

Abstract
Anti-apoptotic Bcl-2 family proteins are overexpressed in a wide spectrum of cancers and have become well validated therapeutic targets. Cancer cells display survival dependence on individual or subsets of anti-apoptotic proteins that could be effectively targeted by multimodal inhibitors. We designed a 2,5-substituted benzoic acid scaffold that displayed equipotent binding to Mcl-1 and Bfl-1. Structure based design was guided by several solved co-crystal structures with Mcl-1, leading to the development of compound 24, which binds both Mcl-1 and Bfl-1 with Ki values of 100 nM and shows appreciable selectivity over Bcl-2/Bcl-xL. The selective binding profile of 24 was translated to on-target cellular activity in model lymphoma cell lines. These studies lay a foundation for developing more advanced dual Mcl-1/Bfl-1 inhibitors that have potential to provide greater single agent efficacy and broader coverage to combat resistance in several types of cancer than selective Mcl-1 inhibitors alone.
Graphical Abstract
Introduction
Cancer cells advantageously hijack apoptotic machinery to promote malignancy and therapeutic resistance.1 The Bcl-2 family of proteins govern intfrinsic apoptosis and are frequently dysregulated in a wide array of cancers in a manner that favors cell survival.2 The Bcl-2 protein family consists of a delicate balance of anti-apoptotic (Bcl-2, Bcl-xL, Mcl-1, Bfl-1/A1, and Bcl-w) and two subgroups of pro-apoptotic proteins, the BH3-only (e.g., Bim, Puma, Bad, and Noxa) and effector (Bax and Bak) proteins, which all share at least one of the four Bcl-2 homology (BH) domains.3 These pro- and anti- apoptotic proteins operate via a network of antagonistic protein-protein interactions defined by differential binding selectivity for one another. For example, Bim acts as a pan inhibitor of all the anti-apoptotic proteins, while Bad is selective for Bcl-2/BclxL/Bcl-w and Noxa binds only Mcl-1 and Bfl-1.4
The varying selectivity of the BH3-only proteins highlights both the feasibility of developing selective small molecule inhibitors and the biological relevance of doing so.5 Several of the anti-apoptotic proteins have been successfully drugged by small molecules known as BH3 mimetics, named for their mimicry of BH3-only protein binding.6 The natural gossypol molecule was discovered as a pan Bcl-2 family inhibitor7 and was developed into various analogues, such as TW-37,8 that displayed efficacy in several types of cancer. ABT-263,9 an orally available analogue of its predecessor, ABT-737,10 is a small molecule that mimics the selectivity of the Bad BH3-only protein (Bcl-2/Bcl-xL/Bcl-w inhibitor). ABT-263 is being evaluated in clinical trials with promise in solid tumors, but must be closely monitored for on-target dose-limiting thrombocytopenia due to the inhibition of Bcl-xL in platelet cells.11 Aside from the on-target toxicity concerns, Bcl-xL remains an important therapeutic target, especially in solid tumors, therefore development of selective Bcl-xL inhibitors remains an attractive approach.12,13 The selective Bcl-2 inhibitor, ABT-199 or venetoclax, is the first BH3 mimetic with granted FDA approval for its success in treating chronic lymphocytic leukemia and is currently being evaluated in numerous clinical trials for various indications.14,15 The need for selective Mcl-1 inhibitors emerged from the observance of Mcl-1 as a prominent resistance factor to other BH3 mimetics, as well as to many targeted, chemo-, and radio- therapies.16 This need was addressed by selective Mcl-1 inhibitors developed by our group17–19 and others, both in academia20–26 and industry27–31. The work on selective Mcl-1 inhibitors has culminated to several compounds now being evaluated in Phase I clinical trials for the treatment of hematologic malignancies as a single agent (ClinicalTrials.gov; NCT02675452, NCT03465540, NCT02979366, NCT02992483, NCT03218683) and in combination with venetoclax (ClinicalTrials.gov; NCT03672695). There is still a need, however, to effectively target Bfl-1 with drug-like molecules, as the main successes have come from selective peptides.32–34 We and others, using biochemical screening assays, have identified hit small molecule inhibitors of Bfl-1 which require further optimization.19,35 A recent study identified bicyclic stapled peptides as dual Mcl-1/Bfl-1 inhibitors with cellular activity and demonstrated an opportunity to develop Noxa-mimetic type inhibitors.36
Bfl-1 is considered an “underdog” in the Bcl-2 family, being one of the less studied anti-apoptotic proteins, but there is a growing body of evidence suggesting its value as a therapeutic target, particularly in cancers such as leukemia, lymphoma, and melanoma.37,38 Mcl-1 target validation is well established in a wider spectrum of cancers39 and shares structural and functional features with Bfl-1, along with the shared selective binding partner, Noxa.40–42 The close relationship between Mcl-1 and Bfl-1 has been evaluated by phylogenetic analysis, separating the two from the other anti-apoptotic proteins.43 The concurrent genetic silencing of Mcl-1 and Bfl-1 led to enhanced apoptosis of melanoma cell lines, while leaving non-malignant skin cells unharmed.44 Mcl-1 and Bfl-1 have both been identified as resistant factors to other BH3 mimetics in lymphoma45,46 and to MAPK inhibitors in melanoma,47,48 which was circumvented by genetic modulation in each case. Additionally, concurrent overexpression of Mcl-1 and Bfl-1 in poorly differentiated thyroid cancers was identified to be responsible for de novo therapeutic resistance and was overcome with the pan Bcl-2 inhibitor, obatoclax.49 Thus, compounds that selectively target both Mcl-1 and Bfl-1 hold clinical promise in treating several types of hematological and solid cancers.
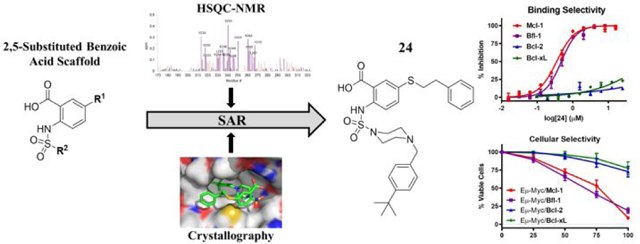
Our recent work utilizing an integrated high throughput and virtual screening approach yielded several scaffolds that showed binding to Mcl-1 and Bfl-1, including compound 19 (here referred to as 19SR).19,50 Our screening strategy using the Noxa pharmacophore to funnel high throughput hits likely selected for molecular scaffolds that display a dual Mcl-1/Bfl-1 binding profile.19 In this manuscript, we describe the further development of this hit molecule, 19SR, by re-designing the chemical core structure (Figure 1A). The difuryl-triazine core was replaced with a 2,5-substituted aromatic benzoic acid core, allowing variable chemical functionalization possibilities. The resulting molecule 1 displayed equipotent binding to Mcl-1 and Bfl-1, with selectivity over Bcl-2 and Bcl-xL (Figure 1A). Guided by structural information obtained via HSQC-NMR and crystallography, the 2,5-substituded benzoic acid class of inhibitors was developed and led to compound 24 with 15-fold improved binding to both Mcl-1 and Bfl-1, while maintaining a selective profile against other anti-apoptotic proteins. The obtained biological data suggests that both Mcl-1 and Bfl-1 are selectively targeted by 24, by binding to the endogenous proteins and inducing cell death in engineered lymphoma cells that depend on these two proteins for their survival. This class of molecules displays a Noxa-like, dual selective binding profile that provides key structural information which could lead to the development of more advanced dual Mcl-1/Bfl-1 inhibitors.
Figure. 1. Design of compound 1 and its binding site characterization to Mcl-1 and Bfl-1 with HSQC-NMR spectroscopy and computational docking.

A) Chemical structures and binding affinity of compounds 19SR19 and 1, against Bcl-2 family anti-apoptotic proteins (distinct structural features color coded). B) HSQC-NMR chemical shift perturbations of Mcl-1 residues in the presence of 1. C) Predicted binding pose of 1 in the BH3 binding site of Mcl-1 (PDB ID: 4HW2 was used for docking); key residue shifts highlighted corresponding to shift intensity noted in HSQC-NMR experiments. D) Superimposed 15N-HSQC spectra of Bfl-1 in the absence (blue) and presence (red) of compound 1 with close up view of select residue shifts. E) Predicted binding pose of 1 to Bfl-1 (PDB ID: 3MQP used for docking) with highlighted residues shifted in HSQC-NMR spectra.
Results & Discussion
Structure-based design of 2,5-substituted benzoic acid dual inhibitors
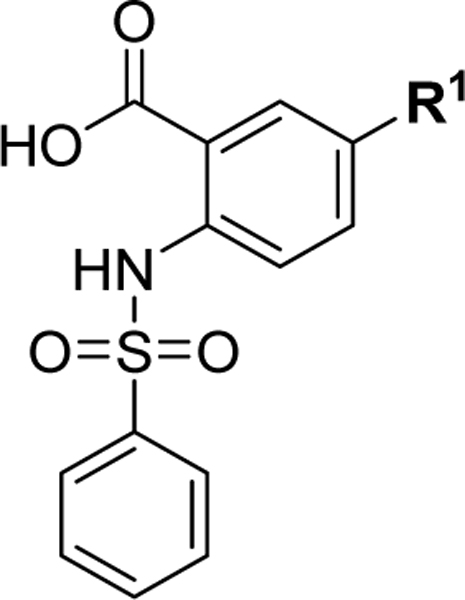
We have recently reported a class of small molecule Mcl-1 inhibitors with a difuryl-triazine core scaffold, based on the validated hit molecule 19SR identified by integrated high throughput and virtual screening strategy.19 The structure-activity relationship (SAR) studies of this series of compounds provided two important findings for their binding to the Mcl-1 protein: the conserved hydrogen bond with Arg263 formed with one of the furan rings and substituting the amide with a flexible carbon linker. In addition to our own findings, we analyzed reported promising Mcl-1 lead compounds, in particular two distinct structural classes derived from biphenyl sulfonamide and salicylic acid cores discovered in an NMR-based fragment screen.27 Comparing these scaffolds to our difuryl-triazine core provided us with additional insights in redesigning the hit molecule 19SR19 to increase the functionalization of this core and synthesize a more diverse molecule library (Figure 1A). These biphenyl sulfonamide and salicylic acid fragments, as well as most of the reported classes of Mcl-1 inhibitors,17,23,28,30,51 have a carboxylic group in their core structures which anchor them into the BH3 binding groove through the conserved hydrogen bond with the Mcl-1 Arg263 residue. Based on this well-known interaction, the triazine core was redesigned as a 2,5- substituted benzoic acid where one of the furan rings was replaced with a carboxyl group to preserve and increase the strength of the essential hydrogen bond with Arg263. The 2,5- substituents were inspired from our SAR data for the difuryl-triazine analogues19 and the available structural information for the aryl sulfonamide and salicylic acid based inhibitors.27 Thus, the second furan ring at the position 2 of 19SR was replaced with a phenylsulfonamide group, while the 5- thiol substituent was preserved, where the amide was replaced with a methylene linker to increase the flexibility of the substituent at this position, resulting in compound 1 (Figure 1A). We were pleased to determine that the new compound 1 shows equipotent binding to both Mcl-1 and Bfl-1 proteins with 2- and 3-fold improvement, respectively, in comparison with 19SR (Figure 1A and Table 1). Importantly, compound 1 has >20-fold selectivity to Bcl-2 and Bcl-xL, with no binding observed in the tested concentrations.
Table 1.
SAR of substituents and linkers of the 2-, 5- substituted benzoic acid core
 | |||
|---|---|---|---|
| Compound | R1 | Mcl-1 Ki (μM) | Bfl-1 Ki (μM) |
| 1 | 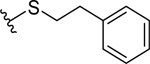 |
1.5 ± 0.3 | 1.6 ± 0.2 |
| 2 | H | 47.8 ± 3.9 | >100 |
| 3 | 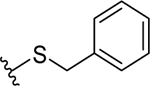 |
2.6 ± 0.4 | 3.0 ± 0.4 |
| 4 | 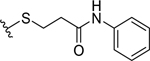 |
13.1 ± 2.4 | 14.8 ± 2.7 |
| 5 | 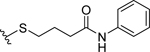 |
32.8 ± 4.9 | >70 |
| 6 | 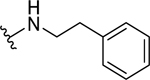 |
4.7 ± 0.7 | 3.8 ± 1.1 |
| 7 | 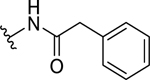 |
70.4 ± 5.4 | >70 |
| 8 | 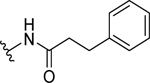 |
19.0 ± 1.2 | 62.0 ± 1.8 |
| 9 | 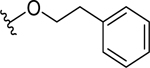 |
6.1 ± 1.0 | 10.6 ± 1.2 |
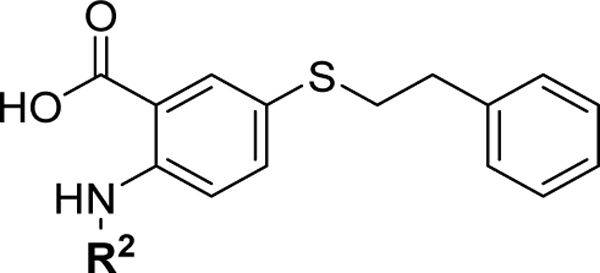 | |||
| Compound | R2 | Mcl-1 Ki (μM) | Bfl-1 Ki (μM) |
| 10 | H | 3.4 ± 0.4 | 3.7 ± 0.4 |
| 11 | 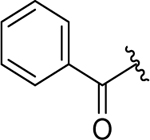 |
2.7 ± 0.6 | 4.6 ± 0.5 |
The direct binding of 1 to the Mcl-1 and Bfl-1 proteins was confirmed by HSQC-NMR spectroscopy and the obtained spectra showed concentration-dependent perturbations of the backbone amide residues (Figure 1B and 1D). The analysis of the HSQC chemical shift changes of 1 in complex with Mcl-1 showed that the residues forming the Mcl-1 BH3-binding groove were primarily affected and provided strong evidence that it binds at the same site where the conserved BH3-only proteins interact with the Mcl-1 protein. To gain structural understanding of the ligand-protein interactions, we predicted the binding model of 1 in complex with Mcl-1 by molecular docking. The predicted model revealed that the phenethylthio moiety occupies the p2 pocket and participates in hydrophobic interactions with the Leu267, Val253, Val243 and Leu235 residues (Figure 1C), which showed significant chemical shift perturbations (Figure 1B). The carboxyl group forms a hydrogen bond with the Arg263, mimicking the conserved aspartate in the pro-apoptotic BH3 only proteins, confirmed by the HSQC-NMR spectrum of the Mcl-1:1 complex where Arg263 showed a significant chemical shift perturbation. The predicted model showed that the phenylsulfonamide is placed above the p3 pocket, making weak interactions with Thr266, Ala277, and Phe228 (Figure 1C). In a similar way, the HSQC-NMR studies with the Bfl-1 protein showed that compound 1 binds to Bfl-1 through several key residue perturbations (Figure 1D). Along with a noticeable shift of Arg88, which corresponds to Arg263 of Mcl-1, the residues from the hydrophobic p2 pocket displayed significant perturbations, including Ala94, Phe95, and Ile98, as well as additional identified shifts by Val44 and Lys46 on the α3 helix. The docking model of compound 1 to Bfl-1 (Figure 1E) predicts similar binding as to Mcl-1, with the 5-phenethylthio moiety occupying the p2 pocket and interaction with Arg88 through the acid group. The phenylsulfonamide substituent appears to be pointed toward the α4 helix and solvent exposed. Taken together, biophysical HSQC-NMR experiments in combination with in silico docking studies strongly suggest that compound 1 binds to the BH3-binding groove of both the Mcl-1 and Bfl-1 proteins. This dual selective binding profile of compound 1 matches that of the endogenous pro-apoptotic Noxa protein. Since Bfl-1 is an emerging therapeutic target and there is an unmet need for small molecules that inhibit this protein, we sought to further explore the SAR of its inhibition together with Mcl-1.
SAR investigation of 2,5-substituted benzoic acid-based Mcl-1/Bfl-1 dual inhibitors
The first series of compounds was synthesized to explore the importance and binding affinity contribution of the 5-phenethylthio (R1) and 2-phenylsulfonamide (R2) substituents to the Mcl-1 and Bfl-1 proteins (Table 1). Deletion of the phenethylthio substituent in compound 2 resulted in an over 30- and 60-fold decrease in binding affinity to Mcl-1 and Bfl-1, respectively, demonstrating that this group significantly contributes to the Mcl-1/Bfl-1 binding potency. These findings are consistent with the predicted binding model showing that the phenethylthio substituent is accommodated into the p2 pockets of both Mcl-1 and Bfl-1, maintaining a network of hydrophobic interactions (Figures 1C and 1E). One carbon reduction of the aliphatic chain at the R1 position to a phenyl methylthio substituent resulted in compound 3 with similar binding affinities to Mcl-1 and Bfl-1. Several compounds were synthesized to investigate the importance of the ethylthio linker, its length and nature. Reducing the flexibility of the ethyl chain by introducing an amide group in compound 4, resulted in 10-fold decreased binding to both, Mcl-1 and Bfl-1, consistent with the SAR studies of the difuryl-triazine series, based on the 19SR hit molecule.19 Extending the carbon chain in compound 5 resulted in further significant decreases in binding to both Mcl-1 and Bfl-1. Replacement of the sulfur with an amine linker, as in 6, resulted in a 6-fold loss in binding to Mcl-1. Replacing the sulfur with an amide linker having different carbon chain lengths, as in 7 and 8, resulted in a substantial loss of binding affinity to Mcl-1 and an even greater loss in Bfl-1 binding. Furthermore, a 5- ether analogue was synthesized to compare against the sulfur and amine linked compounds to find that compound 9 displayed a 4- and 6- fold affinity decrease for Mcl-1 and Bfl-1, respectively. Importantly, deletion of the R2 phenylsulfonamide substituent in compound 10 or changing the sulfonamide to an amide linker in compound 11 did not have significant effect on binding (Table 1). These results together with the predicted binding model demonstrate the importance of the phenethylthio substituent in preserving the carbon chain length and its flexibility, as well as keeping the sulfur linker at the 5- substitution site of the core benzoic acid. In addition, it seems that the sulfur linker is necessary to preserve equipotent binding to Mcl-1/Bfl-1, as the affinities deviate when a direct amide linker is used (7 and 8).
