

Summary
UDP-glucose:glycoprotein glucosyltransferase (UGGT) flags misfolded glycoproteins for ER retention. We report crystal structures of full-length Chaetomium thermophilum UGGT (CtUGGT), two CtUGGT double-cysteine mutants, and its TRXL2 domain truncation (CtUGGT-ΔTRXL2). CtUGGT molecular dynamics (MD) simulations capture extended conformations and reveal clamping, bending, and twisting inter-domain movements. We name “Parodi limit” the maximum distance on the same glycoprotein between a site of misfolding and an N-linked glycan that can be reglucosylated by monomeric UGGT in vitro, in response to recognition of misfold at that site. Based on the MD simulations, we estimate the Parodi limit as around 70–80 Å. Frequency distributions of distances between glycoprotein residues and their closest N-linked glycosylation sites in glycoprotein crystal structures suggests relevance of the Parodi limit to UGGT activity in vivo. Our data support a “one-size-fits-all adjustable spanner” UGGT substrate recognition model, with an essential role for the UGGT TRXL2 domain.
Keywords: UGGT, glycoprotein folding, negative-stain EM, molecular dynamics, Parodi limit, misfolding, misfold sensing, X-ray diffraction, re-glucosylation, GT24 domain
Graphical abstract
Highlights
-
•
UGGT MD simulations widen the conformational range observed in crystal structures
-
•
The UGGT TRXL2 domain is essential for enzymatic activity
-
•
A misfold site is closer than the Parodi limit to a glycan in UGGT clients in vitro
-
•
N-Glycan distributions suggest evolution optimizes glycoprotein surface coverage
Modenutti et al. carry out molecular dynamics simulations of UGGT, the enzyme surveying correct folding of glycoproteins, and propose a “one-size-fits all adjustable spanner” UGGT:substrate recognition model. The UGGT TRXL2 domain is essential for its function. The size of UGGT likely dictates restraints on the evolution of N-linked glycosylation sites.
Introduction
A wonderfully efficient protein-folding machinery in the ER of eukaryotic cells ensures that only correctly folded glycoproteins can exit the ER, proceed to the Golgi, and from there continue along the secretory pathway toward their cellular or extracellular destinations (Vincenz-Donnelly and Hipp, 2017). The stringency of this ER quality control (ERQC) system is of great advantage to healthy cells. It allows time for complex glycoproteins to fold in the ER and prevents premature secretion of incompletely folded species. In the background of a misfold-inducing missense mutation in a secreted glycoprotein gene, the resulting misfolded glycoprotein is either retained in the ER by ERQC or degraded by the ER-associated degradation (ERAD) machinery (Amara et al., 1992). ERQC-mediated ER retention and ERAD degradation of glycoprotein mutants bear particularly unfortunate consequences when the mutation induces a minor folding defect but does not abrogate the function of the glycoprotein (“responsive mutant”). In these cases ERQC/ERAD cause disease by blocking the secretion of the glycoprotein mutant, even though its residual activity would be beneficial to the organism (see for example Parodi et al., 2014).
Central to ERQC is the ER-resident 170-kDa enzyme UDP-glucose:glycoprotein glucosyltransferase (UGGT). The enzyme selectively reglucosylates a misfolded glycoprotein on one of its N-glycans and promotes its association with the ER lectins calnexin and calreticulin, thus mediating its ER retention. More than 25 years after the discovery of UGGT (Parodi, 2007; Parodi et al., 2014), recent structural and functional work has uncovered the protein's multi-domain architecture and provided preliminary evidence of its inter-domain conformational flexibility (Calles-Garcia et al., 2017; Roversi et al., 2017; Satoh et al., 2017). Here, we use molecular dynamics (MD) to further characterize UGGT's inter-domain flexibility and present recently obtained CtUGGT crystal structures and activity data. We define and give a numerical estimate of the “Parodi limit,” the maximum distance between a site of misfolding and an N-linked glycan that can be reglucosylated by monomeric UGGT on the same glycoprotein in vitro in response to recognition of misfold at that site. The MD trajectories are discussed in the light of all the available structural and functional data, supporting a one-size-fits-all model of UGGT promiscuity, with an essential role for the UGGT TRXL2 domain.
Results
The CtUGGTKif crystal structure adds to the landscape sampled by previously observed UGGT conformations
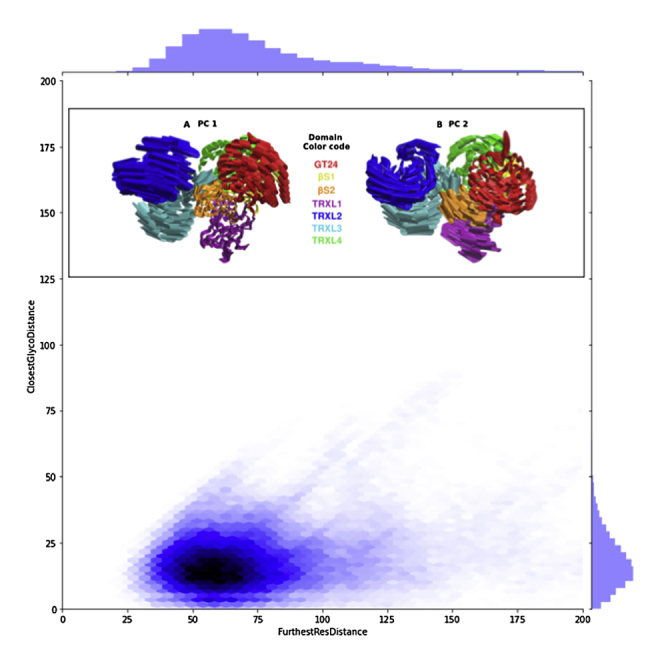
The full-length Chaetomium thermophilum UGGT (CtUGGT) crystal structures revealed four DsbA-like domains (TRXL1–4) arranged in a long arc, terminating in two β sandwiches (βS1 and βS2) tightly clasping the glucosyltransferase family 24 (GT24) domain (Figures 1A and 1B) (Roversi et al., 2017). These UGGT domains are labeled as thioredoxin-like (TRXL) but strictly speaking, the UGGT TRXL2-4 domains belong to a modified version of the thioredoxin fold, the DsbA-like fold: βαβ-αααα-αββα (Pfam DSBA family PF01323). This fold has an extra four-helical subdomain capping one side of a thioredoxin domain βαβ-αββα (Kozlov and Gehring, 2020). The UGGT-TRXL1 domain has a slightly altered and unique topology, with the four-helical subdomain inserted before the thioredoxin one: αααα-βαβ-αββα (Kryshtafovych et al., 2018). The wild-type protein crystallized in three different conformations, called “closed” (PDB: 5N2J, Figure 1A, and gray with purple TRXL2 and TRXL3 domains in Figure 1C), “open” (PDB: 5MZO, Figure 1B and gray with green TRXL2 and TRXL3 domains in Figure 1C), and “intermediate” (PDB: 5MU1, gray with yellow TRXL2 and TRXL3 domains in Figure 1C) (Roversi et al., 2017). Additionally, the mutant CtUGGTD611C/G1050C, engineered to form an extra disulfide bridge between the TRXL2 and βS2 domains, was trapped in a “closed-like” conformation (PDB: 5NV4, gray with orange TRXL2 and TRXL3 domains in Figure 1C). Those four CtUGGT structures mainly differ in the spatial organization of domains TRXL2 and TRXL3 (respectively blue and cyan in Figures 1A, 1B, and 2, and Video S1). The TRXL2 domain is rotated by different amounts with respect to the rest of the protein and adopts different degrees of proximity to it. The TRXL3 domain instead appears in the same relative conformation in all structures, except for the “open” one, in which the TRXL3 and TRXL1 domains move apart, leading to the opening of a cleft between them (Figures 1B and 2A).
Figure 1.

Crystal structures of CtUGGT and the first two principal components of the joint MD simulations
(A and B) Structural comparison between (A) CtUGGT in “closed” (PDB: 5N2J) and (B) “open” (PDB: 5MZO) conformations, colored domain by domain: TRXL1 (residues 45–220), magenta; TRXL2 (residues 414–656), blue; TRXL3 (residues 667–880), cyan; TRXL4 (residues 275–410; 897–950), green; βS1 (residues 28–36; 225–242; 957–1037), yellow; βS2 (residues 1,039–1,149), orange; GT24 (residues 1,197–1,475), red.
(C) Superimpositions of all four CtUGGT X-ray structures available prior to this publication; domains colored in gray (GT24, βS1, βS2, and TRXL4) represent the relatively rigid portion of the molecule (RMSDCα less than 0.750 Å), which was used to align the structures. TRXL2 and TRXL3 domains are colored as follows: purple, “closed” conformation (PDB: 5N2J); orange, D611C-G1050C mutant also known as “closed-like” conformation (PDB: 5NV4); yellow, “intermediate” conformation (PDB: 5MU1); green, “open” conformation (PDB: 5MZO).
(D) Superimposition of the “open” conformation (TRXL2 and TRXL3 domains in green) with the recently reported “new-intermediate” CtUGGTKif conformation (TRXL2 and TRXL3 domains in magenta) (PDB: 6TRF).
(E and F) The first two principal components (PCs) of the joint MDs. Domains colored as in (A) and (B).
Figures made in VMD (Cross et al., 2009).
Figure 2.

Main UGGT motions and conformational coordinates
(A) Simplified representation of CtUGGT overall movements. “Clamping” movement between domains TRXL3 and TRXL1; “bending” movement between TRXL2 and the core comprising domains GT24-βS1-βS2-TRXL4; “twisting” movement of TRXL2 with respect to TRXL3. The gray area represents the strong structural inter-domain orientation invariance of the TRXL4-βS1-βS2-GT24 domains.
(B–D) Conformational coordinates (CCs) for describing CtUGGT conformational states: TRXL1 (magenta), TRXL2 (blue), and TRXL3 (cyan). (B) Along “CC1,” the “clamping” coordinate measures the openness of the cleft between the TRXL1 and TRXL3 domains. (C) Along “CC2,” the “bending” coordinate measures the distance between the TRXL2 and GT24 domains across the central saddle. (D) Along “CC3,” the “twisting” coordinate changes with the relative orientation of the TRXL2 and TRXL3 domains.
(B), (C), and (D) made in VMD (Cross et al., 2009).
Five crystal structures of CtUGGT, in surface representation, with domains TRXL2 and TRXL3 in color, and the rest of the protein in gray
We describe here a fifth full-length CtUGGT structure (hereafter CtUGGTKif, PDB: 6TRF), obtained from recombinant protein purified from mammalian cells treated with the mannosidase inhibitor kifunensine. We used this mannosidase inhibitor to prevent elaboration of N-linked glycans along the secretory pathway and ensure homogeneous high-mannose glycosylation, in the hope of obtaining better diffracting crystals. Despite carrying mostly high-mannose glycans as expected (see http://doi.org/10.5281/zenodo.3608191), CtUGGTkif yielded a crystal which diffracted only to 4.1 Å. Overall the thermal motion is rather high (<B> = 270 Å2, see Table S3). This is likely due to loose crystal packing. Alternatively, kifunensine has inhibited ERAD mannosidases and our crystal may have grown from a mixture of molecules bearing minor folding defects: the high B factors in this case would be modeling static disorder. In addition to the three regions that are usually disordered in CtUGGT crystal structures (namely the TRXL4 loop, residues 246–276; the flexible linker between BS2 and the GT24 domain, residues 1,153–1,192; and the residues between the last helix and the ER retrieval motif at the C terminus, residues 1,474–1,510), this structure has disordered regions in the center of the TRXL2 domain (missing residues 461–505); and at the boundary between TRXL3 and TRXL4 (missing residues 862–886). The resolution of the CtUGGTKif crystal structure is nevertheless sufficient to reveal that the molecule adopts a so far unobserved conformation, which we label “new-intermediate.” Figure 1D shows a superimposition of the “open” conformation (TRXL2 and TRXL3 domains in green) with the “new-intermediate” conformation (TRXL2 and TRXL3 domains in magenta). The CtUGGTkif “new-intermediate” conformation combines a TRXL1-TRXL3 distance as found in the “open” conformation and a TRXL2/TRXL3 relative orientation similar to the one found in the “closed-like” conformation. In what follows, we refer to the volume at the center of the UGGT molecule (between the βS1-βS2:GT24 portion and the TRXL2 domain) as the “central saddle;” the volume between the TRXL1 and TRXL3 domains are referred to as the “cleft” (Figure 2A).
To establish a framework for the discussion of UGGT inter-domain motions, we define here three collective conformational coordinates (CCs) (Figures 2A–2D). “CC1,” or “clamping,” measures the distance between the centers of mass of the TRXL1 and TRXL3 domains and the openness of the cleft between them (Figure 2B). “CC2,” or “bending,” measures the angle between the centers of mass of the TRXL1, TRXL2, and TRXL3 domains, and the proximity of the TRXL2 and GT24 domains across the central saddle (Figure 2C). Lastly, “CC3,” or “twisting,” measures the dihedral angle between the Cα atoms of residues CtUGGT Y518, F466, T863, and I735 (the first two residues in the TRXL2 and the last two in the TRXL3 domain). Thus, the extent of UGGT twisting informs on the relative orientation of the TRXL2 and TRXL3 domains (Figure 2D).
Table 1 reports the values of the CCs for the conformations observed in CtUGGT X-ray structures. The TRXL1:TRXL3 domain clamp is open in the “new-intermediate” CtUGGTKif structure (CC1 = 43.2 Å). In the same structure, the TRXL2:TRXL3 domain pair twist adopts a middle-of-the-range value (CC3 = 3.2°). The pair of CC1/clamping and CC3/twisting values for the “new-intermediate” CtUGGTKif structure—compared with the values of CC1 and CC3 in previously determined CtUGGT structures—suggest that UGGT clamping and twisting motions may be to an extent independent of one another.
Table 1.
