

Abstract
α-Synuclein (α-Syn) is a key pathogenic protein in α-synucleinopathies including Parkinson disease (PD) and Dementia with Lewy Bodies. The aggregation of α-Syn is believed to be deleterious and a critical step leading to neuronal dysfunction and death. One of the factors that may contribute to the initial steps of this aggregation is crosslinking through transglutaminase 2 (TG2). We previously demonstrated that overexpression of TG2 exacerbates α-Syn toxicity in mice and yeast by increasing the higher-order species of α-Syn. Herein, we investigated whether deletion of the TG2 encoding gene could mitigate the toxicity of α-Syn in vivo. Compared with α-Syn transgenic (SynTg) mice, TG2 null /α-Syn transgenic mice (TG2KO/SynTg) exhibited a reduced amount of phosphorylated α-Syn aggregates and fewer proteinase K-resistant α-Syn aggregates in sections of brain tissue. Neuritic processes that are depleted in SynTg mice compared to wild-type mice were preserved in double TG2KO/SynTg mice. Additionally, the neuroinflammatory reaction to α-Syn was attenuated in TG2KO/SynTg animals. These neuropathological markers of diminished α-Syn toxicity in the absence of TG2 were associated with better motor performance on the rotarod and balance beam. These results suggest that deleting TG2 reduces the toxicity of α-Syn in vivo and improves the behavioral performance of SynTg mice. Accordingly, these findings collectively support pharmacological inhibition of TG2 as a potential disease modifying therapeutic strategy for α-synucleinopathies.
Keywords: TG2, α-synuclein, protein aggregation, neuroinflammation, Parkinson disease
INTRODUCTION
Parkinson disease (PD) and Dementia with Lewy Bodies (DLB) are common neurodegenerative disorders characterized pathologically by intraneuronal aggregates of α-Synuclein (α-Syn) in Lewy bodies and Lewy neurites (Jenner and Olanow, 1998; Spillantini et al., 1998). α-Syn is a small, 140 amino acid intrinsically disordered protein (Lee and Trojanowski, 2006) that is prone to self-aggregate and forms fibrils in neuropathological hallmark inclusions in response to diverse exogenous and endogenous factors (Goldberg and Lansbury, 2000). This aggregation of α-Syn is believed to be a critical step leading to neuronal cell death (Goedert, 2001; Cookson, 2009). Thus, preventing α-Syn aggregation at an early stage is of therapeutic interest in α-synucleinopathies.
Transglutaminases (TGs) are a family of enzymes that catalyze a calcium-dependent formation of epsilon-(gamma-glutamyl) lysine isodipeptide bonds and result in a covalent linkage between two peptide molecules (Greenberg et al., 1991). Transglutaminase 2 (TG2) is one member of this family, which is expressed broadly in the mammalian and human brain in both neurons and astrocytes (Kim et al., 1999; Lesort et al., 1999). Several lines of evidence suggest that TG2 plays a pathogenic role in PD and DLB. In in vitro and cell culture studies, TG2 catalyzes the formation of high molecular weight α-Syn aggregates in a calcium dependent manner (Junn et al., 2003), and increased transamidation of α-Syn by TG2 is found in the 1-methyl-4-phenylpyridine (MPP(+)) toxicity model in SH-SY5Y cells (Verhaar et al., 2011; Grosso et al., 2014). In human studies, compared with control subjects, a significant increase in TG2 protein and mRNA expression is found in the substantia nigra of PD patients (Citron et al., 2002; Andringa et al., 2004; Wilhelmus et al., 2011) as well as increased TG2 protein levels in their cerebrospinal fluid (Vermes et al., 2004). Postmortem immunohistochemical and immunoblot studies have also shown the presence of isodipeptide bonds formed by TG2 co-localizing with α-Syn in Lewy bodies in both PD and DLB affected brains (Citron et al., 2002; Junn et al., 2003). Additionally, several pathogenic aberrations found in neurodegenerative disease brains, including oxidative stress, elevated calcium, and ATP depletion can activate TG2 (Grosso and Mouradian, 2012).
Based on this evidence, TG2 may impact the pathogenesis of PD and related disorders by contributing to α-Syn misfolding. However, whether TG2 depletion can mitigate the toxicity of α-Syn in vivo has not yet been demonstrated. In the present study, we show that genetic deletion of the TG2 gene in mice attenuates the accumulation of α-Syn aggregates, protects neurons from the toxicity of α-Syn overexpression, improves neuronal integrity, and reduces the associated neuroinflammation, leading to improved behavioral performance.
EXPERIMENTAL PROCEDURES
Animals
C57BL/6J mice were obtained from the Jackson Laboratories (Bar Harbor, ME). TG2 knockout mice (Nanda et al., 2001) and human wild-type α-synuclein transgenic mice under the control of the murine Thy-1 promoter (Rockenstein et al., 2002) are described previously. To create TG2KO/SynTg double modified mice, α-Synuclein transgenic (SynTg) female mice were crossbred with male TG2KO mice. The TG2KO line was maintained by breeding TG2KO mice with WT mice of the C57B/6 background. Genotypes were determined by PCR of tail DNA. Only male mice were used in this study. Mice were sacrificed, and brains were collected from 6 to 9 month old animals. All housing, breeding, and procedures were performed according to the NIH Guide for the Care and Use of Experimental Animals and approved by the Rutgers-Robert Wood Johnson Medical School Institutional Animal Care and Use Committee.
