

Summary
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is a curative therapy for hematological malignancies, due to graft-versus-leukemia (GVL) activity mediated by alloreactive donor T cells. However, graft-versus-host disease (GVHD) is also mediated by these cells. Here, we assessed the effect of attenuating TCR-mediated SLP76:ITK interaction in GVL vs. GVHD effects after allo-HSCT. CD8+ and CD4+ donor T cells from mice expressing a Y145F mutation in SLP-76 did not cause GVHD but preserved GVL effects against B-ALL cells. SLP76Y145FKI CD8+ and CD4+ donor T cells also showed less inflammatory cytokine production and migration to GVHD target organs. We developed a novel peptide to specifically inhibit SLP76:ITK interactions, resulting in decreased phosphorylation of PLCγ1 and ERK, decreased cytokine production in human T cells, and separation of GVHD from GVL effects. Altogether, our data suggest that inhibiting SLP76:ITK interaction could be a therapeutic strategy to separate GVHD from GVL effects after allo-HSCT treatment.
Subject areas: Molecular Biology, Immunology
Graphical abstract
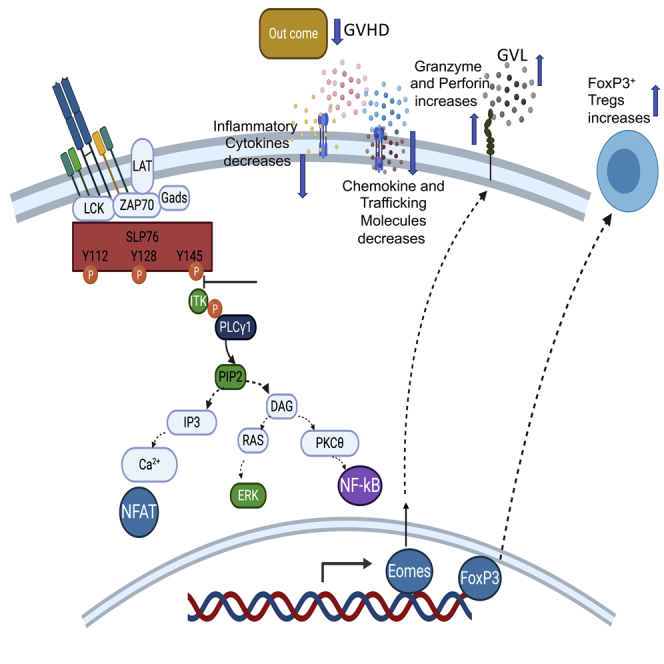
For a Figure360 author presentation of this figure, see https://doi.org/10.1016/j.isci.2021.102286.
Highlights
-
•
SLP76Y145FKI donor T cells exhibit minimal GVHD but maintain GVL activity
-
•
Inhibiting SLP76:ITK signaling by a novel peptide reduces GVHD while retaining GVL
Molecular Biology; Immunology
Introduction
Graft-versus-host disease (GVHD) is primarily orchestrated by mature donor T cells (Breems and Lowenberg, 2005). Mature T cells in the graft facilitate stem cell engraftment and, most importantly, ensure the therapeutic graft-versus-leukemia (GVL) effect (Breems and Lowenberg, 2005; Tugues et al., 2018). However, these alloreactive T cells also facilitate the unwanted effect of GVHD (Bastien et al., 2012). Standard immunosuppressive therapy for GVHD is not optimal because it leaves patients susceptible to opportunistic infections such as Cytomegalovirus and relapse of the malignancy being treated (Bleakley et al., 2012) (Ferrara, 2014). The pathophysiology of GVHD depends upon interactions between donor T cells and host antigen-presenting cells (APCs). T cell receptor (TCR)-mediated activation of donor T cells by APCs is critical for both GVHD and GVL effects (Guinan et al., 1999). Following TCR activation and expansion in secondary lymphoid organs, the alloreactive T cells migrate to target organs and cause tissue damage by producing inflammatory cytokines and by exhibiting cytotoxicity against healthy tissues (Ferrara, 2014). Thus, GVHD can be treated by interfering with T cell activation and proliferation using calcineurin inhibitors (cyclosporine, tacrolimus), mTOR inhibitors (sirolimus), and antiproliferative agents (methotrexate, cyclophosphamide, or mycophenolate) (Reddy and Ferrara, 2008; Baxter and Hodgkin, 2002). However, while alleviating GVHD, global inhibition of T cell activation also negates the beneficial GVL effect. Thus, specific signaling pathways, which can be targeted to allow GVL effects to occur while inhibiting GVHD, need to be identified. T cell signaling requires a multimolecular proximal signaling complex, which consists of adapter proteins such as SLP76 (Koretzky et al., 2006; Kambayashi et al., 2009). SLP is involved in phosphorylation of phospholipase C-gamma isoforms by IL-2-inducible T cell kinase (ITK) in T cells (Su et al., 1999). ITK is a critical mediator of TCR signaling (Bunnell et al., 2000).
SLP-76 activates ITK through its N-terminal tyrosine at the position Y145 (Bogin et al., 2007; Jordan et al., 2006). When phosphorylated, the tyrosine residue 145 (Y145) of SLP76 binds to and activates the Tec family tyrosine kinase ITK (Bogin et al., 2007). Thus, a Y→F mutation at Y145 of SLP76 leads to defective TCR-mediated ITK activation (Jordan et al., 2006). The SLP76 Y145 and ITK interaction is involved in signaling pathways that lead to cytokine production by T cell populations, as well as in regulating the development of a distinct, innate-type cytokine-producing T cell population in the thymus (Atherly et al., 2006), referred to as innate memory phenotype (IMP) T cells. CD4+ and CD8+ T cells from SLP76Y145FKI mice express significantly higher CD122, CD44, and Eomes compared to T cells from WT mice on a basal, unstimulated level (Mammadli et al., 2020). Since the activation, expansion, cytokine production, and migration of alloreactive donor T cells to target organs are hallmarks of GVHD (Henden and Hill, 2015; Lynch Kelly et al., 2015), efforts to understand signaling pathways that allow separation from GVL will enable us to develop target-specific therapeutic modulators. Both CD4+ and CD8+ T cells with CD44 low (CD44lo) are considered naive cells. CD4+ and CD8+ T cells with CD44 high (CD44hi) are considered antigen-experienced and activated cells. Both CD4+ and CD8+ T cells from SLP76Y145F- and ITK-deficient mice express a higher proportion of cells with CD44hi and CD122hi (Mammadli et al., 2020). Both CD4+ and CD8+ T cells with CD44hi and CD122hi from SLP76Y145FKI- and ITK-deficient mice arise in the thymus during development, unlike memory CD44hi CD4+ and CD8+ T cells that mainly arise in the periphery of WT mice in response to foreign antigens or because of homeostatic proliferation (Mammadli et al., 2020). Experimental studies in CD44hi and CD44lo cells arising in the periphery of WT mice show conflicting results (Dutt et al., 2011; Zheng et al., 2009; Loschi et al., 2015; Anderson, 2003; Huang et al., 2019). In this report, we show that WT CD8+CD44lo T cells induce severe GVHD, and that while WT CD8+CD44hi T cells induce less GVHD, as has been reported, they eventually cause GVHD (Zhang et al., 2005; Huang et al., 2019). In contrast, SLP76Y145FKI CD8+CD44lo T cells are much less likely to induce GVHD, and SLP76Y145FKI CD8+CD44hi T cells do not cause GVHD but maintain a significant GVL effect (Mammadli et al., 2020). Both CD8+ and CD4+ SLP76Y145FKI T cells exhibit attenuated TCR signaling and an IMP as indicated by expression of high levels of CD44 and CD122, and CD8+ SLP76Y145FKI T cells also express higher levels of the transcription factor Eomes (Huang et al., 2014; Carty et al., 2014). Our data suggest that IMP phenotype may not be enough to separate the wanted effect of GVL from the unwanted GVHD effect. We also show that disruption of the SLP76Y145/ITK interaction allows T cells to differentiate GVHD from GVL effects. Proinflammatory cytokines play a key role in the development of GVHD pathophysiology (Holler, 2002), and we further show that both CD4+ and CD8+ T cells from SLP76Y145FKI mice have reduced proinflammatory cytokine production, both on a serum level and a cellular level. Next, we examined how CD8+ T cells from SLP76Y145FKI mice maintained GVL effects. We also observed that about 70–80% CD8+ T cells from SLP76 Y145FKI and ITK-deficient mice express Eomes, and we found these Eomes-expressing cells to be critical for GVL effects. We further provided evidence that Eomes-deficient WT or SLP76Y145FKI T cells did not mount a cytotoxic response against primary leukemia cells, both in vitro and in vivo (Cheng et al., 2016). Disrupting SLP76Y145 and ITK signaling in T cells also led to defects in migration to GVHD target organs.
Finally, to make our findings clinically relevant, we developed a novel peptide inhibitor, named SLP76145pTYR, that disrupts the interaction between SLP76 and ITK (Stritesky et al., 2012). We show that SLP76145pTYR specifically inhibits the phosphorylation of ITK and downstream signaling molecules, including PLCγ1 and ERK, in both human and mouse T cells (Kim et al., 2009). Furthermore, treating T cells with SLP76pTYR enhances the development of FoxP3+ mouse regulatory T cells, while significantly reducing IFN-γ (Lu and Waller, 2009) and TNF-α (Mancusi et al., 2018) production by T cells from primary healthy human blood samples. Finally, SLP76145pTYR significantly reduced GVHD pathophysiology but maintained GVL function in a murine allogeneic hematopoietic stem cell transplantation (allo-HSCT) major mismatch model. Our studies therefore identify a novel, specific inhibitor capable of separating GVHD and GVL after allo-HSCT, with potential benefits for other T cell-mediated diseases as well (Summary Figure).
