

Abstract
Background
The etiology of male breast cancer (MBC) is poorly understood. In particular, the extent to which the genetic basis of MBC differs from female breast cancer (FBC) is unknown. A previous genome-wide association study of MBC identified 2 predisposition loci for the disease, both of which were also associated with risk of FBC.
Methods
We performed genome-wide single nucleotide polymorphism genotyping of European ancestry MBC case subjects and controls in 3 stages. Associations between directly genotyped and imputed single nucleotide polymorphisms with MBC were assessed using fixed-effects meta-analysis of 1380 cases and 3620 controls. Replication genotyping of 810 cases and 1026 controls was used to validate variants with P values less than 1 × 10–06. Genetic correlation with FBC was evaluated using linkage disequilibrium score regression, by comprehensively examining the associations of published FBC risk loci with risk of MBC and by assessing associations between a FBC polygenic risk score and MBC. All statistical tests were 2-sided.
Results
The genome-wide association study identified 3 novel MBC susceptibility loci that attained genome-wide statistical significance (P < 5 × 10–08). Genetic correlation analysis revealed a strong shared genetic basis with estrogen receptor–positive FBC. Men in the top quintile of genetic risk had a fourfold increased risk of breast cancer relative to those in the bottom quintile (odds ratio = 3.86, 95% confidence interval = 3.07 to 4.87, P = 2.08 × 10–30).
Conclusions
These findings advance our understanding of the genetic basis of MBC, providing support for an overlapping genetic etiology with FBC and identifying a fourfold high-risk group of susceptible men.
Male breast cancer (MBC) is rare, accounting for less than 1% of all breast cancer diagnoses. A greater proportion of MBCs than female breast cancers (FBCs) are of the estrogen receptor (ER)–positive subtype (>95% MBC vs 75% FBC), suggesting that MBC may comprise a more homogeneous group of tumors than FBC. Although there is a paucity of data regarding the etiology of MBC, family history and genetic susceptibility are important risk factors (1,2). Approximately 10% of cases are attributable to inherited mutations in BRCA2 (1). Conversely, mutations in BRCA1 are observed in only a small number of cases, suggesting differences in the underlying genetic etiologies of MBC and FBC (1).
Common germline variants influence susceptibility to MBC (3–5). Our previous genome-wide association study (GWAS) of MBC identified single nucleotide polymorphisms (SNPs) at 14q24.1 and 16q12.1 that were associated with susceptibility at genome-wide levels of statistical significance (4). Moreover, although these loci were also associated with FBC susceptibility (6,7), they conferred greater risks of breast cancer in men than women (14q24.1 odds ratio [OR] = 1.57 vs 1.07 and 16q12.1 OR = 1.50 vs 1.22 for MBC and FBC, respectively) (4), lending weight to findings from population-based family history studies that suggest a greater contribution of genetic variation to MBC than FBC predisposition (2).
In this study, we pooled individual-level data from our GWAS (4) with 2 additional case-control datasets to identify novel MBC risk variants, to illuminate better the genetic basis of MBC and to enable comparisons between determinants of polygenic predisposition to MBC and FBC.
Methods
Study Populations
Cases for discovery analysis were primarily from the Breast Cancer Now Male Breast Cancer Study, a population-based case-control study of MBC in England and Wales (UK-BCN-MBCS, n = 1210). Additional cases were from UK studies at the University of Leeds (UK-UoL, n = 31) and the University of Cambridge (UK-UoC, n = 138), and a US study at City of Hope (US-CoH, n = 113). The UK-UoL, UK-UoC, US-CoH, and 540 of the UK-BCN-MBCS cases were analyzed previously (4). Here we have added an additional 670 MBC cases from the UK-BCN-MBCS. To estimate autosomal SNP genotype frequencies from the general population, we used male and female controls from the 1958 British Birth Cohort (UK-58BC, n = 2663), male controls from the UK-BCN-MBCS (n = 264), and female controls from the UK Generations Study (UK-GS, n = 698) (Supplementary Table 1, available online) (8). The inclusion of female controls was predicated on the observation that autosomal SNPs do not differ in frequency between males and females sampled from the same ancestral population so that GWAS of sexually differentiated traits, such as breast cancer, need not be restricted to selection of same-sex controls (9). Descriptions of each of the studies used for discovery analysis are provided in the Supplementary Methods (available online). For validation of promising associations, we used 810 cases and 1026 controls of European ancestry that were assembled internationally for our previous GWAS (Supplementary Table 1, available online) (4). All sample collection was undertaken with informed consent and ethical approval.
Genotyping, Quality Control, and Imputation
Discovery analysis samples, genotyping arrays, and SNPs or samples excluded during quality control are summarized in Supplementary Figure 1 (available online). Genotyping was performed using Illumina (San Diego, CA) Infinium OmniExpress 710K BeadChips (UK-BCN-MBCS, UK-UoL, UK-UoC, and US-CoH cases), Infinium OncoArray 500K BeadChips (UK-BCN-MBCS cases and UK-GS controls), and Infinium Global Screening Array 640K BeadChips (UK-BCN-MBCS cases and controls). UK-58BC controls were genotyped using Infinium 1.2M BeadChips. Replication genotyping was performed using either Agena (San Diego, CA) iPLEX chemistry or KASP assays (LGC, Hoddesdon, UK).
Samples were excluded based on genotyping completion rate (<95.0%), relatedness (identity by descent [IBD] first- and second-degree relatives), and genetically determined non-European ancestry. SNPs were excluded according to call rates (<95.0%), minor allele frequency (MAF, <2.0%), and genotype deviation from Hardy-Weinberg proportions (P < 1 × 10–05). SNP data from cases genotyped using Infinium OmniExpress 710K BeadChips were harmonized with UK-58BC control data yielding 486 160 SNPs. Cases and controls genotyped using Infinium OncoArray 500K BeadChips were similarly harmonized, as were those using Infinium Global Screening Array 640K BeadChips. Genome-wide imputation was performed for each GWAS dataset using 1KGP Phase 3 reference data. Haplotypes were prephased using SHAPEIT2 (10) and imputation was performed using IMPUTE2 (11). Imputed SNPs with INFO scores less than 0.80 and/or MAFs less than 2.0% were excluded. After quality control, 8 074 073 SNPs were available for analysis.
