

Abstract
Abstract
The non-structural protein (nsp)-3 of SARS-CoV2 coronavirus is sought to be an essential target protein which is also named as papain-like protease (PLpro). This protease cleaves the viral polyprotein, but importantly in human host it also removes ubiquitin-like interferon-stimulated gene 15 protein (ISG15) from interferon responsive factor 3 (IRF3) protein which ultimately downregulates the production of type I interferon leading to weakening of immune response. GRL0617 is the most potent known inhibitor for PLpro that was initially developed for SARS outbreak of 2003. The PLpro of SARS-CoV and CoV2 share 83% sequence identity but interestingly have several identical conserved amino acids that suggests GRL0617 to be an effective inhibitor for PLpro of SARS-CoV2. GRL0617 is a naphthalene-based molecule and interacts with Tyr268 of SARS-CoV2-PLpro (and Tyr269 of SARS-CoV-PLpro). To identify PLpro inhibitors, we prepared a library of secondary metabolites from fungi with aromatic nature and docked them with PLpro of SARS-CoV and SARS-CoV2. We found six hits which interacts with Tyr268 of SARS-CoV2-PLpro (and Tyr269 of SARS-CoV-PLpro). More surprisingly the top hit, Fonsecin, has naphthalene moiety in its structure, which recruits Tyr268 of SARS-CoV2-PLpro (and Tyr269 of SARS-CoV-PLpro) and has binding energy at par with control (GRL0617). Molecular dynamics (MD) simulation showed Fonsecin to interact with Tyr268 of SARS-CoV2-PLpro more efficiently than control (GRL0617) and interacting with a greater number of amino acids in the binding cleft of PLpro.
Graphic abstract

Keywords: Papain-like protease (PLpro), SARS-CoV2 novel coronavirus, Fungal metabolites, Molecular dynamics simulation, GRL0617
Introduction
Since the inception of the pandemic rose by novel Severe Acute Respiratory Syndrome Virus-Coronavirus 2 (SARS-CoV2), people of scientific and clinical society are aggregately attempting to discover an immunization or to build-up a technique to battle this life threatening virus, SARS-CoV2 [1–3]. Global impact of SARS-CoV2 has been severe, and it is hard to come over with the situation of pandemic even after a year of its onset. There is no focused drug to stop the spread and replication of this viral particle in human host [4, 5]. According to the details of the World health organization (WHO), worldwide ~25 million people have been affected by the infection of SARS-CoV2 of which ~0.85 million deaths have occurred (https://covid19.who.int/). According to the description of the International Committee on Taxonomy of Viruses (ICTV), this pandemic causing infection is hereditarily alike the SARS-CoV infection causing the flare-up of 2003; thus, the identified virus was assigned as SARS-CoV2 on eleventh of February 2020 [6]. Albeit evolutionary related, the two infection strains are very unique in their strength of causing disease, where the latest identified novel CoV2 is known to be quickly contracting and shows lower mortality, notwithstanding, its high infectivity which has made this viral infection spread all around the world in an exceptionally brief timeframe. It causes a respiratory or gastrointestinal disease with symptoms like fever, pneumonia, short-windedness in major populaces [7]. It is unavoidable to deploy a vaccine regimen or a helpful drug that can function as a counteractant or that can debilitate the virus causing a decrease in its spread. To the other side of discovering the vaccine, there exists community of researchers on alternative strategy that are focusing to develop an anti-viral drug by targeting either of several structural and non-structural proteins with an agenda for developing inhibitors (i) to impede viral passage into the host cells, and (ii) to forestall viral replication [3]. For these purposes, an aggregate of twelve proteins that can serve as a target is sought, belonging to two major families of: (i) Structural Proteins and (ii) Non-structural protein. Structural proteins, namely spike protein (S-protein), envelop small membrane protein (E-protein), membrane protein (M-protein), nucleocapsid protein (N-protein), are significant for accomplishing the virulent viral physical conformation. Bending or misfolding of these proteins is focused on essentially lessening the harmfulness and infectivity of the virus. Non-structural proteins (nsp), namely main protease (Mpro) (nsp-5), papain-like protease (PLpro) (nsp-3), nsp-10, nsp-11 nsp-13, nsp-14, nsp-15 and nsp-16, are needed for replication and bundling of the virus particle into the capsid protein coat [5, 6, 8]. Thus, these proteins are viewed as targets on the grounds that hindering their functionality can stop viral load increase in host cell [6, 8, 9].
PLpro, encoded by nsp3 gene is one of the two known CoV proteases and it functions to cleave nsp1, nsp2, and nsp3 from the viral polyprotein which is a prerequisite for viral genome transcription and replication. Apart from its role as a viral protease, PLpro from SARS-CoV antagonizes cellular ubiquitination and ISGylation. Ubiquitination is a process where ubiquitin (Ub) chains are added to lysine residues of a protein and it is a part of post-translational protein modification which regulates its activity, notably via its targeting to proteasomal degradation [10, 11]. Like ubiquitination, ISGylation is a process where interferon-stimulated gene 15 (ISG15) is conjugated to a target protein which is induced by IFN-I and regulates their functions. Both ubiquitination and ISGylation play important roles in the regulation of innate immune responses to viral infection, and it may therefore not be surprising to observe that multiple viruses have evolved different strategies to antagonize these pathways (Fig. 1). The function of PLpro of SARS-CoV and SARS-CoV2 is more or less identical with few differences in deISGylating and deubiquitylating activities. SARS-CoV2-PLpro is proven to be more efficient at cleaving ISG15 than Ub [12]. Detailed investigation outlined a shift in substrate specificity between the PLpro’s from SARS-CoV and SARS-CoV2 where the later prefers to cleave ISG15. Amino acid residue, Leu76 of SARS-CoV-PLpro facilitates a contact with Ile44 of Ub via hydrophobic interaction, the corresponding residue on SARS-CoV2-PLpro is Thr75 which has lower affinity for Ub. Further, deISGylating by SARS-CoV2-PLpro is not well understood but it is hypothesized that Val66 faces the hydrophobic surface on the N-terminal ubiquitin fold domain of ISG15 is observed to have improved activity [12].
Fig. 1.

Illustration depicting function of IRF3 in production of INF1 and its inhibition by PLpro along with showing other functions involved in viral replication
Like a double-edged sword, PLpro promoting viral replication and inhibiting innate immune responses (Fig. 1) had attracted researchers by being qualified as an attractive target for drug development. Previously described SARS-CoV-PLpro inhibitor, GRL0617 can effectively inhibit PLpro of SARS-CoV2 [13]. In vitro studies on lung cancer cell line stimulated with IFN-I has proven that GRL0617 can effectively inhibit the deubiquitination and deISGylation activities of SARS-CoV2-PLpro and restores the IFN-I response. Moreover, it have been also proved that 100 μM dose of GRL0617 can strongly inhibit SARS-CoV2 replication [12]. In silico studies have revealed that naphthalene-based GRL0617 interacts with Tyr268 of SARS-CoV2-PLpro and Tyr269 of SARS-CoV-PLpro. Moreover, GRL0617 is ineffective as an inhibitor against PLpro of Middle East respiratory syndrome (MERS)-CoV. This is because of the presence of amino acid residue, Thr instead of Tyr at this conserved position (269 in case of SARS-CoV and 268 in case of SARS-CoV2) [12].
