysfunction. Hypertension. 2009;54:619–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kassan M, Galan M, Partyka M, Trebak M, Matrougui K. Interleukin-10 released by cd4(+)cd25(+) natural regulatory t cells improves microvascular endothelial function through inhibition of nadph oxidase activity in hypertensive mice. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:2534–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kuczma M, Podolsky R, Garge N, Daniely D, Pacholczyk R, Ignatowicz L, Kraj P. Foxp3-deficient regulatory t cells do not revert into conventional effector cd4+ t cells but constitute a unique cell subset. J Immunol. 2009;183:3731–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kim JM, Rasmussen JP, Rudensky AY. Regulatory t cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197 [DOI] [PubMed] [Google Scholar]
- 192.Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, Nakayama M, Rosenthal W, Bluestone JA. Instability of the transcription factor foxp3 leads to the generation of pathogenic memory t cells in vivo. Nature immunology. 2009;10:1000–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Birjandi SZ, Palchevskiy V, Xue YY, Nunez S, Kern R, Weigt SS, Lynch JP 3rd, Chatila TA, Belperio JA. Cd4(+)cd25(hi)foxp3(+) cells exacerbate bleomycin-induced pulmonary fibrosis. Am J Pathol. 2016;186:2008–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Boveda-Ruiz D, D’Alessandro-Gabazza CN, Toda M, et al. Differential role of regulatory t cells in early and late stages of pulmonary fibrosis. Immunobiology. 2013;218:245–254 [DOI] [PubMed] [Google Scholar]
- 195.Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, Prabhu SD. Dysfunctional and proinflammatory regulatory t-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation. 2019;139:206–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Mohr A, Malhotra R, Mayer G, Gorochov G, Miyara M. Human foxp3+ t regulatory cell heterogeneity. Clinical & Translational Immunology. 2018;7:e1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Panduro M, Benoist C, Mathis D. Tissue tregs. Annu Rev Immunol. 2016;34:609–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Caillon A, Mian MOR, Fraulob-Aquino JC, Huo KG, Barhoumi T, Ouerd S, Sinnaeve PR, Paradis P, Schiffrin EL. Gammadelta t cells mediate angiotensin ii-induced hypertension and vascular injury. Circulation. 2017;135:2155–2162 [DOI] [PubMed] [Google Scholar]
- 199.Ebringer A, Doyle AE. Raised serum igg levels in hypertension. Br Med J. 1970;2:146–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kristensen BO. Increased serum levels of immunoglobulins in untreated and treated essential hypertension. I. Relation to blood pressure. Acta Med Scand. 1978;203:49–54 [DOI] [PubMed] [Google Scholar]
- 201.Suryaprabha P, Padma T, Rao UB. Increased serum igg levels in essential hypertension. Immunol Lett. 1984;8:143–145 [DOI] [PubMed] [Google Scholar]
- 202.Chan CT, Sobey CG, Lieu M, et al. Obligatory role for b cells in the development of angiotensin ii-dependent hypertension. Hypertension. 2015;66:1023–1033 [DOI] [PubMed] [Google Scholar]
- 203.Khamis RY, Hughes AD, Caga-Anan M, et al. High serum immunoglobulin g and m levels predict freedom from adverse cardiovascular events in hypertension: A nested case-control substudy of the anglo-scandinavian cardiac outcomes trial. EBioMedicine. 2016;9:372–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Dingwell LS, Shikatani EA, Besla R, et al. B-cell deficiency lowers blood pressure in mice. Hypertension. 2019;73:561–570 [DOI] [PubMed] [Google Scholar]
- 205.Chen Y, Dale BL, Alexander MR, Xiao L, Ao M, Pandey AK, Smart CD, Davis GK, Madhur MS. Class switching and high affinity igg production by b cells is dispensable for the development of hypertension in mice. Cardiovasc Res. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hamaguchi Y, Uchida J, Cain DW, Venturi GM, Poe JC, Haas KM, Tedder TF. The peritoneal cavity provides a protective niche for b1 and conventional b lymphocytes during anti-cd20 immunotherapy in mice. J Immunol. 2005;174:4389–4399 [DOI] [PubMed] [Google Scholar]
- 207.Rauch M, Tussiwand R, Bosco N, Rolink AG. Crucial role for baff-baff-r signaling in the survival and maintenance of mature b cells. PLoS One. 2009;4:e5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wolfe F, Mitchell DM, Sibley JT, et al. The mortality of rheumatoid arthritis. Arthritis and rheumatism. 1994;37:481–494 [DOI] [PubMed] [Google Scholar]