Statistical Analysis
For each GWAS dataset, tests of association between imputed SNPs and MBC status were performed, assuming a log-additive model, using SNPTEST v2.5 (12). Quantile–quantile plots showed no evidence of overdispersion (λ = 0.99-1.05; Supplementary Figure 2, available online). Combined analysis of each dataset was performed using fixed-effects inverse variance-weighted meta-analysis (Supplementary Figure 3, available online) (13). Heterogeneity was assessed using Cochran’s Q-test and I2 statistics. Sensitivity analyses of the effect estimates when US-CoH cases or UK-GS and UK-58BC female controls were omitted were consistent with the main results (Supplementary Tables 2 and 3, available online). For replication analysis, effects under a log-additive model were estimated by performing multiple logistic regression, adjusted for study, using the Genotype Libraries and Utilities package (14). Bayesian false discovery probabilities were calculated to assess the noteworthiness of FBC predisposition SNP associations with MBC assuming that the cost of a false nondiscovery was 4 times that of a false discovery (giving a noteworthiness cutoff value of 0.80) and that the odds ratio lies between 0.83 and 1.2 with probability 0.95 (15). These comparisons were restricted to 172 published loci with FBC P less than 5 × 10–08 (16). To compare the MBC odds ratios with those of FBC, we assumed both sets of odds ratios were log-normally distributed and that the difference between the log odds ratios was normally distributed with mean 0 and variance equal to the sum of the squared standard errors of the 2 estimates to obtain a χ2 statistic. All statistical tests were 2-sided, and P less than .05 was used as the cut point for statistical significance unless otherwise stated.
Heritability and Genetic Correlation
The heritability of MBCwas estimated assuming a continuous underlying liability and an MBC population prevalence of 0.1% using linkage disequilibrium score regression (LDSC) (17). LDSC was used to calculate the genetic correlation () between MBC and FBC using summary statistics from 122 977 FBC cases and 105 974 controls in the Breast Cancer Association Consortium (16). Subtype-specific genetic correlations between MBC and both ER-positive and ER-negative FBC also used Breast Cancer Association Consortium data (n = 69 501 ER-positive and n = 21 468 ER-negative cases). To assess cross-cancer genetic correlations with other hormonally driven cancers, we used summary statistics from 79 148 prostate cancer cases and 61 106 controls in the Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome Consortium (PRACTICAL) (18) and 22 406 invasive epithelial ovarian cancer cases and 40 941 controls in the Ovarian Cancer Association Consortium (OCAC) (19).
Polygenic Risk Score Analysis
A 313-SNP FBC polygenic risk score (PRS) (20) was calculated using effect estimates for overall, ER-positive, and ER-negative FBC, standardized such that the PRS distribution in controls (2663 male and female individuals from the UK-58BC) had a mean = 0 and SD = 1. The 313-SNP FBC PRS includes 305 SNPs that were associated with overall FBC at P less than 1 × 10–05 plus 6 additional SNPs that were associated with ER-positive FBC and 2 rare variants in the BRCA2 and CHEK2 genes (20). To enable comparison with FBC, we derived PRS for 1671 female cases from UK-GS (9). Logistic regression was used to estimate risk of MBC by quintiles and per SD increase in the PRS.
Gene Expression and Expression Quantitative Trait Locus (eQTL) Analysis
eQTL analyses were performed using Genotype-Tissue Expression Project (GTEx) gene expression data on normal breast samples from 157 males and 107 females (21). Associations between log10-normalized gene counts of candidate target genes and SNP genotypes were assessed using linear regression with and without interaction terms for genotype and sex. Linear regression was used to assess associations between gene expression of putative target genes and sex.
Results
Heritability of MBC and Genetic Correlation With FBC
After quality control, the 3 GWAS datasets yielded SNP genotypes at 8 074 073 loci in 1380 MBC cases and 3620 controls. The heritability of MBC attributable to common SNPs was 0.09 (standard error [SE] = 0.06) on the liability scale, which is accordant with published estimates for FBC (22). Using cross-trait LDSC, we observed strong between MBC and FBC (= 0.83, SE = 0.30, P = .005). Consistent with the predominance of ER-positive tumors in MBC, genetic correlation was stronger between MBC and ER-positive FBC ( = 0.82, SE = 0.30, P = .005) than ER-negative FBC ( = 0.47, SE = 0.24, P = .047). Predicated on evidence of pleiotropy between breast cancer and other hormonally driven epithelial tumors (23), we estimated genetic correlation between MBC and both prostate and ovarian cancer. Although there was no evidence of genetic correlation with prostate cancer ( = 0.01, SE = 0.11, P = .90), there was borderline evidence of a moderate genetic correlation with ovarian cancer ( = 0.55, SE = 0.29, P = .06).
GWAS and Validation Analysis
Combined analysis of the GWAS datasets detected a novel genome-wide statistically significant association (P < 5 × 10–08) between SNP rs9371545 at 6q25.1 and risk of MBC (P = 1.63 × 10–08; Table 1; Supplementary Table 4, available online) and validated the associations at 14q24.1 (rs1022979 P = 1.53 × 10–16) and 16q12.1 (rs35850695 P = 1.57 × 10–11; Supplementary Table 4, available online). We observed promising associations (P < 1 × 10–06) at 11q13.3 (rs78540526 and rs554219; Table 1; Supplementary Table 4, available online) and 15q24 (rs4407020; Supplementary Table 4, available online). Replication genotyping of 810 cases and 1026 controls provided support for rs78540526 and rs554219 (Table 1; Supplementary Table 4; Supplementary Figure 4, available online) but not rs4407020 (Supplementary Table 4, available online) in a joint analysis with the discovery data (rs78540526 P = 1.06 × 10–11, rs554219 P = 2.86 × 10–11). Similar to the loci at 14q24.1 and 16q12.1, the 6q25.1 and 11q13.1 SNPs were also associated with predisposition to FBC but had larger risk effects in males (Table 1).