Results
Disruption of ITK: SLP76 Y145 signaling allows tumor clearance without inducing GVHD
We recently showed that ITK is differentially required for GVHD and GVL (Mammadli et al., 2020). Therefore, we tested whether this distinction depends on the interaction of ITK with SLP76. T cell signaling requires a multimolecular proximal signaling complex that includes adapter proteins such as SLP76 (Koretzky et al., 2006; Kambayashi et al., 2009). When SLP76 is phosphorylated on tyrosine residue 145 (Y145), it binds to and activates the Tec family tyrosine kinase ITK (Bogin et al., 2007). Thus, a Y→F mutation at Y145 of SLP-76 leads to defective TCR-mediated ITK activation (Jordan et al., 2006), with effects on signaling pathways that lead to cytokine production by T cell populations. Given the role of SLP76 in regulating ITK signaling downstream of the TCR, we tested whether GVL effects would remain intact when allo-BMT was performed with T cells from SLP76 Y145FKI mice. To induce GVHD, we used MHC-mismatched donors and recipients, with T cell-depleted bone marrow (TCDBM) from B6.PL-Thy1a/CyJ (Thy1.1) mice, donor T cells from C57BL/6 (B6) WT or SLP76Y145FKI mice (MHC haplotype b), and lethally irradiated BALB/c (MHC haplotype d) mice as recipients. Recipient mice were injected intravenously with 10X106 wild-type (WT) TCDBM cells along with 2×106 FACS-sorted donor T cells (1X106 CD8+ and 1X106 CD4+). 2X105 luciferase-expressing primary B-cell acute lymphoblastic leukemia (B-ALL)-luc blast cells as previously described (Cheng et al., 2016) were mixed with TCDBM and CD4 and CD8 T cells, and intravenously injected into recipient BALB/c mice by tail vein. B-ALL is a primary B cell acute lymphoblastic leukemia, syngeneic to BALB/c mice and allogeneic to C57BL/6 (B6) mice. B-ALL cells were mixed with donor T cells and TCDBM right before injection. Recipient BALB/c mice were monitored for cancer cell growth using IVIS bioluminescence imaging for over 60 days (Figure 1A). Although leukemia cell growth was observed in mice given T cell-depleted BM but no T cells, leukemia cell growth was not seen in mice transplanted with CD4+ and CD8+ T cells from either WT or SLP76Y145FKI mice. As expected, mice transplanted with WT CD4+ and CD8+ T cells suffered from GVHD, while mice transplanted with SLP76 Y145FKI CD4+ and CD8+ T cells displayed minimal signs of GVHD and survived for >65 days post-HSCT and tumor challenge (Figure 1). Most animals transplanted with SLP76Y145FKI T cells survived for more than 65 days post-allo-HSCT (Figure 1B), with significantly better survival and reduced clinical scores compared to those transplanted with WT T cells (scored based on weight, posture, activity, fur texture, and skin integrity as previously described (Cooke et al., 1996) (Figures 1C and 1D)). BALB/c mice transplanted with SLP76Y145FKI T cells showed only residual tumor cell growth (as measured by bioluminescence), indicating that the donor cells maintained GVL functions similar to WT T cells (Figure 1E). Donor CD8+ T cells are more potent than CD4+ T cells in mediating GVL effects, but both CD4+ and CD8+ T cells mediate severe GVHD in mice and humans (Amir et al., 2012; Yu et al., 2006; Wu et al., 2013). To determine whether CD4+ T cell-intrinsic SLP76:ITK signaling might be sufficient to induce GVHD, we repeated the same experiments using purified CD4+ T cells from either WT or SLP76Y145FKI mice in the MHC-mismatch mouse model of allo-HSCT (B6→BALB/c) (Figures S1A–S1C). Recipients of WT CD4+ T cells exhibited worse survival than mice receiving TCDBM cells alone (Figure S1A). In contrast, recipients of TCDBM mixed with SLP76Y145FKI CD4+ T cells had greatly reduced mortality and clinical scores compared to those given WT CD4+ T cells (Figure S1C), indicating that CD4+ T cell-intrinsic SLP76:ITK signaling can contribute to the severity of GVHD.
Figure 1.

Disruption of ITK → SLP76 Y145 signaling allows tumor clearance without inducing GVHD
1X106 purified CD8+ T cells and 1X106 purified CD4+ T cells from WT or SLP76 Y145FKI mice were mixed at a 1:1 ratio and transplanted with 2X105 B-ALL cells and 10 × 106 T cell-depleted bone marrow (TCDBM) cells transplanted into irradiated BALB/c mice. Host BALB/c mice were imaged using the IVIS 50 system three times a week.
(A) Group one received 10 × 106 T cell depleted bone marrow cells (TCDBM) only. Group one mice are used as negative controls while imaging other groups that have luciferase-expressing primary leukemia cells. Group two received 10X106TCDBM with 2X105 B-ALL-luc cells (TCDBM + B-ALL luc). The third group was transplanted with 10X106TCDBM cells and 1X106 purified WT CD8+ and 1X106 CD4+ T cells (1:1 ratio) along with 2X105 B-ALL -luc cells (TCDBM + B-ALL-luc + WT CD8+ and CD4+). Group four received 10X106TCDBM cells and 1X106 purified CD8+ and 1X106 CD4+ T cells (1:1 ratio from SLP76 Y145FKI) along with 2X105 B-ALL-luc B-ALL-luc cells (TCDBM + B-ALLluc + SLP76Y145F CD8+CD4).
(B–D) (B) We monitored the survival of recipient animals, (C) body weight changes, and (D) clinical score for 65 days post-BMT. For weight changes and clinical score, one representative of 2 independent experiments is shown (n = 3 mice/group for BM alone; n = 5 experimental mice/group for all three groups).
(E) We have quantitated tumor growth via luciferase bioluminescence. Statistical analysis for survival and the clinical score was performed using the log rank test and two-way ANOVA, respectively. Note: Controls are naive for cancer, but transplanted with 10 × 106T cell depleted bone marrow alone (TCDBM)and used as a negative control for BLI. See also Figures S1 and S2.
To test whether the IMP observed in SLP76Y145FKI T cells is necessary to separate the effects of GVL from GVHD, irradiated recipient BALB/c (MHC haplotype d) mice were transplanted with sorted CD8+CD44hi (expressing the IMP) or CD8+CD44lo (control) T cells from WT or SLP76Y145FKI mice (Figure S2A). Before transplantation into recipient mice, donor T cells were examined for CD44 expression before and after sorting based on CD44 expression. Recipient mice were injected with 10X106 WT C57Bl/6 TCDBM cells, with or without 1X106 FACS-sorted CD8+ CD44loCD122lo or CD8+CD44hiCD122hi (IMP) T cells from WT or SLP76Y145FKI mice (Figure S2B). Recipient mice were challenged with 1X105 primary B-ALL-luc (Cheng et al., 2016) tumor cells and monitored for survival, weight changes, clinical score, and tumor burden (monitored by bioluminescence imaging twice a week) for at least 60 days (Figure S2B). We found that WT CD8+CD44loCD122lo T cells cleared the tumor, but recipients developed acute GVHD, while WT CD8+CD44hiCD122hi IMP T cells cleared the tumor but exhibited delayed induction of GVHD. In contrast, CD8+CD44hiCD122hi IMP (Mammadli et al., 2020) T cells from SLP76Y145FKI mice cleared the tumor, but recipients did not develop GVHD, while CD8+CD44loCD122lo T cells from SLP76Y145F mice cleared the tumor effectively, but three out of 10 mice developed GVHD. These data indicate that WT donor CD8+CD44hiCD122hi IMP T cells exhibited delayed GVHD but eventually caused GVHD, while SLP76Y145FKI CD8+CD44hiCD122hi IMP T cells induce GVL but avoid the induction of GVHD (Figures S2B–S2F).
SLP76Y145FKI T cells produce less proinflammatory cytokines and exhibit reduced proliferation
Proinflammatory cytokine production by donor T cells is considered to be one of the hallmarks of GVHD (D'Aveni et al., 2015). To assess whether CD4+ or CD8+ T cells with attenuated TCR signaling produce inflammatory cytokines similar to CD4+ or CD8+ T cells from WT mice, we transplanted 1X106 CD4+ or CD8+ T cells in separate experiments from either WT mice or SLP76Y145FKI mice to irradiated BALB/c mice as recipients. At day seven post-transplantation, recipient BALB/c mice were sacrificed, and serum was obtained and assessed for levels of the proinflammatory cytokines IL-33, IL-1α, IFN-γ, TNF-α, and IL-17A by multiplex ELISA (Figures 2A and 2B). We discovered that recipient animals transplanted with CD4+ or CD8+ T cells (in separate experiments) from SLP76Y145FKI mice had significantly less production of proinflammatory cytokines compared to recipient mice transplanted with CD4+ or CD8+ T cells from WT mice (Figures 2A and 2B). To examine the sources of these observed proinflammatory cytokines, we restimulated spleen cells from recipient mice that were transplanted with CD4+ or CD8+ T cells from WT or SLP76Y145FKI mice in separate experiments. Spleen cells were restimulated with anti-CD3 and anti-CD28 in the presence of Brefeldin A, and donor CD4+ and CD8+ T cells were gated by flow cytometry using anti-H2Kb antibodies (expressed by donor cells), anti-CD3, anti-CD4, and anti-CD8. We observed that donor CD4+ and CD8+ T cells from SLP76Y145FKI mice produced significantly less IFN-γ and TNF-α compared to donor CD4+ and CD8+ T cells from WT mice. Thus, these data confirm that the changes in proinflammatory cytokine serum levels result from changes in production by the donor T cells, and that T cells from TCR-attenuated mice produce less inflammatory cytokines (Figures 2C–2E). To examine whether SLP76Y145FKI T cells are capable of producing cytokines in general, spleen cells from recipient mice transplanted with either WT or SLP76Y145FKI T cells were stimulated with PMA/ionomycin (to bypass the proximal signaling defect (Figure S3)), or left unstimulated for 6 hr in the presence of Brefeldin A, followed by the analysis of IFN-γ and TNF-α production. SLP76Y145FKI T cells were capable of producing IFN-γ and TNF-α when T cell signaling was bypassed by re-stimulation with PMA and ionomycin (Figure S3); however, compared to WT T cells, they produced significantly less inflammatory cytokines when stimulated via TCR (anti-CD3 and anti-CD28) (Figures 2C–2E). Next, we determined whether the reduction in cytokine production by SLP76Y145FKI donor T cells was due to cell-intrinsic or cell-extrinsic factors. We mixed purified SLP76Y145FKI CD8+ and CD4+ T cells with purified WT CD8+ or CD4+ T cells separately at a 1:1 ratio, and transplanted the mixed cells into irradiated BALB/c mice as described above. The congenic markers CD45.1 (WT C57BL/6) and CD45.2 (SLP76Y145FKI) were used to distinguish donor cells from the different strains of mice within the same recipient. On day 7, donor T cells were isolated from recipient mice using flow cytometry (anti-H2Kb) and examined for IFN-γ and TNF-α expression as described above. We found that WT donor CD8+ and CD4+ T cells (CD45.1) produced higher levels of inflammatory cytokines than SLP76Y145FKI donor CD8+ and CD4+ T cells (CD45.2), suggesting that the reduced cytokine production observed by SLP76Y145FKI donor T cells is T cell-intrinsic (Figure 2F).