Based on these results, we synthesized a series of inhibitors where the R1 phenethylthio group was fixed at the 5- position and proceeded to probe the SAR of the substituents at the R2 position (Table 2). Extending the phenyl group away from the sulfonamide linker by two carbons (compound 12) resulted in no changes in binding affinity. Bioisosteric replacement of the phenyl group with the thiophene moiety, as in 13, gave a similar Mcl-1 binding affinity with a 2-fold decrease in Bfl-1 binding. Introducing a small hydrophobic substituent at the para position of the phenyl ring, such as bromine in compound 14, led to a 2-fold improvement over 1, alluding to the possibility of introducing more hydrophobicity at this site. Indeed, substituting the phenyl for a bulkier naphthalene ring resulted in compound 15, with 6- and 3- fold increases in binding potency to Mcl-1 and Bfl-1, respectively. Introducing a biphenyl substituent in compound 16, which was beneficial in our previously reported series of Mcl-1 inhibitors,17 proved again to be a favorable moiety for targeting these protein-protein interactions (PPIs) and led to 15-fold improvements in binding affinity to Mcl-1 (Ki = 0.09 ± 0.02 μM), as well as to Bfl-1 (Ki = 0.15 ± 0.02 μM). Replacing the distal phenyl ring with a saturated cyclohexyl group in compound 17 resulted in similar potent binding to both Mcl-1 and Bfl-1 (Ki = 0.087 ± 0.03 μM and Ki = 0.15 ± 0.03 μM, respectively). It is well known that anti-apoptotic proteins, including Mcl-1 and Bfl-1, have a prominent hydrophobic groove, thus it is required for ligands to have high lipophilicity to achieve potent affinity to the BH3 binding site. However, compounds with high binding affinity that is primarily driven by lipophilic interactions are likely to be less selective, with potential off-target effects and toxicity. Replacing flat aromatic rings with 3D heteroatom containing rings has been shown to improve many qualities of small molecules, including increasing solubility and decreasing promiscuity.52,53 In an effort to balance the lipophilic properties of the newly designed dual inhibitors, and improve physicochemical properties, the R2 proximal phenyl ring in 16 was replaced with the heterocyclic pyridine (18), piperidine (19), and piperazine (20) moieties. Compounds 18 and 19 both displayed slightly decreased binding affinities, but improved aqueous solubility compared to 16 (Supporting Information, Figure S1). Compound 20 showed a 5- to 6-fold decrease in binding to Mcl-1 and Bfl-1, compared to 16, which can be explained by the presence of hydrophilic piperazine in the predominantly hydrophobic environment of the Mcl-1 binding pocket. Introducing a methylene spacer between the piperazine moiety and the phenyl substituent in compound 21 and a para-fluoro-phenyl derivative, 22, showed similar binding affinities to Mcl-1 and Bfl-1 as the parent compound 20. Importantly, as expected, compounds 20 and 22 showed improved solubility and calculated physicochemical properties in comparison with 16 and 17 (Supporting Information, Figure S1). This improved solubility allowed us to successfully obtain complex structures with the Mcl-1 protein, discussed below. To verify the importance of the carboxyl acid interaction with the conserved Arg263 and Arg88 residues on Mcl-1 and Bfl-1, respectively, we tested several esterified analogues of compounds in Table 2 and as expected no binding was observed to both Mcl-1 and Bfl-1 at the tested concentrations (Supporting Information, Table S1).
Table 2.
SAR of 5-(phenethylthio)-2-(arylsulfonamido)benzoic acid compounds
 | |||
|---|---|---|---|
| Compound | R2 | Mcl-1 Ki (μM) | Bfl-1 Ki (μM) |
| 12 | 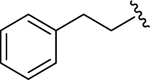 |
1.7 ± 0.5 | 1.7 ± 0.3 |
| 13 | 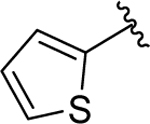 |
2.0 ± 0.2 | 3.4 ± 0.3 |
| 14 | 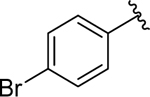 |
0.81 ± 0.2 | 0.81 ± 0.2 |
| 15 | 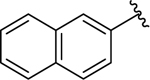 |
0.22 ± 0.03 | 0.57 ± 0.1 |
| 16 | 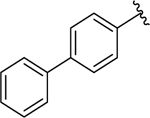 |
0.090 ± 0.02 | 0.15 ± 0.02 |
| 17 | 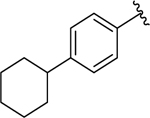 |
0.087 ± 0.03 | 0.17 ± 0.04 |
| 18 | 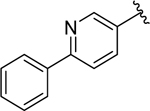 |
0.20 ± 0.04 | 0.28 ± 0.05 |
| 19 | 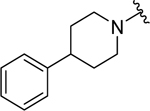 |
0.33 ± 0.06 | 0.39 ± 0.1 |
| 20 | 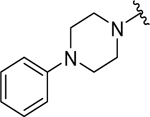 |
0.57 ± 0.1 | 0.76 ± 0.1 |
| 21 | 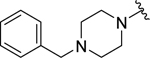 |
0.84 ± 0.1 | 1.0 ± 0.2 |
| 22 | 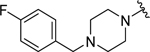 |
0.77 ± 0.2 | 0.90 ± 0.2 |
Crystallography studies and structure based design of optimized inhibitors
To better understand the key binding elements of these inhibitors and to obtain molecular insight into the Mcl-1 protein-inhibitor interactions, co-crystal structures of 15, 20, and 22 bound to Mcl-1 were solved to 2.74 Å, 2.55 Å, and 2.10 Å resolution, respectively (Figure 2). The 15 co-crystal structure contains four copies of Mcl-1 present in the asymmetric unit with each protein molecule containing one 15 molecule in the BH3 binding site. In the Mcl-1:20 complex structure there is one Mcl-1 protein per asymmetric unit and the Mcl-1:22 co-crystal structure produced a crystal with four molecules in the asymmetric unit.
Figure 2. Co-crystal structures of compounds 15, 20 and 22 with Mcl-1.

A) Overlay of the Cα traces of Mcl-1 in complex with 15 (PDB ID: 6U63) (green), 20 (PDB ID: 6U64) (blue), 22 (PDB ID: 6U65) (orange), and apo-MBP-Mcl-1 (PDB ID: 4WMS) (gray) depicting movement in the α4-helix (circled in red) upon ligand binding. X-ray crystallography complex of several inhibitors bound to Mcl-1 oriented 90° clockwise with respect to A. Mcl-1 co-crystal structures with compounds B) 15, C) 20 and D) 22. The side chains of selected interacting Mcl-1 residues are labeled. Dashed lines represent hydrogen bonds.
It has previously been reported that the Mcl-1 BH3 binding site adapts its shape with bound ligands and the pockets become larger and deeper, especially in the p2 pocket.23,27,54 Thus, using the apo-MBP-Mcl-1 (PDB ID: 4WMS), the obtained 3D complex structures were compared and analyzed for conformational changes with respect to different binding partners (Figure 2). The structures were aligned to 22 using the SSM algorithm in Coot55 and the resulting root mean square deviations (RMSD) for each structural pair is 1.193 Å for apo, 0.725 Å for 15, and 0.775 Å for 20. As expected, the most notable difference between the aligned structures is the position of the α4 helix (Figure 2A). In the apo structure, the final turn of the helix, residues 254–256, is disordered leading to its larger deviation in the BH3 binding site. The displacement of this helix opens up the canonical BH3 binding groove to accommodate the ligand in the hydrophobic p2 pocket.23 Among the three complex structures, all Mcl-1 inhibitors have an anchoring hydrogen bond between their carboxyl group and the Arg263 side chain of Mcl-1, mimicking the interaction observed between Arg263 and the conserved aspartic acid in pro-apoptotic proteins. The common aromatic 2,5-substituted benzoic acid scaffold is placed in the p2/p3 pockets junction. Although, all three ligands bind Mcl-1 in a U-shaped binding mode with the Arg263-carboxyl group interaction acting as the main anchoring point, 15 binds in a different way in comparison with 20 and 22.
In the Mcl-1:15 co-crystal structure, the R1 phenethylthio moiety is pointed towards the p2 pocket interacting with the hydrophobic side chains of α3 helix (e.g. Met231) and the side chain of Met250 on the α4 helix (Figure 2B), similarly as in the proposed binding model for the initially designed compound 1 (Figure 1C). Additional hydrophobic interactions with the ligand were observed with Val253 and Phe270 from the p2 pocket, as well as with Mcl-1 residue Leu235 from the p1 pocket. The R2 naphthalene moiety in 15 is located on top of the p3 hydrophobic pocket, partially stabilized by the π- π stacking with the phenyl group of 15 as well as through hydrophobic interactions with Met231. The orientation of the side chain of Met231 causes the p3 pocket to close up, resembling the reported three-dimensional structures of complexes formed between the Mcl-1 BH3 peptides (e.g. Bim and Noxa), but in contrast to some of the previously reported Mcl-1 inhibitor co-crystal structures.20,21,23
Surprisingly, compounds 20 and 22 differ in their binding modes as compared to the Mcl1 complex structure with 15 (Figure 2C–D). 20 and 22, which have different R2 substitutions on the aromatic core scaffold, bind to the Mcl-1 BH3 binding site in a flipped U-shaped conformation. In addition to the flipped binding conformation, the benzene core scaffold occupies an altered position compared to the Mcl-1:15 co-crystal structure. It is positioned perpendicularly to the BH3 binding site. The R2 piperazine-containing substituents in compounds 20 and 22 occupy the Mcl-1 p2 hydrophobic binding pocket, while the phenethylthio moiety is directed towards the p1 pocket. The phenylpiperazine-sulfonamido substituent in 20 interacts with the side chains of Leu267 and Val253 from the p2 pocket and Leu235 from the p1 pocket through hydrophobic interactions with the distal phenyl group. The addition of the CH2 linker in the R2 substituent of 22 is providing additional length and flexibility of the terminal 4-fluoro phenyl moiety, placing it deeper in the p2 binding site in comparison with 20, and forming hydrophobic interactions with the side chains of Phe270, Met250, and Leu290. The R1 phenethylthio moiety in both complexes with 20 and 22, is located adjacent to the Mcl-1 α3 helix and has hydrophobic interactions with the sulfur-containing side chain of Met231 as well as the side chain of Leu235 and Val253 (Figure 2C–D). The folded conformation of compound 20 is also partially stabilized by the π- π stacking of its two distal phenyl groups.
The structural findings demonstrate that the occupancy of the p2 hydrophobic pocket and the binding mode of these compounds depend on the nature and size of the introduced substituents into the aromatic 2,5-substituted benzoic acid scaffold. The different binding modes and orientation of investigated ligands demonstrate the complex nature of molecular recognition governing the binding of small molecules to Mcl-1 and the challenges that generally face structure-based design efforts of protein-protein interactions inhibitors.
Based on the experimentally determined binding modes of these inhibitors and knowing that optimal occupancy of the p2 hydrophobic pocket is essential for improved binding affinity to Mcl-1 protein,23 compounds 23 and 24 were synthesized by introducing a bulky tert-butyl group in the para position on the distal phenyl group of the R2 substituent (Table 3). Indeed, both compounds, 23 and 24, displayed an 8-fold increase in the binding affinity to Mcl-1 with Ki values of 73 nM and 94 nM, respectively, compared to their counterparts 20 and 21, which lack the tert-butyl. Importantly, tighter binding affinities of 23 and 24 were also determined for Bfl-1 protein with Ki values of 84 nM and 100 nM, respectively. Similar improvement was also identified in comparison with 22 which has a fluorine at the same position. This finding confirmed the importance of the hydrophobic interactions in the p2 pocket and optimizing its occupancy. To further validate the direct binding of compounds 23 and 24 to the Mcl-1 and Bfl-1 proteins, we employed the biophysical method of bio-layer interferometry (BLI). Using immobilized biotin-labeled anti-apoptotic proteins, 23 and 24 were tested and demonstrated dose dependent binding against Mcl-1 with Kd values of 490 nM and 670 nM, respectively, calculated by using steady state analysis (Figures 3A and 3B; Supporting Information, Figure S2). Compounds 23 and 24 gave similar Kd values for Bfl-1, 920 nM and 1,270 nM, respectively (Figures 3C and 3D; Supporting Information, Figure S2). To support the FP obtained SAR data, compound 22, with about 9-fold weaker binding affinity to both proteins (Table 2), showed similar decrease in the Kd values determined by BLI (Supporting Information, Figure S2).
Table 3.
Optimized compounds designed to accommodate the p2 pocket of Mcl-1 and Bfl-1with terminal R2 tert-butyl moiety.
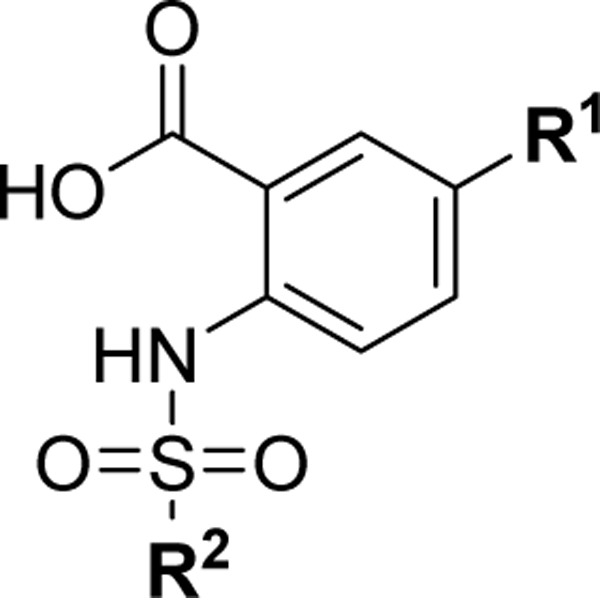 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | Mcl-1 Ki (μM) | Bfl-1 Ki (μM) |
| 23 | 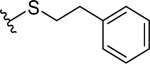 |
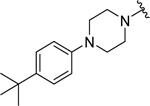 |
0.073 ± 0.02 | 0.084 ± 0.04 |
| 24 | 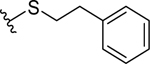 |
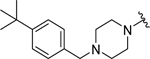 |
0.094 ± 0.01 | 0.10 ± 0.02 |
| 25 | 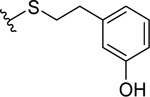 |
 |
0.29 ± 0.01 | 0.25 ± 0.05 |
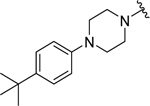
| 26 | 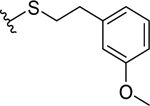 |
 |
0.074 ± 0.01 | 0.10 ± 0.04 |
| 27 | 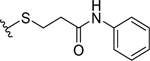 |
 |
0.95 ± 0.08 | 1.1 ± 0.4 |
Figure 3. Determination of the binding affinity by BLI.

Sensorgrams for Mcl-1 binding with compounds A) 23 (2.5, 1.25, 0.625, 0.3125, and 0.156 μM) and B) 24 (2.5, 1.25, 0.625, 0.3125, and 0.156 μM). Corresponding sensorgrams for Bfl-1 binding with compounds C) 23 (2.5, 1.25, 0.625, 0.3125, and 0.156 μM) and D) 24 (3, 1.5, 0.75, 0.375, and 0.1875 μM). Kd values were calculated from steady state analysis, available in Supporting Information, Figure S2. Figures are representative of ≥ 3 independent experiments.
We successfully obtained the crystal structure of compound 24 in complex with Mcl-1 (Figure 3A), showing a similar binding pose as 22, with the Arg263-carboxyl group interaction acting as the main anchoring point and aromatic scaffold interacting with Met231. The tert-butyl phenyl moiety is deeply accommodated into the p2 pocket, which becomes larger and more opened in complex with 24. The tert-butyl group forms hydrophobic interactions with the side chains of Met250, Ile237, Val243 and Leu290, while the phenyl group interacts with the Phe270 which constitutes the bottom of the p2 pocket. Overall the tert-butyl phenyl group shifts towards the α4 helix and distorts it between residues 245–247. The R1 phenethylthio substituent is directed towards the p1 pocket and the distal phenyl ring makes hydrophobic interactions with Leu235, leading to opening and merging of the p1 and p2 pockets.
To investigate the contribution and interactions of the R1 substituent, several compounds were synthesized by modifying the distal phenyl ring as well as the flexibility of the methylene linker. Adding a polar hydroxyl group on the R1 phenethylthio substituent illustrates the necessity to preserve hydrophobicity at this site, as compound 25 gives a 3- to 4- fold decrease in binding affinity. Methylation of the phenolic hydroxyl group in 26 restored the binding, confirming the unfavorable interactions of the polar hydroxyl group. Furthermore, we tested the importance of the linker geometry of the R1 substituent. Introducing an amide linker in compound 27 resulted in more than a 10-fold decrease in the binding affinity against both Mcl-1 and Bfl-1. We successfully solved the complex structure of 27 with the Mcl-1 protein (Figure 4B). This binding pose is very similar to the Mcl-1:24 co-crystal structure, showing the Arg263-carboxyl group interaction and the tert-butyl phenyl piperazine moiety deeply accommodated in the p2 pocket, forming the hydrophobic interactions with Val243, Leu246 and Leu290 residues. However, the thioethylamide phenyl substituent is solvent exposed and lacks interactions with the p1 pocket. These results confirm the contribution of the R1 substituent by directly interacting with the Mcl-1 protein, as well as partially stabilizing of the folded conformation of these inhibitors by the π- π stacking between the two distal phenyl groups in the R1 and R2 substituents.
Figure 4. Co-crystal structures of compounds 24 and 27 bound to Mcl-1.

A) Mcl-1:24 x-ray complex structure (PDB ID: 6U6F). B) Mcl-1:27 x-ray complex structure (PDB ID: 6U67), water molecule involved in hydrogen binding appears as a red sphere. Key residues on protein are labeled and hydrogen bonds depicted as black dashed lines.
Elucidation of dual inhibitors binding to Bfl-1
Recently, the apo-Bfl-1 structure, as well as complex structures of Bfl-1 and BH3 peptides have been reported.33,56 The alignment of the Bfl-1 structures bound to peptides available in the PDB database shows a predominantly uniform conformation of the targeted binding site (Supporting Information, Figure S3). As members of the Bcl-2 protein family, Mcl-1 and Bfl-1 contain four BH domains with conserved tertiary structure forming the canonical hydrophobic BH3 binding site where the pro-apoptotic proteins bind.3 The structures of apo Bfl-1 and Mcl-1 are largely similar, although Mcl-1 and Bfl-1 display marginal overall sequence identities of approximately 20%. The alignment of the Mcl-1 structure with the Bfl-1 protein showed that the highest degree of sequence similarity is found in their BH3 binding pocket, with several corresponding Mcl-1/ Bfl-1 residue pairs involved in interactions with BH3 domain and inhibitors, including Arg263/Arg88, Met250/Met75, Val274/Leu99 and Phe270/Phe95, indicated comparable overall topology of both binding sites (Supporting Information, Figure S3). The most conserved region identified in the loop between α4 and α5 (residues 81–84 in Bfl-1 and 256–259 in Mcl-1) and the N-terminus of helix α5 (residues 85–91 in Bfl-1 and 260–266 in Mcl-1). In addition, the size of the BH3-binding pocket is very similar in apo structures of Mcl-1 and Bfl-1. However, as it was established from numerous Mcl-1 protein crystallography studies, including experiments presented in this work, the amino acid conformations in Mcl-1 binding pocket can be greatly affected by the ligand binding, in particular the α4 helix adjusted its side chain conformations in response to different binding partners (Figure 2A). Thus, a similar conformational change of the Bfl-1 BH3 binding site can also be predicted upon ligand binding. Indeed, a recently reported complex structure of a stapled peptide inhibitor displayed a conformational change upon binding in comparison with the apo-structure by opening the binding groove by displacement of α2, α3 and α4 helices.33 Thus, it was not surprising when in our first docking attempts using the complex structure of Noxa BH3 peptide bound to Bfl-1 (PDB ID: 3MQP), we were not able to obtain reasonable binding modes. In order to take the conformational change of the protein into consideration, we used our co-crystal structure of the Mcl-1 in complex with compound 24 and aligned it with the Noxa-bound Bfl-1 protein (PDB ID: 3MQP). In this way we determined the initial coordinates and conformation of compound 24 bound to the Bfl-1 protein, followed by energy minimization of the produced complex. After the minimization, the most notable conformational change observed was the side chain movements of the Phe71, Met75, and Phe95 residues, which enabled a wider opening of the Bfl-1 p2 pocket to accommodate the compound (Supporting Information, Figure S3). Subsequent re-docking of compounds 23 and 24 into the resulted Bfl-1 structure, followed by minimization, provided the predicted binding mode, displayed in Figure 5A. The predicted binding of compounds 23 and 24 resembles poses observed in the Mcl-1 co-crystal complex structures with other inhibitors from this series. As expected, Arg88 served as the main anchoring residue. In a similar way, the R2 substituent was predicted to be buried deep into the Bfl-1 p2 pocket through a network of hydrophobic interactions. The tert-butyl group makes contacts with Val122, Ala67, Ile118, Phe121, and Leu99, and the distal phenyl group with Phe71, Ile92 and Phe95. Interestingly the R1 phenethylthio moiety was oriented towards the α4 helix of the BH3 binding groove for both compounds 23 and 24, making hydrophobic interactions between the side chain methylene groups of Lys77 and the distal aromatic ring. The predicted models of Bfl-1 in complex with compounds 23 and 24 suggest that the ligands can accommodate the BH3 pocket by rotating certain binding-site side-chains leading to the opening of a hydrophobic cavity in particular the p2 pocket.
