

Abstract
With 28 potential N-glycosylation sites, human carcinoembryonic antigen (CEA) bears an extreme amount of N-linked glycosylation, and approximately 60% of its molecular mass can be attributed to its carbohydrates. CEA is often overexpressed and released by many solid tumors, including colorectal carcinomas. CEA displays an impressive heterogeneity and variability in sugar content; however, site-specific distribution of carbohydrate structures has not been reported so far. The present study investigated CEA samples purified from human colon carcinoma and human liver metastases and enabled the characterization of 21 out of 28 potential N-glycosylation sites with respect to their occupancy. The coverage was achieved by a multienzymatic digestion approach with specific enzymes, such as trypsin, endoproteinase Glu-C, and the nonspecific enzyme, Pronase, followed by analysis using sheathless CE-MS/MS. In total, 893 different N-glycopeptides and 128 unique N-glycan compositions were identified. Overall, a great heterogeneity was found both within (micro) and in between (macro) individual N-glycosylation sites. Moreover, notable differences were found on certain N-glycosylation sites between primary adenocarcinoma and metastatic tumor in regard to branching, bisection, sialylation, and fucosylation. Those features, if further investigated in a targeted manner, may pave the way toward improved diagnostics and monitoring of colorectal cancer progression and recurrence. Raw mass spectrometric data and Skyline processed data files that support the findings of this study are available in the MassIVE repository with the identifier MSV000086774 [DOI: 10.25345/C5Z50X].
Keywords: carcinoembryonic antigen, CEACAM5, glycosylation, colorectal cancer, glycopeptide, bottom-up proteomics, glycomics, CE-MS
1. Introduction
With more than half of all secretory and cellular human proteins being glycosylated, protein glycosylation is assumed to be the most common and highly complex post-translational modification (PTM).1 Glycoproteins play a role in an astonishing variety of cellular processes, in particular, cell–cell interaction, recognition, signaling, and adhesion processes on the cell surfaces. For instance, protein N-glycosylation regulates and fine-tunes critical immune response mechanisms and plays a major role in tumor recognition and antitumor responses.2,3 An assembly error, as minor as a single monosaccharide misplacement, may strongly impact the function of a glycoprotein and, in turn, the cell phenotype.4 Moreover, alterations of the N-glycan profile can actively contribute to tumor development and growth,5 as well as to the metastatic phenotype formation of the tumor cells.6 Therefore, the common acceptance of glycans and glycoproteins as cancer biomarkers is not surprising and keeps driving the cancer glycomics field forward.7
Currently, various analytical platforms are being used to study protein glycosylation. Among them, matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS),8,9 and electrospray ionization mass spectrometry (ESI-MS), coupled online or off-line, with liquid chromatography (LC)9,10 and capillary electrophoresis (CE)11,12 show considerable popularity. In-depth site-specific characterization of highly glycosylated proteins poses an extra challenge for an analytical method that can only be well-resolved on a glycopeptide level through bottom-up PTM mapping of the enzymatic glycoprotein hydrolysates.13,14 Whereas LC-MS is an acknowledged and well-established candidate for this task, CE-MS with a sheathless interface has advantages with respect to sensitivity and analyte coverage in bottom-up LC-MS analysis of glycopeptides.15,16 The excellent performance currently shown by sheathless CE-MS is due to the specific design of a novel interface that was first described by Moini.17 The use of this interface for glycopeptide analysis has been successfully demonstrated on moderately glycosylated proteins, paving the way toward more complex glycopeptidome separations and analysis.18,19
Human carcinoembryonic antigen (CEA) (UniProt entry P06731, CEACAM5_HUMAN) is a highly N-glycosylated protein of approximately 180 kDa with sugars accounting for about half of its molecular mass.20 CEA can be found in normal human colonic epithelial cells and is reported to be upregulated in tumor forming and colonic adenocarcinogenic cell lines;21,22 elevated CEA levels are valued as a progression and outcome biomarker in several forms of human cancers,23 including colorectal cancer (CRC).24 Continuous measurement of CEA serum levels is clinically used for the postoperative and post-therapy recurrence monitoring of CRC patients. Nonetheless, patients with no elevation of CEA in disease progression have been shown to maintain these low levels in case of recurrence.25 Another study also demonstrated a poor predictive value and low reliability of the CEA test for detecting treatable recurrences at an early stage.26 The CEA test is also used for initial clinical diagnostics of CRC, but its predictive value as a standalone biomarker in cancer screening or detection proves rather poor.27 As CEA is predominantly produced by the tumor cells and has been shown to tightly participate in the tumor metastatic events,28 the expectation is that the overly abundant and considerably heterogeneous glycosylation could be reflective of cancer development and progression. While some attempts have already been undertaken to profile CEA glycosylation and analyze its relationship with cancer29−31 and other biological processes,32 so far, its glycan structural and site-specific heterogeneity in the cancer context has remained largely unaddressed. Compared to the analysis of released N-glycans, the bottom-up approach allows to characterize the protein N-glycan pool with respect to the site occupancy. The same considerations are also relevant to its biomarker potential, as glycopeptides are gaining an increased attention in diagnostics of cancer and other diseases and are investigated as target substrates in cancer immunotherapy and immunodiagnostics.33−35
In this study we used a sheathless CE-MS to analyze CEA N-glycopeptides obtained after independent enzymatic digestions with trypsin, endoproteinase Glu-C, and Pronase. CEA samples retrieved from three different sources (two purified from human colon carcinoma and one purified from human liver metastases) were enzymatically hydrolyzed and subsequently analyzed by sheathless CE-MS/MS. In total, 21 out of 28 potential N-glycosylation sites, as well as their site-associated dominant N-glycan sets could be identified. The comparison between colon and metastatic CEA N-glycomes in terms of N-glycan classes and their structural features hints toward different N-glycosylation trends in primary and metastatic CRC tumors.
2. Materials and Methods
2.1. Chemicals, Reagents, and Samples
All the chemicals were of analytical reagent grade or higher. Proponan-2-ol (iPrOH), methanol (MeOH), ammonium bicarbonate (ABC), and sodium hydroxide (NaOH) were purchased from Merck (Darmstadt, Germany). Water (LC-MS grade), acetonitrile (MeCN) (LC-MS grade), glacial acetic acid (HAc), hydrochloric acid (HCl), DL-dithiothreitol (DTT), iodoacetamide (IAA), ammonium acetate (NH4Ac), formic acid (FA), TPCK-treated trypsin from bovine pancreas, and Pronase from Streptomyces griseus were purchased from Sigma-Aldrich (Steinheim, Germany). Endoproteinase Glu-C from Staphylococcus aureus V8 was supplied by Promega Corporation (Madison, WI, USA). CEA purified from human colon carcinoma was obtained from MyBioSource, Inc. (CEA1; San Diego, CA, USA) and Fitzgerald Industries International (CEA2; Acton, MA, USA). CEA purified from human liver metastases of colorectal carcinoma cells was obtained from Lee BioSolutions, Inc. (CEA3; Maryland Heights, MO, USA).
2.2. Digestion of CEA
Equal amounts of each CEA sample (10 μg) were taken for all the digestion procedures and diluted with 10 μL of 25 mM ABC buffer (1 μg/μL of protein). The protein disulfide bridges were reduced with 1 μL of 22 mM (tryptic) or 55 mM DTT (Glu-C or Pronase) for 30 min at 60 °C. After the sample was cooled to room temperature, 1 μL of 72 mM (tryptic) or 180 mM IAA (Glu-C or Pronase) was added. After addition of the alkylation reagent the samples were left in the dark for 30 min. Prior to adding the enzyme to the sample, the reaction was inhibited with 1 μL of 78 mM DTT and left for 30 min at room temperature. TPCK-treated trypsin (0.5 μg/μL), Glu-C (0.2 μg/μL) or Pronase (0.2 μg/μL) was added to the sample in an enzyme:substrate ratio of 1:10 w/w (trypsin) or 1:20 w/w (Glu-C and Pronase). Finally, incubation was performed overnight at 37 °C. Each enzymatic digestion was performed once for each CEA sample.