In the present study, a 100 naturally occurring fungal secondary metabolites with aromatic moiety were collected with an aim to identify the analogue of naphthalene-based GRL0617. All the fungal metabolites were screened based on their ability to interact with (i) Tyr268 of SARS-CoV2-PLpro and Tyr269 of SARS-CoV-PLpro (ii) in silico binding energy, (iii) formation of hydrogen bonds and (iv) hydrophobic interactions with PLpro through molecular docking. We found six hits which can interact with Tyr268 of SARS-CoV2-PLpro (and Tyr269 of SARS-CoV-PLpro). More surprisingly the top hit, Fonsecin, has naphthalene moiety in its structure and can recruit Tyr268 of SARS-CoV2-PLpro (and Tyr269 of SARS-CoV-PLpro) and had binding energy at par with control (GRL0617). Molecular dynamics (MD) simulation for 100 ns showed Fonsecin to interact with Tyr268 of SARS-CoV2-PLpro more efficiently than control (GRL0617) involving interaction with greater number of amino acids in the binding cleft of PLpro. Moreover, ADMET (Absorption–Distribution–Metabolism–Excretion–Toxicity) properties of the Fonsecin along with all the screened hits were performed and were compared with GRL0617.
Materials and methods
Retrieving and comparing PLpro’s from SARS-CoV, SARS-CoV2 and MERS-CoV
Based on the quality and sequences PLpro’s from SARS-CoV, SARS-CoV2 and MERS-CoV were retrieved from PDB with respective id’s as 3E9S, 7CMD, and 4RNA. This was performed to be sure that the proteins used further for docking were having same sequence, structure, and no mutation. The agenda here is to ensure the presence of Tyr at position 269 for 3E9S and 268 for 7CMD as per the literature and this conserved amino acid to be Thr for 4RNA.
Preparation of receptor and ligands
PLpro of SARS-CoV and SARS-CoV2 with PDB id’s 3E9S and 7CMD, respectively, were co-crystalized with GRL0617. The coordinates of GRL0617 binding on 3E9S and 7CMD were determined and prior to docking studies all the co-crystallized residues were also removed. Using DockPrep tool in UCSF Chimera version 1.14, the protein structure was prepared by assigning the hydrogen atoms, charges and performing energy minimization [14]. The charges were assigned as per the AM1-BCC method which quickly and efficiently generates high-quality atomic charges for protein and the charges were computed using ANTECHAMBER algorithm [15]. The energy minimization was performed using 1000 steepest descent steps with 0.02 Å step size and an update interval of 10. All the steps mentioned above were performed using UCSF Chimera version 1.14 [16].
All the ligands used for the in silico interaction assays are fungal secondary metabolites and were retrieved from PubChem. Before performing the molecular docking, the ligands were optimized by addition of hydrogen and energy minimization was performed using Gasteiger algorithm [17] in structure editing wizard of UCSF Chimera version 1.14, which works on the chemoinformatic principle of electronegativity equilibration and the files were saved in mol2 format.
Molecular docking
Receptor-ligand docking analysis was performed using AutoDock Vina [18], and the program was executed as an add-on in UCSF Chimera version 1.14. Cavity of the co-crystallized ligand (GRL0617) in the crystal structure of PLpro was identified and its coordinates were used to dock 100 fungal metabolites identified from multiple literature and libraries. Known inhibitor for PLpro, GRL0617 was taken into consideration as positive reference control.
In the AutoDock Vina algorithm, the following parameters were set as: (i) number of binding modes-10; (ii) exhaustiveness of search-8 and (iii) maximum energy difference-3 kcal/mol. Out of all the possible poses suggested by AutoDock Vina, the pose showing maximum hydrogen bonds and binding energy (kcal/mol) as represented in the ViewDock window were chosen. They were further analysed in BIOVIA Discovery Studio (DS) visualizer for hydrogen bond formation by the functional groups of ligands with amino acids. DS also suggested other supporting hydrophobic interactions made by the GRL0617 and fungal metabolites. The metabolite compound making interaction with Tyr268 of SARS-CoV2-PLpro (and Tyr269 of SARS-CoV-PLpro) with highest binding affinity, having maximum number of interactions with amino acids, was chosen for further MD simulation analysis. The six best metabolites were screened based on their ability to interact with PLpro’s of SARS-CoV and SARS-CoV2 as the GRL0617 is known to interact with both the PLpro’s in identical fashion. The best metabolite that can interact with both the PLpro’s efficiently like GRL0617 was chosen further validation using MD simulations.
MM/GBSA calculations
The binding free energy change calculations were performed using molecular mechanics generalized Born surface area (MM/GBSA) calculation [19–22]. The docked complexes were minimized by using local optimization feature in Prime wizard of Maestro (Schrödinger Release 2018–4). The OPLS-2005 force field was employed to determine the binding energy for a set of receptor and ligand. The following equation was used to calculate the binding free energy:
| 1 |
where, ΔEMM is the variance between the minimized energy of the protein–ligand complexes, while ΔGSolv is the variation between the GBSA solvation energy of the protein–ligand complexes and the sum of the solvation energies for the protein and ligand. In ΔGSA contains some of the surface area energies in the protein and ligand and the difference in the surface area energies for the complexes. The minimization of the docked complexes was done using a local optimization feature of prime.
Molecular dynamic (MD) simulation
The simulation of the SARS-CoV2-PLpro in the presence of GRL0617 and Fonsecin was performed in two sets of experiments using Desmond (Schrödinger Release 2018–4). Fonsecin has best docking score when compared to other fungal metabolites. MD simulation was carried out for the SARS-CoV2-PLpro-Fonsecin and SARS-CoV2-PLpro-GRL0617 complexes, where the later was taken as control. These complexes were prepared using a protein preparation wizard to allow complex relaxation. The addition of hydrogens, water removal, bond orders assignment, fill in missing side chains and loops with optimization of hydrogen bond assignment (sampling of water orientations and use of pH 7.0) was done. The ligands were prepared using LigPrep of Maestro which adds hydrogen atoms, generates tautomer, ionization states, ring conformations, and produces minimized 3D structures. Whereas the protein was prepared by performing restrained minimization using OPLS-2005 force field. The system for simulation was built keeping solvent model as TIP3P, the boundaries were defined with the box shape of orthorhombic with the dimension of 10 Å × 10 Å × 10 Å. This was then followed by neutralization by Cl− or Na+ counter ions. Steepest descent energy minimization was performed, and the simulation was proceeded for 100 ns with NPT (constant Number of particles, Pressure, and Temperature) with 300 K and 1.01 bar, constant volume, Smooth Particle Mesh Ewald (PME) method. On completion of simulation, the trajectories were analysed in simulation interaction diagram wizard which computes trajectories for root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), Ligand–protein contact profiles, for Ligand and Protein modifications.