Figure 5. Molecular docking and HSQC-NMR studies of the optimized compounds with Bfl-1 protein.

A) Docking model of compounds 23 and 24 to Bfl-1 (PBD ID: 3MQP was used for docking); residues with chemical-shift perturbations upon 23 binding are highlighted in red. B) Superposition of 15N HSQC spectra of labeled Bfl-1, free and in complex with compound 23 in different ratio (DMSO = blue; 1:1 = green; 1:2 = red). Close up view of select residues with noticeable chemical shifts.
Currently there are no reported co-crystal structures of the Bfl-1 protein with small molecules in the literature. Our attempts to determine the complex structure between our inhibitors and Bfl-1 were unsuccessful. Thus, we proceeded with Bfl-1 HSQC-NMR studies to provide further evidence to support the molecular docking and predicted binding mode of the analogues in this series. We were successful in obtaining HSQC-NMR data demonstrating that compound 23 binds and interacts with Bfl-1 protein (Figure 5B). By performing a titration of different concentration ratios between compound 23 and the Bfl-1 protein, we identified key residue perturbations that substantiate our evidence of Bfl-1 binding. The important anchoring hydrogen bond interaction with Arg88 was confirmed by chemical shifts with this residue, along with Phe95 and Ala94 in the p2 pocket. Future structural studies of Bfl-1 in complex with these compounds are needed in order to provide in-depth and empirical insights into how this class of inhibitors bind Bfl-1.
Binding selectivity to anti-apoptotic Bcl-2 family proteins
The overall goal of the design and development of these inhibitors was to have molecules that bind to Mcl-1 and Bfl-1 with selectivity over Bcl-2 and Bcl-xL. Several key inhibitors were tested for binding against the homologous Bcl-2 and Bcl-xL anti-apoptotic proteins (Table 4). The initially designed molecule 1 displayed substantial selectivity with Ki values for Bcl-2/Bcl-xL greater than 25 μM. Optimized compounds 15, 20, and 27 maintained at least 10-fold selectivity, while 22 gave Ki values for Bcl-2/Bcl-xL greater than 25 μM. Along with the increase in potency for Mcl-1/Bfl-1, compounds 23 and 26 maintained at least a 15-fold selectivity over Bcl-2/Bcl-xL. Compound 24 displayed the greatest selectivity window with Ki values against Bcl-2 and Bcl-xL both being greater than 25 μM and was selected for further biological evaluation.
Table 4.
Binding selectivity profiles of select compounds to anti-apoptotic proteins
| Compound | Mcl-1 Ki (μM) | Bfl-1 Ki (μM) | Bcl-2 Ki (μM) | Bcl-xL Ki (μM) |
|---|---|---|---|---|
| 1 | 1.5 ± 0.3 | 1.6 ± 0.2 | >25 | >25 |
| 15 | 0.22 ± 0.03 | 0.57 ± 0.1 | 5.6 ± 0.7 | 5.2 ± 1.1 |
| 20 | 0.57 ± 0.1 | 0.76 ± 0.1 | 11.7 ± 3.1 | 6.6 ± 1.1 |
| 22 | 0.77 ± 0.2 | 0.90 ± 0.2 | >25 | >25 |
| 23 | 0.073 ± 0.02 | 0.084 ± 0.04 | 1.2 ± 0.4 | 3.5 ± 0.5 |
| 24 | 0.094 ± 0.01 | 0.10 ± 0.02 | >25 | >25 |
| 26 | 0.074 ± 0.01 | 0.10 ± 0.04 | 1.2 ± 0.5 | 6.7 ± 1.6 |
| 27 | 0.95 ± 0.08 | 1.1 ± 0.4 | 16.1 ± 0.9 | >25 |
Biological evaluation of dual inhibitors
The results presented thus far were focused on binding to recombinant proteins and in order to validate the engagement of Mcl-1 and Bfl-1 in more biologically relevant systems we evaluated the cellular activity of the most potent compounds. To confirm the endogenous target engagement and PPI disruption capabilities of this class of inhibitors, we employed a biotin-streptavidin pull down assay using a biotin-labeled Bim peptide (BL-Bim) and whole cell lysate from the M14 melanoma cell line. The M14 cell line was used as it contains high endogenous levels of all anti-apoptotic proteins, particularly Bfl-1, and melanoma cell lines in general are known to display functional dependence on Mcl-1 and Bfl-1.44,57 As shown in Figure 6A, Mcl-1 and Bfl-1 were pulled down by BL-Bim and the unlabeled Bim peptide served as a positive control to disrupt interactions of BL-Bim and the anti-apoptotic proteins. Incubation with 100 μM of compound 24 resulted in complete disruption of Mcl-1 and Bfl-1 complexes with BL-Bim, while incubation with 100 μM of its methyl ester analogue, 30r, didn’t show any effect, consistent with the FP binding data (Supporting Information, Table S1). The dose dependent and selective PPI disruption capabilities of compound 24 were also evaluated by the pulldown assay (Figure 6B). Compound 24 induced dose-dependent disruption of interactions between BL-Bim and endogenous Mcl-1 and Bfl-1, without affecting the complexes between BL-Bim and Bcl-2 or Bcl-xL. Compound 23 was also evaluated under the same pulldown assay conditions and showed similar results (Supporting Information, Figure S4). To demonstrate that compounds can disrupt PPIs with different pro-apoptotic proteins, we also used biotin labeled Noxa BH3 peptide. As expected all tested compounds, 15, 20, 22, and 23, disrupted the interactions between BL-Noxa and endogenous Mcl-1 with compound 23 being the most potent corresponding with their binding affinities (Supporting Information, Figure S5). These results validate the ability of this class of inhibitors to bind endogenous Mcl-1 and Bfl-1 and selectively disrupt PPIs.
Figure 6. Biological evaluation of compound 24.

A) Biotin-streptavidin pulldown assay using lysate from the M14 melanoma cell line and biotinylated Bim peptide (BL-Bim) after treatment with 100 μM of compound 24 and its methyl ester analogue, 30r. B) Dose dependent and selective disruption of BL-Bim and Mcl-1/Bfl-1 interactions by compound 24. Complexes of BL-Bim and endogenous anti-apoptotic proteins were analyzed by western blot. C) Eμ-Myc lymphoma cell lines (Mcl-1, Bfl-1, Bcl-2, and Bcl-xL) treated for 24 hours with compounds 24 and 30r, cell death analyzed by flow cytometry. Significance determined by two-tailed t-test compared with DMSO (0 μM) controls of respective cell lines (*, p < 0.05; **, p < 0.005). Data represents mean and SEM from 3 independent experiments.
The target selectivity of compound 24 was further confirmed using the reported Eμ-Myc lymphoma cell lines, which were engineered to overexpress individual anti-apoptotic Bcl-2 proteins with a strong survival dependence on these targets, making them excellent tools for testing and characterizing inhibitors of Bcl-2 family proteins.58 Positive controls were used to validate selective sensitivity of these cell lines to reported BH3 mimetics12,14,28 (Supporting Information, Figure S6). Lymphoma cells overexpressing Mcl-1, Bfl-1, Bcl-2, or Bcl-xL were treated with 24 at various concentrations and analyzed for cell death by flow cytometry. Eμ-Myc cells overexpressing Mcl-1 and Bfl-1 showed dose-dependent increase in cell death in response to compound 24 treatment, with significant effect starting from 50 μM. Importantly, at this concentration, 24 is disrupting about 50% of the PPIs between the pro-apoptotic BH3-only BL-Bim peptide and endogenous Mcl-1 and Bfl-1, but not with other anti-apoptotic proteins (Figures 6B). Consistent with these results, cells overexpressing Bcl-2 and Bcl-xL were minimally affected even at the highest tested concentration, 100 μM, further demonstrating the selective targeting of Mcl-1 and Bfl-1. It is important to note the difference between binding affinity and cellular potency of 24, which can be attributed to several different factors. For example, it is known that the endogenous BH3-only proteins bind to anti-apoptotic proteins with high affinity, which requires a sub-nanomolar binding affinity of small molecules in order to disrupt these interactions and produce relevant biological activity.54,59 Another possible limiting factor is the potential binding to serum proteins, which has been reported as an issue for BH3 mimetics.14,30,60 To further evaluate on-target cellular activity of 24, the negative control, compound 30r, was tested and did not affect cell viability up to 100 μM (Figure 6C). Furthermore, compound 23, which has similar binding affinity against Mcl-1 and Bfl-1 as 24, and only 10- to 15-fold selectivity over Bcl-2 and Bcl-xL, displayed significant cell killing of Mcl-1 and Bfl-1 lymphoma cell lines as expected, but also shows effect on Bcl-2 and Bcl-xL cell lines at the highest doses, consistent with its binding profile (Supporting Information, Figure S4 and Table 4). In addition, compounds 15, 20, and 22 didn’t show significant cellular activity at 100 μM (Supporting Information, Figure S7), supporting that the increased potency of 23 and 24 contributes to cellular activity. Overall, this biological data confirms that the selective binding profile of 24 translates to cellular activity by selectively targeting endogenous Mcl-1 and Bfl-1 in an equipotent manner.
Mcl-1/Bfl-1 dual inhibitor synthesis
The series of the 2,5-substituted benzoic acid dual inhibitors was synthesized through a novel route (Scheme 1) which allowed facile access to a variety of analogs with variations at R1, R2, and the linker region.
Scheme 1.

aSynthetic route for compound 1 and analogues
aReagents and conditions: (i) HS(CH2)nX, Pd2(dba)3, Xantphos, i-Pr2NEt, 1,4-dioxane, reflux, 16 h; (ii) R2SO2Cl, pyridine, CH2Cl2, rt, 16 h (Tables 1, 2), or R2SO2Cl, pyridine, 100 °C, 14 h (Table 2), or 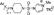 , CH3CN, 80 °C, 16 h (Table 2, 3); (iii) LiOH, MeOH/THF/H2O, rt, 16 h (iv) PhCOCl, K2CO3, CH2Cl2, rt, 16 h (Table 1).
, CH3CN, 80 °C, 16 h (Table 2, 3); (iii) LiOH, MeOH/THF/H2O, rt, 16 h (iv) PhCOCl, K2CO3, CH2Cl2, rt, 16 h (Table 1).
The commercially available starting material, methyl 2-amino-5-iodobenzoate (28), was subjected to palladium catalyzed cross-coupling reaction using conditions previously reported to provide desired intermediates 29 (29a-d).61 Reactions of the amines with appropriate sulfonyl- or acid- chlorides or 3-methyl-1-((piperazin-1-yl)sulfonyl)-1H-imidazol-3-iums62 provided the penultimate compounds 30 (30a-s), which were subsequently hydrolyzed using lithium hydroxide. Purification by reverse-phase HPLC afforded the target compounds (1, 3-5, 12-24, 26, 27) with more than 95% purity (Tables 1, 2, 3). Compound 25, a corresponding hydroxy analogue of 26 was obtained by reacting the intermediate of 26 with excess of boron tribromide. The readily available compound 2 was synthesized by reacting methyl 2-aminobenzoate with sulfonyl chloride followed by lithium hydroxide-mediated hydrolysis.63 The same intermediate 29a was utilized to assemble compounds 10 and 11 (Scheme 1). The synthesis of compounds 6, 7 and 8 (Table 1) was completed starting from methyl 2-amino-5-nitrobenzoate (Scheme 2). Synthesis of the 5-ether linked compound 9 (Table 1) is outlined in Scheme 3.
Scheme 2.

bSynthesis of compounds 6–8
bReagents and conditions: (v) benzenesulfonyl chloride, NaH, THF, 0 °C to rt, 16 h; (vi) iron powder, AcOH/EtOH, reflux, 4 h; (vii) (2-chloroethyl)benzene, K2CO3, DMF, 120 °C, 16 h, or Ph(CH2)n=1, 2COCl, Et3N, DMAP, CH2Cl2, rt, 14–16 h (viii) LiOH, MeOH/THF/H2O, rt, 16 h.
Scheme 3.

cSynthesis of compound 9
cReagents and conditions: (ix) (2-chloroethyl)benzene, K2CO3, DMF, 90 °C, 18 h; (x) iron powder, AcOH/EtOH, 80 °C, 4 h; (xi) benzenesulfonyl chloride, NaH, THF, 0 °C to rt, 16 h; (xii) LiOH, MeOH/THF/H2O, rt, 16 h.
Conclusions
Antagonizing the function of anti-apoptotic proteins constitutes a validated and attractive new paradigm in anticancer therapy and this work contributes to the progress of drugging the Bcl-2 protein family. It is known that cancer cells depend on individual and subsets of Bcl-2 family anti-apoptotic proteins, therefore, development of selective and multimodal inhibitors have therapeutic relevance. Building from our previous reports, we designed a new 2,5-subsitiuted benzoic acid scaffold that enabled further SAR exploration of dual Mcl-1/Bfl-1 inhibition. The binding mode of the designed compound 1 was validated by HSQC-NMR and was shown to have several hydrophobic contacts with key pockets in the BH3 binding groove of Mcl-1 and Bfl-1, anchored by a hydrogen bond between the carboxyl group and Arg263/Mcl-1 or Arg88/Bfl-1. A series of inhibitors was developed by 2,5-substitutions of the benzoic acid core to explore the binding optimization. Designed compounds comprise the first discovered class of small molecules showing selective inhibition of Mcl-1/Bfl-1 over the remaining anti-apoptotic proteins. While balancing hydrophobic driven increases in potency and hydrophilicity that favored physicochemical properties, several improved molecules were co-crystallized with Mcl-1 and provided valuable structural information that drove further development. The resulting optimized molecule, 24, was 20-fold more potent for both Mcl-1 and Bfl-1 than compound 1. Selectivity over Bcl-2/Bcl-xL was preserved and was demonstrated on the cellular level by the ability of 24 to selectively bind endogenous Mcl-1 and Bfl-1 and to kill Mcl-1 and Bfl-1 dependent cells. This work contributes to the set of known Bcl-2 family inhibitors and provides small molecules that mimic the binding profile of Noxa as dual Mcl-1/Bfl-1 selective inhibitors. Further optimized dual Mcl-1/Bfl-1 selective inhibitors will have biologically and clinically relevant utility as single agents and in combination for treatment of cancers where Mcl-1 and Bfl-1 have been implicated in their pathogenesis and chemoresistance, including leukemia, lymphoma, and melanoma.
Materials and Methods
1. Chemistry and characterization of compounds
All reactions were performed under anhydrous conditions, unless otherwise stated. Reagents were used as supplied without further purification. Reactions were monitored by TLC using precoated silica gel 60 F254 plates. Silica gel chromatography was performed with silica gel (40–60 μm, 60A) obtained from Acros Organics. Purities of final compounds were assessed by analytical reverse-phase HPLC performed with method: Shimadzu system with a Restek Ultra C18 (4.6 mm × 150 mm, 5 μm particle size) column with the gradient 20% ACN/water (5 min), 20−100% ACN/water (24 min), 100%ACN/water (5 min) flow = 1 mL/min. Semi-preparative HPLC was performed on a Shimadzu system with a Restek Ultra C18 (21.2 mm × 150 mm, 5μm particle size) column. All NMR spectra were obtained in Actone-d6, Methanol-d4, DMSO-d6 or CDCl3 and results were recorded at on a Bruker 300 MHz Ultrashield or 400 MHz Ascend or on a Varian 400 MHz or 500 MHz instruments. High-resolution mass spectrometry (HRMS) analysis was performed on an Agilent Q-TOF system. All final compounds were purified to >95% purity.
Representative Procedure for Palladium Catalyzed Cross Coupling Reaction
Methyl 2-amino-5-(phenethylthio)benzoate (29a).
Synthesized using reported procedure with modification.25 To a solution of methyl 2-amino-5-iodobenzoate (28, 831 mg, 3 mmol) in dry 1,4-dioxane (30 mL) at ambient temperature under the atmosphere of nitrogen were added N, N-diisopropylethylamine (775 mg, 6 mmol), tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3, 137 mg, 0.15 mmol) and 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (Xantphos, 173 mg, 0.3 mmol) respectively. After stirring the brown mixture at the same temperature for 15 minutes, 2-phenylethanethiol (415 mg, 3 mmol) was added and the reaction mixture was heated to reflux for 16 h. The mixture was allowed to cool down to room temperature. The reaction mixture was filtered using celite and the residue was washed with dichloromethane. The filtrate was concentrated under reduced pressure to give crude oil that was purified by column chromatography (Hexane/EtOAc, 85:15) to afford 29a (707 mg, 2.46 mmol, 82%) as a yellow oil that solidified upon standing. 1H NMR (300 MHz, CDCl3) δ 7.99 (s, 1H), 7.37–7.14 (m, 6H), 6.62 (d, J = 6.0 Hz, 1H), 5.80 (s, 2H), 3.87 (s, 3H), 3.04–2.99 (m, 2H), 2.87–2.82 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 168.0, 149.9, 140.4, 138.8, 135.9, 128.5, 128.4, 126.3, 120.8, 117.4, 111.1, 51.6, 37.8, 35.9.
Representative Procedure for Coupling Reaction of Aryl Amines with Sulfonyl Chlorides
Methyl 5-(phenethylthio)-2-(phenylsulfonamido)benzoate (30a).
To a solution ethyl 2-amino-5-(phenethylthio)benzoate (29a, 100 mg, 0.35 mmol) in dry CH2Cl2 (15 mL) were added, respectively, pyridine (83 mg, 0.085 mL, 1.05 mmol) and benzenesulfonyl chloride (93 mg, 0.53 mmol) under the atmosphere of nitrogen at ambient temperature. The mixture was stirred at the same temperature for 16 h. The reaction mixture was concentrated under reduced pressure to give crude oil which followed by column chromatography (Hexane/EtOAc, 85:15) afforded 30a (131 mg, 0.31 mmol, 88%) as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 10.51 (s, 1H), 7.89–7.82 (m, 3H), 7.64 (d, J = 9.0 Hz, 1H), 7.53–7.41 (m, 4H), 7.30–7.13 (m, 5H), 3.87 (s, 3H), 3.12–3.07 (m, 2H), 2.88–2.83 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 167.7, 139.7, 139.2, 138.6, 135.9, 133.1, 132.3, 130.9, 129.0, 128.5, 128.5, 127.2, 126.5, 120.0, 116.7, 52.6, 35.8, 35.5.