Immunohistochemistry and immunofluorescence
Mice were perfused transcardially with PBS, and brains were removed and fixed in 10% formalin (Sigma-Aldrich) at 4 °C overnight. Brains were sectioned using a Leica VT1000 S vibratome at 40 μm thickness in the coronal plane through the entirety of the brain from the frontal association cortex through the pons, and serial sections were collected as sets with the same interval. Sections were then selected from this bank of tissue for each staining marker. For cortical and striatal studies, sections were selected at approximately Bregma 0.98 in the Paxinos and Franklin Mouse Brain Atlas using the anterior commissure and corpus callosum as landmarks to select equivalent sections across animals. For immunohistochemistry, free-floating sections were pretreated differently before adding antibodies depending on staining conditions. For regular immunohistochemistry, sections were blocked in 5% BSA (Sigma-Aldrich) following incubation with 3% hydrogen peroxide (Sigma-Aldrich) to inhibit endogenous peroxidase activity. For proteinase K treatment, samples were incubated in 88% formic acid (Thermo Fisher Scientific) for 10 min for antigen retrieval and then incubated in 10 μg/ml proteinase K (Sigma-Aldrich) for 10 min before being blocked in 5% BSA. After pretreatments, sections were incubated with α-synuclein antibody (#610787, BD bioscience) at 4 °C overnight and with biotinylated secondary antibody (Sigma-Aldrich) for 1 h at room temperature. Vectastain elite ABC kit (Vector Laboratories, Burlingame, CA, USA) and 3.3’-diaminobenzidine (Sigma-Aldrich) were used for amplification and color development. Images were captured using a Nikon Eclipse 55i microscope and NIS Elements D3.2 software (Nikon, Tokyo, Japan). For immunofluorescence staining of microtubule-associated protein 2 (MAP2), sections were blocked with 5% goat serum (Sigma-Aldrich) and 0.2 % Triton X-100 (Sigma-Aldrich) in PBS. Sections were then incubated with primary antibody overnight at 4 °C and fluorescent secondary antibody for 1 h at room temperature. Images were captured using Carl Zeiss Axiovert 200 microscope. For cortical studies, images were taken of the outer layers of the motor cortex. ImageJ was used to threshold stained areas and to automatically calculate the number of defined regions and total optical density (OD). Primary antibodies used were anti-α-synuclein (#610787, BD Bioscience), anti-phospho-Ser129-α-Syn (#015–25191, WAKO), anti-glial fibrillary acidic protein (GFAP) (Dako Carpinatria, CA, USA), anti-Ionized calcium-binding adaptor molecule 1 (Iba-1, a marker of microglial activation as an indication of neuroinflammation) (#019–19741, WAKO), and anti-microtubule-associated protein 2 (MAP2, a marker of neuritic processes as a reflection of neuronal integrity) (Santa Cruz Biotechnology).
Behavioral assessments
Behavioral assessments were performed at 6 months of age. The rotarod test was done as described previously (Lee et al., 2015). Briefly, mice were placed on a rotating cylinder (diameter = 4.5 cm) with a coarse surface for firm grip and tested for 3 trials with an accelerating speed of 0.2 rpm/s, increasing from 4 to 40 rpm. A cutoff time of 3 min and an inter-trial interval of 60 min were used.
Latency on the rod before falling was measured. The balance beam test was as described before (Quinn et al., 2008). In brief, mice were habituated to a dark goal box for 3 min and then trained to walk across a narrow beam to reach that box. The following day, three consecutive trials were done, and the time taken to cross the beam to reach the goal box with each trial was recorded for each mouse.
Statistical analysis
The results are presented as box-plots where the bottom and the top of the box are the first and third quartiles, respectively, and the whiskers above and below the box indicate the 95th and 5th percentiles. The median is indicated as a horizontal line. Data are analyzed by one-way factorial analysis of variance (ANOVA) followed by Tukey’s Multiple Comparison Test when comparing all four groups. Unpaired t test was used when only SynTg and TG2KO/SynTg mice were compared for p-α-Syn staining and proteinase K-resistant α-Syn staining. Significance was determined at p < 0.05. Statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software, Inc., San Diego, CA).
RESULTS
Genetic deletion of TG2 prevents the formation of α-Syn aggregates in the mouse brain
Given that TG2 crosslinks α-Syn and promotes its aggregation (Junn et al., 2003; Grosso et al., 2014), we first assessed whether deletion of the TG2 gene impacts the formation of these aggregates in the brains of SynTg mice using immunohistochemical stains. As α-Syn aggregates in both human α-synucleinopathies and in SynTg mouse brains are characteristically hyperphosphorylated at serine 129 and are resistant to clearance by proteinase K (Neumann et al., 2002), antibody to phosphorylated α-Syn (p-α-Syn) and staining for α-Syn after digestion with proteinase K were employed to address this question. As expected, brain sections from TG2KO/SynTg mice had lower phosphorylated α-Syn intensity in the cortex, by as much as half of that detected in SynTg mice (Fig. 1A, B). Brains of wild-type and TG2KO mice had no p-α-Syn immunoreactive neurons (Fig. 1A). Similarly, the number of proteinase K-resistant α-Syn aggregates in the striatum of TG2KO/SynTg mice was 42% of that detected in SynTg mice (Fig. S1A, B). No punctate aggregates were found in WT or TG2KO mice following this protease digestion (Fig. S1A). These results suggest that deletion of TG2 prevents the formation of misfolded pathogenic species of α-Syn in vivo.