Table 1.
Three novel risk variants with P less than 5 × 10−08 identified from analysis of GWAS and replication data and their odds ratios for FBC
| Cytoband | SNP | Allelesb | Stage | Control MAF | Case MAF | MBC OR (95% CI) | P c | FBC OR (95% CI)d | P HET e | I2 |
|---|---|---|---|---|---|---|---|---|---|---|
| 6q25.1 | rs9371545a | G/A | GWAS | 0.07 | 0.10 | 1.60 (1.36 to 1.89) | 1.63 × 10–08 | — | — | — |
| rs9383938a | G/T | Replication | 0.09 | 0.11 | 1.30 (1.04 to 1.63) | .02 | — | — | — | |
| Joint | — | — | 1.47 (1.30 to 1.67) | 2.93 × 10−09 | 1.11 (1.08 to 1.14) | 9.56 × 10–06 | 94.9 | |||
| 11q13.3 | rs554219 | C/G | GWAS | 0.12 | 0.16 | 1.42 (1.24 to 1.62) | 1.86 × 10−07 | — | — | — |
| Replication | 0.11 | 0.16 | 1.52 (1.25 to 1.84) | 2.65 × 10−05 | — | — | — | |||
| Joint | — | — | 1.45 (1.31 to 1.62) | 2.86 × 10−11 | 1.27 (1.24 to 1.31) | .02 | 82.9 | |||
| 11q13.3 | rs78540526 | C/T | GWAS | 0.07 | 0.10 | 1.58 (1.34 to 1.87) | 7.38 × 10−08 | — | — | — |
| Replication | 0.06 | 0.10 | 1.68 (1.32 to 2.14) | 2.45 × 10−05 | — | — | — | |||
| Joint | — | — | 1.61 (1.40 to 1.85) | 1.06 × 10−11 | 1.39 (1.35 to 1.42) | .04 | 76.2 |
SNP rs9383938 is a proxy for rs9371545 (r2 = 0.90), which failed assay design for replication genotyping. Summary statistics for replication and joint analysis are based on rs9383938. CI = confidence interval; ER = estrogen receptor; FBC = female breast cancer; GWAS = genome-wide association study; MAF = minor allele frequency; MBC = male breast cancer; OR = odds ratio; PHET = P value for heterogeneity; SNP = single nucleotide polymorphism.
Alleles are shown as major and minor alleles based on control frequencies.
MBC P values were derived from fixed-effects inverse variance-weighted meta-analysis (GWAS and joint) and from multiple logistic regression, adjusted for study (replication). All tests were 2-sided.
Odds ratios for ER-positive FBC from (16).
P value for Cochran’s Q-test for heterogeneity between the MBC and FBC odds ratios.
– Control and case MAFs were not calculated for meta-analyzed SNPs at the joint analysis stage. FBC odd ratios, PHET, and I2 are not applicable for GWAS and replication stages because published FBC odd ratios were compared only with the MBC odd ratios estimated in joint analysis of our GWAS and replication studies.
Associations Between FBC Predisposition SNPs and Risk of MBC
We next evaluated the MBC associations of 172 published FBC risk SNPs (16) (Supplementary Table 5, available online). Thirty-five SNPs (20.3%) had P less than .05 and consistent directions of effect with FBC; 33 remained noteworthy using BFDP analysis (15). Eight loci had statistically significant differences in their odds ratios for MBC and FBC (false discovery rate [FDR] < 0.10; Table 2; Supplementary Table 6, available online). At 6q25.1 and 14q24.1, rs9397437 and rs2588809 had odds ratios that were greater for MBC than FBC, and rs2981578 at 10q26.13 had an odds ratio that was greater for FBC. The directions of the odds ratios for rs4233486 at 1p34.2, rs12710696 at 2p24.1, rs13066793 at 3p12.1, rs3215401 at 5p15.33, and rs10816625 at 9q31.2 were opposite to FBC.
Table 2.
FBC predisposition SNPs with FDR adjusted P less than .10 that confer statistically significantly different risk effects in males and females
| Cytoband | SNP | Allelesa | MBC OR (95% CI) | FBC OR (95% CI)b | P c | P FDR |
|---|---|---|---|---|---|---|
| 1p34.2 | rs4233486 | T/C | 1.12 (1.02 to 1.23) | 0.97 (0.95 to 0.98) | .003 | .09 |
| 2p24.1 | rs12710696 | C/T | 0.87 (0.79 to 0.95) | 1.03 (1.01 to 1.04) | 7.75 × 10−04 | .03 |
| 3p12.1 | rs13066793 | A/G | 1.28 (1.08 to 1.51) | 0.94 (0.91 to 0.97) | 4.68 × 10−04 | .03 |
| 5p15.33 | rs3215401 | A/AG | 1.10 (1.00 to 1.21) | 0.93 (0.91 to 0.95) | 7.63 × 10−04 | .03 |
| 6q25 | rs9397437 | G/A | 1.58 (1.34 to 1.87) | 1.17 (1.14 to 1.21) | 4.15 × 10−04 | .03 |
| 9q31.2 | rs10816625 | A/G | 0.85 (0.71 to 1.03) | 1.11 (1.07 to 1.15) | .004 | .09 |
| 10q26.13 | rs2981578 | T/C | 1.08 (0.99 to 1.18) | 1.23 (1.21 to 1.25) | .004 | .09 |
| 14q24.1 | rs2588809 | C/T | 1.59 (1.41 to 1.78) | 1.06 (1.03 to 1.08) | 1.25 × 10−10 | 2.15 × 10−08 |
Alleles are shown as major and minor alleles based on control frequencies. CI = confidence interval; FBC = female breast cancer; FDR = false discovery rate; MBC = male breast cancer; OR = odds ratio; SNP = single nucleotide polymorphism.
Odds ratios for FBC from (16).
P value for statistical significance of the difference between MBC and FBC log odds ratios.