Representative Procedure for Lithium Hydroxide Hydrolysis
5-(Phenethylthio)-2-(phenylsulfonamido)benzoic acid (1).
To a solution of 30a (81 mg, 0.19 mmol) in tetrahydrofuran (THF, 3 mL) and methanol (6 mL) was added the solution of LiOH (23 mg, 0.96 mmol) in water (3 mL) drop wise. The reaction mixture was stirred for 16 h at ambient temperature. After completion of the reaction, the reaction mixture was acidified with HCl solution (1 M). The resulting mixture was extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were dried (Na2SO4) and filtered. The filtrate was concentrated under reduced pressure to give crude product that was purified to afford analytical sample 1 (66 mg, 0.16 mmol, 84%) as a yellow solid. 1H NMR (300 MHz, Acetone-d6) δ 7.97 (s, 1H), 7.88 (d, J = 9.0 Hz, 2H), 7.72–7.53 (m, 5H), 7.30–7.17 (m, 5H), 3.22–3.17 (m, 2H), 2.90–2.85 (m, 2H); 13C NMR (75 MHz, Acetone-d6) δ 169.1, 140.1, 139.4, 138.9, 135.9, 133.3, 132.2, 131.1, 129.3, 128.5, 128.4, 127.1, 126.3, 119.6, 116.8, 35.2, 35.0; HRMS (ESI) calcd for C21H20NO4S2 [M + H+] 414.0834, found 414.0821.
Methyl 2-amino-5-(benzylthio)benzoate (29b).
Synthesized using the procedure for 29a except using phenylmethanethiol as the starting material, which afforded the title compound as a yellow oil that solidified upon standing. 1H NMR (300 MHz, CDCl3) δ 7.87 (s, 1H), 7.28–7.14 (m, 6H), 6.51 (d, J = 9.0 Hz, 1H), 5.78 (s, 2H), 3.93 (s, 2H), 3.84 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 168.0, 150.1, 139.5, 138.1, 136.8, 128.9, 128.3, 126.9, 120.4, 117.1, 111.0, 51.5, 41.8.
Methyl 5-(benzylthio)-2-(phenylsulfonamido)benzoate (30b).
Synthesized using the procedure for 30a except using 29b as the starting material, which afforded the title compound as a yellow oil that solidified upon standing. 1H NMR (300 MHz, CDCl3) δ 10.52 (s, 1H), 7.83–7.80 (m, 3H), 7.61–7.15 (m, 10H), 4.01 (s, 2H), 3.84 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 167.7, 139.2, 139.0, 137.1, 137.0, 133.6, 133.1, 130.3, 129.0, 128.8, 128.5, 127.3, 127.2, 119.7, 116.4, 52.5, 39.8.
5-(Benzylthio)-2-(phenylsulfonamido)benzoic acid (3).
Synthesized using the procedure for 1 except using 30b as the starting material, which afforded the title compound as a yellow solid. 1H NMR (300 MHz, Acetone-d6) δ 7.94–7.85 (m, 3H), 7.65–7.49 (m, 5H), 7.24–7.20 (m, 5H), 4.14 (s, 2H); 13C NMR (100 MHz, Acetone-d6) δ 169.4, 139.4, 139.3, 137.5, 136.4, 133.5, 133.3, 130.5, 129.3, 128.9, 128.3, 127.1, 119.3, 38.6; HRMS (ESI) calcd for C20H18NO4S2 [M + H+] 400.0677, found 400.0670.
Methyl 2-amino-5-((3-oxo-3-(phenylamino)propyl)thio)benzoate (29c).
Synthesized according to the representative procedure for 29a using 28 and 3-mercapto-N-phenylpropanamide as the starting materials. 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 1.8 Hz, 1H), 7.71 (s, 1H), 7.47 (d, J = 8.0 Hz, 2H), 7.37 – 7.31 (m, 1H), 7.27 (m, 2H), 7.08 (t, J = 7.4 Hz, 1H), 6.59 (d, J = 8.4 Hz, 1H), 5.56 (s, 2H), 3.09 (t, J = 7.2 Hz, 2H), 2.54 (t, J = 7.2 Hz, 2H).
Methyl 5-((3-oxo-3-(phenylamino)propyl)thio)-2-(phenylsulfonamido)benzoate (30c).
Synthesized using 29c according to the procedure employed for 30a. 1H NMR (400 MHz, CDCl3) δ 10.59 (s, 1H), 7.95 (d, J = 2.2 Hz, 1H), 7.88 – 7.83 (m, 2H), 7.63 (d, J = 8.8 Hz, 1H), 7.58 – 7.53 (m, 2H), 7.51 – 7.44 (m, 5H), 7.32–7.30 (m, 2H), 7.11 (t, J = 7.4 Hz, 1H), 3.86 (s, 3H), 3.23 (t, J = 7.2 Hz, 2H), 2.62 (t, J = 7.2 Hz, 2H).
5-((3-Oxo-3-(phenylamino)propyl)thio)-2-(phenylsulfonamido)benzoic acid (4).
Synthesized by hydrolyzing intermediate 30c using the representative procedure for 1. 1H NMR (500 MHz, Acetone-d6) δ 7.99 (d, J = 2.2 Hz, 1H), 7.92 – 7.86 (m, 2H), 7.69 (d, J = 8.8 Hz, 1H), 7.63–7.61 (m, 4H), 7.56 (t, J = 7.6 Hz, 2H), 7.27 (t, J = 8.0 Hz, 2H), 7.04 (t, J = 7.4 Hz, 1H), 3.26 (t, J = 7.2 Hz, 2H), 2.67 (t, J = 7.2 Hz, 2H).
Methyl 2-amino-5-((4-oxo-4-(phenylamino)butyl)thio)benzoate (29d).
Synthesized according to the representative procedure for 29a using 28 and 4-mercapto-N-phenylbutanamide as the starting materials. 1H NMR (300 MHz, CDCl3) δ 7.96 (d, J = 2.0 Hz, 1H), 7.57 – 7.45 (m, 3H), 7.37 – 7.25 (m, 4H), 7.08 (t, J = 7.2 Hz, 1H), 6.60 (d, J = 8.6 Hz, 1H), 6.04 (s, 2H), 3.84 (s, 3H), 2.84 (t, J = 6.8 Hz, 2H), 2.48 (t, J = 6.8 Hz, 2H), 2.01 – 1.93 (m, 2H).
Methyl 5-((4-oxo-4-(phenylamino)butyl)thio)-2-(phenylsulfonamido)benzoate (30d).
Synthesized using 29d according to the procedure employed for 30a. 1H NMR (300 MHz, CDCl3) δ 10.55 (s, 1H), 7.95 (s, 1H), 7.89 – 7.78 (m, 3H), 7.60 (d, J = 8.8 Hz, 1H), 7.51 (d, J = 2.2 Hz, 1H), 7.49 (s, 2H), 7.42–7.40 (m, 3H), 7.26 (t, J = 7.8 Hz, 2H), 7.06 (t, J = 7.2 Hz, 1H), 3.81 (s, 3H), 2.92 (t, J = 7.2 Hz, 2H), 2.47 (t, J = 7.2 Hz, 2H), 2.00 – 1.91 (m, 2H).
5-((4-Oxo-4-(phenylamino)butyl)thio)-2-(phenylsulfonamido)benzoic acid (5).
Synthesized by hydrolyzing intermediate 30d using the representative procedure employed for 1. 1H NMR (400 MHz, Acetone-d6) δ 9.19 (s, 1H), 8.01 (s, 1H), 7.88 (d, J = 7.8 Hz, 2H), 7.68–7.65 (m, 3H), 7.62 – 7.46 (m, 4H), 7.28 (t, J = 7.6 Hz, 2H), 7.04 (t, J = 7.4 Hz, 1H), 3.01 (t, J = 7.2 Hz, 2H), 2.54 (t, J = 7.2 Hz, 2H), 1.97 – 1.90 (m, 2H).
Methyl 5-nitro-2-(phenylsulfonamido)benzoate (32).
To a solution of methyl 2-amino-5-nitrobenzoate (31) (500 mg, 2.55 mmol) in dry THF (30 mL) cooled to 0 °C under the atmosphere of nitrogen, NaH (60%, 204 mg, 5.10 mmol) was added in portions and the mixture was stirred at the same temperature for 1 h. Benzenesulfonyl chloride (901 mg, 5.10 mmol) was added slowly. After stirring at the same temperature for 5 minutes, the reaction flask was replaced to ambient temperature and stirred for 16 h. The mixture was quenched with saturated NH4Cl (20 mL) slowly. The resulting mixture was extracted with Et2O (3 × 10 mL). The combined organic layers were concentrated under reduced pressure and the crude was purified by column chromatography (Hexane/EtOAc, 8:2) to afford (32, 551 mg, 3.27 mmol, 64%) as a yellow solid. All data corroborated with those of the previously reported sample.64
Methyl 5-amino-2-(phenylsulfonamido)benzoate (33).
Synthesized using reported procedure with modification 65. To a mixture of iron powder (321 mg, 5.75 mmol) in AcOH (10 mL) and dry EtOH (10 mL) was added 32 (388 mg, 1.15 mmol) and the mixture was heated to reflux for 4 h. After cooling, the mixture was diluted with H2O (50 mL) and was extracted in Et2O (3 × 30 mL). The combined organic layers were dried (Na2SO4) and filtered. The filtrate was concentrated under reduced pressure and the crude was purified by column chromatography (Hexane/EtOAc, 4:6) to afford 33 (267 mg, 0.87 mmo, 75%) as a yellow solid. H NMR (300 MHz, CDCl3) δ 9.71 (s, 1H), 7.68 (d, J = 6.0 Hz, 2H), 7.54–7.34 (m, 4H), 7.12 (d, J = 3.0 Hz, 1H), 6.82 (dd, J = 9.0, 3.0 Hz, 1H), 3.72–3.68 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 167.7, 143.1, 138.8, 132.7, 130.6, 128.6, 127.2, 123.7, 121.0, 119.2, 116.1, 52.2.
Methyl 5-(phenethylamino)-2-(phenylsulfonamido)benzoate (34a).
A mixture of 33 (184 mg, 0.60 mmol) and (2-chloroethyl)benzene (211 mg, 1.5 mmol) in dry DMF (20 mL) was added K2CO3 (691 mg, 5.0 mmol) under the atmosphere of nitrogen. The mixture was heated at 120 °C for 16 h. After cooling, the mixture was diluted with ice-cooled H2O (20 mL) and was extracted in Et2O (3 × 20 mL). The combined organic layers were dried (Na2SO4 and filtered. The filtrate was concentrated under reduced pressure and the crude was purified by column chromatography (Hexane/EtOAc, 1:1) to afford 34a (211 mg, 0.51 mmol, 86%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.62–7.12 (m, 11H), 6.71–6.64 (m, 2H), 3.92–3.69 (m, 6H), 3.03–2.85 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 166.6, 146.5, 139.7, 138.6, 133.1, 132.2, 131.8, 128.8, 128.6, 128.4, 127.7, 127.4, 126.3, 117.7, 117.0, 53.6, 52.0, 35.6.
5-(Phenethylamino)-2-(phenylsulfonamido)benzoic acid (6).
Synthesized using the procedure for 1 except using 34a as the starting material, which afforded the title compound as a yellow solid. H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 7.4 Hz, 2H), 7.55 (t, J = 7.4 Hz, 1H), 7.43 (t, J = 7.8 Hz, 2H), 7.29–7.16 (m, 4H), 7.09 (d, J = 6.8 Hz, 2H), 6.69 (dd, J = 8.4, 2.6 Hz, 1H), 6.57 (d, J = 8.4 Hz, 1H), 3.77 (dd, J = 110.5, 12.5 Hz, 2H), 3.04 – 2.70 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 168.1, 146.1, 138.7, 138.3, 133.6, 132.6, 130.8, 128.8, 128.7, 128.4, 127.8, 127.6, 126.4, 118.2, 117.7, 53.5, 35.1; HRMS (ESI) calcd for C21H21N2O4S [M + H+] 397.1222, found 397.1218.
Methyl 5-(2-phenylacetamido)-2-(phenylsulfonamido)benzoate (34b).
In a flame-dried round-bottomed flask, aniline 33 (76 mg, 0.25 mmol), triethylamine (30 mg, 0.3 mmol) and DMAP (6 mg, 0.05 mmol) were dissolved in dry dichloromethane (DCM, 5 ml) under the atmosphere of nitrogen at ambient temperature. Phenylacetyl chloride (46 mg, 0.3 mmol) was added drop wise. The reaction mixture was stirred at the same temperature for 16 h whereby reaction completed as indicated by TLC. The reaction mixture was diluted with DCM (15 mL) and was washed, respectively, with water, 1 M HCl solution, 5% sodium bicarbonate solution and brine. The organic layers were dried (Na2SO4) and filtered. The filtrate was concentrated under reduced pressure to give crude product that was used for the next step without further purification.
5-(2-Phenylacetamido)-2-(phenylsulfonamido)benzoic acid (7).
Synthesized by hydrolyzing crude intermediate 34b using the representative procedure as for 1. 1H NMR (500 MHz, Acetone-d6) δ 8.33 (d, J = 2.4 Hz, 1H), 7.85 (dd, J = 9.0, 2.4 Hz, 1H), 7.80 (d, J = 7.6 Hz, 2H), 7.66 (d, J = 9.0 Hz, 1H), 7.59 (t, J = 7.6 Hz, 1H), 7.51 (t, J = 7.8 Hz, 2H), 7.36 (d, J = 7.2 Hz, 2H), 7.30 (t, J = 7.8 Hz, 2H), 7.23 (t, J = 7.2 Hz, 1H), 3.68 (s, 2H).
Methyl 5-(3-phenylpropanamido)-2-(phenylsulfonamido)benzoate (34c).
This compound was synthesized using the procedure employed for 34b and used for the next step without further purification.
5-(3-Phenylpropanamido)-2-(phenylsulfonamido)benzoic acid (8).
Synthesized by hydrolyzing the crude intermediate 34c using the representative procedure as for 1. 1H NMR (500 MHz, Acetone-d6) δ 8.32 (d, J = 2.4 Hz, 1H), 7.84 (dd, J = 9.0, 2.6 Hz, 1H), 7.82 – 7.78 (m, 2H), 7.66 (d, J = 9.0 Hz, 1H), 7.60 (t, J = 7.4 Hz, 1H), 7.51 (t, J = 7.8 Hz, 2H), 7.28 – 7.23 (m, 4H), 7.19 – 7.13 (m, 1H), 2.97 (t, J = 7.8 Hz, 2H), 2.67 (t, J = 7.8 Hz, 2H).
Methyl 2-nitro-5-phenethoxybenzoate (9a).
A mixture of methyl 5-hydroxy-2-nitrobenzoate (500 mg, 2.536 mmol), (2-chloroethyl)benzene (714 mg, 5.074 mmol) and K2CO3 (876 mg, 6.340 mmol) in DMF (8 mL) was heated at 90 °C for 18 h. After cooling to ambient temperature, the mixture was diluted with EtOAc and the layer was washed with water three times. The organic layer was concentrated under reduced pressure and the crude oil was purified by chromatography (Hexane/EtOAc, 85:15) to afford 9a (763 mg, 2.532 mmol, 100%) as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 8.02 (d, J = 9.0 Hz, 1H), 7.36 – 7.24 (m, 5H), 7.04–6.98 (m, 2H), 4.27 (t, J = 6.8 Hz, 2H), 3.92 (s, 3H), 3.13 (t, J = 6.8 Hz, 2H).
Methyl 2-amino-5-phenethoxybenzoate (9b).
Synthesized using the procedure discussed for 33 by heating a mixture of 9a (760 mg, 2.523 mmol) and Fe powder (704 mg, 12.614 mmol) in a mixture of AcOH (15 mL) and dry EtOH (15 mL) at 80 °C for 4 h. 1H NMR (300 MHz, CDCl3) δ 7.36 – 7.20 (m, 6H), 6.93 (dd, J = 8.8, 3.0 Hz, 1H), 6.59 (d, J = 8.8 Hz, 1H), 5.41 (s, 2H), 4.10 (t, J = 7.2 Hz, 2H), 3.84 (s, 3H), 3.06 (t, J = 7.2 Hz, 2H).
Methyl 5-phenethoxy-2-(phenylsulfonamido)benzoate (9c).
Synthesized according to the procedure discussed for 30a using intermediate 9b. 1H NMR (300 MHz, CDCl3) δ 10.00 (s, 1H), 7.74–7.64 (m, 3H), 7.53 – 7.22 (m, 9H), 7.04 (dd, J = 9.2, 3.0 Hz, 1H), 4.10 (t, J = 7.0 Hz, 2H), 3.78 (s, 3H), 3.06 (t, J = 7.0 Hz, 2H).
5-Phenethoxy-2-(phenylsulfonamido)benzoic acid (9).
Synthesized according to the procedure discussed for 1 using intermediate 9c. 1H NMR (400 MHz, THF-d8) δ 10.56 (s, 1H), 7.81 – 7.61 (m, 3H), 7.51 – 7.43 (m, 1H), 7.41–7.36 (m, 3H), 7.31 – 7.20 (m, 4H), 7.18–7.16 (m, 1H), 7.07 (dd, J = 9.0, 3.0 Hz, 1H), 4.09 (t, J = 7.0 Hz, 2H), 3.01 (t, J = 7.0 Hz, 2H); 13C NMR (100 MHz, THF-d8) δ 170.4, 155.7, 141.1, 139.4, 135.2, 133.6, 129.9, 129.8, 129.3, 128.2, 127.3, 122.6, 122.0, 119.2, 116.9, 70.0, 36.6.
2-Amino-5-(phenethylthio)benzoic acid (10).
Synthesized using the representative procedure for 1 except using 29a as the starting material, which afforded the title compound as a yellow solid. 1H NMR (300 MHz, CDCl3) δ 8.07 (s, 1H), 7.41–7.15 (m, 6H), 6.63 (d, J = 9.0 Hz, 1H), 3.06–3.01 (m, 2H), 2.89–2.84 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 173.1, 150.5, 140.3, 139.7, 136.5, 126.3, 121.2, 117.6, 109.9, 37.7, 35.9; HRMS (ESI) calcd for C15H15NO2S [M + H+] 274.0902, found 274.0895.
Methyl 2-benzamido-5-(phenethylthio)benzoate (30e).
To a solution of 29a (100 mg, 0.35 mmol) in dry dichloromethane (15 mL) under the atmosphere of nitrogen were added K2CO3 (770mg, 5.57 mmol) followed by the drop wise addition of the solution of benzoyl chloride (150 mg, 1.07 mmol) in DCM (2 mL). The resulting mixture was stirred at ambient temperature for 16 h. The reaction mixture was filtered, concentrated under reduced pressure to give crude oil that was purified by chromatography (Hexane/EtOAc, 9:1) to afford 30e (121 mg, 0.31 mmol, 89%). 1H NMR (400 MHz, CDCl3) δ 11.97 (s, 1H), 8.91 (d, J = 8.8 Hz, 1H), 8.09 (d, J = 2.2 Hz, 1H), 8.06 – 8.01 (m, 2H), 7.56–7.54 (m, 4H), 7.31–7.30 (m, 2H), 7.25 – 7.16 (m, 3H), 3.97 (s, 3H), 3.19 – 3.13 (m, 2H), 2.95 – 2.88 (m, 2H).