ADMET analysis
The pkCSM—pharmacokinetics server [23] was used to predict the ADMET properties of the GRL0617 and top six screened hits. It predicted both physiochemical and pharmacological properties. SMILES (Simplified Molecule Input Line Entry Specification) of the compounds were retrieved from PubChem and uploaded to pkCSM—pharmacokinetics server. It computed in vivo Absorption parameters like; Water solubility in buffer system (SK atomic types, mg/L), in vivo Caco2 cell permeability (Human colorectal carcinoma), Human intestinal absorption (HIA, %), in vivo P-glycoprotein inhibition and in vivo skin permeability (logKp, cm/hour). Metabolic parameters were determined using in vivo Cytochrome P450 2C19 inhibition, in vivo Cytochrome P450 2C9 inhibition, in vivo Cytochrome P450 2D6 inhibition, in vivo Cytochrome P450 2D6 substrate, in vivo Cytochrome P450 3A4 inhibition and in vivo Cytochrome P450 3A4 substrate. Distribution property included tests like, Blood–Brain Barrier (BBB) penetration, Lipinski’s Rule (Rule of Five), Central Nervous System (CNS) permeability. To access the toxicity of compounds under study, a range of important endpoints including, Acute algae toxicity, Ames test, 2 years carcinogenicity bioassay in mouse, 2 years carcinogenicity bioassay in rat, in vivo Ames test result in TA100 strain (Metabolic activation by rat liver homogenate) were computed. Excretion again is a very important parameter and as many drugs often withdrawn at clinical trial stages due to their poorer renal clearance. In this study, we included Total Renal clearance and Renal OCT2 Substrate to identify Excretion efficacy of the proposed metabolite.
Results
PLpro’s from SARS-CoV, SARS-CoV2 and MERS-CoV and understanding interaction of GRL0617
PLpro of SARS-CoV (3E9S) and SARS-CoV2 (7CMD) showed 83% similarity. While PLpro of SARS-CoV (3E9S) and MERS-CoV (4RNA) share only 30% similarity which was deduced by performing multiple sequence alignment using ClustalW (Fig. 2). The alignment showed that Tyr to be present at position 269 for 3E9S and 268 for 7CMD as per the literature and this conserved amino acid to be Thr for 4RNA. When the proteins 3E9S and 7CMD were superimposed, there were two inferences (i) both the proteins were superimposable and structurally identical (ii) the poses of native co-crystallized ligand GRL0617 of both the proteins were identical and were superimposed too along with the protein (Fig. 2).
Fig. 2.

a Superimposition of PLpro of SARS-CoV and SARS-CoV2 b Multiple sequence alignment of sequences of PLpro’s of MERS-CoV, SARS-CoV and SARS-CoV2
For PLpro of SARS-CoV, specifically, the amide group of the inhibitor forms hydrogen bonds with the side chain of Asp165 and the backbone nitrogen of Gln270. Asp165 is highly conserved among the ubiquitin-specific protease (USP) family of deubiquitinating enzymes. Most contacts between SARS-CoV-PLpro and inhibitor GRL0617 are hydrophobic in nature. The 1-naphthyl group is partly solvent-exposed but forms hydrophobic interactions with the aromatic rings of Tyr265 (Tyr264 for CoV2) and Tyr269 (Tyr268 for CoV2) and with the side chains of Pro248 (Pro247 for CoV2) and Pro249 (Pro248 for CoV2). These residues line the pocket and accommodate the leucine at the P4 position of PLpro substrates. The (R)-methyl group, attached to the stereocenter of the inhibitor, points directly into the interior of the protein between Tyr265 (Tyr264 for CoV2) and Thr302 (Thr301 for CoV2), where it is accommodated by a cavity that is mostly polar in nature. The other ring substituent, -NH2 at the R3 position of GRL0617, extends from the opening of the cleft where it is surrounded by a series of polar groups, including the side chain oxygens Gln270 (Gln269 for CoV2) and the hydroxyl of Tyr269 (Tyr268 for CoV2), any of which could serve as a hydrogen bond acceptor. Interaction of GRL0617 with PLpro of SARS-CoV is shown in Fig. 3 and with that of SARS-CoV2 is shown in Fig. 4.
Fig. 3.

Interaction profile of GRL0617 and top six fungal metabolites docked with SARS-CoV-PLpro (PDB: 3E9S)
Fig. 4.

Interaction profile of GRL0617 and top six fungal metabolites docked with SARS-CoV2-PLpro (PDB: 7CMD)
Docking fungal metabolites with PLpro of SARS-CoV and SARS-CoV2
Total of six fungal metabolites, namely Fonsecin, Pyranonigrin-B, Nigerloxin, Flaviolin, Tensidol A and Ochratoxin Beta showed effective binding with Tyr269 for SARS-CoV-PLpro and Tyr268 for SARS-CoV2-PLpro, had multiple types of interactions with amino acids and showed binding energy close to or at par with GRL0617. For PLpro of SARS-CoV, binding energy of these fungal metabolites in kcal/mol are −7.25, −6.76, −6.62, −6.55, −6.34 and −6.03, respectively. For PLpro of SARS-CoV2, their binding energy in kcal/mol is −7.11, −6.26, −6.12, −6.01, −5.61 and −5.23, respectively. Interactions of these metabolites with PLpro of SARS-CoV are shown in Fig. 3 and with that of SARS-CoV2 are shown in Fig. 4. Surprisingly, two fungal metabolites Fonsecin and Flaviolin share a common structural feature with GRL0617, that is to possess the naphthalene moiety in its structure. For PLpro of SARS-CoV, Fonsecin strongly interacts with Tyr269 making hydrogen bond as well as two Pi-Pi T-shaped hydrophobic interactions. It further interacts with Asp165 and Leu163 by making hydrogen bonds. Further it also interacts with Pro249 and Try265 in same fashion as done by GRL0617.