- 209.Park YB, Ahn CW, Choi HK, Lee SH, In BH, Lee HC, Nam CM, Lee SK. Atherosclerosis in rheumatoid arthritis: Morphologic evidence obtained by carotid ultrasound. Arthritis Rheum. 2002;46:1714–1719 [DOI] [PubMed] [Google Scholar]
- 210.Chung CP, Oeser A, Raggi P, Gebretsadik T, Shintani AK, Sokka T, Pincus T, Avalos I, Stein CM. Increased coronary-artery atherosclerosis in rheumatoid arthritis: Relationship to disease duration and cardiovascular risk factors. Arthritis Rheum. 2005;52:3045–3053 [DOI] [PubMed] [Google Scholar]
- 211.Manzi S, Meilahn EN, Rairie JE, Conte CG, Medsger TA, Jr., Jansen-McWilliams L, D’Agostino RB, Kuller LH. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: Comparison with the framingham study. Am J Epidemiol. 1997;145:408–415 [DOI] [PubMed] [Google Scholar]
- 212.Rho YH, Chung CP, Oeser A, et al. Inflammatory mediators and premature coronary atherosclerosis in rheumatoid arthritis. Arthritis Rheum. 2009;61:1580–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Somers EC, Zhao W, Lewis EE, Wang L, Wing JJ, Sundaram B, Kazerooni EA, McCune WJ, Kaplan MJ. Type i interferons are associated with subclinical markers of cardiovascular disease in a cohort of systemic lupus erythematosus patients. PLoS One. 2012;7:e37000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Budman DR, Steinberg AD. Hypertension and renal disease in systemic lupus erythematosus. Arch Intern Med. 1976;136:1003–1007 [PubMed] [Google Scholar]
- 215.Petri M, Spence D, Bone LR, Hochberg MC. Coronary artery disease risk factors in the johns hopkins lupus cohort: Prevalence, recognition by patients, and preventive practices. Medicine (Baltimore). 1992;71:291–302 [DOI] [PubMed] [Google Scholar]
- 216.Tselios K, Gladman DD, Su J, Urowitz M. Impact of the new american college of cardiology/american heart association definition of hypertension on atherosclerotic vascular events in systemic lupus erythematosus. Ann Rheum Dis. 2020;79:612–617 [DOI] [PubMed] [Google Scholar]
- 217.Panoulas VF, Metsios GS, Pace AV, John H, Treharne GJ, Banks MJ, Kitas GD. Hypertension in rheumatoid arthritis. Rheumatology (Oxford). 2008;47:1286–1298 [DOI] [PubMed] [Google Scholar]
- 218.Panoulas VF, Douglas KM, Milionis HJ, et al. Prevalence and associations of hypertension and its control in patients with rheumatoid arthritis. Rheumatology (Oxford). 2007;46:1477–1482 [DOI] [PubMed] [Google Scholar]
- 219.Mathis KW, Wallace K, Flynn ER, Maric-Bilkan C, LaMarca B, Ryan MJ. Preventing autoimmunity protects against the development of hypertension and renal injury. Hypertension. 2014;64:792–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Taylor EB, Barati MT, Powell DW, Turbeville HR, Ryan MJ. Plasma cell depletion attenuates hypertension in an experimental model of autoimmune disease. Hypertension. 2018;71:719–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mathis KW, Venegas-Pont M, Masterson CW, Stewart NJ, Wasson KL, Ryan MJ. Oxidative stress promotes hypertension and albuminuria during the autoimmune disease systemic lupus erythematosus. Hypertension. 2012;59:673–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Patrick DM, Visitacion ND, Ormseth MJ, et al. Isolevuglandins promote autoimmunity and hypertension in systemic lupus erythematosus. medRxiv. 2020:2020.2002.2010.20021741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with covid-19 in wuhan, china: A retrospective cohort study. Lancet. 2020;395:1054–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wu A, Good C, Downs JR, Fine MJ, Pugh MJ, Anzueto A, Mortensen EM. The association of cardioprotective medications with pneumonia-related outcomes. PloS one. 2014;9:e85797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Mortensen EM, Nakashima B, Cornell J, et al. Population-based study of statins, angiotensin ii receptor blockers, and angiotensin-converting enzyme inhibitors on pneumonia-related outcomes. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2012;55:1466–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Sparks MA, South A, Welling P, et al. Sound science before quick judgement regarding ras blockade in covid-19. Clin J Am Soc Nephrol. 2020;15:714–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Trump S, Lukassen S, Anker MS, et al. Hypertension delays viral clearance and exacerbates airway hyperinflammation in patients with covid-19. Nat Biotechnol. 2020 [DOI] [PubMed] [Google Scholar]