At each of the 172 loci, we investigated whether any variants correlated (r2 ≥ 0.10) with a published FBC susceptibility SNP were more statistically significantly associated with MBC in our GWAS than the FBC SNP itself (Supplementary Table 5, available online). We identified 4 such SNPs with P less than 1 × 10–05, at 6q25.1, 10p12.31, and 11q13.3, which we genotyped alongside the corresponding lead FBC predisposition SNPs in our replication samples and analyzed jointly with the discovery data (Figure 1; Table 3;Supplementary Table 5, available online). At 6q25.1, rs9383938 (P = 2.93 × 10–09) was correlated with FBC SNP rs9397437 (P = 5.29 × 10–09, r2 = 0.83) but was nominally more statistically significantly associated with MBC. SNP rs146723925 was correlated (r2 = 0.89) with a second FBC risk locus at 6q25.1 demarcated by rs3757322 and was more statistically significantly associated with MBC in the discovery data, but failed assay design for replication genotyping. However, rs3757322 surpassed the genome-wide statistical significance threshold following joint analysis (P = 6.23 × 10–09), and conditional analyses indicated that rs3757322 and rs9383938 tag independent causal alleles at 6q25.1 (Supplementary Table 7, available online). At 10p12.31, rs2183271 (P = 2.69 × 10–07) was several orders of magnitude more strongly associated with MBC than lead FBC SNP rs7072776 (P = 2.46 × 10–04, r2 = 0.68), and the effect of rs7072776 was strongly dependent on rs2183271 (Figure 1; Table 3;Supplementary Tables 5 and 7, available online). At 11q13.3, rs78540526 (P = 1.06 × 10–11) was correlated with FBC SNP rs75915166 (P = 7.71 × 10–08, r2 = 0.63; Figure 1; Table 3;Supplementary Table 5, available online). rs75915166 and a second variant at 11q13.3, rs554219, have been reported to independently influence risk of FBC (24). Analysis of these SNPs conditioned on rs78540526 did not provide compelling evidence for independence in MBC (P = .64 and .03, respectively). However, rs554219 (P = 4.74 × 10–05; Supplementary Table 7, available online) and rs78540526 (P = 5.55 × 10–05) were associated with MBC after conditioning on rs75915166.
Figure 1.

Regional association plots for 6q25.1 (A), 10p12.31 (B), and 11q13.3 (C) male breast cancer risk loci. Each point represents an individual single nucleotide polymorphism (SNP) sorted on the x-axis by physical position based on National Center for Biotechnology Information build 37 of the human genome and plotted by −log10P value on the y-axis. Recombination rates, estimated using HapMap data, are plotted in blue. For each region, the published female breast cancer predisposition SNP is plotted as a circle alongside the variant most strongly associated with male breast cancer, plotted as a diamond. In instances where there are multiple independent predisposition loci at the same genomic region, pairs of SNPs are grouped by color. Lighter colors represent the genome-wide association study P value and darker colors the joint P value for the top SNPs. All statistical tests were 2-sided.
Table 3.
Four FBC predisposition loci at which variants correlated at r2 ≥ 0.10 with a published FBC susceptibility SNP were more statistically significantly associated with MBC than the lead FBC SNP
| Cytoband | SNPa | Allelesb | Stage | Control MAF | Case MAF | OR (95% CI) | P c |
|---|---|---|---|---|---|---|---|
| 6q25.1 | rs3757322 | T/G | GWAS | 0.33 | 0.37 | 1.23 (1.12 to 1.35) | 1.11 × 10−05 |
| Replication | 0.33 | 0.39 | 1.32 (1.15 to 1.52) | 9.53 × 10−05 | |||
| Joint | — | — | 1.26 (1.16 to 1.36) | 6.23 × 10−09 | |||
| rs146723925 | GAA/G | GWAS | 0.35 | 0.39 | 1.23 (1.13 to 1.35) | 7.73 × 10−06 | |
| r 2 = 0.89 | Replication | — | — | Failed assay design | — | ||
| Joint | — | — | — | — | |||
| 6q25.1 | rs9397437 | G/A | GWAS | 0.07 | 0.10 | 1.58 (1.34 to 1.87) | 4.22 × 10−08 |
| Replication | 0.07 | 0.10 | 1.33 (1.05 to 1.69) | .02 | |||
| Joint | — | — | 1.50 (1.31 to 1.71) | 5.29 × 10−09 | |||
| rs9383938 | G/T | GWAS | 0.08 | 0.11 | 1.60 (1.36 to 1.89) | 1.63 × 10−08 | |
| r 2 = 0.83 | Replication | 0.09 | 0.11 | 1.30 (1.04 to 1.63) | .02 | ||
| Joint | — | — | 1.47 (1.30 to 1.67) | 2.93 × 10−09 | |||
| 10p12.31 | rs7072776 | G/A | GWAS | 0.27 | 0.30 | 1.17 (1.06 to 1.29) | .002 |
| Replication | 0.28 | 0.31 | 1.15 (0.99 to 1.33) | .06 | |||
| Joint | — | — | 1.16 (1.07 to 1.26) | 2.46 × 10−04 | |||
| rs2183271 | T/C | GWAS | 0.36 | 0.41 | 1.24 (1.13 to 1.36) | 3.50 × 10−06 | |
| r 2 = 0.68 | Replication | 0.36 | 0.40 | 1.18 (1.02 to 1.35) | .02 | ||
| Joint | — | — | 1.22 (1.13 to 1.31) | 2.69 × 10−07 | |||
| 11q13.3 | rs75915166 | C/A | GWAS | 0.06 | 0.08 | 1.52 (1.25 to 1.83) | 1.64 × 10−05 |
| Replication | 0.05 | 0.08 | 1.56 (1.19 to 2.04) | .001 | |||
| Joint | — | — | 1.53 (1.31 to 1.79) | 7.71 × 10−08 | |||
| rs78540526 | C/T | GWAS | 0.07 | 0.10 | 1.58 (1.34 to 1.87) | 7.38 × 10−08 | |
| r 2 = 0.63 | Replication | 0.06 | 0.10 | 1.68 (1.32 to 2.14) | 2.45 × 10−05 | ||
| Joint | — | — | 1.61 (1.40 to 1.85) | 1.06 × 10−11 |
For each locus the MBC effect estimates and association statistics for the lead FBC SNP are shown, followed by the estimates, correlation coefficient and association statistics for the correlated variant that was more strongly associated with MBC. CI = confidence interval; FBC = female breast cancer; GWAS = genome-wide association study; MAF = minor allele frequency; MBC = male breast cancer; OR = odds ratio; SNP = single nucleotide polymorphism.