Values of the conformational coordinates for the conformations observed in CtUGGT X-ray structures and in extreme MD conformations
| CtUGGT PDB ID | Conformation | CC1 (Å) | CC2 (°) | CC3 (°) |
|---|---|---|---|---|
| 5MZO | open | 41.5 | 123.9 | −13.1 |
| 5MU1 | intermediate | 37.5 | 133.0 | −13.4 |
| 6TRF | intermediate | 43.2 | 102.8 | 3.7 |
| 5N2J | closed | 36.9 | 115.5 | 22.2 |
| CtUGGTD611C/G1050C, 5NV4 | closed | 37.1 | 116.4 | −0.7 |
| CtUGGTS180C/T742C, 6TRT | closed | 35.1 | 119.1 | 48.8 |
| MD “W” | closed | 40.3 | 88.7 | 9.5 |
| MD “X” | open | 47.0 | 125.0 | −9.7 |
| MD “Y” | open | 38.6 | 148.8 | −2.6 |
| MD “Z” | open | 37.7 | 143.8 | 15.9 |
UGGT's motions can be described in simple terms as two rigid groups of domains moving with respect to each other
Next, we asked whether the conformational landscape spanned by CtUGGT full-length crystal structures can be extended by in silico MD. We performed 250-ns long MD simulations starting from four of the CtUGGT crystal structures (Figures 1E, 1F, 3, and S3; Videos S2 and S3). Compared with the set of crystal structures, UGGT MD trajectories do indeed span a wider conformational landscape. Principal components (PCs, also called essential modes [Capece et al., 2008]) were computed from the four individual MD trajectories and from the fusion of all four MDs into a single trajectory. Overall, UGGT's motions can be described in simple terms as two rigid groups of domains moving with respect to one another. One group is formed by domains TRXL2-TRXL3 and the other is formed by domains TRXL1-TRXL4-βS1-βS2-GT24—the latter group is enclosed in a gray circle in Figure 2A. The interface between domains TRXL3 and TRXL4 acts as a hinge region between the two domain groups.
Figure 3.

CtUGGT projected MD trajectories
Projections of individual MD trajectories and their respective X-ray starting structures onto the full conformational landscape as described by the first and second PCs, colored as a function of time. Domains colored as in Figures 1A and 1B.
(A) MD trajectory projection starting from the “closed structure.”
(B) MD trajectory projection starting from the “intermediate structure.”
(C) MD trajectory projection starting from the “open structure.”
(D) MD trajectory projection starting from the “new-intermediate structure.”
In red, we list a few CtUGGT structures representative of extreme values of the conformational coordinates, as identified within the MD conformational landscape (see Figure 4A). Figures made in RStudio (Team, 2016).
The first PC of the joint MD simulation describes the transition between “open” and “closed” states and follows domain TRXL2 bending toward domain βS2 across the central saddle, with TRXL3 and TRXL1 clamping together across the cleft at the same time. Domains colored as in Figure 2.
The second PC of the joint MD simulation describes a movement in which the TRXL2 domain rotates with respect to TRXL3, and the βS2, TRXL1, and TRXL4 domains also undergo motion. Domains colored as in Figure 2.
The first two principal components of the joint MD simulation (PC1 and PC2) are illustrated in Figures 1E, 1F, and 3. PC1 and PC2 suffice to parameterize most of the observed motion. PC1 describes the transition between “closed” and “open” states and follows domain TRXL2 bending toward domain βS2 across the central saddle, with TRXL3 and TRXL1 clamping together across the cleft at the same time (Figures 1E and 3; Video S2). Figures 3B and 3C show that the MD simulations starting from the “intermediate” and “open” crystal structures both move significantly along PC1 and visit both “open” and “closed” states. The MD simulation starting from the “intermediate” structure drifts to the “closed” state and beyond, reaching very open conformations (Figure 3B). The MD simulation starting from the “open” structure shows a back-and-forth movement along PC1 (Figure 3C).
PC2 describes a movement in which the TRXL2 domain rotates with respect to TRXL3 (Figures 1F and 3; Video S3), with the βS2, TRXL1, and TRXL4 domains also undergoing motion. The motion encoded by PC2 is well represented in the MD starting from the “new-intermediate” CtUGGTKif structure, whose projection in Figure 3D also shows a considerable degree of back-and-forth movement.
Does the UGGT catalytic domain detach from the rest of the molecule?
The CtUGGT βS1-βS2:GT24 portion of the molecule behaving as one relatively rigid structure throughout the MD simulations is hardly a surprise. The βS1-βS2:GT24 interface buries a 1,400-Å2 surface, with a calculated −7.1 kcal/mol solvation free energy gain (Krissinel, 2015). The βS1-βS2:GT24 interface is supported by 16 hydrogen bonds, five salt bridges, and 11 hydrophobic interactions, involving 86 residues overall (Figure S1A). The PISA server Complex Formation Significance Score is 1.0 (Krissinel, 2015), suggesting that the contacts in the CtUGGT βS1-βS2:GT24 interface are sufficient to support the physiological nature of the observed Nterm:Cterm inter-domain structure. The solvation free energy gain computed by the same server has a p value of 0.326. p < 0.5 indicates interfaces with higher than average hydrophobicity, implying that the interface is likely interaction specific (Krissinel, 2015).
The tight association we observe between the GT24 and βS1-βS2 domains is at odds with a hypothesis formulated on the basis of negative-stain electron microscopy (EM) and atomic force microscopy (AFM) of Thermomyces dupontii UGGT (TdUGGT) (Satoh et al., 2017; Satoh and Kato, 2018). That study proposed that the UGGT GT24 domain assumes a number of different relative orientations with respect to the rest of the molecule, enabled by the flexible linker between the βS2 and GT24 domains. No full-length crystal structure is available for TdUGGT. Based on sequence conservation, the GT24:βS1-βS2 interface of TdUGGT is also likely to be stable: of the 48 residues in the UGGT βS1-βS2:GT24 interface, 44 are conserved between TdUGGT and CtUGGT, and none of the four residue differences would likely abrogate contributions to the GT24:βS1-βS2 interface (see Figure S1A). Hydrogen-deuterium exchange mass spectrometry (HDX-MS) data measured in solution for Drosophila melanogaster UGGT (DmUGGT) (Calles-Garcia et al., 2017) also support solvent inaccessibility of the residues buried in the DmUGGT βS1-βS2:GT24 interface (Figure S1B). These data taken together prompt the hypothesis that the GT24 and βS1-βS2 domains constitute a rigid group in TdUGGT also (and, by extension, in UGGTs across all eukaryotes), just as observed in our full-length CtUGGT structures and MD simulations.
In the absence of a full-length TdUGGT crystal structure, the only information about the relative orientation of TdUGGT GT24 and βS1-βS2 domains comes from a 25-Å negative-stain EM reconstruction of TdUGGT in complex with an anti-TdUGGT antibody fragment (Fab) (Satoh et al., 2017; Satoh and Kato, 2018). To check whether the TdUGGT negative-stain EM reconstruction is compatible with a model in which GT24 and βS1-βS2 domains also form a rigid group, we generated a full-length TdUGGT homology model. We also selected a representative Fab structure from the Protein Data Bank (PDB). We then fitted the TdUGGT and Fab models both to the 25-Å negative-stained EM map for the complex of TdUGGT with its Fab and (separately) to its enantiomeric mirror image (Robert and Gouet, 2014). The TdUGGT:Fab models fitted to the original and inverted hands have been deposited in the PDB-DEV database (accession code PDBDEV_00000054). The correlation coefficients between the 25-Å negative-stained EM map and the TdUGGT:Fab models are around 90% for the fits to the original (Figures S2A–S2C) and the inverted hand map (Figures S2D–S2F) for both TdUGGT and Fab models. In the fitted models, the Fab contacts the 440–460 portion of TdUGGT domain TRXL2, in agreement with the published Fab epitope (residues TdUGGT 29–468) (Satoh et al., 2017; Satoh and Kato, 2018). In conclusion, the 25-Å negative-stained EM map of the complex of TdUGGT with its Fab can be fitted by a full-length TdUGGT model without invoking any detachment of the catalytic domain from the βS1-βS2 region, contrary to what is stated in Satoh et al. (2017) and Satoh and Kato (2018).
UGGT inter-domain conformational mobility spans a wide range of conformations
As shown in Figure 3, MD simulations take CtUGGT beyond the space sampled by the X-ray structures. In particular, the MD simulations starting from the “open,” “intermediate,” and “new-intermediate” CtUGGTKif structures reach conformations with extreme PC values (Figure 4A and Table 1). Most notably, the structure labeled “W” in Figure 4 represents an extreme version of a closed state. It reveals that the seven UGGT domains can converge to a conformation of very compact overall shape. At the opposite end of the UGGT conformational landscape, structures labeled “X,” “Y,” and “Z” resemble extreme open-like states. Structure “X” in particular presents a notable opening of the TRXL1-TRXL3 cleft along the clamping motion described by CC1, while also showing a considerable degree of twisting along CC3. In contrast to “X,” structures “Y” and “Z” both exhibit a clamped cleft, but at extreme CC2 values. These MD conformations suggest that UGGT is able to push the bending motion even further than observed in the “open” structure while at the same time retaining a clamped cleft.
Figure 4.

MD snapshots with extreme PC values
The UGGT “one-size-fits-all adjustable spanner” model.
(A) A few CtUGGT structures representative of extreme values of the conformational coordinates (CCs), as identified within the MD conformational landscape. W, “clamped, bent, and twisted shut” (small values of CC1, CC2, and CC3). X, “clamped open” and “twisted open” (large CC1 and CC3 values); Y and Z, “clamped shut” (smaller values of CC1) but “bent open” (large CC2 values). Domains colored as in Figures 1A and 1B.
(B) Two CtUGGT conformations in complex with experimentally validated substrates of different sizes. The bright-green region shows the active site. Upper panel: crambin in complex with CtUGGT “closed” crystal structure, conformation, MD-derived structure CtUGGT “W” of (A). Lower panel: exo-(1,3)-β-glucanase in complex with MD-derived structure CtUGGT “Y” of (A).
Figures made in VMD (Cross et al., 2009).
Measurements of the central saddle surface area in the observed UGGT MD conformations, from the most compact structure, “W,” to the most open structure, “Y,” span the range 8,600–11,300 Å2, with average values around 9,200–9,700 Å2 for most conformations (Video S4). Substrate glycoproteins with a “radius of gyration” (ROG) ⪟15 Å and around 150–200 residues or less would snugly fit in the central saddle of compact or middle-of-the-range UGGT conformers (Table S1 and Figure 4B, upper panels). In contrast, for binding of larger substrates (15 Å ⪟ ROG ⪟ 23 Å, and 200–500 residues), an opening of the central saddle would be needed (Table S1 and Figure 4B, lower panels).
Snapshots from the MD simulations of CtUGGT (gray), from a closed to an open conformation, with the residues contributing to the saddle surface colored according to the domain to which they belong (see Figure 2).
In vitro monomeric UGGT activity implies the existence of the Parodi limit
Importantly, irrespective of the misfolded glycoprotein substrate, the finite size of UGGT puts an upper limit to the maximum distance between a site of misfold and an N-linked glycan that monomeric UGGT can reglucosylate on the same glycoprotein substrate in vitro. We propose the name “Parodi limit,” in honor of Armando J. Parodi (Parodi, 2007), for the maximum distance between a site of misfolding and an N-linked glycan that can be reglucosylated by monomeric UGGT in vitro, in response to recognition of misfold at that site. On the basis of our CtUGGT MD simulations at 300 K and on the conformational mobility of Man9GlcNAc2 N-linked glycans (Mackeen et al., 2009), we estimate the Parodi limit to be in the region of 70–80 Å.
The relevance of this limit to UGGT in vivo activity remains to be elucidated. If UGGT acts as a monomer in vivo and does not rely on partner proteins in the ER to recognize its clients, the Parodi limit would impose evolutionary pressure on glycoproteins to evolve glycosylation sites within the same distance from their folding “Achilles' heels.” To probe this hypothesis, we have analyzed a sample of 1,244 glycoproteins structures in the PDB. The frequency distributions of the distance between every amino acid and its closest (and second-closest) N-linked glycan in these proteins are illustrated in Figure 5, together with the frequency distribution of aminoacid-aminoacid distances in the same structures (as a control). It is apparent that 99.61% of residues in these glycoproteins are closer to an N-glycosylation site than the Parodi limit and that this cannot be explained simply in terms of average glycoprotein size (Figures 5A–5D).
Figure 5.

Frequency distributions of distances between glycoprotein residues and their nearest and second-nearest N-linked glycan
Frequency distributions calculated over a sample of 1,244 glycoprotein crystal structures in the PDB.
(A) Blue and orange: frequency distribution of the distance from every amino acid to its closest and second-closest N-linked glycan, respectively; green: frequency distribution of all unique aminoacid-aminoacid distances in the same structures.
(B) Cumulative distributions of the distances in (A) (same color codes).
(C) Frequency distribution of the distance from every amino acid to its closest N-linked glycan versus frequency distribution of all unique aminoacid-aminoacid distances.
(D) Frequency distribution of the distance from every amino acid to its second-closest N-linked glycan versus frequency distribution of all unique aminoacid-aminoacid distances.
(E) Correlation between the distances to the closest and second-closest glycan.
(F) Cumulative distribution of the radius of gyration (ROG) of the glycoproteins. Red lines indicate ROG higher than or equal to that in 50% of the structures.
UGGT activity depends on its inter-domain conformational mobility
In UGGT-mediated reglucosylation assays of urea-misfolded bovine thyroglobulin, both CtUGGTN796C/G1118C and CtUGGTD611C/G1050C mutants had lower activity than wild-type CtUGGT, while CtUGGTN796C/G1118C had a higher catalytic activity and a lower melting temperature than CtUGGTD611C/G1050C (Roversi et al., 2017). Due to the extra disulfide bridge, CtUGGTN796C/G1118C cannot attain the “open” state, while CtUGGTD611C/G1050C can attain neither the “open” nor the “intermediate” conformation. As evidenced in Figure S3A, the MD trajectory starting from the CtUGGTD611C/G1050C structure shows significantly restricted mobility along the first PC, confirming that the extra disulfide bridge in CtUGGTD611C/G1050C tethers the TRXL2 and βS2 domains in a closed conformation. Along the second PC, CtUGGTD611C/G1050C moves further than the other double Cys mutants. The CtUGGTN796C/G1118C mutant, on the other hand, still retains most of its mobility, being able to explore a similar conformational space as those observed for wild-type CtUGGT (Figure S3B). Taken together, these results suggest that the “bending” motion is important for reglucosylation of this particular substrate.