- 228.Shah ASV, Stelzle D, Lee KK, et al. Global burden of atherosclerotic cardiovascular disease in people living with hiv: Systematic review and meta-analysis. Circulation. 2018;138:1100–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Althoff KN, Gebo KA, Moore RD, et al. Contributions of traditional and hiv-related risk factors on non-aids-defining cancer, myocardial infarction, and end-stage liver and renal diseases in adults with hiv in the USA and canada: A collaboration of cohort studies. Lancet HIV. 2019;6:e93–e104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Armah KA, Chang CC, Baker JV, et al. Prehypertension, hypertension, and the risk of acute myocardial infarction in hiv-infected and -uninfected veterans. Clin Infect Dis. 2014;58:121–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tenorio AR, Zheng Y, Bosch RJ, et al. Soluble markers of inflammation and coagulation but not t-cell activation predict non-aids-defining morbid events during suppressive antiretroviral treatment. J Infect Dis. 2014;210:1248–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Burdo TH, Lo J, Abbara S, Wei J, DeLelys ME, Preffer F, Rosenberg ES, Williams KC, Grinspoon S. Soluble cd163, a novel marker of activated macrophages, is elevated and associated with noncalcified coronary plaque in hiv-infected patients. J Infect Dis. 2011;204:1227–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Hofman FM, Wright AD, Dohadwala MM, Wong-Staal F, Walker SM. Exogenous tat protein activates human endothelial cells. Blood. 1993;82:2774–2780 [PubMed] [Google Scholar]
- 234.Bandera A, Masetti M, Fabbiani M, Biasin M, Muscatello A, Squillace N, Clerici M, Gori A, Trabattoni D. The nlrp3 inflammasome is upregulated in hiv-infected antiretroviral therapy-treated individuals with defective immune recovery. Front Immunol. 2018;9:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Reis KG, Desderius B, Kingery J, Kirabo A, Makubi A, Myalla C, Lee MH, Kapiga S, Peck RN. Blood pressure, t cells, and mortality in people with hiv in tanzania during the first 2 years of antiretroviral therapy. J Clin Hypertens (Greenwich). 2020;22:1554–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Ding RX, Goh WR, Wu RN, Yue XQ, Luo X, Khine WWT, Wu JR, Lee YK. Revisit gut microbiota and its impact on human health and disease. J Food Drug Anal. 2019;27:623–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Tang WH, Kitai T, Hazen SL. Gut microbiota in cardiovascular health and disease. Circ Res. 2017;120:1183–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Yan Q, Gu Y, Li X, et al. Alterations of the gut microbiome in hypertension. Front Cell Infect Microbiol. 2017;7:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Ferguson JF, Aden LA, Barbaro NR, et al. High dietary salt-induced dendritic cell activation underlies microbial dysbiosis-associated hypertension. JCI Insight. 2019;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kim S, Goel R, Kumar A, et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin Sci (Lond). 2018;132:701–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Li J, Zhao F, Wang Y, et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Ferguson JF, Aden LA, Barbaro NR, et al. High dietary salt-induced dc activation underlies microbial dysbiosis-associated hypertension. JCI insight. 2019;4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Robles-Vera I, Toral M, de la Visitacion N, et al. Probiotics prevent dysbiosis and the rise in blood pressure in genetic hypertension: Role of short-chain fatty acids. Mol Nutr Food Res. 2020;64:e1900616. [DOI] [PubMed] [Google Scholar]
- 244.Wang L, Zhu Q, Lu A, Liu X, Zhang L, Xu C, Liu X, Li H, Yang T. Sodium butyrate suppresses angiotensin ii-induced hypertension by inhibition of renal (pro)renin receptor and intrarenal renin-angiotensin system. J Hypertens. 2017;35:1899–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Chen L, Sun M, Wu W, et al. Microbiota metabolite butyrate differentially regulates th1 and th17 cells’ differentiation and function in induction of colitis. Inflamm Bowel Dis. 2019;25:1450–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Sun M, Wu W, Chen L, et al. Microbiota-derived short-chain fatty acids promote th1 cell il-10 production to maintain intestinal homeostasis. Nat Commun. 2018;9:3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Marques FZ, Nelson E, Chu PY, et al. High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation. 2017;135:964–977 [DOI] [PubMed] [Google Scholar]
