

Abstract
The mineral composition of bovine milk plays an important role in determining its nutritional and cheese-making value. Concentrations of the main minerals predicted from mid-infrared spectra produced during milk recording, combined with cow genotypes, provide a unique opportunity to decipher the genetic determinism of these traits. The present study included 1 million test-day predictions of Ca, Mg, P, K, Na, and citrate content from 126,876 Montbéliarde cows, of which 19,586 had genotype data available. All investigated traits were highly heritable (0.50–0.58), with the exception of Na (0.32). A sequence-based genome-wide association study (GWAS) detected 50 QTL (18 affecting two to five traits) and positional candidate genes and variants, mostly located in non-coding sequences. In silico post-GWAS analyses highlighted 877 variants that could be regulatory SNPs altering transcription factor (TF) binding sites or located in non-coding RNA (mainly lncRNA). Furthermore, we found 47 positional candidate genes and 45 TFs highly expressed in mammary gland compared to 90 other bovine tissues. Among the mammary-specific genes, SLC37A1 and ANKH, encoding proteins involved in ion transport were located in the most significant QTL. This study therefore highlights a comprehensive set of functional candidate genes and variants that affect milk mineral content.
Subject terms: Animal breeding, Gene regulation, Genetic association study
Introduction
Bovine milk contains many essential nutrients, such as lactose (~ 48 g/L), fatty acids (~ 37 g/L), proteins (~ 34 g/L), and minerals (~ 9 g/L). Although less abundant than other solid components of milk, the major minerals—potassium (K), calcium (Ca), phosphorus (P), sodium (Na), and magnesium (Mg)—have an important effect on both human health and the cheese-making process. In humans, all of these minerals are necessary for many vital functions and therefore for the maintenance of good health. Dairy products can represent an important source of minerals in the human diet, especially of well-assimilated Ca1. In milk, minerals are found either in solution (soluble fraction) or in colloidal form (insoluble fraction). Some minerals are exclusively found in the soluble fraction (e.g., K and Na) while others exist in both fractions (e.g., Ca, P, and Mg). In the soluble fraction, Ca, P, and Mg exist in different forms, including ions and salts (phosphates and citrates), while in colloidal form, they are associated with casein molecules in the micelles and play a role in the structure and stability of these assemblages during the cheesemaking process2. Higher mineral concentrations are therefore associated with improved coagulation properties of milk3,4 and could enhance the nutritional value for human consumers.
Despite the potential benefits to human nutrition and milk processing, little is known about the genetic factors that influence milk mineral composition, mainly because the determination of mineral content via reference analyses is costly and time-consuming. A number of studies have reported genetic variation in milk mineral composition5–11, but to our knowledge, only two studies have conducted genome-wide association studies (GWAS) to investigate the genomic regions associated with these traits9,12. Both of those prior studies used 777 k SNPs and a relatively small sample of cows. As an alternative to reference analyses, mid-infrared (MIR) spectrometry can predict various milk components, including mineral fractions13–15, quickly and cheaply. Because of these advantages, milk MIR spectra are routinely recorded and stored. The combination of this technology with i) the widespread genotyping of cows for genomic selection, ii) the availability of whole-genome sequence (WGS) data from the 1000 Bull Genomes Project16, and iii) ever-increasing knowledge of the bovine genome17,18 creates the possibility of large-scale, high-resolution analyses for identification of the genes and variants that affect the mineral content of milk. We have previously applied this approach—whole-genome sequence-based GWAS combined with MIR predictions—to investigate milk protein composition19 and cheese-making traits20, and in both cases we succeeded in highlighting functional candidate genes. In particular, the genes SLC37A1 and ANKH were strongly linked with milk quality; both encode transmembrane proteins involved in ion transport and are therefore likely to have an effect on milk mineral composition. However, in these genes, as well as in other genes we identified, the variants with the most significant effects were mostly found in non-coding regions with limited annotation, which made it difficult to distinguish the causal variant. This pattern is quite general and many studies have reported the major role of regulatory variants in the architecture of complex traits21. To address this challenge, further investigation of non-coding regions is needed, particularly with respect to binding sites of transcription factors and non-coding RNA which could regulate gene expression.
The main objective of this study was to identify the best candidate genes and variants that might affect the content of Ca, P, Mg, K, Na, and citrate in milk, as predicted from MIR spectra. For this, we first conducted a GWAS on imputed WGS data of 19,586 Montbéliarde cows, and then performed post-GWAS analyses using different sources of annotation data to further refine our results.
Results
We analyzed six traits predicted from MIR spectra in Montbéliarde cows, representing the mineral (Ca, P, Mg, K, and Na) and citrate content of milk. MIR prediction equations originated from the Optimir project14,15. The accuracies of these MIR predictions, as assessed by the coefficient of determination (R2) in a validation population (Table 1), ranged from 0.68 to 0.90, with the exception of Na (0.44).
Table 1.
Mean, standard deviation (SD), and accuracy (R2), estimated by cross-validation, of mid-infrared (MIR) predictions for concentrations of minerals and citrate in milk from Montbéliarde cows.
| Trait | Abbrev | Mean | SD | R2 MIR prediction |
|---|---|---|---|---|
| Calcium, in mg kg−1 of milk | Ca | 1161.4 | 92.6 | 0.82 |
| Phosphorus, in mg kg−1 of milk | P | 1007.0 | 77.5 | 0.75 |
| Magnesium, in mg kg−1 of milk | Mg | 1473.8 | 104.5 | 0.77 |
| Potassium, in mg kg−1 of milk | K | 100.5 | 7.2 | 0.68 |
| Sodium, in mg kg−1 of milk | Na | 341.7 | 44.5 | 0.44 |
| Citrate, in g kg−1 of milk | Citrate | 8.27 | 1.49 | 0.90 |
Heritability and genetic correlation estimates for mineral and citrate content
Genetic parameters, i.e. heritabilities (h2) and genetic correlations (rg), were estimated for milk mineral and citrate content, as predicted from more than 1 million test-day records from 126,873 cows (Table 2). At the test-day level, heritability estimates were moderate for Na content (h2 = 0.32) but higher for other minerals (h2 = 0.50 to 0.56) and citrate (h2 = 0.48). Na was negatively and poorly correlated with other minerals (-0.23 ≤ rg ≤ − 0.02) and citrate (rg = − 0.15), while the levels of most other minerals were generally positively correlated (0.11 ≤ rg ≤ 0.60); the exception was the relationship between Ca and K (rg = − 0.22). Values of the genetic correlation between citrate and mineral levels in milk ranged quite broadly, depending on the mineral: null with K (rg = 0.01 with K), slightly negative with Na (rg = − 0.15) and P (rg = − 0.16), and highly positive with Ca (rg = 0.57) and Mg (rg = 0.59).
Table 2.
Estimates of heritability (in bold, diagonal) and genetic correlation (above the diagonal) for the concentrations of minerals and citrate in milk (SE < 0.01).
| Ca | P | Mg | K | Na | Citrate | |
|---|---|---|---|---|---|---|
| Ca | 0.50 | 0.34 | 0.60 | − 0.22 | − 0.20 | 0.57 |
| P | 0.56 | 0.58 | 0.39 | − 0.23 | − 0.16 | |
| Mg | 0.52 | 0.11 | − 0.02 | 0.59 | ||
| K | 0.53 | − 0.05 | 0.01 | |||
| Na | 0.32 | − 0.15 | ||||
| Citrate | 0.48 |
QTL identified in GWAS
We conducted single-trait GWASs on imputed WGSs from 19,586 cows (12,907,802 variants) and primarily identified 96 trait x region combinations with significant effects (-log10(P) ≥ 8.4) on milk mineral and citrate composition by applying the procedure described in the Methods section (Fig. 1). In two genomic regions (a 12Mbp-region on BTA1 and a 27Mbp-region on BTA20), the single marker analyses identified multiple trait x region combinations (10 and 15, respectively). Therefore, conditional analyses including the most significant variant identified in each region was applied to decipher if significant variants were due to linkage disequilibrium (LD) with the same causal mutation or to the presence of multiple causal mutations. These analyses led to the exclusion of 4 and 9 trait x region combinations on BTA1 and BTA20, respectively, resulting in 83 independent QTL. These QTL corresponded to 50 genomic regions located on Bos taurus (BTA) autosomes 1, 2, 3, 4, 5, 6, and 7 (Table 3) and 11, 12, 13, 14, 15, 17, 18, 19, 20, 21, 22, 24, 25, 27, 28, and 29 (Table 4). Of these, 18 regions had effects on several traits (2 to 5), while 32 affected only one trait. The four regions with the most significant effects (-log10(P) ≥ 50) each affected four to five traits; these were located on BTA 1 (~ 142.8 Mbp, for P, K, Mg, and Na; Fig. 2), 20 (~ 58.2 Mbp, for citrate, Mg, K, and Ca; Fig. 2), 6 (~ 45.3 Mbp, for K, Ca, P, and Na), and 5 (~ 116.4 Mbp, for Ca, Mg, P, citrate, and Na). Ten other regions with smaller but still highly significant effects (20 ≤ − log10(P) < 50) were located on BTA 5 (~ 119.0 Mbp), 6 (~ 43.3, 48.1, and 85.6 Mbp), 11 (~ 103.2 Mbp), 17 (~ 50.8 Mbp), 19 (~ 60.6 Mbp), 20 (~ 55.9 and 60.8 Mbp), and 22 (~ 32.8 Mbp); these also generally affected multiple milk components. All remaining significant QTL regions (8.4 ≤ − log10(P) < 20) were spread across 22 different autosomes. Overall, 6 to 21 different QTL were identified for each trait, and their cumulative effects explained from 19.8% to 45.5% of the genetic variance of a given trait (Table 5). The most significant QTL, located on BTA1 and BTA20, explained more than 23% of the genetic variance of P and citrate, respectively.