To probe the functional role of the “clamping” motion uncovered in the present analysis, we engineered four double-cysteine CtUGGT mutants, CtUGGTG178C/A786C, CtUGGTG177C/A786C, CtUGGTG179C/T742C, and CtUGGTS180C/T742C, all designed to form disulfide bridges across the TRXL1 and TRXL3 domains, clamping shut the cleft between them. The CtUGGTG178C/A786C failed to express and was not studied any further. The presence of the engineered disulfide bridges in the remaining three mutants was confirmed by mass spectrometry (Figure S4). The crystal structures of CtUGGTG177C/A786C and CtUGGTS180C/T742C were determined to about 4.6-Å resolution. Both crystal structures show the TRXL3 domain tethered to the TRXL1 domain by the extra disulfide bridge (Figure 6A). We tested the in vitro activity of the three “clamped-shut” double Cys mutants (in addition to the activity of the wild type and the already published CtUGGTD611C/G1050C) on urea-misfolded bovine thyroglobulin. Despite their structural similarity, the CtUGGTS180C/T742C and CtUGGTG177C/A786C mutants differ significantly in their ability to reglucosylate urea-misfolded bovine thyroglobulin: the former is more active than wild-type CtUGGT while the latter has activity similar to that of the wild type (Figure 6B).
Figure 6.

CtUGGT double Cys and truncation mutants
(A) The TRXL1 (magenta) and TRXL3 (cyan) domains in the crystal structures of CtUGGTG177C/A786C (PDB: 6TS8, dark colors) and CtUGGTS180C/T742C (PDB: 6TRT, lighter colors). The disulfide bonds are in sphere representation.
(B) Reglucosylating activity of CtUGGT double Cys mutants and wild-type (WT) CtUGGT against urea-misfolded bovine thyroglobulin (mean values and standard deviation over three independent replicas).
(C) Crystal structure of CtUGGT-ΔTRXL2 (PDB: 6TS2, copy “A,” solid colors) overlaid onto wild-type CtUGGT (“open” conformation, PDB: 5MZO, semi-transparent). Domains colored as in Figures 1A and 1B.
(D) Activity of CtUGGT-ΔTRXL2 and CtUGGT-ΔTRXL3 against urea-misfolded bovine thyroglobulin, compared with wild-type (WT) CtUGGT (mean values and standard deviation over three independent replicas).
(A) and (C) made in PyMOL (Rigsby and Parker, 2016).
CtUGGT-mediated reglucosylation of urea-misfolded bovine thyroglobulin requires the TRXL2 domain
To assay the contributions of individual UGGT TRXL domains to UGGT reglucosylating activity, we cloned three mutants of CtUGGT, each lacking one of the TRXL1-3 domains: CtUGGT-ΔTRXL1, lacking residues 42–224; CtUGGT-ΔTRXL2, lacking residues 417–650; and CtUGGT-ΔTRXL3, lacking residues 666–870. CtUGGT-ΔTRXL1 did not express and was not studied further. CtUGGT-ΔTRXL2 and CtUGGT-ΔTRXL3 expressed and were purified. CtUGGT-ΔTRXL2 also yielded crystals, enabling crystal structure determination by X-ray diffraction to 5.7-Å resolution. At this resolution, the CtUGGT-ΔTRXL2 crystal structure most closely resembles the “closed” structure (root-mean-square deviationCα [RMSDCα] 1.32 Å with PDB: 5NV4, over 975 residues) apart from a minor rearrangement of the TRXL3 domain, which moves away from the rest of the truncated molecule (Figure 6C). CtUGGT-ΔTRXL2 and CtUGGT-ΔTRXL3 reglucosylation activity assays against urea-misfolded bovine thyroglobulin detect impaired reglucosylation activity upon deletion of TRXL3 and complete loss of activity upon deletion of TRXL2 (Figure 6D).
Discussion
Since the discovery of UGGT in 1989 (Parodi, 2007; Trombetta et al., 1989), UGGT activity studies have used a range of glycoprotein substrates (Trombetta et al., 1989; Taylor et al., 2004; Ritter and Helenius, 2000; Ritter et al., 2005), small-size glycosylated synthetic compounds (Totani et al., 2006, 2009), and chemically synthesized misfolded glycoproteins (Izumi et al., 2016a, 2016b; Kiuchi et al., 2018). In addition to glycoprotein monomers, UGGT also surveys the quaternary structure of glycoprotein oligomers and larger multi-glycoprotein complexes (Keith et al., 2005; Zhang et al., 2011; Gardner and Kearse, 1999). A comprehensive list of physiological UGGT substrate glycoproteins has not been compiled, and the molecular detail of UGGT:substrate interactions remains uncharacterized. Yet it is apparent that the enzyme is highly promiscuous. The UGGT2 isoform (only present in higher eukaryotes) is competent in reglucosylating glycopeptides (Takeda et al., 2016), suggesting duplication of the UGGT gene and evolution of two isoforms with separate pools of misfolded glycoprotein substrates. If this is the case, the “UGGT1-ome” and “UGGT2-ome” (defined as full lists of clients of UGGT1 and UGGT2, respectively [Tax et al., 2019]) would contain distinct (although possibly overlapping) sets of substrate glycoproteins. Still, each substrate glycoprotein potentially presents a unique relative orientation and distance between the site of misfold and the N-linked glycan receiving the glucose. How each UGGT isoform can reglucosylate misfolded glycoproteins of such a wide variety of different sizes and shapes therefore constitutes a major open question.
Our MD simulations of CtUGGT confirm that—despite its tightly woven topology (Roversi et al., 2017)—the enzyme is indeed quite flexible. More importantly, our analysis of the observed UGGT MD conformational landscape establishes the framework necessary to discuss the enzyme's dynamics. The molecule's inter-domain conformational motions can be described in terms of three simple CCs: the relative movement between domains TRXL3 and TRXL1, resulting in the opening and closing of the cleft between them (“clamping,” along CC1); the movement restricted to TRXL2 moving closer or further away from the relatively rigid core composed of domains GT24-βS1-βS2-TRXL4 (“bending,” along CC2); and the rotation of TRXL2 with respect to TRXL3 (“twisting,” along CC3). The three motions are to some extent independent of each other. These observations open the way to the cloning, expression, and purification of Cys quadruple mutants such as CtUGGTG177C/A786C, D611C/G1050C: these mutants would block the molecule in a clamped and bent closed conformation across the cleft and the central saddle, respectively. Equivalent mutants would likely aid structural studies of mammalian UGGTs, which so far have resisted structural determination (Parodi et al., 2014).
UGGTs recombinantly expressed or tissue-purified from eukaryotic cells have all so far revealed cleavage in the flexible linker between the folding sensor N-terminal portion and the catalytic GT24 domain (see a survey in Roversi et al., 2017). The one study speculating large relative movements between the two portions of the UGGT molecule thanks to this flexible linker (Satoh et al., 2017; Satoh and Kato, 2018) was based on bacterially expressed T. dupontii UGGT, TdUGGT. The GT24 and βS1-βS2 domains of TdUGGT in that study can indeed be pulled apart by AFM: this experiment likely induces mechanical denaturation, breaking the interface between these domains in a non-physiological manner. Here, we consulted all the available biochemical and structural evidence to test the hypothesis that the UGGT βS1-βS2 and GT24 domains also constitute a rigid unit in TdUGGT. We analyzed all crystal structures of full-length UGGTs and their mutants, their MD trajectories, HDX-MS data for DmUGGT (Calles-Garcia et al., 2017), and the 25-Å negative-stained EM map for the complex between TdUGGT and an anti-TdUGGT Fab (Satoh et al., 2017; Satoh and Kato, 2018). We found no evidence suggesting separation of the βS1-βS2 and GT24 domains on either side of the cleaved flexible linker. Claims to the contrary in Satoh et al. (2017) and Satoh and Kato (2018) were likely due to difficulties in docking the N-terminal (PDB: 5Y7O) and C-terminal portions (PDB: 5H18) of TdUGGT separately into the negative-stain EM map, without any higher-resolution knowledge of a full-length UGGT structure. It is possible that the energy needed for disrupting the UGGT GT24:βS1-βS2 interface be supplied for example by ATP hydrolysis. Of course, if the cleavage at the UGGT flexible linker (observed in all eukaryotically expressed UGGT to date) is physiological, the two portions of the molecule would then fly apart upon disassociation.
Based on the data available, UGGT's promiscuity is not likely dependent on the flexible linker between the catalytic domain and the N-terminal misfold sensing region. Rather, it appears to be underpinned by the motions uncovered by the MD simulations. The question remains regarding UGGT's reported ability to survey not only folding of small- and medium-size glycoprotein monomers but also the quaternary structure of glycoprotein oligomers and larger multi-glycoprotein complexes (Keith et al., 2005; Zhang et al., 2011; Gardner and Kearse, 1999). The UGGT inter-domain movements (as uncovered by our MD simulations) extend beyond what was observed in the crystal structures. Indeed, in silico modeling suggests that the extended UGGT conformations sampled by MD could accommodate glycoprotein substrates of different sizes. Such extended conformations would enable UGGT reglucosylation across a wide range of distances between an N-glycosylation site and a site of misfold.
UGGT is active in vitro as a monomer (as judged by its size-exclusion chromatography elution volume [Roversi et al., 2017]). Activity of monomeric UGGT in vitro implies the existence of an upper bound to the distance between a site of misfold on a UGGT substrate and the closest N-linked glycan the enzyme can reglucosylate on the same substrate in vitro. We introduce the term “Parodi limit” for this upper bound, in honor of Armando J. Parodi (Parodi, 2007). On the basis of the most “open” conformations observed in the MD simulations carried out on CtUGGT at 300 K, we estimate the Parodi limit to be close to or less than 70–80 Å. The current lack of knowledge about the UGGT site(s) of misfold recognition, the considerable conformational mobility of the N-linked glycan, and the unknown dependence of the UGGT conformational landscape on the temperature make it difficult to put an estimated error on this value. Functional data from UGGT-mediated reglucosylation of a series of rigid, misfolded UGGT glycoprotein substrates (each bearing one recombinantly engineered N-linked glycosylation site at a specific distance from a single site of misfold common to all substrates in the series) would enable experimental estimation of the Parodi limit. Ideally, one such series of artificial N-linked glycosylation sites at varying distances from a single site of misfold would have to be engineered for a number of different substrate glycoprotein scaffolds in order to minimize the dependency of the Parodi limit estimation from a given substrate series and enable estimation of a standard error on that value.
In vivo/in cellula, it is of course possible that UGGT misfolded glycoprotein recognition can be mediated by UGGT dimers/multimers or aided by UGGT ER partner proteins. One such candidate is the ER HSP70 BiP ATPase, which is found in ER multiprotein complexes with UGGT1 (Kastritis et al., 2017; Meunier et al., 2002). The physiological relevance of the Parodi limit to UGGT's activity in vivo therefore remains to be investigated. Existence of this limit in vivo would in turn imply evolutionary pressure on N-glycosylation sites to develop at accessible distances from the portions of a glycoprotein that are most prone to folding difficulties (i.e., the folding glycoprotein's Achilles’ heels). We made a first preliminary attempt at checking the distance distributions of N-linked glycans from glycoprotein residues, examining them in the light of our current estimation of the Parodi limit. In the absence of detailed knowledge on UGGT-omes, a sample of 1,244 glycoprotein structures in the PDB were analyzed. The analysis assumes glycoprotein crystal structures to be representative of the sizes/shapes of UGGT misfolded clients: UGGT efficiently reglucosylates only misfolded rather than fully unfolded glycoproteins. The majority of glycoproteins in this sample have at least one N-linked glycan within the Parodi limit from every amino acid, irrespective of glycoprotein size. These observations support the hypothesis that the constraints imposed by UGGT structure exert evolutionary pressure on the distribution of N-linked glycosylation sites in UGGT clients.
When it comes to correlating UGGT inter-domain conformational mobility with its activity, among the CtUGGT double-cysteine mutants tested so far the CtUGGTD611C/G1050C mutant described in Roversi et al. (2017) is the least active in reglucosylating urea-misfolded bovine thyroglobulin. This observation is compatible with MD trajectory of this mutant being the most severely limited one across our simulations. The extra disulfide bridge engineered in this mutant joins the βS2-TRXL2 domains, giving rise to the hypothesis that during the enzyme:substrate encounter, a portion of misfolded thyroglobulin may be accommodated in the UGGT central saddle between these domains. The CtUGGT crystal and cryo-EM structures (Roversi et al., 2017), and our MD simulations, all highlight the TRXL2 domain as the most mobile in the molecule, supporting this hypothesis. The total loss of activity of the CtUGGT-ΔTRXL2 construct in the reglucosylation of urea-misfolded bovine thyroglobulin also points to a critical role for the TRXL2 domain. In the light of these data, it is possible that TRXL2 motions adjust the size or vary the surface area of UGGT's central saddle, making the bending motion a crucial one for activity against this substrate.
As to the UGGT clamping motion, we tested two a priori rather similar double-cysteine mutants, CtUGGTS180C/T742C and CtUGGTG177C/A786C, both designed to clamp the TRXL1-TRXL3 domains shut. Surprisingly, the two mutants differ in their ability to reglucosylate urea-misfolded bovine thyroglobulin, with the former mutant more active than (and the latter mutant having similar activity to) wild-type CtUGGT. These observations point to the possibility that each misfolded glycoprotein substrate may depend to a different degree on a different subset of UGGT inter-domain conformational degrees of freedom. In the light of these data, dissection of the UGGT structure/activity relationship will profit from a number of reglucosylation assays using the same set of UGGT mutants on different glycoprotein substrates.
The CtUGGT-ΔTRXL3 mutant appears to be competent in reglucosylating urea-misfolded bovine thyroglobulin. Full kinetic characterization would be needed to further test the observed lower activity of the CtUGGT-ΔTRXL3 mutant in terms of slower rate and/or lower substrate affinity. In contrast, CtUGGT-ΔTRXL2 is completely inactive against urea-misfolded bovine thyroglobulin. Substrate recruitment via TRXL2 movements would not require complete burial of the misfolded glycoprotein into the central saddle of the molecule: UGGT would minimally need to establish contact with the portion of substrate containing the misfold. This is plausible for relatively big substrates such as transferrin (77 kDa, radius of gyration = 29.7 Å) or urea-misfolded bovine thyroglobulin (670 kDa, a long, presumably snake-like chain of eleven 60-amino-acid compact domains, no structure available). For smaller substrates, such as glycopeptides or synthetic fluorescent probes, more closed UGGT conformations, bringing TRXL2 toward βS2 and the GT24 domain across the central saddle, may be needed.
Apart from TRXL2, other untested UGGT regions potentially harboring exposed hydrophobic patches are the CtUGGT TRXL4 disordered region (CtUGGT 243–285), the flexible linker around the endo-proteolysis site between the βS2 and GT24 domains (CtUGGT 1,153–1,195), and the residues between the last helix and the ER retrieval motif at the C terminus (CtUGGT 1,474–1,510) (Roversi et al., 2017). Experiments described in a recent report ascribe intrinsic refoldase activity to UGGT (Wang et al., 2020) and await being reproduced. Again, structural and functional data from a range of UGGT mutants and glycoprotein substrates will be required to further test these hypotheses and fully dissect the UGGT structure-function relationship.