2-Benzamido-5-(phenethylthio)benzoic acid (11).
Synthesized using the procedure for 1 except using 30e as the starting material, which afforded the title compound as a white solid. 1H NMR (300 MHz, Acetone-d6) δ 12.21 (s, 1H), 8.94 (d, J = 9.0 Hz, 1H), 8.18 (s, 1H), 8.04 (d, J = 9.0 Hz, 2H), 7.75–7.56 (m, 4H), 7.34–7.19 (m, 5H), 3.29–3.24 (m, 2H), 2.98–2.93 (m, 2H); 13C NMR (75 MHz, Acetone-d6) δ 169.5, 164.8, 140.6,140.2, 135.9, 134.9, 132.4, 132.1, 130.0, 128.9, 128.6, 128.4, 127.2, 126.3, 120.7, 116.0, 35.4, 35.3; HRMS (ESI) calcd for C22H20NO3S [M + H+] 378.1164, found 378.1154.
Methyl 5-(phenethylthio)-2-(2-phenylethylsulfonamido)benzoate (30f).
Synthesized using the procedure for 30a except using 2-phenylethanesulfonyl chloride as the starting material, which afforded the title compound as a yellow oil that solidified upon standing. 1H NMR (300 MHz, CDCl3) δ 10.36 (s, 1H), 8.01 (d, J = 3.0 Hz, 1H), 7.70 (d, J = 9.0 Hz, 1H), 7.51 (dd, J = 9.0, 2.0 Hz, 1H), 7.32–7.08 (m, 10H), 3.93 (s, 3H), 3.44–3.38 (m, 2H), 3.17–3.09 (m, 4H), 2.93–2.88 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 167.8, 139.7, 139.1, 137.2, 136.5, 132.9, 130.4, 128.8, 128.5, 128.5, 128.2, 126.9, 126.5, 118.6, 115.7, 53.3, 52.7, 36.0, 35.6, 29.6.
5-(Phenethylthio)-2-(2-phenylethylsulfonamido)benzoic acid (12).
Synthesized using the representative procedure for 1 except using 30f as the starting material, which afforded the title compound as a yellow oil that solidified upon standing. 1H NMR (300 MHz, CDCl3) δ 10.05 (s, 1H), 8.09 (d, J = 2.0 Hz, 1H), 7.71 (d, J = 9.0 Hz, 1H), 7.56 (dd, J = 9.0, 3.0 Hz, 1H), 7.33–7.08 (m, 10H), 3.50–3.44 (m, 2H), 3.20–3.12 (m, 4H), 2.96–2.90 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 171.7, 139.6, 139.4, 137.4, 137.0, 133.3, 131.0, 128.8, 128.6, 128.5, 128.2, 127.0, 126.6, 118.4, 114.5, 53.3, 35.8, 35.5, 29.6; HRMS (ESI) calcd for C23H24NO4S2 [M + H+] 442.1147, found 442.1134.
Methyl 5-(phenethylthio)-2-(thiophene-2-sulfonamido)benzoate (30g).
Synthesized using the procedure for 30a except using thiophene-2-sulfonyl chloride as the starting material, which afforded the title compound as a yellow oil that solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 10.59 (s, 1H), 7.92 (s, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.59–6.99 (m, 9H), 3.89 (s, 3H), 3.12 (t, J = 8.0 Hz, 2H), 2.88 (t, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 167.7, 139.9, 139.7, 138.3, 135.8, 132.8, 132.5, 132.2, 131.4, 128.5, 128.5, 127.3, 126.6, 120.2, 116.9, 52.7, 35.7, 35.5.
5-(Phenethylthio)-2-(thiophene-2-sulfonamido)benzoic acid (13).
Synthesized using the procedure for 1 except using 30g as the starting material, which afforded the title compound as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 10.46 (s, 1H), 8.02 (s, 1H), 7.73–6.99 (m, 10H), 3.14 (t, J = 8.0 Hz, 2H), 2.89 (t, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 171.6, 139.6, 139.6, 138.6, 136.5, 133.1, 132.8, 131.8, 128.6, 128.5, 127.4, 126.6, 120.0, 116.1, 35.5, 35.5; HRMS (ESI) calcd for C19H18NO4S3 [M + H+] 420.0398, found 420.0396.
Methyl 2-(4-bromophenylsulfonamido)-5-(phenethylthio)benzoate (30h).
Synthesized using the procedure for 30a except using 4-bromobenzene-1-sulfonyl chloride as the starting material, which afforded the title compound as a yellow oil that solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 10.51 (s, 1H), 7.89 (s, 1H), 7.70–7.15 (m, 11H), 3.88 (s, 3H), 3.11 (t, J = 8.0 Hz, 2H), 2.87 (t, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 167.7, 139.7, 138.3, 138.2, 135.8, 132.3, 132.2, 131.5, 128.7, 128.5, 128.5, 128.2, 126.6, 120.1, 116.8, 52.7, 35.7, 35.5.
2-(4-Bromophenylsulfonamido)-5-(phenethylthio)benzoic acid (14).
Synthesized using the procedure for 1 except using 30h as the starting material, which afforded the title compound as a yellow oil that solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 10.26 (s, 1H), 8.01 (s, 1H), 7.74–7.16 (m, 11H), 3.15 (t, J = 8.0 Hz, 2H), 2.90 (t, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 171.7, 139.6, 138.6, 138.1, 136.8, 132.8, 132.5, 131.9, 128.7, 128.6, 128.5, 128.5, 126.6, 119.7, 115.4, 35.5, 35.4; HRMS (ESI) calcd for C21H19BrNO4S2 [M + H+] 491.9939, found 491.9920.
Methyl 2-(naphthalene-2-sulfonamido)-5-(phenethylthio)benzoate (30i).
Synthesized using the procedure for 30a except using naphthalene-2-sulfonyl chloride as the starting material, which afforded the title compound as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 10.66 (s, 1H), 8.44 (s, 1H), 7.91–7.08 (m, 14H), 3.82 (s, 3H), 3.07–3.02 (m, 2H), 2.83–2.78 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 167.7, 139.6, 138.5, 136.0, 135.8, 134.8, 132.2, 131.8, 130.7, 129.4, 129.2, 128.9, 128.8, 128.4, 128.4, 127.8, 127.5, 126.4, 122.1, 119.7, 116.5, 52.5, 35.6, 35.4.
2-(Naphthalene-2-sulfonamido)-5-(phenethylthio)benzoic acid (15).
Synthesized using the procedure for 1 except using 30i as the starting material, which afforded the title compound as a yellow oil that solidified upon standing. 1H NMR (300 MHz, Acetone-d6) δ 11.12 (br, s, 1H), 8.58 (s, 1H), 8.10–7.54 (m, 9H), 7.22–7.16 (m, 5H), 3.14–3.09 (m, 2H), 2.82–2.77 (m, 2H); 13C NMR (75 MHz, Acetone-d6) δ 169.2, 140.0, 139.0, 136.3, 135.6, 135.0, 132.3, 132.0, 131.0, 129.7, 129.3, 129.2, 128.9, 128.5, 128.3, 127.9, 127.8, 126.3, 122.1, 119.5, 116.7, 35.2, 34.9; HRMS (ESI) calcd for C25H22NO4S2 [M + H+] 464.0990, found 464.0978.
Methyl 2-([1,1’-biphenyl]-4-ylsulfonamido)-5-(phenethylthio)benzoate (30j).
Synthesized using the procedure for 30a except using biphenyl-4-sulfonyl chloride as the starting material, which afforded the title compound as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 10.56 (s, 1H), 7.91–7.88 (m, 3H), 7.71–7.12 (m, 14H), 3.86 (s, 3H), 3.12–3.07 (m, 2H), 2.88–2.83 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 167.7, 145.9, 139.7, 139.0, 138.6, 137.7, 135.9, 132.3, 130.9, 129.0, 128.5, 128.5, 128.4, 127.7, 127.6, 127.2, 126.5, 119.9, 116.6, 52.6, 35.7, 35.5.
2-([1,1’-Biphenyl]-4-ylsulfonamido)-5-(phenethylthio)benzoic acid (16).
Synthesized using the representative procedure for 1 except using 30j as the starting material, which afforded the title compound as a yellow oil that solidified upon standing. 1H NMR (300 MHz, Acetone-d6) δ 7.99–7.92 (m, 3H), 7.82 (d, J = 9.0 Hz, 2H), 7.73 (d, J = 9.0 Hz, 1H), 7.68–7.64 (m, 2H), 7.61 (dd, J = 9.0, 3.0 Hz, 1H), 7.50–7.38 (m, 3H), 7.27–7.13 (m, 5H), 3.22–3.14 (m, 2H), 2.90–2.81 (m, 2H); 13C NMR (75 MHz, Acetone-d6) δ 169.2, 145.7, 140.0, 139.0, 138.8, 138.0, 135.7, 132.3, 131.1, 129.0, 128.6, 128.5, 128.3, 127.8, 127.6, 127.2, 126.3, 119.5, 116.8, 35.2, 35.0; HRMS (ESI) calcd for C27H24NO4S2 [M + H+] 490.1147, found 490.1134.
Methyl 2-(4-cyclohexylphenylsulfonamido)-5-(phenethylthio)benzoate (30k).
Synthesized using the procedure for 30a except using 4-cyclohexylbenzene-1-sulfonyl chloride as the starting material, which afforded the title compound as a yellow oil that solidified upon standing. 1H NMR (300 MHz, CDCl3) δ 10.48 (s, 1H), 7.89–7.13 (m, 12H), 3.86 (s, 3H), 3.12–3.07 (m, 2H), 2.88–2.83 (m, 2H), 2.51 (s, 1H), 1.82–1.76 (m, 5H), 1.39–1.32 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 167.7, 153.8, 139.7, 138.8, 136.5, 136.0, 132.4, 130.4, 128.5, 128.4, 127.5, 127.3, 126.5, 119.8, 116.5, 52.6, 44.4, 35.8, 35.5, 33.9, 26.5, 25.8.
2-(4-Cyclohexylphenylsulfonamido)-5-(phenethylthio)benzoic acid (17).
Synthesized using the procedure for 1 except using 30k as the starting material, which afforded the title compound as a yellow oil that solidified upon standing. 1H NMR (300 MHz, Acetone-d6) δ 10.95 (br, s, 1H), 7.98–7.18 (m, 12H), 3.21–3.16 (m, 2H), 2.88–2.83 (m, 2H), 2.56 (s, 1H), 1.78–1.68 (m, 5H), 1.43–1.20 (m, 5H); 13C NMR (75 MHz, Acetone-d6) δ 169.0, 153.9, 140.1, 139.2, 136.8, 135.8, 132.4, 130.8, 128.5, 128.4, 127.7, 127.3, 126.3, 119.4, 116.5, 44.2, 35.2, 35.0, 33.7, 26.4, 25.6; HRMS (ESI) calcd for C27H30NO4S2 [M + H+] 496.1616, found 496.1606.
Methyl 5-(phenethylthio)-2-((6-phenylpyridine)-3-sulfonamido)benzoate (30l).
Synthesized according to the procedure discussed for 30a using intermediate 29a and 6-phenylpyridine-3-sulfonyl chloride. 1H NMR (400 MHz, CDCl3) δ 10.68 (s, 1H), 9.10 (dd, J = 2.4, 0.8 Hz, 1H), 8.17 (dd, J = 8.4, 2.4 Hz, 1H), 8.06 – 7.99 (m, 2H), 7.92 (d, J = 2.4 Hz, 1H), 7.82 (dd, J = 8.4, 0.8 Hz, 1H), 7.75 (d, J = 8.8 Hz, 1H), 7.53 – 7.47 (m, 4H), 7.31–7.28 (m, 2H), 7.25–7.21 (m, 1H), 7.19–7.17 (m, 2H), 3.90 (s, 3H), 3.17 – 3.08 (m, 2H), 2.94 – 2.84 (m, 2H).
5-(Phenethylthio)-2-((6-phenylpyridine)-3-sulfonamido)benzoic acid (18).
Synthesized according to the procedure discussed for 1 using intermediate 30l. 1H NMR (400 MHz, THF-d8) δ 11.12 (s, 1H), 9.10 – 9.02 (m, 1H), 8.19 (dd, J = 8.4, 2.4 Hz, 1H), 8.11–8.08 (m, 2H), 7.97–7.94 (m, 2H), 7.76 (d, J = 8.8 Hz, 1H), 7.53 (dd, J = 8.8, 2.4 Hz, 1H), 7.47 – 7.37 (m, 3H), 7.24 – 7.09 (m, 5H), 3.13–3.09 (m, 2H), 2.84 – 2.81 (m, 2H); 13C NMR (101 MHz, THF-d8) δ 170.3, 161.4, 148.9, 141.1, 139.5, 138.4, 136.7, 136.3, 135.3, 133.4, 132.5, 131.1, 129.6, 129.3, 129.2, 128.2, 127.1, 120.7, 120.7, 118.3, 36.5, 36.1.
Methyl 5-(phenethylthio)-2-((4-phenylpiperidine)-1-sulfonamido)benzoate (30m).
A mixture of 29a (60 mg, 0.209 mmol) and 4-phenylpiperidine-1-sulfonyl chloride (71 mg, 0.273 mmol) in pyridine (3 mL) was heated at 100 °C for 14 h. The resulting mixture was concentrated under reduced pressure and purified by chromatography (Hexane/EtOAc, 9:1) to afford 30m (41 mg, 0.080 mmol, 38%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 10.42 (s, 1H), 8.05 (d, J = 2.2 Hz, 1H), 7.67 (d, J = 8.8 Hz, 1H), 7.54 (dd, J = 8.8, 2.2 Hz, 1H), 7.33 – 7.11 (m, 10H), 4.04 – 3.87 (m, 5H), 3.17–3.12 (m, 2H), 2.94 – 2.85 (m, 4H), 2.61–2.51 (m, 1H), 1.90–1.86 (m, 2H), 1.75–1.61 (m, 2H).
5-(Phenethylthio)-2-((4-phenylpiperidine)-1-sulfonamido)benzoic acid (19).
Synthesized according to the procedure discussed for 1 using intermediate 30m. 1H NMR (300 MHz, THF-d8) δ 10.74 (s, 1H), 8.10 (d, J = 2.2 Hz, 1H), 7.72 (d, J = 8.8 Hz, 1H), 7.59 (dd, J = 8.8, 2.2 Hz, 1H), 7.31 – 7.06 (m, 10H), 3.90–3.86 (m, 2H), 3.18–3.13 (m, 2H), 2.90 – 2.79 (m, 4H), 2.59- – 2.51 (m, 1H), 1.82 – 1.56 (m, 4H).
Representative Procedure for Intermediates 30n-s
Methyl 5-(phenethylthio)-2-(1-phenylpiperazine-4-sulfonamido)benzoate (30n).
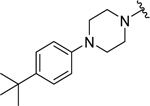
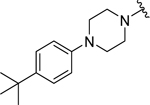
Synthesized using reported procedure with modification.26 To a round-bottom-flask under nitrogen were added 1-(1H-imidazol-1-ylsulfonyl)-1H-imidazole (80 mg, 0.40 mmol) and dry CH2Cl2 (10 mL) at 0 °C. Methyl trifluoromethanesulfonate (MeOTf, 72 mg, 0.44 mmol) was added and the mixture was stirred for 3 h. The solvent was removed under reduced pressure and re-dissolved in dry CH3CN (10 mL) at ambient temperature. 1-Phenylpiperazine (65 mg, 0.40 mmol) was added and then the mixture was stirred for 16 h. The reaction mixture was concentrated under reduced pressure. The crude product was dissolved in dry CH2Cl2 (10 mL) at 0 °C. MeOTf (72 mg, 0.44 mmol) was added and then the mixture was stirred for an additional 2 h. The solvent was removed under reduced pressure and re-dissolved in dry CH3CN (10 mL) at ambient temperature. Methyl 2-amino-5-(phenethylthio)benzoate (29a, 57.5 mg, 0.20 mmol) was added and then the mixture was heated to reflux for 16 h. The reaction mixture was concentrated under reduced pressure to give crude oil that was purified by column chromatography (Hexane/EtOAc, 7:3) to afford 30n (75 mg, 0.15 mmol, 73%, over 2 steps) as a yellow oil that solidified upon standing. 1H NMR (300 MHz, CDCl3) δ 10.45 (s, 1H), 8.02 (d, J = 2.0 Hz, 1H), 7.68 (d, J = 9.0 Hz, 1H), 7.51 (dd, J = 9.0, 3.0 Hz, 1H), 7.31–7.16 (m, 7H), 6.92–6.86 (m, 3H), 3.94 (s, 3H), 3.44–3.41 (m, 4H), 3.19–3.11 (m, 6H), 2.92–2.87 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 168.0, 150.6, 139.8, 139.8, 136.4, 132.7, 129.8, 129.2, 128.5, 128.5, 126.5, 120.8, 119.2, 116.8, 115.4, 52.7, 49.1, 46.3, 36.1, 35.6.
5-(Phenethylthio)-2-(1-phenylpiperazine-4-sulfonamido)benzoic acid (20).
Synthesized using the procedure for 1 except using 30n as the starting material, which afforded the title compound as a yellow solid after HPLC purification. HPLC (tR = 23.27 min), purity >99%. 1H NMR (400 MHz, CDCl3) δ 8.09 (s, 1H), 7.65–6.95 (m, 12H), 3.49–3.46 (m, 4H), 3.23–3.12 (m, 6H), 2.92–2.88 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 169.7, 149.5, 139.8, 139.4, 136.1, 133.2, 130.0, 129.3, 128.4, 126.4, 122.0, 119.1, 117.3, 116.0, 49.7, 45.9, 35.9, 35.5; HRMS (ESI) calcd for C25H28N3O4S2 [M + H+] 498.1521, found 498.1516.
Methyl 2-(1-benzylpiperazine-4-sulfonamido)-5-(phenethylthio)benzoate (30o).
Synthesized using the procedure for 30n except using 1-benzylpiperazine as the starting material, which afforded the title compound as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 10.54 (s, 1H), 8.01 (s, 1H), 7.53–7.18 (m, 12H), 4.13 (s, 2H), 3.93 (s, 3H), 3.62 (br, 4H), 3.18–3.12 (m, 6H), 2.91(t, J = 6.0 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 168.1, 139.7, 138.4, 136.0, 132.3, 131.3, 130.9, 130.3, 129.4, 128.5, 127.8, 126.5, 119.3, 115.9, 60.9, 52.8, 50.2, 43.5, 35.7, 35.5.
2-(1-Benzylpiperazine-4-sulfonamido)-5-(phenethylthio)benzoic acid (21).
Synthesized using the procedure for 1 except using 30o as the starting material, which afforded the title compound as a white solid after HPLC purification. HPLC (tR = 18.13 min), purity >99%. 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 2.0 Hz, 1H), 7.57 (d, J = 8.8 Hz, 1H), 7.41–7.17 (m, 11H), 4.09 (s, 2H), 3.50–3.11 (m, 10H), 2.90 (t, J = 7.8 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 171.2, 139.9, 138.3, 134.8, 132.7, 131.5, 131.0, 130.2, 129.3, 128.5, 128.5, 127.8, 126.5, 120.1, 119.1, 60.8, 50.5, 43.8, 35.6, 35.5; HRMS (ESI) calcd for C26H30N3O4S2 [M + H+] 512.1678, found 512.1673.