Figure 1.

–log10(P) value of the effect of variants on milk mineral (Ca, P, Mg, K, and Na) or citrate content plotted against their position on Bos taurus autosomes.
Table 3.
QTL identified for milk mineral and citrate content on Bos taurus (BTA) autosomes 1 to 10 (when a QTL region affected multiple traits, the most significantly affected trait is indicated in bold).
| Region | QTL | Trait | BTA | Confidence Interval of the QTL | Variant with the most significant effect (frequency p and effect b of the ALT allele) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| From (bp) | To (bp) | # variants | # genes | Position (bp) | ID | p | R2 | −log10P | b | SE | Name of gene(s) | Impact | ||||
| 1 | 1 | P | 1 | 135,384,680 | 136,262,601 | 8 | 3 | 136,262,601 | rs210630730 | 0.199 | 0.69 | 11.5 | 8.57 | 1.23 | TMEM108 | Intron |
| 2 | 4 | Mg | 1 | 142,826,156 | 142,837,365 | 9 | 1 | 142,834,737 | rs109459130 | 0.576 | 0.85 | 64.1 | 1.48 | 0.09 | SLC37A1 | Intron |
| 2 | 5 | P | 1 | 142,826,156 | 142,835,551 | 7 | 1 | 142,834,737 | rs109459130 | 0.576 | 0.85 | 219.3 | 30.2 | 0.95 | SLC37A1 | Intron |
| 2 | 6 | K | 1 | 142,826,156 | 142,838,477 | 31 | 1 | 142,835,551 | rs109717634 | 0.613 | 1.00 | 138.1 | 22.4 | 0.89 | SLC37A1 | Intron |
| 2 | 7 | Na | 1 | 142,826,156 | 142,835,551 | 7 | 1 | 142,835,551 | rs109717634 | 0.613 | 1.00 | 19.5 | − 3.77 | 0.41 | SLC37A1 | Intron |
| 3 | 9 | Mg | 1 | 145,114,056 | 146,804,023 | 64 | 3 | 146,775,338 | rs43048006 | 0.630 | 0.33 | 10.3 | − 0.70 | 0.11 | – | – |
| 4 | 11 | Na | 2 | 5,738,340 | 7,483,560 | 104 | 7 | 5,739,439 | rs209271012 | 0.111 | 0.86 | 10.4 | 3.85 | 0.58 | LOC104971101 | lncRNA |
| 5 | 12 | Na | 2 | 131,111,763 | 131,742,987 | 206 | 9 | 131,197,793 | rs133533873 | 0.672 | 0.99 | 13.8 | 2.84 | 0.37 | ALPL | Intron |
| 6 | 13 | Na | 3 | 15,459,107 | 15,540,579 | 7 | 6 | 15,470,670 | rs110073735 | 0.026 | 0.92 | 8.9 | − 5.20 | 0.86 | EFNA1 / LOC107132270 | Intron / lncRNA |
| 7 | 14 | Ca | 4 | 48,711,128 | 48,835,496 | 22 | 4 | 48,711,294 | rs381111559 | 0.291 | 0.59 | 13.0 | − 10.5 | 1.42 | SLC26A4 | Intron |
| 8 | 15 | Na | 4 | 75,153,659 | 77,188,370 | 14 | 7 | 77,147,308 | rs386097640 | 0.024 | 0.87 | 9.5 | 6.65 | 1.06 | GCK | Intron |
| 8 | 16 | Ca | 4 | 77,147,308 | 77,257,775 | 9 | 7 | 77,173,366 | rs133824313 | 0.025 | 0.98 | 9.1 | 14.8 | 2.40 | GCK | Intron |
| 8 | 17 | Mg | 4 | 77,037,103 | 77,188,370 | 8 | 4 | 77,173,366 | rs133824313 | 0.025 | 0.98 | 11.1 | 1.34 | 0.20 | GCK | Intron |
| 9 | 18 | Ca | 4 | 91,807,789 | 92,192,288 | 182 | 6 | 92,192,288 | rs42451422 | 0.028 | 0.95 | 9.5 | 13.4 | 2.13 | LRRC4 / SND1 | Intron / Intron |
| 10 | 19 | Ca | 5 | 108,256,181 | 109,217,271 | 87 | 11 | 109,141,258 | rs383141568 | 0.570 | 0.82 | 8.4 | 5.73 | 0.98 | BID | Intron |
| 11 | 20 | Ca | 5 | 111,637,039 | 111,848,859 | 47 | 7 | 111,745,513 | rs384814366 | 0.231 | 0.94 | 16.6 | − 9.38 | 1.11 | MRTFA = MKL1 | Intron |
| 12 | 21 | Ca | 5 | 116,403,262 | 116,463,104 | 48 | 8 | 116,429,840 | rs382268272 | 0.049 | 0.58 | 60.5 | − 34.8 | 2.11 | LOC101903383 | lncRNA |
| 12 | 22 | Na | 5 | 116,403,262 | 116,907,862 | 72 | 17 | 116,429,922 | rs110716509 | 0.050 | 0.67 | 9.3 | 5.05 | 0.81 | LOC101903383 | lncRNA |
| 12 | 23 | Citrate | 5 | 116,403,262 | 116,468,630 | 50 | 8 | 116,439,760 | rs110048000 | 0.042 | 0.56 | 9.5 | − 0.25 | 0.04 | PPARA | Intron |
| 12 | 24 | Mg | 5 | 116,403,806 | 116,463,104 | 36 | 6 | 116,448,403 | rs110659119 | 0.048 | 0.97 | 24.2 | − 1.38 | 0.13 | PPARA | Intron |
| 12 | 25 | P | 5 | 116,420,172 | 117,323,874 | 28 | 5 | 116,448,403 | rs110659119 | 0.048 | 0.97 | 19.5 | − 13.5 | 1.46 | PPARA | Intron |
| 13 | 26 | Ca | 5 | 119,066,644 | 119,069,138 | 25 | 1 | 119,068,337 | rs134583661 | 0.046 | 0.95 | 48.4 | − 24.8 | 1.69 | – | Intergenic |
| 13 | 27 | Mg | 5 | 119,066,644 | 119,069,138 | 25 | 1 | 119,068,337 | rs134583661 | 0.046 | 0.95 | 20.1 | − 1.29 | 0.14 | – | Intergenic |
| 13 | 28 | P | 5 | 119,066,644 | 119,069,138 | 25 | 1 | 119,068,337 | rs134583661 | 0.046 | 0.95 | 17.9 | − 13.2 | 1.50 | – | Intergenic |
| 14 | 29 | K | 6 | 40,012,262 | 40,518,049 | 18 | 2 | 40,024,540 | rs43610452 | 0.064 | 0.99 | 11.7 | − 11.6 | 1.66 | SLIT2 | Intron |
| 15 | 30 | P | 6 | 42,855,236 | 43,032,274 | 17 | 3 | 42,870,371 | rs42610629 | 0.523 | 0.97 | 9.2 | 5.59 | 0.91 | – | Intergenic |
| 16 | 31 | K | 6 | 43,297,568 | 43,322,676 | 12 | 1 | 43,322,676 | rs43463132 | 0.778 | 0.97 | 40.7 | 13.2 | 0.98 | – | Intergenic |
| 17 | 32 | P | 6 | 45,018,531 | 45,401,570 | 78 | 4 | 45,056,055 | rs378008740 | 0.713 | 0.76 | 30.6 | 12.7 | 1.09 | – | Intergenic |
| 17 | 33 | K | 6 | 45,018,882 | 45,401,570 | 66 | 4 | 45,328,581 | rs135848932 | 0.174 | 1.00 | 120.0 | − 27.4 | 1.17 | SEL1L3 | Intron |
| 17 | 34 | Ca | 6 | 45,328,392 | 45,411,116 | 17 | 4 | 45,393,335 | rs134399380 | 0.183 | 0.91 | 8.6 | 7.24 | 1.22 | – | Intergenic |
| 17 | 35 | Na | 6 | 45,215,035 | 45,401,570 | 17 | 4 | 45,401,485 | rs108972810 | 0.177 | 0.99 | 10.2 | − 3.39 | 0.52 | SMIM20 / LOC112447060 | Upstream BS DOF5.3 / lncRNA |
| 18 | 36 | K | 6 | 47,687,737 | 49,094,600 | 79 | 1 | 48,145,964 | rs43461552 | 0.336 | 0.92 | 26.9 | − 10.6 | 0.98 | – | Intergenic |
| 19 | 37 | K | 6 | 50,277,420 | 51,315,936 | 82 | 2 | 51,311,430 | rs110769289 | 0.555 | 0.53 | 9.1 | − 5.72 | 0.93 | – | Intergenic |
| 20 | 38 | K | 6 | 56,960,135 | 56,970,541 | 6 | 1 | 56,968,022 | rs134533189 | 0.091 | 0.41 | 9.4 | − 8.37 | 1.34 | RELL1 | Intron |
| 21 | 39 | Na | 6 | 85,409,619 | 86,104,900 | 312 | 15 | 85,512,651 | rs110772556 | 0.660 | 0.98 | 8.6 | 2.79 | 0.47 | – | Intergenic |
| 21 | 40 | Mg | 6 | 85,587,260 | 85,599,456 | 41 | 2 | 85,594,649 | rs209134503 | 0.481 | 0.85 | 29.8 | − 1.09 | 0.10 | ODAM | Synonymous |
| 21 | 41 | Ca | 6 | 85,406,891 | 85,599,456 | 44 | 5 | 85,594,717 | rs135113545 | 0.485 | 0.79 | 17.0 | − 10.6 | 1.24 | ODAM | Intron |
| 21 | 42 | P | 6 | 85,564,407 | 87,276,896 | 1004 | 6 | 87,033,142 | rs110121930 | 0.613 | 1.00 | 12.3 | − 7.88 | 1.09 | – | Intergenic |
| 22 | 43 | Mg | 6 | 88,380,434 | 88,436,951 | 4 | 1 | 88,390,406 | rs133413027 | 0.252 | 0.99 | 9.8 | − 0.56 | 0.09 | – | Intergenic |
| 23 | 44 | Citrate | 6 | 117,331,679 | 117,456,476 | 30 | 7 | 117,407,851 | rs43489995 | 0.306 | 0.30 | 10.4 | 0.22 | 0.03 | TMEM175 / DGKQ | Downstream / Synonymous |
| 23 | 45 | Ca | 6 | 117,331,679 | 117,457,454 | 42 | 6 | 117,442,167 | – | 0.407 | 0.41 | 19.8 | 12.7 | 1.37 | GAK | Intron |
| 24 | 46 | Na | 7 | 1,055,458 | 1,365,056 | 387 | 3 | 1,057,563 | rs210648426 | 0.346 | 0.91 | 14.3 | − 2.85 | 0.36 | RASGEF1C | Intron |
| 25 | 47 | Ca | 7 | 37,353,946 | 39,223,460 | 24 | 16 | 39,194,875 | rs41567828 | 0.068 | 0.99 | 9.1 | 12.8 | 2.08 | LOC107132625 | lncRNA |
| 25 | 48 | P | 7 | 38,917,914 | 40,541,631 | 10 | 5 | 40,541,609 | rs1116258782 | 0.009 | 0.22 | 16.9 | 31.7 | 3.71 | – | Intergenic |
| 25 | 49 | Ca | 7 | 39,528,343 | 42,832,365 | 179 | 16 | 41,321,781 | rs209051255 | 0.059 | 0.76 | 11.6 | 18.4 | 2.63 | ENSBTAG00000053872 | Upstream BS FOXD3 |
| 26 | 50 | Ca | 7 | 43,090,012 | 44,848,268 | 250 | 19 | 44,836,642 | rs210107035 | 0.068 | 0.98 | 17.1 | 21.6 | 2.51 | FSTL4 / LOC101902643 | Intron / lncRNA |
Table 4.
