

Abstract
Dr. Irvine Page proposed the Mosaic Theory of Hypertension in the 1940s advocating that hypertension is the result of many factors that interact to raise blood pressure and cause end-organ damage. Over the years, Dr. Page modified his paradigm, and new concepts regarding oxidative stress, inflammation, genetics, sodium homeostasis and the microbiome have arisen that allow further refinements of the Mosaic Theory. A constant feature of this approach to understanding hypertension is that the various nodes are interdependent, and that these almost certainly vary between experimental models and between individuals with hypertension. This review discusses these new concepts and provides an introduction to other reviews in this compendium of Circulation Research.
Subject Terms: Autonomic Nervous System, Hypertension, Pathophysiology, Vascular Biology
Keywords: integrative physiology, history, autonomic, hypertension, pathophysiology, reactive oxygen species, renal function, vascular function
Introduction: The state of clinical hypertension in 2021:
According to the 2017 guidelines of the American Heart Association, almost one half of the adult population in the United States has hypertension,1 and the Global Burden of Disease study ranks elevated systolic pressure as the leading cause of lost years of quality life.2 Despite advances in medical therapy, hypertension remains a major risk factor for stroke, heart failure, renal failure, atherosclerosis and we now recognize dementia. In large part this is because almost one half of individuals with hypertension are not aware that they have this disease, and hypertension is not adequately treated in about one half of those in whom the diagnosis is made.3
Clinical hypertension can be grouped into two broad categories. Primary (or essential) hypertension represents between 85 to 95% of human cases and has an unidentified etiology. In contrast, secondary hypertension is caused by identifiable underlying conditions, including renal artery stenosis, pheochromocytoma, adrenal adenoma or single gene mutations. Historically, most patients were screened for secondary causes. However, it is now clear that this is not cost effective and that in the absence of compelling clinical findings (abdominal bruit, hypokalemia, clinical symptoms of a pheochromocytoma) or a pressing need to improve treatment (drug resistant hypertension) patients are generally treated without such an evaluation.
Pathophysiological/historical concepts:
The elusive nature of primary hypertension and the precise mechanisms underlying an elevation of blood pressure, even in cases of secondary hypertension and in models of experimental hypertension, have remained a focus of debate and research for more than a century. Beginning with seminal observations by Robert Tigerstedt,4 Franz Volhard,5, 6 and subsequently refined and elegantly published by Goldblatt,7 it became apparent that the ischemic or underperfused kidney releases a substance or substances that can raise the blood pressure in recipient animals. These early findings set the groundwork for Irvine Page and Braun Menendez in 1939 to report their discovery of a potent vasoconstrictor and pro-hypertensive agent isolated from renal extracts that they named hypertensin and angiotonin, respectively.8, 9 They ultimately agreed to call it angiotensin.10
Irvine Page is among the most prominent pioneers of hypertension research and is notable for his enduring contributions. In his initial experimental observations with Oscar Helmer, he discovered that when preparations of renin were exposed to plasma containing what he termed “renin activator” a pressor substance was formed that could raise blood pressure up to 300 mmHg. He named this substance “angiotonin”.11 It would have been easy to assume that this elevation of blood pressure was entirely due to intense vasoconstriction. However, his further observations suggested this was an inadequate explanation. In a classic paper in 1949, he summarized evidence that hypertension could be mediated by the central nervous system, cardiovascular factors, endocrine factors and perturbations of renal function.12 He provided evidence that the hypertension induced by acute injections of “angiotonin” could be reversed by ganglionic blockade with tetraethylammonium. In a subsequent study, Dr. Page and colleagues reported that low rates of angiotensin II infusion had minimal or no effect on blood pressure in the first hours after starting the infusion, but increased blood pressure by 30 mmHg 24 hours later,13 suggesting the importance of mechanisms beyond immediate vasoconstriction. His discovery of this “slow pressor” response to angiotensin was, in itself, a major contribution to the field and has given subsequent investigators a valuable model of human hypertension. He reported further that these infusions of low dose angiotensin II caused the blood pressure to become highly labile, such that minimal environmental stimulation would cause enormous increases in pressure, accompanied by increases in heart rate. These increases in blood pressure were exacerbated by tyramine (that releases norepinephrine from nerve terminals) and were blocked by guanethidine (that prevents norepinephrine release). Page concluded from these highly insightful studies that “angiotensin causes hypertension by an indirect action mediated by the central nervous system, an action independent of its vasoconstrictor action”.13 He observed that renin was not elevated in the chronic phase of many cases of human hypertension and surmised that human hypertension has complex origins, and therefore presumably was multifactorial. In his 1949 paper, he chose to describe this multifactorial nature by the term “Mosaic”, pointing out that “… many mechanisms are more or less involved. Elevated blood pressure is the resultant of multiple forces acting on the variety of tissues which compose the circulatory apparatus.” In a subsequent review, Dr. Page presented a now iconic octagonal diagram (Figure 1A), that has been modified and often reproduced to illustrate his concept that hypertension is caused by multiple factors, including neural and chemical perturbations, alterations of vascular caliber and elastance, cardiovascular reactivity, blood volume and viscosity.14 Dr. Page subsequently revised the Mosaic Theory to include more general terms (Figure 1B),15 however the principle of his Mosaic Theory has prevailed to the present day.16, 17 It is fascinating to ponder that the factors composing his Mosaic Theory reflect an ongoing central discussion about the importance of the kidney (volume and cardiac output), the vasculature (elastance, vascular caliber and reactivity) and the central nervous system (reactivity and neural). In the past two decades, there have been numerous refinements of our understanding of the nodes in Figure 1A and 1B, and entirely new concepts. In this review we will begin by considering time honored topics: the roles of the kidney, the vasculature and the central nervous system and a prevailing effect of aldosterone in hypertension. We will then discuss new concepts that have emerged in the past two decades that Dr. Page did not consider in his writings, but likely influence every node in the Mosaic diagram. Many of these topics are covered in greater depth by other articles in this compendium.
Figure 1:
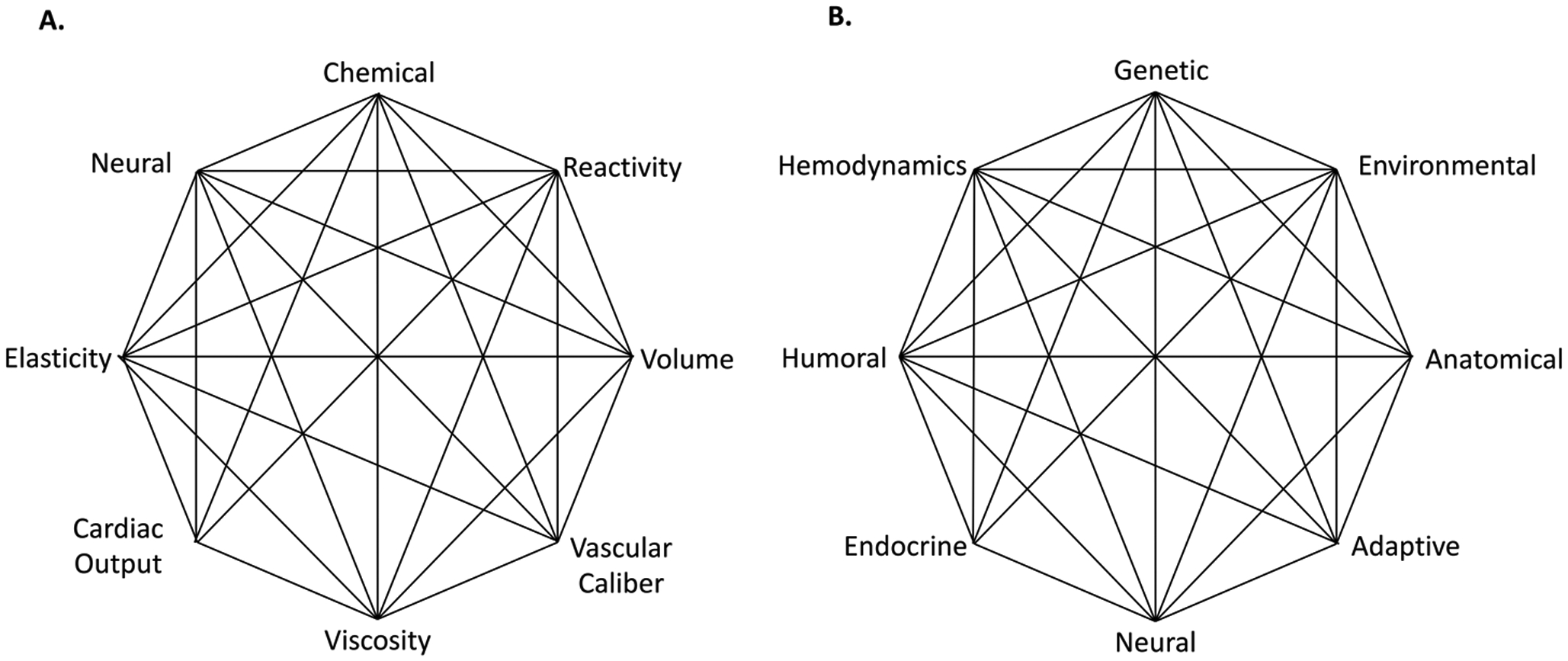
The original (A) and revised (B) Mosaic Theories proposed by Page.
The role of the kidney and body fluid volumes in hypertension:
The kidney is an important node in Dr. Page’s Mosaic Theory. Figure 1A refers to volume and cardiac output. Cardiac output is the product of heart rate and cardiac stroke volume. Stroke volume is dependent on venous return and is governed in large part by the kidneys and their modulation of volume homeostasis. Guyton built on these early concepts of Irvine Page to elaborate a complex mathematic model of the dynamic interactions between many of these Mosaic components.18 The model predicted that the renal pressure natriuresis mechanism regulated the body fluids and thereby exerted a primary role in setting the level of blood pressure, while the central nervous system and vascular function, amongst others, provided important modulation. The critical role of the kidney has been illustrated repeatedly by studies in which the kidneys of hypertensive animals raise blood pressure, or permit hypertension, when transplanted into normal animals, dating to seminal work by Fasciola et al.19 Dahl et al performed cross transplantation of kidneys between salt sensitive and salt resistant rats and showed that hypertension followed the kidney.20 Moreover, the transplant of kidneys from normotensive donors into patients with renal failure from hypertensive nephrosclerosis has led in some cases to a slow reversal of their hypertension and some of the accompanying organ damage.21 Crowley et al subsequently reported that transplantation of kidneys from mice lacking the angiotensin type 1 receptor protected wild type recipient mice from angiotensin II-induced hypertension, while transplantation of wild type kidneys into AT1R ko mice restored their ability to develop hypertension upon angiotensin II infusion.22 This refinement relates a critical component of hypertension from angiotensin II to activation of its principal receptor within the kidneys, and provides further mechanisms extending the theories of Page and Guyton.
The kidney has at least 4 major roles in hypertension. One is the production of renin, an aspartic protease that cleaves angiotensinogen to angiotensin I, which in turn is acted on by the angiotensin I-converting enzyme to generate angiotensin II. Renin is the rate-limiting step for activation of the circulating renin-angiotensin system (RAS) and its synthesis and secretion by the kidney is tightly regulated.23 Renin is initially synthesized as the inactive precursor pro-renin, which is converted to active renin upon binding to the pro-renin receptor (PRR). Under normal conditions, renin is produced almost exclusively by specialized juxtaglomerular cells of the afferent arteriole. It is released in response to a reductions in perfusion pressure or delivery of sodium chloride to macula densa cells, or by an increase in sympathetic stimulation.24 Prolonged stimulation of the renal sympathetic nerves transforms a subset of vascular smooth muscle cells into renin producing cells in the afferent arteriole. This transformation is governed by the transcription factor SRY-related HMG-box gene (Sox)-6.25 Principal cells of the collecting duct also can produce renin. This is mediated in part by a loss of nitric oxide production in various pathophysiological conditions.26 While renin is normally released almost exclusively from the juxtaglomerular apparatus of the kidney, the proximal tubules can become an important site of renin production during oxidative stress or in response to increased glomerular protein filtration.27 Drs. Gomez and Sequeira-Lopez will discuss these and other features of renin biology in their review of this compendium.
Mice lacking the PRR in the collecting duct have a reduced hypertensive response to angiotensin II infusion, in part due to reduced sodium resorption in this segment.28 Direct actions of renin per se in clinical settings remain controversial. Treatment with the direct renin antagonist Aliskiren paradoxically worsened outcomes in humans with heart failure,29 and a recent meta-analysis reported no improvement in outcomes in hypertension and a lower certainty of blood pressure reduction for Aliskiren than for ACE inhibition.30 However, these disappointing reports may reflect pharmacokinetic defects of Aliskiren.31 Moreover, renin is a dual purpose enzyme that both metabolizes angiotensinogen and acts as a complement C3 convertase.32, 33 It has long been known that circulating levels of C3 predict the subsequent development of hypertension,34 likely by promoting innate and adaptive immune responses.35 Therefore, a role may yet emerge for improved direct renin inhibitors in the treatment of hypertension and for reducing complement C3 activation.