2.3. Sheathless CE-MS/MS
CE experiments were carried out on a SCIEX/Beckman Coulter CESI 8000 system (SCIEX, Framingham, MA) equipped with a temperature-controlled sample tray and a power supply delivering up to 30 kV. A 91 cm long (LT) × 30 μm i.d. × 150 μm o.d. bare fused-silica capillary (Silica Surface OptiMS cartridge, SCIEX) with the high sensitivity porous sprayer in the outlet tip was used for all the separations. A 10% v/v HAc water solution (pH 2.3) was used as a background electrolyte (BGE). Prior to each sample injection, the capillary was rinsed at pressure of 5 psi with 0.1 M NaOH (2.5 min), water (4 min), 0.1 M HCl (2.5 min), water (4 min), and BGE (4 min). An online sample preconcentration by transient isotachophoresis (t-ITP) was achieved by diluting the CEA digests to a final concentration of 100 mM NH4Ac at pH 4.0, which acted as a leading electrolyte solution (final concentration of digested protein 0.40 μg/μL).36 Samples were hydrodynamically injected in three different amounts: 1 psi for 60 s, 5 psi for 60 s and 8 psi for 60 s. In all experiments, sample injection was preceded by a water dip and followed by a postplug BGE injection (both at 0.5 psi for 25 s) to enhance t-ITP stacking and to prevent sample loss. A separation voltage of 20 kV (normal polarity, anode at the capillary inlet) was applied for all electrophoretic separations, the temperature was set for 25 °C. Multiple analysis (n = 3) were carried out for every digested CEA sample per injected volume.
The CE instrument was hyphenated to an Impact HD UHR-QqTOF-MS (Bruker Daltonics, Bremen, Germany) via a CESI OptiMS Bruker MS adapter kit (SCIEX) that allowed an optimal positioning of the capillary porous tip in front of the mass spectrometer nanospray shield (Bruker Daltonics). All experiments were carried out using dopant enriched nitrogen gas (DEN-gas).19 For this purpose, an in-house made polymer cone was slid onto the housing of the porous tip, allowing for a coaxial sheath flow of the DEN-gas around the nano-ESI emitter (MeCN was used as a dopant). Under optimized conditions, CE-MS/MS experiments were carried out in ESI positive mode using the following parameters: glass capillary voltage at 1200 V, drying gas temperature at 150 °C, drying gas flow rate at 1.2 L/min, nebulizer gas pressure at 0.2 bar, quadrupole ion energy at 3.0 eV and collision cell energy at 7.0 eV. MS data was acquired between m/z 200 and 2000 with a spectral acquisition rate of 1 Hz. MS/MS spectra were acquired in a data dependent mode with an absolute threshold of 4548 counts and active exclusion. Specific m/z values that were already acquired three times were excluded and released after 0.8 min, unless the precursor had a five times higher intensity than the observed in the previous acquisitions. Raw CE-MS/MS data are available via the MassIVE repository with identifier MSV000086774 [DOI: 10.25345/C5Z50X].
2.4. Data Analysis
Manual interpretation of CE-MS/MS spectra were performed in DataAnalysis 4.3 (Bruker Daltonics, Build 110.102.1532). All mass spectra were recalibrated internally with sodium acetate clusters detected at the beginning of the electrophoretic runs. Carbohydrate moieties of the N-glycopeptides were deduced from the fragmentation spectra by manual annotation on the basis of general glycan fragmentation rules37 and/or basic rules of the N-glycan biosynthetic pathway.38 Glycoforms were labeled with glycan net compositions specifying number of hexoses (Hex), N-acetylglucosamines (HexNAc), fucoses (Fuc), and N-acetylneuraminic acids (NeuAc); these monosaccharide abbreviations will be further used throughout this manuscript. Exemplary annotations of MS/MS N-glycopeptide spectra are provided in Supporting Information, Figure S1. N-Glycopeptides were included based upon their exact mass (±10 ppm), signal-to-noise (S/N; >9), and migration order. Briefly, all MS/MS scans were screened for oxonium ions (m/z 204.087, 366.140, and 292.103, singly charged HexNAc1, Hex1HexNAc1, and NeuAc1 fragments, respectively). The peptide mass was deduced by subtracting 203.079 (neutral loss of HexNAc) from the characteristically intense Y1 ion (corresponding to [peptide + HexNAc + H]+) and then matched against in silico digests of CEA (trypsin and Glu-C) to determine the amino acid sequence and N-glycosylation site position. In addition, the presence of low-intensity peptide b-ions was used to further confirm the amino acid sequence. The glycan composition could be assigned based upon the presence of other Y-ions in the spectra as well as by the neutral loss of (combined) monosaccharides from the precursor mass. In case of Pronase, deduced peptide Mr values were matched to theoretical Mr values of randomly cleaved peptides from CEA generated by the FindPept tool (http://www.expasy.org/tools/findpept.html, with a mass accuracy value of 10 ppm).39 Through introducing some minor adaptations of the default data processing pipeline of Skyline software (MacCoss Lab Software, version 20.1.0.76), nonfragmented N-glycopeptides in the MS1 spectra were identified as well as to confirm the identity of the manually annotated fragmented species. Briefly, all MS/MS-confirmed glycan compositions, their expected derivatives inferred from the N-glycan biosynthetic pathway and extra glycan species found in the glycoprofile of tumor-associated CEA in the literature29 were loaded into Skyline software as possible modifications for all the samples (in total 128 compositions (Supporting Information, Table S1), 89 of these were MS/MS-confirmed by at least one glycopeptide in at least one sample type (CEA1, CEA2, or CEA3)). In addition, a peptide mass list was created for every identified peptide backbone of every manually detected N-glycopeptide. Upon the basis of this mass list in combination with all possible glycan compositions, a full assignment was provided by Skyline for the tryptic and Glu-C digestions. Within a single data processing step, improved peak assignment and quantification of the relative peak area of all compounds could be achieved with Skyline. All assigned N-glycopeptides by Skyline were visually evaluated based upon peak shapes of the detected protonated molecular ions of a compound, ppm error (±10 ppm) as well as on the fitness and quality of the isotopic patterns (dot product value >0.8), only those that passed these criteria were chosen for quantitation. After this, generated peak areas were normalized per sample and per N-glycosylation site to obtain their relative abundances (percentage of all glycoform peak areas detected per site); these values were used for further analyses and visualization. Despite Skyline software tool not being primarily designed for glycopeptide profiling and annotation, its above-mentioned features appeared to be very useful for this application. Within a single data processing step Skyline can improve peak assignment and quantify the relative area of all compounds. All resulting N-glycopeptides were supplied with a graphical structural representation of the attached glycan (Supporting Information, Tables S2 and S3). A representation of the glycopeptide annotation and quantitation pipeline highlighting the Bruker DataAnalysis and Skyline software environments and features are shown in Supporting Information, Figure S2. Skyline processed data are available via the MassIVE repository with identifier MSV000086774 [DOI: 10.25345/C5Z50X].