QTL identified for milk mineral and citrate content on Bos taurus (BTA) autosomes 11 to 29 (when a QTL affected multiple traits, the most significantly affected trait is indicated in bold).
| Region | QTL | Trait | BTA | Confidence Interval of the QTL | Variant with the most significant effect (frequency p and effect b of the ALT allele) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| From (bp) | To (bp) | # variants | # genes | Position (bp) | ID | p | R2 | −log10P | b | SE | Name of gene(s) | Impact | ||||
| 27 | 51 | K | 11 | 13,798,363 | 15,368,263 | 563 | 7 | 14,405,285 | rs110177775 | 0.102 | 0.96 | 10.3 | 8.04 | 1.22 | – | Intergenic |
| 28 | 52 | Citrate | 11 | 103,239,082 | 103,277,147 | 279 | 5 | 103,244,306 | rs435710868 | 0.514 | 0.85 | 23.9 | 0.21 | 0.02 | ENSBTAG00000048091 | Downstream |
| 28 | 53 | P | 11 | 103,239,082 | 103,277,147 | 278 | 6 | 103,250,796 | rs379692402 | 0.473 | 0.72 | 25.0 | − 11.8 | 1.13 | – | Intergenic |
| 29 | 54 | Na | 12 | 68,052,340 | 69,715,887 | 87 | 7 | 68,475,505 | rs133520200 | 0.257 | 1.00 | 9.2 | − 2.91 | 0.47 | – | Intergenic |
| 30 | 55 | Na | 12 | 73,666,737 | 73,725,910 | 47 | 1 | 73,674,445 | rs136814628 | 0.680 | 0.99 | 8.5 | 2.53 | 0.43 | HS6ST3 | Intron |
| 31 | 56 | K | 13 | 54,405,085 | 54,637,805 | 45 | 8 | 54,431,192 | rs133004626 | 0.623 | 0.95 | 10.2 | − 6.48 | 0.99 | – | Intergenic |
| 32 | 57 | Citrate | 14 | 238,942 | 613,906 | 35 | 15 | 453,437 | rs384162250 | 0.512 | 1.00 | 11.0 | 0.10 | 0.01 | PPP1R16A | Upstream |
| 32 | 58 | K | 14 | 232,311 | 639,312 | 79 | 20 | 515,185 | rs460225555 | 0.501 | 0.96 | 14.8 | 5.60 | 0.70 | VPS28 | Downstream |
| 32 | 59 | P | 14 | 271,581 | 758,897 | 64 | 20 | 577,322 | rs133788084 | 0.212 | 0.21 | 13.8 | 12.6 | 1.64 | FBXL6 | Upstream |
| 32 | 60 | Ca | 14 | 284,571 | 700,497 | 27 | 7 | 666,982 | rs133196323 | 0.025 | 0.34 | 13.0 | 32.2 | 4.31 | MROH1 | Intron |
| 33 | 61 | Na | 15 | 52,059,631 | 52,851,538 | 466 | 6 | 52,461,335 | rs210668186 | 0.218 | 0.60 | 9.2 | − 2.65 | 0.43 | FCHSD2 | Intron |
| 34 | 62 | K | 17 | 50,508,807 | 50,988,913 | 227 | 6 | 50,825,796 | rs474259058 | 0.055 | 0.95 | 21.3 | 15.1 | 1.56 | BRI3BP | Intron |
| 35 | 63 | Ca | 18 | 10,563,963 | 10,833,324 | 11 | 4 | 10,833,324 | rs137549452 | 0.045 | 0.91 | 11.3 | − 12.9 | 1.86 | KLHL36 / COTL1 | Upstream / Upstream |
| 36 | 64 | Ca | 19 | 56,448,441 | 56,609,243 | 340 | 8 | 56,524,233 | rs207749796 | 0.335 | 0.87 | 16.1 | 7.39 | 0.89 | FADS6 | Intron |
| 37 | 65 | Na | 19 | 60,550,831 | 60,870,548 | 95 | 2 | 60,555,853 | rs435138644 | 0.783 | 0.61 | 23.6 | 4.40 | 0.43 | – | Intergenic |
| 37 | 66 | Ca | 19 | 60,009,085 | 60,870,548 | 41 | 1 | 60,561,566 | rs41923848 | 0.843 | 0.73 | 11.2 | − 7.19 | 1.05 | – | Intergenic |
| 38 | 67 | Ca | 20 | 31,888,449 | 31,888,449 | 1 | 1 | 31,888,449 | rs385640152 | 0.009 | 0.91 | 17.0 | − 25.1 | 2.92 | GHR / LOC112443004 | Missense / lncRNA |
| 39 | 68 | Mg | 20 | 31,888,449 | 31,888,449 | 1 | 2 | 31,888,449 | rs385640152 | 0.009 | 0.91 | 13.9 | − 1.85 | 0.24 | GHR / LOC112443004 | Missense / lncRNA |
| 39 | 69 | P | 20 | 31,888,449 | 31,888,449 | 1 | 1 | 31,888,449 | rs385640152 | 0.009 | 0.91 | 10.3 | − 17.0 | 2.59 | GHR / LOC112443004 | Missense / lncRNA |
| 40 | 75 | Ca | 20 | 58,185,895 | 58,207,145 | 21 | 1 | 58,189,663 | rs110048176 | 0.170 | 1.00 | 25.3 | 13.1 | 1.24 | ENSBTAG00000048498 | lncRNA |
| 40 | 76 | Mg | 20 | 58,185,895 | 58,388,462 | 65 | 3 | 58,189,663 | rs110048176 | 0.170 | 1.00 | 117.2 | 2.34 | 0.10 | ENSBTAG00000048498 | lncRNA |
| 40 | 77 | Citrate | 20 | 58,185,895 | 58,388,462 | 81 | 3 | 58,204,929 | rs109956167 | 0.166 | 0.90 | 187.2 | 0.73 | 0.02 | ENSBTAG00000048498 | lncRNA |
| 40 | 78 | K | 20 | 58,185,895 | 58,388,849 | 69 | 2 | 58,386,888 | rs134021638 | 0.054 | 0.92 | 26.3 | 17.4 | 1.61 | ANKH | Intron |
| 41 | 81 | Citrate | 20 | 63,223,010 | 64,759,735 | 87 | 2 | 63,223,010 | rs109390768 | 0.595 | 0.98 | 14.9 | 0.14 | 0.02 | – | Intergenic |
| 41 | 82 | Mg | 20 | 63,223,010 | 64,753,386 | 13 | 1 | 63,223,010 | rs109390768 | 0.595 | 0.98 | 9.0 | 0.47 | 0.08 | – | Intergenic |
| 42 | 85 | Mg | 21 | 39,683,083 | 41,254,513 | 144 | 2 | 39,933,138 | rs134950504 | 0.070 | 0.99 | 9.3 | − 0.85 | 0.14 | – | Intergenic |
| 43 | 86 | P | 22 | 28,516,349 | 30,130,198 | 35 | 3 | 30,130,198 | rs108953480 | 0.966 | 0.90 | 8.6 | 11.4 | 1.92 | FOXP1 / ENSBTAG00000052330 | Upstream / lncRNA |
| 44 | 87 | P | 22 | 32,774,174 | 32,787,867 | 10 | 2 | 32,786,684 | rs208411747 | 0.040 | 0.92 | 20.6 | − 14.5 | 1.53 | FAM19A4 | Intron |
| 44 | 88 | Mg | 22 | 32,772,562 | 33,329,180 | 45 | 4 | 32,786,854 | rs209198296 | 0.046 | 0.82 | 13.3 | − 1.03 | 0.14 | FAM19A4 | Intron |
| 45 | 89 | K | 24 | 49,837,381 | 50,315,415 | 274 | 8 | 50,074,695 | rs448650804 | 0.324 | 0.61 | 11.1 | 5.18 | 0.76 | – | Intergenic |
| 46 | 90 | P | 24 | 58,272,345 | 58,396,158 | 102 | 4 | 58,306,855 | rs380879212 | 0.362 | 0.56 | 12.8 | 5.26 | 0.71 | LMAN1 | Intron |
| 47 | 91 | Ca | 25 | 20,480,835 | 20,482,817 | 2 | 1 | 20,480,835 | rs109069510 | 0.888 | 0.99 | 8.4 | − 8.01 | 1.36 | HS3ST2 | Intron |
| 48 | 92 | Mg | 27 | 36,511,563 | 36,592,556 | 65 | 6 | 36,522,002 | rs378026790 | 0.488 | 0.96 | 15.2 | 0.63 | 0.08 | GPAT4 | Upstream |
| 49 | 93 | K | 28 | 6,518,121 | 6,534,988 | 2 | 2 | 6,518,121 | rs42033936 | 0.224 | 0.98 | 13.0 | − 6.44 | 0.86 | KCNK1 | Intron |
| 49 | 94 | Na | 28 | 5,987,406 | 6,889,024 | 58 | 3 | 6,518,121 | rs42033936 | 0.224 | 0.98 | 10.8 | − 2.70 | 0.40 | KCNK1 | Intron |
| 50 | 95 | Ca | 29 | 9,193,304 | 9,907,275 | 162 | 7 | 9,517,882 | rs380735416 | 0.163 | 0.41 | 13.1 | 8.67 | 1.16 | PICALM | Upstream |
| 50 | 96 | Na | 29 | 8,731,208 | 10,144,903 | 486 | 11 | 9,923,696 | rs385315120 | 0.166 | 0.87 | 14.8 | − 4.28 | 0.54 | TMEM126B | Downstream |
Figure 2.