A second major role of the kidney in hypertension is to reset or alter the pressure diuresis and natriuresis. As originally described by Guyton and co-workers, a rise in blood pressure causes a brisk diuresis and natriuresis, thereby returning blood pressure towards normal values. Guyton defined the relationship between blood pressure with sodium and water excretion as the pressure-natriuresis curve and reasoned that there must be a rightward resetting of this curve for hypertension to be sustained.36 While the mechanisms for pressure natriuresis and its resetting are multifactorial, substantial evidence implicates increases in renal interstitial pressure and alterations of the sodium transporters such as the sodium hydrogen exchanger 3 (NHE3) and the sodium–phosphate cotransporter isoform 2 in the proximal tubule.37 Acute hypertension retracts these from the luminal cell membrane to the base of the apical microvilli in the proximal tubule where they cannot participate in sodium reabsorption. Likewise, an increase in BP can relocate the thiazide-sensitive sodium chloride co-transporter in the distal tubule. However, during chronic hypertension, these transporters are translocated to the apical microvilli, where they enhance sodium reabsorption and likely sustain hypertension.38 The sodium glucose linked transporter type 2 (SGLT2) in the proximal tubule is also upregulated by angiotensin II acting on AT1 receptors during renovascular hypertension39 where it is functionally linked to NHE3 via the microtubule-associated protein 340 to enhance proximal Na+ reabsorption during ROS stimulation.41 Perfusion of an SGLT2 inhibitor into the proximal tubule inhibits Na+:H+ exchange.42 Since NHE3 transports more Na+ than SGLT2, this functional interaction between the two is likely to contribute to the surprisingly robust 27% inhibition of proximal Na+ and fluid reabsorption by the SGLT2 inhibitor dapagliflozin in diabetic rats.43 This may underlie the reported effects of SGLT2 inhibitors to reduce the plasma volume in heart failure and the blood pressure in hypertension.44,45 Multiple other sodium transport mechanisms are similarly affected as has been expertly reviewed elsewhere.38
Shifts in transporter location and function can be mediated by angiotensin II, inflammatory cytokines, loss of nitric oxide and adrenergic stimulation. It is important to note that shifts in pressure natriuresis are often not reflected by overt changes in renal function as measured by usual clinical parameters such as blood urea nitrogen, creatinine or creatinine clearance. Moreover, renal Na+ retention in hypertension is not usually accompanied by overt signs of fluid overload.
A third, recently recognized role of the kidney in hypertension is to modulate systemic sympathetic tone by generating reflex signals via renal afferent nerves. Approximately 90% of renal nerves are efferent nerves sending sympathetic signals to the kidney, thereby enhancing tubular sodium resorption, renin release and vasomotor tone depending on the intensity of nerve traffic.46 However, a smaller proportion of renal nerves are afferents that generate signals from within the kidney and transmit them to the brainstem where they can initiate reflexes that promote increases in efferent SNS tone and induce hypertension.47 More than 20 years ago, Campese and colleagues reported that the hypertension in rats with 5/6 nephrectomy was reduced markedly after severing renal afferent nerve traffic by bilateral spinal dorsal rhizotomy.48 This was accompanied by a marked reduction in norepinephrine turnover in the posterior and lateral hypothalamus. In a subsequent study, this group reported that injection of a small bolus of phenol into one pole of the kidney also increased renal afferent nerve activity and norepinephrine production by the posterior hypothalamus led to hypertension. These events were abrogated by renal denervation, suggesting that excitation of renal afferent nerves played a critical role. These findings are in keeping with other observations that the hypertension associated with cyclosporine administration in renal allograft recipients is in part mediated by afferent nerve activation.49 More recently, it has been recognized that the renal afferent nerves have a special role in salt-dependent hypertension. Banek et al reported that renal deafferentation reduced hypertension in rats with DOCA-salt hypertension.50 The ability of renal afferent nerves to initiate an increase in global sympathetic outflow might explain the pleiotropic effects of renal denervation.51 In angiotensin II-induced hypertension, renal efferent, rather than afferent nerves seem to have a predominant role.52, 53
Although the precise sites in the kidney where afferent nerves are activated remains undefined, recent studies have thrown light on some of the underlying mechanisms. A high salt diet in the context of both acute54 or chronic47 causes the expected reduction in the circulating RAS but paradoxically activates the intrarenal RAS via increased ROS and activates renal afferent nerve traffic to reflexly increase blood pressure.
A fourth important role of the kidney in hypertension is to serve as a site of immune activation. As discussed below, angiotensin II-induced hypertension activates antigen presenting dendritic cells in the kidney that migrate to secondary lymphoid organs to activate T cells which in turn return back to the kidney.53 This process is blocked by renal denervation, suggesting that efferent renal nerves promote neoantigen formation in the kidney in hypertension. Single cell sequencing has shown that the kidney, as opposed to blood vessels or the spleen, accumulates T cells with an oligoclonal T cell receptor population, supporting the role of the kidney in antigen formation.55
The role of the vasculature in hypertension:
Several of the nodes in Dr. Page’s Mosaic diagram refer to potential vascular mechanisms, including vascular caliber, reactivity, elasticity and indirectly, cardiac output. Systemic vascular resistance is almost uniformly enhanced in adults with hypertension, and many common agents used for the treatment of hypertension are vasodilators. Historically, Guyton proposed that in response to an initial expansion of blood volume and elevation of cardiac output, “systemic autoregulation” caused vasoconstriction and a further increase in blood pressure.56 This leads to additional pressure natriuresis and returned the blood volume to normal, but at the expense of sustained hypertension. Somewhat in keeping with this scenario, Fagard and Staessen reported an increased cardiac output in hypertensive individuals less than 25 years of age but a normal cardiac output in older subjects.57 In addition to systemic autoregulation, that is a physiological response, it is now clear that there are long-term pathophysiological manifestations of hypertension that increase vascular tone and resistance. In the following paragraphs we will review some new observations about how vascular function is altered and contributes to hypertension. We will attempt to relate these to older concepts embodied in Page’s Mosaic theory.
There are at least 4 vascular perturbations that occur in and contribute to hypertension. The first is an enhanced milieu of vasoconstrictor hormones including angiotensin II, catecholamines and vasopressin, coupled with alterations in vascular function that promote vasoconstriction and diminish vasodilatation. Studies of isolated vessels have demonstrated that both conduit vessels and arterioles from hypertensive animals display enhanced vasoconstriction to a variety of agents. Of particular interest are genes that modulate G-protein coupled receptor (GPCR) signaling, including the regulators of G protein signaling (RGS) proteins, that induce GTP hydrolysis to terminate G-protein signaling. The RGS proteins 1 and 2 are linked to GPCRs that enhance responses to vasoconstrictors including thromboxane, angiotensin II and norepinephrine. Almost 20 years ago, Heximer et al showed that mice lacking RGS2, expressed predominantly in vascular smooth muscle, develop baseline hypertension that is dependent on the angiotensin type 1 receptor.58 Additional studies showed that vascular smooth muscle cells from these mice displayed augmented calcium transients, with delayed declines to baseline following agonist stimulation. A key role for RGS2 was highlighted in subsequent work showing that RGS2 in the renal vasculature was critical for promoting hypertension.59 More recently, Patel et al defined an important role of RGS1 in vascular control in atherosclerotic ApoE deficient mice where the knockout of RGS1 evoked a marked increase in blood pressure and vasoconstriction to phenylephrine, thromboxane and angiotensin II.60 Bone marrow transplant studies demonstrated that this phenotype was not mediated by hematopoietic cells. Rather, these investigators showed that mRNA expression of RGS1 is attenuated by chronic angiotensin II infusion and suggested that hypertension leads to a loss of this important modulatory pathway. In contrast, RGS5 seems to have modest to no effects on blood pressure by itself but activates RhoA stress fiber formation and a contractile phenotype of vascular smooth muscle.61 RhoA is a critical activator of Rho Kinase that targets contractile proteins including the myosin light chain in vascular smooth muscle to promote vasoconstriction.62 Endothelial Rho kinase phosphorylates NO synthase on threonine 495 to inhibit NO production, which also increases vasoconstriction.63 The Rho kinase inhibitor Fasudil is approved for use in Japan and China for the treatment of cerebral vasospasm and pulmonary hypertension and has blood pressure lowering effects in genetic and salt induced forms of experimental hypertension.64
Hypertension is also associated with impaired vasodilatation. For many years it has been recognized that endothelium-dependent vasodilatation and nitric oxide signaling are reduced in hypertension, as recently reviewed.65 There are fundamental differences in the mechanisms underlying endothelium-dependent vasodilatation between larger conduit arteries, branch arteries and resistance arterioles. Larger vessels predominantly employ nitric oxide while smaller arteries and arterioles are also modulated by endothelium-dependent hyperpolarization and vasodilator prostaglandins. In larger vessels, defects in NO synthesis and bioavailability include loss of critical co-factors for NO synthase including L-arginine and tetrahydrobiopterin,66, 67 inhibition of NOS activity by ADMA and oxidative inactivation of NO.68 It should be stressed that alterations of NO production extend beyond the vasculature and have implications for renal and CNS dysfunction in hypertension. In smaller arteries and arterioles endothelium-dependent vasodilatation is mediated by hyperpolarization, but the factors responsible for this have largely defied identification. In the past decade, there has been increasing interest the role of direct communications or gap junctions that permit transmission of signals between the endothelium and vascular smooth muscle cells. Early studies using intracellular labeling showed polarized unidirectional movement of fluorescent dyes from the endothelium to adjacent smooth muscle cells.69 These dyes were shown to traverse myoendothelial junctions (MEJ)s that were subsequently identified as holes in the internal elastic membrane lying between the endothelium and vascular smooth muscle. MEJs are particularly common in smaller arterioles,70 and are sites of signaling events and signaling molecules, including calcium pulsars, the inositol 3 phosphate receptor and calcium sensitive potassium channels.71 Endothelium-dependent hyperpolarization is transmitted via MEJs directly to vascular smooth muscle cells. MEJs also facilitate the transfer of nitric oxide generated by NOS that is expressed at these junctions. The NO travels from the endothelium to the vascular smooth muscle where it regulates connexin43 to modulate the permeability of the MEJs.72 MEJs also contains hemoglobin alpha, that controls the passage of NO from the endothelium to the vascular smooth muscle cells in a redox-dependent fashion.73 Intriguingly, a peptide mimic that disrupts the binding of NO to hemoglobin alpha lowers blood pressure abruptly in wild type animals and reverses experimental hypertension caused by angiotensin II infusion.74 Importantly, the density of MEJs is reduced and their local IP3-stimulated calcium signals are blunted in mesenteric arterioles of Spontaneously Hypertensive Rats, indicating that these signaling loci are deficient in hypertensive resistance vessels.75 In keeping with this, mice lacking the MEJ component Connexin40 exhibit hypertension and reduced endothelium-dependent vasodilatation in response to the potassium channel activator SKA-31. Likewise, endothelial specific expression of a mutant Connexin40, that causes electrical impairment of gap junctions, leads to exercise-induced hypertension.76 Recently it has been shown that chronic sympathetic stimulation in the setting of hypertension due to experimental chronic kidney disease decreases the expression of Connexin43, leading to disruption of MEJ function and enhanced vasoconstriction.77 These various lines of evidence strongly support a role of MEJs in modulation of microvascular tone and abnormalities thereof in hypertension.
Perturbations of both microvascular and large vessel structure represent a second vascular contribution to hypertension. More than 60 years ago, Folkow et al proposed that vascular smooth muscle hypertrophy and concomitant narrowing of the arteriolar lumen increases systemic vascular resistance.78 This phenomenon has been detected in the retinal arterioles by fundoscopic examination in clinical hypertension79 where a reduced arteriole to venule ratio increases the odds of developing hypertension. This indicates that arteriolar remodeling can precede, and possibly predispose, to hypertension.80 The application of artificial intelligence and machine learning methods to quantitate parameters of retinal arterial morphology have further demonstrated that retinal artery morphology correlates with cardiovascular outcomes.81 There are multifactorial mechanisms of arteriolar remodeling in hypertension.82 These include the direct hypertrophic/growth-promoting actions of angiotensin II and catecholamines, oxidative signaling,83 and inflammatory cytokines released by immune cells. Many of these enhance the expression or activity of matrix metalloproteinases that promote cell migration, hypertrophy and reorganization.
A related alteration of vascular function that likely contributes to hypertension is stiffening of large conduit arteries, and in particular the proximal aorta, via mechanisms that continue to be investigated. A healthy aorta distends during systole and recoils in diastole. This leads to a reduction in the systolic pressure while maintaining diastolic pressure and perfusion. The aorta stiffens in common conditions, including aging, diabetes, obesity and tobacco use, thereby reducing this critical Windkessel function.84–87 Increased pulse wave velocity, reflecting aortic stiffening, has been reported in population studies to precede hypertension by several years and is a harbinger of stroke, myocardial infarction, and renal failure.88 Aortic stiffening can be both a cause and a consequence of hypertension. It develops well before the onset of hypertension in spontaneously hypertensive rats,89 in diet-induced obesity,90 and in two models of chronic vascular oxidative stress.91 In contrast, Wolinsky reported that aortic collagen increases following the onset of Goldblatt hypertension,92 and we found that experimental hypertension in mice causes aortic stiffening. Mechanical stretch of cultured aortic murine fibroblasts increases their expression of collagens I, III and V. Wu et al found that aortic stiffening occurs before blood pressure elevation in two models of chronic vascular oxidative stress.91 It is thus likely that aortic stiffening and hypertension act in concert, with one leading to the other in a feed-forward fashion.93
While the mechanisms by which aortic stiffening causes end organ damage are not completely understood, a consequence is enhanced propagation of pressure into the microcirculation, leading to barotrauma in target organs, in part via increasing cyclical stretch of microvascular endothelial cells. Indeed, in an elegant analysis of the components of the pulse wave, an increased forward pressure wave was associated with an increased risk for incident cardiovascular disease.94 Drs. Boutouyrie, Chowienczyk and Mitchell discuss vascular stiffening in greater detail in their review of this issue.