Fig. 1.
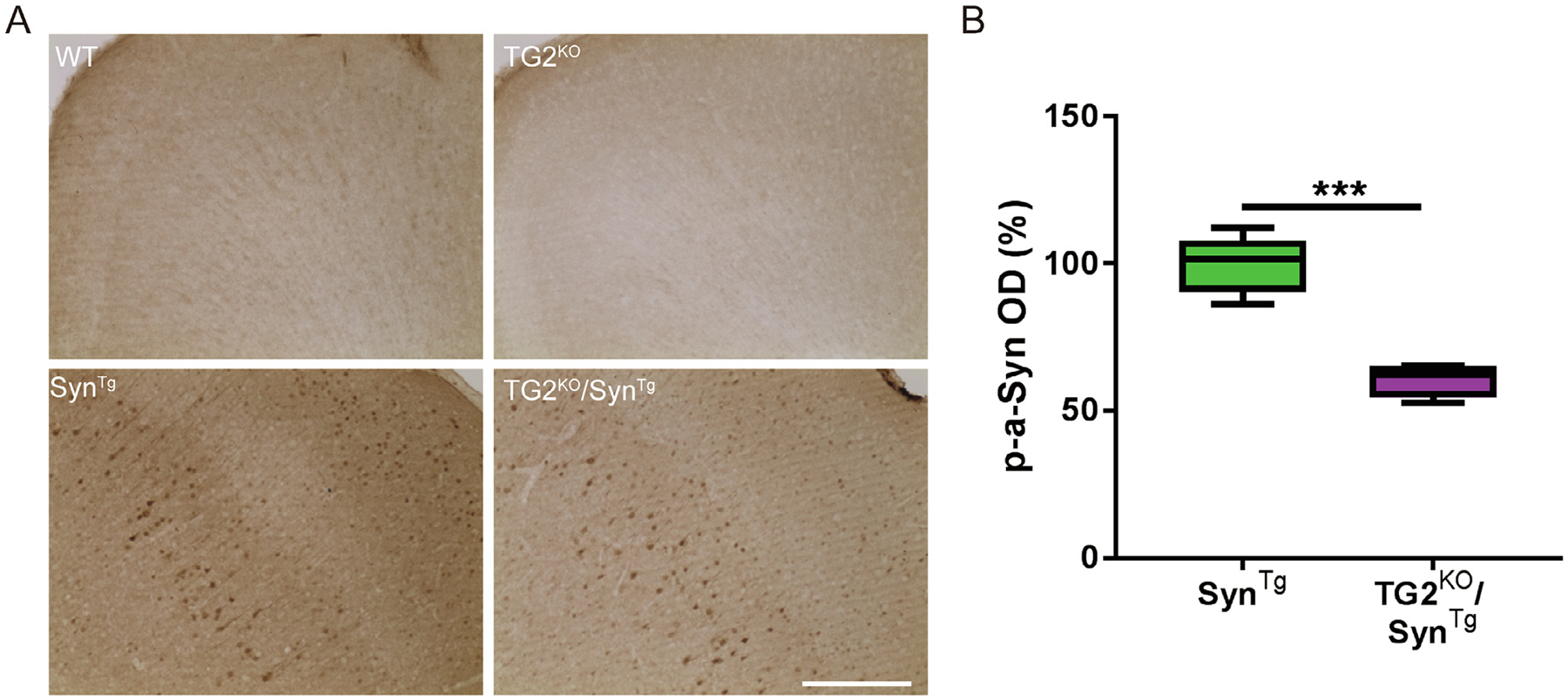
TG2 deletion decreases the formation of phosphorylated α-Syn in the mouse brain. (A) Representative images of p-α-Syn staining in the cortex of mice. (B) Quantification of p-α-Syn staining intensity in (A) (n: SynTg = 6; TG2KO/SynTg = 5). In the box-plots, the bottom and top of the box are the first and third quartiles, respectively, and the whiskers above and below the box indicate the 95th and 5th percentiles. The median is indicated as a horizontal line. ***P < 0.001, two-tailed unpaired t test. Scale bar = 50 μm.
TG2 depletion improves neuronal integrity that is lost in SynTg mice
We previously reported that TG2 exacerbates the neuronal toxicity of α-Syn in vivo (Grosso et al., 2014).To investigate whether TG2 deletion prevents the deleterious effect of α-Syn overexpression, MAP2 staining was assessed next. SynTg mice have substantial depletion of MAP2 immunoreactivity in the cortex (Fig. 2A), suggestive of disrupted nerve fibers and reduced dendritic complexity (Harada et al., 2002; Koob et al., 2010; Lee et al., 2011). On the other hand, TG2 deletion prevented the degradation of neuritic complexity in SynTg mice (Fig. 2A, B). These observations suggest that TG2 depletion protects against α-Syn induced neuronal toxicity.
Fig. 2.