The molecular forces supporting UGGT-mediated glycoprotein misfold recognition have been generally hypothesized to be hydrophobic interactions (Caramelo et al., 2003). Our observation that the face of the TRXL2 domain overlooking the central saddle bears distinct patches of hydrophobic residues conserved across UGGT1 sequences (Roversi et al., 2017) supports a model of hydrophobic-mediated misfold recognition. Dependency of misfold recognition on disordered portions of a misfold checkpoint enzyme was observed for the mouse ERAD mannosidase (EDEM), which also preferentially acts on misfolded glycoproteins and was proved to undergo constant ERAD degradation itself (Marin et al., 2012). The hypothesis that UGGT works by having evolved an intrinsically misfolded portion, with which the enzyme would interact with substrate glycoprotein misfolded regions, would in turn imply that UGGT may reglucosylate itself. If this is the case, UGGT would also be likely subjected to constant ERAD demannosylation and degradation. The fact that UGGT bears demannosylated glycans—a hallmark of ERAD (Daikoku et al., 2014, 2015)—is compatible with this hypothesis. If indeed the ERAD and ERQC checkpoint enzymes recognize misfolded glycoproteins via an intrinsically misfolded domain (“it takes one to know one” [Tax et al., 2019]), the associated biochemical costs of this strategy may be the price eukaryotic cells pay in order to afford UGGT/EDEMs as broad-specificity glycoprotein misfolding checkpoints. In vitro and in cellula experiments to test these ideas are in progress.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E.coli DH5-α | New England Bioscience | Cat# C2987I |
| E.coli BL21 | New England Bioscience | Cat# C2530H |
| XL10-Gold Ultracompetent cells | Agilent | Cat# 200317 |
| Chemicals, peptides, and recombinant proteins | ||
| Q5® Hot Start High-Fidelity 2X Master Mix | New England Biolabs | Cat# M0494S |
| NEBuilder® HiFi DNA Assembly Master Mix | New England Biolabs | Cat# E2621S |
| Gibson Assembly Kit | New England Biolabs | Cat# E2611 |
| Bovine Thyroglobulin | Sigma-Aldrich | Cat# T1001 |
| KLD Enzyme Mix | New England Biolabs | Cat# M0554S |
| KLD Reaction Buffer | New England Biolabs | Cat# M0554S |
| QIAprep Spin Miniprep Kit (Qiagen) | QIAGEN | Cat# 27104 |
| EndoFree Plasmid Kits | QIAGEN | Cat# 12362 |
| QIAquick gel extraction kit | QIAGEN | Cat# 28706 |
| CutsmartTM buffer | New England Biolabs | Cat# B7204S |
| AgeI | New England Biolabs | Cat# R0552S |
| KpnI | New England Biolabs | Cat# R0142S |
| PNGase F | New England Biolabs | Cat# P0704S |
| PNGase glycobuffer 2 | New England Biolabs | Cat# B7002S |
| Denaturing buffer | New England Biolabs | Cat# B1704S |
| Anthranilic Acid | Sigma-Aldrich | Cat# A89855 |
| OptiPRO™ SFM | ThermoFisher Scientific | Cat# 12309019 |
| FreeStyleTM 293 Expression Medium | ThermoFisher Scientific | Cat# 12338001 |
| FreeStyleTM MAX transfection reagent | ThermoFisher Scientific | Cat# 16447100 |
| SOC Media | New England Biolabs | Cat# B9020S |
| Kifunensine | Cayman Chemical | Cat# 109944-15-2 |
| Carbenicillin | Sigma-Aldrich | Cat# C1389 |
| CtUGGT | Ref. (Roversi et al., 2017) | N/A |
| CtUGGTS180C/T742C protein | This paper | N/A |
| CtUGGTKif protein | This paper | N/A |
| CtUGGT-ΔTRXL2 protein | This paper | N/A |
| CtUGGTG177C/A786C protein | This paper | N/A |
| Imidazole | Honeywell Fluka | Cat# 56750 |
| HEPES | Sigma-Aldrich | Cat# H3375 |
| MORPHEUS Crystallisation Screen | Molecular Dimensions | Cat# MD1–47 |
| MORPHEUS2 Crystallisation Screen | Molecular Dimensions | Cat# MD1-92 |
| JCSG+ Crystallisation Screen | Molecular Dimensions | Cat# MD1–40 |
| Critical commercial assays | ||
| Sequencing Grade Modified Trypsin | Promega | Cat# V5111 |
| Deposited data | ||
| CtUGGTG1050C/D611C mutant | (Roversi et al., 2017) | PDB ID: 5NV4 |
| CtUGGT ‘closed’ | (Roversi et al., 2017) | PDB ID: 5N2J |
| CtUGGT ‘open’ | (Roversi et al., 2017) | PDB ID: 5MZO |
| CtUGGT ‘intermediate’ | (Roversi et al., 2017) | PDB ID: 5MU1 |
| CtUGGTS180C/T742C mutant | This paper | PDB ID: 6TRT |
| CtUGGTKif mutant | This paper | PDB ID: 6TRF |
| CtUGGT-ΔTRXL2 mutant | This paper | PDB ID: 6TS2 |
| CtUGGTG177C/A786C mutant | This paper | PDB ID: 6TS8 |
| TdUGGT:Fab model | This paper | PDBDEV 00000054 |
| Experimental models: cell lines | ||
| HEK FreeStyleTM 293F cells | ThermoFisher Scientific | Cat# R79007 |
| Oligonucleotides | ||
| Primers for Gibson Assembly | See Table S4 | N/A |
| Primers for mutagenesis to obtain CtUGGT_ΔTRXL1 | See Table S4 | N/A |
| Primers for mutagenesis to obtain CtUGGT_ΔTRXL2 | See Table S4 | N/A |
| Primers for mutagenesis to obtain CtUGGT_ΔTRXL3 | See Table S4 | N/A |
| Primers for mutagenesis to obtain CtUGGTG177C mutation | See Table S4 | N/A |
| Primers for mutagenesis to obtain CtUGGTV178C mutation | See Table S4 | N/A |
| Primers for mutagenesis to obtain CtUGGTS180C mutation | See Table S4 | N/A |
| Primers for mutagenesis to obtain CtUGGTT742C mutation | See Table S4 | N/A |
| Primers for mutagenesis to obtain CtUGGTA786C mutation | See Table S4 | N/A |
| Recombinant DNA | ||
| CtUGGT-pHLsec plasmid | Ref. (Roversi et al., 2017) | N/A |
| CtUGGTS180C/T742C -pHLsec plasmid | This paper | N/A |
| CtUGGTKif -pHLsec plasmid | This paper | N/A |
| CtUGGT-ΔTRXL2 -pHLsec plasmid | This paper | N/A |
| CtUGGTG177C/A786C -pHLsec plasmid | This paper | N/A |
| Software and algorithms | ||
| Empower | Waters Inc. | Version 3.0 |
| AMBER suite | https://ambermd.org/ | Version18 |
| Clustal omega | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Version 1.2.4 |
| Modeller | http://salilab.org/modeller/ | Version 9.19 |
| VMD | (Cross et al., 2009) | Version 1.9.3 |
| RStudio Desktop | RStudio Team (2020). RStudio: Integrated Development for R. RStudio, PBC, Boston, MA URL http://www.rstudio.com/. (RStudio Team, 2016) | Version 1.2.5042 |
| MassMatrix Suite 1.3.3 | https://massmatrix.bio/ | Version 2.4.2 |
| MSconvert from the ProteoWizard toolbox | http://proteowizard.sourceforge.net/ | Version 3.3.19172-57d620127 |
| autoBUSTER | (Blanc et al., 2004) | Version 2.10.3 |
| Phaser | (McCoy et al., 2007) | Version 2.8.3 |
| autoPROC | (Vonrhein et al., 2011) | Version 1.0.5 |
| Coot | (Emsley et al., 2010) | Version 0.9 |
| Refmac5 | (Nicholls et al., 2012) | Version 5.8.0258 |
| Molrep | (Vagin and Teplyakov, 2010) | Version 11.7.02 |
| Chimera | (Pettersen et al., 2004) | Version 1.14 |
| GraphPad Prism | GraphPad Software, San Diego, California USA, www.graphpad.com | Version 8.0.0 for Windows |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Pietro Roversi (pr159@leicester.ac.uk).
Materials availability
Plasmids generated in this study will be made available on request by the Lead Contact with a completed Materials Transfer Agreement (MTA).
Data and code availability
The X-ray crystallographic data and atomic models have been deposited at the Protein Data Bank. The accession number for the CtUGGTKif crystal structure reported in this paper is PDB: 6TRF. The accession number for the CtUGGT-ΔTRXL2 crystal structure reported in this paper is PDB: 6TS2. The accession number for the CtUGGTS180C/T742C crystal structure reported in this paper is PDB: 6TRT. The accession number for the CtUGGTG177C/A786C crystal structure reported in this paper is PDB: 6TS8. The models for the TdUGGT:Fab complex (fitted in the original hand and the inverted hand of the negative stain EM reconstruction) have been deposited in PDB-DEV. The accession number for the TdUGGT:Fab complex reported in this paper is PDBDEV: 00000054.
A list of pieces of software used in this study can be found in the Key Resources Table.
Experimental model and subject details
Bacteria
Commercial E. coli DH-5α chemically competent cells and BL21 cells were purchased from New England Biolabs (NEB) and handled as per manufacturer’s instructions (Cat# C2987I and Cat# C2530H). XL10-Gold Ultracompetent cells were purchased from Agilent and handled as per manufacturer’s instructions (Cat# 200317).
Cell lines
Human epithelial kidney FreeStyle 293F cells (ThermoFisher Scientific) were cultured in FreeStyle 293 Media (ThermoFisher Scientific) in Erlenmeyer flasks with 0.2 μm vent caps (Corning) shaking at 135 revolutions per min (rpm) in a 37 °C incubator kept at 8% CO2.
Methods details
Cloning
All DNA primers were purchased from Sigma. Details of the cloning of full-length CtUGGT are described in (Roversi et al., 2017). All CtUGGT mutants were generated starting from the gene of CtUGGT inserted in Litmus28i (an optimal vector for mutagenesis experiments), using Q5® Hot Start High-Fidelity 2X Master Mix (New England Biolabs - NEB) following manufacturer instructions; briefly: 12.5 μL of Q5® Hot Start High-Fidelity 2X Master Mix (New England Biolabs) were added to 1.25 μL of each forward and reverse primer at 10 μM, 1 μL of CtUGGT:Litmus28i DNA at 1 ng/μL and 9 μL of nuclease-free water, obtaining a 25 μL final volume. To generate the double cysteine mutants, the DNA obtained from the first mutation was used as starting material for the second mutation insertion. PCR amplification was then performed with a personalised protocol for each mutant, as described in detail further on in this section. Kinase, Ligase & DpnI (KLD) treatment: 1 μL of PCR product was mixed with 5 μL of 2X KLD Reaction buffer, 1 μL of 10X KLD Enzyme Mix (both from NEB) and 3 μL of nuclease-free water. The mixture was incubated at room temperature for 5 minutes. The KLD reaction mixture was used to transform E. coli DH-5α chemically competent cells (NEB) using the following protocol: 5 μL of KLD reaction mix were added to a tube of thawed DH-5α competent E. coli cells on ice, and mixed gently for a few seconds; after transformation, the bacteria were incubated on ice for 30 minutes, heat shocked at 42 °C for 30 seconds and incubated on ice again for 5 minutes. 950 μL of SOC media (New England Biolabs) were added to a final volume of 1 mL and the mixture was incubated for 1 hour at 37 °C with gentle shaking at 200/300 rpm. 100 μL of the bacteria were spread onto a pre-warmed (37 °C) LB agar culture plate containing carbenicillin (Sigma-Aldrich, 0.1 mg/mL). The plate was incubated at 37 °C overnight. Colony-PCR was performed on DNA from various colonies by using T7_F (5’-TAATACGACTCACTATAGGG-3’) and T7_R (5’-GCTAGTTATTGCTCAGCGG-3’) primers and the DNA obtained was loaded on a 1% w/v agarose gel and run for 50 minutes at 150 V. Analysis of this gel allowed identification of colonies with amplified DNA of the appropriate size; cells from colonies containing an amplified product of the desired size were used to inoculate 5 mL LB supplemented with 0.1 mg/mL carbenicillin. Following overnight incubation at 37°C, plasmid mini-preps were performed using the QIAprep Spin Miniprep Kit (Qiagen) according to the manufacturer's instructions. Glycerol stocks were obtained by mixing 16% glycerol with 84% bacteria, and freezing and storing at -80 °C. The DNA obtained was sequenced with the appropriate primers. The glycerol stock was used to inoculate 5 mL LB supplemented with 0.1 mg/mL carbenicillin and incubated over night at 37 °C. This culture was then used to inoculate 200 mL LB supplemented with 0.1 mg/mL carbenicillin and the bacteria were incubated at 37 °C and 110 rpm. Upon reaching an OD600nm of 2.0, the cells were spun down at 3320xg for 18 minutes. The pellets were resuspended, and the plasmid maxi-preps were performed to purify the DNA using EndoFree Plasmid Kits (Qiagen), following the recommended protocol. The mutants in Litmus28i were subsequently cloned into the pHLsec expression vector (Aricescu et al., 2006) to contain a hexa-His Tag at the C-terminus. DNA for pHLsec was linearised using AgeI and KpnI restriction enzymes (NEB) at 37 °C for 16 hours in CutsmartTM buffer (NEB). The restriction digest was then run on a 0.8% w/v agarose gel at 150 V for 1 hour. The linearised vector was cut from the gel and purified with a QIAquick gel extraction kit (Qiagen). PCR was then performed on each CtUGGT mutant in Litmus 28i as follow: 1 μL of DNA (1 ng/μL) added at 25 μL of Q5® Hot Start High-Fidelity 2X Master Mix (NEB), 2.5 μL of each forward (pHLsec_CtUGGT_F: 5'-GGTTGCGTAGCTGAAACCGGTCAAGTCGCAGCCTCTCCA-3') and reverse (pHLsec_CtUGGT_R: 5'-GATGGTGGTGCTTGGTACCCTCCCGAACCGTCTTGAC-3') primers and 19 μL of nuclease-free water. PCR protocol: step 1: 98 °C for 30 seconds; step 2: 98 °C for 10 seconds; step 3: 62 °C for 30 seconds; step 4: 70 °C for 150 seconds; step 2-4 were repeated 35 times; step 5: 72 °C for 2 minutes. The PCR products were run on a 0.8% w/v agarose gel at 150 V for 1 hour and the amplified insert was cut from the gel and purified with the same QIAquick gel extraction kit. A Gibson Assembly was then performed using the gel-purified PCR-amplified CtUGGT mutant insert mixed with gel-purified linearised pHLsec at a ratio of 3:1 with NEBuilder® HiFi DNA Assembly Master Mix (NEB) using manufacturer suggested protocol, for 1 hour at 50 °C. 2 μl of this ligation product was added to 50 μl XL10-Gold Ultracompetent cells (Agilent), following the transformation guideline protocol. The cells were then plated on 0.1 mg/mL carbenicillin agar plates and incubated over night at 37 °C. Colony-PCR was performed on DNA from various colonies (using pHLsec_F and pHLsec_R primers) and the DNA obtained was run on a 1% w/v agarose gel for 50 minutes at 150 V. Analysis of this gel allowed identification of colonies with amplified DNA of the appropriate size; mini-prep, glycerol stock, DNA sequencing and maxi-prep were performed to obtain CtUGGT mutant pHLsec plasmid DNA.