On the other hand, Flaviolin also interacts by making interactions with all the key amino acids of PLpro that are Pro248, Pro249, Tyr265, Tyr269 and Tyr274. Like GRL0617, Flaviolin also interacts with Tyr269 by making two Pi-Pi T-shaped hydrophobic interactions and by making Pi-alkyl interaction with Pro248 and Pro249. For PLpro of SARS-CoV2, Fonsecin strongly interacts with Tyr268 by making two Pi-Pi T-shaped hydrophobic interactions. It further interacts with Asp164, Asn267 and Tyr273 by making Carbon hydrogen bonds. Further it also interacts with Pro248, Pro247 and Try264 in same fashion as done by GRL0617. On the other hand, Flaviolin also interacts by making interactions with all the key amino acids of PLpro that are Leu162, Tyr268 and Glu167 whereby making in total of four hydrogen bonds. However, in this case unlike PLpro of SARS-CoV, Flaviolin fails to make hydrophobic interactions that are necessary for imparting inhibitory effect. Therefore, despite possessing naphthalene moiety, that essentially Pi-Pi T-shaped hydrophobic interaction with Tyr268, fails to interact for this protein. Though structurally different, Nigerloxin effectively interacts with all the essential amino acids to impart inhibitory effect on PLpro which includes Leu163, Glu168, Gly164, Asp165, Tyr265, Tyr274 and Tyr269 by which it makes a total of two hydrogen bonds and nine other hydrophobic interactions for SARS-CoV. For PLpro of SARS-CoV2, Nigerloxin interacts Asp164, Leu162, Gly163, Tyr268, Tyr273, Tyr264 and Gln269 by making total of four hydrogen bonds and seven hydrophobic interactions. Despite effective interaction and docking score shown by Nigerloxin, it does not possess has naphthalene moiety and so it cannot be considered analogue of GRL0617. The structural features of all these screened fungal metabolites in comparison with GRL0617 are presented in Table 1.
Table 1.
Structures and chemical properties of screened fungal aromatic metabolites
| Compound | Structure | Molecular weight | LogP | #Rotatable bonds | #Acceptors | #Donors | Surface area |
|---|---|---|---|---|---|---|---|
| GRL-0617 (Control) |
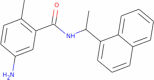
|
304.393 | 4.22142 | 3 | 2 | 2 | 135.681 |
| Fonsecin |
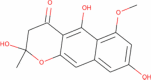
|
290.271 | 1.9333 | 1 | 6 | 3 | 119.703 |
| Pyranonigrin-B |
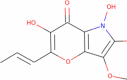
|
253.21 | 1.2847 | 2 | 7 | 3 | 100.719 |
| Nigerloxin |
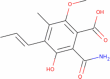
|
265.265 | 1.53942 | 4 | 4 | 3 | 109.661 |
| Flaviolin |
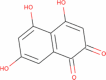
|
206.153 | 0.7621 | 0 | 5 | 3 | 0.7621 |
| Tensidol A |
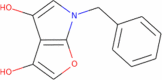
|
229.235 | 2.6938 | 2 | 4 | 2 | 97.337 |
| Ochratoxin Beta |
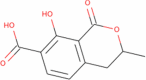
|
222.196 | 1.1918 | 1 | 4 | 2 | 91.275 |
MM/GBSA calculations
The energy released (ΔGBind) due to bond formation, or rather interaction of the ligand with protein is in the form of binding energy and it determines the stability of any given protein–ligand complex. The free energy of a favourable reaction is negative. The binding free energy change profiles of all the top six fungal metabolites in comparison with reference GRL0617 are represented in Table 2. The interaction of GRL0617 with the PLpro’s of both the SARS-CoV and SARS-CoV2 is sought to be occurring highly spontaneously as the ΔGBind for both the cases is less than −60.00 kcal/mol. The next best ligand in terms of binding free energy change is Fonsecin, where ΔGBind for both the cases is less than −50.00 kcal/mol. Of all the least spontaneous to interact with PLpro’s is predicted to be Pyranonigrin-B which shows ΔGBind equal to −24.64 kcal/mol, though this does not necessary mean that interaction is not favourable, the value in the negative defines the interaction to occur when given chance. In addition to the total energy, the contributions to the total energy from different components such as Coulomb energy, Hydrogen-bonding correction, Lipophilic energy, Pi-pi packing correction and Van der Waals energy are provided in Table 2.
Table 2.
MM/GBSA binding free energy change profiles of ligands with PLpro of SARS-CoV and SARS-CoV2 for docked complexes
| Ligand | ΔGBind (Kcal/mol) | ΔGCoulomb (Kcal/mol) | ΔGHbond (Kcal/mol) | ΔGLipo (Kcal/mol) | ΔGPacking (Kcal/mol) | ΔGvdW (Kcal/mol) |
|---|---|---|---|---|---|---|
| Ligands interacting with SARS-CoV-PLpro (PDB-3E9S) | ||||||
| GRL-0617 | −67.06 | −24.83 | −2.45 | −25.08 | −3.32 | −44.25 |
| Fonsecin | −54.22 | −20.52 | −1.56 | −22.45 | −3.52 | −47.34 |
| Pyranonigrin-B | −24.64 | −14.12 | −1.16 | −16.33 | −1.23 | −32.15 |
| Nigerloxin | −37.34 | −19.34 | −2.01 | −17.23 | −2.67 | −22.45 |
| Flaviolin | −32.66 | −30.92 | −1.72 | −6.23 | −1.66 | −39.62 |
| Tensidol A | −29.32 | −16.66 | −2.03 | −10.09 | −1.09 | −27.87 |
| Ochratoxin Beta | −34.54 | −10.34 | −1.04 | −9.82 | −1.56 | −25.49 |
| Ligands interacting with SARS-CoV2-PLpro (PDB-7CMD) | ||||||
| GRL-0617 | −63.12 | −23.41 | −2.38 | −24.66 | −3.12 | −46.24 |
| Fonsecin | −50.82 | −19.56 | −1.34 | −23.71 | −3.46 | −45.30 |
| Pyranonigrin-B | −26.43 | −15.67 | −0.28 | −15.92 | −1.08 | −33.78 |
| Nigerloxin | −35.56 | −16.43 | −0.81 | −16.88 | −2.06 | −25.76 |
| Flaviolin | −30.45 | −29.51 | −2.04 | −5.79 | −1.54 | −36.22 |
| Tensidol A | −28.21 | −14.74 | −2.11 | −9.11 | −1.13 | −24.74 |
| Ochratoxin Beta | −30.63 | −11.93 | −0.78 | −10.92 | −1.42 | −30.60 |
Meaning of abbreviations used in the table are as follows: Coulomb—Coulomb energy, Hbond—Hydrogen-bonding correction, Lipo—Lipophilic energy, Packing—Pi-Pi packing correction, vdW—Van der Waals energy
MD simulations
Owing to structural similarity, docking scores and interactions, Fonsecin was screened to be the best hit and its interaction profile by performing MD simulations for 100 ns were compared to that of the profile of GRL0617. For this, the MD simulations were performed in two sets. The control set consisted of complex SARS-CoV2-PLpro-GRL0617 and test set was SARS-CoV2-PLpro-Fonsecin.