Alleles are shown as major and minor alleles based on control frequencies.
MBC P values were derived from fixed-effects inverse variance-weighted meta-analysis (GWAS and joint) and from multiple logistic regression, adjusted for study (replication). All tests were 2-sided.
- Control and case MAFs were not calculated for meta-analyzed SNPs at the joint analysis stage.
FBC PRS Association With MBC
Because our data supported a strong genetic correlation between MBC and FBC, we assessed whether a recent 313-SNP FBC PRS (20) was associated with breast cancer risk in our study. The odds ratio per SD increase in the PRS was 1.55 (95% confidence interval = 1.45 to 1.66, P = 3.54 × 10–37; Table 4). Men in the top quintile of genetic risk had an almost fourfold increased risk of breast cancer (OR = 3.86, 95% confidence interval = 3.07 to 4.87, P = 2.08 × 10–30) compared with men in the bottom quintile. We examined MBC associations with the 313-SNP PRS incorporating weightings for ER-positive or ER-negative FBC. Risk estimates for the ER-positive PRS were similar to the overall PRS; the ER-negative PRS was less strongly associated with MBC risk (Table 4), consistent with our genetic correlation analysis. The PRS distribution in male cases was similar to that of FBC cases (Figure 2).
Table 4.
Association between 313-SNP PRSs and MBC risk
| SNP weightsa | Quintile | No. of controlsb | Male |
Female |
||||
|---|---|---|---|---|---|---|---|---|
| No. of cases | OR (95% CI) | P | No. of cases | OR (95% CI) | P | |||
| Overall FBC | 1st | 533 | 124 | 1.00 (Referent) | 165 | 1.00 (Referent) | ||
| 2nd | 532 | 227 | 1.83 (1.43 to 2.35) | 1.92 × 10−06 | 251 | 1.52 (1.21 to 1.92) | 3.35 × 10−04 | |
| 3rd | 533 | 244 | 1.97 (1.54 to 2.52) | 8.07 × 10−08 | 340 | 2.06 (1.65 to 2.57) | 1.53 × 10−10 | |
| 4th | 532 | 306 | 2.47 (1.94 to 3.15) | 1.72 × 10−13 | 357 | 2.17 (1.74 to 2.70) | 5.67 × 10−12 | |
| 5th | 533 | 479 | 3.86 (3.07 to 4.87) | 2.08 × 10−30 | 558 | 3.38 (2.74 to 4.18) | 1.17 × 10−29 | |
| Trendc | 2663 | 1380 | 1.55 (1.45 to 1.66) | 3.54 × 10−37 | 1671 | 1.51 (1.42 to 1.61) | 4.58 × 10−37 | |
| ER-positive FBC | 1st | 533 | 120 | 1.00 (Referent) | 167 | 1.00 (Referent) | ||
| 2nd | 532 | 229 | 1.91 (1.49 to 2.46) | 4.37 × 10−07 | 254 | 1.52 (1.21 to 1.92) | 3.17 × 10−04 | |
| 3rd | 533 | 243 | 2.03 (1.58 to 2.60) | 2.97 × 10−08 | 312 | 1.87 (1.49 to 2.33) | 3.95 × 10−08 | |
| 4th | 532 | 307 | 2.56 (2.01 to 3.27) | 3.01 × 10−14 | 393 | 2.36 (1.90 to 2.93) | 1.02 × 10−14 | |
| 5th | 533 | 481 | 4.01 (3.17 to 5.06) | 1.91 × 10−31 | 545 | 3.26 (2.64 to 4.03) | 4.10 × 10−28 | |
| Trendc | 2663 | 1380 | 1.55 (1.45 to 1.66) | 3.54 × 10−37 | 1671 | 1.50 (1.41 to 1.60) | 1.27 × 10−36 | |
| ER-negative FBC | 1st | 533 | 175 | 1.00 (Referent) | 201 | 1.00 (Referent) | ||
| 2nd | 532 | 204 | 1.17 (0.92 to 1.48) | .20 | 244 | 1.22 (0.97 to 1.52) | .08 | |
| 3rd | 533 | 280 | 1.60 (1.28 to 2.00) | 3.85 × 10−05 | 354 | 1.76 (1.43 to 2.17) | 1.39 × 10−07 | |
| 4th | 532 | 302 | 1.73 (1.39 to 2.16) | 1.28 × 10−06 | 368 | 1.83 (1.49 to 2.26) | 1.43 × 10−08 | |
| 5th | 533 | 419 | 2.39 (1.93 to 2.96) | 1.06 × 10−15 | 504 | 2.51 (2.05 to 3.07) | 6.56 × 10−19 | |
| Trendc | 2663 | 1380 | 1.37 (1.29 to 1.47) | 6.92 × 10−21 | 1671 | 1.38 (1.29 to 1.47) | 1.02 × 10−23 | |
Weights for the 313 SNPs in the PRS for overall, ER-positive, and ER-negative FBC were obtained from (20). CI = confidence interval; FBC = female breast cancer; ER = estrogen receptor; GWAS = genome-wide association study; MBC = male breast cancer; OR = odds ratio; PRS = polygenic risk score; SNP = single nucleotide polymorphism; UK-58BC = 1958 British Birth Cohort.
2663 males and females from the UK-58BC were used as UK population representative controls in the PRS analysis.
Odds ratio per SD increase in the PRS.
Figure 2.