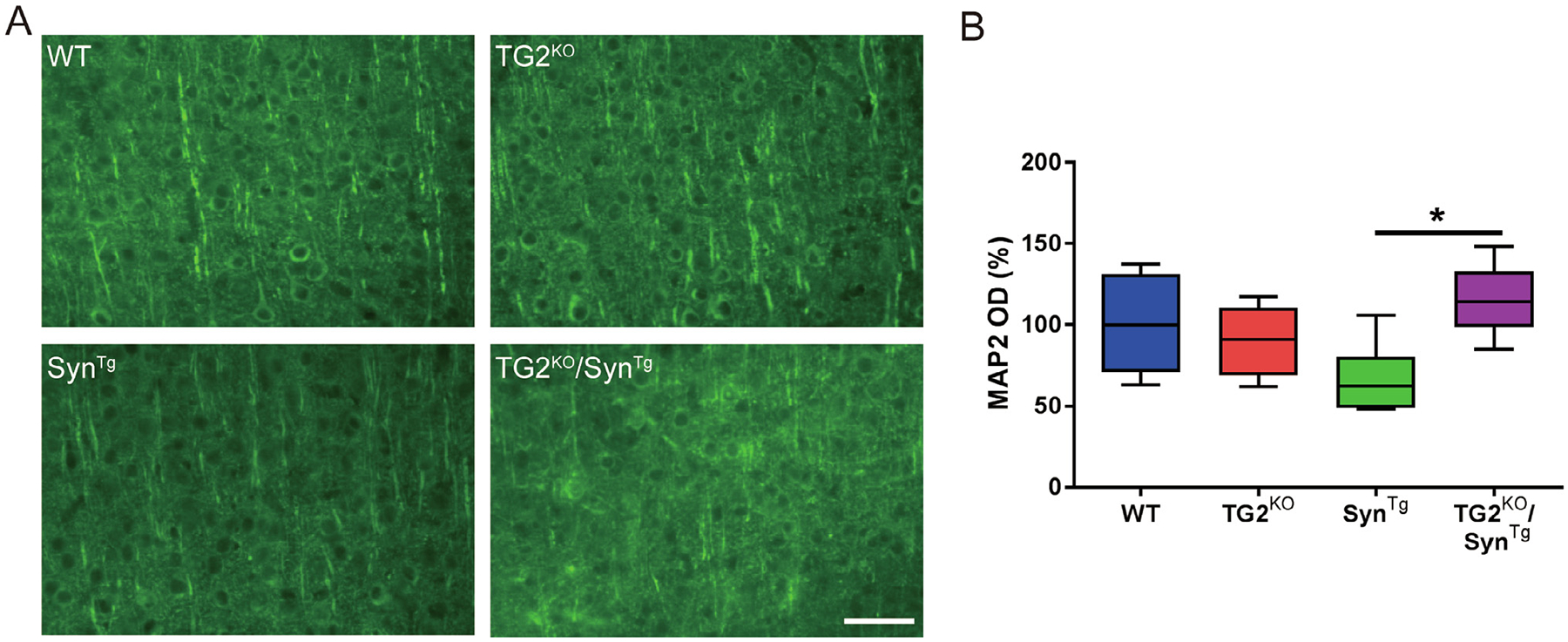
TG2 deletion protects against the neuronal toxicity of α-Syn. (A) Representative images of MAP2 staining in the cortex of mice. (B) Quantification of immunofluorescence staining of MAP2 in (A) (n: WT = 6; TG2KO = 6; SynTg = 6; TG2KO/SynTg = 5). In the box-plots, the bottom and top of the box are the first and third quartiles, respectively, and the whiskers above and below the box indicate the 95th and 5th percentiles. The median is indicated as a horizontal line. *P < 0.05, one-way ANOVA. Scale bar = 50 μm.
TG2 deletion attenuates the neuroinflammation in SynTg mice
Neuroinflammation is one of the neuropathological features of PD and DLB (Tansey and Goldberg, 2010; Surendranathan et al., 2018) as well as in models of α-synucleinopathy including SynTg mice (Lee et al., 2011). We previously showed that TG2 aggravates the neuroinflammation in the brains of SynTg mice (Grosso et al., 2014). To investigate whether deleting TG2 has the opposite effect, immunohistochemistry for the astrocytic marker GFAP (Fig. 3A) and the microglial marker Iba1 (Fig. 3B) were performed. Consistent with our previous findings (Lee et al., 2011; Grosso et al., 2014; Lee et al., 2015; Yan et al., 2018), SynTg mice had a markedly increased GFAP positive signal in the cortex compared with WT and TG2KO mice, while genetic deletion of TG2 significantly decreased GFAP signal induced by overexpression of α-Syn (Fig. 3A, C). Similarly, SynTg mice had significantly increased staining of the microglial marker Iba1 compared to the other 3 genotypes including TG2KO/SynTg mice (Fig. 3B, D). These findings suggest that TG2 deletion can mitigate the neuroinflammatory response to α-Syn.
Fig. 3.