A fourth role of the vasculature in hypertension is to serve as a source and target of immune activation. This is in part mediated by a cross talk between the endothelium and immune cells that is covered in depth in the section on immune mechanisms below.
Finally, there is an impaired vessel wall defense against thrombosis in hypertension that has been related to increased endothelial expression of tissue factor and the vascular cell adhesion molecule 1. This enhances platelet-dependent leukocyte adhesion and is driven by thrombin factor XI and the platelet Factor XI receptor.95 Inhibition of Factor XI in angiotensin-infused rats and mice prevents platelet thrombin formation, vascular leukocyte infiltration, endothelial dysfunction, and hypertension. Further studies of the mechanisms that activate thrombosis in hypertension may provide insight into stroke, atherosclerosis and end organ damage. Drs. Webb, Eguchi and Tostes cover the role of the vasculature in greater depth in their review.
The central nervous system in hypertension:
Dr. Page’s revised diagram (Figure 1B) included the term “neural”. Indeed Page demonstrated that angiotensin II caused hypertension predominantly by sympathetic neural activation.13 Measures of muscle sympathetic nerve activity, norepinephrine spillover and heart rate variability have suggested that humans with hypertension commonly have increased sympathetic outflow and enhanced catecholamine mediated vasoconstriction.96 Angiotensin II and ROS in the brain stem nuclei enhance vascular resistance by in part by inhibition of the microvascular endothelium-dependent hyperpolarizing factor that normally reduces vascular tone.77 In keeping with these central vascular effects, ganglionic blockade acutely lowers blood pressure in humans with hypertension to a variable extent,97 particularly in those who are obese.98
The many mechanisms whereby sympathetic tone modulates hypertension include enhanced vasoconstriction and vascular remodeling, renal renin production via beta 1 adrenergic receptors in the juxtaglomerular apparatus and enhanced renal sodium resorption and inflammation.
The central pathways involved in hypertension have been reviewed elsewhere.99 An important initiating site is the laminal terminalis, located anterior to the 3rd ventricle, which contains the subfornical organ (SFO), the median preoptic eminence and the organum vasculosum of the lamina terminalis (OVLT). The SFO and OVLT are circumventricular organs that have a poorly formed blood brain barrier that permit blood born angiotensin II and sodium to activate neuronal firing. Indeed, the OVLT is responsive to even modest increases in sodium to initiate salt-sensitive hypertension.100 Xiao et al recently identified a role of the prostaglandin EP3 receptor in this response, leading to a cascade of ROS production in renal arterioles, isoLG accumulation in dendritic cells and immune activation,101 thereby providing a contemporary example of the mosaic interaction between neural regulation, vasculature and inflammation.
In addition to efferent signals promoting hypertension, there is ample evidence that afferent pathways are likewise involved. In addition to renal afferent activation, discussed above, afferent nerves from adipose tissue are triggered by a high fat diet to reflexively increase BP and insulin resistance.102 Another source of afferent nerve stimulation is the heart. Infusion of saline into cardiac transplant recipients with no evidence of cardiac dysfunction failed to reduce their sympathetic nervous system activity, arginine vasopressin levels or their renin, angiotensin and aldosterone levels and led to hypertension.103, 104 This hypertensive response was linked to a failure to suppress angiotensin II during blood volume expansion and was prevented by angiotensin converting enzyme inhibition.105
A summary of some of the roles and perturbations of renal, vascular, endocrine and neural control in hypertension are depicted in figure 2. These are organ specific features of the Mosaic Theory and hypertension pathophysiology that remain the focus of ongoing research. In the past 2 decades new factors and phenomena have emerged that likely affect the mosaic factor in a global fashion. These include oxidative signaling, inflammation and the microbiome. In addition, a new role of sodium has been identified. These are briefly considered in the following paragraphs.
Figure 2:
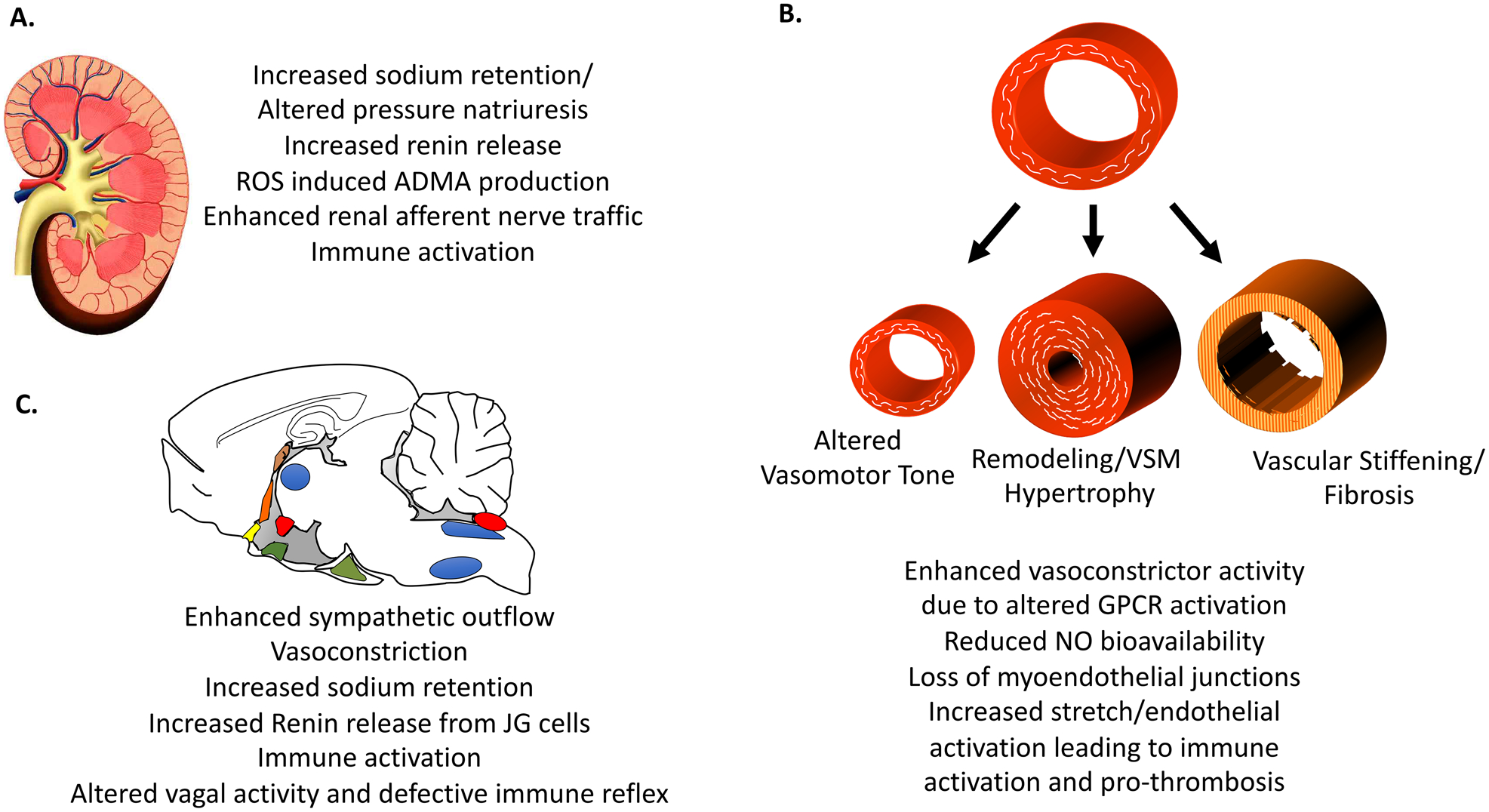
Perturbations of the kidney (A), vasculature (B) and central nervous system (C) contributing to hypertension.
Aldosterone, the mineralocorticoid receptor and their ubiquitous role in the Mosaic Theory:
A major development since the initial Mosaic theory is the evolving understanding of aldosterone in hypertension. Aldosterone is produced by the zona granulosa cells of the adrenal gland in response to angiotensin II and elevations of extracellular potassium. Until recently, the sole effect of aldosterone was thought to enhance sodium reabsorption in the collecting duct and aldosterone blockade was felt to predominantly promote diuresis. It has become obvious that the mineralocorticoid receptor (MR) is expressed in numerous organs and cells, including the heart, blood vessels, immune cells and in the brain. Studies using cre-lox technology have shown important roles of many of these extra-renal sites in hypertension. As an example, mice with vascular smooth muscle deletion of MR have decreased blood pressure with aging and their vessels exhibit diminished vascular myogenic tone, agonist-dependent contraction, vascular superoxide production and expression and L-type calcium channel activity.106 Likewise, Jia et al showed that endothelial cell MR deficiency prevents aortic stiffening caused by feeding a western diet, and established a role for the endothelial MR receptor in modulating endothelial sodium channel activation, oxidative stress and endothelial nitric oxide synthase activation.107 Sun et al used Cre-Lox technology to eliminate the MR in all T cells and showed that this abrogated T cell interferon gamma production and the hypertension caused by chronic infusion of ang II. These authors showed that the MR interacted with other transcription factors to mediate IFNγ production and that its deletion from T cells attenuated fibrosis in the aorta and kidneys, reduced albuminuria and preserved endothelium-dependent vasodilatation. A role for the MR in macrophage activation has been reviewed elsewhere.108
These myriad effects of the MR receptor are important because at least 5% of all cases of hypertension are due to primary hyperaldosteronism and this is likely much higher among those with resistant hypertension. Brown et al recently reported that urinary aldosterone levels (in the sodium loaded state) parallel the severity of hypertension across all subjects,109 suggesting that hyperaldosteronism is much more prevalent than previously considered, even among those without severe hypertension.
The role of ROS in hypertension:
It was first observed in the 1990s that scavenging superoxide could lower blood pressure in rats with hypertension due to chronic ang II infusion and in spontaneously hypertensive rats.110–113 It has subsequently been recognized that several enzymatic sources of ROS are activated in tissues of hypertensive animals and humans, including the nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, the mitochondria and uncoupled NO synthase. ROS likely contribute to almost each node of Dr. Page’s diagram. Superoxide and other related ROS can oxidatively inactivate nitric oxide and oxidize tetrahydrobiopterin, a critical co-factor for NO synthase.114, 115 ROS can also enhance vasoconstriction and thereby alter vascular caliber and reactivity. ROS are important for vascular stiffening and for remodeling of microarterioles.83, 116 In the kidney ROS enhance renal sodium reabsorption,117 and thus enhance volume and cardiac output. They have also been implicated in activation of matrix metalloproteinases and thus promote tissue remodeling.118 ROS likewise increase neuronal firing in critical brain centers, including the subfornical organ,119, 120 and brain stem centers,121 thus altering neural control.
Griendling et al showed in the 1990s that angiotensin II could stimulate superoxide production in cultured vascular smooth muscle cells and in vivo by activating an enzyme similar to the phagocytic NADPH oxidase.113, 122 The catalytic subunit of the NADPH oxidase is Nox2 and homologs of this are activated in vascular smooth muscle cells, the endothelium, renal tubular cells and in the central nervous system in hypertensive animal models. There is clearly an interaction of ROS derived from the NADPH oxidase and with other enzymatic sources, such that ROS from one can trigger ROS production by others in a feed-forward fashion.
While early studies emphasized the role of oxidative stress in vascular cells, the kidney and its vasculature develop oxidative stress with increased NADPH oxidase and reduced SOD activity both during angiotensin II infusion and during high salt intake. High salt intake enhances renal expression of NADPH oxidase and NOX2 and reduces SOD1 and SOD2,123 whereas angiotensin II infusion increases renal NADPH/p22phox and reduces SOD 3 expression.124 Renal ROS can activate renal afferent nerves,125 and increase renal and circulating levels of asymmetric dimethyl arginine (ADMA),126 by reducing its metabolism by dimethylarginine dimethylaminohydrolase and increasing its generation by protein arginine methyltransferase.66 Thus, an increase in circulating ADMA is one means whereby ROS in the kidney may enhance peripheral vasoconstriction. ADMA inhibits endothelial function and flow-mediated vasodilation and activation of the SNS during high salt intake could contribute to the failure of salt-sensitive subjects to reduce their peripheral vascular resistance during increases in cardiac output evoked by a high salt intake. In the kidney, the proximal tubular cells take up oxidatively modified albumin that induces a robust local activation of the RAS via NADPH oxidase, protein kinase C, nuclear factor kappa beta NF-κB and activator protein-1 (AP-1).27 These together increase proximal tubular angiotensin II that initiates renal inflammation and fibrosis. This proximal tubule ROS pathway provides a potential link between hypertension, systemic oxidative stress and proteinuria and the activation of the intrarenal RAS. These events damage the kidney and can thereby perpetuate salt-sensitivity and worsening hypertension.