3. Results and Discussion
3.1. Analysis of CEA N-Glycopeptides by Sheathless CE-MS/MS
CEA N-glycopeptides were generated using three different proteases and analyzed by CE-MS/MS. Representative extracted ion electropherograms (EIEs) of several N-glycoforms related to N-glycosylation site N650 (tryptic peptide backbone ITPNNN650GTYACFVSNLATGR) are illustrated in Figure 1A. A broad range of glycans was observed varying in the overall composition of monosaccharide units, their structural assembly and charge (presence of terminal NeuAc). The glycan portions were found to strongly affect the electrophoretic mobility of the N-glycopeptides. The number of negatively charged NeuAc units determined the formation of distinct glycoform clusters per peptide backbone. Semiempirical models can be used to relate the molecular mass (Mr) and the charge (q) of a peptide to its electrophoretic mobility (me) and to predict the electrophoretic migration behavior. Recently, it has been shown that the classical polymer model in which q/Mr1/2 is proportional to me can be applied to predict the electrophoretic mobility of N-glycopeptides.40 An illustration of this model is shown in Figure 1C, where for a range of N-glycoforms the theoretical Mr, q, and q/Mr1/2 values are provided next to their experimentally obtained migration time values, which are inversely proportional to me. The relationship observed between migration times and q/Mr1/2 was in accordance with the classical polymer model (Figure 1B). By investigating these models, structural modifications, charge characteristics and conformations of peptides can be studied. In our study these plots were constructed for all the detected N-glycoforms with the same peptide backbone as a sanity check of the glycopeptide structure assignment which was primarily based on MS/MS data.
Figure 1.
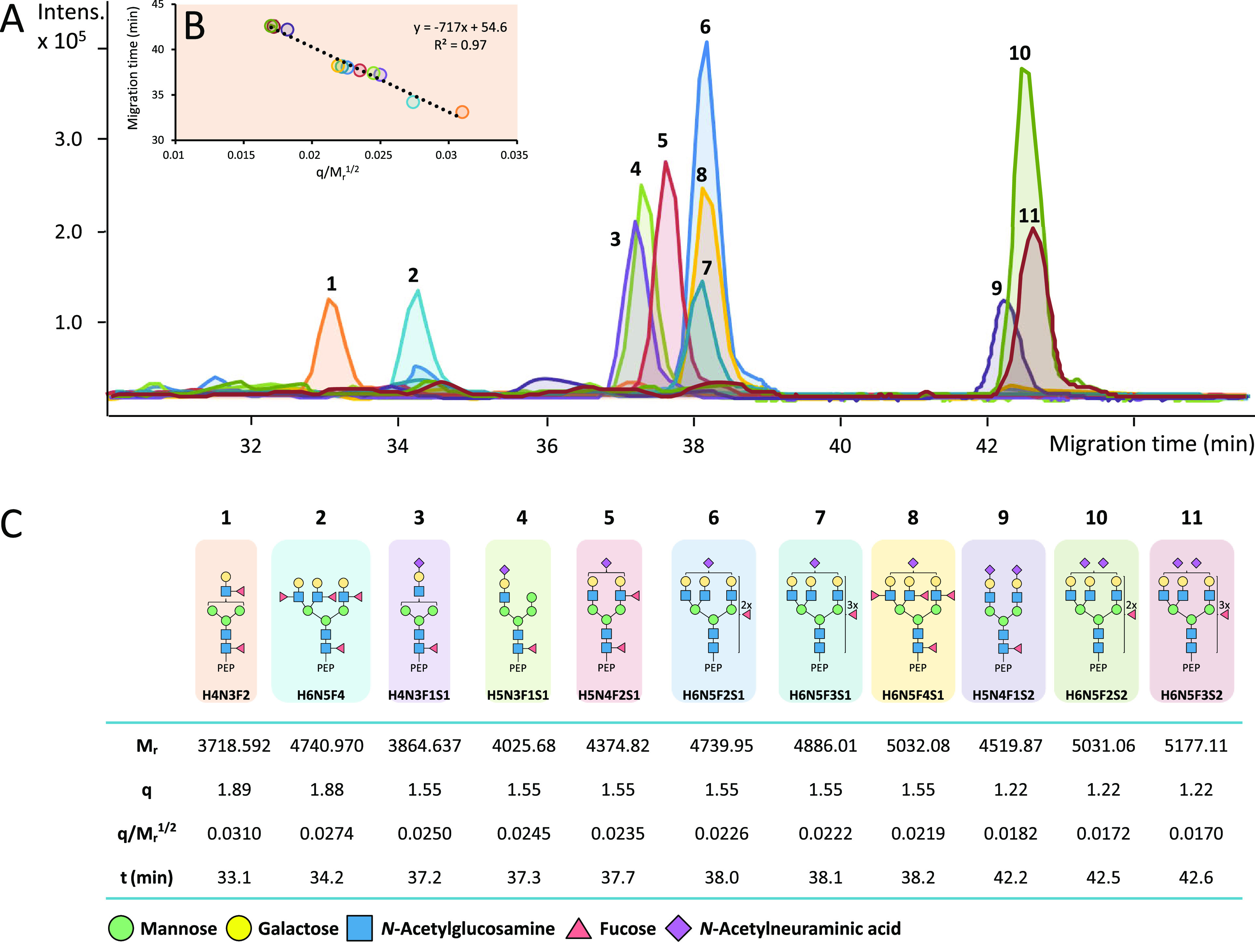
Experimentally observed linearity of electrophoretic behavior exemplified with N-glycopeptides sharing ITPNNNGTYACFVSNLATGR peptide backbone (N-glycosylation site N650). Experimental migration times of the N-glycopeptides in the electropherogram (A) were fitted to a linear regression line (B) with the classical polymer semiempirical model (q/Mr1/2).40 The table (C) showcases theoretical Mr, q, and q/Mr1/2 values and experimentally obtained migration time values, which are proportional to me. Blue square: N-acetylglucosamine (N), green circle: mannose (H), yellow circle: galactose (H), red triangle: fucose (F), pink diamond: N-acetylneuraminic acid (S).
3.2. Sequence Coverage and Characterization of CEA (Glyco)Peptides
Figure 2 shows the amino acid sequence of CEA and its potential 28 N-glycosylation sites (marked in bold red). In addition to the numerous N-glycosylation sites, CEA was expected to have a considerable heterogeneity in the sugar content per site of the protein, hence the analysis and structural elucidation of N-glycopeptides from CEA enzymatic digests was not straightforward. Taking these factors into account, digestion with several proteases or mixtures of proteases was considered, as it was previously demonstrated to increase sequence coverage and improve the PTM characterization.41,42 Three different proteolytic enzymes were used to generate three distinct protein digests for each CEA sample. Trypsin and Glu-C are specific serine proteases that cleave C-terminal peptide bonds of lysine (K) and arginine (R), or aspartic (D) and glutamic acid (E), respectively. Spectra of the MS/MS analysis of trypsin and Glu-C digests are often sufficient to characterize moderately glycosylated proteins.41 In particular, Glu-C is able to generate the peptide backbones that are less likely to be covered by trypsin, as the trypsin-specific cleavage sites that have D and E amino acids in close proximity to R amino acid have lower probability of being hydrolyzed.43 However, the abundant glycosylation on CEA may account for inefficient cleaving in PTM-rich protein regions and large glycopeptides with multiple glycosylation sites, thereby impairing the annotation. Another issue are the nonglycosylated peptides in proteolytic digests suppressing the glycopeptide ionization in ESI-MS and leading to substantial sensitivity reduction for glycopeptides.41 Pronase—a nonspecific mixture of proteolytic enzymes—was reported to help in circumventing these limitations.44 Pronase produces smaller peptide moieties (typically 1 to 8 amino acids) and usually cleaves nonglycosylated peptides to single amino acids, which reduces signal suppression of N-glycopeptides.45 However, since nonspecific proteases generate peptides rather haphazardly, CE-MS/MS data sets from Pronase digests were harder to interpret and, most importantly, the resulting N-glycopeptides could not be reliably quantified. Therefore, these N-glycopeptides were viewed from an exploratory perspective only, namely for providing a better overview of the total glycoprofile and enriching the putative modifications list. When combining the information obtained with the different enzymes, 21 out of the potential 28 N-glycosylation sites were covered. These peptide backbones are highlighted on the CEA sequence in Figure 2. Some very short N-glycopeptides from Pronase digests were annotated (Supporting Information, Table S3) but not mapped, due to the lack of specificity of the sequence motif and could be assigned to multiple glycosylation sites. Additionally, the peptide mass fingerprint data of all samples were searched against the human Uniprot database with Mascot Daemon (v.2.5.1, Matrix Science); the resulting peptide coverage is illustrated in Supporting Information, Figure S3). Albeit the total peptide coverage appears to be rather low, this could be explained by the high glycosylation degree of CEA. The Mascot matching algorithm is not designed to map glycosylated peptides, moreover, glycans are likely to cause steric hindrance for the proteases attempting to reach the cleavage site. From Glu-C digests a rather long peptide (Ala174–Asp227) was found as an unmodified sequence only, although it is harboring N-glycosylation specific sequons some of which are glycosylated and characterized from our other data sets (e.g., Pronase digests). That underscored the proteoform variability in N-glycosylation for these sites, i.e., a certain portion of the glycoprotein does not bear glycan modifications on these putative sites.