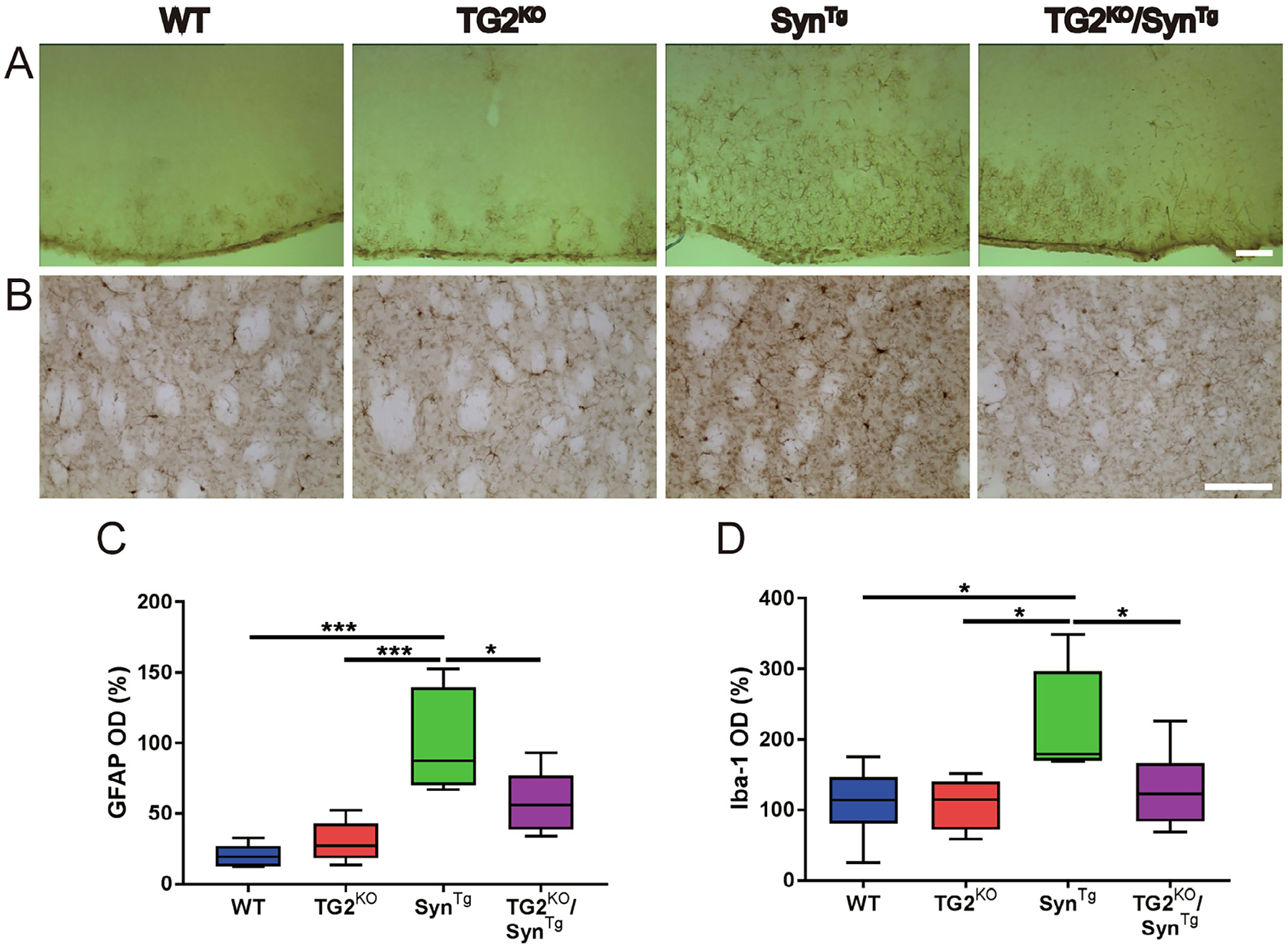
TG2 deletion prevents the neuroinflammatory response to α-Syn. (A) Representative immunohistochemical images of cortical sections from each of the four mouse lines stained for GFAP. (B) Representative images of striatal sections stained for Iba-1. (C) Quantification of immunohistochemical staining of GFAP in (A) (n: WT = 6; TG2KO = 6; SynTg = 6; TG2KO/SynTg = 5). (D) Quantification of immunohistochemical staining of Iba-1 in (B) (n: WT = 6; TG2KO = 6; SynTg = 6; TG2KO/SynTg = 6). In the box-plots, the bottom and top of the box are the first and third quartiles, respectively, and the whiskers above and below the box indicate the 95th and 5th percentiles. The median is indicated as a horizontal line. *P < 0.05; *** P < 0.001, one-way ANOVA. Scale bar = 50 μm.
Deleting TG2 prevents the behavioral deficits of SynTg mice
To determine if the neuropathological markers of protection associated with TG2 deletion correlate with improved motor behavior, performance on the balance beam test and the rotarod were evaluated at 6 months of age. Both tasks were chosen because they reflect nigrostriatal function (Rozas and Labandeira García, 1997; Quinn et al., 2008). As expected, SynTg mice showed significantly impaired ability to stay on the rotarod compared to WT and TG2KO mice, whereas the performance of double modified TG2KO/SynTg mice was significantly better (Fig. 4A). A similar profile of differences in the balance beam test was found. SynTg mice had worse performance than WT and TG2KO mice, while TG2KO/SynTg mice demonstrated less severe impairment compared to SynTg mice (Fig. 4B). These behavioral improvements in TG2KO/SynTg mice are consistent with the histopathologic data in these animals.
Fig. 4.