Figure 2.

SLP76Y145FKI donor CD8+ and CD4+ T cells exhibit reduced cytokine production and reduced proliferation
1X106 purified CD8+ or CD4+ T cells from C57Bl/6 WT or C57Bl/6 SLP76Y145FKI (MHC haplotype b) mice were transplanted into irradiated. BALB/c (MHC haplotype d) mice in separate experiments.
(A and B) At day seven post-allo-HSCT, recipient BALB/c mice transplanted with either CD8+ or CD4+ T cells were euthanized, and ELISA was performed to determine serum cytokines (IL-33, IL1α, IFN-γ, TNF-α, and IL-17A) from recipient mice.
(C and D) Donor CD4+ or CD8+ T cells were examined for IFN-γ and TNF-α by intracellular staining after stimulation with anti-CD3/anti-CD28, as determined by flow cytometry.
(E) Donor CD8+ or CD4+ T cells from several experiments were examined for IFN-γ and TNF-α as above.
(F) Flow cytometry analysis of purified CD4+ or CD8+WT and SLP76Y145FKI T cells that were mixed at a 1:1 ratio for transplantation into irradiated BALB/c mice. At day seven donor T cells were stimulated with anti-CD3/anti-CD28 and then were gated for expression of H-2Kb, CD45.1, CD 45.2. on WT T cells and SLP76Y145FKI cells and intracellular expression of IFN-γ and TNF-α analyzed by flow cytometry. Combined data from two independent experiments is shown, and the p value for each experiment is shown.
(G) Purified CD8+ or CD4+ WT or SLP76Y145FKI donor T cells were transplanted into irradiated BALB/c mice. On day seven, donor T cells were analyzed for donor CD8+ or CD4+ T cell proliferation by examining BrdU incorporation by flow cytometry.
(H) Purified CD8+ or CD4+ T cells from WT or SLP76Y145FKI mice were mixed at a 1:1 WT:SLP76Y145FKI ratio and transplanted into irradiated BALB/c mice. At day 7, splenic donor T cells were gated for the expression of H-2Kb, CD45.1, and CD45.2 and analyzed for BrdU incorporation. See also Figure S3.
Next, we examined whether SLP76Y145FKI donor T cells proliferated similarly to WT donor CD4+ and CD8+ T cells. Lethally irradiated recipient BALB/c mice were transplanted as mentioned above, with either WT or SLP76Y145FKI donor CD4+ or CD8+ T cells. Recipient mice were injected with BrdU as described, and seven days post-allotransplantation, recipient mice were sacrificed and examined for proliferation by BrdU incorporation. SLP76Y145FKI donor T cells showed reduced proliferation compared to WT donor T cells Figure 2G. To determine if the reduced proliferation by SLP76Y145FKI donor T cells was due to cell-intrinsic mechanisms, we mixed sort-purified SLP76Y145FKI and WT CD4+ and CD8+ at a 1:1 ratio, followed by transplantation as described above in (Figure 2G). Interestingly, no difference was observed in BrdU incorporation by WT and SLP76Y145FKI donor CD4+ and CD8+ T cells in spleens of recipient mice in the mixed transplant models, indicating that the reduced proliferation of donor SLP76Y145FKI T cell proliferation was due to cell-extrinsic effects (Figure 2H).
Eomes is required for cytotoxicity and GVL effect by both SLP76Y145FKI and WT T cells
The IMP (IMP: CD44hiCD122hiEomeshi) (Mammadli et al., 2020; Huang et al., 2013) of SLP76Y145FKI CD8+ and CD4+ T cells arises in the thymus during development, as opposed to memory CD8+ and CD4+ T cells that are also CD44hi, but largely arise in the periphery of WT mice in response to foreign antigens or due to homeostatic proliferation (Weinreich et al., 2010). We examined pre-transplanted CD4+ and CD8+ T cells for the CD44 and CD122 phenotype, and CD8+ T cells for CD44, CD122, and Eomes expression, and observed that SLP76Y145FKI T cells expressed higher levels of CD44, CD122, and Eomes compared to CD8+ T cells from WT mice (Figures 3A and 3B).
Figure 3.

Eomes is required for cytotoxicity and GVL effect by both WT and SLP76Y145FKI T cells
(A and B) Purified WT and SLP76Y145FKI CD8+ and CD4+ T cells were examined for expression of CD44, CD122, and Eomes by flow cytometry.
(C) Purified donor CD8+ T cells from either WT or SLP76 Y145FKI Eomes-sufficient, Eomes-deficient, or Eomes-flox control mice were transplanted into irradiated BALB/c (MHC haplotype d) mice. On day seven, donor T cells were purified as described and used in an ex vivo cytotoxicity assay against B-ALL-luc cells at 40:1, 20:1, and 10:1 ratios.
(D) 1X106 purified WT or SLP76Y145FKI. Eomes-sufficient, Eomes-deficient, or Eomes-flox control CD8+ T cells and 1X106 purified CD4+ T cells were mixed and transplanted along with 2X105 B-ALL-luc cells and 10 × 106 T cell-depleted bone marrow TCDBM cells into irradiated BALB/c mice. Host BALB/c mice were imaged using IVIS 3 times a week. Group one received 10 × 106 T TCDBM alone. Group two received 10X106TCDBM along with 2X105 B-ALL-luc cells (TCDBM + B-ALLluc). Group three was transplanted with 10X106TCDBM and 1X106 purified WT CD8+ T cells +1X106 CD4+ T cells, and 2X105 B-ALL-luc cells (TCDBM + B-ALLluc+ WT CD8+CD4). Group four received 10X106TCDBM and 1X106 purified CD8+ T cells +1X106 CD4+ T cells from SLP76 Y145FKI Eomes-sufficient mice along with 2X105 B-ALL-luc cells (TCDBM + B ALLluc+ SLP75Y145FKI CD8+CD4). Group five received 10X106TCDBM and 1X106 CD8+ T cells +1X106 CD4+ purified T cells from SLP76 Y145FKI Eomes-deficient mice along with 2X105 B-ALL-luc cells (TCDBM + B-ALLluc + SLP75Y145FKI Eomes-cKO CD8+CD4). Group six received 10X106TCDBM and 1X106 CD8+ T cells +1X106 CD4+ purified T cells from WT Eomes-deficient mice along with 2X105 B-ALL-luc cells (TCDBM + B-ALLluc + WT Eomes cKO CD8+CD4).
(E–G) (E) The mice were monitored for survival, (F) body weight changes, and (G) clinical score for 50 days post-BMT. For weight changes and clinical score, one representative of 2 independent experiments is shown (n = 3 mice/group for BM alone; n = 5 experimental mice/group for all three groups). The survival groups are a combination of all experiments.
(H) We have quantitated luciferase bioluminescence of tumor growth. Statistical analysis for survival and the clinical score was performed using log calculation. Two-way ANOVA was used for statistical analysis and results were confirmed by students t-test, p values are presented. Note: Controls are naive for tumor but transplanted with 10 × 106T cell depleted bone marrow alone (TCDBM) and used as a negative control for BLI. See also Figure S4.
To further investigate the role of Eomes in tumor clearance and cytotoxic function, we crossed SLP76Y145FKI mice with Eomesflox/flox mice and crossed these offspring with CD4cre to delete Eomes specifically in both CD4+ and CD8+ T cells (Jordan et al., 2008; Carty et al., 2014; Pikovskaya et al., 2016) (SLP76Y145FKI Eomes conditional knockout, SLP76Y145FKI cKO). To obtain ex vivo activated cells, we performed similar allo-HSCT experiments as described above and used WT or SLP76Y145FKI CD8+ T cells with or without Eomes expression. Seven days post-transplant, donor CD8+ T cells were sorted as previously described by H2Kb positivity, and in vitro cytotoxicity assays were performed at a 40:1, 20:1, and 10:1 ratio (effector: target). We observed that donor T cells lacking Eomes from either SLP76Y145FKI or WT mice could not kill tumor targets (Figure 3C). Next, we examined the role of Eomes in the allo-HSCT model. Lethally irradiated BALB/c mice were injected intravenously with 10X106 WT T cell-depleted BM cells along with 1X106 FACS-sorted CD8+ and CD4+ T cells from either WT mice or SLP76Y145FKI Eomesflox/flox (SLP76Y145FKI Eomes cKO) mice, with or without CD4cre (to delete Eomes specifically in CD4+ and CD8+ T cells), along with 2X105 luciferase-expressing B-ALL-luc blast cells as described (Cheng et al., 2016). Recipient animals transplanted with WT T cells cleared the tumor cells but developed acute GVHD (Figure 3D). Recipient animals transplanted with Eomes sufficient (Eomesflox/flox mice without CD4cre) SLP76Y145FKI T cells cleared the tumor without showing signs of GVHD (Figure 3D). These animals were monitored for survival (Figure 3E) and weight loss (Figure 3F). Recipient animals were also evaluated for clinical score 2–3 times per week by a scoring system that sums changes in 6 clinical parameters: (1) weight loss, (2) posture, (3) activity, (4) fur texture, (5) diarrhea, and (6) skin integrity (Cooke et al., 1996). Animals that lost ≥30% of their initial body weight were euthanized (Figure 3G). The bioluminescence data were analyzed and quantified with Living Image Software (Xenogen) and Igor Pro (Wave Metrics, Lake Oswego, OR) (Figure 3H). Notably, recipient animals transplanted with Eomes-deficient (Eomesflox/flox mice with CD4cre, called SLP76Y145FKI Eomes cKO) T cells could not clear the tumor, and all died from tumor burden. Recipient animals transplanted with WT Eomes-deficient (Eomesflox/flox mice with CD4cre, called WT Eomes cKO) T cells developed severe GVHD and were also unable to clear transplanted leukemia cells. We have confirmed the deletion of Eomes using the Eomesflox/flox CD4 cre mice by flow cytometry (Figure S4). These data provided further evidence that Eomes is required for the GVL effect.