After performing MD simulations, the root-mean-square deviation (RMSD) appraisal was performed which is utilized to quantify the normal change in dislodging of an of particles for a specific frame as for a reference constant frame. It is determined for all frames of trajectory. The plots in Fig. 5 depict RMSD movements in the portions of the protein (left Y-axis). The docked pose of ligand and protein in the complex is considered as the reference frame and then the movement for this original alignment during MD simulation is gauged by aligning all the protein frames concerning time. Checking the RMSD of the protein can give knowledge into its auxiliary 3D structural movement on a graph during the simulation. RMSD examination can demonstrate if the simulation has equilibrated—its changes towards the finish of the recreation are around some thermal energetically stable conformation. Changes in the range of 1–4 Å are completely satisfactory for little, globular proteins. However, this range of value widens as the size of the protein increases. For the complex of SARS-CoV2-PLpro-GRL0617 (Fig. 5a), the protein backbone hovers the value of RMSD not exceeding 2.5 Å; for the SARS-CoV2-PLpro-Fonsecin complex (Fig. 5b), the value stays well under 2.5 Å as well and. Ligand RMSD (right Y-axis, plots of Fig. 5) suggests the stability of ligand posture concerning the docked position of the ligand in the binding cleft of the protein. 'Lig fit Prot' suggests the RMSD of a ligand for protein backbone. For this, the values slightly larger than the protein’s RMSD are considered satisfactory but if the values observed are significantly larger than the RMSD of the protein, then it is likely that the ligand acquires a different stable position than the original posture. For SARS-CoV2-PLpro-GRL0617 (Fig. 5a), the Lig fit Prot stays around 3.0 Å throughout simulation. For SARS-CoV2-PLpro-Fonsecin (Fig. 5b), the Lig fit Prot value stays below 2.4 Å but between the time frame of 60 to 80 ns the RMSD slightly increases, and then stabilizes after 80 ns, suggesting the Fonsecin changing poses and then stabilizing. Lig Fit Lig suggests how much the ligands moves/vibrates from the still docked pose. Here, for Fonsecin, this value is much lower than shown by GRL0617, suggesting the ligand being very stable for the given docked pose. The root-mean-square fluctuation (RMSF) is useful for portraying confined changes along the protein chain (Fig. 6). In the graph, the peaks demonstrate regions of the protein that vary the most throughout the simulation. Ordinarily, the tails (N-and C-terminal) change the maximum than other internal regions of the protein. Secondary regions of proteins like alpha helices and beta strands are generally more inflexible and rigid than the unstructured regions and hence vacillate not exactly like loop forming portions of protein. Alpha-helical and beta-strand areas are featured in red and blue foundations, separately. These districts are characterized by helices or strands that endure over 70% of the whole re-enactment. Protein deposits that contact ligand is set apart with green-hued vertical bars. The RMSF of the protein can likewise be related to the exploratory x-beam B-factor (right Y-hub). Because of the distinction between the RMSF and B-factor definitions, balanced correspondence ought not to be normal. Notwithstanding, the reproduction results should resemble the crystallographic information. It is seen that both buildings SARS-CoV2-PLpro-GRL0617 (Fig. 6a) and SARS-CoV2-PLpro-Fonsecin (Fig. 6b) (i) protein interacts with both the ligands and (ii) trends of RMSF and B-factor definitions correspond similarly.
Fig. 5.

MD simulation Protein–ligand interaction root-mean-square deviation (RMSD) profile of a SARS-CoV2-PLpro-GRL0617 b SARS-CoV2-PLpro-Fonsecin
Fig. 6.

MD simulation Protein–ligand interaction root-mean-square fluctuation (RMSF) profile of a SARS-CoV2-PLpro-GRL0617 b SARS-CoV2-PLpro-Fonsecin
Protein interactions with the ligand can be monitored throughout the simulation. These interactions can be categorized by type and summarized, as shown in Fig. 7 for the SARS-CoV2-PLpro-GRL0617 complex and in Fig. 8 for SARS-CoV2-PLpro-Fonsecin. Protein–ligand interactions are categorized into four types: Hydrogen Bonds, Hydrophobic, Ionic and Water Bridges. Every connection type contains more explicit subtypes, which can be investigated through the 'Simulation Interactions Diagram' board. The stacked bar outlines are standardized throughout the direction: for instance, an estimation of 0.8 recommends that 80% of the simulation time the collaboration is kept up. Qualities over 1.0 are conceivable as some protein build-up may make numerous contacts of the same subtype with the ligand. In Fig. 7, it is observed that results of docking are corroborated for GRL0617 where it interacts with Leu162, Asp164, Pro247, Tyr264, Tyr268, Gln269 and Tyr273. Values for Tyr268 is above 1.0 suggests its constant interaction with GRL0617 which is represented in Fig. 7b, by two Pi-Pi staking bonds been constantly been persistent during the course of simulation. Similarly, a very strong interaction by Fonsecin is represented in Fig. 8, where its interaction score with Tyr268 is above 1.2 and it even forms hydrogen bond along with Pi-Pi stacking during simulation (Fig. 8b). A timetable portrayal of the associations and contacts (Hydrogen bonds, Hydrophobic, Ionic, Water spans) is appeared in the Fig. 9a for SARS-CoV2-PLpro-GRL0617 complex and Fig. 9b for SARS-CoV2-PLpro-Fonsecin complex. These figures portray which deposits communicate with the ligand in every direction outline. A few residues make more than one explicit contact with the ligand, which is shown by a hazier shade of orange, as indicated by the scale to one side of the plot. The plots corroborate the findings of docking suggesting the interactions proposed to be formed during docking between ligand and amino acids, are being made by same amino acids during simulations.
Fig. 7.

Protein–Ligand interaction profile during the course of MD simulation of SARS-CoV2-PLpro-GRL0617 complex a interaction profile of crucial interacting amino acids b Ligand interaction diagram showing percent of total time a particular interaction is involved in
Fig. 8.

Protein–Ligand interaction profile during the course of MD simulation of SARS-CoV2-PLpro-Fonsecin complex a interaction profile of crucial interacting amino acids b Ligand interaction diagram showing percent of total time a particular interaction is involved in
Fig. 9.

Timeline representation of the interactions of ligand with amino acids for the complex a SARS-CoV2-PLpro-GRL0617 b SARS-CoV2-PLpro-Fonsecin
ADMET analysis
All the ADMET properties of the top six screened compounds along with GRL0617 are depicted in Table 3. For a compound to be classified as an oral drug, it is important to predict its mobility through the intestinal epithelial layers of cells that predicts the bioavailability. The theoretical model makes the use of Caco-2 permeability and its value higher than 0.90 means the compound has high permeability. Under present study, all the compounds show the Caco-2 permeability values in positive integer suggesting them to be absorbing through intestinal epithelial layers. Especially, GRL0617 and Tensidol have value above 0.90 which makes them efficient to be absorbed from the gut when consumed orally. Intestinal absorption (human) value is another parameter that calculates the absorption of the drug from human gut when administered orally. Fonsecin, Tensidol A and GRL061 have this value above 90% suggesting good absorption. While Nigerloxin has this value of 31% which is the lowest of all. The next important parameter is skin permeability, where it was observed that all the compounds under study have values smaller than −2.5 log Kp, which means these compounds have poor permeability. The ATP-binding cassette (ABC) transporter is important for transport of molecules through cell membrane and P-glycoprotein is its component needed for efficient transport, and its value ‘yes’ predicts the compound to pass cell membrane through ABC transporter. Here, all the five compounds are predicted to pass through the cell membrane via ABC transporters. Total diffusion of drug in total blood volume is determined by “volume of Distribution (VDss)” and its value below −0.15 logVDss suggest poor diffusion while values higher than 0.45 LogVDss suggests faster and higher equal distribution of drug in the total blood volume, however, all the compounds here show poor VDss values suggesting their poor diffusion in blood. The ability of compound to travel to the brain is given by the values of Blood–Brain barrier (BBB) permeability. When the logBB values are greater than 0.3, they can pass BBB. Values of compounds under study predicted that all the compounds may not be able to cross BBB. Further, where none of the compounds out of five is predicted to show CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4 inhibition as analysis obtained from metabolism prediction. Renal excretion rate of all the compounds under study was different and no compound predicted to show AMES toxicity which important to predict as this test suggest drug’s property of mutagenicity. None of the compound shows Hepatotoxicity and Skin Sensitisation. Potassium flux in heart is controlled by hERG I and II, improper flux of potassium by these transporters can cause QT syndrome where the Q and T peaks of heart electrocardiogram gets altered. The impact of screened compounds under present study on hERG I and II transporters is shown in Table 3 along with other essential ADMET properties.