Of interest is an emerging role of the mitochondria as a source of ROS in hypertension. Recent studies suggest that aging and hypertension reduce the function of Sirtuin3 (Sirt3), a histone deacetylase that modulates the acetylation status of SOD2. Mice lacking Sirt3 are protected against hypertension and its associated vascular disfunction in response to angiotensin II infusion, and this is prevented by mitochondrial-targeted antioxidants, including mitoTempol and mitoEbselen.127 These alterations of the mitochondria in hypertension likely have myriad effects on cellular metabolism and function that contribute to the pathophysiology of hypertension through yet undefined mechanisms.
Therapeutically, scavenging ROS is not clinically employed, and there is evidence that high dose vitamin antioxidants can be harmful, possibly because ROS can have important signaling function.128 Targeted scavenging of ROS using agents like mitoTempol, or treatment with agents that inhibit untoward activation of ROS forming enzymes are viable future options. Indeed, one of the major benefits of angiotensin II blockade might be due to inhibition of ROS formation. This topic is reviewed in depth in this compendium by Drs. Griendling and Touyz.
Inflammatory and immune mechanisms in hypertension:
It has been recognized for more than 50 years that inflammatory cells accumulate in the kidney and blood vessels of hypertensive humans and animals with experimental hypertension. It is now recognized that virtually every type of immune cell, including those of innate and adaptive immunity, contribute to hypertension. These transmigrate into the interstitium of the kidney and blood vessels where they release potent cytokines, ROS and metalloproteinases that modulate renal and vascular function and structure. It is likely that these mediators affect virtually every node of Dr. Page’s Mosaic diagram. A notable example is the cytokine interleukin 17A (IL-17A) produced by a subset of T cells, innate lymphocytes, histiocytes and renal tubular cells. IL-17A affects renal tubular handling of sodium and seems to modulate pressure natriuresis.129 This cytokine stimulates vascular superoxide production,130 causes inhibitory phosphorylation of the endothelial NOS,131 and therefore reduces the caliber of blood vessels. Over the long term IL-17A promotes vascular fibrosis and impairs vascular elasticity.132 In contrast, regulatory T cells and anti-inflammatory cytokines such as IL-10 have antihypertensive effects mediated , in part by counteracting IL-17A.133, 134
The manner by which immune cells are activated in hypertension is a matter of ongoing investigation. Accumulating data suggest that ROS in antigen presenting cells promote oxidation of fatty acids to form highly reactive isolevuglandin (isoLG) products that covalently modify protein lysines.135 These altered proteins are antigenic and activate both CD8+ and CD4+ T cells.
There is substantial evidence that sympathetic outflow promotes immune activation in hypertension. Lesions of the forebrain that prevent sympathetic outflow reduce T cell activation,119, 136 while manipulations that increase sympathetic outflow promote T cell activation.137 Recent data suggest that renal sympathetic tone modulates trafficking of immune cells to and from the kidney and the homing of memory T cells to the bone marrow, where they can be reactivated upon future hypertensive challenges.53, 138 In the vasculature, T cells accumulate in the perivascular fat and adventitia of larger vessels in hypertension, likely via expression of chemokines such as RANTES and MCP-1.139, 140 This adventitial inflammatory response likely triggers collagen deposition, described above. There is also an immune reflex, whereby local tissue signals trigger a vagally mediated anti-inflammatory response.141 This reflex seems to be miswired to promote inflammation in prehypertensive animals.142
Hypertension is associated with endothelial activation. It has long been recognized that the activated endothelium promotes rolling, adhesion and transmigration of leukocytes. As monocytes transmigrate the endothelium, they are transformed into inflammatory macrophages and dendritic cells that emerge as activated monocytes with enhanced potential to produce cytokines and activate T cells.143, 144 Endothelial cell stretch, oxidative stress and loss of nitric oxide in hypertension can all enhance endothelial/leukocyte interaction and increase the endothelial activation of monocytes. Human monocytes activated in this way by the endothelium produce copious quantities of IL-6, IL-23, IL-1β and stimulate autologous T cells potently.145 In addition, endothelial transmigration likely promotes myeloid cell accumulation in the corticomedullary junction of the kidney, where supraphysiological levels of sodium may provide a stimulus to activate monocytes.146
This vascular/immune cross talk again reflects the mosaic nature of hypertension, whereby a primary perturbation, in this case of endothelial (vascular) function, in turn generates mediators that promote inflammation that likely affects multiple other organs and cells. In this compendium, Drs. Madhur, Kirabo and colleagues provide an in-depth review that discusses more fully the role of inflammation in hypertension.
Genetic influences:
Genetic alterations are atop Dr. Page’s revised Mosaic diagram (Figure 1B). Several single gene mutations, including those responsible for Liddle’s syndrome, pseudohyperaldosteronism, aldosterone producing adenomas, glucocorticoid remedial hypertension and missense mutations of the mineralocorticoid receptor have been identified to be responsible for highly heritable Mendelian forms of hypertension.147 While these single gene mutations have provided insight into the pathogenesis of hypertension, they are rare and do not explain the high prevalence of familial hypertension commonly observed in the clinic. In contrast, single nucleotide polymorphisms (SNPs) that often occur in non-protein coding regions of the genome and usually do not alter protein function are common. Genome wide association studies in the last decade have identified more than 1000 SNPs that associate with hypertension.148 While individual SNPs have small effects on blood pressure, when multiple SNPS are analyzed as a polygenic risk score (PRS), they can account for as much as 13 mmHg of blood pressure variability. Vaura et al recently showed such a PRS is predictive of early onset hypertension in a progressive fashion, such that those with the highest 2.5% of PRS have an almost 3-fold risk of developing hypertension, while a low PRS is protective.149 Refinements of the PRS promise to contribute to our future understanding and treatment of hypertension. The genetics in hypertension will be reviewed in detail by Dr. Caufield a subsequent section of this compendium.
Novel concepts related to salt and sodium intake:
Dr. Page’s revised Mosaic Diagram (Figure 1B) included the environment, which includes diet, and in particular salt intake. There are numerous large epidemiological studies linking high levels of dietary sodium intake with risk for hypertension.150 Conversely, reducing dietary salt or administration of diuretic agents such as thiazides are effective treatments for elevated blood pressure.151, 152 In human populations, sodium-sensitivity, defined as an exaggerated change in blood pressure in response to extremes of dietary salt intake, is a relatively common phenotype associated with increased risk for hypertension and cardiovascular events.153 The blood pressure of about one quarter of normal subjects and one half of hypertensive increases with salt intake.154 While salt-sensitive hypertension entails a renal defect in salt excretion, the increase in BP with salt intake is caused by increased peripheral resistance that requires a communication between the kidneys and the peripheral vasculature. Indeed, enhanced vasoconstriction and endothelial dysfunction,155 rather than a primary increase in renal Na+ retention, can initiate salt-sensitive hypertension.156 Salt-sensitive subjects are unable to increase peripheral vascular resistance appropriately during a low salt intake,157 or to reduce their peripheral vascular resistance during a high salt intake,158 thereby causing their blood pressure to be unusually dependent on salt intake. There is an accompanying failure of salt-sensitive subjects to suppress the SNS during salt loading,159 accompanied by an increase in circulating ADMA during salt loading only in salt-sensitive subjects.158
Classic Guytonian models suggest that a defect in sodium excretion by the kidney is the primary basis for hypertension, with impaired elimination of sodium during high salt feeding leading directly to expanded extracellular fluid volume promoting increased blood pressure.18 Studies in recent years indicate that the impact of dietary sodium intake upon blood pressure is complex, beyond the simple construct of intra-vascular volume expansion balanced by excretory functions of the kidney. For example, it is now apparent that during high salt feeding, sodium accumulates in the interstitium of the skin and other tissues where it may be stored at hypertonic concentrations in complexes with proteoglycans, acting as a reservoir and buffering the impact of sodium accumulation on intravascular volume and blood pressure.160 This interstitial sodium storage can be visualized and quantified in humans using 23Na MRI.161 Accumulation of sodium in tissues increases with high salt intake or aging, and has been associated with hypertension and increased cardiovascular risk.162 In the sub-dermal space, sodium can also stimulate macrophages, triggering expression of TonEBP, a transcription factor regulating the expression of osmo-protective genes including vascular endothelial growth factor (VEGF)-C, a potent inducer of lymphangiogenesis.160 It has been suggested that these immunomodulatory actions of salt deposited in skin may have evolved to subserve a barrier function to prevent invasion by micro-organisms, but in the modern world where dietary salt is ubiquitous, this may now be a maladaptive response.163 Other recent studies indicate that very high levels of sodium intake can trigger archetypal metabolic changes aimed at conserving water, with a side effect of raising blood pressure.164
Elevated concentrations of tissue sodium can also activate immune cells that contribute to hypertension. Modest elevations of sodium can stimulate T cells to produce the cytokine IL-17A,165, 166 which as mentioned above, promotes vascular remodeling, endothelial dysfunction and renal sodium retention. Sodium entry via an amiloride sensitive sodium channel can also drive dendritic cell activation,167 and promotes transformation of human monocytes to a dendritic cell-like phenotype that produce cytokines like IL-6, TNFα and IL-1β.146 Upon entry of sodium into myeloid cells calcium activation of the NADPH oxidase ensues leading to ROS formation and the generation of isoLG adducts, which as discussed above act as neoantigens in these cells.
In both T cells and myeloid cells, the effect of sodium is mediated by the serum glucocorticoid kinase-1 (SGK1). Wu et al. showed that high-salt feeding stimulated Th17 cells in the central nervous system and mesenteric lymph nodes and exacerbated experimental allergic encephalitis (EAE), a model of multiple sclerosis mediated by IL-17A.165 Interestingly, mice with T cell SGK1 deficiency exhibited reduced EAE incidence and severity that was not exacerbated by high-salt feeding. Norlander et al showed that deletion of SGK1 in T cells blunts the hypertensive response to angiotensin II and DOCA-salt challenge in mice and abrogates renal and vascular inflammation in these models.168 In this study, a T cell sodium-potassium-2 chloride cotransporter 1 was found to mediate the salt-induced increase in SGK1 and the IL-23 receptor (IL-23R), which in turn promoted IL-17A production. Likewise, deletion of this kinase specifically in antigen presenting cells markedly reduces renal inflammation and hypertension in a salt sensitive model of hypertension.169 In this myeloid cell subset, SGK1 promotes assembly of the alpha and gamma subunits of the epithelial sodium channel, which further promotes sodium entry. These studies suggest that diuretic agents previously thought to act only on the kidney might prove useful in reducing salt-driven inflammation in hypertension.
The microbiome as an emerging modulator of blood pressure:
Along with effects on fluid homeostasis and immune activation, sodium as a dietary constituent may promote hypertension by modifying gut microbiota.170 Indeed, a number of recent studies have highlighted the potential of the microbiome to influence blood pressure.171–173 Advances in unbiased sequencing techniques have allowed precise identification and assessment of commensural bacterial species in the gut that cannot be cultured, transforming understanding of the role of the microbiome in health and disease. Such studies have provided evidence for contributions of the microbiome in the pathogenesis of a number of diseases, including hypertension.174, 175 This work has also identified conditioning effects of diet and drugs on the composition of the gut microbiota and highlighted the potential of these disturbances to affect host responses including blood pressure regulation.
In healthy individuals, the gut microbiome is typically in a stable state of eubiosis and in relative equilibrium with the surrounding community.176 By contrast, significant alteration in the microbiome (dysbiosis) has been observed in range of disorders including hypertension. For example, in humans with hypertension and pre-hypertension, characteristic alterations in the microbiome have been observed.177, 178 Dysbiosis has also been seen in experimental models of hypertension such as Dahl salt-sensitive and spontaneously hypertensive rats,179, 180 and in mice chronically administered angiotensin II or DOCA + high salt diet.181 Fecal transfer from hypertensive mice to germ free mice sensitizes the latter to hypertension, providing strong evidence for a role of the microbiome.178 In hypertension, alterations in the gut microbiome have been associated with inflammation, structural changes in the intestinal wall, and enhanced gut permeability.182 Altered intestinal permeability may facilitate egress of mediators such as microbial products, hormones, and immune cells into the circulation where they can impact peripheral and central BP control mechanisms.