SLP76Y145/ITK signaling is required for T cell migration to the GVHD target tissues
GVHD involves early migration of alloreactive donor T cells into the target organs, followed by T cell expansion and tissue destruction (Ferrara, 2014). Modulation of alloreactive T cell trafficking has been suggested to play a significant role in ameliorating experimental GVHD (Lu et al., 2010). Therefore, we examined the trafficking of donor T cells to GVHD target tissues, as previously described (Lu et al., 2010). Irradiated BALB/c recipient mice were injected with CD8+ and CD4+ T cells from C57Bl/6 background SLP76Y145FKI (CD45.2+) and WT B6LY5 (CD45.1+) mice mixed at a 1:1 ratio (Figure 4A), and seven days post-transplantation, recipient mice were examined for the presence of donor CD8+ and CD4+ T cells in the spleen, lymph nodes, liver, and small intestines (SIs). While the WT: SLP76Y145FKI T cell ratio for both CD8+ and CD4+ cells remained approximately 1:1 in the spleen and lymph nodes (Figures 4B and 4C), this ratio in the liver and small intestine was significantly elevated, suggesting that SLP76Y145FKI CD8+ and CD4+ T cells were defective in migration to and expansion in those tissues (Figures S5A and S5B). Using histological staining for H&E, we also observed significant leukocyte infiltration into GVHD target organs such as liver, skin, and SI (Cho et al., 2020), in WT T cell recipients but not in SLP76Y145FKI T cell recipients (Figure 4D).
Figure 4.

SLP76Y145/ITK signaling is required for T cell migration to the GVHD target tissues
(A) Irradiated BALB/c mice were allo-transplanted and injected with FACS-sorted CD8+ or CD4+ T cells mixed at a 1:1 ratio from WT B6.SJL (Ly5 CD45.1) and WT C57B16 (CD45.2) mice. We also transplanted FACS-sorted CD8+ or CD4+ T cells from B6.SJL (Ly5 CD45.1) and SLP75Y145FKI (C57B16 in background, CD45.2) mice at a 1:1 ratio. FACS analysis of sorted T cells pre-transplant is shown.
(B and C) On day seven post-transplantation, the spleen, liver, and small intestine (SI) from recipients were examined for donor CD4 and CD8+ T cells by H2Kb+ CD45.1+ (LY5) or CD45.2+(B6). We also examined donor CD4 or CD8+ T cells from WT mice by H2Kb+ CD45.1+, and from SLP75Y145FKI mice by H2Kb+ and CD45.2+. The ratio of WT: SLP75Y145FKI CD8+ and CD4+ T cells in the organs was determined.
(D) We transplanted either CD4+ or CD8+ T cells separately into irradiated BALB/c mice, in separate experiments. At day 7 post-allo-HSCT, liver and small intestines were examined by H&E staining (10X) magnification).
(E and F) On Day 7 post-allo-HSCT, donor CD8+ or CD4+ T cells from separate experiments were isolated and examined for the expression of CXCr3, CX3r1, CXCr1, CCR12, s1pR1, CrTAM, CXCR6, CCR9, CXCR5, and CXCr4 using q-PCR. p values were calculated using two-way ANOVA and Student's t test, p values are listed.
(G) Irradiated BALB/c mice were BM-transplanted and injected with CD8+ T cells and CD4+ T cells from luciferase-expressing WT or SLP75Y145FKI mice (C57Bl/6 background). On day 7 post-allo-HSCT, recipient BALB/c mice were injected with D-luciferin. Spleen, lymph nodes, liver, and small intestine were examined for donor CD8+ T cells or CD4+ T cells by luciferase expression. One representative of 2 independent experiments is shown (n = 3 mice/group for control, n = 5 mice for WT, and n = 5 mice for SLP75Y145FKI. p values were calculated using two-way ANOVA and Student's t test, p values are listed). See also Figures S5 and S6.
Pro-inflammatory conditioning treatment may promote donor T cell migration into GVHD target tissues (Wysocki et al., 2004; Seif et al., 2017). We therefore examined if the observed defect in trafficking was due to chemokine receptor expression on donor T cells. Irradiated BALB/c recipient mice were injected with WT and SLP76Y145FKI CD8+ and CD4+ T cells as described previously, and at seven days post-transplantation, donor CD4+ and CD8+ T cells were FACS sorted from the recipient using H2Kb expression. Purified donor CD4+ and CD8+ T cells were examined for chemokine receptor expression by qPCR. Indeed, we found that expression of chemokine receptors and other molecules that play a critical role in T cell migration (CXCR3, CX3r1, CXCR1, CCR12, s1pR1, CrTAM, CXCR6, CCR9, CXCR5, CXCr4) were significantly reduced in SLP76Y145FKI CD8+ and CD4+ T cells at day 7 post-transplantation compared to WT CD8+ and CD4+ T cells (Figures 4E and 4F).
As an alternative approach, we tracked both CD8+ and CD4+ T cells in allo-BMT mice by using donor CD8+ and CD4+ T cells from WT and SLP76Y145FKI mice that also express luciferase, which could be monitored by bioluminescence (Negrin and Contag, 2006). We observed that both CD8+ and CD4+ donor T cells from SLP76Y145FKI mice had significantly impaired residency in GVHD target organs—including the liver and SI—compared to WT, despite no differences in spleen and lymph nodes (Figure 4G). Luciferase bioluminescence was quantified for secondary lymphoid organs (spleen and lymph nodes) and GVHD target organs (SI and liver) (Figures S6A and S6B). These data suggest that SLP76Y145FKI CD8+ and CD4+ T cells display attenuated chemokine receptor expression, which correlates with defective migration to GVHD target organs and reduced target organ pathology.
Novel peptide inhibitor SLP76pTYR specifically targets ITK signaling and enhances Treg cell development
Since T cells from SLP76Y145FKI mice can separate GVHD from GVL, we sought to determine whether disruption of ITK signaling with pharmacological agents would have a similar effect. When we used several commercially available small molecule inhibitors, including 10n (Carson et al., 2015; Riether et al., 2009), CTA056 (Guo et al., 2012), and GSK2250665A (Alder et al., 2013), we observed that these small molecules also inhibit several other kinases including mTOR and AKT, suggesting that these molecules were not specific (Figures S7A–S7F). Thus, we sought to design a novel inhibitor that would explicitly target the SLP76-ITK interaction and signaling by preventing the SH2 domain of ITK from docking onto SLP76 at tyrosine 145. Since evolution usually selects residues at specific protein:protein interfaces for certain properties (Pletneva et al., 2006), we analyzed the physico-chemical and structural properties of the interface of the SH2 domain and SLP76-pY145 in our quest to generate a potent inhibitor of the ITK-SH2 domain: SLP76-pY145 interaction (Figures 5A and 5B). We examined the Y145 region of SLP76 to design a short peptide that can inhibit ITK via competitive binding. In addition, to avoid the unintended non-specific binding of our peptide to the more than hundred other SH2 domains (and/or to other unexpected targets) in vivo (Andersen et al., 2019), we incorporated as many distinctive features of the SLP76 region around the pY145 as possible. We used the previously published atomic-resolution NMR spectroscopy structures of the SH2 domain of ITK, free and in complex with a short peptide containing a pTyr residue (Pletneva et al., 2006), as a guide (Figure 5A). The SH2 domain of ITK contains a complementary electrostatic surface, because the phosphotyrosine binding pocket, as well as the surrounding surface groove, is highly positively charged, suggesting that electrostatics most likely will play a vital role in this interaction (Figure 5B). These efforts led to the design of a novel SLP76pTYR peptide predicted to bind to the ITK SH2 domain and prevent ITK from docking onto SLP76 at the tyrosine at 145 position (Figure 5C). BLASTing (Altschul et al., 1990) the peptide sequence of our novel peptide SLP76145pTYR against the non-redundant human proteome showed minimal identity with other proteins, suggesting that the interaction between SLP76pTYR and the SH2 domain is unique, and most likely will be specific toward ITK signaling. The SLP76pTYR construct consists of two components (Figure 5C): (i) amino acid residues 132 to 155 of SLP76 with phosphorylated tyrosine at position 145 and (ii) a TAT peptide sequence (GRKKRRQRRRPQ) for cell membrane penetration (i.e. 132NEEEEAPVEDDADpYEPPPSNDEEA155-TAT). To test the effect of this construct to inhibit the interaction of ITK and SLP76, we cultured T cells with a FITC-conjugated SLP76pTYR peptide, vehicle, or nonspecific peptide, and examined the cells for FITC uptake using microscopy and flow cytometry. We observed that significant numbers of cells were positive for FITC following treatment with the SLP76pTYR peptide (Figures 5D–5F). The peptide was localized in specific locations in the cell as observed by imaging the cells in a single focal plane near the cover glass. This result would be expected if the peptide were binding to ITK in signalosomes in the cell (Figure 5D).
Figure 5.

Development of a novel peptide that disrupts the interaction between SLP76 and ITK
(A) NMR spectroscopy structure of murine ITK SH2 domain showing its complex with a peptide containing a pTyr residue (PDB code:2ETZ) that was previously solved by Pletneva et al. (46). The SH2 domain is rendered in surface representation (wheat), while the peptide derived from residues 143–148 of SLP76 with a sequence (143ADpYEPP148) is shown in stick model. In contrast, our SLP76pTYR inhibitor consists of residues 132–155 of SLP76.
(B) Electrostatic profile is shown, calculated using the APBS plugin in Pymol.
(C) Top: Organization of the domain architecture of full-length ITK showing the c-terminal Kinase domain, Src-homology 2 (SH2), the Src-homology 3 (SH3) domains, the intrinsically disordered proline-rich region (PRR), and the N-termimal Pleckstrin homology (PH) and Tec homology (TH) domains. Bottom: Organization of the domain architecture of full-length SLP76 adaptor protein showing the N-terminal SAM domain, the intrinsically disordered region containing phosphotyrosines pY112, pY128, pY145, which are followed by a proline-rich domain (PRD) and a C-terminal SH2 domain. Bottom: Design of the novel peptide, SLP76pTYR with an N-terminal FITC to monitor the peptide in cells and a C-terminal TAT sequence (GRKKRRQRRRPQ TAT sequences) for cell membrane permeability. Also shown are the amino acid sequence of residues 132–155 of SLP76, which was used to design the SLP76pTYR peptide inhibitor.
(D) T cells were examined for percentage FITC positive by fluorescence microscopy. A single cell in focal plane near the cover glass was imaged.
(E) Primary cells cultured with SLP76pTYR or the non-specific peptide were washed and examined for FITC expression by flow cytometry.