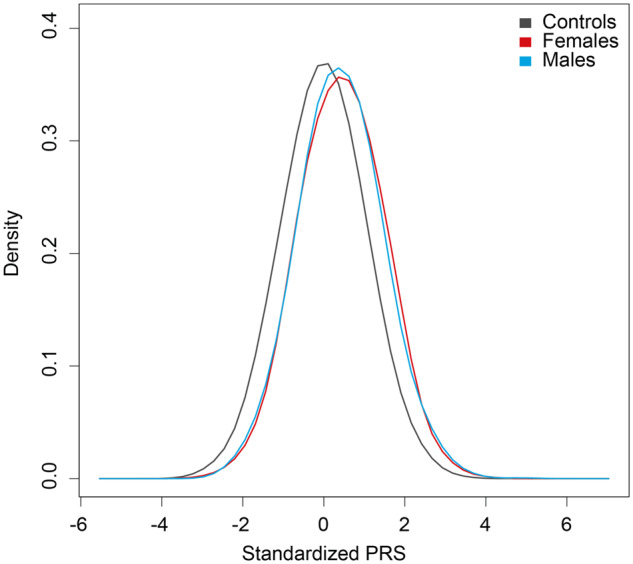
Distributions of the 313-single nucleotide polymorphism polygenic risk scores (PRSs) in 1380 male breast cancer cases, 1671 female breast cancer cases, and 2663 controls. PRSs were standardized to mean = 0, SD = 1 using 2663 controls from the 1958 British Birth Cohort. The mean PRS was 0.44 in males and 0.41 in females.
Candidate Target Gene Expression in Male and Female Breast Tissue
Functional studies have identified putative target genes for 5 of the 8 FBC predisposition loci that had statistically significant differences in their odds ratios for FBC and MBC: TERT at 5p15.33, ESR1 and CCDC170 at 6q25.1, KLF4 at 9q31.2, FGFR2 at 10q26.13, and ZFP36L1 at 14q24.1 (25–30). By examining GTEx multi-tissue eQTL analyses (31), we suggest that CITED4 (rs4233486 P = 2.36 × 10–11) and VGLL3 (rs13066793 P = 1.21 × 10–07) are candidate target genes at the loci mapping to 1p34.2 and 3p12.1 (21). CITED4 encodes Cbp/p300-interacting transactivator 4, a transcriptional coactivator that is induced during lactogenic differentiation of breast epithelial cells and is involved in milk secretion (32), and VGLL3 encodes transcription cofactor vestigial-like protein 3 and may act as a tumor suppressor gene in high-grade serous ovarian carcinoma (33).
We hypothesized that variation in the basal gene expression levels of predisposition SNP target genes in male and female breast tissue might partly explain the different MBC and FBC risks observed at these loci. To investigate, we evaluated GTEx RNA-sequencing data from 157 males and 107 females. Four candidate target genes had statistically significant sex-biased tissue expression (Supplementary Figure 5, available online). CITED4 at 1p34.2 (P = 3.00 × 10–25) and FGFR2 at 10q26.13 (P = 3.24 × 10–10) had higher expression in female than male breast tissue, whereas KLF4 at 9q31.2 (P = 9.10 × 10–10) and CCD170 at 6q25.1 (P = 2.80 × 10–04) were more highly expressed in males than females.
We also assessed eQTL associations between the lead SNPs at these loci and their candidate target genes using GTEx data from breast tissue (n = 264). The risk allele of rs13066793 at 3p12.1 was associated with reduced expression of VGLL3 (P = .02; Supplementary Figure 6, available online), albeit the association was not statistically significant after adjusting for multiple comparisons. SNP rs3757322 at 6q25.1 was borderline associated with expression of CCDC170 (P = .06), and this association varied according to sex (P = .02; Supplementary Figure 7, available online). There was no evidence of breast-specific eQTL associations with target genes at the other loci, which could reflect limited power to detect subtle differences in gene expression.
Discussion
We have performed the largest genetic association study of MBC to date by conducting, as is usual in GWAS, genome-wide imputation and meta-analysis of existing (4) and newly generated genotyping data. We identified 3 novel MBC predisposition loci that attained genome-wide levels of statistical significance, of which 2 mapped to 6q25.1 and 1 mapped to 11q13.3, bringing the total number of confirmed predisposition loci to 5. Notably, each of these loci is also associated with risk of FBC, and almost 20% of confirmed FBC susceptibility SNPs showed evidence of association with MBC predisposition. To date, no low-penetrance alleles have been identified that are exclusively associated with MBC but not FBC. Although our study does not rule out the possibility that such loci exist, it does suggest that the magnitudes of their effects will be small. Although differences between the frequencies of pathogenic BRCA1 and BRCA2 mutations have led to the suggestion that MBC and FBC have distinct genetic etiologies, our genetic correlation analysis provides evidence of a shared genetic basis for MBC and ER-positive FBC. Interestingly, we detected borderline evidence (P = .06) of a cross-cancer genetic correlation between MBC and ovarian cancer, consistent with a recently reported genetic correlation between FBC and ovarian cancer (22).
Lecarpentier et al., recently demonstrated that a FBC PRS is associated with breast cancer risk in male BRCA1/2 mutation carriers (34). We show here for the first time, to our knowledge, that a FBC PRS is associated with MBC risk in men from the general population. The odds ratio per SD increase in the PRS for males is almost identical to that of unselected females and is greater than that of male BRCA1/2 mutation carriers (20,34,35). Although risk stratification among the general population using a PRS is unfeasible given both the rarity of MBC and level of risk differentiation, the striking similarity between the PRS distributions of MBC and FBC cases suggests that a larger number of FBC predisposition variants than were detected by our study probably influence susceptibility to MBC.
Although our genetic correlation analysis indicated that MBC shares a pronounced genetic basis with ER-positive FBC, there are distinctions. For example, we observed several MBC associations among SNPs that confer greater risks of ER-negative than ER-positive FBC, including SNP rs9371545 at 6q25.1 and the BRCA2 truncating variant rs11571833. Conversely, several SNPs that are most strongly associated with ER-positive FBC were not associated with MBC, including rs11249433 at 1p11.2, rs34005590 at 2q35, and rs2981578 at 10q26.13. Although this may be a consequence of power, the ER-positive FBC odds ratio for rs2981578 is large and should be detectable in our study. We hypothesize that the underlying etiological mechanisms affected by SNPs that had statistically significant differences in their odds ratios for MBC and FBC might be influenced by sex-specific differences in expression or activity of their target genes or by different endogenous factors in males and females. The comparatively lower expression of FGFR2 in male than female breast tissue, as observed in GTEx data, could explain the lack of an MBC association with rs2981578 at the 10q26.13 locus, despite it being among the most strongly associated SNPs with ER-positive FBC.
