

Abstract
Self-assembling peptides are being applied both in the biomedical area and as building blocks in nanotechnology. Their applications are closely linked to their modes of self-assembly, which determine the functional nanostructures that they form. This work brings together two structural elements that direct nanoscale self-association in divergent directions: proline as a β-breaker and the β-structure-associated diphenylalanine motif, into a single tripeptide sequence. Amino acid chirality was found to resolve the tension inherent to these conflicting self-assembly instructions. Stereoconfiguration determined the ability of each of the eight possible Pro-Phe-Phe stereoisomers to self-associate into diverse nanostructures, including nanoparticles, nanotapes, or fibrils, which yielded hydrogels with gel-to-sol transition at a physiologically relevant temperature. Three single-crystal structures and all-atom molecular dynamics simulations elucidated the ability of each peptide to establish key interactions to form long-range assemblies (i,e., stacks leading to gelling fibrils), medium-range assemblies (i.e., stacks yielding nanotapes), or short-range assemblies (i.e., dimers or trimers that further associated into nanoparticles). Importantly, diphenylalanine is known to serve as a binding site for pathological amyloids, potentially allowing these heterochiral systems to influence the fibrillization of other biologically relevant peptides. To probe this hypothesis, all eight Pro-Phe-Phe stereoisomers were tested in vitro on the Alzheimer’s disease-associated Aβ(1–42) peptide. Indeed, one nonfibril-forming stereoisomer effectively inhibited Aβ fibrillization through multivalent binding between diphenylalanine motifs. This work thus defined heterochirality as a useful feature to strategically develop future therapeutics to interfere with pathological processes, with the additional value of resistance to protease-mediated degradation and biocompatibility.
Keywords: peptide, self-assembly, chirality, amyloid, hydrogels, proline, d-amino acids
Simple peptides that self-assemble into functional nanostructures have been attracting great interest for their use in medicine and nanotechnology.1−4 They are extremely versatile building blocks capable of generating diverse nanomorphologies that are often based on β-sheets.5,6 However, the uncontrolled growth of β-sheets and, more generally, peptide assemblies, remains an unsolved general challenge,7 and approaches are continuously developed to address it. The vast majority of these exploit solvent-induced effects,8−10 or chemical modification of the building blocks to attain morphological diversity9,11 or to control supramolecular behavior by means of chemical switches.12,13
Proline (Pro) is a well-known β-breaker moiety that interferes with β-sheet formation and may be used to limit β-sheet growth,14 which instead can be favored by the incorporation of the diphenylalanine (Phe-Phe) motif.15,16 The incorporation of both Pro and Phe in short self-assembling sequences17,18 thus encodes two conflicting sets of self-assembly instructions, leading to outcomes that are difficult to predict a priori. An elegant computational study by Tuttle and colleagues identified Pro-Phe-Phe as the most aggregating-prone sequence among all possible 8000 combinations of 20 l-natural amino acids in tripeptides.19 Their simulations were based on hydrophobicity-related parameters, and subsequent experimental data evidenced a β-sheet-like spectroscopic signature for l-Pro-Phe-Phe aggregates suspended in water, despite the presence of Pro.19 Bera et al. noted the surprising rearrangement, over the course of several days, of l-Pro-Phe-Phe aggregates into nanofibers with a distinctive α-helix spectroscopic signature.18 It is worth noting that the l-Ala-Phe-Phe analog displayed instead spectra reminiscent of β-sheets, confirming the β-breaker role played by Pro.18
The incorporation of d-amino acids into l-peptides can lead to unexpected effects on conformation, self-assembly, and even therapeutic activity, as recently reviewed.20 Research is revealing the use of heterochirality to fine-tune peptide self-assembly into a variety of nanostructures, including twisted nanofibrils,21,22 nanotapes,23,24 nanotubes,25,26 2D-sheets,27 or gels.28,29d-amino acids can inhibit pathological amyloid aggregation, as with d-Phe30 on l-Phe fibrillization linked to phenylketonuria,31 or d-Trp-Aib (Aib = aminoisobutyrric acid) on Alzheimer’s disease-related Aβ peptide.32 Peptide inhibitors that combine a recognition motif with β-breaking Pro demonstrated promising activity in vitro and in vivo.14 Yet, their use as therapeutics has been hampered by their susceptibility to enzymatic hydrolysis and, consequently, short half-lives.33 In addition, peptides longer than two-to-three amino acids are costly to produce on a large scale, and it is thus preferable to shorten the sequence as much as possible for applications.
This study explores the introduction of d- and l-amino acids in the sequence Pro-Phe-Phe (Chart 1) to resolve the conflict between the contradictory assembly directing effects of Pro as β-breaker and Phe-Phe as β-sheet-associated motif, enabling the emergence of functional nanostructures. To this end, the self-organizing behavior of the eight distinct stereoisomers of Pro-Phe-Phe was probed, and they were tested in vitro as inhibitors of aggregation of the Aβ(1–42) peptide, which is associated with Alzheimer’s disease. The most promising compound was also probed for resistance against protease-mediated hydrolysis and cytocompatibility in cell culture.
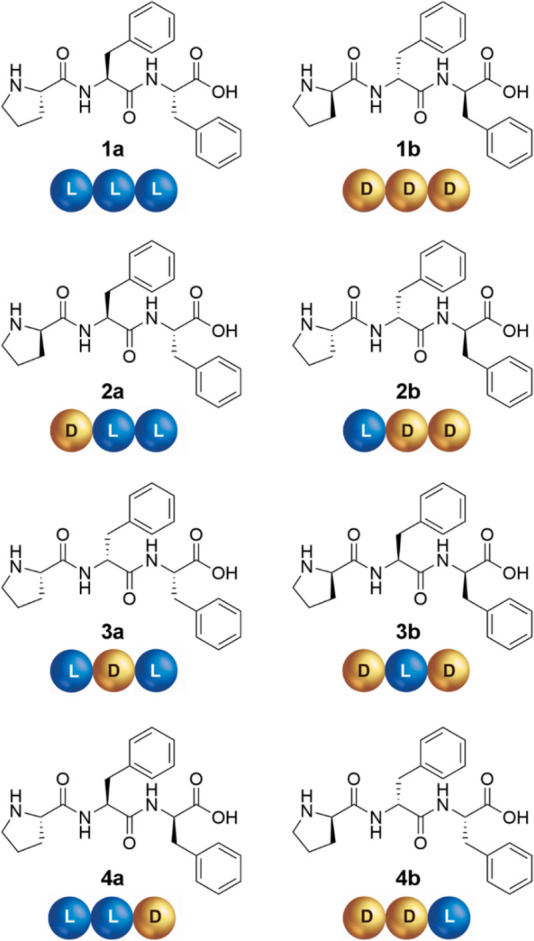
Chart 1. Pro-Phe-Phe Stereoisomers by Enantiomer Pairs (a/b) and Their Cartoon Keycode (Each Amino Acid is Represented as Either a Blue (l) or Gold (d) Sphere).
Results and Discussion
Self-Assembly of Pro-Phe-Phe Stereoisomers into Hydrogels
The eight stereoisomers of Pro-Phe-Phe were prepared by solid-phase peptide synthesis.34 Self-assembly studies were performed in phosphate buffer, following a pH switch from alkaline to neutral. Since enantiomeric pairs display the same supramolecular behavior in achiral environments, we report below only the data for one compound (a) for each pair (a/b). Interestingly, among the four stereoisomers 1–4, only 2a was observed to stack (Figure 1A) so as to form self-supportive hydrogels (Figure 1B). All-atom molecular dynamics (MD, Figure 1A) simulations in explicit water solvent confirmed a kinked backbone for the peptides when assembled, and their ability to give rise to parallel stacks reminiscent of β-sheets (see Movie S1 and Section S6 of the Supporting Information). The amphipathic character of the assembly, with the net segregation between hydrophilic and hydrophobic components, allowed the formation of dry surfaces involving Phe aromatic residues (Figure 1A). Oscillatory rheometry time sweeps at the minimum gelling concentration (mgc, 24 mM) revealed immediate gelation leading to a G′ of 4 kPa at plateau (Figure 1B). The hydrogel network was rather fragile, requiring only a stress of 6 Pa to break it (see Supporting Information, Section S3). The higher mgc and the lower elastic modulus G′, relative to the analogous Val-Phe-Phe DLL/LDD enantiomers,24 suggested self-assembly was partially hindered by the β-breaker proline. This hypothesis was also supported by Thioflavin T (ThT) fluorescence, which was reduced in the case of 2a relative to its DLL analogue Val-Phe-Phe (see Supporting Information, Section S11). ThT staining is a standard method to quantify amyloids, as this dye is known to bind laterally to fibrils composed of at least four consecutive β-strands of l-peptides, leading to fluorescence.35
Figure 1.