Figure 2.

Human carcinoembryonic antigen (CEACAM5_HUMAN) sequence. Putative N-glycosylation sites are indicated in bold red. Glycopeptides that were detected after specific proteases digestion are highlighted in green (Glu-C) and orange (trypsin). The glycopeptide coverage by a nonspecific protease (Pronase) digestion is shown in blue. The total molecular mass of the glycosylated protein is reported to vary between 150 000 and 200 000 Da; the mass calculations are based on the most prominent mass (180 000 Da).21
3.3. Site-Specific N-Glycoprofile of CEA
Supporting Information, Table S2 represents all the discovered N-glycopeptides that were quantified (trypsin and Glu-C digestion) and Supporting Information, Table S3 provides those that were only structurally characterized (Pronase digestion), all grouped by N-glycosylation site for the three CEA samples. To ease the interpretation of the trends in differential N-glycosylation patterns and structural features of the glycoprofiles, glycosylation traits (namely glycosylation type, as well as antennarity, fucosylation, and sialylation present on all glycans (total) and within the complex types (complex)) were mathematically derived from the data sets with relative abundance values (Supporting Information, Figure S4, derived traits calculation is provided in Supporting Information, Table S4). Moreover, to obtain a better representation of the most characteristic N-glycan species, the top 10 N-glycans (based upon their relative abundance) were selected per quantifiable N-linked glycosylation site and plotted as histograms in Figure 3 and Supporting Information, Figure S5.
Figure 3.
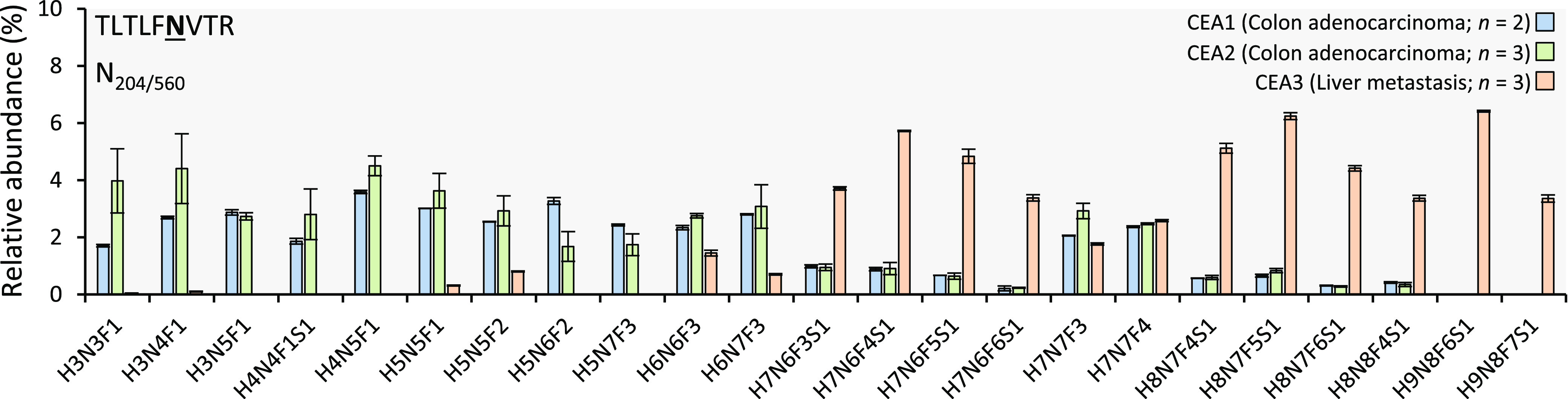
Relative abundances of the 10 most abundant glycoforms of the quantitatively characterized N-glycosylation site N204/560 (CEA1 is plotted in blue, CEA2 in green and CEA3 in orange). Peak area values were normalized by N-glycosylation site for all samples (as percentage of all glycoforms peak areas detected per site). The top 10 most abundant glycoforms of all N-glycosylation sites quantitatively characterized can be found in Supporting Information, Figure S4. Error bars indicate the standard deviation. H: hexose, N: N-acetylhexosamine, F: fucose and S: N-acetyl neuraminic acid.
Overall, the tryptic digestion allowed the identification of a total number of 489 different N-glycopeptides across the three CEA sample types, corresponding to eight N-glycosylation sites (N197/N553, N204/N560, N375, N580, N650, and N665). Unfortunately, no differentiation could be made between N-glycopeptides belonging to sites N197 and N553 or N204 and N560, as they presented the same peptide sequences after digestion with trypsin (LQLSNGN197/553R and TLTLFN204/560VTR, respectively). Some N-glycosylation sites were expected, but not found in the data (e.g., N152, N208, N246, N480). This may be caused by glycans shielding the theoretical tryptic cleavage sites, which resulted in large peptide moieties (aa ≥ 20). Furthermore, certain peptide sequences carried several potential N-glycosylation sites. Hence, if all of these sites were occupied, the complexity of the glycopeptide would complicate the detection and identification of these sites. Interestingly, site N197/553 was represented as two charge variants: LQLSNGN197/553R and its deamidated version; LQLSDGN197/553R, which has not been reported yet. It is well-known that asparagine (N) can undergo spontaneous deamidation both in vivo and in vitro and the rate increases under temperature and pH typical for tryptic digestion.46 Therefore, this modification could happen as a result of peptide degradation during sample preparation. However, no other asparagine degradation was observed for (glyco)peptides in any of the samples. The peak areas and shapes, observed for both charge variants, were quite similar in value and appearance, and they migrated one after another as expected. Moreover, the glycan subsets of the charge variants overlapped significantly, but not fully (Supporting Information, Table S2 and Figure S5). Taking into account that the peptide sequence is shared between the two N-glycosylation sites, it was possible to assume that the two peptides originated from different parts of the protein, but further research is needed to confirm this suggestion.