–log10(P) values of the effects of variants on milk mineral (Ca, P, Mg, K, and Na) or citrate content plotted against their position on Bos taurus autosomes 1 and 20.
Table 5.
Number of QTL and the percentage of genetic variance explained for each trait.
| Trait | Number of QTL | % of genetic variance explained by the QTL | ||
|---|---|---|---|---|
| Total | Min | Max | ||
| Ca | 21 | 33.1 | 0.43 | 5.0 |
| P | 14 | 41.7 | 0.27 | 23.3 |
| Mg | 13 | 31.0 | 0.42 | 10.6 |
| K | 14 | 42.2 | 0.66 | 13.6 |
| Na | 15 | 19.8 | 0.52 | 2.6 |
| Citrate | 6 | 32.9 | 0.76 | 23.1 |
Annotation of GWAS peaks
The sizes of the confidence intervals (CI) of QTL ranged from 1 bp to 3.3 Mbp; each CI contained between 1 and 1004 variants with significant effects (99.3 on average). While only ca. 6% of the tested variants were included on SNP chips, these chip variants (mainly HD) were disproportionately represented among the variants ranked in the top 10 in QTL peaks, and accounted for nearly 43% of the variants ranked first in the peaks, i.e. “top 1” variants (Fig. 3). For less than one-third of the QTL detected (24/83), the variant with the most significant effect was not located in a gene. Depending on the set of significant variants under consideration (all, top 100, top 50, top 10, or top 1), between 25.1% (top 1 variants) and 28.8% (all variants in the CI of the QTL) of variants were intergenic, while 60.5% (top 1) to 65.7% (top 10) of variants were located in genes, including upstream and downstream regions (Table 6). In all QTL, we therefore identified at least one positional candidate gene (1 to 20 per QTL, 5.3 on average). In total, 271 different genes were found within the CIs of the 83 QTL. In the four QTL regions with the most significant effects, the top-ranked variants were located in the genes SLC37A1 (BTA1), ANKH (BTA20), SEL1L3 (BTA6), and PPARA (BTA5); and the top-ranked variants in the remaining QTL were located in dozens of other genes (Tables 3 and 4). The majority of the variants located in genes were intronic, and were only rarely found in coding regions: only 1%, approximately, of variants were non-synonymous, i.e. 84 variants within the CIs of the QTL, and 55, 24, 4, and 1 among the top 100, top 50, top 10 and top 1 variants, respectively (Table 6). Out of these non-synonymous variants located in coding regions of genes, 4 are serious candidates because they were found among the 10 most significant variants of the QTL peaks: G446S in SLC26A4 (ranked 2nd for the QTL found on BTA4 at 48.7Mbp for Ca), V298M in ENSBTAG00000050954 (ranked 10th for the QTL found on BTA7 at 40.3Mbp for P), F267V in ARHGAP39 (ranked 5th for the QTL found on BTA14 at 0.4Mbp for K and Citrate) and F257Y in GHR (ranked 1st for the QTL found on BTA20 at 31.9Mbp for P, Ca and Mg). We also found a relatively high proportion of variants located in non-coding RNA (7.9% of all variants in the CIs of the QTL and 13.6% of top 1 variants). As presented in Table 6, these variants were mostly located in long non-coding RNA (lncRNA), less frequently in micro RNA (miRNA) and small nuclear RNA (snRNA), and very rarely in small nucleolar RNA (small Cajal body-specific RNA, scaRNA), and transfer RNA (tRNA).
Figure 3.
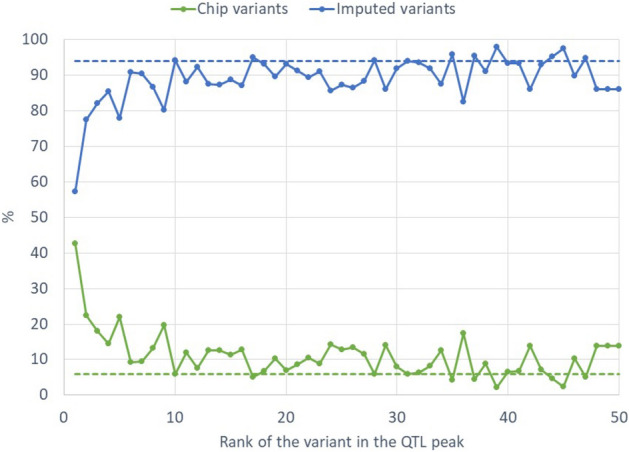
Percentages of chip variants (50 K, HD, or “research” SNPs, in green) and imputed variants (in blue) found among the top 50 variants of QTL peaks. Dashed lines represent the total percentage of chip variants (6%, in green) and imputed variants (94%, in blue) of all variants that were tested in the GWAS.
Table 6.
Annotation of variants located within confidence intervals of the QTL (a single variant could have different annotations).
| Annotation | ALL | Top 100 | Top 50 | Top 10 | Top 1 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | N | % | N | % | |
| Intergenic | 3705 | 28.8 | 1785 | 27.3 | 1062 | 26.0 | 269 | 25.1 | 29 | 25.9 |
| Total protein coding | 5575 | 63.3 | 2745 | 61.8 | 1792 | 62.9 | 494 | 65.7 | 49 | 60.5 |
| Intron | 3223 | 36.6 | 1427 | 32.1 | 980 | 34.4 | 267 | 35.5 | 33 | 40.7 |
| Downstream | 1203 | 11.7 | 635 | 12.1 | 368 | 11.1 | 71 | 8.0 | 4 | 4.2 |
| Upstream | 948 | 9.2 | 562 | 10.7 | 378 | 11.4 | 136 | 15.3 | 9 | 9.4 |
| 3′ UTR | 102 | 1.0 | 40 | 0.76 | 23 | 0.69 | 5 | 0.56 | 0 | 0 |
| Missense | 84 | 0.81 | 55 | 1.1 | 24 | 0.78 | 4 | 0.68 | 1 | 1.0 |
| Synonymous | 76 | 0.74 | 38 | 0.73 | 28 | 0.84 | 10 | 1.1 | 3 | 3.1 |
| 5′ UTR | 61 | 0.59 | 44 | 0.84 | 27 | 0.81 | 13 | 1.5 | 0 | 0 |
| Splice region | 11 | 0.11 | 5 | 0.10 | 1 | 0.03 | 0 | 0 | 0 | 0 |
| Splice acceptor | 1 | 0.01 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Frameshift | 1 | 0.01 | 0 | 0.00 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total non-coding RNA | 693 | 7.9 | 487 | 11.0 | 315 | 11.1 | 69 | 9.2 | 11 | 13.6 |
| lncRNA | 552 | 8.8 | 367 | 11.4 | 237 | 11.2 | 68 | 12.1 | 11 | 18.3 |
| miRNA | 69 | 1.1 | 50 | 1.5 | 42 | 2.0 | 1 | 0.18 | 0 | 0.00 |
| scaRNA | 4 | 0.06 | 4 | 0.12 | 4 | 0.19 | 0 | 0.00 | 0 | 0.00 |
| snRNA | 67 | 1.1 | 65 | 2.0 | 31 | 1.5 | 0 | 0.00 | 0 | 0.00 |
| tRNA | 1 | 0.02 | 1 | 0.03 | 1 | 0.05 | 0 | 0.00 | 0 | 0.00 |
| Total | 10,311 | 100 | 5234 | 100 | 3327 | 100 | 888 | 100 | 96 | 100 |
We then completed annotation of the QTL (1) identifying putative regulatory SNPs (rSNPs) located within transcription factor binding sites (TFBSs) according to the procedure described in the Methods section, and (2) using transcriptomic data available on the Cattle Gene Atlas website, http://cattlegeneatlas.roslin.ed.ac.uk/.