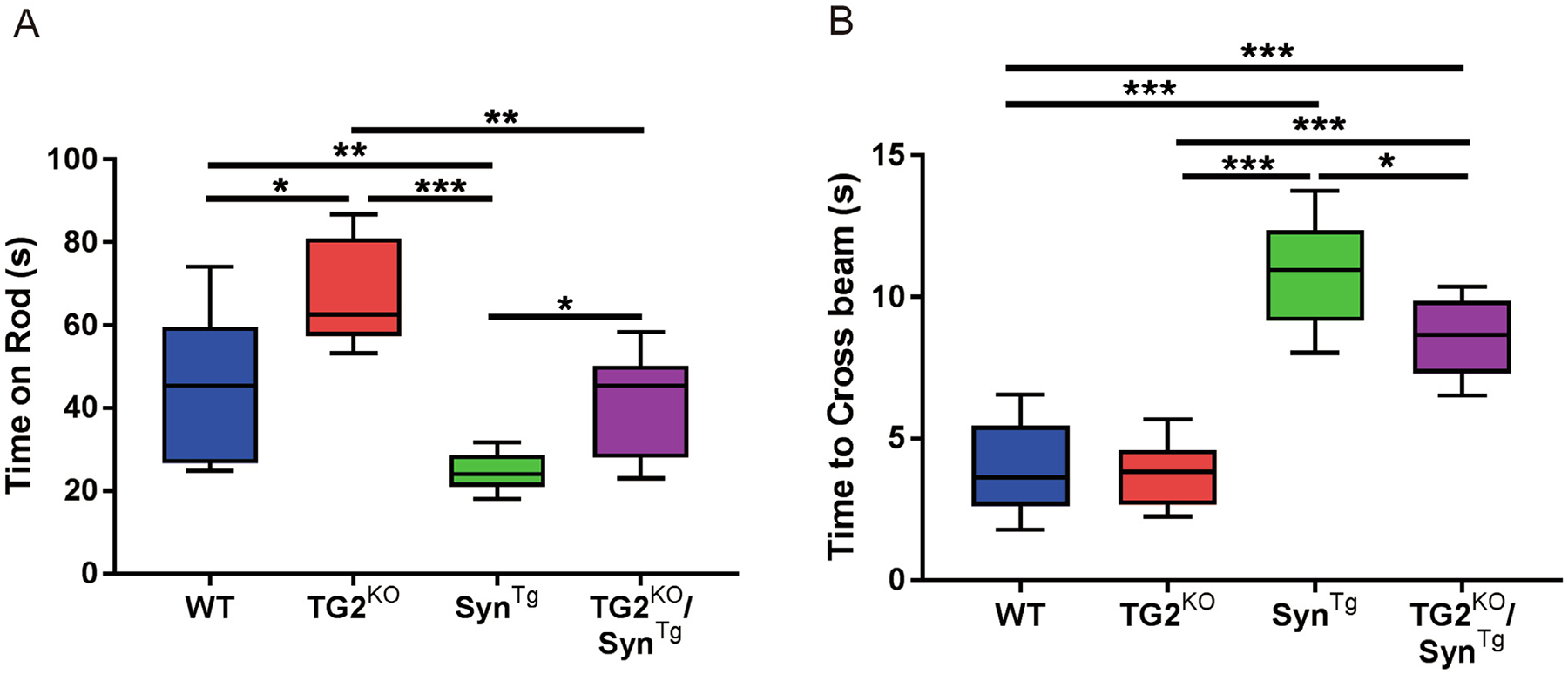
TG2 deletion prevents the behavioral deficits of SynTg mice. (A) Performance on the rotarod (n: WT = 11; TG2KO = 6; SynTg = 9; TG2KO/SynTg = 10). (B) Time taken to cross the balance beam (n: WT = 11; TG2KO = 6; SynTg = 9; TG2KO/SynTg = 6). In the box-plots, the bottom and top of the box are the first and third quartiles, respectively, and the whiskers above and below the box indicate the 95th and 5th percentiles. The median is indicated as a horizontal line. *P < 0.05; **P < 0.01; ***P < 0.001, one-way ANOVA.
DISCUSSION
The present findings demonstrate that deleting the TG2 gene in SynTg mice results in reduced α-Syn aggregation in the brain, preserved neuronal integrity and less intense neuroinflammation, as well as better motor performance. These findings together suggest that deletion of TG2 attenuates the toxicity of α-Syn.
α-Syn plays a key role in both familial and sporadic forms of PD based on several lines of evidence from genetic, neuropathologic and cellular/molecular studies (Lee and Trojanowski, 2006; Bridi and Hirth, 2018). Due to its natively unfolded conformation, α-Syn tends to self-aggregate and accumulate in Lewy bodies and Lewy neurites (Paik et al., 1998; Goldberg and Lansbury, 2000). In vitro experiments have shown that α-Syn can form aggregates and assemble into elongated filaments (Giasson et al., 1999; Uversky et al., 2002; Yang et al., 2019), which is consistent with the high molecular weight (HMW) α-Syn found in α-Syn transgenic mice (Grosso et al., 2014; Lee et al., 2015). The fact that TG2 augments the accumulation of these HMW species has been demonstrated in both in vitro and in vivo studies (Junn et al., 2003; Grosso et al., 2014). The present study shows that deletion of TG2 prevents the formation of α-Syn aggregates in synTg mice where the protein is overexpressed in neurons. This finding complements our previous observation that over-expression of TG2 enhances α-Syn aggregation in the mouse brain (Grosso et al., 2014), thus further supporting the conclusion that TG2 promotes α-Syn aggregation in vivo.