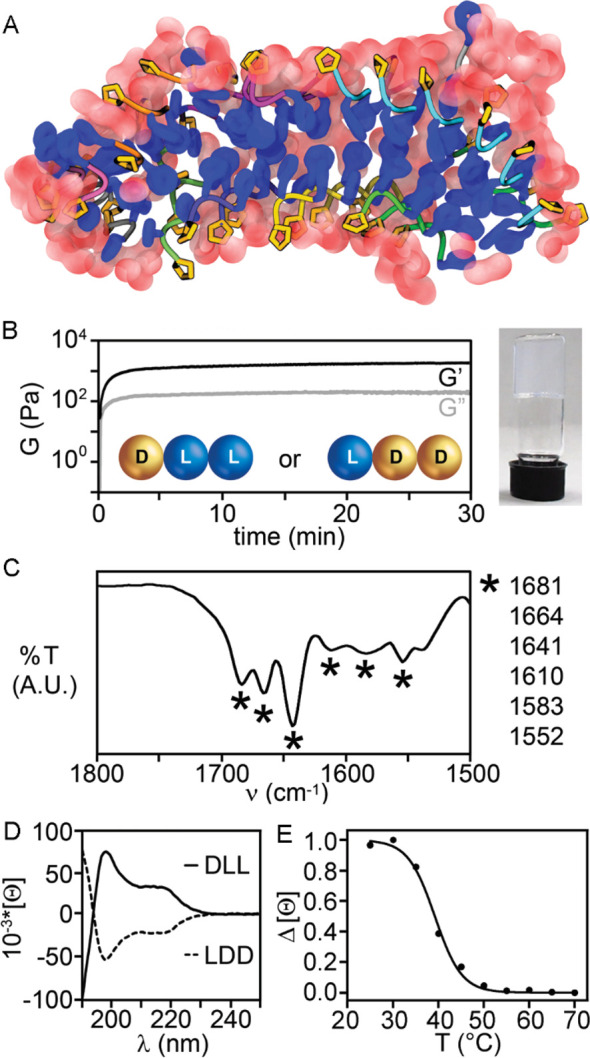
(A) All-atom molecular dynamics (MD) revealing the formation of stacks of kinked peptides for 2a with Phe residues (blue) facing each other to define water-excluding hydrophobic surfaces, and Pro residues (gold) facing the outer surface to interact with water (red spheres). (B) Rheology time sweep and photograph of 2a hydrogel. (C) ATR-IR of the gel of 2a. (D) CD spectra of 2a and 2b. (E) CD heating ramp for 2a.
Attenuated total reflection Fourier transformed infrared (ATR-FT-IR) spectroscopy on the assembled gel (Figure 1C) revealed amide I band frequencies reminiscent of β-structure signatures, such as sheets and turns, at 1610 and 1681 cm–1, as well as disordered structures at 1641 cm–1. The insertion of d-Pro in l-peptides is a well-known strategy to promote turns.36 However, an analogous effect in a sequence as short as a tripeptide is far from sure, particularly as the term “turn” implies longer sequences.37 Circular dichroism (CD) spectra (Figure 1D) of the enantiomers were mirror images, as expected. Two peaks were observed at 198 and 219 nm, with an overall signature analogous to those of heterochiral tripeptides Phe-Xaa-Phe (Xaa = Ala, Val, Leu, etc.).38 In those cases, in silico and experimental data indicated that the signal corresponded to populations of different conformations in solution. The most stable ones, which were amplified during assembly, had a backbone conformation similar to that observed for β-structures, with torsion angles of their central residue falling in the β-sheet region of the Ramachandran plot.38 We inferred an analogous case here, where the peptides adopted a variety of conformations in solution, yet it was those leading to self-assembly that were predominant in the FT-IR spectra of the gel. Reversible gel-to-sol transition occurred at the physiologically relevant onset temperature of 39 °C, which is convenient for potential use in drug delivery, and these data were confirmed by CD heating ramps (Figure 1E). Hyperthermia-induced drug release is a hot area of research for the topical delivery of chemotherapeutics to treat a variety of tumors, while diminishing the side effects normally associated with systemic use.39,40 In particular, if a drug was embedded in a supramolecular hydrogel, its release could be controlled and sustained over time upon thermo-induced hydrogel disassembly by local heating.41 The gelator concentration could also be used as a convenient variable to control release kinetics; for instance, when immersed in a hot bath at 40 °C, hydrogels formed by 2a at 24 and 30 mM disassembled into a clear solution in 25 and 90 min, respectively.
Nanostructure Morphology
Transmission electron microscopy (TEM) and atomic force microscopy (AFM) images revealed diverse nanostructures formed by the four enantiomeric pairs (Figure 2 and Supporting Information, Sections S4 and S5), thus confirming a directing effect of stereoconfiguration on nanoscale assembly. The homochiral sequence 1a (Figure 2A) formed mainly amorphous aggregates, and occasionally microcrystals, in agreement with recent reports.18,19 Both the gelling 2a (DLL, Figure 2B) and the nongelling 4a (LLD, Figure 2D) assembled into rigid nanotapes, which arose from the alignment of individual nanofibrils with a mean diameter of 1.6 ± 0.2 nm (see Supporting Information, Sections S4 and S5), which was compatible with two or more layers of peptides as shown in Figure 1A for 2a). Multicopy all-atom MD simulations of DLL 2a peptides in water indicated the formation of fibrils, thus shedding further light upon the assembly process as described above (see Supporting Information, Section S6).
Figure 2.

TEM (left) and AFM (right) images. (A) 1a aggregates; (B) 2a bundles of nanofibrils; (C) 3a nanoparticles; (D) 4a nanotapes. Full-size images and further details can be found in the Supporting Information Sections S4 and S5.
Notably, 3a (LDL, Figure 2C) formed spherical nanoparticles that AFM height profiles revealed to be 8 ± 3 nm in diameter (counts = 100), with only rare instances of short fibrils of analogous size as 2a, compatible with two or more layers of peptides (see the Supporting Information, Sections S4 and S5). For comparison, Val-Phe-Phe (LDL stereoisomer) under analogous conditions formed gelling nanotapes that displayed ThT fluorescence.24 By contrast, in the case of 3a, the total absence of ThT fluorescence, which normally arises from the binding of the dye onto fibrils composed of at least four consecutive β-strands, confirmed the disruptive role exerted by Pro (see the Supporting Information, Section S11).35
Dynamic light scattering (DLS) was used to investigate the corresponding size distribution of 3a nanoparticles in solution (see the Supporting Information, Section S7). The hydrodynamic size increased over time; freshly prepared samples consisted of a mixture of smaller (∼18 nm) and larger (∼98 nm) nanoparticles that over 3 h further developed into a monodisperse distribution of 146 nm sized particles (PDI = 0.13).
Time-dependent growth of spherical nanostructures was formerly observed for self-assembling tetrapeptides composed of Pro and Phe.17 The discrepancy between DLS and AFM data reflected the differences between wet and dry samples; overall the in silico and experimental analyses agreed on the tendency of 3a toward the formation of stable spherical nanoparticles with uniform distribution of size that could be changed by controlling the hydration level (i.e., wet or dry).
MD Simulations Elucidated the Divergent Supramolecular Behavior
Multiple all-atom MD simulations of hundreds to 1000 peptides were carried out to shed light on the structural basis of the divergent supramolecular behavior, noted especially for the gelator 2a and the nanoparticle-forming 3a (Figure 3). In the case of 2a, nanofibrils (Figure 3A and the Supporting Information, Section S6) arose from several instances of parallel stacks of peptides. Figure 3B shows a snapshot from the last part of the MD simulation, when sheet-like structures formed by up to ten peptides were clearly visible (shown as ribbons ending with proline in orange, and colored differently). For 2a, the assembling nature of Phe prevailed over Pro β-breaker, as a continuous network of hydrogen bonds between amides held together parallel stacks (Figure 3C), involving mainly Phe residues. This organization allowed a net segregation between hydrophobic (Phe) and hydrophilic (Pro) residues, so that an amphiphilic supramolecular architecture could arise to allow for higher-order assemblies.38 In this manner, Phe residues could face each other to establish dry, steric zippers (as shown in Figure 1A), while Pro residues and the hydrophilic peptide backbones could interact with water to stabilize the hydrogel.
Figure 3.

Representative conformations of morphologies of hundreds of 2a (A–C) and 3a (D–E) peptides as extracted from multiple all-atom MD simulations extending over 1 μs. (A, D) Assembly of 216 DLL 2a (A) and 3a (D) peptides. The surface colored by atom type (C, N, and O in gray, blue, and red, respectively) is the one in the primary simulation box. That in gray extends along the x direction, while the two other in yellow or green represent adjacent images. (B) Compound 2a (DLL) formed fibrils from the association of parallel stacks (colored ribbons with Pro in orange). (C) Amphiphilic parallel stacks of 2a displaying a continuous network of hydrogen bonds (dashed black lines) between peptide amide groups. (E) 3a (LDL) self-associated into disordered clusters of ∼7 nm nanoparticles, where hydrogen bonding stabilized mainly antiparallel dimers and trimers.
In the case of 3a (Figure 3D,E), the β-breaker nature of Pro prevailed over the assembling tendency of Phe, as most interactions were limited to antiparallel pairs or triplets of peptides, and no elongated and stable nanostructure arose during the simulations. In the antiparallel arrangement, more hydrogen bonds and salt bridges could be established, often involving Pro residues (Figure 3E). However, as a result, no net segregation between hydrophilic and hydrophobic components persisted, and this could explain the impossibility to establish higher-order assemblies, in contrast with gelling 2a. Indeed, 3a dimers and trimers associated disorderly into ∼7 nm wide nanoparticles, in agreement with the absence of ThT fluorescence, and the microscopy and DLS data described above.