In the CIs of 40 of the 83 QTL, we identified 184 variants that overlapped with the TFBSs of 438 transcription factors (TFs), i.e. putative rSNPs (see Supplementary Table S1). These 438 TFs had 67 unique target genes among the 271 positional candidates identified in the QTL. Each TF targeted from 1 to 5 positional candidate genes, and 110 TFs had binding sites (BSs) at multiple loci. One TF (ASCL2) targeted 5 genes (BHLHE23, DGKQ, RMND5B, bta-let-7a-3, and bta-mir-2443), while 2, 21, and 85 TFs targeted 4, 3, and 2 genes, respectively. Two top 1 variants, rs108972810 (~ 45.4 Mbp in the upstream region of SMIM20 on BTA6, for Na) and rs209051255 (~ 41.3 Mbp in the upstream region of ENSBTAG00000053872 on BTA7, for Ca) were in the TFBSs for the TFs DOF and FOXD3, respectively. In total, for 22 QTL, top 10 variants were found in the TFBSs of 19 different target genes (including CSN2, PAEP, GPAT4, BRI3BP, and PPARA), which are potentially regulated by 47 TFs.
We then assessed the degree to which these positional candidate genes and TFs demonstrated tissue-specific expression. Of the 271 genes and 438 TFs associated with milk-mineral QTL, 203 and 160, respectively, were present in the Cattle Gene Atlas, which contains expression data from 91 different tissues or cell lines. Using the model described in the Methods section, we assessed the overexpression or tissue specificity of these genes by evaluating the t-statistic and the P value, Pt, associated with the effect of the tissue under consideration (see Supplementary Tables S2 and S3). Among the 203 positional candidate genes and 160 TFs with a putative regulatory effect on these genes, we calculated the number that were overexpressed in each tissue type (Pt < 10−4). The profiles obtained for both categories of genes, presented in Fig. 4, were quite similar. The tissues or cell types in which we found the highest number of genes associated with milk mineral composition were white blood cells (51 candidate genes and 59 TFs) and mammary gland (47 candidate genes and 45 TFs). In each tissue, we identified the most-specific genes by retaining the top 10% with respect to t-statistic values, i.e. 20 candidate genes and 16 TFs. Among the candidate genes that were most specific to mammary gland, we found the genes encoding the main milk proteins (CSN1S1, CSN1S2, CSN2, CSN3, and PAEP), and SLC37A1 and ANKH, which are located within the QTL that had the most significant effects in the present study. The ASCL2 TF, which potentially regulates five genes located in the CIs of the QTL, was one of the most mammary-gland-specific TFs. Among genes specific to white blood cells, we found COTL1, MKL1, and FOXP1. SLC37A1 was not among the 20 most-specific genes but it was ranked 22nd. In the upstream or intronic regions of these four genes, we identified the top-ranked variant of four different QTL, located on BTA18, BTA5, BTA22, and BTA1, respectively. Furthermore, we identified 27 rSNPs for which both TF and target gene were highly specific to the mammary gland; these combinations were composed of 26 unique TFs and 15 unique target genes (see Supplementary Table S4).
Figure 4.

Counts of significantly expressed genes (P < 10−4) in 91 tissues, showing (a) positional candidate genes and (b) transcription factors identified as putative regulators of these genes.
Focus on the SLC37A1 and ANKH gene regions
As mentioned above, two QTL—located on BTA1 (142.8 Mbp) and BTA20 (58.2 Mbp) in the vicinity of the SLC37A1 and ANKH genes, respectively—had very strong effects on milk mineral and citrate content. The SLC37A1 region affected P, K, Mg, and Na levels while the ANKH region had effects on citrate, Mg, K, and Ca (Fig. 2). These two regions alone affected all six milk components analyzed in this study. They explained a high proportion of the genetic variance in P (23.3%), citrate (23.1%), Mg (17.9%, i.e. 7.3% for SLC37A1 and 10.6% for ANKH), and K (15.4%, i.e. 13.6% for SLC37A1 and 1.8% for ANKH), but had much more moderate effects on Na (2.6%) and Ca (2.2%).
In the SLC37A1 region, the variant with the most significant effect was located in an intron; it was an imputed variant (R2 = 0.85) for P and Mg (rs109459130 at 142,834,737 bp) and a chip variant (HD SNP) for K and Na (rs109717634 at 142,835,551 bp). For both variants, which were located 814 bp apart, the most frequent allele (0.58 and 0.61, respectively) increased the amount of all minerals affected by this region.
In the ANKH region, we identified three different top 1 variants, depending on the trait. For K, the top variant was rs134021638 (at 58,386,888 bp, imputed with R2 = 0.92), located in an intronic region of ANKH; for Ca, Mg and citrate, the top variants were located in an lncRNA ENSBTAG00000048498 (rs110048176 at 58,189,663 bp, HD SNP for Ca and Mg and rs109956167 at 58,204,929 bp, imputed variant with R2 = 0.90 for citrate). ENSBTAG00000048498 spans from 58,186,301 to 58,224,434 bp and is located around 83 kbp upstream of the ANKH gene (58,307,527–58,477,497 bp). In all cases, the alleles responsible for an increase in citrate and mineral content were not the most common, with a MAF of 0.05 for the ANKH variant (rs134021638) and 0.17 for the two other variants.
In both regions, we examined the top-ranked variant for the most-affected traits, i.e. rs109459130 and rs109956167, and tested the interaction effects of their genotypes on milk mineral and citrate content. The results, presented in Fig. 5, revealed no significant interaction for any of the traits analyzed (P ≥ 0.02), suggesting that the effects of the two regions on milk composition were additive.
Figure 5.

SLC37A1 and ANKH genotypes and genotype interaction effects on milk mineral (Ca, P, Mg, K, and Na) or citrate content.
Discussion
To the best of our knowledge, this study is unique because it is the first GWAS of imputed whole-genome sequences based on the most-recent bovine reference genome with such a large population of cattle (19,586 cows); and it is the first attempt to investigate milk mineral content with a sequence-based GWAS, which here assessed a very large panel of genomic polymorphisms (12.9 million).
Using this approach, we identified 83 QTL that explained a substantial part of the genetic variance (up to 42.2%) for mineral and citrate content in cows’ milk; in each QTL region, we then identified functional candidate genes and variants. Our results build on those of two previous studies that used GWAS to investigate the mineral composition of milk—based on mineral content measured with reference methods and HD 777 k SNP genotypes—in small populations of Holstein (3719 and 44412) or Jersey (3219) cows. The study of Buitenhuis et al.9 was dedicated to mineral composition while Kemper et al.12 attempted to identify QTL that overlapped between milk production and composition traits. Only two QTL were shared between the two studies: a region located on BTA1 that affected P, for which both studies identified SLC37A1 as the best functional candidate gene, and a region located near the DGAT1 gene on BTA14 that affected Ca and P. In our study, we also identified these two regions, together with dozens of other regions located throughout the bovine genome.
By considering a very large dataset (more than 1 million test-day records of 126,873 cows), we estimated heritability values for mineral and citrate composition to be moderate to high, i.e. higher5,7,8,10 than or similar6,9 to those previously reported in the literature. Furthermore, we found strong genetic correlations among Ca, P, and Mg and among Ca, Mg, and citrate, which was consistent with the studies of Toffanin et al.5 and Denholm et al.8. Among minerals, Na had both the lowest heritability value (0.32 vs 0.48–0.56 for the other minerals) and the smallest percentage of genetic variance explained by its associated QTL (19.8% vs 33.1–42.2% for the other minerals). Ca, P, and Mg shared more QTL than any other group of minerals: 7, 6, and 6 QTL were shared between Ca and P, Ca and Mg, and P and Mg, respectively, while 4 QTL had effects on all three. In contrast, despite the high degree of genetic correlation between Ca, Mg, and citrate, only 2 QTL were shared among these three traits; however, one of these was a QTL located on BTA20 (~ 58.2 Mbp) that explained 2.2%, 10.6%, and 23.1% of the genetic variance of Ca, Mg, and citrate, respectively. Overall, estimates of genetic correlations were consistent with QTL results, which probably reflects the common biological pathway of these minerals in milk. In colloidal form, Ca, P, and Mg are associated with caseins in the micelles while in the soluble fraction, Ca and Mg are associated with citrate. Further studies, using random regression models fitted across lactation, could investigate the pattern of genetic relationships between the different milk minerals during lactation and thus provide a better understanding of the underlying biological mechanisms.
The resolution of our study was high enough to identify a single or a few positional candidate genes in most of the 83 QTL we identified. In each of these regions, though, the variant with the most significant effect was not necessarily located in a gene. As an example, we detected the overrepresentation of chip SNPs at the top of the QTL peaks (42.7% of the top 1 variants), and the majority of these SNPs were located in intergenic regions. Chip SNPs are directly genotyped or have a higher imputation accuracy than the surrounding variants, which probably enabled more precise estimation of their effects. To identify the best candidate variant(s) for each QTL, we therefore considered not only the variant with the most significant effect, but the set of variants ranked at the top of the peak that were located in or close to genes. In most cases, we found that these variants were located in non-coding regions of genes; for example, 15.3% and 12.1% of all top 10 variants were located in upstream regions of genes or in lncRNA, respectively, i.e. in putative regulatory regions. To further refine our results, we consulted in silico annotations of rSNPs in upstream regions of genes as well as existing knowledge regarding genomic regions that are transcribed into non-coding RNA. Moreover, to support the putative roles of these variants, we also considered the tissue specificity of candidate genes and transcription factors. Unfortunately, the expression dataset, which was based on Ensembl release 94, did not contain all of the genes located in QTL regions. Here we present some examples of QTL in which the putative causal mutation was located in a regulatory region, with particular attention to functional candidate genes previously associated with milk composition and the QTL with the most significant effects in this study.