The pathology of PD is associated with a neuroinflammatory reaction, and α-Syn-induced neuroinflammation is well documented. Microglia are activated with elevated inflammatory cytokines in SynTg animals under the control of the pan-neuronal Thy-1 promoter (Grosso et al., 2014; Lee et al., 2015) or the dopamine neuron specific tyrosine hydroxylase promoter (Richfield et al., 2002). Localized overexpression of α-Syn using Adeno-Associated Virus vector-mediated delivery also activates microglia (Sanchez-Guajardo et al., 2010). In addition to microglial activation, overexpression of α-Syn in mice induces astrocytic activation (Lee et al., 2011; Kurz et al., 2012). We previously demonstrated that TG2 overexpression aggravates the neuroinflammatory effects of α-Syn in vivo, including activation of microglia and astrocytes (Grosso et al., 2014). Here, we demonstrate that deleting TG2 attenuates the neuroinflammation in SynTg mice, which strongly supports the involvement of TG2 in α-Syn induced neuroinflammation.
Impaired neuronal function and morphology in SynTg mice indicate α-Syn toxicity, which is detected by depletion of MAP2 and impaired motor performance (Harada et al., 2002; Koob et al., 2010; Lee et al., 2011). Exacerbation of α-Syn-mediated neuronal toxicity due to TG2 over-expression was demonstrated in our previous study (Grosso et al., 2014). The reciprocal finding in the present study shows that depletion of TG2 prevents the neurotoxicity of α-Syn by maintaining neuronal integrity and minimizing the motor behavioral deficits of SynTg mice. Thus, the present results confirm that TG2 impacts the toxicity of α-Syn in vivo.
In conclusion, our data provide evidence that deleting TG2 mitigates the toxicity of α-Syn and its downstream neuropathologic consequences. These findings extend and support our previous study that shows TG2 exacerbates α-Syn toxicity in mice and yeast (Grosso et al., 2014). Thus, the experimental evidence from two studies collectively suggests that inhibiting TG2 is a plausible disease modifying therapeutic strategy for PD and related α-synucleinopathies.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Eliezer Masliah (then at the University of California, San Diego) for providing Thy-1-α-synuclein transgenic mice (SynTg). This study was supported by a grant (ID 12350) from the Michael J. Fox Foundation for Parkinson’s Research to M.M.M. who is the William Dow Lovett Professor of Neurology and is supported by grants from the National Institutes of Health (NIH) (AT006868, NS073994, NS096032, and NS101134) and the American Parkinson Disease Association. E.J. is supported by the NIH (NS070898 and NS095003) and the State of New Jersey.
Abbreviations:
- ANOVA
analysis of variance
- DLB
dementia with lewy bodies
- GFAP
glial fibrillary acidic protein
- HMW
high molecular weight
- Iba-1
ionized calcium-binding adaptor molecule 1
- MAP2
microtubule-associated protein 2
- MPP(+)
1-methyl-4-phenylpyridine
- PD
Parkinson disease
- p-α-Syn
phosphorylated α-Syn
- SynTg
α-Syn transgenic
- TG2
transglutaminase 2
- TG2KO
TG2 null
- α-Syn
α-Synuclein
Footnotes
APPENDIX A. SUPPLEMENTARY DATA
Supplementary data to this article can be found online at https://doi.org/10.1016/j.neuroscience.2020.05.047.
REFERENCES
- Andringa G, Lam KY, Chegary M, Wang X, Chase TN, Bennett MC (2004) Tissue transglutaminase catalyzes the formation of alpha-synuclein crosslinks in Parkinson’s disease. FASEB J 18:932–934. [DOI] [PubMed] [Google Scholar]
- Bridi JC, Hirth F (2018) Mechanisms of alpha-synuclein induced synaptopathy in Parkinson’s disease. Front Neurosci 12:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron BA, Suo Z, SantaCruz K, Davies PJ, Qin F, Festoff BW (2002) Protein crosslinking, tissue transglutaminase, alternative splicing and neurodegeneration. Neurochem Int 40:69–78. [DOI] [PubMed] [Google Scholar]
- Cookson MR (2009) alpha-Synuclein and neuronal cell death. Mol Neurodegener 4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Uryu K, Trojanowski JQ, Lee VM (1999) Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem 274:7619–7622. [DOI] [PubMed] [Google Scholar]
- Goedert M (2001) Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci 2:492–501. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Lansbury PT Jr (2000) Is there a cause-and-effect relationship between alpha-synuclein fibrillization and Parkinson’s disease? Nat Cell Biol 2:E115–119. [DOI] [PubMed] [Google Scholar]
- Greenberg CS, Birckbichler PJ, Rice RH (1991) Transglutaminases: multifunctional cross-linking enzymes that stabilize tissues. FASEB J 5:3071–3077. [DOI] [PubMed] [Google Scholar]
- Grosso H, Mouradian MM (2012) Transglutaminase 2: biology, relevance to neurodegenerative diseases and therapeutic implications. Pharmacol Ther 133:392–410. [DOI] [PubMed] [Google Scholar]
- Grosso H, Woo JM, Lee KW, Im JY, Masliah E, Junn E, Mouradian MM (2014) Transglutaminase 2 exacerbates alpha-synuclein toxicity in mice and yeast. FASEB J 28:4280–4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A, Teng J, Takei Y, Oguchi K, Hirokawa N (2002) MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J Cell Biol 158:541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P, Olanow CW (1998) Understanding cell death in Parkinson’s disease. Ann Neurol 44:S72–84. [DOI] [PubMed] [Google Scholar]