Cloning of CtUGGTA786C. Mutation of the CtUGGT into CtUGGTA786C was carried out starting from the gene of CtUGGT inserted in Litmus28i as described before, using forward (A786C_F: 5'-CGCTTACGACtgtTCTCTAGCCAAC-3') and reverse (A786C_R: 5'-ACATCTGGTTCGAACTCG-3') primers. PCR amplification: step 1: 98 °C for 30 s; step 2: 98 °C for 10 s; step 3: 60 °C for 20 s; step 4: 72 °C for 135 s. Steps 2-4 were repeated 25 times; step 5: 72 °C for 2 minutes. KLD treatment and E.coli transformation was performed as described before, and later mini and maxi-prep, as detailed above. CtUGGTA786C:Litmus28i plasmid DNA, 3 mL at 400 ng/μL were obtained.
Cloning of CtUGGTG177C/A786C. To obtain the double mutant CtUGGTG177C/A786C, the second mutation G177C was introduced starting from the gene of CtUGGTA786C in Litmus28i as described above, using forward (G177C_F: 5'-TCGGAAGTTTtgcGTTGGTTCCC-3') and reverse (G177C_R: 5'-TCAAATGGCAGTGTCCGC-3') primers. PCR protocol: step 1: 98 °C for 30 seconds; step 2: 98 °C for 10 seconds; step 3: 66 °C for 30 seconds; step 4: 72 °C for 135 seconds; steps 2-4 were repeated 25 times. Step 5: 72 °C for 2 minutes. After KLD treatment (see above) E. coli DH-5α chemically competent cells were transformed with the DNA as described previously. Mini and maxi-prep, as detailed above, yielded 3 mL of CtUGGTG177C/A786C:Litmus28i plasmid DNA at 700 ng/μL. The insert was then linearised, cloned into pHLsec vector (by Gibson Assembly) as described above, and after mini- and then maxi-prep, 3 mL of CtUGGTG177C/A786C:pHLsec plasmid DNA at 300 ng/μL were obtained.
Cloning of CtUGGTV178C/A786C. To obtain the double mutant CtUGGTV178C/A786C, the second mutation V178C was introduced starting from the gene of CtUGGTA786C in Litmus28i as described above, using forward (V178C_F: 5'-GAAGTTTGGCtgtGGTTCCCGTG-3') and reverse (V178C_R: 5'-CGATCAAATGGCAGTGTC-3') primers. PCR protocol: step 1: 98 °C for 30 seconds; step 2: 98 °C for 10 seconds; step 3: 60 °C for 30 seconds; step 4: 72 °C for 135 seconds; steps 2-4 were repeated 25 times. Step 5: 72 °C for 2 minutes. After KLD treatment (see above) E. coli DH-5α chemically competent cells were transformed with the DNA as described previously. Mini and maxi-prep, as detailed above, yielded 3 mL of CtUGGTV178C/A786C:Litmus28i plasmid DNA at 500 ng/μL. The insert was then linearised, cloned into pHLsec vector (by Gibson Assembly) as described above, and after mini- and then maxi-prep, 3 mL of CtUGGTV178C/A786C:pHLsec plasmid DNA at 700 ng/μL were obtained.
Cloning of CtUGGTS180C/T742C. Mutation of the CtUGGT into CtUGGTT742C was effected starting from the gene of CtUGGT inserted in Litmus28i as described before, using forward (T742C_F: 5'-TCCCAAGGATtgcTCACGTTCCC-3') and reverse (T742C_R: 5'-TTGTGGACAATGTCCAAC-3') primers properly designed. PCR amplification: step 1: 98 °C for 30 s; step 2: 98 °C for 10 s; step 3: 59 °C for 20 s; step 4: 72 °C for 135 s. Steps 2-4 were repeated 25 times; step 5: 72 °C for 2 minutes. KLD treatment and E.coli transformation were performed as described before, and later mini and maxi-prep, as detailed above. CtUGGTT742C:Litmus28i plasmid DNA, 3 mL at 500 ng/μL were obtained. To obtain the double mutant CtUGGTS180C/T742C, the second mutation S180C was introduced starting from the gene of CtUGGTT742C in Litmus28i as described above, using forward (S180C_F: 5'-TGGCGTTGGTtgcCGTGATGTGA-3') and reverse (S180C_R: 5'-AACTTCCGATCAAATGGCAGTGTC-3') primers. PCR protocol: step 1: 98 °C for 30 seconds; step 2: 98 °C for 10 seconds; step 3: 68 °C for 30 seconds; step 4: 72 °C for 135 seconds; steps 2-4 were repeated 25 times. Step 5: 72 °C for 2 minutes. After KLD treatment (see above) E. coli DH-5α chemically competent cells were transformed with the DNA as described previously. Mini and maxi-prep, as detailed above, yielded 3 mL of CtUGGTS180C/T742C:Litmus28i plasmid DNA at 500 ng/μL. The insert was then linearised, cloned into pHLsec vector (by Gibson Assembly) as described above, and after mini- and then maxi-prep, 3 mL of CtUGGTS180C/T742C:pHLsec plasmid DNA at 700 ng/μL were obtained.
Cloning of CtUGGT-ΔTRXL1. The CtUGGT-ΔTRXL1 construct lacks residues CtUGGT 42-224. The deletion of the CtUGGT TRXL1 domain was performed starting from the gene of CtUGGT in Litmus28i as described before, using forward (Δ1_F: 5'-GAGTCTCTGTCCGTCAATGG-3') and reverse (Δ1_R: 5'-AGAGGGGAAAGCGGCTTT-3') primers properly designed. PCR protocol: step 1: 98 °C for 30 seconds; step 2: 98 °C for 10 seconds; step 3: 65 °C for 20 seconds; step 4: 72 °C for 130 seconds; step 2-4 were repeated 25 times; step 5: 72 °C for 2 minutes. KLD treatment and E.coli transformation were performed as described before, and later mini and maxi-prep, as detailed above. CtUGGT-ΔTRXL1:Litmus28i plasmid DNA, 3 mL at 400 ng/μL were obtained. The insert was then linearised, cloned into pHLsec vector (by Gibson Assembly) as described above, and after mini- and then maxi-prep, 3 mL of CtUGGT-ΔTRXL1:pHL-sec plasmid DNA at 500 ng/μL were obtained.
Cloning of CtUGGT-ΔTRXL2. The CtUGGT-ΔTRXL2 construct lacks residues CtUGGT 417-650. The deletion of the CtUGGT TRXL2 domain was performed starting from the gene of CtUGGT in Litmus28 as described before, using forward (Δ2_F: 5’-GCCCTATCAAGACGGAAC-3’) and reverse (Δ2_R: 5’-AAATCTCCGGGGCTCGTC-3’) primers. PCR protocol: step 1: 98 °C for 30 seconds; step 2: 98 °C for 10 seconds; step 3: 64 °C for 20 seconds; step 4: 72 °C for 180 seconds; step 2-5 were repeated 25 times; step 6: 72 °C for 2 minutes. KLD treatment and E.coli transformation were performed as described before, and later mini and maxi-prep, as detailed above. CtUGGT-ΔTRXL2:Litmus28i plasmid DNA, 3 mL at 300 ng/μL were obtained. The insert was then linearised, cloned into pHLsec vector (by Gibson Assembly) as described above, and after mini- and then maxi-prep, 3 mL of CtUGGT-ΔTRXL2:pHL-sec plasmid DNA at 300 ng/μL were obtained.
Cloning of CtUGGT-ΔTRXL3. The CtUGGT-ΔTRXL3 construct lacks residues CtUGGT 666-870. The deletion of the CtUGGT TRXL3 domain was performed starting from the gene of CtUGGT in Litmus28i as described before, using forward (Δ3_F: 5'-ATTTCGGATCTCCCACAG-3') and reverse (Δ3_R: 5'-GTTCTTGTCTTCGGGGAAAATG-3') primers properly designed. PCR protocol: step 1: 98 °C for 30 seconds; step 2: 98 °C for 10 seconds; step 3: 62 °C for 20 seconds; step 4: 72 °C for 130 seconds; step 2-4 were repeated 25 times; step 5: 72 °C for 2 minutes. KLD treatment and E.coli transformation was performed as described before, and later mini and maxi-prep, as detailed above. CtUGGT-ΔTRXL3:Litmus28i plasmid DNA, 3 mL at 500 ng/μL were obtained. The insert was then linearised, cloned into pHLsec vector (by Gibson Assembly) as described above, and after mini- and then maxi-prep, 3 mL of CtUGGT-ΔTRXL3:pHL-sec plasmid DNA at 800 ng/μL were obtained.
Protein expression and purification
All CtUGGT mammalian expression plasmids were transfected into FreeStyleTM 293-F Cells. Human epithelial kidney (HEK) FreeStyleTM 293-F Cells (ThermoFisher Scientific) at 106 cells/mL suspended in FreeStyleTM 293 Expression Medium (ThermoFisher Scientific) were transfected using the FreeStyleTM MAX 293 Expression System (ThermoFisher Scientific). For 50 mL culture, the manufacturer suggested protocol was used: 62.5 μL of FreeStyleTM MAX Reagent (ThermoFisher Scientific) and 62.5 μg of plasmid DNA were each diluted separately into 1 mL of OptiPROTM SFM reduced serum medium (ThermoFisher Scientific), then mixed, incubated for 10 min at room temperature and finally added to the cell suspension. Transfected cells were left shaking in 500 mL Erlenmeyer flasks with 0.2 μm vent caps (Corning) shaking at 135 revolutions per min (rpm) in a 37 °C incubator kept at 8% CO2. All the DNA plasmids contained signal sequences to ensure protein secretion into the supernatant. The cells were harvested and supernatants were separated from cells by centrifugation for 15 minutes at 4 °C and 3,000g. Constructs were purified as follow. The supernatants were added of phosphate buffer saline (PBS) to get 1X to final volume, of imidazole to 5mM final concentration and the pH was adjusted to 7.4-7.6 by adding the needed volume of 2M NaOH solution. The supernatants were filtered by vacuum through a 0.22 μm filter. All protein constructs were purified using Immobilised Metal Affinity Chromatography (IMAC) with a 1 mL HisTrap HP Ni IMAC column (GE Healthcare) (unless otherwise specified) and then size exclusion chromatography (SEC) with a HiLoad Superdex 200 16/60 column (GE Healthcare), unless otherwise specified. A HisTrap HP Ni IMAC column (GE Healthcare) already equilibrated with binding buffer (1x PBS, 5 mM imidazole and pH adjusted to 7.4-7.6 with the needed volume of 2M NaOH solution) was loaded with the cells’ supernatant treated as described above. The IMAC column was washed with 20 column volumes (cV) of binding buffer and protein eluted with a linear gradient over 20 cV from 0 % to 100 % of elution buffer (1x PBS, 400-500 mM imidazole, pH adjusted to 7.4-7.6 with the needed volume of 2M NaOH solution) at 2 mL/min. Peak fractions were pooled and concentrated using a PES membrane centrifugal ultrafiltration device (Sartorius), with the appropriate MW cut-off, to a maximum 5 mL volume. This concentrated protein sample was then loaded to the HiLoad Superdex 200 16/60 SEC column (GE Healthcare) equilibrated against the appropriate SEC buffer (see below). Peak fractions were pooled and concentrated as before; protein concentration measured by loading 1.5 μL of sample on a NanoDrop 1000 spectrophotometer (Thermo Scientific). The calculated ϵ280 (from http://protcalc.sourceforge.net/) was then used to estimate the protein concentration. Protein aliquots were frozen in liquid N2 and stored at -80 °C. SDS-PAGE of SEC fractions was used to assess purity. All SEC chromatography was run at 1 mL/min flow rate on ÄKTA Pure (room temperature) or ÄKTA Start (4 °C) systems (GE Healthcare) unless otherwise specified. CtUGGTKif was expressed in 300 mL (2x150 mL) of HEK293F cells, supplementing the media with 5 μM kifunensine (Cayman Chemical Company). Transfected cells were left shaking at 135 rpm in 0.5 L Erlenmeyer flasks with 0.2 μm vent caps, at 37 °C and 8% CO2 incubator, for 6 days. Size exclusion chromatography was performed with SEC buffer: 20 mM Na-HEPES pH 7.5, 150 mM NaCl. The final concentration of CtUGGTKif (1 mL volume) was 7.24 mg/mL. CtUGGTS180C/T742C was expressed in 400 mL of HEK293F cells. Transfected cells were left shaking at 135 rpm in 0.5 L Erlenmeyer flasks with 0.2 μm vent caps, at 37 °C and 8% CO2 incubator, for 4 days. The IMAC step used a 5 mL HisTrap column. Eluted fractions were analysed using SDS-PAGE and the protein was detected in the flow-through, having apparently failed to bind to the IMAC column. We can only speculate that the His-tag is either proteolysed or sequestered to the surface of the mutant in the IMAC binding buffer conditions. The 660 mL of flow-through was re-filtered through a 1 μm filter, then a 0.45 μm filter, then a 0.22 μm filter. It was diluted to 1 L with H2O and the pH adjusted to 8.5 with 2M NaOH solution, and thereafter loaded onto a HiPrep Q HP 16/60 anion exchange column equilibrated in buffer A: K2HPO4/KH2PO4 20 mM pH 8.5, flowing at 4 mL/min. The column turned pink - probably because the pH indicator from the HEK293F cells medium is anionic at pH 8.5. The column was washed with 250 mL of buffer A and the protein eluted using buffer B (buffer A supplemented with NaCl to a final concentration of 1 M), in three steps: (i) 3.5 cV of 25% buffer B; (ii) 3.5 cV of 50% buffer B; (iii) 3.5 cV of 100% buffer B; 15 mL fractions were collected. Protein containing fractions were pooled and the 30 mL sample concentrated to 5 mL, then exchanged to 20 mM MES pH 6.5, 50 mM NaCl in two 150 KDa MWCO spin concentrators. The 5 mL of CtUGGTS180C/T742C in 20 mM MES pH 6.5, 50 mM NaCl were injected onto a HiLoad Superdex 200 16/60 column equilibrated in the same buffer, and run at 1 mL/min, collecting 1.5 mL fractions. Protein containing fractions were pooled and concentrated to 6.28 mg/mL (V=800 μL). CtUGGTG177C/A786C was expressed in 200 mL of HEK293F cells. Transfected cells were left shaking at 135 rpm in 0.5 L Erlenmeyer flasks with 0.2 μm vent caps, at 37 °C and 8% CO2 incubator, for 4 days. The cells’ supernatant was processed as previously described and run on a 5 mL HisTrap HP column (GE Life Sciences). Fractions were pooled and concentrated with a centrifugal concentrator before loading on a HiLoad Superdex 200 16/60 column in SEC buffer: 20 mM HEPES pH 7.2, 120 mM NaCl. Eluted fractions were analysed by SDS-PAGE and concentrated as before. The final concentration of CtUGGTG177C/A786C construct (0.8 mL volume) was 7.91 mg/mL. CtUGGT-ΔTRXL1 was expressed in 200 mL of HEK293F cells, harvested after 4 days of incubation. The construct was purified as per above. The final concentration of CtUGGT-ΔTRXL1 construct (0.1 mL volume) was 0.4 mg/mL. CtUGGT-ΔTRXL2 was expressed in 150 mL of HEK293F cells. Transfected cells were left shaking at 135 rpm in 0.5 L Erlenmeyer flasks with 0.2 μm vent caps, at 37 °C and 8% CO2 incubator, for 3 days. The protein was purified from the cells’ supernatant as described. IMAC elution fractions were pooled and concentrated and injected in two 5 mL batches onto the SEC column (GE Healthcare), with SEC buffer: 20 mM HEPES pH 7.2, 120 mM NaCl. Elution fractions containing the protein were then pooled based on SDS-PAGE analysis. CtUGGT-ΔTRXL3 was expressed in 200 mL of HEK293F cells, harvested after 4 days of incubation. The construct was purified as per above. The final concentration of CtUGGT-ΔTRXL3 construct (0.2 mL volume) was 12.80 mg/mL.