(F) Quantification of the FITC expression for (E). We used two-way ANOVA for statistical analysis and confirmed our statistical finding by Student's t-test was performed. See also Figure S7.
ITK deficiency is known to enhance the development of regulatory T cells (Tregs) (Elmore et al., 2020; Owen et al., 2019) so we tested the peptide inhibitor to determine whether inhibition of the ITK:SLP76Y145 interactions using SLP76pTYR would induce Tregs. First, we examined the frequency of Tregs by expression of CD4+ and FoxP3+ (Figure 6A). Next, total mouse T cells were stimulated with anti-CD3 (Sugie et al., 2004) in the presence of either SLP76pTYR, non-specific peptide, or vehicle alone for 5 to 24 hr, and cells were harvested and examined for the presence of Tregs (CD4+CD25+FoxP3+). We observed significantly enhanced differentiation of Treg cells in T cell cultures treated with SLP76pTYR peptide compared to vehicle alone or nonspecific peptide (Figures 6B and 6C). Next, we stimulated mouse T cells with anti-CD3 and anti-CD28 for 5 min, in the presence of SLP76pTYR or vehicle alone, and observed a reduction in ITK phosphorylation. We did not observe a reduction in non-stimulated phosphorylation ITK, nor did we observe any reduction for non-stimulated total ITK or stimulated total ITK. Similarly, we only observed a reduction in stimulated phosphorylation PLCγ-1; we did not observe any reduction with non-stimulated phospho-PLCγ-1 or either stimulated or non-stimulated PLCγ-1. We also observed similar effects on ERK as those we observed in ITK and PLCγ-1(Figures 6D and S8A–S8C). We did not observe any differences in the phosphorylation of PI3K or AKT, either stimulated and non-stimulated, or on total PI3K and AKT (Figures 6D, S8D, and S8E). Next, we investigated the effects of SLP76pTYR peptide on human PBMC samples from healthy human patients. T cells from these patients were stimulated with anti-CD3 (OKT3) (Karimi et al., 2005) for 5 min in the presence of SLP76pTYR or vehicle alone. We observed reduced phosphorylation of PLCγ1 and ERK, in stimulated T cells (Figure 6E). We did not observe any differences in non-stimulated phospho PLCγ1 and ERK (Figures 6E, S8F, and S8G). We also did not observe any differences in total AKT or either stimulated or unstimulated phospho AKT. We also did not observe any differences in stimulated or unstimulated total human AKT (Figures 6E and S8H), providing evidence that the SLP76pTYR peptide has an impact on signaling pathways downstream of SLP76 in both mouse and human T cells. Notably, SLP76pTYR peptide exhibited minimal off-target effects against other kinases, including PI3K and AKT (Figures S8A–S8H). It is possible that PI3K and AKT lie downstream of ITK, but that the specific pathways affected by the disruption of the SLP76:ITK interaction does not affect the activation of PI3K and AKT. These data provide further evidence that our peptide affects early T cells signaling (Figure 6E). Next, we investigated the ability of SLP76pTYR peptide to affect the production of proinflammatory cytokines in human PBMCs. T cells from healthy human were stimulated with anti-CD3 (OKT3) and anti-CD28 in the presence of SLP76pTYR or vehicle alone, along with Brefeldin A. We also examined T cells incubated with SLP76pTYR or vehicle alone in the presence of PMA + Ionomycin and Brefeldin A for 6 hr. Our data show that T cells stimulated with anti-CD3/anti-CD28 had significantly reduced IFN-γ and TNF-α production when incubated in the presence of SLP76pTYR compared with the vehicle alone (Figure 6F).
Figure 6.

Novel peptide SLP76pTYR specifically targets SLP76:ITK signaling and enhances Treg cell development
(A) Murine CD4+ T cells were examine for total CD4+ and FOXP3 expression prior to treatment with SLP76pTYR. n = 3, and one representative experiment is shown.
(B) Total T cells stimulated in the presence of SLP76pTYR, nonspecific peptide, or vehicle alone were examined for total CD4 cells that are FoxP3+. n = 3, and one representative experiment is shown.
(C) Quantification of three experiments as in (A).
(D) Cell lysates were obtained from mouse T cells stimulated with anti-CD3 and anti-CD28 in the presence of SLP76pTYR, or vehicle alone. Lysate from stimulated cells and non-stimulated cells were examined for phosphorylated ITK, total ITK (size 50–75kDa), phosphorylated PLCγ1, total PLCγ1 (size ~155kDa), phosphorylated ERK, total ERK (size ~42kDa), phosphorylated PI3K, total PI3K, (size ~85kDa), phosphorylated AKT, and total AKT (size ~60kDa). n = 3 and one representative experiment is shown.
(E) Cell lysates from human T cells, non-stimulated or stimulated with OKT3 for 5min in the presence of SLP76pTYR or vehicle alone, were examined for phosphorylated pPLCγ1 and total PLCγ1 on stimulated and non-stimulated T cells. Cell lysate from stimulated and non-stimulated cells were examine for pERK and total ERK. Lysates from stimulated and non-stimulated were also examined for phosphorylation and total AKT. n = 3 and one representative experiment is shown.
(F) Primary human T cells from PBMCs were stimulated with anti-CD3 and anti-CD28, or with PMA/Ionomycin, for 6 hr in the presence of vehicle alone or SLP76pTYR in the presence of Brefeldin A (BFA) (Webb et al., 2015). Intracellular IFN-γ and TNF-α expression by CD8+ and CD4+ T cells was determined by flow cytometry. For statistical analysis we used two-way ANOVA and Student's t test. p values are presented. See also Figure S8.
Inhibition of SLP76-ITK interaction and signaling by the peptide SLP76pTYR allows tumor clearance without inducing GVHD
Next, we examined whether our peptide can inhibit SLP76:ITK interaction in vivo and separate GVL from GVHD as proof of principle for the approach. WT CD8+ and CD4+ T cells were mixed at a 1:1 ratio and transduced with a retrovirus carrying SLP76pTYR-pCherry or empty vector. Lethally irradiated recipient BALB/c mice were transplanted with 10 × 106 T cell-depleted BM (TCDBM) as described, alone or together with WT CD8+ and CD4+ T cells transduced with SLP76pTYR-pCherry or empty vector-carrying virus. Recipient mice were also given 1x105 primary B-ALL-luc tumor cells as described (Cheng et al., 2016). While tumor growth was observed in TCDBM-transplanted mice that did not receive donor T cells, tumor growth was not seen in mice transplanted with either untransduced T cells, or T cells transduced with either empty viruses or SLP76pTYR-pCherry carrying viruses. Notably, mice transplanted with untransduced T cells or T cells transduced with empty virus suffered from GVHD, while mice transplanted with T cells transduced with SLP76pTYR-pCherry carrying virus survived for >40 days post-HSCT without tumor growth, with minimal signs of GVHD (Figures 7A–7E). Tumor growth was observed in only 1 out of 9 mice in the group that received the T cells transduced with SLP76pTYR-pCherry carrying virus (Figure 7F).
Figure 7.

Inhibition of T cells by the peptide SLP76pTYR allows tumor clearance without inducing GVHD
(A) Purified WT CD8+ and CD4+ T cells were mixed (1X106 total) at a 1:1 ratio, and transduced with viruses containing SLP76pTYR or empty vector, then transplanted along with 1X105 B-ALL- luc cells and 5 × 106 T cell-depleted bone marrow cells into irradiated BALB/c mice. Host BALB/c mice were imaged using IVIS 200 3 times a week. Group one received 10 × 106 T cell-depleted bone marrow alone (TCDBM). Group two received 10X106TCDBM along with 1X105 B-ALL-luc cells (TCDBM + B-ALLluc). The third group was transplanted with 10X106TCDBM and a 1:1 ratio of purified WT CD8+ and CD4+ T cells (1X106 total) along with 1X105 B-ALL-luc cells (TCDBM + B-ALLluc + WT CD8+CD4). Group four received 10X106TCDBM and a 1:1 ratio of purified WT CD8+ and CD4+ T cells (1X106 total) transduced with control viruses along with 2X105 B-ALL-luc cells (TCDBM + B-ALLluc + Empty CD8+CD4). Group five received 10X106TCDBM, a 1:1 ratio of purified WT CD8+ and CD4+ T cells (1X106 each) transduced with SLP76pTYR-carrying viruses, and with 1X105 B-ALL-luc cells (TCDBM + B-ALLluc + SLP76pTYR virus CD8+CD4).
(B–D) (B) The mice were monitored for survival, (C) body weight changes, and (D) clinical score for 40 days post BMT. For weight changes and clinical score, one representative of 2 independent experiments is shown (n = 3 mice/group for BM alone; n = 5 experimental mice/group for all three group).
(E) Quantitated luciferase bioluminescence of tumor growth.
(F) Tumor incidence for each of the experimental groups.
Statistical analysis for survival and clinical score was performed using log rank test and two-way ANOVA, respectively. Note: Controls are naive for tumor, but transplanted with 10 × 106T cell depleted bone marrow alone (TCDBM) and used as a negative control for BLI.
Altogether, these data demonstrate that disruption of SLP76:ITK signaling can separate GVHD from GVL. Inhibition of ITK signaling by SLP76pTYR, by specifically targeting the SLP76 and ITK interaction, allows tumor clearance and minimizes development of GVHD. Finally, our novel peptide inhibitor of ITK is specific and has the potential to be used in a clinical setting for T cell-mediated disorders. (Summary Figure)
Discussion
This report shows that targeting the SLP76:ITK interaction and its downstream factors significantly suppresses GVHD pathogenesis while maintaining GVL effects in an allo-HSCT model. Since GVHD is primarily caused by donor T cells, modulating donor T cells by specifically targeting kinase activity will enable us to separate GVHD from GVL. Our data suggest that the SLP76:ITK signaling pathway could represent a potential target for the separation of GVHD and GVL responses after allo-HSCT.