- Junn E, Ronchetti RD, Quezado MM, Kim SY, Mouradian MM (2003) Tissue transglutaminase-induced aggregation of alpha-synuclein: Implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A 100:2047–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Grant P, Lee JH, Pant HC, Steinert PM (1999) Differential expression of multiple transglutaminases in human brain. Increased expression and cross-linking by transglutaminases 1 and 2 in Alzheimer’s disease. J Biol Chem 274:30715–30721. [DOI] [PubMed] [Google Scholar]
- Koob AO, Ubhi K, Paulsson JF, Kelly J, Rockenstein E, Mante M, Adame A, Masliah E (2010) Lovastatin ameliorates α-synuclein accumulation and oxidation in transgenic mouse models of α-synucleinopathies. Exp Neurol 221:267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz A, May C, Schmidt O, Muller T, Stephan C, Meyer HE, Gispert S, Auburger G, et al. (2012) A53T-alpha-synuclein-overexpression in the mouse nigrostriatal pathway leads to early increase of 14-3-3 epsilon and late increase of GFAP. J Neural Transm (Vienna) 119:297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Chen W, Junn E, Im JY, Grosso H, Sonsalla PK, Feng X, Ray N, et al. (2011) Enhanced phosphatase activity attenuates alpha-synucleinopathy in a mouse model. J Neurosci 31:6963–6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Trojanowski JQ (2006) Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron 52:33–38. [DOI] [PubMed] [Google Scholar]
- Lee KW, Woo JM, Im JY, Park ES, He L, Ichijo H, Junn E, Mouradian MM (2015) Apoptosis signal-regulating kinase 1 modulates the phenotype of alpha-synuclein transgenic mice. Neurobiol Aging 36:519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesort M, Chun W, Johnson GV, Ferrante RJ (1999) Tissue transglutaminase is increased in Huntington’s disease brain. J Neurochem 73:2018–2027. [PubMed] [Google Scholar]
- Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM (2001) Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem 276:20673–20678. [DOI] [PubMed] [Google Scholar]
- Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, Müller V, Odoy S, et al. (2002) Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J Clin Invest 110:1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik SR, Lee JH, Kim DH, Chang CS, Kim YS (1998) Self-oligomerization of NACP, the precursor protein of the non-amyloid beta/A4 protein (A beta) component of Alzheimer’s disease amyloid, observed in the presence of a C-terminal A beta fragment (residues 25–35). FEBS Lett 421:73–76. [DOI] [PubMed] [Google Scholar]
- Quinn LP, Crook B, Hows ME, Vidgeon-Hart M, Chapman H, Upton N, Medhurst AD, Virley DJ (2008) The PPARγ agonist pioglitazone is effective in the MPTP mouse model of Parkinson’s disease through inhibition of monoamine oxidase B. Br J Pharmacol 154:226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, Wuertzer C, Gainetdinov RR, Caron MG, Di Monte DA, Federoff HJ (2002) Behavioral and neurochemical effects of wild-type and mutated human α-synuclein in transgenic mice. Exp Neurol 175:35–48. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E (2002) Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res 68:568–578. [DOI] [PubMed] [Google Scholar]
- Rozas G, Labandeira García JL (1997) Drug-free evaluation of rat models of parkinsonism and nigral grafts using a new automated rotarod test. Brain Res 749:188–199. [DOI] [PubMed] [Google Scholar]
- Sanchez-Guajardo V, Febbraro F, Kirik D, Romero-Ramos M (2010) Microglia acquire distinct activation profiles depending on the degree of α-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLOS ONE 5 e8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci 95:6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surendranathan A, Su L, Mak E, Passamonti L, Hong YT, Arnold R, Vázquez Rodríguez P, Bevan-Jones WR, et al. (2018) Early microglial activation and peripheral inflammation in dementia with Lewy bodies. Brain 141:3415–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS (2010) Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis 37:510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN, Li J, Souillac P, Millett IS, Doniach S, Jakes R, Goedert M, Fink AL (2002) Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem 277:11970–11978. [DOI] [PubMed] [Google Scholar]
- Verhaar R, Jongenelen CA, Gerard M, Baekelandt V, Van Dam AM, Wilhelmus MM, Drukarch B (2011) Blockade of enzyme activity inhibits tissue transglutaminase-mediated transamidation of alpha-synuclein in a cellular model of Parkinson’s disease. Neurochem Int 58:785–793. [DOI] [PubMed] [Google Scholar]
- Vermes I, Steur EN, Jirikowski GF, Haanen C (2004) Elevated concentration of cerebrospinal fluid tissue transglutaminase in Parkinson’s disease indicating apoptosis. Mov Disord 19:1252–1254. [DOI] [PubMed] [Google Scholar]
- Wilhelmus MM, Verhaar R, Andringa G, Bol JG, Cras P, Shan L, Hoozemans JJ, Drukarch B (2011) Presence of tissue transglutaminase in granular endoplasmic reticulum is characteristic of melanized neurons in Parkinson’s disease brain. Brain Pathol 21:130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R, Zhang J, Park HJ, Park ES, Oh S, Zheng H, Junn E, Voronkov M, et al. (2018) Synergistic neuroprotection by coffee components eicosanoyl-5-hydroxytryptamide and caffeine in models of Parkinson’s disease and DLB. Proc Natl Acad Sci U S A 115: E12053–E12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Williams JK, Yan R, Mouradian MM, Baum J (2019) Increased dynamics of α-synuclein fibrils by β-synuclein leads to reduced seeding and cytotoxicity. Sci Rep 9:17579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.