Table 3.
ADMET properties of screened aromatic fungal metabolites
| Property | Model name | Predicted value | Unit | ||||||
|---|---|---|---|---|---|---|---|---|---|
| GRL0617 | Fonsecin | Pyranonigrin-B | Nigerloxin | Flaviolin | Tensidol A | Ochratoxin beta | |||
| Absorption | Water solubility | −4.678 | −2.993 | −2.931 | −2.357 | −2.042 | −3.422 | −1.775 | Numeric (log mol/L) |
| Absorption | Caco2 permeability | 1.302 | 0.278 | 0.136 | 0.141 | 0.215 | 1.23 | 0.734 | Numeric (log Papp in 10–6 cm/s) |
| Absorption | Intestinal absorption (human) | 92.815 | 93.197 | 68.066 | 31.249 | 62.173 | 93.918 | 86.31 | Numeric (% Absorbed) |
| Absorption | Skin Permeability | −2.785 | −2.736 | −2.737 | −2.735 | −2.771 | −2.728 | −2.732 | Numeric (log Kp) |
| Absorption | P-glycoprotein substrate | Yes | Yes | No | No | No | No | No | Categorical (Yes/No) |
| Absorption | P-glycoprotein I inhibitor | No | No | No | No | No | No | No | Categorical (Yes/No) |
| Absorption | P-glycoprotein II inhibitor | Yes | No | No | No | No | No | No | Categorical (Yes/No) |
| Distribution | VDss (human) | 0.086 | 0.625 | 0.558 | −1.416 | 0.313 | 0.187 | −1.731 | Numeric (log L/kg) |
| Distribution | Fraction unbound (human) | 0 | 0.158 | 0.646 | 0.559 | 0.685 | 0.368 | 0.508 | Numeric (Fu) |
| Distribution | BBB permeability | 0.055 | −0.754 | −1.192 | −0.982 | −0.776 | 0.475 | −0.064 | Numeric (log BB) |
| Distribution | CNS permeability | −1.604 | −3.088 | −3.404 | −2.686 | −3.294 | −3.111 | −2.55 | Numeric (log PS) |
| Metabolism | CYP2D6 substrate | No | No | No | No | No | No | No | Categorical (Yes/No) |
| Metabolism | CYP3A4 substrate | Yes | No | No | No | No | No | No | Categorical (Yes/No) |
| Metabolism | CYP1A2 inhibitor | Yes | No | No | No | No | Yes | No | Categorical (Yes/No) |
| Metabolism | CYP2C19 inhibitor | Yes | No | No | No | No | No | No | Categorical (Yes/No) |
| Metabolism | CYP2C9 inhibitor | Yes | No | No | No | No | No | No | Categorical (Yes/No) |
| Metabolism | CYP2D6 inhibitor | No | No | No | No | No | No | No | Categorical (Yes/No) |
| Metabolism | CYP3A4 inhibitor | Yes | No | No | No | No | No | No | Categorical (Yes/No) |
| Excretion | Total Clearance | 0.221 | 0.407 | 0.114 | 0.556 | 0.425 | 0.207 | 0.541 | Numeric (log ml/min/kg) |
| Excretion | Renal OCT2 substrate | No | No | No | No | No | No | No | Categorical (Yes/No) |
| Toxicity | AMES toxicity | Yes | No | No | No | No | Yes | No | Categorical (Yes/No) |
| Toxicity | Max. tolerated dose (human) | −0.043 | 0.432 | 0.316 | 1.15 | 0.29 | −0.036 | 0.595 | Numeric (log mg/kg/day) |
| Toxicity | hERG I inhibitor | No | No | No | No | No | No | No | Categorical (Yes/No) |
| Toxicity | hERG II inhibitor | Yes | No | No | No | No | No | No | Categorical (Yes/No) |
| Toxicity | Oral Rat Acute Toxicity (LD50) | 2.472 | 1.777 | 2.047 | 2.294 | 1.544 | 2.318 | 2.529 | Numeric (mol/kg) |
| Toxicity | Oral Rat Chronic Toxicity (LOAEL) | 0.462 | 2.205 | 2.23 | 2.694 | 2.688 | 0.719 | 2.66 | Numeric (log mg/kg_bw/day) |
| Toxicity | Hepatotoxicity | No | No | Yes | No | No | Yes | No | Categorical (Yes/No) |
| Toxicity | Skin Sensitisation | No | No | No | No | No | No | No | Categorical (Yes/No) |
| Toxicity | T. pyriformis toxicity | 0.529 | 0.421 | 0.27 | 0.285 | 0.288 | 0.42 | 0.282 | Numeric (log ug/L) |
| Toxicity | Minnow toxicity | 1.936 | 2.571 | 2.097 | 1.762 | 2.952 | 2.622 | 2.035 | Numeric (log mM) |
Discussion
Coronavirus (CoVs) comprises of a huge virus group and is broadly spread amongst human and other animals. CoVs have regular indistinguishable basic highlights. All these viruses possess a positive strand RNA as their core genetic material and have S-protein, E-protein, M-protein, and N-protein. Analysing hereditary analysis, in an aggregate of 7 significant branches CoVs are bifurcated as (i) Alpha Coronavirus, (ii) OC43 (Beta Coronavirus), (iii) MERS-CoV (the Beta Coronavirus that causes Middle East Respiratory Syndrome, or MERS), (iv) HKU1 (Beta Coronavirus), (v) SARS-CoV (Beta Coronavirus causing the serious intense respiratory disorder, or SARS) and (vi) SARS-CoV2 (the novel Coronavirus that causes COVID-19) and (vii) Gamma Coronavirus. All these CoVs does not contaminate people, to taint human, the CoV ought to have a sort of S-protein that can cooperate with human ACE2, to incite viral layer combination with human cell. Till date, CoVs, for example, Avian Infectious Bronchitis Virus (IBV), Bovine Coronavirus (BCV), Canine Coronavirus (CCoV), Feline Infectious Peritonitis Virus (FIPV), Human Coronaviruses (HCoVs), Porcine Hemagglutinating Encephalomyelitis Virus (HEV), Porcine Transmissible Gastroenteritis Virus (TGEV), and Turkey Coronavirus are very much recognized [24]. A large portion of the CoVs have S-protein that can explicitly associate with a specific host but cross infectivity is uncommon [25–27]. However, in the year 2003, the flare-up of SARS-CoV was unique as the genuine host of the infection was bat which crosses infected people. Once more, in the year 2005, a comparative flare-up was set apart by MERS-CoV, where its actual host was Camel which cross contaminated human. The recurrence at which such diseases are happening is expanding a colossal well-being danger. Such cross infectivity brought about by CoV has raised another health alert with the latest COVID-19 being the most serious episodes throughout the entire existence of CoVs [7, 28].