The Glu-C digest resulted in 150 unique N-glycopeptides and yielded the identification of three additional N-glycosylation sites (N152/N508, N466) compared to the tryptic digest and overlapped in one site (N580). However, two of these sites (N152/N508) could not be reliably quantified due to peptide backbone variability from the nonspecific Glu-C cleavages (Supporting Information, Table S3). Interestingly, the site that was detected in both trypsin and Glu-C digests (N580), revealed much less glycoforms in the trypsin data than in the Glu-C data (albeit fully overlapping). This further points toward the possible interference of abundant and bulky N-glycan modifications with the performance of certain enzymes, in line with literature.43,47
With regard to Pronase, the digests revealed in total of 254 N-glycopeptides. Due to the nonspecificity of the enzyme, a partial overlap in coverage was found between the Pronase generated N-glycopeptide pool and the glycopeptide sets produced by the specific enzymes. Nonetheless, 10 new sites were confirmed (N104, N208, N256, N274, N330, N351, N432, N480, N529, N612) and Pronase digest contributed to the characterization of 21 N-glycosylation sites in total.
Throughout the identified and quantified N-glycosylation sites, different classes of N-glycans were observed on the same site, including some high-mannose and hybrid types but mainly complex structures (bi-, tri-, and tetra-antennary structures). Some nonquantifiable sites (N152/508, N351, N256 in Supporting Information, Table S3) appeared to be populated only with high-mannose type structures. The rest of identified N-glycopeptides from nonquantifiable data were a mixture of different N-glycan types, though the majority of these were strongly overlapping in sequence position. Therefore, more high-mannose only sites could exist on the CEA structure.
Figure 4 gives an overview of the site-specific N-glycosylation of the three CEA samples. The overall resulting N-glycosylation site coverage is rather comprehensive, albeit the B1 protein domain was covered quite poorly. From the Pronase digest, small high-mannose type glycoforms structures could be assigned to this sequence area (Supporting Information, Table S3), which is most likely caused by steric hindrance from the glycans shielding binding site for the specific proteases. Figure 4 gives the N-glycan structures with the highest relative abundance per CEA sample analyzed. The N-glycosylation sites (indicated in red) are covered from the Pronase digests and, as these could not be reliably quantified, were not accompanied by the most abundant glycan structures. Visual evaluation prompts two outcomes: distal part (C-terminal) area of CEA is easier to reach by specific proteolytic enzymes and it harbors more potential differential traits of increasing malignancy potential of the tumor cells. In particular, the highest levels of fucosylation and, especially, sialylation, were observed for the liver metastases CEA sample (CEA3), which presented a higher degree of metastatic involvement (Figure 3 and Supporting Information, Figures S4 and S5).
Figure 4.

Schematic representation of human carcinoembryonic antigen (CEACAM5_HUMAN) domain structure with the N-glycosylation sites revealed by this study marked for the different type of CEA samples. The CEA domains are depicted as barrels and N-glycosylation sites (shown as double dots) are mapped along the structure. The black double dots are identified N-glycosylation sites in this study (tryptic or Glu-C digest) and are supplied with the most abundant N-glycan composition per all three types of CEA analyzed (side panels). The N-glycans highlighted in orange (N197 and N553) and in green (N204 and N560) share the same peptide backbone, hence the discovered glycan populations are shared. Confidently characterized by nonspecific digestion data sites are shown as red double dots (Pronase) and are not accompanied by the most abundant glycan structures due to the lack of reliable quantification. Undiscovered N-glycosylation sites are shown as gray double dots. All identified N-glycopeptides can be found in Supporting Information, Table S2 (quantified) and S3 (nonquantified)). Blue square: N-acetylglucosamine (N), green circle: mannose (H), yellow circle: galactose (H), red triangle: fucose (F), pink diamond: N-acetylneuraminic acid (S).
Looking closer at the overall N-glycan population for the three different types of CEA samples analyzed, a characteristic cancer glycosylation pattern emerges. To start with, a notable expression of truncated/paucimannosidic and high-mannose structures was observed in all three sources analyzed in this study. This feature is hypothesized to raise from incomplete N-glycan processing in the early stage of CRC.48,49 As could be seen from the glycosylation type panels in Supporting Information, Figure S4, the location of these structures has no predilection toward any particular area of the protein. Despite the vast presence of incomplete structures, they are not very abundant and moderately contribute to the overall CEA glycoprofile.
Bisection and branching appear to have a controversial role in cancer glycobiology. A fair amount of bisecting complex-type structures represents an advantage in immune evasion and metastatic potential50 and impacts several cancer-assisting biological pathways.51 Increased branching is also viewed as both cancer-associated and oncogenesis-promoting.48,51 It is widely accepted that high bisection levels are likely to coincide with low degrees of branching as bisecting GlcNAc expression is governed by GNT3 enzyme that effectively inhibits the activities of other GlcNAc transferases.52 Surprisingly, this study revealed that both branching and bisection levels are relatively high in all CEA samples and particularly high for CEA3 (liver metastasis). Furthermore, in many cases both features were combined within the same glycan structure which, to the best of our knowledge, is uncommon for cancer-related phenotypes or CEA glycosylation features.29 Notable differences in antennarity between primary colon carcinoma (CEA1 and CEA2) and metastatic tumor tissue (CEA3) samples were found on sites N204/560, N375, N466, N650 and N655 (Supporting Information, Figure S4). Peculiarly, while it is typical for mammalian cells to terminate GlcNAc residues with galactose units, the detected highly branched tetra-antennary structures often had their antennae only partially occupied by galactose (i.e., incomplete galactosylation), indicating lowered levels of substrate or β4GalT enzyme dysregulation. Similar findings of lowered galactosylation in tumor-associated CEA N-glycome were obtained before, by lectin microarray analysis, and the authors speculated about its role in immune response involving CEA.53 The same study also reported overall increased levels of GalNAc in tumor-associated CEA, which could also explain and/or contribute to undergalactosylation of antennae. Alongside with high branching, CEA N-glycans showed proclivity toward (poly-)LacNAc elongation on highly branched structures, which is also a known CRC feature associated with poor prognosis.48 This signature difference between primary and metastatic samples is the most notable in the most distal part of CEA sequence (sites N650 and N655) and is not reflected much along the other sites.
Sialylation changes are commonly found in cancer biology and have potential for diagnosis and therapy.54−56 Particularly, increased sialylation has often been associated with cancer invasiveness, and elevated serum total sialylation (TSA), and particularly high level of sialylation were found for liver metastases.57,58 Induced by dysregulation of sialidases and sialyltransferases aberrant NeuAc patterns play a role in immune evasion and cancer associated inflammation. CRC is no exception, as increased sialylation is shown to contribute to postoperative recurrence,59 therapeutic resistance and metastatic spread.60 The CEA samples exhibited an overall moderate degree of sialylation (even highly branched N-glycans never harbored more than two NeuAcs), while elevated sialylation levels were found for the CEA3 sample on sites N204/560, N375, N580, N650, and N665 (Supporting Information, Figure S4). Total sialylation was rather in line with complex-type sialylation levels indicating low contribution of hybrid-type N-glycans in the overall sialylation pool. NeuAc modifications (e.g., methylation, acetylation and sulfation) were not included in the scope of this work. It is also worth mentioning that NeuAc linkage isomers (α-2,3 and α-2,6 and α-2,9 variants) were proven to be an important discriminatory trait in several cancers, including CRC.60 In this study, no NeuAc linkage information could be obtained, but, acknowledging the importance of the trait, experimental considerations for linkage specificity evaluation will be included in follow-up research.