The adapter protein SLP76 plays an essential role in regulating T cell activation downstream of the TCR by assembling a multimolecular signaling complex that includes ITK. The phosphorylation of SLP76 at Y145 leads to the activation and recruitment of ITK, which phosphorylates PLCγ1, leading to its activation, mobilization of calcium, and activation of the NFAT transcription factor (Sahu and August, 2009). T cells that carry a Y145F mutation in SLP76 fail to phosphorylate and activate PLCγ1 in response to TCR stimulation (Jordan et al., 2008). Although T cells expressing the SLP76 Y145F mutation exhibit signaling defects downstream of TCR stimulation, not all T cell functions are lost when ITK recruitment and activation is defective. For example, SLP76Y145FKI mice can clear acute LCMV infection (Smith-Garvin et al., 2010). The ability of T cells from SLP76Y145FKI mice to induce GVL without GVHD indicates that the SLP76:ITK pathway controls these functions. Several groups have reported that both naive CD44lo CD4+ and CD8+ T cells can induce lethal GVHD, while CD44hi T cells do not (Dutt et al., 2011). Since a high proportion of CD4+ and CD8+ T cells from SLP76 Y145F mice are CD44hi and CD122hi and express higher levels of Eomes (IMP phenotype) (Weinreich et al., 2010), we investigated the capacity of this IMP population to induce GVHD and GVL compared to CD8+ and CD4+ CD44loCD122lo T cell populations in our allo-HSCT model. We found that WT CD8+ and CD4+, CD44loCD122lo T cells induced acute GVHD, while CD8+ and CD4+ with IMP phenotype had reduced ability to do so, with significantly delayed induction of GVHD. However, animals eventually succumbed to the symptoms of GVHD, confirming previous reports (Zhang et al., 2005). Although the SLP76Y145F mice exhibit defects in the development of other T cells in addition to the IMP cells, such as iNKT (Gerth and Mattner, 2019; Muro et al., 2019) cells and gamma delta T cells (Muro et al., 2019; Navas et al., 2017), we have specifically focused on the role of CD8+ and CD4+ T cells in GVL vs. GVHD. We also noted that both CD8+ and CD4+ SLP76Y145FKI T cells with IMP phenotype as well as those lacking the IMP phenotype exhibit GVL with reduced capacity to induce GVHD. It is likely that the altered signaling experienced by the cells with the SLP76Y145FKI mutation allows these cells to be able to have anti-tumor activity in a T cell-intrinsic manner.
Recently, several convergent lines of evidence have suggested that inflammatory cytokines act as mediators of acute GVHD (Lynch Kelly et al., 2015; Holler, 2002; Ju et al., 2005). Therefore, we investigated whether donor T cells with attenuated TCR signaling might reduce cytokine storm mediated by donor T cells. Our data showed that both CD8+ and CD4+ T cells from WT C57Bl/6 mice (MHC haplotype b) produced significantly higher cytokines both on a serum and a cellular level when transplanted into BALB/c mice. In contrast, both donor CD8+ and CD4+ T cells from SLP76 Y145FKI C57Bl/6 mice produced significantly less cytokines on both a serum and a cellular level. Both donor CD8+ and CD4+ T cells from SLP76 Y145FKI also exhibited reduced expression of chemokine receptors compared to WT donor T cells. The defective migration of donor CD8+ and CD4+ SLP76Y145FKI T cells likely contributes to the attenuation of GVHD. The retention of T cells in secondary lymphoid organs by FTY720-mediated inhibition of S1PR1 also ameliorates GVHD while maintaining GVL effects (Villarroel et al., 2014; Liu et al., 2012).The chemokine receptor CXCR3 has an important role in the migration of effector T cells in GVHD model (Duffner et al., 2003). CX3r1, CXCr1, CCR12, CrTAM, CXCR6, CCR9, CXCR5, and CXCR4 have been shown to play a significant role in donor T cell migration to GVHD target organs (Barrett, 2015; Castor et al., 2012; Hsiao et al., 2020). In a clinical study, blockade of these chemokine receptors by a small molecule antagonist led to a reduction in GVHD with no significant difference in relapse rates, suggesting that blocking T cell migration to target tissues could reduce GVHD severity without compromising the beneficial GVL effect (Vadakekolathu and Rutella, 2017). Activated alloreactive CD8+ T cells upregulate the expression of CX3CR1 and CXCR6 after allo-HSCT (Duffner et al., 2003; Vadakekolathu and Rutella, 2017), and these receptors are important for the homing of CD8+ T cells to the liver and intestines. Thus, CXCR6 deficiency or blockade of the CXCR3 and CXCR6 ligands attenuates GVHD (Duffner et al., 2003), and importantly, the GVL effect is still maintained under these conditions (Sato et al., 2005). Thus, blocking T cell migration by chemokine receptor blockade could be beneficial in the treatment of GVHD after allo-HSCT. We have also demonstrated as a proof of concept that specific targeting of the SLP76: ITK interactions can be achieved to potentially differentially modulate GVL and GVHD by pharmacologic agents. Rather than directly inhibiting the activity of the kinase domain of ITK, which could result in complete blockage of all ITK kinase activity in T cells (and potentially non-specifically affect other tyrosine kinases), we developed a strategy to specifically disrupt the SLP76-pY145-mediated activation of ITK function in T cells. The interaction between ITK and SLP76 involves the phospho-tyrosines at position pY145 and the proline-rich domain (PRD) of SLP76 docking onto the SH2 and SH3 domains of ITK, respectively. This multivalent anchoring of ITK on the different SLP76 docking sites results in distinct downstream signaling effects (Grasis et al., 2010). Previous work from Grasis et al. has shown that blocking the interaction between the PRD of SLP76 and SH3 domain of ITK, using a peptide mimicking the PRD of SLP76, inhibited the TCR-induced association between ITK and SLP-76 and the transphosphorylation of ITK, as well as actin polarization at the T-cell contact site and expression of Th2 cytokines (Grasis et al., 2010). Based on our findings that SLP76Y145FKI mutant T cells can mediate tumor clearance through GVL without inducing unwanted GVHD effects, we took advantage of this fact and blocked the SLP76 pY145-mediated docking of ITK through its SH2 domain. Thus, converting Y145 to F145 in SLP76 or preventing SH2 docking by our novel SLP76145pTYR peptide does not entirely abolish the interaction between SLP76 and ITK, but significantly affects ITK kinase activity and results in severe defects in specific downstream signaling pathways (Andreotti et al., 2010). Targeting this specific interaction would therefore retain signaling pathways that maintain GVL effects but ameliorate GVHD. When we utilized the SLP76pTYR peptide to specifically target ITK signaling, we observed specific effects only on ITK signaling, without significant effects on other tyrosine kinases. Furthermore, SLP76pTYR inhibition of ITK signaling also enhances Tregs frequency in vitro, confirming the peptide’s ability to affect ITK signaling and T cell effector functions. We recently reported similar effects using a model of ITK deficiency (Mammadli et al., 2020)
This SLP76pTYR peptide significantly reduced IFN-γ and TNF-α production by TCR stimulated primary human T cells isolated from PBMCs. Treatment of murine donor T cells with SLP76pTYR before transfer resulted in tumor clearance without inducing GVHD. Future therapies involving our novel SLP76pTYR peptide inhibitor and small molecule inhibitors could potentially be an effective strategy for enhancing GVL while avoiding GVHD. Thus, more selective ITK inhibition using our SLP76pTYR peptide could be beneficial in the treatment of autoimmune diseases while maintaining T cell effector functions.
Limitations of the study
Currently, this study's limitation is the use of our novel peptide SLP76pTYR, which can only be used in vitro or as a construct to transduce T cells. We are working with structural and medicinal chemists to make our peptide smaller and more stable, allowing it to be used for in vivo studies. From our in vitro studies, our peptide does reproduce the effects seen in SLP76Y145FKI mice.
Resource availability
Lead contact
-
(1)
for SLP76Y145FKI mice permission from lead contact Dr. Martha S. Jordan (University of Pennsylvania) (jordanm@mail.med.upenn.edu) is required by Material transfer Agreement (MTA) rules.
-
(2)
ITK-deficient mice permission from lead contact Dr. Avery August, Department of Microbiology and Immunology Cornell University College of Veterinary Medicine Ithaca, NY 14853 (aa749@cornell.edu) by MTA rules.
-
(3)
SLP76pTYR is patented (P38257) by The Research Foundation For The State University of New York, at SUNY Upstate Medical University Registration No. 43,497. At this point, this material is under further consideration and might not be available immediately.
-
(4)
For all other reagents please contact Mobin Karimi, Department of Microbiology and Immunology. MTA rules will be applied. SUNY Upstate Medical University,766 Irving Ave Weiskotten Hall Suite 2281, Syracuse, NY 13210 (karimim@upstate.edu)
Materials availability
All resources are available to anyone by request. SLP76pTYR is under further consideration and might not be available immediately.
Data and code availability
No software products, custom code, or algorithms were developed for this manuscript.
Methods
All methods can be found in the accompanying transparent methods supplemental file.
Acknowledgments
We thank all members of the Karimi and August laboratories for helpful discussions. This research was funded in part by a grant from the National Blood Foundation Scholar Award to (MK) and the National Institutes of Health (NIH LRP #L6 MD0010106 and K22 (AI130182) to MK. Upstate Medical University Cancer grant (1146249-1-75632) to MK.
Author contributions
M.M., W.H., R.H., J.R., S.W., H.X., A.B., A.M., A.W., and M.K. performed experiments; W.T., Y.C., and T.G. provided valuable reagents; R.H. assisted with data analysis, experimental design, scientific discussion, and editing the manuscript; and W.H., A.A., A.B., and M.K. designed experiments, analyzed the data, and wrote the manuscript.
Declaration of interests
A.A. receives research support from 3M Corporation. W.H. receives research support from Mega Robo Technologies. The other authors declare no conflicts of interest.
Published: April 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102286.