Looking through compounds that can restrain SARS-CoV2 is of the most elevated need for the scientific fraternity. To commercialize such compounds, molecules from natural plant and microbe are being looked for, because of their low harmfulness, simple extraction procedures and being effectively acknowledged by individuals. Such medications likewise have more limited periods of preliminaries. As of late, our research group proposed Pyranonigrin A and Flaviolin to have the possibilities to interact with Mpro and hinder its viral replication capacity [9]. Another gathering of specialists has comparably recognized a concoction from tea plants that have the possibilities to associate with Mpro and restrain its activity [29, 30]. The subsequent methodology is to check the accessible commercial drugs to market against the SARS-CoV2. There may be an exceptionally high possibility that current medications may interfere with the biochemical cycles of the virus and restrain it. Such a methodology is called drug repurposing. Utilizing drug repurposing, Procainamide, Tetrahydrozoline, Levamisole was distinguished as medications that can meddle with the papain-like protease of SARS-CoV2 [31]. Such methodology utilizes molecular docking and MD simulations as the centre strategies for the in silico examination, and by utilizing these techniques several lead compounds are recognized to have potential to meddle with the biochemistry and life cycle of SARS-CoV2 as of late. There are a few spaces of life sciences where the approaches of docking and MD reproductions have been of an incredible use [32–34]. Comparable examinations utilizing docking and MD reproductions are likewise performed for accessing the potency of hydroxychloroquine with various targets of SARS-CoV2 [35].
There are several efforts been made to find the inhibitors for PLpro [10, 36]. In one study by Klemm and colleagues, targeted 3,727 unique approved drugs to satisfy the purpose of drug repurposing showed Remdesivir and Hydroxychloroquine to interact with PLpro and showed promising results even under in vitro assays [10]. In another study, Elekofehinti and colleagues targeted about fifty thousand (50,000) compounds from natural origin from IBS database (https://www.ibscreen.com/natural.shtml) to find inhibitors for PLpro, their best compounds were STOCK1N-69160 [(S)-2-((R)-4-((R)-2-amino-3-methylbutanamido)-3-(4-chlorophenyl)butanamido)propanoic acid hydrochloride] and STOCK1N-69160 [(S)-2-((R)-4-((R)-2-amino-3-methylbutanamido)-3-(4-chlorophenyl) butanamido)propanoic acid hydrochloride] which could at maximum make only one Pi-Pi stacked bonds with Tyr268. Under current study the fungal metabolite, Fonsecin showed much better interaction with Tyr268. Moreover, their research did not made use of any positive control like GRL0617 to compare their results. Efforts are also made to identify the PLpro inhibitors using QSAR based data mining of diverse molecules followed by QSAR based virtual screening [37]. In a comprehensive review by France colleagues, identified published inhibitors for PLpro and they are as follows, luteolin, Ribavirin, valganciclovir, thymidine, chloramphenicol, cefamandole, tigecycline, chlorphenesin carbamate, levodropropizine, ritonavir, lopinavir, darunavir, omeprazole, methicillin, and tolazamide; these were all identified using drug repurposing approaches. In the same review, it is mentioned that GRL0617 is the most potent inhibitor identified till date for PLpro’s of SARS-CoV and SARS-CoV2 [36]. This supports our hypothesis to choose GRL0617 as the reference inhibitor for performing in silico study. With restricted extension to work with SARS-CoV2, as it requires Biosafety Level 4 (BSL4) arrangement, increasingly more exploration through in silico approach with docking and MD simulations is developing as the foundation of the computational examination, regardless of such examination ailing in vitro investigations, gigantic volumes of information demonstrating the collaborations of a functioning drug compound or phytochemical with the viral protein is brought to the analysts, which can come very handy for those having facility of performing in vitro examinations for approving computational estimates at last sparing a ton of time.
There exists a strong history where fungal metabolites have proven to be the boon for humans in combating the bane of viruses and bacteria. Antibiotics, flavouring agents, sedatives, cholesterol-lowering drugs, etc. have been isolated from fungi and are being commercialized. Apart from the compounds that are commercialized, there exists large number of other fungal metabolites that are well characterized structurally and there is a huge scope that few of these might interact with PLpro. With this rationale, the fungal metabolites were looked for pursuing this research. There are reports where fungal metabolites have shown as anti-viral traits. The anti-viral compounds from fungal origin are vividly described where the promising anti-viral compounds portrayed belonged to the chemical class of Indole alkaloids, Non-ribosomal peptides, Polyketides, Terpenoids [38]. Under current study, the reference inhibitor GRL0617 is aromatic in nature and possess naphthalene moiety that is key for its inhibitory effect on PLpro. The goal was to find structurally similar compound from natural source that can act as analogue of GRL0617. The lead fungal metabolite identified is Fonsecin. This is a naphthopyrone pigment and was first isolated in the year 1962 from a mutant of Aspergillus fonsecaeus [39]. This compound is characterized by a rich deposit of yellow pigment in the vesicles and primary sterigmata of fungi. As per the original article published in 1962, the crude pigment can be easily extracted with ethyl acetate from the dried mycelium of fungi. Fractional precipitation and recrystallization from ethyl acetate-petroleum ether mixtures can give pure substance in the form of bright yellow irregular prisms and hence its purification from fungi is relatively simple [39]. Herewith, we propose Fonsecin to interact with Tyr268 of SARS-CoV2-PLpro and can inhibit its function as deduced using docking and Molecular dynamics.
Acknowledgements
Authors are thankful to Department of Microbiology and Biotechnology, School of Sciences, Gujarat University, DST-FIST Sponsored Department, for providing necessary facilities to perform experiments. Authors are also thankful to UGC (Start-up Research Grant) for providing fund under the F.30-521/2020(BSR), Under Secretary FD-III Section, University Grant Commission, New Delhi 110002. We acknowledge GSBTM, DST, Government of Gujarat for providing Bioinformatics Node facility and Finishing School support. We acknowledge GUJCOST, DST, Government of Gujarat for Super-computing facility provision.
Declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Animal and Human Participants
This article does not contain any studies with human participants or animals performed by any of the authors.