MD simulations revealed how and why the fibrillization of Pro-Phe-Phe peptides occurred. When the stereoconfiguration allowed segregation of assembling Phe and β-breaker Pro residues into amphiphilic architectures, the former could zip into higher-order assemblies, whereas the latter could interact with water to gel. On the contrary, when stereoconfiguration impeded the establishment of such supramolecular order, only short-range self-organization persisted, leading to the formation of discrete nanostructures. We anticipate a similar behavior also in the case of the scrambled sequence Phe-Pro-Phe, which displayed an amyloid-like behavior in the case of the homochiral peptide.18
Conformation of Nongelling Tripeptides
Enantiomeric pairs displayed mirror-image CD spectra, as expected (Figures 1D and 4A). In particular, the CD signature of homochiral Pro-Phe-Phe (1a/1b) was very similar to that of heterochiral 2a/2b; we thus inferred an analogous mixture of conformations in solution, of which those with a spectroscopic signature typical of β-structures were the most stable in fresh samples, in agreement with the literature,38 and also confirmed by IR spectra (see Supporting Information Section S8).19 The CD spectra displayed the same features also in the case of 4a/4b, albeit with lower signal intensity, especially at 198 nm. FT-IR spectra of 4a were dominated by two maxima at 1624 and 1680 cm–1, which typically arise from amyloids and turns, respectively. The presence of ThT fluorescence both in the case of 2a and 4a (see Supporting Information, Section S11) also suggested a degree of similarity in the conformations adopted by the two isomers.
Figure 4.
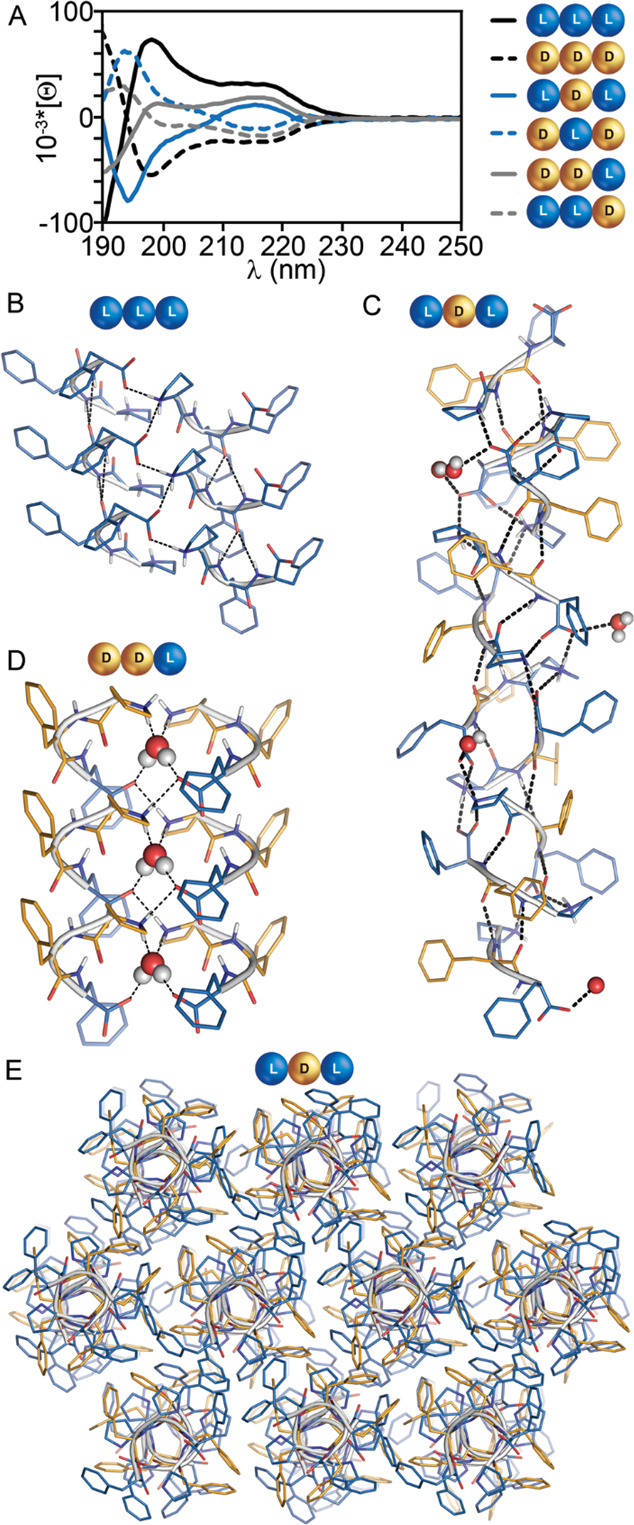
(A) Mirror-image CD spectra of enantiomer pairs 1a/1b, 3a/3b, and 4a/4b. (B–E) Single-crystal XRD structures of 1a (B), CCDC 2021319, revealing parallel stacks (side view); 3a ((C) stack side view, (E) stacks top view), CCDC 2026055, revealing a superhelical structure; 4b (D), CCDC 2021318, revealing parallel stacks of peptide pairs enclosing one hydrogen-bound water molecule.
Interestingly, the nanoparticle-forming enantiomeric pair 3a/3b showed a more complex CD signal, with sign inversion at 209 nm, reminiscent of β-turns.42 The amide I region of the FT-IR spectra was dominated by two maxima at 1687 and 1655 cm–1, corresponding to a region in which the characteristic bands of turns appear.43 Overall, for all tripeptides the sign of the CD signal in the 210–220 nm range appeared to be dictated by the chirality of the C-terminal amino acid, in agreement with other,44 but not all,24 reports on short peptides, suggesting the amino acid sequence also played a role.45
Single-Crystal X-ray Diffraction
X-ray diffraction (XRD) analyses of 1a (CCDC 2021319) and 3a (CCDC 2026055) revealed that both stereoisomers crystallized in the P21 space group. Whereas the crystal structure of 1a had only one crystallographically independent peptide molecule, 3a showed a much more complex architecture, with 10 independent molecules in the unit cell. Analysis of the backbone torsion angles of the central residue highlighted a different conformation for the peptides 1a and 3a, with a pair of angles analogous to those of α-helices18 for 1a and to those of inverted β-sheets for 3a, owing to the non-natural chirality of the central residue (see Supporting Information, Section S9). As expected from their different conformation, hydrogen bonding patterns in the crystals were significantly different.
In 1a, a single three-centered hydrogen bonding interaction connected parallel peptides, forming a stack along the a crystallographic direction (Figure 4B). Stacks were held together in the crystal through further hydrophilic interactions, i.e., salt bridges involving the charged C- and N-termini of peptides belonging to different stacks. No solvent molecules were present in the unit cell of the crystal. We infer that the tendency of 1a to form dry stacks devoid of water drove its precipitation from solution, as described above in the microscopy section.
Interestingly, 3a revealed an unusual superhelical arrangement responsible for holding tripeptides in amphipathic stacks (side view in Figure 4C), along the ac diagonal of the unit cell. In the crystal packing, multiple stacks bundled thanks to hydrophobic interactions between Phe residues at their interface (top view in Figure 4E). In crystals of 3a, backbone amide groups of each peptide interacted with two neighboring molecules, forming two stronger hydrogen bonds with one of them (O···N distances between 2.8 and 2.9 Å) and two weaker hydrogen bonds with the other (O···N distances between 2.9 and 3.1 Å, Figure 4C). Additional salt-bridge interactions (distances lower than 2.8 Å) were present between neighboring peptides but also between alternate peptides. A careful analysis of the electron density on the N-terminal proline residues confirmed the zwitterionic form for the peptide in the crystal and revealed the presence of water molecules interacting with the charged termini. Thus, water-mediated interactions stabilized peptide–peptide interactions throughout the network (see the Supporting Information, Section S9). The crystallization of 3a under different experimental conditions (i.e., methanol/water or phosphate buffer) suggested a structural role for these solvent molecules in the formation of the supramolecular organization, as their position was superimposable in the isomorphic structures. We infer that the presence of ordered water molecules every three to four peptides along the stack played a key role in stabilizing long-range assembly, as observed in the crystalline state where water was not mobile. In contrast, in solution, only rare instances occurred of short fibrils as shown by TEM, whose size was compatible with a double layer of peptides that could self-associate through the hydrophobic face (as shown in the view down the stacks in Figure 4E).
Removal of structural water from the crystal would thus destabilize elongated structures that extend beyond three to four peptide molecules, so that mainly short-range assemblies in the form of dimers and trimers would remain, as shown by all-atom MD simulations (Figure 3E). Hydrophobicity-driven self-association into nanoparticles would then proceed in a disordered manner, thus impeding the establishment of long-range assembly as required to form superstructures, which span a length scale that goes well beyond that of their building blocks.
Ordered water molecules served to organize the solid-state structure of 4b (CCDC 2021318), which crystallized in the C2 space group. In this case, one solvent molecule was held between two peptide molecules by strong H-bonds with donor–acceptor distances between 2.65 and 2.75 Å (Figure 4D). Hydrophobic interactions between peptide molecules facilitated the stacking of dimers to generate the observed crystal packing. As in the case of 3a, the crystal packing was generated by the bundling of stacks only through hydrophobic interactions of the aromatic moieties. However, in contrast with 3a, water was confined in a hydrophilic region defined by the peptide backbones, thus allowing for a higher level of segregation between hydrophilic and hydrophobic components. We hypothesized that the observed 4a nanotapes in solution arose from the lateral association of peptide stacks, with an intermediate-range supramolecular assembly relative to 2a and 3a. Overall, the three X-ray structures revealed how different amino acid chiralities could be used to fine-tune supramolecular organization.