Along with associations between hypertension and gut dysbiosis, there is a direct relationship between diet and the composition of gut microbiota with potential to affect propensity for hypertension. For example, increased dietary salt intake causes significant alterations in the composition of microbiota and has been associated with accumulation of pro-inflammatory TH17 cells discussed above.170 Sodium is not the only dietary constituent with potential to impact blood pressure through effects on the microbiome. Multiple studies have shown that diets enriched in fruit, vegetables, and fiber can lower blood pressure and reduce risk for hypertension.183 Moreover, dietary fiber increases bacterial populations in the gut responsible for generating short-chain fatty acids (SCFAs) through fermentation.184 In the human circulation, SCFAs are almost exclusively of microbial origin,185 and have been implicated as a key pathway whereby microbiota influence host physiology,186 perhaps through a family of nutrient-sensing GCPRs expressed in vascular smooth muscle cells, the autonomic nervous system and renal juxtaglomerular cells influencing renin release.171, 187 Thus, the gut microbiome can influence blood pressure control through pathways affecting many of Page’s Mosaic components, raising the possibility that pre- and pro-biotics, antibiotics, and specific dietary formulations could be used to modify gut microbiome and its metabolites to prevent and treat hypertension. The potential impact of the microbiome in hypertension will be reviewed by Drs. Muller and Luft.
Summary:
These considerations of the Mosaic Theory and beyond reflect the prescient nature of Dr. Page’s original concept, depicted in Figures 1A and 1B, which is notable for the interdependence of the various components, such that a change in one can almost certainly perturb the others. As research has evolved, a newer Mosaic diagram could be constructed as shown in figure 3, emphasizing the interplay of factors like inflammation, oxidative stress, the microbiome with alterations of renal, neural and vascular function. It should be stressed that the role of these and other factors almost certainly vary from one hypertensive human to the next or upon the experimental model studied. Importantly, another factor that should be considered is that blood pressure elevation can influence hypertension by initially causing baroreflex activation and pressure natriuresis (to lower blood pressure) and ultimately by producing pathophysiological effects that progressively raise blood pressure in a feed-forward fashion. These features of the Mosaic Theory will undoubtedly guide hypertension research in the foreseeable future.
Figure 3:
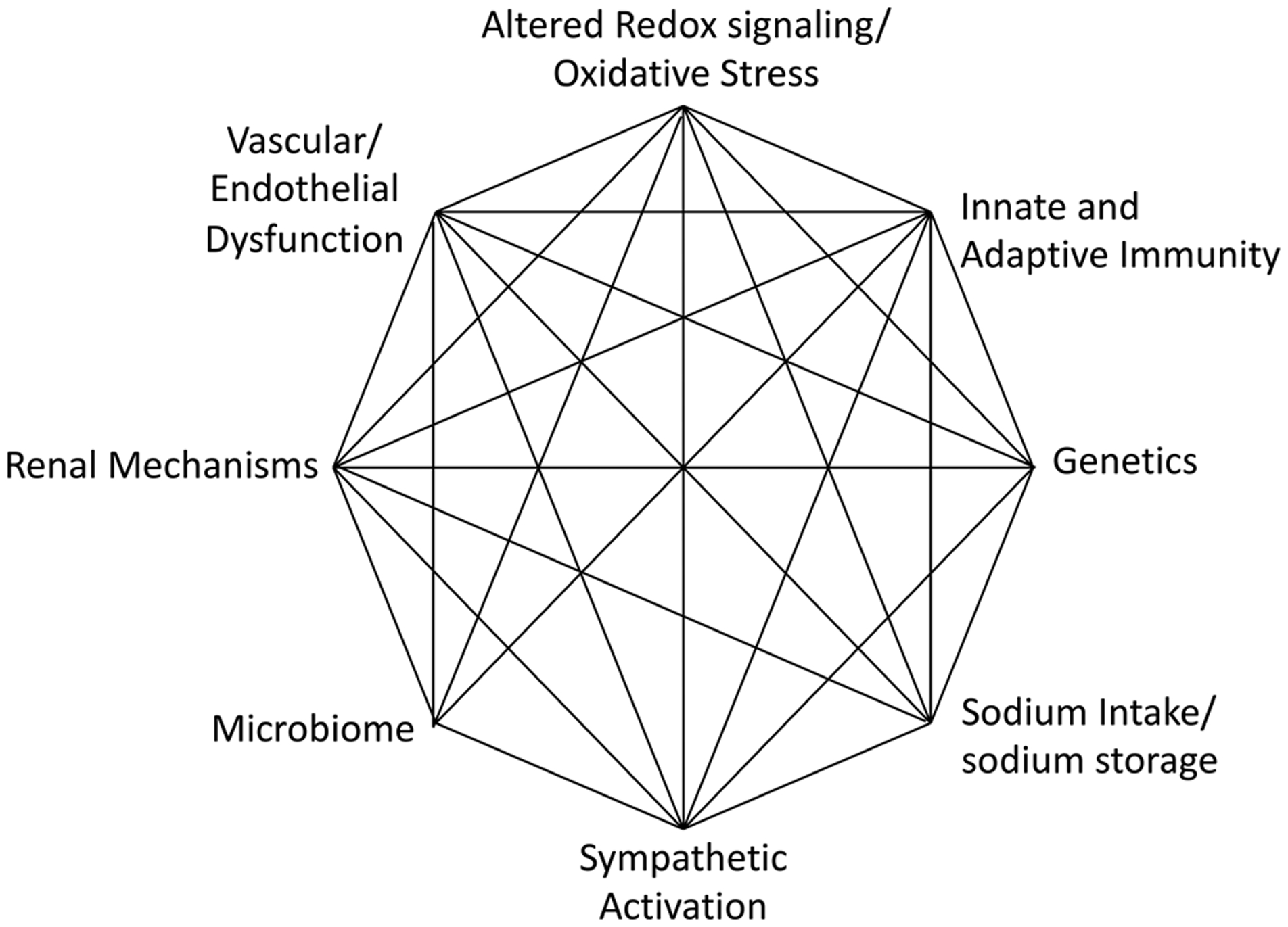
A revised Mosaic Theory incorporating new understanding of cellular, environmental and genetic mechanisms.
Sources of Funding:
Supported by NIH HL5R35HL140016, P01HL129941, UL1TR001881 and T32HL144446 to DGH, by the Singapore National Medical Research Council grant OFLCG/001/201 to TMC and by NIH grants RO-1 HL-134511 and R21-HD101340 and by funds from the Walter Family Chair of Cardiovascular Research, the Smith-Kogod Family Foundation, the Gildenhorn-Speisman Family Foundation and the Georgetown University Hypertension Research Center to CSW.
Non-standard abbreviations and acronyms:
- ACE
angiotensin I-converting enzyme
- ADMA
asymmetric dimethyl arginine
- AP-1
activator protein-1
- ApoE
apolipoprotein E
- AT1R
angiotensin type 1 receptor
- BP
blood pressure
- CNS
central nervous system
- DOCA
Deoxycorticosterone acetate
- EAE
experimental allergic encephalitis
- EP3
E prostanoid receptor type 3
- GPCR
G-protein coupled receptor
- IL
interleukin
- isoLG
isolevuglandin
- MCP1
monocyte chemoattractant protein 1
- MEJ
myoendothelial junction
- MR
mineralocorticoid receptor
- NADPH
nicotinamide adenine dinucleotide phosphate
- Na+:H+
sodium:hydrogen
- 23Na MRI
magnetic resonance imaging of the 23sodium isotope
- NHE3
Sodium hydrogen exchanger 3
- NO
nitric oxide
- NOS
nitric oxide synthase
- Nox
NADPH oxidase catalytic subunit
- OVLT
organum vasculosum of the lateral terminalis
- PRR
pro-renin receptor
- PRS
polygenic risk score
- RAS
renin angiotensin system
- RANTES
Regulated upon Activation, Normal T Cell Expressed and Presumably Secreted
- RhoA
rat sarcoma virus homologous protein A
- RGS
regulator of G protein signaling
- ROS
reactive oxygen species
- SCFAs
short-chain fatty acids
- SFO
subfornical organ
- SGK1
serum glucocorticoid kinase 1
- SGLT2
sodium glucose linked transporter type 2
- Sirt3
sirtuin3
- SNPs
single nucleotide polymorphisms
- SNS
sympathetic nervous system
- SOD
superoxide dismutase
- Sox6
SRY-related HMG-box gene 6
- TonEBP
Tonicity-responsive enhancer binding protein
- TNFα
tumor necrosis factor alpha
- VEGF
vascular endothelial growth factor
Footnotes
Disclosures: The authors have no conflicts of interest relevant to this manuscript.
LITERATURE CITED
- 1.Whelton PK, Carey RM, Aronow WS, Casey DE Jr., Collins KJ, Dennison Himmelfarb C, DePalma SM, Gidding S, Jamerson KA, Jones DW, MacLaughlin EJ, Muntner P, Ovbiagele B, Smith SC Jr., Spencer CC, Stafford RS, Taler SJ, Thomas RJ, Williams KA Sr., Williamson JD and Wright JT Jr. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2017. [Google Scholar]
- 2.Collaborators GBDRF. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396:1223–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mills KT, Bundy JD, Kelly TN, Reed JE, Kearney PM, Reynolds K, Chen J and He J. Global Disparities of Hypertension Prevalence and Control: A Systematic Analysis of Population-Based Studies From 90 Countries. Circulation. 2016;134:441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tigerstedt R and Bergman P. Niere und Kreislauf. Scand Arch Physiol 1898;8:223–271. [Google Scholar]
- 5.Vollhard H. S. Franz: Roter und blasser Hochdruck aus heutiger sicht Med Klin 1973;68. [PubMed] [Google Scholar]
- 6.Luft FC and Dietz R. Franz Volhard in historical perspective. Hypertension. 1993;22:253–6. [DOI] [PubMed] [Google Scholar]
- 7.Goldblatt H, Lynch J, Hanzal RF and Summerville WW. Studies on Experimental Hypertension : I. The Production of Persistent Elevation of Systolic Blood Pressure by Means of Renal Ischemia. J Exp Med. 1934;59:347–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braun-Menendez E, Fasciolo JC, Leloir LF and Munoz JM. The substance causing renal hypertension. J Physiol. 1940;98:283–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Page IH. On the Nature of the Pressor Action of Renin. J Exp Med. 1939;70:521–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braun-Menendez E and Page IH. Suggested Revision of Nomenclature--Angiotensin. Science. 1958;127:242. [DOI] [PubMed] [Google Scholar]
- 11.Page IH and Helmer OM. A Crystalline Pressor Substance (Angiotonin) Resulting from the Reaction between Renin and Renin-Activator. J Exp Med. 1940;71:29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Page IH. Pathogenesis of arterial hypertension. J Am Med Assoc. 1949;140:451–8. [DOI] [PubMed] [Google Scholar]
- 13.McCubbin JW, DeMoura RS, Page IH and Olmsted F. Arterial hypertension elicited by subpressor amounts of angiotensin. Science. 1965;149:1394–5. [DOI] [PubMed] [Google Scholar]
- 14.Page IH. The nature of arterial hypertension. Arch Intern Med. 1963;111:103–15. [DOI] [PubMed] [Google Scholar]
- 15.Page IH. The mosaic theory 32 years later. Hypertension. 1982;4:177. [DOI] [PubMed] [Google Scholar]
- 16.Lip S and Padmanabhan S. Genomics of Blood Pressure and Hypertension: Extending the Mosaic Theory Toward Stratification. Can J Cardiol. 2020;36:694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrison DG. The Mosaic Theory Revisited: Common Molecular Mechanisms Coordinating Diverse Organ and Cellular Events in Hypertension. Journal of the American Society of Hypertension. 2013;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guyton AC, Coleman TG, Cowley AW Jr., Liard JF, Norman RA Jr. and Manning RD Jr. Systems analysis of arterial pressure regulation and hypertension. Ann Biomed Eng. 1972;1:254–81. [DOI] [PubMed] [Google Scholar]
- 19.Fasciolo JC, Houssay BA and Taquini AC. The blood-pressure raising secretion of the ischaemic kidney. J Physiol. 1938;94:281–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahl LK, Heine M and Thompson K. Genetic influence of renal homografts on the blood pressure of rats from different strains. Proc Soc Exp Biol Med. 1972;140:852–6. [DOI] [PubMed] [Google Scholar]
- 21.Curtis JJ, Luke RG, Dustan HP, Kashgarian M, Whelchel JD, Jones P and Diethelm AG. Remission of essential hypertension after renal transplantation. N Engl J Med. 1983;309:1009–15. [DOI] [PubMed] [Google Scholar]
- 22.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH and Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103:17985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guessoum O, de Goes Martini A, Sequeira-Lopez MLS and Gomez RA. Deciphering the Identity of Renin Cells in Health and Disease. Trends Mol Med. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castrop H, Hocherl K, Kurtz A, Schweda F, Todorov V and Wagner C. Physiology of kidney renin. Physiol Rev. 2010;90:607–73. [DOI] [PubMed] [Google Scholar]
- 25.Saleem M, Hodgkinson CP, Xiao L, Gimenez-Bastida JA, Rasmussen ML, Foss J, Payne AJ, Mirotsou M, Gama V, Dzau VJ and Gomez JA. Sox6 as a new modulator of renin expression in the kidney. Am J Physiol Renal Physiol. 2020;318:F285–F297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Curnow AC, Gonsalez SR, Gogulamudi VR, Visniauskas B, Simon EE, Gonzalez AA, Majid DSA, Lara LS and Prieto MC. Low Nitric Oxide Bioavailability Increases Renin Production in the Collecting Duct. Front Physiol. 2020;11:559341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao W, Zhou QG, Nie J, Wang GB, Liu Y, Zhou ZM and Hou FF. Albumin overload activates intrarenal renin-angiotensin system through protein kinase C and NADPH oxidase-dependent pathway. J Hypertens. 2011;29:1411–21. [DOI] [PubMed] [Google Scholar]
- 28.Prieto MC, Reverte V, Mamenko M, Kuczeriszka M, Veiras LC, Rosales CB, McLellan M, Gentile O, Jensen VB, Ichihara A, McDonough AA, Pochynyuk OM and Gonzalez AA. Collecting duct prorenin receptor knockout reduces renal function, increases sodium excretion, and mitigates renal responses in ANG II-induced hypertensive mice. Am J Physiol Renal Physiol. 2017;313:F1243–F1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMurray JJ, Krum H, Abraham WT, Dickstein K, Kober LV, Desai AS, Solomon SD, Greenlaw N, Ali MA, Chiang Y, Shao Q, Tarnesby G, Massie BM and Investigators AC. Aliskiren, Enalapril, or Aliskiren and Enalapril in Heart Failure. N Engl J Med. 2016;374:1521–32. [DOI] [PubMed] [Google Scholar]
- 30.Wang GM, Li LJ, Tang WL and Wright JM. Renin inhibitors versus angiotensin converting enzyme (ACE) inhibitors for primary hypertension. Cochrane Database Syst Rev. 2020;10:CD012569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wal P, Wal A, Rai AK and Dixit A. Aliskiren: An orally active renin inhibitor. J Pharm Bioallied Sci. 2011;3:189–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Békássy ZD, Kristoffersson AC, Rebetz J, Tati R, Olin AI and Karpman D. Aliskiren inhibits renin-mediated complement activation. Kidney Int. 2018;94:689–700. [DOI] [PubMed] [Google Scholar]
- 33.Perez-Gomez MV and Ortiz A. Aliskiren and the dual complement inhibition concept. Clin Kidney J. 2020;13:35–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engström G, Hedblad B, Berglund G, Janzon L and Lindgärde F. Plasma levels of complement C3 is associated with development of hypertension: a longitudinal cohort study. J Hum Hypertens. 2007;21:276–82. [DOI] [PubMed] [Google Scholar]