Amid the glycan traits discussed above and illustrated in Supporting Information, Figure S4, fucosylation was the most striking feature of the CEA N-glycome in the analyzed samples. Aberrant increase in fucosylation—arising from a large pool of GDP-fucose donors, abnormal expression of enzymes, and substrate availability—is reputed to be a red flag in many oncological diseases (brain, colorectal, breast, liver, lung, and other cancers) and it is involved in multiple stages of cancer biology (proliferation, survival, multidrug resistance, invasion and metastasis) and sustained inflammatory processes.61,62 Whereas upregulation of core fucosylation (governed by FUT8 enzyme overexpression) has been reported to directly promote epithelial-mesenchymal transformation (EMT) and dedifferentiation (i.e., metastatic potential),62 terminal fucosylation is partaking in the generation of Lewis antigens. Upon interaction with certain selectins, Lewis type antigens (on both immune cells and especially cancer cells) promote angiogenesis, reform tumor microenvironment, and, paradoxically, enhance cancer progression via chronic activation of innate immune cell. Additionally, elevated fucose levels are linked to the cancer cell stemness. The possible roles of fucosylated antigens in cancer biology were recently reviewed by Blanas and colleagues in full details.63 Sialyl-Lewis antigens (sialylated version of Lewis antigens) are also impacting the CRC progression on multiple stages of development.48 However, these will not be engaged in the current discussion due to rather moderate partaking of sialylation in the CEA glycoprofiles observed in this study. CEA-associated-glycan pool observed in this study appeared to be overabundantly fucosylated with regards to both core and terminal fucosylation: on average about half of all glycans from all quantifiable N-glycosylation sites carried at least one fucose. The maximum number of fucose monosaccharides in a single glycan composition could reach up to seven units. As an illustration, the MS/MS-confirmed structure in H9N8F7S1 on site N204/560 (CEA3 sample, Supporting Information, Table S2) can be revised. Interestingly, almost all N-glycans associated with the sites N204/560 and N466 carried at least one fucose within the structure regardless of the CEA source. Nonetheless, Supporting Information, Figure S4 demonstrate that, on average, the CEA3 sample exhibits a higher level of fucosylation than CEA1 and CEA2, when considering both the abundances of fucosylated glycans as well as number of fucose units per glycans. That is particularly evident for sites N375, N580, and N650, where primary tumors showed significant levels of nonfucosylated N-glycan species. Moreover, for sites N204/560 and N466 of the CEA3 sample a higher amount of fucosylated determinants and oligofucosylated N-glycans was observed accounting for more Lewis antigens.
Overall, notable differences in one or more of the analyzed N-glycan traits per N-glycosylation site could be observed per CEA sample type (primary tumor vs metastatic site; Figure 3, Supporting Information, Figures S4 and S5).
4. Concluding Remarks
In this study, CEA N-glycosylation sites were characterized using N-glycopeptide profiles obtained after enzymatic digestion with trypsin, Glu-C, and Pronase and analysis by sheathless CE-MS/MS. Complementary information was obtained through the use of the three above-mentioned enzymes, hence allowing an improved N-glycosylation site coverage and the identification of most of the potential N-glycosylation sites (21 out of 28), their degree of occupancy, and their site-specific dominant N-glycan types (893 different N-glycopeptide glycoforms were identified with a total of 128 unique glycan compositions). Overall, the CEA N-glycoprofile follows the previously reported cancer-associated patterns, while exhibiting its distinct features: simultaneous increased bisection and branching, incomplete galactosylation or (poly)LacNAc elongation on highly branched structures, moderate levels of sialylation, and extremely high levels of fucosylation. For a better understanding of CEA glycosylation pattern heterogeneity, and to confirm our findings, an average glycosylation profile using a glycomics approach could be explored in the foreseeable future. Nonetheless, the N-glycome profile seems to be a less promising source of biomarkers. This is especially due to the low abundance of CEA and high abundance of other glycoproteins present in complex biological samples that may skew the obtained N-glycome profiles of CEA, even for immunopurified samples. It should be noted that this study presents an exploratory perspective on the N-glycosylation heterogeneity of CEA, and, due to the small sample set, only initial observations about the biological differences in glycosylation between primary and metastatic CRC can be made. These findings should be validated on a larger set of samples to establish novel biomarkers. To bring an example, the distal part of the CEA sequence (sites N580, N650, N655 from tryptic digest, and site N466 from Glu-C digest) exhibits the most potential in tumor biological status discrimination (colon primary carcinoma versus liver metastases) with regards to bisection, antennarity, fucosylation, and sialylation traits. The presented multienzyme sheathless CE-MS/MS bottom-up strategy shows potential to provide important biologic information on how N-glycosylation may influence CEA processing in cancer biogenesis. Furthermore, this approach may be successfully translated to the characterization of other highly glycosylated and complex endogenous glycoprotein biomarkers or glycoprotein biopharmaceuticals.
Acknowledgments
This study was supported by the European Union (Horizon 2020 Framework Programme; Marie Curie European Training Network, GlyCoCan project, Grant Number 676421 and Seventh Framework Programme HighGlycan and IBD-BIOM, Grant Numbers 278535 and 305479), a grant Spanish Ministry of Economy and Competitiveness (RTI2018-097411-B-I00), a Horizon Programme Zenith project of The Netherlands Genomic Initiative (Project No. 93511033) and SCIEX. L.P. acknowledges the Spanish Ministry of Economy and Competitiveness for a FPI fellowship (BES-2012-061127) and a short stay fellowship (EEBB-I-16-11506). We thank Rob Haselberg for his input on the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00875.
Figure S1: CE-MS/MS fragmentation spectra; Figure S2: Data assessment by DataAnalysis and Skyline; Figure S3: Protein coverage; Figure S4: Derived traits per glycosylation site; Figure S5: Relative abundance of the top 10 glycoforms per glycosylation site (PDF)
Table S1: Glycan modification, Skyline software; Table S2: Quantified data; Table S3: Nonquantifiable data; Table S4: Derived trait calculation (XLSX)
Author Contributions
∥ L.P. and V.K. are shared first authors.