Methyl 2-(1-(4-fluorobenzyl)piperazine-4-sulfonamido)-5-(phenethylthio)benzoate (30p).
Synthesized using the procedure for 30n except using 1-(4-fluorobenzyl)piperazine as the starting material, which afforded the title compound as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 10.53 (s, 1H), 8.01 (s, 1H), 7.53–7.08 (m, 11H), 4.09 (s, 2H), 3.94 (s, 3H), 3.62 (br, 4H), 3.17–3.13 (m, 6H), 2.92 (t, J = 8.0 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 168.1, 163.7 (d, J = 250.0 Hz), 139.8, 138.4, 136.1, 132.9 (d, J = 8.75 Hz), 132.3, 131.4, 128.5, 128.5, 126.5, 119.4, 116.5 (d, J = 21.25 Hz), 115.9, 109.9, 60.2, 52.82, 50.2, 43.6, 35.7, 35.5.
2-(1-(4-Fluorobenzyl)piperazine-4-sulfonamido)-5-(phenethylthio)benzoic acid (22).
Synthesized using the procedure for 1 except using 30p as the starting material, which afforded the title compound as a yellow oil that solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 10.85 (s, 1H), 8.01 (s, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.43–7.01 (m, 10H), 4.06 (s, 2H), 3.51 (br, 4H), 3.15–3.12 (m, 6H), 2.90(t, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 171.2, 163.7 (d, J = 250.0 Hz), 139.9, 138.3, 135.0, 133.0 (d, J = 8.0 Hz), 132.7, 131.6, 128.5, 126.5, 123.9, 120.1, 118.8, 116.5 (d, J = 21.0 Hz), 60.0, 50.5, 43.8, 35.6, 35.5; HRMS (ESI) calcd for C26H29FN3O4S2 [M + H+] 530.1584, found 530.1579.
Methyl 2-((4-(4-(tert-butyl)phenyl)piperazine)-1-sulfonamido)-5-(phenethylthio) benzoate (30q).
Synthesized using the procedure for 30n except using 1-(4-(tert-butyl)phenyl)piperazine as the starting material, which afforded the title compound as a yellow solid. 1H NMR (300 MHz, CDCl3) δ 10.45 (s, 1H), 8.02 (d, J = 2.2 Hz, 1H), 7.68 (d, J = 8.8 Hz, 1H), 7.53–7.49 (m, 1H), 7.32–7.17 (m, 7H), 6.83 (d, J = 8.8 Hz, 2H), 3.95 (s, 3H), 3.44–3.41 (m, 4H), 3.16–3.11 (m, 6H), 2.92–2.87 (m, 2H), 1.27 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 168.1, 148.3, 143.8, 140.0, 136.6, 132.9, 129.9, 128.7, 126.7, 126.2, 119.4, 116.7, 115.5, 52.8, 49.4, 46.5, 36.2, 35.8, 34.1, 31.5.
2-((4-(4-(tert-butyl)phenyl)piperazine)-1-sulfonamido)-5-(phenethylthio)benzoic acid (23).
Synthesized using the procedure for 1 except using 30q as the starting material, which afforded the title compound as a brown oil that solidified upon standing. HPLC (tR = 25.81 min), purity >95%. 1H NMR (300 MHz, CDCl3) δ 10.71 (s, 1H), 7.95 (d, J = 2.3 Hz, 1H), 7.79 (d, J = 8.8 Hz, 1H), 7.53–7.49 (m, 1H), 7.35–7.17 (m, 7H), 7.00 (d, J = 8.7 Hz, 2H), 3.52–3.49 (m, 4H), 3.27–3.24 (m, 4H), 3.17–3.11 (m, 2H), 2.94–2.88 (m, 2H), 1.29 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 170.8, 146.8, 146.3, 140.0, 136.4, 133.1, 130.5, 128.7, 126.6, 126.4, 119.6, 118.0, 115.7, 50.7, 46.1, 36.0, 35.7, 34.3, 31.4; HRMS (ESI) calcd for C29H36N3O4S2 [M + H+] 554.2147, found 554.2136.
Methyl 2-((4-(4-(tert-butyl)benzyl)piperazine)-1-sulfonamido)-5-(phenethylthio)benzoate (30r).
Synthesized using the procedure for 30n except using 1-(4-(tert-butyl)benzyl)piperazine as the starting material, which afforded the title compound as a yellow oil. HPLC (tR = 22.07 min), purity >95%. 1H NMR (300 MHz, CDCl3) δ 10.40 (s, 1H), 8.02 (s, 1H), 7.64 (d, J = 9 Hz, 1H), 7.49 (d, J = 9 Hz, 1H), 7.35–7.27 (m, 5H), 7.24–7.16 (m, 4H), 3.94 (s, 3H), 3.45 (s br, 2H), 3.29 (s br, 4H), 3.18–3.09 (m, 2H), 2.94–2.87 (m, 2H), 2.46 (s br, 4H), 1.30 (s, 9H).
2-((4-(4-(Tert-butyl)benzyl)piperazine)-1-sulfonamido)-5-(phenethylthio)benzoic acid (24).
Synthesized using the procedure for 1 except using 30r as the starting material, which afforded the title compound as a white solid after HPLC purification. HPLC (tR = 22.47 min), purity >95%. 1H NMR (400 MHz, Acetone-d6) δ 8.06 (d, J = 2.0 Hz, 1H), 7.64 (d, J = 8.4 Hz, 1H), 7.59 (dd, J = 8.8, 2.4 Hz, 1H), 7.45 (s, 4H), 7.30–7.24 (m, 4H), 7.22–7.17 (m, 1H), 4.31 (s, 2H), 3.60 (s br, 4H), 3.29 (s br, 4H), 3.24–3.19 (m, 2H), 2.94–2.90 (m, 2H), 1.29 (s, 9H); 13C NMR (125 MHz, Acetone-d6) δ 170.5, 153.4, 141.1, 140.1, 136.3, 133.4, 131.8, 131.4, 129.5, 129.3, 127.7, 127.2, 126.7, 120.5, 118.2, 60.2, 51.3, 44.5, 36.2, 36.1, 35.3, 31.5; HRMS (ESI) calcd for C30H38N3O4S2 [M+H+] 568.2304, found 568.2295.
2-((4-(4-(Tert-butyl)phenyl)piperazine)-1-sulfonamido)-5-((3-hydroxyphenethyl)thio) benzoic acid (25).
This compound was synthesized from the intermediate for 26 using commonly employed procedure for boron tribromide (BBr3). The reaction was carried out with 5 equivalents of boron tribromide (−78 °C-rt, overnight) which afforded compound 25. 1H NMR (500 MHz, acetone) δ 8.10 (d, J = 2.0 Hz, 1H), 7.74 (d, J = 8.8 Hz, 1H), 7.66 (dd, J = 8.8, 2.2 Hz, 1H), 7.25 (d, J = 8.8 Hz, 2H), 7.07 (t, J = 7.8 Hz, 1H), 6.87 (d, J = 8.8 Hz, 2H), 6.74–6.67(m, 3H), 3.40 – 3.37 (m, 4H), 3.21–3.17 (m, 2H), 3.15–3.13 (m, 4H), 2.85–2.80 (m, 2H), 1.25 (s, 9H); HRMS (ESI) calcd for C29H36N3O5S2 [M+H+] 570.2096, found 570.2094.
2-((4-(4-(Tert-butyl)phenyl)piperazine)-1-sulfonamido)-5-((3-methoxyphenethyl)thio) benzoic acid (26).
Synthesized using the representative procedure as for 1. 1H NMR (500 MHz, Acetone-d6) δ 8.09 (d, J = 2.2 Hz, 1H), 7.73 (d, J = 8.6 Hz, 1H), 7.67 (dd, J = 8.8, 2.2 Hz, 1H), 7.25 (d, J = 8.8 Hz, 2H), 7.17 (t, J = 7.8 Hz, 1H), 6.86 (d, J = 8.8 Hz, 2H), 6.83 (s, 1H), 6.80 (d, J = 7.6 Hz, 1H), 6.76 (dd, J = 8.2, 2.2 Hz, 1H), 3.76 (s, 3H), 3.41 – 3.35 (m, 4H), 3.24 – 3.20 (m, 2H), 3.16 – 3.11 (m, 4H), 2.90 – 2.86 (m, 2H), 1.25 (s, 9H); HRMS (ESI) calcd for 30H38N3O5S2 [M+H+] 584.2253, found 584.2251.
Methyl 2-((4-(4-(tert-butyl)phenyl)piperazine)-1-sulfonamido)-5-((3-oxo-3-(phenylamino)propyl)thio)benzoate (30s).
Synthesized using the procedure for 30n. 1H NMR (400 MHz, CDCl3) δ 10.45 (s, 1H), 8.11 (d, J = 2.0 Hz, 1H), 7.73 (d, J = 8.8 Hz, 1H), 7.71 (dd, J = 8.8, 2.0 Hz, 1H), 7.63 (d, J = 7.8 Hz, 2H), 7.27 (t, J = 8.0 Hz, 4H), 7.03 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.8 Hz, 2H), 3.89 (s, 3H), 3.42 – 3.37 (m, 4H), 3.28 (t, J = 7.2 Hz, 2H), 3.19 – 3.14 (m, 4H), 2.70 (t, J = 7.2 Hz, 2H), 1.25 (s, 9H).
2-((4-(4-(Tert-butyl)phenyl)piperazine)-1-sulfonamido)-5-((3-oxo-3-(phenylamino)propyl)thio) benzoic acid (27).
Synthesized by hydrolyzing intermediate 30s. 1H NMR (500 MHz, Acetone-d6) δ 8.11 (d, J = 2.0 Hz, 1H), 7.74 (d, J = 8.8 Hz, 1H), 7.71 (dd, J = 8.8, 2.0 Hz, 1H), 7.63 (d, J = 7.8 Hz, 2H), 7.28–7.26 (m, 4H), 7.03 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.8 Hz, 2H), 3.42 – 3.37 (m, 4H), 3.28 (t, J = 7.0 Hz, 2H), 3.19 – 3.14 (m, 4H), 2.71–2.69 (m, 2H), 1.25 (s, 9H). 13C NMR (125 MHz, Acetone- d6) δ 169.1, 168.8, 148.4, 142.9, 140.2, 139.2, 136.6, 133.2, 129.3, 128.6, 125.7, 123.2, 119.3, 119.0, 116.5, 115.6, 49.1, 46.2, 36.4, 33.6, 30.8, 29.7.
2. Expression and purification of anti-apoptotic proteins
DNA sequences containing human Mcl-1 (residues 171–323 and residues 171–320), human Bfl-1 (residues 1–151), the isoform 2 construct of the human Bcl-2 [Bcl2–2 construct for protein production starts with the Bcl2 sequence of 1–34 aa, followed by the BclxL sequence of 35–50 aa, and ends with the Bcl2 sequence of 92–202aa.], and Bcl-xL (human Bcl-xL protein, which has an internal deletion of the 45–85 amino acid residues and a C-terminal truncation for the amino acid residues 212–233) were cloned into an N-terminal His6-TEV vector. All constructs were transformed into Rosetta2 DE3 cells. Cultures were grown in Terrific Broth at 37 °C, induced with 0.4 mM IPTG and expressed overnight at 20 °C.
Frozen cell pellets of Mcl-1 were sonicated in 20 mM HEPES pH 7.0, 200 mM NaCl, 0.1% βME with Leupeptin/Aprotinin mixture and pelleted at 17,000 rpm for 45 min. The soluble fraction was applied to a Ni-NTA resin (Qiagen) for 1 h at 4 °C, the matrix washed with 20 mM HEPES pH 7.0 and 200 mM NaCl and the protein eluted with 20 mM HEPES pH 7.0, 200 mM NaCl and 500 mM imidazole. The protein was subsequently applied to a Superdex 75 column (GE Healthcare) pre-equilibrated with 20 mM HEPES pH 7.0 and 150 mM NaCl. For crystallography, the tag was removed prior to gel filtration by dialysis against 20 mM HEPES pH 7.0, 150 mM NaCl and 0.1% βME overnight in the presence of TEV protease.
Bfl-1 cells were lysed with sonication in 50 mM Tris pH 7.5, 200 mM NaCl. The lysate was cleared and loaded onto a Ni-NTA column. The column was washed with lysis buffer containing 10 mM imidazole, then protein eluted in lysis buffer containing 500 mM imidazole. Glycerol was added to the eluate to a final concentration 10%. The tag was cleaved in the presence of TEV protease during an overnight dialysis against 50 mM Tris 7.5, 150 mM NaCl, 0.1% βME and 10% glycerol. The protein was diluted with 20 mM Tris, pH 7.5, 0.1% βME and 10% glycerol then loaded onto a Source Q (GE Healthcare). The protein that eluted in the flow through was concentrated and applied to a Superdex 75 (GE Healthcare) preequilibrated with 25 mM HEPES pH 7.5, 300 mM NaCl, 1 mM DTT and 10% glycerol. Protein was concentrated to 1 mg/mL and stored at −80 °C.
To purify Bcl-2 and Bcl-xL proteins, cells were lysed in 25 mM Tris pH 8.5, 200 mM NaCl (Bcl-2), and 50 mM Tris pH 7.5, 200 mM NaCl (Bcl-xL) and applied to a Ni-NTA resin (QIAGEN) pre-equilibrated with lysis buffer containing 10 mM imidazole, then eluted with lysis buffer containing 500 mM imidazole. The His-tag was removed during dialysis overnight against buffer (25 mM Tris pH 8.5, 150 mM NaCl and 0.1% βME [Bcl-2] and 50 mM Tris 7.5, 150 mM NaCl, 0.1% βME [Bcl-xL]) containing TEV protease. Each protein was loaded onto Source 15Q column (GE Healthcare) and eluted with a 0 – 1 M NaCl gradient. Concentrated protein was further purified on a gel filtration column (Superdex 75, Amersham Biosciences) pre-equilibrated with 25 mM Tris pH 8.5, 150 mM NaCl, 0.1% βME (Bcl-2) or 20 mM Tris pH 7.5, 150 mM NaCl, 0.1% βME (Bcl-xL). Mcl-1, Bfl-1, Bcl-2, and Bcl-xL were used in fluorescent polarization binding assay. Protein used for HSQC-NMR studies (His-tagged Mcl-1 and His-Bfl-1) were prepared and purified the same way with the exception that the bacteria were grown on M9 minimal media supported with 3 g/L of 13C-glucose and/or 1 g/L of (15NH4)2SO4.
3. Heteronuclear single quantum correlation (HSQC) NMR spectroscopy
The Mcl-1 HSQC-NMR studies were performed as previously reported.17,19 HSQC NMR experiments with Bfl-1 proceeded as follows, NMR spectra were acquired in NMR Buffer (20 mM NaH2PO4/Na2HPO4 pH 7.0, 150 mM NaCl, 0.5 mM DTT, and 0.5 mM TECP) supplemented with 10% D2O and 4% DMSO using 5mm D2O matched Shigemi NMR tubes. 15N labeled Bfl1-His (50 μM) was incubated with compounds 1 and 23. All NMR data were collected on a Bruker Avance NEO 600 MHz spectrometer at 298K with a TCl-H&F/C/N cryoprobe. A transverse relaxation optimized spectroscopy (TROSY) with a solvent suppression pulse sequence was used to acquire all HSQC data. NMRPipe66 and Sparky (Goddard and Kneller, Sparky 3, University of California, San Francisco) were used for all NMR data processing and analysis. Assignments of Bfl-1 residues were obtained from the printed HSQC as it was not deposited online by previous studies.67
4. Fluorescence polarization (FP) binding assays
The FP binding assay was optimized for each individual anti-apoptotic protein. The following two fluorescent labeled Bid BH3-only peptides were used as competitive probes: a) Flu-Bid, a fluorescein tagged Bid peptide and b) FAM-Bid, a 5-FAM (Abgent, #SP2121) labeled Bid peptide (Supporting Information, Table S2). The buffer for all experiments was composed of 20 mM phosphate (pH 7.4), 50 mM NaCl, 1 mM EDTA, 0.05% Pluronic F68 with a final DMSO concentration of 4%. All experiments were performed in black 96-well plates (Corning #3792) and analyzed using a Synergy H1 Hybrid BioTek plate reader at an excitation wavelength of 485 nm and an emission wavelength of 530 nm.
Dissociation constant (Kd) values were determined by protein saturation experiments where fixed concentrations of the fluorescent probes were mixed with increasing concentrations of the protein and polarization was measured to monitor saturation. Kd values were calculated by fitting the sigmoidal dose-dependent FP increases as a function of protein concentrations using GraphPad Prism 7.0 software. Based on this optimization of peptide probe to protein binding, 2 nM Flu-Bid was used in Mcl-1 assays and 2nM FAM-Bid was used for Bcl-2 and Bcl-xL, while 1 nM FAM-Bid was used for Bfl-1assays. The following Kd values were determined: 3.24 ± 0.06 nM (Mcl-1), 0.424 ± 0.04 nM (Bfl-1), 18.44 ± 1.40 nM (Bcl-2), and 20.04 ± 2.34 nM (Bcl-xL). Based on saturation curves (Supporting Information, Figure S8), the following protein concentrations were used in all FP assays: 10 nM Mcl-1, 2 nM Bfl-1, 60 nM Bcl-2, and 80 nM Bcl-xL.
Competitive binding assays were performed in a final volume of 125 uL and were incubated for three hours at room temperature before analyzing. IC50 values were determined by nonlinear regression fitting of the competition curves (GraphPad Prism 7.0 Software) and converted into Ki values as previously described.68 Positive controls consisting of BH3-only peptide binding to the anti-apoptotic proteins were used to validate the specificity of the assay (Supporting Information, Figure S8). Representative binding inhibition curves corresponding to Mcl-1 and Bfl-1 binding data of the tested inhibitors (Tables 1–3) are provided in Supporting Information, Figure S9. Raw data of IC50 and Ki value replicates used to calculate averages presented in Tables 1–3 are provided in Supporting Information, Table S3.
5. Crystallography of Mcl-1 and compounds
For the Mcl-1:20 complex, prior to crystallization, protein (residues 171–320) was concentrated to 10.66 mg/mL in 20 mM HEPES pH 7.0, 50 mM NaCl and incubated in a 1:1.5 molar ratio with compound 20 at 4 °C for 48 hours. The final DMSO concentration in the protein-ligand sample was 6%. The complex was crystallized in a sitting drop vapor diffusion experiment against well solution of 30% PEG 3350, 0.2 mM MgCl2 and 0.1 M ADA pH 6.5. The drops consisted of 1.5 uL protein-complex, 0.3 uL of 30% 6-aminohexanoic acid and 1.2 uL of well solution. Crystals grew within 1 day at 20 °C and were cryoprotected in 30% PEG 3350 prior to data collection.
To crystallize Mcl-1:15, Mcl-1:27 and Mcl-1:22 complexes, protein (residues 171–323) was concentrated to 10 mg/mL and incubated with compound 1:1.5 ratio for 24 hours at 4 °C. The complex of Mcl-1:24 was prepared by incubating Mcl-1 at 0.5 mg/mL with compound at 1:4 molar ratio in the presence of 5% DMSO for 12 hours and concentrated to 10 mg/mL. Mcl-1:15 and Mcl-1:27 grew crystals from drops containing equal volumes of protein and well solution setup against 23 % PEG 3350, 0.1 M Tris pH 8.0 and 20% (w/v) PEG-1000, 0.1 M Tris pH 7.0, respectively. Mcl-1:22 and Mcl-1:24 grew crystals from drops containing equal volumes of protein and well solution setup against 18% PEG 3350, 200 mM NH4 Acetate, 100 mM Bis-Tris pH 6.5. All crystals were cryoprotected in well solution containing 25% ethylene glycol.