- 35.Wenzel UO, Bode M, Kurts C and Ehmke H. Salt, inflammation, IL-17 and hypertension. Br J Pharmacol. 2019;176:1853–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guyton AC. Renal function curve--a key to understanding the pathogenesis of hypertension. Hypertension. 1987;10:1–6. [DOI] [PubMed] [Google Scholar]
- 37.Wilcox CS, Sterzel RB, Dunckel PT, Mohrmann M and Perfetto M. Renal interstitial pressure and sodium excretion during hilar lymphatic ligation. Am J Physiol. 1984;247:F344–51. [DOI] [PubMed] [Google Scholar]
- 38.McDonough AA and Nguyen MT. Maintaining Balance Under Pressure: Integrated Regulation of Renal Transporters During Hypertension. Hypertension. 2015;66:450–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bautista R, Manning R, Martinez F, Avila-Casado Mdel C, Soto V, Medina A and Escalante B. Angiotensin II-dependent increased expression of Na+-glucose cotransporter in hypertension. Am J Physiol Renal Physiol. 2004;286:F127–33. [DOI] [PubMed] [Google Scholar]
- 40.Huber G and Matus A. Microtubule-associated protein 3 (MAP3) expression in non-neuronal tissues. J Cell Sci. 1990;95 (Pt 2):237–46. [DOI] [PubMed] [Google Scholar]
- 41.Panico C, Luo Z, Damiano S, Artigiano F, Gill P and Welch WJ. Renal proximal tubular reabsorption is reduced in adult spontaneously hypertensive rats: roles of superoxide and Na+/H+ exchanger 3. Hypertension. 2009;54:1291–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pessoa TD, Campos LC, Carraro-Lacroix L, Girardi AC and Malnic G. Functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J Am Soc Nephrol. 2014;25:2028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomson SC, Rieg T, Miracle C, Mansoury H, Whaley J, Vallon V and Singh P. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol. 2012;302:R75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Griffin M, Rao VS, Ivey-Miranda J, Fleming J, Mahoney D, Maulion C, Suda N, Siwakoti K, Ahmad T, Jacoby D, Riello R, Bellumkonda L, Cox Z, Collins S, Jeon S, Turner JM, Wilson FP, Butler J, Inzucchi SE and Testani JM. Empagliflozin in Heart Failure: Diuretic and Cardiorenal Effects. Circulation. 2020;142:1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilcox CS. Antihypertensive and Renal Mechanisms of SGLT2 (Sodium-Glucose Linked Transporter 2) Inhibitors. Hypertension. 2020;75:894–901. [DOI] [PubMed] [Google Scholar]
- 46.DiBona GF. Neural control of the kidney: functionally specific renal sympathetic nerve fibers. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1517–24. [DOI] [PubMed] [Google Scholar]
- 47.Cao W, Li A, Wang L, Zhou Z, Su Z, Bin W, Wilcox CS and Hou FF. A Salt-Induced Reno-Cerebral Reflex Activates Renin-Angiotensin Systems and Promotes CKD Progression. J Am Soc Nephrol. 2015;26:1619–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campese VM and Kogosov E. Renal afferent denervation prevents hypertension in rats with chronic renal failure. Hypertension. 1995;25:878–82. [DOI] [PubMed] [Google Scholar]
- 49.Moss NG, Powell SL and Falk RJ. Intravenous cyclosporine activates afferent and efferent renal nerves and causes sodium retention in innervated kidneys in rats. Proc Natl Acad Sci U S A. 1985;82:8222–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Banek CT, Knuepfer MM, Foss JD, Fiege JK, Asirvatham-Jeyaraj N, Van Helden D, Shimizu Y and Osborn JW. Resting Afferent Renal Nerve Discharge and Renal Inflammation: Elucidating the Role of Afferent and Efferent Renal Nerves in Deoxycorticosterone Acetate Salt Hypertension. Hypertension. 2016;68:1415–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Donazzan L, Mahfoud F, Linz D, Ewen S, Ukena C and Bohm M. Novel and nonpharmacologic approaches to cardio-protection in hypertension. Curr Hypertens Rep. 2014;16:430. [DOI] [PubMed] [Google Scholar]
- 52.Foss JD, Fiege J, Shimizu Y, Collister JP, Mayerhofer T, Wood L and Osborn JW. Role of afferent and efferent renal nerves in the development of AngII-salt hypertension in rats. Physiol Rep. 2018;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, Takahashi T, Loperena R, Foss JD, Mernaugh RL, Chen W, Roberts J 2nd, Osborn JW, Itani HA and Harrison DG. Renal Denervation Prevents Immune Cell Activation and Renal Inflammation in Angiotensin II-Induced Hypertension. Circ Res. 2015;117:547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cao W, Li A, Li J, Wu C, Cui S, Zhou Z, Liu Y, Wilcox CS and Hou FF. Reno-Cerebral Reflex Activates the Renin-Angiotensin System, Promoting Oxidative Stress and Renal Damage After Ischemia-Reperfusion Injury. Antioxid Redox Signal. 2017;27:415–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, Wu J, Goldstein A, Arendshorst WJ, Madhur MS, Chen W, Li CI, Shyr Y and Harrison DG. Oligoclonal CD8+ T Cells Play a Critical Role in the Development of Hypertension. Hypertension. 2014;64:1108–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coleman TG, Granger HJ and Guyton AC. Whole-body circulatory autoregulation and hypertension. Circ Res. 1971;28:Suppl 2:76–87. [DOI] [PubMed] [Google Scholar]
- 57.Fagard R and Staessen J. Relation of cardiac output at rest and during exercise to age in essential hypertension. Am J Cardiol. 1991;67:585–9. [DOI] [PubMed] [Google Scholar]
- 58.Heximer SP, Knutsen RH, Sun X, Kaltenbronn KM, Rhee MH, Peng N, Oliveira-dos-Santos A, Penninger JM, Muslin AJ, Steinberg TH, Wyss JM, Mecham RP and Blumer KJ. Hypertension and prolonged vasoconstrictor signaling in RGS2-deficient mice. J Clin Invest. 2003;111:445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gurley SB, Griffiths RC, Mendelsohn ME, Karas RH and Coffman TM. Renal actions of RGS2 control blood pressure. J Am Soc Nephrol. 2010;21:1847–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patel J, Chuaiphichai S, Douglas G, Gorvin CM and Channon KM. Vascular wall regulator of G-protein signalling-1 (RGS-1) is required for angiotensin II-mediated blood pressure control. Vascul Pharmacol. 2018;108:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arnold C, Demirel E, Feldner A, Genove G, Zhang H, Sticht C, Wieland T, Hecker M, Heximer S and Korff T. Hypertension-evoked RhoA activity in vascular smooth muscle cells requires RGS5. FASEB J. 2018;32:2021–2035. [DOI] [PubMed] [Google Scholar]
- 62.Shimokawa H, Sunamura S and Satoh K. RhoA/Rho-Kinase in the Cardiovascular System. Circ Res. 2016;118:352–66. [DOI] [PubMed] [Google Scholar]
- 63.Sugimoto M, Nakayama M, Goto TM, Amano M, Komori K and Kaibuchi K. Rho-kinase phosphorylates eNOS at threonine 495 in endothelial cells. Biochem Biophys Res Commun. 2007;361:462–7. [DOI] [PubMed] [Google Scholar]
- 64.Behuliak M, Bencze M, Vaneckova I, Kunes J and Zicha J. Basal and Activated Calcium Sensitization Mediated by RhoA/Rho Kinase Pathway in Rats with Genetic and Salt Hypertension. Biomed Res Int. 2017;2017:8029728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li Q, Youn JY and Cai H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J Hypertens. 2015;33:1128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wilcox CS. Asymmetric dimethylarginine and reactive oxygen species: unwelcome twin visitors to the cardiovascular and kidney disease tables. Hypertension. 2012;59:375–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harrison DG, Chen W, Dikalov S and Li L. Regulation of endothelial cell tetrahydrobiopterin pathophysiological and therapeutic implications. Adv Pharmacol. 2010;60:107–32. [DOI] [PubMed] [Google Scholar]
- 68.Touyz RM. Oxidative stress and vascular damage in hypertension. Curr Hypertens Rep. 2000;2:98–105. [DOI] [PubMed] [Google Scholar]
- 69.Little TL, Xia J and Duling BR. Dye tracers define differential endothelial and smooth muscle coupling patterns within the arteriolar wall. Circ Res. 1995;76:498–504. [DOI] [PubMed] [Google Scholar]
- 70.Sandow SL and Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ Res. 2000;86:341–6. [DOI] [PubMed] [Google Scholar]
- 71.Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI and Nelson MT. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci U S A. 2008;105:9627–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Straub AC, Billaud M, Johnstone SR, Best AK, Yemen S, Dwyer ST, Looft-Wilson R, Lysiak JJ, Gaston B, Palmer L and Isakson BE. Compartmentalized connexin 43 s-nitrosylation/denitrosylation regulates heterocellular communication in the vessel wall. Arterioscler Thromb Vasc Biol. 2011;31:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Straub AC, Lohman AW, Billaud M, Johnstone SR, Dwyer ST, Lee MY, Bortz PS, Best AK, Columbus L, Gaston B and Isakson BE. Endothelial cell expression of haemoglobin alpha regulates nitric oxide signalling. Nature. 2012;491:473–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Straub AC, Butcher JT, Billaud M, Mutchler SM, Artamonov MV, Nguyen AT, Johnson T, Best AK, Miller MP, Palmer LA, Columbus L, Somlyo AV, Le TH and Isakson BE. Hemoglobin alpha/eNOS coupling at myoendothelial junctions is required for nitric oxide scavenging during vasoconstriction. Arterioscler Thromb Vasc Biol. 2014;34:2594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wilson C, Zhang X, Buckley C, Heathcote HR, Lee MD and McCarron JG. Increased Vascular Contractility in Hypertension Results From Impaired Endothelial Calcium Signaling. Hypertension. 2019;74:1200–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morton SK, Chaston DJ, Howitt L, Heisler J, Nicholson BJ, Fairweather S, Broer S, Ashton AW, Matthaei KI and Hill CE. Loss of functional endothelial connexin40 results in exercise-induced hypertension in mice. Hypertension. 2015;65:662–9. [DOI] [PubMed] [Google Scholar]
- 77.Cao W, Wu L, Zhang X, Zhou J, Wang J, Yang Z, Su H, Liu Y, Wilcox CS and Hou FF. Sympathetic Overactivity in CKD Disrupts Buffering of Neurotransmission by Endothelium-Derived Hyperpolarizing Factor and Enhances Vasoconstriction. J Am Soc Nephrol. 2020;31:2312–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Folkow B, Grimby G and Thulesius O. Adaptive structural changes of the vascular walls in hypertension and their relation to the control of the peripheral resistance. Acta Physiol Scand. 1958;44:255–72. [DOI] [PubMed] [Google Scholar]
- 79.Lehmann MV and Schmieder RE. Remodeling of retinal small arteries in hypertension. Am J Hypertens. 2011;24:1267–73. [DOI] [PubMed] [Google Scholar]
- 80.Wong TY, Klein R, Sharrett AR, Duncan BB, Couper DJ, Klein BE, Hubbard LD, Nieto FJ and Atherosclerosis Risk in Communities S. Retinal arteriolar diameter and risk for hypertension. Ann Intern Med. 2004;140:248–55. [DOI] [PubMed] [Google Scholar]
- 81.Poplin R, Varadarajan AV, Blumer K, Liu Y, McConnell MV, Corrado GS, Peng L and Webster DR. Prediction of cardiovascular risk factors from retinal fundus photographs via deep learning. Nat Biomed Eng. 2018;2:158–164. [DOI] [PubMed] [Google Scholar]
- 82.Heagerty AM, Heerkens EH and Izzard AS. Small artery structure and function in hypertension. J Cell Mol Med. 2010;14:1037–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li L, Feng D, Luo Z, Welch WJ, Wilcox CS and Lai EY. Remodeling of Afferent Arterioles From Mice With Oxidative Stress Does Not Account for Increased Contractility but Does Limit Excessive Wall Stress. Hypertension. 2015;66:550–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vaitkevicius PV, Fleg JL, Engel JH, O’Connor FC, Wright JG, Lakatta LE, Yin FC and Lakatta EG. Effects of age and aerobic capacity on arterial stiffness in healthy adults. Circulation. 1993;88:1456–62. [DOI] [PubMed] [Google Scholar]
- 85.Agnoletti D, Lieber A, Zhang Y, Protogerou AD, Borghi C, Blacher J and Safar ME. Central hemodynamic modifications in diabetes mellitus. Atherosclerosis. 2013;230:315–21. [DOI] [PubMed] [Google Scholar]
- 86.Safar ME, Czernichow S and Blacher J. Obesity, arterial stiffness, and cardiovascular risk. J Am Soc Nephrol. 2006;17:S109–11. [DOI] [PubMed] [Google Scholar]