Supplemental information
References
- Alder C.M., Ambler M., Campbell A.J., Champigny A.C., Deakin A.M., Harling J.D., Harris C.A., Longstaff T., Lynn S., Maxwell A.C. Identification of a novel and selective series of itk inhibitors via a template-hopping strategy. ACS Med. Chem. Lett. 2013;4:948–952. doi: 10.1021/ml400206q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Amir A.L., Hagedoorn R.S., Van Luxemburg-Heijs S.A., Marijt E.W., Kruisselbrink A.B., Frederik Falkenburg J.H., Heemskerk M.H. Identification of a coordinated CD8 and CD4 T cell response directed against mismatched HLA Class I causing severe acute graft-versus-host disease. Biol. Blood Marrow Transpl. 2012;18:210–219. doi: 10.1016/j.bbmt.2011.10.018. [DOI] [PubMed] [Google Scholar]
- Andersen T.C.B., Kristiansen P.E., Huszenicza Z., Johansson M.U., Gopalakrishnan R.P., Kjelstrup H., Boyken S., Sundvold-Gjerstad V., Granum S., Sorli M. The SH3 domains of the protein kinases ITK and LCK compete for adjacent sites on T cell-specific adapter protein. J. Biol. Chem. 2019;294:15480–15494. doi: 10.1074/jbc.RA119.008318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K. Broadening the spectrum of patient groups at risk for transfusion-associated GVHD: implications for universal irradiation of cellular blood components. Transfusion. 2003;43:1652–1654. doi: 10.1111/j.0041-1132.2003.00631.x. [DOI] [PubMed] [Google Scholar]
- Andreotti A.H., Schwartzberg P.L., Joseph R.E., Berg L.J. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb. Perspect. Biol. 2010;2:a002287. doi: 10.1101/cshperspect.a002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherly L.O., Lucas J.A., Felices M., Yin C.C., Reiner S.L., Berg L.J. The Tec family tyrosine kinases Itk and Rlk regulate the development of conventional CD8+ T cells. Immunity. 2006;25:79–91. doi: 10.1016/j.immuni.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Barrett A.J. A new checkpoint in the path to GVHD? How bedside-to-bench stem cell transplant studies can inform human GVHD biology. J. Leukoc. Biol. 2015;97:213–215. doi: 10.1189/jlb.5CE0814-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastien J.P., Roy J., Roy D.C. Selective T-cell depletion for haplotype-mismatched allogeneic stem cell transplantation. Semin. Oncol. 2012;39:674–682. doi: 10.1053/j.seminoncol.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Baxter A.G., Hodgkin P.D. Activation rules: the two-signal theories of immune activation. Nat. Rev. Immunol. 2002;2:439–446. doi: 10.1038/nri823. [DOI] [PubMed] [Google Scholar]
- Bleakley M., Turtle C.J., Riddell S.R. Augmentation of anti-tumor immunity by adoptive T-cell transfer after allogeneic hematopoietic stem cell transplantation. Expert Rev. Hematol. 2012;5:409–425. doi: 10.1586/ehm.12.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogin Y., Ainey C., Beach D., Yablonski D. SLP-76 mediates and maintains activation of the Tec family kinase ITK via the T cell antigen receptor-induced association between SLP-76 and ITK. Proc. Natl. Acad. Sci. U S A. 2007;104:6638–6643. doi: 10.1073/pnas.0609771104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breems D.A., Lowenberg B. Autologous stem cell transplantation in the treatment of adults with acute myeloid leukaemia. Br. J. Haematol. 2005;130:825–833. doi: 10.1111/j.1365-2141.2005.05628.x. [DOI] [PubMed] [Google Scholar]
- Bunnell S.C., Diehn M., Yaffe M.B., Findell P.R., Cantley L.C., Berg L.J. Biochemical interactions integrating Itk with the T cell receptor-initiated signaling cascade. J. Biol. Chem. 2000;275:2219–2230. doi: 10.1074/jbc.275.3.2219. [DOI] [PubMed] [Google Scholar]
- Carson C.C., Moschos S.J., Edmiston S.N., Darr D.B., Nikolaishvili-Feinberg N., Groben P.A., Zhou X., Kuan P.F., Pandey S., Chan K.T. IL2 inducible T-cell kinase, a novel therapeutic target in melanoma. Clin. Cancer Res. 2015;21:2167–2176. doi: 10.1158/1078-0432.CCR-14-1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carty S.A., Koretzky G.A., Jordan M.S. Interleukin-4 regulates eomesodermin in CD8+ T cell development and differentiation. PLoS One. 2014;9:e106659. doi: 10.1371/journal.pone.0106659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castor M.G., Pinho V., Teixeira M.M. The role of chemokines in mediating graft versus host disease: opportunities for novel therapeutics. Front. Pharmacol. 2012;3:23. doi: 10.3389/fphar.2012.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Chikwava K., Wu C., Zhang H., Bhagat A., Pei D., Choi J.K., Tong W. LNK/SH2B3 regulates IL-7 receptor signaling in normal and malignant B-progenitors. J. Clin. Invest. 2016;126:1267–1281. doi: 10.1172/JCI81468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H.S., Ha S., Shin H.M., Reboldi A., Hall J.A., Huh J.R., Usherwood E.J., Berg L.J. CD8(+) T cells require ITK-mediated TCR signaling for migration to the intestine. Immunohorizons. 2020;4:57–71. doi: 10.4049/immunohorizons.1900093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke K.R., Kobzik L., Martin T.R., Brewer J., Delmonte J., Jr., Crawford J.M., Ferrara J.L. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88:3230–3239. [PubMed] [Google Scholar]
- D'Aveni M., Rossignol J., Coman T., Sivakumaran S., Henderson S., Manzo T., Santos E Sousa P., Bruneau J., Fouquet G., Zavala F. G-CSF mobilizes CD34+ regulatory monocytes that inhibit graft-versus-host disease. Sci. Transl. Med. 2015;7:281ra42. doi: 10.1126/scitranslmed.3010435. [DOI] [PubMed] [Google Scholar]
- Duffner U., Lu B., Hildebrandt G.C., Teshima T., Williams D.L., Reddy P., Ordemann R., Clouthier S.G., Lowler K., Liu C. Role of CXCR3-induced donor T-cell migration in acute GVHD. Exp. Hematol. 2003;31:897–902. doi: 10.1016/s0301-472x(03)00198-x. [DOI] [PubMed] [Google Scholar]
- Dutt S., Baker J., Kohrt H.E., Kambham N., Sanyal M., Negrin R.S., Strober S. CD8+CD44(hi) but not CD4+CD44(hi) memory T cells mediate potent graft antilymphoma activity without GVHD. Blood. 2011;117:3230–3239. doi: 10.1182/blood-2010-10-312751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore J.P., Mcgee M.C., Nidetz N.F., Anannya O., Huang W., August A. Tuning T helper cell differentiation by ITK. Biochem. Soc. Trans. 2020;48:179–185. doi: 10.1042/BST20190486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara J.L. Blood and marrow transplant clinical trials network: progress since the state of the science symposium 2007. Biol. Blood Marrow Transpl. 2014;20:149–153. doi: 10.1016/j.bbmt.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth E., Mattner J. The role of adaptor proteins in the biology of natural killer T (NKT) cells. Front. Immunol. 2019;10:1449. doi: 10.3389/fimmu.2019.01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasis J.A., Guimond D.M., Cam N.R., Herman K., Magotti P., Lambris J.D., Tsoukas C.D. In vivo significance of ITK-SLP-76 interaction in cytokine production. Mol. Cell. Biol. 2010;30:3596–3609. doi: 10.1128/MCB.01657-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinan E.C., Boussiotis V.A., Neuberg D., Brennan L.L., Hirano N., Nadler L.M., Gribben J.G. Transplantation of anergic histoincompatible bone marrow allografts. N. Engl. J. Med. 1999;340:1704–1714. doi: 10.1056/NEJM199906033402202. [DOI] [PubMed] [Google Scholar]
- Guo W., Liu R., Ono Y., Ma A.H., Martinez A., Sanchez E., Wang Y., Huang W., Mazloom A., Li J. Molecular characteristics of CTA056, a novel interleukin-2-inducible T-cell kinase inhibitor that selectively targets malignant T cells and modulates oncomirs. Mol. Pharmacol. 2012;82:938–947. doi: 10.1124/mol.112.079889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henden A.S., Hill G.R. Cytokines in graft-versus-host disease. J. Immunol. 2015;194:4604–4612. doi: 10.4049/jimmunol.1500117. [DOI] [PubMed] [Google Scholar]
- Holler E. Cytokines, viruses, and graft-versus-host disease. Curr. Opin. Hematol. 2002;9:479–484. doi: 10.1097/00062752-200211000-00002. [DOI] [PubMed] [Google Scholar]
- Hsiao M., Tatishchev S., Khedro T., Yaghmour B., O'connell C., Yaghmour G. First report of severe acute graft-versus-host disease after allogeneic stem cell transplant in a patient with myelodysplastic syndrome treated with atezolizumab: literature review. World J. Oncol. 2020;11:112–115. doi: 10.14740/wjon1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Hu J., August A. Cutting edge: innate memory CD8+ T cells are distinct from homeostatic expanded CD8+ T cells and rapidly respond to primary antigenic stimuli. J. Immunol. 2013;190:2490–2494. doi: 10.4049/jimmunol.1202988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Huang F., Kannan A.K., Hu J., August A. ITK tunes IL-4-induced development of innate memory CD8+ T cells in a gammadelta T and invariant NKT cell-independent manner. J. Leukoc. Biol. 2014;96:55–63. doi: 10.1189/jlb.1AB0913-484RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Mo W., Jiang J., Chao N.J., Chen B.J. Donor allospecific CD44(high) central memory T cells have decreased ability to mediate graft-vs.-host disease. Front. Immunol. 2019;10:624. doi: 10.3389/fimmu.2019.00624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan M.S., Sadler J., Austin J.E., Finkelstein L.D., Singer A.L., Schwartzberg P.L., Koretzky G.A. Functional hierarchy of the N-terminal tyrosines of SLP-76. J. Immunol. 2006;176:2430–2438. doi: 10.4049/jimmunol.176.4.2430. [DOI] [PubMed] [Google Scholar]
- Jordan M.S., Smith J.E., Burns J.C., Austin J.E., Nichols K.E., Aschenbrenner A.C., Koretzky G.A. Complementation in trans of altered thymocyte development in mice expressing mutant forms of the adaptor molecule SLP76. Immunity. 2008;28:359–369. doi: 10.1016/j.immuni.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju X.P., Xu B., Xiao Z.P., Li J.Y., Chen L., Lu S.Q., Huang Z.X. Cytokine expression during acute graft-versus-host disease after allogeneic peripheral stem cell transplantation. Bone Marrow Transpl. 2005;35:1179–1186. doi: 10.1038/sj.bmt.1704972. [DOI] [PubMed] [Google Scholar]