Inhibition of Aβ(1–42) Fibrillization
Given the recognized β-breaker character of Pro and the therapeutic avenues offered by the incorporation of d-amino acids in short sequences, the inhibitory activity of the eight stereoisomers toward fibrillization of the amyloid-forming Aβ(1–42) peptide was assessed starting with a ThT fluorescence assay.46 On the basis of the structural analysis described above, stereoisomers 3a/3b are inferred to be the best inhibitors, as their stereoconfiguration directed noncovalent interactions so that the disassembling nature of Pro prevailed over the assembling nature of Phe.
Among the eight stereoisomers, 3a was the best inhibitor, with activity analogous to d-Trp-Aib32 that served as a positive control (see Supporting Information Section S10). This dipeptide inhibited the ability of Aβ(1–42) to form toxic assemblies especially when incubated in a large molar excess of 40:1 or more in vitro.32 When tested on PC12 cells, the toxic effects induced by Aβ(1–42) were reduced with a maximum effect when d-Trp-Aib was used in 10-fold excess relative to Aβ(1–42) over a range of experiments that varied the Aβ(1–42):d-Trp-Aib ratio from 10:1 to 1:10.32 The design of this dipeptide aimed at overcoming the limited bioavailability of natural l-peptide inhibitors, such as Leu-Pro-Phe-Phe-Asp that was previously identified as effective inhibitor of Aβ(1–42) fibrillization when used in a 20-fold molar excess, in pioneering work by Soto and colleagues.47 Aromatic residues were demonstrated to play a key role in amyloid hydrophobic association and were identified as an effective target to impede formation of toxic oligomers and higher-order assemblies.48−51
Dose–response assays revealed that 11:1 3a tripeptide molecules per Aβ(1–42) were required to achieve the EC50, and further 3a concentration increase led to a maximum of 56% inhibition (Figure 5A). It is worth noting that the inhibitor high concentrations were relevant to the experimental protocol for ease of detection, rather than to the physiological levels of Aβ(1–42), or the envisioned therapeutic levels.32 These data, which fall within the range of inhibitor:Aβ concentration ratios that proved effective for similar short peptides,32,47,52 suggested multiple binding of the inhibitor to Aβ(1–42), as confirmed by CD spectra that showed a reduced β-sheet signature for Aβ(1–42) when 3a was present in a molar excess (Figure 5B and Supporting Information Section S10). A 250 ns all-atom MD simulation of Aβ(1–42) and 20 molecules of 3a in water solution revealed up to seven molecules of 3a binding to the Phe-Phe motif of Aβ(1–42), as expected by design (Figure 5C), and up to 16 binding to a single Aβ(1–42) peptide.
Figure 5.
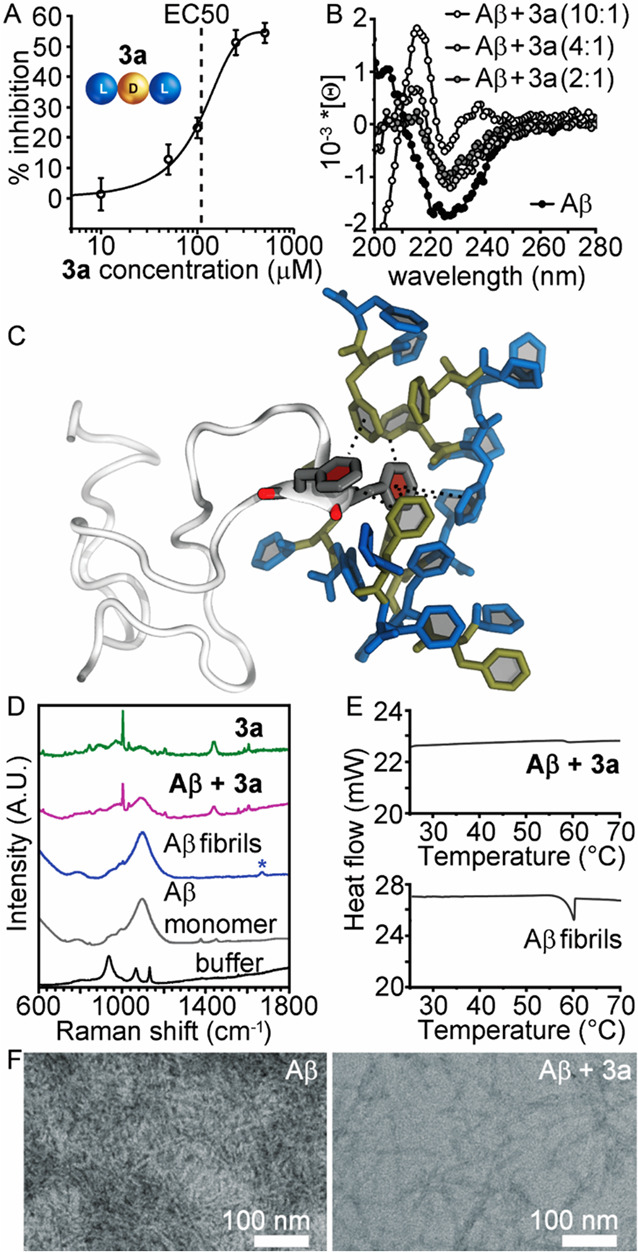
Aβ(1–42) fibrillization inhibition by 3a. (A) ThT assay dose–response curve (Aβ(1–42) at 10 μM). (B) CD spectra using Aβ(1–42) at 15 μM. (C) Snapshot extracted from a 250 ns all-atom MD simulation of LDL 3a (l-/d-amino acids in blue/gold) and Aβ(1–42) in gray revealing binding on the Phe-Phe motif as expected by design (interactions shown as dotted black lines). (D) Raman spectra using Aβ(1–42) at 10 μM (* denotes the amyloid diagnostic signal). (E) DSC data (Aβ(1–42) at 100 μM). (F) TEM micrographs.
Fibrillization inhibition was also confirmed by Raman spectroscopy, as the diagnostic amyloid signal53,54 in the amide I region at 1670 cm–1 (denoted with a blue star in Figure 5D) rose with fibrillization but it was absent when Aβ(1–42) was incubated with 3a. Under analogous conditions, differential scanning calorimetry (DSC, Figure 5E) qualitatively confirmed inhibition,55 as the exothermic peak in the thermogram centered at 57.3 ± 2.1 °C was significantly reduced in the presence of 3a. Nanomorphological analysis supported these findings as TEM micrographs of Aβ(1–42) showed a dense network of amyloid fibrils with widths of 8.6 ± 1.8 nm, in agreement with previous reports,56,57 whereas incubation with 3a led to fewer and shorter fibrils (Figure 5F).
Enzyme Stability and Biocompatibility of LDL 3a
Another critical aspect for the development of amyloidosis inhibitors is the rapid enzymatic processing in vivo undergone by natural l-peptides. Therefore, we tested the stability of 3a when incubated with a large excess of proteinase K, with l-homochiral 1a serving as a control. The latter was quantitatively hydrolyzed already after 1 h of incubation while only 34% of LDL 3a was digested after 48 h (Figure 6A). These results confirmed the stabilizing effect against proteases of even one d-amino acid strategically positioned at the center of the tripeptide sequence, thus allowing us to address one of the main limitations of peptide-based therapeutics. However, the limited enzyme-processability of heterochiral sequences may also raise concerns over their biocompatibility. To allay these concerns, keratinocytes or fibroblast cells were cultured in the presence of increasing concentrations of 3a, and cell viability was assessed by MTT or live/dead assays (Figure 6B,C). Only at the highest concentration tested were evident signs of cytotoxicity noted, with low cell viability, density, and altered morphology, as cells appeared more elongated with reduced volume. All amyloidosis inhibition tests were performed at lower concentrations in the micromolar range, however, at which no toxicity was detected in cell culture. Furthermore, the vast majority of xenobiotics are expected to exhibit side effects at millimolar concentrations. Overall, these data suggest that heterochiral short peptides are a promising starting point for the development of amyloid inhibitors.
Figure 6.