Author Contributions
M.W. and G.S.M.L.K. conceptually designed the work. L.P. performed the experiments. L.P., V.K., M.W., and G.S.M.L.K. performed the analysis and interpretation of the data and wrote the manuscript. All authors read and revised the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Khoury G. A.; Baliban R.C.; Floudas C.A. Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci. Rep. 2011, 10.1038/srep00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamczyk B.; Tharmalingam T.; Rudd P. M. Glycans as cancer biomarkers. Biochim. Biophys. Acta, Gen. Subj. 2012, 1820 (9), 1347–1353. 10.1016/j.bbagen.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Lau K. S.; Dennis J. W. N-Glycans in cancer progression. Glycobiology 2008, 18 (10), 750–760. 10.1093/glycob/cwn071. [DOI] [PubMed] [Google Scholar]
- Freeze H. H. Understanding Human Glycosylation Disorders: Biochemistry Leads the Charge. J. Biol. Chem. 2013, 288 (10), 6936–6945. 10.1074/jbc.R112.429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W. K.; Oon C. E. How glycosylation aids tumor angiogenesis: An updated review. Biomed. Pharmacother. 2018, 103, 1246–1252. 10.1016/j.biopha.2018.04.119. [DOI] [PubMed] [Google Scholar]
- Häuselmann I.; Borsig L. Altered tumor-cell glycosylation promotes metastasis. Front. Oncol. 2014, 4, 4. 10.3389/fonc.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kailemia M. J.; Park D.; Lebrilla C. B. Glycans and glycoproteins as specific biomarkers for cancer. Anal. Bioanal. Chem. 2017, 409 (2), 395–410. 10.1007/s00216-016-9880-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H. J.; et al. High-Throughput Quantitative Analysis of Total N-Glycans by Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. Anal. Chem. 2012, 84 (7), 3453–3460. 10.1021/ac203440c. [DOI] [PubMed] [Google Scholar]
- Wuhrer M. Glycomics using mass spectrometry. Glycoconjugate J. 2013, 30 (1), 11–22. 10.1007/s10719-012-9376-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuhrer M.; et al. Glycoproteomics based on tandem mass spectrometry of glycopeptides. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2007, 849 (1–2), 115–128. 10.1016/j.jchromb.2006.09.041. [DOI] [PubMed] [Google Scholar]
- Amon S.; Zamfir A. D.; Rizzi A. Glycosylation analysis of glycoproteins and proteoglycans using capillary electrophoresis-mass spectrometry strategies. Electrophoresis 2008, 29 (12), 2485–2507. 10.1002/elps.200800105. [DOI] [PubMed] [Google Scholar]
- Gimenez E.; et al. Capillary electrophoresis time-of-flight mass spectrometry for a confident elucidation of a glycopeptide map of recombinant human erythropoietin. Rapid Commun. Mass Spectrom. 2011, 25 (16), 2307–2316. 10.1002/rcm.5114. [DOI] [PubMed] [Google Scholar]
- An H. J.; et al. Determination of N-glycosylation sites and site heterogeneity in glycoproteins. Anal. Chem. 2003, 75 (20), 5628–5637. 10.1021/ac034414x. [DOI] [PubMed] [Google Scholar]
- Wohlgemuth J.; et al. Quantitative site-specific analysis of protein glycosylation by LC-MS using different glycopeptide-enrichment strategies. Anal. Biochem. 2009, 395 (2), 178–188. 10.1016/j.ab.2009.08.023. [DOI] [PubMed] [Google Scholar]
- Faserl K.; et al. Optimization and Evaluation of a Sheathless Capillary Electrophoresis-Electrospray Ionization Mass Spectrometry Platform for Peptide Analysis: Comparison to Liquid Chromatography-Electrospray Ionization Mass Spectrometry. Anal. Chem. 2011, 83 (19), 7297–7305. 10.1021/ac2010372. [DOI] [PubMed] [Google Scholar]
- Heemskerk A. A. M.; Deelder A. M.; Mayboroda O. A. CE-ESI-MS for bottom-up proteomics: Advances in separation, interfacing and applications. Mass Spectrom. Rev. 2016, 35 (2), 259–271. 10.1002/mas.21432. [DOI] [PubMed] [Google Scholar]
- Moini M. Simplifying CE-MS operation. 2. Interfacing low-flow separation techniques to mass spectrometry using a porous tip. Anal. Chem. 2007, 79 (11), 4241–4246. 10.1021/ac0704560. [DOI] [PubMed] [Google Scholar]
- Dominguez-Vega E.; et al. Capillary electrophoresis-tandem mass spectrometry as a highly selective tool for the compositional and site-specific assessment of multiple peptide-deamidation. Anal. Chim. Acta 2017, 982, 122–130. 10.1016/j.aca.2017.06.021. [DOI] [PubMed] [Google Scholar]
- Kammeijer G. S. M.; et al. Dopant Enriched Nitrogen Gas Combined with Sheathless Capillary Electrophoresis-Electrospray Ionization-Mass Spectrometry for Improved Sensitivity and Repeatability in Glycopeptide Analysis. Anal. Chem. 2016, 88 (11), 5849–56. 10.1021/acs.analchem.6b00479. [DOI] [PubMed] [Google Scholar]
- Mottola-Hartshorn C.; Lorenzoni P.; Ceccarini C. Biosynthesis and Glycosylation of the Carcinoembryonic Antigen. Biochem. J. 1988, 255 (3), 943–948. 10.1042/bj2550943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarström S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin. Cancer Biol. 1999, 9 (2), 67–81. 10.1006/scbi.1998.0119. [DOI] [PubMed] [Google Scholar]
- Lopez N. E.; Peterson C. Y. Advances in Biomarkers: Going Beyond the Carcinoembryonic Antigen. Clinics in Colon and Rectal Surgery 2016, 29 (3), 196–204. 10.1055/s-0036-1584289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K.; et al. Diagnostic value of CEA and CYFRA 21–1 tumor markers in primary lung cancer. Lung Cancer 2013, 80 (1), 45–49. 10.1016/j.lungcan.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Becerra A. Z.; et al. Evaluating the Prognostic Role of Elevated Preoperative Carcinoembryonic Antigen Levels in Colon Cancer Patients: Results from the National Cancer Database. Annals of Surgical Oncology 2016, 23 (5), 1554–1561. 10.1245/s10434-015-5014-1. [DOI] [PubMed] [Google Scholar]
- Su B. B.; Shi H.; Wan J. Role of serum carcinoembryonic antigen in the detection of colorectal cancer before and after surgical resection. World Journal of Gastroenterology 2012, 18 (17), 2121–2126. 10.3748/wjg.v18.i17.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen C. G.; et al. The diagnostic accuracy of carcinoembryonic antigen to detect colorectal cancer recurrence - A systematic review. Int. J. Surg 2016, 25, 134–44. 10.1016/j.ijsu.2015.11.065. [DOI] [PubMed] [Google Scholar]
- Hall C.; et al. A Review of the Role of Carcinoembryonic Antigen in Clinical Practice. Annals of Coloproctology 2019, 35 (6), 294–305. 10.3393/ac.2019.11.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H.; Lee S.W. The Roles of Carcinoembryonic Antigen in Liver Metastasis and Therapeutic Approaches. Gastroenterol. Res. Pract. 2017, 2017, 1. 10.1155/2017/7521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.; Zhan T.; Liu Y.; Li Q.; Wu H.; Ji D.; Li Y. Glycomic profiling of carcinoembryonic antigen isolated from human tumor tissue. Clin. Proteomics 2015, 10.1186/s12014-015-9088-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannicht C.; et al. N-glycosylation of the carcinoembryonic antigen related cell adhesion molecule, C-CAM, from rat liver: detection of oversialylated bi- and triantennary structures. Glycobiology 1999, 9 (9), 897–906. 10.1093/glycob/9.9.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeland E.; et al. Differential glycosylation of MUC1 and CEACAM5 between normal mucosa and tumour tissue of colon cancer patients. Int. J. Cancer 2012, 131 (1), 117–128. 10.1002/ijc.26354. [DOI] [PubMed] [Google Scholar]