Protein crystallisation and cryoprotection
CtUGGTKif. A CtUGGTKif crystal grew in a sitting drop from protein at 7.24 mg/mL, in condition 34 of the MORPHEUS screen (Molecular Dimensions, (Gorrec, 2009) [0.09 M NPS: 0.03 M Sodium nitrate, 0.03 M Sodium phosphate dibasic, 0.03M Ammonium sulphate; 0.1 M buffer System 3: Tris Bicine pH 8.5; 8.530% v/v; Precipitant Mix 2: 40% v/v Glycerol, 20% w/v PEG 4000] mixed in protein:mother liquor ratio 100 nL:100 nL. The crystal grew at 18 °C and it was flash-cooled in liquid N2. Details described in the Open Laboratory Notebook at https://doi.org/10.5281/zenodo.3608191. CtUGGT-ΔTRXL2. A CtUGGT-ΔTRXL2 crystal grew at 18 °C from protein concentrated to 6.5 mg/mL and mixed in 133:67 nL protein:mother liquor ratio with solution 2 of the JCSG+ crystallisation screen (Molecular Dimensions, (Newman et al., 2005)) in a sitting drop: 0.1 M sodium citrate pH 5.5, 20% w/v PEG 3,000. The crystal was cryo-protected with 20% glycerol in mother liquor and cryo-cooled with liquid nitrogen. CtUGGTS180C/T742C. Crystal growth, cryoprotection and X-ray data collection are described in the Open Laboratory Notebook at https://doi.org/10.5281/zenodo.1345671. Briefly: the P3212 CtUGGTS180C/T742C crystal grew from sitting drop from protein at 6.28 mg/mL in HEPES 20 mM pH 6.5, 50 mM NaCl, 5 mM UDP-Glc, 1 mM CaCl2 mixed in protein:mother liquor ratio 100 nL:100 nL with condition 57 of the MORPHEUS2 screen (Gorrec, 2015) [2 mM Lanthanides, 0.1 M buffer System 6 (1.0 M, pH 8.5 at 20 °C, Gly-Gly, AMPD), 36 % v/v Precipitant Mix 5 (30% w/v PEG 3000, 40% v/v 1, 2, 4-Butanetriol, 2% w/v NDSB 256)]. The crystal grew between day 57 and day 71, at 18 °C. The crystal was flash-cooled in liquid N2. The P212121 CtUGGTS180C/T742C crystal grew from mixing protein at OD280=7.29 in HEPES 20 mM pH 6.5, 50 mM NaCl, 5 mM UDP-Glc, 1 mM CaCl2 with condition 14 of the JCSG+ screen (0.2 M Sodium thiocyanate, 20% w/v PEG 3350) in protein:mother liquor ratio 133 nL:66 nL. The crystal grew in a sitting drop two days at 18 °C and broke into smaller pieces upon fishing. The crystal was cryoprotected in liquid N2 after quick transfer to a solution obtained by mixing 2 μL of ethylene glycol (EG) in 8 μL of mother liquor (i.e. 20% EG). CtUGGTG177C/A786C: Initial CtUGGTG177C/A786C crystals grew from a solution of mother liquor: 16.54% w/v PEG 4,000, 0.03 M citric acid pH 5.3, 0.07 M citric acid pH 6.0, 12.75% v/v isopropanol. The crystals initially diffracted to 25 Å only and it was decided to dehydrate them by re-equilibrating the crystallization drop against a PEG 6,000-containing mother liquor reservoir: 13 μL of mother liquor were taken out of the 50 μL in the reservoir, replaced with 13 μL of a solution of 50% w/V PEG 6,000 in mother liquor, and the plate re-sealed. After undergoing dehydration for a week, one crystal was flash frozen in liquid N2 for data collection.
UGGT-mediated re-glucosylation of urea-misfolded bovine thyroglobulin (UDT)
Bovine thyroglobulin (Sigma-Aldrich) was denatured with urea following the protocol in (Trombetta et al., 1989), briefly here: 20 mg of thyroglobulin (TG) powder was dissolved in 1 ml of ‘Column buffer’ using a vortex mixer and spun at 12000xg for 1 min to ensure that mixing occurred. The thyroglobulin was then injected onto a SuperoseTM 6 10/300 Increase gel filtration column (GE Life Sciences) which had been equilibrated with the ‘Column buffer’. SDS-PAGE analysis of the elution profile was used to select fractions containing pure protein, which were pooled and spin-concentrated to 2 ml. The thyroglobulin was then dialysed against 200 ml of ‘Denaturation buffer’ at room temperature for 36 hours (with fresh buffer changes at 12, 70 and 24 h) to achieve a dialysis exchange factor of 1,000,000x. After 36 h, the thyroglobulin was dialysed against 200 ml of ‘Renaturation buffer’ for 96 h at 4 °C (with fresh buffer changes at 48, 60 and 84 h). The urea-treated thyroglobulin was diluted to 1 ml with ‘Column buffer’, spin-filtered and run down a SuperoseTM 6 10/300 gel filtration column equilibrated with ‘Column buffer’. After SDS-PAGE analysis, fractions containing pure protein were pooled and concentrated. The pooled fractions gave the UDT used in the activity assays. Each reaction mixture contained 100 μg of UDT, 86 μM UDP-Glucose, 8.6 mM CaCl2, 8.6 mM Tris-HCl pH 8.0 and 45 pmol of CtUGGT enzyme. The reaction mixtures were set up at 37 °C. Each reaction was 70 μL to start with, in triplicate. 10 μL aliquots were taken at each time point (5’, 15’, 30’, 1 h, 2 h and O/N), and the re-glucosylation quenched by addition to each 10 μL aliquot of 1 μL of PNAGaseF denaturing buffer, then heating for 10 min at 90 °C. Then 5 μL of 10X PNGase glycobuffer 2 (NEB), 5 μL of NP40 10%, 1 μL of PNGase F (NEB) at 1 mg/mL and 27 μL of water were added to each sample for the overnight digestion with PNGase F. The N-linked glycan were labelled with anthranilic acid (2-AA) (Sigma-Aldrich), purified by adsorption to Speed-amide SPE columns and detected by normal-phase high-performance liquid chromatography, see (Caputo et al., 2016). The amount of re-glucosylation was measured in comparison to control by measuring the peak area of the PNGase F released 2-AA-labelled species Man9GlcNAc2 and Glc1Man9GlcNAc2 using Waters Empower software. This allows the % of glucosylation to be determined as the amount of Glc1-species (Peak Area Glc1Man9GlcNAc2) expressed as a fraction of the total of the two species (Peak Area of Glc1Man9GlcNAc2 + Man9GlcNAc2).
X-ray diffraction data collection and processing
CtUGGTKif: diffraction data were collected on I04@DLS, at a wavelength λ=0.9763 Å, beam size 80x20μm, 0.2° oscillation. Batches 2,3: plate set at 2.9 Å max resolution; batches 4,5, plate set at 3.5 Å max resolution. Batch 2: 450 images, 0.10 s exposure, Transmission T=70%. Batches 3,4: 500 images, 0.20 s exposure, T=100%. Batch 5: 350 images, 0.50 s exposure, T=100%. Recentring followed after each exposure. CtUGGT-ΔTRXL2: data were collected on I04@DLS, at a wavelength λ=0.97950 Å, beam size 43×30 μm, 0.15° oscillation, 1200 images, 0.02 s/image and T=100%; plate set at 4.5 Å max resolution. CtUGGTS180C/T742C: data were collected on I24@DLS, at a wavelength λ=0.96860 Å, beam size 50×50 μm, 0.10° oscillation, 1800 images, 0.10 s/image and T=30%; plate set at 3.5 Å max resolution. CtUGGTG177C/A786C: data were collected on I04@DLS, at wavelength λ=0.97949 Å, beam size 19×10 μm, 0.10° oscillation, 1800 images, 0.10 s/image and T=100%; plate set at 4.5 Å max resolution.
All datasets were processed with the autoPROC suite of programs (Vonrhein et al., 2011). Table S2 contains the data processing statistics.
Crystal structure determination and refinement
CtUGGTKif (PDB ID 6TRF): Phaser (McCoy et al., 2007) was run in all primitive orthorhombic space groups searching for one copy of PDB ID 5NV4 from which TRXL2 was removed (declaring a RMSDCα of 2.0 Å - Phaser refined it to 0.77 Å). The results were clearly best in P212121 (RF Z-score 7.0; TF Z-score: 10.4; Refined TFZ-equiv: 16.3; LLG: 114; Refined TF Z-score: 16.3, Refined LLG: 208. wR=0.58). The first map obtained in autoBUSTER (Blanc et al., 2004) from this MR model (which lacks TRXL2) showed strong density for the TRXL2 domain. The TRXL2 domain was added by superposing PDB ID 5NV4 onto the model, and real-space fitting the domain to the Fo-Fc map in CCP4-coot (Emsley et al., 2010). The structure was refined in autoBUSTER with one TLS body per domain with external restraints (Smart et al., 2012) to PDB ID 5NV4.
CtUGGT-ΔTRXL2 (PDB ID 6TS2): Molecular replacement with the program CCP4-Molrep was initially attempted using CtUGGT PDB entry 5NV4 with the TRXL2 domain removed. Electron density for the TRXL3 domain (residues 667-879) was poor. This suggested that upon deletion of TRXL2, the relative orientation of TRXL3 with respect to the rest of the protein was also changed. The TRXL3 domain was therefore also cut from the search model. Three copies of this model were placed with CCP4-Molrep. A first round of refinement was carried out in autoBUSTER with one TLS body per domain, and one rigid body per domain, with automated NCS restraints and external secondary structure restraints to the deposited 5NV4 structure (R=35.0%, Rfree=37.6%). The phases showed positive difference density in regions close to the loose ends of the search model on either side of TRXL3 for copies A, B, C, suggesting that indeed the deletion of TRXL2 caused TRXL3 to rearrange. Two copies of the TRXL3 domain were then placed with CCP4-Molrep, clearly belonging to two of the molecules in the asymmetric unit. An additional search for a third TRXL3 copy gave a convincing solution that did not appear to belong to the three molecules so far placed, highlighting the possible presence of a fourth copy in the asymmetric unit. This model comprising two copies of CtUGGT-ΔTRXL2, a CtUGGT-ΔTRXL2-ΔTRXL3 model and a TRXL3 domain for a fourth copy was subject to refinement with the same protocol as above (R=31.9% Rfree=33.3%). After this refinement, electron density for the missing TRXL3 domain and the remaining domains of the fourth copy of the molecule was visible in the map. One of the CtUGGT-ΔTRXL2 molecules was superposed onto the fourth copy’s TRXL3 domain, followed by rigid body fitting of the bulk of the final copy in Coot (Emsley et al., 2010). The final model was refined in autoBUSTER with one set of TLS thermal motion tensors per domain and non-crystallographic symmetry and external restraints to the PDB ID 5NV4 structure.