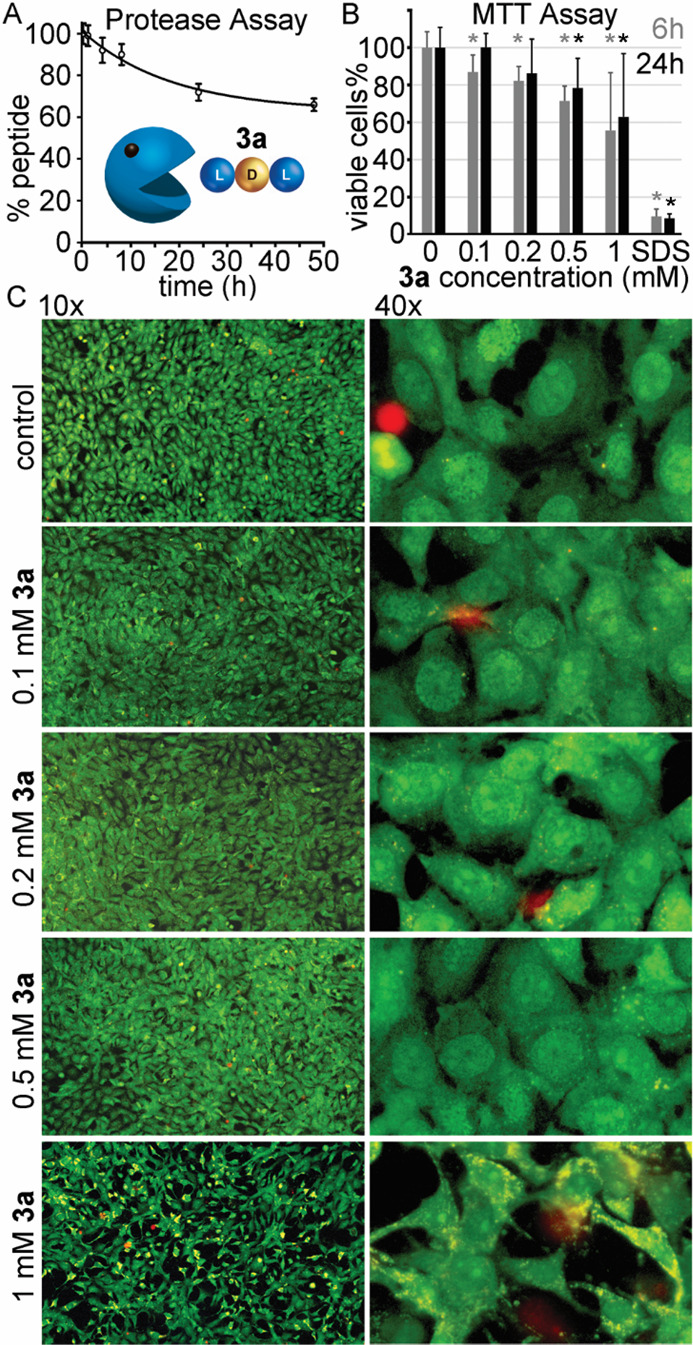
(A) Protease-digestion assay for 3a. (B) MTT assay for 3a on keratinocytes. * denotes p < 0.05 (t test). (C) Live–dead fluorescence microscopy images of fibroblasts cultured in the presence of increasing concentrations of 3a.
Conclusions
The presence of proline affects, but does not impede, assembly of unprotected tripeptides based on the Phe-Phe motif. With the additional variable of amino acid stereoconfiguration, it is indeed possible to direct the self-organization of Pro-Phe-Phe peptides that contain conflicting assembly instructions, so as to achieve a diversity of discrete nanostructures, such as nanotapes or nanoparticles, or even to obtain a self-supporting hydrogel. Furthermore, this soft material undergoes a gel-to-sol transition at a physiologically relevant temperature, which is convenient for the future development of smart biomaterials. Single-crystal XRD data and all-atom MD simulations revealed fine details of supramolecular packing for all of the peptides studied and were in agreement with spectroscopy and microscopy data, thus significantly advancing the understanding of these systems for their future application in biomedical nanotechnology.
The presence of divergent programming elements in a peptide sequence as short as three amino acids thus offers the opportunity to direct supramolecular behavior, based on the strategic use of heterochirality. When self-assembly prevails, long-range assembly can be achieved for fibrillization to occur and superstructures can organize effectively into a network holding together a macroscopic hydrogel, as demonstrated with peptides 2a/2b. When disassembly prevails, only short-range assembly is attained, and the disruptive nature of Pro can be exploited to extend it on fibrillating biological targets, as demonstrated on Alzheimer’s related Aβ(1–42) in vitro and shown effectively for 3a. Finally, a balance between the divergent effects can stabilize intermediate-range assembly, as noted for the nanotape-forming 4a/4b.
Homochirality thus represents a fundamental principle that governs the structure and function of living organisms, allowing heterochirality to serve as a strategic tool for the design of orthogonal systems to achieve structures and functions at the interface with living beings. This work has shown how the combination of simple elements with divergent supramolecular behaviors can be an effective approach to achieve structural and functional diversity for the design of biocompatible soft materials and therapeutics, allowing avenues to be envisaged for a broader scope of applications, spanning from artificial enzymes to innovative approaches directing protein-mediated cell fate.
Materials and Methods
Chemical Synthesis
2-Chlorotrytil and Fmoc-phenylalanine preloaded resin, O-benzotriazole-N,N,N,N′-tetramethyluronium hexafluorophosphate (HBTU), 1-hydroxy-7-azabenzotriazole (HOAt), and Fmoc-d-phenylalanine were purchased from GL Biochem (Shanghai) Ltd. All of the other chemicals and solvents of analytical grade were purchased from Merck. Tripeptides were synthesized on solid phase according to established procedures.34 The crude oils were purified by reverse-phase HPLC on an Agilent 1260 Infinity system equipped with a gradient pump, C-18 column (Kinetex, 5 μm, 100 Å, 250 × 10 mm, Phenomenex), autosampler, and photodiode array detector. The gradient used consisted of acetonitrile (MeCN)/water with 0.1% TFA with the following program: t = 0–2 min, 25% MeCN; t = 16 min, 95% MeCN; t = 16–18 min, 95% MeCN (Rt = 10–12 min). The compounds were then freeze-dried and characterized by ESI-MS, 1H NMR, and 13C NMR analyses. 1H NMR spectra were recorded at 400 MHz and 13C NMR spectra were recorded at 100 MHz on a Varian Innova Instrument with chemical shift reported as ppm (in DMSO or acetonitrile with tetramethylsilane as internal standard). LC-MS data were acquired on an Agilent 6120 LC-MS system with a C-18 analytical column (Zorbax SB-C18 Rapid Resolution HT 2.1 mm × 50 mm, 1.8 μm) with a single quadrupole ESI system. Flow was 0.5 mL/min. The gradient used consisted of acetonitrile (MeCN)/water with 0.1% formic acid with the following program: t = 0–2 min, 5% MeCN; t = 20 min, 95% MeCN (+0.1% formic acid). Spectroscopic data and HPLC retention times are reported in the Supporting Information, Sections S1 and S2.
Self-Assembly of Tripeptides
Each peptide was dissolved in phosphate buffer (sodium phosphate, 0.1 M; pH ∼ 11.9) by sonication at room temperature; then the solution was diluted with an equal volume of phosphate buffer (0.1 M sodium phosphate; pH ∼ 5.9) to reach pH neutrality. Self-assembly of peptide zwitterions thus occurred at physiological pH 7.4 and a concentration of 20 mM (except for the hydrogel at 24 mM). Note: complete dissolution of the peptide and fine pH control are important to achieve correct hydrogelation.
TEM
TEM micrographs were acquired on a Jeol instrument, JEM 2100, Japan, at 100 kV. TEM grids (copper-grid-supported lacey carbon film) were first rendered hydrophilic by a 20 min treatment in a UV–ozone cleaner (UV-Ozone Procleaner Plus). After 1 h of self-assembly, sample aliquots (∼20 μL) were deposited on top of a copper grid for 30 s (200 mesh, copper, carbon only or lacey carbon). Next, water was drawn from the sample and the grid was placed for 30 s in contact onto a drop of 2% aqueous potassium phosphotungstate at pH 7.2. Finally, the grid was dried first at room temperature and then in vacuo. Image analysis was performed using ImageJ software.
AFM
Samples were spread on a 1 cm2 mica substrate, and after 1 h of self-assembly, they were dried in vacuo for 24 h. AFM analysis was carried out on a Nanoscope V microscope (Digital Instruments Metrology Group, model MMAFMLN) in tapping mode at room temperature in air, using a standard μmasch SPM probe (HQ:NSC15/AIBS) with tip height of 12–18 μm and a cone angle < 40° (resonant frequency, 320 kHz; force constant ∼ 40 N m–1). Gwyddion software was used for image analysis.
DLS
The hydrodynamic size distributions were measured on a Fritsch, Analysette 12 DynaSizer (Germany) supported with NanoQ V2.5 software (Cordouan Technologies). DLS is equipped with variable laser power (from 1 to 65 mW) operating at 657 nm and a scattering detector at 135°. The peptide powder was dissolved in corresponding buffers immediately before measurements. A quantity of 50 μL of nondiluted sample was measured in an ultrathin layer (20 μm) to avoid multiple scattering events. All measurements were performed in triplicate at room temperature (25 °C).
Rheology
Hydrogel 2a was measured on a Malvern Kinexus Ultra Plus Rheometer (Alfatest) with a 2 cm steel parallel plate at 25 °C (Peltier, Alfatest). Samples were prepared in situ and analyzed with a gap of 1 mm. Time sweeps were recorded for 1 h at 1 Hz and 1 Pa; frequency sweeps were recorded at 1 Pa and stress sweeps at 1 Hz. Each analysis was repeated at least 3 times, and representative measures are reported in the Supporting Information, Section S3.
Circular Dichroism
Analyses were carried out in a 0.1 mm quartz cell using a Jasco J-815 spectropolarimeter, with 1 s integrations, step size of 1 nm, bandwidth of 1 nm, from 185 to 280 nm at 25 °C. Samples were prepared directly in the cuvette. For the hydrogel tripeptide 2a, a temperature ramp up to 85 °C was applied after 1 h of gelation. Aβ(1–42) was monomerized for 2 h in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, 500 μM); then, HFIP was evaporated under an argon flow and Aβ left overnight under vacuum. Each sample contained 15 μM Aβ(1–42) and the tripeptide 3a at different concentrations (2:1, 4:1, or 10:1 relative to Aβ) in PBS 20 mM (pH 7.4). Controls containing only 15 μM Aβ(1–42) and tripeptide 3a at different concentrations were also measured. Samples were incubated for 48 h in a thermomixer (150 rpm) at 37 °C; spectra were recorded for freshly prepared samples and after 24 and 48 h of incubation. Two independent experiments were performed. The data shown are the average of the five accumulations. Further data are shown in the Supporting Information, Section S10.