- Roda G.; et al. Characterizing CEACAM5 interaction with CD8alpha and CD1d in intestinal homeostasis. Mucosal Immunol. 2014, 7 (3), 615–24. 10.1038/mi.2013.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerciello F.; Choi M.; Nicastri A.; Bausch-Fluck D.; Ziegler A.; Vitek O.; Felley-Bosco E.; Stahel R.; Aebersold R.; Wollscheid B. Identification of a seven glycopeptide signature for malignant pleural mesothelioma in human serum by selected reaction monitoring. Clin. Proteomics 2013, 10.1186/1559-0275-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancera-Arteu M.; Lleshi N.; Sanz-Nebot V.; Gimenez E.; Benavente F. Analysis of glycopeptide biomarkers by on-line TiO2 solid-phase extraction capillary electrophoresis-mass spectrometry. Talanta 2020, 209, 120563. 10.1016/j.talanta.2019.120563. [DOI] [PubMed] [Google Scholar]
- Zhu J. H.; et al. Glycopeptide Biomarkers in Serum Haptoglobin for Hepatocellular Carcinoma Detection in Patients with Nonalcoholic Steatohepatitis. J. Proteome Res. 2020, 19 (8), 3452–3466. 10.1021/acs.jproteome.0c00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk A. A.; et al. Ultra-low flow electrospray ionization-mass spectrometry for improved ionization efficiency in phosphoproteomics. Anal. Chem. 2012, 84 (10), 4552–9. 10.1021/ac300641x. [DOI] [PubMed] [Google Scholar]
- Wuhrer M.; Deelder A. M.; Hokke C. H. Protein glycosylation analysis by liquid chromatography-mass spectrometry. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2005, 825 (2), 124–133. 10.1016/j.jchromb.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Stanley P.; Taniguchi N.; Aebi M.. N-Glycans. In Essentials of Glycobiology; Cold Spring Harbor: New York, 2015; pp 99–111. [Google Scholar]
- Gattiker A.; et al. FindPept, a tool to identify unmatched masses in peptide mass fingerprinting protein identification. Proteomics 2002, 2 (10), 1435–1444. . [DOI] [PubMed] [Google Scholar]
- Barroso A.; et al. Modelling the electrophoretic migration behaviour of peptides and glycopeptides from glycoprotein digests in capillary electrophoresis-mass spectrometry. Anal. Chim. Acta 2015, 854, 169–177. 10.1016/j.aca.2014.10.038. [DOI] [PubMed] [Google Scholar]
- Dalpathado D. S.; Desaire H. Glycopeptide analysis by mass spectrometry. Analyst 2008, 133 (6), 731–8. 10.1039/b713816d. [DOI] [PubMed] [Google Scholar]
- Giansanti P.; et al. Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat. Protoc. 2016, 11 (5), 993–1006. 10.1038/nprot.2016.057. [DOI] [PubMed] [Google Scholar]
- Šlechtová T.; et al. Insight into Trypsin Miscleavage: Comparison of Kinetic Constants of Problematic Peptide Sequences. Anal. Chem. 2015, 87 (15), 7636–43. 10.1021/acs.analchem.5b00866. [DOI] [PubMed] [Google Scholar]
- Lee J.; et al. Designation of fingerprint glycopeptides for targeted glycoproteomic analysis of serum haptoglobin: insights into gastric cancer biomarker discovery. Anal. Bioanal. Chem. 2018, 410 (6), 1617–1629. 10.1007/s00216-017-0811-y. [DOI] [PubMed] [Google Scholar]
- Zauner G.; et al. Protein glycosylation analysis by HILIC-LC-MS of Proteinase K-generated N- and O-glycopeptides. J. Sep. Sci. 2010, 33 (6–7), 903–910. 10.1002/jssc.200900850. [DOI] [PubMed] [Google Scholar]
- Hao P.; Ren Y.; Alpert A. J.; Sze S. K. Detection, Evaluation and Minimization of Nonenzymatic Deamidation in Proteomic Sample Preparation. Mol. Cell. Proteomics 2011, 10 (10), O111.009381. 10.1074/mcp.O111.009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proc J. L.; et al. A Quantitative Study of the Effects of Chaotropic Agents, Surfactants, and Solvents on the Digestion Efficiency of Human Plasma Proteins by Trypsin. J. Proteome Res. 2010, 9 (10), 5422–5437. 10.1021/pr100656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst S.; Wuhrer M.; Rombouts Y. Glycosylation Characteristics of Colorectal Cancer. Adv. Cancer Res. 2015, 126, 203–256. 10.1016/bs.acr.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Sethi M. K.; et al. In-depth N-glycome profiling of paired colorectal cancer and non-tumorigenic tissues reveals cancer-, stage- and EGFR-specific protein N-glycosylation. Glycobiology 2015, 25 (10), 1064–1078. 10.1093/glycob/cwv042. [DOI] [PubMed] [Google Scholar]
- Lee L. Y.; et al. Comprehensive N-Glycome Profiling of Cultured Human Epithelial Breast Cells Identifies Unique Secretome N-Glycosylation Signatures Enabling Tumorigenic Subtype Classification. J. Proteome Res. 2014, 13 (11), 4783–4795. 10.1021/pr500331m. [DOI] [PubMed] [Google Scholar]
- Pearce O. M. T. Cancer glycan epitopes: biosynthesis, structure and function. Glycobiology 2018, 28 (9), 670–696. 10.1093/glycob/cwy023. [DOI] [PubMed] [Google Scholar]
- Taniguchi N.; Kizuka Y. Glycans and cancer: role of N-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv. Cancer Res. 2015, 126, 11–51. 10.1016/bs.acr.2014.11.001. [DOI] [PubMed] [Google Scholar]
- Zhao Q.; Zhan T.; Deng Z.; Li Q.; Liu Y.; Yang S.; Ji D.; Li Y. Glycan analysis of colorectal cancer samples reveals stage-dependent changes in CEA glycosylation patterns. Clin. Proteomics 2018, 15, 9. 10.1186/s12014-018-9182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull C.; et al. Targeting aberrant sialylation in cancer cells using a fluorinated sialic acid analog impairs adhesion, migration, and in vivo tumor growth. Mol. Cancer Ther. 2013, 12 (10), 1935–46. 10.1158/1535-7163.MCT-13-0279. [DOI] [PubMed] [Google Scholar]
- Hao C.; Zhang G.; Zhang L. Serum CEA levels in 49 different types of cancer and noncancer diseases. Prog. Mol. Biol. Transl Sci. 2019, 162, 213–227. 10.1016/bs.pmbts.2018.12.011. [DOI] [PubMed] [Google Scholar]
- Pearce O. M.; Laubli H. Sialic acids in cancer biology and immunity. Glycobiology 2016, 26 (2), 111–28. 10.1093/glycob/cwv097. [DOI] [PubMed] [Google Scholar]
- Plucinsky M. C.; et al. Total and lipid-associated serum sialic acid levels in cancer patients with different primary sites and differing degrees of metastatic involvement. Cancer 1986, 58 (12), 2680–5. . [DOI] [PubMed] [Google Scholar]
- Vajaria B. N.; et al. Sialylation: an Avenue to Target Cancer Cells. Pathol. Oncol. Res. 2016, 22 (3), 443–7. 10.1007/s12253-015-0033-6. [DOI] [PubMed] [Google Scholar]
- Park J. J.; Lee M. Increasing the alpha 2, 6 sialylation of glycoproteins may contribute to metastatic spread and therapeutic resistance in colorectal cancer. Gut Liver 2013, 7 (6), 629–41. 10.5009/gnl.2013.7.6.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi M. K.; et al. Comparative N-glycan profiling of colorectal cancer cell lines reveals unique bisecting GlcNAc and alpha-2,3-linked sialic acid determinants are associated with membrane proteins of the more metastatic/aggressive cell lines. J. Proteome Res. 2014, 13 (1), 277–88. 10.1021/pr400861m. [DOI] [PubMed] [Google Scholar]
- Li J.; et al. Unmasking Fucosylation: from Cell Adhesion to Immune System Regulation and Diseases. Cell Chem. Biol. 2018, 25 (5), 499–512. 10.1016/j.chembiol.2018.02.005. [DOI] [PubMed] [Google Scholar]
- Shan M.; et al. Fucosylation in cancer biology and its clinical applications. Prog. Mol. Biol. Transl Sci. 2019, 162, 93–119. 10.1016/bs.pmbts.2019.01.002. [DOI] [PubMed] [Google Scholar]
- Blanas A.; Sahasrabudhe N. M.; Rodriguez E.; van Kooyk Y.; van Vliet S. J. Fucosylated Antigens in Cancer: An Alliance toward Tumor Progression, Metastasis, and Resistance to Chemotherapy. Front. Oncol. 2018, 8, 39. 10.3389/fonc.2018.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.