- Kambayashi T., Larosa D.F., Silverman M.A., Koretzky G.A. Cooperation of adapter molecules in proximal signaling cascades during allergic inflammation. Immunol. Rev. 2009;232:99–114. doi: 10.1111/j.1600-065X.2009.00825.x. [DOI] [PubMed] [Google Scholar]
- Karimi M., Cao T.M., Baker J.A., Verneris M.R., Soares L., Negrin R.S. Silencing human NKG2D, DAP10, and DAP12 reduces cytotoxicity of activated CD8+ T cells and NK cells. J. Immunol. 2005;175:7819–7828. doi: 10.4049/jimmunol.175.12.7819. [DOI] [PubMed] [Google Scholar]
- Kim M., Jung J., Lee K. Roles of ERK, PI3 kinase, and PLC-gamma pathways induced by overexpression of translationally controlled tumor protein in HeLa cells. Arch. Biochem. Biophys. 2009;485:82–87. doi: 10.1016/j.abb.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Koretzky G.A., Abtahian F., Silverman M.A. SLP76 and SLP65: complex regulation of signalling in lymphocytes and beyond. Nat. Rev. Immunol. 2006;6:67–78. doi: 10.1038/nri1750. [DOI] [PubMed] [Google Scholar]
- Liu W., Ren H.Y., Dong Y.J., Wang L.H., Yin Y., Li Y., Qiu Z.X., Cen X.N., Shi Y.J. Bortezomib regulates the chemotactic characteristics of T cells through downregulation of CXCR3/CXCL9 expression and induction of apoptosis. Int. J. Hematol. 2012;96:764–772. doi: 10.1007/s12185-012-1195-6. [DOI] [PubMed] [Google Scholar]
- Loschi M., Porcher R., Peffault De Latour R., Vanneaux V., Robin M., Xhaard A., Sicre De Fontebrune F., Larghero J., Socie G. High number of memory t cells is associated with higher risk of acute graft-versus-host disease after allogeneic stem cell transplantation. Biol. Blood Marrow Transpl. 2015;21:569–574. doi: 10.1016/j.bbmt.2014.12.009. [DOI] [PubMed] [Google Scholar]
- Lu S.X., Holland A.M., Na I.K., Terwey T.H., Alpdogan O., Bautista J.L., Smith O.M., Suh D., King C., Kochman A. Absence of P-selectin in recipients of allogeneic bone marrow transplantation ameliorates experimental graft-versus-host disease. J. Immunol. 2010;185:1912–1919. doi: 10.4049/jimmunol.0903148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Waller E.K. Dichotomous role of interferon-gamma in allogeneic bone marrow transplant. Biol. Blood Marrow Transpl. 2009;15:1347–1353. doi: 10.1016/j.bbmt.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch Kelly D., Lyon D.E., Ameringer S.A., Elswick R.K. Symptoms, cytokines, and quality of life in patients diagnosed with chronic graft-versus-host disease following allogeneic hematopoietic stem cell transplantation. Oncol. Nurs. Forum. 2015;42:265–275. doi: 10.1188/15.ONF.265-275. [DOI] [PubMed] [Google Scholar]
- Mammadli M., Huang W., Harris R., Sultana A., Cheng Y., Tong W., Pu J., Gentile T., Dsouza S., Yang Q. Targeting interleukin-2-inducible T-cell kinase (ITK) differentiates GVL and GVHD in allo-HSCT. Front. Immunol. 2020;11:593863. doi: 10.3389/fimmu.2020.593863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancusi A., Piccinelli S., Velardi A., Pierini A. The effect of TNF-alpha on regulatory T cell function in graft-versus-host disease. Front. Immunol. 2018;9:356. doi: 10.3389/fimmu.2018.00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro R., Takayanagi H., Nitta T. T cell receptor signaling for gammadeltaT cell development. Inflamm. Regen. 2019;39:6. doi: 10.1186/s41232-019-0095-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas V.H., Cuche C., Alcover A., Di Bartolo V. Serine phosphorylation of SLP76 is dispensable for T cell development but modulates helper T cell function. PLoS One. 2017;12:e0170396. doi: 10.1371/journal.pone.0170396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrin R.S., Contag C.H. In vivo imaging using bioluminescence: a tool for probing graft-versus-host disease. Nat. Rev. Immunol. 2006;6:484–490. doi: 10.1038/nri1879. [DOI] [PubMed] [Google Scholar]
- Owen D.L., Mahmud S.A., Sjaastad L.E., Williams J.B., Spanier J.A., Simeonov D.R., Ruscher R., Huang W., Proekt I., Miller C.N. Thymic regulatory T cells arise via two distinct developmental programs. Nat. Immunol. 2019;20:195–205. doi: 10.1038/s41590-018-0289-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikovskaya O., Chaix J., Rothman N.J., Collins A., Chen Y.H., Scipioni A.M., Vivier E., Reiner S.L. Cutting edge: eomesodermin is sufficient to direct type 1 innate lymphocyte development into the conventional NK lineage. J. Immunol. 2016;196:1449–1454. doi: 10.4049/jimmunol.1502396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletneva E.V., Sundd M., Fulton D.B., Andreotti A.H. Molecular details of Itk activation by prolyl isomerization and phospholigand binding: the NMR structure of the Itk SH2 domain bound to a phosphopeptide. J. Mol. Biol. 2006;357:550–561. doi: 10.1016/j.jmb.2005.12.073. [DOI] [PubMed] [Google Scholar]
- Reddy P., Ferrara J.L.M. StemBook; 2008. Mouse Models of Graft-Versus-Host Disease. [PubMed] [Google Scholar]
- Riether D., Zindell R., Kowalski J.A., Cook B.N., Bentzien J., Lombaert S.D., Thomson D., Kugler S.Z., Jr., Skow D., Martin L.S. 5-Aminomethylbenzimidazoles as potent ITK antagonists. Bioorg. Med. Chem. Lett. 2009;19:1588–1591. doi: 10.1016/j.bmcl.2009.02.012. [DOI] [PubMed] [Google Scholar]
- Sahu N., August A. ItK inhibitors in inflammation and immune-mediated disorders. Curr. Top. Med. Chem. 2009;9:690–703. doi: 10.2174/156802609789044443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T., Thorlacius H., Johnston B., Staton T.L., Xiang W., Littman D.R., Butcher E.C. Role for CXCR6 in recruitment of activated CD8+ lymphocytes to inflamed liver. J. Immunol. 2005;174:277–283. doi: 10.4049/jimmunol.174.1.277. [DOI] [PubMed] [Google Scholar]
- Seif F., Khoshmirsafa M., Aazami H., Mohsenzadegan M., Sedighi G., Bahar M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017;15:23. doi: 10.1186/s12964-017-0177-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Garvin J.E., Burns J.C., Gohil M., Zou T., Kim J.S., Maltzman J.S., Wherry E.J., Koretzky G.A., Jordan M.S. T-cell receptor signals direct the composition and function of the memory CD8+ T-cell pool. Blood. 2010;116:5548–5559. doi: 10.1182/blood-2010-06-292748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stritesky G.L., Jameson S.C., Hogquist K.A. Selection of self-reactive T cells in the thymus. Annu. Rev. Immunol. 2012;30:95–114. doi: 10.1146/annurev-immunol-020711-075035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y.W., Zhang Y., Schweikert J., Koretzky G.A., Reth M., Wienands J. Interaction of SLP adaptors with the SH2 domain of Tec family kinases. Eur. J. Immunol. 1999;29:3702–3711. doi: 10.1002/(SICI)1521-4141(199911)29:11<3702::AID-IMMU3702>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Sugie K., Jeon M.S., Grey H.M. Activation of naive CD4 T cells by anti-CD3 reveals an important role for Fyn in Lck-mediated signaling. Proc. Natl. Acad. Sci. U S A. 2004;101:14859–14864. doi: 10.1073/pnas.0406168101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugues S., Amorim A., Spath S., Martin-Blondel G., Schreiner B., De Feo D., Lutz M., Guscetti F., Apostolova P., Haftmann C. Graft-versus-host disease, but not graft-versus-leukemia immunity, is mediated by GM-CSF-licensed myeloid cells. Sci. Transl. Med. 2018;10:eaat8410. doi: 10.1126/scitranslmed.aat8410. [DOI] [PubMed] [Google Scholar]
- Vadakekolathu J., Rutella S. T-cell manipulation strategies to prevent graft-versus-host disease in haploidentical stem cell transplantation. Biomedicines. 2017;5:33. doi: 10.3390/biomedicines5020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarroel V.A., Okiyama N., Tsuji G., Linton J.T., Katz S.I. CXCR3-mediated skin homing of autoreactive CD8 T cells is a key determinant in murine graft-versus-host disease. J. Invest. Dermatol. 2014;134:1552–1560. doi: 10.1038/jid.2014.2. [DOI] [PubMed] [Google Scholar]
- Webb J.R., Milne K., Nelson B.H. PD-1 and CD103 are widely coexpressed on prognostically favorable intraepithelial CD8 T cells in human ovarian cancer. Cancer Immunol. Res. 2015;3:926–935. doi: 10.1158/2326-6066.CIR-14-0239. [DOI] [PubMed] [Google Scholar]
- Weinreich M.A., Odumade O.A., Jameson S.C., Hogquist K.A. T cells expressing the transcription factor PLZF regulate the development of memory-like CD8+ T cells. Nat. Immunol. 2010;11:709–716. doi: 10.1038/ni.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T., Young J.S., Johnston H., Ni X., Deng R., Racine J., Wang M., Wang A., Todorov I., Wang J., Zeng D. Thymic damage, impaired negative selection, and development of chronic graft-versus-host disease caused by donor CD4+ and CD8+ T cells. J. Immunol. 2013;191:488–499. doi: 10.4049/jimmunol.1300657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki C.A., Burkett S.B., Panoskaltsis-Mortari A., Kirby S.L., Luster A.D., Mckinnon K., Blazar B.R., Serody J.S. Differential roles for CCR5 expression on donor T cells during graft-versus-host disease based on pretransplant conditioning. J. Immunol. 2004;173:845–854. doi: 10.4049/jimmunol.173.2.845. [DOI] [PubMed] [Google Scholar]
- Yu X.Z., Albert M.H., Anasetti C. Alloantigen affinity and CD4 help determine severity of graft-versus-host disease mediated by CD8 donor T cells. J. Immunol. 2006;176:3383–3390. doi: 10.4049/jimmunol.176.6.3383. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Joe G., Hexner E., Zhu J., Emerson S.G. Alloreactive memory T cells are responsible for the persistence of graft-versus-host disease. J. Immunol. 2005;174:3051–3058. doi: 10.4049/jimmunol.174.5.3051. [DOI] [PubMed] [Google Scholar]
- Zheng H., Matte-Martone C., Jain D., Mcniff J., Shlomchik W.D. Central memory CD8+ T cells induce graft-versus-host disease and mediate graft-versus-leukemia. J. Immunol. 2009;182:5938–5948. doi: 10.4049/jimmunol.0802212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No software products, custom code, or algorithms were developed for this manuscript.