GPAT4 (glycerol-3-phosphate acyltransferase 4) was the best candidate gene for the QTL on BTA 27 with effects on Mg. Although it was not the most notable QTL in terms of its effect on minerals, this region, and this gene in particular, have been highlighted by previous studies as affecting milk composition (fat, protein, and lactose content)19,22–24. In these studies, five variants in linkage disequilibrium—four in the upstream region and one in the 5′ UTR region of GPAT4—were highlighted as the best candidate causal variants. In our study, these variants were ranked in the top 5 in the GWAS peak; the variants ranked 3rd (rs209479876) and 5th (rs209855549) alter the WRKY48 and TWI transcription factor binding sites (TFBSs), respectively, and are more likely to be the causal variants. Daetwyler et al.23 also highlighted rs209479876 as the best causal variant in this region because of the high probability that it overlaps a TFBS. We also confirmed that GPAT4 was overexpressed in the mammary glands (P = 3.10−12) compared to 90 other tissues or cell types, but expression data were not available for the TFs associated with this gene.
For the QTL identified on BTA11 at ~ 103.2 Mbp with effects on P and citrate, the top two variants in the peak for P (the most-affected trait) were intergenic. Instead, the variants ranked 3rd to 7th were located in the upstream region of the PAEP (progestogen-associated endometrial protein) gene; the 5th-ranked variant, rs110710904, probably alters a binding site of the Macho-1 transcription factor (P = 0.02). PAEP, which is one of the most mammary-gland-specific genes (Pt = 1.8.10−37), encodes β-lactoglobulin, the most abundant whey protein in cow milk. Two non-synonymous variants in this gene, rs109625649 and rs110066229, were previously highlighted as the causal mutations underlying β-lactoglobulin concentration in milk25. In our study, these were respectively ranked 157th and 171st in the peak, i.e. far below the top-ranked variants upstream. These results corroborate previous reports that these two putatively causative missense mutations did not explain all the effects of the region on milk composition19,20 and highlight an rSNP as a likely causative variant.
BRI3BP (BRI3 binding protein), located on BTA 17 at ~ 50.8 Mbp, has been previously associated with de novo short chain fatty acid synthesis in bovine milk26,27. In our study, this region appeared to have effects on the K content of milk, and we found that BRI3BP was significantly overexpressed in mammary gland compared to the 90 other tissues investigated (P = 3.9.10−8). The first seven variants in the peak were located in an intronic region of BRI3BP. The 8th (rs477456528) and 9th (rs440703666) variants, instead, were located in the upstream region of the gene and rs440703666 probably alter the binding site of the transcription factor SPL8. This variant thus represents a very attractive functional candidate in this region.
In the four regions with the most significant effects on milk minerals, we identified SLC37A1 (BTA 1), ANKH (BTA 20), SEL1L3 / SMIM20 (BTA 6), and PPARA (BTA 5) as the best candidate genes.
Five of the six traits analyzed in this study were affected by a QTL region at ~ 116.4 Mbp on BTA5. Here, a variant located at 116,438,773 bp (1000G_80994138) ranked 2nd for four traits. This variant was located in the upstream region of PPARA (peroxisome proliferator activated receptor alpha) and overlapped the binding sites for the transcription factors E2F4, RSC3, RSC30, TDA9, and TFDP1. In this QTL region, top 1 variants were located either in an intronic region of PPARA (citrate, Mg, and P) or in an lncRNA, LOC101903383 (Ca and Na). PPARA encodes the PPAR-α transcription factor, a key regulator of lipid metabolism belonging to the superfamily of PPAR hormone receptors28; expression of this TF in mammary gland was previously found to be associated with milk fatty-acid composition in dairy cows29,30. Here, we did not detect any mammary-gland specificity of PPARA but we did observe that this gene was expressed in this tissue. It is therefore possible that polymorphisms in the binding sites of this TF or in the lncRNA LOC101903383, located 1.3 kbp upstream of PPARA, could regulate the expression of PPARA and be responsible for the strong effects of this region on milk composition.
On BTA6 at ~ 45.3 Mbp, we identified a QTL with very strong significant effects on K and to a lesser extent on P, Na, and Ca. Within the confidence interval of this QTL, we found 80 different potential variants, of which the majority (69) were intergenic, 9 were located in an intronic region of SEL1L3 (SEL1L family member 3), and 2 were in the upstream region of SMIM20 (small integral membrane protein 20). However, when we examined only the top 10 variants for the different traits, the number of distinct variants shrank to 18 (9 intergenic, 7 in SEL1L3, and 2 in SMIM20). Of these, two (rs108972810 at 45,401,485 bp and rs136498639 at 45,401,570 bp) were located in the upstream region of SMIM20 and overlapped a TFBS. SEL1L3 was previously identified as a candidate gene in a QTL region with effects on bovine milk protein composition19. Instead, to the best of our knowledge, this study is the first to propose SMIM20 as a candidate gene for milk composition. However, the functional link between these genes and milk composition has yet to be established, as we found both genes to be endometrium-specific and underexpressed in mammary glands compared to the 90 other tissues investigated.
For milk mineral composition, the two best functional candidate genes highlighted by our analysis were SLC37A1 (solute carrier family 37, member A1) and ANKH (inorganic pyrophosphate transport regulator). These two genes were previously found to be overexpressed in mammary glands relative to 17 other types of tissue31, and here we found both among the top 10% of mammary-gland-specific genes (Pt = 3.4.10−26 and 6.6.10−16, respectively). This suggests that the main function of these genes occurs in epithelial cells of this tissue. SLC37A1 and ANKH both encode transmembrane proteins involved in ion transport and have been found to play a role in inorganic anion transport20. Variants located in both genes explained a large degree of the genetic variation in milk mineral content. Earlier studies also linked SLC37A1 with mineral content, in particular that of P9,12, while both genes have been implicated in determining milk protein composition in Holstein, Montbéliarde, and Normande cows19 and cheese-making traits in the same Montbéliarde cows we analyzed here20. These results are consistent with what is known about the association of minerals with casein molecules in micelles; milk mineral composition is strongly related to milk protein composition2 and therefore to cheese-making traits3,4.
In SLC37A1, 7, 31, 9, and 7 variants were located in the confidence intervals of QTL for P, K, Mg, and Na, respectively. These variants (32 of which were unique) were overwhelmingly located in introns of the gene, in a 12.3-kbp-region from 142,826,156 to 142,838,477 bp. Among these 32 variants, we were not able to distinguish the best candidate based on annotation, but two variants, 814 bp apart, were particularly highly ranked for all traits. Specifically, rs109459130, at 142,834,737 bp, was ranked 1st, 2nd, 1st, and 3rd for P, K, Mg, and Na, respectively, while rs109717634, at 142,835,551 bp, was ranked 2nd, 1st, 3rd, and 1st for P, K, Mg, and Na, respectively. Of these, the first (rs109459130) appeared to be the better candidate because it was top-ranked for the most affected trait, P, and because it was imputed (R2 = 0.85), while the second variant was an HD SNP (R2 = 0.997).
In the region of the ANKH gene, we found 81, 65, 69, and 21 variants in the confidence intervals of QTL for citrate, Mg, K, and Ca, respectively. These represented 82 unique variants, all located either in intronic regions of the ANKH gene (59 variants between 58,344,839 and 58,388,849 bp) or in the lncRNA ENSBTAG00000048498 (23 variants between 58,185,895 and 58,212,187 bp). The same variant was ranked first for both Mg and Ca. Two top-1 variants were located in the lncRNA ENSBTAG00000048498—rs109956167 (at 58,204,929 bp) for citrate and rs110048176 (at 58,189,663 bp) for Mg and Ca—while the top-ranked variant for K was located in ANKH (rs134021638 at 58,386,888 bp). We propose rs109956167 as the best candidate causal variant because i) it was ranked 1st for the most-affected trait (citrate), ii) it was imputed, in contrast to rs110048176 which was from the HD chip, and iii) it was the only variant present in the top 10 for all traits affected by this region (4th for Mg, 6th for Ca, and 9th for K). We hypothesize that the lncRNA ENSBTAG00000048498, which is 83 kbp downstream of the closest gene, ANKH, may affect milk mineral content by regulating the expression of ANKH and thus the amount of protein available for ion transport.
When we examined the best candidate variants in the regions of SLC37A1 (rs109459130) and ANKH (rs109956167), we found that alleles of these genes did not have significant interactions with respect to the six milk composition traits analyzed in this study and that, combined together, these two regions explained a large part of the genetic variance in milk mineral content. The allele that increased mineral content was the most frequent allele of SLC37A1 (allele A, frequency = 0.58), while it was the least frequent allele in the ANKH region (allele C, frequency = 0.17).
Compared to previous studies, our GWAS-based investigation of milk mineral composition was conducted at the whole-genome level, after imputation with the most recent reference genome and using data from a large population of animals. This approach led to the identification of a large number of genomic regions that explain a great deal of the genetic variance of the traits studied here. Moreover, post-GWAS investigations conducted using different annotation datasets enabled the identification of candidate causal genes and the prioritization of candidate variants in these genomic regions. In most situations, exonic variants were not proposed as the best candidates, although they cannot be excluded in four QTL regions, including GHR. In contrast, the best candidate variants often were putative regulatory variants. Although the functional causative effect of the candidate variants remains to be demonstrated, this study highlights a ‘short list’ of candidate variants, in particular rs109459130 (SLC37A1) and rs109956167 (ANKH), whose favorable alleles can be feasibly selected to increase the mineral content (Ca, P, Mg, and K) of milk and therefore improve both its nutritional and cheese-making qualities in Montbéliarde cows.
Methods
Ethics statement
Milk samples were analyzed with MIR spectrometry during routine milk recording in commercial herds of Montbéliarde cows in the Franche-Comté region (France). We did not perform any experiments on animals and no ethical approval was required.
Animals and phenotypes
The original dataset was generated by the From’MIR project and is described in detail by Sanchez et al.11,20. It comprised 6,670,769 mid-infrared (MIR) spectra of milk samples from 410,622 Montbéliarde cows. The concentrations of five milk minerals (Ca, P, Mg, K, and Na) and citrate were predicted from MIR spectra using equations developed by the Optimir project14,15 (Table 1). As previously described11,20, to ensure that the dataset was homogeneous (and less computationally demanding), we retained only the first-lactation records with at least seven test-day records per cow for estimating variance components (1,100,238 test-day records from 126,873 cows11). To estimate environmental effects used to derive the phenotypes included in GWAS, at least three test-day records per cow were required, corresponding to a dataset of 1,442,371 test-day records from 189,817 cows20.