Authors Approval
All authors have seen and approved the manuscript.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Chen Y, Guo Y, Pan Y, Zhao ZJ. Structure analysis of the receptor binding of 2019-nCoV. Biochem Biophys Res Commun. 2020 doi: 10.1016/j.bbrc.2020.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song Z, Xu Y, Bao L, et al (2019) From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses 11 [DOI] [PMC free article] [PubMed]
- 3.Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartz DA, Graham AL (2020) Potential maternal and infant outcomes from coronavirus 2019-NCOV (SARS-CoV-2) infecting pregnant women: lessons from SARS, MERS, and other human coronavirus infections. Viruses 12 [DOI] [PMC free article] [PubMed]
- 5.Joshi RS, Jagdale SS, Bansode SB, et al. Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1760137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rao P, Shukla A, Parmar P, et al. Reckoning a fungal metabolite, Pyranonigrin A as a potential Main protease (Mpro) inhibitor of novel SARS-CoV-2 virus identified using docking and molecular dynamics simulation. Biophys Chem. 2020 doi: 10.1016/j.bpc.2020.106425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kong R, Yang G, Xue R, et al (2020) COVID-19 docking server: an interactive server for docking small molecules, peptides and antibodies against potential targets of COVID-19. J arXiv Prepr [DOI] [PMC free article] [PubMed]
- 9.Rao P, Shukla A, Parmar P, et al. Proposing a fungal metabolite-Flaviolin as a potential inhibitor of 3CLpro of novel coronavirus SARS-CoV-2 identified using docking and molecular dynamics. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1813202. [DOI] [PubMed] [Google Scholar]
- 10.Klemm T, Ebert G, Calleja DJ, et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020;39:1–17. doi: 10.15252/embj.2020106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McClain CB, Vabret N. SARS-CoV-2: the many pros of targeting PLpro. Signal Transduct Target Ther. 2020;5:1–2. doi: 10.1038/s41392-020-00335-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin D, Mukherjee R, Grewe D, et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587:657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu Z, Huang B, Tang J, et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat Commun. 2021;12:488. doi: 10.1038/s41467-020-20718-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krivov GG, Shapovalov MV, Dunbrack RL., Jr Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Wang W, Kollman PA, Case DA. Antechamber: an accessory software package for molecular mechanical calculations. J J Am Chem Soc. 2001;222:U403. [Google Scholar]
- 16.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera: a visualization system for exploratory research and analysis. J J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 17.Gasteiger J, Jochum C. An algorithm for the perception of synthetically important rings. J Chem Inf. 1979;19:43–48. [Google Scholar]
- 18.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang W, Donini O, Reyes CM, Kollman PA. Biomolecular simulations: recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu Rev Biophys Biomol Struct. 2001;30:211–243. doi: 10.1146/annurev.biophys.30.1.211. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Hou T, Xu X. Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Curr Comput Aided Drug Des. 2006;2:287–306. doi: 10.2174/157340906778226454. [DOI] [Google Scholar]
- 21.Kollman PA, Massova I, Reyes C, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 22.Massova I, Kollman PA. Combined molecular mechanical and continuum solvent approach (MM- PBSA/GBSA) to predict ligand binding. Perspect Drug Discov Des. 2000;18:113–135. doi: 10.1023/A:1008763014207. [DOI] [Google Scholar]
- 23.Pires DEV, Blundell TL, Ascher DB. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shukla A, Parmar P, Kapoor G, et al. Curse of La Corona: unravelling the scientific and psychological conundrums of the 21st century pandemic. Mol Divers. 2021;1:3. doi: 10.1007/s11030-020-10167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren Z, Yan L, Zhang N, et al. The newly emerged SARS-Like coronavirus HCoV-EMC also has an “Achilles’’ heel": current effective inhibitor targeting a 3C-like protease”. Protein Cell. 2013;4:248–250. doi: 10.1007/s13238-013-2841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar A, Mehta V, Raj U, et al. Computational and in-vitro validation of natural molecules as potential acetylcholinesterase inhibitors and neuroprotective agents. Curr Alzheimer Res. 2018;16:116–127. doi: 10.2174/1567205016666181212155147. [DOI] [PubMed] [Google Scholar]
- 27.Yang H, Bartlam M, Rao Z. Drug design targeting the main protease, the Achilles heel of coronaviruses. Curr Pharm Des. 2006;12:4573–4590. doi: 10.2174/138161206779010369. [DOI] [PubMed] [Google Scholar]
- 28.Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhardwaj VK, Singh R, Sharma J, et al. Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1766572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhardwaj VK, Purohit R. Targeting the protein-protein interface pocket of Aurora-A-TPX2 complex: rational drug design and validation. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1772109. [DOI] [PubMed] [Google Scholar]
- 31.Arya R, Das A, Prashar V, Kumar M (2020) Potential inhibitors against papain-like protease of novel coronavirus (SARS-CoV-2) from FDA approved drugs
- 32.Parmar P, Shukla A, Rao P, et al (2020) The rise of gingerol as anti-QS molecule: darkest episode in the LuxR-mediated bioluminescence saga. 103823 [DOI] [PubMed]
- 33.Shukla A, Parmar P, Rao P, et al. Twin peaks: presenting the antagonistic molecular interplay of curcumin with LasR and LuxR quorum sensing pathways. Curr Microbiol. 2020 doi: 10.1007/s00284-020-01997-2. [DOI] [PubMed] [Google Scholar]
- 34.Kumar SP, Patel CN, Rawal RM, Pandya HA. Energetic contributions of amino acid residues and its cross-talk to delineate ligand-binding mechanism. Proteins Struct Funct Bioinforma. 2020;88:1207–1225. doi: 10.1002/prot.25894. [DOI] [PubMed] [Google Scholar]
- 35.Mukherjee S, Dasgupta S, Adhikary T, et al. Structural insight to hydroxychloroquine-3C-like proteinase complexation from SARS-CoV-2: inhibitor modelling study through molecular docking and MD-simulation study. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1804458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.France A, Cilia H, Miclot T, et al. Molecular basis of SARS-CoV-2 infection and rational design of potential antiviral agents: modeling and simulation approaches. Cite This J Proteome Res. 2020;19:4291–4315. doi: 10.1021/acs.jproteome.0c00779. [DOI] [PubMed] [Google Scholar]
- 37.Amin SA, Ghosh K, Gayen S, Jha T. Chemical-informatics approach to COVID-19 drug discovery: Monte Carlo based QSAR, virtual screening and molecular docking study of some in-house molecules as papain-like protease (PLpro) inhibitors. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1780946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Linnakoski R, Reshamwala D, Veteli P, et al (2018) Antiviral agents from fungi: diversity, mechanisms and potential applications. 9:2325 [DOI] [PMC free article] [PubMed]
- 39.Galmarini OL, Stodola FH, Raper KB, Fennell DI. Fonsecin, a naphthopyrone pigment from a mutant of Aspergillus fonsecaeus. Nature. 1962;195:502–503. doi: 10.1038/195502a0. [DOI] [Google Scholar]