CtUGGTS180C/T742C (PDB ID 6TRT): CCP4-Molrep was run against the CtUGGTS180C/T742C data in P3112 and P3212 searching with a copy of PDB ID 5NV4 from which all three TRXL1,2,3 were removed (leaving only TRXL4, BS1,BS2 and GT24 domains). The results were clearly better in P3212 (P3212 has wR=0.606, Score=0.435, TF/sigma=10.35, Contrast=9.21 versus P3112 wR=0.637, Score=0.372, TF/sigma=5.61, Contrast=4.76). The first electron density map obtained in autoBUSTER (Blanc et al., 2004) from this MR model shows strong density for the TRXL3 domain and the Ca2+ site in the GT24 domain. The TRXL3 domain was added by superposing PDB ID 5NV4 onto the model, and real-space fitting the domain to the Fo-Fc map in CCP4-coot (Emsley et al., 2010). After one more round of refinement, the TRXL1 domain was added in the same way. Finally, the TRXL2 domain was added by molecular replacement with CCP4-Molrep in presence of the rest of the structure. The structure was refined in autoBUSTER (Blanc et al., 2004) with one TLS body per domain, one rigid body per domain, with external restraints (Smart et al., 2012) to PDB ID 5NV4. Fo-Fc residuals on two sites (the catalytic site and a crystal contact between TRXL2 and one of its symmetry mates) suggested a lanthanide ion from the crystallisation mix (which contains Y3+, Tb3+, Er3+, Yb3+). The ions are likely either Er3+ or Tb3+, which are known to substitute for Ca2+ and Mn2+ in protein coordination sites (Kastritis et al., 2017). At the wavelength of data collection, λ=0.96861 Å, f'Er3+=-1.7235 e- and f''Er3+=8.2682 e-, while f'Tb3+=-1.046 e- and f''Tb3+=6.9753 e-. Peaks at +9.4 and +7.4 sigmas are indeed visible at these two sites in the anomalous Fourier difference map. The ions were modelled as Tb3+, with a Tb3+-O distance of 2.4 ± 0.3 Å (coordinating residues: site 1: D1302, D1304, D1435; site 2: E774 from a symmetry mate, E713, E716 and D818).
CtUGGTG177C/A786C (PDB ID 6TS8): The CtUGGTG177C/A786C crystal structure was initially phased by Molecular Replacement with Molrep searching in space group P43 for two copies of PDB ID 5NV4 from which TRXL1 and TRXL2 were removed. The first map obtained in autoBUSTER (Blanc et al., 2004) from this MR model showed density for the missing domains, which were added by superposing PDB ID 5NV4 onto the model, and real-space fitting the TRXL1 and TRXL2 domains to the Fo-Fc map in CCP4-coot (Emsley et al., 2010). The structure was refined in autoBUSTER with one TLS body per domain with external and automated NCS restraints (Smart et al., 2012) to PDB ID 5NV4. Portions of the catalytic domain are disordered in the crystal and could not be traced.
Table S3 reports the Rfactors and geometry statistics for all models after the final refinements.
Homology modelling of the Drosophila melanogaster (DmUGGT) structure
Modeller (Webb and Sali, 2016) was used to homology model the DmUGGT structure, after aligning the sequences of the DmUGGT and CtUGGT proteins with Clustal Omega (Sievers and Higgins, 2018), using PDB ID 5NV4 as a template, and enforcing the disulfide bonds DmUGGT C109:C123, C1348:C1441, C1437:C1455.
Fitting of the TdUGGT:FAb structure in the negative stain EM map
The crystal structures of TdUGGT catalytic domain (PDB ID 5H18, residues 1190-1466) and TdUGGT N-terminal portion (PDB ID 5Y7O, residues 29-1042) were aligned with the full-length CtUGGT intermediate structure (PDB ID 5MU1, residues 1190-1466) in Coot (Emsley et al., 2010). Modeller (Webb and Sali, 2016) was then used to complete the TdUGGT structure, homology modelling the missing portions 158-165; 251-282; 403-414; 684-693; 738-741; 756-759; 1038-1150; 1380-1384 (after aligning the sequences of the TdUGGT (http://genome.fungalgenomics.ca Thermomyces dupontii NRRL 2155 Talth1p4_002475) and CtUGGT proteins with Clustal Omega (Sievers and Higgins, 2018)). The disulfide bonds TdUGGT C141:C153, C1322:C1415, C1411:C1429 were enforced. The fits of TdUGGT and anti- TdUGGT Fab models to the negative stain EM map (EMDB accession number EMD-30386) and to its inverse hand were carried out in Chimera (Pettersen et al., 2004) . Both for original and inverse hand map, the TdUGGT homology model was first aligned manually with the map, low-pass filtered to a resolution of 25 Å, then fitted to the EM map using the Fit in map tool in Chimera. After fitting the TdUGGT model, a Fab model from PDB ID 1FGN was fitted to the map with the same Fit in map tool in Chimera, again after low-pass filtering the PDB model to 25 Å. Final real space CCs in the original and inverse hand maps: oriCCTdUGGT=0.89; oriCCFab=0.90; invCCTdUGGT=0.90; invCCFab=0.90. The two models were deposited in PDB-DEV, accession code PDBDEV_00000054.
Frequency distributions of distances from glycoprotein residues to N-linked glycosylation sites
Starting with a list of 1372 Uniprot entries annotated as human glycoprotein for which some structural information is available, we selected 1244 Protein Data Bank (PDB) entries that contain structure for more than 90% of the primary sequence and include structural information for at least one N-linked glycan. For each entry, the distances from every residue atom and atoms in N-glycosylated asparagine residues were computed, and the distances to the closest and second-closest glycans were histogrammed. As a control for glycoprotein size, we also histogrammed all aminoacid-aminoacid distances within each same structure. The same data were sorted numerically to produce cumulative distributions.
Computational simulations
System Preparation. We used as initial structures the four available CtUGGT structures , which we call 'open' (PDB ID 5MZO); 'intermediate' (PDB ID 5MU1); 'closed' (PDB ID 5N2J); the mutant D611C-G1050C ‘closed-like’ (PDB ID 5NV4); and the newly determined ‘new-intermediate’ CtUGGTKif (PDB ID 6TRF). Starting from each structure, we performed 250 ns all-atom Molecular Dynamics (MD) simulations using the AMBER force field and package (Maier et al., 2015) and analyzed the resulting dynamics and conformational landscape using Principal Component (PC) analysis. For each system, all non-protein molecules (carbohydrates and ions) were removed from the crystal structure. Gap regions (residues 242-273, 1152-1187 and 1334-1342) were completed and refined using Modeller (Eswar et al., 2008). Standard protonation states were assigned to titratable residues (Asp and Glu are negatively charged; Lys and Arg are positively charged). Histidine protonation was assigned favoring formation of hydrogen bonds in the crystal structure. The complete protonated systems were then solvated by a truncated cubic box of TIP3P waters, ensuring that the distance between the biomolecule surface and the box limit was at least 10 Å.
MD simulations. The systems were prepared with the tleap module from the AMBER package using the ff14SB/TIP3P force fields for amino acid/water molecules, respectively (Maier et al., 2015). Each system was first optimized using a conjugate gradient algorithm for 5000 steps, followed by 150 ps. Long constant volume MD equilibration, in which the first 100 ps were used to gradually raise the temperature of the system from 0 to 300 K (integration step = 0.0005 ps/step). The heating was followed by a 300 ps long constant temperature and constant pressure MD simulation to equilibrate the system density (integration step = 0.001 ps/step). During these temperature and density equilibration processes, the protein alpha-carbon atoms were constrained by 5 kcal/mol/Å force constant using a harmonic potential centered at each atom starting position. Next, a second equilibration MD of 500 ps was performed, in which the integration step was increased to 2 fs and the force constant for restrained alpha-carbons was decreased to 2 kcal/mol/Å, followed by 20 ns long MD simulation with no constraints. Finally, a 250 ns long MD simulation was carried out with no constraints and the 'Hydrogen Mass Repartition' method, which allows an integration step of 4 fs. The results of the latter step are described in this paper.
All simulations were performed using the pmemd.cuda algorithm from the AMBER package. Pressure and temperature were kept constant using the Monte-Carlo barostat and Langevin thermostat, respectively, using default coupling parameters. All simulations were performed with a 10 Å cutoff for nonbonded interactions, and periodic boundary conditions using the Particle Mesh Ewald summation method for long-range electrostatic interactions. The SHAKE algorithm was applied to all hydrogen-containing bonds in all simulations with an integration step equal or higher than 2 fs.
PC calculations. All trajectory processing and PC calculations were performed with the CPPTRAJ (Roe and Cheatham, 2013) module of the AMBER package. For each individual MD, PCs of the alpha-carbons were computed over an ensemble of 6000 trajectory frames representing the 250 ns long trajectories.
Mass spectroscopy: tryptic peptides
Protein samples were digested in-solution with sequencing grade trypsin (Promega). Briefly: samples were treated in 100 mM iodo-acetamide for 1 hour in dark to alkylate any free cysteines followed by denaturing with 8M urea for 40 min. The samples were further diluted with 50 mM ammonium bicarbonate to reduce the Urea concentration to 1 M. 1uL of 300ng/uL trypsin solution was added to each sample and incubated at 37 °C overnight. The resulting samples were directly analysed by LC-MS. Tryptic peptides of CtUGGT, and the double mutants CtUGGTG177C/A786C, CtUGGTG179C/T742C and CtUGGTS180C/T742C were separately run on an Dionex UltiMate3000 RSLC (Thermo Scientific) and electrosprayed directly into a Q Exactive mass spectrometer (Thermo Fischer Scientific) through an Flex ion-electrospray ion source (Thermo Fischer Scientific). Peptides were trapped on a C18 PepMap trapping column (μ-Precolumn, 300 μM I.D. x 5 mm, 100 μm particle size, 100 Å, Thermo Scientific) at a flow rate 10 μL/min. The trapping buffer was 0.05% v/v trifluoroacetic acid (TFA) in water (LC-MS grade). Samples were then separated using a C18, 75 μm x 25 cm (Acclaim PepMap nanoViper, part number 164941, 2.0 μm particle size, 100 Å, Thermo Scientific) analytical column (with mobile phases: 0.1% Formic acid in water (A) and 0.1% formic acid in acetonitrile (B)) at a flow rate of 300 nL/min, and the following gradients: minutes (mins) 0-5.5: 2% B; mins 5.5-10: 8% B; mins 10-40: 45% B; mins 40-41: 95% B; mins 41–46: 95% B; mins 46-60: 2% B.
Data were acquired in Data Dependent Mode (DDA) using the settings: chromatographic peak width: 20 s; resolution: 70,000; AGC target: 3x106; maximum IT (injection time): 100 ms; scan range: 300 to 2000 m/z; ddMS2 resolution: 17,500; AGC target: 5x104; maximum IT: 100 ms; loop count: 10 (i.e. Top 10); isolation width: 4.0 m/z; fixed first mass: 120.0 m/z. Data dependent (dd) settings: minimum AGC target: 5.0x103; intensity threshold: 5.0 x104; charge exclusion: 1; peptide match: preferred; exclude isotope: on; dynamic exclusion: 30.0 s. A normalized Collision energy (NCE) of 27 was used for the fragmentation of peptides in a high-energy collision dissociation (HCD) cell and the s-lens setting in the tune file was changed to 70.
Data analysis (crosslinking and protein identification): MassMatrix (version 2.4.2) was used for data analysis to find S-S cross linking and protein/peptide identification (Xu and Freitas, 2009). A customized database, containing the sequences of the proteins of interest, was used to perform searches. MS data were converted into .mgf format using MSconvert from the ProteoWizard toolbox (Chambers et al., 2012). Search parameters were as follows: maximum number of missed cleavages = 4; fixed modification = none; variable modifications: CAMC- Iodoacetamide derivative (Carbamidomethyl) of C and OxiM- Oxidation of M; disulphide bonds were considered as the crosslink (Cys-Cys, -2.02 Da); mass accuracy filter = 20 ppm for precursor ions; MS2 tolerance = 0.02 Da (values as per the MassMatrix user’s protocol) (Xu and Freitas, 2009). The quality of a peptide match is mainly evaluated by three statistical scores: pp, pp2, pptag. A peptide match with max (pp, pp2) > 2.7 and pptag > 1.3 is considered to be significant with p value < 0.05 (as documented in (Xu and Freitas, 2009)).
Structural analysis
Structural figures were made using PyMOL (Rigsby and Parker, 2016) and Chimera (Pettersen et al., 2004). Structural movies were made using VMD (Cross et al., 2009).
Quantification and statistical analysis
X-ray diffraction data merging was carried out in autoPROC (Vonrhein et al., 2011) within the XDS (Kabsch, 2010) and Aimless (Evans and Murshudov, 2013) programs (default statistical analysis as per the references). The re-glucosylation experiments were carried out on n=3 independent samples per data point and the mean and standard deviation evaluated as documented in the user guide of GraphPad Prism, version 8.0.0 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com. When evaluating statistical significance of mass spectrometry data in MassMatrix, the quality of a peptide match is mainly evaluated by three statistical scores: pp, pp2, pptag. A peptide match with max (pp, pp2) > 2.7 and pptag > 1.3 is considered to be significant with p value <0.05 (Xu and Freitas, 2009).
Acknowledgments
We thank Julio J. Caramelo, Alessandro T. Caputo, Bevin Gangadharan, and Raymond Dwek for helpful discussions, and the members of N.Z.'s laboratory for assistance in the lab. Y.S. Albrecht designed the CtUGGT full-length wild-type sequencing primers. E. Lowe and the staff at beamlines I03, I04, and I24 at the Diamond Light Source, Harwell, England, UK assisted with X-ray data collection. T. Satoh and K. Kato kindly provided us with their 25-Å negative-stain EM reconstruction of the complex between TdUGGT and its Fab. P.R. is the recipient of an LISCB Wellcome Trust ISSF award, grant reference 204801/Z/16/Z. G.T. is funded by a Wellcome Trust Seed Award in Science to P.R., grant reference 214090/Z/18/Z. A.L. and R.I. are recipients of Italian Government PhD Studentships. S.R. is the recipient of an undergraduate Erasmus Studentship. N.Z. is a Fellow of Merton College, Oxford.
Author contributions
Conceptualization, C.P.M., J.I.B.C, R.I., D.S.A., P.R., M.A.M., and N.Z.; Methodology, all authors; Investigation, all authors; Writing – Original Draft, P.R.; Writing – Review & Editing, all authors; Funding Acquisition, P.R., M.A.M., A.S., and N.Z.; Resources, P.R., M.A.M., A.S., and N.Z.; Supervision, P.R., C.P.M., D.S.A., S.V., L.M., M.A.M., A.S., and N.Z.
Declaration of interests
The authors declare no competing interests.
Published: December 21, 2020
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.str.2020.11.017.
Contributor Information
Marcelo A. Martí, Email: marti.marcelo@gmail.com.
Nicole Zitzmann, Email: nicole.zitzmann@bioch.ox.ac.uk.
Pietro Roversi, Email: pr159@leicester.ac.uk.