- 87.Yun M, Li S, Sun D, Ge S, Lai CC, Fernandez C, Chen W, Srinivasan SR and Berenson GS. Tobacco smoking strengthens the association of elevated blood pressure with arterial stiffness: the Bogalusa Heart Study. J Hypertens. 2015;33:266–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vasan RS, Short MI, Niiranen TJ, Xanthakis V, DeCarli C, Cheng S, Seshadri S and Mitchell GF. Interrelations Between Arterial Stiffness, Target Organ Damage, and Cardiovascular Disease Outcomes. J Am Heart Assoc. 2019;8:e012141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van Gorp AW, Schenau DS, Hoeks AP, Boudier HA, de Mey JG and Reneman RS. In spontaneously hypertensive rats alterations in aortic wall properties precede development of hypertension. Am J Physiol Heart Circ Physiol. 2000;278:H1241–7. [DOI] [PubMed] [Google Scholar]
- 90.Weisbrod RM, Shiang T, Al Sayah L, Fry JL, Bajpai S, Reinhart-King CA, Lob HE, Santhanam L, Mitchell G, Cohen RA and Seta F. Arterial stiffening precedes systolic hypertension in diet-induced obesity. Hypertension. 2013;62:1105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KR, Xiao L, Chen W, Mernaugh RL, Cai H, Bernstein KE, Goronzy JJ, Weyand CM, Curci JA, Barbaro NR, Moreno H, Davies SS, Roberts LJ 2nd, Madhur MS and Harrison DG. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wolinsky H Response of the rat aortic media to hypertension. Morphological and chemical studies. Circ Res. 1970;26:507–22. [DOI] [PubMed] [Google Scholar]
- 93.Humphrey JD, Harrison DG, Figueroa CA, Lacolley P and Laurent S. Central Artery Stiffness in Hypertension and Aging: A Problem with Cause and Consequence. Circulation Research. 2016;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cooper LL, Rong J, Benjamin EJ, Larson MG, Levy D, Vita JA, Hamburg NM, Vasan RS and Mitchell GF. Components of hemodynamic load and cardiovascular events: the Framingham Heart Study. Circulation. 2015;131:354–61; discussion 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kossmann S, Lagrange J, Jackel S, Jurk K, Ehlken M, Schonfelder T, Weihert Y, Knorr M, Brandt M, Xia N, Li H, Daiber A, Oelze M, Reinhardt C, Lackner K, Gruber A, Monia B, Karbach SH, Walter U, Ruggeri ZM, Renne T, Ruf W, Munzel T and Wenzel P. Platelet-localized FXI promotes a vascular coagulation-inflammatory circuit in arterial hypertension. Sci Transl Med. 2017;9. [DOI] [PubMed] [Google Scholar]
- 96.Head GA. The sympathetic nervous system in hypertension: assessment by blood pressure variability and ganglionic blockade. J Hypertens. 2003;21:1619–21. [DOI] [PubMed] [Google Scholar]
- 97.Diedrich A, Jordan J, Tank J, Shannon JR, Robertson R, Luft FC, Robertson D and Biaggioni I. The sympathetic nervous system in hypertension: assessment by blood pressure variability and ganglionic blockade. J Hypertens. 2003;21:1677–86. [DOI] [PubMed] [Google Scholar]
- 98.Gamboa A, Figueroa R, Paranjape SY, Farley G, Diedrich A and Biaggioni I. Autonomic Blockade Reverses Endothelial Dysfunction in Obesity-Associated Hypertension. Hypertension. 2016;68:1004–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.DeLalio LJ, Sved AF and Stocker SD. Sympathetic Nervous System Contributions to Hypertension: Updates and Therapeutic Relevance. Can J Cardiol. 2020;36:712–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kinsman BJ, Simmonds SS, Browning KN and Stocker SD. Organum Vasculosum of the Lamina Terminalis Detects NaCl to Elevate Sympathetic Nerve Activity and Blood Pressure. Hypertension. 2017;69:163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xiao L, Itani HA, do Carmo LS, Carver LS, Breyer RM and Harrison DG. Central EP3 (E Prostanoid 3) Receptors Mediate Salt-Sensitive Hypertension and Immune Activation. Hypertension. 2019;74:1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cao W, Shi M, Wu L, Li J, Yang Z, Liu Y, Wilcox CS and Hou FF. Adipocytes initiate an adipose-cerebral-peripheral sympathetic reflex to induce insulin resistance during high-fat feeding. Clin Sci (Lond). 2019;133:1883–1899. [DOI] [PubMed] [Google Scholar]
- 103.Braith RW, Mills RM Jr., Wilcox CS, Convertino VA, Davis GL, Limacher MC and Wood CE. Fluid homeostasis after heart transplantation: the role of cardiac denervation. J Heart Lung Transplant. 1996;15:872–80. [PubMed] [Google Scholar]
- 104.Braith RW, Mills RM Jr., Wilcox CS, Davis GL and Wood CE. Breakdown of blood pressure and body fluid homeostasis in heart transplant recipients. J Am Coll Cardiol. 1996;27:375–83. [DOI] [PubMed] [Google Scholar]
- 105.Braith RW, Mills RM, Wilcox CS, Mitchell MJ, Hill JA and Wood CE. High dose angiotensin-converting enzyme inhibition prevents fluid volume expansion in heart transplant recipients. J Am Coll Cardiol. 2000;36:487–92. [DOI] [PubMed] [Google Scholar]
- 106.McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, Chambon P, Hill MA, Dorrance AM, Mendelsohn ME and Jaffe IZ. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med. 2012;18:1429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jia G, Habibi J, Aroor AR, Martinez-Lemus LA, DeMarco VG, Ramirez-Perez FI, Sun Z, Hayden MR, Meininger GA, Mueller KB, Jaffe IZ and Sowers JR. Endothelial Mineralocorticoid Receptor Mediates Diet-Induced Aortic Stiffness in Females. Circ Res. 2016;118:935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bene NC, Alcaide P, Wortis HH and Jaffe IZ. Mineralocorticoid receptors in immune cells: emerging role in cardiovascular disease. Steroids. 2014;91:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brown JM, Siddiqui M, Calhoun DA, Carey RM, Hopkins PN, Williams GH and Vaidya A. The Unrecognized Prevalence of Primary Aldosteronism: A Cross-sectional Study. Ann Intern Med. 2020;173:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schnackenberg CG, Welch WJ and Wilcox CS. Normalization of blood pressure and renal vascular resistance in SHR with a membrane-permeable superoxide dismutase mimetic: role of nitric oxide. Hypertension. 1998;32:59–64. [DOI] [PubMed] [Google Scholar]
- 111.Schnackenberg CG and Wilcox CS. Two-week administration of tempol attenuates both hypertension and renal excretion of 8-Iso prostaglandin f2alpha. Hypertension. 1999;33:424–8. [DOI] [PubMed] [Google Scholar]
- 112.Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA and Harrison DG. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation. 1997;95:588–93. [DOI] [PubMed] [Google Scholar]
- 113.Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK and Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, Tarpey M, Fukai T and Harrison DG. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103:1282–8. [DOI] [PubMed] [Google Scholar]
- 115.Somers MJ, Mavromatis K, Galis ZS and Harrison DG. Vascular superoxide production and vasomotor function in hypertension induced by deoxycorticosterone acetate-salt. Circulation. 2000;101:1722–8. [DOI] [PubMed] [Google Scholar]
- 116.Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KR, Xiao L, Chen W, Mernaugh RL, Cai H, Bernstein KE, Goronzy JJ, Weyand CM, Curci JA, Barbaro NR, Moreno H, Davies SS, Roberts LJ 2nd, Madhur MS and Harrison DG. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. 2016;126:50–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Araujo M and Wilcox CS. Oxidative stress in hypertension: role of the kidney. Antioxid Redox Signal. 2014;20:74–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rajagopalan S, Meng XP, Ramasamy S, Harrison DG and Galis ZS. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest. 1996;98:2572–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lob HE, Schultz D, Marvar PJ, Davisson RL and Harrison DG. Role of the NADPH oxidases in the subfornical organ in angiotensin II-induced hypertension. Hypertension. 2013;61:382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang HW, Huang BS, White RA, Chen A, Ahmad M and Leenen FH. Mineralocorticoid and angiotensin II type 1 receptors in the subfornical organ mediate angiotensin II - induced hypothalamic reactive oxygen species and hypertension. Neuroscience. 2016;329:112–21. [DOI] [PubMed] [Google Scholar]
- 121.Carvalho-Galvao A, Guimaraes DD, De Brito Alves JL and Braga VA. Central Inhibition of Tumor Necrosis Factor Alpha Reduces Hypertension by Attenuating Oxidative Stress in the Rostral Ventrolateral Medulla in Renovascular Hypertensive Rats. Front Physiol. 2019;10:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Griendling KK, Minieri CA, Ollerenshaw JD and Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–8. [DOI] [PubMed] [Google Scholar]
- 123.Kitiyakara C, Chabrashvili T, Chen Y, Blau J, Karber A, Aslam S, Welch WJ and Wilcox CS. Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J Am Soc Nephrol. 2003;14:2775–82. [DOI] [PubMed] [Google Scholar]
- 124.Welch WJ, Chabrashvili T, Solis G, Chen Y, Gill PS, Aslam S, Wang X, Ji H, Sandberg K, Jose P and Wilcox CS. Role of extracellular superoxide dismutase in the mouse angiotensin slow pressor response. Hypertension. 2006;48:934–41. [DOI] [PubMed] [Google Scholar]
- 125.Ye S, Zhong H, Yanamadala S and Campese VM. Oxidative stress mediates the stimulation of sympathetic nerve activity in the phenol renal injury model of hypertension. Hypertension. 2006;48:309–15. [DOI] [PubMed] [Google Scholar]
- 126.Jayachandran I, Sundararajan S, Venkatesan S, Paadukaana S, Balasubramanyam M, Mohan V and Manickam N. Asymmetric dimethylarginine (ADMA) accelerates renal cell fibrosis under high glucose condition through NOX4/ROS/ERK signaling pathway. Sci Rep. 2020;10:16005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dikalova AE, Itani HA, Nazarewicz RR, McMaster WG, Flynn CR, Uzhachenko R, Fessel JP, Gamboa JL, Harrison DG and Dikalov SI. Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circ Res. 2017;121:564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Irazabal MV and Torres VE. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L, Dale BL, Iwakura Y, Hoover RS, McDonough AA and Madhur MS. Interleukin-17A Regulates Renal Sodium Transporters and Renal Injury in Angiotensin II-Induced Hypertension. Hypertension. 2016;68:167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ and Harrison DG. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ and Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97:696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, Madhur MS, Chen W and Harrison DG. Inflammation and Mechanical Stretch Promote Aortic Stiffening in Hypertension Through Activation of p38 Mitogen-Activated Protein Kinase. Circ Res. 2014;114:616–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kassan M, Galan M, Partyka M, Trebak M and Matrougui K. Interleukin-10 released by CD4(+)CD25(+) natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler Thromb Vasc Biol. 2011;31:2534–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Matrougui K, Abd Elmageed Z, Kassan M, Choi S, Nair D, Gonzalez-Villalobos RA, Chentoufi AA, Kadowitz P, Belmadani S and Partyka M. Natural regulatory T cells control coronary arteriolar endothelial dysfunction in hypertensive mice. Am J Pathol. 2011;178:434–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J 2nd and Harrison DG. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. 2014;124:4642–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ and Harrison DG. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res. 2010;107:263–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lob HE, Marvar PJ, Guzik TJ, Sharma S, McCann LA, Weyand C, Gordon FJ and Harrison DG. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension. 2010;55:277–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xiao L, do Carmo LS, Foss JD, Chen W and Harrison DG. Sympathetic Enhancement of Memory T-Cell Homing and Hypertension Sensitization. Circ Res. 2020;126:708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Guzik TJ, Skiba DS, Touyz RM and Harrison DG. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc Res. 2017;113:1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mikolajczyk TP, Nosalski R, Szczepaniak P, Budzyn K, Osmenda G, Skiba D, Sagan A, Wu J, Vinh A, Marvar PJ, Guzik B, Podolec J, Drummond G, Lob HE, Harrison DG and Guzik TJ. Role of chemokine RANTES in the regulation of perivascular inflammation, T-cell accumulation, and vascular dysfunction in hypertension. FASEB J. 2016;30:1987–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–9. [DOI] [PubMed] [Google Scholar]