FT-IR
Spectra were recorded on a Varian 660-IR spectrometer in ATR mode (Ge crystal) with 80 scans, 4 cm–1 resolution. Each sample was spread onto a clean piece (∼1 cm2) of silicon wafer, and after 1 h of self-assembly it was dried in vacuo for 24 h.
Crystallization
Single crystals were grown by dissolving each peptide in 0.8 mL of MeOH inside a reservoir with 3 mL of a mixture of methanol and water. Peptides 1a/1b and 3a/3b were dissolved at 2.5 mM in methanol and the reservoir contained methanol/water (40:60). Peptides 4a/4b were dissolved at 1 mM methanol, and the reservoir contained methanol/water (50:50). Peptides 3a/3b also crystallized, dissolved at 10 mM in 10 mM PBS over a month.
Single-Crystal X-ray Diffraction
Single crystals of a single enantiomer for each crystallized pair were analyzed by X-ray diffraction at the XRD1 beamline of the Elettra synchrotron (Trieste, Italy). Crystals were collected from their mother solution using a nylon loop and stored in liquid nitrogen, with poly(ethylene glycol) (for 1a and 3a) or glycerol (for 4b) as cryoprotectant. Data collections were performed on the frozen crystals, kept at 100 K by a stream of nitrogen, with the rotating crystal technique. Data were indexed and integrated using either the software Mosflm58 (for 1a and 3a) or the XDS package59 (for 4b), and scaled using AIMLESS software.60 For all structures the phase problem was solved using direct methods, with software SHELX-T61 (for 1a), SIR 201462 (for 3a), or SHELXS63 (for 4b). Refinement cycles were conducted with SHELXL-14,64 by full matrix least-squares methods on F2.
Thioflavin T Test
ThT assay on the tripeptides was performed as previously described, using a concentration for each peptide of 20 mM, or 24 mM for 2a/2b, and 6 and 24 h as time points for self-assembly.26 For the Aβ(1–42) fibrillization inhibition studies, Aβ(1–42) was monomerized for 2 h in HFIP (0.5 mM); then HFIP was evaporated under an argon flow and left overnight under vacuum. Then, it was dissolved in DMSO at 0.3 mM and stored at −20 °C. Tripeptide solutions were prepared at 10 mM in DMSO and diluted to 2 mM with PBS 10 mM, being the final concentration in the plates, 0.2 mM. Thioflavin T solution was prepared at 25 μM in glycine–NaOH buffer (pH 8.5). The experiment was performed in a 96-well flat black clear-bottom plate (final Aβ(1–42) concentration of 10 μM and final volume per well = 150 μL). The plate was incubated at 37 °C with a shaking speed of 150 rpm, covered with aluminum foil. The bottom fluorescence was measured using a Tecan Infinite M1000 Pro, excitation 446 nm and emission 490 nm, respectively, for freshly prepared samples and after 24 and 48 h of incubation. Controls contained only Aβ(1–42), only tripeptides, or d-Trp-Aib. A sample containing only ThT was used as reference. Three independent experiments were carried out in triplicate.
Raman Spectroscopy
Raman spectra were recorded with an Invia Renishaw microspectrometer (50) equipped with He–Ne laser at 532 nm. At least 25 spectra (resolution 1 cm–1) per sample were collected to assess their homogeneity. Compound 3a (0.2 mM) was added 20:1 to Aβ(1–42) for 24 h, placed atop a 1 cm2 piece of a silicon wafer, and then dried in vacuo prior to analysis.
DSC
Aβ(1–42) was dissolved in 0.1 M aqueous ammonia solution at 1 mg/mL. Then, it was diluted with PBS 10 mM to 100 μM, with or without 3a (2 mM in PBS 10 mM), and samples were incubated at 37 °C for 48 h. Data were acquired on a Q100 (TA Instruments). For every measurement, 50 μL of each sample was transferred to the DSC pan and they were hermetically closed with their lids. DSC scans were performed using the following program: (1) isotherm at 20 °C for 1 min; (2) heating ramp from 20 to 80 °C at 5 °C/min; (c) isotherm at 80 °C for 5 min; (d) cooling to 20 °C at 5 °C/min. Measurements were repeated at least in triplicates for each independent sample.
Protease Assay
A 0.25 mL aliquot of peptide solution in DMSO (10 mM) was prepared from each peptide in 15 mL Falcon tubes, and 4.75 mL of 50 mM sodium phosphate buffer (pH 7.4) with a large excess (5 mg) of recombinant proteinase K (Sigma-Aldrich) was added. The tubes were incubated at 37 °C, 60 rpm, and at the selected time points, 0.2 mL of 1 M NaOH was added to 0.5 mL of the solution to inhibit further protease activity prior to HPLC analysis. The experiment was carried out twice in triplicate.
Molecular Models
Model structures of zwitterionic tripeptides were built upon the experimental structure (3a) or generated using the AmberTools19 package (D. A. Case et al. AMBER 2019 (University of California, San Francisco)) and the VMD1.9.3 software65 through in-house tcl scripts (2a). Multicopy MD simulations of the self-assembly process for 216 2a or 3a tripeptides were performed as previously described38 (see Supporting Information Section S6). To further investigate the molecular determinants of 2a fibrillization, an additional set of simulations was performed with 1,000 such peptides. The initial structures were generated by placing the center of mass of one peptide repeatedly on the points of a 10 × 10 × 10 (2a) or 6 × 6 × 6 (2a/3a) grid of 15 Å spaced points. Initial orientations of peptides were randomized, and the systems were solvated with water molecules (concentration ∼0.25 M). To investigate the molecular interactions occurring in solvent between 3a and Aβ(1–42), we performed a MD simulation of Aβ(1–42)66 interacting with 20 peptides in water solution. See Supporting Information Section S6 for further details.
Cell Culture
A 0.2 μm filtration setup was used to sterilize all solutions. MTT assays were performed as previously described.26 For live/dead assays, NIH3T3 fibroblasts (10,000 cells per well, 30 μL of DMEM + 10% fetal serum albumin, and 2% antimycotic and antibiotic from GIBCO) were added to the microwells of an uncoated μ-Slide for Angiogenesis (Ibidi, Germany) and cultured at 37 °C, 5% CO2 for 24 h, according to the manufacturers’ instructions. A stock solution of 3a (4 mg/mL in 10 mM PBS, pH 7.4) was diluted to the desired concentration (0.1, 0.2, 0.5, or 1 mM) and added to the wells. After 24 h, cell viability was assayed with acridine orange (5 μL/well of a 20 μM solution in 50 mM PBS) and propidium iodide (4 μL/well of a 30 μM solution in 50 mM PBS). After 15 min at 37 °C, cells were imaged with a Leica microscope (DFC450C; software LASV4.13) with fluorescence green filter (excitation, 450–490 nm; emission > 520 nm) with 10× and 40× objectives. Each condition was repeated in triplicate and the experiment twice. The t test was performed with Excel by comparing each condition against its control without peptide.