Estimation of genetic parameters
First lactation data were analyzed using bivariate repeatability animal models applied to all pairs of traits. (Co)variance components were estimated using the AI-REML algorithm as implemented in Wombat software32, with the following linear animal model:
| 1 |
where is the vector of test-day observations; a ~ is the vector of random additive genetic effects, p ~ is the vector of random permanent environmental effects, and ~ is the vector of random residual effects.X, Z, and W are incidence matrices, A is the relationship matrix among individuals calculated from the pedigree (traced back over four generations and containing 315,661 animals), and I the identity matrix. , and are 2 × 2 matrices of additive genetic, permanent environmental, and residual variances-covariances, respectively. The vector included the fixed effects of the herd x test-day x spectrometer combination, stage of lactation, and season of calving.
Genotypes and imputation to whole-genome sequences
A subset of 19,586 cows for which MIR spectra were available had been genotyped for the purpose of genomic selection using the BovineSNP50 (50 K, 6505 cows) or the EuroG10K BeadChip (Illumina Inc., San Diego, 13,081 cows); the latter contains generic SNPs and a research add-on for causal or predictive SNPs for traits of interest in cattle. Missing genotypes were imputed using FImpute software33 for the 53,469 autosomal SNPs (50 K and “research” SNPs, 50 K+) that passed all quality control filters (individual call rate > 95%; SNP call rate > 90%; minor allele frequency (MAF) > 1% in at least one major French dairy cattle breed; genotype frequencies in HW equilibrium with P > 10−4). Whole-genome sequences (WGSs) were then imputed in two steps. In the first step, 777 K high-density and “research” SNPs (HD+) were imputed with FImpute software using a within-breed reference set of 522 Montbéliarde bulls genotyped with the Illumina BovineHD BeadChip (Illumina Inc., San Diego, CA)34. Finally, allele dosages of the WGSs were imputed using Minimac software35 and WGS variants of 1479 Bos taurus animals from the 7th run of the 1000 Bull Genomes Project, representing 17 cattle breeds and including 63 Montbéliarde bulls. This 2-step strategy was found to be the most accurate36 (eg, Boowman & Veerkamp, 2014) because (1) the first step takes advantage of the high number of HD genotypes of influential bulls and is performed with very limited loss in accuracy34 (Hozé et al., 2013) ; (2) the number of sequenced Montbéliarde bulls was too limited for an accurate imputation to sequence when used alone; and (3) linkage disequilibrium is partially conserved across breeds at the HD level (~ several kb), making use of close breeds sequence data very beneficial. WGS variants were selected following the protocol defined by the 1000 Bull Genomes consortium16,23, as described in Boussaha et al.37. Short reads were filtered for quality and aligned to the ARS-UCD1.2 reference sequence17, and small genomic variations (SNPs and InDels) were detected using SAMtools 0.0.1838. Raw variants were then filtered to produce a dataset of 25,050,323 variants. The precision of imputation from HD+ to WGS was assessed using R2 values calculated with Minimac software35. Only variants with R2 ≥ 0.20 and MAF ≥ 0.005 were retained for association analyses, i.e. 12,907,802 variants, with a mean R2 of 0.67 and MAF of 0.19.
GWAS
We performed single-trait association analyses between all 12,907,802 polymorphic variants and each of six milk mineral and citrate traits, described in Table 1. All association analyses were performed using the mlma option of GCTA software (version 1.24), which applies a mixed linear model that includes the variant to be tested39:
| 2 |
where is the vector of so-called yield deviations, i.e. test-day records adjusted for non-genetic effects with the mixed linear model (1) using Genekit software40 and averaged per cow; is the overall mean; is the additive fixed effect of the variant to be tested for association; is the vector of imputed allele dosages, ranging from 0 to 2; is the vector of random polygenic effects, with the genomic relationship matrix (GRM) calculated using the HD SNP genotypes (which offer both high density and accuracy of imputation), and the polygenic variance, estimated based on the null model and then assumed as known while testing for the association between each variant and the trait of interest; and is the vector of random residual effects, with I the identity matrix and the residual variance. Association was tested using a t-statistic calculated by dividing the variant effect estimate by its standard error.
In order to correct for multiple testing, the Bonferroni correction was applied to take into account all 12.9 million independent tests. The 5% genome-wide threshold of significance therefore corresponded to a nominal P-value of 4 × 10−9 (-log10(P) = 8.4) per test. When a given trait was significantly affected by multiple variants, variants that were located less than 2 million base-pairs (Mbp) apart were grouped together to define QTL, considering the variants belonged to the same QTL region. The bounds of the confidence intervals (CIs) of each region were then determined based on the positions of variants that were included in the upper third of the QTL peak.
In regions where multiple neighboring QTL were identified (BTA1 and BTA20), conditional analyses were carried out using the cojo option of GCTA39 in order to conclude if multiple significant variants in a genomic region were due to LD with the same causal mutation or to the presence of multiple causal mutations. Association analyses were performed by including in the model the most significant variant as a fixed effect and by testing all variants in these neighboring QTL that were not in strong LD with the conditional variant (r2 < 0.9).
Functional annotations
Genomic regions and variants were annotated with FAANGMine v1.1 (https://faangmine.elsiklab.missouri.edu/), developed by the Functional Annotation of ANimal Genomes initiative18 and which integrates the ARS-UCD1.2 bovine reference genome with a variety of external data sources, including RefSeq from NCBI (https://www.ncbi.nlm.nih.gov) and Ensembl (https://www.ensembl.org) gene sets.
The ability of genetic variants to alter transcription factor binding sites (TFBSs) was predicted with a custom script that used TFBS models from the JASPAR (JASPAR CORE 2018 collection41), HOCOMOCO (version v1042), and TRANSFAC (version v3.2 public43) databases. These databases contain curated sets of transcription factor binding models represented as Position Weight Matrices (PWM), which are derived from published collections of experimentally defined eukaryote TFBSs. Only vertebrate PWMs were downloaded for use in our study.
Gene overexpression or specificity in different tissues was determined using gene expression patterns of 24,616 genes (Ensembl release 94) available in the Cattle Gene Atlas, which contains 723 RNA-seq datasets representing 91 tissues and cell types, classified into 17 biological categories (http://cattlegeneatlas.roslin.ed.ac.uk/). To assess the expression specificity of each gene in a given type of tissue (by excluding tissues in the same biological category), we applied the following linear model as described in Fang et al.44:
| 3 |
where is the vector of expression level in the tissues, assessed by the scaled log2FPKM (Fragments Per Kilobase per Million mapped reads); is the overall mean; is the vector of the variable with value 1 for samples of the tested tissue and − 1 for samples outside the same category; is the corresponding tissue effect; is the incidence matrix related to the corresponding covariables effects , including age, sex and study effects; is the residual effect. Model (3) was implemented adapting R scripts available on the Cattle Gene Atlas website. For each gene, a t-statistic was computed by dividing the tissue effect by its standard error. A gene was considered to be overexpressed in a tissue if the probability associated with the t-statistic (Pt) was lower than 10−4. To determine the tissue specificity of genes, we ranked the genes in each type of tissue by their t-statistics; the top 10% were considered to be tissue-specific.
SLC37A1-ANKH genotype interactions
We tested putative interaction effects between the genes SLC37A1 and ANKH on milk mineral and citrate content with the following mixed linear model:
| 4 |
where as defined in (2); is the overall mean; and are the fixed effects of the genotypes of the best candidate variant in the SLC37A1 and ANKH regions, respectively; represents the interaction between the two genotypes;, and are incidence matrices related to the individual effects of the SLC37A1 and ANKH genotypes and their interaction, respectively; is the vector of random sire effects and P the corresponding incidence matrix; and is the vector of random residual effects. All effects were tested using t-statistics computed in the MIXED procedure of SAS software.
Supplementary Information
Acknowledgements
This study was funded by the French Ministry of Agriculture, Agro-food, and Forests; the French Dairy Interbranch Organization (CNIEL); the Regional Union of Protected Designation Cheeses of Franche-Comté (URFAC); and the Regional Council of Bourgogne-Franche-Comté, through the project From’MIR. Chip genotypes originated from the genomic selection program of the breeding company UMOTEST. The authors gratefully acknowledge the breeders who participated in the From’MIR project; colleagues from the Conseil-Elevage 25-90 who coordinated data collection; and the contribution of the 1000 Bull Genomes consortium.
Author contributions
M.P.S. estimated genetic parameters; performed imputation, GWAS, and post-GWAS analyses; and drafted the first version of the manuscript. D.R. and M.C. managed rSNP analyses. M.Bo. managed sequence analyses of the 1000 Bull Genomes Project. C.H. contributed to imputation analyses. M.Br., A.D.B., P.G., and D.B. designed and managed the From’MIR project. D.B. supervised all analyses. All authors have read and approved the manuscript.
Data availability
The data (genotypes and phenotypes) that enabled the findings of this study were made available by UMOTEST, CEL25-90, and GENIATEST. However, restrictions apply to the availability of these commercial data: they were used under license for the current study, and are not publicly available.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-87078-1.