Diffraction data were collected on LS-CAT 21-ID-D (17) and 21-ID-G (15, 27, 22, and 24) beamlines at the Advanced Photon Source at Argonne National Laboratory and processed with HKL2000.69 The structures were solved via molecular replacement in Molrep70 using an in-house Mcl-1:Bim structure missing helix 237–257 as the search model. Iterative rounds of electron density fitting and refinement were completed using Coot55 and Buster71 respectively. The coordinates and geometric restraints for each inhibitor were created from smiles using Grade71 with the qm+mogul option. The coordinates were validated with Molprobity.72 Data collection and refinement statistics are listed in Supporting Information, Table S4. Difference electron density maps (Supporting Information, Figure S10) showed one inhibitor bound in the BH3 binding pocket of each protein chain of 15, 20, 22, and 24. Several additional inhibitor compounds acting as additives enabling crystal packing were also seen in 15, 20, and 24 structures. In the case of 27, the compound bound differently to the two protein chains with multiple chains in each binding site. In the Mcl-1:20 structure, residues 171 and 197–202 are missing from the single protein chain in the asymmetric unit. In the Mcl-1:15 crystal, residues 171, 197–200 and 322–323 are missing from chain A; residues 171, 193–201 and 322–323 are missing in chain B; residues 194–202, 238–240 and 321–323 are missing in chain C; and residues 171, 198–201 and 321–323 are missing from chain D. In the Mcl-1:27 crystal, residues 172–321 and residues 171–321, plus one residue of the N-terminal tag were fit into density for the A and B chains, respectively. For Mcl-1:22, residues 172–320 are present in protein chains A, B and D; chain C contains residues 171–321 plus one N-terminal residue of the tag. For Mcl-1:24, residues A (171–321), B (171–321) plus three residues of N-terminal tag, C (172–322) are present.
6. Bio-layer interferometry (BLI)
Mcl-1 and Bfl-1 proteins were biotinylated using the Thermo EZ-link Sulpho-NHS-LC-biotin biotinylation kit (CAT# 21435). Protein and biotin were mixed in a 1:1 molar ratio in PBS on ice for 2 hours, followed by dialysis in PBS buffer. BLI assays were performed on a ForteBio Octet Red96 instrument. Biotinylated anti-apoptotic proteins (10 μg/mL) were immobilized and saturated on Super Streptavidin Dip and Read Biosensors (ForteBio, #18–5065). All experiments were performed at 30 °C in the same buffer used for FP binding assays and were conducted in 96-well microplates (Greiner bio-one, #655209). Buffer was used for custom, baseline, and dissociation steps, while buffer containing serially diluted compounds with 5% DMSO was used for association. Association and dissociation cycles were fixed at 10 minutes each. Kinetic data was collected and processed with the Data Analysis software provided by ForteBio. All experiments were analyzed with referencing by subtraction of the DMSO and buffer-only wells. Plotting the response nm values of the binding sensorgrams with their respective compound concentration allowed for steady state analysis and was used to calculate Kd values.
7. Pulldown assay
Cell lysate from the M14 melanoma and 2LMP breast cancer cell lines was obtained from harvesting cells and lysing via sonication in CHAPS buffer [10 mM HEPES (pH 7.4), 2.5 mM EDTA, 150 mM NaCl, 1% (w/v) CHAPS] with protease inhibitor. Cell lysate (2 mg/mL for M14 and 1 mg/L for 2LMP) was pre-cleared with streptavidin-agarose beads and incubated on a tube rotator overnight at 4 °C with BL-Bim (0.2 μM) or BL-Noxa (0.1 μM) and either DMSO, free Bim peptide, or small molecule inhibitors; sequences of biotinylated peptides provided in Supporting Information, Table S2. Protein-peptide complexes were pulled down with streptavidin-agarose beads for 2 hrs at 4 °C. Beads were washed three times with CHAPS buffer and protein was eluted by boiling in 5X SDS loading dye. Samples were then run on a 4–20% Tris-Glycine gel and analyzed by western blot with Mcl-1 (Thermo Fisher # AHO0102), Bfl-1 (Cell Signaling #14093), Bcl-xL (Cell Signaling #2764), and Bcl-2 (Cell Signaling #15071) antibodies. The ThermoFisher iBright FL1000 imager was used to expose and quantify the western blot images. All uncropped western blot images are provided in Supporting Information, Figure S11.
8. Eμ-Myc lymphoma cell lines
The retrovirally transduced lymphoma cells isolated from Eμ-Myc transgenic mice were gifts from Ricky W. Johnstone at the University of Melbourne, Melbourne, Australia and were cultured as previously described.58 The Mcl-1, Bfl-1, Bcl-2, and Bcl-xL Eμ-Myc cells were seeded in 24-well plates at 0.5×106 cells/well. Cells were treated with compounds for 24 hrs then stained with violet LIVE/DEAD Fixable Dead Cell Stain Kit (Invitrogen, #L34964), according to manufacturers’ protocol. The percentage of fluorescent positive cells was determined by flow cytometry and calculated using WinList 3.0; statistical analysis was performed using a two-tailed T-test, comparing to the corresponding DMSO controls
9. Molecular docking
Molecular docking calculations were performed using GOLD molecular docking tool.73 The binding site was defined as a 8 Å cavity around the corresponding ligand in the Mcl-1 binding site. Hydrogen atoms were added to the Mcl-1 protein using GOLD, with default settings of the amino acid protonation pattern. Side chains of amino acids were treated as rigid entities. Compounds were docked 10 times into the defined binding site by applying the following parameters of the GOLD genetic algorithm (GA) (population size = 100, selection pressure = 1.1, no. of operations = 100000, no. of islands = 5, niche size = 2, migrate = 10, mutate = 95, crossover = 95) and different scoring functions available in GOLD. The best agreement between the crystallized and docked pose was obtained using the ChemPLP scoring function present in GOLD. The docking software tool GOLD was validated and for this purpose, the literature-reported bound conformation of a ligand in the Mcl-1 active site (PDB ID: 4HW2) was extracted from its active site and subsequently docked (Supporting Information, Figure S12). The docking calculation reproduced the experimental binding mode with the RMSD of 0.4 Å. We additionally reproduced the experimental binding mode of our structure 22 using its corresponding co-crystal Mcl-1 (PDB ID: 6U65) structure with comparable precision (Supporting Information, Figure S12). These settings were also used in the molecular docking calculations of the selected compounds from our synthesized series.
For the molecular docking of compounds 1, 23, and 24 into the Bfl-1 protein PDB structure 3MQP was used. The preformed alignment with Mcl-1:24 co-crystal structure yielded the initial Bfl-1: 24 complex and subsequent complex minimization are described in the Results and Discussion section. Protein preparation, binding site definition and GOLD docking settings were taking analogously to the previous Mcl-1 docking calculations. Docking calculations were visualized using LigandScout software.74
Supplementary Material
Figure S1. Evaluation of compound solubility and associated methods
Figure S2. BLI steady state binding curves
Figure S3. Bfl-1 protein alignments and minimization
Figure S4. Biological evaluation of compound 23
Figure S5. Pulldown assay using BL-Noxa
Figure S6. Eμ-Myc cell based assay positive controls
Figure S7. Eμ-Myc Lymphoma cell data for compounds 15, 20, and 22
Figure S8. FP assay development and positive controls
Figure S9. Mcl-1 and Bfl-1 FP binding curves
Figure S10. Omit electron density maps of co-crystal structures
Figure S11. Uncropped western blots
Figure S12. Molecular docking validation
Table S1. Binding data of esterified compounds
Table S2. Sequences of BH3-only protein chemical tools
Table S3. Replicate IC50 and Ki data from Mcl-1 and Bfl-1 FP binding assays
Table S4. Crystallography data collection and refinement statistics
Acknowledgments
This work was supported by the NIH R01 CA-149442, CA-217141, and HL-13982401, R21 NS056915 grants, Harrington Discovery Award, AACR Bayer Innovation and Discovery Award, and the MTRAC Fast Forward Medical Innovation Award to Z.N.-C.; NIH Training Program in Translational Research (T32-GM113900) and Rackham Merit Fellowship to K.J.K.; Advanced Proteome Informatics of Cancer (T32 CA140044) to A.S.A.M.; the Bilateral research grant (BI/US/18–20-068) funded by the Slovenian Research Agency between National Institute of Chemistry, Slovenia and University of Michigan, as well as the P1–0012 Core research Grant from Slovenian Research Agency funded the research visit of A.P. to the University of Michigan in the lab of Z.N.-C.. Z.N.-C. and J.A.S. are grateful for the support of this work from NIH P30 CA046592. We kindly thank W. Clay Brown from the U-M Center for Structural Biology for providing expression vectors. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02–06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817).
Abbreviations Used
- Mcl-1
myeloid cell leukemia 1
- Bfl-1
BCL2 related protein A1
- Bcl-2
B-cell lymphoma 2
- Bcl-xL
B-cell lymphoma-extra large
- BH3
Bcl-2 homology domain 3
- Bid
BH3 interacting-domain death agonist
- Bim
Bcl-2 interacting mediator of cell death
- FP
fluorescence polarization
- BLI
bio-layer interferometry
- PPI
protein-protein interaction
Footnotes
Supporting Information
Additional data figures and tables regarding biochemical, biophysical, crystallographic, computational, and biological evaluation of compounds (Figures S1–S12 and Tables S1–S4).
Molecular Formula Strings of dual Mcl-1/Bfl-1 inhibitors
PDB ID Codes
The following co-crystal structures have been deposited in the PDB: 6U63 (Mcl-1:15); 6U64 (Mcl-1:20); 6U65 (Mcl-1:22); 6U6F (Mcl-1:24); 6U67 (Mcl-1:27). Authors will release the atomic coordinates and experimental data upon article publication.
References
- (1).Hanahan D; Weinberg RA Hallmarks of Cancer: The next Generation. Cell 2011, 144 (5), 646–674. [DOI] [PubMed] [Google Scholar]
- (2).Yip KW; Reed JC Bcl-2 Family Proteins and Cancer. Oncogene 2008, 27 (50), 6398–6406. [DOI] [PubMed] [Google Scholar]
- (3).Czabotar PE; Lessene G; Strasser A; Adams JM Control of Apoptosis by the BCL-2 Protein Family: Implications for Physiology and Therapy. Nat. Rev. Mol. Cell Biol. 2014, 15 (1), 49–63. [DOI] [PubMed] [Google Scholar]
- (4).Opferman JT Attacking Cancer’s Achilles Heel: Antagonism of Anti-Apoptotic BCL-2 Family Members. FEBS J. 2016, 283 (14), 2661–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chen L; Willis SN; Wei A; Smith BJ; Fletcher JI; Hinds MG; Colman PM; Day CL; Adams JM; Huang DCS Differential Targeting of Prosurvival Bcl-2 Proteins by Their BH3-Only Ligands Allows Complementary Apoptotic Function. Mol. Cell 2005, 17 (3), 393–403. [DOI] [PubMed] [Google Scholar]
- (6).Delbridge ARD; Strasser A. The BCL-2 Protein Family, BH3-Mimetics and Cancer Therapy. Cell Death Differ. 2015, 22 (7), 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kitada S; Leone M; Sareth S; Zhai D; Reed JC; Pellecchia M. Discovery, Characterization, and Structure−Activity Relationships Studies of Proapoptotic Polyphenols Targeting B-Cell Lymphocyte/Leukemia-2 Proteins. J. Med. Chem. 2003, 46 (20), 4259–4264. [DOI] [PubMed] [Google Scholar]
- (8).Wang G; Nikolovska-Coleska Z; Yang C-Y; Wang R; Tang G; Guo J; Shangary S; Qiu S; Gao W; Yang D; Meagher J; Stuckey J; Krajewski K; Jiang S; Roller PP; Abaan HO; Tomita Y; Wang S. Structure-Based Design of Potent Small-Molecule Inhibitors of Anti-Apoptotic Bcl-2 Proteins. J. Med. Chem. 2006, 49 (21), 6139–6142. [DOI] [PubMed] [Google Scholar]
- (9).Tse C; Shoemaker AR; Adickes J; Anderson MG; Chen J; Jin S; Johnson EF; Marsh KC; Mitten MJ; Nimmer P; Roberts L; Tahir SK; Xiao Y; Yang X; Zhang H; Fesik S; Rosenberg SH; Elmore SW ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res 2008, 68 (9), 3421–3428. [DOI] [PubMed] [Google Scholar]
- (10).Oltersdorf T; Elmore SW; Shoemaker AR; Armstrong RC; Augeri DJ; Belli BA; Bruncko M; Deckwerth TL; Dinges J; Hajduk PJ; Joseph MK; Kitada S; Korsmeyer SJ; Kunzer AR; Letai A; Li C; Mitten MJ; Nettesheim DG; Ng S; Nimmer PM; O’Connor JM; Oleksijew A; Petros AM; Reed JC; Shen W; Tahir SK; Thompson CB; Tomaselli KJ; Wang B; Wendt MD; Zhang H; Fesik SW; Rosenberg SH. An Inhibitor of Bcl-2 Family Proteins Induces Regression of Solid Tumours. Nature 2005, 435 (7042), 677–681. [DOI] [PubMed] [Google Scholar]
- (11).Sarosiek KA; Letai A. Directly Targeting the Mitochondrial Pathway of Apoptosis for Cancer Therapy Using BH3 Mimetics - Recent Successes, Current Challenges and Future Promise. FEBS J. 2016, 283 (19), 3523–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Tao Z-F; Hasvold L; Wang L; Wang X; Petros AM; Park CH; Boghaert ER; Catron ND; Chen J; Colman PM; Czabotar PE; Deshayes K; Fairbrother WJ; Flygare JA; Hymowitz SG; Jin S; Judge RA; Koehler MFT; Kovar PJ; Lessene G; Mitten MJ; Ndubaku CO; Nimmer P; Purkey HE; Oleksijew A; Phillips DC; Sleebs BE; Smith BJ; Smith ML; Tahir SK; Watson KG; Xiao Y; Xue J; Zhang H; Zobel K; Rosenberg SH; Tse C; Leverson JD; Elmore SW; Souers AJ Discovery of a Potent and Selective BCL-X L Inhibitor with in Vivo Activity. ACS Med. Chem. Lett 2014, 5 (10), 1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lessene G; Czabotar PE; Sleebs BE; Zobel K; Lowes KN; Adams JM; Baell JB; Colman PM; Deshayes K; Fairbrother WJ; Flygare JA; Gibbons P; Kersten WJA; Kulasegaram S; Moss RM; Parisot JP; Smith BJ; Street IP; Yang H; Huang DCS; Watson KG Structure-Guided Design of a Selective BCL-XL Inhibitor. Nat. Chem. Biol. 2013, 9 (6), 390–397. [DOI] [PubMed] [Google Scholar]
- (14).Souers AJ; Leverson JD; Boghaert ER; Ackler SL; Catron ND; Chen J; Dayton BD; Ding H; Enschede SH; Fairbrother WJ; Huang DCS; Hymowitz SG; Jin S; Khaw SL; Kovar PJ; Lam LT; Lee J; Maecker HL; Marsh KC; Mason KD; Mitten MJ; Nimmer PM; Oleksijew A; Park CH; Park C-M; Phillips DC; Roberts AW; Sampath D; Seymour JF; Smith ML; Sullivan GM; Tahir SK; Tse C; Wendt MD; Xiao Y; Xue JC; Zhang H; Humerickhouse RA; Rosenberg SH; Elmore SW ABT-199, a Potent and Selective BCL-2 Inhibitor, Achieves Antitumor Activity While Sparing Platelets. Nat. Med. 2013, 19 (2), 202–208. [DOI] [PubMed] [Google Scholar]
- (15).Roberts AW; Davids MS; Pagel JM; Kahl BS; Puvvada SD; Gerecitano JF; Kipps TJ; Anderson MA; Brown JR; Gressick L; Wong S; Dunbar M; Zhu M; Desai MB; Cerri E; Heitner Enschede S; Humerickhouse RA; Wierda WG; Seymour JF Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374 (4), 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Quinn BA; Dash R; Azab B; Sarkar S; Das SK; Kumar S; Oyesanya RA; Dasgupta S; Dent P; Grant S; Rahmani M; Curiel DT; Dmitriev I; Hedvat M; Wei J; Wu B; Stebbins JL; Reed JC; Pellecchia M; Sarkar D; Fisher PB Targeting Mcl-1 for the Therapy of Cancer. Expert Opin. Investig. Drugs 2011, 20 (10), 1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Abulwerdi FA; Liao C; Mady AS; Gavin J; Shen C; Cierpicki T; Stuckey JA; Showalter HDH; Nikolovska-Coleska Z. 3-Substituted-N-(4-Hydroxynaphthalen-1-Yl)Arylsulfonamides as a Novel Class of Selective Mcl-1 Inhibitors: Structure-Based Design, Synthesis, SAR, and Biological Evaluation. J. Med. Chem. 2014, 57 (10), 4111–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Abulwerdi F; Liao C; Liu M; Azmi AS; Aboukameel A; Mady ASA; Gulappa T; Cierpicki T; Owens S; Zhang T; Sun D; Stuckey JA; Mohammad RM; Nikolovska-Coleska Z. A Novel Small-Molecule Inhibitor of Mcl-1 Blocks Pancreatic Cancer Growth in Vitro and in Vivo. Mol. Cancer Ther. 2014, 13 (3), 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mady ASA; Liao C; Bajwa N; Kump KJ; Abulwerdi FA; Lev KL; Miao L; Grigsby SM; Perdih A; Stuckey JA; Du Y; Fu H; Nikolovska-Coleska Z. Discovery of Mcl-1 Inhibitors from Integrated High Throughput and Virtual Screening. Sci. Rep. 2018, 8 (1), 10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Burke JP; Bian Z; Shaw S; Zhao B; Goodwin CM; Belmar J; Browning CF; Vigil D; Friberg A; Camper DV; Rossanese OW; Lee T; Olejniczak ET; Fesik SW Discovery of Tricyclic Indoles That Potently Inhibit Mcl-1 Using Fragment-Based Methods and Structure-Based Design. J. Med. Chem. 2015, 58 (9), 3794–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Pelz NF; Bian Z; Zhao B; Shaw S; Tarr JC; Belmar J; Gregg C; Camper DV; Goodwin CM; Arnold AL; Sensintaffar JL; Friberg A; Rossanese OW; Lee T; Olejniczak ET; Fesik SW Discovery of 2-Indole-Acylsulfonamide Myeloid Cell Leukemia 1 (Mcl-1) Inhibitors Using Fragment-Based Methods. J. Med. Chem. 2016, 59 (5), 2054–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lee T; Bian Z; Zhao B; Hogdal LJ; Sensintaffar JL; Goodwin CM; Belmar J; Shaw S; Tarr JC; Veerasamy N; Matulis SM; Koss B; Fischer MA; Arnold AL; Camper DV; Browning CF; Rossanese OW; Budhraja A; Opferman J; Boise LH; Savona MR; Letai A; Olejniczak ET; Fesik SW Discovery and Biological Characterization of Potent Myeloid Cell Leukemia-1 Inhibitors. FEBS Lett. 2017, 591 (1), 240–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Friberg A; Vigil D; Zhao B; Daniels RN; Burke JP; Garcia-Barrantes PM; Camper D; Chauder BA; Lee T; Olejniczak ET; Fesik SW Discovery of Potent Myeloid Cell Leukemia 1 (Mcl-1) Inhibitors Using Fragment-Based Methods and Structure-Based Design. J. Med. Chem. 2013, 56 (1), 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chen L; Wilder PT; Drennen B; Tran J; Roth BM; Chesko K; Shapiro P; Fletcher S. Structure-Based Design of 3-Carboxy-Substituted 1,2,3,4-Tetrahydroquinolines as Inhibitors of Myeloid Cell Leukemia-1 (Mcl-1). Org. Biomol. Chem. 2016, 14 (24), 5505–5510. [DOI] [PubMed] [Google Scholar]