Supplemental information
References
- Amara J.F., Cheng S.H., Smith A.E. Intracellular protein trafficking defects in human disease. Trends Cell Biol. 1992;2:145–149. doi: 10.1016/0962-8924(92)90101-r. [DOI] [PubMed] [Google Scholar]
- Aricescu A.R., Lu W., Jones E.Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. D Biol. Crystallogr. 2006;62:1243–1250. doi: 10.1107/S0907444906029799. [DOI] [PubMed] [Google Scholar]
- Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S.M., Bricogne G. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2210–2221. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]
- Calles-Garcia D., Yang M., Soya N., Melero R., Ménade M., Ito Y., Vargas J., Lukacs G.L., Kollman J.M., Kozlov G., Gehring K. Single-particle electron microscopy structure of UDP-glucose:glycoprotein glucosyltransferase suggests a selectivity mechanism for misfolded proteins. J. Biol. Chem. 2017;292:11499–11507. doi: 10.1074/jbc.M117.789495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capece L., Estrin D.A., Marti M.A. Dynamical characterization of the heme NO oxygen binding (HNOX) domain. Insight into soluble guanylate cyclase allosteric transition. Biochemistry. 2008;47:9416–9427. doi: 10.1021/bi800682k. [DOI] [PubMed] [Google Scholar]
- Caputo A.T., Alonzi D.S., Marti L., Reca I.-B., Kiappes J.L., Struwe W.B., Cross A., Basu S., Lowe E.D., Darlot B. Structures of mammalian ER α-glucosidase II capture the binding modes of broad-spectrum iminosugar antivirals. Proc. Natl. Acad. Sci. U S A. 2016;113:E4630–E4638. doi: 10.1073/pnas.1604463113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramelo J.J., Castro O.A., Alonso L.G., De Prat-Gay G., Parodi A.J. UDP-Glc:glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc. Natl. Acad. Sci. U S A. 2003;100:86–91. doi: 10.1073/pnas.262661199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers M.C., Maclean B., Burke R., Amodei D., Ruderman D.L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012;30:918–920. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross S., Kuttel M.M., Stone J.E., Gain J.E. Visualisation of cyclic and multi-branched molecules with VMD. J. Mol. Graph. Model. 2009;28:131–139. doi: 10.1016/j.jmgm.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daikoku S., Seko A., Ito Y., Kanie O. Glycan structure and site of glycosylation in the ER-resident glycoprotein, uridine 5'-diphosphate-glucose: glycoprotein glucosyltransferases 1 from rat, porcine, bovine, and human. Biochem. Biophys. Res. Commun. 2014;451:356–360. doi: 10.1016/j.bbrc.2014.07.095. [DOI] [PubMed] [Google Scholar]
- Daikoku S., Seko A., Son S.-H., Suzuki K., Ito Y., Kanie O. The relationship between glycan structures and expression levels of an endoplasmic reticulum-resident glycoprotein, UDP-glucose: glycoprotein glucosyltransferase 1. Biochem. Biophys. Res. Commun. 2015;462:58–63. doi: 10.1016/j.bbrc.2015.04.105. [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N., Eramian D., Webb B., Shen M.-Y., Sali A. Protein structure modeling with modeller. Methods Mol. Biol. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- Evans P.R., Murshudov G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013;69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner T.G., Kearse K.P. Modification of the T cell antigen receptor (TCR) complex by UDP-glucose:glycoprotein glucosyltransferase. TCR folding is finalized convergent with formation of alpha beta delta epsilon gamma epsilon complexes. J. Biol. Chem. 1999;274:14094–14099. doi: 10.1074/jbc.274.20.14094. [DOI] [PubMed] [Google Scholar]
- Gorrec F. The MORPHEUS II protein crystallization screen. Acta Crystallogr. F Struct. Biol. Commun. 2015;71:831–837. doi: 10.1107/S2053230X1500967X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrec F. The MORPHEUS protein crystallization screen. J. Appl. Crystallogr. 2009;42:1035–1042. doi: 10.1107/S0021889809042022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi M., Komaki S., Okamoto R., Seko A., Takeda Y., Ito Y., Kajihara Y. Synthesis of misfolded glycoprotein dimers through native chemical ligation of a dimeric peptide thioester. Org. Biomol. Chem. 2016;14:6088–6094. doi: 10.1039/c6ob00928j. [DOI] [PubMed] [Google Scholar]
- Izumi M., Oka Y., Okamoto R., Seko A., Takeda Y., Ito Y., Kajihara Y. Synthesis of Glc1Man9-glycoprotein probes by a misfolding/enzymatic glucosylation/misfolding sequence. Angew. Chem. Int. Ed. Engl. 2016;55:3968–3971. doi: 10.1002/anie.201511491. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastritis P.L., O'Reilly F.J., Bock T., Li Y., Rogon M.Z., Buczak K., Romanov N., Betts M.J., Bui K.H., Hagen W.J. Capturing protein communities by structural proteomics in a thermophilic eukaryote. Mol. Syst. Biol. 2017;13:936. doi: 10.15252/msb.20167412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith N., Parodi A.J., Caramelo J.J. Glycoprotein tertiary and quaternary structures are monitored by the same quality control mechanism. J. Biol. Chem. 2005;280:18138–18141. doi: 10.1074/jbc.M501710200. [DOI] [PubMed] [Google Scholar]
- Kiuchi T., Izumi M., Mukogawa Y., Shimada A., Okamoto R., Seko A., Sakono M., Takeda Y., Ito Y., Kajihara Y. Monitoring of glycoprotein quality control system with a series of chemically synthesized homogeneous native and misfolded glycoproteins. J. Am. Chem. Soc. 2018;140:17499–17507. doi: 10.1021/jacs.8b08653. [DOI] [PubMed] [Google Scholar]
- Kozlov G., Gehring K. Calnexin cycle – structural features of the ER chaperone system. FEBS J. 2020;287:4322–4340. doi: 10.1111/febs.15330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E. Stock-based detection of protein oligomeric states in jsPISA. Nucleic Acids Res. 2015;43:W314–W319. doi: 10.1093/nar/gkv314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryshtafovych A., Albrecht R., Baslé A., Bule P., Caputo A.T., Carvalho A.L., Chao K.L., Diskin R., Fidelis K., Fontes C.M.G.A. Target highlights from the first post-PSI CASP experiment (CASP12, May-August 2016) Proteins. 2018;86:27–50. doi: 10.1002/prot.25392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackeen M.M., Almond A., Deschamps M., Cumpstey I., Fairbanks A.J., Tsang C., Rudd P.M., Butters T.D., Dwek R.A., Wormald M.R. The conformational properties of the Glc3Man unit suggest conformational biasing within the chaperone-assisted glycoprotein folding pathway. J. Mol. Biol. 2009;387:335–347. doi: 10.1016/j.jmb.2009.01.043. [DOI] [PubMed] [Google Scholar]
- Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theor. Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin M.B., Ghenea S., Spiridon L.N., Chiritoiu G.N., Petrescu A.-J., Petrescu S.-M. Tyrosinase degradation is prevented when EDEM1 lacks the intrinsically disordered region. PLoS One. 2012;7:e42998. doi: 10.1371/journal.pone.0042998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier L., Usherwood Y.-K., Chung K.T., Hendershot L.M. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell. 2002;13:4456–4469. doi: 10.1091/mbc.E02-05-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman J., Egan D., Walter T.S., Meged R., Berry I., Ben Jelloul M., Sussman J.L., Stuart D.I., Perrakis A. Towards rationalization of crystallization screening for small- to medium-sized academic laboratories: the PACT/JCSG+ strategy. Acta Crystallogr. D Biol. Crystallogr. 2005;61:1426–1431. doi: 10.1107/S0907444905024984. [DOI] [PubMed] [Google Scholar]
- Nicholls R.A., Long F., Murshudov G.N. Low-resolution refinement tools in REFMAC5. Acta Crystallogr. D Biol. Crystallogr. 2012;68(Pt 4):404–417. doi: 10.1107/S090744491105606X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parodi A.J. How I became a biochemist. TBMB. 2007;59:361–363. [Google Scholar]
- Parodi A.J., Caramelo J.J., D’Alessio C. UDP-glucose: glycoprotein glucosyltransferase 1,2 (UGGT1,2) In: Taniguchi N., Honke K., Fukuda M., editors. Handbook of Glycosyltransferases and Related Genes. Springer; 2014. pp. 15–30. [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Rigsby R.E., Parker A.B. Using the PyMOL application to reinforce visual understanding of protein structure. Biochem. Mol. Biol. Educ. 2016;44:433–437. doi: 10.1002/bmb.20966. [DOI] [PubMed] [Google Scholar]
- Ritter C., Helenius A. Recognition of local glycoprotein misfolding by the ER folding sensor UDP-glucose:glycoprotein glucosyltransferase. Nat. Struct. Biol. 2000;7:278–280. doi: 10.1038/74035. [DOI] [PubMed] [Google Scholar]
- Ritter C., Quirin K., Kowarik M., Helenius A. Minor folding defects trigger local modification of glycoproteins by the ER folding sensor GT. EMBO J. 2005;24:1730–1738. doi: 10.1038/sj.emboj.7600645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X., Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe D.R., Cheatham T.E. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theor. Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Roversi P., Marti L., Caputo A.T., Alonzi D.S., Hill J.C., Dent K.C., Kumar A., Levasseur M.D., Lia A., Waksman T. Interdomain conformational flexibility underpins the activity of UGGT, the eukaryotic glycoprotein secretion checkpoint. Proc. Natl. Acad. Sci. U S A. 2017 doi: 10.1073/pnas.1703682114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T., Kato K. Structural aspects of ER glycoprotein quality-control system mediated by glucose tagging. Adv. Exp. Med. Biol. 2018;1104:149–169. doi: 10.1007/978-981-13-2158-0_8. [DOI] [PubMed] [Google Scholar]
- Satoh T., Song C., Zhu T., Toshimori T., Murata K., Hayashi Y., Kamikubo H., Uchihashi T., Kato K. Visualisation of a flexible modular structure of the ER folding-sensor enzyme UGGT. Sci. Rep. 2017;7:12142. doi: 10.1038/s41598-017-12283-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F., Higgins D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018;27:135–145. doi: 10.1002/pro.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart O.S., Womack T.O., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., Bricogne G. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D Biol. Crystallogr. 2012;68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda Y., Seko A., Fujikawa K., Izumi M., Kajihara Y., Ito Y. Effects of domain composition on catalytic activity of human UDP-glucose:glycoprotein glucosyltransferases. Glycobiology. 2016;26:999–1006. doi: 10.1093/glycob/cww069. [DOI] [PubMed] [Google Scholar]
- Tax G., Lia A., Santino A., Roversi P. Modulation of ERQC and ERAD: a broad-spectrum spanner in the works of cancer cells? J. Oncol. 2019;2019:8384913–8384914. doi: 10.1155/2019/8384913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S.C., Ferguson A.D., Bergeron J.J.M., Thomas D.Y. The ER protein folding sensor UDP-glucose glycoprotein–glucosyltransferase modifies substrates distant to local changes in glycoprotein conformation. Nat. Struct. Mol. Biol. 2004;11:128–134. doi: 10.1038/nsmb715. [DOI] [PubMed] [Google Scholar]
- RStudio Team . RStudio, Inc.; 2016. RStudio: Integrated Development for R. [Google Scholar]
- Totani K., Ihara Y., Matsuo I., Ito Y. Substrate specificity analysis of endoplasmic reticulum glucosidase II using synthetic high mannose-type glycans. J. Biol. Chem. 2006;281:31502–31508. doi: 10.1074/jbc.M605457200. [DOI] [PubMed] [Google Scholar]
- Totani K., Ihara Y., Tsujimoto T., Matsuo I., Ito Y. The recognition motif of the glycoprotein-folding sensor enzyme UDP-glc:glycoprotein glucosyltransferase. Biochemistry. 2009;48:2933–2940. doi: 10.1021/bi8020586. [DOI] [PubMed] [Google Scholar]
- Trombetta S.E., Bosch M., Parodi A.J. Glucosylation of glycoproteins by mammalian, plant, fungal, and trypanosomatid protozoa microsomal membranes. Biochemistry. 1989;28:8108–8116. doi: 10.1021/bi00446a022. [DOI] [PubMed] [Google Scholar]
- Vagin A., Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010;66(Pt 1):22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- Vincenz-Donnelly L., Hipp M.S. The endoplasmic reticulum: a hub of protein quality control in health and disease. Free Radic. Biol. Med. 2017;108:383–393. doi: 10.1016/j.freeradbiomed.2017.03.031. [DOI] [PubMed] [Google Scholar]
- Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011;67:293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N., Seko A., Takeda Y., Ito Y. Glycan dependent refolding activity of ER glucosyltransferase (UGGT) Biochim. Biophys. Acta Gen. Subj. 2020;1864:129709. doi: 10.1016/j.bbagen.2020.129709. [DOI] [PubMed] [Google Scholar]
- Webb B., Sali A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2016;86:2.9.1–2.9.37. doi: 10.1002/cpps.20. [DOI] [PubMed] [Google Scholar]
- Xu H., Freitas M.A. MassMatrix: a database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics. 2009;9:1548–1555. doi: 10.1002/pmic.200700322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Wearsch P.A., Zhu Y., Leonhardt R.M., Cresswell P. A role for UDP-glucose glycoprotein glucosyltransferase in expression and quality control of MHC class I molecules. Proc. Natl. Acad. Sci. U S A. 2011;108:4956–4961. doi: 10.1073/pnas.1102527108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Five crystal structures of CtUGGT, in surface representation, with domains TRXL2 and TRXL3 in color, and the rest of the protein in gray
The first PC of the joint MD simulation describes the transition between “open” and “closed” states and follows domain TRXL2 bending toward domain βS2 across the central saddle, with TRXL3 and TRXL1 clamping together across the cleft at the same time. Domains colored as in Figure 2.
The second PC of the joint MD simulation describes a movement in which the TRXL2 domain rotates with respect to TRXL3, and the βS2, TRXL1, and TRXL4 domains also undergo motion. Domains colored as in Figure 2.
Snapshots from the MD simulations of CtUGGT (gray), from a closed to an open conformation, with the residues contributing to the saddle surface colored according to the domain to which they belong (see Figure 2).
Data Availability Statement
The X-ray crystallographic data and atomic models have been deposited at the Protein Data Bank. The accession number for the CtUGGTKif crystal structure reported in this paper is PDB: 6TRF. The accession number for the CtUGGT-ΔTRXL2 crystal structure reported in this paper is PDB: 6TS2. The accession number for the CtUGGTS180C/T742C crystal structure reported in this paper is PDB: 6TRT. The accession number for the CtUGGTG177C/A786C crystal structure reported in this paper is PDB: 6TS8. The models for the TdUGGT:Fab complex (fitted in the original hand and the inverted hand of the negative stain EM reconstruction) have been deposited in PDB-DEV. The accession number for the TdUGGT:Fab complex reported in this paper is PDBDEV: 00000054.
A list of pieces of software used in this study can be found in the Key Resources Table.