Acknowledgments
We acknowledge Elettra Sincrotrone Trieste for providing access to its synchrotron radiation facilities, and we thank M. Polentarutti, N. Demitri, and G. Bais for assistance in using beamline XRD1. This research used the Savio computational cluster resource provided by the Berkeley Research Computing program at the University of California, Berkeley (supported by the UC Berkeley Chancellor, Vice Chancellor for Research, and Chief Information Officer). A.V.V. thanks A. Bosin for assistance with the GPU cluster at the University of Cagliari. We acknowledge funding from the Italian Ministry of Education and Research (MIUR) through the SIR program (Personal Grant No. RBSI14A7PL to S.M.), Ramón Areces Foundation (fellowship to A.M.G.), the European Social Fund, Operational Programme 2014–2020 (Axis 3 – Education and Training, Specific Programme No. 26 – TALENTS3 Fellowship “MAGIC SPY” to S.K.). We acknowledge the financial support from the Slovenian Research Agency (ARRS) for Research Core Funding No. P2-0089, Project No. J1-7302, and the CENN Nanocenter (Slovenia) for TEM access.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.0c09386.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Lampel A.; Ulijn R. V.; Tuttle T. Guiding Principles for Peptide Nanotechnology through Directed Discovery. Chem. Soc. Rev. 2018, 47, 3737–3758. 10.1039/C8CS00177D. [DOI] [PubMed] [Google Scholar]
- Du X.; Zhou J.; Shi J.; Xu B. Supramolecular Hydrogelators and Hydrogels: From Soft Matter to Molecular Biomaterials. Chem. Rev. 2015, 115, 13165–13307. 10.1021/acs.chemrev.5b00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamley I. W. Small Bioactive Peptides for Biomaterials Design and Therapeutics. Chem. Rev. 2017, 117, 14015–14041. 10.1021/acs.chemrev.7b00522. [DOI] [PubMed] [Google Scholar]
- Krieg E.; Bastings M. M. C.; Besenius P.; Rybtchinski B. Supramolecular Polymers in Aqueous Media. Chem. Rev. 2016, 116, 2414–2477. 10.1021/acs.chemrev.5b00369. [DOI] [PubMed] [Google Scholar]
- Reynolds N. P. Amyloid-Like Peptide Nanofibrils as Scaffolds for Tissue Engineering: Progress and Challenges. Biointerphases 2019, 14, 040801 10.1116/1.5098332. [DOI] [PubMed] [Google Scholar]
- Kim S.; Kim J. H.; Lee J. S.; Park C. B. Beta-Sheet-Forming, Self-Assembled Peptide Nanomaterials towards Optical, Energy, and Healthcare Applications. Small 2015, 11, 3623–3640. 10.1002/smll.201500169. [DOI] [PubMed] [Google Scholar]
- Besenius P.; Portale G.; Bomans P. H. H.; Janssen H. M.; Palmans A. R. A.; Meijer E. W. Controlling the Growth and Shape of Chiral Supramolecular Polymers in Water. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 17888. 10.1073/pnas.1009592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Liu K.; Yan L.; Wang A.; Bai S.; Yan X. Trace Solvent as a Predominant Factor to Tune Dipeptide Self-Assembly. ACS Nano 2016, 10, 2138–2143. 10.1021/acsnano.5b06567. [DOI] [PubMed] [Google Scholar]
- Brown N.; Lei J.; Zhan C.; Shimon L. J. W.; Adler-Abramovich L.; Wei G.; Gazit E. Structural Polymorphism in a Self-Assembled Tri-Aromatic Peptide System. ACS Nano 2018, 12, 3253–3262. 10.1021/acsnano.7b07723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charalambidis G.; Georgilis E.; Panda M. K.; Anson C. E.; Powell A. K.; Doyle S.; Moss D.; Jochum T.; Horton P. N.; Coles S. J.; Linares M.; Beljonne D.; Naubron J.-V.; Conradt J.; Kalt H.; Mitraki A.; Coutsolelos A. G.; Balaban T. S. A Switchable Self-Assembling and Disassembling Chiral System Based on a Porphyrin-Substituted Phenylalanine–Phenylalanine Motif. Nat. Commun. 2016, 7, 12657. 10.1038/ncomms12657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karikis K.; Butkiewicz A.; Folias F.; Charalambidis G.; Kokotidou C.; Charisiadis A.; Nikolaou V.; Nikoloudakis E.; Frelek J.; Mitraki A.; Coutsolelos A. G. Self-Assembly of (Boron-Dipyrromethane)-Diphenylalanine Conjugates Forming Chiral Supramolecular Materials. Nanoscale 2018, 10, 1735–1741. 10.1039/C7NR08667A. [DOI] [PubMed] [Google Scholar]
- Sequeira M. A.; Herrera M. G.; Dodero V. I. Modulating Amyloid Fibrillation in a Minimalist Model Peptide by Intermolecular Disulfide Chemical Reduction. Phys. Chem. Chem. Phys. 2019, 21, 11916–11923. 10.1039/C9CP01846H. [DOI] [PubMed] [Google Scholar]
- Spitzer D.; Rodrigues L. L.; Straßburger D.; Mezger M.; Besenius P. Tuneable Transient Thermogels Mediated by a pH- and Redox-Regulated Supramolecular Polymerization. Angew. Chem., Int. Ed. 2017, 56, 15461–15465. 10.1002/anie.201708857. [DOI] [PubMed] [Google Scholar]
- Estrada L. D.; Soto C. Disrupting β-Amyloid Aggregation for Alzheimer Disease Treatment. Curr. Top. Med. Chem. 2007, 7, 115–126. 10.2174/156802607779318262. [DOI] [PubMed] [Google Scholar]
- Reches M.; Gazit E. Casting Metal Nanowires within Discrete Self-Assembled Peptide Nanotubes. Science 2003, 300, 625–627. 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- Marchesan S.; Vargiu A. V.; Styan K. E. The Phe-Phe Motif for Peptide Self-Assembly in Nanomedicine. Molecules 2015, 20, 19775–19788. 10.3390/molecules201119658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi K. B.; Verma S. Sequence Shuffle Controls Morphological Consequences in a Self-Assembling Tetrapeptide. J. Pept. Sci. 2008, 14, 118–126. 10.1002/psc.955. [DOI] [PubMed] [Google Scholar]
- Bera S.; Mondal S.; Xue B.; Shimon L. J. W.; Cao Y.; Gazit E. Rigid Helical-Like Assemblies from a Self-Aggregating Tripeptide. Nat. Mater. 2019, 18, 503–509. 10.1038/s41563-019-0343-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederix P. W.; Scott G. G.; Abul-Haija Y. M.; Kalafatovic D.; Pappas C. G.; Javid N.; Hunt N. T.; Ulijn R. V.; Tuttle T. Exploring the Sequence Space for (Tri-)Peptide Self-Assembly to Design and Discover New Hydrogels. Nat. Chem. 2015, 7, 30–37. 10.1038/nchem.2122. [DOI] [PubMed] [Google Scholar]
- Melchionna M.; Styan K. E.; Marchesan S. The Unexpected Advantages of Using D-Amino Acids for Peptide Self-Assembly into Nanostructured Hydrogels for Medicine. Curr. Top. Med. Chem. 2016, 16, 2009–2018. 10.2174/1568026616999160212120302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Zhou P.; Wang J.; Zhao Y.; Ma H.; Lu J. R.; Xu H. Left or Right: How Does Amino Acid Chirality Affect the Handedness of Nanostructures Self-Assembled from Short Amphiphilic Peptides?. J. Am. Chem. Soc. 2017, 139, 4185–4194. 10.1021/jacs.7b00847. [DOI] [PubMed] [Google Scholar]
- Cringoli M. C.; Romano C.; Parisi E.; Waddington L. J.; Melchionna M.; Semeraro S.; De Zorzi R.; Gronholm M.; Marchesan S. Bioadhesive Supramolecular Hydrogel from Unprotected, Short D,L-Peptides with Phe-Phe and Leu-Asp-Val Motifs. Chem. Commun. 2020, 56, 3015–3018. 10.1039/C9CC09947F. [DOI] [PubMed] [Google Scholar]
- Clover T. M.; O’Neill C. L.; Appavu R.; Lokhande G.; Gaharwar A. K.; Posey A. E.; White M. A.; Rudra J. S. Self-Assembly of Block Heterochiral Peptides into Helical Tapes. J. Am. Chem. Soc. 2020, 142, 19809–19813. 10.1021/jacs.9b09755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesan S.; Styan K. E.; Easton C. D.; Waddington L.; Vargiu A. V. Higher and Lower Supramolecular Orders for the Design of Self-Assembled Heterochiral Tripeptide Hydrogel Biomaterials. J. Mater. Chem. B 2015, 3, 8123–8132. 10.1039/C5TB00858A. [DOI] [PubMed] [Google Scholar]
- Méndez-Ardoy A.; Bayón-Fernández A.; Yu Z.; Abell C.; Granja J. R.; Montenegro J. Spatially Controlled Supramolecular Polymerization of Peptide Nanotubes by Microfluidics. Angew. Chem., Int. Ed. 2020, 59, 6902–6908. 10.1002/anie.202000103. [DOI] [PubMed] [Google Scholar]
- Kralj S.; Bellotto O.; Parisi E.; Garcia A. M.; Iglesias D.; Semeraro S.; Deganutti C.; D’Andrea P.; Vargiu A. V.; Geremia S.; De Zorzi R.; Marchesan S. Heterochirality and Halogenation Control Phe-Phe Hierarchical Assembly. ACS Nano 2020, 14, 16951–16961. 10.1021/acsnano.0c06041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insua I.; Montenegro J. 1D to 2D Self Assembly of Cyclic Peptides. J. Am. Chem. Soc. 2020, 142, 300–307. 10.1021/jacs.9b10582. [DOI] [PubMed] [Google Scholar]
- Méndez-Ardoy A.; Granja J. R.; Montenegro J. pH-Triggered Self-Assembly and Hydrogelation of Cyclic Peptide Nanotubes Confined in Water Micro-Droplets. Nanoscale Horiz. 2018, 3, 391–396. 10.1039/C8NH00009C. [DOI] [PubMed] [Google Scholar]