- (25).Lanning ME; Yu W; Yap JL; Chauhan J; Chen L; Whiting E; Pidugu LS; Atkinson T; Bailey H; Li W; Roth BM; Hynicka L; Chesko K; Toth EA; Shapiro P; MacKerell AD; Wilder PT; Fletcher S. Structure-Based Design of N-Substituted 1-Hydroxy-4-Sulfamoyl-2-Naphthoates as Selective Inhibitors of the Mcl-1 Oncoprotein. Eur. J. Med. Chem. 2016, 113, 273–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Doi K; Li R; Sung SS; Wu H; Liu Y; Manieri W; Krishnegowda G; Awwad A; Dewey A; Liu X; Amin S; Cheng C; Qin Y; Schonbrunn E; Daughdrill G; Loughran TP; Sebti S; Wang HG Discovery of Marinopyrrole A (Maritoclax) as a Selective Mcl-1 Antagonist That Overcomes ABT-737 Resistance by Binding to and Targeting Mcl-1 for Proteasomal Degradation. J. Biol. Chem. 2012, 287 (13), 10224–10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Petros AM; Swann SL; Song D; Swinger K; Park C; Zhang H; Wendt MD; Kunzer AR; Souers AJ; Sun C. Fragment-Based Discovery of Potent Inhibitors of the Anti-Apoptotic MCL-1 Protein. Bioorg. Med. Chem. Lett. 2014, 24 (6), 1484–1488. [DOI] [PubMed] [Google Scholar]
- (28).Kotschy A; Szlavik Z; Murray J; Davidson J; Maragno AL; Le Toumelin-Braizat G; Chanrion M; Kelly GL; Gong J-N; Moujalled DM; Bruno A; Csekei M; Paczal A; Szabo ZB; Sipos S; Radics G; Proszenyak A; Balint B; Ondi L; Blasko G; Robertson A; Surgenor A; Dokurno P; Chen I; Matassova N; Smith J; Pedder C; Graham C; Studeny A; Lysiak-Auvity G; Girard A-M; Gravé F; Segal D; Riffkin CD; Pomilio G; Galbraith LCA; Aubrey BJ; Brennan MS; Herold MJ; Chang C; Guasconi G; Cauquil N; Melchiore F; Guigal-Stephan N; Lockhart B; Colland F; Hickman JA; Roberts AW; Huang DCS; Wei AH; Strasser A; Lessene G; Geneste O. The MCL1 Inhibitor S63845 Is Tolerable and Effective in Diverse Cancer Models. Nature 2016, 538 (7626), 477–482. [DOI] [PubMed] [Google Scholar]
- (29).Caenepeel S; Brown SP; Belmontes B; Moody G; Keegan KS; Chui D; Whittington DA; Huang X; Poppe L; Cheng AC; Cardozo M; Houze J; Li Y; Lucas B; Paras NA; Wang X; Taygerly JP; Vimolratana M; Zancanella M; Zhu L; Cajulis E; Osgood T; Sun J; Damon L; Egan RK; Greninger P; McClanaghan JD; Gong J; Moujalled D; Pomilio G; Beltran P; Benes CH; Roberts AW; Huang DCS; Wei A; Canon J; Coxon A; Hughes PE AMG 176, a Selective MCL1 Inhibitor, Is Effective in Hematological Cancer Models Alone and in Combination with Established Therapies. Cancer Discov. 2018, 8 (12), CD-18–0387. [DOI] [PubMed] [Google Scholar]
- (30).Bruncko M; Wang L; Sheppard GS; Phillips DC; Tahir SK; Xue J; Erickson S; Fidanze S; Fry E; Hasvold L; Jenkins GJ; Jin S; Judge RA; Kovar PJ; Madar D; Nimmer P; Park C; Petros AM; Rosenberg SH; Smith ML; Song X; Sun C; Tao Z-F; Wang X; Xiao Y; Zhang H; Tse C; Leverson JD; Elmore SW; Souers AJ Structure-Guided Design of a Series of MCL-1 Inhibitors with High Affinity and Selectivity. J. Med. Chem. 2015, 58 (5), 2180–2194. [DOI] [PubMed] [Google Scholar]
- (31).Tron AE; Belmonte MA; Adam A; Aquila BM; Boise LH; Chiarparin E; Cidado J; Embrey KJ; Gangl E; Gibbons FD; Gregory GP; Hargreaves D; Hendricks JA; Johannes JW; Johnstone RW; Kazmirski SL; Kettle JG; Lamb ML; Matulis SM; Nooka AK; Packer MJ; Peng B; Rawlins PB; Robbins DW; Schuller AG; Su N; Yang W; Ye Q; Zheng X; Secrist JP; Clark EA; Wilson DM; Fawell SE; Hird AW Discovery of Mcl-1-Specific Inhibitor AZD5991 and Preclinical Activity in Multiple Myeloma and Acute Myeloid Leukemia. Nat. Commun. 2018, 9 (1), 5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Huhn AJ; Guerra RM; Harvey EP; Bird GH; Walensky LD Selective Covalent Targeting of Anti-Apoptotic BFL-1 by Cysteine-Reactive Stapled Peptide Inhibitors. Cell Chem. Biol. 2016, 23 (9), 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Harvey EP; Seo H-S; Guerra RM; Bird GH; Dhe-Paganon S; Walensky LD Crystal Structures of Anti-Apoptotic BFL-1 and Its Complex with a Covalent Stapled Peptide Inhibitor. Structure 2018, 26 (1), 153–160.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Guerra RM; Bird GH; Harvey EP; Dharia NV; Korshavn KJ; Prew MS; Stegmaier K; Walensky LD Precision Targeting of BFL-1/A1 and an ATM Co-Dependency in Human Cancer. Cell Rep. 2018, 24 (13), 3393–3403.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mathieu A-L; Sperandio O; Pottiez V; Balzarin S; Herlédan A; Elkaïm JO; Fogeron M-L; Piveteau C; Dassonneville S; Deprez B; Villoutreix BO; Bonnefoy N; Leroux F. Identification of Small Inhibitory Molecules Targeting the Bfl-1 Anti-Apoptotic Protein That Alleviates Resistance to ABT-737. J. Biomol. Screen. 2014, 19 (7), 1035–1046. [DOI] [PubMed] [Google Scholar]
- (36).de Araujo A. D; Lim J; Wu K-C; Xiang Y; Good AC; Skerlj R; Fairlie DP Bicyclic Helical Peptides as Dual Inhibitors Selective for Bcl2A1 and Mcl-1 Proteins. J. Med. Chem. 2018, 61 (7), 2962–2972. [DOI] [PubMed] [Google Scholar]
- (37).Vogler M. BCL2A1: The Underdog in the BCL2 Family. Cell Death Differ. 2012, 19 (1), 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hind CK; Carter MJ; Harris CL; Chan HTC; James S; Cragg MS Role of the Pro-Survival Molecule Bfl-1 in Melanoma. Int. J. Biochem. Cell Biol. 2015, 59, 94–102. [DOI] [PubMed] [Google Scholar]
- (39).Akgul C. Mcl-1 Is a Potential Therapeutic Target in Multiple Types of Cancer. Cell. Mol. Life Sci. 2009, 66 (8), 1326–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Simmons MJ; Fan G; Zong W-X; Degenhardt K; White E; Gélinas C. Bfl-1/A1 Functions, Similar to Mcl-1, as a Selective TBid and Bak Antagonist. Oncogene 2008, 27 (10), 1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Barile E; Marconi GD; De SK; Baggio C; Gambini L; Salem AF; Kashyap MK; Castro JE; Kipps TJ; Pellecchia M. HBfl-1/HNOXA Interaction Studies Provide New Insights on the Role of Bfl-1 in Cancer Cell Resistance and for the Design of Novel Anticancer Agents. ACS Chem. Biol. 2017, 12 (2), 444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Adams JM; Cory S. The Bcl-2 Apoptotic Switch in Cancer Development and Therapy. Oncogene 2007, 26 (9), 1324–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Lanave C; Santamaria M; Saccone C. Comparative Genomics: The Evolutionary History of the Bcl-2 Family. Gene 2004, 333, 71–79. [DOI] [PubMed] [Google Scholar]
- (44).Senft D; Berking C; Graf SA; Kammerbauer C; Ruzicka T; Besch R. Selective Induction of Cell Death in Melanoma Cell Lines through Targeting of Mcl-1 and A1. PLoS One 2012, 7 (1), e30821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Esteve-Arenys A; Valero JG; Chamorro-Jorganes A; Gonzalez D; Rodriguez V; Dlouhy I; Salaverria I; Campo E; Colomer D; Martinez A; Rymkiewicz G; Pérez-Galán P; Lopez-Guillermo A; Roué G. The BET Bromodomain Inhibitor CPI203 Overcomes Resistance to ABT-199 (Venetoclax) by Downregulation of BFL-1/A1 in in Vitro and in Vivo Models of MYC+/BCL2+ Double Hit Lymphoma. Oncogene 2018, 37 (14), 1830–1844. [DOI] [PubMed] [Google Scholar]
- (46).Yecies D; Carlson NE; Deng J; Letai A. Acquired Resistance to ABT-737 in Lymphoma Cells That up-Regulate MCL-1 and BFL-1. Blood 2010, 115 (16), 3304–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Haq R; Yokoyama S; Hawryluk EB; Jönsson GB; Frederick DT; McHenry K; Porter D; Tran T-N; Love KT; Langer R; Anderson DG; Garraway LA; Duncan LM; Morton DL; Hoon DSB; Wargo JA; Song JS; Fisher DE BCL2A1 Is a Lineage-Specific Antiapoptotic Melanoma Oncogene That Confers Resistance to BRAF Inhibition. Proc. Natl. Acad. Sci. 2013, 110 (11), 4321–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Fofaria NM; Frederick DT; Sullivan RJ; Flaherty KT; Srivastava SK Overexpression of Mcl-1 Confers Resistance to BRAFV600E Inhibitors Alone and in Combination with MEK1/2 Inhibitors in Melanoma. Oncotarget 2015, 6 (38), 40535–40556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Champa D; Russo MA; Liao X-H; Refetoff S; Ghossein RA; Di Cristofano A. Obatoclax Overcomes Resistance to Cell Death in Aggressive Thyroid Carcinomas by Countering Bcl2a1 and Mcl1 Overexpression. Endocr. Relat. Cancer 2014, 21 (5), 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Du Y; Nikolovska-Coleska Z; Qui M; Li L; Lewis I; Dingledine R; Stuckey JA; Krajewski K; Roller PP; Wang S; Fu H. A Dual-Readout F2 Assay That Combines Fluorescence Resonance Energy Transfer and Fluorescence Polarization for Monitoring Bimolecular Interactions. Assay Drug Dev. Technol. 2011, 9 (4), 382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Tron AE; Belmonte MA; Adam A; Aquila BM; Boise LH; Chiarparin E; Cidado J; Embrey KJ; Gangl E; Gibbons FD; Gregory GP; Hargreaves D; Hendricks JA; Johannes JW; Johnstone RW; Kazmirski SL; Kettle JG; Lamb ML; Matulis SM; Nooka AK; Packer MJ; Peng B; Rawlins PB; Robbins DW; Schuller AG; Su N; Yang W; Ye Q; Zheng X; Secrist JP; Clark EA; Wilson DM; Fawell SE; Hird AW Discovery of Mcl-1-Specific Inhibitor AZD5991 and Preclinical Activity in Multiple Myeloma and Acute Myeloid Leukemia. Nat. Commun. 2018, 9 (1), 5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Aldeghi M; Malhotra S; Selwood DL; Chan AWE Two- and Three-Dimensional Rings in Drugs. Chem. Biol. Drug Des. 2014, 83 (4), 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Meanwell NA Improving Drug Candidates by Design: A Focus on Physicochemical Properties As a Means of Improving Compound Disposition and Safety. Chem. Res. Toxicol. 2011, 24 (9), 1420–1456. [DOI] [PubMed] [Google Scholar]
- (54).Belmar J; Fesik SW Small Molecule Mcl-1 Inhibitors for the Treatment of Cancer. Pharmacol. Ther. 2015, 145, 76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Emsley P; Cowtan K. Coot : Model-Building Tools for Molecular Graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60 (12), 2126–2132. [DOI] [PubMed] [Google Scholar]
- (56).Herman MD; Nyman T; Welin M; Lehtiö L; Flodin S; Trésaugues L; Kotenyova T; Flores A; Nordlund P. Completing the Family Portrait of the Anti-Apoptotic Bcl-2 Proteins: Crystal Structure of Human Bfl-1 in Complex with Bim. FEBS Lett. 2008, 582 (25–26), 3590–3594. [DOI] [PubMed] [Google Scholar]
- (57).Lee EF; Harris TJ; Tran S; Evangelista M; Arulananda S; John T; Ramnac C; Hobbs C; Zhu H; Gunasingh G; Segal D; Behren A; Cebon J; Dobrovic A; Mariadason JM; Strasser A; Rohrbeck L; Haass NK; Herold MJ; Fairlie WD BCL-XL and MCL-1 Are the Key BCL-2 Family Proteins in Melanoma Cell Survival. Cell Death Dis. 2019, 10 (5), 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Whitecross KF; Alsop AE; Cluse LA; Wiegmans A; Banks K-M; Coomans C; Peart MJ; Newbold A; Lindemann RK; Johnstone RW Defining the Target Specificity of ABT-737 and Synergistic Antitumor Activities in Combination with Histone Deacetylase Inhibitors. Blood 2009, 113 (9), 1982–1991. [DOI] [PubMed] [Google Scholar]
- (59).Leverson JD; Zhang H; Chen J; Tahir SK; Phillips DC; Xue J; Nimmer P; Jin S; Smith M; Xiao Y; Kovar P; Tanaka A; Bruncko M; Sheppard GS; Wang L; Gierke S; Kategaya L; Anderson DJ; Wong C; Eastham-Anderson J; Ludlam MJC; Sampath D; Fairbrother WJ; Wertz I; Rosenberg SH; Tse C; Elmore SW; Souers AJ Potent and Selective Small-Molecule MCL-1 Inhibitors Demonstrate on-Target Cancer Cell Killing Activity as Single Agents and in Combination with ABT-263 (Navitoclax). Cell Death Dis. 2015, 6 (1), e1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Zhao B; Sensintaffar J; Bian Z; Belmar J; Lee T; Olejniczak ET; Fesik SW Structure of a Myeloid Cell Leukemia-1 (Mcl-1) Inhibitor Bound to Drug Site 3 of Human Serum Albumin. Bioorganic Med. Chem. 2017, 25 (12), 3087–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Itoh T; Mase T. A General Palladium-Catalyzed Coupling of Aryl Bromides/Triflates and Thiols. Org. Lett. 2004, 6 (24), 4587–4590. [DOI] [PubMed] [Google Scholar]
- (62).Beaudoin S; Kinsey KE; Burns JF Preparation of Unsymmetrical Sulfonylureas from N,N′-Sulfuryldiimidazoles. J. Org. Chem. 2003, 68 (1), 115–119. [DOI] [PubMed] [Google Scholar]
- (63).Wydysh EA; Medghalchi SM; Vadlamudi A; Townsend CA Design and Synthesis of Small Molecule Glycerol 3-Phosphate Acyltransferase Inhibitors. J. Med. Chem. 2009, 52 (10), 3317–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Comess KM; Erickson SA; Henkin J. Sulfonamides Having Antiangiogenic and Anticancer Activity, Patent US7491718., 2003.
- (65).Krolski ME; Renaldo AF; Rudisill DE; Stille JK Palladium-Catalyzed Coupling of 2-Bromoanilines with Vinylstannanes. A Regiocontrolled Synthesis of Substituted Indoles. J. Org. Chem. 1988, 53 (6), 1170–1176. [Google Scholar]
- (66).Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A. NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes. J. Biomol. NMR 1995, 6 (3), 277–293. [DOI] [PubMed] [Google Scholar]
- (67).Godoi PHC; Wilkie-Grantham RP; Hishiki A; Sano R; Matsuzawa Y; Yanagi H; Munte CE; Chen Y; Yao Y; Marassi FM; Kalbitzer HR; Matsuzawa SI; Reed JC Orphan Nuclear Receptor NR4A1 Binds a Novel Protein Interaction Site on Anti-Apoptotic B Cell Lymphoma Gene 2 Family Proteins. J. Biol. Chem. 2016, 291 (27), 14072–14084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Nikolovska-Coleska Z; Wang R; Fang X; Pan H; Tomita Y; Li P; Roller PP; Krajewski K; Saito NG; Stuckey JA; Wang S. Development and Optimization of a Binding Assay for the XIAP BIR3 Domain Using Fluorescence Polarization. Anal. Biochem. 2004, 332 (2), 261–273. [DOI] [PubMed] [Google Scholar]
- (69).Otwinowski Z; Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (70).Vagin A; Teplyakov A; IUCr. Molecular Replacement with MOLREP. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 (1), 22–25. [DOI] [PubMed] [Google Scholar]
- (71).Smart OS; Womack TO; Flensburg C; Keller P; Paciorek W; Sharff A; Vonrhein C; Bricogne G. Exploiting Structure Similarity in Refinement: Automated NCS and Target-Structure Restraints in BUSTER. Acta Crystallogr. D. Biol. Crystallogr. 2012, 68 (Pt 4), 368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Chen VB; Arendall WB; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity : All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 (1), 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Jones G; Willett P; Glen RC; Leach AR; Taylor R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267 (3), 727–748. [DOI] [PubMed] [Google Scholar]
- (74).Wolber G; Langer T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45 (1), 160–169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Evaluation of compound solubility and associated methods
Figure S2. BLI steady state binding curves
Figure S3. Bfl-1 protein alignments and minimization
Figure S4. Biological evaluation of compound 23
Figure S5. Pulldown assay using BL-Noxa
Figure S6. Eμ-Myc cell based assay positive controls
Figure S7. Eμ-Myc Lymphoma cell data for compounds 15, 20, and 22
Figure S8. FP assay development and positive controls
Figure S9. Mcl-1 and Bfl-1 FP binding curves
Figure S10. Omit electron density maps of co-crystal structures
Figure S11. Uncropped western blots
Figure S12. Molecular docking validation
Table S1. Binding data of esterified compounds
Table S2. Sequences of BH3-only protein chemical tools
Table S3. Replicate IC50 and Ki data from Mcl-1 and Bfl-1 FP binding assays
Table S4. Crystallography data collection and refinement statistics