References
- 1.Zamberlin S, Antunac N, Havranek J, Samarzija D. Mineral elements in milk and dairy products. Mljekarstvo. 2012;62:111–125. [Google Scholar]
- 2.Gaucheron F. The minerals of milk. Reprod. Nutr. Dev. 2005;45:473–483. doi: 10.1051/rnd:2005030. [DOI] [PubMed] [Google Scholar]
- 3.Toffanin V, De Marchi M, Lopez-Villalobos N, Cassandro M. Effectiveness of mid-infrared spectroscopy for prediction of the contents of calcium and phosphorus, and titratable acidity of milk and their relationship with milk quality and coagulation properties. Int. Dairy J. 2015;41:68–73. doi: 10.1016/j.idairyj.2014.10.002. [DOI] [Google Scholar]
- 4.Malacarne M, et al. Influence of micellar calcium and phosphorus on rennet coagulation properties of cows milk. J. Dairy Res. 2014;81:129–136. doi: 10.1017/S0022029913000630. [DOI] [PubMed] [Google Scholar]
- 5.Toffanin V, et al. Genetic parameters for milk mineral content and acidity predicted by mid-infrared spectroscopy in Holstein-Friesian cows. Animal. 2015;9:775–780. doi: 10.1017/S1751731114003255. [DOI] [PubMed] [Google Scholar]
- 6.van Hulzen KJE, Sprong RC, van der Meer R, van Arendonk JAM. Genetic and nongenetic variation in concentration of selenium, calcium, potassium, zinc, magnesium, and phosphorus in milk of Dutch Holstein-Friesian cows. J. Dairy Sci. 2009;92:5754–5759. doi: 10.3168/jds.2009-2406. [DOI] [PubMed] [Google Scholar]
- 7.Visentin G, et al. Genetic (co)variances between milk mineral concentration and chemical composition in lactating Holstein-Friesian dairy cows. Animal. 2019;13:477–486. doi: 10.1017/S1751731118001507. [DOI] [PubMed] [Google Scholar]
- 8.Denholm S, et al. Phenotypic and genetic analysis of milk and serum element concentrations in dairy cows. J. Dairy Sci. 2019;102:11180–11192. doi: 10.3168/jds.2019-16960. [DOI] [PubMed] [Google Scholar]
- 9.Buitenhuis B, Poulsen N, Larsen L, Sehested J. Estimation of genetic parameters and detection of quantitative trait loci for minerals in Danish Holstein and Danish Jersey milk. BMC Genet. 2015;16:52. doi: 10.1186/s12863-015-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonfatti V, Vicario D, Lugo A, Carnier R. Genetic parameters of measures and population-wide infrared predictions of 92 traits describing the fine composition and technological properties of milk in Italian Simmental cattle. J. Dairy Sci. 2017;100:5526–5540. doi: 10.3168/jds.2016-11667. [DOI] [PubMed] [Google Scholar]
- 11.Sanchez MP, et al. Genetic parameters for cheese-making properties and milk composition predicted from mid-infrared spectra in a large dataset of Montbéliarde cows. J. Dairy Sci. 2018;101:10048–10061. doi: 10.3168/jds.2018-14878. [DOI] [PubMed] [Google Scholar]
- 12.Kemper KE, et al. Leveraging genetically simple traits to identify small-effect variants for complex phenotypes. BMC Genom. 2016;17:1–9. doi: 10.1186/s12864-016-3175-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Marchi M, Toffanin V, Cassandro M, Penasa M. Invited review: Mid-infrared spectroscopy as phenotyping tool for milk traits. J. Dairy Sci. 2014;97:1171–1186. doi: 10.3168/jds.2013-6799. [DOI] [PubMed] [Google Scholar]
- 14.Gengler N, et al. Capitalizing on fine milk composition for breeding and management of dairy cows. J. Dairy Sci. 2016;99:4071–4079. doi: 10.3168/jds.2015-10140. [DOI] [PubMed] [Google Scholar]
- 15.Soyeurt H, et al. Potential estimation of major mineral contents in cow milk using mid-infrared spectrometry. J. Dairy Sci. 2009;92:2444–2454. doi: 10.3168/jds.2008-1734. [DOI] [PubMed] [Google Scholar]
- 16.Bouwman AC, et al. Meta-analysis of genome-wide association studies for cattle stature identifies common genes that regulate body size in mammals. Nat. Genet. 2018;50:362–367. doi: 10.1038/s41588-018-0056-5. [DOI] [PubMed] [Google Scholar]
- 17.Rosen BD, et al. De novo assembly of the cattle reference genome with single-molecule sequencing. GigaScience. 2020;9:2giaa021. doi: 10.1093/gigascience/giaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giuffra, E., Tuggle, C.K., & Consortium, FAANG. Functional annotation of animal genomes (FAANG): Current achievements and roadmap. Annu. Rev. Anim. Biosci.7, 65–88 (2019). [DOI] [PubMed]
- 19.Sanchez MP, et al. Within-breed and multi-breed GWAS on imputed whole-genome sequence variants reveal candidate mutations affecting milk protein composition in dairy cattle. Genet. Sel. Evol. 2017;49:68. doi: 10.1186/s12711-017-0344-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanchez MP, et al. Sequence-based GWAS, network and pathway analyses reveal genes co-associated with milk cheese-making properties and milk composition in Montbéliarde cows. Genet. Sel. Evol. 2019;51:34. doi: 10.1186/s12711-019-0473-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Albert F, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 2015;16:197–212. doi: 10.1038/nrg3891. [DOI] [PubMed] [Google Scholar]
- 22.Littlejohn M, et al. Expression variants of the lipogenic AGPAT6 gene affect diverse milk composition phenotypes in Bos taurus. PLoS ONE. 2014;9:e85757. doi: 10.1371/journal.pone.0085757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daetwyler HD, et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 2014;46:858–867. doi: 10.1038/ng.3034. [DOI] [PubMed] [Google Scholar]
- 24.Pausch H, et al. Meta-analysis of sequence-based association studies across three cattle breeds reveals 25 QTL for fat and protein percentages in milk at nucleotide resolution. BMC Genom. 2017;18:1–11. doi: 10.1186/s12864-017-4263-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ganai NA, Bovenhuis H, van Arendonk JA, Visker MH. Novel polymorphisms in the bovine beta-lactoglobulin gene and their effects on beta-lactoglobulin protein concentration in milk. Anim. Genet. 2009;40:127–133. doi: 10.1111/j.1365-2052.2008.01806.x. [DOI] [PubMed] [Google Scholar]
- 26.Boichard D, et al. Genetic determinism of milk composition in fatty acids and proteins in ruminants, and selection potential. INRAE Prod. Anim. 2014;27:283–298. doi: 10.20870/productions-animales.2014.27.4.3074. [DOI] [Google Scholar]
- 27.Knutsen T, et al. Unravelling genetic variation underlying de novo-synthesis of bovine milk fatty acids. Sci. Rep. 2018;8:2179. doi: 10.1038/s41598-018-20476-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schoonjans K, Staels B, Auwerx J. The peroxisome proliferator activated receptors (PPARs) and their effects on lipid metabolism and adipocyte differentiation. Biochim. Biophys. Acta. 1996;1302:93–109. doi: 10.1016/0005-2760(96)00066-5. [DOI] [PubMed] [Google Scholar]
- 29.Fougère H, Bernard L. Effect of diets supplemented with starch and corn oil, marine algae, or hydrogenated palm oil on mammary lipogenic gene expression in cows and goats: A comparative study. J. Dairy Sci. 2019;102:768–779. doi: 10.3168/jds.2018-15288. [DOI] [PubMed] [Google Scholar]
- 30.Bernard L, Toral PG, Chilliard Y. Comparison of mammary lipid metabolism in dairy cows and goats fed diets supplemented with starch, plant oil, or fish oil. J. Dairy Sci. 2017;100:9338–9351. doi: 10.3168/jds.2017-12789. [DOI] [PubMed] [Google Scholar]
- 31.Chamberlain A, et al. Extensive variation between tissues in allele specific expression in an outbred mammal. BMC Genom. 2015;16:993. doi: 10.1186/s12864-015-2174-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer K. WOMBAT—A tool for mixed model analyses in quantitative genetics by restricted maximum likelihood (REML) J. Zhejiang Univ-Sc. B. 2007;8:815–821. doi: 10.1631/jzus.2007.B0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sargolzaei M, Chesnais J, Schenkel F. A new approach for efficient genotype imputation using information from relatives. BMC Genom. 2014;15:478. doi: 10.1186/1471-2164-15-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoze C, et al. High-density marker imputation accuracy in sixteen French cattle breeds. Genet. Sel. Evol. 2013;45:33. doi: 10.1186/1297-9686-45-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bouwman AC, Veerkamp RF. Consequences of splitting whole-genome sequencing effort over multiple breeds on imputation accuracy. BMC Genet. 2014;15:1–9. doi: 10.1186/s12863-014-0105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boussaha M, et al. Construction of a large collection of small genome variations in French dairy and beef breeds using whole-genome sequences. Genet. Sel. Evol. 2016;48:87. doi: 10.1186/s12711-016-0268-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J, Lee S, Goddard M, Visscher P. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ducrocq V. Genekit, BLUP software. INRA; 2011. [Google Scholar]
- 41.Sandelin A, Alkema W, Engstrom P, Wasserman W, Lenhard B. JASPAR: An open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khamis A, et al. A novel method for improved accuracy of transcription factor binding site prediction. Nucleic Acids Res. 2018;46:e72. doi: 10.1093/nar/gky237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knüppel A, Dietze P, Lehnberg W, Frech K, Wingender E. TRANSFAC retrieval program: A network model database of eukaryotic transcription regulating sequences and proteins. J. Comput. Biol. 1994;1:191–198. doi: 10.1089/cmb.1994.1.191. [DOI] [PubMed] [Google Scholar]
- 44.Fang L, et al. Comprehensive analyses of 723 transcriptomes enhance genetic and biological interpretations for complex traits in cattle. Genome Res. 2020;30:790–801. doi: 10.1101/gr.250704.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data (genotypes and phenotypes) that enabled the findings of this study were made available by UMOTEST, CEL25-90, and GENIATEST. However, restrictions apply to the availability of these commercial data: they were used under license for the current study, and are not publicly available.