- 248.Wilck N, Matus MG, Kearney SM, et al. Salt-responsive gut commensal modulates th17 axis and disease. Nature. 2017;551:585–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Hu J, Luo H, Wang J, et al. Enteric dysbiosis-linked gut barrier disruption triggers early renal injury induced by chronic high salt feeding in mice. Exp Mol Med. 2017;49:e370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Rescigno M, Di Sabatino A. Dendritic cells in intestinal homeostasis and disease. J Clin Invest. 2009;119:2441–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Fryar CD, Ostchega Y, Hales CM, Zhang G, Kruszon-Moran D. Hypertension prevalence and control among adults: United states, 2015–2016. NCHS Data Brief. 2017:1–8 [PubMed] [Google Scholar]
- 252.Weinberger MH. Salt sensitivity is associated with an increased mortality in both normal and hypertensive humans. J Clin Hypertens (Greenwich). 2002;4:274–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Gillis EE, Sullivan JC. Sex differences in hypertension: Recent advances. Hypertension. 2016;68:1322–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Wang P, Deger MS, Kang H, Ikizler TA, Titze J, Gore JC. Sex differences in sodium deposition in human muscle and skin. Magn Reson Imaging. 2017;36:93–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Siedner MJ, Zanni M, Tracy RP, Kwon DS, Tsai AC, Kakuhire B, Hunt PW, Okello S. Increased systemic inflammation and gut permeability among women with treated hiv infection in rural uganda. The Journal of Infectious Diseases. 2018;218:922–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Okello S, Kim JH, Sentongo RN, Tracy R, Tsai AC, Kakuhikire B, Siedner MJ. Blood pressure trajectories and the mediated effects of body mass index and hiv-related inflammation in a mixed cohort of people with and without hiv in rural uganda. J Clin Hypertens (Greenwich). 2019;21:1230–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.He J, Gu D, Chen J, et al. Gender difference in blood pressure responses to dietary sodium intervention in the gensalt study. J Hypertens. 2009;27:48–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Markle JG, Frank DN, Mortin-Toth S, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science. 2013;339:1084–1088 [DOI] [PubMed] [Google Scholar]
- 259.Org E, Mehrabian M, Parks BW, Shipkova P, Liu X, Drake TA, Lusis AJ. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes. 2016;7:313–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Arnold AP, Cassis LA, Eghbali M, Reue K, Sandberg K. Sex hormones and sex chromosomes cause sex differences in the development of cardiovascular diseases. Arterioscler Thromb Vasc Biol. 2017;37:746–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Rettew JA, Huet-Hudson YM, Marriott I. Testosterone reduces macrophage expression in the mouse of toll-like receptor 4, a trigger for inflammation and innate immunity. Biol Reprod. 2008;78:432–437 [DOI] [PubMed] [Google Scholar]
- 262.Rettew JA, Huet YM, Marriott I. Estrogens augment cell surface tlr4 expression on murine macrophages and regulate sepsis susceptibility in vivo. Endocrinology. 2009;150:3877–3884 [DOI] [PubMed] [Google Scholar]
- 263.Edwards JM, Roy S, Tomcho JC, et al. Microbiota are critical for vascular physiology: Germ-free status weakens contractility and induces sex-specific vascular remodeling in mice. Vascul Pharmacol. 2020;125–126:106633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Vinturache AE, Gyamfi-Bannerman C, Hwang J, Mysorekar IU, Jacobsson B, Preterm Birth International C. Maternal microbiome - a pathway to preterm birth. Semin Fetal Neonatal Med. 2016;21:94–99 [DOI] [PubMed] [Google Scholar]
- 265.Wang J, Gu X, Yang J, Wei Y, Zhao Y. Gut microbiota dysbiosis and increased plasma lps and tmao levels in patients with preeclampsia. Front Cell Infect Microbiol. 2019;9:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Xue P, Zheng M, Gong P, et al. Single administration of ultra-low-dose lipopolysaccharide in rat early pregnancy induces tlr4 activation in the placenta contributing to preeclampsia. PLoS One. 2015;10:e0124001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Turbeville HR, Sasser JM. Preeclampsia beyond pregnancy: Long-term consequences for mother and child. Am J Physiol Renal Physiol. 2020;318:F1315–F1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Moore RE, Townsend SD. Temporal development of the infant gut microbiome. Open Biol. 2019;9:190128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Walker RW, Clemente JC, Peter I, Loos RJF. The prenatal gut microbiome: Are we colonized with bacteria in utero? Pediatr Obes. 2017;12 Suppl 1:3–17 [DOI] [PMC free article] [PubMed] [Google Scholar]