The principal limitation of our study was its relatively small size compared with typical cancer GWAS. Consequently, it had limited capacity to detect MBC predisposition loci that are associated with small risk effects, and much larger studies will be needed for their discovery. The merits, or otherwise, of continually striving to identify polygenic determinants of disease susceptibility that confer relatively small effects have been debated extensively, particularly because they may have limited clinical usefulness in the short term. However, all statistically robust genetic associations (even those with small effects) are underpinned by risk alleles that perturb biological processes, of which some might harbor effective targets for therapeutic intervention (36), thus justifying efforts that could lead to their detection. The subsequent illumination of the target genes and pathways that underlie risk associations in MBC will also likely be difficult, not least because of a paucity of cell line models derived from male breast tumors for functional analysis. In conclusion, our findings indicate several elements of shared genetic basis for susceptibility to MBC and FBC, provide further support for a polygenic component to MBC susceptibility, and advance our understanding of the genetics of MBC development.
Funding
This work was supported by Programme Grants from Breast Cancer Now, including Programme Funding to the Breast Cancer Now Toby Robins Research Centre, the Institute of Cancer Research, and by funding from Queen’s University Belfast to NO. We acknowledge National Institute for Health Research (NIHR) funding to the Royal Marsden Hospital Biomedical Research Centre (BRC). This work made use of data and samples generated by the 1958 British Birth Cohort under grant G0000934 from the UK Medical Research Council and grant 068545/Z/02 from the Wellcome Trust. The Finnish Male Breast Cancer Study acknowledges funding from the Finnish Breast Cancer Group, the Finnish Oncology Association, and the Salonoja Foundation. LO acknowledges funding from the Italian Association for Cancer Research (Fondazione AIRC IG2018-21389). DP acknowledges funding from the Istituto Toscano Tumori, Florence, Italy. SEARCH is funded by a grant from Cancer Research UK (C490/A10124) and is supported by the University of Cambridge NIHR Biomedical Research Centre. AMD has been funded by Cancer Research UK grant CA8197/A10865 and by the Joseph Mitchell Fund. DTB acknowledges funding by Cancer Research UK (Programme Award C588/A10589). MG-D and JEC acknowledge the support of the Department of Pathology of the CHUS Santiago Hospital, the Galician Foundation of Genomic Medicine, and the Program Grupos Emergentes, Cancer Genetics Unit of CHUVI, Instituto de Salud Carlos III. HO and IH acknowledge funding from the Swedish Cancer Society and The Swedish Medical Research Council. The collection of samples and epidemiological data for the US cases was supported by US ARMY Grant DAMD-17–96-I-6266 and US National Institutes of Health (NIH) grant R01CA74415. SLN was partially supported by the Morris and Horowitz Families Endowed Professorship at the City of Hope. The Genotype-Tissue Expression Project (GTEx) was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by the National Cancer Institute, the National Human Genome Research Institute, the National Heart, Lung and Blood Institute, the National Institute on Drug Abuse, the National Institute of Mental Health, and the National Institute of Neurological Disorders and Stroke.
Notes
Role of the funder: The funders had no role in the design of the study; the collection, analysis, and interpretation of the data; the writing of the manuscript; and the decision to submit the manuscript for publication.
Conflicts of interest: AA is a cofounder of Tango Therapeutics, Azkarra Therapeutics, Ovibio Corporation; a consultant for SPARC, Bluestar, ProLynx, Earli, Cura, GenVivo and GSK; a member of the SAB of Genentech and GLAdiator; receives grant / research support from SPARC and AstraZeneca; holds patents on the use of PARP inhibitors held jointly with AstraZeneca from which he has benefitted financially (and may do so in the future).
Role of the authors: SM: Formal analysis; Investigation; Methodology; Writing—review and editing. EP: Formal analysis; Investigation; Writing—review and editing. KT: Formal analysis; Methodology; Resources; Writing—review and editing. MEJ: Resources; Writing—review and editing. OF: Investigation; Writing—review and editing. MP: Investigation; Writing—review and editing. TW: Investigation; Writing—review and editing. KT: Investigation; Writing—review and editing. RC: Data curation; Investigation; Writing—review and editing. kConFab Consortium: Resources. AT: Resources. PJ: Resources. SB: Resources. HF: Resources. HN: Resources. JM: Resources. EF: Resources. YL: Resources. DP: Resources. GM: Resources. IZ: Resources. LO: Resources. VS: Resources. AH: Resources. MJH: Resources. SN: Resources. MK: Resources. MG-D: Resources. JEC: Resources. HO: Resources. IH: Resources. ES: Resources. VG: Resources. DFE: Resources. PP: Resources. AMD: Resources. DTB: Resources. SLN: Resources. LS: Resources. AA: Conceptualization; Funding acquisition. MGC: Conceptualization; Investigation; Methodology. RH: Conceptualization; Investigation; Methodology. AS: Conceptualization; Data curation; Funding acquisition; Methodology; Project administration; Resources; Supervision; Writing—review and editing.