- 142.Harwani SC, Chapleau MW, Legge KL, Ballas ZK and Abboud FM. Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ Res. 2012;111:1190–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, Ivanov S, Duan Q, Bala S, Condon T, van Rooijen N, Grainger JR, Belkaid Y, Ma’ayan A, Riches DW, Yokoyama WM, Ginhoux F, Henson PM and Randolph GJ. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Randolph GJ, Beaulieu S, Lebecque S, Steinman RM and Muller WA. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science. 1998;282:480–3. [DOI] [PubMed] [Google Scholar]
- 145.Loperena R, Van Beusecum JP, Itani HA, Engel N, Laroumanie F, Xiao L, Elijovich F, Laffer CL, Gnecco JS, Noonan J, Maffia P, Jasiewicz-Honkisz B, Czesnikiewicz-Guzik M, Mikolajczyk T, Sliwa T, Dikalov S, Weyand CM, Guzik TJ and Harrison DG. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: roles of STAT3, interleukin 6 and hydrogen peroxide. Cardiovasc Res. 2018;114:1547–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Barbaro NR, Van Beusecum J, Xiao L, do Carmo L, Pitzer A, Loperena R, Foss JD, Elijovich F, Laffer CL, Montaniel KR, Galindo CL, Chen W, Ao M, Mernaugh RL, Alsouqi A, Ikizler TA, Fogo AB, Moreno H, Zhao S, Davies SS, Harrison DG and Kirabo A. Sodium activates human monocytes via the NADPH oxidase and isolevuglandin formation. Cardiovasc Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lifton RP, Gharavi AG and Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–56. [DOI] [PubMed] [Google Scholar]
- 148.Cabrera CP, Ng FL, Nicholls HL, Gupta A, Barnes MR, Munroe PB and Caulfield MJ. Over 1000 genetic loci influencing blood pressure with multiple systems and tissues implicated. Hum Mol Genet. 2019;28:R151–R161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vauraa F, Kaukoa a, Suvilaa K, Havulinnac A, Marsd N, Salomaac V, Chenge S and Niiranena T. Polygenic Risk Scores Predict Hypertension Onset and Cardiovascular Risk. Hypertension. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.He FJ and MacGregor GA. A comprehensive review on salt and health and current experience of worldwide salt reduction programmes. J Hum Hypertens. 2009;23:363–84. [DOI] [PubMed] [Google Scholar]
- 151.He FJ, Li J and Macgregor GA. Effect of longer term modest salt reduction on blood pressure: Cochrane systematic review and meta-analysis of randomised trials. BMJ. 2013;346:f1325. [DOI] [PubMed] [Google Scholar]
- 152.Officers A, Coordinators for the ACRGTA and Lipid-Lowering Treatment to Prevent Heart Attack T. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA. 2002;288:2981–97. [DOI] [PubMed] [Google Scholar]
- 153.Elijovich F, Weinberger MH, Anderson CA, Appel LJ, Bursztyn M, Cook NR, Dart RA, Newton-Cheh CH, Sacks FM, Laffer CL, American Heart Association P, Public Education Committee of the Council on H, Council on Functional G, Translational B and Stroke C. Salt Sensitivity of Blood Pressure: A Scientific Statement From the American Heart Association. Hypertension. 2016;68:e7–e46. [DOI] [PubMed] [Google Scholar]
- 154.Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension. 1996;27:481–90. [DOI] [PubMed] [Google Scholar]
- 155.Choi HY, Park HC and Ha SK. Salt Sensitivity and Hypertension: A Paradigm Shift from Kidney Malfunction to Vascular Endothelial Dysfunction. Electrolyte Blood Press. 2015;13:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Morris RC Jr., Schmidlin O, Sebastian A, Tanaka M and Kurtz TW. Vasodysfunction That Involves Renal Vasodysfunction, Not Abnormally Increased Renal Retention of Sodium, Accounts for the Initiation of Salt-Induced Hypertension. Circulation. 2016;133:881–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Laffer CL, Scott RC 3rd, Titze JM, Luft FC and Elijovich F Hemodynamics and Salt-and-Water Balance Link Sodium Storage and Vascular Dysfunction in Salt-Sensitive Subjects. Hypertension. 2016;68:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Schmidlin O, Forman A, Leone A, Sebastian A and Morris RC Jr. Salt sensitivity in blacks: evidence that the initial pressor effect of NaCl involves inhibition of vasodilatation by asymmetrical dimethylarginine. Hypertension. 2011;58:380–5. [DOI] [PubMed] [Google Scholar]
- 159.Campese VM, Romoff MS, Levitan D, Saglikes Y, Friedler RM and Massry SG. Abnormal relationship between sodium intake and sympathetic nervous system activity in salt-sensitive patients with essential hypertension. Kidney Int. 1982;21:371–8. [DOI] [PubMed] [Google Scholar]
- 160.Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, Park JK, Beck FX, Muller DN, Derer W, Goss J, Ziomber A, Dietsch P, Wagner H, van Rooijen N, Kurtz A, Hilgers KF, Alitalo K, Eckardt KU, Luft FC, Kerjaschki D and Titze J. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15:545–52. [DOI] [PubMed] [Google Scholar]
- 161.Kopp C, Linz P, Wachsmuth L, Dahlmann A, Horbach T, Schofl C, Renz W, Santoro D, Niendorf T, Muller DN, Neininger M, Cavallaro A, Eckardt KU, Schmieder RE, Luft FC, Uder M and Titze J. (23)Na magnetic resonance imaging of tissue sodium. Hypertension. 2012;59:167–72. [DOI] [PubMed] [Google Scholar]
- 162.Kopp C, Linz P, Dahlmann A, Hammon M, Jantsch J, Muller DN, Schmieder RE, Cavallaro A, Eckardt KU, Uder M, Luft FC and Titze J. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension. 2013;61:635–40. [DOI] [PubMed] [Google Scholar]
- 163.Jantsch J, Schatz V, Friedrich D, Schroder A, Kopp C, Siegert I, Maronna A, Wendelborn D, Linz P, Binger KJ, Gebhardt M, Heinig M, Neubert P, Fischer F, Teufel S, David JP, Neufert C, Cavallaro A, Rakova N, Kuper C, Beck FX, Neuhofer W, Muller DN, Schuler G, Uder M, Bogdan C, Luft FC and Titze J. Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage-driven host defense. Cell Metab. 2015;21:493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kitada K, Daub S, Zhang Y, Klein JD, Nakano D, Pedchenko T, Lantier L, LaRocque LM, Marton A, Neubert P, Schroder A, Rakova N, Jantsch J, Dikalova AE, Dikalov SI, Harrison DG, Muller DN, Nishiyama A, Rauh M, Harris RC, Luft FC, Wassermann DH, Sands JM and Titze J. High salt intake reprioritizes osmolyte and energy metabolism for body fluid conservation. J Clin Invest. 2017;127:1944–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A and Kuchroo VK. Induction of pathogenic T17 cells by inducible salt-sensing kinase SGK1. Nature. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN and Hafler DA. Sodium chloride drives autoimmune disease by the induction of pathogenic T17 cells. Nature. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, Mernaugh RL, Itani HA, Loperena R, Chen W, Dikalov S, Titze JM, Knollmann BC, Harrison DG and Kirabo A. Dendritic Cell Amiloride-Sensitive Channels Mediate Sodium-Induced Inflammation and Hypertension. Cell Rep. 2017;21:1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Norlander AE, Saleh MA, Pandey AK, Itani HA, Wu J, Xiao L, Kang J, Dale BL, Goleva SB, Laroumanie F, Du L, Harrison DG and Madhur MS. A salt-sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end-organ damage. JCI Insight. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Van Beusecum JP, Barbaro NR, McDowell Z, Aden LA, Xiao L, Pandey AK, Itani HA, Himmel LE, Harrison DG and Kirabo A. High Salt Activates CD11c(+) Antigen-Presenting Cells via SGK (Serum Glucocorticoid Kinase) 1 to Promote Renal Inflammation and Salt-Sensitive Hypertension. Hypertension. 2019;74:555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, Haase S, Mahler A, Balogh A, Marko L, Vvedenskaya O, Kleiner FH, Tsvetkov D, Klug L, Costea PI, Sunagawa S, Maier L, Rakova N, Schatz V, Neubert P, Fratzer C, Krannich A, Gollasch M, Grohme DA, Corte-Real BF, Gerlach RG, Basic M, Typas A, Wu C, Titze JM, Jantsch J, Boschmann M, Dechend R, Kleinewietfeld M, Kempa S, Bork P, Linker RA, Alm EJ and Muller DN. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Marques FZ, Mackay CR and Kaye DM. Beyond gut feelings: how the gut microbiota regulates blood pressure. Nat Rev Cardiol. 2018;15:20–32. [DOI] [PubMed] [Google Scholar]
- 172.Thornton AM and Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Raizada MK, Joe B, Bryan NS, Chang EB, Dewhirst FE, Borisy GG, Galis ZS, Henderson W, Jose PA, Ketchum CJ, Lampe JW, Pepine CJ, Pluznick JL, Raj D, Seals DR, Gioscia-Ryan RA, Tang WHW and Oh YS. Report of the National Heart, Lung, and Blood Institute Working Group on the Role of Microbiota in Blood Pressure Regulation: Current Status and Future Directions. Hypertension. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tang WHW, Li DY and Hazen SL. Dietary metabolism, the gut microbiome, and heart failure. Nat Rev Cardiol. 2019;16:137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Forsburg SL and Guarente L. Communication between mitochondria and the nucleus in regulation of cytochrome genes in the yeast Saccharomyces cerevisiae. Annu Rev Cell Biol. 1989;5:153–80. [DOI] [PubMed] [Google Scholar]
- 176.Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Li J, Zhao F, Wang Y, Chen J, Tao J, Tian G, Wu S, Liu W, Cui Q, Geng B, Zhang W, Weldon R, Auguste K, Yang L, Liu X, Chen L, Yang X, Zhu B and Cai J. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ferguson JF, Aden LA, Barbaro NR, Van Beusecum JP, Xiao L, Simmons AJ, Warden C, Pasic L, Himmel LE, Washington MK, Revetta FL, Zhao S, Kumaresan S, Scholz MB, Tang Z, Chen G, Reilly MP and Kirabo A. High dietary salt-induced dendritic cell activation underlies microbial dysbiosis-associated hypertension. JCI Insight. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mell B, Jala VR, Mathew AV, Byun J, Waghulde H, Zhang Y, Haribabu B, Vijay-Kumar M, Pennathur S and Joe B. Evidence for a link between gut microbiota and hypertension in the Dahl rat. Physiol Genomics. 2015;47:187–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM, Zadeh M, Gong M, Qi Y, Zubcevic J, Sahay B, Pepine CJ, Raizada MK and Mohamadzadeh M. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65:1331–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Marques FZ, Nelson E, Chu PY, Horlock D, Fiedler A, Ziemann M, Tan JK, Kuruppu S, Rajapakse NW, El-Osta A, Mackay CR and Kaye DM. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation. 2017;135:964–977. [DOI] [PubMed] [Google Scholar]
- 182.Santisteban MM, Qi Y, Zubcevic J, Kim S, Yang T, Shenoy V, Cole-Jeffrey CT, Lobaton GO, Stewart DC, Rubiano A, Simmons CS, Garcia-Pereira F, Johnson RD, Pepine CJ and Raizada MK. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ Res. 2017;120:312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, Obarzanek E, Conlin PR, Miller ER 3rd, Simons-Morton DG, Karanja N, Lin PH and Group DA-SCR. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N Engl J Med. 2001;344:3–10. [DOI] [PubMed] [Google Scholar]
- 184.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ and Turnbaugh PJ. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, Xavier RJ, Teixeira MM and Mackay CR. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Koh A, De Vadder F, Kovatcheva-Datchary P and Backhed F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell. 2016;165:1332–1345. [DOI] [PubMed] [Google Scholar]
- 187.Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, Brunet I, Wan LX, Rey F, Wang T, Firestein SJ, Yanagisawa M, Gordon JI, Eichmann A, Peti-Peterdi J and Caplan MJ. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci U S A. 2013;110:4410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]