- Kieffer M.; Garcia A. M.; Haynes C. J. E.; Kralj S.; Iglesias D.; Nitschke J. R.; Marchesan S. Embedding and Positioning of Two Fe(II)4L4 Cages in Supramolecular Tripeptide Gels for Selective Chemical Segregation. Angew. Chem., Int. Ed. 2019, 58, 7982–7986. 10.1002/anie.201900429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V.; Rai R. K.; Arora A.; Sinha N.; Thakur A. K. Therapeutic Implication of L-Phenylalanine Aggregation Mechanism and Its Modulation by D-Phenylalanine in Phenylketonuria. Sci. Rep. 2015, 4, 3875. 10.1038/srep03875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler-Abramovich L.; Vaks L.; Carny O.; Trudler D.; Magno A.; Caflisch A.; Frenkel D.; Gazit E. Phenylalanine Assembly into Toxic Fibrils Suggests Amyloid Etiology in Phenylketonuria. Nat. Chem. Biol. 2012, 8, 701–706. 10.1038/nchembio.1002. [DOI] [PubMed] [Google Scholar]
- Frydman-Marom A.; Rechter M.; Shefler I.; Bram Y.; Shalev D. E.; Gazit E. Cognitive-Performance Recovery of Alzheimer’s Disease Model Mice by Modulation of Early Soluble Amyloidal Assemblies. Angew. Chem., Int. Ed. 2009, 48, 1981–1986. 10.1002/anie.200802123. [DOI] [PubMed] [Google Scholar]
- Adessi C.; Soto C. Converting a Peptide into a Drug: Strategies to Improve Stability and Bioavailability. Curr. Med. Chem. 2002, 9, 963–978. 10.2174/0929867024606731. [DOI] [PubMed] [Google Scholar]
- Iglesias D.; Melle-Franco M.; Kurbasic M.; Melchionna M.; Abrami M.; Grassi M.; Prato M.; Marchesan S. Oxidized Nanocarbons-Tripeptide Supramolecular Hydrogels: Shape Matters!. ACS Nano 2018, 12, 5530–5538. 10.1021/acsnano.8b01182. [DOI] [PubMed] [Google Scholar]
- Wu C.; Biancalana M.; Koide S.; Shea J. E. Binding Modes of Thioflavin-T to the Single-Layer Beta-Sheet of the Peptide Self-Assembly Mimics. J. Mol. Biol. 2009, 394, 627–633. 10.1016/j.jmb.2009.09.056. [DOI] [PubMed] [Google Scholar]
- Struthers M. D.; Cheng R. P.; Imperiali B. Design of a Monomeric 23-Residue Polypeptide with Defined Tertiary Structure. Science 1996, 271, 342–345. 10.1126/science.271.5247.342. [DOI] [PubMed] [Google Scholar]
- Richardson J. S.The Anatomy and Taxonomy of Protein Structure. Advances in Protein Chemistry; Academic Press: New York, 1981; Vol. 34, pp 167–339. [DOI] [PubMed] [Google Scholar]
- Garcia A. M.; Iglesias D.; Parisi E.; Styan K. E.; Waddington L. J.; Deganutti C.; De Zorzi R.; Grassi M.; Melchionna M.; Vargiu A. V.; Marchesan S. Chirality Effects on Peptide Self-Assembly Unraveled from Molecules to Materials. Chem. 2018, 4, 1862–1876. 10.1016/j.chempr.2018.05.016. [DOI] [Google Scholar]
- Seynhaeve A. L. B.; Amin M.; Haemmerich D.; van Rhoon G. C.; Ten Hagen T. L. M. Hyperthermia and Smart Drug Delivery Systems for Solid Tumor Therapy. Adv. Drug Delivery Rev. 2020, 163–164, 125–144. 10.1016/j.addr.2020.02.004. [DOI] [PubMed] [Google Scholar]
- Jain A.; Tiwari A.; Verma A.; Jain S. K. Ultrasound-Based Triggered Drug Delivery to Tumors. Drug Delivery Transl. Res. 2018, 8, 150–164. 10.1007/s13346-017-0448-6. [DOI] [PubMed] [Google Scholar]
- Yu S.; He C.; Chen X. Injectable Hydrogels as Unique Platforms for Local Chemotherapeutics-Based Combination Antitumor Therapy. Macromol. Biosci. 2018, 18, 1870030 10.1002/mabi.201870030. [DOI] [PubMed] [Google Scholar]
- Brahms S.; Brahms J. Determination of Protein Secondary Structure in Solution by Vacuum Ultraviolet Circular Dichroism. J. Mol. Biol. 1980, 138, 149–178. 10.1016/0022-2836(80)90282-X. [DOI] [PubMed] [Google Scholar]
- Goormaghtigh E.; Ruysschaert J.-M.; Raussens V. Evaluation of the Information Content in Infrared Spectra for Protein Secondary Structure Determination. Biophys. J. 2006, 90, 2946–2957. 10.1529/biophysj.105.072017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.; Li B.; Huang Z.; Li Y.; Yang Y. Terminal Is Important for the Helicity of the Self-Assemblies of Dipeptides Derived from Alanine. Langmuir 2013, 29, 6013–6017. 10.1021/la400910g. [DOI] [PubMed] [Google Scholar]
- Lin S.; Li Y.; Li B.; Yang Y. Control of the Handedness of Self-Assemblies of Dipeptides by the Chirality of Phenylalanine and Steric Hindrance of Phenylglycine. Langmuir 2016, 32, 7420–7426. 10.1021/acs.langmuir.6b01874. [DOI] [PubMed] [Google Scholar]
- Streich C.; Akkari L.; Decker C.; Bormann J.; Rehbock C.; Müller-Schiffmann A.; Niemeyer F. C.; Nagel-Steger L.; Willbold D.; Sacca B.; Korth C.; Schrader T.; Barcikowski S. Characterizing the Effect of Multivalent Conjugates Composed of Aβ-Specific Ligands and Metal Nanoparticles on Neurotoxic Fibrillar Aggregation. ACS Nano 2016, 10, 7582–7597. 10.1021/acsnano.6b02627. [DOI] [PubMed] [Google Scholar]
- Soto C.; Sigurdsson E. M.; Morelli L.; Asok Kumar R. A.; Castaño E. M.; Frangione B. β-Sheet Breaker Peptides Inhibit Fibrillogenesis in a Rat Brain Model of Amyloidosis: Implications for Alzheimer’s Therapy. Nat. Med. 1998, 4, 822–826. 10.1038/nm0798-822. [DOI] [PubMed] [Google Scholar]
- Wu C.; Lei H.; Duan Y. The Role of Phe in the Formation of Well-Ordered Oligomers of Amyloidogenic Hexapeptide (NFGAIL) Observed in Molecular Dynamics Simulations with Explicit Solvent. Biophys. J. 2005, 88, 2897–2906. 10.1529/biophysj.104.055574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia G. G.; Cavalli A.; Pellarin R.; Caflisch A. The Role of Aromaticity, Exposed Surface, and Dipole Moment in Determining Protein Aggregation Rates. Protein Sci. 2004, 13, 1939–1941. 10.1110/ps.04663504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makin O. S.; Atkins E.; Sikorski P.; Johansson J.; Serpell L. C. Molecular Basis for Amyloid Fibril Formation and Stability. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 315–320. 10.1073/pnas.0406847102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar A. P.; DuBay K. F.; Zurdo J.; Chiti F.; Vendruscolo M.; Dobson C. M. Prediction of “Aggregation-Prone” and “Aggregation-Susceptible” Regions in Proteins Associated with Neurodegenerative Diseases. J. Mol. Biol. 2005, 350, 379–392. 10.1016/j.jmb.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Horsley J. R.; Jovcevski B.; Wegener K. L.; Yu J.; Pukala T. L.; Abell A. D. Rationally Designed Peptide-Based Inhibitor of Aβ42 Fibril Formation and Toxicity: A Potential Therapeutic Strategy for Alzheimer’s Disease. Biochem. J. 2020, 477, 2039–2054. 10.1042/BCJ20200290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn J. D.; Lee J. C. Raman Fingerprints of Amyloid Structures. Chem. Commun. 2018, 54, 6983–6986. 10.1039/C8CC03217C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn J. D.; McGlinchey R. P.; Walker R. L.; Lee J. C. Structural Features of α-Synuclein Amyloid Fibrils Revealed by Raman Spectroscopy. J. Biol. Chem. 2018, 293, 767–776. 10.1074/jbc.M117.812388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasahara K.; Naiki H.; Goto Y. Kinetically Controlled Thermal Response of β2-Microglobulin Amyloid Fibrils. J. Mol. Biol. 2005, 352, 700–711. 10.1016/j.jmb.2005.07.033. [DOI] [PubMed] [Google Scholar]
- Yamin G.; Ruchala P.; Teplow D. B. A Peptide Hairpin Inhibitor of Amyloid β-Protein Oligomerization and Fibrillogenesis. Biochemistry 2009, 48, 11329–11331. 10.1021/bi901325g. [DOI] [PubMed] [Google Scholar]
- Xiong N.; Dong X.-Y.; Zheng J.; Liu F.-F.; Sun Y. Design of LVFFARK and LVFFARK-Functionalized Nanoparticles for Inhibiting Amyloid β-Protein Fibrillation and Cytotoxicity. ACS Appl. Mater. Interfaces 2015, 7, 5650–5662. 10.1021/acsami.5b00915. [DOI] [PubMed] [Google Scholar]
- Battye T. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. iMOSFLM: A New Graphical Interface for Diffraction-Image Processing with MOSFLM. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, D67, 271–281. 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, D66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. How Good Are My Data and What Is the Resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, D69, 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. SHELXT - Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr., Sect. A: Found. Adv. 2015, A71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burla M. C.; Caliandro R.; Carrozzini B.; Cascarano G. L.; Cuocci C.; Giacovazzo C.; Mallamo M.; Mazzone A.; Polidori G. Crystal Structure Determination and Refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. 10.1107/S1600576715001132. [DOI] [Google Scholar]
- Sheldrick G. A Short History of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, A64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. Crystal Structure Refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, C71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Wälti M. A.; Ravotti F.; Arai H.; Glabe C. G.; Wall J. S.; Böckmann A.; Güntert P.; Meier B. H.; Riek R. Atomic-Resolution Structure of a Disease-Relevant Aβ(1–42) Amyloid Fibril. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4976–E4984. 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.