Acknowledgments: We thank the subjects who participated in the study. For UK-BCN-MBCS sample collection, we are grateful to the administrative and data management staff of the ICR Aetiological Epidemiology Team for help and advice; to the research nurses for collection of blood samples and questionnaire data from participants; to the UK-GS team for coordinating collection of controls; and to the cancer registries of England and Wales for providing information on eligible participants. The Study of Epidemiology and Risk Factors in Cancer Heredity (SEARCH) thanks the SEARCH team. We wish to thank H. Thorne, E. Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics and the Clinical Follow Up Study [funded from 2001–2009 by the Australian National Health and Medical Research Council (NHMRC) and currently by the National Breast Cancer Foundation and Cancer Australia, 628333] for their contributions to this resource and the many families who contribute to kConFab. We thank the staff at the Finnish Male Breast Cancer Study for their contributions. We thank the staff of the Department of Pathology of CHUS Santiago for their contributions to the Spanish study. Finally, we thank the consultants who cared for the study participants for their advice and help. The BCN Male Breast Cancer Study was approved by the South Eastern Research Ethics Committee
Data availability statement
The summary data underlying this article will be shared on reasonable request to the corresponding author.
Supplementary Material
References
- 1. Basham VM, Lipscombe JM, Ward JM, et al. BRCA1 and BRCA2 mutations in a population-based study of male breast cancer. Breast Cancer Res. 2001;4(1):R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bevier M, Sundquist K, Hemminki K.. Risk of breast cancer in families of multiple affected women and men. Breast Cancer Res Treat. 2012;132(2):723–728. [DOI] [PubMed] [Google Scholar]
- 3. Orr N, Cooke R, Jones M, et al. Genetic variants at chromosomes 2q35, 5p12, 6q25.1, 10q26.13, and 16q12.1 influence the risk of breast cancer in men. PLoS Genet. 2011;7(9):e1002290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Orr N, Lemnrau A, Cooke R, KConFab Consortium, et al. Genome-wide association study identifies a common variant in RAD51B associated with male breast cancer risk. Nat Genet. 2012;44(11):1182–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Silvestri V, Rizzolo P, Scarno M, et al. Novel and known genetic variants for male breast cancer risk at 8q24.21, 9p21.3, 11q13.3 and 14q24.1: results from a multicenter study in Italy. Eur J Cancer. 2015;51(16):2289–2295. [DOI] [PubMed] [Google Scholar]
- 6. Easton DF, Pooley KA, Dunning AM, The SEARCH orators, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447(7148):1087–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thomas G, Jacobs KB, Kraft P, et al. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1). Nat Genet. 2009;41(5):579–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Swerdlow AJ, Jones ME, Schoemaker MJ, et al. The Breakthrough Generations Study: design of a long-term UK cohort study to investigate breast cancer aetiology. Br J Cancer. 2011;105(7):911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boraska V, Jeroncic A, Colonna V, et al. Genome-wide meta-analysis of common variant differences between men and women. Hum Mol Genet. 2012;21(21):4805–4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Delaneau O, Zagury JF, Marchini J.. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods. 2013;10(1):5–6. [DOI] [PubMed] [Google Scholar]
- 11. Howie BN, Donnelly P, Marchini J.. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5(6):e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marchini J, Howie B, Myers S, et al. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39(7):906–913. [DOI] [PubMed] [Google Scholar]
- 13. Willer CJ, Li Y, Abecasis GR.. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yeager M, Orr N, Hayes RB, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39(5):645–649. [DOI] [PubMed] [Google Scholar]
- 15. Wakefield J. A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am J Hum Genet. 2007;81(2):208–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Michailidou K, Lindstrom S, Dennis J, et al. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551(7678):92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bulik-Sullivan B, Finucane HK, Anttila V, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schumacher FR, Al Olama AA, Berndt SI, et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet. 2018;50(7):928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Phelan CM, Kuchenbaecker KB, Tyrer JP, et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat Genet. 2017;49(5):680–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mavaddat N, Michailidou K, Dennis J, et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am J Hum Genet. 2019;104(1):21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang X, Finucane HK, Schumacher FR, et al. Shared heritability and functional enrichment across six solid cancers. Nat Commun. 2019;10(1):431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kar SP, Beesley J, Amin Al Olama A, et al. Genome-wide meta-analyses of breast, ovarian, and prostate cancer association studies identify multiple new susceptibility loci shared by at least two cancer types. Cancer Discov. 2016;6(9):1052–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. French JD, Ghoussaini M, Edwards SL, et al. Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long-range enhancers. Am J Hum Genet. 2013;92(4):489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baxter JS, Leavy OC, Dryden NH, et al. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nat Commun. 2018;9(1):1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bojesen SE, Pooley KA, Johnatty SE, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45(4):371–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dunning AM, Michailidou K, Kuchenbaecker KB, et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat Genet. 2016;48(4):374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li Q, Seo JH, Stranger B, et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell. 2013;152(3):633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meyer KB, O’Reilly M, Michailidou K, et al. Fine-scale mapping of the FGFR2 breast cancer risk locus: putative functional variants differentially bind FOXA1 and E2F1. Am J Hum Genet. 2013;93(6):1046–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Orr N, Dudbridge F, Dryden N, et al. Fine-mapping identifies two additional breast cancer susceptibility loci at 9q31.2. Hum Mol Genet. 2015;24(10):2966–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sul JH, Han B, Ye C, et al. Effectively identifying eQTLs from multiple tissues by combining mixed model and meta-analytic approaches. PLoS Genet. 2013;9(6):e1003491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sornapudi TR, Nayak R, Guthikonda PK, et al. Comprehensive profiling of transcriptional networks specific for lactogenic differentiation of HC11 mammary epithelial stem-like cells. Sci Rep. 2018;8(1):11777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gambaro K, Quinn MC, Wojnarowicz PM, et al. VGLL3 expression is associated with a tumor suppressor phenotype in epithelial ovarian cancer. Mol Oncol. 2013;7(3):513–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lecarpentier J, Silvestri V, Kuchenbaecker KB, et al. Prediction of breast and prostate cancer risks in male BRCA1 and BRCA2 mutation carriers using polygenic risk scores. J Clin Oncol. 2017;35(20):2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mavaddat N, Pharoah PD, Michailidou K, et al. Prediction of breast cancer risk based on profiling with common genetic variants. J Natl Cancer Inst. 2015;107(5):djv036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Price AL, Spencer CC, Donnelly P.. Progress and promise in understanding the genetic basis of common diseases. Proc R Soc B. 2015;282(1821):20151684. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The summary data underlying this article will be shared on reasonable request to the corresponding author.